 Eliana Toren
Eliana Toren KaLia S. Burnette
KaLia S. Burnette Ronadip R. Banerjee
Ronadip R. Banerjee Chad S. Hunter
Chad S. Hunter Hubert M. Tse
Hubert M. Tse
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 October 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.756548
This article is part of the Research Topic Immunopathology of Type 1 Diabetes View all 21 articles
Type 1 diabetes (T1D) is an autoimmune disease characterized by autoreactive T cell-mediated destruction of insulin-producing pancreatic beta-cells. Loss of beta-cells leads to insulin insufficiency and hyperglycemia, with patients eventually requiring lifelong insulin therapy to maintain normal glycemic control. Since T1D has been historically defined as a disease of immune system dysregulation, there has been little focus on the state and response of beta-cells and how they may also contribute to their own demise. Major hurdles to identifying a cure for T1D include a limited understanding of disease etiology and how functional and transcriptional beta-cell heterogeneity may be involved in disease progression. Recent studies indicate that the beta-cell response is not simply a passive aspect of T1D pathogenesis, but rather an interplay between the beta-cell and the immune system actively contributing to disease. Here, we comprehensively review the current literature describing beta-cell vulnerability, heterogeneity, and contributions to pathophysiology of T1D, how these responses are influenced by autoimmunity, and describe pathways that can potentially be exploited to delay T1D.
Type 1 diabetes (T1D) is a chronic autoimmune disease in which loss of beta-cell mass and subsequent insulin-insufficiency leads to hyperglycemia. T1D is linked to secondary complications including cardiovascular disease, kidney disease, and neuropathy (1). T1D is the most common form of diabetes in children, comprising approximately 75% of new diabetes diagnoses in patients under 19 years of age (2). Nonetheless, T1D is not a disease only of the young. Epidemiological studies now show that the incidence of autoimmune diabetes in adults (age 30 – 49 years) is at least as high as young adults (age 15 – 19 years) (3). Incidence of T1D is between 4 and 41 per 100,000 in the United States, but interestingly there is significant geographic variation in incidence rates worldwide. Asian countries have relatively lower rates of T1D, while Switzerland, Finland, Norway, the UK, and Sardinia have among the highest rates, with values greater than 20 per 100,000 people (4). Even considering the large geographic variability, overall new diagnoses are on the rise in both childhood and adult populations.
T1D is a multifactorial disease; both genetic and environmental factors contribute to risk. While incompletely understood, putative environmental triggers include microbial infections, neonatal nutrition status/weight, and exposure to certain toxins, such as nitrates (5). Following a triggering event in genetically-susceptible individuals, immune effector cells infiltrate the pancreas and activate inflammatory pathways to mediate targeted destruction of insulin-producing beta-cells. Since the early 1970’s, when the genetic connection between human leukocyte antigen (HLA) and T1D was first discovered, pathogenesis of T1D was largely defined by autoimmunity and the selective presentation of islet autoantigens. Strictly defining T1D by an immunological mechanism, however, does not acknowledge any potential role for the beta-cell itself in promoting disease pathology. Mounting evidence indicates the beta-cell is more than just a passive target in the development of T1D: the lack of long-term success with immune intervention therapies, the existence of islet autoimmunity without T1D development, and the persistence of beta-cells after diagnosis and T1D progression, all provide evidence that the beta-cell is an active participant along with the immune system in T1D pathogenesis (6, 7).
In this review, we will focus on the beta-cell in both healthy and T1D environments. We will explore inherent beta-cell heterogeneity and vulnerabilities, contributions to the local inflammatory environment, and how the beta-cell response to metabolic stress can perpetuate disease. Shifting focus from the beta-cell as a passive target to an active participant in disease progression will help identify novel therapeutic approaches, potentially leveraging these unique beta-cell responses and susceptibilities for both treatment and prevention of T1D.
In 1985, Dr. Gian Franco Bottazzo’s lecture titled “Death of a Beta Cell: Homicide or Suicide?” posed the idea of beta-cell fragility (8). Dr. Bottazzo questioned whether beta-cells were innocent bystanders of immune attack or contributors to their own destruction (9). Beta-cells must rapidly respond to glucose fluctuations by secreting the appropriate amount of insulin to maintain euglycemia, a taxing process that, even in healthy cells, makes them vulnerable to stressors such as inflammation and nutrition excess. The metabolic demand associated with tightly regulated insulin secretion, paired with a highly vascularized environment, reduced antioxidant defense mechanisms, and sensitivity to proinflammatory cytokines, makes beta-cells uniquely susceptible to autoimmune-mediated destruction (Figure 1).
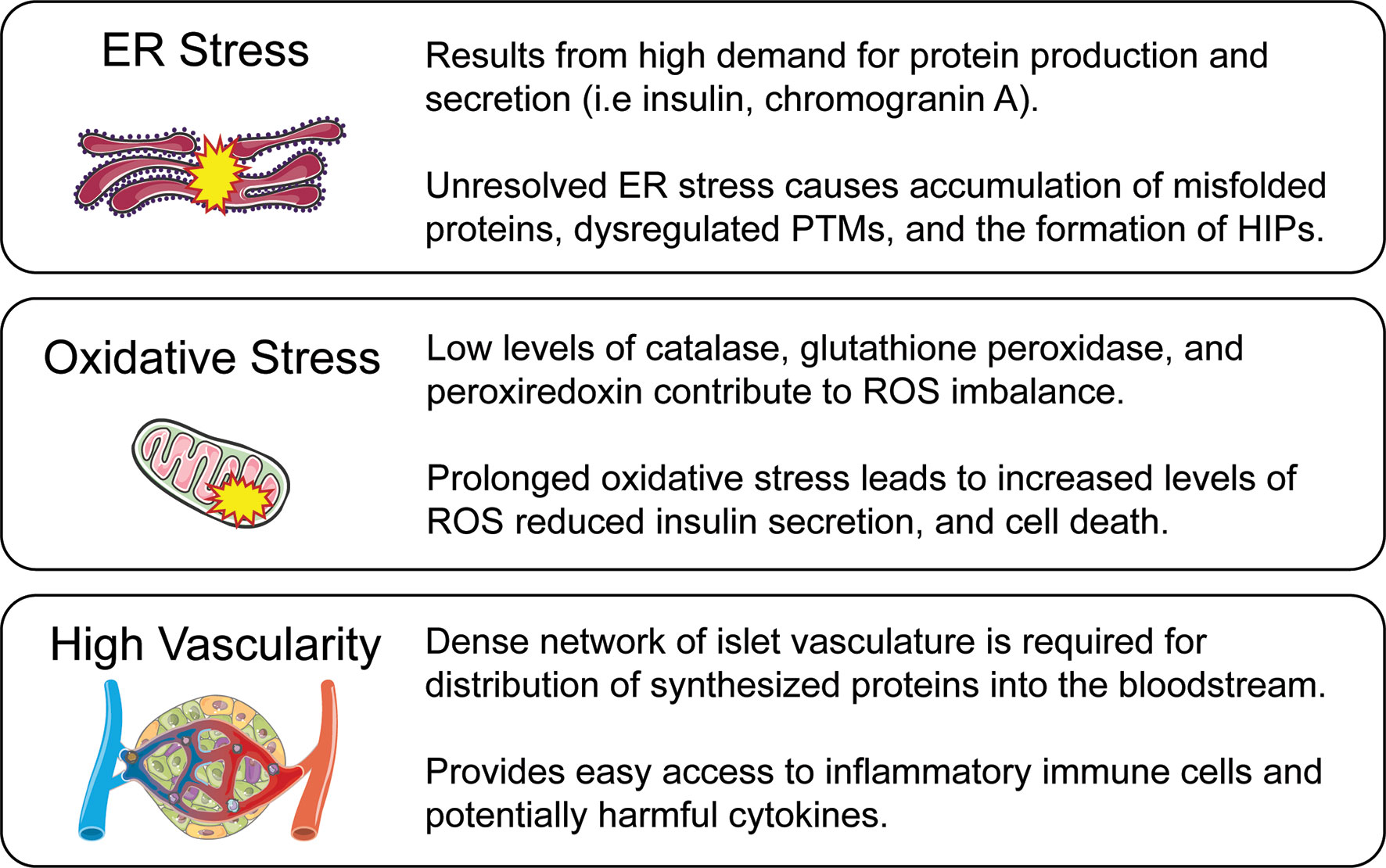
Figure 1 Beta-cell vulnerabilities. While autoimmunity is a major driver of T1D pathogenesis, innate features of beta-cell biology make it a complicit partner in disease progression. These beta-cell characteristics are a result of normal beta-cell function while also active contributors to disease amplification. ER stress is caused by the high protein production and secretory demand of the beta-cell, but in excess leads to misfolded protein response and the generation of HIPs through PTMs. Oxidative stress is caused by an imbalance between the generation of ROS and their detoxification by antioxidants. The reduced antioxidant capabilities of the beta-cell can lead to impaired function and cell death. A densely vascularized environment is required for secretion of insulin and other peptides directly into the bloodstream, but creates a direct dialogue between the beta-cell and potentially harmful immune cells and inflammatory cytokines which may further lead the cell toward stress and apoptosis.
The beta-cell is responsible for producing and secreting multiple secretory granule proteins including insulin, chromogranin-A (ChgA), and islet amyloid polypeptide (IAPP). The endoplasmic reticulum (ER) is the site of protein production and relies heavily on Ca2+ concentrations to maintain the environment needed for proper protein synthesis and folding (Figure 2A) (10). Insulin secretory demand makes the beta-cell particularly vulnerable to exceeding ER protein folding capacity, which leads to the accumulation of misfolded proteins and a disruption of ER homeostasis (Figure 2F) (11). This physiological state is termed ER stress (12). Prolonged efforts by the cell to correct misfolded proteins can lead to unregulated changes in enzyme activity, reduced beta-cell function, and induction of apoptosis (13–16). To meet the metabolic demands of glucose-stimulated insulin secretion (GSIS), the beta-cell requires a tightly-coupled process with cellular metabolism to properly maintain euglycemia (17). In brief, glucose is transported into the beta-cell via the glucose transporter 2 (Glut2) in rodents (GLUT1 and 3 in humans), converted to pyruvate, and shuttled into the mitochondria where it is used for ATP production (18). Changes in the ATP to ADP ratio lead to beta-cell depolarization, Ca2+ influx, and insulin release (19–21). Insulin mRNA is translated at the ER following nutrient stimulation, which in rodents can signal up to a 10-fold increase in insulin synthesis at a rate of 1 million molecules per minute (22, 23).
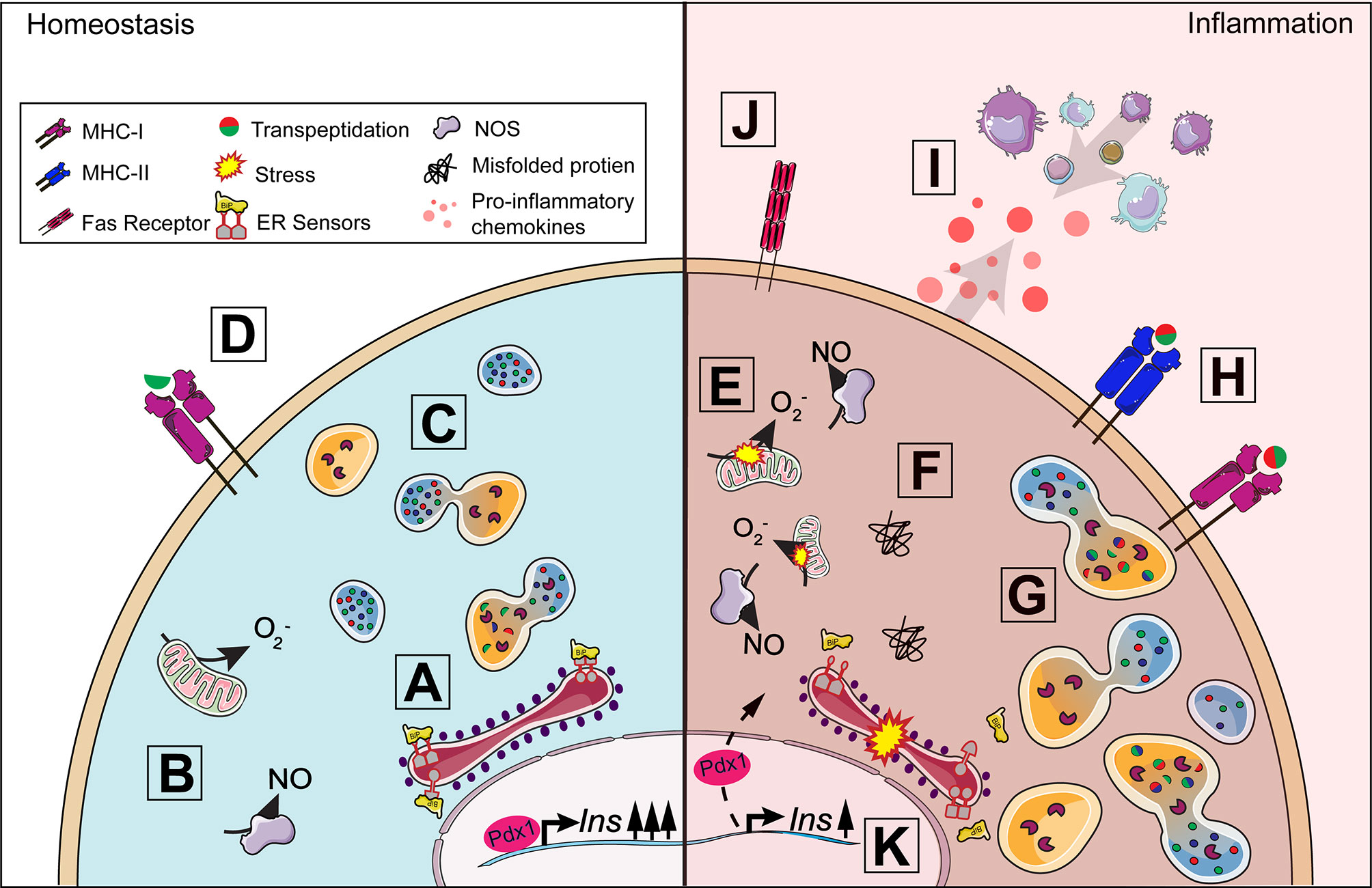
Figure 2 Beta-cell response to inflammation. Under homeostasis conditions, insulin production is tightly coupled with cellular metabolism including protein synthesis in the endoplasmic reticulum (ER) (A) and mitochondrial function (B). When insulin secretory granule proteins are in excess, they can be broken down and recycled by crinophagy, a process by which granules fuse with lysosomes (C). Some peptides from this degradation process are presented on MHC-I (D) and, in healthy cells, should not lead to activation. A proinflammatory environment around the islet exacerbates ER and oxidative stress (E) contributing to the dysregulation of multiple processes in the beta-cell. The accumulation of misfolded proteins can result in the activation of the unfolded protein response (F) and increase lysosomal degradation of insulin secretory granule proteins (G). Protein degradation under stress can lead to the production of neo-antigens, such as hybrid insulin peptides, through transpeptidation. ER and oxidative stress results in the upregulation of MHC-I and the unique expression of MHC-II (H) by the beta-cell allowing for increased presentation of potential neo-antigens to T cells. Fas receptor expression (J) makes the beta-cell vulnerable to Fas-mediated apoptosis. The release of chemokines (I) from the beta-cell further contributes to immune cell recruitment and the development of insulitis. Insulin production can be affected as disturbances in cellular homeostasis can lead to the translocation of Pdx1 from the nucleus to the cytoplasm, decreasing insulin production (K).
To meet these high demands, beta-cells have an extensive ER with multiple chaperones to aid in protein folding, packaging, and secretion. However, high protein synthesis puts a significant amount of stress on the ER. The unfolded protein response (UPR) is triggered when an excessive amount of misfolded proteins accumulate in the ER, which can be caused by overnutrition, increased reactive oxygen species (ROS), or proinflammatory cytokines (24, 25). Three major sensors of the UPR are protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 alpha (IRE1α) and activating transcription factor 6 (ATF6) (13, 26). In an unstressed state, these sensors are bound to the ER chaperone binding immunoglobulin protein (BiP) (Figure 2A). Accumulation of misfolded proteins leads to the dissociation of BiP from the three UPR sensors (Figure 2F) (27, 28). Together, the UPR sensors alleviate ER stress by attenuating global protein synthesis to reduce the load of unfolded proteins, increasing chaperone synthesis to guide protein degradation or refolding misfolded proteins, and synthesizing lipids to increase ER volume (29).
In addition to protein synthesis, the ER is also responsible for storage of intracellular Ca2+ and therefore, regulates calcium-dependent signaling within the cell, such as protein folding and enzymatic function (13). ER stress disrupts intracellular Ca2+ balance influencing multiple processes, including activation of cytosolic post-translational modification (PTM) enzymes by facilitating their translocation into subcellular compartments. This imparts downstream changes in gene expression, protein conformation, and enzyme activity (30–33). Dysregulation of PTM enzymes has been linked to the development of rheumatoid arthritis, celiac disease, and T1D (34–38). In T1D, this includes citrullinating peptidyl arginine deiminase (PAD) enzymes and tissue transglutaminase 2 (tTG2) deaminating enzyme (13, 39). PAD, tTG2, and similar enzymes can alter the binding affinity of peptide epitopes, such as insulin, to major histocompatibility complex (MHC) class II, resulting in increased CD4 T cell activation (36, 40). Inhibition of systemic PAD enzymes in NOD mice can protect against diabetes progression, suggesting a role in T1D initiation (41).
Stress-induced PTMs can also result in the creation of neo-antigens in peripheral tissues for which the thymus has not established tolerance. Many T1D neo-antigens generated from PTMs have been identified (42). Some PTMs can lead to non-functional protein products resulting from alternately spliced RNA called defective ribosomal products (DRiPs) (43). Increased expression of DRiPs from insulin have been measured in beta-cells in response to ER stress and can be recognized by T cells from patients with T1D (44, 45). Hybrid insulin peptides (HIPs) are another group of neo-antigens generated from transpeptidation, a PTM where insulin peptides are covalently linked to other beta-cell granule peptides including insulin C-peptide, IAPP, and ChgA (Figure 2G) (46, 47). HIPs are not only recognized by autoreactive CD4 T cells in mouse models of T1D, but CD4+ T cells from patients with T1D recognized HIPs as well, signifying their potential role in disease initiation and progression (48–50). Our understanding of how neo-antigens are generated and contribute to the development of autoreactivity in T1D is currently unknown. Future studies are warranted to further define how ER stress and subsequent downstream disruptions induced by the secretory demands of the beta-cell can influence autoreactive T cell responses and beta-cell vulnerability in T1D (Figure 1).
Oxidative stress occurs when there is an imbalance between ROS generation and antioxidant activity (51). Superoxide is primarily a byproduct of normal cellular metabolism that is generated in the mitochondria and cytoplasm (Figure 2B) and is an initiating free radical that can result in the formation of other reactive species such as hydrogen peroxide (H2O2), hydroxyl radical, and peroxynitrite (52). Free radicals are highly reactive and can induce cellular damage, but antioxidants including superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), peroxiredoxins, thioredoxin, and glutathione protect the cell by detoxifying these reactive species (53, 54). SOD dismutates superoxide to molecular oxygen and H2O2, a less destructive oxidant and signaling molecule. H2O2 regulates insulin secretion by activating the second messenger c-Jun N-terminal Kinase (JNK) (55). This leads to decreased insulin production through the translocation of the transcription factor pancreatic and duodenal homeobox 1 (Pdx1) from the nucleus to the cytoplasm, resulting in decreased Insulin transcription (56) (Figure 2K). H2O2 is further converted to oxygen and water by catalase, GPx, and peroxiredoxin. Increased levels of H2O2 can form extremely reactive hydroxyl radicals through Fenton reactions with free iron present in the cytoplasm, which has downstream negative effects on intracellular calcium levels, protein synthesis, glycosylation, and redox status (53). Beta-cells, however, have decreased antioxidant levels and therefore, are highly susceptible to free radical-mediated damage (Figure 2E). Rodent and human beta-cells have reduced transcriptional and protein levels of cytosolic copper/zinc (Cu/Zn) SOD1, mitochondrial manganese (Mn) SOD2, catalase, and GPx, which can result in exacerbated levels of superoxide, H2O2, hydroxyl radical, and peroxynitrite that are implicated in beta-cell death in T1D (57–61). The inability to properly restore cellular homeostasis due to the negative effects of oxidative and ER stress can induce apoptosis in insulin-secreting beta-cells. Increased beta-cell apoptosis has been measured in patients with T1D and NOD mice when compared to healthy controls (14, 62–66). In addition to oxidative stress, the beta-cell is also impacted by the islet microenvironment in which it is closely associated.
Importantly for T1D pathology, islets are highly vascularized. This provides an interface by which immune cells, even from distant sites, can gain local access to pancreatic islets (67, 68). Islets contain a glomerular-like network of fenestrated capillaries that comprise about 8-10% of islet volume. Islet capillary density is estimated to be 10 times higher than that of the exocrine pancreas and is driven by high local production of VEGF-A (69, 70). This rich vascularization and high islet blood flow is autonomously regulated through complex interactions between hormones, metabolites, and the nervous system. While islet blood flow is innately required for and coupled to insulin sensing and release, extensive vasculature also makes the beta-cell uniquely poised for interactions with the immune system.
The dense islet vasculature network facilitates activated immune cell trafficking across the vascular endothelium into the islet (Figure 1). This causes a local inflammatory microenvironment which in turn, further increases permeability, facilitating access even for naïve T cells (71). Interestingly, this “open” environment remains, even after reversal of diabetes with anti-CD3 treatment. In addition to naïve T cell infiltration, activated immune cells that are primed locally in the pancreatic lymph nodes (pLNs) can also cross the vascular endothelium. pLNs may contribute to T1D pathogenesis as drainage from the pancreas and local gut regions provides a crossroad for the immune cells traveling between these compartments (67, 72). The interplay between the microbiome, the immune system, and a “leaky gut” has been implicated as a key factor in T1D pathogenesis (73, 74). Toll-like receptors (TLRs) are a family of innate pattern recognition receptors important for microbial clearance by the immune system. Many TLRs signal through the MyD88 adapter protein. NOD mice deficient in MyD88 exhibit microbiota-dependent protection from autoimmunity development (75). Specific manipulation of TLR expression and microbiota composition can further regulate disease progression or prevention (76). Sex-specific autoimmune risk can also be influenced by microbiome manipulation (77–79). With new data unveiling the importance of the microbiome for the gut immune environment and shaping peripheral tolerance, the relationship between pLNs, islet vasculature, and immune cell trafficking is increasingly relevant to beta-cell vulnerability and T1D pathogenesis (80, 81). In addition to facilitating interactions between islet cells and immune cells, the islet vasculature also sensitizes the beta-cell to the damaging effects of circulating proinflammatory cytokines.
Beta-cells are sensitive to cytokine-mediated damage. Cytokines can alter crucial beta-cell characteristics including insulin secretion, mitochondrial function, and intracellular calcium stores (82, 83). Inflammatory cytokines including tumor necrosis factor alpha (TNFα), interferon gamma (IFNγ), and interleukin-1 beta (IL-1β), cause beta-cell dysfunction by impairing ATP production, inducing DNA damage, and promoting apoptosis (Figure 3) (84, 85). Proinflammatory cytokine exposure inhibits GSIS due to the limited availability of ATP in both rodent and human islets, as well as in beta-cell lines (86–89). Islet exposure to cytokines triggers nuclear factor κB (NFκB) induction of inducible nitric oxide synthase (iNOS), which increases NO formation in the beta-cell (87). NO has temporal effects on beta-cell responses, as early and transient levels of NO (less than 24 hours) facilitate the repair of cytokine-induced DNA damage by inhibiting the activation of the DNA damage response and preventing the induction of apoptosis (90). However, prolonged exposure to NO can induce beta-cell death due to DNA damage, UPR activation, and decreased mitochondrial oxidation (i.e., ATP production) (87, 91). In cultured islets, pre-exposure treatment with NO inhibitors, such as aminoguanidine, attenuates cytokine-mediated beta-cell death (92). Cytokine-mediated beta-cell death becomes exacerbated in an inflammatory microenvironment in the pancreas, creating a positive feedback loop resulting in more inflammation, stress, vulnerability, and eventually cell death (Figure 3). Unfortunately, clinical trials with anti-cytokine therapies such as Anakinra, an IL-1 receptor antagonist, were not efficacious in delaying T1D, suggesting a more complex interaction between cytokines and beta-cells in vivo (92). Nonetheless, not every beta-cell is lost in T1D, nor do they all respond negatively to ER and/or oxidative stress, indicating an intrinsic beta-cell heterogeneity in response to disease promoting factors.
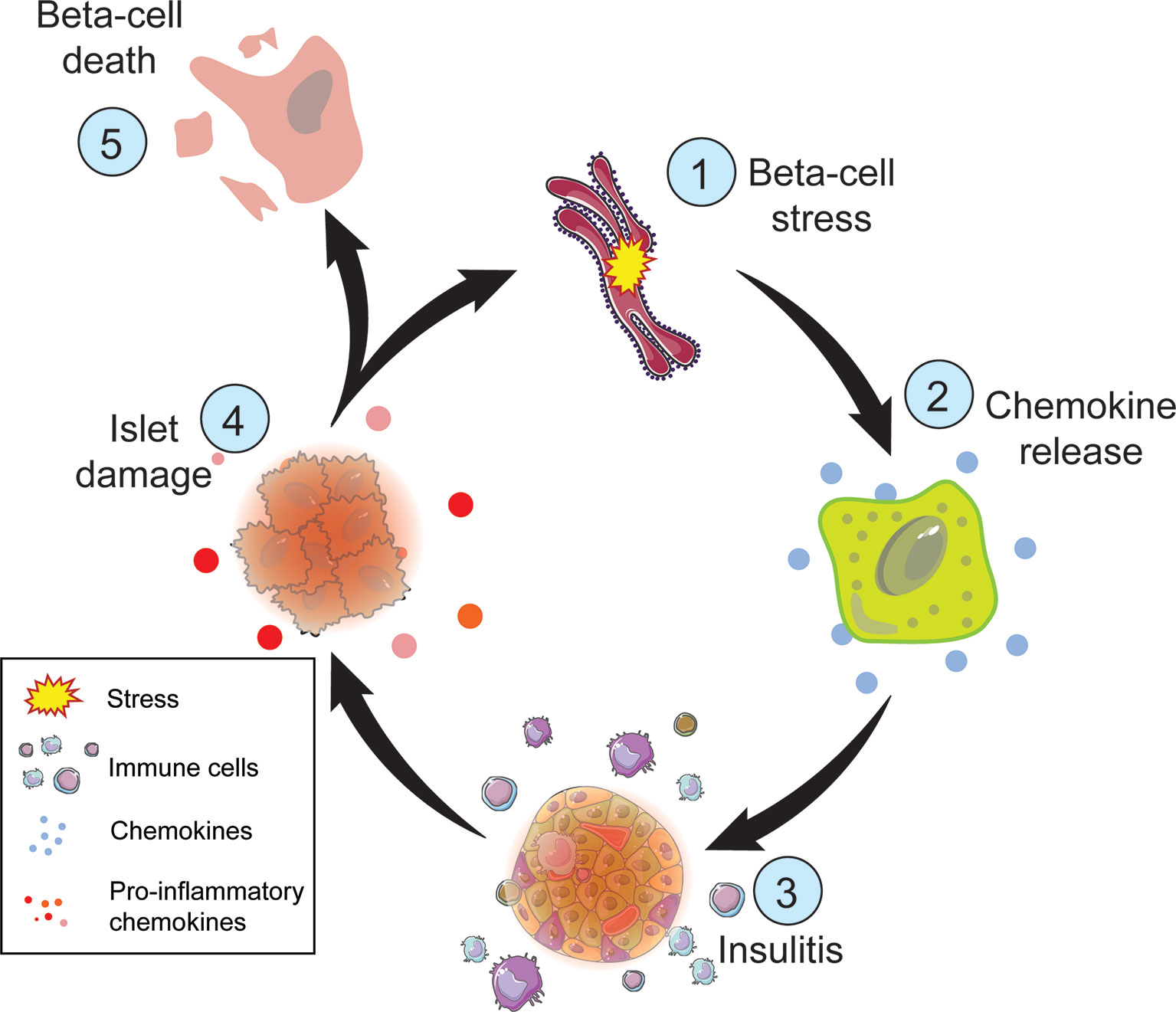
Figure 3 Steps of beta-cell death in T1D. Metabolic demand of nutrient challenge results in ER and oxidative stress (1) followed by chemokine release by the beta-cell (2). Chemokines attract immune cells (3), such as macrophages and T cells, which can damage the islet (4) directly though T cell interactions and indirectly through the release of inflammatory cytokines and reactive oxygen species. Cellular damage exacerbates ER and oxidative stress perpetuating this cycle. The inability to restore cellular homeostasis will result in beta-cell death (5).
It is challenging to fully understand the response and contribution of the beta-cell to the T1D disease state without understanding beta-cell heterogeneity. Many groups have focused on determining whether different subtypes of beta-cells exist, and if so, how they might differ in functional ways such as proliferative and secretory capacities (93–95). Identifying subpopulations, and then understanding their inter- and intra-islet communication, has uncovered a level of complexity and diversity not previously appreciated. We will briefly explore the recent findings regarding functional and transcriptional heterogeneity of the beta-cell and discuss potential impacts on susceptibility to T1D.
Evidence for functional beta-cell heterogeneity in calcium flux, metabolism, ion channel conductance, and insulin secretion has been appreciated for almost 30 years (96, 97). More recently, this functional diversity has been specifically defined by many research groups into beta-cell subpopulations (Table 1). Using novel cell surface markers (ST8SIA1 and CD9) identified by immunizing mice with human islets, four human beta-cell subtypes with unique basal and GSIS responses were defined as β1-4 (93). All subtypes contain insulin granules but exhibit variable functionality and abundance; β1 is the most abundant and glucose responsive, while β4 is the rarest and least responsive, with highest basal insulin secretion. Interestingly there are no correlations found between subtype ratios and sex, age, or obesity, but subtype abundance is altered and much more variable in Type 2 diabetes (T2D). For example, β3 and β4 subsets are unusually overrepresented in T2D islets, but abundance of these two subsets varied much more than in control non-diabetic tissue. It is unknown if β1-4 cells exist in unique patterns before or after T1D diagnosis and disease progression.

Table 1 Selected pancreatic beta-cell subtypes. Heterogeneity in beta-cell response has led to the identification of beta-cell subtypes. These subtypes may vary in spatial location within the islet, speed of response to a stimulus, and secretory capacity. The names of beta-cell subtypes, description of characteristics, and whether they were identified in mouse or human pancreata are defined below.
In 2016, Johnston et al. discovered specialized beta-cells they termed “hub” cells that exert disproportionate control over blood glucose (94). Hub cells, comprising 1-10% of islet beta-cell mass, are metabolically active, exhibit evidence of transcriptional immaturity (low or absent Pdx1 and Nkx6.1 transcription factor levels), and are hypothesized to act as a pacemaker within the islet. Supporting this model, calcium tracing showed that surrounding cells, termed “followers”, respond to glucose stimulation slightly after the hub cell. Using optogenetic and pharmacological techniques, silencing hubs caused desynchrony in the calcium-induced response of beta-cells. The Huising group reported on a beta-cell subtype located in the islet periphery that is both transcriptionally and functionally immature, called the “virgin” beta-cell (95). Virgin cells lack the maturity marker urocortin 3 (UCN3) and are incapable of sensing glucose or proper calcium influx. Using lineage tracing, they found that these cells are transdifferentiation intermediates between alpha- and beta-cells. This work defined a “neogenic niche” of new beta-cells originating from an alpha-cell lineage, establishing a plasticity between these cell types that had also been suggested by models of extreme beta-cell loss (100). The hub versus virgin subpopulations can be compared to flattop+ and flattop– populations, where this novel marker differentiates between a mature, functional beta-cell population (flattop+) and an immature and highly proliferative one (flattop-) (101).
In 2020, the Benninger lab defined yet another subpopulation that is functionally distinct from those previously described (102). These cells are termed “first-responder” beta-cells, as defined by calcium dynamics. They found that first-phase response time of beta-cells is spatially organized and dependent on the physical distance from the first-responder cell. How these sub-populations of cells as defined by these specific markers may be unique or overlapping remains to be determined.
The implementation of single-cell transcriptomics has allowed exploration of beta-cell heterogeneity on a scale that was not previously possible. The Kubicek lab published the first application of single-cell transcriptomics to human islet cells in 2016 (103); while the number of beta-cells identified was extremely low, they showed transcriptome heterogeneity for genes such as DLK1, a T1D associated gene by GWAS (Genome-Wide Association Study) which will be discussed later in more detail. That same year the Kaestner lab published single-cell mass cytometric analysis of human islets and found heterogeneity in markers including Ki67, identifying four distinct beta-cell subpopulations (104). Several groups have since published large-scale single-cell transcriptome analyses of human and mouse beta-cells and identified unique subpopulations. These populations display differential levels of various beta-cell characteristics including maturity (UCN3), aging (IGF1R), and ER stress response (SRXN1, SQSTM1) (105, 106). Genes involved in ER/oxidative stress have been some of the strongest distinguishers of subpopulation clusters in various studies, with ER stress markers also correlating to proliferation and reduced beta-cell function (107–109). As discussed earlier, beta-cell ER stress is extremely relevant to an understanding of T1D contribution and response. Perhaps certain populations of proliferative-ER stressed beta-cells are the first to be lost during T1D pathogenesis.
For years, technical hurdles to interrogating mRNA in islets due to the digestive enzymes present in the surrounding exocrine pancreas limited our understanding of transcriptional dynamics and heterogeneity in the beta-cell. Dr. Shalev Itzkovitz at the Weitzmann Institute developed methods to visualize the dynamics of beta-cell mRNAs. His group designed an optimized single-molecule FISH (fluorescence in situ hybridization) protocol that allowed for assessment of transcriptional heterogeneity within the beta-cell population (110). This method revealed a subpopulation of “extreme” beta-cells that contain high levels of insulin and secretory factor mRNAs (IAPP, ChgA), but interestingly, low levels of insulin protein. The investigators suggested this may impart a specialization for basal insulin secretion. Additionally, beta-cell mRNAs displayed a uniquely polarized pattern, with elevated Insulin mRNA concentration in apical ER enriched compartments of the cell. The ratio of extreme beta-cells was increased in db/db diabetic mice, potentially facilitating increased requirements for basal insulin. This work gives rise to another unappreciated aspect of beta-cell heterogeneity: transcriptional heterogeneity. Future studies are needed to explore the proportion of extreme beta-cells in T1D and how this type of transcriptional variability may affect vulnerability to autoimmune recognition and attack.
The relationship between these different beta-cell subpopulations is still being defined, as the markers discussed above may represent unique or overlapping populations. For example, each of these functional and transcriptional subpopulations represents a unique niche that may have specific susceptibilities or contributions to T1D. Is a hub cell the same as a β1 or flattop+ cell? What are the differences and are they physiologically relevant? What is the relationship between these cell types and how do those relationships change with age and nutrition state? Understanding the relationship between functionality, heterogeneity, and vulnerability will provide a deeper understanding of T1D etiology, potentially setting the stage for more effective therapeutic strategies.
Therapeutic strategies for preventing or treating T1D have historically focused on modulating the immune response to the beta-cell. Emerging strategies that instead focus on beta-cell dysfunction through manipulation of ER, oxidative, or cytokine-induced cell stressors may prove to be beneficial, as the beta-cell itself actively contributes to inflammatory responses in T1D. The following section will discuss beta-cell contributions to the T1D inflammatory environment, which may represent optimal targets for future combinatorial therapies.
Chemokines are a family of small molecules involved in lymphoid physiology, pathology, and hemopoietic cell migration (111, 112). Chemokines can be broadly separated into two categories: constitutive and inducible (113). Constitutive chemokines perform homeostatic functions involving non-inflammatory leukocyte trafficking, while inducible chemokines are produced in response to inflammation to recruit activated leukocytes to the sites of damage or stress (114). Multiple chemokines, detailed below, are secreted by the beta-cell and contribute to immune infiltration into the islet (Figure 2I). These secreted factors make the beta-cell a target for immune invasion and destruction.
The chemokine C-C ligand 2 (CCL2) also known as monotype chemoattractant protein (MCP)-1 is an inducible chemokine involved in monocyte, NK cell, and T cell recruitment during inflammation (115–117). Human and NOD islets cultured with proinflammatory cytokines IL-1β and IFNγ can induce CCL2 production (118–120). Beta-cells express Ccl2 in an NF-κB−dependent manner and can be induced in vivo by environmental triggers such as viral infections leading to inflammation and macrophage recruitment (121, 122). Macrophages are the first and most abundant immune cell to infiltrate the islet during the progression of T1D in NOD mice and have also been identified in islets from patients with recent-onset T1D (115, 123, 124). CCL2 may be responsible for this influx in macrophages, as transgenic overexpression of Ccl2 in murine beta-cells results in increased monocyte recruitment, insulitis, and islet destruction (115). Binding of CCL2 to its receptor C-C chemokine receptor-2 (CCR2) in macrophages leads to the production of proinflammatory cytokines and chemokines such as TNFα, IL-1β, IL-12, and CXCL10 to exacerbate the inflammatory environment of the islet (125). Prolonged exposure to proinflammatory cytokines leads to ER stress, oxidative stress, and cell death (115) (Figure 3).
CC ligand 5 (CCL5) also called RANTES (regulated on activation, normal T cell expressed and secreted) is a chemoattractant for T cells, eosinophils, and monocytes involved in inflammatory responses. CCL5 has been measured in rodent islets and from cell sorted beta-cells in response to inflammatory cytokines TNFα, IL-1β, and IFNγ (126, 127). Increased expression of CCR5, one of the cognate receptors for CCL5, was detected on T cells from patients with T1D and NOD mice (128, 129). Blocking CCR5 using neutralizing antibodies in 2-month-old NOD mice (after islet infiltration, but before overt diabetes) inhibits future immune infiltration and prevents development of diabetes (126).
C-X-C motif chemokine ligand 10 (CXCL10) also called IP-10 (IFNγ-induced protein 10) is a chemokine secreted by many cell types including monocytes, neutrophils, and endothelial cells (130, 131). CXCL10 is increased in the serum and tissues of patients with various autoimmune diseases including T1D (99, 132). Human islets, murine islets, and NIT-1 NOD beta-cells secrete CXCL10 when cultured with pro-inflammatory cytokines IL-1β and IFNγ (127). CXCL10 binds the seven transmembrane G protein coupled receptor CXC receptor 3 (CXCR3), expressed on both immune and non-immune cells (133, 134). In lymphocytes, CXCR3 mediates chemotaxis, while in non-lymphocytes CXCR3 regulates tissue repair, proliferation, and angiogenesis (135, 136). Mice lacking CXCR3 and infected with lymphocytic choriomeningitis virus-WE strain (LCMV-WE), an established model to study T1D, exhibited a delay in insulitis, while overexpressing CXCL10 in mouse islets accelerated LCMV-induced diabetes (127, 137, 138). As predicted, using neutralizing antibodies to block CXCL10 also decreased T cell trafficking to the islet and abrogated diabetes development (139–141). In culture, the NIT-1 NOD beta-cell line was found to secrete CXCL10 in response to inflammatory cytokines IL-1β, TNFα, and IFNγ (127). These data suggest that elevated CXCL10 secretion by the beta-cell may occur early in T1D progression. CXCL10 not only contributes to immune cell recruitment but is also directly toxic to beta-cells (142). In addition to CXCR3, CXCL10 also binds Toll-like receptor-4 (TLR4), a pattern recognition receptor involved in the immune response to microbial pathogens (143). The CXCL10:TLR4 signaling pathway in beta-cells leads to cleavage and translocation of activated protein activated kinase 2 (PAK-2) into the nucleus, contributing to apoptotic signaling within the cell (144, 145). Islets from C57BL/6 Tlr4-/- knockout mice are protected against CXCL10-induced damage. Therefore, CXCL10 released by the beta-cell contributes to cell death by attracting activated immune cells and inducing apoptosis within the beta-cell.
Beta-cells produce proinflammatory CCL2, CCL5, and CXCL10 chemokines when exposed to inflammatory conditions or environmental triggers and can perpetuate the recruitment of immune cells to initiate insulitis. Once present, these immune cells can damage the beta-cell by synthesizing ROS, proinflammatory cytokines, and expressing receptors that can directly mediate beta-cell death (Figure 2).
Of the infiltrating cells causing insulitis in T1D, T cells are the major destroyer of beta-cells, with both CD4 and CD8 T cells being required to effectively transfer disease (146–148). CD4 and CD8 T cells have different roles in disease development (149). When activated, CD4 T cells or T “helper” cells influence the activation of surrounding immune cells through the production of pro- or anti-inflammatory cytokines (149). Human CD4 T cells conventionally recognize peptides presented on HLA-II molecules expressed by antigen-presenting cells (APCs), but the expression of HLA-II has also been detected on beta-cells from patients with T1D (150). CD8 T cells recognize peptides presented on MHC-I on mouse cells and HLA-I on human cells (Figure 2D). Islets biopsied from patients with T1D displayed HLA-I hyperexpression, which warrants their susceptibility to CD8 T cell-mediated destruction (151). The activation of CD8 T cells leads to the differentiation of CD8 T cells to become cytolytic T lymphocytes (CTLs) resulting in the directed release of cytotoxic cytokines, cytolytic granules, and Fas ligand (FasL)-mediated death of the target cell. Beta-cells from diabetic patients not only express HLA-I/II molecules, but also the Fas receptor (CD95/Apo-1) (152–154) (Figure 2J). Fas/FasL signaling is suggested to play a role in T1D pathology as NOD mice deficient in Fas do not develop inflammation or diabetes (155). Fas-deficient mice are also protected against adoptive transfer of splenocytes from diabetic NOD mice. In rodent and human islets, the expression of Fas receptor in beta-cells is induced by proinflammatory cytokines IL-1α, IL-1β, IFNγ, and the upregulation of iNOS, as sequestering NO in the beta-cell decreases Fas expression (156, 157). Fas/FasL signaling in the beta-cell leads to apoptosis via the activation of caspase 8 and the mitochondrial pathway of apoptosis (158). Beta-cell-derived proinflammatory chemokines, HLA-I/II (or MHC-I/II) molecules, and Fas/FasL receptors can perpetuate T1D disease progression by promoting immune cell recruitment, T cell activation, and subsequent beta-cell destruction. Since autoantibodies can be detected in circulation for years prior to disease onset (159) and patients from the Medalist study (discussed below) retain a portion of insulin-secreting beta-cells, these observations provide evidence that at least some beta-cell populations may possess mechanisms to evade the immune response.
In addition to the intrinsic, “baseline” heterogeneity of beta-cells, heterogeneity of disease progression within islets from individual patients, and heterogeneity of disease progression amongst patients with T1D are becoming apparent through longitudinal clinical studies and new analytical techniques examining T1D animal models. The Joslin Medalist Study of T1D patients with disease duration of 50 years or longer revealed that some insulin producing beta-cells persist, ostensibly even after years in a chronic inflammatory environment (160). This highlights that some level of heterogeneity is present in the T1D islet, supporting that certain beta-cell populations may be protected from autoimmune destruction. The expansion of single-cell transcriptomics has contributed to our understanding of cell populations dynamics, but whether it be mouse or human, almost all published studies have used either healthy or T2D islets.
Exciting work in the past few years has given rise to the idea that disease-specific beta-cell heterogeneity may arise during T1D progression, with certain populations that are more vulnerable than others to autoimmune-mediated death. The Herold lab was one of the first to identify distinct cell populations in T1D with their discovery of a low granularity beta-cell population termed “bottom” cells in the NOD mouse model (161) (Table 1). They found that this non-glucose-responsive population emerges prior to hyperglycemia and immune infiltration and expands over time, comprising over 50% of the beta-cell population by 12 weeks of age. The bottom cells express “stemness” markers and were found to be less sensitive to treatment with cytokines and immune infiltrates compared to their “top” counterparts, suggesting they may evade immune attack (161). As we continue to understand disease etiology more deeply, these resistant populations may provide a novel target for treatment.
The discovery of distinct T1D endotypes associated with age of diagnosis has recently contributed to our knowledge of the T1D beta-cell (162). Using immunohistochemical analysis of pancreas samples from patients diagnosed under the age of 30, Leete et al. found a distinct pattern of insulin/proinsulin localization in the beta-cell that is not present in non-T1D controls. Specifically, they found high insulin/proinsulin colocalization in patients who were diagnosed under 13, and even more consistently in patients diagnosed before 7 years of age. Similar subtypes had been described regarding insulitis, with two discrete histological profiles associating strongly with age of diagnosis (163, 164). The authors postulate that discovery of these histologically distinct phenotypes points to disease endotypes that could even be described as T1DE1 and T1DE2 and may require different immunotherapeutic options based on age of diagnosis. While this work is not necessarily beta-cell specific, the heterogenous nature of disease that the field continues to uncover further points to the importance of understanding beta-cell heterogeneity and response to autoimmunity. We propose that an understanding of beta-cell dynamics prior to, during, and after immune-cell infiltration in T1D will be vital to development of therapies that can not only combat T1D development, but perhaps even precede and bypass it.
T1D pathogenesis involves both genetic and environmental triggers and susceptibilities. Thus, examining genetic associations that link specific loci to T1D vulnerability has been a major area of research focus. Surprisingly, the Wellcome Trust Case Control Consortium (WTCCC) established GWAS found relatively few novel risk loci for T1D. It was not until the T1D Genetics Consortium (T1DGC) conducted a meta-analysis that approximately 41 distinct susceptibility loci were identified (165). Fine mapping of these loci using ImmunoChIP established credible sets of single nucleotide polymorphisms (SNPs), most of which are found in non-coding DNA regulatory regions, including tissue-specific enhancers (166). Most of our understanding of the identified SNPs has been centered around the HLA loci that are strongly associated with the disease and T cell autoreactivity. While these studies of the immune arm of pathogenesis are invaluable, not only do SNPs in the INSULIN (INS) gene remain one of the highest risks (167), but approximately 60% of all T1D susceptibility genes are expressed in the islet (168). These data further support the concept that the beta-cell has a larger role in its own destruction than previously appreciated, and that genetic susceptibility is not solely based on the status of the immune system. Below we describe beta-cell-associated SNPs and what is known thus far about their T1D implications.
As mentioned above, after the HLA locus, the 5’ upstream region of the INS locus is the genomic region with the strongest association with T1D risk (167). Specifically, it is the INS-VNTR (variable number tandem repeat) locus that confers susceptibility differences. The VNTR alleles are defined by two classes: class I (26-63 repeats) and class III (140-200 repeats). The shorter class I VNTRs confer a 2-5-fold increase in T1D risk while the longer class III allele is protective against T1D, this is thought to be due to effects on proinsulin expression in the thymus (169). Class III VNTRs are associated with increased INSULIN transcription in the thymus during induction of central immune tolerance. The authors proposed that these increased thymic insulin levels may promote negative selection of insulin-specific T cells, ultimately leading to a protective effect on T1D susceptibility. Class I VNTRs result in decreased INSULIN transcription in the thymus and potentially allow insulin-specific autoreactive T cells to escape from the thymus due to defects in central tolerance and negative selection. This class I versus III allele-specific mechanism illustrates the complexity of T1D risk, and while this susceptibility locus can absolutely be seen as a beta-cell associated SNP, the proposed mechanisms that have been defined thus far are still largely immune system focused. So, despite the 100 years since the discovery of insulin, large gaps in our understanding of how it is involved in the response of the beta-cell itself in T1D remain.
Due to the scarcity of studies of beta-cell contributions to T1D, many associations have been made between beta-cell death and failure in T1D and T2D. Remarkably, GWAS indicates there are very few susceptibility loci associated with both maladies. Variations of the Kruppel-like zinc finger transcription factor GLIS3 are one of the few that have been strongly associated with both T1 and T2D (170). Also a known MODY (Maturity Onset Diabetes of the Young) gene, GLIS3 is expressed predominantly in the pancreas, thyroid, and kidney. While there have been some discrepancies in the literature regarding the exact timing of expression, in GLIS3-EGFP knock-in mice, GLIS3 mainly co-expresses with Sox9 in bipotent pancreatic islet progenitor cells and is absent from the acinar progenitors at embryonic day (E)13.5. This pattern correlates with its importance in development of the pancreatic endocrine lineage and seeming negligibility for the exocrine portion of the pancreas. The association between GLIS3 and T1D was first identified in European populations but has more recently been recapitulated in a Pakistani cohort (171, 172). Interestingly, a GLIS3 variant (A908V) is associated with T1D resistance in Japanese patients (173). Considering the thymic expression of GLIS3, the authors propose that perhaps this variant induces central or peripheral immune tolerance more efficiently than the wild-type variant, but more studies are needed to understand this mechanism. Regarding the molecular mechanisms that underlie the associations of GLIS3 and diabetes, little was known until multiple groups independently generated both global and beta-cell-specific GLIS3 knockout models (174–176). Not only do GLIS3-/- mice die within the first few days of life, but their islet area is approximately 15% that of littermate controls (177). Insulin production was reduced by 80%, making it difficult to assess GSIS in these mice. These groups also found that the endocrine progenitor gene, Ngn3, is a GLIS3 target, and that GLIS3 physically and functionally interacts with the beta-cell transcription factor Pdx1 to regulate insulin transcription. Interestingly, GLIS3 overexpression leads to an upregulation of Ngn3 mRNA in ductal cells, further supporting the role of GLIS3 in pancreatic islet progenitor specification. Pancreatic progenitors, as well as the adult acinar compartment, seem to be unperturbed in GLIS3 knockouts showing an islet progenitor specificity to its role during development. These mechanisms do not tie directly to T1D association but understanding the role of GLIS3 in beta-cell identity may help unveil the mechanisms involved in both T1 and T2D disease susceptibility.
C-type lectin domain family 16, member A (CLEC16a) is a gene locus associated with T1D, multiple sclerosis, and adrenal dysfunction (98, 178, 179). Though genetic associations have been long established, until the work of Soleimanpour et al., a molecular basis for how CLEC16A might increase T1D was unknown. Interestingly, these investigators found that mouse Clec16a interacts with Nrdp1 (an E3 ubiquitin ligase) and has roles in normal GSIS in the beta-cell (180). Pancreatic Clec16a deletion causes reduced ATP levels and mitochondrial oxygen consumption, establishing the factor as a novel regulator of beta-cell mitophagy. Additionally, patients with the T1D-associated SNP in the CLEC16A gene exhibit reductions in CLEC16A expression and perturbed insulin secretion.
These observations of impaired insulin and glucose homeostasis, along with ER-stress in their mouse model, are some of the few providing insight into the non-immune related mechanisms of T1D (180). ER-stress and perturbations in first-phase insulin release are among the earliest signs of T1D, predating immune infiltration and insulitis (181, 182). The role of Clec16a in these processes not only highlights its crucial role in beta-cell function, but also establishes it as a potential player in the first steps of beta-cell vulnerability in T1D.
Delta-like 1 (DLK1), also known as DLL1 or Pref-1 (preadipocyte factor 1), is a transmembrane protein that belongs to the Delta-Notch signaling family. Both mouse and human Dlk1 are known to be subject to genomic imprinting, and Dlk1 is paternally inherited, with the maternal gene being silenced during development. This becomes potentially interesting considering the sexual discordance in inheritance risk in T1DM, as risk of transmission to offspring is 1.7 fold higher from diabetic fathers than mothers (183). However, Wurst et al. found that, in the case of gestational diabetes mellitus (GDM), serum Dlk1 levels were not significantly different between diabetic and control patients (184). Mouse Dlk1 is expressed highly and ubiquitously during development in the embryo and placenta, starting around E11.5, but becomes downregulated in most adult tissues. Adult Dlk1 expression becomes restricted to the beta-cell, bone marrow, pituitary, and adrenal glands. Some evidence suggests that Dlk1 may help undifferentiated cells maintain their pluripotent state, working as a growth factor to maintain proliferation. In preadipocytes, Dlk1 must be downregulated for differentiation to occur (185). Rodent models have remained somewhat controversial, as Dlk1 null mice have partially penetrant neonatal lethality and complex adult and developmental phenotypes, yet conditional loss of function models in various tissues using floxed mice failed to recapitulate null phenotypes (186, 187). Dlk1 beta-cell knockout mice were found to be fully viable with normal islet architecture up to six weeks of age, though glycemic control was not assessed. More thorough analyses of Dlk1 in the beta-cell, including insulin secretion and glucose tolerance, are required to fully understand if and how it may contribute to both function and potentially pathogenesis of T1D.
The genetic basis of T1D pathogenesis is complicated and still poorly understood. A majority of these studies have been conducted using data exclusively from Caucasian patients, and inclusion of multi-ethnic populations is required for a more complete and accurate understanding of genetic variants. The few studies using African-ancestry participants have already yielded unique haplotypes and signatures (188, 189). Additionally, as mentioned above, non-coding DNA regulatory regions make up a majority of T1D associated SNPs, which suggests that genetic variation may be impacting regulatory functions rather than gene-coding abilities (165, 166). Gene expression can be controlled via long-range interactions, with regulatory elements impacting genes that are hundreds of kilobases away. Understanding these potential interactions requires employment of techniques such as chromatin conformation capture, building a more complete picture of how these SNPs may regulate distant genes through physical contact with non-adjacent promoters. Recently, the use of chromatin-accessibility quantitative trait loci (caQTL) and fine mapping analysis expanded the genetic variants and loci associated with T1D and provided novel molecular targets to investigate (190). Whether it be immune or beta-cell related, understanding these “true” gene targets is a vital steppingstone in leveraging this genetic information to develop diagnostic and therapeutic solutions to T1D.
Therapeutic strategies for preventing T1D in high-risk patients have often focused on modulating the immune response to the beta-cell. Newer strategies include methods that focus on the beta cell: reduction of beta-cell dysfunction through the manipulation of ER, oxidative, or cytokine induced cell death. Additionally, functional beta-cell mass replacement strategies through alternative sources, such as stem cells, are being exploited in the field. These strategies, however, will likely have limited clinical utility until autoimmune destruction of the beta-cell replacement can be avoided. Therefore, combinatorial therapeutic programs will likely be required to truly prevent or reverse T1D. Here we describe the state of a few current beta-cell focused therapeutics.
Development of compounds targeting ER-stress pathways are being explored to prevent beta-cell death in early onset T1D. The three UPR sensors PERK, ATF6, and IRE1α regulate apoptosis and thus make a promising target for reducing ER stress and subsequent death in the beta-cell (191). Tauroursdoxycholic acid (TUDCA), a naturally occurring bile acid, can reduce ER-stress by inhibiting the dissociation of BiP from PERK, preventing cell death (192). In a multiple low-dose STZ C57BL/6 mouse model of beta-cell death, TUDCA improved glucose tolerance, increased beta-cell mass, and improved glycemia compared to control diabetic mice (193). The benefits of TUDCA and other UPR chaperones continue to be investigated for their ability to prevent ER-stress induced apoptosis in T1D (194).
An ongoing clinical trial using imatinib mesylate (brand name Gleevec), a tyrosine kinase inhibitor, shows promising results in targeting beta-cell ER stress (195). The efficacy of imatinib for the treatment of various immune-mediated diseases is currently being tested. Initially found to abrogate type 2 diabetes in db/db mice, imatinib treatment in the NOD mouse was able to reverse autoimmune diabetes (196, 197). By blunting IRE1α RNase hyperactivity, imatinib reduces beta-cell apoptosis and preserves physiological function. In humans, a clinical trial found imatinib preserved beta-cell function at 12 months in adults with recent-onset T1D (195). Ongoing studies will investigate dose and duration of therapy as well as safety and efficacy for use in children.
Considering the major role of oxidative stress in T1D pathogenesis, therapies designed to improve antioxidant defenses in beta-cells are another promising avenue for clinical use. Thioredoxin interacting protein (TXNIP), a thioredoxin (TRX) inhibitor of the peroxiredoxin/thioredoxin detoxification pathway has demonstrated clinical potential in both animal models and initial clinical trials (198). The binding of TXNIP to TRX promotes oxidative stress by preventing peroxide clearance. TXNIP is elevated in patients with T1D and T2D (199). In vivo overexpression of TXNIP in mouse beta-cells induces apoptosis, while inhibition is protective against STZ-induced diabetes (200). Anti-diabetic agents including insulin and metformin, were found to augment TXNIP degradation through activation of adenosine monophosphate activated protein kinase (AMPK), supporting the idea that TXNIP may be a viable clinical target (201). Verapamil, which blocks voltage-gated calcium channels, decreased TXNIP and enhanced beta-cell survival in both human and rodent islets (202). A clinical trial in which Verapamil was administered (along with insulin therapy) promoted patient beta-cell function and lowered exogenous insulin requirements (203). TXNIP is expressed in multiple cell types throughout the body and additional clinical trials (NCT04545151, NCT04233034) are ongoing to explore the efficacy of TXNIP inhibition in protecting beta-cells and affecting autoimmune responses in patients with T1D.
For both imatinib, an FDA approved anti-leukemia drug, and verapamil, a widely used anti-hypertensive, the ability to repurpose drugs already on the market and with established safety profiles for T1D is an attractive way to expedite the often lengthy and costly process of bringing drugs to market.
The development of human stem cells for clinical use may provide a long-term solution to T1D without the challenge of organ shortage and HLA mismatch. While for years now, stem cell-derived insulin-producing cells can be generated and studied in the lab, improvements in cell viability, identity, and reproducibility may be needed before they can be applied as a safe and affordable therapy (204). Beta-cell differentiation from multiple cell sources have been attempted, the most promising of which seem to be induced pluripotent stem cell (iPSC)-derived beta-cells (205). Clinical trials are in progress investigating the efficacy of treating T1D with one version of iPSC derived cells designed by Viacyte, Inc. and CRISPR (Clustered Regularly Interspaced Short Palindromic Repeat) Therapeutics (NCT03163511).
Combining iPSC-derived cells with CRISPR gene editing technology allows the potential to correct monoallelic mutations in genes, such as those that cause MODY diabetes. Optimal for T1D treatment, some therapies are also focused on creating “stealthy” beta-cells and islets, that can be specifically engineered to evade immune recognition. One gene of interest is renalase (Rnls), which encodes for an FAD-dependent amine oxidase enzyme that was identified in NIT-1 cells. Rnls deletion elicits beta-cell protection against autoimmune attack. NIT-1 cells carrying the Rnls mutation improved graft survival when transplanted into diabetic NOD mice (206). Using CRISPR, human iPSCs were generated lacking RNLS. These RNLS-/- iPSCs could be successfully differentiated and exhibited normal insulin secretion in vitro. Thus far, the in vivo function of these cells has not been reported. Similarly, some groups have recently generated iPSC lines that are “hypoimmune” by inactivating MHC class I and II genes and overexpressing protective marker CD47 (207, 208). These stem cells evade immune rejection in fully competent recipients, while maintaining their pluripotency. Hypoimmune stem cells have been used to treat pulmonary and cardiovascular disease and have major implications for universal transplantation (209). The widespread availability of techniques such as single-cell RNA sequencing can bolster the design of iPSC derived beta-cells by identifying the gene expression repertoire needed to obtain the appropriate distribution of cell types within the islet (210).
An exciting new technology that may aid in our understanding of the T1D beta-cell is Patch-Seq, a powerful method that can link single-cell transcriptomes with electrophysiology measurements (211). Groups using this technique in the beta-cell may be able to uncover various levels of beta-cell heterogeneity and link it to functionality in both healthy and T1D contexts. The Yoshihara group is also working on combining concepts with their work reproducing disease with 3D organoids engineered to model immune invasion (212). Overexpression of PD-L1 in human islet organoids was able to protect xenografts from immune invasion and restore glucose homeostasis for 50 days in immune competent mice. Similarly, the Melero-Martin group has used a combinatorial approach to incorporate the importance of islet vasculature in their studies. Their “vascular organoids” include microvessels that become perfused during transplantation and even reduce the islet requirement for transplantation, highlighting the importance of vascular cell types for ideal glucose regulation (213). More combinatorial therapies should be explored as we continue to uncover the nuances of the beta-cell and immunological interface of T1D pathology.
We have highlighted the crosstalk between pancreatic beta-cells and the immune system in T1D and potential mechanisms by which innate beta-cell characteristics contribute to T1D initiation and progression. Beta-cells display an increased vulnerability to destruction and can also perpetuate inflammatory and autoimmune responses in a destructive positive feedback loop. Despite recent advances in technologies such as single-cell sequencing and the optimization of differentiation protocols for stem cell-derived beta-cells, we still have an incomplete understanding regarding the dynamics of beta-cell biology in T1D. In particular, the relationship between the transcription factors involved in beta-cell heterogeneity that can influence immune evasion versus immune susceptibility need to be further defined. Beta-cell vulnerability to oxidative stress needs to be further explored, as redox-dependent signaling pathways influence numerous facets of beta-cell biology including the differentiation of beta-cell subtypes in T1D. Many of the aforementioned emerging technologies have been examined in T2D in the islet but have not been studied in T1D. Despite the challenges, more studies of human islets before, during, and after autoimmunity in T1D should be performed to improve our understanding of beta-cells that can resist immune destruction, and therefore our ability to design more effective treatments. Could an exploitation of beta-cell populations that are less vulnerable prevent or delay T1D onset? Perhaps as we understand these “resistant” populations more fully, therapies can target and pharmacologically expand them.
ET, KB, HT, CH, and RB outlined, wrote, and edited the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Our work was supported by the following NIH grant awards R01 DK127497 (HT), R01 DK126456 (HT and CH); R01 DK111483 (CH); F31 DK120217 (ET), T32 GM008111 (KB), and R01 DK120761 (RB). We apologize to any researchers if we inadvertently omitted any of your research articles in this review due to space limitations. Figures created using Servier Medical Art.

1. Sekercioglu N, Lovblom LE, Bjornstad P, Lovshin JA, Lytvyn Y, Boulet G, et al. Risk Factors for Diabetic Kidney Disease in Adults With Longstanding Type 1 Diabetes: Results From the Canadian Study of Longevity in Diabetes. Ren Fail (2019) 41:427–33. doi: 10.1080/0886022X.2019.1614057
2. Patterson CC, Karuranga S, Salpea P, Saeedi P, Dahlquist G, Soltesz G, et al. Worldwide Estimates of Incidence, Prevalence and Mortality of Type 1 Diabetes in Children and Adolescents: Results From the International Diabetes Federation Diabetes Atlas, 9th Edition. Diabetes Res Clin Pract (2019) 157:107842. doi: 10.1016/j.diabres.2019.107842
3. Bruno G, Runzo C, Cavallo-Perin P, Merletti F, Rivetti M, Pinach S, et al. Incidence of Type 1 and Type 2 Diabetes in Adults Aged 30-49 Years: The Population-Based Registry in the Province of Turin, Italy. Diabetes Care (2005) 28:2613–9. doi: 10.2337/diacare.28.11.2613
4. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and Incidence of Type 1 Diabetes in the World: A Systematic Review and Meta-Analysis. Health Promot Perspect (2020) 10:98–115. doi: 10.34172/hpp.2020.18
5. Rewers M, Ludvigsson J. Environmental Risk Factors for Type 1 Diabetes. Lancet (2016) 387:2340–8. doi: 10.1016/S0140-6736(16)30507-4
6. Oram RA, Jones AG, Besser RE, Knight BA, Shields BM, Brown RJ, et al. The Majority of Patients With Long-Duration Type 1 Diabetes Are Insulin Microsecretors and Have Functioning Beta Cells. Diabetologia (2014) 57:187–91. doi: 10.1007/s00125-013-3067-x
7. Pugliese A. Insulitis in the Pathogenesis of Type 1 Diabetes. Pediatr Diabetes (2016) 17(Suppl 22):31–6. doi: 10.1111/pedi.12388
8. Bottazzo GF. Lawrence Lecture. Death of a Beta Cell: Homicide or Suicide? Diabetes Med (1986) 3:119–30. doi: 10.1111/j.1464-5491.1986.tb00722.x
9. Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, et al. How Does Type 1 Diabetes Develop?: The Notion of Homicide or Beta-Cell Suicide Revisited. Diabetes (2011) 60:1370–9. doi: 10.2337/db10-1797
10. Zhang IX, Raghavan M, Satin LS. The Endoplasmic Reticulum and Calcium Homeostasis in Pancreatic Beta Cells. Endocrinology (2020) 161:1–14. doi: 10.1210/endocr/bqz028
11. Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1alpha Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death Under Irremediable ER Stress. Cell Metab (2012) 16:250–64. doi: 10.1016/j.cmet.2012.07.007
12. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator Ire1. Front Mol Biosci (2019) 6:11. doi: 10.3389/fmolb.2019.00011
13. Marre ML, James EA, Piganelli JD. Beta Cell ER Stress and the Implications for Immunogenicity in Type 1 Diabetes. Front Cell Dev Biol (2015) 3:67. doi: 10.3389/fcell.2015.00067
14. Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal Beta-Cell Apoptosis: A Trigger for Autoimmune Diabetes? Diabetes (2000) 49:1–7. doi: 10.2337/diabetes.49.1.1
15. O'Brien BA, Harmon BV, Cameron DP, Allan DJ. Apoptosis Is the Mode of Beta-Cell Death Responsible for the Development of IDDM in the Nonobese Diabetic (NOD) Mouse. Diabetes (1997) 46:750–7. doi: 10.2337/diab.46.5.750
16. Augstein P, Elefanty AG, Allison J, Harrison LC. Apoptosis and Beta-Cell Destruction in Pancreatic Islets of NOD Mice With Spontaneous and Cyclophosphamide-Accelerated Diabetes. Diabetologia (1998) 41:1381–8. doi: 10.1007/s001250051080
17. McClenaghan NH. Physiological Regulation of the Pancreatic {Beta}-Cell: Functional Insights for Understanding and Therapy of Diabetes. Exp Physiol (2007) 92:481–96. doi: 10.1113/expphysiol.2006.034835
18. Ding L, Han L, Dube J, Billadeau DD. WASH Regulates Glucose Homeostasis by Facilitating Glut2 Receptor Recycling in Pancreatic Beta-Cells. Diabetes (2019) 68:377–86. doi: 10.2337/db18-0189
19. Layden BT, Durai V, Lowe WL Jr. G-Protein-Coupled Receptors, Pancreatic Islets, and Diabetes. Nat Educ (2010) 3.
20. Klec C, Ziomek G, Pichler M, Malli R, Graier WF. Calcium Signaling in Ss-Cell Physiology and Pathology: A Revisit. Int J Mol Sci (2019) 20:1–24. doi: 10.3390/ijms20246110
21. Marasco MR, Linnemann AK. Beta-Cell Autophagy in Diabetes Pathogenesis. Endocrinology (2018) 159:2127–41. doi: 10.1210/en.2017-03273
22. Schuit FC, In't Veld PA, Pipeleers DG. Glucose Stimulates Proinsulin Biosynthesis by a Dose-Dependent Recruitment of Pancreatic Beta Cells. Proc Natl Acad Sci USA (1988) 85:3865–9. doi: 10.1073/pnas.85.11.3865
23. Scheuner D, Kaufman RJ. The Unfolded Protein Response: A Pathway That Links Insulin Demand With Beta-Cell Failure and Diabetes. Endocr Rev (2008) 29:317–33. doi: 10.1210/er.2007-0039
24. Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, et al. Islet Beta-Cell Endoplasmic Reticulum Stress Precedes the Onset of Type 1 Diabetes in the Nonobese Diabetic Mouse Model. Diabetes (2012) 61:818–27. doi: 10.2337/db11-1293
25. Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, Wakeham MC, et al. Initiation and Execution of Lipotoxic ER Stress in Pancreatic Beta-Cells. J Cell Sci (2008) 121:2308–18. doi: 10.1242/jcs.026062
26. Lenghel A, Gheorghita AM, Vacaru AM, Vacaru AM. What Is the Sweetest UPR Flavor for the Beta-Cell? That Is Question. Front Endocrinol (Lausanne) (2020) 11:614123. doi: 10.3389/fendo.2020.614123
27. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol Cell Biol (2003) 23:7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003
28. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic Interaction of BiP and ER Stress Transducers in the Unfolded-Protein Response. Nat Cell Biol (2000) 2:326–32. doi: 10.1038/35014014
29. Fonseca SG, Gromada J, Urano F. Endoplasmic Reticulum Stress and Pancreatic Beta-Cell Death. Trends Endocrinol Metab (2011) 22:266–74. doi: 10.1016/j.tem.2011.02.008
30. Han JA, Park SC. Transglutaminase-Dependent Modulation of Transcription Factor Sp1 Activity. Mol Cells (2000) 10:612–8. doi: 10.1007/s10059-000-0612-5
31. Fok JY, Mehta K. Tissue Transglutaminase Induces the Release of Apoptosis Inducing Factor and Results in Apoptotic Death of Pancreatic Cancer Cells. Apoptosis (2007) 12:1455–63. doi: 10.1007/s10495-007-0079-3
32. Jang B, Shin HY, Choi JK, Nguyen du PT, Jeong BH, Ishigami A, et al. Subcellular Localization of Peptidylarginine Deiminase 2 and Citrullinated Proteins in Brains of Scrapie-Infected Mice: Nuclear Localization of PAD2 and Membrane Fraction-Enriched Citrullinated Proteins. J Neuropathol Exp Neurol (2011) 70:116–24. doi: 10.1097/NEN.0b013e318207559e
33. Bruggeman Y, Sodre FMC, Buitinga M, Mathieu C, Overbergh L, Kracht MJL. Targeting Citrullination in Autoimmunity: Insights Learned From Preclinical Mouse Models. Expert Opin Ther Targets (2021) 25:269–81. doi: 10.1080/14728222.2021.1918104
34. Lee HJ, Joo M, Abdolrasulnia R, Young DG, Choi I, Ware LB, et al. Peptidylarginine Deiminase 2 Suppresses Inhibitory {Kappa}B Kinase Activity in Lipopolysaccharide-Stimulated RAW 264.7 Macrophages. J Biol Chem (2010) 285:39655–62. doi: 10.1074/jbc.M110.170290
35. Gundemir S, Colak G, Tucholski J, Johnson GV. Transglutaminase 2: A Molecular Swiss Army Knife. Biochim Biophys Acta (2012) 1823:406–19. doi: 10.1016/j.bbamcr.2011.09.012
36. Marre ML, McGinty JW, Chow IT, DeNicola ME, Beck NW, Kent SC, et al. Modifying Enzymes Are Elicited by ER Stress, Generating Epitopes That Are Selectively Recognized by CD4(+) T Cells in Patients With Type 1 Diabetes. Diabetes (2018) 67:1356–68. doi: 10.2337/db17-1166
37. Marre ML, Profozich JL, Coneybeer JT, Geng X, Bertera S, Ford MJ, et al. Inherent ER Stress in Pancreatic Islet Beta Cells Causes Self-Recognition by Autoreactive T Cells in Type 1 Diabetes. J Autoimmun (2016) 72:33–46. doi: 10.1016/j.jaut.2016.04.009
38. McGinty JW, Marre ML, Bajzik V, Piganelli JD, James EA. T Cell Epitopes and Post-Translationally Modified Epitopes in Type 1 Diabetes. Curr Diabetes Rep (2015) 15:90. doi: 10.1007/s11892-015-0657-7
39. Russo L, Marsella C, Nardo G, Massignan T, Alessio M, Piermarini E, et al. Transglutaminase 2 Transamidation Activity During First-Phase Insulin Secretion: Natural Substrates in INS-1e. Acta Diabetol (2013) 50:61–72. doi: 10.1007/s00592-012-0381-6
40. Sidney J, Vela JL, Friedrich D, Kolla R, von Herrath M, Wesley JD, et al. Low HLA Binding of Diabetes-Associated CD8+ T-Cell Epitopes Is Increased by Post Translational Modifications. BMC Immunol (2018) 19:12. doi: 10.1186/s12865-018-0250-3
41. Sodre FMC, Bissenova S, Bruggeman Y, Tilvawala R, Cook DP, Berthault C, et al. Peptidylarginine Deiminase Inhibition Prevents Diabetes Development in NOD Mice. Diabetes (2021) 70:516–28. doi: 10.2337/db20-0421
42. Rodriguez-Calvo T, Johnson JD, Overbergh L, Dunne JL. Neoepitopes in Type 1 Diabetes: Etiological Insights, Biomarkers and Therapeutic Targets. Front Immunol (2021) 12:667989. doi: 10.3389/fimmu.2021.667989
43. Pugliese A. Autoreactive T Cells in Type 1 Diabetes. J Clin Invest (2017) 127:2881–91. doi: 10.1172/JCI94549
44. Thomaidou S, Zaldumbide A, Roep BO. Islet Stress, Degradation and Autoimmunity. Diabetes Obes Metab (2018) 20(Suppl 2):88–94. doi: 10.1111/dom.13387
45. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity Against a Defective Ribosomal Insulin Gene Product in Type 1 Diabetes. Nat Med (2017) 23:501–7. doi: 10.1038/nm.4289
46. Baker RL, Rihanek M, Hohenstein AC, Nakayama M, Michels A, Gottlieb PA, et al. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes (2019) 68:1830–40. doi: 10.2337/db19-0128
47. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T Cells in Type 1 Diabetes Recognize Epitopes Formed by Peptide Fusion. Science (2016) 351:711–4. doi: 10.1126/science.aad2791
48. Baker RL, Jamison BL, Haskins K. Hybrid Insulin Peptides Are Neo-Epitopes for CD4 T Cells in Autoimmune Diabetes. Curr Opin Endocrinol diabetes Obes (2019) 26:195–200. doi: 10.1097/MED.0000000000000490
49. Wiles TA, Powell R, Michel R, Beard KS, Hohenstein A, Bradley B, et al. Identification of Hybrid Insulin Peptides (HIPs) in Mouse and Human Islets by Mass Spectrometry. J Proteome Res (2019) 18:814–25. doi: 10.1021/acs.jproteome.8b00875
50. Reed B, Crawford F, Hill RC, Jin N, White J, Krovi SH, et al. Lysosomal Cathepsin Creates Chimeric Epitopes for Diabetogenic CD4 T Cells via Transpeptidation. J Exp Med (2021) 218:1–17. doi: 10.1084/jem.20192135
51. Betteridge DJ. What Is Oxidative Stress? Metabolism (2000) 49:3–8. doi: 10.1016/S0026-0495(00)80077-3
52. Newsholme P, Rebelato E, Abdulkader F, Krause M, Carpinelli A, Curi R. Reactive Oxygen and Nitrogen Species Generation, Antioxidant Defenses, and Beta-Cell Function: A Critical Role for Amino Acids. J Endocrinol (2012) 214:11–20. doi: 10.1530/JOE-12-0072
53. Lei XG, Vatamaniuk MZ. Two Tales of Antioxidant Enzymes on Beta Cells and Diabetes. Antioxid Redox Signal (2011) 14:489–503. doi: 10.1089/ars.2010.3416
54. Veal EA, Day AM, Morgan BA. Hydrogen Peroxide Sensing and Signaling. Mol Cell (2007) 26:1–14. doi: 10.1016/j.molcel.2007.03.016
55. Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, et al. ROS Signaling, Oxidative Stress and Nrf2 in Pancreatic Beta-Cell Function. Toxicol Appl Pharmacol (2010) 244:77–83. doi: 10.1016/j.taap.2009.05.025
56. Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. Beta-Cell Pdx1 Expression Is Essential for the Glucoregulatory, Proliferative, and Cytoprotective Actions of Glucagon-Like Peptide-1. Diabetes (2005) 54:482–91. doi: 10.2337/diabetes.54.2.482
57. Elsner M, Gehrmann W, Lenzen S. Peroxisome-Generated Hydrogen Peroxide as Important Mediator of Lipotoxicity in Insulin-Producing Cells. Diabetes (2011) 60:200–8. doi: 10.2337/db09-1401
58. Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation Between Antioxidant Enzyme Gene Expression and Antioxidative Defense Status of Insulin-Producing Cells. Diabetes (1997) 46:1733–42. doi: 10.2337/diab.46.11.1733
59. Grankvist K, Marklund SL, Taljedal IB. CuZn-Superoxide Dismutase, Mn-Superoxide Dismutase, Catalase and Glutathione Peroxidase in Pancreatic Islets and Other Tissues in the Mouse. Biochem J (1981) 199:393–8. doi: 10.1042/bj1990393
60. Drews G, Krippeit-Drews P, Dufer M. Oxidative Stress and Beta-Cell Dysfunction. Pflugers Arch (2010) 460:703–18. doi: 10.1007/s00424-010-0862-9
61. Miki A, Ricordi C, Sakuma Y, Yamamoto T, Misawa R, Mita A, et al. Divergent Antioxidant Capacity of Human Islet Cell Subsets: A Potential Cause of Beta-Cell Vulnerability in Diabetes and Islet Transplantation. PloS One (2018) 13:e0196570. doi: 10.1371/journal.pone.0196570
62. Walter U, Franzke A, Sarukhan A, Zober C, von Boehmer H, Buer J, et al. Monitoring Gene Expression of TNFR Family Members by Beta-Cells During Development of Autoimmune Diabetes. Eur J Immunol (2000) 30:1224–32. doi: 10.1002/1521-4141(200004)30:4<1224::AID-IMMU1224>3.0.CO;2-B
63. Tomita T. Immunocytochemical Localization of Cleaved Caspase-3 in Pancreatic Islets From Type 1 Diabetic Subjects. Islets (2010) 2:24–9. doi: 10.4161/isl.2.1.10041
64. Marroqui L, Dos Santos RS, Floyel T, Grieco FA, Santin I, Op de Beeck A, et al. TYK2, a Candidate Gene for Type 1 Diabetes, Modulates Apoptosis and the Innate Immune Response in Human Pancreatic Beta-Cells. Diabetes (2015) 64:3808–17. doi: 10.2337/db15-0362
65. Wilcox NS, Rui J, Hebrok M, Herold KC. Life and Death of Beta Cells in Type 1 Diabetes: A Comprehensive Review. J Autoimmun (2016) 71:51–8. doi: 10.1016/j.jaut.2016.02.001
66. Tomita T. Apoptosis of Pancreatic Beta-Cells in Type 1 Diabetes. Bosn J Basic Med Sci (2017) 17:183–93. doi: 10.17305/bjbms.2017.1961
67. Mallone R, Eizirik DL. Presumption of Innocence for Beta Cells: Why Are They Vulnerable Autoimmune Targets in Type 1 Diabetes? Diabetologia (2020) 63:1999–2006. doi: 10.1007/s00125-020-05176-7
68. Jansson L, Barbu A, Bodin B, Drott CJ, Espes D, Gao X, et al. Pancreatic Islet Blood Flow and Its Measurement. Ups J Med Sci (2016) 121:81–95. doi: 10.3109/03009734.2016.1164769
69. Henderson JR, Moss MC. A Morphometric Study of the Endocrine and Exocrine Capillaries of the Pancreas. Q J Exp Physiol (1985) 70:347–56. doi: 10.1113/expphysiol.1985.sp002920
70. Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, et al. VEGF-Dependent Plasticity of Fenestrated Capillaries in the Normal Adult Microvasculature. Am J Physiol Heart Circ Physiol (2006) 290:H560–76. doi: 10.1152/ajpheart.00133.2005
71. Magnuson AM, Thurber GM, Kohler RH, Weissleder R, Mathis D, Benoist C. Population Dynamics of Islet-Infiltrating Cells in Autoimmune Diabetes. Proc Natl Acad Sci USA (2015) 112:1511–6. doi: 10.1073/pnas.1423769112
72. Costa FR, Francozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut Microbiota Translocation to the Pancreatic Lymph Nodes Triggers NOD2 Activation and Contributes to T1D Onset. J Exp Med (2016) 213:1223–39. doi: 10.1084/jem.20150744
73. Dedrick S, Sundaresh B, Huang Q, Brady C, Yoo T, Cronin C, et al. The Role of Gut Microbiota and Environmental Factors in Type 1 Diabetes Pathogenesis. Front Endocrinol (Lausanne) (2020) 11:78. doi: 10.3389/fendo.2020.00078
74. Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased Intestinal Permeability Precedes Clinical Onset of Type 1 Diabetes. Diabetologia (2006) 49:2824–7. doi: 10.1007/s00125-006-0465-3
75. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate Immunity and Intestinal Microbiota in the Development of Type 1 Diabetes. Nature (2008) 455:1109–13. doi: 10.1038/nature07336
76. Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota Regulates Type 1 Diabetes Through Toll-Like Receptors. Proc Natl Acad Sci USA (2015) 112:9973–7. doi: 10.1073/pnas.1508740112
77. Markle JG, Frank DN, Adeli K, von Bergen M, Danska JS. Microbiome Manipulation Modifies Sex-Specific Risk for Autoimmunity. Gut Microbes (2014) 5:485–93. doi: 10.4161/gmic.29795
78. Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender Bias in Autoimmunity Is Influenced by Microbiota. Immunity (2013) 39:400–12. doi: 10.1016/j.immuni.2013.08.013
79. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex Differences in the Gut Microbiome Drive Hormone-Dependent Regulation of Autoimmunity. Science (2013) 339:1084–8. doi: 10.1126/science.1233521
80. Stewart CJ, Ajami NJ, O'Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal Development of the Gut Microbiome in Early Childhood From the TEDDY Study. Nature (2018) 562:583–8. doi: 10.1038/s41586-018-0617-x
81. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The Human Gut Microbiome in Early-Onset Type 1 Diabetes From the TEDDY Study. Nature (2018) 562:589–94. doi: 10.1038/s41586-018-0620-2
82. Dula SB, Jecmenica M, Wu R, Jahanshahi P, Verrilli GM, Carter JD, et al. Evidence That Low-Grade Systemic Inflammation Can Induce Islet Dysfunction as Measured by Impaired Calcium Handling. Cell Calcium (2010) 48:133–42. doi: 10.1016/j.ceca.2010.07.007
83. Ramadan JW, Steiner SR, O'Neill CM, Nunemaker CS. The Central Role of Calcium in the Effects of Cytokines on Beta-Cell Function: Implications for Type 1 and Type 2 Diabetes. Cell Calcium (2011) 50:481–90. doi: 10.1016/j.ceca.2011.08.005
84. Miani M, Barthson J, Colli ML, Brozzi F, Cnop M, Eizirik DL. Endoplasmic Reticulum Stress Sensitizes Pancreatic Beta Cells to Interleukin-1beta-Induced Apoptosis via Bim/A1 Imbalance. Cell Death Dis (2013) 4:e701. doi: 10.1038/cddis.2013.236
85. Gurzov EN, Ortis F, Cunha DA, Gosset G, Li M, Cardozo AK, et al. Signaling by IL-1beta+IFN-Gamma and ER Stress Converge on DP5/Hrk Activation: A Novel Mechanism for Pancreatic Beta-Cell Apoptosis. Cell Death Differ (2009) 16:1539–50. doi: 10.1038/cdd.2009.99
86. Zhang S, Kim KH. TNF-Alpha Inhibits Glucose-Induced Insulin Secretion in a Pancreatic Beta-Cell Line (INS-1). FEBS Lett (1995) 377:237–9. doi: 10.1016/0014-5793(95)01272-9
87. Collier JJ, Burke SJ, Eisenhauer ME, Lu D, Sapp RC, Frydman CJ, et al. Pancreatic Beta-Cell Death in Response to Pro-Inflammatory Cytokines Is Distinct From Genuine Apoptosis. PloS One (2011) 6:e22485. doi: 10.1371/journal.pone.0022485
88. Dickerson MT, Bogart AM, Altman MK, Milian SC, Jordan KL, Dadi PK, et al. Cytokine-Mediated Changes in K(+) Channel Activity Promotes an Adaptive Ca(2+) Response That Sustains Beta-Cell Insulin Secretion During Inflammation. Sci Rep (2018) 8:1158. doi: 10.1038/s41598-018-19600-x
89. El Muayed M, Billings LK, Raja MR, Zhang X, Park PJ, Newman MV, et al. Acute Cytokine-Mediated Downregulation of the Zinc Transporter ZnT8 Alters Pancreatic Beta-Cell Function. J Endocrinol (2010) 206:159–69. doi: 10.1677/JOE-09-0420
90. Oleson BJ, Corbett JA. Dual Role of Nitric Oxide in Regulating the Response of Beta Cells to DNA Damage. Antioxid Redox Signal (2018) 29:1432–45. doi: 10.1089/ars.2017.7351
91. Oleson BJ, Broniowska KA, Naatz A, Hogg N, Tarakanova VL, Corbett JA. Nitric Oxide Suppresses Beta-Cell Apoptosis by Inhibiting the DNA Damage Response. Mol Cell Biol (2016) 36:2067–77. doi: 10.1128/MCB.00262-16
92. Akerfeldt MC, Howes J, Chan JY, Stevens VA, Boubenna N, McGuire HM, et al. Cytokine-Induced Beta-Cell Death Is Independent of Endoplasmic Reticulum Stress Signaling. Diabetes (2008) 57:3034–44. doi: 10.2337/db07-1802
93. Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, et al. Human Islets Contain Four Distinct Subtypes of Beta Cells. Nat Commun (2016) 7:11756. doi: 10.1038/ncomms11756
94. Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab (2016) 24:389–401. doi: 10.1016/j.cmet.2016.06.020
95. van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dolleman S, et al. Virgin Beta Cells Persist Throughout Life at a Neogenic Niche Within Pancreatic Islets. Cell Metab (2017) 25:911–926 e6. doi: 10.1016/j.cmet.2017.03.017
96. Salomon D, Meda P. Heterogeneity and Contact-Dependent Regulation of Hormone Secretion by Individual B Cells. Exp Cell Res (1986) 162:507–20. doi: 10.1016/0014-4827(86)90354-X
97. Hiriart M, Ramirez-Medeles MC. Functional Subpopulations of Individual Pancreatic B-Cells in Culture. Endocrinology (1991) 128:3193–8. doi: 10.1210/endo-128-6-3193
98. Skinningsrud B, Husebye ES, Pearce SH, McDonald DO, Brandal K, Wolff AB, et al. Polymorphisms in CLEC16A and CIITA at 16p13 Are Associated With Primary Adrenal Insufficiency. J Clin Endocrinol Metab (2008) 93:3310–7. doi: 10.1210/jc.2008-0821
99. Lee EY, Lee ZH, Song YW. CXCL10 and Autoimmune Diseases. Autoimmun Rev (2009) 8:379–83. doi: 10.1016/j.autrev.2008.12.002
100. Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of Adult Pancreatic Alpha-Cells to Beta-Cells After Extreme Beta-Cell Loss. Nature (2010) 464:1149–54. doi: 10.1038/nature08894
101. Bader E, Migliorini A, Gegg M, Moruzzi N, Gerdes J, Roscioni SS, et al. Identification of Proliferative and Mature Beta-Cells in the Islets of Langerhans. Nature (2016) 535:430–4. doi: 10.1038/nature18624
102. Kravets V, Dwulet JM, Schleicher WE, Hodson DJ, Davis AM, Piscopio RA, et al. Functional Architecture of the Pancreatic Islets: First Responder Cells Drive the First-Phase [Ca2+] Response. bioRxiv (2020) 2020.12.22.424082. doi: 10.1101/2020.12.22.424082
103. Li J, Klughammer J, Farlik M, Penz T, Spittler A, Barbieux C, et al. Single-Cell Transcriptomes Reveal Characteristic Features of Human Pancreatic Islet Cell Types. EMBO Rep (2016) 17:178–87. doi: 10.15252/embr.201540946
104. Wang YJ, Golson ML, Schug J, Traum D, Liu C, Vivek K, et al. Single-Cell Mass Cytometry Analysis of the Human Endocrine Pancreas. Cell Metab (2016) 24:616–26. doi: 10.1016/j.cmet.2016.09.007
105. Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-Cell Population Structure. Cell Syst (2016) 3:346–60.e4. doi: 10.1016/j.cels.2016.08.011
106. Aguayo-Mazzucato C, van Haaren M, Mruk M, Lee TB Jr., Crawford C, Hollister-Lock J, et al. Beta Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab (2017) 25:898–910.e5. doi: 10.1016/j.cmet.2017.03.015
107. Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst (2016) 3:385–94.e3. doi: 10.1016/j.cels.2016.09.002
108. Zeng C, Mulas F, Sui Y, Guan T, Miller N, Tan Y, et al. Pseudotemporal Ordering of Single Cells Reveals Metabolic Control of Postnatal Beta Cell Proliferation. Cell Metab (2017) 25:1160–75.e11. doi: 10.1016/j.cmet.2017.04.014
109. Qiu WL, Zhang YW, Feng Y, Li LC, Yang L, Xu CR. Deciphering Pancreatic Islet Beta Cell and Alpha Cell Maturation Pathways and Characteristic Features at the Single-Cell Level. Cell Metab (2018) 27:702. doi: 10.1016/j.cmet.2018.01.017
110. Farack L, Golan M, Egozi A, Dezorella N, Bahar Halpern K, Ben-Moshe S, et al. Transcriptional Heterogeneity of Beta Cells in the Intact Pancreas. Dev Cell (2019) 48:115–25.e4. doi: 10.1016/j.devcel.2018.11.001
111. Lira SA, Furtado GC. The Biology of Chemokines and Their Receptors. Immunol Res (2012) 54:111–20. doi: 10.1007/s12026-012-8313-7
113. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation Between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell (2019) 35:885–900.e10. doi: 10.1016/j.ccell.2019.05.004
114. Griffith JW, Sokol CL, Luster AD. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu Rev Immunol (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
115. Martin AP, Rankin S, Pitchford S, Charo IF, Furtado GC, Lira SA. Increased Expression of CCL2 in Insulin-Producing Cells of Transgenic Mice Promotes Mobilization of Myeloid Cells From the Bone Marrow, Marked Insulitis, and Diabetes. Diabetes (2008) 57:3025–33. doi: 10.2337/db08-0625
116. Gschwandtner M, Derler R, Midwood KS. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front Immunol (2019) 10:2759. doi: 10.3389/fimmu.2019.02759
117. Daly C, Rollins BJ. Monocyte Chemoattractant Protein-1 (CCL2) in Inflammatory Disease and Adaptive Immunity: Therapeutic Opportunities and Controversies. Microcirculation (2003) 10:247–57. doi: 10.1080/mic.10.3-4.247.257
118. Chen MC, Proost P, Gysemans C, Mathieu C, Eizirik DL. Monocyte Chemoattractant Protein-1 Is Expressed in Pancreatic Islets From Prediabetic NOD Mice and in Interleukin-1 Beta-Exposed Human and Rat Islet Cells. Diabetologia (2001) 44:325–32. doi: 10.1007/s001250051622
119. Sarkar SA, Lee CE, Victorino F, Nguyen TT, Walters JA, Burrack A, et al. Expression and Regulation of Chemokines in Murine and Human Type 1 Diabetes. Diabetes (2012) 61:436–46. doi: 10.2337/db11-0853
120. Cowley MJ, Weinberg A, Zammit NW, Walters SN, Hawthorne WJ, Loudovaris T, et al. Human Islets Express a Marked Proinflammatory Molecular Signature Prior to Transplantation. Cell Transplant (2012) 21:2063–78. doi: 10.3727/096368911X627372
121. Burke SJ, Stadler K, Lu D, Gleason E, Han A, Donohoe DR, et al. IL-1beta Reciprocally Regulates Chemokine and Insulin Secretion in Pancreatic Beta-Cells via NF-Kappab. Am J Physiol Endocrinol Metab (2015) 309:E715–26. doi: 10.1152/ajpendo.00153.2015
122. Burke SJ, Goff MR, Updegraff BL, Lu D, Brown PL, Minkin SC Jr., et al. Regulation of the CCL2 Gene in Pancreatic Beta-Cells by IL-1beta and Glucocorticoids: Role of MKP-1. PloS One (2012) 7:e46986. doi: 10.1371/journal.pone.0046986
123. Jansen A, Homo-Delarche F, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA. Immunohistochemical Characterization of Monocytes-Macrophages and Dendritic Cells Involved in the Initiation of the Insulitis and Beta-Cell Destruction in NOD Mice. Diabetes (1994) 43:667–75. doi: 10.2337/diab.43.5.667
124. Wang YJ, Traum D, Schug J, Gao L, Liu C, Consortium H, et al. Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab (2019) 29:769–83.e4. doi: 10.1016/j.cmet.2019.01.003
125. Xu R, Li Y, Yan H, Zhang E, Huang X, Chen Q, et al. CCL2 Promotes Macrophages-Associated Chemoresistance via MCPIP1 Dual Catalytic Activities in Multiple Myeloma. Cell Death Dis (2019) 10:781. doi: 10.1038/s41419-019-2012-4
126. Carvalho-Pinto C, Garcia MI, Gomez L, Ballesteros A, Zaballos A, Flores JM, et al. Leukocyte Attraction Through the CCR5 Receptor Controls Progress From Insulitis to Diabetes in Non-Obese Diabetic Mice. Eur J Immunol (2004) 34:548–57. doi: 10.1002/eji.200324285
127. Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Hollander GA, et al. Beta Cells Are Responsible for CXCR3-Mediated T-Cell Infiltration in Insulitis. Nat Med (2002) 8:1414–20. doi: 10.1038/nm1202-792
128. Hanifi-Moghaddam P, Kappler S, Seissler J, Muller-Scholze S, Martin S, Roep BO, et al. Altered Chemokine Levels in Individuals at Risk of Type 1 Diabetes Mellitus. Diabetes Med (2006) 23:156–63. doi: 10.1111/j.1464-5491.2005.01743.x
129. Jamali Z, Nazari M, Khoramdelazad H, Hakimizadeh E, Mahmoodi M, Karimabad MN, et al. Expression of CC Chemokines CCL2, CCL5, and CCL11 Is Associated With Duration of Disease and Complications in Type-1 Diabetes: A Study on Iranian Diabetic Patients. Clin Lab (2013) 59:993–1001. doi: 10.7754/Clin.Lab.2012.120810
130. Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-Specific Chemokine Receptor CXCR3: Regulation, Chemokine Binding and Gene Localization. Eur J Immunol (1998) 28:3696–705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W
131. Vazirinejad R, Ahmadi Z, Kazemi Arababadi M, Hassanshahi G, Kennedy D. The Biological Functions, Structure and Sources of CXCL10 and Its Outstanding Part in the Pathophysiology of Multiple Sclerosis. Neuroimmunomodulation (2014) 21:322–30. doi: 10.1159/000357780
132. Nigi L, Brusco N, Grieco GE, Licata G, Krogvold L, Marselli L, et al. Pancreatic Alpha-Cells Contribute Together With Beta-Cells to CXCL10 Expression in Type 1 Diabetes. Front Endocrinol (Lausanne) (2020) 11:630. doi: 10.3389/fendo.2020.00630
133. Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, et al. Chemokine Receptor Specific for IP10 and Mig: Structure, Function, and Expression in Activated T-Lymphocytes. J Exp Med (1996) 184:963–9. doi: 10.1084/jem.184.3.963
134. Luster AD, Ravetch JV. Biochemical Characterization of a Gamma Interferon-Inducible Cytokine (IP-10). J Exp Med (1987) 166:1084–97. doi: 10.1084/jem.166.4.1084
135. Nagaoka K, Nojima H, Watanabe F, Chang KT, Christenson RK, Sakai S, et al. Regulation of Blastocyst Migration, Apposition, and Initial Adhesion by a Chemokine, Interferon Gamma-Inducible Protein 10 kDa (IP-10), During Early Gestation. J Biol Chem (2003) 278:29048–56. doi: 10.1074/jbc.M300470200
136. Yates-Binder CC, Rodgers M, Jaynes J, Wells A, Bodnar RJ, Turner T. An IP-10 (CXCL10)-Derived Peptide Inhibits Angiogenesis. PloS One (2012) 7:e40812. doi: 10.1371/journal.pone.0040812
137. Van Belle TL, Taylor P, von Herrath MG. Mouse Models for Type 1 Diabetes. Drug Discov Today Dis Models (2009) 6:41–5. doi: 10.1016/j.ddmod.2009.03.008
138. Christoffersson G, Flodstrom-Tullberg M. Mouse Models of Virus-Induced Type 1 Diabetes. Methods Mol Biol (2020) 2128:93–105. doi: 10.1007/978-1-0716-0385-7_7
139. Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MB. Among CXCR3 Chemokines, IFN-Gamma-Inducible Protein of 10 kDa (CXC Chemokine Ligand (CXCL) 10) But Not Monokine Induced by IFN-Gamma (CXCL9) Imprints a Pattern for the Subsequent Development of Autoimmune Disease. J Immunol (2003) 171:6838–45. doi: 10.4049/jimmunol.171.12.6838
140. Shimada A, Oikawa Y, Yamada Y, Okubo Y, Narumi S. The Role of the CXCL10/CXCR3 System in Type 1 Diabetes. Rev Diabetes Stud (2009) 6:81–4. doi: 10.1900/RDS.2009.6.81
141. Morimoto J, Yoneyama H, Shimada A, Shigihara T, Yamada S, Oikawa Y, et al. CXC Chemokine Ligand 10 Neutralization Suppresses the Occurrence of Diabetes in Nonobese Diabetic Mice Through Enhanced Beta Cell Proliferation Without Affecting Insulitis. J Immunol (2004) 173:7017–24. doi: 10.4049/jimmunol.173.11.7017
142. Schulthess FT, Paroni F, Sauter NS, Shu L, Ribaux P, Haataja L, et al. CXCL10 Impairs Beta Cell Function and Viability in Diabetes Through TLR4 Signaling. Cell Metab (2009) 9:125–39. doi: 10.1016/j.cmet.2009.01.003
143. Lee JH, Kim B, Jin WJ, Kim HH, Ha H, Lee ZH. Pathogenic Roles of CXCL10 Signaling Through CXCR3 and TLR4 in Macrophages and T Cells: Relevance for Arthritis. Arthritis Res Ther (2017) 19:163. doi: 10.1186/s13075-017-1353-6
144. Arnush M, Heitmeier MR, Scarim AL, Marino MH, Manning PT, Corbett JA. IL-1 Produced and Released Endogenously Within Human Islets Inhibits Beta Cell Function. J Clin Invest (1998) 102:516–26. doi: 10.1172/JCI844
145. Saitoh N, Awaya A, Sakudo A, SungWook S, Saeki K, Matsumoto Y, et al. Serum Thymic Factor Prevents LPS-Induced Pancreatic Cell Damage in Mice via Up-Regulation of Bcl-2 Expression in Pancreas. Microbiol Immunol (2004) 48:629–38. doi: 10.1111/j.1348-0421.2004.tb03471.x
146. Kanazawa Y, Komeda K, Sato S, Mori S, Akanuma K, Takaku F. Non-Obese-Diabetic Mice: Immune Mechanisms of Pancreatic Beta-Cell Destruction. Diabetologia (1984) 27 Suppl:113–5. doi: 10.1007/BF00275663
147. Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In Situ Characterization of Autoimmune Phenomena and Expression of HLA Molecules in the Pancreas in Diabetic Insulitis. N Engl J Med (1985) 313:353–60. doi: 10.1056/NEJM198508083130604
148. Slattery R. Transgenic Approaches to Understanding the Role of MHC Genes in Insulin Dependent Diabetes Mellitus. II. The Non-Obese Diabetic (NOD) Mouse. Baillieres Clin Endocrinol Metab (1991) 5:449–54. doi: 10.1016/S0950-351X(05)80141-7
149. Roep BO. The Role of T-Cells in the Pathogenesis of Type 1 Diabetes: From Cause to Cure. Diabetologia (2003) 46:305–21. doi: 10.1007/s00125-003-1089-5
150. Russell MA, Redick SD, Blodgett DM, Richardson SJ, Leete P, Krogvold L, et al. HLA Class II Antigen Processing And Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet Beta-Cells From Donors With Type 1 Diabetes. Diabetes (2019) 68:988–1001. doi: 10.2337/db18-0686
151. Richardson SJ, Rodriguez-Calvo T, Gerling IC, Mathews CE, Kaddis JS, Russell MA, et al. Islet Cell Hyperexpression of HLA Class I Antigens: A Defining Feature in Type 1 Diabetes. Diabetologia (2016) 59:2448–58. doi: 10.1007/s00125-016-4067-4
152. Leiter EH, Christianson GJ, Serreze DV, Ting AT, Worthen SM. MHC Antigen Induction by Interferon Gamma on Cultured Mouse Pancreatic Beta Cells and Macrophages. Genetic Analysis of Strain Differences and Discovery of an "Occult" Class I-Like Antigen in NOD/Lt Mice. J Exp Med (1989) 170:1243–62. doi: 10.1084/jem.170.4.1243
153. Stassi G, De Maria R, Trucco G, Rudert W, Testi R, Galluzzo A, et al. Nitric Oxide Primes Pancreatic Beta Cells for Fas-Mediated Destruction in Insulin-Dependent Diabetes Mellitus. J Exp Med (1997) 186:1193–200. doi: 10.1084/jem.186.8.1193
154. Moriwaki M, Itoh N, Miyagawa J, Yamamoto K, Imagawa A, Yamagata K, et al. Fas and Fas Ligand Expression in Inflamed Islets in Pancreas Sections of Patients With Recent-Onset Type I Diabetes Mellitus. Diabetologia (1999) 42:1332–40. doi: 10.1007/s001250051446
155. Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, et al. Requirement of Fas for the Development of Autoimmune Diabetes in Nonobese Diabetic Mice. J Exp Med (1997) 186:613–8. doi: 10.1084/jem.186.4.613
156. Sia C, Hanninen A. Apoptosis in Autoimmune Diabetes: The Fate of Beta-Cells in the Cleft Between Life and Death. Rev Diabetes Stud (2006) 3:39–46. doi: 10.1900/RDS.2006.3.39
157. Amrani A, Verdaguer J, Thiessen S, Bou S, Santamaria P. IL-1alpha, IL-1beta, and IFN-Gamma Mark Beta Cells for Fas-Dependent Destruction by Diabetogenic CD4(+) T Lymphocytes. J Clin Invest (2000) 105:459–68. doi: 10.1172/JCI8185
158. Loweth AC, Williams GT, James RF, Scarpello JH, Morgan NG. Human Islets of Langerhans Express Fas Ligand and Undergo Apoptosis in Response to Interleukin-1beta and Fas Ligation. Diabetes (1998) 47:727–32. doi: 10.2337/diabetes.47.5.727
159. Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in Diabetes. Diabetes (2005) 54 Suppl 2:S52–61. doi: 10.2337/diabetes.54.suppl_2.S52
160. Keenan HA, Sun JK, Levine J, Doria A, Aiello LP, Eisenbarth G, et al. Residual Insulin Production and Pancreatic Ss-Cell Turnover After 50 Years of Diabetes: Joslin Medalist Study. Diabetes (2010) 59:2846–53. doi: 10.2337/db10-0676
161. Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. Beta Cells That Resist Immunological Attack Develop During Progression of Autoimmune Diabetes in NOD Mice. Cell Metab (2017) 25:727–38. doi: 10.1016/j.cmet.2017.01.005
162. Leete P, Oram RA, McDonald TJ, Shields BM, Ziller C, T.S. TEAM, et al. Studies of Insulin and Proinsulin in Pancreas and Serum Support the Existence of Aetiopathological Endotypes of Type 1 Diabetes Associated With Age at Diagnosis. Diabetologia (2020) 63:1258–67. doi: 10.1007/s00125-020-05115-6
163. Leete P, Willcox A, Krogvold L, Dahl-Jorgensen K, Foulis AK, Richardson SJ, et al. Differential Insulitic Profiles Determine the Extent of Beta-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes (2016) 65:1362–9. doi: 10.2337/db15-1615
164. Arif S, Leete P, Nguyen V, Marks K, Nor NM, Estorninho M, et al. Blood and Islet Phenotypes Indicate Immunological Heterogeneity in Type 1 Diabetes. Diabetes (2014) 63:3835–45. doi: 10.2337/db14-0365
165. Grant SFA, Wells AD, Rich SS. Next Steps in the Identification of Gene Targets for Type 1 Diabetes. Diabetologia (2020) 63:2260–9. doi: 10.1007/s00125-020-05248-8
166. Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine Mapping of Type 1 Diabetes Susceptibility Loci and Evidence for Colocalization of Causal Variants With Lymphoid Gene Enhancers. Nat Genet (2015) 47:381–6. doi: 10.1038/ng.3245
167. Bell GI, Horita S, Karam JH. A Polymorphic Locus Near the Human Insulin Gene Is Associated With Insulin-Dependent Diabetes Mellitus. Diabetes (1984) 33:176–83. doi: 10.2337/diab.33.2.176
168. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The Human Pancreatic Islet Transcriptome: Expression of Candidate Genes for Type 1 Diabetes and the Impact of Pro-Inflammatory Cytokines. PloS Genet (2012) 8:e1002552. doi: 10.1371/journal.pgen.1002552
169. Pugliese A, Zeller M, Fernandez A Jr., Zalcberg LJ, Bartlett RJ, Ricordi C, et al. The Insulin Gene Is Transcribed in the Human Thymus and Transcription Levels Correlated With Allelic Variation at the INS VNTR-IDDM2 Susceptibility Locus for Type 1 Diabetes. Nat Genet (1997) 15:293–7. doi: 10.1038/ng0397-293
170. Wen X, Yang Y. Emerging Roles of GLIS3 in Neonatal Diabetes, Type 1 and Type 2 Diabetes. J Mol Endocrinol (2017) 58:R73–85. doi: 10.1530/JME-16-0232
171. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-Wide Association Study and Meta-Analysis Find That Over 40 Loci Affect Risk of Type 1 Diabetes. Nat Genet (2009) 41:703–7. doi: 10.1038/ng.381
172. Kiani AK, John P, Bhatti A, Zia A, Shahid G, Akhtar P, et al. Association of 32 Type 1 Diabetes Risk Loci in Pakistani Patients. Diabetes Res Clin Pract (2015) 108:137–42. doi: 10.1016/j.diabres.2015.01.022
173. Awata T, Yamashita H, Kurihara S, Morita-Ohkubo T, Miyashita Y, Katayama S, et al. A Low-Frequency GLIS3 Variant Associated With Resistance to Japanese Type 1 Diabetes. Biochem Biophys Res Commun (2013) 437:521–5. doi: 10.1016/j.bbrc.2013.06.102
174. Kang HS, Kim YS, ZeRuth G, Beak JY, Gerrish K, Kilic G, et al. Transcription Factor Glis3, A Novel Critical Player in the Regulation of Pancreatic Beta-Cell Development and Insulin Gene Expression. Mol Cell Biol (2009) 29:6366–79. doi: 10.1128/MCB.01259-09
175. Yang Y, Chang BH, Samson SL, Li MV, Chan L. The Kruppel-Like Zinc Finger Protein Glis3 Directly and Indirectly Activates Insulin Gene Transcription. Nucleic Acids Res (2009) 37:2529–38. doi: 10.1093/nar/gkp122
176. Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N, et al. A Murine Model of Neonatal Diabetes Mellitus in Glis3-Deficient Mice. FEBS Lett (2009) 583:2108–13. doi: 10.1016/j.febslet.2009.05.039
177. Yang Y, Chang BH, Yechoor V, Chen W, Li L, Tsai MJ, et al. The Kruppel-Like Zinc Finger Protein GLIS3 Transactivates Neurogenin 3 for Proper Fetal Pancreatic Islet Differentiation in Mice. Diabetologia (2011) 54:2595–605. doi: 10.1007/s00125-011-2255-9
178. Hakonarson H, Grant SF, Bradfield JP, Marchand L, Kim CE, Glessner JT, et al. A Genome-Wide Association Study Identifies KIAA0350 as a Type 1 Diabetes Gene. Nature (2007) 448:591–4. doi: 10.1038/nature06010
179. C. International Multiple Sclerosis Genetics. The Expanding Genetic Overlap Between Multiple Sclerosis and Type I Diabetes. Genes Immun (2009) 10:11–4. doi: 10.1038/gene.2008.83
180. Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, et al. The Diabetes Susceptibility Gene Clec16a Regulates Mitophagy. Cell (2014) 157:1577–90. doi: 10.1016/j.cell.2014.05.016
181. Chase HP, Cuthbertson DD, Dolan LM, Kaufman F, Krischer JP, Schatz DA, et al. First-Phase Insulin Release During the Intravenous Glucose Tolerance Test as a Risk Factor for Type 1 Diabetes. J Pediatr (2001) 138:244–9. doi: 10.1067/mpd.2001.111274
182. In't Veld P, Lievens D, De Grijse J, Ling Z, van der Auwera B, Pipeleers-Marichal M, et al. Screening for Insulitis in Adult Autoantibody-Positive Organ Donors. Diabetes (2007) 56:2400–4. doi: 10.2337/db07-0416
183. Harjutsalo V, Reunanen A, Tuomilehto J. Differential Transmission of Type 1 Diabetes From Diabetic Fathers and Mothers to Their Offspring. Diabetes (2006) 55:1517–24. doi: 10.2337/db05-1296
184. Wurst U, Ebert T, Kralisch S, Stumvoll M, Fasshauer M. Serum Levels of the Adipokine Pref-1 in Gestational Diabetes Mellitus. Cytokine (2015) 71:161–4. doi: 10.1016/j.cyto.2014.10.015
185. Wang Y, Kim KA, Kim JH, Sul HS. Pref-1, a Preadipocyte Secreted Factor That Inhibits Adipogenesis. J Nutr (2006) 136:2953–6. doi: 10.1093/jn/136.12.2953
186. Appelbe OK, Yevtodiyenko A, Muniz-Talavera H, Schmidt JV. Conditional Deletions Refine the Embryonic Requirement for Dlk1. Mech Dev (2013) 130:143–59. doi: 10.1016/j.mod.2012.09.010
187. Moon YS, Smas CM, Lee K, Villena JA, Kim KH, Yun EJ, et al. Mice Lacking Paternally Expressed Pref-1/Dlk1 Display Growth Retardation and Accelerated Adiposity. Mol Cell Biol (2002) 22:5585–92. doi: 10.1128/MCB.22.15.5585-5592.2002
188. Onengut-Gumuscu S, Chen WM, Robertson CC, Bonnie JK, Farber E, Zhu Z, et al. Type 1 Diabetes Risk in African-Ancestry Participants and Utility of an Ancestry-Specific Genetic Risk Score. Diabetes Care (2019) 42:406–15. doi: 10.2337/dc18-1727
189. Noble JA, Johnson J, Lane JA, Valdes AM. HLA Class II Genotyping of African American Type 1 Diabetic Patients Reveals Associations Unique to African Haplotypes. Diabetes (2013) 62:3292–9. doi: 10.2337/db13-0094
190. Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen WM, Santa Cruz DF, Yang H, et al. Fine-Mapping, Trans-Ancestral and Genomic Analyses Identify Causal Variants, Cells, Genes and Drug Targets for Type 1 Diabetes. Nat Genet (2021) 53:962–71. doi: 10.1038/s41588-021-00880-5
191. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Sci Transl Med (2013) 5:206ra138. doi: 10.1126/scitranslmed.3006767
192. Yoon YM, Lee JH, Yun SP, Han YS, Yun CW, Lee HJ, et al. Tauroursodeoxycholic Acid Reduces ER Stress by Regulating of Akt-Dependent Cellular Prion Protein. Sci Rep (2016) 6:39838. doi: 10.1038/srep39838
193. Bronczek GA, Vettorazzi JF, Soares GM, Kurauti MA, Santos C, Bonfim MF, et al. The Bile Acid TUDCA Improves Beta-Cell Mass and Reduces Insulin Degradation in Mice With Early-Stage of Type-1 Diabetes. Front Physiol (2019) 10:561. doi: 10.3389/fphys.2019.00561
194. Bilekova S, Sachs S, Lickert H. Pharmacological Targeting of Endoplasmic Reticulum Stress in Pancreatic Beta Cells. Trends Pharmacol Sci (2021) 42:85–95. doi: 10.1016/j.tips.2020.11.011
195. Gitelman SE, Bundy BN, Ferrannini E, Lim N, Blanchfield JL, DiMeglio LA, et al. Imatinib Therapy for Patients With Recent-Onset Type 1 Diabetes: A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Diabetes Endocrinol (2021) 9:502–14. doi: 10.1016/S2213-8587(21)00139-X
196. Han MS, Chung KW, Cheon HG, Rhee SD, Yoon CH, Lee MK, et al. Imatinib Mesylate Reduces Endoplasmic Reticulum Stress and Induces Remission of Diabetes in Db/Db Mice. Diabetes (2009) 58:329–36. doi: 10.2337/db08-0080
197. Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, et al. Targeting ABL-IRE1alpha Signaling Spares ER-Stressed Pancreatic Beta Cells to Reverse Autoimmune Diabetes. Cell Metab (2017) 25:883–97.e8. doi: 10.1016/j.cmet.2017.03.018
198. Stancill JS, Broniowska KA, Oleson BJ, Naatz A, Corbett JA. Pancreatic Beta-Cells Detoxify H2O2 Through the Peroxiredoxin/Thioredoxin Antioxidant System. J Biol Chem (2019) 294:4843–53. doi: 10.1074/jbc.RA118.006219
199. Malone CF, Emerson C, Ingraham R, Barbosa W, Guerra S, Yoon H, et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov (2017) 7:1450–63. doi: 10.1158/2159-8290.CD-17-0177
200. Minn AH, Hafele C, Shalev A. Thioredoxin-Interacting Protein Is Stimulated by Glucose Through a Carbohydrate Response Element and Induces Beta-Cell Apoptosis. Endocrinology (2005) 146:2397–405. doi: 10.1210/en.2004-1378
201. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: From Mechanisms of Action to Therapies. Cell Metab (2014) 20:953–66. doi: 10.1016/j.cmet.2014.09.018
202. Xu G, Chen J, Jing G, Shalev A. Preventing Beta-Cell Loss and Diabetes With Calcium Channel Blockers. Diabetes (2012) 61:848–56. doi: 10.2337/db11-0955
203. Ovalle F, Grimes T, Xu G, Patel AJ, Grayson TB, Thielen LA, et al. Verapamil and Beta Cell Function in Adults With Recent-Onset Type 1 Diabetes. Nat Med (2018) 24:1108–12. doi: 10.1038/s41591-018-0089-4
204. Millman JR, Xie C, Van Dervort A, Gurtler M, Pagliuca FW, Melton DA. Generation of Stem Cell-Derived Beta-Cells From Patients With Type 1 Diabetes. Nat Commun (2016) 7:11463. doi: 10.1038/ncomms11463
205. de Klerk E, Hebrok M. Stem Cell-Based Clinical Trials for Diabetes Mellitus. Front Endocrinol (Lausanne) (2021) 12:631463. doi: 10.3389/fendo.2021.631463
206. Cai EP, Ishikawa Y, Zhang W, Leite NC, Li J, Hou S, et al. Genome-Scale In Vivo CRISPR Screen Identifies RNLS as a Target for Beta Cell Protection in Type 1 Diabetes. Nat Metab (2020) 2:934–45. doi: 10.1038/s42255-020-0254-1
207. Han X, Wang M, Duan S, Franco PJ, Kenty JH, Hedrick P, et al. Generation of Hypoimmunogenic Human Pluripotent Stem Cells. Proc Natl Acad Sci USA (2019) 116:10441–6. doi: 10.1073/pnas.1902566116
208. Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, et al. Hypoimmunogenic Derivatives of Induced Pluripotent Stem Cells Evade Immune Rejection in Fully Immunocompetent Allogeneic Recipients. Nat Biotechnol (2019) 37:252–8. doi: 10.1038/s41587-019-0016-3
209. Deuse T, Tediashvili G, Hu X, Gravina A, Tamenang A, Wang D, et al. Hypoimmune Induced Pluripotent Stem Cell-Derived Cell Therapeutics Treat Cardiovascular and Pulmonary Diseases in Immunocompetent Allogeneic Mice. Proc Natl Acad Sci USA (2021) 118:1–11. doi: 10.1073/pnas.2022091118
210. Augsornworawat P, Millman JR. Single-Cell RNA Sequencing for Engineering and Studying Human Islets. Curr Opin BioMed Eng (2020) 16:27–33. doi: 10.1016/j.cobme.2020.06.003
211. Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, et al. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab (2020) 31:1017–31.e4. doi: 10.1016/j.cmet.2020.04.005
212. Yoshihara E, O'Connor C, Gasser E, Wei Z, Oh TG, Tseng TW, et al. Immune-Evasive Human Islet-Like Organoids Ameliorate Diabetes. Nature (2020) 586:606–11. doi: 10.1038/s41586-020-2631-z
Keywords: beta-cell, beta-cell heterogeneity, pancreatic islet, autoimmunity, ER stress, oxidative stress, Type 1 Diabetes
Citation: Toren E, Burnette KS, Banerjee RR, Hunter CS and Tse HM (2021) Partners in Crime: Beta-Cells and Autoimmune Responses Complicit in Type 1 Diabetes Pathogenesis. Front. Immunol. 12:756548. doi: 10.3389/fimmu.2021.756548
Received: 10 August 2021; Accepted: 13 September 2021;
Published: 07 October 2021.
Edited by:
F. Susan Wong, Cardiff University, United KingdomReviewed by:
Stephan Kissler, Joslin Diabetes Center and Harvard Medical School, United StatesCopyright © 2021 Toren, Burnette, Banerjee, Hunter and Tse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hubert M. Tse, aHRzZUB1YWIuZWR1; Chad S. Hunter, aHVudGVyY3NAdWFiLmVkdQ==; Ronadip R. Banerjee, cnJiYW5lcmplZUBqaG1pLmVkdQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.