 Ali Ataya
Ali Ataya Vijaya Knight
Vijaya Knight Brenna C. Carey
Brenna C. Carey Elinor Lee
Elinor Lee Elizabeth J. Tarling
Elizabeth J. Tarling Tisha Wang
Tisha Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 November 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.752856
Autoantibodies to multiple cytokines have been identified and some, including antibodies against granulocyte-macrophage colony-stimulating factor (GM-CSF), have been associated with increased susceptibility to infection. High levels of GM-CSF autoantibodies that neutralize signaling cause autoimmune pulmonary alveolar proteinosis (aPAP), an ultrarare autoimmune disease characterized by accumulation of excess surfactant in the alveoli, leading to pulmonary insufficiency. Defective GM-CSF signaling leads to functional deficits in multiple cell types, including macrophages and neutrophils, with impaired phagocytosis and host immune responses against pulmonary and systemic infections. In this article, we review the role of GM-CSF in aPAP pathogenesis and pulmonary homeostasis along with the increased incidence of infections (particularly opportunistic infections). Therefore, recombinant human GM-CSF products may have potential for treatment of aPAP and possibly other infectious and pulmonary diseases due to its pleotropic immunomodulatory actions.
Autoantibodies (AAbs) against cytokines are detectable in the plasma of both healthy and diseased individuals, but the role and potential pathology of anti-cytokine autoantibodies are not fully known (1). Cytokine-specific AAbs are considered naturally occurring immune regulators as well as pathogenic instigators. AAbs to multiple cytokines have been identified and some, including antibodies against granulocyte-macrophage colony-stimulating factor (GM-CSF), have been associated with increased susceptibility to infection. High levels of AAbs that neutralize GM-CSF signaling cause autoimmune pulmonary alveolar proteinosis (aPAP), an ultrarare autoimmune disease characterized by accumulation of excess surfactant in the alveoli, leading to pulmonary insufficiency. This review article summarizes the role of GM-CSF in aPAP pathogenesis and pulmonary homeostasis along with the increased incidence of infections (particularly opportunistic infections).
Inborn errors of immunity involving mutations affecting selected cytokines or their signaling pathways can increase susceptibility to infections. Anti-cytokine AAbs can produce the same effects as pathogenic gene mutations or variants causing inborn errors of immunity, a phenomenon known as “autoimmune phenocopies of primary immunodeficiencies” (2). To date, associations of anti-cytokine antibodies and infections have been identified for antibodies against GM-CSF, multiple interleukins, granulocyte colony-stimulating factor (G-CSF), tumor necrosis factor-alpha (TNF-α), and various types of interferon (3–13).
AAbs are usually found complexed with cytokines, and in some (but not all) cases these AAbs are associated with cytokine-neutralizing activity (1, 8, 14). Their exact function is unclear, but they might serve to negatively regulate or neutralize cytokine activity through sequestration and subsequent degradation of autoantibody–cytokine complexes (8, 15). Such complexes could still confer biological activity via Fc-mediated effects, assuming the autoantibodies do not interfere with the cytokine’s biological activity (15). Alternatively, these complexes might exert agonistic effects by serving as cytokine reservoirs, thus prolonging their half-life.
Factors driving development of anti-cytokine AAbs in healthy individuals are not clearly understood. Exposure to Aspergillus can produce AAbs that cross-react with IFN-γ (due to shared epitopes) and neutralize IFN-γ function, suggesting that molecular mimicry may play a role in the generation of these antibodies (16). Patients with high levels of neutralizing IFN-γ AAbs are, therefore, at increased risk of mycobacterial infection, which is severe in the majority of cases; such patients may also be at risk for severe Salmonella and varicella zoster infections. Neutralizing anti–IFN-α and anti–IFN-ω antibodies also have been detected in patients with hypomorphic recombination-activating genes (RAG) mutations, which could inhibit the antiviral immune response and predispose patients to infections and infection-related complications (17). More recently, AAbs against IFN-α and IFN-ω have been found to be associated with cases of severe COVID-19 (18). Similarly, AAbs to IL-6 can increase the risk of severe pyogenic infections (2). IL-6 AAbs have been reported in a patient with recurrent staphylococcal cellulitis, suggesting impaired IL-6–mediated immunity may have contributed to staphylococcal disease (19). Patients with primary immunodeficiency autoimmune polyendocrine syndrome type I (APS-I) suffer from chronic mucocutaneous candidiasis (CMC) and are shown to have increased serum titers of AAbs to IL−17A/F and IL−22 (10). Defects in these signaling pathways may impair immunity and contribute to development of CMC in these patients.
Anti-cytokine AAbs have been described for many other autoimmune diseases including rheumatoid arthritis, systemic sclerosis, multiple sclerosis, Felty’s syndrome, systemic lupus erythematosus, and sarcoidosis; however, their frequency and role have been extensively debated (20, 21). Recently, AAbs to various cytokines, including GM-CSF, were identified in some patients with postherpetic neuralgia, which were hypothesized to cause an autoimmune immunodeficiency syndrome leading to uncontrolled varicella zoster virus reactivation and nerve damage (22). While the precise role of anti-cytokine AAbs in these diseases is less clear, the presence of GM-CSF AAbs in aPAP and their role in its pathogenesis have been firmly established.
Autoimmune PAP (previously known as idiopathic PAP) accounts for approximately 90% of all PAP cases (23). The remainder consist of secondary PAP, which is associated with various disorders (e.g., chronic inflammatory syndromes, immunodeficiencies, certain hematological diseases and solid tumors, immunoglobulin A deficiency); hereditary PAP that arises from mutations in genes for CSF2RA and CSF2RB; and congenital PAP, associated with mutations in genes for surfactant proteins (SFTPA, SFTPB, SFTPC), ATP-binding cassette subfamily A member 3 (ABCA3), and thyroid transcription factor-1 (TTF1).
Autoimmune PAP is characterized by high concentrations of GM-CSF AAbs that bind to GM-CSF, decreasing its level and biological activity (24). Defective GM-CSF signaling leads to functional deficits in multiple cell types, including macrophages and neutrophils, with impaired phagocytosis and cytokine production (25, 26). GM-CSF regulates the maturation, differentiation, and proliferation of monocytes and macrophages, as well as clearance of surfactant from alveolar spaces (25, 27, 28). Loss of GM-CSF signaling, as seen in aPAP and in mice functionally deficient in GM-CSF, interferes with terminal differentiation and function of alveolar macrophages (11, 27, 29, 30). This loss of alveolar macrophage functional activity leads to reduced clearance of lung surfactant, ineffective host immune defense, and respiratory failure, all of which can facilitate the proliferation and dissemination of extrapulmonary pathogens. Surfactant accumulation in PAP was shown to arise from reduced catabolism in alveolar macrophages and not increased surfactant production; surfactant metabolism in type II alveolar epithelial cells (AEC-II) was not affected (31, 32).
High-resolution CT imaging of the lungs shows diffuse bilateral alveolar infiltrates, with patchy ground-glass opacities in a characteristic “crazy paving” pattern (24). Bronchoalveolar lavage fluid is often thick and milky-appearing with granular periodic acid-Schiff–positive acidophilic acellular material, large foamy alveolar macrophages containing phospholipoprotein, and phagocytosomes. Patients with aPAP — even those who are otherwise immunocompetent — are known to be at increased risk of infection, especially due to opportunistic intracellular microorganisms. Infections can precede or follow development of aPAP clinical symptoms, or occur concurrently (33). However, the precise role and interactions between infections and aPAP are not yet fully understood.
Increased systemic GM-CSF AAbs as well as localized (pulmonary) GM-CSF AAbs are found in all patients with aPAP. Elevated serum GM-CSF AAb levels diagnostic of aPAP are considered sufficient to effectively neutralize all GM-CSF activity, suggesting that aPAP is functionally equivalent to knockout mice deficient in GM-CSF production (Csf2-/-) (34, 35). Low levels or weak neutralizing activity of GM-CSF AAbs may be sufficient to allow for partial alveolar macrophage function in some patients, whereas high-titer AAbs suppress these functions and lead to development of aPAP (2).
GM-CSF is an immunomodulatory cytokine produced by multiple cell types including macrophages, lymphocytes, endothelial cells, fibroblasts, and alveolar epithelial cells. It acts to stimulate the proliferation and differentiation of hematopoietic stem cells into granulocytes and macrophages, including the terminal differentiation of alveolar macrophages, and to promote the growth, activation, and survival of myeloid cells (36). GM-CSF also enhances neutrophil function and increases their adherence, phagocytosis, and bactericidal activity (26). In the lungs, GM-CSF is produced by AEC-II and binds to the heterodimeric GM-CSF receptor (GM-CSFR) expressed on AEC-II and alveolar macrophages (Figure 1) (37, 38). GM-CSF acts by initially binding to the GM-CSF receptor alpha (α) chain and recruiting the GM-CSF receptor beta (β) chain to trigger intracellular signaling, including downstream phosphorylation and activation of signal transducer and activator of transcription 5 (STAT5), and expression of transcription factors including PU.1 and peroxisome proliferator-activated receptor-γ (PPARγ) (Table 1) (25, 30, 37, 38). GM-CSF also modulates lipid homeostasis, increasing ABCG1 expression and inducing efflux of cholesterol from alveolar macrophages and its subsequent catabolism (56, 57).
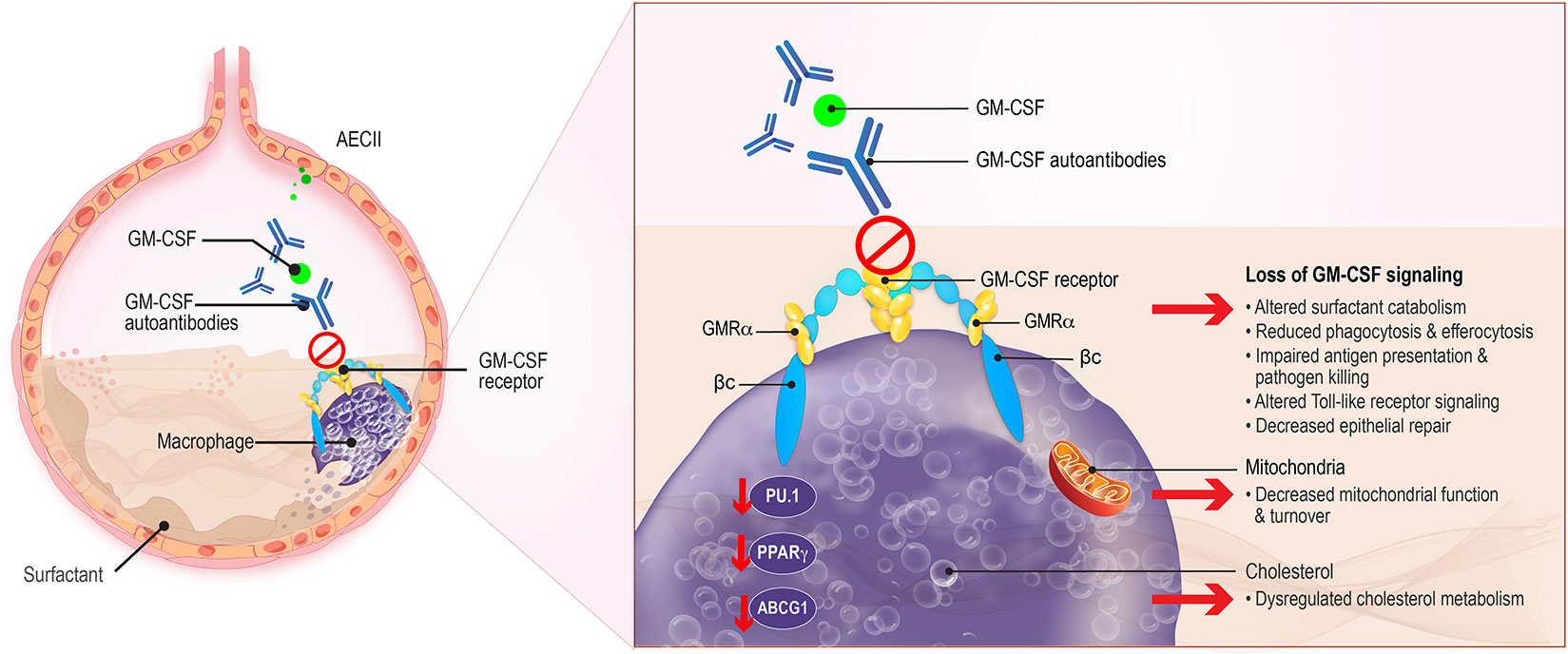
Figure 1 Macrophage Dysregulation in aPAP. GM-CSF, produced by alveolar epithelial cells type II (AECII) which binds to the GM-CSF receptor on AECII and macrophages, contributes to surfactant homeostasis and host defense against pulmonary microbial infections. Surfactant production is normally balanced by its catabolism by alveolar macrophages. Uptake and efflux of pulmonary surfactant from alveolar macrophages is normally regulated by GM-CSF. The presence of GM-CSF autoantibodies in aPAP interferes with GM-CSF regulation of surfactant, leading to its accumulation in alveoli. Other critical macrophage functions such as antigen presentation, phagocytosis, and Toll-like receptor signaling, are also affected. Additionally, key regulators of phospholipid metabolism such as the transcription factor PU.1 and ABCG1 are modified, leading to altered fatty acid and phospholipid catabolism and changes in amino acid biosynthesis. βc, GM-CSF receptor common β-subunit; GMRα, GM-CSF receptor subunit-α.
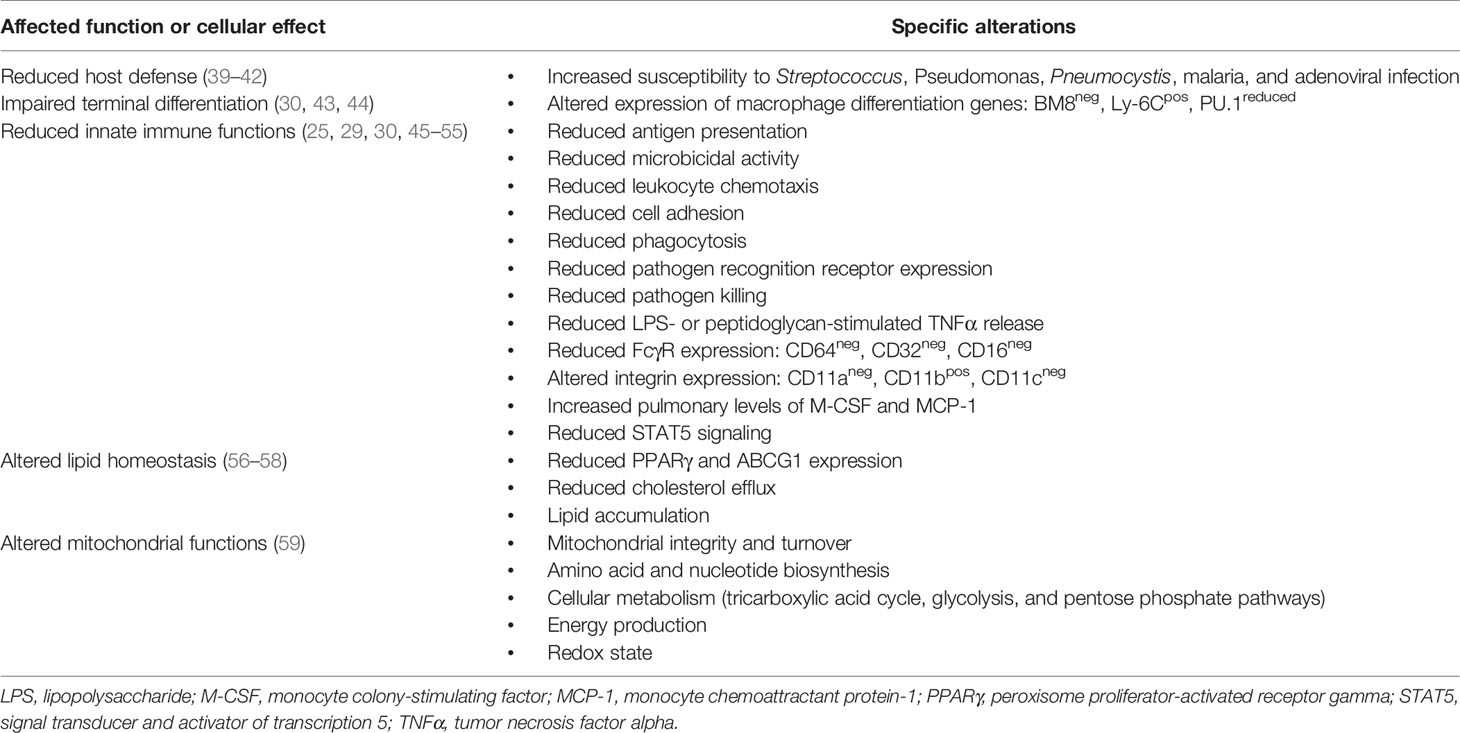
Table 1 Effects of GM-CSF deficiency on alveolar macrophages in autoimmune pulmonary alveolar proteinosis.
The effects of GM-CSF on macrophage function are extensive and include enhancing macrophage antigen presentation capacity and antibody-mediated phagocytosis via complement (29, 60). Macrophage microbicidal capacity, as well as leukocyte chemotaxis and adhesion, are also enhanced. GM-CSF induces the production of multiple cytokines including IL-6, IL-12p70, IL-23, and TNF-α (61, 62). Recent data from studies of GM-CSFR β-chain–deficient (Csf2rb–/–) mice suggest GM-CSF is essential for mitochondrial function and turnover, including fatty acid β-oxidation (59). GM-CSF also stimulates ATP production, tricarboxylic acid cycle activity, oxidative phosphorylation, and regulates other critical metabolic functions, thus helping to maintain cellular metabolism and cell growth (59).
Given the key role of neutrophils in phagocytosis and intracellular killing of a variety of pathogens (Figure 2), inhibition of neutrophil function due to decreased GM-CSF activity could also have a significant impact on local and systemic infections (26). Neutrophils from patients with aPAP demonstrate reduced baseline functions including phagocytic ability, bacterial cell adhesion, and oxidative burst compared with normal controls, as well as reduced GM-CSF priming. This neutrophil inhibition was reproduced in vitro by incubating normal neutrophils with GM-CSF AAbs, and by injection of GM-CSF antibodies into wild-type mice (26). These results support the pivotal role of GM-CSF in regulating antimicrobial functions of neutrophils and explain, in part, its effects on elimination of intracellular pathogens. The results also suggest the potential for studying GM-CSF therapy in the treatment of serious infections associated with aPAP.
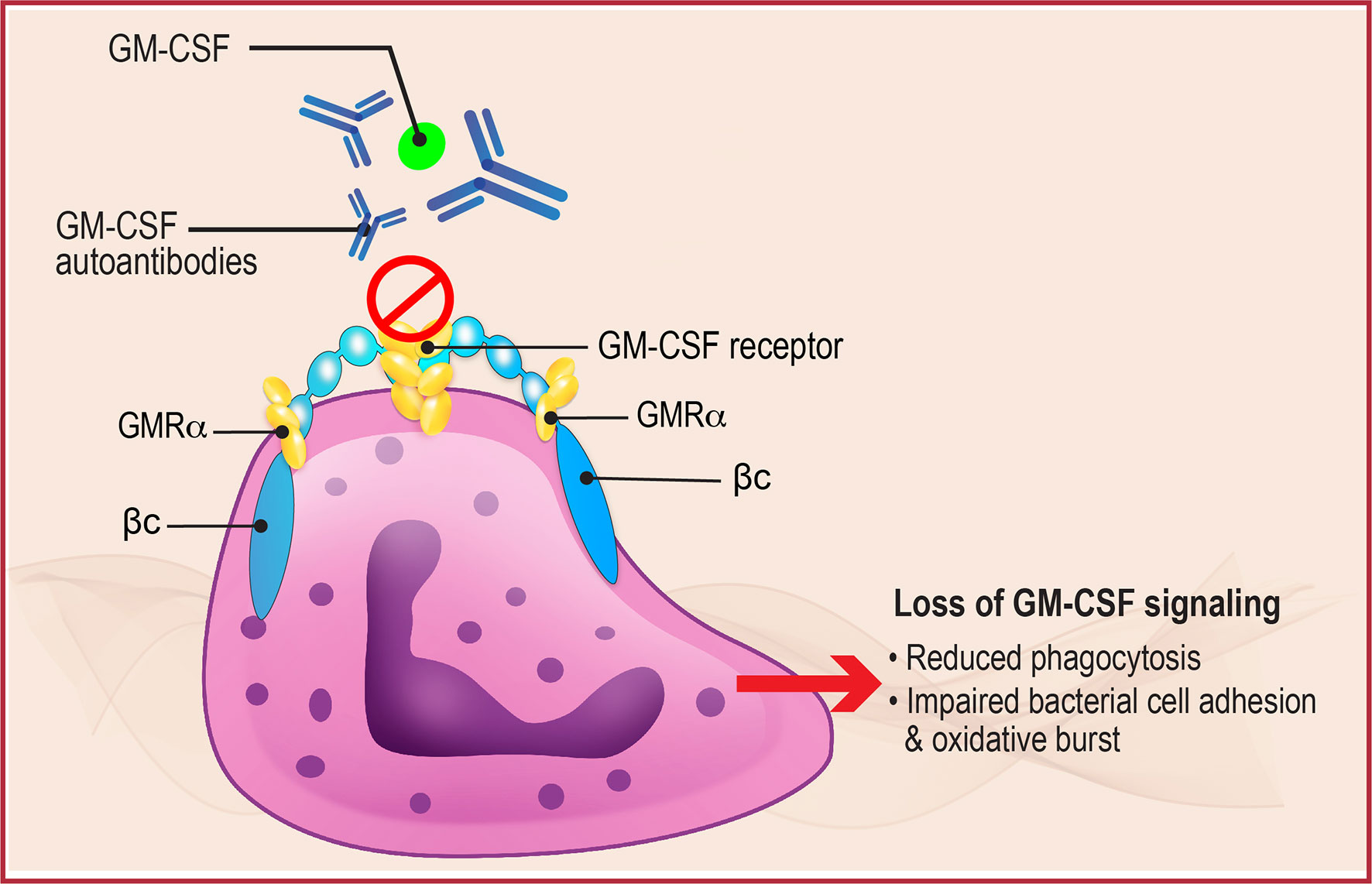
Figure 2 Neutrophil Dysregulation in aPAP. GM-CSF normally binds to the GM-CSF receptor, present on the surface of neutrophils (shown here) and alveolar macrophages, to initiate downstream signaling that regulates multiple functions including phagocytosis, bacterial cell adhesion, and oxidative burst. In aPAP, high levels of GM-CSF autoantibodies bind to GM-CSF preventing binding and receptor activation, thus inhibiting receptor signaling and leading to neutrophil and macrophage dysfunction. βc, GM-CSF receptor common β-subunit; GMRα, GM-CSF receptor subunit-α.
Damiani et al. have detailed in a separate review multiple host GM-CSF–driven functions that counter intracellular pathogens (45). For example, GM-CSF–mediated zinc (Zn) sequestration via metallothioneins has been demonstrated in vitro and in vivo, in mice and human macrophages, to drive phagocyte antimicrobial effector function (63). GM-CSF is also known to drive macrophage polarization. Infections trigger a complex sequence of inflammatory and immune events that vary according to type of pathogen, immune cell involvement, cytokine production, and time to resolution. Intracellular pathogens and bacterial proteins such as lipopolysaccharide can induce production of GM-CSF and IFN-γ to drive M1 macrophage polarization (46, 62). Such cells, which respond mainly during the early stages of infection, are responsible for production of chemokines and proinflammatory cytokines contributing to the differentiation and activation of T helper cells that aid in fighting infections. In contrast, certain parasitic, fungal, and helminthic infections cause M2 polarization, which are more anti-inflammatory and function primarily in tissue remodeling and homeostasis.
Alveolar macrophages and alveolar epithelial cells work in tandem to respond to insults. GM-CSF helps to maintain the alveolar epithelium and pulmonary immune system under both physiological and pathological conditions, including infection.
Early preclinical and translational studies demonstrated the pivotal role of GM-CSF in the regulation of pulmonary surfactant homeostasis and alveolar macrophage immune function. Mice deficient in GM-CSF (Csf2-/-) were generated via targeted ablation of the GM-CSF gene (32, 64). The absence of GM-CSF resulted in impaired alveolar macrophage function, surfactant accumulation, and lung histopathology similar to PAP. This pathology could be reversed by pulmonary administration of exogenous GM-CSF, adenoviral expression of GM-CSF, or retroviral vector expression of PU.1 transcription factor (25, 65, 66). Overexpression of a GM-CSF transgene in the lung epithelium of Csf2-/- mice restores normal lung histology and function as well as pulmonary surfactant homeostasis (31). To generate mice that overexpress GM-CSF in the lung, a construct of the human surfactant protein C promoter directing expression of mouse GM-CSF cDNA was injected into ova fertilized with sperm from Csf2-/- mice. Offspring were backcrossed to Csf2-/- mice (64). Alveolar macrophages from patients with aPAP were determined not to be deficient in either the production of GM-CSF or GM-CSFR function; however, they have a decreased bioavailability of GM-CSF due to the presence of GM-CSF AAbs and negative regulation by IL-10 (67, 68).
Under certain circumstances, chronic overexpression of pulmonary GM-CSF may result in adverse effects. Exposure to cigarette smoke, which increases the risk of pulmonary diseases such as lung cancer, pulmonary emphysema, and desquamative interstitial pneumonia (DIP), has been shown to stimulate pulmonary GM-CSF expression and to increase the number of alveolar macrophages or alter their function (69–72). Using a transgenic mouse model, chronic overexpression of pulmonary GM-CSF led to spontaneous activation and progressive accumulation of alveolar macrophages, increased metalloprotease expression, and parenchymal lung damage, resulting in development of emphysema, secondary polycythemia, and increased mortality (73). Chronic pulmonary GM-CSF expression was thus able to reproduce the features of DIP (74, 75). Of note, wild-type mice subjected to chronic cigarette smoke also showed increased pulmonary GM-CSF and alveolar macrophage accumulation, although these studies used high-level smoke exposure for a prolonged duration. These data suggest chronic, continuous enhanced GM-CSF expression might play a role in the pathogenesis of smoking-related DIP. GM-CSF may act more like a pulmonary hormone to regulate alveolar macrophage number and function, and chronic hyperactivation could result in progressive lung damage and onset of DIP (73).
Csf2-/- mice have been invaluable in deciphering the pathogenesis underlying the development of aPAP. Such mice recapitulate the pathological findings in PAP, have defective macrophage and neutrophil function, and exhibit compromised clearance of surfactant. As a result, Csf2-/- mice show increased susceptibility to a variety of pulmonary and systemic infections including streptococcal, Pseudomonas aeruginosa, Pneumocystis carinii, malaria, and adenovirus infections (39–41, 76). The deficiency in GM-CSF activity, leading to decreased clearance of surfactant and reduced immune function, provides a mechanistic basis for the increased rates of infection and infection-related mortality frequently seen in patients with aPAP (42).
Loss of functional GM-CSF in Csf2-/- mice and patients with aPAP is also associated with altered lipid homeostasis, including dysregulated cholesterol update and efflux. This is due in part to a deficiency in PPARγ and ABCG1 activity that regulate lipid metabolism, as well as decreased expression of the master transcription factor PU.1 that reduces surfactant catabolism (30, 56, 77). Together, these result in an accumulation of lipids within alveolar macrophages and build-up of lipid-laden pulmonary surfactant. In both GM-CSF and PPARγ knockout animal models and in patients with aPAP, extracellular deposition of lipids in pulmonary alveoli and bronchoalveolar lavage fluid can be seen (29, 77). This extracellular lipid accumulation could lead to decreased access to and function of immune cells, further contributing to the persistence and growth of infections and slowing the removal of pathogens and cellular debris.
Clinical lab evaluation of serum GM-CSF AAbs is routinely performed using a standardized ELISA, with a reported sensitivity and specificity of 100% for the diagnosis of aPAP (35). GM-CSF AAb levels are much higher in the sera of patients with aPAP compared with patients who have other pulmonary diseases or healthy controls (58). The presence of GM-CSF AAbs is instrumental in confirming the diagnosis of aPAP; however, the presence of pulmonary manifestations is necessary for a clinical diagnosis. Asymptomatic individuals with elevated GM-CSF AAbs may potentially be at risk of developing the clinical manifestations and complications of aPAP.
GM-CSF AAbs can be detected in patients with cancer and selected inflammatory conditions, albeit at low levels (15, 35, 78). GM-CSF AAbs also are measurable in healthy individuals but at levels 10- to 100-fold less than that observed in aPAP. Such antibodies in healthy individuals are usually found to be complexed with GM-CSF and thus are biologically less active. Routine clinical testing for GM-CSF AAbs is available only through specialized laboratories in the United States (Cincinnati Children’s Hospital Medical Center, National Jewish Health), Japan, Germany, and China (more information regarding testing is available from the PAP Foundation and EuPAPNet) (79). It should be emphasized that although diagnostic for aPAP, GM-CSF AAb levels do not reliably correlate with disease severity (15, 35, 78).
A study of patients with aPAP found that GM-CSF AAb levels were significantly lower at baseline or gradually declined over time in patients who spontaneously improved compared with those who did not (58). Decreases in GM-CSF AAb levels in serum and/or bronchoalveolar lavage fluid correlate with clinical response to aPAP therapy in some studies (24, 80–82) but not in others (83, 84). The reasons for these disparate findings are not fully understood but could be due in part to intra-patient variation or differences in AAb quantitation methodology. These results, if confirmed in larger studies, suggest that levels of serum GM-CSF AAbs might reflect disease persistence for individual patients even if they are not prognostic across the entire aPAP population. Further research is needed to better understand the role of GM-CSF AAb levels in the aPAP patient population.
In addition to standardized ELISA assays, AAbs to GM-CSF, IFN-γ, IL-2, and other cytokines have been detected using alternative techniques such as bead multiplex assay (17); however, such research-based approaches are generally not used for routine clinical diagnosis. Similarly, novel approaches such as immune-mass cytometry are being explored but not yet developed for detection of GM-CSF AAbs.
Patients with symptoms of PAP but a low level or absence of GM-CSF AAbs (effectively excluding the diagnosis of aPAP) should be assessed for GM-CSF functional signaling and serum GM-CSF concentration (Figure 3) (79).
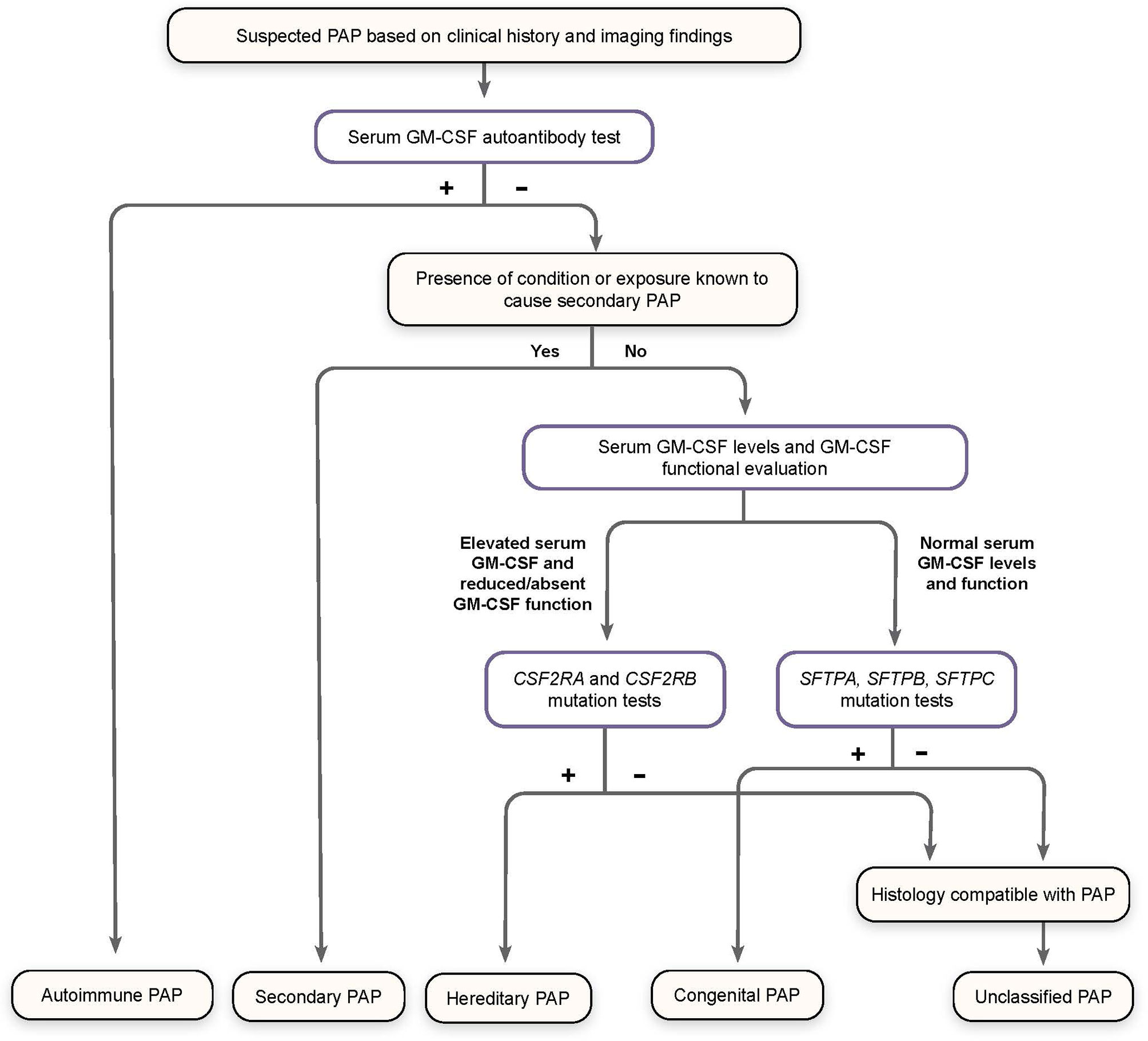
Figure 3 Algorithm for Diagnosis of Pulmonary Alveolar Proteinosis (PAP). Patients suspected of having PAP on the basis of clinical history and imaging findings should undergo screening for serum GM-CSF autoantibodies. Elevated autoantibody levels confirm a diagnosis of autoimmune PAP. Patients with a known condition or exposure associated with secondary PAP who do not have increased GM-CSF autoantibodies are diagnosed accordingly. Otherwise, patients should be screened for serum GM-CSF levels and GM-CSF function signaling by flow cytometry, as well as genomic analysis for mutations affecting GM-CSF receptor genes (CSF2RA and CSF2RB) and those related to surfactant production (SFTPA, SFTPB, SFTPC), to confirm a diagnosis of hereditary, congenital, or unclassified PAP, all of which are exceedingly rare. (Adapted from “Pulmonary alveolar proteinosis,” by BC Trapnell, 2019, Nat Rev Dis Primers, 5:16:8.).
Alterations in GM-CSF signaling can be detected by flow cytometry functional assays based on the ability of GM-CSF to stimulate cell-surface CD11b expression and intracellular STAT5 phosphorylation. The resulting CD11b stimulation index can effectively discriminate between healthy individuals and those with aPAP who exhibit significantly reduced induction of cell surface CD11b expression. This method has a reported sensitivity and specificity of 100%, aiding in the diagnosis of aPAP (85). Quantification of intracellular phosphorylated STAT5 (pSTAT5) in whole blood or peripheral blood mononuclear cells can similarly discriminate between healthy individuals and those with aPAP (8). Routine clinical testing for GM-CSF functional signaling is available only through specialized laboratories in the United States (Cincinnati Children’s Hospital Medical Center and National Jewish Health) (Table 2). In the future, functional assays may also have potential for therapeutic monitoring and to inform treatment decisions.
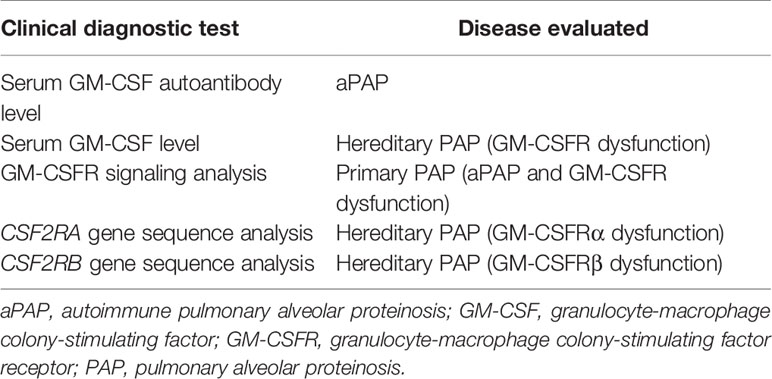
Table 2 Clinical diagnostic testing for pulmonary alveolar proteinosis syndrome.
Serum GM-CSF should be measured for patients with normal GM-CSF AAb levels who do not have obvious causes of secondary PAP (79). Elevated serum GM-CSF concentrations (>10 pg/mL) are present in patients with hereditary PAP but are generally undetectable in normal individuals. This could be because GM-CSF induces more paracrine effects and may be limited by tissue specificity, or due to the fact that in healthy subjects more than 99% of serum GM-CSF is bound and neutralized by endogenous GM-CSF AAbs and therefore may not be detected by standard assays for GM-CSF AAb levels (47). GM-CSF levels also can be increased as a result of infection or systemic inflammation in the absence of hereditary PAP (86–89). If GM-CSF AAb levels, serum GM-CSF levels, and functional tests prove normal with no known cause of secondary PAP, genomic analysis can be used to rule out congenital PAP (79). Histopathology of lung biopsies as a last resort may confirm PAP by demonstrating the presence of granular periodic acid-Schiff–positive material and foamy alveolar macrophages, although this does not specify the etiology of the PAP and is discouraged as a routine diagnostic aid given the invasive nature of the procedure.
In summary, detection of GM-CSF AAb levels, along with functional assays and determination of serum GM-CSF levels, can confirm or rule out a suspected diagnosis of aPAP and may also aid in monitoring disease persistence.
Infections are common in patients with aPAP. Additionally, individuals with GM-CSF AAbs can develop unusual pulmonary and extrapulmonary infections at rates exceeding expected complications of their lung disease (90). The onset of PAP may occur concurrently with, or be temporally distinct from, opportunistic infections (33).
Approximately 4% to 6% of PAP patients present with opportunistic infections, typically Nocardia, mycobacteria, and fungal infections (Table 3) (23, 33, 42). This predisposition to infection is systemic, rather than confined to the lungs. Although organisms common in community- and hospital-acquired infections have been reported in PAP (e.g., Streptococcus, Klebsiella, Hemophilus, Staphylococcus, Pseudomonas, Serratia, Proteus, Escherichia), more often reports have identified opportunistic microbial pathogens (27, 90). In a study of 212 Japanese patients with aPAP, 12 (5.7%) were found to have concurrent infections (primarily aspergillosis, atypical mycobacteria, and Mycobacterium tuberculosis), of which at least half were pulmonary infections (23). Superinfection with H1N1 influenza also has been reported, which can exacerbate aPAP (91). While the outcome of these infections is determined by a complex set of interacting pathogen and host factors, such infections are often associated with significant morbidity and mortality. As previously outlined, anti-cytokine antibodies including those against GM-CSF are increasingly appreciated as a cause of acquired immune dysregulation syndromes (90).

Table 3 Opportunistic pathogens commonly seen in patients with autoimmune pulmonary alveolar proteinosis.
Data suggest the downstream effects of GM-CSF AAbs in the context of aPAP have broad immunological effects on monocytes, macrophages, and neutrophils (25, 26). Understanding the immunologic effects of GM-CSF on Toll-like receptor activation, phagocytosis, bactericidal activity, oxidative burst, and cell adhesion in neutrophils and macrophages provide a physiologic rationale for the infectious pathology associated with GM-CSF AAbs. These broad effects can be deleterious for host control of certain pathogens.
Disruption in GM-CSF signaling (due to the presence of AAbs) suppresses alveolar macrophage function and facilitates pulmonary infections. Defective alveolar macrophage function could also contribute to extrapulmonary dissemination of infections seen in aPAP. GM-CSF is known to regulate alveolar macrophage production of multiple factors involved in local and systemic response to infection including prostaglandins, leukotrienes, and oxygen radicals (43). This dysregulation of GM-CSF signaling inhibits the inflammatory response to lipopolysaccharide (LPS) associated with many pulmonary bacterial infections (92). Alveolar macrophages from Csf2-/- mice have been shown to exhibit reductions in multiple immune functions including cell adhesion, phagocytosis, toll-receptor signaling, bacterial killing, and expression of pathogen-associated molecular pattern recognition receptors, all of which can contribute to an increase in infections (29). Studies of adenovirus infection of alveolar macrophages indicate GM-CSF acts through the master transcriptional regulator PU.1 to inhibit their infection, prevent lysosomal translocation, and promote virion destruction (48). In mice, neutralization of GM-CSF by administration of anti–GM-CSF antibodies exacerbated pulmonary histoplasmosis infection, indicating GM-CSF regulates antimicrobial defenses against this and possibly other opportunistic infections (93).
The lungs are the predominant site for aPAP-related infections. However, extrapulmonary infections also can occur, in some cases concurrent with respiratory infections. In aPAP, deficient pulmonary macrophages may result in an ineffective pulmonary barrier, thereby facilitating dissemination beyond the lung (11).
Nocardiosis is an opportunistic infection that typically occurs in patients who are immunocompromised, especially those with defects in phagocytosis (94). While generally limited to pulmonary or cutaneous infections, dissemination is possible, including to the central nervous system (CNS). In a review of published studies of opportunistic infections in patients with aPAP, 61% were due to Nocardia (42). This pathogen induces GM-CSF–mediated STAT5 phosphorylation in monocytes, indicating that this pathway is important in host immune response to this organism (95). Conversely, the presence of neutralizing GM-CSF AAbs, which in a separate study were detected in 5 of 7 patients with disseminated/extrapulmonary CNS nocardiosis, could abrogate the effects of GM-CSF and thus promote susceptibility and dissemination of Nocardia and perhaps other opportunistic infections (11).
As with nocardiosis, cryptococcemia is rare for those individuals with intact immunity; however, impairment of first-line antimicrobial defense mechanisms — as seen in aPAP — may contribute to progression to disseminated disease (96). The presence of GM-CSF AAbs appears to increase risk for CNS infection with C. gattii although not with C. neoformans (97).
The occurrence of both localized and disseminated infections indicates that the immune dysfunction and predisposition to infection observed in aPAP occur at the systemic level and are not limited to only the respiratory system. In support of this, Csf2-/- mice also are predisposed to both pulmonary and extrapulmonary infections by a wide array of bacterial, fungal, and viral pathogens. These mice exhibit immunological defects that result in decreased ability to eliminate a variety of bacterial, viral, parasitic, and opportunistic infections (41, 44, 98, 99). Such mice also exhibit increased susceptibility to experimentally-induced autoimmune disorders, defects in the vascular extracellular matrix, reproductive effects, and increased mortality (100, 101). The widespread, multisystemic nature of these effects in the Csf2-/- mouse model, in which immunological defects are expressed at the whole-animal level, suggests aPAP is manifested by a state of systemic functional GM-CSF deficiency rather than simply an isolated pulmonary syndrome.
Based on the efficacy and safety seen to date and its known activation of both the innate and adaptive immune responses, rhu GM-CSF may concurrently aid in elimination of infectious pathogens by enhancing the host immune response in patients with aPAP, in addition to overcoming GM-CSF neutralization. Of note, three forms of rhu GM-CSF are described in the literature: sargramostim (yeast-derived), molgramostim (bacteria-derived), and regramostim (Chinese hamster ovary cell–derived); however, sargramostim is the only form commercially available. The efficacy of inhaled rhu GM-CSF, particularly following whole-lung lavage, indicates that this approach may serve as a useful therapy in aPAP (102–104). Preliminary data also support the potential use of long-term rhu GM-CSF inhalation therapy, with responses seen in patients with aPAP who failed to respond or relapsed following repeated whole-lung lavage procedures (103, 105–107).
While preliminary data does not seem to support that administration of inhaled rhu GM-CSF leads to an increase in GM-CSF AAb levels, current assays measure only the concentration of “free” GM-CSF AAb (i.e., AAb not complexed with circulating GM-CSF or bound to cells). If administration of exogenous GM-CSF saturates available GM-CSF AAbs, it could hinder the ability to accurately measure AAb levels. Currently, the concentration of GM-CSF needed to saturate such AAbs in vivo is not known, but this is an area of active investigation.
Patients with GM-CSF AAbs and aPAP are known to be at increased risk of infection, especially from opportunistic microorganisms. Deficiencies in the immune functions of alveolar macrophages and neutrophils, such as impaired phagocytosis and cytokine production, and increased surfactant accumulation are believed to enhance the risk of infection, even in otherwise immunocompetent patients. In addition to localized pulmonary infections, patients with GM-CSF neutralizing AAbs are also at risk for disseminated infections by opportunistic intracellular pathogens and of subsequent development of aPAP (11, 97). The precise role and interactions between infections and GM-CSF in patients with aPAP are not fully understood, however.
In some autoimmune diseases and infections, the level of certain cytokine AAbs may correlate with disease severity and progression. For those disease conditions in which levels of anti-cytokine AAbs do correlate with changes in clinical symptoms, such as lupus and rheumatoid arthritis, AAb levels might serve as surrogate markers of disease progression and/or response to therapy (20, 108, 109). This does not appear to be the case with GM-CSF AAb levels in aPAP (110), however, suggesting that differences exist in the functions and/or concentrations of individual cytokines in the pathogenesis of specific diseases. More studies are also necessary to better elucidate the role of GM-CSF AAb levels in individual patients as their disease progresses and/or improves.
Greater use of testing for GM-CSF AAbs in general has been recommended to improve the noninvasive detection and diagnosis of aPAP (111, 112). Such screening is particularly important since up to one third of patients with aPAP may be asymptomatic at diagnosis (23). GM-CSF AAbs were detected, for example, in 5 of 7 adults who had CNS nocardiosis but were otherwise healthy (11). Screening for aPAP is not routinely performed due to the low incidence of this disease, but AAb assessment may be considered in selected patients with abnormal chest radiology results and unusual infections. Wider testing for GM-CSF AAbs might also allow for monitoring of patients who are at increased risk of developing aPAP and other immunodeficiencies.
It should be noted that diagnosis of aPAP takes several months, often 18 months or longer after initiation of symptoms (111). Part of this lag in diagnosis could stem from the delay of an aPAP evaluation following diagnosis and treatment of a classically-associated opportunistic infection such as Nocardia, especially in the absence of other apparent comorbidities. Awareness of this lag could provide additional rationale for clinicians to evaluate GM-CSF AAb levels in all patients with opportunistic infections who otherwise appear immunocompetent to confirm or rule out an underlying diagnosis of aPAP.
Given the increased risk of infections (particularly opportunistic infections) with aPAP, it is imperative that such patients be monitored for possible infections and infectious complications. Routine preventive vaccinations for influenza and pneumococcal infection are also important (113). Although standardized clinical practice guidelines for PAP are lacking, the increased susceptibility to organisms such as Pneumocystis jiroveci and Nocardia spp. suggests that trimethoprim/sulfamethoxazole prophylaxis could be considered for all patients with significant disease (24). Such prophylaxis has been used successfully by other investigators for patients with nocardiosis and concurrent aPAP or primary immunodeficiency (6, 11, 114).
Patients with certain chronic autoimmune disorders such as rheumatoid arthritis and psoriasis can have increased levels of GM-CSF in serum and affected tissues, suggesting a possible role for this cytokine in the pathogenesis of these conditions and providing the rationale for therapeutic targeting of GM-CSF using exogenous GM-CSF monoclonal antibodies (115, 116). The role of GM-CSF in the immune response against aPAP-associated infections raises the question, however, of why more opportunistic infections are not seen in patients who receive anti–GM-CSF therapy in patients with rheumatoid arthritis and psoriasis. Results from preclinical studies suggest a possible explanation. In vitro, at least three non–cross-competing antibodies are required to generate stable immune complexes with GM-CSF and trigger their Fc-dependent degradation (34). Treatment using a single GM-CSF monoclonal antibody would not be sufficient to induce immune complex formation and subsequent degradation of GM-CSF that could increase risk of infection.
This review highlights opportunities for enhanced awareness of testing for GM-CSF AAbs in immunocompetent individuals with certain infections. More research is needed on the intrapatient correlation of GM-CSF AAb levels and severity of disease to help guide treatment. Further study is warranted on optimal treatment strategies and the potential use of sargramostim in patients with high GM-CSF AAb levels, aPAP, and concurrent infection.
All authors contributed to co-development of the manuscript including conceptualization, selection of content for inclusion, and review, editing, and revising of content. All authors approved submission for publication and accept accountability for the content.
This work was supported by Partner Therapeutics, Inc.
AA serves on the Medical and Scientific Advisory Board of the PAP Foundation. VK received support for attending workshops, meetings, and education in leadership roles for Workshop in Primary Immunodeficiencies, College of American Pathologists, and Clinical Immunology Society, and served as President/Past President of the Association of Medical Laboratory Immunologists. BCC’s institution received an NIH-funded R01 grant related to the content of the manuscript. Outside of the current work, BC is a consultant to Partner Therapeutics, Inc. and serves on the Board of Directors and as Secretary/Treasurer of the PAP Foundation. EL’s institution received NIH-funded grants related to the content of the manuscript. EL has served as a consultant to Guidepoint Global and has received travel support from the PAP Foundation and the Rare Lung Disease Consortium. TW’s institution received a fellowship grant funded by Partner Therapeutics, Inc. TW received consultant fees from Partner Therapeutics for research outside of the current work; participated on an Advisory Board for IQVIA; and serves on the Board of Directors, Medical and Scientific Advisory Board, and as Vice President and Clinical Director of the PAP Foundation.
The authors declare this study received funding from Partner Therapeutics, Inc. The funder had the following involvement with the study: Professional medical writing, graphic artist, and publication fees for this manuscript.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Pillar Medical Communications and Larry Rosenberg, PhD, for assistance with the preparation of our manuscript.
1. von Stemann JH, Rigas AS, Thørner LW, Rasmussen DGK, Pedersen OB, Rostgaard K, et al. Prevalence and Correlation of Cytokine-Specific Autoantibodies With Epidemiological Factors and C-Reactive Protein in 8,972 Healthy Individuals: Results From the Danish Blood Donor Study. PloS One (2017) 12(6):e0179981. doi: 10.1371/journal.pone.0179981
2. Ku CL, Chi CY, von Bernuth H, Doffinger R. Autoantibodies Against Cytokines: Phenocopies of Primary Immunodeficiencies? Hum Genet (2020) 139(6-7):783–94. doi: 10.1007/s00439-020-02180-0
3. Fomsgaard A, Svenson M, Bendtzen K. Auto-Antibodies to Tumour Necrosis Factor Alpha in Healthy Humans and Patients With Inflammatory Diseases and Gram-Negative Bacterial Infections. Scand J Immunol (1989) 30(2):219–23. doi: 10.1111/j.1365-3083.1989.tb01204.x
4. Humbert L, Cornu M, Proust-Lemoine E, Bayry J, Wemeau JL, Vantyghem MC, et al. Chronic Mucocutaneous Candidiasis in Autoimmune Polyendocrine Syndrome Type 1. Front Immunol (2018) 9:2570. doi: 10.3389/fimmu.2018.02570
5. Knight V, Merkel PA, O’Sullivan MD. Anticytokine Autoantibodies: Association With Infection and Immune Dysregulation. Antibodies (Basel) (2016) 5(1):1–20. doi: 10.3390/antib5010003
6. Lafont E, Conan PL, Rodriguez-Nava V, Lebeaux D. Invasive Nocardiosis: Disease Presentation, Diagnosis and Treatment - Old Questions, New Answers? Infect Drug Resist (2020) 13:4601–13. doi: 10.2147/idr.S249761
7. Meager A, Wadhwa M, Bird C, Dilger P, Thorpe R, Newsom-Davis J, et al. Spontaneously Occurring Neutralizing Antibodies Against Granulocyte-Macrophage Colony-Stimulating Factor in Patients With Autoimmune Disease. Immunology (1999) 97(3):526–32. doi: 10.1046/j.1365-2567.1999.00806.x
8. Merkel PA, Lebo T, Knight V. Functional Analysis of Anti-Cytokine Autoantibodies Using Flow Cytometry. Front Immunol (2019) 10:1517. doi: 10.3389/fimmu.2019.01517
9. Peichl P, Pursch E, Bröll H, Lindley IJ. Anti-IL-8 Autoantibodies and Complexes in Rheumatoid Arthritis: Polyclonal Activation in Chronic Synovial Tissue Inflammation. Rheumatol Int (1999) 18(4):141–5. doi: 10.1007/s002960050073
10. Puel A, Döffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies Against IL-17a, IL-17F, and IL-22 in Patients With Chronic Mucocutaneous Candidiasis and Autoimmune Polyendocrine Syndrome. J Exp Med (2010) 207(2):291–7. doi: 10.1084/jem.20091983
11. Rosen LB, Rocha Pereira N, Figueiredo C, Fiske LC, Ressner RA, Hong JC, et al. Nocardia-Induced Granulocyte Macrophage Colony-Stimulating Factor is Neutralized by Autoantibodies in Disseminated/Extrapulmonary Nocardiosis. Clin Infect Dis (2015) 60(7):1017–25. doi: 10.1093/cid/ciu968
12. Svenson M, Hansen MB, Ross C, Diamant M, Rieneck K, Nielsen H, et al. Antibody to Granulocyte-Macrophage Colony-Stimulating Factor is a Dominant Anti-Cytokine Activity in Human IgG Preparations. Blood (1998) 91(6):2054–61. doi: 10.1182/blood.V91.6.2054
13. Svenson M, Poulsen LK, Fomsgaard A, Bendtzen K. IgG Autoantibodies Against Interleukin 1 Alpha in Sera of Normal Individuals. Scand J Immunol (1989) 29(4):489–92. doi: 10.1111/j.1365-3083.1989.tb01149.x
14. Kärner J, Pihlap M, Ranki A, Krohn K, Trebusak Podkrajsek K, Bratanic N, et al. IL-6-Specific Autoantibodies Among APECED and Thymoma Patients. Immun Inflamm Dis (2016) 4(2):235–43. doi: 10.1002/iid3.109
15. Watanabe M, Uchida K, Nakagaki K, Kanazawa H, Trapnell BC, Hoshino Y, et al. Anti-Cytokine Autoantibodies are Ubiquitous in Healthy Individuals. FEBS Lett (2007) 581(10):2017–21. doi: 10.1016/j.febslet.2007.04.029
16. Barcenas-Morales G, Jandus P, Döffinger R. Anticytokine Autoantibodies in Infection and Inflammation: An Update. Curr Opin Allergy Clin Immunol (2016) 16(6):523–9. doi: 10.1097/aci.0000000000000316
17. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-Spectrum Antibodies Against Self-Antigens and Cytokines in RAG Deficiency. J Clin Invest (2015) 125(11):4135–48. doi: 10.1172/jci80477
18. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies Against Type I IFNs in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):1–12. doi: 10.1126/science.abd4585
19. Puel A, Picard C, Lorrot M, Pons C, Chrabieh M, Lorenzo L, et al. Recurrent Staphylococcal Cellulitis and Subcutaneous Abscesses in a Child With Autoantibodies Against IL-6. J Immunol (2008) 180(1):647–54. doi: 10.4049/jimmunol.180.1.647
20. Cappellano G, Orilieri E, Woldetsadik AD, Boggio E, Soluri MF, Comi C, et al. Anti-Cytokine Autoantibodies in Autoimmune Diseases. Am J Clin Exp Immunol (2012) 1(2):136–46.
21. Katayama K, Hirose M, Arai T, Hatsuda K, Tachibana K, Sugawara R, et al. Clinical Significance of Serum Anti-Granulocyte-Macrophage Colony-Stimulating Factor Autoantibodies in Patients With Sarcoidosis and Hypersensitivity Pneumonitis. Orphanet J Rare Dis (2020) 15(1):272. doi: 10.1186/s13023-020-01546-x
22. Bayat A, Burbelo PD, Browne SK, Quinlivan M, Martinez B, Holland SM, et al. Anti-Cytokine Autoantibodies in Postherpetic Neuralgia. J Transl Med (2015) 13:333. doi: 10.1186/s12967-015-0695-6
23. Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N, et al. Characteristics of a Large Cohort of Patients With Autoimmune Pulmonary Alveolar Proteinosis in Japan. Am J Respir Crit Care Med (2008) 177(7):752–62. doi: 10.1164/rccm.200708-1271OC
24. Wang T, Lazar CA, Fishbein MC. Lynch JP, 3rd. Pulmonary Alveolar Proteinosis. Semin Respir Crit Care Med (2012) 33(5):498–508. doi: 10.1055/s-0032-1325160
25. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF Regulates Alveolar Macrophage Differentiation and Innate Immunity in the Lung Through PU.1. Immunity (2001) 15(4):557–67. doi: 10.1016/s1074-7613(01)00218-7
26. Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK, et al. GM-CSF Autoantibodies and Neutrophil Dysfunction in Pulmonary Alveolar Proteinosis. N Engl J Med (2007) 356(6):567–79. doi: 10.1056/NEJMoa062505
27. Carey B, Trapnell BC. The Molecular Basis of Pulmonary Alveolar Proteinosis. Clin Immunol (2010) 135(2):223–35. doi: 10.1016/j.clim.2010.02.017
28. Dranoff G. The Cloning of GM-CSF. J Immunol (2017) 198(7):2519–21. doi: 10.4049/jimmunol.1700182
29. Trapnell BC, Whitsett JA. GM-CSF Regulates Pulmonary Surfactant Homeostasis and Alveolar Macrophage-Mediated Innate Host Defense. Annu Rev Physiol (2002) 64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847
30. Bonfield TL, Farver CF, Barna BP, Malur A, Abraham S, Raychaudhuri B, et al. Peroxisome Proliferator-Activated Receptor-Gamma is Deficient in Alveolar Macrophages From Patients With Alveolar Proteinosis. Am J Respir Cell Mol Biol (2003) 29(6):677–82. doi: 10.1165/rcmb.2003-0148OC
31. Ikegami M, Jobe AH, Huffman Reed JA, Whitsett JA. Surfactant Metabolic Consequences of Overexpression of GM-CSF in the Epithelium of GM-CSF-Deficient Mice. Am J Physiol (1997) 273(4):L709–14. doi: 10.1152/ajplung.1997.273.4.L709
32. Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, et al. Involvement of Granulocyte-Macrophage Colony-Stimulating Factor in Pulmonary Homeostasis. Science (1994) 264(5159):713–6. doi: 10.1126/science.8171324
33. Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic Infections in Patients With Pulmonary Alveolar Proteinosis. J Infect (2012) 65(2):173–9. doi: 10.1016/j.jinf.2012.03.020
34. Piccoli L, Campo I, Fregni CS, Rodriguez BM, Minola A, Sallusto F, et al. Neutralization and Clearance of GM-CSF by Autoantibodies in Pulmonary Alveolar Proteinosis. Nat Commun (2015) 6:7375. doi: 10.1038/ncomms8375
35. Uchida K, Nakata K, Carey B, Chalk C, Suzuki T, Sakagami T, et al. Standardized Serum GM-CSF Autoantibody Testing for the Routine Clinical Diagnosis of Autoimmune Pulmonary Alveolar Proteinosis. J Immunol Methods (2014) 402(1-2):57–70. doi: 10.1016/j.jim.2013.11.011
36. Ushach I, Zlotnik A. Biological Role of Granulocyte Macrophage Colony-Stimulating Factor (GM-CSF) and Macrophage Colony-Stimulating Factor (M-CSF) on Cells of the Myeloid Lineage. J Leukoc Biol (2016) 100(3):481–9. doi: 10.1189/jlb.3RU0316-144R
37. Cakarova L, Marsh LM, Wilhelm J, Mayer K, Grimminger F, Seeger W, et al. Macrophage Tumor Necrosis Factor-Alpha Induces Epithelial Expression of Granulocyte-Macrophage Colony-Stimulating Factor: Impact on Alveolar Epithelial Repair. Am J Respir Crit Care Med (2009) 180(6):521–32. doi: 10.1164/rccm.200812-1837OC
38. Rosas M, Gordon S, Taylor PR. Characterisation of the Expression and Function of the GM-CSF Receptor Alpha-Chain in Mice. Eur J Immunol (2007) 37(9):2518–28. doi: 10.1002/eji.200636892
39. LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA. GM-CSF-Deficient Mice Are Susceptible to Pulmonary Group B Streptococcal Infection. J Clin Invest (1999) 103(4):563–9. doi: 10.1172/JCI5212
40. Paine R 3rd, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, et al. Granulocyte-Macrophage Colony-Stimulating Factor in the Innate Immune Response to Pneumocystis Carinii Pneumonia in Mice. J Immunol (2000) 164(5):2602–9. doi: 10.4049/jimmunol.164.5.2602
41. Seymour JF. Extra-Pulmonary Aspects of Acquired Pulmonary Alveolar Proteinosis as Predicted by Granulocyte-Macrophage Colony-Stimulating Factor-Deficient Mice. Respirology (2006) 11(Suppl):S16–22. doi: 10.1111/j.1440-1843.2006.00801.x
42. Seymour JF, Presneill JJ. Pulmonary Alveolar Proteinosis: Progress in the First 44 Years. Am J Respir Crit Care Med (2002) 166(2):215–35. doi: 10.1164/rccm.2109105
43. Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary Alveolar Proteinosis, a Primary Immunodeficiency of Impaired GM-CSF Stimulation of Macrophages. Curr Opin Immunol (2009) 21(5):514–21. doi: 10.1016/j.coi.2009.09.004
44. Berclaz PY, Zsengeller Z, Shibata Y, Otake K, Strasbaugh S, Whitsett JA, et al. Endocytic Internalization of Adenovirus, Nonspecific Phagocytosis, and Cytoskeletal Organization Are Coordinately Regulated in Alveolar Macrophages by GM-CSF and PU.1. J Immunol (2002) 169(11):6332–42. doi: 10.4049/jimmunol.169.11.6332
45. Damiani G, McCormick TS, Leal LO, Ghannoum MA. Recombinant Human Granulocyte Macrophage-Colony Stimulating Factor Expressed in Yeast (Sargramostim): A Potential Ally to Combat Serious Infections. Clin Immunol (2020) 210:108292. doi: 10.1016/j.clim.2019.108292
46. Atri C, Guerfali FZ, Laouini D. Role of Human Macrophage Polarization in Inflammation During Infectious Diseases. Int J Mol Sci (2018) 19(6):1–15. doi: 10.3390/ijms19061801
47. Uchida K, Nakata K, Suzuki T, Luisetti M, Watanabe M, Koch DE, et al. Granulocyte/macrophage-Colony-Stimulating Factor Autoantibodies and Myeloid Cell Immune Functions in Healthy Subjects. Blood (2009) 113(11):2547–56. doi: 10.1182/blood-2009-05-155689
48. Carey B, Staudt MK, Bonaminio D, van der Loo JC, Trapnell BC. PU.1 Redirects Adenovirus to Lysosomes in Alveolar Macrophages, Uncoupling Internalization From Infection. J Immunol (2007) 178(4):2440–7. doi: 10.4049/jimmunol.178.4.2440
49. Bhattacharya P, Thiruppathi M, Elshabrawy HA, Alharshawi K, Kumar P, Prabhakar BS. GM-CSF: An Immune Modulatory Cytokine That can Suppress Autoimmunity. Cytokine (2015) 75(2):261–71. doi: 10.1016/j.cyto.2015.05.030
50. Golde DW, Territo M, Finley TN, Cline MJ. Defective Lung Macrophages in Pulmonary Alveolar Proteinosis. Ann Intern Med (1976) 85(3):304–9. doi: 10.7326/0003-4819-85-3-304
51. Gonzalez-Rothi RJ, Harris JO. Pulmonary Alveolar Proteinosis. Further Evaluation of Abnormal Alveolar Macrophages. Chest (1986) 90(5):656–61. doi: 10.1378/chest.90.5.656
52. Kushner BH, Cheung NK. Absolute Requirement of CD11/CD18 Adhesion Molecules, FcRII and the Phosphatidylinositol-Linked FcRIII for Monoclonal Antibody-Mediated Neutrophil Antihuman Tumor Cytotoxicity. Blood (1992) 79(6):1484–90. doi: 10.1182/blood.V79.6.1484.1484
53. Rubio MA, Lopez-Rodriguez C, Nueda A, Aller P, Armesilla AL, Vega MA, et al. Granulocyte-Macrophage Colony-Stimulating Factor, Phorbol Ester, and Sodium Butyrate Induce the CD11c Integrin Gene Promoter Activity During Myeloid Cell Differentiation. Blood (1995) 86(10):3715–24. doi: 10.1182/blood.V86.10.3715.bloodjournal86103715
54. Subramaniam R, Hillberry Z, Chen H, Feng Y, Fletcher K, Neuenschwander P, et al. Delivery of GM-CSF to Protect Against Influenza Pneumonia. PloS One (2015) 10(4):e0124593. doi: 10.1371/journal.pone.0124593
55. Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF)-Induced STAT5 Activation and Target-Gene Expression During Human Monocyte/Macrophage Differentiation. J Leukoc Biol (2002) 71(3):511–9.
56. Thomassen MJ, Barna BP, Malur AG, Bonfield TL, Farver CF, Malur A, et al. ABCG1 is Deficient in Alveolar Macrophages of GM-CSF Knockout Mice and Patients With Pulmonary Alveolar Proteinosis. J Lipid Res (2007) 48(12):2762–8. doi: 10.1194/jlr.P700022-JLR200
57. Sallese A, Suzuki T, McCarthy C, Bridges J, Filuta A, Arumugam P, et al. Targeting Cholesterol Homeostasis in Lung Diseases. Sci Rep (2017) 7(1):10211. doi: 10.1038/s41598-017-10879-w
58. Nishimura M, Yamaguchi E, Takahashi A, Asai N, Katsuda E, Ohta T, et al. Clinical Significance of Serum Anti-GM-CSF Autoantibody Levels in Autoimmune Pulmonary Alveolar Proteinosis. Biomark Med (2018) 12(2):151–9. doi: 10.2217/bmm-2017-0362
59. Wessendarp M, Watanabe M, Liu S, Stankiewicz T, Ma Y, Shima K, et al. Role of GM-CSF in Regulating Metabolism and Mitochondrial Functions Critical to Macrophage Proliferation. Mitochondrion (2021) S1567-7249(21)00149-5. doi: 10.1016/j.mito.2021.10.009
60. Dougan M, Dranoff G, Dougan SK. GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity (2019) 50(4):796–811. doi: 10.1016/j.immuni.2019.03.022
61. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-Macrophage Colony-Stimulating Factor (CSF) and Macrophage CSF-Dependent Macrophage Phenotypes Display Differences in Cytokine Profiles and Transcription Factor Activities: Implications for CSF Blockade in Inflammation. J Immunol (2007) 178(8):5245–52. doi: 10.4049/jimmunol.178.8.5245
62. Hamilton TA, Zhao C, Pavicic PG Jr, Datta S. Myeloid Colony-Stimulating Factors as Regulators of Macrophage Polarization. Front Immunol (2014) 5:554. doi: 10.3389/fimmu.2014.00554
63. Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS Jr. Granulocyte Macrophage-Colony Stimulating Factor Induced Zn Sequestration Enhances Macrophage Superoxide and Limits Intracellular Pathogen Survival. Immunity (2013) 39(4):697–710. doi: 10.1016/j.immuni.2013.09.006
64. Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmonary Epithelial Cell Expression of GM-CSF Corrects the Alveolar Proteinosis in GM-CSF-Deficient Mice. J Clin Invest (1996) 97(3):649–55. doi: 10.1172/JCI118461
65. Reed JA, Ikegami M, Cianciolo ER, Lu W, Cho PS, Hull W, et al. Aerosolized GM-CSF Ameliorates Pulmonary Alveolar Proteinosis in GM-CSF-Deficient Mice. Am J Physiol (1999) 276(4):L556–63. doi: 10.1152/ajplung.1999.276.4.L556
66. Zsengellér ZK, Reed JA, Bachurski CJ, LeVine AM, Forry-Schaudies S, Hirsch R, et al. Adenovirus-Mediated Granulocyte-Macrophage Colony-Stimulating Factor Improves Lung Pathology of Pulmonary Alveolar Proteinosis in Granulocyte-Macrophage Colony-Stimulating Factor-Deficient Mice. Hum Gene Ther (1998) 9(14):2101–9. doi: 10.1089/hum.1998.9.14-2101
67. Thomassen MJ, Raychaudhuri B, Bonfield TL, Malur A, Abraham S, Barna BP, et al. Elevated IL-10 Inhibits GM-CSF Synthesis in Pulmonary Alveolar Proteinosis. Autoimmunity (2003) 36(5):285–90. doi: 10.1080/0891693031000152688
68. Thomassen MJ, Yi T, Raychaudhuri B, Malur A, Kavuru MS. Pulmonary Alveolar Proteinosis is a Disease of Decreased Availability of GM-CSF Rather Than an Intrinsic Cellular Defect. Clin Immunol (2000) 95(2):85–92. doi: 10.1006/clim.2000.4859
69. Vlahos R, Bozinovski S, Chan SP, Ivanov S, Linden A, Hamilton JA, et al. Neutralizing Granulocyte/Macrophage Colony-Stimulating Factor Inhibits Cigarette Smoke-Induced Lung Inflammation. Am J Respir Crit Care Med (2010) 182(1):34–40. doi: 10.1164/rccm.200912-1794OC
70. Basilico P, Cremona TP, Oevermann A, Piersigilli A, Benarafa C. Increased Myeloid Cell Production and Lung Bacterial Clearance in Mice Exposed to Cigarette Smoke. Am J Respir Cell Mol Biol (2016) 54(3):424–35. doi: 10.1165/rcmb.2015-0017OC
71. Harris JO, Swenson EW, Johnson JE 3rd. Human Alveolar Macrophages: Comparison of Phagocytic Ability, Glucose Utilization, and Ultrastructure in Smokers and Nonsmokers. J Clin Invest (1970) 49(11):2086–96. doi: 10.1172/JCI106426
72. Richens TR, Linderman DJ, Horstmann SA, Lambert C, Xiao YQ, Keith RL, et al. Cigarette Smoke Impairs Clearance of Apoptotic Cells Through Oxidant-Dependent Activation of RhoA. Am J Respir Crit Care Med (2009) 179(11):1011–21. doi: 10.1164/rccm.200807-1148OC
73. Suzuki T, McCarthy C, Carey BC, Borchers M, Beck D, Wikenheiser-Brokamp KA, et al. Increased Pulmonary GM-CSF Causes Alveolar Macrophage Accumulation. Mechanistic Implications for Desquamative Interstitial Pneumonitis. Am J Respir Cell Mol Biol (2020) 62(1):87–94. doi: 10.1165/rcmb.2018-0294OC
74. Craig PJ, Wells AU, Doffman S, Rassl D, Colby TV, Hansell DM, et al. Desquamative Interstitial Pneumonia, Respiratory Bronchiolitis and Their Relationship to Smoking. Histopathology (2004) 45(3):275–82. doi: 10.1111/j.1365-2559.2004.01921.x
75. Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA. Desquamative Interstitial Pneumonia and Respiratory Bronchiolitis-Associated Interstitial Lung Disease. Chest (2005) 127(1):178–84. doi: 10.1378/chest.127.1.178
76. Ballinger MN, Paine R 3rd, Serezani CH, Aronoff DM, Choi ES, Standiford TJ, et al. Role of Granulocyte Macrophage Colony-Stimulating Factor During Gram-Negative Lung Infection With Pseudomonas Aeruginosa. Am J Respir Cell Mol Biol (2006) 34(6):766–74. doi: 10.1165/rcmb.2005-0246OC
77. Baker AD, Malur A, Barna BP, Ghosh S, Kavuru MS, Malur AG, et al. Targeted PPARγ Deficiency in Alveolar Macrophages Disrupts Surfactant Catabolism. J Lipid Res (2010) 51(6):1325–31. doi: 10.1194/jlr.M001651
78. Bendtzen K, Svenson M, Hansen MB. GM-CSF Autoantibodies in Pulmonary Alveolar Proteinosis. N Engl J Med (2007) 356(19):2001. doi: 10.1056/NEJMc070650
79. Salvaterra E, Campo I. Pulmonary Alveolar Proteinosis: From Classification to Therapy. Breathe (Sheff) (2020) 16(2):200018. doi: 10.1183/20734735.0018-2020
80. Arai T, Hamano E, Inoue Y, Ryushi T, Nukiwa T, Sakatani M, et al. Serum Neutralizing Capacity of GM-CSF Reflects Disease Severity in a Patient With Pulmonary Alveolar Proteinosis Successfully Treated With Inhaled GM-CSF. Respir Med (2004) 98(12):1227–30. doi: 10.1016/j.rmed.2004.08.011
81. Ohashi K, Sato A, Takada T, Arai T, Nei T, Kasahara Y, et al. Direct Evidence That GM-CSF Inhalation Improves Lung Clearance in Pulmonary Alveolar Proteinosis. Respir Med (2012) 106(2):284–93. doi: 10.1016/j.rmed.2011.10.019
82. Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, et al. Inhaled Granulocyte/Macrophage-Colony Stimulating Factor as Therapy for Pulmonary Alveolar Proteinosis. Am J Respir Crit Care Med (2010) 181(12):1345–54. doi: 10.1164/rccm.200906-0978OC
83. Ioachimescu OC, Kavuru MS. Pulmonary Alveolar Proteinosis. Chron Respir Dis (2006) 3(3):149–59. doi: 10.1191/1479972306cd101rs
84. Schoch OD, Schanz U, Koller M, Nakata K, Seymour JF, Russi EW, et al. BAL Findings in a Patient With Pulmonary Alveolar Proteinosis Successfully Treated With GM-CSF. Thorax (2002) 57(3):277–80. doi: 10.1136/thorax.57.3.277
85. Kusakabe Y, Uchida K, Hiruma T, Suzuki Y, Totsu T, Suzuki T, et al. A Standardized Blood Test for the Routine Clinical Diagnosis of Impaired GM-CSF Signaling Using Flow Cytometry. J Immunol Methods (2014) 413:1–11. doi: 10.1016/j.jim.2014.07.009
86. Ahmed O. CAR-T-Cell Neurotoxicity: Hope is on the Horizon. Blood (2019) 133(20):2114–6. doi: 10.1182/blood-2019-03-900985
87. Gupta S, Weitzman S. Primary and Secondary Hemophagocytic Lymphohistiocytosis: Clinical Features, Pathogenesis and Therapy. Expert Rev Clin Immunol (2010) 6(1):137–54. doi: 10.1586/eci.09.58
88. Reghunathan R, Jayapal M, Hsu LY, Chng HH, Tai D, Leung BP, et al. Expression Profile of Immune Response Genes in Patients With Severe Acute Respiratory Syndrome. BMC Immunol (2005) 6:2. doi: 10.1186/1471-2172-6-2
89. Tugues S, Amorim A, Spath S, Martin-Blondel G, Schreiner B, De Feo D, et al. Graft-Versus-Host Disease, But Not Graft-Versus-Leukemia Immunity, Is Mediated by GM-CSF-Licensed Myeloid Cells. Sci Transl Med (2018) 10(469):1–12. doi: 10.1126/scitranslmed.aat8410
90. Browne SK, Holland SM. Immunodeficiency Secondary to Anticytokine Autoantibodies. Curr Opin Allergy Clin Immunol (2010) 10(6):534–41. doi: 10.1097/ACI.0b013e3283402b41
91. Albogami SM, Touman AA. Viral Pneumonia and Pulmonary Alveolar Proteinosis: The Cause and the Effect, Case Report. AME Case Rep (2019) 3:41. doi: 10.21037/acr.2019.09.04
92. Bozinovski S, Jones J, Beavitt SJ, Cook AD, Hamilton JA, Anderson GP. Innate Immune Responses to LPS in Mouse Lung Are Suppressed and Reversed by Neutralization of GM-CSF via Repression of TLR-4. Am J Physiol Lung Cell Mol Physiol (2004) 286(4):L877–85. doi: 10.1152/ajplung.00275.2003
93. Deepe GS Jr, Gibbons R, Woodward E. Neutralization of Endogenous Granulocyte-Macrophage Colony-Stimulating Factor Subverts the Protective Immune Response to Histoplasma Capsulatum. J Immunol (1999) 163(9):4985–93.
94. Dorman SE, Guide SV, Conville PS, DeCarlo ES, Malech HL, Gallin JI, et al. Nocardia Infection in Chronic Granulomatous Disease. Clin Infect Dis (2002) 35(4):390–4. doi: 10.1086/341416
95. Rosen LB, Freeman AF, Yang LM, Jutivorakool K, Olivier KN, Angkasekwinai N, et al. Anti-GM-CSF Autoantibodies in Patients With Cryptococcal Meningitis. J Immunol (2013) 190(8):3959–66. doi: 10.4049/jimmunol.1202526
96. Elsegeiny W, Marr KA, Williamson PR. Immunology of Cryptococcal Infections: Developing a Rational Approach to Patient Therapy. Front Immunol (2018) 9:651. doi: 10.3389/fimmu.2018.00651
97. Saijo T, Chen J, Chen SC, Rosen LB, Yi J, Sorrell TC, et al. Anti-Granulocyte-Macrophage Colony-Stimulating Factor Autoantibodies Are a Risk Factor for Central Nervous System Infection by Cryptococcus Gattii in Otherwise Immunocompetent Patients. mBio (2014) 5(2):e00912–14. doi: 10.1128/mBio.00912-14
98. Riopel J, Tam M, Mohan K, Marino MW, Stevenson MM. Granulocyte-Macrophage Colony-Stimulating Factor-Deficient Mice Have Impaired Resistance to Blood-Stage Malaria. Infect Immun (2001) 69(1):129–36. doi: 10.1128/iai.69.1.129-136.2001
99. Zhan Y, Lieschke GJ, Grail D, Dunn AR, Cheers C. Essential Roles for Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and G-CSF in the Sustained Hematopoietic Response of Listeria Monocytogenes-Infected Mice. Blood (1998) 91(3):863–9. doi: 10.1182/blood.V91.3.863
100. Plenz G, Eschert H, Beissert S, Arps V, Sindermann JR, Robenek H, et al. Alterations in the Vascular Extracellular Matrix of Granulocyte Macrophage Colony-Stimulating Factor (GM-CSF) -Deficient Mice. FASEB J (2003) 17(11):1451–7. doi: 10.1096/fj.02-1035com
101. Robertson SA, Roberts CT, Farr KL, Dunn AR, Seamark RF. Fertility Impairment in Granulocyte-Macrophage Colony-Stimulating Factor-Deficient Mice. Biol Reprod (1999) 60(2):251–61. doi: 10.1095/biolreprod60.2.251
102. Campo I, Mariani F, Paracchini E, Kadija Z, Zorzetto M, Tinelli C, et al. Inhaled Sargramostim and Whole Lung Lavage (WLL) as Therapy of Autoimmune Pulmonary Alveolar Proteinosis (aPAP). Eur Respir J (2016) 48(suppl 60):PA3870. doi: 10.1183/13993003.congress-2016.PA3870
103. Papiris SA, Tsirigotis P, Kolilekas L, Papadaki G, Papaioannou AI, Triantafillidou C, et al. Long-Term Inhaled Granulocyte Macrophage-Colony-Stimulating Factor in Autoimmune Pulmonary Alveolar Proteinosis: Effectiveness, Safety, and Lowest Effective Dose. Clin Drug Investig (2014) 34(8):553–64. doi: 10.1007/s40261-014-0208-z
104. Yamamoto H, Yamaguchi E, Agata H, Kandatsu N, Komatsu T, Kawai S, et al. A Combination Therapy of Whole Lung Lavage and GM-CSF Inhalation in Pulmonary Alveolar Proteinosis. Pediatr Pulmonol (2008) 43(8):828–30. doi: 10.1002/ppul.20856
105. Robinson TE, Trapnell BC, Goris ML, Quittell LM, Cornfield DN. Quantitative Analysis of Longitudinal Response to Aerosolized Granulocyte-Macrophage Colony-Stimulating Factor in Two Adolescents With Autoimmune Pulmonary Alveolar Proteinosis. Chest (2009) 135(3):842–8. doi: 10.1378/chest.08-1317
106. Tazawa R, Inoue Y, Arai T, Takada T, Kasahara Y, Hojo M, et al. Duration of Benefit in Patients With Autoimmune Pulmonary Alveolar Proteinosis After Inhaled Granulocyte-Macrophage Colony-Stimulating Factor Therapy. Chest (2014) 145(4):729–37. doi: 10.1378/chest.13-0603
107. Tazawa R, Nakata K, Inoue Y, Nukiwa T. Granulocyte-Macrophage Colony-Stimulating Factor Inhalation Therapy for Patients With Idiopathic Pulmonary Alveolar Proteinosis: A Pilot Study; and Long-Term Treatment With Aerosolized Granulocyte-Macrophage Colony-Stimulating Factor: A Case Report. Respirology (2006) 11(Suppl):S61–4. doi: 10.1111/j.1440-1843.2006.00811.x
108. Graudal NA, Svenson M, Tarp U, Garred P, Jurik AG, Bendtzen K. Autoantibodies Against Interleukin 1alpha in Rheumatoid Arthritis: Association With Long Term Radiographic Outcome. Ann Rheum Dis (2002) 61(7):598–602. doi: 10.1136/ard.61.7.598
109. Hellmich B, Csernok E, Schatz H, Gross WL, Schnabel A. Autoantibodies Against Granulocyte Colony-Stimulating Factor in Felty’s Syndrome and Neutropenic Systemic Lupus Erythematosus. Arthritis Rheum (2002) 46(9):2384–91. doi: 10.1002/art.10497
110. Watanabe M, Uchida K, Nakagaki K, Trapnell BC, Nakata K. High Avidity Cytokine Autoantibodies in Health and Disease: Pathogenesis and Mechanisms. Cytokine Growth Factor Rev (2010) 21(4):263–73. doi: 10.1016/j.cytogfr.2010.03.003
111. Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, et al. Pulmonary Alveolar Proteinosis. Nat Rev Dis Primers (2019) 5(1):16. doi: 10.1038/s41572-019-0066-3
112. McCarthy C, Carey B, Trapnell BC. Blood Testing for Differential Diagnosis of Pulmonary Alveolar Proteinosis Syndrome. Chest (2019) 155(2):450–2. doi: 10.1016/j.chest.2018.11.002
113. Kumar A, Abdelmalak B, Inoue Y, Culver DA. Pulmonary Alveolar Proteinosis in Adults: Pathophysiology and Clinical Approach. Lancet Respir Med (2018) 6(7):554–65. doi: 10.1016/s2213-2600(18)30043-2
114. Berthoux C, Mailhe M, Vély F, Gauthier C, Mège JL, Lagier JC, et al. Granulocyte Macrophage Colony-Stimulating Factor-Specific Autoantibodies and Cerebral Nocardia With Pulmonary Alveolar Proteinosis. Open Forum Infect Dis (2021) 8(2):ofaa612. doi: 10.1093/ofid/ofaa612
115. Papp KA, Gooderham M, Jenkins R, Vender R, Szepietowski JC, Wagner T, et al. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) as a Therapeutic Target in Psoriasis: Randomized, Controlled Investigation Using Namilumab, a Specific Human Anti-GM-CSF Monoclonal Antibody. Br J Dermatol (2019) 180(6):1352–60. doi: 10.1111/bjd.17195
116. Taylor PC, Saurigny D, Vencovsky J, Takeuchi T, Nakamura T, Matsievskaia G, et al. Efficacy and Safety of Namilumab, a Human Monoclonal Antibody Against Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) Ligand in Patients With Rheumatoid Arthritis (RA) With Either an Inadequate Response to Background Methotrexate Therapy or an Inadequate Response or Intolerance to an Anti-TNF (Tumour Necrosis Factor) Biologic Therapy: A Randomized, Controlled Trial. Arthritis Res Ther (2019) 21(1):101. doi: 10.1186/s13075-019-1879-x
Keywords: autoantibodies, autoimmune pulmonary alveolar proteinosis, sargramostim, Nocardia, Cryptococcus, Aspergillus, Histoplasma, granulocyte-macrophage colony-stimulating factor (GM-CSF)
Citation: Ataya A, Knight V, Carey BC, Lee E, Tarling EJ and Wang T (2021) The Role of GM-CSF Autoantibodies in Infection and Autoimmune Pulmonary Alveolar Proteinosis: A Concise Review. Front. Immunol. 12:752856. doi: 10.3389/fimmu.2021.752856
Received: 03 August 2021; Accepted: 04 November 2021;
Published: 22 November 2021.
Edited by:
Anna Shcherbina, Dmitry Rogachev National Research Center of Pediatric Hematology, Oncology and Immunology, RussiaReviewed by:
Cormac McCarthy, University College Dublin, IrelandCopyright © 2021 Ataya, Knight, Carey, Lee, Tarling and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali Ataya, QWxpLkF0YXlhQG1lZGljaW5lLnVmbC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.