
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 08 November 2021
Sec. Vaccines and Molecular Therapeutics
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.752168
 Enrico König1†
Enrico König1† Assunta Gagliardi2†
Assunta Gagliardi2† Ilary Riedmiller1
Ilary Riedmiller1 Chiara Andretta1
Chiara Andretta1 Michele Tomasi1
Michele Tomasi1 Carmela Irene1
Carmela Irene1 Luca Frattini1
Luca Frattini1 Ilaria Zanella1
Ilaria Zanella1 Francesco Berti3
Francesco Berti3 Alberto Grandi2,4
Alberto Grandi2,4 Elena Caproni1
Elena Caproni1 Laura Fantappiè2
Laura Fantappiè2 Guido Grandi1*
Guido Grandi1*Modification of surface antigens and differential expression of virulence factors are frequent strategies pathogens adopt to escape the host immune system. These escape mechanisms make pathogens a “moving target” for our immune system and represent a challenge for the development of vaccines, which require more than one antigen to be efficacious. Therefore, the availability of strategies, which simplify vaccine design, is highly desirable. Bacterial Outer Membrane Vesicles (OMVs) are a promising vaccine platform for their built-in adjuvanticity, ease of purification and flexibility to be engineered with foreign proteins. However, data on if and how OMVs can be engineered with multiple antigens is limited. In this work, we report a multi-antigen expression strategy based on the co-expression of two chimeras, each constituted by head-to-tail fusions of immunogenic proteins, in the same OMV-producing strain. We tested the strategy to develop a vaccine against Staphylococcus aureus, a Gram-positive human pathogen responsible for a large number of community and hospital-acquired diseases. Here we describe an OMV-based vaccine in which four S. aureus virulent factors, ClfAY338A, LukE, SpAKKAA and HlaH35L have been co-expressed in the same OMVs (CLSH-OMVsΔ60). The vaccine elicited antigen-specific antibodies with functional activity, as judged by their capacity to promote opsonophagocytosis and to inhibit Hla-mediated hemolysis, LukED-mediated leukocyte killing, and ClfA-mediated S. aureus binding to fibrinogen. Mice vaccinated with CLSH-OMVsΔ60 were robustly protected from S. aureus challenge in the skin, sepsis and kidney abscess models. This study not only describes a generalized approach to develop easy-to-produce and inexpensive multi-component vaccines, but also proposes a new tetravalent vaccine candidate ready to move to development.
Mutation-driven modification of surface antigens and differential expression of adhesion, colonization and virulence factors are among the most frequent strategies pathogens adopt to escape the host immune system. These escape mechanisms not only make pathogens a “moving target” for our immune system but also represent a challenge for vaccine development since, to provide a sufficiently broad coverage, vaccines must be formulated with more than one, sometime several, antigens. For instance, the most recent pneumococcal and Human Papilloma Virus vaccines include up to 15 glycoconjugates and five variants of the L1 protein, respectively, while the acellular Bordetella pertussis vaccine and the Meningococcus B vaccine both contain five distinct virulence factors (1).
The need to formulate vaccines with more than one component complicates the production processes and increases the production costs quite substantially. Therefore, the availability of platforms, which simplify vaccine design, is highly desirable, particularly to allow broad vaccination coverage in low income countries.
Outer Membrane Vesicles (OMVs) have emerged as a promising vaccine platform which have been already exploited for human use (2). OMVs are particularly attractive for their built-in adjuvanticity, which avoids the need of additional adjuvants to elicit antigen-specific immune responses (3). Moreover, OMVs can be easily purified: OMV purification essentially consists in the separation of the biomass from the culture supernatant and in the use of tangential flow ultrafiltration to purify and concentrate the released vesicles from the latter (4). Finally, OMVs can be decorated with foreign proteins/polypeptides by genetic manipulation of the OMV-producing strains (5–7) and it has been extensively shown that immunization with engineered OMVs induce potent antigen-specific immune responses (8, 9).
OMV engineering has been proven for single antigens and therefore the preparation of multi-component vaccines requires the purification and the subsequent combination of individual OMVs (9). Although OMVs are very easy to produce, the co-expression of more than one antigen in the same OMVs would simplify the production of multivalent vaccines additionally. However, so far data on if and how OMVs can be engineered with multiple full-length antigens are limited. We previously published the decoration of OMVs with a string of immunogenic epitopes (10) and Daleke-Schermerhorn et al. (11) used the Hbp autotransporter to deliver to the OMV surface protein fusions constituted by up to three small size antigens/protein domains.
In this work, we have investigated the possibility to decorate OMVs with more than one antigen and we describe a strategy to co-express four antigens in the same OMVs. We also show that the immunization of mice with four-antigen OMVs elicit functional immune responses against each engineered antigen.
To test the feasibility and the effectiveness of our multivalent OMV approach, we focused our attention on Staphylococcus aureus. This pathogen appears to be an attractive model since it expresses more than 35 immune evasion molecules and various virulence factors (12) and it is generally accepted that to be effective a vaccine should contain a selected combination of antigens (9, 13, 14). Moreover, once phagocytosed, S. aureus has the ability to avoid killing, thus using phagocytes as “Trojan Horses” to disseminate itself inside the host (15, 16). Therefore, to counteract the ability of S. aureus to survive inside host cells, a vaccine should elicit a Th1/Th17-skewed adaptive immune response and strong innate immunity, a property that is intrinsic to OMVs.
Here we describe the co-expression, in the proteome-minimized OMVs released by E. coli BL21(DE3)Δ60 (17), of ClfAY338A, LukE, SpAKKAA and HlaH35L, four well characterized virulent factors shown to induce protection in different animal models. The vaccine (CLSH-OMVsΔ60) elicits antigen-specific antibodies with functional activity, as judged by their capacity to promote opsonophagocytosis and to inhibit Hla-mediated hemolysis, LukED-mediated leukocyte killing, and ClfA-mediated S. aureus binding to fibrinogen. Mice vaccinated with CLSH-OMVsΔ60 are robustly protected from S. aureus challenge in the skin, sepsis and kidney abscess models.
This study provides a generalized approach to develop easy-to-produce and inexpensive multi-component vaccines. Moreover, considering that the four selected virulence factors are among the most promising protective antigens for S. aureus, this study proposes a new tetravalent vaccine candidate ready to move to development.
Oligonucleotides (primers) were designed using Serial Cloner and Benchling. Densitometry was performed with Image Studio Lite Version 5.2 (Licor).
E. coli strains (BL21(DE3) derivatives, DH5α, TOP10, HK100) were routinely cultured in lysogeny broth (LB Miller; Sigma-Aldrich) supplemented with ampicillin (100 mg/L), chloramphenicol (25 mg/L) or a combination of both, when necessary. For recombinant protein expression in OMVs, cultures were supplemented with 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at OD600 between 0.5 and 0.7 and OMVs were collected after 3 hours from induction. Fermentation was carried out using an ez-Control bioreactor (Applikon) at 30°C until OD600 0.5, then growth continued at 25°C, pH 6.8 ( ± 0.2), dO2 > 30%, 280-500 rpm. At OD600 1.0, the culture was induced with 0.1 mM IPTG together with a feed consisting in ampicillin (50 mg/L) and/or chloramphenicol (12.5 mg/L), glycerol (15 g/L), and MgSO4 (0.25 g/L).
Plasmid derivatives of the pET21b(+) backbone encoding FhuD2, SpAKKAA, LukE, HlaH35L, ClfAY338A were already described (9, 17), while derivatives of the pACYC backbone were constructed ex novo using PIPE-PCR (18). Briefly, the coding sequence of the target protein was PCR amplified from its respective template pET21b(+) plasmid (I-PIPE) using the primers reported in Supplementary Table 1. These primers feature overhangs that are complementary to the 5’- and 3’-ends of a linearized “recipient” pACYC derivative. The latter plasmid was linearized by PCR (V-PIPE) using the primer couples listed in Supplementary Table 1. The PCR products of both reactions were digested with DpnI (New England BioLabs), combined (2 μl each) and used to transform chemically competent E. coli HK100 cells. Positive clones identified by colony PCR using GoTaq® Green Master Mix (Promega) and gene specific primers (Supplementary Table 1) were subjected to sequence analysis (Eurofins) before using the resultant plasmids (Supplementary Table 2) for transformation of the OMV producer strain.
The PIPE-PCR method (18) was also used to construct plasmids encoding chimera. Here, the coding sequence of the C-terminal protein of the chimera was PCR amplified from its respective template plasmid (I-PIPE) using the primers reported in Supplementary Table 1. These primers feature overhangs that, at the 5’-end, encode a short linker sequence (translated as GGGGS) complementary to the 3’-end of a “recipient” pET21b(+) derivative carrying the Lpp leader sequence followed by the N-terminal antigen of the chimera, while at the 3’-end the overhangs carry the stop codon and are complementary to the 5’-end of the recipient plasmid. The recipient plasmids were linearized by PCR (V-PIPE) excluding the stop codon and introducing the same linker sequence stated above using the primer couples listed in Supplementary Table 1. The PCR products were mixed together as described above and the resultant plasmids (Supplementary Table 2) were used for transforming the OMV producer strain.
FhuD2, LukE, SpAKKAA and HlaH35L S. aureus proteins were purified as previously described (9).
For ClfAY338A, the gene coding for the wild-type N1N2N3 portion (amino acid 40 to 559) of the antigen was amplified from the genomic DNA of S. aureus Newman strain. After genomic amplification, the Y338A mutation was inserted using PIPE-PCR. In particular, the gene was first amplified using primer couples 32 + 37 and 36 + 33 (Supplementary Table 1) and then the PCR product was used as template for the final PCR with the primer 32 + 33. The resulting gene (clfAY338A) was then cloned in a pET15 plasmid (containing His6-tag and TEV cleavage site coding sequences) downstream from a T7 inducible promoter and expressed in E. coli BL21(DE3) strain. Bacterial biomass was produced using an ez-Control bioreactor (Applikon Biotechnology) and purification of the recombinant protein was performed using an ÄKTA purifier System (General Electric Healthcare) for IMAC and size-exclusion chromatography and 10 mM Tris + 100 mM NaCl and PBS (both pH 8.0), respectively, as buffer.
OMVs were purified as previously described (9, 17, 19). Purification from cultures in bioreactor was carried out using Tangential Flow Filtration (ÄKTA flux system, GE Healthcare) with a Hollow Fiber cartridge UFB-500-C-3MA (GE Healthcare). Protein content of OMVs was quantified using DC™ Protein Assay (Bio-Rad) and the quality of OMVs was monitored by SDS-PAGE loading OMVs (normalized by μg) on Any kD™ Criterion™ TGX Stain-Free™ Protein Gel (BioRad) stained with ProBlue Safe Stain (Giotto Biotech). The size of purified OMVs was determined using NanoSight NS300 (Malvern Panalytical Ltd) as previously described (17).
OMVs (100 μg of proteins) were diluted in PBS, ice cold Triton X-114 was added to 1% final concentration and the OMV-containing solution was incubated at 4°C for 1 hour under shaking. The solution was then heated at 37°C for 10 minutes and the aqueous phase was separated from the detergent phase by centrifugation at 13,000 g for 10 minutes. Proteins in both phases were then precipitated by standard chloroform/methanol procedure, separated by SDS-PAGE electrophoresis and the protein of interest visualized by Western blot. Western blot analysis was performed as previously reported (9). Antibodies against each antigen were obtained from Genscript by immunizing rabbits with the following antigen-specific synthetic peptides: LukE: NEFVTPDGKKSAHD, SpAKKAA: AKKLNDAQAPKADN, HlaH35L: GTNTKDKWIDRSSE, ClfAY338A: IDKPVVPEQPDEPG. All synthetic peptides were conjugated to KLH protein.
Animal experiments were carried out in accordance with experimental protocols that were reviewed and approved by the Animal Ethical Committees of the University of Trento and Toscana Life Sciences (Siena, Italy) and by the Italian Ministry of Health.
For antibody titers, five-week old CD1 female mice were immunized intraperitoneally (i.p., 100 μl final volume) and intramuscularly (i.m., 50 μl final volume) three times every two weeks with 20 μg of CLSH-OMVsΔ60 formulated either with or without Alum hydroxide (2 mg/ml). Blood was collected from anesthetized mice through cardiac puncture at day 35 and serum was obtained from blood through centrifugation at 2,000 rpm for 10 minutes.
For challenge studies, mice were immunized i.p. at day 0, 14 and 28 with either 20 μg of “empty” OMVsΔ60 or 20 μg of CLSH-OMVsΔ60 in the presence of 2 mg/ml Alum. Alum was included in the vaccine formulation to use conditions similar to Bexsero, a vaccine which contains OMVs and has been approved for human use (2). For the sepsis model of infection two weeks after the third immunization mice were i.p. challenged with 3×108 CFUs of S. aureus Newman strain. Mice were monitored daily for a 7-day period. Animals health was evaluated using a 1 to 4 pain scale. A value of 4 was given to mice with: loss of weight >15%, very rough hair coat, impaired mobility. A score of 3 was given to mice with loss of weight approximately 15% and rough hair coat, while scores of 2 and 1 were given to mice with a loss of weight between 6% and 14% or 0% and 5%, respectively. All procedures were approved by the National Health Institution and the Ethical Committee and for human reasons animals were sacrificed at symptoms of sickness as recommended by 3Rs rules (‘‘Refinement, Reduction, Replacement’’ policy towards the use of animals for scientific procedures_ 99/167/EC, Council Decision of 25/1/99).
Experiments using the renal abscess and skin infection model were performed as previously described (13). Briefly, for the renal model mice were challenged intravenously (i.v.) 10 days after the third immunization with 1×107 CFUs of S. aureus Newman strain, while in the skin infection model mice were challenged subcutaneously (s.c.) 14 days after the third immunization with 5×107 CFUs of S. aureus Newman strain.
ELISA assays on mice sera collected after immunizations were performed as previously described (9) using alkaline phosphatase-conjugated secondary antibodies that recognize murine total IgG (Sigma-Aldrich), IgG2a or IgG1 (both Biolegend).
The OPK assay was performed following previously published protocols (20–22). In detail, S. aureus Newman strain was grown in tryptic soy broth (TSB) under aerobic and shaking conditions (170 r.p.m.) at 37°C starting from OD600 0.05 for 1 hour. Then, bacteria were harvested by centrifugation and diluted in Reaction Buffer (RB, DMEM + BSA 1%) and 500 CFUs/15µl were incubated with previously serially diluted immune sera for 45 minutes at room temperature (RT) under shaking (550 r.p.m.). Human promyelocytic leukemia HL-60 cells (ATCC CCL-240), differentiated into neutrophils by adding 0.78% (v/v) dimethylformamide (DMF) and incubating cells at 37°C and 5% CO2 for 5 days, were adjusted to 3×105 cells in 15µl RB, and Low-Tox Guinea Pig complement (Cederlane) diluted in RB to 4% (15µl) was added. As negative controls we included the following: i) Bacteria + RB without (w/o) serum + HL-60 cells + complement, ii) Bacteria + RB w/o serum and w/o HL-60 cells + complement, iii) Bacteria + RB w/o serum + HL-60 cells + heat-inactivated complement (HC), and iv) the same as in iii) + tested serum at the lowest dilution (1:100). The reaction mixtures were incubated at 37°C with shaking (550 r.p.m.). After 90 minutes (T90), the reactions were stopped by incubation on ice for 5 minutes and addition of 15 µl 1% Triton X-100 to facilitate cell lysis and bacterial release to the supernatant. After 3 minutes of incubation at 37°C with agitation, 10 µl of each mixture were diluted in RB (1:5) and plated on tryptic soy agar (TSA) plates. Bacterial CFUs were counted manually after an overnight incubation at 37°C. Bacterial OPK for each sample was calculated with respect to the CFUs of the negative control containing bacteria + HL-60 + complement without serum according the following formula: [CFUs of the negative control at T90 - CFUs of each sample at T90]/CFUs of the negative control at T90.
Flat-bottom 96-well plates (Nunc MaxiSorp™) were coated with 10 μg/ml human fibrinogen (Merck) overnight at 4°C. The plates were blocked with BSA 5% (p/v) for 2 hours at 37°C and then washed 3 times with PBS. S. aureus Newman strain (1×107 CFUs), which was grown in TSB to mid-log phase under aerobic and shaking conditions at 37°C, was pre-incubated with serially diluted immune sera for 30 minutes at RT and then transferred to the fibrinogen-coated plates. After 1-hour incubation at 37°C, adherent cells were washed once with PBS, fixed with formaldehyde 2.5% (v/v) for 30 minutes at RT and stained with crystal violet (CV) 0.5% (v/v) for 10 minutes at RT. Bacteria were then washed once with PBS and the plates were allowed to air-dry. CV was solubilized using acetic acid 30% (v/v) for 15 minutes at RT. Then, the crystal violet/acetic acid solution was transferred from each well to a separate well in a flat-bottom 96-well plate. Absorbance was read at 595 nm using a SpectraMax M2 Microplate reader (Molecular Devices). Adherence of tested samples was given as a percentage of values measured in wells lacking serum (100% adherence), and the percentage of inhibition of binding to fibrinogen was calculated by subtracting the adherence percentage values from 100.
LukE neutralization was assayed using the Cell proliferation kit II (XTT, Sigma-Aldrich). HL-60 cells were cultured in flask in RPMI medium plus 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin, 2mM glutamine, 1mM Sodium pyruvate and non-essential amino acids at 37°C and 5% CO2. Then, HL-60 cells were differentiated into neutrophils by adding 0.78% (v/v) dimethylformamide (DMF) and incubating cells at 37°C and 5% CO2 for 5 days. Subsequently, 5×105 differentiated HL-60 cells in a volume of 10 μl/well of RPMI culture medium were dispensed into flat-bottom 96-well plates (Corning, Corning, NY USA) and incubated with either rLukE, or rLukD (Abcam) as negative control or LukED (200 nM in 40 μl/well of RPMI culture medium) in presence of diluted immune sera (50 μl/well), for 24 hours at 37°C. Cell viability was then measured after 16 hours incubation with XTT Assay reagent (50 μl/well; Sigma-Aldrich) according to manufacturer’s instructions by reading the absorbance at 470nm using a SpectraMax M2 Microplate reader (Molecular Devices).
Hla neutralization assay was performed as previously described (9).
To test the possibility of designing a multivalent vaccine constituted by a single preparation of E. coli–derived OMVs engineered with four heterologous proteins, we focused our attention on SpAKKAA, ClfAY338A, HlaH35L and LukE antigens.
SpA substantially contributes to S. aureus virulence and toxicity. By binding to the Fcγ domain of antibodies, surface-exposed SpA interferes with the association of the C1q component of the complement classical pathway, and this results in the inhibition of bacterial opsonophagocytosis and killing (OPK) by phagocytic cells (23). Moreover, by cross-linking VH3 clonal B cell receptors, secreted SpA acts as a B cell super-antigen promoting the production of all VH3 antibodies, irrespectively of their antigen specificity (24). Finally, SpA is responsible for an anaphylactic syndrome due to its binding to the VH3 region of IgG and IgE antibodies associated to basophils and mast cells (25). It was elegantly shown that immunization with the SpAKKAA, a mutant no longer capable of binding Fcγ and VH3, induces antibodies which (i) inhibit SpA binding to Ig, (ii) promote OPK in mouse, guinea pig and human blood (23, 26), and (iii) protect animals from bacteremia (27).
Clumping Factor A (ClfA) is a surface-exposed virulence factor expressed in most S. aureus isolates and whose primary function is to allow the adhesion of S. aureus to fibrinogen (28–32). Since fibrinogen is ubiquitous in the host, ClfA promotes a rapid attachment of bacteria to tissues and organs, thus playing a relevant role in the initiation of infection (33). Inhibiting the fibrinogen binding capacity of ClfA has been proposed as a way to prevent pathogenesis. Indeed, immunization of monkeys and human volunteers with the N1N2N3 portion of ClfA carrying the Y338A mutation (30) elicited high titers of antibodies which prevented S. aureus adhesion to fibrinogen (32).
S. aureus α-hemolysin (Hla) (also known as α-toxin) is a pore-forming β-barrel toxin (34, 35), which assembles in a heptameric structure forming a pore of 2-nm in diameter into the plasma membrane (35). The toxin is expressed by almost all clinical isolates (36) and its level of expression correlates with increased severity of Skin and Soft Tissue Infections (SSTIs) and pneumonia in humans (37, 38). Hla causes dermonecrotic skin injury by interacting with ADAM10, a zinc-dependent metalloprotease that cleaves E-cadherin and destabilizes the epithelial barrier upon toxin binding (39, 40). The key role of Hla in S. aureus pathogenicity is highlighted by experiments showing that the intraperitoneal injection of 10 μg of purified Hla is sufficient to kill mice (41) and the passive transfer of neutralizing monoclonal antibodies is highly protective against S. aureus infection in different animal models (42, 43). Finally, vaccination with the nontoxigenic HlaH35L mutant (44) protects mice against S. aureus-mediated pneumonia (37), lethal sepsis, organ dissemination (45) and skin infection (13).
Human S. aureus isolates produce five additional pore-forming toxins known as leukocidins (46). Leukocidins are constituted by two subunits, the host cell targeting S component and the polymerization F component. The major target of leukotoxins are immune cells of myeloid and lymphoid lineages, suggesting that these toxins have evolved to inhibit both innate and adaptive immunity. Among the five leukocidins, LukED appears to be particularly important in that it kills virtually all immune cells, including neutrophils, monocytes, macrophages, dendritic cells, T cells, erythrocytes and NK cells (46). The toxin is encoded in the stable S. aureus pathogenicity island vSaβ53, which is present in about 70% of all clinical isolates (47). Because of their key role in pathogenesis, inactivated leukocidins are considered to be key targets for both therapeutic and prophylactic intervention (46, 48).
Based on the above, a vaccine including SpAKKAA, HlaH35L, ClfAY338A and LukE should elicit immune responses that, in the presence of innate immune responses promoted by the OMVs, should synergize and confer protection against S. aureus infections. Therefore, we selected these four antigens to attempt the construction of a multivalent vaccine based on a single OMV preparation.
Supported by preliminary data indicating that even high molecular weight heterologous antigens can be efficiently incorporated in OMVs, we created two chimeras carrying the lipoprotein leader sequence of Lpp at their 5’-ends (9) by fusing ClfAY338A-LukE genes and SpAKKAA-HlaH35L genes. The gene fusions were cloned in both pET21b(+) and pACYC, two compatible plasmids, which differ in copy number. The plasmids were used to transform the hyper-vesiculating strain E. coli BL21(DE3)Δ60 recently created in our laboratories (17). OMVs were purified from the culture supernatants and analyzed by SDS-PAGE. As shown in Figure 1A, both chimeras accumulated in the vesicular compartment at a level of 7% of total OMV proteins, as determined by densitometry scanning. Interestingly, the different copy numbers of pET21b(+) and pACYC did not substantially influence the level of accumulation in the vesicular compartment, suggesting that the rate limiting step of chimera compartmentalization in OMVs was largely determined by the translation and/or transport machineries.
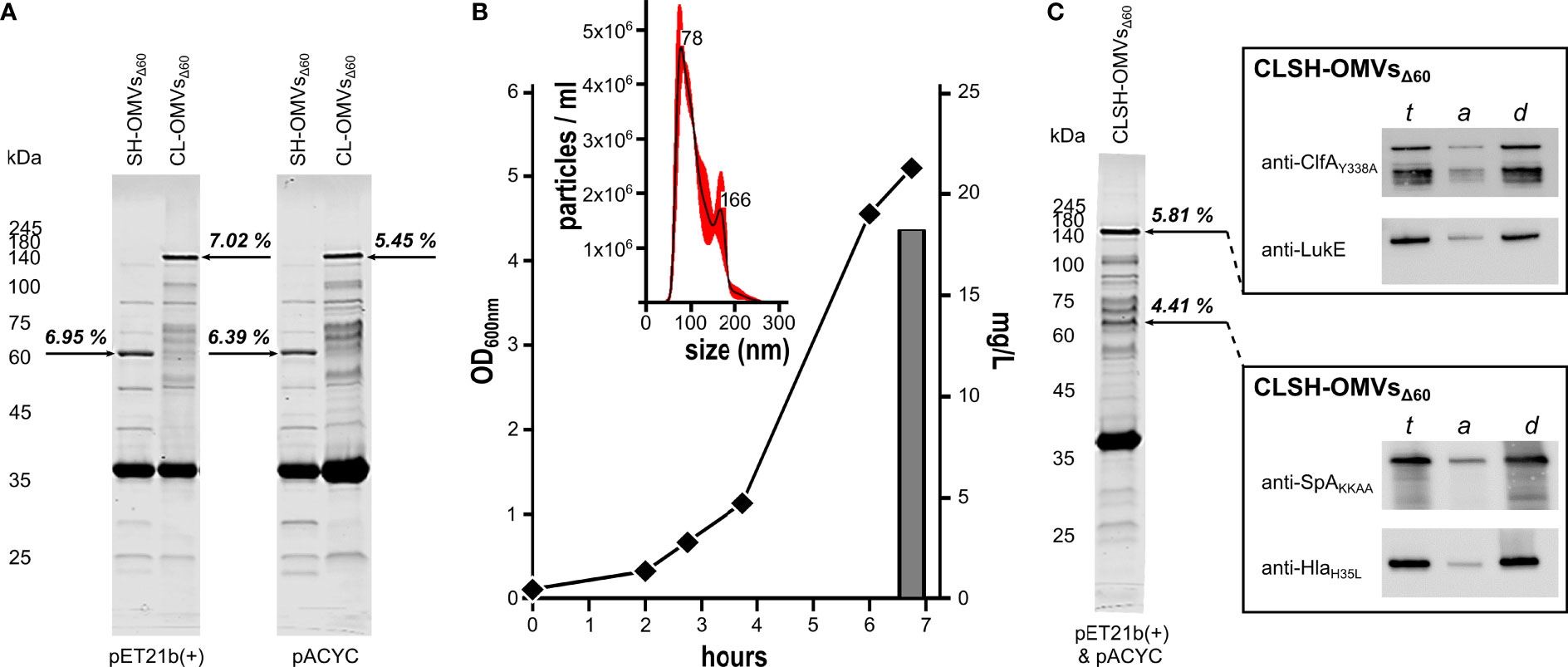
Figure 1 Expression of Staphylococcal antigen fusions in OMVs from E. coli BL21(DE3)Δ60. (A) OMVs from E. coli BL21(DE3)Δ60 strains expressing heterologous antigen fusions SpAKKAA-HlaH35L (SH-OMVsΔ60) and ClfAY338A-LukE (CL-OMVsΔ60) as lipoproteins using either pET21b(+) or pACYC as plasmid backbone were purified from culture supernatants as described in Materials and Methods. Aliquots (10 μg of total OMV proteins) were analyzed by SDS-PAGE. (B) Growth curve, OMV production yield and particle size analysis of CLSH-OMVsΔ60 grown in a 3-L bioreactor (see Materials and Methods for details). The size of CLSH-OMVsΔ60 was determined using a NanoSight NS300 (Malvern Panalytical). The graph shows the average of three independent measurements and standard error as calculated by the manufacturer’s software. Numbers indicate the particle size of main peaks. (C) OMVs from E. coli BL21(DE3)Δ60 co-expressing the two heterologous antigen fusions ClfAY338A-LukE and SpAKKAA-HlaH35L as lipoproteins (CLSH-OMVsΔ60) and analysis of protein lipidation by Triton X-114 extraction of OMV proteins. ClfAY338A-LukE and SpAKKAA-HlaH35L were co-expressed using pET21b(+) and pACYC, respectively, and purified and analyzed by SDS-PAGE as stated for panel (A) For analysis of protein lipidation, CLSH-OMVsΔ60 expressing heterologous protein fusions in the membrane were dissolved by adding 1% Triton X-114 at 4°C and subsequently aqueous and detergent phases were partitioned by centrifugation. Unfractionated total proteins from CLSH-OMVsΔ60 (t), hydrophilic proteins in the aqueous phase (a), and hydrophobic proteins in the detergent phase (d) were precipitated with chloroform-methanol and separated by SDS-PAGE. Finally, proteins were transferred onto nitrocellulose filters and the presence of the heterologous antigen fusions in either the aqueous or detergent phase was detected by Western blot using antigen-specific antibodies. The numbers next to each arrow (A, C) represent the percentage of each recombinant chimera over total OMV protein content as estimated by densitometry using Image Studio Lite Ver 5.2.
pET21b(+) and pACYC plasmids have compatible origins of replication. This prompted us to test whether the co-transformation of E. coli BL21(DE3)Δ60 with two recombinant plasmids, one having the pACYC scaffold and the other the pET21b(+) scaffold, could lead to the co-expression of the two chimeras in the OMVs. E. coli BL21(DE3)Δ60 was co-transformed with the plasmid couple pET(ClfAY338A-LukE)/pACYC(SpAKKAA-HlaH35L). The recombinant strain was grown in liquid culture using a bioreactor, and after 4-hour induction of protein expression with 0.1 mM IPTG, the OMVs, named CLSH-OMVsΔ60, were purified from the culture supernatant (Figure 1B). Particles sizes and protein content of CLSH-OMVsΔ60 were analyzed by NanoSight NS300 and SDS-PAGE, respectively. CLSH-OMVsΔ60 had a mean size of 111.6 +/- 2.1 nm, in line with the size of “empty” OMVsΔ60 previously reported (17) (Figure 1B). As shown in Figure 1C, two protein bands corresponding to the molecular mass of the two chimeras were visible among total OMV proteins. Based on densitometry analysis, the two co-expressed chimeras accumulated in similar amounts, corresponding to 4.4% and 5.8% of total proteins from SpAKKAA-HlaH35L-OMVsΔ60 and ClfAY338A-LukE-OMVsΔ60, respectively.
The fusion of the two chimeras to the lipoprotein leader sequence of Lpp should promote their N-terminal lipidation and subsequent compartmentalization in the OMV membrane (9). To indirectly demonstrate the presence of the acyl groups at the N-terminus of the two chimeras, CLSH-OMVsΔ60 were solubilized at 4°C with a 1% water solution of Triton X-114 and subsequently the sample was warmed to 37°C to partition Triton X-114 into two phases: a detergent-rich ‘hydrophobic’ phase and a detergent-poor ‘hydrophilic’ phase. Membrane proteins, including lipoproteins, typically partition into the hydrophobic phase (49). Aliquots from both aqueous and organic phases were analyzed by Western Blot using antibodies specific for the corresponding S. aureus antigens. As shown in Figure 1C, the two chimeras compartmentalized in the hydrophobic phase.
Next, we tested whether the immunization with CLSH-OMVsΔ60 could elicit antigen-specific antibody responses. Mice were immunized intraperitoneally (i.p.) three times, two weeks apart, with 20 μg of CLSH-OMVsΔ60 + Alum and we determined the antibody titers against each antigen by ELISA using plates coated with the respective purified recombinant proteins. As shown in Figure 2A, immunization elicited antibodies specific for each of the four S. aureus antigens present in the CLSH-OMVsΔ60, while in sera from mice immunized with “empty” OMVs no antigen-specific antibodies were detected (Supplementary Figure 1).
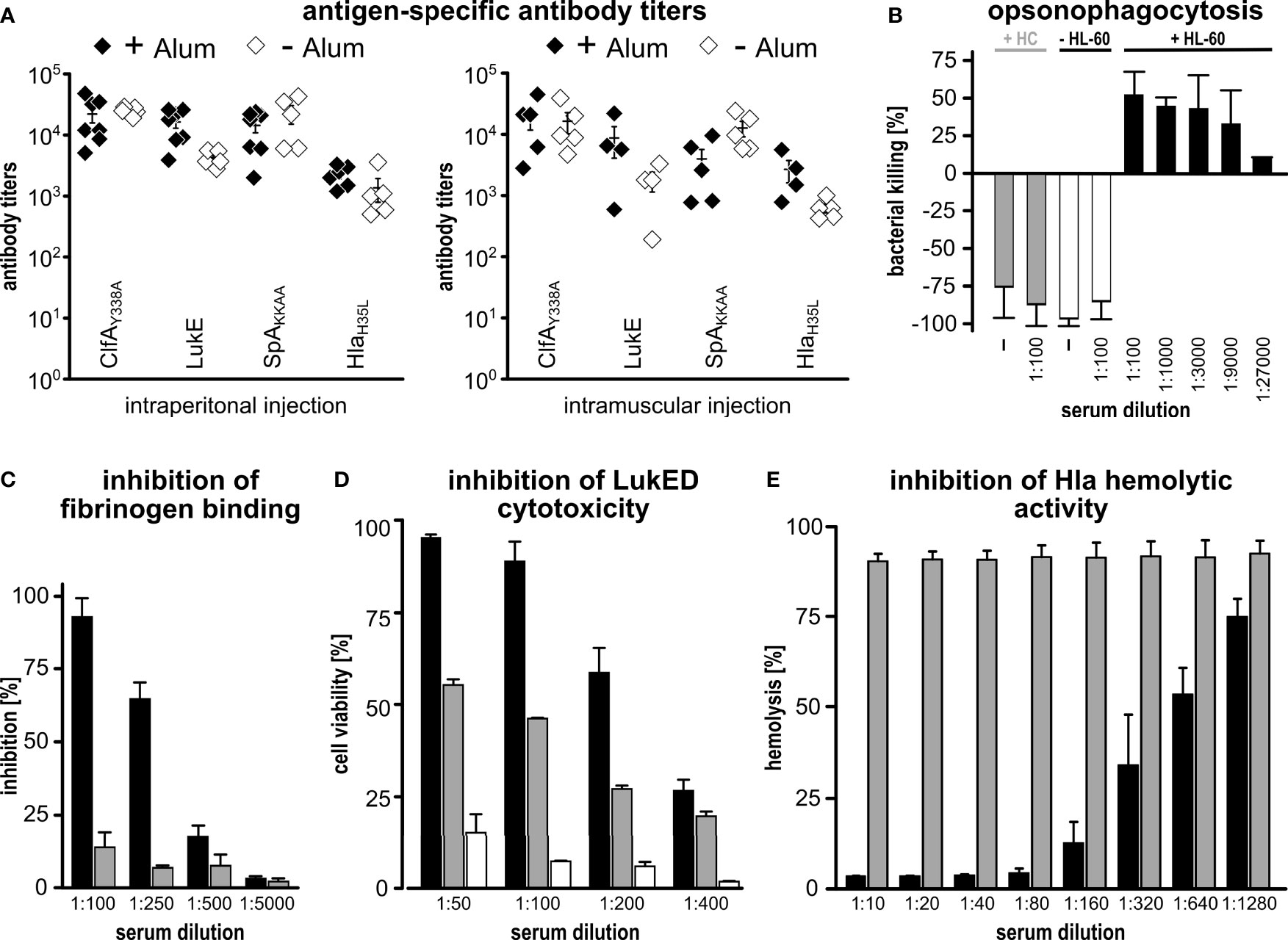
Figure 2 Immunogenicity of CLSH-OMVsΔ60. (A) Antigen-specific antibody titers. Groups of 8 female CD1 mice were immunized i.p. (left panel) or i.m. (right panel) 3 times at 2-week intervals with 20 μg of CLSH-OMVsΔ60 formulated with or without Alum (filled and open symbols, respectively). Sera were collected 7 days after the last immunization and IgG titers were analyzed by ELISA using plates coated with the respective purified recombinant protein (300 ng/well). Each data point represents the antibody titer from a single mouse. Mean ± s.e.m. is shown. (B) Opsonophagocytosis killing (OPK). 500 CFUs of S. aureus Newman strain grown in TSB at 37°C were incubated with serially diluted immune sera for 45 minutes at RT. Then, 3×105 HL-60 cells differentiated into neutrophils and guinea pig complement were added to each reaction for 90 minutes at 37°C (T90). Bacterial killing refers to the CFUs of the negative control (bacteria + HL-60 cells + complement without serum) set as 0% killing and was calculated as follows: [CFUs of the negative control at T90 - CFUs of each sample at T90]/CFUs of the negative control at T90]. Data are reported as mean ± s. d. of two independent experiments. HC: heat-inactivated complement. (C) Inhibition of fibrinogen binding. S. aureus Newman strain (1×107 CFUs/well) was pre-incubated for 30 minutes at RT with serially diluted pooled sera from mice immunized with either 20 μg of “empty” OMVsΔ60 (grey bars) or 20 μg of CLSH-OMVsΔ60 (black bars). Mixtures were transferred to a 96-well plate previously coated with human fibrinogen (10 µg/ml) and incubated for 1 hour at 37°C. Adherent bacteria were fixed with formaldehyde and then stained with crystal violet. Adherence to fibrinogen was calculated as a percentage of values measured in control wells lacking serum (= 100%), and inhibition of binding to fibrinogen was calculated by subtracting the adherence percentage values from the control (= 100% - x %). The graph shows the mean ± s. d. of three independent experiments. (D) Inhibition of leukocidin-mediated cytotoxicity. HL-60 cells differentiated into neutrophils (5×105/well) were incubated for 24 hours at 37°C with LukED (200 nM) in presence of serially two-fold diluted sera from mice immunized with 20 μg of CLSH-OMVsΔ60 (black bars), “empty” OMVsΔ60 (grey bars) or with Alum (white bars). For assessment of cell viability, the XTT Assay reagent was added and after 16-hour incubation at 37°C the absorbance was read at 470 nm. Data are reported as mean ± s. d. of two independent experiments. (E) Inhibition of Hla-mediated hemolysis. Serially two-fold diluted sera from mice immunized with either 20 μg of “empty” OMVsΔ60 (grey bars) or 20 μg of CLSH-OMVsΔ60 (black bars) were pre-incubated with recombinant Hla for 20 minutes at RT and then with rabbit erythrocytes for 30 minutes at 37°C. Hla hemolytic activity was calculated as percentage of hemolytic activity obtained for rabbit erythrocytes incubated with water (100% hemolysis). The graph shows the mean ± s. d. of three independent experiments.
We also tested the role of Alum and the route of immunization in determining the level of antibody titers. To this aim, we repeated the i.p. immunization in the absence of Alum, and we immunized mice intramuscularly (i.m.) with 20 μg of CLSH-OMVsΔ60 in the presence or absence of Alum. Sera from immunized mice were analyzed by ELISA (Figure 2A). In the presence of Alum, the route of immunization did not substantially influence the antibody titers, with the exception for SpAKKAA antibody titers, which were approximately 3.5-fold lower when the vaccine was administered intramuscularly. As far as the influence of Alum is concerned, only a marginal difference was observed when mice were immunized i.p., while titers against HlaH35L and LukE were enhanced by Alum in i.m.-immunized mice. Finally, IgG titers against SpAKKAA appeared to be higher in the absence of Alum when the i.m. route was used.
Next we evaluated the IgG isotypes as an indication of the type of immune response (Th1 vs. Th2-skewed response) induced by CLSH-OMVsΔ60 immunization. The sera from animals immunized i.p. in the presence of Alum were analyzed by ELISA using anti-mouse anti-IgG2a and anti-IgG1 antibodies as secondary antibodies and coating plates with ClfAY338A and SpAKKAA, the two N-terminal antigens of the chimeras. As shown in Supplementary Figure 2, and in line with what previously observed (8), IgG2a were abundantly present in the sera of mice immunized with CLSH-OMVsΔ60, indicating that the immunization elicited a Th1-skewed response.
Finally, we tested whether the antigen-specific antibodies induced by immunization with CLSH-OMVsΔ60 had functional activities. First, we analyzed the capacity of differentiated HL-60 cells to kill the S. aureus Newman strain in the presence of different dilutions of mouse sera and guinea pig complement. As shown in Figure 2B, the sera from mice immunized with CLSH-OMVsΔ60 rescued the HL-60-mediated bacterial killing in a dose-dependent manner.
Next, we tested whether the same sera could inhibit the biological activity of ClfA, LukED and Hla. To follow ClfA inhibition, plates were coated with fibrinogen and the binding of S. aureus Newman strain was analyzed in the presence or absence of sera from CLSH-OMVsΔ60-immunized mice. In the case of LukED, a toxin which can kill activated HL-60 cells, anti-CLSH-OMVsΔ60 sera and HL-60 cells were incubated together with purified LukED. Finally, the inhibition of the hemolytic activity was tested by incubating the sera with purified Hla and rabbit erythrocytes. As shown in Figures 2C–E, the pool of mouse sera from the CLSH-OMVsΔ60-immunized group inhibited the activities of all three virulence factors. In the case of LukED inhibition, “empty” OMVsΔ60 gave some neutralization, but well below the inhibition of sera from CLSH-OMVΔ60-immunized mice. This effect could be due to the presence of some cross-reacting antibodies elicited by OMVsΔ60.
Finally, we followed the protective activity of CLSH-OMVsΔ60 immunization in mice challenged with S. aureus Newman strain. In particular, three challenge models were used: the sepsis model, the skin model and the kidney abscess model. For the sepsis model, mice were immunized three times with 20 μg of CLSH-OMVsΔ60 and 14 days after the last immunization the animals received 3×108 colony forming units (CFUs) of bacteria (i.p.). Animal health was followed every day over a period of seven days using a 1 to 4 pain scale (see Material and Methods). Animals that reached the pain value of 4 were sacrificed since they reached the “near mortality point” and would have died within 24 hours. On the other hand, those animals that maintained a score lower than 4, fully recovered within 7 days. Figure 3A reports the data of mice immunized with Alum alone, “empty” OMVsΔ60 and CLSH-OMVsΔ60. As shown in the figure, immunization with “empty” OMVsΔ60 conferred a certain level of protection, with 50% of the animals that did not reach pain value 4 and survived. By contrast, 100% protection was observed in the group of mice immunized with CLSH-OMVsΔ60 and none of the animals experienced a pain value higher than 2. The protective activity of the CLSH-OMVsΔ60 was also tested in the skin infection model and in the kidney abscess model. As shown in Figures 3B, C, CLSH-OMVsΔ60 vaccination was highly protective in both models. In particular, differently from Alum-immunized and “empty” OMVsΔ60-immunized animals, mice immunized with CLSH-OMVsΔ60 and subsequently s.c. challenged with 5×107 CFUs developed only a mild and transient skin abscess with no dermonecrotic lesions. The differences between vaccinated and control animals was statistically significant (p < 0.001) as evaluated by means of repeated ANOVA measures using the R base function aov(). As far as the kidney abscess model is concerned, according to which 107 CFUs were given intravenously and bacteria were counted in the kidneys four days after challenge, the CFUs counts in 13 out of 16 immunized mice were below the detection limit (102 CFUs). As previously shown (9), immunization with “empty” OMVsΔ60 also resulted in a substantial level of protection (62.5%), in line with the protective role of an innate-type immune response induced by the OMVs.
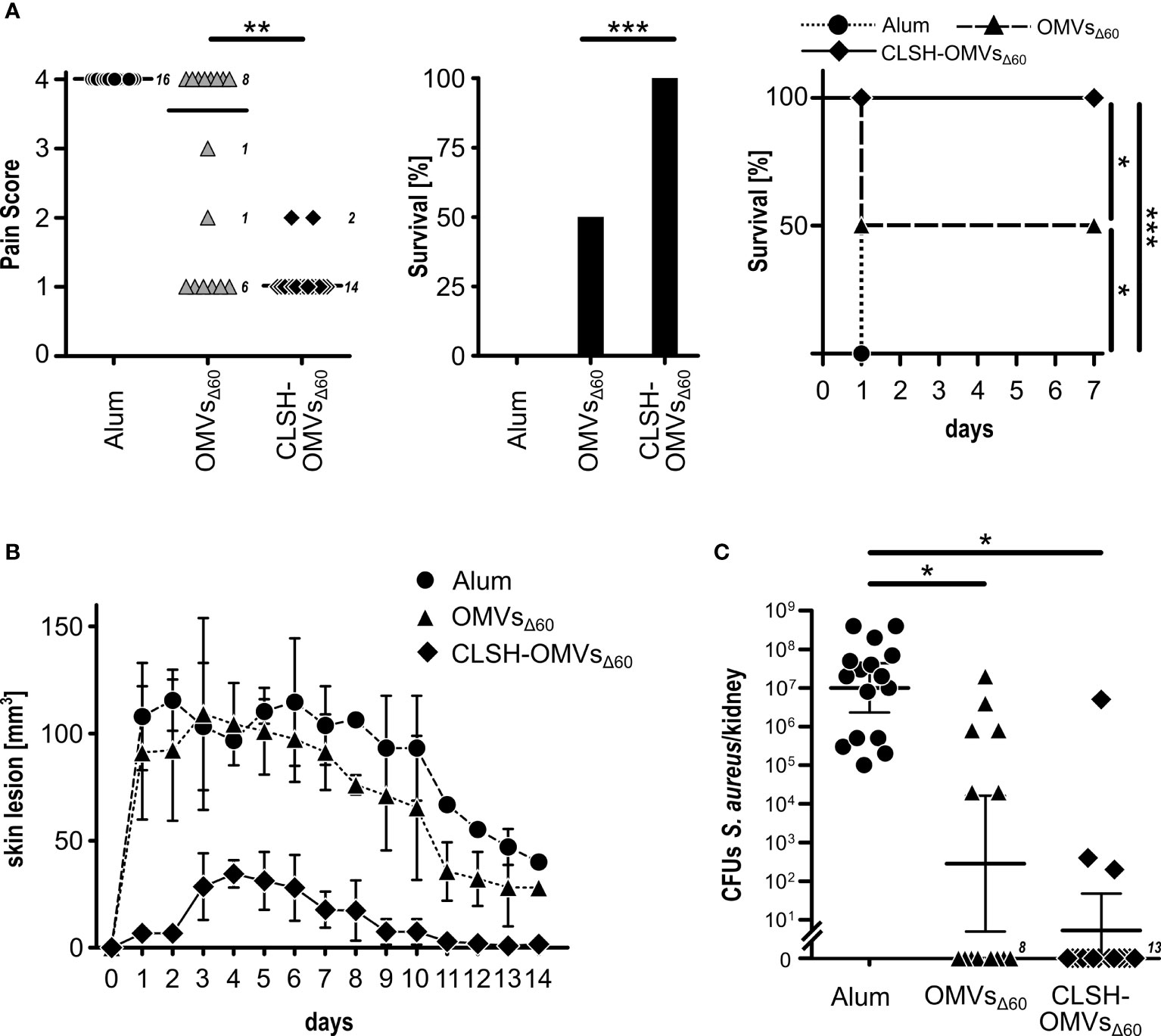
Figure 3 In vivo protective activity of OMVsΔ60 expressing S. aureus antigens. (A) Sepsis model. Groups of 16 female CD1 mice were immunized three times at 2-week intervals with Alum alone (circles), “empty” OMVsΔ60 (triangles) or CLSH-OMVsΔ60 (diamonds), formulated in Alum. After 2 weeks, mice were infected i.p. with 3×108 CFUs of S. aureus Newman strain. Animal health was monitored over a period of 7 days, assigning a “pain score” from 1 to 4. Animals which reached a pain score = 4 were sacrificed. The three graphs show the pain score (left), survival at day 7 (center) and the survival over time (Kaplan-Meier curve) (right). See text for definition of “survival”. Statistical analysis for the pain score plot was performed using Mann-Whitney test. Median is shown. **P = 0.0015. Statistical analysis for the Kaplan–Meier plot was performed using the log rank test. *P = 0.0253; ***P = 0.0001. (B) Skin model. Groups of 16 CD1 female mice were immunized three times at 2-week intervals with Alum alone (circles), “empty” OMVsΔ60 (triangles) and CLSH-OMVsΔ60 (diamonds). At day 14 after the third immunization, mice were s.c. infected with 5×107 CFUs of S. aureus Newman strain. Abscess size was monitored once a day for 14 days. Mean ± s. d. is shown. Skin abscess areas of vaccinated and control animals were tested for significance by means of repeated ANOVA measures using the R base function aov(). (C) Renal abscess model. Groups of 16 CD1 female mice were immunized three times at 2-week intervals with Alum alone (circles), “empty” OMVsΔ60 (triangles) and CLSH-OMVsΔ60 (diamonds). Ten days after the last immunization, mice were infected i.v. with a sub-lethal dose of S. aureus Newman strain (1×107 CFUs) and 4 days afterward, mice were sacrificed, kidneys collected and homogenized in PBS, and aliquots were plated on agar media for CFUs determination. Statistical analysis was performed using two-tailed Student’s t-test. Geometric mean ± 95% confidence interval is shown. *P < 0.05.
The results described above demonstrate the feasibility of co-expressing the two chimeras SpAKKAA-HlaH35L and ClfAY338A-LukE in the same OMVs preparation. For a broad applicability of the technology it would be important to prove that OMVs can be successfully decorated with other chimera combinations. Therefore, we created three additional chimeras by combining LukE and HlaH35L with FhuD2, another virulence factor involved in S. aureus pathogenesis (50), and by fusing together the two toxins LukE and HlaH35L. Three plasmids were generated pET(FhuD2-LukE), pACYC(FhuD2-HlaH35L) and pET(LukE-HlaH35L). The plasmids were used to transform E. coli BL21(DE3)Δ60 strain and one transformant colony of each transformation was used to produce OMVs. As shown in Figure 4A, the three chimeras efficiently accumulated in the vesicular compartment. Next, we co-transformed the strain with the plasmid couples pET(FhuD2-LukE)/pACYC(SpAKKAA-HlaH35L) and pET(ClfAY338A-LukE)/pACYC(FhuD2-HlaH35L) and the successful compartmentalization of the two chimera couples in FLSH-OMVsΔ60 and CLFH-OMVsΔ60 was confirmed by SDS-PAGE (Figure 4A). Finally, we used CLFH-OMVsΔ60 to immunize mice and we analyzed the antibody titers against all antigens. As shown in Figure 4B, the engineered OMVs elicited antibodies specific for all four chimera antigens.
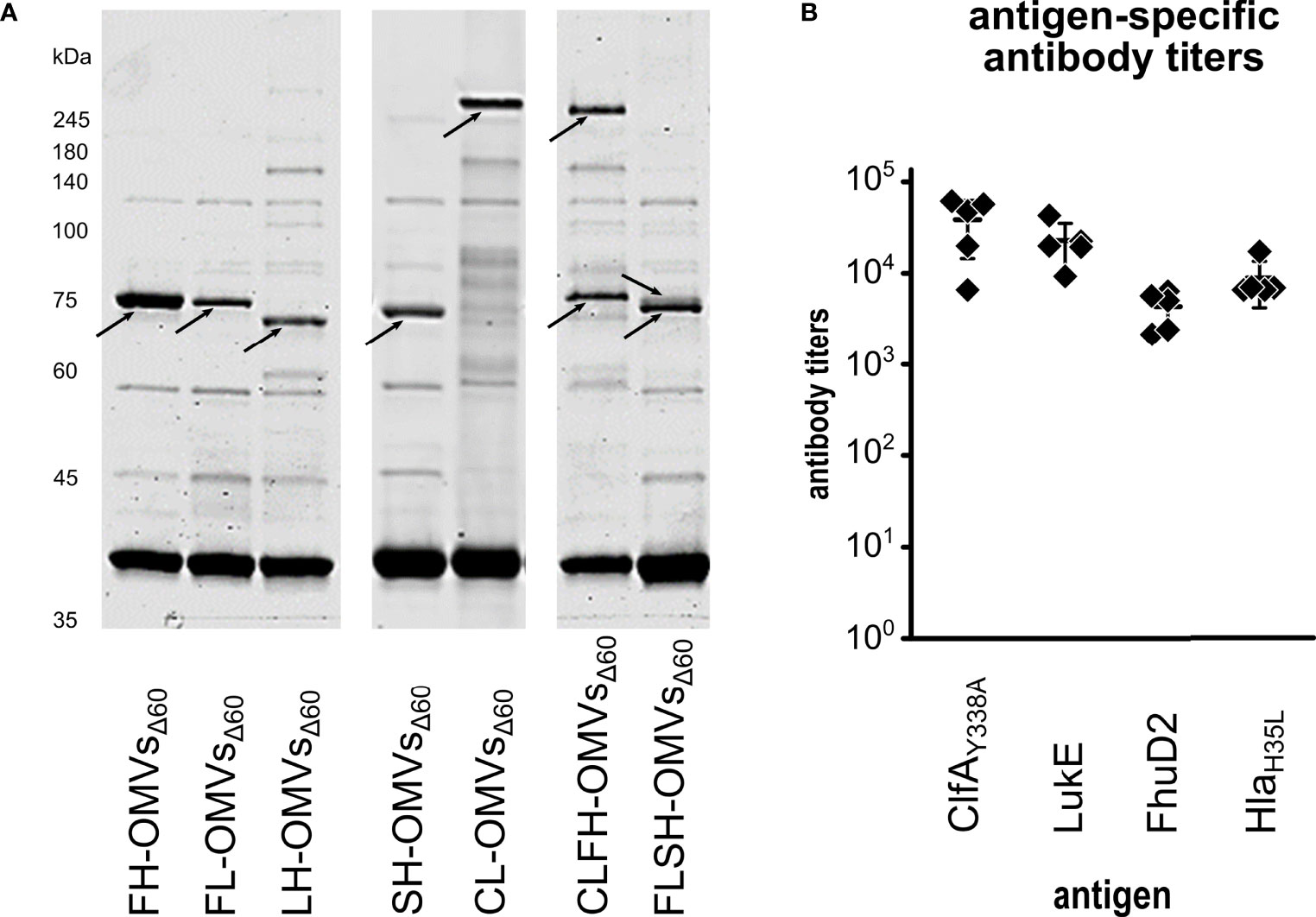
Figure 4 Expression of chimeras in OMVsΔ60. (A) E. coli BL21(DE3)Δ60 was transformed with plasmids expressing the following chimeras: FhuD2-HlaH35L, FhuD2-LukE, LukE-HlaH35L, SpAKKAA-HlaH35L and ClfAY338A-LukE. One recombinant clone from each transformation was inoculated in liquid culture and the corresponding OMVs, named FH-OMVsΔ60, FL-OMVsΔ60, LH-OMVsΔ60, SH-OMVsΔ60 and CL-OMVsΔ60, respectively, were purified from each culture supernatants. Purified OMVs were analyzed by SDS-PAGE (10 μg total OMV proteins) (left panel). The right panel reports the SDS-PAGE analysis of OMVs from E. coli BL21(DE3)Δ60 co-transformed with plasmids expressing either ClfAY338A-LukE and FhuD2-HlaH35L (CLFH-OMVsΔ60) or FhuD2-LukE and SpAKKAA-HlaH35L (FLSH-OMVsΔ60) using pET21b(+) and pACYC, respectively (see Materials and Methods for details). (B) A group of five mice were immunized three times two weeks apart with CLFH-OMVsΔ60. After two weeks from the last immunization sera were collected and pooled and the ELISA titers were measured coating the plates with recombinant proteins.
On the basis of these experiments we concluded that our OMV engineering strategy has a broad applicability and can be exploited for the preparation of multi-component vaccines against one or more pathogens.
One of the key issues in vaccinology is to find ways to make vaccine production as economic and easy as possible to guarantee that vaccines can reach out even the poorest people and to allow low income countries to establish their own production plans and distribute vaccines to their population.
In this respect, vaccines based on OMVs engineered with heterologous antigens appear to be attractive. Their built-in adjuvanticity and the ease with which they can be produced by using a single ultrafiltration step from bacterial culture supernatants make their production process inexpensive and simple. However, vaccines against recalcitrant pathogens usually require more than one antigen. This complicates the production process, even in the case of the OMV-based vaccines. Multi-component vaccines would require the purification of more than one engineered OMVs and their subsequent combination to make the final formulation (9).
In this work we describe a new strategy to co-express four antigens in the same OMVs, thus making the preparation of multi-component vaccines simple and inexpensive.
Using four important S. aureus virulence factors as a model, we first showed that OMVs can be engineered with two antigens by creating two-protein chimeras. Such chimeras can be efficiently expressed in the membrane of OMVs exploiting the lipoprotein transport machinery. From an immunological standpoint, protein fusions are attractive since each antigen of the fusion takes advantage of the additional T cell help provided by the T cell epitopes of the partner. This usually results in enhanced antigen-specific immunogenicity and antibody titers (51).
Next, we showed that if two chimeras are expressed from two plasmids with compatible origins of replication in the same OMV-producing strain, vesicles containing both chimeras can be obtained. In line with the evidence that the loading capacity of OMVsΔ60 is limited and cannot exceed a certain level (17), the expression of each co-expressed chimera was slightly reduced with respect to the same chimera expressed alone. However, since our OMV-producing strain has the unique property to release vesicles with high yield and improved loading capacity, being deprived of 60 endogenous proteins, the amount of each chimeric protein (approx. 5% of total OMV proteins) was sufficiently high to guarantee excellent immunogenicity. The immunization with 20 μg of CLSH-OMVsΔ60 elicited antibody titers against each antigen similar to what obtained with the combination of OMVs (5 μg each, 20 μg of total OMVs proteins) expressing the single antigens (9). Since we previously showed that 5 μg of OMVs engineered with single antigens are sufficient to reach the plateau of antigen-specific antibody titers, 20 μg of CLSH-OMVsΔ60 should represent an appropriate dose to elicit optimal immune responses.
To demonstrate the broad applicability of the approach, we created three additional chimeras and we showed that all of them could be successfully expressed in OMVs. Moreover, by using the two plasmid expression strategy, we confirmed that two chimeras could be efficiently co-expressed in the same OMVs and that such OMVs elicited immune responses against all four antigens. Therefore, we believe that our approach represents a step forward to make multi-component vaccines, including protective antigens from the same pathogen as well as from different pathogens.
A second important result of our work is the development of a new S. aureus vaccine candidate ready to move to development.
S. aureus is a Gram-positive organism, which colonizes the anterior nares and the skin of approximately one third of individuals at any time (16). Usually colonization is asymptomatic. However, in patients with immunological or barrier defects, S. aureus can become invasive and responsible for severe diseases (52). Moreover, highly pathogenic strains have recently emerged, many of which are resistant to most antibiotics and have the ability to cause diseases in otherwise healthy individuals (53). In the USA, approximately 3.4 million community-acquired diseases and 340.000 hospital acquired-diseases occur annually, leading to more than 30.000 deaths (54).
When S. aureus becomes invasive, it expresses more than 35 immune evasion molecules and various virulence factors (12). Moreover, once phagocytosed, S. aureus has the ability to avoid killing, thus using phagocytes as “Trojan Horses” to disseminate itself inside the host (15, 16). This peculiarity makes the development of an effective vaccine particularly challenging. Three Phase III S. aureus vaccine trials have been reported (55–57). However, the efficacy data of these vaccines, all formulated without adjuvants, were largely disappointing. The vaccine failures can be attributed to an inappropriate antigen selection in terms of both quality and quantity. In this respect, the redundancy of virulent factors expressed by the different S. aureus isolates represents a serious challenge in establishing which antigens are necessary and sufficient to elicit a broadly protective immunity. Moreover, to counteract the ability of S. aureus to survive inside host cells, a vaccine should elicit a Th1/Th17-skewed adaptive immune response and a strong innate immunity, the latter necessary to enhance the killing capacity of phagocytic cells. To achieve this, a vaccine should combine protective antigens with a proper adjuvant and, to keep innate immunity constantly alerted, the vaccine should be administered repeatedly, particularly when the risk of infection increases, for instance during hospitalization.
We believe that CLSH-OMVsΔ60 have many properties that make them attractive as vaccine candidate.
The vaccine elicits both opsonophagocytic antibodies and functional antibodies against four important virulence factors. Opsonophagocytosis is considered to be an important prerequisite of S. aureus vaccines (55–57), and the sera from mice immunized with CLSH-OMVsΔ60 have the ability of killing S. aureus in vitro in the presence of phagocytic cells and complement. Such killing activity is likely to be mediated by the recognition of SpA and ClfA, which are both surface-exposed proteins. Moreover, anti-CLSH-OMVsΔ60 antibodies inhibited the cytotoxic activity of Hla and LukED, two potent toxins that S. aureus releases to damage tissues and to kill different subsets of immune cells. Finally, the sera from immunized animals neutralized the fibrinogen-mediated adhesion of S. aureus to tissues and, although not directly tested, are expected to inhibit the SpA-specific binding to immunoglobulins as previously described (23, 26, 27).
The single OMV vaccine here described appears to provide a protection which is at least as good as the protection we observed with our five-OMVs formulation, which included five vesicles each engineered with one of the antigens HlaH35L, LukE, SpAKKAA, FhuD2 and CsaA1 (9). Interestingly, our new candidate shares three antigens with the previously published vaccine (9), but differs for the replacement of Csa1A and FhuD2 with ClfAY338A. This data would corroborate the notion that ClfA is an important virulence factor whose inhibition is particularly relevant in the early stage of infection (32).
A second important property of our CLSH-OMVsΔ60 vaccine is its capacity to elicit a Th1/Th17-skewed adaptive immunity and a strong innate immunity. According to our opinion, this is an important feature since, to be efficiently neutralized, S. aureus, which has the unique capacity to survive within phagocytic cells, should encounter a full-blown adaptive and innate immunity at the time of the host invasion. This would imply that CLSH-OMVsΔ60 should be administered whenever the risk of infection increases, as it is for instance the case of hospitalized patients awaiting surgery.
The role of vaccine-induced innate immunity is gaining increasing attention in the light of the evidence that certain infections and vaccinations can induce a broad protection against other pathogens through innate immune mechanisms (58, 59). Strikingly, innate immunity appears to have memory characteristics (referred to as “trained innate immunity”), a feature previously thought to be an exquisite property of adaptive immunity [for an excellent review, see (60)]. The role and the duration of OMV-induced innate immunity in humans is certainly a topic of great interest that should be addressed in future clinical trials.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the Animal Ethical Committees of the University of Trento, the Animal Ethics Committees of Toscana Life Sciences and the Italian Ministry of Health.
GG conceived and designed the study, analyzed data and wrote the manuscript. EK, IR, CA, and CI: OMV engineering. EK, IR, CA, LF, IZ, and EC: OMV production and purification. EK, AGr, MT, LFa, and AG: OMV immunogenicity. MT and LFa: mouse models. AG, AGr and FB: OPK assay. AG and AGr: neutralization assays for ClfA, LukE and Hla. EK, AG, and MT: ELISA. EK and AG: cloning and purification of recombinant proteins. EK and AG edited manuscript and Figures. All authors contributed to the article and approved the submitted version.
The work has been financially supported by the Advanced European Research Council grant OMVAC 340915 and the Advanced European Research Council grant Vaccibiome 834634 (both assigned to GG).
Author FB was employed by company GlaxoSmithKline Vaccines. GG, EK, IZ, CI, and LFa are coinventors of a patent on OMVs. AGr and GG are involved in a biotech company interested in exploiting the OMV platform.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors wish to thank Riccardo Corbellari, Marialucia Massaro, Chiara Lago and Rubens Begaj for their contribution in the construction of many plasmids to co-express single antigens during their internships. Furthermore, we gratefully thank Dr. Silvia Valensin, Dr. Erika Bellini and Dr. Antonella De Rosa for their support in animal studies. We also highly appreciate the help of Dr. Leonardo Caproni (Institute of Life Sciences, Scuola Universitaria Superiore Sant’Anna, Pisa) for the statistical analysis of the skin infection model.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.752168/full#supplementary-material
Supplementary Figure 1 | Analysis of antigen-specific antibody titer curves. Sera from mice immunized i.p. with CLSH-OMVsΔ60 (squares) or “empty” OMVsΔ60 (triangles) both with Alum were tested for antibody titers by ELISA using plates coated with recombinant proteins (300 ng/well). Each data point represents the media (n = 8) of absorbance at 405 nm at the respective serum dilution. Mean ± s.e.m. is shown.
Supplementary Figure 2 | Antigen-specific antibody titers of IgG isotypes. Sera from mice immunized i.p. with CLSH-OMVsΔ60 + Alum were tested for antibody titers of IgG isotypes (IgG2a and IgG1) by ELISA using plates coated with recombinant ClfAY338A and SpAKKAA (300 ng/well). Each data point represents the antibody titer from a single mouse. Geometric mean ± 95% CI is shown.
1. Plotkin SA, Orenstein WA, Offit PA, Edwards KM eds. Vaccines. 7th ed. Philadelphia: Elsevier (2018).
2. McCarthy PC, Sharyan A, Moghaddam LS. Meningococcal Vaccines: Current Status and Emerging Strategies. Vaccines (2018) 6:12. doi: 10.3390/vaccines6010012
3. Ellis TN, Kuehn MJ. Virulence and Immunomodulatory Roles of Bacterial Outer Membrane Vesicles. Microbiol Mol Biol Rev (2010) 74:81–94. doi: 10.1128/MMBR.00031-09
4. Klimentovà J, Stulík J. Methods of Isolation and Purification of Outer Membrane Vesicles From Gram-Negative Bacteria. Microbiol Res (2015) 170:1–9. doi: 10.1016/j.micres.2014.09.006
5. Kesty NC, Kuehn MJ. Incorporation of Heterologous Outer Membrane and Periplasmic Proteins Into Escherichia Coli Outer Membrane Vesicles. J Biol Chem (2004) 279:2069–76. doi: 10.1074/jbc.M307628200
6. Baker JL, Chen L, Rosenthal JA, Putnam D, DeLisa MP. Microbial Biosynthesis of Designer Outer Membrane Vesicles. Curr Opin Biotechnol (2014) 29:76–84. doi: 10.1016/j.copbio.2014.02.018
7. Gerritzen MJH, Martens DE, Wijffels RH, van der Pol L, Stork M. Bioengineering Bacterial Outer Membrane Vesicles as Vaccine Platform. Biotechnol Adv (2017) 35:565–74. doi: 10.1016/j.biotechadv.2017.05.003
8. Fantappiè L, de Santis M, Chiarot E, Carboni F, Bensi G, Jousson O, et al. Antibody-Mediated Immunity Induced by Engineered Escherichia Coli OMVs Carrying Heterologous Antigens in Their Lumen. J Extracell Vesicles (2014) 3:24015. doi: 10.3402/jev.v3.24015
9. Irene C, Fantappiè L, Caproni E, Zerbini F, Anesi A, Tomasi M, et al. Bacterial Outer Membrane Vesicles Engineered With Lipidated Antigens as a Platform for Staphylococcus Aureus Vaccine. Proc Natl Acad Sci USA (2019) 116:21780–8. doi: 10.1073/pnas.1905112116
10. Grandi A, Tomasi M, Zanella I, Ganfini L, Caproni E, Fantappiè L, et al. Synergistic Protective Activity of Tumor-Specific Epitopes Engineered in Bacterial Outer Membrane Vesicles. Front Oncol (2017) 7:253. doi: 10.3389/fonc.2017.00253
11. Daleke-Schermerhorn MH, Felix T, Soprova Z, ten Hagen-Jongman CM, Vikström D, Majlessi L, et al. Decoration of Outer Membrane Vesicles With Multiple Antigens by Using an Autotransporter Approach. Appl Environ Microbiol (2014) 80:5854–65. doi: 10.1128/AEM.01941-14
12. Foster TJ. Immune Evasion by Staphylococci. Nat Rev Microbiol (2005) 3:948–58. doi: 10.1038/nrmicro1289
13. Bagnoli F, Fontana MR, Soldaini E, Mishra RPN, Fiaschi L, Cartocci E, et al. Vaccine Composition Formulated With a Novel TLR7-Dependent Adjuvant Induces High and Broad Protection Against Staphylococcus Aureus. Proc Natl Acad Sci USA (2015) 112:3680–5. doi: 10.1073/pnas.1424924112
14. Scully IL, Timofeyeva Y, Illenberger A, Lu P, Liberator PA, Jansen KU, et al. Performance of a Four-Antigen Staphylococcus Aureus Vaccine in Preclinical Models of Invasive S. Aureus Disease. Microorganisms (2021) 9:177. doi: 10.3390/microorganisms9010177
15. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal Manipulation of Host Immune Responses. Nat Rev Microbiol (2015) 13:529–43. doi: 10.1038/nrmicro3521
16. Bagnoli F, Rappuoli R, Grandi G eds. Staphylococcus Aureus. Cham: Springer International Publishing AG (2017).
17. Zanella I, König E, Tomasi M, Gagliardi A, Frattini L, Fantappiè L, et al. Proteome-Minimized Outer Membrane Vesicles From Escherichia Coli as a Generalized Vaccine Platform. J Extracell Vesicles (2021) 10:e12066. doi: 10.1002/jev2.12066
18. Klock HE, Lesley SA. The Polymerase Incomplete Primer Extension (PIPE) Method Applied to High-Throughput Cloning and Site-Directed Mutagenesis. Methods Mol Biol (2009) 498:91–103. doi: 10.1007/2F978-1-59745-196-3_6
19. Fantappiè L, Irene C, De Santis M, Armini A, Gagliardi A, Tomasi M, et al. Some Gram-Negative Lipoproteins Keep Their Surface Topology When Transplanted From One Species to Another and Deliver Foreign Polypeptides to the Bacterial Surface. Mol Cell Proteomics (2017) 16:1348–64. doi: 10.1074/mcp.M116.065094
20. Fattom A, Schneerson R, Szu SC, Vann WF, Shiloach J, Karakawa WW, et al. Synthesis and Immunologic Properties in Mice of Vaccines Composed of Staphylococcus Aureus Type 5 and Type 8 Capsular Polysaccharides Conjugated to Pseudomonas Aeruginosa Exotoxin a. Infect Immun (1990) 58:2367–74. doi: 10.1128/iai.58.7.2367-2374.1990
21. Fattom A, Shiloach J, Bryla D, Fitzgerald D, Pastan I, Karakawa WW, et al. Comparative Immunogenicity of Conjugates Composed of the Staphylococcus Aureus Type 8 Capsular Polysaccharide Bound to Carrier Proteins by Adipic Acid Dihydrazide or N-Succinimidyl-3-(2-Pyridyldithio)Propionate. Infect Immun (1992) 60:584–9. doi: 10.1128/iai.60.2.584-589.1992
22. Fattom A, Fuller S, Propst M, Winston S, Muenz L, He D, et al. Safety and Immunogenicity of a Booster Dose of Staphylococcus Aureus Types 5 and 8 Capsular Polysaccharide Conjugate Vaccine (StaphVAX ®) in Hemodialysis Patients. Vaccine (2004) 23:656–63. doi: 10.1016/j.vaccine.2004.06.043
23. Kim HK, Falugi F, Thomer L, Missiakas DM, Schneewind O. Protein A Suppresses Immune Responses During Staphylococcus Aureus Bloodstream Infection in Guinea Pigs. MBio (2015) 6:e02369–14. doi: 10.1128/mBio.02369-14
24. Goodyear CS, Silverman GJ. Staphylococcal Toxin Induced Preferential and Prolonged In Vivo Deletion of Innate-Like B Lymphocytes. Proc Natl Acad Sci USA (2004) 101:11392–7. doi: 10.1073/pnas.0404382101
25. Shi M, Chen X, Sun Y, Kim HK, Schneewind O, Missiakas D. A Protein A Based Staphylococcus Aureus Vaccine With Improved Safety. Vaccine (2021) 39:3907–15. doi: 10.1016/j.vaccine.2021.05.072
26. Kim HK, Emolo C, DeDent AC, Falugi F, Missiakas DM, Schneewind O. Protein A-Specific Monoclonal Antibodies and Prevention of Staphylococcus Aureus Disease in Mice. Infect Immun (2012) 80:3460–70. doi: 10.1128/IAI.00230-12
27. Kim HK, Cheng AG, Kim H-Y, Missiakas DM, Schneewind O. Nontoxigenic Protein A Vaccine for Methicillin-Resistant Staphylococcus Aureus Infections in Mice. J Exp Med (2010) 207:1863–70. doi: 10.1084/jem.20092514
28. Mcdevitt D, Nanavaty T, Housh-Pompeo K, Bell E, Turner N, Mcintire L, et al. Characterization of the Interaction Between the Staphylococcus Aureus Clumping Factor (ClfA) and Fibrinogen. Eur J Biochem (1997) 247:416–24. doi: 10.1111/j.1432-1033.1997.00416.x
29. Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. Genetic Requirements for Staphylococcus Aureus Abscess Formation and Persistence in Host Tissues. FASEB J (2009) 23:3393–404. doi: 10.1096/fj.09-135467
30. Josefsson E, Higgins J, Foster TJ, Tarkowski A. Fibrinogen Binding Sites P336 and Y338 of Clumping Factor A are Crucial for Staphylococcus Aureus Virulence. PloS One (2008) 3:e2206. doi: 10.1371/journal.pone.0002206
31. Hawkins J, Kodali S, Matsuka YV, McNeil LK, Mininni T, Scully IL, et al. A Recombinant Clumping Factor A-Containing Vaccine Induces Functional Antibodies to Staphylococcus Aureus That are Not Observed After Natural Exposure. Clin Vaccine Immunol (2012) 19:1641–50. doi: 10.1128/CVI.00354-12
32. Anderson AS, Scully IL, Buurman ET, Eiden J, Jansen KU. Staphylococcus Aureus Clumping Factor a Remains a Viable Vaccine Target for Prevention of S. Aureus Infection. MBio (2016) 7:e00225. doi: 10.1128/mBio.00225-16
33. McAdow M, Kim HK, DeDent AC, Hendrickx APA, Schneewind O, Missiakas DM. Preventing Staphylococcus Aureus Sepsis Through the Inhibition of its Agglutination in Blood. PloS Pathog (2011) 7:e1002307. doi: 10.1371/journal.ppat.1002307
34. Bhakdil S, Tranum-Jensen J. Alpha-Toxin of Staphylococcus Aureus. Microbiol Rev (1991) 55:733–51. doi: 10.1128/mr.55.4.733-751.1991
35. Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of Staphylococcal α-Hemolysin, a Heptameric Transmembrane Pore. Science (1996) 274:1859–66. doi: 10.1126/science.274.5294.1859
36. Berube B, Wardenburg J. Staphylococcus Aureus α-Toxin: Nearly a Century of Intrigue. Toxins (Basel) (2013) 5:1140–66. doi: 10.3390/toxins5061140
37. Wardenburg JB, Schneewind O. Vaccine Protection Against Staphylococcus Aureus Pneumonia. J Exp Med (2008) 205:287–94. doi: 10.1084/jem.20072208
38. DeLeo FR, Kennedy AD, Chen L, Wardenburg JB, Kobayashi SD, Mathema B, et al. Molecular Differentiation of Historic Phage-Type 80/81 and Contemporary Epidemic Staphylococcus Aureus. Proc Natl Acad Sci USA (2011) 108:18091–6. doi: 10.1073/pnas.1111084108
39. Wilke GA, Wardenburg JB. Role of a Disintegrin and Metalloprotease 10 in Staphylococcus Aureus α-Hemolysin - Mediated Cellular Injury. Proc Natl Acad Sci USA (2010) 107:13473–8. doi: 10.1073/pnas.1001815107
40. Inoshima N, Wang Y, Wardenburg JB. Genetic Requirement for ADAM10 in Severe Staphylococcus Aureus Skin Infection. J Invest Dermatol (2012) 132:1513–6. doi: 10.1038/jid.2011.462
41. Bernheimer AW. Staphylococcal Alpha Toxin. Ann N Y Acad Sci (1965) 128:112–23. doi: 10.1111/j.1749-6632.1965.tb11633.x
42. Vu TTT, Nguyen NTQ, Tran VG, Gras E, Mao Y, Jung DH, et al. Protective Efficacy of Monoclonal Antibodies Neutralizing Alpha-Hemolysin and Bicomponent Leukocidins in a Rabbit Model of Staphylococcus Aureus Necrotizing Pneumonia. Antimicrob Agents Chemother (2020) 64:e02220–19. doi: 10.1128/AAC.02220-19
43. Rouha H, Badarau A, Visram ZC, Battles MB, Prinz B, Magyarics Z, et al. Five Birds, One Stone: Neutralization of α-Hemolysin and 4 Bi-Component Leukocidins of Staphylococcus Aureus With a Single Human Monoclonal Antibody. MAbs (2015) 7:243–54. doi: 10.4161/19420862.2014.985132
44. Menzies BE, Kernodle DS. Site-Directed Mutagenesis of the Alpha-Toxin Gene of Staphylococcus Aureus: Role of Histidines in Toxin Activity In Vitro and in a Murine Model. Infect Immun (1994) 62:1843–7. doi: 10.1128/iai.62.5.1843-1847.1994
45. Rauch S, DeDent AC, Kim HK, Wardenburg JB, Missiakas DM, Schneewind O. Abscess Formation and Alpha-Hemolysin Induced Toxicity in a Mouse Model of Staphylococcus Aureus Peritoneal Infection. Infect Immun (2012) 80:3721–32. doi: 10.1128/IAI.00442-12
46. Spaan AN, Van Strijp JAG, Torres VJ. Leukocidins: Staphylococcal Bi-Component Pore-Forming Toxins Find Their Receptors. Nat Rev Microbiol (2017) 15:435–47. doi: 10.1038/nrmicro.2017.27
47. Baba T, Kuwahara-Arai K, Uchiyama I, Takeuchi F, Ito T, Hiramatsu K. Complete Genome Sequence of Macrococcus Caseolyticus Strain JSCS5402, Reflecting the Ancestral Genome of the Human-Pathogenic Staphylococci. J Bacteriol (2009) 191:1180–90. doi: 10.1128/JB.01058-08
48. Miller LS, Fowler VG, Shukla SK, Rose WE, Proctor RA. Development of a Vaccine Against Staphylococcus Aureus Invasive Infections: Evidence Based on Human Immunity, Genetics and Bacterial Evasion Mechanisms. FEMS Microbiol Rev (2019) 44:123–53. doi: 10.1093/femsre/fuz030
49. Bordier C. Phase Separation of Integral Membrane Proteins in Triton X-114 Solution. J Biol Chem (1981) 256:1604–7. doi: 10.2116/bunsekikagaku.26.252
50. Mishra RPN, Mariotti P, Fiaschi L, Nosari S, MacCari S, Liberatori S, et al. Staphylococcus Aureus FhuD2 Is Involved in the Early Phase of Staphylococcal Dissemination and Generates Protective Immunity in Mice. J Infect Dis (2012) 206:1041–9. doi: 10.1093/infdis/jis463
51. Giuliani MM, Adu-Bobie J, Comanducci M, Aricò B, Savino S, Santini L, et al. A Universal Vaccine for Serogroup B Meningococcus. Proc Natl Acad Sci USA (2006) 103:10834–9. doi: 10.1073/pnas.0603940103
52. Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. Staphylococcus Aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin Microbiol Rev (2015) 28:603–61. doi: 10.1128/CMR.00134-14
53. Foster TJ. Antibiotic Resistance in Staphylococcus Aureus. Current Status and Future Prospects. FEMS Microbiol Rev (2017) 41:430–49. doi: 10.1093/femsre/fux007
54. Chambers HF, DeLeo FR. Waves of Resistance: Staphylococcus Aureus in the Antibiotic Era. Nat Rev Microbiol (2009) 7:629–41. doi: 10.1038/nrmicro2200
55. Fattom A, Matalon A, Buerkert J, Taylor K, Damaso S, Boutriau D. Efficacy Profile of a Bivalent Staphylococcus Aureus Glycoconjugated Vaccine in Adults on Hemodialysis: Phase III Randomized Study. Hum Vaccines Immunother (2015) 11:632–41. doi: 10.4161/hv.34414
56. Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, et al. Effect of an Investigational Vaccine for Preventing Staphylococcus Aureus Infections After Cardiothoracic Surgery: A Randomized Trial. JAMA - J Am Med Assoc (2013) 309:1368–78. doi: 10.1001/jama.2013.3010
57. Anderson AS, Miller AA, Donald RGK, Scully IL, Nanra JS, Cooper D, et al. Development of a Multicomponent Staphylococcus Aureus Vaccine Designed to Counter Multiple Bacterial Virulence Factors. Hum Vaccines Immunother (2012) 8:1585–94. doi: 10.4161/hv.21872
58. Netea MG, Quintin J, van der Meer JWM. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe (2011) 9:355–61. doi: 10.1016/j.chom.2011.04.006
59. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
Keywords: Staphylococcus aureus, outer membrane vesicles (OMVs), chimeric proteins, multivalent vaccines, OMV engineering
Citation: König E, Gagliardi A, Riedmiller I, Andretta C, Tomasi M, Irene C, Frattini L, Zanella I, Berti F, Grandi A, Caproni E, Fantappiè L and Grandi G (2021) Multi-Antigen Outer Membrane Vesicle Engineering to Develop Polyvalent Vaccines: The Staphylococcus aureus Case. Front. Immunol. 12:752168. doi: 10.3389/fimmu.2021.752168
Received: 02 August 2021; Accepted: 14 October 2021;
Published: 08 November 2021.
Edited by:
Lee Mark Wetzler, Boston University, United StatesReviewed by:
Jean Claire Lee, Brigham and Women’s Hospital and Harvard Medical School, United StatesCopyright © 2021 König, Gagliardi, Riedmiller, Andretta, Tomasi, Irene, Frattini, Zanella, Berti, Grandi, Caproni, Fantappiè and Grandi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guido Grandi, Z3VpZG8uZ3JhbmRpQHVuaXRuLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.