
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 15 November 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.760451
This article is part of the Research Topic Microbe-Driven Malignancies: Mechanisms, Prevention, and Treatment View all 8 articles
 Yi-Ru Lai1,2†
Yi-Ru Lai1,2† Yu-Fang Chang1,2
Yu-Fang Chang1,2 Jason Ma1,2†Cheng-Hsun Chiu1,2,3
Jason Ma1,2†Cheng-Hsun Chiu1,2,3 Ming-Ling Kuo1,2,4*
Ming-Ling Kuo1,2,4* Chih-Ho Lai1,2,3,5,6*
Chih-Ho Lai1,2,3,5,6*Cytolethal distending toxin (CDT), one of the most important genotoxins, is produced by several gram-negative bacteria and is involved in bacterial pathogenesis. Recent studies have shown that bacteria producing this peculiar genotoxin target host DNA, which potentially contributes to development of cancer. In this review, we highlighted the recent studies focusing on the idea that CDT leads to DNA damage, and the cells with inappropriately repaired DNA continue cycling, resulting in cancer development. Understanding the detailed mechanisms of genotoxins that cause DNA damage might be useful for targeting potential markers that drive cancer progression and help to discover new therapeutic strategies to prevent diseases caused by pathogens.
Bacterial genotoxins are toxins that trigger single-strand breaks (SSBs) or double-strand breaks (DSBs) on DNA in target host cells, and are functionally homologous to mammalian type I deoxyribonuclease (DNase I), resulting in the activation of DNA damage response (DDR) (1). These responses subsequently lead to cell senescence, apoptosis, or genomic instability, which favors tumor initiation and progression. Three bacterial virulence factors are now characterized as genotoxins: cytolethal distending toxin (CDT) in gram-negative bacteria, typhoid toxin produced by Salmonella enterica serovar Typhi, and colibactin produced by the phylogenetic group B2 Escherichia coli (2). CDT was discovered in E. coli by Johnson and Lior in 1987 (3), and similar toxin activities were found in two other enteric pathogens, Shigella spp (3). and Campylobacter spp (4). CDT is capable of modulating eukaryotic cell cycle by pausing the G2/M transition, and was thus further defined as cyclomodulins (5). To clarify the possible virulence factors in enteric pathogens, cloning and gene sequencing were performed within different strains of E. coli and three open reading frames (ORFs) in an operon were identified, known as cdtA, cdtB, and cdtC (6, 7). Among the proteins encoded by these genes, CdtB was demonstrated to harbor nuclease activity (8–10). CdtA and CdtC are required for delivering CdtB into target cells, which allows for CdtB to translocate into the nucleus and cause DNA damage (11, 12). The catalytic activity of CdtB in target cells can activate DDR, which increases genomic instability, disturbs the cell cycle, and establishes a chronic proinflammatory environment (13). Since these characteristics are closely associated with cancer development, it is proposed that bacterial infections play a role in the neoplastic process. This review highlights the current state of knowledge on the interaction of CDT with host DNA and its role in tumor progression.
Cancer risk is generally attributed to hereditary, genetic, environmental, and lifestyle factors (14). The contribution of infectious agents to cancer development is often underappreciated. In fact, more than 16% of cancer cases are related to infectious agents worldwide (15). Persistent infection-induced chronic inflammation, which is likely to be associated with the secretion of virulence factors, ultimately facilitates oncogenic processes in hosts (16–18). Bacterial toxins disrupt cellular signals, including cell proliferation, cell cycle progression, and DNA repair, and dysregulation of either of which is intimately intertwined with oncogenesis (19). For instance, Helicobacter pylori, remaining the most notorious pathogen to cause cancer, has been identified as a group 1 carcinogen by the International Agency for Research on Cancer since 1994 (20). It can secrete cytotoxin-associated gene A (CagA), which empowers cells with numerous cancerous traits, including cell death resistance, adherence junctional defects, and genomic instability after its entry into gastric epithelial cells via type IV secretion system (20). In addition, Salmonella species produce AvrA protein, a deubiquitinase that inhibits β-catenin ubiquitination to promote colonic epithelial cell proliferation (21). Enterotoxigenic Bacteroides fragilis secretes B. fragilis toxin (BFT), a zinc-dependent metalloprotease that can induce colitis and colorectal cancer (CRC) in multiple intestinal neoplasia (Min) mice (22, 23). Notably, with the growing number of studies on microbiota, genotoxin-producing bacteria have also been identified as potential carcinogens (24–27). In addition, other gastrointestinal tract-dwelling pathogens reported to produce CDT, including C. jejuni and Helicobacter hepaticus, may also have an impact on carcinogenesis (28–31). Collectively, the above studies indicate that bacteria together with their virulence factors not only cause infectious diseases, but also promote cancer development (Table 1). Therefore, the mechanisms involved in cancer development caused by bacteria and their toxins deserve further investigation.
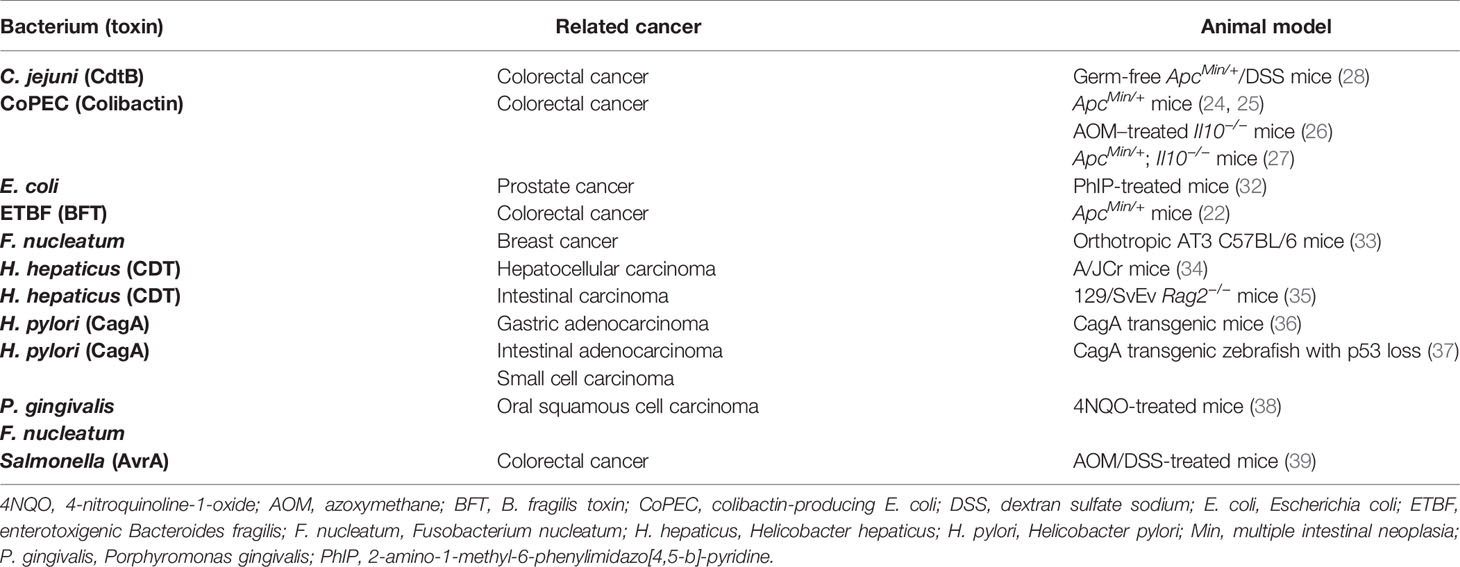
Table 1 The relationships between bacterial pathogens, virulence factors, and cancers in the animal models.
Among the bacterial genotoxins, CDT is the first to be characterized and shown to cause DSBs (8, 40). In most CDT-harboring bacteria, the gene cluster encoding CDT subunits, consisting of adjacent or slightly overlapping cdtA, cdtB, and cdtC, is located on the chromosome (41). Special exceptions occur in some E. coli strains, in which the operon is found on a large conjugative plasmid called pVir (42). The location of the cdt cluster differs in the genomes of different species but is well conserved within the same species (43). In most cases, the expression of all three genes is indispensable for CDT toxicity (44), although the identified cdt is mainly composed of three ORFs (cdtA, cdtB, and cdtC), apart from Salmonella enterica serovar Typhi (S. Typhi). Notably, S. Typhi cdt contains a conserved cdtB, whereas cdtA and cdtC are substituted by genes encoding two homologs of the pertussis toxins, referred to as pertussis-like toxins A and B (PltA and PltB) (45).
Studies have analyzed the prevalence of CDT production in different bacterial species, including C. jejuni, A. actinomycetemcomitans, H. ducreyi, etc., from clinical specimens, and revealed that the majority of these species produce stable amounts of CDT (46–49). CDT is a prominent virulence factor of CDT-producing bacteria and aids in effective tissue colonization, thereby promoting potent infection by breaking down host defense (50). The dampened host defense mainly results from: (i) disrupted epithelial barrier, which is caused by CDT-induced cell cycle arrest and subsequent cell death in epithelial cells (51); and (ii) impaired host immunity, which is caused by the extreme sensitivity of lymphocytes to CDT cytotoxicity and altered macrophage functions (52, 53). To perform such sophisticated tasks, the CDT must first enter the cells to exert its activity. CDT holotoxin contains one active subunit (CdtB), which requires two binding subunits (CdtA and CdtC) to facilitate its transport through the cell membrane (54). The homology of CDT subunits varies among different bacterial species, and the pairwise identity of CdtA and CdtC ranges from 19% to 95%. CdtB appears to be the most conserved, with 45% sequence identity, even between the least-related CDTs (55). As the active component, CdtB has been demonstrated to share striking similarity with the DNase I protein family (56). At the sequence level, CdtB possesses the essential residues responsible for DNase I enzymatic activities, including residues important in active site and Mg2+-binding site (40). At the 3D structure level, CdtB exhibits the canonical characteristics of DNase-like protein: stranded β-sandwich flanked with α-helix and loops (57). Thus, the final destination for CdtB is the cell nucleus, where it can induce DSBs and immediately trigger DNA damage-dependent checkpoint activation (58, 59). Subsequently, stalling of cell cycle progression occurs at G1/S or G2/M transition to block cell division and allow for DNA repair (60).
Binding of CDT holotoxin to the cell membrane primarily depends on CdtA and CdtC. These two subunits adopt a ricin-like lectin structure, forming an aromatic patch and a deep groove on the protein surface, which play key roles in cell surface recognition and association with specific membrane components (61, 62). Despite the specific receptor unidentified, several studies have highlighted the requirement of lipid rafts (sphingolipid- and cholesterol-rich regions on the membrane) for CdtA and CdtC binding to the cell membrane (63–65). Combined with the fact that the deep groove in the holotoxin structure is rather hydrophobic, it is implied that the binding subunits may contain a cholesterol recognition amino acid consensus sequence (CRAC)-like region. Indeed, in further motif analysis, the CRAC site has been identified in CdtC (64, 66). Many molecular mechanisms remain unclear for the subsequent internalization and intracellular transport processes. In addition, it is suggested that these pathways may vary among bacterial species and are also influenced by a broad range of cell types being intoxicated by CDT (67). Nevertheless, a general concept is that after the binding of CDT holotoxin, CdtA remains on the membrane, while CdtB and CdtC are internalized into the cytosol. Only CdtB is delivered to other subcellular compartments and ultimately to the nucleus (56, 68). Upon reaching the nucleus, CdtB exerts its DNase activity to cause DNA damage, which is possibly SSBs with low-dose treatment, and DSBs with high-dose treatment (69). The triggered DDR is dominantly orchestrated by phosphatidylinositol 3-kinase (PI3K)-like protein kinase ataxia telangiectasia mutated (ATM) (70, 71). Activation of ATM simultaneously causes the phosphorylation of histone H2AX (γH2AX) as well as the recruitment of Mre11-Rad50-Nbs1 (MRN) complex, which provides a platform for DNA repair, and sets off checkpoint responses via the phosphorylation of CHK2 and p53, resulting in cell cycle arrest thus inhibiting cell proliferation (Figure 1) (72). In most cases, DNA damage becomes way too devastating, and the repair system fails to rescue the situation, which consequently leads to cell death (73, 74) or senescence (75–77). However, in a few cases the intoxicated cells bypass death; these cells escape the built-in carcinogenesis barrier of cell death/senescence and are likely to develop a tendency for cancer formation, including genomic instability, heightened mutation frequency, and anchorage-independent cell growth (13, 78, 79).
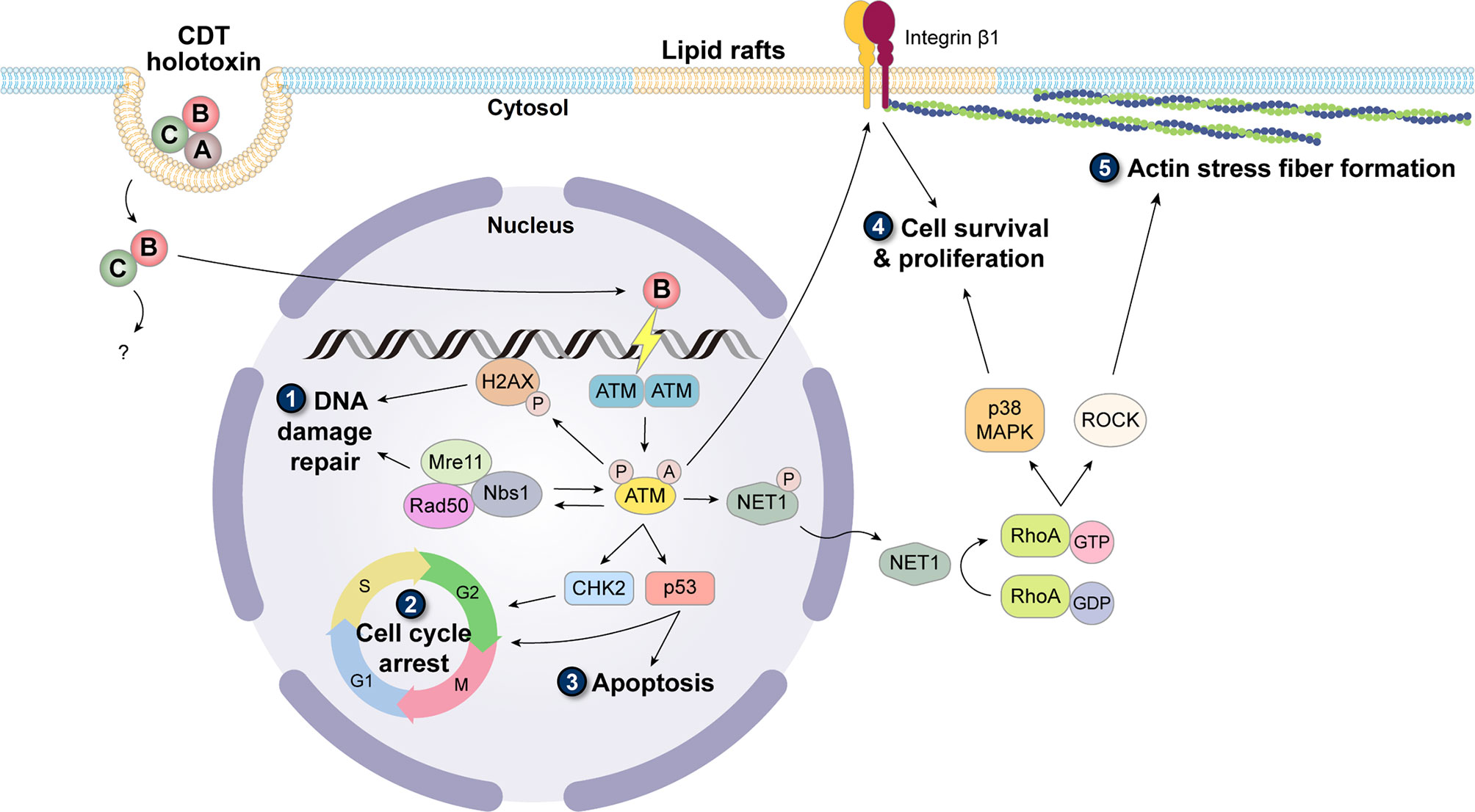
Figure 1 Carcinogenic induction caused by CDT. Binding of CdtA and CdtC to lipid rafts facilitates the entry of CdtB and CdtC. In the cytoplasm, CdtB dissociates with CdtC and translocates into the nucleus alone. As a DNase, CdtB damages the host DNA and immediately triggers the activation of ATM, which is involved in the formation of monomer and biochemical modifications including phosphorylation and acetylation. (1) Activated ATM targets H2AX, phosphorylated on Ser139 (γH2AX), and initiates the cascade of DDR signaling pathway. (2) To allow for DNA repair, with the co-signaling of MRN complex, ATM also activates CHK2 and p53 to stall cell cycle progression. (3) When the DNA damage is too devastating, the cells are prone to undergo apoptosis. However, the cells with misrepaired DNA can continue cycling, thereby accumulating mutations to cause genomic instability. Activated ATM also causes dephosphorylation of Net1, which is activated and translocated to the cytoplasm. Net1 switches the inactive GDP-bound form to the active GTP-bound form of RhoA. The downstream region of RhoA mainly diverges into two pathways: (4) one activates p38 MAPK and further promotes cell survival and proliferation, and (5) the other activates ROCK and induces the formation of actin stress fibers. In addition, stress fibers are often anchored on the focal adhesion complex constituting integrin, of which the inside-out activation signal can be transduced by ATM. Together, these cellular responses triggered by CDT are related to the acquisition of cancer hallmarks.
Since the discovery of genotoxin, its DNA-damaging activity has long been considered a powerful cell-killing strategy. However, in recent years, its role in pathogenesis has started to appear in a completely different perspective. The connection between genotoxins and cancer has been assessed through both gut microbiota analysis and epidemiology profiling. The research revealed a higher prevalence of cdt and pks (gene encoding colibactin)-positive E. coli in the gut microbiome of patients with inflammatory bowel disease and CRC than in the non-cancer group (26, 80). Moreover, a mutational pattern characteristic of colibactin exposure was found to be enriched in the sequencing data of two independent cohorts of primary CRC tumors and CRC metastases (81). Several in vivo studies also validated the potential of genotoxin to increase the risk of malignancy. A/JCr mice developed hepatic dysplastic nodules after chronic infection with Helicobacter hepaticus (34). H. hepaticus infection causes chronic hepatitis; however, the progression of inflammation toward dysplasia was found to be associated with the presence of CDT, which upregulates a subset of proinflammatory mediators, and increases hepatocyte proliferation as well as mRNA expression of anti-apoptotic proteins (34). Moreover, invasive carcinoma can be detected in susceptible mice exposed to H. hepaticus but not in those exposed to the isogenic cdtB mutant (35). The study partly explained that CDT affects Stat-3 signaling, thereby promoting oncogenic processes. Similarly, persistent infection with CDT-harboring C. jejuni resulted in tumor formation in ApcMin/+ mice fed with 1% dextran sulfate sodium (DSS) (28). The developed tumor number and tumor size were significantly reduced when the infecting bacteria possessed mutated cdtB. Additionally, human colonic epithelial cells with defective genes commonly observed in CRC models are prone to micronucleus formation and anchorage-independent cell growth after CDT treatment (79). Collectively, these findings indicate that the cell response to genotoxin intoxication appears to be detrimental, but not necessarily destructive.
To elucidate the detailed mechanisms behind this phenomenon, numerous studies have been conducted in recent years. Generally, CDT-intoxicated cells tend to enter cell cycle arrest as soon as the DNA damage takes place; however, a proportion of cells that manage to tolerate DNA damage induced by CDT and persist cycling have been identified, and further analysis indicated that these cells showed signatures of malignant transformation (13). As a consequence of dampened DDR and the slowing of replication fork velocity, the genetic stability and integrity are disrupted, which was observed through elevated fragile sites expression and chromosome aberrations (13, 82). As DNA lesions continue to accumulate, they increase the risk of mutation occurrence and are more likely to lead the cells down the path of pro-cancerous progression. Additionally, unrepaired DNA lesions can lead to micronuclei formation after cell division, which causes a proinflammatory response once micronuclei are sensed as cytosolic DNA and triggers the cGAS-STING pathway (83).
On the other hand, the master regulator of CDT-triggered DDR, ATM, transduces not only the DNA repair signal, but also activates a survival pathway involving p38 mitogen-activated protein kinase (MAPK) and integrin β1 (13, 78, 84). As its name indicates, CDT causes the distension morphology of the intoxicated cells, which has been examined to be associated with the formation of actin stress fiber (85). This phenomenon raised the possibility that there might be an intriguing crosstalk between DNA damage and cytoskeleton arrangement. Later studies identified a small GTPase (RhoA) as a crucial molecule in this potential signaling pathway (59). RhoA mainly participates in the coordination of actin cytoskeleton reorganization and focal adhesion, which may contribute to tumor invasion and metastasis (86). It can be activated by a nuclear-localized guanine nucleotide exchange factor (GEF) Net1 (87), the activation of which requires dephosphorylation at the inhibitory site Ser152 (88). The detailed molecular mechanism of how the Net1/RhoA response is triggered remains obscure; however, the participation of ATM and flap structure-specific endonuclease 1 (FEN1) has been implied (59, 89). The downstream pathway of RhoA diverges into p38 MAPK and Rho-associated kinase (ROCK) signaling. Aside from a plethora of reports concerning the proinflammatory effect of p38 MAPK (90–92), it has also been reported that sustained p38 MAPK is vital for cell survival under genotoxic stress (13, 93, 94). In parallel, ROCK signaling manipulates the stress fiber formation and cellular contractility (95). Moreover, ATM signaling can act as an inside-out activation signal for integrin β1, a membrane-bound receptor that transduces the signal favoring cell survival and proliferation. Accordingly, abolishing this signaling pathway compromises the ability of the intoxicated cells to avoid anchorage-independent cell death (78).
Furthermore, a recent study showed that disruption of the intestinal structural barrier facilitates dissemination of the gut bacteria, which can be delivered to the liver through intestinal capillaries and the portal vein. Bacteria in the liver recruit immune cells and promote the formation of an inflammatory environment, likely establishing a premetastatic niche (96). This phenomenon suggests a role for CDT in the process of metastasis. Theoretically, upon infection with CDT-harboring bacteria and the secretion of CDT, a group of cells become intoxicated by CDT. Most of these cells die, which damages the integrity of the intestinal barrier, whereas a small proportion survives; these cells become tumor cells and travel with the bacteria to the liver. In the inflammatory environment promoted by the bacteria, tumor cells settle in the premetastatic niche and, thus, favor distant metastasis formation.
Extensive studies have explored how CDT has been linked to a variety of diseases, and most of them reported the proinflammatory nature of this special bacterial genotoxin, which potentiates the carcinogenic property of CDT. CDT-induced genotoxic stress not only fuels the inflammatory response but also disrupts the structural barrier by inducing epithelial cell death. However, a small portion of the intoxicated cells outrun cell cycle arrest and continue to proliferate with incorrectly repaired or unrepaired DNA lesions. As more DNA lesions accumulate, it enhances the mutation frequency, interferes with genomic stability, and develops tumor initiation. The connection between the infection of CDT-harboring bacteria and cancer development has been reported in several animal studies, which demonstrated that with the help of CDT, bacteria-induced inflammatory response can be further depraved to malignancy formation (summarized in Figure 1).
This review emphasized the importance of recent findings regarding the genotoxicity of CDT associated with cancer formation. However, the direct link between toxin action and intracellular delivery and its clinical relevance remains largely unclear. Various crucial issues must be addressed: (i) Whether long-term persistent infection of CDT-producing bacteria is related to an increased risk of cancer progression in the host should be evaluated in clinical cases. (ii) The concentration of genotoxin produced by bacteria that can naturally cause oncogenesis in vivo is unclear. (iii) Although delivery of CdtB into the nucleus and triggering of DNA damage have been demonstrated, the molecular mechanism and intracellular trafficking pathways of the various CDTs produced by different bacterial species remain to be clarified. It is crucial to explore the detailed mechanism of CDT function using in vivo models or in clinical studies. Further investigations are required to provide pivotal insights into the mechanisms underlying the interplay between genotoxicity and cancer development. This may aid in developing novel strategies to combat diseases caused by pathogens along with their virulence factors.
Conception or design of this work: C-HC, M-LK, and C-HL. Writing the manuscript: Y-RL, Y-FC, and JM. Y-RL and JM were equally contributed to this work. All authors contributed to the article and approved the submitted version.
This research was supported by Taiwan Ministry of Science and Technology (108-2911-I-005-509, 109-2911-I-005-503, 109-2320-B-182-025-MY3, and 109-2320-B-182-029-MY3), Chang Gung Memorial Hospital (CMRPD1I0061-3, CMRPD1J0021-3, CMRPD1K0361, and BMRPE90), and Tomorrow Medical Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank the editor and reviewers for the editorial assistance and their valuable comments.
1. Guerra L, Guidi R, Frisan T. Do Bacterial Genotoxins Contribute to Chronic Inflammation, Genomic Instability and Tumor Progression? FEBS J (2011) 278(23):4577–88. doi: 10.1111/j.1742-4658.2011.08125.x
2. Grasso F, Frisan T. Bacterial Genotoxins: Merging the DNA Damage Response Into Infection Biology. Biomolecules (2015) 5(3):1762–82. doi: 10.3390/biom5031762
3. Johnson WM, Lior H. Production of Shiga Toxin and a Cytolethal Distending Toxin (CLDT) by Serogroups of Shigella Spp. FEMS Microbiol Lett (1987) 48(1-2):235–8. doi: 10.1111/j.1574-6968.1987.tb02548.x
4. Johnson WM, Lior H. A New Heat-Labile Cytolethal Distending Toxin (CLDT) Produced by Campylobacter Spp. Microb Pathog (1988) 4(2):115–26. doi: 10.1016/0882-4010(88)90053-8
5. Nougayrede JP, Taieb F, De Rycke J, Oswald E. Cyclomodulins: Bacterial Effectors That Modulate the Eukaryotic Cell Cycle. Trends Microbiol (2005) 13(3):103–10. doi: 10.1016/j.tim.2005.01.002
6. Pickett CL, Cottle DL, Pesci EC, Bikah G. Cloning, Sequencing, and Expression of the Escherichia Coli Cytolethal Distending Toxin Genes. Infect Immun (1994) 62(3):1046–51. doi: 10.1128/iai.62.3.1046-1051.1994
7. Scott DA, Kaper JB. Cloning and Sequencing of the Genes Encoding Escherichia Coli Cytolethal Distending Toxin. Infect Immun (1994) 62(1):244–51. doi: 10.1128/iai.62.1.244-251.1994
8. Elwell CA, Dreyfus LA. DNase I Homologous Residues in CdtB Are Critical for Cytolethal Distending Toxin-Mediated Cell Cycle Arrest. Mol Microbiol (2000) 37(4):952–63. doi: 10.1046/j.1365-2958.2000.02070.x
9. Frisan T, Cortes-Bratti X, Thelestam M. Cytolethal Distending Toxins and Activation of DNA Damage-Dependent Checkpoint Responses. Int J Med Microbiol (2002) 291(6-7):495–9. doi: 10.1078/1438-4221-00158
10. Cortes-Bratti X, Frisan T, Thelestam M. The Cytolethal Distending Toxins Induce DNA Damage and Cell Cycle Arrest. Toxicon (2001) 39(11):1729–36. doi: 10.1016/S0041-0101(01)00159-3
11. Lara-Tejero M, Galan JE. CdtA, CdtB, and CdtC Form a Tripartite Complex That is Required for Cytolethal Distending Toxin Activity. Infect Immun (2001) 69(7):4358–65. doi: 10.1128/IAI.69.7.4358-4365.2001
12. Cortes-Bratti X, Chaves-Olarte E, Lagergard T, Thelestam M. Cellular Internalization of Cytolethal Distending Toxin From Haemophilus Ducreyi. Infect Immun (2000) 68(12):6903–11. doi: 10.1128/IAI.68.12.6903-6911.2000
13. Guidi R, Guerra L, Levi L, Stenerlow B, Fox JG, Josenhans C, et al. Chronic Exposure to the Cytolethal Distending Toxins of Gram-Negative Bacteria Promotes Genomic Instability and Altered DNA Damage Response. Cell Microbiol (2013) 15(1):98–113. doi: 10.1111/cmi.12034
14. Chen YC, Hunter DJ. Molecular Epidemiology of Cancer. CA Cancer J Clin (2005) 55(1):45–54; quiz 57. doi: 10.3322/canjclin.55.1.45
15. de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global Burden of Cancers Attributable to Infections in 2008: A Review and Synthetic Analysis. Lancet Oncol (2012) 13(6):607–15. doi: 10.1016/S1470-2045(12)70137-7
17. Coussens LM, Werb Z. Inflammation and Cancer. Nature (2002) 420(6917):860–7. doi: 10.1038/nature01322
18. Rosadi F, Fiorentini C, Fabbri A. Bacterial Protein Toxins in Human Cancers. Pathog Dis (2016) 74(1):ftv105. doi: 10.1093/femspd/ftv105
19. Lax AJ. Opinion: Bacterial Toxins and Cancer–a Case to Answer? Nat Rev Microbiol (2005) 3(4):343–9. doi: 10.1038/nrmicro1130
20. Takahashi-Kanemitsu A, Knight CT, Hatakeyama M. Molecular Anatomy and Pathogenic Actions of Helicobacter Pylori CagA That Underpin Gastric Carcinogenesis. Cell Mol Immunol (2020) 17(1):50–63. doi: 10.1038/s41423-019-0339-5
21. Ye Z, Petrof EO, Boone D, Claud EC, Sun J. Salmonella Effector AvrA Regulation of Colonic Epithelial Cell Inflammation by Deubiquitination. Am J Pathol (2007) 171(3):882–92. doi: 10.2353/ajpath.2007.070220
22. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A Human Colonic Commensal Promotes Colon Tumorigenesis via Activation of T Helper Type 17 T Cell Responses. Nat Med (2009) 15(9):1016–22. doi: 10.1038/nm.2015
23. Cheng WT, Kantilal HK, Davamani F. The Mechanism of Bacteroides Fragilis Toxin Contributes to Colon Cancer Formation. Malays J Med Sci (2020) 27(4):9–21. doi: 10.21315/mjms2020.27.4.2
24. Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, et al. Colonization of the Human Gut by E. Coli and Colorectal Cancer Risk. Clin Cancer Res (2014) 20(4):859–67. doi: 10.1158/1078-0432.CCR-13-1343
25. Lucas C, Salesse L, Hoang MHT, Bonnet M, Sauvanet P, Larabi A, et al. Autophagy of Intestinal Epithelial Cells Inhibits Colorectal Carcinogenesis Induced by Colibactin-Producing Escherichia Coli in Apc(Min/+) Mice. Gastroenterology (2020) 158(5):1373–88. doi: 10.1053/j.gastro.2019.12.026
26. Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science (2012) 338(6103):120–3. doi: 10.1126/science.1224820
27. Tomkovich S, Yang Y, Winglee K, Gauthier J, Muhlbauer M, Sun X, et al. Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer Res (2017) 77(10):2620–32. doi: 10.1158/0008-5472.CAN-16-3472
28. He Z, Gharaibeh RZ, Newsome RC, Pope JL, Dougherty MW, Tomkovich S, et al. Campylobacter Jejuni Promotes Colorectal Tumorigenesis Through the Action of Cytolethal Distending Toxin. Gut (2019) 68(2):289–300. doi: 10.1136/gutjnl-2018-317200
29. Gargi A, Reno M, Blanke SR. Bacterial Toxin Modulation of the Eukaryotic Cell Cycle: Are All Cytolethal Distending Toxins Created Equally? Front Cell Infect Microbiol (2012) 2:124. doi: 10.3389/fcimb.2012.00124
30. Fox JG, Li X, Yan L, Cahill RJ, Hurley R, Lewis R, et al. Chronic Proliferative Hepatitis in A/JCr Mice Associated With Persistent Helicobacter Hepaticus Infection: A Model of Helicobacter-Induced Carcinogenesis. Infect Immun (1996) 64(5):1548–58. doi: 10.1128/iai.64.5.1548-1558.1996
31. Young VB, Knox KA, Pratt JS, Cortez JS, Mansfield LS, Rogers AB, et al. In Vitro and In Vivo Characterization of Helicobacter Hepaticus Cytolethal Distending Toxin Mutants. Infect Immun (2004) 72(5):2521–7. doi: 10.1128/IAI.72.5.2521-2527.2004
32. Sfanos KS, Canene-Adams K, Hempel H, Yu SH, Simons BW, Schaeffer AJ, et al. Bacterial Prostatitis Enhances 2-Amino-1-Methyl-6-Phenylimidazo[4,5-B]Pyridine (PhIP)-Induced Cancer at Multiple Sites. Cancer Prev Res (Phila) (2015) 8(8):683–92. doi: 10.1158/1940-6207.CAPR-15-0090
33. Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod-Levi T, et al. Breast Cancer Colonization by Fusobacterium Nucleatum Accelerates Tumor Growth and Metastatic Progression. Nat Commun (2020) 11(1):3259. doi: 10.1038/s41467-020-16967-2
34. Ge Z, Rogers AB, Feng Y, Lee A, Xu S, Taylor NS, et al. Bacterial Cytolethal Distending Toxin Promotes the Development of Dysplasia in a Model of Microbially Induced Hepatocarcinogenesis. Cell Microbiol (2007) 9(8):2070–80. doi: 10.1111/j.1462-5822.2007.00939.x
35. Ge Z, Feng Y, Ge L, Parry N, Muthupalani S, Fox JG. Helicobacter Hepaticus Cytolethal Distending Toxin Promotes Intestinal Carcinogenesis in 129Rag2-Deficient Mice. Cell Microbiol (2017) 19(7). doi: 10.1111/cmi.12728
36. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic Expression of Helicobacter Pylori CagA Induces Gastrointestinal and Hematopoietic Neoplasms in Mouse. Proc Natl Acad Sci USA (2008) 105(3):1003–8. doi: 10.1073/pnas.0711183105
37. Neal JT, Peterson TS, Kent ML, Guillemin K. H. Pylori Virulence Factor CagA Increases Intestinal Cell Proliferation by Wnt Pathway Activation in a Transgenic Zebrafish Model. Dis Model Mech (2013) 6(3):802–10. doi: 10.1242/dmm.011163
38. Harrandah AM, Chukkapalli SS, Bhattacharyya I, Progulske-Fox A, Chan EKL. Fusobacteria Modulate Oral Carcinogenesis and Promote Cancer Progression. J Oral Microbiol (2020) 13(1):1849493. doi: 10.1080/20002297.2020.1849493
39. Lu R, Wu S, Zhang YG, Xia Y, Liu X, Zheng Y, et al. Enteric Bacterial Protein AvrA Promotes Colonic Tumorigenesis and Activates Colonic Beta-Catenin Signaling Pathway. Oncogenesis (2014) 3:e105. doi: 10.1038/onc.2012.545
40. Lara-Tejero M, Galan JE. A Bacterial Toxin That Controls Cell Cycle Progression as a Deoxyribonuclease I-Like Protein. Science (2000) 290(5490):354–7. doi: 10.1126/science.290.5490.354
41. Pickett CL, Whitehouse CA. The Cytolethal Distending Toxin Family. Trends Microbiol (1999) 7(7):292–7. doi: 10.1016/S0966-842X(99)01537-1
42. Johnson TJ, DebRoy C, Belton S, Williams ML, Lawrence M, Nolan LK, et al. Pyrosequencing of the Vir Plasmid of Necrotoxigenic Escherichia Coli. Vet Microbiol (2010) 144(1-2):100–9. doi: 10.1016/j.vetmic.2009.12.022
43. Asakura M, Samosornsuk W, Taguchi M, Kobayashi K, Misawa N, Kusumoto M, et al. Comparative Analysis of Cytolethal Distending Toxin (Cdt) Genes Among Campylobacter Jejuni, C. Coli and C. Fetus Strains. Microb Pathog (2007) 42(5-6):174–83. doi: 10.1016/j.micpath.2007.01.005
44. Lara-Tejero M, Galan JE. Cytolethal Distending Toxin: Limited Damage as a Strategy to Modulate Cellular Functions. Trends Microbiol (2002) 10(3):147–52. doi: 10.1016/S0966-842X(02)02316-8
45. Spano S, Ugalde JE, Galan JE. Delivery of a Salmonella Typhi Exotoxin From a Host Intracellular Compartment. Cell Host Microbe (2008) 3(1):30–8. doi: 10.1016/j.chom.2007.11.001
46. Yamano R, Ohara M, Nishikubo S, Fujiwara T, Kawamoto T, Ueno Y, et al. Prevalence of Cytolethal Distending Toxin Production in Periodontopathogenic Bacteria. J Clin Microbiol (2003) 41(4):1391–8. doi: 10.1128/JCM.41.4.1391-1398.2003
47. Ahmed HJ, Svensson LA, Cope LD, Latimer JL, Hansen EJ, Ahlman K, et al. Prevalence of cdtABC Genes Encoding Cytolethal Distending Toxin Among Haemophilus Ducreyi and Actinobacillus Actinomycetemcomitans Strains. J Med Microbiol (2001) 50(10):860–4. doi: 10.1099/0022-1317-50-10-860
48. Talukder KA, Aslam M, Islam Z, Azmi IJ, Dutta DK, Hossain S, et al. Prevalence of Virulence Genes and Cytolethal Distending Toxin Production in Campylobacter Jejuni Isolates From Diarrheal Patients in Bangladesh. J Clin Microbiol (2008) 46(4):1485–8. doi: 10.1128/JCM.01912-07
49. Ge MC, Kuo SF, Chang SC, Chien CC, You HL, Lu JJ. Antimicrobial Susceptibility and Virulence Surveillance of Campylobacter Spp. Isolated From Patients in Two Tertiary Medical Centers in Taiwan. Front Microbiol (2018) 9:3186. doi: 10.3389/fmicb.2018.03186
50. Scuron MD, Boesze-Battaglia K, Dlakić M, Shenker BJ. The Cytolethal Distending Toxin Contributes to Microbial Virulence and Disease Pathogenesis by Acting as a Tri-Perditious Toxin. Front Cell Infect Microbiol (2016) 6:168. doi: 10.3389/fcimb.2016.00168
51. Jinadasa RN, Bloom SE, Weiss RS, Duhamel GE. Cytolethal Distending Toxin: A Conserved Bacterial Genotoxin That Blocks Cell Cycle Progression, Leading to Apoptosis of a Broad Range of Mammalian Cell Lineages. Microbiology (2011) 157(Pt 7):1851–75. doi: 10.1099/mic.0.049536-0
52. Shenker BJ, Walker LM, Zekavat Z, Ojcius DM, Huang PR, Boesze-Battaglia K. Cytolethal Distending Toxin-Induced Release of Interleukin-1beta by Human Macrophages Is Dependent Upon Activation of Glycogen Synthase Kinase 3beta, Spleen Tyrosine Kinase (Syk) and the Noncanonical Inflammasome. Cell Microbiol (2020) 22(7):e13194. doi: 10.1111/cmi.13194
53. Boesze-Battaglia K, Dhingra A, Walker LM, Zekavat A, Shenker BJ. Internalization and Intoxication of Human Macrophages by the Active Subunit of the Aggregatibacter Actinomycetemcomitans Cytolethal Distending Toxin Is Dependent Upon Cellugyrin (Synaptogyrin-2). Front Immunol (2020) 11:1262. doi: 10.3389/fimmu.2020.01262
54. Pickett CL, Pesci EC, Cottle DL, Russell G, Erdem AN, Zeytin H. Prevalence of Cytolethal Distending Toxin Production in Campylobacter Jejuni and Relatedness of Campylobacter Sp. cdtB Gene. Infect Immun (1996) 64(6):2070–8. doi: 10.1128/iai.64.6.2070-2078.1996
55. Hu X, Nesic D, Stebbins CE. Comparative Structure-Function Analysis of Cytolethal Distending Toxins. Proteins (2006) 62(2):421–34. doi: 10.1002/prot.20767
56. Frisan T. Bacterial Genotoxins: The Long Journey to the Nucleus of Mammalian Cells. Biochim Biophys Acta (2016) 1858(3):567–75. doi: 10.1016/j.bbamem.2015.08.016
57. Pons BJ, Vignard J, Mirey G. Cytolethal Distending Toxin Subunit B: A Review of Structure-Function Relationship. Toxins (Basel) (2019) 11(10):595. doi: 10.3390/toxins11100595
58. Li L, Sharipo A, Chaves-Olarte E, Masucci MG, Levitsky V, Thelestam M, et al. The Haemophilus Ducreyi Cytolethal Distending Toxin Activates Sensors of DNA Damage and Repair Complexes in Proliferating and non-Proliferating Cells. Cell Microbiol (2002) 4(2):87–99. doi: 10.1046/j.1462-5822.2002.00174.x
59. Frisan T, Cortes-Bratti X, Chaves-Olarte E, Stenerlöw B, Thelestam M. The Haemophilus Ducreyi Cytolethal Distending Toxin Induces DNA Double-Strand Breaks and Promotes ATM-Dependent Activation of RhoA. Cell Microbiol (2003) 5(10):695–707. doi: 10.1046/j.1462-5822.2003.00311.x
60. Comayras C, Tasca C, Peres SY, Ducommun B, Oswald E, De Rycke J. Escherichia Coli Cytolethal Distending Toxin Blocks the HeLa Cell Cycle at the G2/M Transition by Preventing Cdc2 Protein Kinase Dephosphorylation and Activation. Infect Immun (1997) 65(12):5088–95. doi: 10.1128/iai.65.12.5088-5095.1997
61. Nesic D, Hsu Y, Stebbins CE. Assembly and Function of a Bacterial Genotoxin. Nature (2004) 429(6990):429–33. doi: 10.1038/nature02532
62. Nesic D, Stebbins CE. Mechanisms of Assembly and Cellular Interactions for the Bacterial Genotoxin CDT. PLoS Pathog (2005) 1(3):e28. doi: 10.1371/journal.ppat.0010028
63. Guerra L, Teter K, Lilley BN, Stenerlow B, Holmes RK, Ploegh HL, et al. Cellular Internalization of Cytolethal Distending Toxin: A New End to a Known Pathway. Cell Microbiol (2005) 7(7):921–34. doi: 10.1111/j.1462-5822.2005.00520.x
64. Lin CD, Lai CK, Lin YH, Hsieh JT, Sing YT, Chang YC, et al. Cholesterol Depletion Reduces Entry of Campylobacter Jejuni Cytolethal Distending Toxin and Attenuates Intoxication of Host Cells. Infect Immun (2011) 79(9):3563–75. doi: 10.1128/IAI.05175-11
65. Eshraghi A, Maldonado-Arocho FJ, Gargi A, Cardwell MM, Prouty MG, Blanke SR, et al. Cytolethal Distending Toxin Family Members are Differentially Affected by Alterations in Host Glycans and Membrane Cholesterol. J Biol Chem (2010) 285(24):18199–207. doi: 10.1074/jbc.M110.112912
66. Boesze-Battaglia K, Brown A, Walker L, Besack D, Zekavat A, Wrenn S, et al. Cytolethal Distending Toxin-Induced Cell Cycle Arrest of Lymphocytes is Dependent Upon Recognition and Binding to Cholesterol. J Biol Chem (2009) 284(16):10650–8. doi: 10.1074/jbc.M809094200
67. Fais T, Delmas J, Serres A, Bonnet R, Dalmasso G. Impact of CDT Toxin on Human Diseases. Toxins (Basel) (2016) 8(7):220. doi: 10.3390/toxins8070220
68. Hontz JS, Villar-Lecumberri MT, Potter BM, Yoder MD, Dreyfus LA, Laity JH. Differences in Crystal and Solution Structures of the Cytolethal Distending Toxin B Subunit: Relevance to Nuclear Translocation and Functional Activation. J Biol Chem (2006) 281(35):25365–72. doi: 10.1074/jbc.M603727200
69. Fedor Y, Vignard J, Nicolau-Travers ML, Boutet-Robinet E, Watrin C, Salles B, et al. From Single-Strand Breaks to Double-Strand Breaks During S-Phase: A New Mode of Action of the Escherichia Coli Cytolethal Distending Toxin. Cell Microbiol (2013) 15(1):1–15. doi: 10.1111/cmi.12028
70. Guerra L, Cortes-Bratti X, Guidi R, Frisan T. The Biology of the Cytolethal Distending Toxins. Toxins (Basel) (2011) 3(3):172–90. doi: 10.3390/toxins3030172
71. Cortes-Bratti X, Karlsson C, Lagergard T, Thelestam M, Frisan T. The Haemophilus Ducreyi Cytolethal Distending Toxin Induces Cell Cycle Arrest and Apoptosis via the DNA Damage Checkpoint Pathways. J Biol Chem (2001) 276(7):5296–302. doi: 10.1074/jbc.M008527200
72. Martin OCB, Frisan T. Bacterial Genotoxin-Induced DNA Damage and Modulation of the Host Immune Microenvironment. Toxins (Basel) (2020) 12(2):63. doi: 10.3390/toxins12020063
73. Ohara M, Hayashi T, Kusunoki Y, Nakachi K, Fujiwara T, Komatsuzawa H, et al. Cytolethal Distending Toxin Induces Caspase-Dependent and -Independent Cell Death in MOLT-4 Cells. Infect Immun (2008) 76(10):4783–91. doi: 10.1128/IAI.01612-07
74. Ceelen LM, Decostere A, Ducatelle R, Haesebrouck F. Cytolethal Distending Toxin Generates Cell Death by Inducing a Bottleneck in the Cell Cycle. Microbiol Res (2006) 161(2):109–20. doi: 10.1016/j.micres.2005.04.002
75. Blazkova H, Krejcikova K, Moudry P, Frisan T, Hodny Z, Bartek J. Bacterial Intoxication Evokes Cellular Senescence With Persistent DNA Damage and Cytokine Signalling. J Cell Mol Med (2010) 14(1-2):357–67. doi: 10.1111/j.1582-4934.2009.00862.x
76. Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-Associated Heterochromatin Foci are Dispensable for Cellular Senescence, Occur in a Cell Type- and Insult-Dependent Manner and Follow Expression of P16(Ink4a). Cell Cycle (2011) 10(3):457–68. doi: 10.4161/cc.10.3.14707
77. Mathiasen SL, Gall-Mas L, Pateras IS, Theodorou SDP, Namini MRJ, Hansen MB, et al. Bacterial Genotoxins Induce T Cell Senescence. Cell Rep (2021) 35(10):109220. doi: 10.1016/j.celrep.2021.109220
78. Levi L, Toyooka T, Patarroyo M, Frisan T. Bacterial Genotoxins Promote Inside-Out Integrin β1 Activation, Formation of Focal Adhesion Complexes and Cell Spreading. PLoS One (2015) 10(4):e0124119. doi: 10.1371/journal.pone.0124119
79. Graillot V, Dormoy I, Dupuy J, Shay JW, Huc L, Mirey G, et al. Genotoxicity of Cytolethal Distending Toxin (CDT) on Isogenic Human Colorectal Cell Lines: Potential Promoting Effects for Colorectal Carcinogenesis. Front Cell Infect Microbiol (2016) 6:34. doi: 10.3389/fcimb.2016.00034
80. Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, et al. High Prevalence of Mucosa-Associated E. Coli Producing Cyclomodulin and Genotoxin in Colon Cancer. PLoS One (2013) 8(2):e56964. doi: 10.1371/journal.pone.0056964
81. Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, et al. Mutational Signature in Colorectal Cancer Caused by Genotoxic Pks(+) E. Coli. Nature (2020) 580(7802):269–73. doi: 10.1038/s41586-020-2080-8
82. Tremblay W, Mompart F, Lopez E, Quaranta M, Bergoglio V, Hashim S, et al. Cytolethal Distending Toxin Promotes Replicative Stress Leading to Genetic Instability Transmitted to Daughter Cells. Front Cell Dev Biol (2021) 9:656795. doi: 10.3389/fcell.2021.656795
83. Pons BJ, Pettes-Duler A, Naylies C, Taieb F, Bouchenot C, Hashim S, et al. Chronic Exposure to Cytolethal Distending Toxin (CDT) Promotes a cGAS-Dependent Type I Interferon Response. Cell Mol Life Sci (2021). doi: 10.1007/s00018-021-03902-x
84. Guerra L, Carr HS, Richter-Dahlfors A, Masucci MG, Thelestam M, Frost JA, et al. A Bacterial Cytotoxin Identifies the RhoA Exchange Factor Net1 as a Key Effector in the Response to DNA Damage. PLoS One (2008) 3(5):e2254. doi: 10.1371/journal.pone.0002254
85. Aragon V, Chao K, Dreyfus LA. Effect of Cytolethal Distending Toxin on F-Actin Assembly and Cell Division in Chinese Hamster Ovary Cells. Infect Immun (1997) 65(9):3774–80. doi: 10.1128/iai.65.9.3774-3780.1997
86. O'Connor K, Chen M. Dynamic Functions of RhoA in Tumor Cell Migration and Invasion. Small GTPases (2013) 4(3):141–7. doi: 10.4161/sgtp.25131
87. Dubash AD, Guilluy C, Srougi MC, Boulter E, Burridge K, García-Mata R. The Small GTPase RhoA Localizes to the Nucleus and Is Activated by Net1 and DNA Damage Signals. PLoS One (2011) 6(2):e17380. doi: 10.1371/journal.pone.0017380
88. Alberts AS, Qin H, Carr HS, Frost JA. PAK1 Negatively Regulates the Activity of the Rho Exchange Factor NET1. J Biol Chem (2005) 280(13):12152–61. doi: 10.1074/jbc.M405073200
89. Guerra L, Guidi R, Slot I, Callegari S, Sompallae R, Pickett CL, et al. Bacterial Genotoxin Triggers FEN1-Dependent RhoA Activation, Cytoskeleton Remodeling and Cell Survival. J Cell Sci (2011) 124(Pt 16):2735–42. doi: 10.1242/jcs.085845
90. Awasthi A, Raju MB, Rahman MA. Current Insights of Inhibitors of P38 Mitogen-Activated Protein Kinase in Inflammation. Med Chem (2021) 17(6):555–75. doi: 10.2174/1573406416666200227122849
91. Suarez-Lopez L, Kong YW, Sriram G, Patterson JC, Rosenberg S, Morandell S, et al. MAPKAP Kinase-2 Drives Expression of Angiogenic Factors by Tumor-Associated Macrophages in a Model of Inflammation-Induced Colon Cancer. Front Immunol (2020) 11:607891. doi: 10.3389/fimmu.2020.607891
92. Burton JC, Antoniades W, Okalova J, Roos MM, Grimsey NJ. Atypical P38 Signaling, Activation, and Implications for Disease. Int J Mol Sci (2021) 22(8):4183. doi: 10.3390/ijms22084183
93. Yang SY, Miah A, Sales KM, Fuller B, Seifalian AM, Winslet M. Inhibition of the P38 MAPK Pathway Sensitises Human Colon Cancer Cells to 5-Fluorouracil Treatment. Int J Oncol (2011) 38(6):1695–702. doi: 10.3892/ijo.2011.982
94. Cheng C, Seen D, Zheng C, Zeng R, Li E. Role of Small GTPase RhoA in DNA Damage Response. Biomolecules (2021) 11(2):212. doi: 10.3390/biom11020212
95. Narumiya S, Thumkeo D. Rho Signaling Research: History, Current Status and Future Directions. FEBS Lett (2018) 592(11):1763–76. doi: 10.1002/1873-3468.13087
Keywords: bacterial genotoxin, cytolethal distending toxin, DNA damage, genotoxicity, cancer development
Citation: Lai Y-R, Chang Y-F, Ma J, Chiu C-H, Kuo M-L and Lai C-H (2021) From DNA Damage to Cancer Progression: Potential Effects of Cytolethal Distending Toxin. Front. Immunol. 12:760451. doi: 10.3389/fimmu.2021.760451
Received: 18 August 2021; Accepted: 21 October 2021;
Published: 15 November 2021.
Edited by:
Traci Testerman, University of South Carolina, United StatesReviewed by:
Saeid Bouzari, Pasteur Institute of Iran (PII), IranCopyright © 2021 Lai, Chang, Ma, Chiu, Kuo and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-Ling Kuo, bWluZ2xpbmdAbWFpbC5jZ3UuZWR1LnR3; Chih-Ho Lai, Y2hsYWlAbWFpbC5jZ3UuZWR1LnR3
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.