
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 November 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.753978
This article is part of the Research Topic Late- and Adult-Onset Primary Immunodeficiencies View all 4 articles
 Frederik Staels1,2
Frederik Staels1,2 Tom Collignon3
Tom Collignon3 Albrecht Betrains4,5
Albrecht Betrains4,5 Margaux Gerbaux1,6Mathijs Willemsen1,6Stephanie Humblet-Baron1
Margaux Gerbaux1,6Mathijs Willemsen1,6Stephanie Humblet-Baron1 Adrian Liston1,7
Adrian Liston1,7 Steven Vanderschueren4,5
Steven Vanderschueren4,5 Rik Schrijvers2,4*
Rik Schrijvers2,4*Inborn errors of immunity (IEI) are a heterogenous group of disorders driven by genetic defects that functionally impact the development and/or function of the innate and/or adaptive immune system. The majority of these disorders are thought to have polygenic background. However, the use of next-generation sequencing in patients with IEI has led to an increasing identification of monogenic causes, unravelling the exact pathophysiology of the disease and allowing the development of more targeted treatments. Monogenic IEI are not only seen in a pediatric population but also in adulthood, either due to the lack of awareness preventing childhood diagnosis or due to a delayed onset where (epi)genetic or environmental factors can play a role. In this review, we discuss the mechanisms accounting for adult-onset presentations and provide an overview of monogenic causes associated with adult-onset IEI.
Inborn errors of immunity (IEI) are a heterogenous group of disorders in which the development and/or function of the immune system is disturbed (1). They result from inborn errors in genes that functionally impact our innate or adaptive immune system (1, 2). As these disorders are genetically driven, a childhood-onset disease and diagnosis before adulthood is expected. However, some patients are diagnosed in adulthood, either because of a lack of awareness preventing childhood diagnosis or due to delayed onset in adulthood. The distinction between the two can also be blurred, with an incremental escalation of symptoms unmasking the underlying IEI at a later age, with retrospective indications of childhood-onset. The majority of adult-onset IEI patients are deemed to have a polygenic etiology, but an increasing number of monogenic adult-onset causes have been identified during the last decade, facilitated by the increased use of next-generation sequencing (NGS) technology (1). Identified and validated culprit mutations in adults by in vitro or in vivo molecular assays often reveal low penetrance germline mutations in families with incomplete penetrance or acquired (somatic) mutations. In some cases, a phenotype will only become apparent when a patient encounters specific environmental triggers (such as a pathogen) for which a disrupted response or defense was present from birth (3). In addition, physicians treating adults are generally less aware of monogenic diseases, especially those of the immune system. Moreover, patients with adult-onset IEI are often seen by various medical specialists because of multisystemic manifestations, which are often not recognized as one disease and treated accordingly. In this review, we discuss the mechanisms accounting for adult-onset presentations and provide an overview of monogenic causes associated with adult-onset IEIs.
Identification of monogenic defects underlying IEI have increased over time due to the widespread availability of whole exome and genome sequencing. The current international union of immunological societies (IUIS) IEI classification published on January 10, 2020 lists 416 human inborn errors of immunity distributed among 10 groups (1). From those, 64 gene defects (15%) have been discovered (or previously characterized and recently validated) from 2018 till 2020. The majority of these genetic defects have a germline origin (Figure 1A), meaning that the mutation is inherited from the father and/or mother. Inheritance can run through an autosomal (AD) or X-linked dominant (one mutation from a (non) affected parent; mother in case of X-linked dominant) or autosomal (AR) or X-linked recessive/compound heterozygous (CH) manner (two identical mutations from each non-affected parent or different mutations in the same gene from each non-affected parent, respectively). In a minority of cases, parents are not (germline) carriers, and a mutation is considered as de novo (Figure 1B). If a mutation is present in the parental gametes or arises during gametogenesis or conception, then every cell originating from the zygote will have the same mutation. When a mutation arises post-zygotically, 2 or more cell populations with different genotypes co-exist within the same organism (mosaicism). Mosaicism can be further divided into three types a) somatic mosaicism (only affecting somatic cells), b) gonadal mosaicism (only affecting gametes) and c) gonosomal mosaicism (affecting both gametes and somatic cells). Only in the case of gonadal and gonosomal mosaicism, a mutation can be inherited by the offspring.
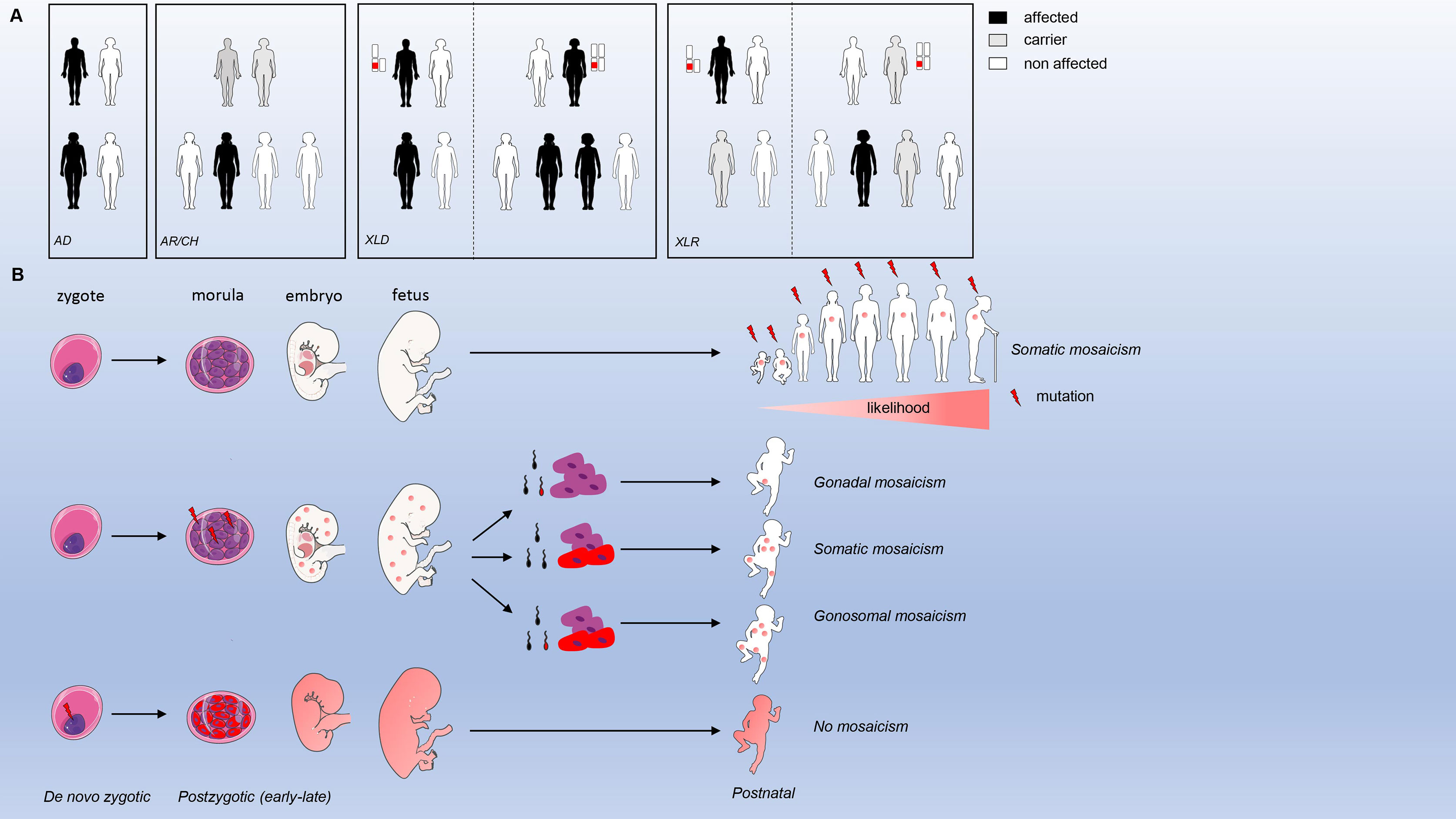
Figure 1 Inheritance modes and mosaicism types. (A) modes of inheritance for germline mutations (B). types of mosaicism and origin.
The genetic mechanisms contributing to the adult-onset phenotype are summarized in Figure 2. Most adult-onset monogenic IEI disease-causing germline mutations are hypomorphic, typically missense mutations (only partially destabilizing functional protein expression) or splice donor/acceptor site mutations (affecting the splicing and processing of mRNA but still allowing for a “leaky” production of the transcript). The impact of the genetic defect is related to the penetrance observed in the affected family, with low penetrance mutations yielding higher chances for adult-onset presentation. For example, in a large Japanese family with X- linked agammaglobulinemia (XLA), a patient (referred to as P2) harboring a splice donor mutation (IVS11+3G>T) in BTK resulting in the skipping of exon 11 still had a leaky expression of normal size BTK transcripts resulting in residual BTK protein expression on B cells and peripheral blood mononuclear cells (PBMCs) (4). Another well-described example can be found in Mendelian susceptibility of mycobacterial disease (MSMD), where patients with AR inherited IFNGR1 defects have a complete abrogated signal with an early-onset presentation, while patients with AD inherited defects can remain asymptomatic for a longer time because the mutation still allows for partially retained IFN-γ signaling activity (3). Another example is autoimmune lymphoproliferative syndrome (ALPS-FAS), where homozygous or CH mutations in the FAS gene are fully penetrant and present early-onset, while AD mutations are less penetrant with a hierarchy according to the location of the mutation (higher penetrance in the intracellular domain compared to the extracellular domain). However, in some cases, there is no association between the pathogenicity or location of a mutation and the penetrance of a disease. This is demonstrated by IEI in families with CTLA4 or NFKB1 haploinsufficiency, where some individuals harbored a complete deleterious variant without a clinical phenotype (5, 6). For NFKB1, healthy members carrying the same loss of function (LOF) variant had a similar reduction in protein expression but slight alterations in their immunophenotype (higher CD21 low B cells compared to affected members), indicating that the cellular penetrance of the same mutation can be different (6). In CTLA4 haploinsufficiency, healthy members with a deleterious mutation had a decreased CTLA4 expression compared to wild-type (WT) healthy controls but increased compared to symptomatically affected family members (5). Therefore, additional disease-modifying factors such as mosaicism, (epi) genetic modifiers, and environmental exposure must play a role in influencing the degree of cellular dysfunction and, thus, the clinical phenotype (7, 8).
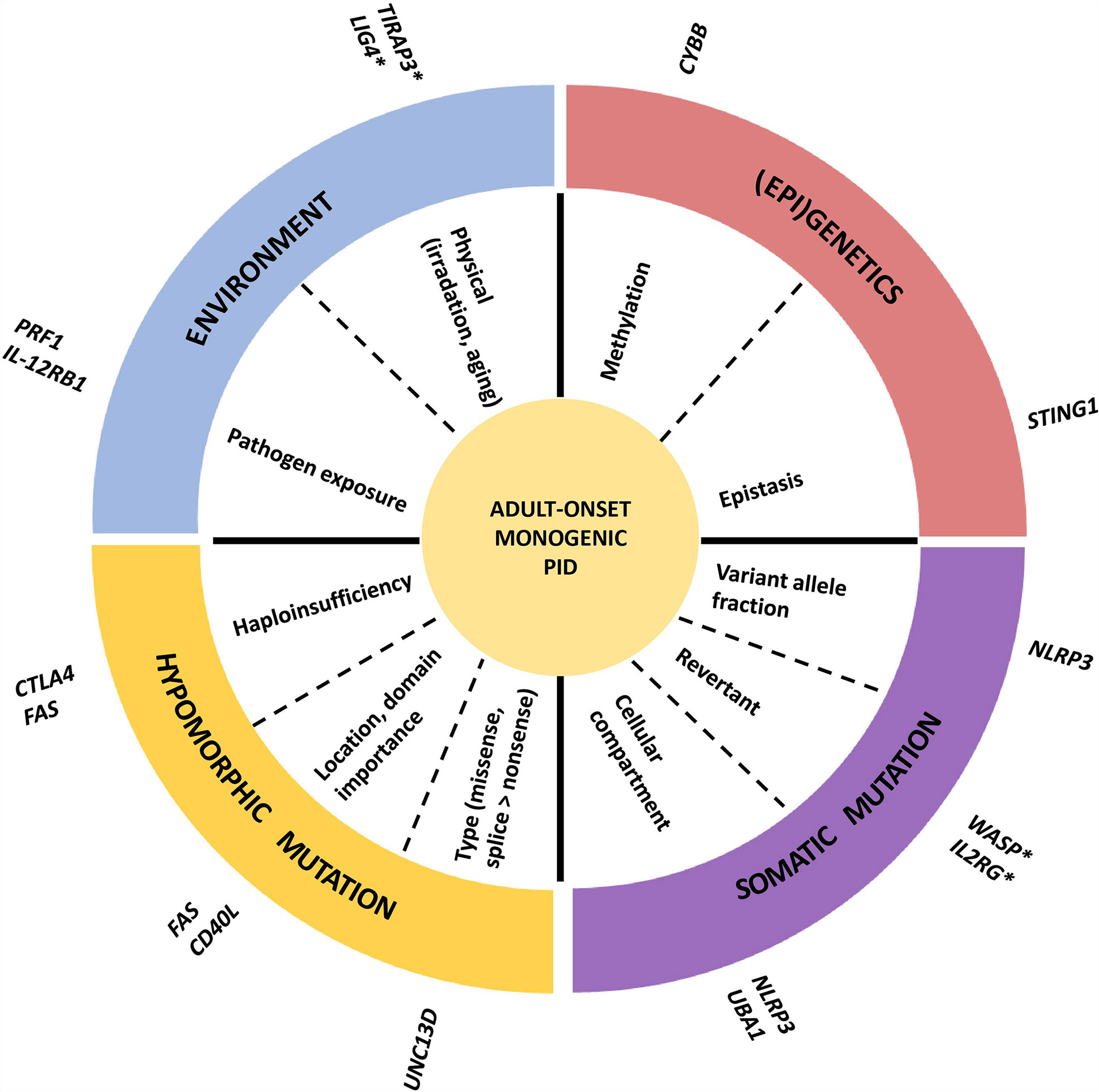
Figure 2 Mechanism of adult-onset IEI. An example of one or two genes is given for every mechanism. *late onset phenotype, but not adult-onset.
Somatic mutations leading to mosaicism are a well-described phenomenon and more common than initially anticipated. A recent study using deep amplicon sequencing found evidence of mosaicism in 30/128 IEI families (23.4%) (9). Mosaicism was most frequently observed in families with de novo mutations, families with a moderate-to-high suspicion of mosaicism, and in monogenic autoinflammatory diseases (9). The likelihood of somatic mosaicism increases with age, and this can contribute to an adult-onset disease presentation, either by itself or in cooperation with an inherited germline mutation. Well described examples of IEI with associated somatic mutations can be found in adult patients with autoinflammatory disorders such as cryopyrin-associated periodic syndrome (CAPS due to gain of function (GOF) mutation in NLRP3), VEXAS (Vacuoles, E1 enzyme, X linked, Autoinflammatory, Somatic, due to hypomorphic mutations in UBA1) syndrome and A20 haploinsufficiency (LOF mutations in TNFAIP3). These patients typically have lineage-restricted (mostly myeloid lineage) somatic mutations, indicating that the culprit mutation most likely arose in the later stages of fetal development or during postnatal life (10–14). The threshold minor allelic frequency (MAF) at which a somatic mutation provokes a phenotype is not determined but the progressive increase in MAF over time seems to correlate with disease severity in some patients as described for CAPS (13). This suggests that reduced penetrance in some mosaic IEI is probably a direct consequence of a gene dosage effect. The cellular compartment where a mosaic mutation resides is equally important, as demonstrated by ALPS patients who only harbored a somatic FAS LOF mutation in double-negative T cells (not detectable in PBMCs by Sanger sequencing) and demonstrated a phenotype comparable to patients with a germline mutation (15). Besides causing an IEI by itself, a somatic variant can also contribute in addition to a germline variant to a late-onset phenotype, as seen in ALPS-FAS (15). In a case study analyzing 17 individuals with ALPS, 5 of them had presenting symptoms after the age of 16 (16). Two of them had a germline variant with predicted haploinsufficiency in FAS and an additional somatic event in the second FAS allele which was hypothesized to account for the delayed disease onset. Another IEI where somatic mosaicism is associated with an adult-onset phenotype is a chronic granulomatous disease (CGD). Wolach et al. describe an 80-year-old woman with symptom onset at the age of 66 caused by a stop mutation (p.Tyr30*) in CYBB encoding gp91phox, important for the oxidative burst response in myeloid cells (17). The mutation was identified in the short-lived blood cells but not in long-lived memory T cells or cheek mucosal cells, suggesting it had occurred later in life. Moreover, leukocytes showed a markedly skewed X inactivation pattern. They hypothesized that this X skewing was caused by another parental gene on the active X chromosome harboring the mutation since no selective advantage has been described for X-CGD mutations in hematopoietic cells. Besides causing disease, somatic mosaicism can also rescue disease, so-called revertant mosaicism. In these cases, a second mutation improves the production of a functional gene product either by giving a selective advantage to WT cells or by directly counteracting the negative impact of the mutant allele on protein function. Although this phenomenon has not yet been reported in adult-onset cases, it is associated with later and milder disease onset as seen in patients with defects in ADA, IL2RG, WASP, SAMD9 and CD3-ζ (18, 19). Finally, epistasis (dependency of the effect of a mutation on the presence or absence of mutations in the same or other genes) can affect disease presentation. In patients with STING associated vasculopathy with onset in infancy (SAVI), caused by GOF mutations in STING, disease-onset and manifestations can be influenced by single nucleotide polymorphisms (SNPs) in STING itself or in other IFN related genes such as IFIH1 (20).
Environmental factors can also contribute to phenotypic variability, both in terms of severity and age of onset. A typical example is seen in patients who develop disseminated mycobacterial disease after Bacillus Calmette-Guérin (BCG) vaccination and, because of these events, are later diagnosed with an inborn error of IFN-γ immunity. These patients were previously healthy because they were never exposed to this specific microorganism (21). Accordingly, it is hypothesized that delayed contact with specific pathogens in patients with an isolated increased susceptibility for this specific pathogen might account for the delayed onset, as in other cases of MSMD and viscerotropic Yellow fever postvaccination (22). Evidence for infections as a trigger of disease onset also comes from murine models. Knock in mice with a GOF mutation in STING (p.N153S) only developed pulmonary fibrosis after infection with gamma herpes virus 68 (γHV68), and this could be prevented by administration of cidofovir, an antiviral drug against γHV68 (23). It is unclear how these data translate to human patients, but adult-onset pulmonary fibrosis has been reported in SAVI (20). Infections can also trigger immune dysregulation, as observed in patients with familial hemophagocytic lymphohistiocytosis (fHLH), most commonly caused by mutations in PRF1, where bouts of autoinflammation often coincide with upper respiratory or gastrointestinal infections (24). Other triggers such as irradiation or chemotherapy can also unmask an underlying DNA repair defect at a later age, as described for patients with Ligase-4 (LIG4) deficiency (25, 26). Age is also an important influencing factor, as humoral immunity can mature and compensate for certain inborn defects in innate immunity. A well-described example is a deficiency in TIRAP3, a key toll-like receptor (TLR) adaptor, caused by recessive LOF mutations. A homozygous LOF mutation was described in a child with severe staphylococcal infection, while older relatives carrying the same mutation were reported to be healthy due to the presence of anti-lipoteichoic acid antibodies targeting S. aureus, which were lacking in the index patient (27).
Finally, epigenetic alterations are often implicated to contribute to incomplete penetrance and late-onset disease. Epigenetic modifications play an important role in shaping our immune system and response (28, 29). This is demonstrated by the identification of genetic defects involved in methylation, histone modification, chromatin remodeling and alteration in non-coding RNA resulting in different immunodeficiency syndromes (29). Progressive X-chromosome skewing (XCI) can cause an X-linked recessive IEI in women bearing a pathogenic mutation on one X chromosome. This is demonstrated by cases of CGD due to mutations in CYBB in adult women (30, 31). Epigenetic changes are well known to influence the transcriptional expression of genes (29). Therefore, it would not be surprising that the difference in epigenome between two individuals with the same pathogenic mutation can impact the final effect of the mutation in terms of gene expression (32). The impact of epigenetics on disease outcome is best studied in monozygotic twins who are genetically identical. A recent study investigating the epigenome of two monozygotic twins discordant for common variable immunodeficiency (CVID) showed a predominant gain of methylation in critical B lymphocyte genes in the patient compared to his healthy sibling (33). Moreover, the epigenome can change over time, as aging is associated with the relaxation of epigenetic control (34).
As a guide, we used the most recent IUIS phenotypical classification list (2019) (35) and updates given thereafter (2). We focused on the following groups: immunodeficiencies affecting cellular and humoral immunity, predominant antibody deficiencies, diseases of immune dysregulation, defects in intrinsic and innate immunity, autoinflammatory disorders, and complement deficiencies because adult-onset phenotypes are most frequently reported within one of these seven groups (Table 1). Only genes for which there is clearly evidence-based functional studies demonstrating the pathogenic effect of disease-causing mutations in vitro or in vivo will be discussed. Risk alleles identified by genome wide association studies but lacking a monogenic genotype-phenotype relationship were omitted from this review. Congenital defects of phagocytosis, combined immunodeficiencies with syndromic features, and bone marrow failure syndromes are also not discussed as those rarely present with adult-onset. Appropriate papers were selected from PubMed using the following search terms: “gene name” AND primary immunodeficiency AND adult OR adult-onset OR adult-onset OR incomplete penetrance. Search results were reviewed by two reviewers (FS and TC) and screened for adult-onset patients (no relevant medical history reported before the age of 18, or explicitly reported as adult-onset in the manuscript). Citation history was screened for additional single case reports or series. An age exception was made for the defects affecting cellular and humoral immunity, for which we included late adolescence onset cases with a clear demonstration of hypomorphic variants or cases with very mild symptoms during childhood and diagnosis in adulthood due to worsening symptoms. The oldest patient for each reviewed disease is depicted in Figure 3.

Table 1 Adult vs non-adult-onset associated genes implicated in PID.
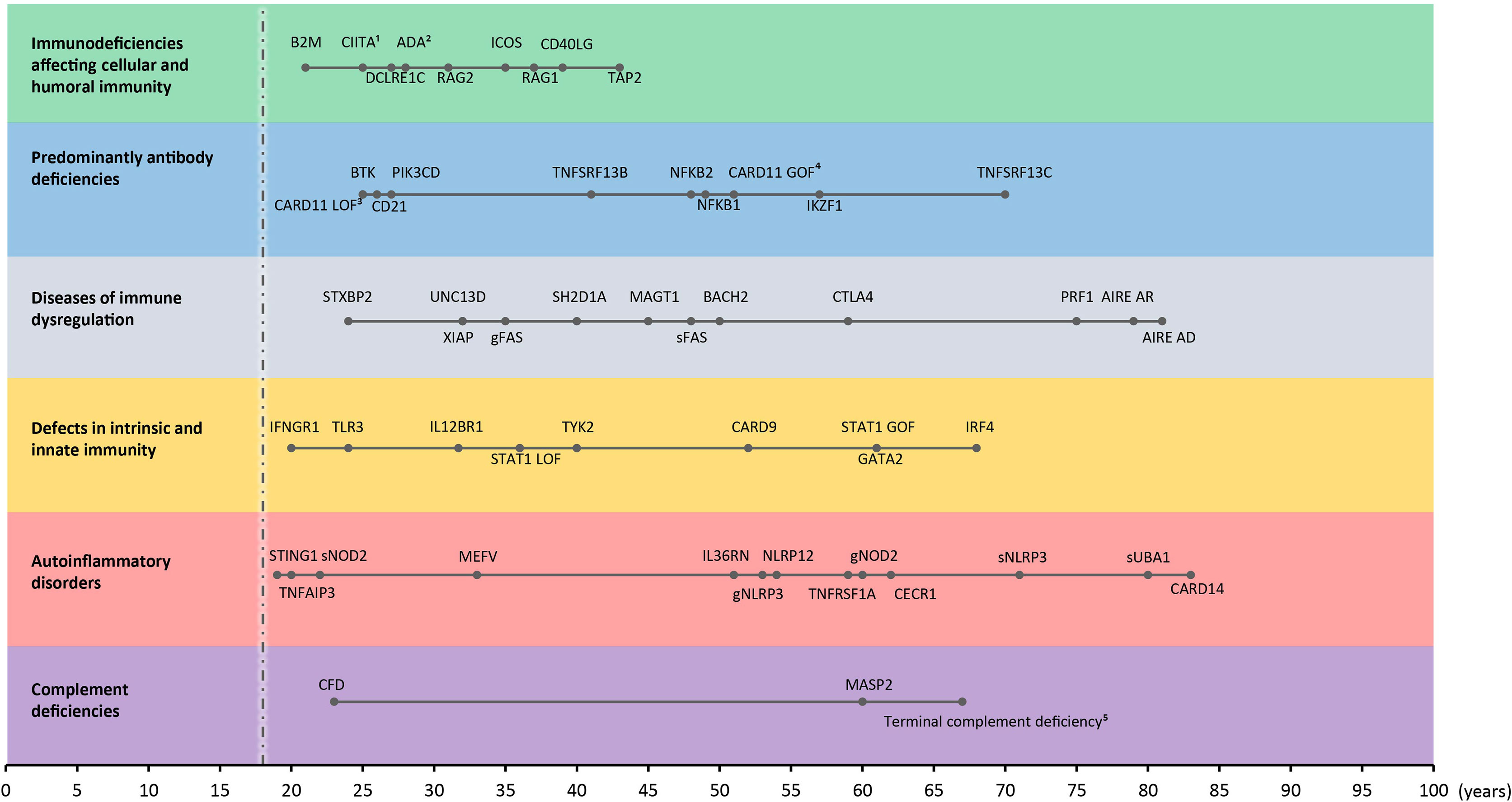
Figure 3 Time of onset in adult IEI. Adult-onset IEI genes with oldest age-of-onset patient reported for every gene (from all reviewed manuscripts) in each category. s: somatic. g: germline. 1onset between 20-30 years, 2immunological abnormalities, 3onset in mid-twenties, 4asymptomatic, 51 years old, 5genetic defect in C5/6/7/8 or 9, mean age of oldest presentations. Dashed line indicates threshold for adult-onset (18-years-old).
Adenosine deaminase (ADA) deficiency is one of the most common forms of severe combined immunodeficiency (SCID) that arises through AR inherited mutations in the ADA gene, encoding an enzyme involved in the purine salvage pathway (36). As a form of SCID, ADA-deficiency typically presents at birth, but it has also been described presenting with a milder phenotype later in childhood (delayed onset) or even in adulthood (adult-onset). Late-onset disease tends to gradually worsen over time (36, 50). The first cases that were classified as adult-onset ADA-deficiency were reported by Shovlin et al. (51). They described two sisters that presented in their mid-30s with recurrent infections and chronic respiratory disease. However, retrospectively the older sister developed idiopathic thrombocytic purpura at 17-years-old and the younger sister had a history of recurrent infections, viral warts and mucocutaneous candidiasis since childhood (51). Both had undetectable ADA activity in erythrocytes (lymphocytes not tested) and a CH mutation (p.R211C and a deletion resulting in the loss of exon1). A third 28-year-old male patient was described by Ozsahin et al. (37). He had no relevant medical history apart from recurrent tonsillitis starting at 10-years-old. He carried a CH mutation (p.R101Q acting as a null allele and p.A215T acting as hypomorphic with 15% of WT activity). ADA activity in lymphocytes was very low, but still detectable. The residual ADA activity potentially explains the late onset and mild phenotype in this case (37). In conclusion, although most commonly presenting as SCID during early life, ADA-deficiency is a genetically and clinically heterogenous disorder with different phenotypes depending on the nature and effect of the genetic mutation. (Table 2)
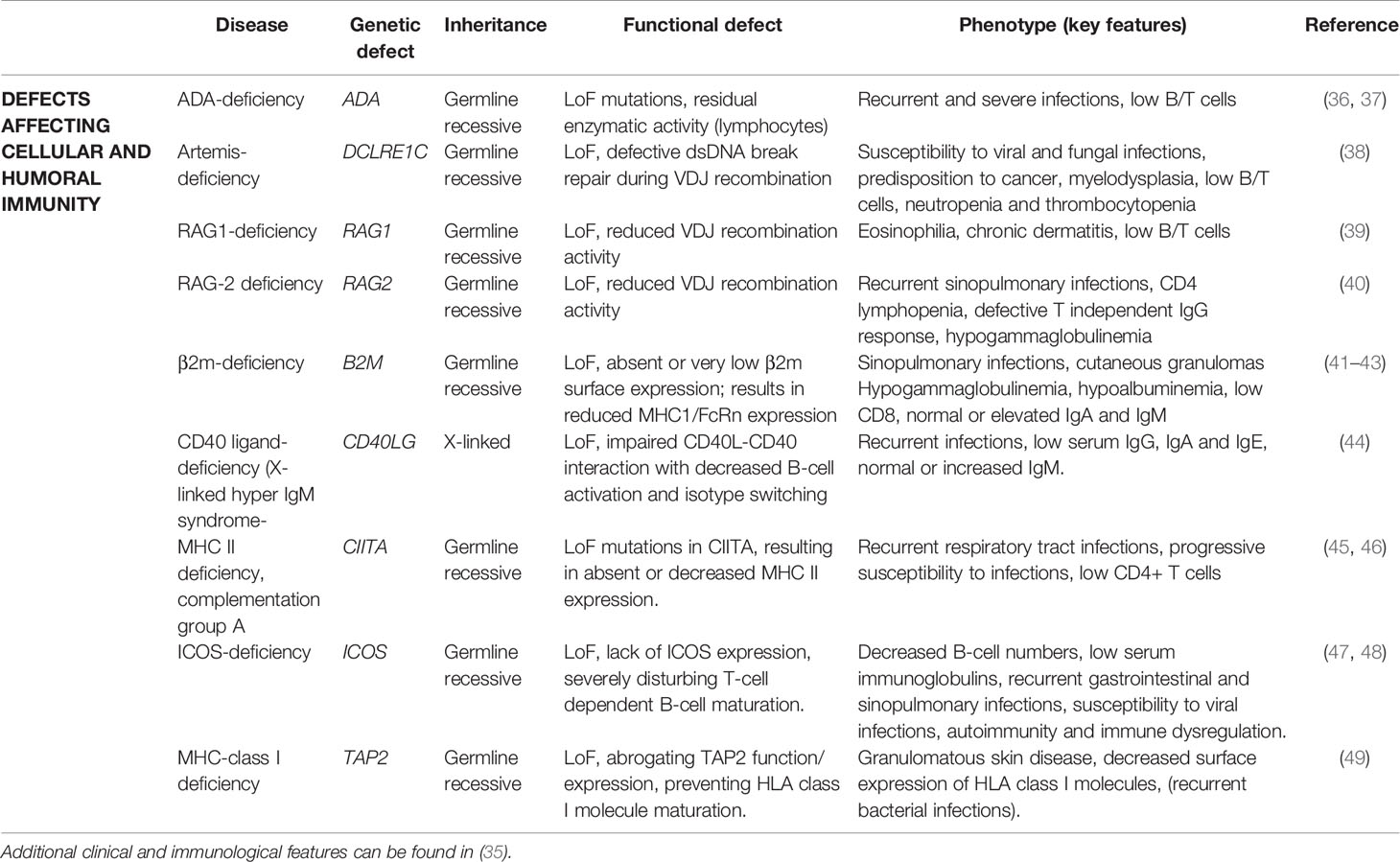
Table 2 Genes associated with adult-onset combined immunodeficiencies.
DLCRE1C encodes Artemis, a protein involved in the repair of double strand DNA breaks (DSB) induced by recombination-activating gene 1 (RAG1) and RAG2 as a part of VDJ recombination (52). Mutations in DCLRE1C result in a form of radiation-sensitive SCID (rs-SCID). Hypomorphic mutations can contribute to later onset disease or leaky SCID, presenting as a combined immunodeficiency at an older age in infancy or childhood (53, 54). One patient reported by Woodbine et al. developed progressive immune dysfunction at 27-years-old (38). She presented with in situ carcinoma in one nipple, and progressed to a phenotype with recurrent viral and fungal infections and myelodysplasia. Skin fibroblasts displayed radiation sensitivity and a defective DSB repair in the G2 phase. Genetic analysis revealed a heterozygous mutation (p.P171R) which caused a 3-fold decrease in Artemis activity in vitro. The other allele did not show any exonic variants, but the gene product was mis-spliced and prone to nonsense mediated decay, so the authors hypothesized the presence of an intronic splicing mutation. The hypomorphic p.P171R variant in combination with an undefined intronic mutation affecting the splicing of Artemis could contribute to the development of a mild and progressive late onset immunodeficiency (38).
RAG1 and RAG2 play a crucial role in inducing DSB during VDJ-recombination, and AR null mutations can lead to SCID. Hypomorphic mutations however can give rise to a spectrum of phenotypes including Omenn syndrome, atypical SCID and combined immunodeficiency (55, 56), that can all exhibit features of immune dysregulation (57). Adult-onset phenotypes are very rare, even in patients with hypomorphic mutations in RAG1 who often have mild disease in childhood. Abraham et al. identified a male patient harboring a heterozygous hypomorphic RAG1 frameshift mutation (p.K86Vfs*33) who presented at 38-years-old with a pruritic skin rash that started two years earlier, eosinophilia and T-cell lymphopenia (39). No other mutation in RAG1 or other SCID-related genes that were known at the time were found. The mutation described is damaging as it is found in homozygous or CH SCID patients or patients with Omenn syndrome. His healthy father was found to carry the same mutation, suggesting that other genetic or environmental modifiers might contribute to the observed phenotype (39). Another case reports an adult patient presenting with progressive multifocal leukoencephalopathy at the age of 37 (58). The patient harbored a CH mutation in RAG1 (p.F478Sfs*14 and p.H375D on one allele and a p.R474C on the other allele). A VDJ recombination assay for p.R474C showed 8% of the normal WT function, while the other mutations were not assessed but predicted to be damaging. In retrospect, the patient already was prone to mild infections during childhood (recurrent respiratory tract and gastroenteritis), but infectious manifestations decreased in puberty.
RAG2 mutations have also been reported to cause an antibody deficiency phenotype (40). A 41-year-old woman with no medical history developed recurrent bronchitis and pneumonia and was diagnosed to have antibody deficiency against bacterial polysaccharide antigens (40). Genetic analysis identified a CH mutation in RAG2 (p.G95R with absent expression and p.S381fs*1 with a C-terminal truncated RAG2). The functional consequence of the frameshift mutation was not assessed because it was assumed to lead at least to a partial LOF effect based on a mouse model, lacking the C terminal domain of RAG2, which showed partially retained protein expression similar to humans but with impaired lymphoid development (59). This could be a plausible explanation for the adult-onset and mild phenotype in the above described patient.
B2M encodes β2-microglobulin (β2m) that composes the light chain of the major histocompatibility complex (MHC) class I molecules and is essential for transport and stabilization of MHC I molecules on the cell surface (41). Furthermore, it also forms a heterodimer with MHC I α chain to form the neonatal Fc receptor (FcRn) which binds IgG and albumin preventing their lysosomal degradation (41, 42). In 1990, two consanguineous siblings were reported suffering from hypoalbuminemia and hypogammaglobulinemia (43). A 34-year-old female patient became symptomatic at 21-years-old, developing a purpuric skin rash and later ulcerative lesions on her leg. She developed thrombocytopenic purpura, and eventually died of sepsis due to bilateral pneumonia. Her 17-year-old brother was asymptomatic, but immunological examination revealed low serum IgG and low albumin levels. Although both siblings were lost to follow up, 35 years later a homozygous LOF B2M mutation (p.A11P, leading to <10% expression β2m compared to WT) was found in DNA isolated from preserved sera (42). β2m is also a component of the neonatal Fc receptor (FcRn). FcRn binds IgG and albumin and is important in regulating their normal concentrations (42), thus deficiency of B2M was the culprit for the hypercatabolic hypoproteinemia in these cases. In 2015, two other consanguineous siblings were identified with a homozygous splice site mutation (c.67+1G>T, resulting in nonsense mediated decay and no protein expression in patient’s lymphocytes) (41). They both displayed hypoalbuminemia and low IgG levels, with normal or elevated IgA or IgM levels. The older sister suffered from granulomatous dermatitis since 9 years of age and developed bronchiectasis. Her brother was clinically asymptomatic but shown to have bronchiectasis on pulmonary computed tomography (CT) scan. Based on these two reports, β2m-deficiency can be characterized as an immunodeficiency with hypogammaglobulinemia and hypoalbuminemia that can range from subclinical phenotype to severe symptoms presenting in late childhood or adolescence.
CD40LG encodes CD40 ligand (CD40L), which is a surface molecule on activated T helper cells that transfers signals through CD40 for B-cell activation, differentiation and isotype switching (60, 61). Mutations in CD40L are associated with an X-linked form of hyper IgM syndrome (HIGM) (60), and are the most commonly identified genetic cause of this disorder (44). HIGM syndrome is characterized by low levels of serum IgG, IgA and IgE, normal or high levels of IgM and recurrent infections (44, 60). In a study of 140 patients with HIGM syndrome, 98 patients were found to carry mutations in CD40LG (44). Of these, 6 patients had a mild clinical phenotype with onset after 14 years of age. Only one patient had adult-onset disease (23 years) harboring a missense mutation (p.T254M, with proven decreased expression of CD40L) affecting the extracellular domain of CD40L. More recently, a 41-year-old Caucasian man, carrying a nonsense mutation (p.R11*, with normal truncated protein expression) with disease onset in the fifth decade was described by Yong et al. (62). He presented with hemiparesis due to cerebral toxoplasmosis, a 2-year history of recurrent impetigo and chest infections (62) and later progressed to develop eosinophilia and pulmonary aspergilloma (63). The milder phenotype was suggested to be the result of a stable expressed mutant with a normal extracellular domain, still enabling CD40-CD40L interaction and partial signaling. His siblings and other family members were also evaluated in a second study (63). An older brother carried the same mutation. Whereas he did not have any symptoms or relevant clinical history, his IgA and IgG levels were abnormally decreased and his IgM was elevated. In two nephews who suffered from recurrent respiratory infections, the mutation was also found. Their IgG levels, however, were higher than those observed in their two uncles, leading to the hypothesis that the p.R11* mutation might contribute to a gradual deterioration of class switching, or that physiological immune senescence could alter the effects of the mutation (63).
CIITA encodes class II trans-activator, that regulates transcription of MHC molecules by functioning both as a transcriptional activator by coordinating DNA binding factors RFX, CREB and NF-Y and as a transcription factor (64). Deficiency of CIITA results in MHC class II deficiency group A, characterized by a total lack of MHC II expression. In a patient presenting in his twenties, Quan et al. identified a homozygous LOF missense mutation (p.F961S) that resulted in absent expression of MHC II molecules (45). His symptoms progressively worsened in his early thirties, and he eventually died of multiple bacterial infections. Another atypical case was found among three siblings that harbored a homozygous missense mutation (p.L469P), with the mutant allele showing residual activity (46). Two of them suffered from mild immunodeficiency characterized by recurrent respiratory tract infections starting in childhood, while the oldest sister was asymptomatic. Apart from two episodes of pneumonia during childhood, she was completely healthy although she lacked MHC II expression. The investigators hypothesized that the mild clinical phenotype may be due to residual CIITA activity (46).
Inducible co-stimulator (ICOS) is a surface molecule on activated T-cells and is predominantly expressed on T-cells in the germinal center of secondary lymphoid tissue (47, 65). It plays an important role in induction of several cytokines, including superinduction of IL-10, and T-cell dependent maturation of B-cells (47, 65, 66). Mutations in ICOS were first discovered as a genetic cause of CVID by the identification of four adult CVID patients carrying homozygous ICOS mutations (c.126-568.del) resulting in a complete lack of ICOS expression (47). A more recent report describing previously published cases expanded the clinical phenotype (48). Six out of 15 ICOS-deficient patients (with age of onset ranging from 1 month to 35 years) presented in adulthood with a variety of symptoms, including recurrent sinopulmonary and gastro-intestinal tract infections, viral infections, signs of autoimmunity and immune dysregulation. They all carried deletions in ICOS that resulted in a frameshift and premature stop codon. Opportunistic infections were associated with early disease onset, and were not observed in patients with adult-onset disease (48). In ICOS deficiency, B cell counts appear to decrease during the course of disease (which might explain an adult-onset phenotype), possibly related to progressive bone marrow output failure (48).
Transporter associated with antigen processing 2 (TAP2) transports antigenic peptides into the ER so that they can be loaded onto human leukocyte antigens (HLA) I molecules (67). TAP2 is the only gene reported in adult-onset cases of MHC class I deficiency. A homozygous LOF intronic mutation at a splice acceptor site (c.1636-1G>A) introducing a frameshift (p.V545Wfs) was found in a 46-year-old male patient who developed granulomatous skin lesions on his leg and his asymptomatic 30-year-old sister (49). The mutation affected the ATP binding site of TAP2 and abrogated its function, thereby inhibiting the maturation of HLA class I molecules. The index patient had 10-15 times lower expression of HLA class I molecules, but still three times higher than in a cell line derived from a previously described TAP2-deficient patient, possibly contributing to the mild or asymptomatic clinical phenotype in these two siblings (49).
X-linked agammaglobulinemia (XLA) is a humoral immunodeficiency found in males, caused by defects in BTK encoding Bruton’s tyrosine kinase, an enzyme involved in B cell maturation by transmitting signals through the pre-B and B cell receptor. Mutations are reported in all domains and result in reduced protein expression or kinase activity. Disease manifestations (recurrent sinopulmonary infections by encapsulated bacteria) mostly occur in infancy (mean age 3.5 years). In exceptional cases, hypomorphic BTK alleles can result in an adult-onset phenotype (Table 3) (68, 69, 81). However, when carefully reviewing medical records, most adults diagnosed with XLA will have an early-onset phenotype (70). Among reported adult-onset patients, one patient had symptom onset at 25 years of age (68) and another was diagnosed at the age of 24 when familial work-up, initiated because a sibling was diagnosed with XLA, revealed hypogammaglobulinemia (69). In most reports, late-onset phenotypes are characterized by partial BTK deficiency on flowcytometry and higher immunoglobulin levels compared to early-onset XLA (70, 81) indicative for the hypomorphic impact of the mutation. Noteworthy, B cell counts should always be tested since BTK deficiency can be misdiagnosed for CVID and early-onset GI disease (82)
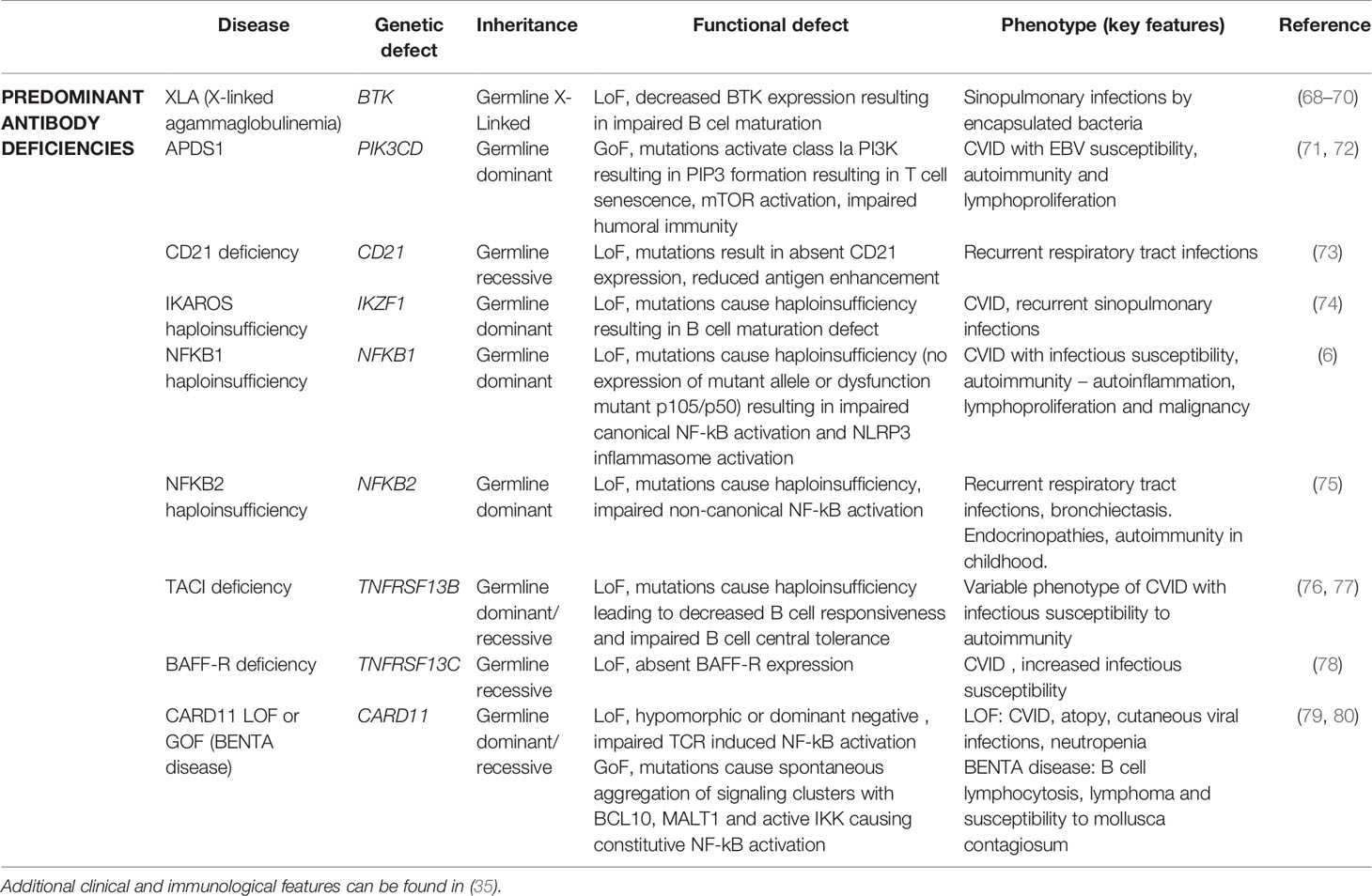
Table 3 Genes associated with adult-onset predominant antibody deficiencies.
GOF mutations in PIK3CD lead to activated phosphoinositide 3-kinase (PI3K) delta syndrome (APDS) type 1. They can be found in patients allegedly categorized as adult-onset CVID with Epstein-Barr virus (EBV) susceptibility, autoimmunity and lymphoproliferation. PIK3CD encodes the catalytic subunit (PI3K delta) of a class Ia PI3K, which is expressed in leukocytes and catalyzes the production of the second messenger phosphatidylinositol (3,4,5)-triphosphate (PIP3). GOF mutations in this catalytic subunit result in excessive production of PIP3, leading to activation of the mechanistic target of rapamycin (mTOR) pathway and other kinase complexes such as BTK and inducible T cell kinase (ITK) (83). Subsequently, this results in T cell senescence, lymphoproliferation and impaired antibody responses (83). Adult diagnosed cases were first observed in a large cohort (n=53) of ADPS patients, although adult-onset phenotypes were considered exceptional (71). Five patients (9.4%) were diagnosed in adulthood, one suffered recurrent respiratory tract infections and a local granulomatous skin reaction in response to BCG vaccination in childhood, one was evaluated for chronic cervical lymphadenopathy, two had bronchiectasis and one was asymptomatic. No further details on the disease onset was provided (71). In the European Society for Immunodeficiencies (ESID) APDS registry compiling 51 APDS1 patients, most had disease-onset before the age of 15, but also here adult-onset cases are reported (72). One APDS1 case developed symptoms at the age of 27, although recurrent upper respiratory infections during adolescence might suggest earlier-onset of disease (84).
A second type of APDS (APDS2) is caused by autosomal recessive LOF mutations in PIK3R1 encoding the regulatory subunit of class Ia PI3K. APDS2 patients present at infancy with severe bacterial infections and autoimmunity, adult-onset cases have not yet been reported.
CD21 is part of the B cell receptor complex. It recognizes complement component 3d (C3d)-opsonized immune complexes and enhances antigen-specific B cell responses. A compound heterozygous mutation, resulting in complete loss of CD21 surface expression, has been described in one adult with recurrent infections, reduced class-switched memory B cells and hypogammaglobulinemia (73). He had frequent childhood respiratory tract infections, resolving after tonsillectomy at the age of 6 years, followed by an asymptomatic period of 20 years.
IKZF1 encodes IKAROS, a transcription factor belonging to the Ikaros zinc finger transcription factor family, essential for the regulation of lymphocyte differentiation, especially of the B cell lineage (85). The first case of heterozygous mutations in IKZF1 causing haploinsufficiency and a phenotype reminiscent of CVID was reported in 2016 (74). Eight out of 29 patients had an adult-onset phenotype (19-57 years, all had infections as presenting symptom) and most of them (n=6) were reported within one family with a 4.7 Mb deletion including IKZF1 on chromosome 7 (in contrast to other families with missense mutations or a very small deletion within the IKZF1 gene). Slowly progressive loss of B cells over time in IKAROS deficiency could provide an explanation for late-onset phenotypes. Aging has been associated with impaired B cell development in the bone marrow, and a mutation in a gene essential for B cell differentiation, such as IKAROS, might accelerate this process (86).
NFKB1 is one of the five nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) proteins, encoding the p105 subunit. This is post-translationally processed into a p50 subunit, functioning as a transcriptional enhancer of NF-κB target genes when heterodimerized with RelA (p65). NFKB1 haploinsufficiency was first described in 2015 in three families with a CVID phenotype and incomplete penetrance. Within the families, the age of onset was highly variable (2 years to 65 years) with diverse phenotypes ranging from asymptomatic to CVID with malignancy, autoimmunity and bronchiectasis. All reported mutations (splice donor, deletion and frameshift) resulted in rapid degradation of the mutant transcript and residual p105/50 was expressed from the WT allele (87). NFKB1 haploinsufficiency is considered one of the most frequent genetic causes of CVID (6). Incomplete penetrance is prototypical and adult-onset disease is frequently observed (6).
Like NFKB1, NFKB2 is one of the five NF-κB proteins, encoding the p100 protein, which is post-translationally processed to a p52 protein. In contrast to p50 which plays a key role in the canonical NF-κB pathway, p52 functions as transcription factor in the non-canonical NF-κB pathway which is important in lymphoid organ development, B and T cell maturation, thymic selection and innate antiviral immunity (75). In a recent review summarizing clinical features on 50 reported patients with NFKB2 haploinsufficiency, 2 were reported as adult-onset (31 and 48 years) and had recurrent respiratory tract infections and bronchiectasis (75).
TNFRSF13B encodes the transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) belonging to the Tumor necrosis factor (TNF) receptor superfamily. This receptor is mainly expressed on B cells and its activation by ligands such as a proliferation-inducing ligand (APRIL) or B-cell activating factor (BAFF) activates a signaling cascade that is involved in B cell differentiation, removal of autoreactive B cells, immunoglobulin production and class switching. Both heterozygous and homozygous LOF mutations have been linked to adult-onset CVID and can be found in 5-10% of CVID patients (76). Heterozygous mutations are associated with autoimmunity and lymphoproliferation, more than biallelic mutations, possibly because B cells are still partially responsive to allow for autoimmune complications (77). Its role as a monogenic cause of IEI has been debated and a role as a modifier gene in CVID postulated (88).
TNFRSF13C encodes the BAFF factor receptor (BAFF-R). Similar to TACI it belongs to the TNF receptor superfamily, and is mainly expressed on B-cells. Upon binding of its ligand, downstream pathways are activated that regulate B cell survival and maturation. BAFF-R deficiency has been described in two adult-onset (37 and 70 years) CVID patients carrying a homozygous LOF deletion in TNFRSF13C resulting in absent expression (78). They presented with respiratory tract infections associated with profound B cell lymphopenia and low number of switched memory B cells.
CARD11 is a scaffold protein that plays a role in TCR and BCR signaling by linking antigen recognition to NF-κB in lymphocytes (79, 89). Mutations in CARD11 can lead to a variety of clinical phenotypes depending on the nature of the mutation (LOF or GOF). In case of LOF, biallelic null mutations result in combined immunodeficiency in childhood. Hypomorphic variants or dominant negative mutations are linked to a milder phenotype that predisposes to a variable immunodeficiency and atopy (79, 89, 90). In a study containing 48 patients with dominant negative CARD11 mutations, three of them were reported with disease onset at 18 years or older (79). One woman had onset at 20 years of age and suffered from neutropenia, hypogammaglobulinemia and indolent LGL. Another patient was 18 years old and apart from atopic disease, she also suffered from bronchiectasis and mollusca contagiosum. Lastly, a female carrying a compound heterozygous mutation was identified with disease onset in her mid-twenties when she developed recurrent bacterial sinopulmonary tract infections and hypogammaglobulinemia. The mutations (p.R47H and p.R187P) carried by the first two patients were shown to exert a dominant negative effect by interfering with WT CARD11, resulting in decreased NF-κB signaling. The last patient carried a compound heterozygous LOF mutation (p.R912Q and p.D1152N). In case of a germline GOF mutation, a B cell lymphoproliferative disorder known as B cell Expansion with NF-κB and T cell Anergy (BENTA) can occur. Patients with BENTA often present with susceptibility to molluscum contagiosum and polyclonal B cell lymphocytosis (91). Although often seen shortly after birth or in childhood, a clinically asymptomatic adult individual has been reported (80).
Familial hemophagocytic lymphohistiocytosis (fHLH) represents a group of immune dysregulation disorders associated with uncontrolled activation of histiocytes and T cells. The pathogenesis of fHLH centers around the impaired cytolytic function of natural killer (NK) cells and cytotoxic T cells, which hinders the clearance of antigens and eventually results in a hyperinflammatory state (i.e. cytokine storm) (118). Classical symptoms include cytopenia, prolonged fevers, hepatosplenomegaly, hypertriglyceridemia, hyperferritinemia and neurological disease. The diagnosis of fHLH can be established using the HLH-2004 criteria (119). Several defects in genes important for cytolytic function have been validated as cause of fHLH and some of them (described below) were unmasked after a first episode in adulthood (Table 4).
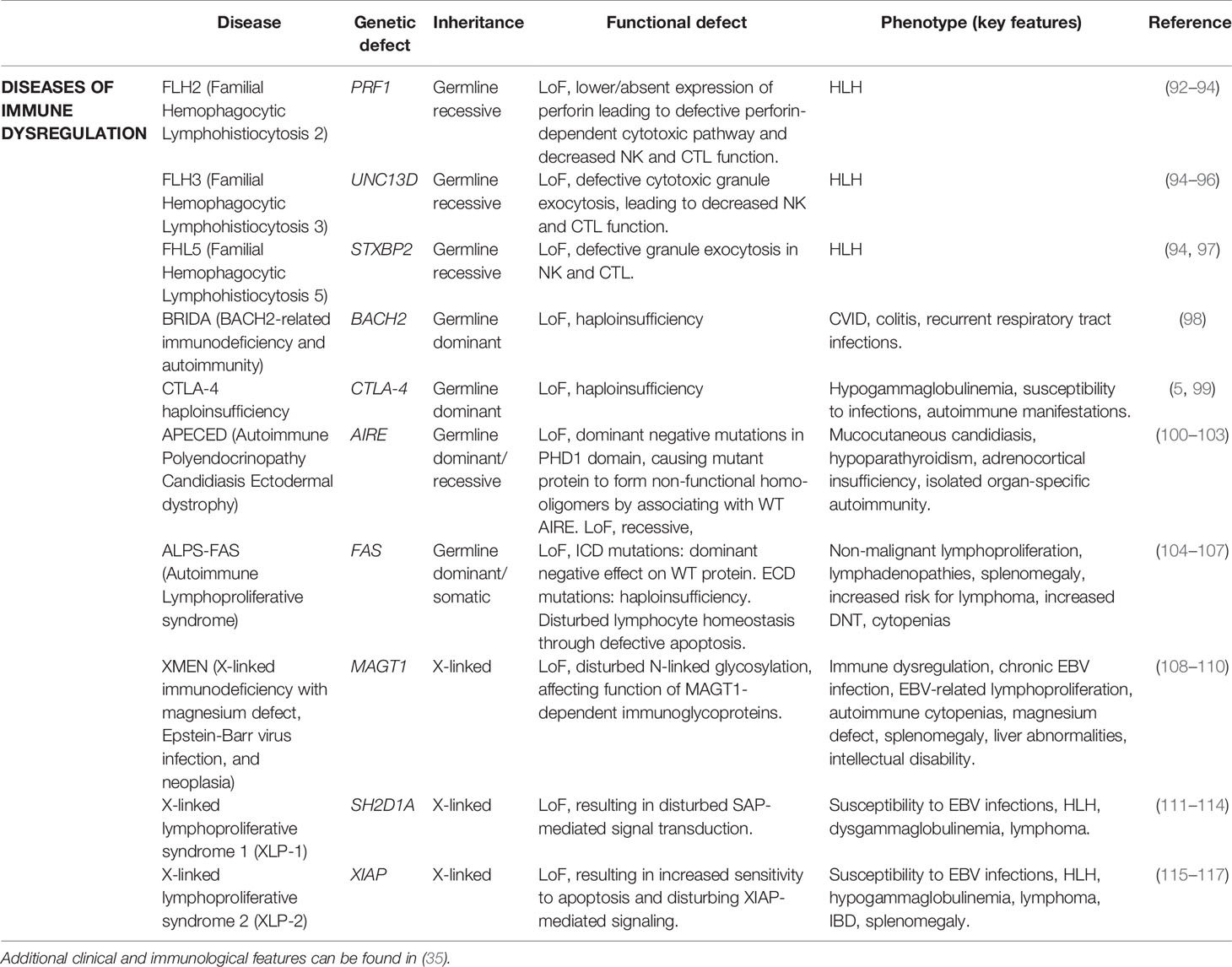
Table 4 Genes associated with adult-onset diseases of immune dysregulation.
PRF1 encodes perforin, highly expressed by cytotoxic T and NK cells. Perforin is a pore forming protein involved in cell death. A case study reports on an adult Japanese patient presenting with a first episode of HLH at the age of 62 years (92). Genetic analysis on PBMCs and nails revealed a germline compound heterozygous PRF1 mutation (p.L364fs and p.V306I). The compound heterozygous mutation was hypomorphic as observed by the decreased but not abolished perforin expression in vitro. Another report from 2002 describes 2 siblings (brother and sister) who presented at the age of 21 and 22, respectively, with a first episode of HLH (93). HLH was preceded by a respiratory tract infection in one sibling and a diagnosis of T cell lymphoblastic lymphoma in the other sibling. Both of them carried a compound heterozygous mutation resulting in a missense (p.A91V) and stop mutation (p.W374*) in PRF1 with decreased perforin expression. Lastly, one of the largest cohorts to date studying 175 adult patients (out of a total of 1531 patients who were referred for suspected HLH), observed genetic defects in PRF1 in 18 of them (age 18-75 years) (94). Of the patients with available perforin expression, only 1 had absent perforin expression, while 6 had low expression and 2 had normal expression (in the presence of an in silico predicted pathogenic heterozygous mutation). In conclusion, adult-onset presentation is associated with hypomorphic PRF1 variants leading to decreased but not abolished perforin activity in most patients and/or requires an additional trigger such as infections or malignancy, similar to secondary HLH.
STXBP2 encodes syntaxin-binding protein 2, which plays a role in the regulation of intracellular granule trafficking in neutrophils, NK cells and mast cells. Almost all reported patients presented during the neonatal period or in early infancy. In a case series by Meeths et al. reporting on 8 patients, the oldest patient presented at the age of 17 and HLH was preceded by an EBV infection (97). She carried compound heterozygous mutations in STXBP2 resulting in a missense (p.S545L) and stop (p.Q432*). NK cell cytolytic activity and degranulation were both impaired. In the cohort described by Zhang et al., one patient had disease onset at 24 years of age. He was found to carry a homozygous mutation (c.1782*12G>A) in the 3’UTR of the STXBP2 gene (94). He had significant decreased NK cell cytotoxic function and perforin activity suggesting a partial defect. Although very rarely reported in general in association with fHLH, mutations in STXBP2 should be considered when assessing adult patients with HLH.
UNC13D encodes the Munc13-4 protein which has a priming function for cytotoxic granules secretion before fusing to the vesicle membrane (95). The first report described patients from 7 families with infancy or adolescence onset of fHLH (1.5 months – 13 years) caused by homozygous or compound heterozygous LOF (frameshift, stop or deletions) mutations in UNC13D. These mutations resulted in defective release of lytic enzymes from T cell receptor activated lymphocytes (95). Rohr et al. described 1 patient with a first HLH episode at 34 years carrying a compound heterozygous mutation (missense and frameshift leading to a premature stop) (96). Zhang et al. identified 7 adult-onset fHLH (18 – 30 years) associated with eight UNC13D variants (5 missense, 2 splice site and 1 intronic) not reported in healthy controls either in heterozygous or compound heterozygous state (94). Two out of three patients (both with a heterozygous c.753 +3G>A splice site mutation) tested for NK cell function in vitro had absent cytotoxic activity. However, for some mutations, in silico scores predicted a benign effect of the mutation (p.E725G, p.S747N) or mutations were located in non-conserved regions (p.R527W, p.S747N), despite being absent in healthy controls. Therefore, given the lack of functional validation, it remains unclear whether all of these mutations are causative for fHLH.
BACH2 is a transcription factor in T- and B-lymphocytes that regulates differentiation and maturation, and is important in the suppression of inflammation (98, 120).
BACH2-related immunodeficiency with autoimmunity (BRIDA) is a recently discovered disorder caused by LOF mutations in the BACH2, resulting in haploinsufficiency (98). Afzali et al. (98) discovered two LOF mutations that have been predicted to disrupt protein stability and prevent dimerization (p.L28K) or interfere with protein localization in the nucleus by aggregation in the cytoplasm (p.E788K). All patients suffered from inflammatory bowel disease-like symptoms and recurrent respiratory tract infections which was attributed to CVID. In two out of three patients (one carrying the p.L28K-mutation and the other carrying the p.E778K-mutation) disease onset was in childhood, while the third patient (harboring the p.E788K-mutation) developed symptoms in the sixth decade. This suggests a variability in clinical phenotype, with the possibility of adult-onset disease (98).
CTLA4 is a surface molecule expressed on T cells, which binds to B7 molecules on antigen presenting cells (APC), thereby competing with the co-stimulatory molecule CD28. As a result, suppressive functions of regulatory T cells (Tregs) are stimulated and proliferation of effector T cells is inhibited (99). CTLA4 haploinsufficiency, caused by LOF mutations in CTLA4, was first described in 2014 in families with AD inherited form of immune dysregulation, characterized by hypogammaglobulinemia, infectious susceptibility and auto-immunity (99, 121). As for most other monogenic diseases caused by haploinsufficiency, penetrance was incomplete and adult-onset symptoms (up to 40 years of age at disease onset) were common (8). Functionally CTLA4 haploinsufficiency led to impaired transendocytosis and suppressive activity of Treg cells, explaining the susceptibility to autoimmunity (99). The largest cohort to date of CTLA4 haploinsufficiency describes 133 patients from 54 different families, of whom 12 (9.0%) had symptoms after the age of 18 years (18-59 years) (5). Both unaffected and affected members had a similar cellular penetrance in vitro, suggesting that additional factors (hitherto not identified) influence the clinical phenotype and disease onset.
Autoimmune polyglandular syndrome type 1 (APS-1) is a disease characterized by a classic triad of chronic mucocutaneous candidiasis (CMC), hypothyroidism and adrenocortical insufficiency (two of three criteria need to be present for a clinical diagnosis). APS-1 is caused by autosomal recessive mutations in AIRE, encoding a protein expressed in thymic medullary epithelial cells which mediates the ectopic expression of tissue restricted proteins to developing T cells. It is essential to regulate self-tolerance and promotes the negative selection of autoreactive T cells, which can cause autoimmunity (122). AIRE deficiency patients reported in large cohorts from different countries (100, 101, 123) often have infancy onset symptoms, but are mostly diagnosed in later life when cumulative autoimmune phenomena alert physicians for monogenic causes. Autoimmune manifestations in adulthood in the context of recessive mutations is very rare and careful history for milder manifestations in infancy is important. In contrast to recessive mutations, dominant negative mutations in AIRE are commonly associated with organ specific autoimmunity (pernicious anemia, vitiligo, autoimmune thyroiditis) presenting in adulthood (for adults range 21-81 years in Oftedal et al.) (102, 103). Some of the mutation carriers are even asymptomatic with or without the presence of autoantibodies (102). Dominant negative mutations are clustered within the plant homeodomain of the AIRE protein, preventing the binding of AIRE to histone H3, thereby negatively impacting its transcription and transactivation activity. The dominant negative effect can be explained by the fact that AIRE functions as a homotetramer. Incomplete penetrance occurs because the formation of WT AIRE tetramer is still possible and can be sufficient to induce self-tolerance in some individuals. In addition, the strength of the dominant negative effect also correlates with the location of the mutation in the PHD domain, accounting for the phenotypical diversity (102).
ALPS is a disease characterized by benign lymphoproliferation, autoimmunity (mostly cytopenias), susceptibility to lymphomas and a high proportion of double negative T cells (DNT) due to a defective lymphocyte homeostasis by inherited defects in apoptosis genes (104). Clinical diagnosis is guided by available criteria (124) and further classification in subtypes is based on genetic analysis (gene and mode of inheritance): ALPS FAS (FAS homozygous or heterozygous, germline), ALPS-sFAS (FAS, somatic), ALPS-FASLG (FASLG, germline), ALPS-CASP10 (CASP10, germline) or ALPS-U (unknown genetic defect but meets diagnostic criteria) (124). To our knowledge, only ALPS-FAS, ALPS-sFAS have been observed in adult-onset presentations.
The majority of ALPS patients have a genetic defect in FAS, encoding a member of the TNF-superfamily, which is expressed as a homotrimer on B and T cells and essential in the regulation of apoptosis (124). In contrast to recessive mutations in FAS, only found in a minority of ALPS patients, heterozygous germline mutations either leading to a mutant FAS protein (with dominant negative action) or decreased FAS protein expression (haploinsufficiency) are more frequently encountered and associate with an early-onset disease (infancy life) but incomplete penetrance (<60%) (105). The penetrance correlates with the location of the mutation in the protein structure (lower in the extracellular compared to intracellular domain, because mutations in the extracellular domain mostly led to haploinsufficiency) (105). In a cohort of 90 patients with ALPS-(s)FAS, 7% of affected patients had adult-onset disease (range 18-35 years) with a milder form of lymphoproliferation (106). These patients had more combined germline and somatic mutations or germline mutations in the extracellular domain compared to patients with early-onset disease. The somatic mutation was either acquired on the second FAS allele or occurred through somatic uniparental disomy (a situation where both chromosomes are derived from 1 parent). Somatic mutations detected in FAS in DNT cells (either dominant negative or haploinsufficiency) were also described in other reports of ALPS-sFAS in adults (43 and 48 years at disease onset) (15, 107).
MAGT1 is located on the X-chromosome and encodes the magnesium transporter 1, that has a dual function as a plasma membrane magnesium transporter and as a subunit of the endoplasmic reticulum localized oligosaccharyltransferase complex (125). Hemizygous LOF mutations can result in a XMEN syndrome (X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia) in males, whereas female carriers are unaffected. The pathogenesis is most likely related to a defective N-glycosylation of key T and NK cell receptors, impairing their function or leading to rapid degradation (108, 109), although defective magnesium influx leading to impaired downstream signaling upon TCR stimulation was also observed in XMEN patients (110). In the first report, adult-onset (45 years) was described in one patient with EBV driven lymphoma (110). Other adult-onset lymphomas have been reported in later reports (109).
X-linked lymphoproliferative syndrome 1 (XLP-1) is a clinically heterogeneous disorder caused by hemizygous mutations in the SH2D1A gene, that is characterized by extreme susceptibility to EBV infection, HLH, dysgammaglobulinemia and malignant lymphoma (111, 126, 127). SH2D1A encodes SAP (signaling lymphocyte activation molecule (SLAM)-associated protein), which is a cytosolic protein that plays an indispensable role in the signal transduction of T cells, NK cells and Natural killer T lymphocytes (NKT) cells (112, 128). The clinical phenotype is very variable, and although pediatric onset is most common, adult-onset with lymphoma or HLH as primary manifestation has been described (111, 113, 114, 129). In a large cohort study of Booth et al. (111), onset of disease in EBV+ patients ranged from 8 months to 40 years (median 4 years), and in EBV- patients from birth to 31 years (median 3.5 years). So even in the absence of EBV infection, disease was observed in adulthood, meaning that other factors than EBV can influence disease onset. Similarly, a recent case report of a 21-year-old male presenting with EBV+ HLH showed presence of a pathogenic variant in SH2D1A (p.E17K), leading to a normally expressed mutant protein with diminished binding to phosphorylated 2B4 receptor (important for NK-cell activation) (114). His siblings carrying the same mutation were unaffected, even after encountering EBV infection. Again, EBV was not a determinant for disease onset. Another report supporting role for genetic confounders comes from Liang et al., describing a 44-year-old female patient presenting with HLH and NK cell leukemia. She harbored a mutation (p.A3S) in the SH domain of SAP, resulting in absent protein expression (129). Targeted sequencing revealed a 28.7% mutant/WT ratio suggestive for a somatic mutation, although this was not confirmed by investigating the presence of the mutation in non-hematopoietic tissue. Somatic mosaicism in combination with X linked skewing (XCI) could explain why this patient had an adult-onset phenotype, but this was not investigated (129).
Another less frequent cause of an XLP (XLP-2) is caused by deficiency of the X-linked inhibitor of apoptosis (XIAP) (115). XIAP is an important inhibitor of apoptosis that is expressed in different hematopoietic cells such as lymphocytes, myeloid cells and NK cells. Cells from XIAP-deficient patients are shown to have increased sensitivity to apoptosis and patients display almost absent numbers of NKT cells (115). Phenotypically it highly resembles XLP1, with the addition that splenomegaly is often the first presenting manifestation (115, 130) and that XIAP deficiency is associated with inflammatory bowel disease (130). Rigaud et al. characterized a potential modifier gene that contributes to development of disease phenotype in patients harboring a hypomorphic mutation in XIAP (p.G466*) (116). In their study, patients carrying a co-segregated CD40-ligand (CD40L) polymorphism (p.G219R) in addition to the XIAP defect developed clinical disease manifestations, whereas patients harboring the hypomorphic XIAP mutation alone remained asymptomatic (116). The CD40L mutation was demonstrated to affect B cell differentiation and class switch recombination. Although non-random X-inactivation favoring the WT allele has been described (115), symptomatic female carriers have been identified (117). Aguilar et al. (117) described two heterozygous female patients carrying hypomorphic mutations in XIAP (p.H220Y and p.G466*) that developed inflammatory bowel disease at an adult age (32 years and 28 years). Both displayed a predominant expression of the mutant allele, suggesting that the hypomorphic nature of their XIAP mutations could have contributed to skewed X-inactivation towards the mutant or that the presence of a second undefined mutation gave a selection advantage (117).
Defects in intrinsic and innate immunity are categorized according to the specific pathogen susceptibility caused by genetic defects. Some of them are rarely or never seen in adults and therefore left out of the scope of this review (ND). Overall, 9 categories exist: a) mendelian susceptibility to mycobacterial disease (MSMD), b) epidermodysplasia verruciformis (ND), c) predisposition to severe viral infection (ND), d) herpes simplex encephalitis (HSE), e) invasive fungal infections, f) chronic mucocutaneous candidiasis, g) TLR signaling pathway deficiency with bacterial susceptibility, h) other inborn errors of immunity related to non-hematopoietic tissues and i) other inborn errors of immunity related to leukocytes (Table 5).
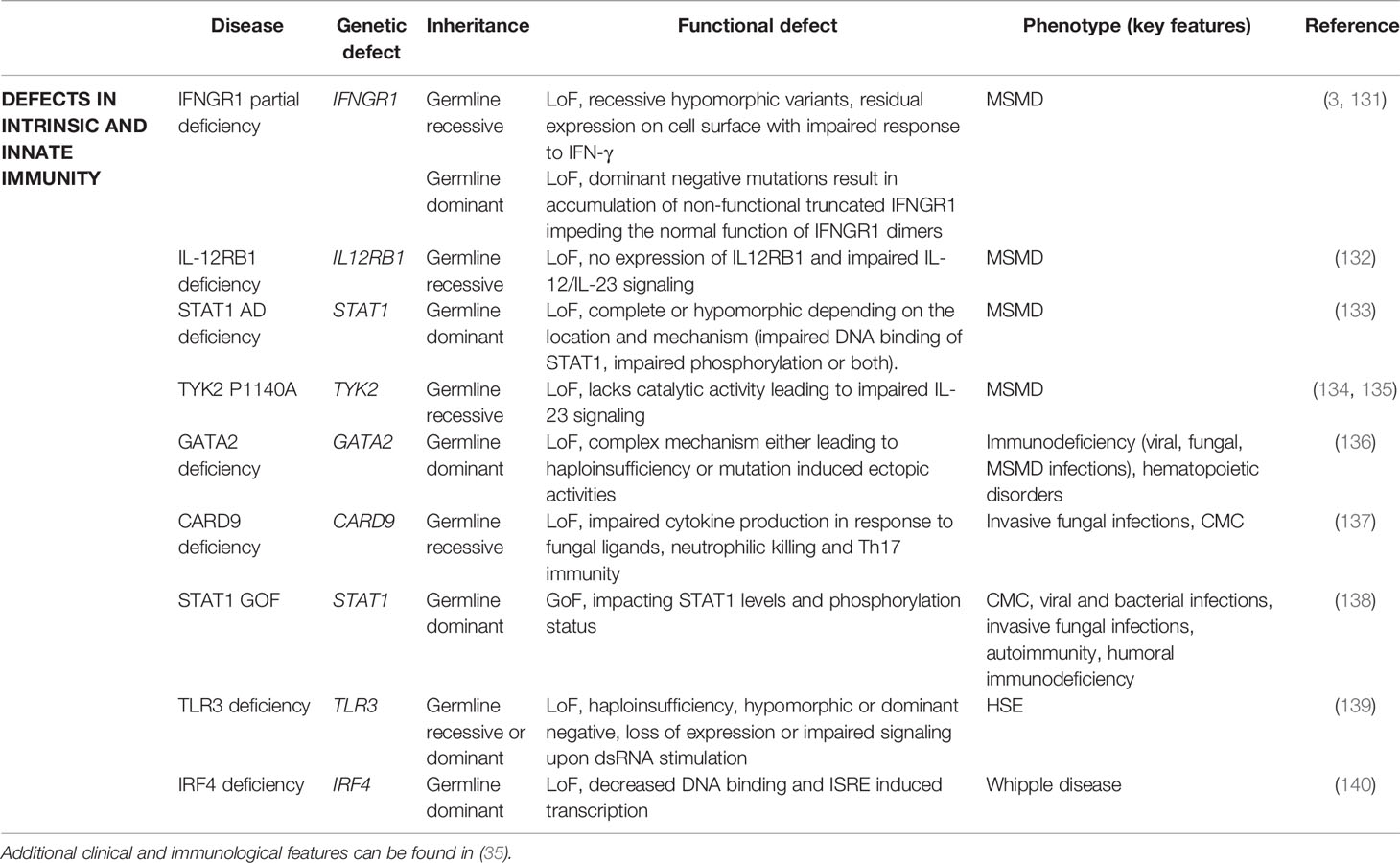
Table 5 Genes associated with adult-onset defects in intrinsic and innate immunity.
MSMD is characterized by predisposition to mycobacterial disease caused by weakly virulent mycobacteria in otherwise healthy individuals. Till present, all genes implied in MSMD play a direct or indirect role in the IFN-γ dependent immunity, crucial to mycobacterial defense.
Mutations in the IFN-γ receptor, composed of a heterodimer of IFNGR1 and IFNGR2, were the first described genetic defects in MSMD (3). Recessive mutations causing a complete deficiency without residual expression cause a severe infancy onset phenotype with life threatening mycobacterial (or some other intracellular bacteria such as Listeria and Salmonella spp.) and viral infections. Hypomorphic recessive LOF mutations in IFNGR1 (although rarely seen in adulthood) or AD LOF mutations in IFNGR1 causing a partial deficiency are associated with a milder phenotype and can be observed in later life (3, 131).
IL12RB1 is both part of the IL12R (in combination with IL12RB2) and of the IL23R (in combination with the IL23R), mediating IL-12 and IL-23 signaling. IL12RB1 deficiency, caused by recessive LOF mutations, is the most common genetic defect in MSMD. One of the largest cohorts studying 141 IL12RB1 deficient patients, observed an age of onset between 1 week to 31.7 years (mean age, 2.4 years, SD ± 4.9 years, range 2 weeks to 31.7 years) (132). Most of the cases were caused by BCG vaccination. Eight of the patients remained asymptomatic at the time of publication, even though the in vitro cellular penetrance was complete. Together this indicates that exposure to a specific pathogen most likely drives the age of onset in this immunodeficiency.
STAT1 is an important transcriptional activator mediating cellular responses to pathogenic organisms, including mycobacteria. Both biallelic LOF mutations (complete or partial deficiency) characterized by severe viral and bacterial infections during infancy and monoallelic LOF or GOF mutations (discussed further) have been described. AD STAT1 deficiency due to LOF mutations has been observed in different kindreds with milder forms of MSMD and incomplete penetrance (133). In a 3 generation Indian kindred with MSMD, the oldest patient with a p.G250A LOF mutation in STAT1 was 36 years at disease onset (141). Mutations can have a complete or partial LOF effect depending on their location and whether DNA binding capacity, STAT1 phosphorylation or both is affected (3). Moreover, for all mutations the effect on IFN-γ signaling is dominant negative in contrast to IFN-α and IFN-β signaling, explaining why most patients do not suffer from severe viral infections (3).
TYK2 is a member of the JAK tyrosine kinase family and associates with multiple type I and II cytokine receptors. Recently, a genome wide study identified a common homozygous SNP in TYK2 (p.P1104A) which is strongly enriched in European populations with a MAF of 4.2%, and is more prevalent in patients with tuberculosis (1%) compared to healthy individuals (0.2%) (134). Functionally this mutation was shown to carry a LOF effect by loss of TYK2 catalytic activity downstream of the IL23 receptor (135). Disease onset was highly variable (ranging from 1 to 40 years). Some patients received the BCG vaccine in early life without further complications and later developed pulmonary tuberculosis in adulthood, suggesting importance of pathogen virulence in addition to genetic susceptibility.
GATA2 encodes a transcriptional regulator of multilineage hematopoiesis. AD inherited LOF mutations can cause GATA2 deficiency syndrome, characterized by immunodeficiency (viral, bacterial, fungal, MSMD), hematopoietic disorders such as myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) and lymphedema, and a highly variable penetrance manifesting from infancy to adult age (142). A possible hypothesis for this variable penetrance is that the trigger of this syndrome might be evoked by somatic mutations in other genes such as RUNX1, ETV6, CEBPA, ASXL1, SETBP1 and STAG2, as they commonly occur together (142). However, clear evidence about their influence on the pathogenesis is currently lacking. A series of 79 patients in France and Belgium was described recently and mycobacterial disease was observed in 8.1% of patients as the first presenting symptom, most of them after the age of 20 (136). In addition, hematological manifestations were often the first manifestation, with 69% presenting with MDS and 9% with AML. The median onset of symptoms in their GATA2 deficiency cohort was 18.6 years, ranging from 0 to 61 years of age.
Toll like receptor 3 (TLR3) recognizes double stranded RNA (dsRNA), which is produced by most viruses including HSV type 1. In a cohort of 120 patients (both children and young adults), 6 pathogenic functionally validated TLR3 variants were found in 6 patients (139). Three out of 6 patients had HSE episodes in adulthood and 1 of them had a first episode at the age of 24 years. LOF mutations can be inherited in a AD or recessive manner, and different mechanisms underly TLR3 deficiency (hypomorphic, haploinsufficiency or dominant negative effect). Not surprisingly the hypomorphic variant (p.R867Q) was found recessively in the patient with an adult-onset phenotype. The TLR3 expression was normal but functionally, the mutation led to an impaired signaling upon Poly(I:C) stimulation of primary fibroblasts and a TLR3-deficient cell line which was transfected with this mutant.
CARD9 encodes an adaptor protein downstream of C-type lectin receptors that recognize fungal components. Mutations in CARD9 – presumed LOF – are associated with predisposition to mucocutaneous and invasive fungal disease (IFD). Functionally they can negatively impact cytokine production in response to fungal ligands, neutrophilic killing and Th17 immunity (137). A recent review on CARD9 deficiency evaluated 58 patients from 39 kindreds with disease causing recessive CARD9 mutations (137). Fungi typically belonged to the phylum Ascomycota (including Candida, Trychophyton, Aspergillus) and most patients were affected by a single fungus Clinical penetrance was complete, although for patients with IFD most cases were adult-onset (median age 18 years, range 3.5-52 years). This is intriguing because in contrast to MSMD, colonization/exposure to fungi such as C. albicans begins very early in life (for C. albicans before the age of one in 50% of cases) (143).
AD GOF STAT1 mutations lead to defective Th1 and Th17 responses with a reduced production of IFN-γ, IL-17 and IL2, thereby leading to a phenotype of CMC, susceptibility to bacterial and viral infection and autoimmunity. The largest cohort till present studied 274 patients from 167 kindreds with STAT1 GOF mutations. Adult-onset was mostly seen in patients without CMC (n=6, range 4-61 year) whereas CMC often presented in early life (n=268, range birth-24 years) (138). Some patients might present first with mild auto-immune features such as autoimmune hypothyroidism, and then later in life during adulthood develop infectious complications such as CMC and severe viral infections with immunophenotypic abnormalities raising a suspicion for STAT1 GOF (144). Others may present in adulthood with IFD as the first manifestation or with multiple auto-immune or autoinflammatory features (such as Takayasu arteritis and inflammatory bowel disease) (145).
IRF4 is a transcription factor with essential functions in lymphocytes including development, antibody affinity maturation and roles in effector T cells. In a family affected by Whipple disease with a mean age of 55 years at onset, a private p.R98W mutation in IRF4 was identified, proven to be LOF in vitro based on its decreased ability to bind DNA and to induce transcription of interferon stimulated response element motif containing promotors compared to WT IRF4 (140). IRF4 deficiency was identified as an AD cause of Whipple disease with (unexplained) incomplete penetrance.
Deficiency of adenosine deaminase 2 (DADA2) was first described in 2014 in 9 patients with intermittent fever, systemic vasculopathy early-onset ischemic stroke (164), caused by recessive mutations in CECR1 encoding ADA2. These mutations cause (near) complete absence of enzyme function resulting in elevated extracellular adenosine leading to dysregulated formation of neutrophilic extracellular traps, neutrophilic activation, polarization of macrophages from M2 to M1 subtype and increased proinflammatory cytokine production (165). Adult-onset cases have been documented in cohorts with idiopathic polyarteritis nodosa (160, 161) without prior manifestations although a thorough history should always be taken for possible features in infancy which retrospectively can be connected to DADA2 (Table 6) (166).
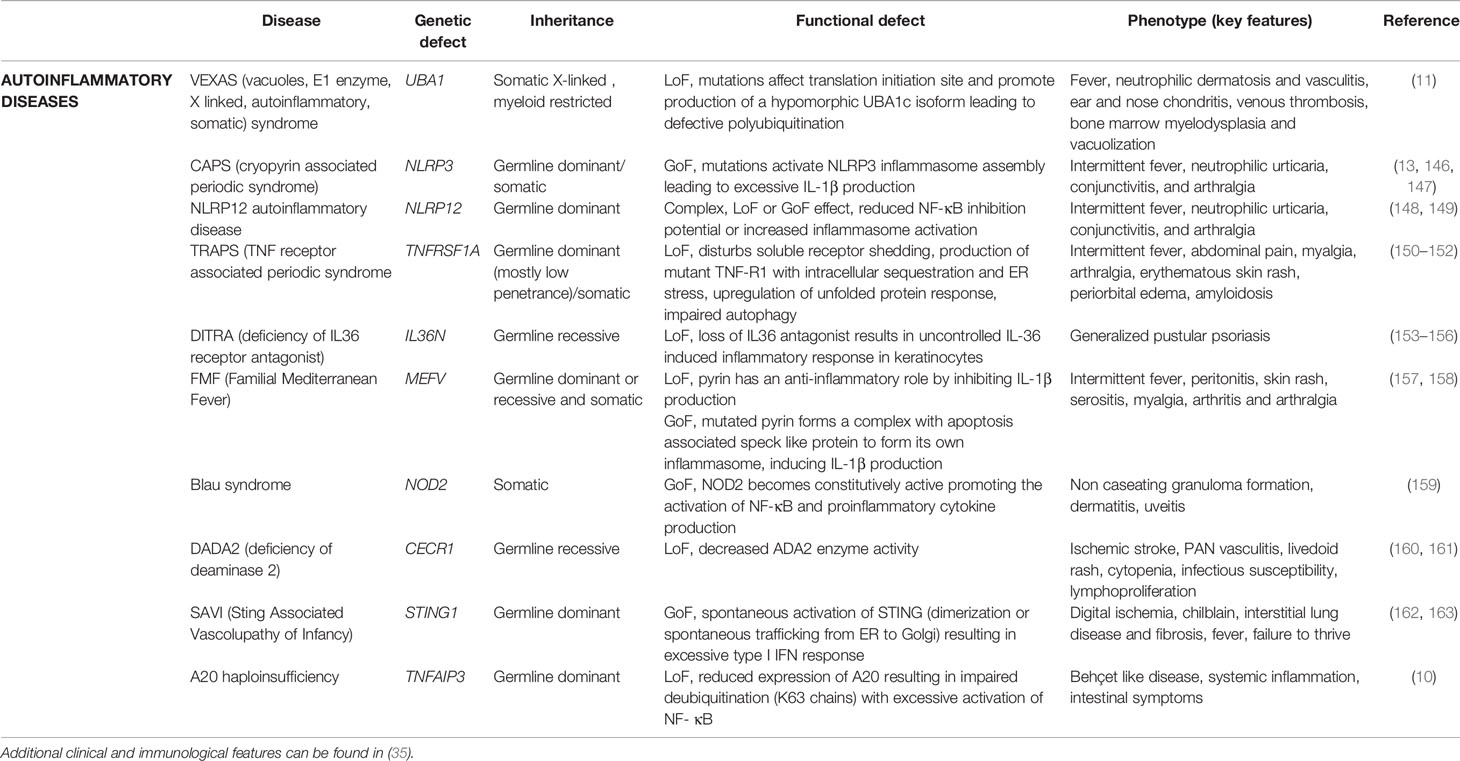
Table 6 Genes associated with adult-onset autoinflammatory diseases.
SAVI is an acronym that stands for STING Associated Vasculopathy of Infancy, described in 2014 (162). It is an AD inherited disease caused by GOF mutations in STING1, causing an uncontrolled activation of the cyclic GMP-AMP synthase (cGAS)-STING cytosolic DNA sensing pathway resulting in excessive type I interferon production (162). Recessive inheritance of GOF mutations has recently been described (167). Clinical manifestations mainly consist of pulmonary (interstitial lung disease, fibrosis), systemic (fever, failure to thrive) or skin manifestations (chilblains, digital ischemia) (163). Although the acronym suggests an infancy onset, adult-onset vasculitis with renal manifestations has been reported in a patient (163). Genetic modifiers such as SNPs in STING1 itself or other interferon related genes such as IFIH1 could impact disease severity (163) and viral exposure in a STING1 GOF mouse model determined the development of pulmonary fibrosis (23).
Familial periodic fever syndrome is the most common monogenic autoinflammatory disorder. It is generally caused by homozygous or CH mutations in MEFV encoding the protein pyrin, although patients with heterozygous mutations are reported where no second hit was found (157). Pyrin interacts with inflammasome components and caspase 1 to induce the production of IL-1β. Whether mutations in MEFV act as GOF or LOF remains a matter of debate and evidence exists to support both hypotheses (168). Up till now 61 (likely) pathogenic mutations have been reported in the Infevers database linked to a phenotype of FMF and most of them validated in vitro. Adult-onset phenotype is commonly seen and differs from childhood onset FMF with regards to genetic and clinical aspects. Adult-onset FMF (onset ≥20 years is associated with a lower prevalence of highly penetrant mutations (e.g. homozygous M694V) and clinical symptoms such as fever, peritonitis, pleuritis, arthritis and erythema are less observed compared to early-onset FMF (169). A somatic, heterozygous myeloid restricted mutation (p.R652H) was claimed to be responsible for late onset FMF in a middle aged Ashkenazi Jewish woman with a prior diagnosis of JAK2 positive polycythemia vera (PV). At the time of PV diagnosis, the MEFV mutation was observed at a very low level by Sanger sequencing on PBMCs, but reanalysis 4 years later when inflammatory symptoms commenced, showed that the MEFV mutation reached a MAF of 46% in PBMCs. This mutation was only observed in co-segregation with the JAK2 mutant suggesting a JAK2 driven clonal expansion of MEFV mutant containing cells (158).
Cryopyrin associated periodic syndrome (CAPS) is a group of diseases related to a defect in the protein cryopyrin (NLRP3). NLRP3 is a key component of the inflammasome functioning as a pattern recognition receptor (PRR) binding pathogen associated molecular patters (PAMP) such as products released from damaged cells (uric acid, extracellular ATP). Upon binding to PAMPs, recruitment of the NLRP3 inflammasome and adapter protein apoptosis associated speck-like protein (ASC) is initiated activating caspase-1 which mediates the production of proinflammatory cytokines such as IL-1β (170). ASC has also been shown to have prionoid activities that propagate inflammation (170). GOF mutations in NLRP3 result in abnormal activation of the inflammasome causing excessive, uncontrolled inflammatory responses. Clinically three phenotypes have been described; familial cold autoinflammatory syndrome (FCAS), Muckle Wells syndrome (MWS) and neonatal onset multisystem inflammatory disease (NOMID). Although initially described in neonates and infants, adult-onset patients have been reported. In one series the median age of onset was 13 (range 4-40) (171) with the oldest case reported having a disease onset at the age of 46 (172). In addition, somatic NLRP3 mosaicism has been identified in adult-onset cases (13, 146). Some of these patients were given the diagnosis of Schnitzler disease prior to genetic diagnosis (147). Clinical presentation includes intermittent febrile episodes, fatigue, headache, neutrophilic urticaria, conjunctivitis, and arthralgia (173).
NLRP12 encodes the protein monarch-1 which mainly functions as a suppressor of the (non)-canonical NF-κB pathway. However, NLRP12 can also be involved in inflammasome signaling and drive caspase-1 activation resulting in proinflammatory cytokine release (174). Several sporadic cases or families with heterozygous NLRP12 mutations, presenting with an autoinflammatory disease highly resembling CAPS, have been described (148, 149). The pathogenesis is complex and seems to depend on the type of the mutation. For example a described nonsense mutation p.R284* was shown to be less effective in suppressing NF-κB activity consistent with a LOF effect. Other missense mutations such as p.R294Q or p.R352C rather directly increase speck formation and caspase-1 signaling suggesting a GOF effect (174). A recent case series, describes adult-onset (range 18-54 years) in 27% (8/29) of studied patients, all harboring a pathogenic missense mutation (p.F402L or p.G448A) (148, 149).
CARD14 functions as a scaffold protein, highly expressed in keratinocytes, that regulates NF-κB signaling. Pathogenic missense variants in CARD14 with a GOF effect resulting in amplified NF-κB responses in keratinocytes can cause a spectrum of skin conditions such as pustular psoriasis, psoriasis vulgaris and familial pytiriasis rubra vulgaris without the presence of systemic symptoms. Incomplete penetrance and considerable disease severity variability and onset (neonatal-83 years) are seen within and between families (175, 176). The degree of NF- κB signaling activity induced by a mutant is probably a determinant for disease onset, as the most severe phenotype of early-onset generalized pustular psoriasis was seen in a patient with a de novo p.E138A variant which was demonstrated to have the highest NF-κB activity in in vitro overexpression experiments compared to other pathogenic CARD14 mutations (176).
IL36 receptor antagonist deficiency is a genetic disorder associated with generalized pustular psoriasis. It was first reported in nine Tunisian families sharing a homozygous missense mutation in IL36RN (p.L27P), leading to a decreased expression and thereby unable to inhibit proinflammatory cytokine production by patient keratinocytes upon stimulation by IL-36. A total of 16 affected individuals of whom 4 developed disease in adulthood were reported (153). Later, other reports on adult- onset pustular psoriasis have been made in association with homozygous complete LOF variants in IL36RN (154–156). The variation in age of onset was therefore attributed to other modifying genes and/or environmental factors, since no partial gene function in vitro was retained.
Blau syndrome is a rare AD autoinflammatory syndrome, characterized by non-caseating granulomatous arthritis, dermatitis and uveitis. It is caused by GOF mutations in NOD2, an intracellular PRR, resulting in a spontaneous activation of NOD2 with downstream activation of NF-κB responsive genes. The classic Blau syndrome was reported once in an adult patient (22 years at onset) (159). He carried gonosomal NOD2 mosaicism (p.R334G) and his both children had early-onset symptoms (11.8 and 30 months). Amplicon based deep sequencing of the NOD2 gene showed a MAF on PBMC of 12.9% in the father and 49-51.5% in the children suggesting a gene dosage or cellular compartment effect. One other patient with somatic mosaicism has been published with a later onset and milder phenotype although this was still with a presentation in infancy (177).
A20 haploinsufficiency is caused by germline heterozygous LOF mutations in TNFAIP3, encoding the deubiquitination enzyme A20. A20 is a critical regulatory unit of the canonical NF-κB pathway, by functioning as an inhibitor of key proinflammatory molecules. The causal link between heterozygous LOF variants in A20 an autoinflammatory disorder with Behçet-like manifestations (aphtous stomatitis, genital ulcers and intestinal symptoms) was described in 2016 (178). Adult-onset cases (oldest age of onset 20 years) have been sporadically reported in families with variable penetrance (10).
TNF receptor associated periodic syndrome (TRAPS) is the second most frequent inherited AD autoinflammatory disease. TNF-R1 expressed on immune cells is activated by TNF-α, allowing the recruitment of several adaptor proteins leading to the formation of complex I which activates NF-κB and transcription of anti-apoptotic and proinflammatory genes. Up till now 103 mutations, most of them located in the extracellular part of the receptor, have been classified as (likely) pathogenic in patients with a compatible phenotype according to the Infevers database. Some of these mutations still remain to be validated by in vitro assays. TRAPS is more complex than other autoinflammatory condition regarding the pathogenesis, since there is not one predominant mechanism. Mutations can have different and multiple impacts on protein function, either by affecting the cleavage of the extracellular domain preventing the release of soluble receptors (‘shedding effect’) attenuating inflammatory response or by expressing a mutant TNF-R1 alongside the WT resulting in a dysregulated inflammatory response (179). Adult-onset phenotypes are associated with low penetrance variants such as p.R92Q or p.P46L (150) but also somatic mosaicism has been described in two cases of adult-onset TRAPS (151, 152). Clinical features may range from atypical images such as isolated recurrent pericarditis to more typical but adult-onset periodic fever syndromes with serositis, myalgia/arthralgia, erythematosus skin lesions, periorbital edema, and, in case of long-standing uncontrolled inflammation, amyloidosis (173).
VEXAS (vacuoles, E1 enzyme, X linked, autoinflammatory, somatic) syndrome is a recently described autoinflammatory disorder that presents in adult males, typically age 45-80 years (11). The underlying genetic defect is caused by somatic mutations in UBA1, an E1 ubiquitin activating enzyme. In a normal situation, two UBA1 isoforms (UBA1a and UBA1b) are produced from two translation sites (M1 and M41). The reported mutations (p.M41V, p.M41L, p.M41T) disrupt the second translation site (M41) of UBA1, resulting in the production of a third isoform (UBA1c) from a third downstream translation site (M67) which is normally not expressed. UBA1c, compared to UBA1a and b, is catalytically impaired resulting in loss of ubiquitylation leading to proteotoxic stress and dysregulated autophagy. Remarkably these mutations are restricted to the myeloid lineage in the periphery. Lymphoid progenitors were shown to carry the mutation, but for an undefined reason, mutated lymphoid progenitors do not further mature and translocate into the peripheral blood. Clinical presentation manifests as fever, nose/ear chondritis, skin disease with vasculitis and neutrophilic infiltration, venous thrombosis and often hematologic abnormalities ranging from isolated macrocytosis to myelodysplastic syndrome on bone marrow biopsy. Vacuolization of myeloid cells was present in all patients.
The complement system is a highly conserved part of our innate immunity, organized in three enzymatic pathways: the classical (CP), alternative (AP) and lectin pathways. Briefly, activation of these pathways results in the cleavage of C3 by a C3 convertase, followed by formation of a C5 convertase which cleaves C5 and initiates the activation of the common terminal pathway where a membrane attack complex (MAC) is formed by complement proteins C5b-9 (185). This MAC is responsible for the lysis of target cells (eg. bacterial, human cancer cells) promoting host defense. Deficiencies in complement are associated with auto-immune diseases such as systemic lupus erythematosus (SLE), frequently seen in defects of the early components of the classical pathway (C1Q, C1R/S, C2, C4) or with susceptibility to meningococcal disease and/or atypical hemolytic uremic syndrome (aHUS) by defects in the terminal pathway components (C5, C6, C7, C8A/B/G, C9) or complement regulators (CFP, CFH, CFD, CFI, CD46). Complement deficiencies in general are rare (~5% of IEIs), and seldomly have a first presentation in adulthood (Table 7) (186). If it occurs, adults mostly present with meningococcal disease or aHUS and genetic defects are found in the terminal component pathway or its regulators (186). For the scope of this review, focusing on immunodeficiency, we focused on genetic defects in adults with infectious susceptibility rather than isolated aHUS (with in rare cases infectious susceptibility) since the latter proportion of patients will likely be seen by a nephrologist. For an overview on C1-esterase inhibitor, encoded by SERPING1, the reader is referred to Busse et al. (183).

Table 7 Genes associated with adult-onset complement deficiencies.
Phenotypically, deficiencies in the components of the MAC complex, are very resembling, presenting with invasive meningococcal disease (186). A nationwide French study enrolled 41 adults (defined as > 15 years) with diverse complement deficiencies and an infectious episode (180). Mean age at diagnosis was 28 years (range 15-67 years), with the highest proportion in group 15-25 years (25%). Importantly, half of the cohort already reported a serious infectious event before diagnosis (unspecified time of delay), so an adult-onset criterium is not always met. Genetic analysis demonstrated that 83% of patients had a terminal pathway deficiency (mostly in C7 and C6, followed by C5 and 8) and in 80% of the cases meningitis was the main clinical symptom. C9 deficiency was not found in this French cohort, because this is almost exclusively seen in patients from Japanese descent (181).
Factor D, encoded by CFD, is a peptidase and a component of the AP. It binds and cleaves factor B to Ba and Bb to promote downstream activation. Deficiency in Factor D, caused by a homozygous p.S42* mutation, was diagnosed in a 23-year-old, previously healthy, woman who presented with meningococcal disease at first presentation (182).
MASP2 encodes mannose binding lectin associated serine protease which forms a multimeric complex with mannose binding lectin and subsequently cleaves components C4 and C2 leading to downstream activation of the complement cascade (187). MASP2 deficiency was first described in 2013 in a patient who presented at the age of 13 years with ulcerative colitis and later in life developed infectious susceptibility (pneumococcal infections), skin involvement (erythema multiforme), progressive lung fibrosis and positive auto-immune antibodies (188). Functional assays showed a non-functional lectin pathway in this patient, caused by a homozygous missense mutation (p.D120G) in MASP2, leading to an abolished expression. Recently, a case of two patients with adult-onset HSE (24 and 60 years) was reported (184). Both of them had a single heterozygous deleterious mutations (p.G634R and p.R203W). Both mutations led to an abnormal protein secretion, a lost ability of auto-activation (p.G634R) or reduced antiviral activity (p.G634R). Furthermore, the authors showed that rare MASP2 variants are enriched among HSE patients compared to healthy controls and that mice deficient in mannose binding lectin (MBL) were more prone to HSE (with lower survival rates and higher viral loads) upon intranasal inoculation (184). Whether MASP2 deficiency is a monogenic cause of IEI or merely a contributing factor is still a matter of debate (189). Healthy individuals who are MASP2 deficient (and have homozygous p.D120G mutations) have been reported. It might be possible that interplay with environmental triggers is important before a phenotype can manifest, or that there is a large redundancy for MASP2 in human defenses (189).
Advances in next generation sequencing has significantly expanded the identification of novel genes involved in IEI over the years, not only in early-onset severe IEI such as SCID but also in milder forms such as antibody deficiencies, innate immune defects, immune dysregulation diseases, autoinflammatory diseases and complement deficiencies that can manifest in adulthood. The spectrum of age of onset during adulthood is highly variable for all forms of IEI, except for the diseases affecting humoral and cellular immunity which usually manifest in the first decades (Figure 3). The elucidation of molecular drivers of IEI has important consequences towards the management of these patients since it can rationalize targeted treatment (eg. JAK-inhibition in interferonopathies, TNF-α inhibitors in DADA2, CTLA4 agonists in CTLA4 haploinsufficiency, sirolimus in ALPS, leniolisib in APDS), bone marrow transplant in some cases, provide prognostic information and inform genetic counselling. Therefore, physicians encountering adult patients with recurrent (common or rare specific) infections, autoinflammatory disorders or lymphoproliferation should be aware of the occurrence of IEI in this population and, if confirmed, consider the possibility of a monogenic driven disease. These patients should be referred to a specialized tertiary center for further diagnostics using targeted gene panels or whole exome/genome sequencing. Mechanisms for late onset disease are not always well understood, but hypomorphic mutations allowing partial protein function, somatic mosaicism, environmental exposure and epigenetics are likely the main contributors.
RS initiated and supervised the study. FS took the lead in drafting the manuscript. FS and TC reviewed the literature. All authors provided critical feedback and helped shaping the manuscript.
FS (11B5520N) is fellow of the Fonds Wetenschappelijk Onderzoek - Vlaanderen National Fund for Scientific Research (FWO). RS is FWO senior clinical investigator fellows (1805518N, respectively) and received funding from KU Leuven C1 (C12/16/024). RS and SV are members of the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases (Project ID No 739543). This work was supported by the VIB Grand Challenges Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
2. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The Ever-Increasing Array of Novel Inborn Errors of Immunity: An Interim Update by the IUIS Committee. J Clin Immunol (2021) 41(3):666–79. doi: 10.1007/s10875-021-00980-1
3. Bustamante J, Boisson-Dupuis S, Abel L, Casanova J-L. Mendelian Susceptibility to Mycobacterial Disease: Genetic, Immunological, and Clinical Features of Inborn Errors of IFN-γ Immunity. Semin Immunol (2014) 26(6):454–70. doi: 10.1016/j.smim.2014.09.008
4. Kaneko H, Kawamoto N, Asano T, Mabuchi Y, Horikoshi H, Teramoto T, et al. Leaky Phenotype of X-Linked Agammaglobulinaemia in a Japanese Family. Clin Exp Immunol (2005) 140(3):520–3. doi: 10.1111/j.1365-2249.2005.02784.x
5. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, Penetrance, and Treatment of 133 Cytotoxic T-Lymphocyte Antigen 4–Insufficient Subjects. J Allergy Clin Immunol (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
6. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-Of-Function Nuclear Factor κb Subunit 1 (NFKB1) Variants are the Most Common Monogenic Cause of Common Variable Immunodeficiency in Europeans. J Allergy Clin Immunol (2018) 142(4):1285–96. doi: 10.1016/j.jaci.2018.01.039
7. Gruber C, Bogunovic D. Incomplete Penetrance in Primary Immunodeficiency: A Skeleton in the Closet. Hum Genet (2020) 139(6):745–57. doi: 10.1007/s00439-020-02131-9
8. Rieux-Laucat F, Casanova J-L. Autoimmunity by Haploinsufficiency. Science (New York NY) (2014) 345(6204):1560. doi: 10.1126/science.1260791
9. Mensa-Vilaró A, Bravo García-Morato M, de la Calle-Martin O, Franco-Jarava C, Martínez-Saavedra MT, González-Granado LI, et al. Unexpected Relevant Role of Gene Mosaicism in Patients With Primary Immunodeficiency Diseases. J Allergy Clin Immunol (2019) 143(1):359–68. doi: 10.1016/j.jaci.2018.09.009
10. Kadowaki T, Ohnishi H, Kawamoto N, Hori T, Nishimura K, Kobayashi C, et al. Haploinsufficiency of A20 Causes Autoinflammatory and Autoimmune Disorders. J Allergy Clin Immunol (2018) 141(4):1485–8.e11. doi: 10.1016/j.jaci.2017.10.039
11. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med (2020) 383(27):2628–38. doi: 10.1056/NEJMoa2026834
12. Zhou Q, Aksentijevich I, Wood GM, Walts AD, Hoffmann P, Remmers EF, et al. Brief Report: Cryopyrin-Associated Periodic Syndrome Caused by a Myeloid-Restricted Somatic NLRP3 Mutation. Arthritis Rheumatol (2015) 67(9):2482–6. doi: 10.1002/art.39190
13. Rowczenio DM, Gomes SM, Aróstegui JI, Mensa-Vilaro A, Omoyinmi E, Trojer H, et al. Late-Onset Cryopyrin-Associated Periodic Syndromes Caused by Somatic NLRP3 Mosaicism—UK Single Center Experience. Front Immunol (2017) 8(1410). doi: 10.3389/fimmu.2017.01410
14. Mensa-Vilaro A, Teresa Bosque M, Magri G, Honda Y, Martínez-Banaclocha H, Casorran-Berges M, et al. Brief Report: Late-Onset Cryopyrin-Associated Periodic Syndrome Due to Myeloid-Restricted Somatic NLRP3 Mosaicism. Arthritis Rheumatol (2016) 68(12):3035–41. doi: 10.1002/art.39770
15. Dowdell KC, Niemela JE, Price S, Davis J, Hornung RL, Oliveira JB, et al. Somatic FAS Mutations are Common in Patients With Genetically Undefined Autoimmune Lymphoproliferative Syndrome. Blood (2010) 115(25):5164–9. doi: 10.1182/blood-2010-01-263145
16. Lambotte O, Neven B, Galicier L, Magerus-Chatinet A, Schleinitz N, Hermine O, et al. Diagnosis of Autoimmune Lymphoproliferative Syndrome Caused by FAS Deficiency in Adults. Haematologica (2013) 98(3):389–92. doi: 10.3324/haematol.2012.067488
17. Wolach B, Scharf Y, Gavrieli R, de Boer M, Roos D. Unusual Late Presentation of X-Linked Chronic Granulomatous Disease in an Adult Female With a Somatic Mosaic for a Novel Mutation in CYBB. Blood (2005) 105(1):61–6. doi: 10.1182/blood-2004-02-0675
18. Van Horebeek L, Dubois B, Goris A. Somatic Variants: New Kids on the Block in Human Immunogenetics. Trends Genet (2019) 35(12):935–47. doi: 10.1016/j.tig.2019.09.005
19. Roucher-Boulez F, Mallet D, Chatron N, Dijoud F, Gorduza DB, Bretones P, et al. Reversion SAMD9 Mutations Modifying Phenotypic Expression of MIRAGE Syndrome and Allowing Inheritance in a Usually De Novo Disorder. Front Endocrinol (Lausanne) (2019) 10:625–. doi: 10.3389/fendo.2019.00625
20. Staels F, Betrains A, Doubel P, Willemsen M, Cleemput V, Vanderschueren S, et al. Adult-Onset ANCA-Associated Vasculitis in SAVI: Extension of the Phenotypic Spectrum, Case Report and Review of the Literature. Front Immunol (2020) 11:575219–. doi: 10.3389/fimmu.2020.575219
21. Reichenbach J, Rosenzweig S, Döffinger R, Dupuis S, Holland SM, Casanova JL. Mycobacterial Diseases in Primary Immunodeficiencies. Curr Opin Allergy Clin Immunol (2001) 1(6):503–11. doi: 10.1097/00130832-200112000-00003
22. Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, et al. Inherited IFNAR1 Deficiency in Otherwise Healthy Patients With Adverse Reaction to Measles and Yellow Fever Live Vaccines. J Exp Med (2019) 216(9):2057–70. doi: 10.1084/jem.20182295
23. Bennion BG, Ingle H, Ai TL, Miner CA, Platt DJ, Smith AM, et al. A Human Gain-Of-Function STING Mutation Causes Immunodeficiency and Gammaherpesvirus-Induced Pulmonary Fibrosis in Mice. J Virol (2019) 93(4):1806–18. doi: 10.1128/JVI.01806-18
24. Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections Associated With Haemophagocytic Syndrome. Lancet Infect Dis (2007) 7(12):814–22. doi: 10.1016/S1473-3099(07)70290-6
25. Riballo E, Doherty AJ, Dai Y, Stiff T, Oettinger MA, Jeggo PA, et al. Cellular and Biochemical Impact of a Mutation in DNA Ligase IV Conferring Clinical Radiosensitivity*. J Biol Chem (2001) 276(33):31124–32. doi: 10.1074/jbc.M103866200
26. Staines Boone AT, Chinn IK, Alaez-Versón C, Yamazaki-Nakashimada MA, Carrillo-Sánchez K, García-Cruz M, et al. Failing to Make Ends Meet: The Broad Clinical Spectrum of DNA Ligase IV Deficiency. Case Series and Review of the Literature. Front Pediatr (2019) 6(426). doi: 10.3389/fped.2018.00426
27. Israel L, Wang Y, Bulek K, Della Mina E, Zhang Z, Pedergnana V, et al. Human Adaptive Immunity Rescues an Inborn Error of Innate Immunity. Cell (2017) 168(5):789–800.e10. doi: 10.1016/j.cell.2017.01.039
28. Dahmer MK, Cornell T, Quasney MW. Genetic and Epigenetic Factors in the Regulation of the Immune Response. Curr Opin Pediatr (2016) 28(3):281–6. doi: 10.1097/MOP.0000000000000356
29. Campos-Sanchez E, Martínez-Cano J, del Pino Molina L, López-Granados E, Cobaleda C. Epigenetic Deregulation in Human Primary Immunodeficiencies. Trends Immunol (2019) 40(1):49–65. doi: 10.1016/j.it.2018.11.005
30. Lewis EM, Singla M, Sergeant S, Koty PP, McPhail LC. X-Linked Chronic Granulomatous Disease Secondary to Skewed X Chromosome Inactivation in a Female With a Novel CYBB Mutation and Late Presentation. Clin Immunol (2008) 129(2):372–80. doi: 10.1016/j.clim.2008.07.022
31. Yamashita Y, Kimura T, Tanaka N, Yazaki M, Itagaki T, Joshita S, et al. Salmonella Enteritidis Cholecystitis With Chronic Granulomatous Disease. IDCases (2018) 12:49–52. doi: 10.1016/j.idcr.2018.03.012
32. Buchbinder D, Nadeau K, Nugent D. Monozygotic Twin Pair Showing Discordant Phenotype for X-Linked Thrombocytopenia and Wiskott-Aldrich Syndrome: A Role for Epigenetics? J Clin Immunol (2011) 31(5):773–7. doi: 10.1007/s10875-011-9561-3
33. Rodríguez-Cortez VC, del Pino-Molina L, Rodríguez-Ubreva J, Ciudad L, Gómez-Cabrero D, Company C, et al. Monozygotic Twins Discordant for Common Variable Immunodeficiency Reveal Impaired DNA Demethylation During Naïve-to-Memory B-Cell Transition. Nat Commun (2015) 6(1):7335. doi: 10.1038/ncomms8335
34. Pal S, Tyler JK. Epigenetics and Aging. Sci Adv (2016) 2(7):e1600584–e. doi: 10.1126/sciadv.1600584
35. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
36. Flinn AM, Gennery AR. Adenosine Deaminase Deficiency: A Review. Orphanet J Rare Dis (2018) 13(1):65. doi: 10.1186/s13023-018-0807-5
37. Ozsahin H, Arredondo-Vega FX, Santisteban I, Fuhrer H, Tuchschmid P, Jochum W, et al. Adenosine Deaminase Deficiency in Adults. Blood (1997) 89(8):2849–55. doi: 10.1182/blood.V89.8.2849
38. Woodbine L, Grigoriadou S, Goodarzi AA, Riballo E, Tape C, Oliver AW, et al. An Artemis Polymorphic Variant Reduces Artemis Activity and Confers Cellular Radiosensitivity. DNA Repair (Amst) (2010) 9(9):1003–10. doi: 10.1016/j.dnarep.2010.07.001
39. Abraham RS, Recher M, Giliani S, Walter JE, Lee YN, Frugoni F, et al. Adult-Onset Manifestation of Idiopathic T-Cell Lymphopenia Due to a Heterozygous RAG1 Mutation. J Allergy Clin Immunol (2013) 131(5):1421–3. doi: 10.1016/j.jaci.2012.09.016
40. Geier CB, Piller A, Linder A, Sauerwein KM, Eibl MM, Wolf HM. Leaky RAG Deficiency in Adult Patients With Impaired Antibody Production Against Bacterial Polysaccharide Antigens. PLoS One (2015) 10(7):e0133220. doi: 10.1371/journal.pone.0133220
41. Ardeniz Ö, Unger S, Onay H, Ammann S, Keck C, Cianga C, et al. β2-Microglobulin Deficiency Causes a Complex Immunodeficiency of the Innate and Adaptive Immune System. J Allergy Clin Immunol (2015) 136(2):392–401. doi: 10.1016/j.jaci.2014.12.1937
42. Wani MA, Haynes LD, Kim J, Bronson CL, Chaudhury C, Mohanty S, et al. Familial Hypercatabolic Hypoproteinemia Caused by Deficiency of the Neonatal Fc Receptor, FcRn, Due to a Mutant Beta2-Microglobulin Gene. Proc Natl Acad Sci USA (2006) 103(13):5084–9. doi: 10.1073/pnas.0600548103
43. Waldmann TA, Terry WD. Familial Hypercatabolic Hypoproteinemia. A Disorder of Endogenous Catabolism of Albumin and Immunoglobulin. J Clin Invest (1990) 86(6):2093–8. doi: 10.1172/JCI114947
44. Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD. Molecular Analysis of a Large Cohort of Patients With the Hyper Immunoglobulin M (IgM) Syndrome. Blood (2005) 105(5):1881–90. doi: 10.1182/blood-2003-12-4420
45. Quan V, Towey M, Sacks S, Kelly AP. Absence of MHC Class II Gene Expression in a Patient With a Single Amino Acid Substitution in the Class II Transactivator Protein CIITA. Immunogenetics (1999) 49(11-12):957–63. doi: 10.1007/s002510050579
46. Wiszniewski W, Fondaneche MC, Le Deist F, Kanariou M, Selz F, Brousse N, et al. Mutation in the Class II Trans-Activator Leading to a Mild Immunodeficiency. J Immunol (2001) 167(3):1787–94. doi: 10.4049/jimmunol.167.3.1787
47. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous Loss of ICOS is Associated With Adult-Onset Common Variable Immunodeficiency. Nat Immunol (2003) 4(3):261–8. doi: 10.1038/ni902
48. Schepp J, Chou J, Skrabl-Baumgartner A, Arkwright PD, Engelhardt KR, Hambleton S, et al. 14 Years After Discovery: Clinical Follow-Up on 15 Patients With Inducible Co-Stimulator Deficiency. Front Immunol (2017) 8:964. doi: 10.3389/fimmu.2017.00964
49. de la Salle H, Saulquin X, Mansour I, Klayme S, Fricker D, Zimmer J, et al. Asymptomatic Deficiency in the Peptide Transporter Associated to Antigen Processing (TAP). Clin Exp Immunol (2002) 128(3):525–31. doi: 10.1046/j.1365-2249.2002.01862.x
50. Hershfield M. Adenosine Deaminase Deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al, editors. GeneReviews(®). Seattle (WA: University of Washington, Seattle Copyright © 1993-2021, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved (1993).
51. Shovlin CL, Hughes JM, Simmonds HA, Fairbanks L, Deacock S, Lechler R, et al. Adult Presentation of Adenosine Deaminase Deficiency. Lancet (1993) 341(8858):1471. doi: 10.1016/0140-6736(93)90910-9
52. Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a Novel DNA Double-Strand Break Repair/V(D)J Recombination Protein, is Mutated in Human Severe Combined Immune Deficiency. Cell (2001) 105(2):177–86. doi: 10.1016/S0092-8674(01)00309-9
53. Felgentreff K, Lee YN, Frugoni F, Du L, van der Burg M, Giliani S, et al. Functional Analysis of Naturally Occurring DCLRE1C Mutations and Correlation With the Clinical Phenotype of ARTEMIS Deficiency. J Allergy Clin Immunol (2015) 136(1):140–50.e7. doi: 10.1016/j.jaci.2015.03.005
54. Lee PP, Woodbine L, Gilmour KC, Bibi S, Cale CM, Amrolia PJ, et al. The Many Faces of Artemis-Deficient Combined Immunodeficiency - Two Patients With DCLRE1C Mutations and a Systematic Literature Review of Genotype-Phenotype Correlation. Clin Immunol (2013) 149(3):464–74. doi: 10.1016/j.clim.2013.08.006
55. Niehues T, Perez-Becker R, Schuetz C. More Than Just SCID–the Phenotypic Range of Combined Immunodeficiencies Associated With Mutations in the Recombinase Activating Genes (RAG) 1 and 2. Clin Immunol (2010) 135(2):183–92. doi: 10.1016/j.clim.2010.01.013
56. Lawless D, Geier CB, Farmer JR, Lango Allen H, Thwaites D, Atschekzei F, et al. Prevalence and Clinical Challenges Among Adults With Primary Immunodeficiency and Recombination-Activating Gene Deficiency. J Allergy Clin Immunol (2018) 141(6):2303–6. doi: 10.1016/j.jaci.2018.02.007
57. Delmonte OM, Villa A, Notarangelo LD. Immune Dysregulation in Patients With RAG Deficiency and Other Forms of Combined Immune Deficiency. Blood (2020) 135(9):610–9. doi: 10.1182/blood.2019000923
58. Schröder C, Baerlecken NT, Pannicke U, Dörk T, Witte T, Jacobs R, et al. Evaluation of RAG1 Mutations in an Adult With Combined Immunodeficiency and Progressive Multifocal Leukoencephalopathy. Clin Immunol (2017) 179:1–7. doi: 10.1016/j.clim.2016.12.013
59. Akamatsu Y, Monroe R, Dudley DD, Elkin SK, Gartner F, Talukder SR, et al. Deletion of the RAG2 C Terminus Leads to Impaired Lymphoid Development in Mice. Proc Natl Acad Sci USA (2003) 100(3):1209–14. doi: 10.1073/pnas.0237043100
60. DiSanto JP, Bonnefoy JY, Gauchat JF, Fischer A, de Saint Basile G. CD40 Ligand Mutations in X-Linked Immunodeficiency With Hyper-IgM. Nature (1993) 361(6412):541–3. doi: 10.1038/361541a0
61. Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39-kDa Protein on Activated Helper T Cells Binds CD40 and Transduces the Signal for Cognate Activation of B Cells. Proc Natl Acad Sci USA (1992) 89(14):6550–4. doi: 10.1073/pnas.89.14.6550
62. Yong PF, Post FA, Gilmour KC, Grosse-Kreul D, King A, Easterbrook P, et al. Cerebral Toxoplasmosis in a Middle-Aged Man as First Presentation of Primary Immunodeficiency Due to a Hypomorphic Mutation in the CD40 Ligand Gene. J Clin Pathol (2008) 61(11):1220–2. doi: 10.1136/jcp.2008.058362
63. Kiani-Alikhan S, Yong PF, Gilmour KC, Grosse-Kreul D, Davies EG, Ibrahim MA. Phenotypic Heterogeneity in a Family With a CD40 Ligand Intracellular Domain Mutation. J Clin Immunol (2012) 32(1):70–7. doi: 10.1007/s10875-011-9607-6
64. Devaiah BN, Singer DS. CIITA and Its Dual Roles in MHC Gene Transcription. Front Immunol (2013) 4:476. doi: 10.3389/fimmu.2013.00476
65. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an Inducible T-Cell Co-Stimulator Structurally and Functionally Related to CD28. Nature (1999) 397(6716):263–6. doi: 10.1038/16717
66. Beier KC, Hutloff A, Dittrich AM, Heuck C, Rauch A, Büchner K, et al. Induction, Binding Specificity and Function of Human ICOS. Eur J Immunol (2000) 30(12):3707–17. doi: 10.1002/1521-4141(200012)30:12<3707::AID-IMMU3707>3.0.CO;2-Q
67. Hanna S, Etzioni A. MHC Class I and II Deficiencies. J Allergy Clin Immunol (2014) 134(2):269–75. doi: 10.1016/j.jaci.2014.06.001
68. Usui K, Sasahara Y, Tazawa R, Hagiwara K, Tsukada S, Miyawaki T, et al. Recurrent Pneumonia With Mild Hypogammaglobulinemia Diagnosed as X-Linked Agammaglobulinemia in Adults. Respir Res (2001) 2(3):188–92. doi: 10.1186/rr56
69. Hashimoto S, Miyawaki T, Futatani T, Kanegane H, Usui K, Nukiwa T, et al. Atypical X-Linked Agammaglobulinemia Diagnosed in Three Adults. Intern Med (1999) 38(9):722–5. doi: 10.2169/internalmedicine.38.722
70. Sigmon JR, Kasasbeh E, Krishnaswamy G. X-Linked Agammaglobulinemia Diagnosed Late in Life: Case Report and Review of the Literature. Clin Mol Allergy (2008) 6(1):5. doi: 10.1186/1476-7961-6-5
71. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical Spectrum and Features of Activated Phosphoinositide 3-Kinase δ Syndrome: A Large Patient Cohort Study. J Allergy Clin Immunol (2017) 139(2):597–606.e4. doi: 10.1016/j.jaci.2016.06.021
72. Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease Evolution and Response to Rapamycin in Activated Phosphoinositide 3-Kinase δ Syndrome: The European Society for Immunodeficiencies-Activated Phosphoinositide 3-Kinase δ Syndrome Registry. Front Immunol (2018) 9:543. doi: 10.3389/fimmu.2018.00543
73. Thiel J, Kimmig L, Salzer U, Grudzien M, Lebrecht D, Hagena T, et al. Genetic CD21 Deficiency is Associated With Hypogammaglobulinemia. J Allergy Clin Immunol (2012) 129(3):801–10.e6. doi: 10.1016/j.jaci.2011.09.027
74. Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B Cells in Patients With Heterozygous Mutations in IKAROS. N Engl J Med (2016) 374(11):1032–43. doi: 10.1056/NEJMoa1512234
75. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and Immunological Phenotype of Patients With Primary Immunodeficiency Due to Damaging Mutations in NFKB2. Front Immunol (2019) 10(297). doi: 10.3389/fimmu.2019.00297
76. Salzer U, Chapel HM, Webster ADB, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B Encoding TACI are Associated With Common Variable Immunodeficiency in Humans. Nat Genet (2005) 37:820. doi: 10.1038/ng1600
77. Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, et al. CVID-Associated TACI Mutations Affect Autoreactive B Cell Selection and Activation. J Clin Invest (2013) 123(10):4283–93. doi: 10.1172/JCI69854
78. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Böhm J, et al. B-Cell Activating Factor Receptor Deficiency is Associated With an Adult-Onset Antibody Deficiency Syndrome in Humans. Proc Natl Acad Sci (2009) 106(33):13945. doi: 10.1073/pnas.0903543106
79. Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM, et al. Hypomorphic Caspase Activation and Recruitment Domain 11 (CARD11) Mutations Associated With Diverse Immunologic Phenotypes With or Without Atopic Disease. J Allergy Clin Immunol (2019) 143(4):1482–95. doi: 10.1016/j.jaci.2018.08.013
80. Buchbinder D, Stinson JR, Nugent DJ, Heurtier L, Suarez F, Sukumar G, et al. Mild B-Cell Lymphocytosis in Patients With a CARD11 C49Y Mutation. J Allergy Clin Immunol (2015) 136(3):819–21.e1. doi: 10.1016/j.jaci.2015.03.008
81. Kanegane H, Futatani T, Wang Y, Nomura K, Shinozaki K, Matsukura H, et al. Clinical and Mutational Characteristics of X-Linked Agammaglobulinemia and its Carrier Identified by Flow Cytometric Assessment Combined With Genetic Analysis. J Allergy Clin Immunol (2001) 108(6):1012–20. doi: 10.1067/mai.2001.120133
82. Barmettler S, Otani IM, Minhas J, Abraham RS, Chang Y, Dorsey MJ, et al. Gastrointestinal Manifestations in X-Linked Agammaglobulinemia. J Clin Immunol (2017) 37(3):287–94. doi: 10.1007/s10875-017-0374-x
83. Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. Pi3kδ and Primary Immunodeficiencies. Nat Rev Immunol (2016) 16(11):702–14. doi: 10.1038/nri.2016.93
84. Lougaris V, Baronio M, Moratto D, Tampella G, Gazzurelli L, Facchetti M, et al. A Novel Monoallelic Gain of Function Mutation in P110δ Causing Atypical Activated Phosphoinositide 3-Kinase δ Syndrome (APDS-1). Clin Immunol (2019) 200:31–4. doi: 10.1016/j.clim.2019.01.003
85. Georgopoulos K, Winandy S, Avitahl N. The Role of the Ikaros Gene in Lymphocyte Development and Homeostasis. Annu Rev Immunol (1997) 15:155–76. doi: 10.1146/annurev.immunol.15.1.155
86. Ma S, Wang C, Mao X, Hao Y. B Cell Dysfunction Associated With Aging and Autoimmune Diseases. Front Immunol (2019) 10:318. doi: 10.3389/fimmu.2019.00318
87. Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-κb1 Subunit P50 in Common Variable Immunodeficiency. Am J Hum Genet (2015) 97(3):389–403. doi: 10.1016/j.ajhg.2015.07.008
88. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, González-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol (2018) 9:636. doi: 10.3389/fimmu.2018.00636
89. Dadi H, Jones TA, Merico D, Sharfe N, Ovadia A, Schejter Y, et al. Combined Immunodeficiency and Atopy Caused by a Dominant Negative Mutation in Caspase Activation and Recruitment Domain Family Member 11 (CARD11). J Allergy Clin Immunol (2018) 141(5):1818–30.e2. doi: 10.1016/j.jaci.2017.06.047
90. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline Hypomorphic CARD11 Mutations in Severe Atopic Disease. Nat Genet (2017) 49(8):1192–201. doi: 10.1038/ng.3898
91. Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital B Cell Lymphocytosis Explained by Novel Germline CARD11 Mutations. J Exp Med (2012) 209(12):2247–61. doi: 10.1084/jem.20120831
92. Koji N, Atsushi N, Takashi K, Yoshikane K, Goichi Y, Katsuto T, et al. Perforin Gene Mutations in Adult-Onset Hemophagocytic Lymphohistiocytosis. Haematologica (2007) 92(7):978–81. doi: 10.3324/haematol.11233
93. Clementi R, Emmi L, Maccario R, Liotta F, Moretta L, Danesino C, et al. Adult Onset and Atypical Presentation of Hemophagocytic Lymphohistiocytosis in Siblings Carryingprf1 Mutations. Blood (2002) 100(6):2266–. doi: 10.1182/blood-2002-04-1030
94. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic Mutations in PRF1, MUNC13-4, and STXBP2 are Associated With Adult-Onset Familial HLH. Blood (2011) 118(22):5794–8. doi: 10.1182/blood-2011-07-370148
95. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is Essential for Cytolytic Granules Fusion and is Mutated in a Form of Familial Hemophagocytic Lymphohistiocytosis (FHL3). Cell (2003) 115(4):461–73. doi: 10.1016/S0092-8674(03)00855-9
96. Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, et al. Atypical Familial Hemophagocytic Lymphohistiocytosis Due to Mutations in UNC13D and STXBP2 Overlaps With Primary Immunodeficiency Diseases. Haematologica (2010) 95(12):2080–7. doi: 10.3324/haematol.2010.029389
97. Meeths M, Entesarian M, Al-Herz W, Chiang SCC, Wood SM, Al-Ateeqi W, et al. Spectrum of Clinical Presentations in Familial Hemophagocytic Lymphohistiocytosis Type 5 Patients With Mutations in STXBP2. Blood (2010) 116(15):2635–43. doi: 10.1182/blood-2010-05-282541
98. Afzali B, Grönholm J, Vandrovcova J, O’Brien C, Sun HW, Vanderleyden I, et al. BACH2 Immunodeficiency Illustrates an Association Between Super-Enhancers and Haploinsufficiency. Nat Immunol (2017) 18(7):813–23. doi: 10.1038/ni.3753
99. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20(12):1410–6. doi: 10.1038/nm.3746
100. Orlova EM, Bukina AM, Kuznetsova ES, Kareva MA, Zakharova EU, Peterkova VA, et al. Autoimmune Polyglandular Syndrome Type 1 in Russian Patients: Clinical Variants and Autoimmune Regulator Mutations. Horm Res Paediatr (2010) 73(6):449–57. doi: 10.1159/000313585
101. Wolff ASB, Erichsen MM, Meager A, Magitta N, Myhre AG, Bollerslev J, et al. Autoimmune Polyendocrine Syndrome Type 1 in Norway: Phenotypic Variation, Autoantibodies, and Novel Mutations in the Autoimmune Regulator Gene. J Clin Endocrinol Metab (2007) 92(2):595–603. doi: 10.1210/jc.2006-1873
102. Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant Mutations in the Autoimmune Regulator AIRE Are Associated With Common Organ-Specific Autoimmune Diseases. Immunity (2015) 42(6):1185–96. doi: 10.1016/j.immuni.2015.04.021
103. Abbott JK, Huoh Y-S, Reynolds PR, Yu L, Rewers M, Reddy M, et al. Dominant-Negative Loss of Function Arises From a Second, More Frequent Variant Within the SAND Domain of Autoimmune Regulator (AIRE). J Autoimmun (2018) 88:114–20. doi: 10.1016/j.jaut.2017.10.010
104. Casamayor-Polo L, López-Nevado M, Paz-Artal E, Anel A, Rieux-Laucat F, Allende LM. Immunologic Evaluation and Genetic Defects of Apoptosis in Patients With Autoimmune Lymphoproliferative Syndrome (ALPS). Crit Rev Clin Lab Sci (2021) 58(4):253–74. doi: 10.1080/10408363.2020.1855623
105. Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, et al. Natural History of Autoimmune Lymphoproliferative Syndrome Associated With FAS Gene Mutations. Blood (2014) 123(13):1989–99. doi: 10.1182/blood-2013-10-535393
106. Neven B, Magerus-Chatinet A, Florkin B, Gobert D, Lambotte O, De Somer L, et al. A Survey of 90 Patients With Autoimmune Lymphoproliferative Syndrome Related to TNFRSF6 Mutation. Blood (2011) 118(18):4798–807. doi: 10.1182/blood-2011-04-347641
107. García García GM, Bureo Dacal JC, Suárez-Varela Pineda S, Elduayen Izaguirre R. Adult Onset Autoimmune Lymphoproliferative Syndrome Due to Somatic FAS Mutation. Internal Med J (2015) 45(4):462–4. doi: 10.1111/imj.12714
108. Blommaert E, Péanne R, Cherepanova NA, Rymen D, Staels F, Jaeken J, et al. Mutations in MAGT1 Lead to a Glycosylation Disorder With a Variable Phenotype. Proc Natl Acad Sci USA (2019) 116(20):9865–70. doi: 10.1073/pnas.1817815116
109. Ravell JC, Matsuda-Lennikov M, Chauvin SD, Zou J, Biancalana M, Deeb SJ, et al. Defective Glycosylation and Multisystem Abnormalities Characterize the Primary Immunodeficiency XMEN Disease. J Clin Invest (2020) 130(1):507–22. doi: 10.1172/JCI131116
110. Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. Second Messenger Role for Mg2+ Revealed by Human T-Cell Immunodeficiency. Nature (2011) 475(7357):471–6. doi: 10.1038/nature10246
111. Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-Linked Lymphoproliferative Disease Due to SAP/SH2D1A Deficiency: A Multicenter Study on the Manifestations, Management and Outcome of the Disease. Blood (2011) 117(1):53–62. doi: 10.1182/blood-2010-06-284935
112. Pasquier B, Yin L, Fondanèche MC, Relouzat F, Bloch-Queyrat C, Lambert N, et al. Defective NKT Cell Development in Mice and Humans Lacking the Adapter SAP, the X-Linked Lymphoproliferative Syndrome Gene Product. J Exp Med (2005) 201(5):695–701. doi: 10.1084/jem.20042432
113. Hervier B, Latour S, Loussouarn D, Rimbert M, De-Saint-Basile G, Picard C, et al. An Atypical Case of X-Linked Lymphoproliferative Disease Revealed as a Late Cerebral Lymphoma. J Neuroimmunol (2010) 218(1-2):125–8. doi: 10.1016/j.jneuroim.2009.10.012
114. Torralba-Raga L, Tesi B, Chiang SCC, Schlums H, Nordenskjöld M, Horne A, et al. Diagnostic Challenges for a Novel SH2D1A Mutation Associated With X-Linked Lymphoproliferative Disease. Pediatr Blood Cancer (2020) 67(4):e28184. doi: 10.1002/pbc.28184
115. Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP Deficiency in Humans Causes an X-Linked Lymphoproliferative Syndrome. Nature (2006) 444(7115):110–4. doi: 10.1038/nature05257
116. Rigaud S, Lopez-Granados E, Sibéril S, Gloire G, Lambert N, Lenoir C, et al. Human X-Linked Variable Immunodeficiency Caused by a Hypomorphic Mutation in XIAP in Association With a Rare Polymorphism in CD40LG. Blood (2011) 118(2):252–61. doi: 10.1182/blood-2011-01-328849
117. Aguilar C, Lenoir C, Lambert N, Bègue B, Brousse N, Canioni D, et al. Characterization of Crohn Disease in X-Linked Inhibitor of Apoptosis-Deficient Male Patients and Female Symptomatic Carriers. J Allergy Clin Immunol (2014) 134(5):1131–41.e9. doi: 10.1016/j.jaci.2014.04.031
118. Sandler RD, Carter S, Kaur H, Francis S, Tattersall RS, Snowden JA. Haemophagocytic Lymphohistiocytosis (HLH) Following Allogeneic Haematopoietic Stem Cell Transplantation (HSCT)—time to Reappraise With Modern Diagnostic and Treatment Strategies? Bone Marrow Transplant (2020) 55(2):307–16. doi: 10.1038/s41409-019-0637-7
119. La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the Management of Hemophagocytic Lymphohistiocytosis in Adults. Blood (2019) 133(23):2465–77. doi: 10.1182/blood.2018894618
120. Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, et al. BACH2 Represses Effector Programs to Stabilize T(reg)-Mediated Immune Homeostasis. Nature (2013) 498(7455):506–10. doi: 10.1038/nature12199
121. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune Dysregulation in Human Subjects With Heterozygous Germline Mutations in CTLA4. Science (New York NY) (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
122. Husebye ES, Anderson MS, Kämpe O. Autoimmune Polyendocrine Syndromes. N Engl J Med (2018) 378(12):1132–41. doi: 10.1056/NEJMra1713301
123. Garelli S, Dalla Costa M, Sabbadin C, Barollo S, Rubin B, Scarpa R, et al. Autoimmune Polyendocrine Syndrome Type 1: An Italian Survey on 158 Patients. J Endocrinol Invest (2021) (11):2493–510. doi: 10.1007/s40618-021-01585-6
124. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised Diagnostic Criteria and Classification for the Autoimmune Lymphoproliferative Syndrome (ALPS): Report From the 2009 NIH International Workshop. Blood (2010) 116(14):e35–40. doi: 10.1182/blood-2010-04-280347
125. Ravell JC, Chauvin SD, He T, Lenardo M. An Update on XMEN Disease. J Clin Immunol (2020) 40(5):671–81. doi: 10.1007/s10875-020-00790-x
126. Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host Response to EBV Infection in X-Linked Lymphoproliferative Disease Results From Mutations in an SH2-Domain Encoding Gene. Nat Genet (1998) 20(2):129–35. doi: 10.1038/2424
127. Purtilo DT, Cassel CK, Yang JP, Harper R. X-Linked Recessive Progressive Combined Variable Immunodeficiency (Duncan’s Disease). Lancet (1975) 1(7913):935–40. doi: 10.1016/S0140-6736(75)92004-8
128. Engel P, Eck MJ, Terhorst C. The SAP and SLAM Families in Immune Responses and X-Linked Lymphoproliferative Disease. Nat Rev Immunol (2003) 3(10):813–21. doi: 10.1038/nri1202
129. Liang JH, Zhu HY, Xu DM, Wang L, Wang Y, Qiao C, et al. A New SH2D1A Mutation in a Female Adult XLP Disease With Hemophagocytic Lymphohistiocytosis and NK-Cell Leukemia. Ann Hematol (2019) 98(12):2829–31. doi: 10.1007/s00277-019-03810-y
130. Latour S, Aguilar C. XIAP Deficiency Syndrome in Humans. Semin Cell Dev Biol (2015) 39:115–23. doi: 10.1016/j.semcdb.2015.01.015
131. Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernández-Pérez L, Chapgier A, et al. Partial Recessive IFN-γr1 Deficiency: Genetic, Immunological and Clinical Features of 14 Patients From 11 Kindreds. Hum Mol Genet (2011) 20(8):1509–23. doi: 10.1093/hmg/ddr029
132. de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting Human IL-12rβ1 Deficiency: A Survey of 141 Patients From 30 Countries. Med (Baltimore) (2010) 89(6):381–402. doi: 10.1097/MD.0b013e3181fdd832
133. Osamu H, Satoshi O, Miyuki T, Reiko K, Mizuka M, Hiroshi K, et al. Heterozygosity for the Y701C STAT1 Mutation in a Multiplex Kindred With Multifocal Osteomyelitis. Haematologica (2013) 98(10):1641–9. doi: 10.3324/haematol.2013.083741
134. Kerner G, Ramirez-Alejo N, Seeleuthner Y, Yang R, Ogishi M, Cobat A, et al. Homozygosity for TYK2 P1104A Underlies Tuberculosis in About 1% of Patients in a Cohort of European Ancestry. Proc Natl Acad Sci USA (2019) 116(21):10430–4. doi: 10.1073/pnas.1903561116
135. Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, et al. Tuberculosis and Impaired IL-23–Dependent IFN-γ Immunity in Humans Homozygous for a Common TYK2 Missense Variant. Sci Immunol (2018) 3(30):eaau8714. doi: 10.1126/sciimmunol.aau8714
136. Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, Ouachee M, et al. Natural History of GATA2 Deficiency in a Survey of 79 French and Belgian Patients. Haematologica (2018) 103(8):1278–87. doi: 10.3324/haematol.2017.181909
137. Corvilain E, Casanova J-L, Puel A. Inherited CARD9 Deficiency: Invasive Disease Caused by Ascomycete Fungi in Previously Healthy Children and Adults. J Clin Immunol (2018) 38(6):656–93. doi: 10.1007/s10875-018-0539-2
138. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 Gain-of-Function Mutations Underlie an Unexpectedly Broad Clinical Phenotype. Blood (2016) 127(25):3154–64. doi: 10.1182/blood-2015-11-679902
139. Lim HK, Seppänen M, Hautala T, Ciancanelli MJ, Itan Y, Lafaille FG, et al. TLR3 Deficiency in Herpes Simplex Encephalitis: High Allelic Heterogeneity and Recurrence Risk. Neurology (2014) 83(21):1888–97. doi: 10.1212/WNL.0000000000000999
140. Guérin A, Kerner G, Marr N, Markle JG, Fenollar F, Wong N, et al. IRF4 Haploinsufficiency in a Family With Whipple’s Disease. eLife (2018) 7:e32340. doi: 10.7554/eLife.32340
141. Bhattad S, Unni J, Varkey S. MSMD in a 3-Generation Multiplex Kindred Due to Autosomal Dominant STAT1 Deficiency. J Clin Immunol (2021) 41(1):259–61. doi: 10.1007/s10875-020-00890-8
142. Bresnick EH, Jung MM, Katsumura KR. Human GATA2 Mutations and Hematologic Disease: How Many Paths to Pathogenesis? Blood Adv (2020) 4(18):4584–92. doi: 10.1182/bloodadvances.2020002953
143. Li J, Vinh DC, Casanova J-L, Puel A. Inborn Errors of Immunity Underlying Fungal Diseases in Otherwise Healthy Individuals. Curr Opin Microbiol (2017) 40:46–57. doi: 10.1016/j.mib.2017.10.016
144. Maeshima K, Ishii K, Shibata H. An Adult Fatal Case With a STAT1 Gain-Of-Function Mutation Associated With Multiple Autoimmune Diseases. J Rheumatol (2019) 46(3):325–7. doi: 10.3899/jrheum.180210
145. Nemoto K, Kawanami T, Hoshina T, Ishimura M, Yamasaki K, Okada S, et al. Impaired B-Cell Differentiation in a Patient With STAT1 Gain-Of-Function Mutation. Front Immunol (2020) 11:557521–. doi: 10.3389/fimmu.2020.557521
146. Verma D, Eriksson P, Sahdo B, Persson A, Ejdebäck M, Särndahl E, et al. Two Adult Siblings With Atypical Cryopyrin-Associated Periodic Syndrome Due to a Novel M299V Mutation in NLRP3. Arthritis Rheum (2010) 62(7):2138–43. doi: 10.1002/art.27489
147. De Koning HD, Schalkwijk J, van der Meer JW, Zeeuwen PL, Neveling K, van Gijn M, et al. PW03-007 - NLRP3 Genetic Variants in Schnitzler’s Syndrome. Pediatr Rheumatol Online J (2013) 11(Suppl 1):A233–A. doi: 10.1186/1546-0096-11-S1-A233
148. Shen M, Tang L, Shi X, Zeng X, Yao Q. NLRP12 Autoinflammatory Disease: A Chinese Case Series and Literature Review. Clin Rheumatol (2017) 36(7):1661–7. doi: 10.1007/s10067-016-3410-y
149. Vitale A, Rigante D, Maggio MC, Emmi G, Romano M, Silvestri E, et al. Rare NLRP12 Variants Associated With the NLRP12-Autoinflammatory Disorder Phenotype: An Italian Case Series. Clin Exp Rheumatol (2013) 31(3 Suppl 77):155–6.
150. Gaggiano C, Vitale A, Obici L, Merlini G, Soriano A, Viapiana O, et al. Clinical Features at Onset and Genetic Characterization of Pediatric and Adult Patients With TNF-α Receptor—Associated Periodic Syndrome (TRAPS): A Series of 80 Cases From the AIDA Network. Mediators Inflamm (2020) 2020:8562485. doi: 10.1155/2020/8562485
151. Kontzias A, Zarabi SK, Calabrese C, Wang Y, Judis L, Yao Q, et al. Somatic Mosaicism in Adult-Onset TNF Receptor-Associated Periodic Syndrome (TRAPS). Mol Genet Genomic Med (2019) 7(8):e791–e. doi: 10.1002/mgg3.791
152. Rowczenio DM, Trojer H, Omoyinmi E, Aróstegui JI, Arakelov G, Mensa-Vilaro A, et al. Brief Report: Association of Tumor Necrosis Factor Receptor–Associated Periodic Syndrome With Gonosomal Mosaicism of a Novel 24-Nucleotide TNFRSF1A Deletion. Arthritis Rheumatol (2016) 68(8):2044–9. doi: 10.1002/art.39683
153. Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei X-Y, Fraitag S, et al. Interleukin-36–Receptor Antagonist Deficiency and Generalized Pustular Psoriasis. N Engl J Med (2011) 365(7):620–8. doi: 10.1056/NEJMoa1013068
154. Zea-Vera AF, Estupiñan-Lopez FE, Cifuentes-Burbano J, Vargas MJ, Bonelo A. Interleukin-36 Receptor Antagonist Deficiency (DITRA) With a Novel IL36RN Homozygous Mutation C.200G > T (P.Cys67Phe) in a Young Colombian Woman. J Clin Immunol (2019) 39(3):261–3. doi: 10.1007/s10875-019-00622-7
155. Sugiura K, Takeichi T, Kono M, Ogawa Y, Shimoyama Y, Muro Y, et al. A Novel IL36RN/IL1F5 Homozygous Nonsense Mutation, P.Arg10X, in a Japanese Patient With Adult-Onset Generalized Pustular Psoriasis. Br J Dermatol (2012) 167(3):699–701. doi: 10.1111/j.1365-2133.2012.10953.x
156. Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, et al. Mutations in IL36RN/IL1F5 are Associated With the Severe Episodic Inflammatory Skin Disease Known as Generalized Pustular Psoriasis. Am J Hum Genet (2011) 89(3):432–7. doi: 10.1016/j.ajhg.2011.07.022
157. Booty MG, Chae JJ, Masters SL, Remmers EF, Barham B, Le JM, et al. Familial Mediterranean Fever With a Single MEFV Mutation: Where is the Second Hit? Arthritis Rheum (2009) 60(6):1851–61. doi: 10.1002/art.24569
158. Shinar Y, Tohami T, Livneh A, Schiby G, Hirshberg A, Nagar M, et al. Acquired Familial Mediterranean Fever Associated With a Somatic MEFV Mutation in a Patient With JAK2 Associated Post-Polycythemia Myelofibrosis. Orphanet J Rare Dis (2015) 10:86–. doi: 10.1186/s13023-015-0298-6
159. Mensa-Vilaro A, Tarng Cham W, Ping Tang S, Chin Lim S, González-Roca E, Ruiz-Ortiz E, et al. Brief Report: First Identification of Intrafamilial Recurrence of Blau Syndrome Due to Gonosomal NOD2 Mosaicism. Arthritis Rheumatol (2016) 68(4):1039–44. doi: 10.1002/art.39519
160. Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 Deficiency (DADA2) as an Unrecognised Cause of Early Onset Polyarteritis Nodosa and Stroke: A Multicentre National Study. Ann Rheum Dis (2017) 76(10):1648–56. doi: 10.1136/annrheumdis-2016-210802
161. Schnappauf O, Sampaio Moura N, Aksentijevich I, Stoffels M, Ombrello AK, Hoffmann P, et al. Sequence-Based Screening of Patients With Idiopathic Polyarteritis Nodosa, Granulomatosis With Polyangiitis, and Microscopic Polyangiitis for Deleterious Genetic Variants in ADA2. Arthritis Rheumatol (2021) 73(3):512–9. doi: 10.1002/art.41549
162. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, et al. Activated STING in a Vascular and Pulmonary Syndrome. N Engl J Med (2014) 371(6):507–18. doi: 10.1056/NEJMoa1312625
163. Staels F, Betrains A, Doubel P, Willemsen M, Cleemput V, Vanderschueren S, et al. Adult-Onset ANCA-Associated Vasculitis in SAVI: Extension of the Phenotypic Spectrum, Case Report and Review of the Literature. Front Immunol (2020) 11(2530). doi: 10.3389/fimmu.2020.575219
164. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-Onset Stroke and Vasculopathy Associated With Mutations in ADA2. N Engl J Med (2014) 370(10):911–20. doi: 10.1056/NEJMoa1307361
165. Kendall JL, Springer JM. The Many Faces of a Monogenic Autoinflammatory Disease: Adenosine Deaminase 2 Deficiency. Curr Rheumatol Rep (2020) 22(10):64. doi: 10.1007/s11926-020-00944-1
166. Betrains A, Staels F, Moens L, Delafontaine S, Hershfield MS, Blockmans D, et al. Diagnosis of Deficiency of Adenosine Deaminase Type 2 in Adulthood. Scand J Rheumatol (2021) 6:1–4. doi: 10.1080/03009742.2021.1881156
167. Lin B, Berard R, Al Rasheed A, Aladba B, Kranzusch PJ, Henderlight M, et al. A Novel STING1 Variant Causes a Recessive Form of STING-Associated Vasculopathy With Onset in Infancy (SAVI). J Allergy Clin Immunol (2020) 146(5):1204–8.e6. doi: 10.1016/j.jaci.2020.06.032
168. Alghamdi M. Familial Mediterranean Fever, Review of the Literature. Clin Rheumatol (2017) 36(8):1707–13. doi: 10.1007/s10067-017-3715-5
169. Yasar Bilge NS, Sari I, Solmaz D, Senel S, Emmungil H, Kilic L, et al. Comparison of Early Versus Late Onset Familial Mediterranean Fever. Int J Rheumatic Dis (2018) 21(4):880–4. doi: 10.1111/1756-185X.13259
170. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The Adaptor ASC has Extracellular and ‘Prionoid’ Activities That Propagate Inflammation. Nat Immunol (2014) 15(8):727–37. doi: 10.1038/ni.2913
171. Hernández-Rodríguez J, Ruíz-Ortiz E, Tomé A, Espinosa G, González-Roca E, Mensa-Vilaró A, et al. Clinical and Genetic Characterization of the Autoinflammatory Diseases Diagnosed in an Adult Reference Center. Autoimmun Rev (2016) 15(1):9–15. doi: 10.1016/j.autrev.2015.08.008
172. Wu D, Shen M. Muckle-Wells Syndrome in Chinese Patients: A Single Center Case Series. Clin Rheumatol (2017) 36(4):965–9. doi: 10.1007/s10067-016-3523-3
173. Betrains A, Staels F, Schrijvers R, Meyts I, Humblet-Baron S, De Langhe E, et al. Systemic Autoinflammatory Disease in Adults. Autoimmun Rev (2021) 20(4):102774. doi: 10.1016/j.autrev.2021.102774
174. Tuncer S, Fiorillo MT, Sorrentino R. The Multifaceted Nature of NLRP12. J Leukoc Biol (2014) 96(6):991–1000. doi: 10.1189/jlb.3RU0514-265RR
175. Aksentijevich I, Schnappauf O. Molecular Mechanisms of Phenotypic Variability in Monogenic Autoinflammatory Diseases. Nat Rev Rheumatol (2021) (7):405–25. doi: 10.1038/s41584-021-00614-1
176. Jordan Catherine T, Cao L, Roberson Elisha DO, Duan S, Helms Cynthia A, Nair Rajan P, et al. Rare and Common Variants in CARD14, Encoding an Epidermal Regulator of NF-Kappab, in Psoriasis. Am J Hum Genet (2012) 90(5):796–808. doi: 10.1016/j.ajhg.2012.03.013
177. de Inocencio J, Mensa-Vilaro A, Tejada-Palacios P, Enriquez-Merayo E, González-Roca E, Magri G, et al. Somatic NOD2 Mosaicism in Blau Syndrome. J Allergy Clin Immunol (2015) 136(2):484–7.e2. doi: 10.1016/j.jaci.2014.12.1941
178. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-Of-Function Mutations in TNFAIP3 Leading to A20 Haploinsufficiency Cause an Early-Onset Autoinflammatory Disease. Nat Genet (2016) 48(1):67–73. doi: 10.1038/ng.3459
179. Savic S, McDermott MF. Tumor Necrosis Factor (TNF) Receptor-Associated Periodic Syndrome (TRAPS). In: Hashkes PJ, Laxer RM, Simon A, editors. Textbook of Autoinflammation. Cham: Springer International Publishing (2019). p. 329–45.
180. Audemard-Verger A, Descloux E, Ponard D, Deroux A, Fantin B, Fieschi C, et al. Infections Revealing Complement Deficiency in Adults: A French Nationwide Study Enrolling 41 Patients. Med (Baltimore) (2016) 95(19):e3548. doi: 10.1097/MD.0000000000003548
181. Horiuchi T, Nishizaka H, Kojima T, Sawabe T, Niho Y, Schneider PM, et al. A Non-Sense Mutation at Arg<sup<95</sup< Is Predominant in Complement 9 Deficiency in Japanese. J Immunol (1998) 160(3):1509.
182. Biesma DH, Hannema AJ, van Velzen-Blad H, Mulder L, van Zwieten R, Kluijt I, et al. A Family With Complement Factor D Deficiency. J Clin Invest (2001) 108(2):233–40. doi: 10.1172/JCI200112023
183. Busse PJ, Christiansen SC. Hereditary Angioedema. N Engl J Med (2020) 382(12):1136–48. doi: 10.1056/NEJMra1808012
184. Bibert S, Piret J, Quinodoz M, Collinet E, Zoete V, Michielin O, et al. Herpes Simplex Encephalitis in Adult Patients With MASP-2 Deficiency. PLoS Pathog (2019) 15(12):e1008168. doi: 10.1371/journal.ppat.1008168
185. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I – Molecular Mechanisms of Activation and Regulation. Front Immunol (2015) 6(262). doi: 10.3389/fimmu.2015.00262
186. Brodszki N, Frazer-Abel A, Grumach AS, Kirschfink M, Litzman J, Perez E, et al. European Society for Immunodeficiencies (ESID) and European Reference Network on Rare Primary Immunodeficiency, Autoinflammatory and Autoimmune Diseases (ERN RITA) Complement Guideline: Deficiencies, Diagnosis, and Management. J Clin Immunol (2020) 40(4):576–91. doi: 10.1007/s10875-020-00754-1
187. Noris M, Remuzzi G. Overview of Complement Activation and Regulation. Semin Nephrol (2013) 33(6):479–92. doi: 10.1016/j.semnephrol.2013.08.001
188. Stengaard-Pedersen K, Thiel S, Gadjeva M, Møller-Kristensen M, Sørensen R, Jensen LT, et al. Inherited Deficiency of Mannan-Binding Lectin–Associated Serine Protease 2. N Engl J Med (2003) 349(6):554–60. doi: 10.1056/NEJMoa022836
Keywords: primary immunodeficiency, genetics, adult-onset, monogenic, mutation, inborn errors of immunity, autoinflammatory disease, common variable immunodeficiency
Citation: Staels F, Collignon T, Betrains A, Gerbaux M, Willemsen M, Humblet-Baron S, Liston A, Vanderschueren S and Schrijvers R (2021) Monogenic Adult-Onset Inborn Errors of Immunity. Front. Immunol. 12:753978. doi: 10.3389/fimmu.2021.753978
Received: 05 August 2021; Accepted: 22 October 2021;
Published: 17 November 2021.
Edited by:
Rosa Bacchetta, Stanford University, United StatesReviewed by:
Alberto Tommasini, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), ItalyCopyright © 2021 Staels, Collignon, Betrains, Gerbaux, Willemsen, Humblet-Baron, Liston, Vanderschueren and Schrijvers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rik Schrijvers, cmlrLnNjaHJpanZlcnNAdXpsZXV2ZW4uYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.