
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 24 September 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.757804
This article is part of the Research Topic Cancer Prevention: Targeting Premalignant Epithelial Neoplasms in the Era of Cancer Immunotherapy and Vaccines View all 10 articles
 Vladimir Roudko1,2,3
Vladimir Roudko1,2,3 Cansu Cimen Bozkus3,4
Cansu Cimen Bozkus3,4 Benjamin Greenbaum5,6
Benjamin Greenbaum5,6 Aimee Lucas7Robert Samstein2,3,8
Aimee Lucas7Robert Samstein2,3,8 Nina Bhardwaj3,4*
Nina Bhardwaj3,4*Defective DNA mismatch repair (dMMR) is associated with many cancer types including colon, gastric, endometrial, ovarian, hepatobiliary tract, urinary tract, brain and skin cancers. Lynch syndrome – a hereditary cause of dMMR – confers increased lifetime risk of malignancy in different organs and tissues. These Lynch syndrome pathogenic alleles are widely present in humans at a 1:320 population frequency of a single allele and associated with an up to 80% risk of developing microsatellite unstable cancer (microsatellite instability – high, or MSI-H). Advanced MSI-H tumors can be effectively treated with checkpoint inhibitors (CPI), however, that has led to response rates of only 30-60% despite their high tumor mutational burden and favorable immune gene signatures in the tumor microenvironment (TME). We and others have characterized a subset of MSI-H associated highly recurrent frameshift mutations that yield shared immunogenic neoantigens. These frameshifts might serve as targets for off-the-shelf cancer vaccine designs. In this review we discuss the current state of research around MSI-H cancer vaccine development, its application to MSI-H and Lynch syndrome cancer patients and the utility of MSI-H as a biomarker for CPI therapy. We also summarize the tumor intrinsic mechanisms underlying the high occurrence rates of certain frameshifts in MSI-H. Finally, we provide an overview of pivotal clinical trials investigating MSI-H as a biomarker for CPI therapy and MSI-H vaccines. Overall, this review aims to inform the development of novel research paradigms and therapeutics.
In the United States, an individual’s lifetime risk for developing cancer is estimated to be as high as 40% (www.cancer.org, “Lifetime Risk of Developing or Dying From Cancer”). Cancer is a genetic disorder in which somatic mutations in specific genes confer a selective growth advantage for tumors. Such mutations can be inherited through the germline, which results in a hereditary predisposition to an early-onset cancer, or can occur sporadically in non-germline carriers. One example is the hereditary nonpolyposis colorectal cancer syndrome known as Lynch syndrome. Defining the genetic causes for Lynch syndrome has uncovered a connection between the cellular machinery that regulates DNA repair, cancer formation and informed clinical approaches to manage the disease.
Lynch syndrome or hereditary non-polyposis colorectal cancer (HNPCC) is characterized by germline inactivation of one allele of genes involved in the mismatch repair system, namely MLH1, MSH2, MSH6, PMS2 or EPCAM and have received prominent clinical and research attention and since 1985 (1, 2). More than 1500 variants of Lynch syndrome alleles have been identified including: retrotransposition and Alu-like element insertion events (3–5), splice site mutations and large exonic deletions (6–8). However, inactivation of MLH1 or PMS2 alleles are the most frequent ones and are associated with approximately 80% of Lynch syndrome cases (9, 10). Lynch-associated genetic abnormalities frequently lead to cancer at ages of 30–40-years and in a broad range of tissues (Figure 1A), including: colon, stomach, brain, pancreas, small intestine, endometrial and urothelial tracts (12–18).
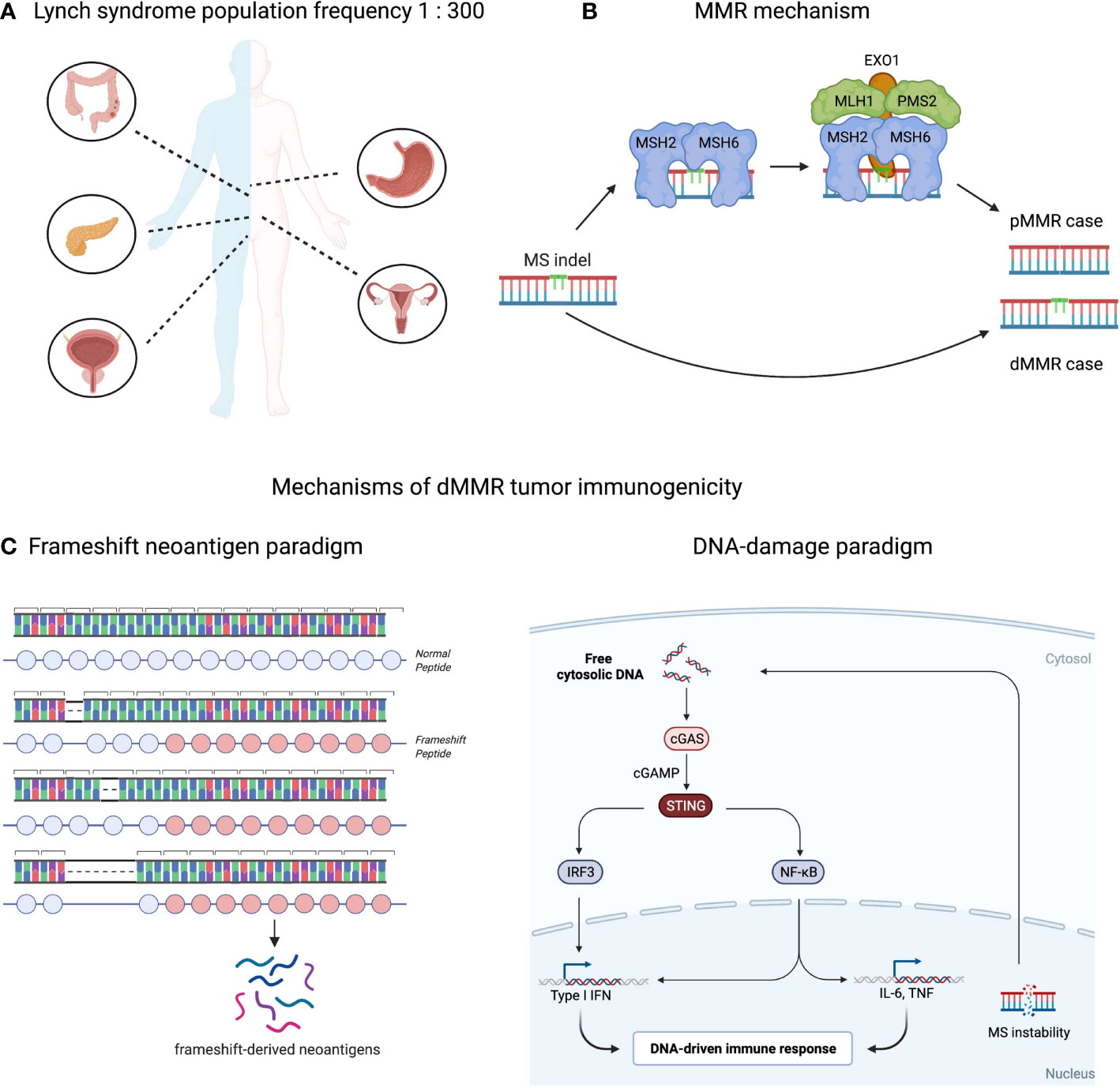
Figure 1 Population, molecular and immunological aspects of mismatch-repair deficient (dMMR) tumors. (A) Predispositions to different cancers conferred by Lynch syndrome condition. Approximately 1 in 300 people in the U.S. has the Lynch Syndrome associated alleles. Carriers have 80% lifetime risk developing cancer including: colorectal, stomach, pancreas, urinary track and prostate for males and urinary, ovary or uterus tracks for females. In total, Lynch syndrome accounts for 2-3% solid tumor cases (11). (B) Mismatch repair (MMR) mechanism. MS indels occurring during DNA replication are repaired by MMR system (proficient MMR). MSH2-MSH3 or MSH2-MSH6 complexes, called MutSα or MutSβ, detect the error and recruit the MLH1-PMS1, MLH1-PMS2 or MLH1-MLH3 (MutLα/β/γ complexes respectively) to bind to the DNA and bring DNA exonuclease with PCNA to the mutation site. The mismatch is then excised and repaired following by DNA resynthesis and re-ligation. These aberrations are left unrepaired in case of MMR deficiency. (C) Two complementary paradigms explaining immune responses in dMMR tumors: neoantigen-driven (left part), and innate immune driven (right part).
Colorectal, stomach and endometrial cancers cumulatively account for the second most common cancer types and one of the leading causes of cancer deaths in developed countries (19). The majority of the solid tumor cases are proficient in mismatch repair (pMMR), however an estimated 14% (95% CI: 10%-19%) of cases are dMMR (20–23). These dMMR cases arise mainly from sporadic, tumor-specific inactivation of MMR pathway (24, 25) however a few cases – 2-3% of all colorectal and endometrial cancer cases (11) – have germline Lynch syndrome alleles as the ones described above (26). In the latter case, cancer onset transpires upon genetic inactivation of the second allele (in the case of the PMS2 allele) or epigenetic silencing of gene expression (in the case of the MLH1 allele) and subsequent acquisition of driver mutations in genes such as APC, KRAS, PI3K, PTEN, BRAF and/or p53 (13, 27–32). Though pMMR and dMMR share similar profiles of tumor drivers, their genomic makeups are different. One of the molecular feature of dMMR tumors is high tumor mutation burden over-represented by somatic indel mutations within short tandem repeats – microsatellites – a molecular signature termed as a high microsatellite instability, or MSI-H (27, 33–35).
Microsatellite extension or shortening in MSI-H tumors happens upon breaking down of the MMR molecular mechanism that controls microsatellite loci length (18, 36). During replication DNA polymerases incorporate deoxyribonucleotides into the growing chain of DNA using one of the paternal DNA strands as a template, thus copying genetic information with high fidelity. However, at microsatellite repeats DNA polymerases can slip from the template upon replication resulting in insertion or deletion of microsatellite units that structurally resembles a “bulge” of non-complemented DNA nucleotides within the parent/daughter DNA double strand helix. The MMR system guards against this type of mutagenesis by detecting and eliminating these bulges. First, MSH2-MSH3 or MSH2-MSH6 complexes, called MutSα or MutSβ, detect the error and recruit the MLH1-PMS1, MLH1-PMS2 or MLH1-MLH3 (MutLα/β/γ complexes respectively) to bind to the DNA and bring DNA exonuclease with PCNA to the mutation site (Figure 1B). The mismatch is then excised and repaired following by DNA resynthesis and re-ligation (33, 37). Since MSI-H cancers are defective in one of the MMR factors, mismatches in malignant cells remain unrepaired and accumulate indel mutations at high rates (18). Depending on where the microsatellite (MS) loci is located in the human genome, the effect of indel mutations differ: if it occurs within a non-coding segment of the genome, the indel mutation may have limited-to-no effect on overall gene expression or function; if it happens in a regulatory or splicing-required segment, the indel might affect linked gene expression; if it occurs within a protein-coding region the indel may result in expression of a truncated protein with novel peptide extension at the C-terminus (38). These cancer-specific peptide extensions called frameshifts can be exploited clinically in a variety of immunotherapy strategies which will be reviewed later.
Multiple biomarker strategies have been developed to detect either Lynch syndrome in the germline or assess penetration of MSI-H phenotype in tumors. Amsterdam II criteria and the Bethesda criteria are two clinically approved methods to diagnose Lynch syndrome in patients, and include a range of molecular tests with familial analysis of disease allele penetration (39). Several protocols exist to characterize MSI-H tumors: immunohistochemistry assay to quantify loss of MMR proteins such as MLH1, MSH2, PMS2 and MSH6, genomic PCR tests to measure MS length variability at the various MS loci and quantification of somatic MS variation from WES/WGS data acquired from matched tumor/normal DNA samples using next generation sequencing and computational tools to calculate standardized MSI-H metrics like the “MSI score” (40–45). The strategies vary widely in terms of recall, sensitivity and specificity; thus, combining multiple protocols may increase the precision of the overall diagnosis (39, 46–48).
The standard of care for MSI-H cancer patients has changed from primarily chemotherapy to include immune checkpoint inhibitors. Approved for pMMR colorectal tumors, standard of care 5-fluorouracil (5-FU)-based regiments turned out to be ineffective in dMMR cases due to the lack of enzymes recognizing DNA damage inflicted by the drug (45, 49). Moreover, dMMR is turned out to be a negative prognostic biomarker for the objective response to 5-FU therapy and overall survival (49). Despite a setback in targeted therapy modalities for dMMR cancers, synthetic lethal interactions hold promise to improve the outcomes. dMMR tumors represent a good example of a system, where simultaneous co-inhibition of two factors may lead to cancer cell apoptosis and death. It has been found, that inhibition of Werner helicase is synthetically lethal with inactive MSH2 or PMS1 – the causative mutations of dMMR cancers. Mechanistically, the helicase is critical to unwind aberrantly replicated DNA and to maintain genome integrity of MSI-H tumors (50, 51). Pharmacological targeting of Werner helicase in dMMR cases still awaits its clinical application.
High tumor mutation burden (TMB) and comparatively strong immune cell infiltration of dMMR tumors sparked interest in applying checkpoint inhibitor (CPI) and other immunotherapy approaches (38, 52–54). Many studies confirmed a strong correlation between objective response rates to CPI and TMB in multiple cancers (55–58). A recent stage II multicohort clinical trial KEYNOTE-158 showed that TMB score is predictive for overall and progression free survival in the adjuvant CPI setting (59). Similarly, a range of clinical studies led by Dr. Diaz Jr. confirmed the strong association between responses to CPI and dMMR tumor status in a tissue-agnostic fashion (60, 61). Further investigation suggested dMMR status or MSI-H score to being predictive to CPI responses in solid tumors (62–64). These results led to the fast-track approval of dMMR as a biomarker for CPI by FDA and initiation of prospective stage II/III clinical trials, e.g., NCT02563002 or NCT04008030, to evaluate dMMR as a predictive biomarker. An interesting observation has arisen suggesting dMMR might be a confounding parameter for TMB biomarker in colorectal cancer patients treated with CPI: dMMR patients, which are often TMB-high due to intrinsic mechanisms of efficient somatic mutagenesis, clinically do better than “TMB-high but pMMR” patients (65). This clinical observation suggests the importance of the biological mechanism generating high somatic mutation load in tumor responsiveness to CPI. For instance, several hypotheses have been suggested to explain high CPI response rates in dMMR mechanistically. One of them is the “frameshift neoantigen” paradigm. Neoantigens are cancer-specific peptides that are usually derived from somatic mutations and presented on MHC-I or MHC-II complexes (66). dMMR tumors are enriched in frameshifts and neoantigens derived from these peptide extensions might confer tumor immunogenicity (67–70). Theoretically, one frameshift mutation may encode a peptide yielding multiple neoantigens with broad MHC-I/II specificities, thereby increasing the probability of (a) epitope presentation, (b) cancer antigen sampling by dendritic cells, (c) T cell recognition and (d) T cell-mediated tumor killing. We and others have shown exceptional immune responses against these frameshift peptides expressed in dMMR tumors, thus supporting the “frameshift neoantigen” paradigm (71–73). Another hypothesis is built around innate immune signaling driven by DNA instability. The aberrant DNA fragments are spilled over from the nucleus during DNA replication and recognized by cGAS-STING as “non-self” inducing Type I interferon and inflammatory NFkB responses (74, 75). In agreement with this hypothesis, recent study by Mowat et al. showed that cGAS-STING driven type I interferon signaling is required for CXCL10/CCL5-dependent T cell recruitment to dMMR tumor site (76). Both “frameshift-neoantigen” and “DNA instability” models are instructive and complementary in explaining the origins of the dMMR tumor immunogenicity (Figure 1C).
dMMR immunotherapies also include the implementation of cancer vaccine strategies in therapeutic and prospective settings (77). Several reasons suggest that such strategies might become successful. First, many dMMR associated somatic mutations are non-random across the genome. Known tumor drivers such as mutations in APC, TP53 or BRAF genes are clonal, positively selected by the developing tumor and overrepresented within dMMR patient samples. Similarly, certain frameshift mutations are found to be under positive selection despite encoding immunogenic neoantigens (78, 79). From a tumor evolution perspective, such “cost” of an immunogenic frameshift can be accepted only if loss-of-function mutation confers a tumor growth advantage. Adaptive resistance against emerged neoantigens can be acquired later through general immune suppression mechanisms like PDL-1 upregulation, infiltration of the TME by myeloid-derived suppressor cells or T regulatory cells, TGF-β expression or other genomically encoded immune-escape mechanisms (80). However, the positively selected frameshifts can be harnessed clinically as shared cancer vaccines (81, 82). Several targets including RNF43fs, TGFBR2fs, ASTE1fs, AIM2fs, have been extensively validated in numerous immunological assays: priming and boosting naïve T cell populations in healthy donors, confirming cytotoxic capacity of CD8+ T cell responses in tumor killing assays and detecting frameshift-specific memory responses within blood and tumor T cell compartments in dMMR cancer patients and Lynch syndrome populations (71, 83–85). The latter is particularly interesting because it suggests that immunological responses against frameshifts are observed in the absence of detectable cancer. In line with these results, a study lead by Dr. Kloor has documented dMMR mutations in crypts and polyps of Lynch syndrome patients which are not cancerous (86). These findings confirm that cancer development and loss of the mismatch repair system are two independent genomic events decoupled in time and may follow each other in any order (32). It also indicates that MS instability can happen early and progress without being recognized by the host immune system, likely because the majority of MS loci – targets of MS instability – are non-coding and spread over the human genome in a random fashion. Alternatively, instability of a protein coding MS locus does not necessarily lead to tumor transformation and might reflect a frequent, hotspot somatic passenger mutation such as those occurring in a healthy dividing cells (87, 88). It might take several cellular divisions and multiple MS instability events to develop a genetic background with an invasive tumor phenotype. Although, complete removal of dMMR cell populations might not be achieved due to the immune “invisibility” of such MS unstable clones, prospective vaccination against frequently observed and/or early detected frameshifts in dMMR cancer or dMMR Lynch syndrome patients is likely to increase the efficiency of immune surveillance of pro-tumorigenic cell populations and potentially delay tumor progression in the at-risk populations. Multiple vaccine formulations including different combinations of shared dMMR-associated frameshifts are currently undergoing safety and immunogenicity tests in clinical trials involving dMMR cancer and Lynch syndrome patients with established tumors (Table 1) (89, 90). It will be exciting to evaluate the clinical outcomes and cancer protection of Lynch syndrome patients in the prospective vaccine setting, similarly to the recently published antigen-agnostic immunomodulatory strategy involving naproxen – inhibitor of prostaglandin signaling (54). Recently published a proof-of-concept study confirmed therapeutic efficiency of a shared frameshift vaccine to delay tumor progression in mouse models of Lynch syndrome (91).
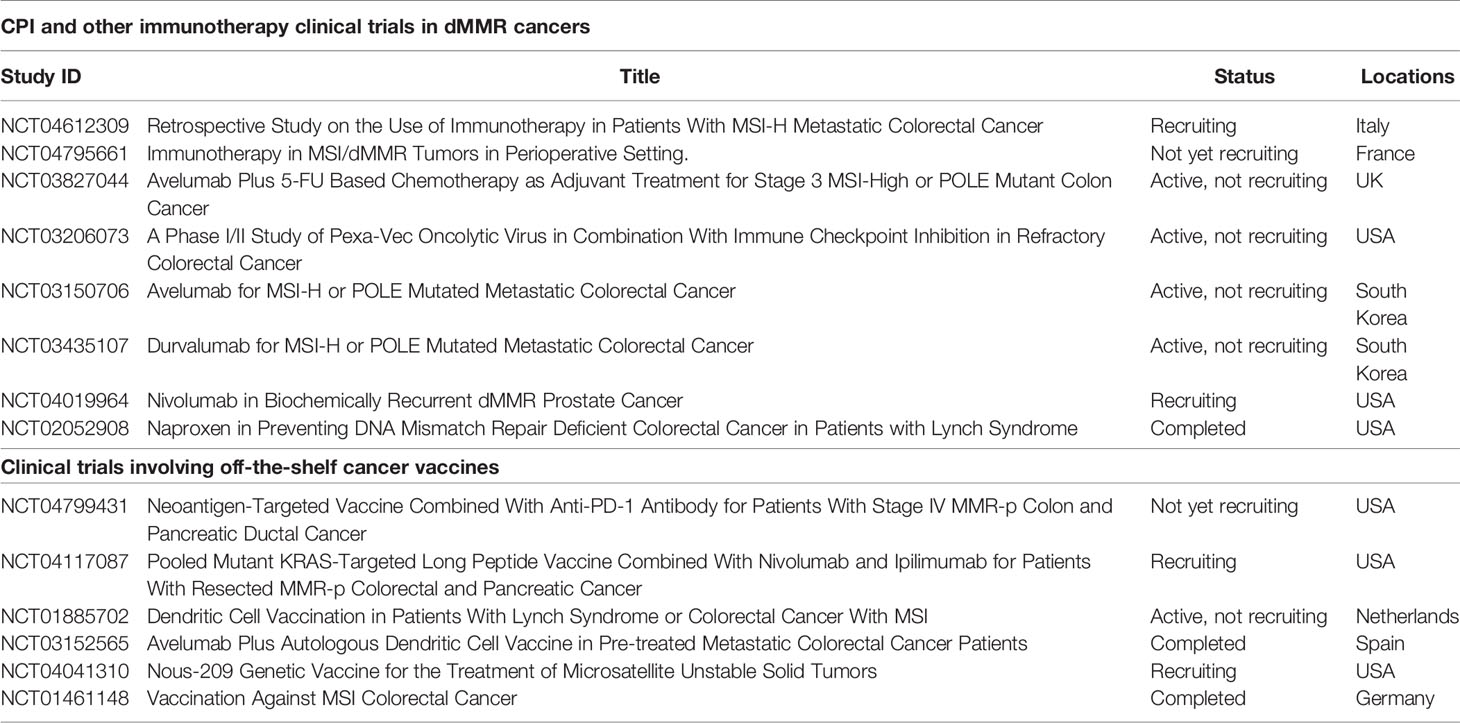
Table 1 List of registered clinical trials of cancer vaccines and/or CPI in dMMR/Lynch syndrome patients.
Several considerations have to be taken during the development and application of a shared dMMR vaccine. General factors, including the platform selection (DNA, RNA, peptide), adjuvant, routes of administration and etc. – have been extensively reviewed elsewhere (92), but here we will discuss tumor intrinsic and potential acquired resistance mechanisms (93). As it has been mentioned above, dMMR tumors have enormous potential to develop somatic mutations through loss of DNA replication fidelity. dMMR tumors can be perceived as a “mutator” machine: cell population with an intrinsic mechanism to sample many different genotypes in a very rapid manner. Administration of external pressure such as through the vaccine-mediated expansion of tumor-specific T cell clones, may promote the development of tumor resistance against the host immune system. Several studies have reported up to 30% frequency of loss-of-function mutations in β-2-microglobulin (B2M) – a gene required for MHC-I antigen presentation and processing; up to 70% frequency of mutations in TGFBR2 – cytokine receptor, rendering tumors non-responsive to TGF-β mediated suppression; up to 80% cumulative frequency of mutations in other genes related to innate and adaptive immune signaling pathways, namely IFN-γ response (JAK1, JAK2) and inflammasome activation (CASP5, AIM2). Other genomic mechanisms include loss of heterozygosity in HLA-I loci which drives tumor escape from CD8+ T cell cytotoxicity (94). Additionally, nonsense-mediated decay of the frameshifted messenger RNA can decrease immunogenicity against frameshift-derived neoantigens due to altered stability of the mutated RNA (95, 96). Non-genetic mechanisms of acquired resistance also can be found in the TME of the dMMR tumors, including: increased Wnt/β-catenin signaling in tumor associated fibroblasts; increased infiltration of Foxp3+ T regs; upregulated expression of immune checkpoints PDL-1 and CTLA-4; as well as CD47 “don’t eat me” signals for macrophages and dendritic cells (93, 97, 98). Interestingly, a retrospective analysis of B2M expression and mutation status in colorectal dMMR cancer patients showed favorable clinical outcomes in patient cohorts despite B2M loss-of-function mutations, counterintuitive to the mechanisms of MHC-I dependance of immune-mediated tumor rejection (99, 100). A recent study by Germano et al. addressed this question and found CD4+ T cells being responsible for tumor rejection and the development of strong immune responses in B2M-null dMMR tumors (101). These and many other disparities between assumed inhibitory mechanisms and clinical outcomes in patients will inform many other mechanisms of therapy response and resistance which might exist in the dMMR cancer setting.
Understanding the trajectories of dMMR tumor genome evolution at the single cell level with and without applied immune pressure will help to describe the landscape of acquired and intrinsic tumor resistance. Knowledge of these mechanisms might inform additional interventions important to include in the shared vaccine formulations such as MHC-II epitopes and/or NK cell engagers or myeloid cell modulation (38). CPI clinical trials conducted in dMMR patients can provide useful insights to address these questions. Discussed previously disparities between observed genomic alterations and immunotherapy clinical outcomes may inform novel mechanisms of immune resistance and response in dMMR tumors. Exploratory genetic and expression analysis of non-responder dMMR patient tumor samples from large-scale phase III CPI clinical trials will be highly informative to address these questions (60). Similarly, large scale sequencing and imaging data mining will be crucial to understand the mechanisms of dMMR tumor and immune cell dynamics. The majority of the detected frameshifts and MS loci indels are subclonal with relatively stable chromosomal copy number. If one specific mutation provides an immune resistance and doing so – growth advantage – why does not it become clonal during tumor evolution? A potential explanation is the uneven spatial tumor clone distribution and cooperativity between different tumor clones which might provide tumor benefit (102). Indeed, one can imagine the tumor surface lined up by clones governing immune resistance protecting other clones growing in the tumor core from infiltrating immune cells. In this “mutual dependency” scenario subclonal protective mutation will be sufficient to gain tumor growth advantage as a whole tumor cell community. Thus, spatially-resolved genomic studies combined with single-cell studies will be extremely informative to gain insight on spatial biomarkers associated with resistance and response to CPI and improve cancer vaccines designs by informing the inclusion of as many frameshifts derived from different tumor clones as possible (103).
In conclusion, we highlight several questions which remain important to address regarding treatment and prevention of MSI tumor in the near future. How many novel MS indels appear per each genome replication in dMMR lesions and/or dMMR Lynch syndrome crypts? What is the probability of acquiring a frameshift expressed at the protein level? Can sequence-based motifs predict the earliest frameshift to appear during dMMR development? Computational modelling leveraging whole genome MSI-H samples will be informative to answer these questions. Which frameshifts generate the most frequent immune responses in vitro and in dMMR cancer/Lynch syndrome patients? Which frameshift combination confers the best protective and cytotoxic potential in different cellular models of dMMR cancer progression? Extensive immunological studies will be very informative to address these points. Finally, characterizing and quantifying tumor intrinsic and acquired mechanisms of resistance from either clinical CPI trials or tumor model studies will be important to find alternative ways of improving therapeutic responses in patients’ populations. Overall, immunotherapeutic development to treat or protect against dMMR tumorigenesis experiences a new spiral of fruitful and exciting research.
VR wrote the manuscript. VR, CC, BG, AL, RS, and NB revised the manuscript. All authors contributed to the article and approved the submitted version.
Support is provided by seed fund from Tisch Cancer Institute at Mount Sinai Hospital.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lynch H, Schuelke G, Kimberling W, Albano W, Lynch J, Biscone K, et al. Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndromes I and II). Biomarker Studies. Cancer (1985) 56:939–51. doi: 10.1002/1097-0142(19850815)56:4<939::AID-CNCR2820560440>3.0.CO;2-T
2. Lynch HT, Kimberling W, Albano W, Lynch JF, Schuelke G, Sandberg AA, et al. Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndromes I and II). Clinical Description of Resource. Cancer (1985) 56:934–8. doi: 10.1002/1097-0142(19850815)56:4<934::AID-CNCR2820560439>3.0.CO;2-I
3. Yamamoto G, Miyabe I, Tanaka K, Kakuta M, Watanabe M. SVA Retrotransposon Insertion in Exon of MMR Genes Results in Aberrant RNA Splicing and Causes Lynch Syndrome. Eur J Hum Genet (2021) 1:680–6. doi: 10.1038/s41431-020-00779-5
4. Salo-mullen YLE, Zhang L. Insertion of an Alu-Like Element in MLH1 Intron 7 as a Novel Cause of Lynch Syndrome. Mol Genet Genomics Med (2020) 8(15):1–7. doi: 10.1002/mgg3.1523
5. Yang C, Li Y, Trottier M, Farrell MP, David EES, Zsofia JG, et al. Insertion of an SVA Element in MSH2 as a Novel Cause of Lynch Syndrome. Genes Chromosomes Cancer (2021) 60(8):1–6. doi: 10.1002/gcc.22950
6. Cui S, Zhang X, Zou R, Ye F, Wang Y, Sun J. MLH1 Exon 12 Gene Deletion Leading to Lynch Syndrome: A Case Report. Oncol Res Treat (2021) 44:414–20. doi: 10.1159/000516659
7. Stella A, Wagner A, Shito K, Lipkin SM, Watson P, Guanti G, et al. A Nonsense Mutation in MLH1 Causes Exon Skipping in Three Unrelated HNPCC Families. Cancer Res (2001) 61:7020–4.
8. Yang C, Sheehan M, Borras E, Cadoo K, Offit K, Zhang L. Characterization of a Germline Splice Site Variant MLH1 C. 678 − 3T > A in a Lynch Syndrome Family. Familial Cancer (2020) 19(4):315–22. doi: 10.1007/s10689-020-00180-7
9. Dong L, Zou S, Jin X, Lu H, Zhang Y, Guo L, et al. Cytoplasmic MSH2 Related to Genomic Deletions in the MSH2/EPCAM Genes in Colorectal Cancer Patients With Suspected Lynch Syndrome. Front Oncol (2021) 11:627460. doi: 10.3389/fonc.2021.627460
10. Chung DC, Rustgi AK. The Hereditary Nonpolyposis Colorectal Cancer Syndrome: Genetics and Clinical Implications. Ann Internal Med (2019) 138(7):560–70. doi: 10.7326/0003-4819-138-7-200304010-00012
11. Boland P, Yurgelun M, Boland R. Recent Progress in Lynch Syndrome and Other Familial Colorectal Cancer Syndromes. CA: A Cancer J Clin Cancer J (2019) 68(3):217–31. doi: 10.3322/caac.21448.Recent
12. Lindner AK, Schachtner G, Tulchiner G, Thurnher M, Untergasser G, Obrist P, et al. Lynch Syndrome: Its Impact on Urothelial Carcinoma. Int J Mol Sci (2021) 22:531. doi: 10.3390/ijms22020531
13. Power DG, Gloglowski E, Lipkin SM. Clinical Genetics of Hereditary Colorectal Cancer. Hematol Oncol Clin (2010) 24:837–59. doi: 10.1016/j.hoc.2010.06.006
14. Carethers JM, Stoffel EM, Carethers JM, Stoffel EM, Gastroenterology D, States U. Advances in Colorectal Cancer Lynch Syndrome and Lynch Syndrome Mimics: The Growing Complex Landscape of Hereditary Colon Cancer. World J Gastroenterol (2015) 21(31):9253–61. doi: 10.3748/wjg.v21.i31.9253
15. Ryan NAJ, Blake D, Cabrera-dandy M, Glaire MA, Evans DG, Crosbie EJ. The Prevalence of Lynch Syndrome in Women With Endometrial Cancer: A Systematic Review Protocol. Systematic Rev (2018) 121:1–6. doi: 10.1186/s13643-018-0792-8
16. Evrard C, Alexandre J. Predictive and Prognostic Value of Microsatellite Instability in Gynecologic Cancer (Endometrial and Ovarian). Cancers (2021) 13:(10)1–15. doi: 10.3390/cancers13102434
17. Sekine M, Enomoto T. Precision Medicine for Hereditary Tumors in Gynecologic Malignancies. J Obstetrics Gynaecol Res (2021) 47:2597–606. doi: 10.1111/jog.14861
18. Salem ME, Bodor JN, Puccini A, Xiu J, Goldberg RM, Grothey A, et al. Relationship Between MLH1, PMS2, MSH2 and MSH6 Gene-Specific Alterations and Tumor Mutational Burden in 1057 Microsatellite Instability-High Solid Tumors. Int J Cancer (2020) 147:2948–56. doi: 10.1002/ijc.33115
19. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. A Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
20. Kim TM, Laird PW, Park PJ. The Landscape of Microsatellite Instability in Colorectal and Endometrial Cancer Genomes. Cell (2013) 58(15):3455–60. doi: 10.1016/j.cell.2013.10.015
21. Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, et al. Proteogenomic Characterization of Human Colon and Rectal Cancer. Nature (2014) 513(7518):382–7. doi: 10.1038/nature13438
22. Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L. Clinical Proteomic Tumor Analysis Consortium. Proteogenomic Characterization of Endometrial Carcinoma. Cell (2020) 180(4):729–48. doi: 10.1016/j.cell.2020.01.026
23. Lorenzi M, Amonkar M, Zhang J, Mehta S, Liaw K. Epidemiology of Microsatellite Instability High (MSI-H) and Deficient Mismatch Repair (dMMR) in Solid Tumors: A Structured Literature Review. Hindawi J Oncol (2020) 2020:1807929. doi: 10.1155/2020/1807929
24. Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ. A Molecular Portrait of Microsatellite Instability Across Multiple Cancers. Nat Commun (2017) 8:1–12. doi: 10.1038/ncomms15180
25. Cunningham JM, Christensen ER, Tester DJ, Burgart LJ, Thibodeau SN. Hypermethylation of the hMLHl Promoter in Colon Cancer With Microsatellite Instability. Cancer Res (1998) 58:3455–60.
26. Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol (2021) 37(4):286–95. doi: 10.1200/JCO.18.00283
27. Veigl M, Kastury L, Olechnowicz J, Ma A, Markowitz S. Biallelic Inactivation of hMLH 1 by Epigenetic Gene Silencing, a Novel Mechanism Causing Human MSI Cancers. PNAS (1998) 95:8698–702. doi: 10.1073/pnas.95.15.8698
28. Ellenson LH. Hmlh1 Promoter Hypermethylation in Microsatellite Instability-Positive Endometrial Carcinoma. Am J Pathol (1999) 155(5):1399–402. doi: 10.1016/S0002-9440(10)65451-X
29. Cunningham JM, Christensen ER, Tester DJ, Burgart LJ, Thibodeau SN, Roche PC. Hypermethylation of the hMLHl Promoter in Colon Cancer With Microsatellite Instability. Cancer Res (1998) 58(15):3455–60.
30. Marchiò C, De Filippo MR, Ng CKY, Piscuoglio S, Soslow RA, Reis-Filho JS, et al. PIKing the Type and Pattern of PI3K Pathway Mutations in Endometrioid Endometrial Carcinomas. Gynecol Oncol (2015) 137(2):321–8. doi: 10.1016/j.ygyno.2015.02.010
31. Getz G, Gabriel SB, Cibulskis K, Lander E, Sivachenko A, Sougnez C, et al. Integrated Genomic Characterization of Endometrial Carcinoma. Nature (2013) 497(7447):67–73. doi: 10.1038/nature12113
32. Ahadova A, Gallon R, Gebert J, Ballhausen A, Endris V, Kirchner M. Three Molecular Pathways Model Colorectal Carcinogenesis in Lynch Syndrome. Int J Cancer (2018) 143:139–50. doi: 10.1002/ijc.31300
33. Sinicrope FA, Sargent DJ. Molecular Pathways: Microsatellite Instability in Colorectal Cancer: Prognostic, Predictive, and Therapeutic Implications. Clin Cancer Res (2012) 18(6):1506–12. doi: 10.1158/1078-0432.CCR-11-1469
34. Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell (2019) 177(4):1035–1049.e19. doi: 10.1016/j.cell.2019.03.030
35. Arzimanoglou II, Lallas T, Osborne M, Barber H, Gilbert F. Microsatellite Instability Differences Between Familial and Sporadic Ovarian Cancers. Carcinogenesis (1996) 17(9):1799–804. doi: 10.1093/carcin/17.9.1799
36. Lipkin SM, Wang V, Jacoby R, Banerjee-basu S, Baxevanis AD, Lynch HT, et al. MLH3: A DNA Mismatch Repair Gene Associated With Mammalian Microsatellite Instability. Nat Genet (2000) 24:27–35. doi: 10.1038/71643
37. Zhao P, Li L, Jiang X, Li Q. Mismatch Repair Deficiency/Microsatellite Instability-High as a Predictor for Anti-PD-1/PD-L1 Immunotherapy Efficacy. J Hematol Oncol (2019) 12(1):1–14. doi: 10.1186/s13045-019-0738-1
38. Kloor M, Von Knebel Doeberitz M. The Immune Biology of Microsatellite-Unstable Cancer. Trends Cancer (2016) 2(3):121–33. doi: 10.1016/j.trecan.2016.02.004
39. Cohen SA, Pritchard CC, Jarvik GP. Lynch Syndrome: From Screening to Diagnosis to Treatment in the Era of Modern Molecular Oncology. Annu Revew Genomics Hum Genet (2019) 20(4):1–15. doi: 10.1146/annurev-genom-083118-015406
40. Delhomme N, Mähler N, Schiffthaler B, Sundell D. Guidelines for RNA-Seq Data Analysis. Epigenesys Protocol (2014), 1–24.
41. Hempelmann JA, Lockwood CM, Konnick EQ, Schweizer MT, Antonarakis ES, Lotan TL, et al. Microsatellite Instability in Prostate Cancer by PCR or Next-Generation Sequencing. J ImmunoTher Cancer (2018) 6(29):1–7. doi: 10.1186/s40425-018-0341-y
42. Yamaguchi K, Kasajima R, Takane K, Hatakeyama S, Shimizu E. Application of Targeted Nanopore Sequencing for the Screening and Determination of Structural Variants in Patients With Lynch Syndrome. J Hum Genet (2021). doi: 10.1038/s10038-021-00927-9
43. Ratovomanana T, Cohen R, Svrcek M, Renaud F, Siret A, Letourneur Q, et al. Performance of Next Generation Sequencing for the Detection of Microsatellite Instability in Colorectal Cancer With Deficient DNA Mismatch Repair. Gastroenterology (2021) 161(3):814–26. doi: 10.1053/j.gastro.2021.05.007
44. Niu B, Ye K, Zhang Q, Lu C, Xie M, Mclellan MD, et al. MSIsensor: Microsatellite Instability Detection Using Paired Tumor-Normal Sequence Data. Bioinformatics (2014) 30(7):1015–6. doi: 10.1093/bioinformatics/btt755
45. Diao Z, Han Y, Chen Y, Zhang R, Li J. The Clinical Utility of Microsatellite Instability in Colorectal Cancer. Crit Rev Oncol/Hematol (2021) 157:103171. doi: 10.1016/j.critrevonc.2020.103171
46. Kurnit KC. Microsatellite Instability in Endometrial Cancer: New Purpose for an Old Test. Cancer (2019) 125(13):1–10. doi: 10.1002/cncr.32058
47. Lu Y, Soong TD, Elemento O. A Novel Approach for Characterizing Microsatellite Instability in Cancer Cells. PloS One (2013) 8(5):1–10. doi: 10.1371/journal.pone.0063056
48. Acosta M--, Marín F, Puliafito B, Bonifaci N, Fernández A, Navarro M, et al. High-Sensitivity Microsatellite Instability Assessment for the Detection of Mismatch Repair Defects in Normal Tissue of Biallelic Germline Mismatch Repair Mutation Carriers. J Med Genet (2020) 57(4):269–73. doi: 10.1136/jmedgenet-2019-106272
49. Hewish M, Lord CJ, Martin SA, Cunningham D, Ashworth A. Mismatch Repair Deficient Colorectal Cancer in the Era of Personalized Treatment. Nat Rev Clin Oncol (2010) 7(4):197–208. doi: 10.1038/nrclinonc.2010.18
50. Picco G, Cattaneo C, van Vliet E, Crisafulli G, Rospo G, Consonni S, et al. Werner Helicase Is a Synthetic-Lethal Vulnerability in Mismatch Repair – Deficient Colorectal Cancer Refractory to Targeted Therapies, Chemotherapy and Immunotherapy. Cancer Discov (2021) 11(8):1923–37. doi: 10.1158/2159-8290.CD-20-1508
51. Chan EM, Shibue T, Mcfarland JM, Gaeta B, Ghandi M, Dumont N, et al. WRN Helicase Is a Synthetic Lethal Target in Microsatellite Unstable Cancers. Nature (2019) 568:551–6. doi: 10.1038/s41586-019-1102-x
52. Gatalica Z, Vranic S, Xiu J, Swensen J. High Microsatellite Instability (MSI-H) Colorectal Carcinoma: A Brief Review of Predictive Biomarkers in the Era of Personalized Medicine. Familial Cancer (2016) 15(3):405–12. doi: 10.1007/s10689-016-9884-6
53. Chalabi M, Fanchi LF, Dijkstra KK, Van Den Berg JG, Snaebjornsson P, Maas M, et al. Neoadjuvant Immunotherapy Leads to Pathological Responses in MMR-Proficient and MMR-Deficient Early-Stage Colon Cancers. Nat Med (2020) 26:566–76. doi: 10.1038/s41591-020-0805-8
54. Reyes-Uribe L, Wu W, Gelincik O, Bommi PV, Francisco-Cruz A, Solis LM, et al. Naproxen Chemoprevention Promotes Immune Activation in Lynch Syndrome Colorectal Mucosa. Gut (2021) 70:555–66. doi: 10.1136/gutjnl-2020-320946
55. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. New Engl J Med (2014) 371(23):2189–99. doi: 10.1056/NEJMoa1406498
56. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
57. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science (2015) 350(6257):207–11. doi: 10.1126/science.aad0095
58. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-Tumor Genomic Biomarkers for PD-1 Checkpoint Blockade – Based Immunotherapy. Science (2018) 3593:eaar3593. doi: 10.1126/science.aar3593
59. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-frommer R, Nakagawa K, et al. Association of Tumour Mutational Burden With Outcomes in Patients With Advanced Solid Tumours Treated With Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol (2020) 21:1353–65. doi: 10.1016/S1470-2045(20)30445-9
60. Andre T, Shiu K-K, Kim T, Jensen B, Jensen L, Punt C, et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. New Engl J Med (2020) 383(23):2207–18. doi: 10.1056/NEJMoa2017699
61. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors With Mismatch-Repair Deficiency. New Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
62. Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin Cancer Res (2016) 22(4):813–20. doi: 10.1158/1078-0432.CCR-15-1678
63. Le DT, Durham JN, Smith KN, Wang H, Bjarne R, Aulakh LK, et al. Mismatch-Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733.Mismatch-repair
64. Danley K, Schmitz K, Ghai R, Sclamberg J, Buckingham L, Burgess K, et al. A Durable Response to Pembrolizumab in a Patient With Uterine Serous Carcinoma and Lynch Syndrome Due to the MSH6 Germline Mutation. Oncol (2021) 312. doi: 10.1002/onco.13832
65. Rousseau B, Foote M, Maron S, Diplas B, Lu S, Argiles G, et al. The Spectrum of Benefit From Checkpoint Blockade in Hypermutated Tumors. New Engl J Med (2021) 384(12):1168–70. doi: 10.1056/NEJMc2031965
66. Roudko V, Greenbaum B, Bhardwaj N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front Immunol (2020) 11:27. doi: 10.3389/fimmu.2020.00027
67. Wagner S, Mullins S C, Linnebacher M. Colorectal Cancer Vaccines: Tumor-Associated Antigens vs Neoantigens. World J Gastroenterol (2018) 24(48):5418–32. doi: 10.1007/s12519-013-0433-1
68. Speetjens FM, Lauwen MM, Franken KL, Rhijn CMJ, Duikeren S, Bres SA, et al. Prediction of the Immunogenic Potential of Frameshift-Mutated Antigens in Microsatellite Instable Cancer. Int J Cancer (2008) 845:838–45. doi: 10.1002/ijc.23570
69. Maletzki C, Schmidt F, Dirks WG, Schmitt M, Linnebacher M. Frameshift-Derived Neoantigens Constitute Immunotherapeutic Targets for Patients With Microsatellite-Instable Haematological Malignancies: Frameshift Peptides for Treating MSI+ Blood Cancers. Eur J Cancer (2013) 49(11):2587–95. doi: 10.1016/j.ejca.2013.02.035
70. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-And-Deletion-Derived Tumour-Specific Neoantigens and the Immunogenic Phenotype: A Pan-Cancer Analysis. Lancet Oncol (2017) 18(8):1009–21. doi: 10.1016/S1470-2045(17)30516-8
71. Roudko V, Bozkus CC, Orfanelli T, McClain C, Carr C, O’Donnel T, et al. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell (2020) 183(6):1634–49. doi: 10.1016/j.cell.2020.11.004
72. Ballhausen A, Przybilla MJ, Jendrusch M, Haupt S, Pfaffendorf E, Seidler F, et al. The Shared Frameshift Mutation Landscape of Microsatellite-Unstable Cancers Suggests Immunoediting During Tumor Evolution. Nat Commun (2020) 11:1–13. doi: 10.1038/s41467-020-18514-5
73. Mandal R, Samstein RM, Lee K, Havel JJ, Wang H, Krishna C, et al. Genetic Diversity of Tumors With Mismatch Repair Deficiency Influences Anti – PD-1 Immunotherapy Response. Science (2019) 491:485–91. doi: 10.1126/science.aau0447
74. Guan J, Lu C, Jin Q, Fu Y-X, Li G-M. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell (2021) 39:109–21. doi: 10.1016/j.ccell.2020.11.004
75. Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-Tumor Immunity. Cancer Cell (2021) 39:96–108. doi: 10.1016/j.ccell.2020.11.006
76. Mowat C, Mosley SR, Namdar A, Schiller D, Baker K. Anti-Tumor Immunity in Mismatch Repair-Deficient Colorectal Cancers Requires Type I IFN – Driven CCL5 and CXCL10. J Exp Med (2021) 218(9). doi: 10.1084/jem.20210108
77. Magnus Von Knebel D, Matthias K. Towards a Vaccine to Prevent Cancer in Lynch Syndrome Patients. Familial Cancer (2013) 12:307–12. doi: 10.1007/s10689-013-9662-7
78. Maruvka YE, Mouw KW, Karlic R, Parasuraman P, Kamburov A, Polak P, et al. Analysis of Somatic Microsatellite Indels Identifies Driver Events in Human Tumors. Nat Biotechnol (2017) 35(10):951–9. doi: 10.1038/nbt.3966
79. Haasl R, Payseur B. Remarkable Selective Constraints on Exonic Dinucleotide Repeats. Evolution (2014) 68(9):2737–44. doi: 10.1111/evo.12460.REMARKABLE
80. Catalano I, Grassi E, Bertotti A, Trusolino L. Immunogenomics of Colorectal Tumors: Facts and Hypotheses on an Evolving Saga. Trends Cancer (2019) 80(18):1–10. doi: 10.1016/J.TRECAN.2019.10.006
81. Leoni G, Alise AMD, Cotugno G, Langone F, Garzia I, De Lucia M, et al. A Genetic Vaccine Encoding Shared Cancer Neoantigens to Treat Tumors With Microsatellite Instability. Cancer Res (2020) 80:3972–82. doi: 10.1158/0008-5472.CAN-20-1072
82. Kloor M, Reuschenbach M, Pauligk C, Karbach J, Rafiyan M-R, Al-Batran S-E, et al. A Frameshift Peptide Neoantigen-Based Vaccine for Mismatch Repair-Deficient Cancers: A Phase I/IIa Clinical Trial. Clin Cancer Res (2020) 1(603):1–31. doi: 10.1158/1078-0432.CCR-19-2706
83. Saterdal I, Gjertsen M, Straten P, Gaudernack G. A TGFbRII Frameshift-Mutation-Derived CTL Epitope Recognised by HLA-A2-Restricted CD8 + T Cells. Cancer Immunol Res (2001) 50:469–76. doi: 10.1007/s002620100222
84. Sæterdal I, Bjørheim J, Lislerud K, Gjertsen MK, Bukholm IK, Olsen OC, et al. Frameshift-Mutation-Derived Peptides as Tumor-Specific Antigens in Inherited and Spontaneous Colorectal Cancer. PNAS (2001) 98(23):13255–60. doi: 10.1073/pnas.231326898
85. Garbe Y, Maletzki C, Linnebacher M. An MSI Tumor Specific Frameshift Mutation in a Coding Microsatellite of MSH3 Encodes for HLA-A0201-Restricted CD8+ Cytotoxic T Cell Epitopes. PloS One (2011) 6(11):2–9. doi: 10.1371/journal.pone.0026517
86. Kloor M, Huth C, Voigt AY, Benner A, Schirmacher P, Doeberitz MVK, et al. Prevalence of Mismatch Repair-Defi Cient Crypt Foci in Lynch Syndrome: A Pathological Study. Lancet Oncol (2012) 13(6):598–606. doi: 10.1016/S1470-2045(12)70109-2
87. Hess JM, Bernards A, Kim J, Miller M, Taylor-Weiner A, Haradhvala NJ, et al. Passenger Hotspot Mutations in Cancer. Cancer Cell (2019) 36(3):675801. doi: 10.1101/675801
88. Tu J, Park S, Yu W, Zhang S, Wu L, Carmon K, et al. The Most Common RNF43 Mutant G659Vfs*41 Is Fully Functional in Inhibiting Wnt Signaling and Unlikely to Play a Role in Tumorigenesis. Sci Rep (2019) 9(1):1–12. doi: 10.1038/s41598-019-54931-3
89. Willis JA, Reyes-Uribe L, Chang K, Lipkin SM, Vilar E. Immune Activation in Mismatch Repair–Deficient Carcinogenesis: More Than Just Mutational Rate. Clin Cancer Res (2019) 24):11–8. doi: 10.1158/1078-0432.ccr-18-0856
90. Alise AMD, Leoni G, Cotugno G, Troise F, Langone F, Fichera I, et al. Adenoviral Vaccine Targeting Multiple Neoantigens as Strategy to Eradicate Large Tumors Combined With Checkpoint Blockade. Nat Commun (2019) 10(2688):1–12. doi: 10.1038/s41467-019-10594-2
91. Gebert J, Gelincik O, Marshall JD, Urban K, Long M, Cortes E, et al. Recurrent Frameshift Neoantigen Vaccine Elicits Protective Immunity With Reduced Tumor Burden and Improved Overall Survival in a Lynch Syndrome Mouse Model. Gastroenterology (2021). doi: 10.1053/j.gastro.2021.06.073
92. Abidi A, Gorris MAJ, Brennan E, Jongmans MCJ, Weijers DD, Kuiper RP, et al. Challenges of Neoantigen Targeting in Lynch Syndrome and Constitutional Mismatch Repair Deficiency Syndrome. Cancers (2021) 13(10):2345. doi: 10.3390/cancers13102345
93. Amodio V, Mauri G, Reilly NM, Sartore-bianchi A, Siena S, Bardelli A, et al. Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers. Cancers (2021) 13:1–33. doi: 10.3390/cancers13112638
94. Montesion M, Murugesan K, Jin DX, Sharaf R, Sanchez N, Guria A, et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discovery (2021) 11:282–92. doi: 10.1158/2159-8290.CD-20-0672
95. Williams DS, Bird MJ, Jorissen RN, Yu YL, Walker F, Hua H, et al. Nonsense Mediated Decay Resistant Mutations Are a Source of Expressed Mutant Proteins in Colon Cancer Cell Lines With Microsatellite Instability. PloS One (2010) 5(12). doi: 10.1371/journal.pone.0016012
96. Litchfield K, Reading J, Lim E, Xu H, Liu P, Al-bakir M, et al. Escape From Nonsense Mediated Decay Associates With Anti-Tumor Immunogenicity. Nat Commun (2020) 11:3800. doi: 10.1038/s41467-020-17526-5
97. Kwon M, An M, Klempner SJ, Lee H, Kim K, Jason K, et al. Determinants of Response and Intrinsic Resistance to PD-1 Blockade in Microsatellite Instability-High Gastric Cancer. Cancer Discovery (2021) 02:2168–85. doi: 10.1158/2159-8290.CD-21-0219
98. Sahin IH, Akce M, Alese O, Shaib W, Lesinski GB, El-Rayes B, et al. Immune Checkpoint Inhibitors for the Treatment of MSI-H/MMR-D Colorectal Cancer and a Perspective on Resistance Mechanisms. Br J Cancer (2019) 121(10):809–18. doi: 10.1038/s41416-019-0599-y
99. Tikidzhieva A, Benner A, Michel S, Formentini A, Link K, Dippold W, et al. Microsatellite Instability and Beta2-Microglobulin Mutations as Prognostic Markers in Colon Cancer: Results of the FOGT-4 Trial. Br J Cancer (2012) 106:1239–45. doi: 10.1038/bjc.2012.53
100. Middha S, Yaeger R, Shia J, Stadler K, King S, Guercio S, et al. Majority of B2M-Mutant and -Deficient Colorectal Carcinomas Achieve Clinical Benefit From Immune Checkpoint Inhibitor Therapy and Are Microsatellite Instability-High. JCO Precis Oncol (2019) 3:1–14. doi: 10.1200/PO.18.00321
101. Germano G, Lu S, Rospo G, Lamba S, Rousseau B, Fanelli S, et al. CD4 T Cell–Dependent Rejection of Beta-2 Microglobulin Null Mismatch Repair – Deficient Tumors. Cancer Discovery (2021) 11(11):1845–59. doi: 10.1158/2159-8290.CD-20-0987
102. Black JRM, Mcgranahan N. Genetic and non-Genetic Clonal Diversity in Cancer Evolution. Nat Rev Cancer (2021) 21:379–92. doi: 10.1038/s41568-021-00336-2
Keywords: Lynch syndrome, MSI-H, dMMR, immunotherapy, cancer vaccine
Citation: Roudko V, Cimen Bozkus C, Greenbaum B, Lucas A, Samstein R and Bhardwaj N (2021) Lynch Syndrome and MSI-H Cancers: From Mechanisms to “Off-The-Shelf” Cancer Vaccines. Front. Immunol. 12:757804. doi: 10.3389/fimmu.2021.757804
Received: 12 August 2021; Accepted: 08 September 2021;
Published: 24 September 2021.
Edited by:
Olivera J. Finn, University of Pittsburgh, United StatesReviewed by:
Arya Biragyn, National Institute on Aging (NIH), United StatesCopyright © 2021 Roudko, Cimen Bozkus, Greenbaum, Lucas, Samstein and Bhardwaj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina Bhardwaj, bmluYS5iaGFyZHdhakBtc3NtLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.