 Jia He1,2†
Jia He1,2† Yue Du1,2†
Yue Du1,2† Gaopeng Li1,2†
Gaopeng Li1,2† Peng Xiao1,2
Peng Xiao1,2 Xingzheng Sun1,2
Xingzheng Sun1,2 Wenjun Song1,2
Wenjun Song1,2 Lihua Lai1,2
Lihua Lai1,2 Meng Xia1,2
Meng Xia1,2 Jianhua Zhang3*
Jianhua Zhang3* Qingqing Wang1,2*
Qingqing Wang1,2*- 1Institute of Immunology, Zhejiang University School of Medicine, Hangzhou, China
- 2The Key Laboratory for Immunity and Inflammatory Diseases of Zhejiang Province, Hangzhou, China
- 3Department of Medical Laboratory, School of Medicine, Shaoxing University, Shaoxing, China
Idiopathic pulmonary fibrosis (IPF) is a group of chronic interstitial pulmonary diseases characterized by an inexorable decline in lung function with limited treatment options. The abnormal expression of transforming growth factor-β (TGF-β) in profibrotic macrophages is linked to severe pulmonary fibrosis, but the regulation mechanisms of TGF-β expression are incompletely understood. We found that decreased expression of E3 ubiquitin ligase Fbxw7 in peripheral blood mononuclear cells (PBMCs) was significantly related to the severity of pulmonary fibrosis in IPF patients. Fbxw7 is identified to be a crucial suppressing factor for pulmonary fibrosis development and progression in a mouse model induced by intratracheal bleomycin treatment. Myeloid cell-specific Fbxw7 deletion increases pulmonary monocyte-macrophages accumulation in lung tissue, and eventually promotes bleomycin-induced collagen deposition and progressive pulmonary fibrosis. Notably, the expression of TGF-β in profibrotic macrophages was significantly upregulated in myeloid cell-specific Fbxw7 deletion mice after bleomycin treatment. C-Jun has long been regarded as a critical transcription factor of Tgfb1, we clarified that Fbxw7 inhibits the expression of TGF-β in profibrotic macrophages by interacting with c-Jun and mediating its K48-linked ubiquitination and degradation. These findings provide insight into the role of Fbxw7 in the regulation of macrophages during the pathogenesis of pulmonary fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a lung disease characterized by fibroblast hyperproliferation and extracellular matrix (ECM) deposition (1), which can cause pulmonary fibrosis and eventually lead to respiratory failure. However, the etiology of IPF remains unclear. Transforming growth factor-β (TGF-β) is one of the best-characterized pro-fibrotic factors (2). In mammals, there are three isoforms of TGF-β (TGF-β1, TGF-β2, and TGF-β3), of which TGF-β1 most closely related to the development of IPF (2). Disturbances of the pulmonary microenvironment induced by TGF-β are critical to promote the recruitment of circulating fibrocytes and bone marrow–derived progenitor cells to the lung (3), and enhance the pro-fibrotic capacities of these cells or induce their differentiation into activated myofibroblasts (4), eventually leading to the typical IPF symptoms like ECM deposition and abnormal collagen accumulation (5). Moreover, TGF-β1 induces macrophage recruitment and stimulates the expression of several pro-inflammatory and profibrogenic cytokines in macrophages, such as TNF-α and IL-1β (5). IPF patients have much higher TGF-β1 mRNA expression in profibrotic macrophages compared to normal volunteers (6, 7). Previous studies have reported that TGF-β1 expression can be induced by cytokines, such as IL-1β, TNF-α (8), IL-13 (9, 10) and IL-24 (11), the increased generation of ROS also induces TGF-β1 production (12). Moreover, the activation of TGF-β1 in fibrotic disorders can be induced by multiple factors, such as matrix metalloproteinases (MMP) 3 and MMP9 (13), thrombospodin-1 (14) and integrins αVβ6 (15). TGF-β1 signaling pathways and their downstream signal transduction mechanisms have been extensively studied, but there are few reports involved in the regulation mechanism of TGF-β1 expression.
Pulmonary macrophages derived from blood monocytes and classified into CD64+SiglecF+ alveolar macrophages (AMs) and CD11b+CD206+ interstitial macrophages (IMs) based on the different anatomical locations (16), both of those macrophage subsets derived from blood monocytes have been known to play a crucial role in IPF pathogenesis. Pulmonary macrophages polarized into alternatively activated anti-inflammatory phenotypes to promote tissue repair and fibrosis after lung injury (17). Aberrant immune function of anti-inflammatory macrophages present profibrogenic phenotypes, which induce the dysfunction of wound repair and accumulate in fibrotic lung, further support fibrogenesis of lung by generating reactive oxygen species (ROS) (6), contributing to collagen synthesis and ECM remodeling. Increased accumulation of profibrotic AMs have been detected in lung of IPF patients (6). During lung fibrogenesis, profibrotic macrophages release soluble factors to create and sustain the profibrotic lung microenvironment (18–20), such as TGF-β1, CCL8 (21), IGF-I (22), PDGF (23), and arginase 1 (24). In contrast, loss of profibrotic macrophages promotes the resolution of established pulmonary fibrosis (25) and the reduction of lung collagen deposition (26). Importantly, in rodents with bleomycin-induced lung fibrosis, pulmonary macrophages have been demonstrated to be the predominant source of profibrogenic mediator TGF-β (27). However, the specific mechanism of TGF-β1 secreted by macrophages in the pathogenesis of IPF is remain poorly understood.
F-box and WD repeat domain–containing 7 (Fbxw7) is a member of the F-box protein family, which is a component of the SKP1-cullin-F-box-protein (SCF) ubiquitin ligase complex. Many studies have shown that Fbxw7 is a tumor suppressor by targeting c-Myc, Notch, MCL1 and c-Jun (28). Fbxw7 mutation was detected in squamous cell carcinoma developed on IPF (29). Fbxw7 also participates in the regulation of Parkinson’s disease (30) and circadian rhythm (31). Moreover, Fbxw7 controls neural stem cell differentiation (32) and myelination (33). Fbxw7 has immunoregulatory capacities, it controls effector T cell polyfunctionality and survival in tumor environment by inhibiting Notch activation (34). Our previous studies have shown that Fbxw7 enhances antiviral innate immune response (35) and aggravate intestinal inflammation (36). These observations implicate that Fbxw7 may act as a transducer of tissue microenvironment, regulating the plastic function of macrophages. In this work, we further investigated the function of Fbxw7 in pulmonary macrophages during the pathogenesis of IPF. Our findings demonstrate that Fbxw7 expression decreased significantly in peripheral blood mononuclear cells (PBMCs) of IPF patients and lung tissues of the mice model of bleomycin-induced fibrosis. Fbxw7 deletion in myeloid cells increased the release of fibrogenic factors and the recruitment of monocytes into lung tissues. In terms of the mechanism, the Fbxw7 deletion promoted the TGF-β expression in macrophages through reducing c-Jun ubiquitination, thereby aggravating the severity of pulmonary fibrosis.
Materials and Methods
Mice Information
Fbxw7fl/fl mice (C57BL/6J background) were obtained from Jackson Laboratories. LysM-Cre mice C57BL/6J were kindly provided by Dr. Ximei Wu (Zhejiang University School of Medicine, Hangzhou, China). LysM+Fbxw7fl/fl mice were generated by crossing Fbxw7f/f mice with LysM-Cre transgenic mice as previously described (36). All mice were bred at the laboratory animal center of Zhejiang University under specific pathogen-free conditions. In all experiments, age-matched (8-12 week) mice were randomized into the experimental or control groups. All animal experiments were performed according to the protocol approved by the Animal Ethics Committee of Zhejiang University and were in compliance with institutional guidelines.
Cell Culture and DNA Transfection
Bone marrow cells were isolated by flushing femurs and tibias of 6- to 8-week-old mice with PBS and differentiated into BMDMs in RPMI-1640 medium with 10% (vol/vol) FCS and 10 ng/ml recombinant mouse M-CSF (R&D, 416-ML). To induce M2 macrophages, BMDMs were stimulated by adding the recombinant mouse IL-4 (30 ng/ml, PeproTech, 214-14). Proteasome inhibitors MG132 (M8699), protein synthesis inhibitor CHX (C4859), and JNK inhibitor SP600125 (420119) were from Sigma-Aldrich. TGF-β neutralizing antibody (MAB1835) and mouse IgG1 isotype control (MAB002) were from R&D systems.
Mouse embryonic fibroblast cell line (MEF) was obtained from American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS; BI). Murine macrophage-like RAW264.7 cells were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS; BI), 100 U/ml penicillin and 100 mg/ml streptomycin in an atmosphere of humidified 5% CO2 at 37°C.
Recombinant vectors encoding mouse c-Jun (NM_010591) were cloned into pcDNA3.1-c-Myc eukaryotic expression vector. The RAW264.7 cells (3 × 105 cells/well, in 24-well plate) were incubated with NATE™ (In vivogen, lyec-nate) for 30 min to enhance the transfection efficiency and transient expression of target plasmid. Then RAW264.7 cells were transfected with c-Jun plasmid (Roche, FuGENE HD Reagent) and treated with IL-4 for the indicated time.
Co-Culture Experiments
MEF cells (2×105) were cultured in the lower chamber and macrophages (5×105) were added to the upper chamber of a 12-well transwell apparatus (0.4 mm pore size, Costar, Cambridge, MA). The MEF were subjected to the further analysis at 48 h after co-culturing.
Induction of Pulmonary Fibrosis
An experimental pulmonary fibrosis model was established using the direct endotracheal injection of bleomycin (37). Briefly, 6- to 8-week-old C57BL/6 mice were anesthetized with pentobarbital by intraperitoneal injections. Neck area of mice was sterilized using povidone-iodine swabs after depilation of hairs, and 1 cm midline incision was made with sterile scissors. Intratracheal administration of 3.5 mg/kg bleomycin hydrochloride (Hisun-Pfizer Pharmaceuticals, Taizhou, China) in sterile PBS, an equal volume of sterile PBS for controls by using 1 ml syringe fitted with a 26 G needle (carefully insert the needle into the visualized trachea and rapidly inject the bleomycin solution during a single inspiration). The incision was closed with surgical sutures after withdrawing the needle. The mice were placed on warmer pads and monitored until fully awake. Mouse lung tissues were sacrificed at 14 days and 21 days post-bleomycin injection for subsequent experiments.
LPS induced pulmonary injury model (38, 39) were established using the direct endotracheal injection of 5 mg/kg LPS (Sigma-Aldrich, St. Louis, Missouri, USA). Mouse lung tissues were sacrificed at 10 days post-LPS injection for subsequent experiments.
Flow Cytometry
Cells were stained with fluorochrome-labeled mAbs and analyzed with an LSRII flow cytometer (BD). Flow cytometric analysis was performed using FlowJo software. The following fluorochrome-labeled mAbs were purchased from Biolegend, Inc. and used according to the manufacturers’ protocols: anti-CD11b-FITC (101205, RRID: AB_312788), anti-CD206-APC (141707, RRID: AB_10896057), anti-Ly6C-APC-Cy7 (128026, RRID: AB_10640120), anti-Ly6G-PE (127608, RRID: AB_1186099), anti-F4/80-Brilliant Violet 421 (123132, RRID: AB_11203717), anti-CD64-PE (139303, RRID: AB_10613467), and anti-Siglec-F-APC (155507, RRID: AB_2750236).
Immunoprecipitation and Immunoblot Analysis
Immunoprecipitation, SDS-PAGE and immunoblot analysis were performed according to a standard protocol as described previously (35). The following antibodies were used according to the manufacturers’ protocols: anti-Fbxw7 (Abcam, ab12292, RRID: AB_442966), anti-Lys48-specific linked polyubiquitin (MilliporeSigma, 05-1307, RRID: AB_1587578), anti-c-Jun (CST, #9165S, RRID: AB_2130165), anti-p65 (CST, #8242, RRID: AB_10859369), anti-p-p65 (CST, #3036, RRID: AB_331281), Collagen-I(Abcam, ab34710, RRID: AB_731684), Collagen-III (Abcam, ab7778, RRID: AB_306066), EGR-1 (CST, #4154S, RRID: AB_2097035). The PVDF membranes were incubated with corresponding primary antibodies followed by horseradish peroxidase-linked secondary antibodies. The images were developed and captured using Chemiluminescence imaging system (ChemiScope 3000 Mini, Clinx Science Instruments Co., Ltd). Relative quantities of target protein were determined comparing to β-actin expression using densitometric analysis (ImageJ 1.52v). The standard deviation was calculated for biological duplicates.
Lung Histology
Lungs from bleomycin-treated or control mice were dissected, fixed in 10% phosphate-buffered formalin, and embedded into paraffin. Tissue sections were stained with H&E for morphological analysis and with Masson’s trichrome for the detection of collagen fibers according to the manufacturers’ instructions (Nanjing Jiancheng, Nanjing, China). The Szapiel’s score was used to evaluate the severity of fibrosis using the following criteria (40): none (0), no evidence of fibrosis; mild (1), <20% of the lung affected; moderate (2), 20%–50% affected; severe (3), >50% affected.
Hydroxyproline Assay
To assess collagen synthesis in the lung, pulmonic hydroxyproline levels were determined using a hydroxyproline assay kit obtained from Jiancheng Bioengineering Institute (Nanjing, China), according to manufacturer’s instructions. Hydroxyproline content was expressed as μg/mg lung wet weight.
Quantitative RT-PCR
Total RNA was extracted from tissues or cells using RNAiso Plus (TakaRa), and then reversely transcribed to cDNA using the PrimeScript RT reagent kit (TaKaRa) with random primers according to the manufacturer’s instructions. Quantitative PCR was performed using KAPA SYBR Green (SYBR Green Fast qPCR Master Mix) on CFX96 Touch™ Real-Time PCR Detection System (BioRad). Expression data were normalized to the mRNA levels of the β-actin gene and calculated using the 2−ΔΔCt method (41). The sequences for primers were listed in Supplementary Table 1.
ELISA
The levels of mouse TGF-β in bronchoalveolar lavage fluid (BALF) or culture supernatants were measured with ELISA kits (Invitrogen, 88-8350-88) according to the manufacturer’s instructions.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism 7.0. All data are shown as mean ± SD. The comparisons of two experimental groups were determined using an unpaired, 2-tailed student’s t-test. Multiple comparisons were assessed by one-way ANOVA with Tukey’s multiple comparisons test. P-values less than 0.05 were considered significant.
Results
Decreased Expression of Fbxw7 Is Related to IPF
We first investigated whether Fbxw7 was related to the development of IPF, the expression level of Fbxw7 from IPF patients was analyzed in the data from Gene Expression Omnibus (GEO) database (GSE70867) (42). The result showed that Fbxw7 mRNA expression in bronchoalveolar lavage (BAL) cells of IPF patients was significantly lower than that in healthy controls (Figure 1A). In another database of microarray expression profiling of PBMCs from IPF patients in the training cohort (GSE132607) (43), Fbxw7 mRNA expression level in 12-months IPF patients was significantly lower than that in baseline, 4-months, and 8-months IPF patients (Figure 1B). We then used the bleomycin-induced mouse model of pulmonary fibrosis to observe the expression of Fbxw7 in lung tissues (Figure 1C) and peripheral blood monocytes (Figure 1D). qRT-PCR results showed that the expression level of Fbxw7 was significantly reduced after 21 days of intratracheal bleomycin administration compared to the PBS controls. Moreover, the expression of Fbxw7 in the macrophages in mice pulmonary fibrosis tissue was markedly decreased compared with those in healthy control subjects detected by immunofluorescence staining (Figure 1E). These results suggest that the decreased expression of Fbxw7 is associated with the formation of pulmonary fibrosis.
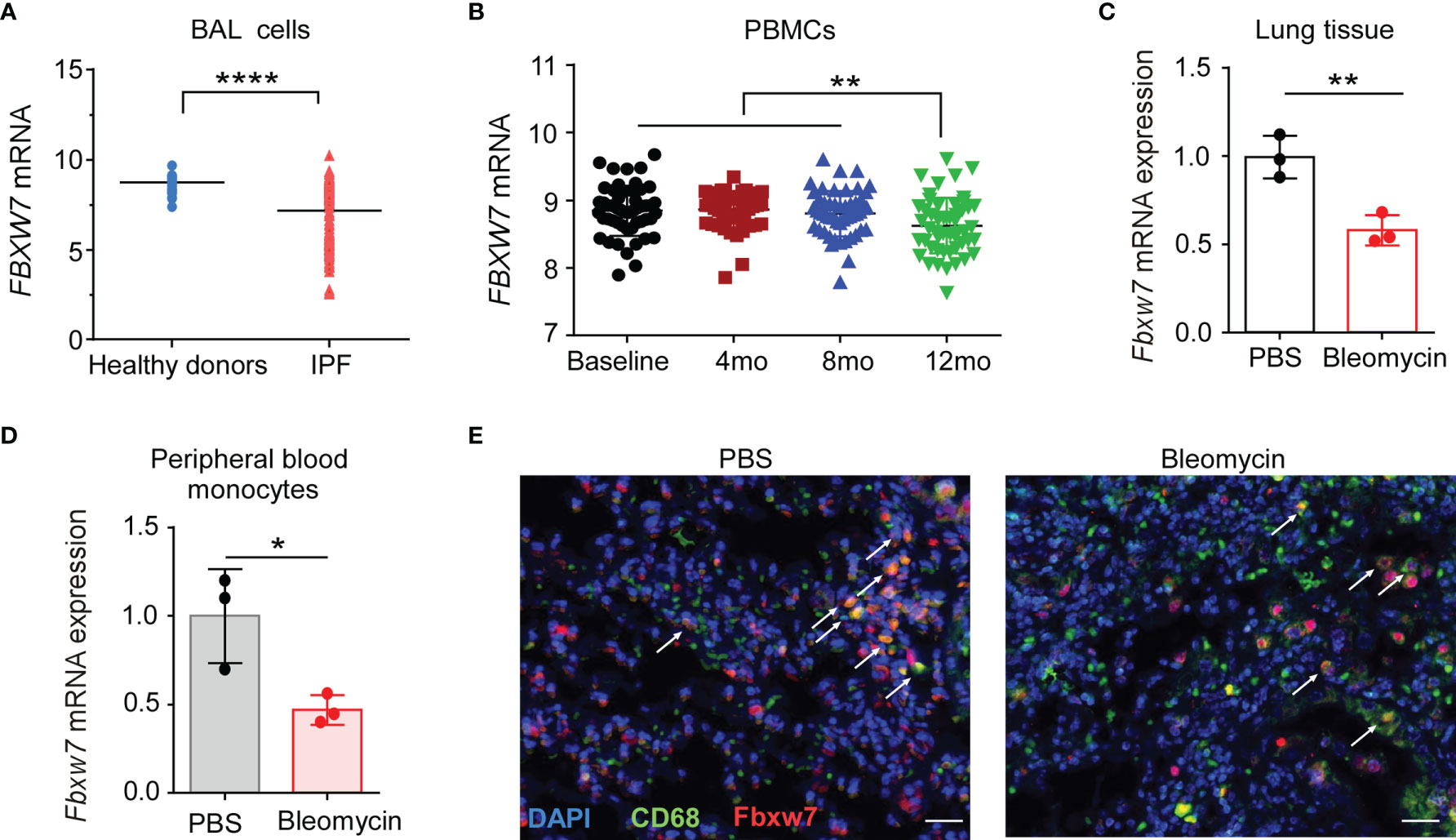
Figure 1 Decreased Fbxw7 expression is related to pulmonary fibrosis. (A) Fbxw7 mRNA expression in bronchoalveolar lavage (BAL) cells from healthy donors (n=48) and from IPF patients (n=176), the data were collected from GEO database (GSE70867), P value was obtained using two-tailed Student’s t test, ****P < 0.0001. (B) Fbxw7 mRNA expression in PBMCs from baseline (n=74), 4 months (4 mo, n=74), 8 months (8 mo, n=68) and 12 months (12 mo, n=60) IPF patients, the data were collected from the GEO database (GSE132607). Statistical significance was assessed by one-way ANOVA with Tukey’s multiple comparisons test, **P < 0.01. qRT-PCR analysis of lung tissues (C) and peripheral blood monocytes (D) obtained from mice after 21 days of bleomycin-induced lung injury and healthy controls. Data are expressed as mean ± SD of biological duplicates (n=3) and are representative of three independent experiments. P value was obtained using two-tailed Student’s t test, *P < 0.05, **P < 0.01. (E) Immunofluorescence staining for FBXW7 (red), CD68 (green), and DAPI for nuclei (blue) in lung tissues obtained from mice after 21 days of bleomycin-induced lung injury and healthy control mice. Scale bars: 20 μm. Data shown are the representative of three independent experiments.
Myeloid Deficiency of Fbxw7 Aggravates Bleomycin-Induced Collagen Deposition in Lung Tissue
Augmented collagen deposition within the interstitium (44, 45) and the remodeling of ECM (46) are recognized as one of the primordial pathophysiological events of IPF. To investigate the role of Fbxw7 in macrophages in the pathogenesis of IPF, we generated LysM-Cre+Fbxw7fl/fl (LysM+Fbxw7fl/fl) mice (Supplementary Figure 1A) and confirmed that myeloid specific deletion of Fbxw7 does not affect the differentiation and development of myeloid cells (Supplementary Figures 1B-F).
LysM+Fbxw7fl/fl mice and their Fbxw7fl/fl littermates were subjected to establish experimental pulmonary fibrosis model. Masson staining of lung tissues showed that collagen deposition was significantly increased in LysM+Fbxw7fl/fl mice 14 days after bleomycin administration, compared with Fbxw7fl/fl mice (Supplemental Figure 2A), and further increased after 21 days (Figure 2A). Szapiel scores also showed that Fbxw7 knockout exacerbated the degree of fibrosis (Supplemental Figure 2B and Figure 2B). The expression of collagen III and α-SMA observed by immune-histochemical staining was also significantly higher in the fibrotic lung tissue of LysM+Fbxw7fl/fl mice compared with Fbxw7fl/fl littermates (Figure 2C). Furthermore, LysM+Fbxw7fl/fl mice had significantly higher levels of hydroxyproline in lung tissues compared with Fbxw7fl/fl mice (Figure 2D).
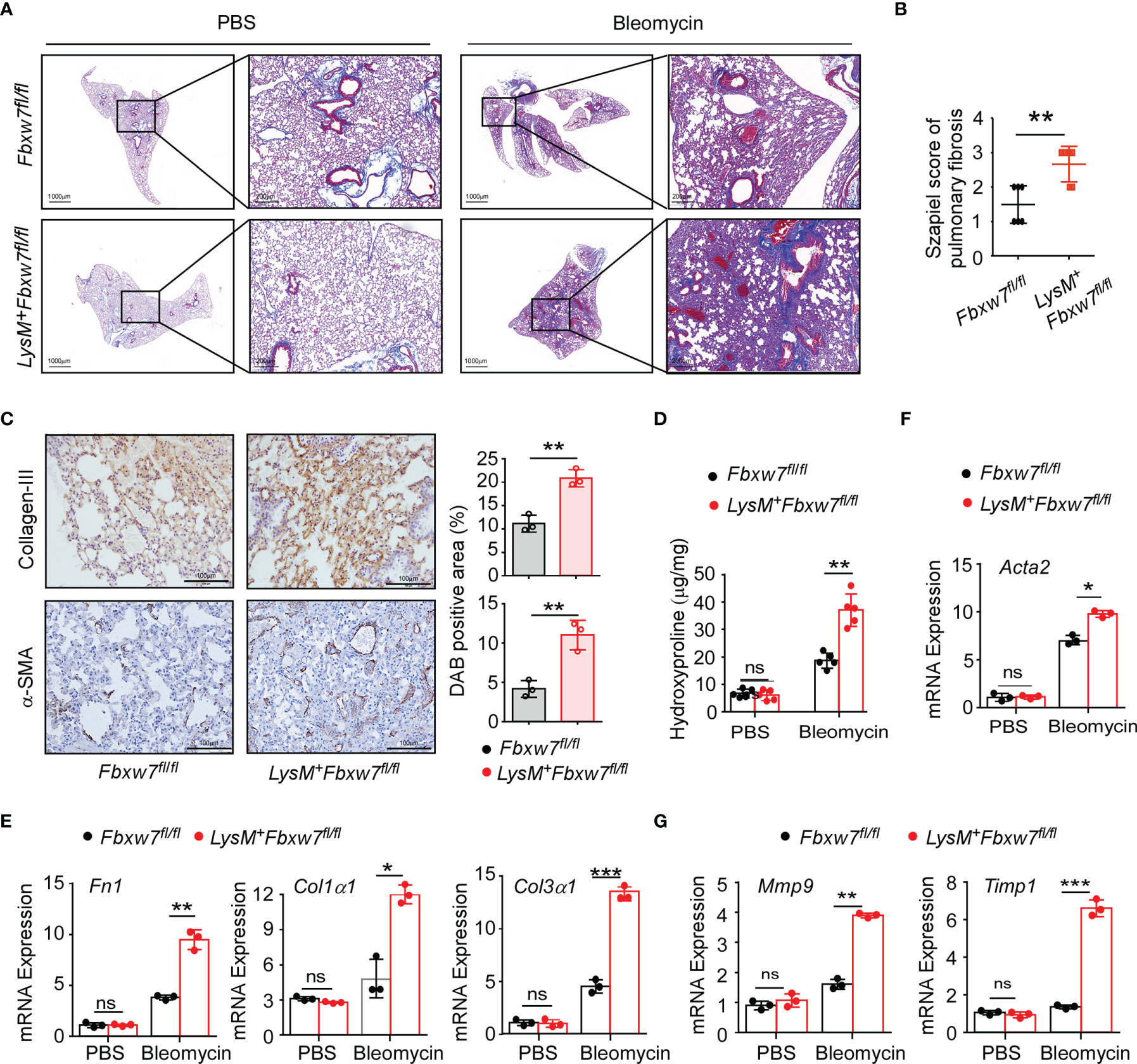
Figure 2 LysM+Fbxw7fl/fl mice show aggravated pulmonary fibrosis. Fbxw7fl/fl and LysM+Fbxw7fl/fl mice were administered bleomycin by direct endotracheal injection to induce pulmonary fibrosis. Three weeks later, lung tissues were isolated for Masson staining and RT-PCR analysis. (A) Masson staining of collagen fiber, scale bars 1000 μm (whole pulmonary section), and 200 μm (detail) and sapiel score of pulmonary tissue (B) obtained from Fbxw7fl/fl and LysM+Fbxw7fl/fl mice. (C) Representative collagen III and α-SMA immunohistochemical staining of lung tissues obtained from Fbxw7fl/fl and LysM+Fbxw7fl/fl mice after 21 days of bleomycin-induced lung injury, scale bars 100 μm. Comparison of DAB staining intensity (Image J) among the groups is shown. Quantification of hydroxyproline contents (D) and relative mRNA expression of collagen genes (E), Acta2 gene (α-SMA) (F), and matrix metalloproteinases (G) in the lungs from Fbxw7fl/fl and LysM+Fbxw7fl/fl mice after bleomycin treatment for 21 days. Data are expressed as mean ± SD of biological duplicates (n≥3) and are the representative of three independent experiments. P values were obtained using two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant.
Moreover, we examined the mRNA expression levels of collagen-related genes in the lung tissues after bleomycin administration. Fibronectin 1 (Fn1), a pro-fibrotic marker, is an important member of the fibronectin family (47). Col1α1 and Col3α1 is component of the ECM in IPF. qRT-PCR results showed that the expression of Fn1 and Col3α1 increased significantly in LysM+Fbxw7fl/fl mice (Supplemental Figure 2C and Figure 2E). Furthermore, as a surface molecular marker of myofibroblasts, the mRNA expression of α-smooth muscle actin (α-SMA or Acta2) was also significantly increased in LysM+Fbxw7fl/fl mice (Supplemental Figure 2D and Figure 2F). Matrix metalloproteinases (MMPs) and their specific inhibitors (tissue inhibitors of metalloproteinases, TIMPs) participate in the formation and degradation of ECM, and the imbalance between MMPs and TIMPs plays a pivotal role in pulmonary fibrosis pathogenesis. qRT-PCR results demonstrated that Fbxw7 deletion significantly increased the expression of Mmp9 and Timp1 (Figure 2G). These results suggest that the myeloid cell specific Fbxw7 knockout mice have increased collagen deposition in their lung tissue that may aggravate the degree of fibrosis induced by bleomycin.
Myeloid Deficiency of Fbxw7 Aggravates Bleomycin-Induced Lung Injury and Monocytes Recruitment
Tissue damage and inflammation are important triggers for pulmonary fibrosis (48). To determine whether Fbxw7 deletion in myeloid cells aggravates lung injury and inflammation, we evaluated the morphological characteristics in mouse lung tissues. The H & E staining results showed that the lung tissues of Lysm+Fbxw7fl/fl mice were more severely damaged 14 days (Supplemental Figure 3A) and 21 days (Figure 3A) after bleomycin administration. Lung tissues of LysM+Fbxw7fl/fl mice appeared to be denser, congestion and edema, as well as loss of alveolar structures compared with Fbxw7fl/fl mice. Moreover, we examined the mRNA expression of surfactant protein-D (Sftpd) and qRT-PCR results showed that myeloid deficiency of Fbxw7 resulted in higher levels of Sftpd in fibrotic lung tissues (Supplemental Figure 3B and Figure 3B).
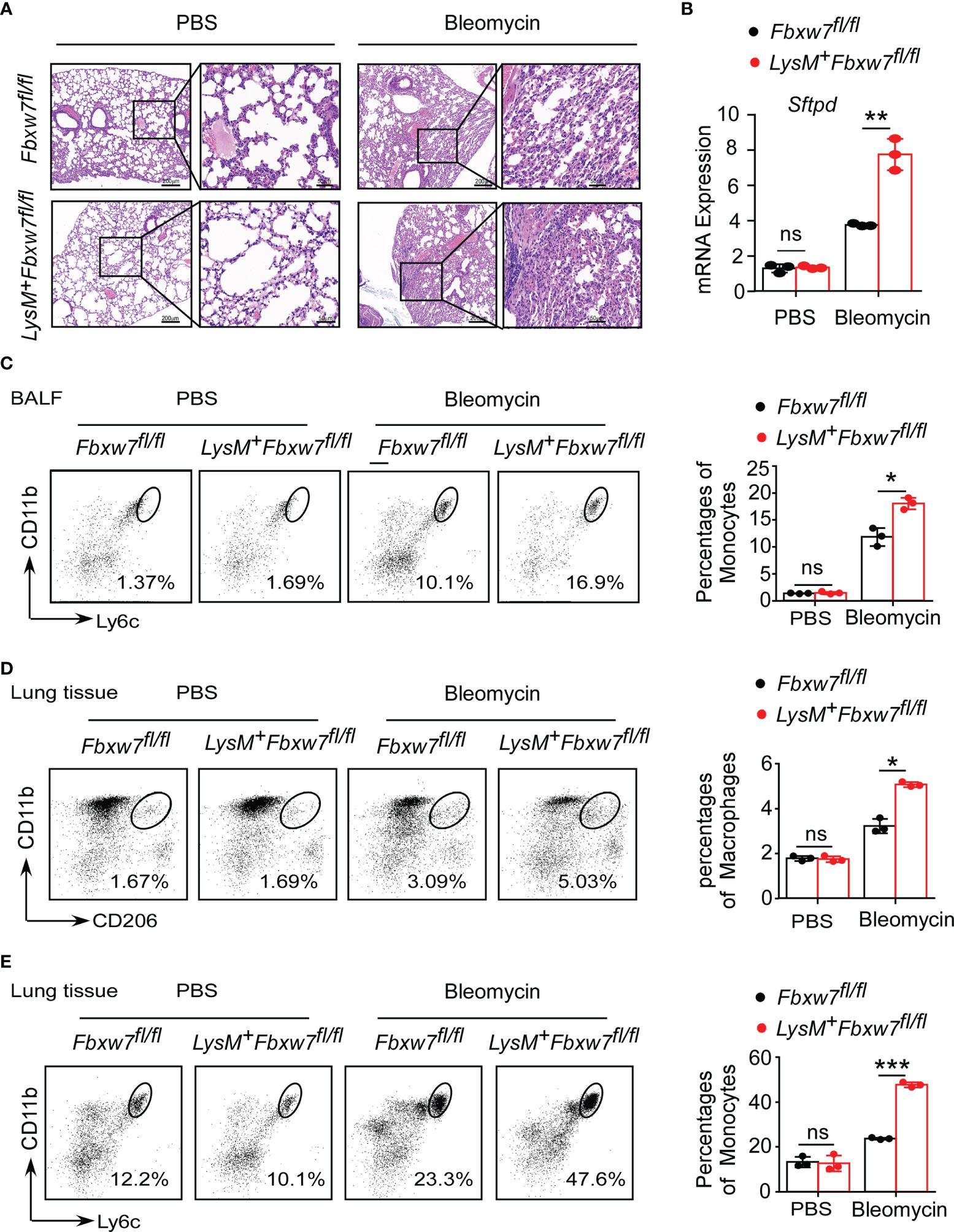
Figure 3 Fbxw7 deficiency increases recruitment of monocytes and macrophages after bleomycin-induced lung injury. Fbxw7fl/fl and LysM+Fbxw7fl/fl mice were administered PBS or bleomycin for 21 days to induce pulmonary fibrosis. (A) H&E staining of lung sections, scale bars 200 μm (whole pulmonary section), and 50 μm (detail). (B) qRT-PCR analysis of relative expression of Sftpd in lung tissue. Flow cytometry and statistical analysis of the percentage of CD11b+Ly6C+ monocytes in bronchoalveolar lavage fluid (BALF) (C), CD11b+CD206+ IMs (D) and CD11b+Ly6C+ monocytes (E) in lung tissues, data are expressed as mean ± SD of biological duplicates (n=3) and are the representative of three independent experiments. P values were obtained using two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant.
Furthermore, LysM+Fbxw7fl/fl mice had higher degree of inflammatory cell infiltration in the alveolar space observed by H & E staining (Figure 3A). Flow cytometric analysis was used to investigate the changes in mononuclear phagocyte subpopulations in bronchoalveolar lavage fluid (BALF) and lung tissue during pulmonary fibrosis development. After 21 days of bleomycin-induced pulmonary fibrosis in mice, Fbxw7 knockout did not increase CD64+SiglecF+ AMs accumulation (Supplemental Figure 3C), but significantly increased CD11b+Ly6C+ monocytes recruitment (Figure 3C) in BALF. Moreover, a single-cell suspension was prepared by digesting lung tissues and analyzed by flow cytometry. Compared with the PBS group, the percentages of CD11b+CD206+ IMs (Figure 3D) and CD11b+Ly6C+ monocytes (Figure 3E) in lung tissues of mice treated with bleomycin were increased. Importantly, LysM+Fbxw7fl/fl mice showed an obviously increased accumulation of IM and monocyte cell populations compared with Fbxw7fl/fl littermates (Figures 3D, E). These results suggest that the deletion of Fbxw7 exacerbates injury-induced lung fibrosis by promoting the recruitment and accumulation of mononuclear phagocytes.
Fbxw7 Deficiency Upregulates TGF-β Expression in Macrophages
Macrophages are the major source of multiple profibrotic mediators in pulmonary fibrosis (49). We next investigated whether myeloid Fbxw7 deletion altered the cytokine profile in fibrotic lung tissues. The qRT-PCR results showed that the mRNA expressions of Tnf-α, and Il1β in lung tissues of LysM+Fbxw7fl/fl mice was significantly higher than that of Fbxw7fl/fl mice 14 days after bleomycin administration, but the expression of Il6 and Il10 was not significantly different (Supplemental Figure 4A). In contrast, LysM+Fbxw7fl/fl mice had significantly higher levels of Il6 and Il1β, but not Tnf-α and Il10, compared with Fbxw7fl/fl mice 21 days after administration of bleomycin (Supplemental Figure 4B). Furthermore, we evaluated cytokines expression in alveolar macrophages and found that the mRNA expressions of Il1β, Tnf-α, Il6 and Il10 were significantly increased after bleomycin administration, however, there were no significant differences in the expression level between LysM+Fbxw7fl/fl mice and Fbxw7fl/fl littermates (Figure 4A). Interestingly, mRNA expression of Tgfb1, the macrophage-derived fibrogenic cytokine, was significantly increased in lung tissues and alveolar macrophages in LysM+Fbxw7fl/fl mice compared to Fbxw7fl/fl littermates (Figure 4B). ELISA further confirmed that the concentration of TGF-β protein in BALF from LysM+Fbxw7fl/fl mice was significantly higher than that from Fbxw7fl/fl littermates (Figure 4C). In lung injury model established by LPS, more severe lung injury, fibrosis associated collagen deposition as well as higher expression of Tgfb1 were found in lung tissue and BAL cells from LysM+Fbxw7fl/fl mice compared to Fbxw7fl/fl littermates (Supplemental Figures 4C, D and Figure 4D). The significantly elevated concentration of TGF-β protein in BALF and serum from LysM+Fbxw7fl/fl mice was observed by ELISA compared with that from Fbxw7fl/fl littermates (Figure 4E). These data suggest that deficiency of Fbxw7 in macrophages exacerbates pulmonary fibrosis, which may be related to the up-regulation of TGF-β expression.
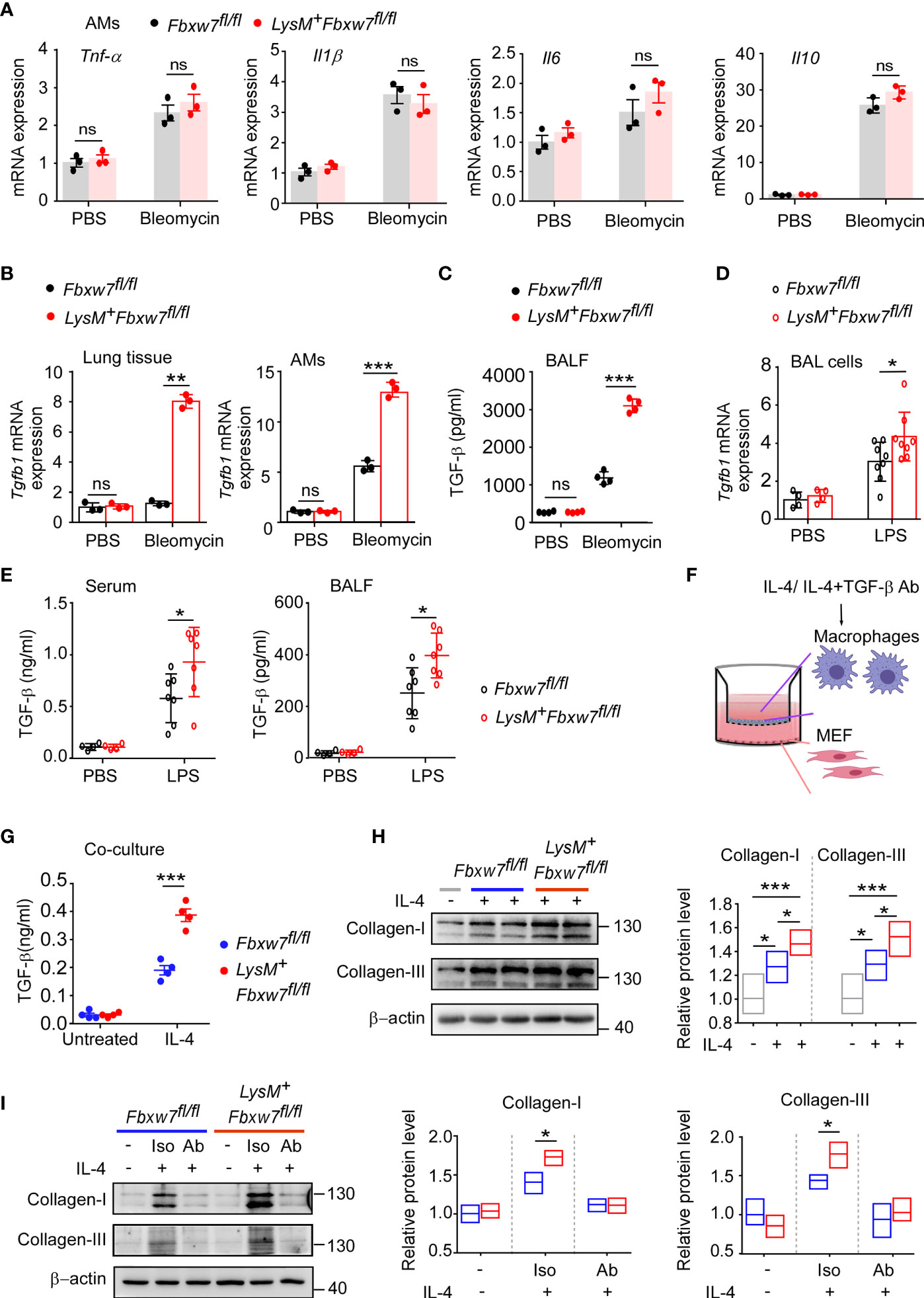
Figure 4 Fbxw7 deletion upregulates TGF-β expression in lung fibrosis model induced by bleomycin. qRT-PCR analysis of Tnf-α, Il1β, Il6, and Il10 relative mRNA expression of AMs (A) after Fbxw7fl/fl and LysM+Fbxw7fl/fl mice were administered PBS or bleomycin for 21 days to induce pulmonary fibrosis. After a 21d period of bleomycin-induced lung injure, qRT-PCR analysis of Tgfb1 relative mRNA expression in lung tissue and AMs (B), TGF-β content in BALF was detected by ELISA (C). Tgfb1 relative mRNA expression in BAL cells was analyzed by qRT-PCR (D), TGF-β content in serum and BALF was detected by ELISA (E) after 10 days of LPS-induced lung injury. Macrophages from Fbxw7fl/fl and LysM+Fbxw7fl/fl mice were stimulated with IL-4 (30 ng/ml) and treated with or without TGF-β neutralizing antibody for 12 h, then co-cultured with MEF for another 48 h. (F) The schematics of the approach. (G) TGF-β in the media of the macrophage co-culture system. Data are expressed as mean ± SD of biological duplicates (n≥3) and are the representative of three independent experiments. P values were obtained using two-tailed Student’s t test, *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant. (H) Representative images of western blot analysis of collagen-I and collagen-III in co-cultured MEF after IL-4 treatment. Densitometric fold change of target proteins based on IL-4 untreated group was calculated. (I) Representative images of western blot analysis of collagen-I and collagen-III in co-cultured MEF after IL-4 with TGF-β neutralizing antibody (Ab) treatment (3 μg/ml). Iso: Isotype. Relative protein levels were presented by densitometric fold change quantified by ImageJ based on Fbxw7fl/f untreated group, the floating bars indicate the min, mean and max value of biological duplicates (n=3), which are the representative of three independent experiments. P values were obtained using One-way ANOVA with Tukey’s Multiple Comparison test (H) or two-tailed Student’s t test (I). *P < 0.05, **P < 0.01, ***P < 0.001.
IL-4, as a Th2 cytokine, was reported to induce TGF-β expression in profibrotic macrophage to activate fibroblasts and promotes extensive tissue fibrosis (50, 51). qRT-PCR confirmed that the expression of Il4 was abundant in fibrotic lung tissues, but the level of Il4 was similar between LysM+Fbxw7fl/fl mice and their Fbxw7fl/fl littermates (Supplemental Figure 4E). Next, we performed a Transwell co-culture assay to evaluate the profibrogenic capability of macrophages from LysM+Fbxw7fl/fl mice and Fbxw7fl/fl littermates after IL-4 treatment in vitro (Figure 4F), the results consistently showed that myeloid Fbxw7 deficiency resulted in higher secretion levels of TGF-β in co-cultured macrophages after IL-4 treatment (Figure 4G), Fbxw7 deficient macrophages treated with IL-4 mediates a higher activation of co-cultured fibroblasts by the examination of the expression levels of collagen-I and collagen-III through western blot (Figure 4H). To further confirm that the enhanced profibrogenic capability of macrophages from LysM+Fbxw7 fl/fl mice is TGF-β-dependent, transwell co-culture assay was performed after IL-4 treatment with or without TGF-β neutralizing antibody. Neutralization of TGF-β abrogated the difference of profibrogenic ability between LysM+Fbxw7fl/fl macrophages and Fbxw7fl/fl macrophages (Figure 4I). Collectively, these data further support that Fbxw7 deletion in myeloid cells can exacerbate bleomycin-induced production of TGF-β and promote the development of pulmonary fibrosis.
Fbxw7 Regulates TGF-β Expression by Mediating the Ubiquitination and Degradation of c-Jun
Transcription factors including early growth response protein 1 (EGR-1) (52, 53) and NF-κB (54) have been reported to be involved in the pathogenesis of fibrosis via enhancing TGF-β dependent signaling. To further elucidate the mechanisms how Fbxw7 regulates TGF-β production in profibrotic macrophages, we investigated the effect of Fbxw7 deletion on the protein levels of EGR-1 and NF-κB by western blot. The protein levels of EGR1 and NF-κB in alveolar macrophages after bleomycin stimulation were not significantly changed between Fbxw7fl/fl and LysM+Fbxw7fl/fl mice, but the level of c-Jun was significantly higher in LysM+Fbxw7fl/fl mice than that in Fbxw7fl/fl mice (Figure 5A). Subsequently, we found significantly higher protein expression of c-Jun in IL-4-stimulated BMDMs after Fbxw7 deletion (Figure 5B). We then overexpressed c-Jun in RAW264.7 cells (Supplemental Figure 5A) and analyzed the expression of TGF-β by qRT-PCR and ELISA, the results showed that the expression level of TGF-β was significantly increased after c-Jun overexpression (Supplemental Figure 5B), indicating that c-Jun expression is sufficient for TGF-β in macrophages. SP600125 is a specific JNK inhibitor that competes with ATP to inhibit the phosphorylation of c-Jun (55). qRT-PCR analysis and ELISA results showed that the mRNA and protein expression of TGF-β increased significantly after the deletion of Fbxw7 (Figures 4C, D), but this effect was abrogated after inhibition of c-Jun phosphorylation by SP600125 (Figure 5C). The MEF cells, co-cultured with peritoneal macrophage from LysM+Fbxw7fl/fl mice or Fbxw7fl/fl littermates after inhibiting the phosphorylation of c-Jun, showed similar protein levels of collagen-I and collagen-III, which were detected by western blot (Supplemental Figures 5C, D). Taken together, Fbxw7 deletion leads to the upregulation of TGF-β expression and severity of pulmonary fibrosis through the regulation of c-Jun.
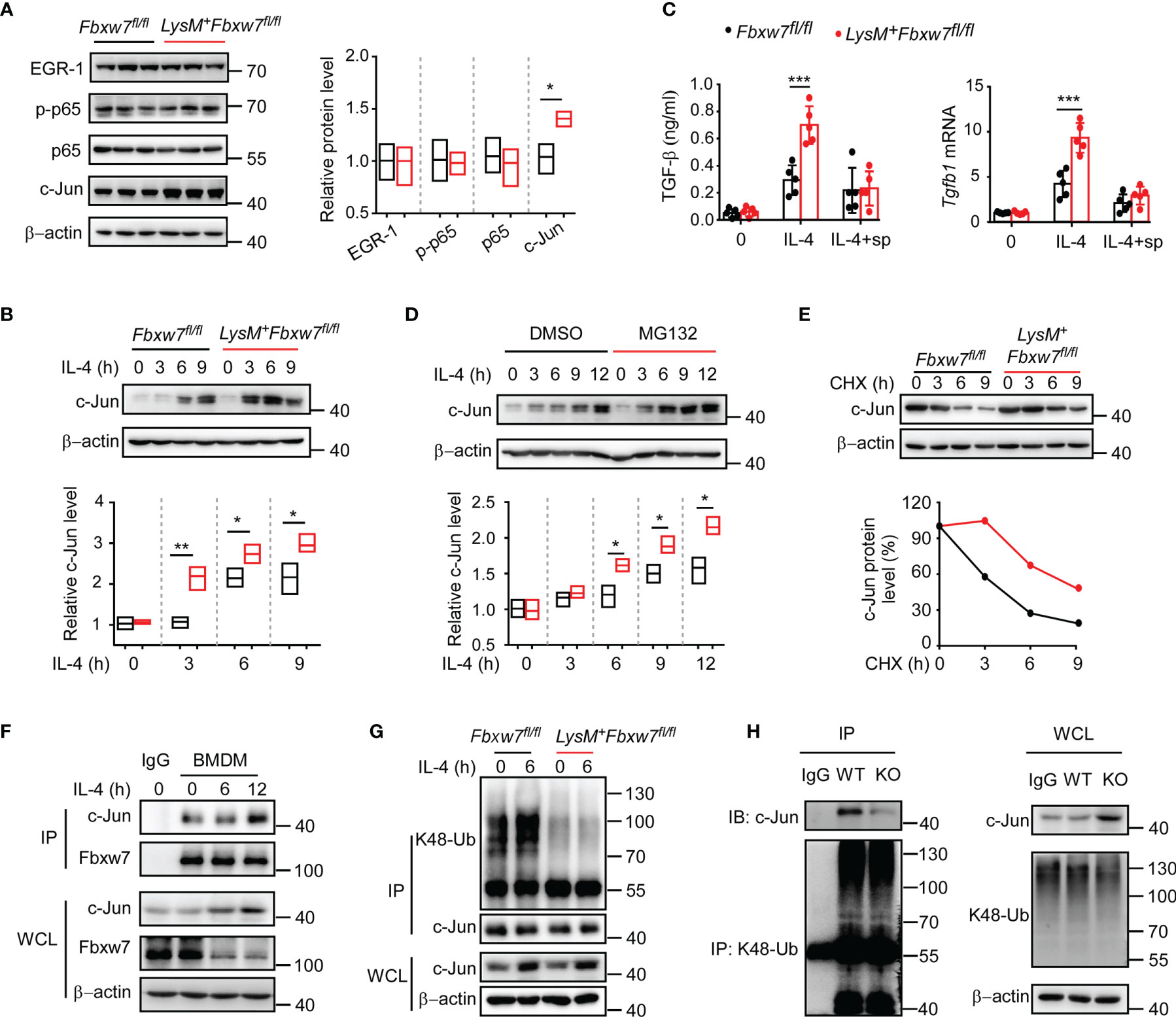
Figure 5 Fbxw7 regulates TGF-β expression via mediating the ubiquitination of c-Jun. (A) Immunoblot analysis of phosphorylated and/or total proteins in lysates of lung tissues obtained from bleomycin-treated Fbxw7fl/fl and LysM+Fbxw7fl/fl mice. Densitometric fold change of target proteins based on Fbxw7fl/fl group was calculated. (B) Immunoblot analysis of c-Jun protein levels in lysates of IL-4 (30 ng/ml) stimulated BMDMs from Fbxw7fl/fl and LysM+Fbxw7fl/fl mice. Densitometric fold change for the relative c-Jun level based on Fbxw7fl/fl untreated group was calculated. (C) Fbxw7fl/fl and LysM+ Fbxw7fl/fl BMDMs stimulated with IL-4 and treated or not with SP600125 (15 μM), qRT-PCR analysis for Tgfb1 relative mRNA expression and ELISA for TGF-β protein expression. Data are expressed as mean ± SD of biological duplicates (n=5) and are the representative of three independent experiments. P values were obtained using two-tailed Student’s t test, ***P < 0.001. (D) Immunoblot analysis of c-Jun expression in BMDMs treated or not with MG132 (20 μM) for 8 hours and stimulated with IL-4 for the indicated durations. Densitometric fold change of c-Jun based on IL-4, MG132 untreated group was calculated. (E) Immunoblot analysis and relative protein levels of c-Jun in lysates of Fbxw7fl/fl and LysM+Fbxw7fl/fl BMDMs treated with CHX (40 μg/ml) for indicated hours after stimulation with IL-4 for 6 h. (F) Coimmunoprecipitation and immunoblot of BMDMs stimulated with IL-4 for indicated hours. (G) Immunoblot analysis of the K48 ubiquitination of c-Jun in Fbxw7fl/fl and LysM+Fbxw7fl/fl BMDMs stimulated with IL-4 for 6 h. (H) Peritoneal macrophages from LysM+Fbxw7fl/fl (WT) and Fbxw7fl/fl (KO) mice were stimulated with IL-4 for 6 h, coimmunoprecipitation was performed with K48-Ub to pull-down ubiquitylated proteins, followed by IB with c-Jun. Relative protein levels were presented by densitometric fold change quantified by ImageJ, the floating bars indicate the min, mean and max value of biological duplicates and are the representative of three independent experiments. P values were obtained using two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001.
It has been demonstrated that the transcription factor c-Jun can be degraded by the ubiquitin-proteasome system as a substrate for Fbxw7 (56). As expected, western blot analysis showed that the degradation of c-Jun protein could be inhibited by protease inhibitor MG132 (Figure 5D). A cycloheximide chase assay showed that Fbxw7 deletion extended the half-life of endogenous c-Jun protein in BMDMs (Figure 5E). Immunoprecipitation assay results showed that Fbxw7 can physically bind to c-Jun protein (Figure 5F) and induce K48-linked polyubiquitination of c-Jun (Figure 5G). Moreover, immunoprecipitation assay was performed by using K48-Ub to pull-down c-Jun and the peritoneal macrophages from LysM+Fbxw7f/f mice showed weaker ability of mediating c-Jun ubiquitination and proteasomal degradation compared with cells from Fbxw7f/f littermates (Figure 5H). This result further revealed that the myeloid Fbxw7 deficiency leads to a significantly accumulation of c-Jun by abolishing its K48 ubiquitination. Collectively, these data confirmed that Fbxw7 promotes TGF-β expression by inducing K48-linked polyubiquitination and proteasome degradation of c-Jun in macrophages.
Discussion
The E3 ubiquitin ligase Fbxw7 has been well characterized as a critical tumor suppressor that can mediate the degradation of various oncoproteins by the ubiquitin-proteasome system (57). Our studies and others have presented compelling evidence that Fbxw7 plays a crucial role in inflammation and antivirus immune response through regulating the function of macrophages (35, 36, 58). In this study, we demonstrated that the deletion of Fbxw7 in the myeloid lineages aggravates bleomycin-induced lung injury, collagen deposition, and monocyte recruitment, and eventually leads to the exacerbation of pulmonary fibrosis in mice. We revealed its underlying mechanism that Fbxw7 deletion will upregulate TGF-β expression by inhibiting the degradation of c-Jun in macrophages. These findings will be beneficial for understanding the role of innate immune response in the pathogenesis of IPF.
Macrophages are the most abundant immune cell population in the lung environment and are involved in tissue repair and lung fibrosis progression, it has also been recognized to play a significant role in driving the fibrogenesis and matrix remodeling. Given the high plasticity of lung macrophages, the relative roles of tissue-resident alveolar macrophages, interstitial macrophages and monocyte-derived infiltrating macrophages in pulmonary fibrosis is complicated (59). High numbers of both interstitial and alveolar macrophages were detected in pulmonary fibrosis (24), the depletion of macrophages, including alveolar and infiltrating macrophage, reduces fibrogenesis (25, 60). Experimental fibrosis leads to the recruitment of bone marrow-derived AMs that are more profibrogenic than their disrupted resident counterparts (61). Selectively targeting alveolar macrophage differentiation without affecting global monocytes or tissue-resident alveolar macrophage may ameliorate fibrosis depletion, while the depletion of tissue-resident AMs cannot alter fibrosis severity (61). In addition, IMs are also thought to arise from monocytes, and highly expression of monocyte-related genes, such as Cd14, Cd163, and Csfr1 (62), activated IMs have been reported to induce myofibroblastic activation and ECM production (24). Furthermore, those more profibrogenic macrophages which play a vital role in airway remodeling in pulmonary fibrosis biases alternatively activated macrophages, so we used IL-4-stimulated BMDMs for in vitro experiments to mimic profibrogenic macrophages in vivo. Our data showed that more monocytes are recruited into the lung tissues and BALF of Fbxw7-KO mice, these monocytes further differentiate into profibrogenic macrophages and then participate in the development of pulmonary fibrosis.
The abundant expression of Fbxw7 mRNA and protein can be detected in most human and mice tissues. The decreased level of Fbxw7 mRNA in IPF may be regulated by many factors, including DNA or RNA silencing. The expression of microRNA-155 (miR-155) has been reported to be correlated to the damage degree of IPF (63), and miR-155 reduces the levels of Fbxw7 mRNA (64). The Fbxw7 promoter can be regulated through epigenetic mechanism during lung fibrosis, including histone modifications. Aberrant elevation of DNA methyltransferase 1 (Dnmt1) can mediate the hypermethylation of Fbxw7 promoter and result in decreased gene level of Fbxw7 (65), subsequently contributes significantly to the development of pulmonary fibrosis (66). Moreover, the expression of enhancer of zeste homolog 2 (Ezh2) is increased in IPF patients, increased Ezh2 recruitment and hypermethylated H3K27 at the promoter of fibrotic related genes have been demonstrated, such as Cox-2 (67) and Cxcl10 (68). We speculate the function of Ezh2 in inducing the increase of H3K27me3 at Fbxw7 promotor and silencing of Fbxw7 gene (34) might also be involved in regulating pulmonary fibrosis. IL-4 has been shown to induce TGF-β expression in profibrotic macrophage to activate fibroblasts and promotes extensive tissue fibrosis (50). In our study, we found IL-4 was highly expressed in lung fibrosis tissue of bleomycin-induced mice, and the expression of Fbxw7 was significantly decreased in bleomycin-induced mice lung issue and in BMDMs after IL-4 stimulation as shown in Figures 1, S4B and 5G. These findings suggest that IL-4 in the profibrotic microenvironment might downregulate Fbxw7 expression and block c-Jun degradation.
TGF-β can be produced by a wide variety of cell types, including lung macrophages, alveolar epithelial cells, endothelial cells, fibroblasts and myofibroblasts. However, lung macrophages are revealed to be the main source of TGF-β (6, 69). In bleomycin induced pulmonary fibrosis, macrophages induce fibroblast differentiation and proliferation by producing TGF-β, thereby exacerbating pulmonary fibrosis (70, 71). The widespread accumulation of collagen in lungs, which was observed in WT mice, disappeared in the myeloid TGF-β deficient mice, which showed similar lung architecture to saline controls (6). Several factors have previously been implicated in the transcriptional induction of Tgfb1, bacterial induction of Tgfb1 mRNA expression in human intestinal epithelial cells is butyrate and SP1-dependent (72). P38/MAPK signal, MEK-1 and JNK are involved in Tgfb1 transcription (73). The higher mRNA expression of Tgfb1 was detected in Egr-1 overexpressed mesangial cell compared with control cells (74). Moreover, p65 and c-Jun were recruited to the promoter of Tgfb1 in A549 cells upon IL-1β stimulation, this result suggesting the role of NF-κB and AP-1 in regulating Tgfb1 expression (75).
C-Jun protein is a component of the dimeric transcription factor activator protein 1 (AP-1) (76), it has been shown to play essential roles in most fibrotic conditions, which can activate multiple signaling pathways, resulting in the expression of many fibrogenesis-associated genes (77). C-Jun N-terminal kinase JNK1 has been reported to promote fibrosis in the lung (78), inhibition of c-Jun phosphorylation in vivo reduced fibrosis and mRNA levels of Tgfb1 in the unilateral ischemia reperfusion injury model (79). C-Jun expression in non-parenchymal liver cells (NPLCs) promotes the fibrogenesis and was particularly correlated with liver fibrosis during non-alcoholic steatohepatitis (80). Moreover, studies have shown that c-Jun can be degraded by the ubiquitin-proteasome system as the substrate of Fbxw7 (81, 82), which was also confirmed by our study. Our data show that deletion of Fbxw7 in macrophages does not affect the levels of P65, EGR-1 and their phosphorylation, but significantly increases the level of AP-1 transcription factor subunit c-Jun.
Notably, the published data from Wang et al. (83) showed that FBXW7 deficiency prevents fibrogenesis by increasing telomere capping, which is contrary to the conclusion of our finding. Several potential possibilities exist for this discrepancy. Wang et al. used a mouse model of Fbw7 conditional knockout in pulmonary alveolar epithelial type 2 (AEC2) stem cells, while we used a model of Fbxw7 conditional knockout in myeloid cells. Those reverse effects of Fbxw7 in AEC2 stem cells and myeloid cells in regulating lung fibrosis indicated that the pathogenesis of IPF is complicated, it involves the abnormal repair of lung tissue injury by alveolar epithelial cells, the disordered differentiation and proliferation of fibroblasts, or the hyperactivated recruitment and activation of innate immune cells. The dysfunction in any cell type can lead to different outcomes of IPF. Moreover, subtle genetic background differences in the strains of the mice used and microbiome differences between the mouse colonies may also contribute to these discrepancies.
In conclusion, our study revealed that Fbxw7 plays a crucial role in regulating pulmonary fibrosis progression. Myeloid-specific deletion of Fbxw7 exacerbates pulmonary fibrosis by inhibiting c-Jun degradation and upregulating TGF-β expression in macrophages.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Medical Experimental Animal Care Commission of Zhejiang University.
Author Contributions
QW, JZ, and JH designed the research. JH, YD, GL, XS, WS, LL, and MX performed experiments and acquired and analyzed data. JZ, JH, and PX drafted the manuscript. QW revised the manuscript. All authors read and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (31870907, 81930041, 91842103), Natural Science Foundation of Zhejiang Province (Z19H100001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YS declared a shared affiliation, with no collaboration, with several of the authors, JH, YD, GL, PX, XS, LL, MX, QW, to the handling editor at the time of review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Prof. Ximei Wu for providing the Lysm-Cre mice. We thank the Core Facilities, Zhejiang University School of Medicine for technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.760138/full#supplementary-material
References
1. Wolters PJ, Blackwell TS, Eickelberg O, Loyd JE, Kaminski N, Jenkins G, et al. Time for a Change: Is Idiopathic Pulmonary Fibrosis Still Idiopathic and Only Fibrotic? Lancet Respir Med (2018) 2:154–60. doi: 10.1016/s2213-2600(18)30007-9
2. Xaubet A, Marin-Arguedas A, Lario S, Ancochea J, Morell F, Ruiz-Manzano J, et al. Transforming Growth Factor-Beta1 Gene Polymorphisms Are Associated With Disease Progression in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2003) 4:431–5. doi: 10.1164/rccm.200210-1165OC
3. Mura M, Porretta MA, Bargagli E, Sergiacomi G, Zompatori M, Sverzellati N, et al. Predicting Survival in Newly Diagnosed Idiopathic Pulmonary Fibrosis: A 3-Year Prospective Study. Eur Respir J (2012) 1:101–9. doi: 10.1183/09031936.00106011
4. King TE Jr., Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting Survival in Idiopathic Pulmonary Fibrosis: Scoring System and Survival Model. Am J Respir Crit Care Med (2001) 7:1171–81. doi: 10.1164/ajrccm.164.7.2003140
5. Fernandez IE, Eickelberg O. The Impact of Tgf-Beta on Lung Fibrosis: From Targeting to Biomarkers. Proc Am Thorac Soc (2012) 3:111–6. doi: 10.1513/pats.201203-023AW
6. Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity (2016) 3:582–96. doi: 10.1016/j.immuni.2016.01.001
7. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The Cytokine Tgf-Beta Promotes the Development and Homeostasis of Alveolar Macrophages. Immunity (2017) 5:903–12 e4. doi: 10.1016/j.immuni.2017.10.007
8. Phillips AO, Topley N, Steadman R, Morrisey K, Williams JD. Induction of Tgf-Beta 1 Synthesis in D-Glucose Primed Human Proximal Tubular Cells by Il-1 Beta and Tnf Alpha. Kidney Int (1996) 5:1546–54. doi: 10.1038/ki.1996.470
9. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary Expression of Interleukin-13 Causes Inflammation, Mucus Hypersecretion, Subepithelial Fibrosis, Physiologic Abnormalities, and Eotaxin Production. J Clin Invest (1999) 6:779–88. doi: 10.1172/JCI5909
10. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 Induces Tissue Fibrosis by Selectively Stimulating and Activating Transforming Growth Factor Beta(1). J Exp Med (2001) 6:809–21. doi: 10.1084/jem.194.6.809
11. Rao LZ, Wang Y, Zhang L, Wu G, Zhang L, Wang FX, et al. Il-24 Deficiency Protects Mice Against Bleomycin-Induced Pulmonary Fibrosis by Repressing Il-4-Induced M2 Program in Macrophages. Cell Death Differ (2021) 4:1270–83. doi: 10.1038/s41418-020-00650-6
12. Sunaga H, Matsui H, Ueno M, Maeno T, Iso T, Syamsunarno MR, et al. Deranged Fatty Acid Composition Causes Pulmonary Fibrosis in Elovl6-Deficient Mice. Nat Commun (2013) 4:2563. doi: 10.1038/ncomms3563
13. Mahalanobish S, Saha S, Dutta S, Sil PC. Matrix Metalloproteinase: An Upcoming Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Pharmacol Res (2020) 152:104591. doi: 10.1016/j.phrs.2019.104591
14. Murphy-Ullrich JE, Suto MJ. Thrombospondin-1 Regulation of Latent Tgf-Beta Activation: A Therapeutic Target for Fibrotic Disease. Matrix Biol (2018) 68-69:28–43. doi: 10.1016/j.matbio.2017.12.009
15. Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, et al. Force Interacts With Macromolecular Structure in Activation of Tgf-Beta. Nature (2017) 7639:55–9. doi: 10.1038/nature21035
16. Byrne AJ, Maher TM, Lloyd CM. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol Med (2016) 4:303–16. doi: 10.1016/j.molmed.2016.02.004
17. Laskin DL, Malaviya R, Laskin JD. Role of Macrophages in Acute Lung Injury and Chronic Fibrosis Induced by Pulmonary Toxicants. Toxicol Sci (2019) 2:287–301. doi: 10.1093/toxsci/kfy309
18. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6chi Monocytes Direct Alternatively Activated Profibrotic Macrophage Regulation of Lung Fibrosis. Am J Respir Crit Care Med (2011) 5:569–81. doi: 10.1164/rccm.201010-1719OC
19. Fernandez IE, Eickelberg O. New Cellular and Molecular Mechanisms of Lung Injury and Fibrosis in Idiopathic Pulmonary Fibrosis. Lancet (2012) 9842:680–8. doi: 10.1016/S0140-6736(12)61144-1
20. Zhang L, Wang Y, Wu G, Xiong W, Gu W, Wang CY. Macrophages: Friend or Foe in Idiopathic Pulmonary Fibrosis? Respir Res (2018) 1:170. doi: 10.1186/s12931-018-0864-2
21. Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A Vicious Circle of Alveolar Macrophages and Fibroblasts Perpetuates Pulmonary Fibrosis via Ccl18. Am J Respir Crit Care Med (2006) 7:781–92. doi: 10.1164/rccm.200509-1518OC
22. Wynes MW, Frankel SK, Riches DW. Il-4-Induced Macrophage-Derived Igf-I Protects Myofibroblasts From Apoptosis Following Growth Factor Withdrawal. J Leukoc Biol (2004) 5:1019–27. doi: 10.1189/jlb.0504288
23. Nagaoka I, Trapnell BC, Crystal RG. Upregulation of Platelet-Derived Growth Factor-A and -B Gene Expression in Alveolar Macrophages of Individuals With Idiopathic Pulmonary Fibrosis. J Clin Invest (1990) 6:2023–7. doi: 10.1172/JCI114669
24. Meziani L, Mondini M, Petit B, Boissonnas A, Thomas de Montpreville V, Mercier O, et al. Csf1r Inhibition Prevents Radiation Pulmonary Fibrosis by Depletion of Interstitial Macrophages. Eur Respir J (2018) 3:1702120. doi: 10.1183/13993003.02120-2017
25. Redente EF, Keith RC, Janssen W, Henson PM, Ortiz LA, Downey GP, et al. Tumor Necrosis Factor-Alpha Accelerates the Resolution of Established Pulmonary Fibrosis in Mice by Targeting Profibrotic Lung Macrophages. Am J Respir Cell Mol Biol (2014) 4:825–37. doi: 10.1165/rcmb.2013-0386OC
26. Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, et al. Tgf-Beta Driven Lung Fibrosis Is Macrophage Dependent and Blocked by Serum Amyloid P. Int J Biochem Cell Biol (2011) 1:154–62. doi: 10.1016/j.biocel.2010.10.013
27. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage Production of Transforming Growth Factor Beta and Fibroblast Collagen Synthesis in Chronic Pulmonary Inflammation. J Exp Med (1989) 3:727–37. doi: 10.1084/jem.170.3.727
28. Sailo BL, Banik K, Girisa S, Bordoloi D, Fan L, Halim CE, et al. Fbxw7 in Cancer: What Has Been Unraveled Thus Far? Cancers (Basel) (2019) 2:246. doi: 10.3390/cancers11020246
29. Guyard A, Danel C, Theou-Anton N, Debray MP, Gibault L, Mordant P, et al. Morphologic and Molecular Study of Lung Cancers Associated With Idiopathic Pulmonary Fibrosis and Other Pulmonary Fibroses. Respir Res (2017) 1:120. doi: 10.1186/s12931-017-0605-y
30. Ekholm-Reed S, Goldberg MS, Schlossmacher MG, Reed SI. Parkin-Dependent Degradation of the F-Box Protein Fbw7beta Promotes Neuronal Survival in Response to Oxidative Stress by Stabilizing Mcl-1. Mol Cell Biol (2013) 18:3627–43. doi: 10.1128/MCB.00535-13
31. Zhao X, Hirota T, Han X, Cho H, Chong LW, Lamia K, et al. Circadian Amplitude Regulation via Fbxw7-Targeted Rev-Erbalpha Degradation. Cell (2016) 7:1644–57. doi: 10.1016/j.cell.2016.05.012
32. Hoeck JD, Jandke A, Blake SM, Nye E, Spencer-Dene B, Brandner S, et al. Fbw7 Controls Neural Stem Cell Differentiation and Progenitor Apoptosis via Notch and C-Jun. Nat Neurosci (2010) 11:1365–72. doi: 10.1038/nn.2644
33. Kearns CA, Ravanelli AM, Cooper K, Appel B. Fbxw7 Limits Myelination by Inhibiting Mtor Signaling. J Neurosci (2015) 44:14861–71. doi: 10.1523/JNEUROSCI.4968-14.2015
34. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer Mediates Effector T Cell Dysfunction by Targeting Micrornas and Ezh2 via Glycolysis Restriction. Nat Immunol (2016) 1:95–103. doi: 10.1038/ni.3313
35. Song Y, Lai L, Chong Z, He J, Zhang Y, Xue Y, et al. E3 Ligase Fbxw7 Is Critical for Rig-I Stabilization During Antiviral Responses. Nat Commun (2017) 8:14654. doi: 10.1038/ncomms14654
36. He J, Song Y, Li G, Xiao P, Liu Y, Xue Y, et al. Fbxw7 Increases Ccl2/7 in Cx3cr1hi Macrophages to Promote Intestinal Inflammation. J Clin Invest (2019) 129:3877–93. doi: 10.1172/JCI123374
37. Liu T, De Los Santos FG, Phan SH. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol Biol (2017) 1627:27–42. doi: 10.1007/978-1-4939-7113-8_2
38. Li Y, Li H, Liu S, Pan P, Su X, Tan H, et al. Pirfenidone Ameliorates Lipopolysaccharide-Induced Pulmonary Inflammation and Fibrosis by Blocking Nlrp3 Inflammasome Activation. Mol Immunol (2018) 99:134–44. doi: 10.1016/j.molimm.2018.05.003
39. Liu YY, Shi Y, Liu Y, Pan XH, Zhang KX. Telomere Shortening Activates Tgf-Beta/Smads Signaling in Lungs and Enhances Both Lipopolysaccharide and Bleomycin-Induced Pulmonary Fibrosis. Acta Pharmacol Sin (2018) 11:1735–45. doi: 10.1038/s41401-018-0007-9
40. Szapiel SV, Elson NA, Fulmer JD, Hunninghake GW, Crystal RG. Bleomycin-Induced Interstitial Pulmonary Disease in the Nude, Athymic Mouse. Am Rev Respir Dis (1979) 4:893–9. doi: 10.1164/arrd.1979.120.4.893
41. Schmittgen TD, Livak KJ. Analyzing Real-Time Pcr Data by the Comparative C(T) Method. Nat Protoc (2008) 6:1101–8. doi: 10.1038/nprot.2008.73
42. Prasse A, Binder H, Schupp JC, Kayser G, Bargagli E, Jaeger B, et al. Data From: BAL Cell Gene Expression is Predictive of Mortality in Idiopathic Pulmonary Fibrosis and Enriched for Genes of Airway Basal Cells. Gene Expression Omnibus (2018), GSE70867.
43. Huang Y, Oldham JM, Ma S, Martinez FJ, Noth I. Data From: Longitudinal Blood Transcriptomic Changes Predict Lung Function Decline in Idiopathic Pulmonary Fibrosis. Gene Expression Omnibus (2019), GSE132607.
44. Desogere P, Tapias LF, Hariri LP, Rotile NJ, Rietz TA, Probst CK, et al. Type I Collagen-Targeted Pet Probe for Pulmonary Fibrosis Detection and Staging in Preclinical Models. Sci Transl Med (2017) 384:eaaf4696. doi: 10.1126/scitranslmed.aaf4696
45. Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, et al. Collagen-Producing Lung Cell Atlas Identifies Multiple Subsets With Distinct Localization and Relevance to Fibrosis. Nat Commun (2020) 1:1920. doi: 10.1038/s41467-020-15647-5
46. Tschumperlin DJ. Matrix, Mesenchyme, and Mechanotransduction. Ann Am Thorac Soc (2015) 12:S24–9. doi: 10.1513/AnnalsATS.201407-320MG
47. Ryu C, Sun H, Gulati M, Herazo-Maya JD, Chen Y, Osafo-Addo A, et al. Extracellular Mitochondrial DNA Is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2017) 12:1571–81. doi: 10.1164/rccm.201612-2480OC
48. Dong J, Ma Q. Type 2 Immune Mechanisms in Carbon Nanotube-Induced Lung Fibrosis. Front Immunol (2018) 9:1120. doi: 10.3389/fimmu.2018.01120
49. He C, Murthy S, McCormick ML, Spitz DR, Ryan AJ, Carter AB. Mitochondrial Cu,Zn-Superoxide Dismutase Mediates Pulmonary Fibrosis by Augmenting H2o2 Generation. J Biol Chem (2011) 17:15597–607. doi: 10.1074/jbc.M110.187377
50. Su S, Zhao Q, He C, Huang D, Liu J, Chen F, et al. Mir-142-5p and Mir-130a-3p Are Regulated by Il-4 and Il-13 and Control Profibrogenic Macrophage Program. Nat Commun (2015) 6:8523. doi: 10.1038/ncomms9523
51. Zhu L, Fu X, Chen X, Han X, Dong P. M2 Macrophages Induce Emt Through the Tgf-Beta/Smad2 Signaling Pathway. Cell Biol Int (2017) 9:960–68. doi: 10.1002/cbin.10788
52. Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, et al. Early Growth Response Gene 1-Mediated Apoptosis Is Essential for Transforming Growth Factor Beta1-Induced Pulmonary Fibrosis. J Exp Med (2004) 3:377–89. doi: 10.1084/jem.20040104
53. Ghosh AK, Bhattacharyya S, Lafyatis R, Farina G, Yu J, Thimmapaya B, et al. P300 Is Elevated in Systemic Sclerosis and Its Expression Is Positively Regulated by Tgf-Beta: Epigenetic Feed-Forward Amplification of Fibrosis. J Invest Dermatol (2013) 5:1302–10. doi: 10.1038/jid.2012.479
54. Freudlsperger C, Bian Y, Contag Wise S, Burnett J, Coupar J, Yang X, et al. Tgf-Beta and Nf-Kappab Signal Pathway Cross-Talk Is Mediated Through Tak1 and Smad7 in a Subset of Head and Neck Cancers. Oncogene (2013) 12:1549–59. doi: 10.1038/onc.2012.171
55. Zhang S, Che D, Yang F, Chi C, Meng H, Shen J, et al. Tumor-Associated Macrophages Promote Tumor Metastasis via the Tgf-Beta/Sox9 Axis in Non-Small Cell Lung Cancer. Oncotarget (2017) 59:99801–15. doi: 10.18632/oncotarget.21068
56. Babaei-Jadidi R, Li N, Saadeddin A, Spencer-Dene B, Jandke A, Muhammad B, et al. Fbxw7 Influences Murine Intestinal Homeostasis and Cancer, Targeting Notch, Jun, and Dek for Degradation. J Exp Med (2011) 2:295–312. doi: 10.1084/jem.20100830
57. Yeh CH, Bellon M, Nicot C. Fbxw7: A Critical Tumor Suppressor of Human Cancers. Mol Cancer (2018) 1:115. doi: 10.1186/s12943-018-0857-2
58. Zhang C, Chen F, Feng L, Shan Q, Zheng GH, Wang YJ, et al. Fbxw7 Suppresses Hmgb1-Mediated Innate Immune Signaling to Attenuate Hepatic Inflammation and Insulin Resistance in a Mouse Model of Nonalcoholic Fatty Liver Disease. Mol Med (2019) 1:29. doi: 10.1186/s10020-019-0099-9
59. Guilliams M, Scott CL. Does Niche Competition Determine the Origin of Tissue-Resident Macrophages? Nat Rev Immunol (2017) 7:451–60. doi: 10.1038/nri.2017.42
60. Osterholzer JJ, Olszewski MA, Murdock BJ, Chen GH, Erb-Downward JR, Subbotina N, et al. Implicating Exudate Macrophages and Ly-6c(High) Monocytes in Ccr2-Dependent Lung Fibrosis Following Gene-Targeted Alveolar Injury. J Immunol (2013) 7:3447–57. doi: 10.4049/jimmunol.1200604
61. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-Derived Alveolar Macrophages Drive Lung Fibrosis and Persist in the Lung Over the Life Span. J Exp Med (2017) 8:2387–404. doi: 10.1084/jem.20162152
62. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am J Respir Cell Mol Biol (2017) 1:66–76. doi: 10.1165/rcmb.2016-0361OC
63. Li P, Zhao GQ, Chen TF, Chang JX, Wang HQ, Chen SS, et al. Serum Mir-21 and Mir-155 Expression in Idiopathic Pulmonary Fibrosis. J Asthma (2013) 9:960–4. doi: 10.3109/02770903.2013.822080
64. Cao S, Wang Y, Li J, Lv M, Niu H, Tian Y. Tumor-Suppressive Function of Long Noncoding Rna Malat1 in Glioma Cells by Suppressing Mir-155 Expression and Activating Fbxw7 Function. Am J Cancer Res (2016) 11:2561–74.
65. Kitade S, Onoyama I, Kobayashi H, Yagi H, Yoshida S, Kato M, et al. Fbxw7 Is Involved in the Acquisition of the Malignant Phenotype in Epithelial Ovarian Tumors. Cancer Sci (2016) 10:1399–405. doi: 10.1111/cas.13026
66. Wei A, Gao Q, Chen F, Zhu X, Chen X, Zhang L, et al. Inhibition of DNA Methylation Derepresses Ppargamma and Attenuates Pulmonary Fibrosis. Br J Pharmacol (2021). doi: 10.1111/bph.15655
67. Coward WR, Feghali-Bostwick CA, Jenkins G, Knox AJ, Pang L. A Central Role for G9a and Ezh2 in the Epigenetic Silencing of Cyclooxygenase-2 in Idiopathic Pulmonary Fibrosis. FASEB J (2014) 7:3183–96. doi: 10.1096/fj.13-241760
68. Coward WR, Brand OJ, Pasini A, Jenkins G, Knox AJ, Pang L. Interplay Between Ezh2 and G9a Regulates Cxcl10 Gene Repression in Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol (2018) 4:449–60. doi: 10.1165/rcmb.2017-0286OC
69. Broekelmann TJ, Limper AH, Colby TV, McDonald JA. Transforming Growth Factor Beta 1 Is Present at Sites of Extracellular Matrix Gene Expression in Human Pulmonary Fibrosis. Proc Natl Acad Sci USA (1991) 15:6642–6. doi: 10.1073/pnas.88.15.6642
70. Toossi Z, Hirsch CS, Hamilton BD, Knuth CK, Friedlander MA, Rich EA. Decreased Production of Tgf-Beta 1 by Human Alveolar Macrophages Compared With Blood Monocytes. J Immunol (1996) 9:3461–8.
71. Young LR, Gulleman PM, Short CW, Tanjore H, Sherrill T, Qi A, et al. Epithelial-Macrophage Interactions Determine Pulmonary Fibrosis Susceptibility in Hermansky-Pudlak Syndrome. JCI Insight (2016) 17:e88947. doi: 10.1172/jci.insight.88947
72. Martin-Gallausiaux C, Beguet-Crespel F, Marinelli L, Jamet A, Ledue F, Blottiere HM, et al. Butyrate Produced by Gut Commensal Bacteria Activates Tgf-Beta1 Expression Through the Transcription Factor Sp1 in Human Intestinal Epithelial Cells. Sci Rep (2018) 1:9742. doi: 10.1038/s41598-018-28048-y
73. Xiao Y, Freire-de-Lima C, Schiemann W, Bratton D, Vandivier R, Henson P, et al. Transcriptional and Translational Regulation of Tgf-Beta Production in Response to Apoptotic Cells. J Immunol (2008) 5:3575–85. doi: 10.4049/jimmunol.181.5.3575
74. Peng W, Huang S, Shen L, Tang Y, Li H, Shi Y. Long Noncoding Rna Nonhsag053901 Promotes Diabetic Nephropathy via Stimulating Egr-1/Tgf-Beta-Mediated Renal Inflammation. J Cell Physiol (2019) 10:18492–503. doi: 10.1002/jcp.28485
75. Lee KY, Ito K, Hayashi R, Jazrawi EP, Barnes PJ, Adcock IM. Nf-Kappab and Activator Protein 1 Response Elements and the Role of Histone Modifications in Il-1beta-Induced Tgf-Beta1 Gene Transcription. J Immunol (2006) 1:603–15. doi: 10.4049/jimmunol.176.1.603
76. Eferl R, Wagner EF. Ap-1: A Double-Edged Sword in Tumorigenesis. Nat Rev Cancer (2003) 11:859–68. doi: 10.1038/nrc1209
77. Wernig G, Chen SY, Cui L, Van Neste C, Tsai JM, Kambham N, et al. Unifying Mechanism for Different Fibrotic Diseases. Proc Natl Acad Sci USA (2017) 18:4757–62. doi: 10.1073/pnas.1621375114
78. Velden JL, Alcorn JF, Guala AS, Badura EC. And Janssen-Heininger Y M. C-Jun N-Terminal Kinase 1 Promotes Transforming Growth Factor-Beta1-Induced Epithelial-To-Mesenchymal Transition via Control of Linker Phosphorylation and Transcriptional Activity of Smad3. Am J Respir Cell Mol Biol (2011) 4:571–81. doi: 10.1165/rcmb.2009-0282OC
79. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial Cell Cycle Arrest in G2/M Mediates Kidney Fibrosis After Injury. Nat Med (2010) 5:535–43. doi: 10.1038/nm.2144
80. Schulien I, Hockenjos B, Schmitt-Graeff A, Perdekamp MG, Follo M, Thimme R, et al. The Transcription Factor C-Jun/Ap-1 Promotes Liver Fibrosis During Non-Alcoholic Steatohepatitis by Regulating Osteopontin Expression. Cell Death Differ (2019) 9:1688–99. doi: 10.1038/s41418-018-0239-8
81. Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The Ubiquitin Ligase Scffbw7 Antagonizes Apoptotic Jnk Signaling. Science (2004) 5662:1374–8. doi: 10.1126/science.1092880
82. Kravchick DO, Karpova A, Hrdinka M, Lopez-Rojas J, Iacobas S, Carbonell AU, et al. Synaptonuclear Messenger Prr7 Inhibits C-Jun Ubiquitination and Regulates Nmda-Mediated Excitotoxicity. EMBO J (2016) 17:1923–34. doi: 10.15252/embj.201593070
Keywords: IPF – idiopathic pulmonary fibrosis, TGF-β, Fbxw7, macrophage, c-Jun
Citation: He J, Du Y, Li G, Xiao P, Sun X, Song W, Lai L, Xia M, Zhang J and Wang Q (2022) Myeloid Fbxw7 Prevents Pulmonary Fibrosis by Suppressing TGF-β Production. Front. Immunol. 12:760138. doi: 10.3389/fimmu.2021.760138
Received: 17 August 2021; Accepted: 06 December 2021;
Published: 05 January 2022.
Edited by:
Haichao Wang, Feinstein Institute for Medical Research, United StatesReviewed by:
Cheng Yang, Nankai University, ChinaYi Sun, Zhejiang University, China
David Wu, University of Chicago, United States
Copyright © 2022 He, Du, Li, Xiao, Sun, Song, Lai, Xia, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingqing Wang, d3FxQHpqdS5lZHUuY24=; Jianhua Zhang, c3h6amgxMDE4QDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship