 Ju Zou1,2†
Ju Zou1,2† Daolin Tang
Daolin Tang Ruochan Chen
Ruochan Chen- 1Department of Infectious Diseases, Xiangya Hospital, Central South University, Changsha, China
- 2Hunan Key Laboratory of Viral Hepatitis, Xiangya Hospital, Central South University, Changsha, China
- 3Department of Surgery, UT Southwestern Medical Center, Dallas, TX, United States
The gasdermin (GSDM) family, a novel group of structure-related proteins, consists of GSDMA, GSDMB, GSDMC, GSDMD, GSDME/DNFA5, and PVJK/GSDMF. GSDMs possess a C-terminal repressor domain, cytotoxic N-terminal domain, and flexible linker domain (except for GSDMF). The GSDM-NT domain can be cleaved and released to form large oligomeric pores in the membrane that facilitate pyroptosis. The emerging roles of GSDMs include the regulation of various physiological and pathological processes, such as cell differentiation, coagulation, inflammation, and tumorigenesis. Here, we introduce the basic structure, activation, and expression patterns of GSDMs, summarize their biological and pathological functions, and explore their regulatory mechanisms in health and disease. This review provides a reference for the development of GSDM-targeted drugs to treat various inflammatory and tissue damage-related conditions.
Introduction and Historical Background
Gasdermins (GSDM) form a gene family with similar structures and include the GSDM family of genes and GSDM-related genes. GSDMs were named based on the terms, “gastro” and “dermato,” as GSDM was found to be predominantly expressed in the gastrointestinal tract and skin. GSDM was first identified on chromosome 11 in a mouse model.
Members of the GSDM family are widely expressed and may exert tissue-specific effects (Table 1), but their content-dependent functions remain largely unknown. For more than 15 years, scientists have been attempting to determine the structures and functions of GSDM family members, with some progress reported to date. Several lines of evidence suggest that the GSDM family plays diverse roles in biological and pathological processes, including in cell differentiation, cell proliferation, cell death, mitochondrial homeostasis, anti-microorganism, inflammation, and tumorigenesis (Figure 1). The direct role of these proteins in facilitating pyroptosis has also been intensively studied in recent years. In this review, we summarize current studies on the GSDM family structure and function and provide perspective on potential therapeutic targets for inflammatory and tissue damage-related conditions.
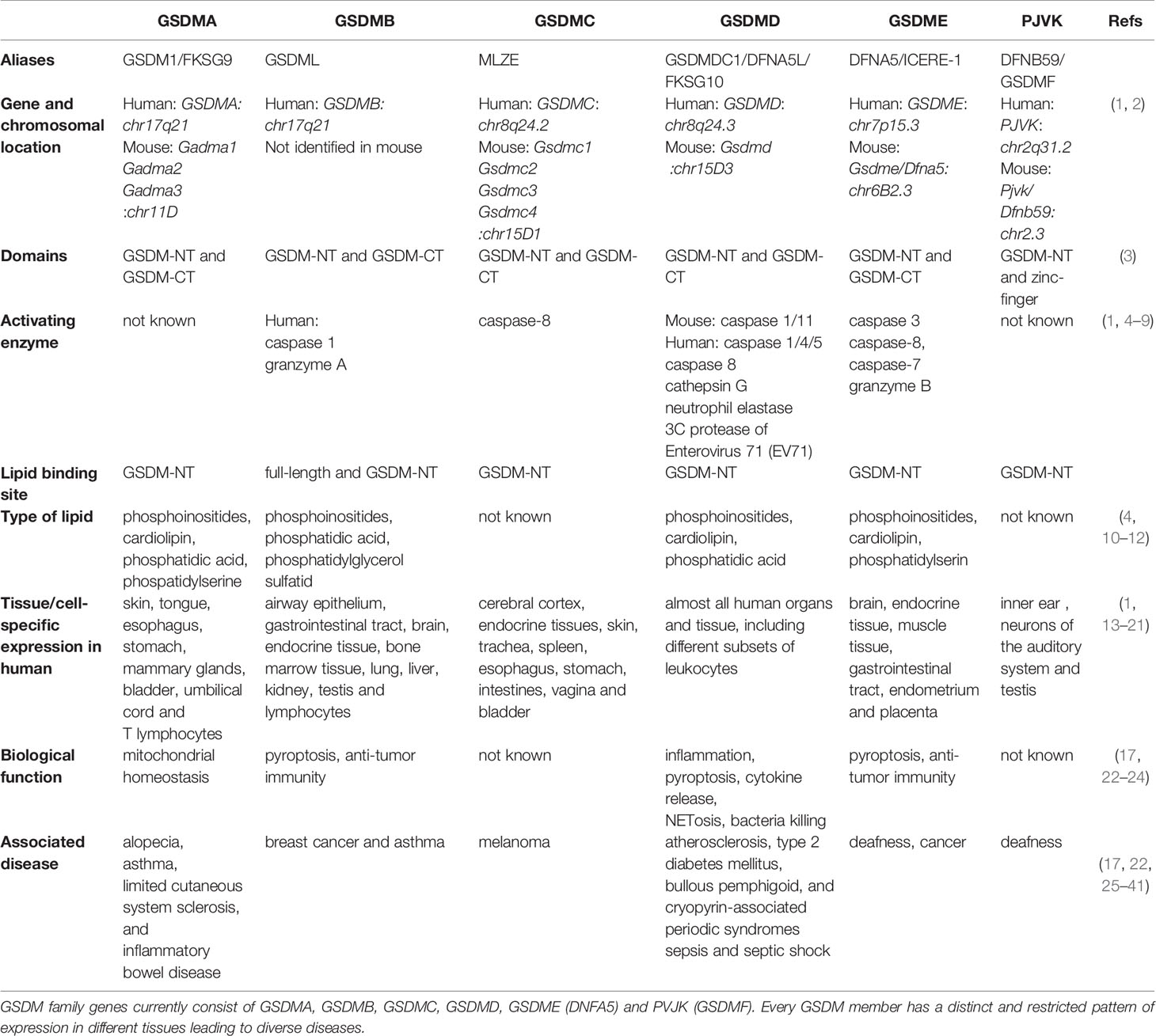
Table 1 GSDM family members, structures, functions, and disease relevance.
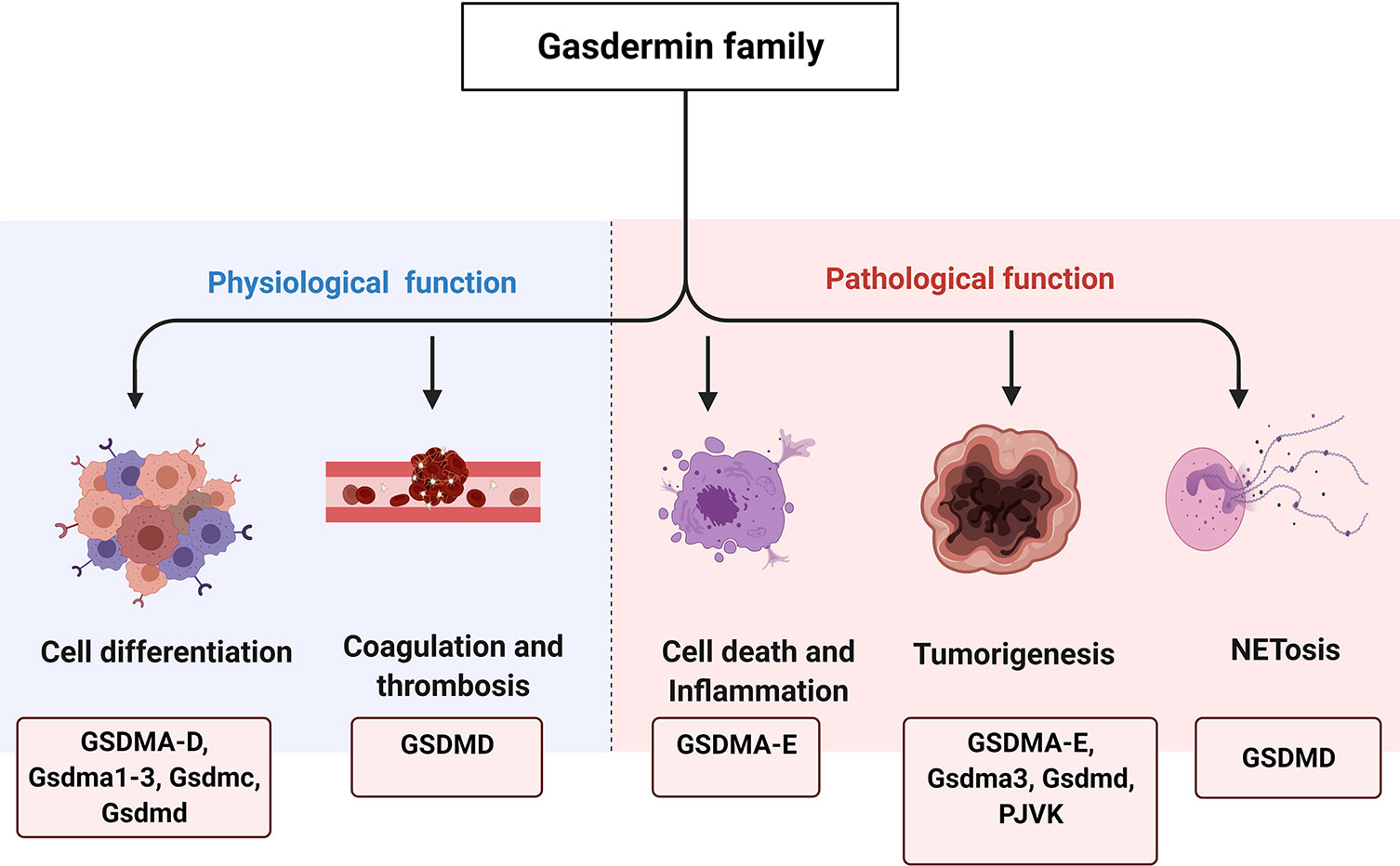
Figure 1 The physiological and pathological functions of GSDM family. GSDM family members are involved in various physiological and pathological processes, such as cell differentiation, coagulation and thrombosis, cell death and inflammation, tumorigenesis as well as NETosis.
GSDM Gene Family in Humans and Mice
History and Nomenclature
In 1998, Van Laer et al. (42) conducted genetic analysis and position cloning to identify an insertion/deletion mutation in deafness autosomal dominant 5 (DFNA5). This gene was associated with non-syndromic hearing impairment in a Dutch family. In addition, the biological information center in Germany used the web-based tool Simple Modular Architecture Research Tool (SMART) to search and analyze a group of proteins with structures similar to that of DFNA5. They identified a new structure region in these proteins and named it as the DFNA5 domain (43). In 2000, Japanese scientists (44) aiming to identify the causative genes of recombination-induced mutation 3 (Rim3) mutation in mice cloned a mice gene consisting of the Dfna5 domain. Rim3 mutant mice were characterized by epidermal abnormalities, such as thickened epidermis, loss of hair follicles, and stratified prickle-cell layers (44). Co-hybridized DNA technology was then utilized to isolate a novel mouse gene sequence, which was cloned from a mouse skin gene library; this mouse gene was named “Gsdm,” a word derived from “gastro” and “dermatome,” as it was predominantly expressed in the stomach and epidermis. Further, Southern blotting revealed the presence of Gsdm in the human genome (44). This prompted an update of the Simple Modular Architecture Research Tool website and renaming of DNFA5 to GSDM (Sanger Center, Pfams/getas PF04598; URL: http://www.sanger.ac.uk/cgi-bin/Pfam/getacc% 3FPF04598). Several other GSDM family members and GSDM-related proteins with homologous structures were identified. Currently, the GSDM family consists of four human genes (gasdemin A [GSDMA], gasdemin B [GSDMB], gasdemin C [GSDMC], and gasdemin D [GSDMD]) and eight mouse genes (Gsdma1, Gsdma2, Gsdma3, Gsdmc1, Gsdmc2, Gsdmc3, Gsdmc4, and Gsdmd) (Table 1). GSDMB is the most divergent member of the GSDM family. Although it is not present in the mouse and rat genomes, felines (cats and tigers) and canines (dogs) contain a Gsdmb orthologue (45). As shown in the study, human GSDMB is cleaved by chloroquine down-regulate caspase-1 (CASP1) and granzyme A at D236 and K244, respectively, whereas dogs, cats and tigers do not possess these key residues. This lack of activation sites in GSDMB in felines and canines indicate that they are deficient in the GSDMB-dependent pyroptosis pathway. Therefore, GSDMB is unique to the human genome to some extent. Moreover, two GSDM-related genes are common to both humans and mice: GSDME, also known as DFNA5, and pejvakin (PJVK), also known as DFNB59 or GSDMF. Based on the divergent expression pattern and mutant-associated phenotypes compared to other GSDM family members, GSDME and PJVK were excluded from the GSDM family in some studies, although they have structures similar to those of the members in the family (46). Additionally, PJVK shows high similarity to GSDME. The nomenclature has been simplified to include all six members from GSDMA to GSDMF in a single GSDM family (4, 47).
Protein Structural Basis of Autoinhibition and Activation
According to functional analysis, the GSDM protein, which contains around 220–480 amino acids, is the most important structure domain of GSDMs (43). Most protein products of GSDM family genes, except for PJVK/GSDMF, have three domains: the GSDM N-terminal (GSDM-NT) domain, linker region, and GSDM C-terminal (GSDM-CT) domain (Figure 2A). Both the N- and C-terminal domains are relatively well-conserved in the family, whereas the linker region is unique to each group (46). Notably, GSDM-NT of PJVK/GSDMF is directly followed by a small C- terminal domain that contains a zinc-finger domain with unknown function, suggesting evolutionary and mechanistic divergence within the GSDM family (3).
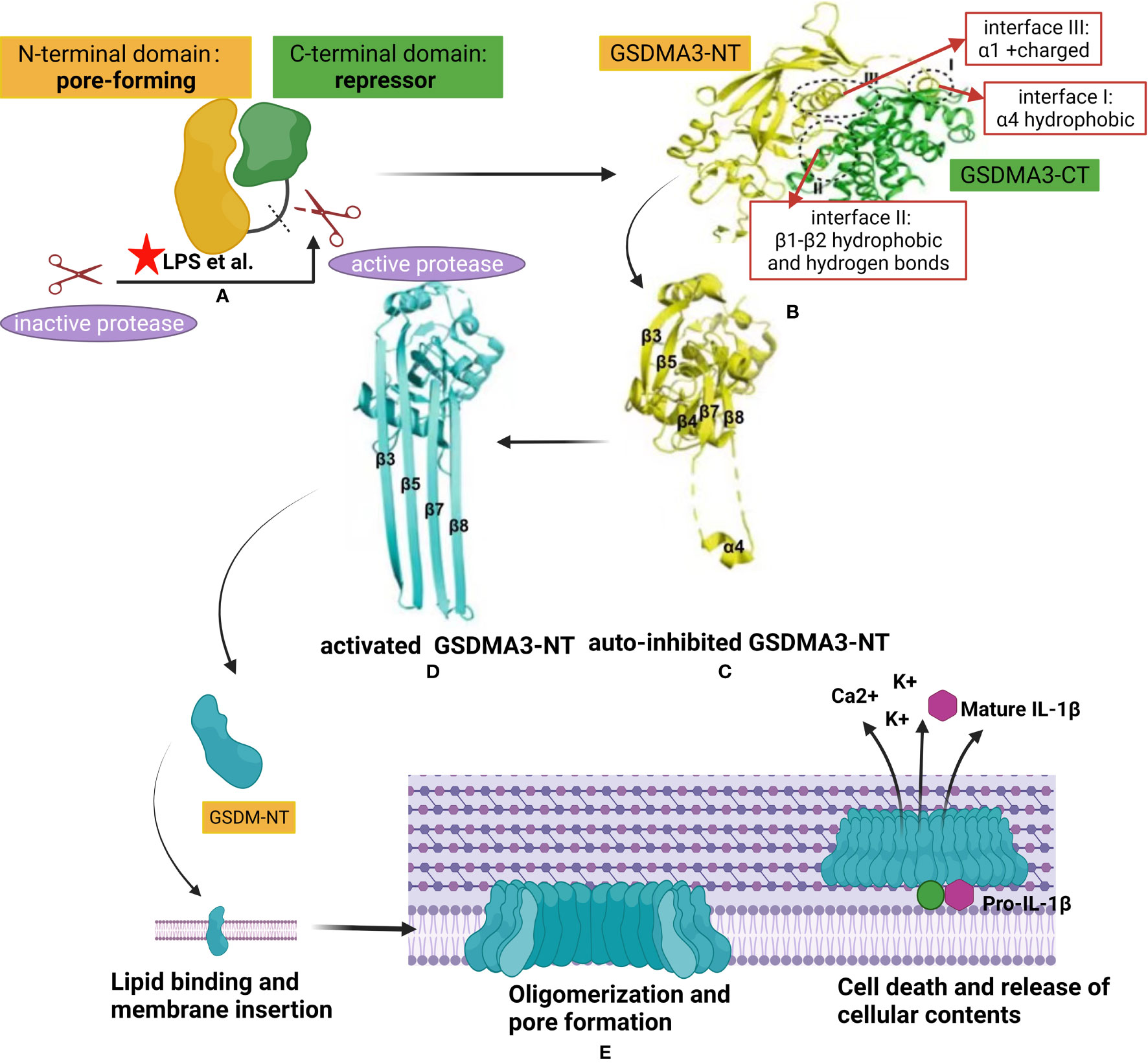
Figure 2 Structural auto-inhibition in GSDM family and mechanism of gasdermin membrane insertion and pore formation. (A) Interdomain interaction between the N-terminal of gasdermin (GSDM-NT) and C- terminal of gasdermin (GSDM-CT), with pore-forming and auto inhibitory characteristics respectively, keeps the full length GSDM protein in an auto-inhibited state. Different proteases, such as inflammatory caspases or granzymes, are activated by danger signals from pathogens like lipopolysaccharide (LPS). The latter was represented by a red pentacle in the image. The linker region of GSDMs is cleaved by the active protease releasing the GSDM-NT from the GSDM-CT. (B) (48) X-ray crystal structure of full-length GSDM and the interfaces mediating the inter-domain interactions (I-III) by forming electrostatic, hydrophobic, and hydrogen bonding. The GSDM-NT and GSDM-CT domains are colored yellow and green, respectively. (C, D) (49) Crystal structure of GSDMA3-NT in auto-inhibited conformation (C) and the pore conformation (D). (E) A proposed universal model showing the pore formation by GSDM family. Once released from the GSDM-CT, the GSDM-NT is recruited to insert in the cell membrane by binding with lipid. Upon membrane binding, the GSDM-N concentrates and starts the oligomerization process forming pores leading to release of cellular contents,including ion flux and mature IL-1β, and finally cell death.
Given the high homogeneity among all GSDM-NT domains, most GSDMs may share a common mechanism of autoinhibition and activation to form pores in the membrane. Structurally, the GSDM-NT domain predominantly comprises β-strands with several α-helices (3). In contrast, GSDM-CT consists of α-helices and folds almost exclusively that form a compact globular conformation (50). The α1 and α4 helices of GSDM-NT, which play important roles in lipid binding and membrane insertion, can fold back on the concave side of GSDM-CT by forming electrostatic, hydrophobic, and hydrogen bonding. For example, Ding et al. reported that the crystal structure of full-length mouse GSDMA3 revealed three main interacting interfaces between the C- and N-terminal domains (3). First, the GSDMA3-CT domain forms a nonpolar surface that interacts with the hydrophobic region on the α4 helix of the GSDMA3-NT domain (Figure 2B, interface I). Second, the GSDMA3-CT domain forms another hydrophobic pocket that interacts with some hydrophobic residues on the β1–β2 loop in the GSDMA3-NT domain. The β1–β2 loop forms four hydrogen bonds with the C-terminal domain (Figure 2B, interface II). Third, the α1 helix in the GSDMA3-NT domain also provides a surface via predominantly charge-charge interactions with some residues in the GSDMA3–CT domain (Figure 2B, interface III). Therefore, inter-domain interactions between the GSDM-NT and GSDM-CT domains maintain the full-length GSDM protein in an auto-inhibited state (49).
In response to activation signals, GSDMs are cleaved in the central linker region by different inflammatory caspases or granzymes, as shown in the Table 1, to generate a 31-kDa GSDM-NT fragment and 22-kDa GSDM-CT fragment (3, 51). GSDM-CT typically functions as a repressor when binding to GSDM-NT; thus, its overexpression inhibits cell death (5). GSDM-NT, when uninhibited by GSDM-CT, binds to membrane lipids and extensively form pores, resulting in cytotoxicity and cell death (3, 13, 51). For GSDMD protein, inflammatory caspases are activated through intracellular lipopolysaccharide (LPS) binding (caspases-11, -4, and -5) or the inflammasome (caspase-1) (Figure 2A). Upon activation, inflammatory caspases cleave the linker region and release the N-terminal domain from the autoinhibitory C-terminal domain, which binds to lipids in the membrane and forms a pore.
The molecular mechanism of pore formation by GSDMs has been confirmed previously (3, 10, 52–54). Structurally, the entire β3–β4–β5 region in the auto-inhibited stated GSDMA3-NT is extended to form the first transmembrane β-hairpin, with β7, β8, and α4 binding the C-terminal domain form the second β-hairpin (Figure 2C). Once the balance of auto-inhibition is disrupted through mechanisms such as cleavage of GSDMD by mammalian caspase 1, mouse caspase 11, or human caspase 4/5, GSDMA3-NT is released from GSDMA3-CT and undergoes conformational changes. Each GSDMA3-NT domain contributes four extended β-strands to pore formation (Figure 2D). The hydrophilic and hydrophobic residues of the four β-strands are alternately arranged, with hydrophobic residues oriented outward to contact the surrounding lipid (3, 49).
The conformational changes leading to pore formation are triggered by binding of the N-terminal domain to membrane phospholipids, with a positively charged pocket and negatively charged group, respectively. The surface of this pocket is fully masked by the GSDM-CT domain in the auto-inhibited full-length of GSDMs (3). GSDMD-NT was reported to tightly bind acidic phospholipids, such as phosphoinositides and cardiolipin, but weakly bind phosphatidic acid and phosphatidylserine, reducing the stability of the target cell membrane (3, 10). The lipid-binding properties of the other GSDMs-NT domain is similar to that of GSDMD, suggesting a common membrane-targeting mechanism in the GSDM family. Notably, the function of the GSDM pore can differ depending upon the target cell type, level of GSDM protein expression, timing of activation, and efficiency of interaction. For example, LPS can elicit GSDMD-dependent release of mature interleukin (IL)-1β from live human monocytes (55). Furthermore, ion fluxes though GSDMs pores have a profound impact on cellular signal pathways because these pores act as large and non-selective membrane channels (Figure 2E).
Interestingly, in the absence of environmental lipids, GSDM-NT and GSDM-CT are connected despite cleavage of the linker region (3). This finding indicates that lipids are required to dissociate GSDM-NT from GSDM-CT. In contrast, GSDM-CT mutations in neighboring residues of GSDM-NT enable full-length GSDMs to form pores, supporting the hypothesis that GSDM-NT and GSDM-CT dissociation is not necessary for pore formation (3). Moreover, full-length GSDMB displays a similar lipid-binding ability, in contrast to the GSDMB-NT domain alone, suggesting that the GSDMB-CT domain does not impair the lipid binding of GSDMB (56). In summary, the precise molecular mechanisms underlying the GSDM-NT/CT interaction and GSMD protein modification and activation require further clarification.
Expression Pattern of GSDM Family Genes in Normal Tissue
Every GSDM member has a distinct and restricted pattern of expression in different tissues. We investigate the RNA and protein expression of GSDM family genes in human and murine tissues by reviewing previous publications and two public databases, the Human Atlas Protein (http://www.proteinatlas.org) and MGI-mouse Gene Expression Database (http://www.informatics.jax.org/expression.shtml). Human GSDMA is mainly expressed in epithelial cells of the skin, tongue, esophagus, stomach, mammary glands, bladder, and umbilical cord (Table 1) (14, 15). Furthermore, GSDMA protein was detected in T lymphocytes (16). Analysis using reverse transcription-polymerase chain reaction indicated that there is a more distinct tissue-specific expression of Gsdma in mice (46). Gsdma1 is expressed in the suprabasal epidermis, cornea, hair follicles, and forestomach (13, 15, 44, 57). Gsdma2 is mainly expressed in the stomach gland. Gsdma3, where Rim3 mutation occurs, is primarily found in the sebaceous gland of the skin (58, 59).
GSDMB, also known as GSDML, is composed of 411 amino acids and is the most divergent member of the GSDMB family. GSDMB is exclusively expressed in humans but not in the mouse and rat genomes. However, a Gsdmb orthologue is reportedly expressed in rodents. The human GSDMB has six splicing variants; their expression is detected in numerous human tissues and cells, such as the airway epithelium, gastrointestinal tract (esophagus, stomach, small intestine, and colon), brain, endocrine tissue, bone marrow tissue, lung, liver, kidney, testis, and lymphocytes (Table 1) (1, 13, 14, 17, 18).
GSDMC, also known as melanoma-derived leucine zipper-containing extranuclear factor, was named after metastatic mice melanoma cells in which its increased expression was observed and the gene was first identified (60). Human GSDMC is expressed in the cerebral cortex (61), endocrine tissues, skin, trachea, spleen, esophagus, stomach, intestines, vagina, and bladder (Table 1) (18). There are four Gsdmc orthologous genes in mice (46), namely, mouse GSDMC1, GSDMC2, and GSDMC4. These orthologous genes are expressed in the stomach, large and small intestines, bladder, and prostate, whereas GSDMC3 expression is restricted to the bladder, prostate, and large intestines (J:171409 GUDMAP Consortium, http://www.gudmap.org. 2004).
GSDMD is best known for its role in mediating pore formation and subsequent pyroptosis. Human DFNA5L/GSDMD, mouse Dfna5l/Gsdmd, and rat Dfna5l/Gsdmd were first identified and characterized via bioinformatics analysis of genomic data (61). Almost all human organs and tissues, including different subsets of leukocytes, express GSDMD mRNA and proteins (Table 1) (16, 18). GSDMD is also widely expressed in the stomach, gut, colon, heart, spleen, liver, lung, and urinary system of mice (62, 63).
The expression patterns of two GSDM-related genes, GSDME and GSDMF, were investigated in human and mice. The mRNA and protein expression of GSDME was detected in a wide range of human tissues and cells, such as the brain, endocrine tissues, muscle tissues, gastrointestinal tract, endometrium, and placenta (Table 1). Mouse GSDME is expressed in the gastrointestinal tract, ears, heart, adipose tissues, endocrine glands, spleen, mammary glands, liver, and lungs (19, 64). GSDME is also expressed in different species of lower vertebrates. For instance, two GSDME orthologous genes, GsdmEa and GsdmEb, were identified in bony fish (56).
PJVK (GSDMF) consists of 352 amino acids; its gene is on chromosome 2q31.1-q31.3. This gene has been implicated in autosomal recessive deafness and has been detected in the inner ear and neurons of the auditory system (20). However, it was initially cloned from the human testis (Table 1). Furthermore, PJVK orthologous genes are present in early chordates and invertebrates, suggesting that GSDM family members evolved from these organisms. Moreover, PJVK shares a high similarity with GSDME. In fact, the RNA expression of PJVK is most abundant in the testis, but it is also widely expressed in other tissues as revealed in various databases (such as Consensus, GTEx, and FANTOM5) (20, 65, 66).
Protein and RNA analyses revealed that GSDM family genes exhibit a unique tissue-specific expression pattern. This pattern appears to be conserved between human and mouse species. A systematic and comparative expression investigation and analysis of human and mouse should be conducted to clarify these patterns.
Physiological and Pathological Function of GSDM Family Genes
Cell Differentiation
Studies of the distinct expression patterns of GSDM family members in the gastrointestinal tissue revealed their potential roles in cell proliferation and differentiation (Figure 1) (67). In the esophageal and gastric epithelium, GSDMA is preferentially expressed in the differentiated pit layers, indicating a potential function in mature epithelial cells. GSDMB mRNA is predominantly expressed in the basal region of the esophagus and in the stomach, where stem cells are typically located. It is also detected in the isthmus or neck of the stomach, where rapidly proliferating precursors of pit cells are harbored. GSDMC is mainly expressed in the differentiated supero-basal region of the esophagus; however, it is not detected in any region of the gastric epithelium. GSDMD is expressed in the differentiating cells of the esophagus and differentiated and differentiating regions of the stomach. In the small intestines and colons of mice, Gsdmc and Gsdmd exhibit a distinct expression pattern, as determined using reverse transcription-polymerase chain reaction (46). These studies indicate that the GSDM family plays a critical role in cell differentiation based on the varying levels of GSDM proteins in epithelial cell in the gastrointestinal tract with various levels of differentiation. However, direct evidence of this role is required.
Mouse homologues of GSDMA, which include Gsdma1, Gsdma2, and Gsdma3, are clustered on chromosome 11 (46). Gsdma3 is predominantly expressed in differentiated epidermal cells of the skin. Histological analysis of the Gsdma3 mutation revealed epidermal cell hyperplasia in the upper hair follicles and an abnormal anagen phase at the first hair cycle. Furthermore, the results of immunohistochemical analysis showed that hyperproliferation and misdifferentiation occurred in the upper follicular epidermis among Gsdma3 mutants (58). These results suggest that Gsdma3 is involved in the proliferation and differentiation of epidermal stem cells. Moreover, hair follicle differentiation defects, such as flattened Cuh, reduced keratin levels in the cortex, and abnormal arrangement of trichohyalin in the inner root sheath, have been observed in Gsdma3 mutant mice with alopecia and excoriation. Furthermore, Gsdma3 and Msx2 are co-expressed in the matrix and inner root sheath, and overexpression of Gsdma3 in the mouse skin promotes activation of Msx2 pathway in vivo (68). In summary, GSDMA3 protein is involved in the Msx2/Foxn1/acidic hair keratin pathway, whereas increased apoptosis is observed in the hair follicles of mice with mutated Gsdma3. These findings suggest that GSDMA3 plays a crucial role in normal hair follicle differentiation (68).
Coagulation and Thrombosis
The development of blood clots and thrombosis is often implicated in various physiological and pathological processes. In response to the presence of LPS or a specific PAMP from Escherichia coli, such as the type III secretion system inner rod protein EprJ, the activation of canonical/non canonical inflammasome mediates GSDMD activation, pyroptosis, and tissue factor release in primary bone marrow-derived macrophages, resulting in systemic coagulation and thrombosis in C57BL/6J mice (Figure 1) (69). Although bacterial infection can induce inflammasome activation and pyroptosis in macrophages, the underlying mechanisms remain unclear. Recently, a study (70) showed that Ca2+ influx, which was mediated by the GSDMD pore, resulted in phosphatidylserine exposure to peripheral leukocytes and splenocytes, thereby leading to life-threatening disseminated intravascular coagulation and endotoxemia in mice. Phosphatidylserine exposure and tissue factor activity was attenuated in murine macrophages cultured with low Ca2+-containing medium. Casp11 deletion, Gsdmd ablation, or phosphatidylserine or tissue factor neutralization significantly prevented LPS-induced disseminated intravascular coagulation in murine models with endotoxemia (70). In the clinical setting, an increase in the biomarkers of GSDMD activation, which include the plasma levels of IL-1A and IL-1B, was significantly correlated with the formation of thrombin-antithrombin complex, a high disseminated intravascular coagulation score, and phosphatidylserine externalization in peripheral leukocytes of septic patients (70). This inflammasome-related coagulation in macrophages is further enhanced by activation of the stimulator of interferon genes pathway (71). Whether other members of GSDMs are involved in coagulation and thrombosis and how they interact with initiators of coagulation require further investigation.
Tumorigenesis
Tumorigenesis is associated with alterations in various cellular processes. GSDMA shows enhanced apoptosis-inducing activity in human gastric pit cells, and its expression and function are regulated by the TGF-β/transcription factor limdomain only 1 signaling pathway (Figure 1). This pathway is associated with the apoptosis of gastric pit cells, as GSDMA is frequently suppressed in esophageal and gastric cancers (57).
Moreover, stable transfection of GSDMB promotes cell proliferation of the ovarian cell line CHO-K1, whereas genetic depletion of GSDMB inhibits the growth of HeLa cells (Figure 1) (72). Consistently, GSDMB is significantly more upregulated in in uterine cervix cancer tissues compared with other precancerous tissues. Additionally, GSDMB is expressed in all examined cases of esophageal and gastric cancers (67). GSDMB is significantly more upregulated in breast carcinoma compared to in normal breast tissue. Moreover, both isoforms of GSDMB, GSDMB1 and GSDMB2, promote invasion and metastasis of the MCF7 breast carcinoma cell line in vitro, whereas silencing of these isoforms in the HCC1954 breast carcinoma cell line significantly decreased migration and invasion (22). In vitro data from MCF7 cells and in vivo data from an intracardiac experimental metastatic mice model suggested that GSDMB interacts with several molecules and signaling pathways, such as Hsp90, Rac-1, and Cdc-42 Rho-GTPases, to induce tumorigenesis (22). GSDMB, which is in amplicons and genomic regions, is often amplified during tumorigenesis. Thus, GSDMB may play a role in tumor progression and metastasis. However, the specific functions of GSDMB in carcinogenesis, cancer metastasis, and progression are unclear.
GSDMC, also known as melanoma-derived leucine zipper-containing extranuclear factor, was originally isolated from mouse melanoma cells and obtained from the mouse melanoma cell B16-BL6 cDNA library (60). The expression levels of mouse GSDMC were positively correlated with the metastatic ability of B16 melanoma cell lines (60). However, it is unclear whether GSDMC is involved in one or more processes in the invasion-metastasis cascade. In addition, whether upregulated GSDMC expression upon acquisition of metastatic potential in melanoma cells is only an accompanying phenomenon is unclear. Nevertheless, knockdown of GSDMC attenuated the proliferation of colorectal cancer cell lines, such as DLD-1 and LoVo cells, indicating a pro-tumorigenic role (73). Furthermore, GSDMC shRNA resulted in a significant decrease in xenograft tumor growth in mice (73). Consistent with the above findings, GSDMC expression was suppressed in several cases of esophageal squamous cell carcinoma, thus supporting that GSDMC is a tumor-suppressor gene (Figure 1) (67). However, it is unknown whether GSMDC acts as a tumor-promoting regulator or an anti-tumor regulator in cancer development; thus, further investigation is required.
In addition to GSDMA, GSDMB, and GSDMC, GSDMD reportedly inhibited proliferation of the gastric cancer cell line MKN28 in a colony formation assay (67). However, a significantly upregulated GSDMD was observed in various types of cancer, such as cervical cancer, liver cancer, ovarian cancer, and pancreatic cancer. The results of these expression assays implicate a role for GSDMD in promoting carcinogenesis. Moreover, GSDMD expression is correlated with CD8+ T cell markers in primary tumors of lung adenocarcinoma, lung squamous cell carcinoma, and melanoma cohorts of The Cancer Genome Atlas (74). The level of GSDMD cleavage was increased in human activated CD8+ T cells, whereas GSDMD gene depletion decreased the cytolytic capacity of CD8+ T cells; thus, GSDMD was required to facilitate an optimal cytotoxic T-lymphocyte response to lung cancer cells (74). Inhibition of GSDMD also attenuated tumor proliferation by promoting intrinsic mitochondrial apoptosis via the EGFR/Akt signaling pathways in non-small cell lung cancer cell lines (PC9 and H1703) (75).
Although the expression and post-translational modification of GSDME in various types of cancer have been examined in several studies, the exact role and underlying molecular mechanisms of these processes remain ambiguous (76, 77). GSDME may be a tumor suppressor, as its expression is decreased in breast, gastric, and colorectal cancers (78–80). Furthermore, GSDME deficiency accelerates the proliferation of gastric, colorectal, and melanoma cancer cell lines (11). Upregulation of GSDME also increased the drug susceptibility of a chemotherapy-resistant melanoma cell line by upregulating caspase-3, which mediated pyroptosis (4).
Cell Death and Inflammation
Several studies have shown that GSDMs induce cell death and inflammation (Figure 1). Particularly, the role of GSDMD in pyroptosis has been widely studied. Pyroptosis is a lytic form of cell death driven by inflammatory caspases and characterized by cell swelling, nuclear condensation, cell membrane disruption, and inflammatory cytokine and DAMP release (81, 82). Inflammatory caspases orchestrate the formation of a multi-protein platform known as the inflammasome, thereby inducing the cleavage and release of proinflammatory cytokines, such as IL-1B and IL-18, from macrophages (83) (Figure 3). In addition to canonical inflammasome activation, a signaling pathway of noncanonical inflammasome regulated by caspase-11/4/5 was identified (25). During infection with gram-negative bacteria, LPS in the outer membrane of the bacteria is imported into the intracellular space of the host cell. It then directly binds to and activates caspase-11 in mice and caspase-4 and -5 in humans (84–87). However, the mechanism underlying the inflammatory caspases’ modulation of the intracellular physiological events remains unclear.
Three groups (5, 6, 88) demonstrated that GSDMD is a fundamental substrate of caspase-1, -11, -4, and -5. A previous study (5) employed genome-wide clustered regularly interspaced palindromic repeat/Cas9 technology and siRNA-mediated knockdown techniques to detect caspase-11- and caspase-1-mediated pyroptosis in the bone marrow macrophages of mice. The researchers identified GSDMD as the major mediator of cytoplasmic LPS-mediated pyroptosis in the macrophages of humans and mice. This finding was supported by another study (6), which employed a forward genetic approach to screen ethyl-N-nitrosourea-mutagenized mice for mutations that diminished induction of the caspase-11-mediated noncanonical signaling pathway. This screening approach also linked GSDMD to intracellular LPS-induced pyroptosis and IL-1B maturation and release (6). In addition, another study (88) identified GSDMD as a molecule that directly interacted with inflammasomes. This was confirmed by analysis of LPS-stimulated macrophage RAW264.7 cell lines and primary cells using quantitative mass spectrometry technology. Apoptosis was shown to be suppressed by pyroptosis in response to LPS and nigericin or to Salmonella typhimurium treatment. These results suggest an interplay between apoptosis and pyroptosis (88). These studies suggest also that GSDMD is a direct substrate of inflammatory caspases (caspase-1, -11, -4, -5) and the executioner of inflammatory cytokine release and pyroptosis, as well as clarify the detailed mechanism of how GSDMD mediates noncanonical inflammasome pathways and pyroptosis. GSDMD is also cleaved by caspase-1, indicating a role for this substrate in caspase-11, -4, and -5-independent canonical inflammasome activation and pyroptosis (89). The results of a study using genome-wide clustered regularly interspaced palindromic repeat/Cas9 involving the human monocyte cell line and ethyl-N-nitrosourea-mutagenized mice showed that the expression and function of GSDMD was positively regulated by the interplay between transcription factors and interferon regulatory factor-1 and -2 (90, 91). In contrast to GSDMD, other members of the GSDM family, such as GSDMA, GSDMB, and GSDMC, are not cleaved by caspase-1 and caspase-11. This may be because of the lack of the 272FLTD275 motif in GSDMD (5). However, overexpression of the gasdermin-N domain of GSDMA3 leads to induction of significant pyroptosis in 293T cells. This suggests that GSDMs are pyroptosis-inducing proteins activated by autoinhibition of their N-terminal domain.
The precise mode of the execution of cell death by the GSDMD N-terminal has been explored. Caspase-matured GSDMD-NT is recruited to bind to the inner lipid membrane and penetrate it by forming pores as the key effector mechanism of pyroptotic cell death (3, 54, 92). Using a cell-free method, the cleaved and recombinant GSDMD-NT were found to bind to liposomes in vitro, indicating that GSDMD mediates its own recruitment to membranes. GSDMD-NT is exclusively associated with lipids of the inner wall of the plasma membrane, explaining why GSDMD only lyses cells from the inside and prevents unwarranted cell death and tissue damage. In addition, GSDMD binds to cardiolipin, a component of prokaryotic and mitochondrial membranes, which may explain its proposed bactericidal activity. Indeed, the recombinant protein of GSDMD-NT but not the full-length protein and GSDMD-CT induces extensive killing of Listeria monocytogenes, Staphylococcus aureus, and E. coli (10). Interestingly, the GSDMD-NT-induced membrane pores also function as conduits for IL-1 family cytokine release and other cytosolic contents, including ions, from cells (93–96). In addition, inhibiting GSDMD-induced pyroptosis using chemicals such as disulfiram or dimethyl fumarate, is a promising strategy for reducing inflammation (97, 98). Furthermore, liquid chromatography-tandem mass spectrometry revealed that dimethyl fumarate and endogenous fumarate reacts with GSDMD at critical cysteine residues to form S-(2-succinyl)-cysteine. These critical cysteine residues include Cys191 in humans and Cys192 in mice. Succination of GSDMD prevents its interaction with caspase 1, which then limits the processing, oligomerization, and capacity to induce death of HEK293T cells (98).
In addition to its role in pyroptosis, GSDMD is a critical mediator of neutrophil extracellular trap (NET) formation and other modes of cell death (Figures 1, 3). Particularly, NET formation is observed in neutrophils, in which neutrophil extracellular traps (NETs) are formed during this type of cell death (99–101). NETs are composed of DNA and non-chromatin molecules, such as histones, HMGB1, and granule proteins. Pore formation in neutrophils induced by GSDMD promotes cell rupture and NET extrusion into the extracellular space. Moreover, neutrophil elastase cleaves GSDMD to produce GSDMD-NT during NET formation. In turn, GSDMD-NT promotes protease activation and nuclear expansion in a feed-forward loop (101). Interestingly, GSDMD induces the formation of pore-induced intracellular pits, which triggers a multifaceted defense against intracellular bacteria by trapping pathogens within the cellular debris (102) (Figure 3). However, further investigation is required to clarify the antibacterial activity of endogenous GSDMD in vivo and in vitro.
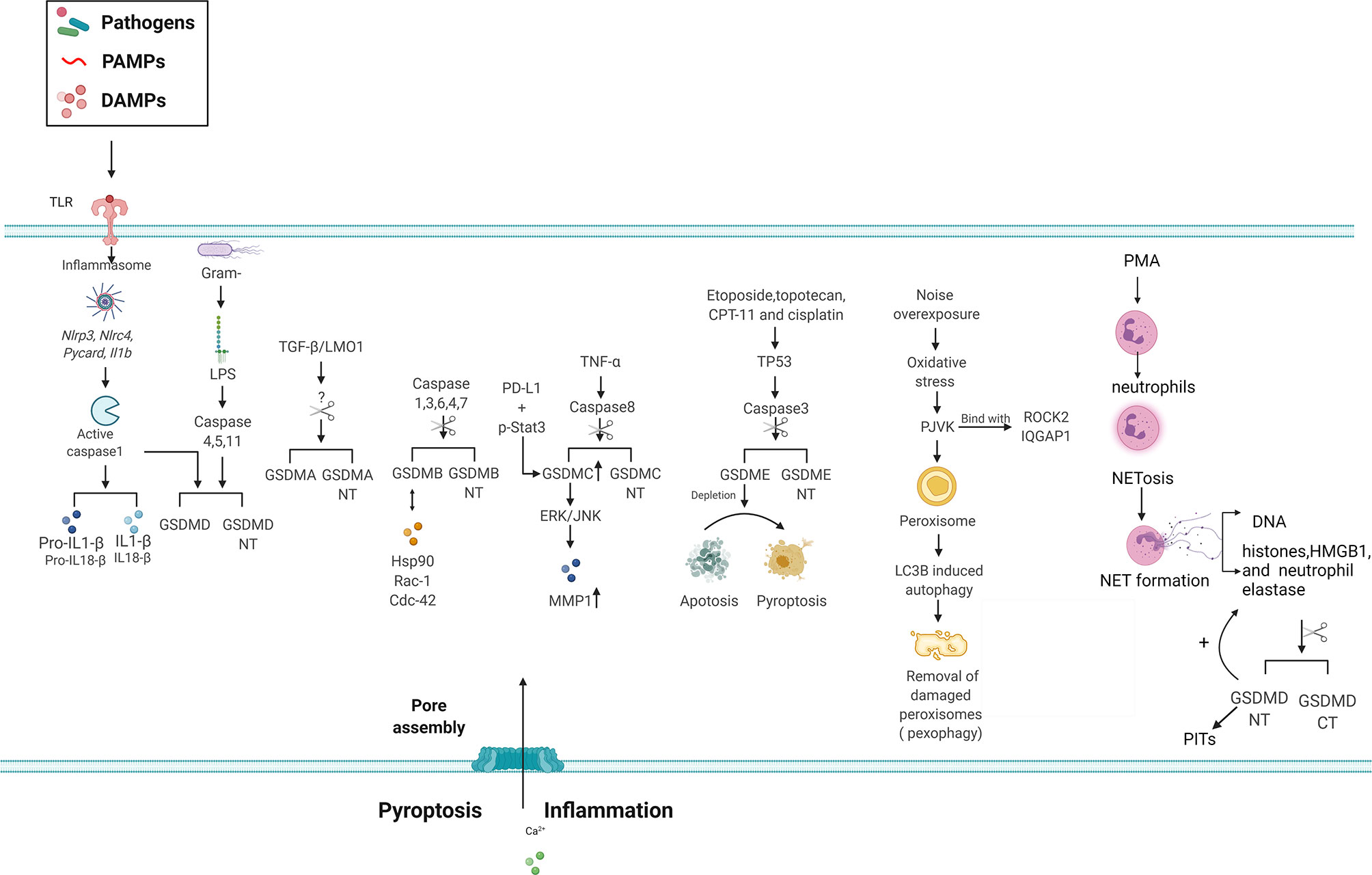
Figure 3 Signaling pathways of GSDM family members in inflammasome signaling, pore formation and cell death. GSDMD is involved in pyroptosis via canonical and non-canonical inflammasome mediated pathways following cleavage by inflammatory caspases. The N-terminal domain of GSDMA-E can bind with membrane lipids of the plasma membrane and form pores, allowing release of the inflammatory cytokines IL-1BIL-1B and IL-18 and induction of pyroptosis. However, the inflammatory caspase responsible for GSDMA cleavage remain elusive. Various stimuli and upstream signals like PD-L1, TP53 and TNF-α, can lead to GSDMs cleavage and subsequent cell death. GSDMs are able to bind with other molecules, such as Hsp90, Rac-1 and ROCK2, and the following function require further investigation. Alternatively, depletion of GSDME can switch apoptosis to pyroptosis in cells. The ability of PJVK to bind membrane lipids or form pores in the plasma membrane is uncertain, however it is implicated autophagy related pexophagy. In addition, GSDMD leads NETosis as the key effector GSDMD is cleaved by neutrophil elastase during NETosis induced by classic stimulants such as PMA, releasing the active GSDMD-N.GSDMD-N, Gasdermin N-terminal; GSDMA, Gasdermin A; GSDMB, Gasdermin B; GSDMC, Gasdermin C; GSDMD, Gasdermin D; GSDME, Gasdermin E; HSP90, heat shock protein 90; IL-1BIL-1B, interleukin-1β; IL-18, interleukin-18; PJVK, Pejvakin; PD-L1, programmed death-ligand1; ROCK2, rho associated coiled-coil containing protein kinase 2; Rac-1, ras-related C3 botulinum toxin substrate 1; Cdc42, cell division cycle 42; IQGAP1, IQ Motif Containing GTPase Activating Protein 1; MMP1, Matrix Metallopeptidase 1; ERK, extracellular regulated protein kinase; JNK,c-Jun N-terminal kinase; TGF-β, transforming growth factor-β; LOM1, LIM Domain Only 1; PMA,phorbol 12-myristate 13-acetate; Netosis, neutrophil extracellular trap formation (NETosis); PITs: pore induced intracellular pits.
Similar to GSDMD-NT, overexpression of GSDMA-NT or GSDMA3-NT in 293T cells may result in pore formation in the plasma membranes and the induction of pyroptosis-like features (3, 5). Upregulation of human GSDMA promotes TGF-β-dependent apoptosis in gastric epithelial cell lines (57), whereas mouse Gsdma3 has been implicated in the TNF-α-mediated apoptosis signal pathway in skin keratinocytes (103). The Gsdma3 Y344H mutant protein and Gsdma3-NT domain display similar pro-autophagic abilities, which is associated with mitochondria and generation of reactive oxygen species in HEK293T and HaCat cells and mouse models. Loss of the conserved self-regulation of Gsdma3 results in autophagy and cell death (51). However, protease cleavage in human and mouse GSDMA and their regulatory mechanisms are not well-understood.
Both the full-length and GSDMB-NT can bind to membrane lipids such as phosphoinositide and glycolipid sulfatide, distinguishing them from other GSDMs (104). GSDMB can be cleaved by caspase-1, -3, -4, -6, and -7 within the pore-performing domain at the 88DNVD91 or D236 sites, which is similar to cleavage of the pore-forming domain in GSDMD (1, 105, 106). However, GSDMB-NT but not the full GSDMB protein induces pyroptosis-like features in human HEK 293T cells (3). This suggests that binding of lipids of the full GSDMB protein leads to the production of GSDMB-NT. Artificially truncated GSDMC-NT also induces pyroptosis. Under hypoxic conditions, p-Stat3 physically interacts with PD-L1 and facilitates its nuclear translocation, which promotes GSDMC/caspase-8 activation, mediates the non-canonical pyroptosis pathway, and induces tumor necrosis in MDA-MB-231 cells (107). The N-terminal domain of GSDMC is sufficient to induce pyroptosis both in vivo and in vitro. However, future studies should focus on the signals that can activate GSDMB and GSDMC and the mechanism underlying cleavage of GSDMB and GSDMC, which mediate cell death.
Recent studies have indicated that GSDME induces tumor cell death by inducing apoptosis and pyroptosis (108–110), which may explain its potential tumor suppressive activity (111, 112). Mutations in intron 7 of GSDME cause sensorineural hearing loss because of skipping of exon 8 at the pre-mRNA level and translation of a C-terminally truncated protein (113). Although the full-length product does not have cytotoxic activity, its truncated form does (112). Moreover, GSDME expression can be induced by the transcription factor TP53 in response to etoposide, a potent inducer of apoptosis (114). The mRNA expression of GSDME is remarkably induced by gamma ray irradiation in the colon of TP53(+/+) mice but not in that of TP53(–/–) mice, suggesting that cooperation between TP53 and GSDME is required to mediate cell death. GSDME is a physiological substrate of caspase-3 when it is stimulated by apoptotic triggers such as etoposide or vesicular stomatitis viral infection. Mechanistically, GSDME is cleaved by caspase-3 at Asp270 to generate a necrotic GSDME-NT domain that translocates to the plasma membrane, where it increases cell membrane permeability. In addition, secondary necrosis and pyroptosis are induced in HeLa cells. Moreover, GSDME specifically requires caspase-3 but not caspase-7 to switch TNF-induced apoptosis to pyroptosis in HeLa cells. Compared to wild-type mice, Gsdme knockout mice are resistant to the toxic effects of chemotherapeutic drugs, such as cisplatin, 5-fluorouracil, and bleomycin (4). These findings indicate that GSDME is a central molecule that regulates apoptotic cell disassembly and progression to secondary necrosis. Furthermore, these findings reveal a molecular mechanism for secondary necrosis (47). Interestingly, it has been suggested that PJVK localizes to the membrane of peroxisomes in inner hair cells (115). Furthermore, it directly recruits the autophagy protein LC3B to induce autophagy-mediated removal of damaged peroxisomes (pexophagy) following oxidative stress caused by noise overexposure (116). PJVK-driven pexophagy is followed by peroxisome proliferation, which protects auditory hair cells from oxidative damage. Two proteins, rho-associated coiled-coil containing protein kinase 2 and the scaffold protein IQGAP1, were shown to bind the C-terminal region of PJVK in a cochlear cDNA library (117). Whether PJVK can be cleaved by a protease and form membrane pores requires further investigation.
Implicated Diseases Associated With GSDM Family Genes
Dermatological Disorders
Mutated Gsdma3, reduced Coat 2, res-denuded, and bareskin in mice have been identified in mice with alopecia (59, 114), revealing Gsdma3 as a mutation hotspot. Spontaneous mouse mutant defolliculated and chemical induced mutant finnegan also harbor several gsdmd3 mutations. Gsdm3 (defolliculated) is a B2 insertion next to the 3′ splice site of exon 7, whereas Gsdm3 (finnegan) is a point mutation in T278P. Different Gsdma3 mutations are associated with different skin-related phenotypes, including aberrant sebaceous gland differentiation, shortened hair shafts, altered catagen stage of the hair cycle, and loss of the hair follicle, which eventually lead to hair loss (Table 1) (26, 118). However, there are no visible developmental skin abnormalities in Gsdma3–/– mice (51), suggesting that these mutations have gain of function effects. GSDMC is also involved in the physiological and pathological processes of skin metabolism. In response to UV irradiation, GSDMC is upregulated in human keratinocytes. Moreover, GSDMC overexpression contributes to skin damage by promoting ERK-JNK-mediated MMP-1 upregulation in primary human skin keratinocytes (119).
Cancer
The expression and function of GSDM family genes are associated with several types of tumors (Table 1). In this section, we focus on the clinical significance of GSDMs in different types of cancer and their correlation with metastasis, early diagnosis, response to treatment, and prognosis. GSDMA is frequently suppressed in human esophageal, gastric, and skin cancer tissues and tumor cell lines. Furthermore, it is associated with cell death and the sensitivity of cancer to treatment (67, 73). However, whether GSDMA is associated with cleavage and pore formation in cancer cells remains unknown.
In contrast, GSDMB overexpression is observed in many types of cancers, such as human gastric cancer, hepatocellular carcinoma, colon cancers, cervical cancer, breast cancer, and their corresponding cancer-derived cell lines (72). In patients with human epidermal growth factor receptor 2-positive breast cancer, increased GSDMB in tumor cells is associated with an increased incidence of metastasis, reduced survival, poor response to human epidermal growth factor receptor 2-targeted therapy, and poor prognosis. GSDMB is co- expressed with ERBB2 (22, 120) in cancer tissues. However, the mechanism underlying cell survival in cells with an upregulated GSDMB remains unknown. Nevertheless, GSDMB-NT induces pyroptosis when overexpressed in cultured cells.
Overexpressions of GSDMC and GSDMD have been observed in various types of cancer (60). However, there is no consensus on the function of GSDMC in cancer, as it exerts pro- and anti-tumor activity in different tumor types. Studies (56, 57, 79, 80) involving large, multicenter cohorts that examined different types of cancer revealed the role of GSDMC in cancer and confirmed the above findings. Chemotherapeutic reagents, such as etoposide, topotecan, CPT-11, and cisplatin, can induce pyroptosis in cancer cells via a GSDME-dependent mechanism, whereas they promote apoptosis in GSDME-negative cells (4). These studies suggest that GSDME is involved in tumorigenesis. The possible mechanism involves inactivation of GSDME via methylation, which leads to decreased apoptosis, contributing to carcinogenesis (121).
Asthma
Asthma is a chronic airway inflammation of the lung tissues that are infiltrated with massive inflammatory cells. This complex clinical syndrome is characterized by increased airway hyperresponsiveness and reversible airway obstruction. Aside from environmental risk factors, up to 75% patients with asthma have a genetic susceptibility background (122). A study of Korean children (27) revealed that the susceptibility to asthma and intermediate asthma phenotypes, such as elevated IgE and bronchial hyperresponsiveness, is associated with GG of GSDMA (rs7212938) and TT of GSDMB (rs7216389) (27). The levels of DNA methylation and gene expression in patients with asthma indicate that GSDMA is a key factor in the disease pathogenesis (28) (Table 1). Hence, GSDMA is crucial in the immune response of the body to disease. Moreover, the interplay between DNA methylation and GSDMA expression plays a crucial role in an individual’s predisposition to asthma (29). Lower DNA methylation at promoter regions of GSDMA in peripheral blood cells of asthmatic individuals and in lymphoblastoid cell lines correspond to asthma-predisposing alleles (29).
GSDMB is highly expressed in the bronchial epithelium of asthmatic human lungs. In addition, it induces TGF-β1 expression (30). Several studies have shown that genetic variation in GSDMB is associated with asthma susceptibility and asthma-related phenotypes (Table 1), such as IgE11 and a change in FEV1 in response to albuterol (105, 120, 123–126). Positive as well as negative correlations have been observed between rs7216389 of GSDMB and asthma; a meta-analysis found moderate evidence of a correlation between GSDMB rs7216389 variants and asthma (127). GSDMB-regulated genes, cytokines, and chemokines, such as TGF-β1, MMP-9, 5-LO, cysteinyl leukotrienes (LTC4/D4/E4), HSP60/70, and CXCLs, contribute significantly to airway inflammation and remodeling in asthma (2). However, additional studies are required to determine whether GSDMB can be cleaved by inflammatory caspases in asthma. Similar to GSDMB, ORM1-like 3 in 17q is associated with asthma (128, 129). Thus, these 2 genes may be co-regulated, as their transcript levels appear to be connected (130). Larger, more comprehensive studies are needed to provide robust evidence of stratification, identify the interaction between genes and gene-environment interactions, and identify the factors important in asthma.
Non-Syndromic Hearing Impairment
In 1998, GSDME (DFNA5) mutation was first identified as a cause of a specific form of progressive, non-syndromic and autosomal dominant hearing loss in a Dutch family (121). Later, many other families suffering from hearing loss were also found to have GSDME (DFNA5) mutations (Table 1) (31). Although these mutations differed at the DNA level, each skipped exon 8 of the GSDME (DFNA5) mRNA transcript, resulting in a frameshift mutation and premature truncation of the protein with cytotoxic activity (121, 131, 132). The mutation in exon 6 of GSDME (DFNA5) is not specific for hearing loss (HL). In fact, it is present in family members with normal hearing (133). These findings were supported by a study of Gsdme (Dfna5)-knockout mice, which did not display HL and suggest that GSDME (DFNA5)-associated HL is an activating and a gain-of-function mutation. The effect is associated with the reported apoptotic inducing capacity of GSDME (DFNA5), in which an increase in apoptosis leads to HL by inducing the death of cells that are crucial for hearing, such as cochlear hair cells (111). Similar to GSDME (DFNA5), mutations in PJVK are associated with HL in both humans and mice (20, 32, 65). Unlike the gain-of-function mutation of GSDME (DFNA5), all known mutations in PJVK are associated with autosomal recessive on syndromic sensorineural HL with or without cochlear dysfunction (32, 134–136). In mice, functional PJVK is necessary to enable existing peroxisomes to proliferate and protect the cochlear sensory hair cells and auditory neurons from noise-induced generation of reactive oxidative species (115). Interestingly, PJVK translocates to the membrane of peroxisomes in the inner hair cells and directly binds to the autophagy protein LC3B to induce autophagy-mediated removal of damaged peroxisomes (pexophagy) following oxidative stress caused by noise overexposure. Knockout of PJVK protects mice from peroxisomal dysfunction and sound-induced pexophagy (116). These results suggest that PJVK-induced pexophagy plays a crucial role in noise-induced peroxisome proliferation. Furthermore, these findings regard defective pexophagy as a cause of noise-induced hearing loss.
Autoimmune- and Inflammation-Driven Diseases
Autoimmune diseases constitute a large group of diseases characterized by a functionally abnormal immune system. Aberrant activation of the immune system results in the production of antibodies against one own cells and tissues, known as autoimmunity. Several studies have implicated GSDMs in autoimmune and inflammation-driven diseases. GSDMA mutants have been linked to limited cutaneous system sclerosis (33, 34) and inflammatory bowel disease (Table 1) (35). The results of transethnic meta-analysis of genome-wide associated studies involving Japanese and European populations with a total of 4,436 cases and 14,751 controls revealed that a missense mutation in GSDMA (rs3894194) is associated with system sclerosis. The rs3894194 was strongly associated with GSDMB and ORM1-like 3 expression and the enhancer activity and enrichment of histone markers (33). RNA sequencing and genome-wide genotyping of monocyte-derived macrophages from patients with system sclerosis revealed that the GSDMA rs3894194 risk variant contributed to several inflammatory pathways and system sclerosis susceptibility, characterized by upregulation of glycolysis, hypoxia, and mTOR signaling and downregulation of the IFN-γ response pathway (137). In contrast to GSDMA, reduced expression of GSDMB increased the susceptibility to inflammatory bowel disease (35). However, another study pointed out that upregulated GSDMB in patients with sepsis and Crohn’s disease promoted caspase-4 activity and GSDMD cleavage (35), revealing a GSDMB-regulated novel regulatory mechanism for GSDMD-induced pyroptosis in inflammatory diseases.
Aberrant inflammasome activation and GSDMD-dependent pyroptosis are important pathogenic mechanisms underlying immune-related and inflammatory diseases, including atherosclerosis, type 2 diabetes mellitus, bullous pemphigoid, and cryopyrin-associated periodic syndromes (Table 1) (36–39). GSDMD-mediated pyroptosis plays a crucial role in the pathogenesis of familial Mediterranean fever in mouse models (138). Infection of familial Mediterranean fever knock-in macrophages with Clostridium difficile resulted in the expression of a chimeric familial Mediterranean fever-associated Mefv (V726A) pyrin-induced pyroptosis and GSDMD-mediated IL-1BIL-1B secretion in vitro. Importantly, GSDMD deletion protected mice from tissue damage. This characterized the autoinflammatory disease model in vivo, highlighting a potential strategy of GSDMD inhibition in inflammasome-driven diseases therapy. Unlike other GSDMs, DFNB59 lacks the cleavable linker domain. Thus, no study has reported that PJVK can form pores in the plasma membranes. Whether PJVK can be activated via inflammatory caspases and mediate pyroptosis requires further analysis.
Sepsis and Septic Shock
Sepsis, as defined by the Third International Consensus Definitions for Sepsis and Septic Shock guidelines, is a life-threatening organ dysfunction caused by a dysregulated host response to infection (139). Septic shock is defined as severe sepsis, in which particularly profound circulatory, cellular, and metabolic abnormalities are associated with a greater risk of mortality than with sepsis alone. LPS, the major structural element of the outer membrane of gram-negative bacteria, triggers strong immune responses. In addition, LPS has been intensively implicated in the pathology of sepsis. Excessive LPS resulting from uncontrolled infection induces both sepsis and septic shock (140). Early studies demonstrated that in innate immunity, extracellular LPS is recognized by Toll-like receptor 4 (TLR4) to stimulate cytokine transcription. Tlr4-/–deficient mice are resistant to endotoxic shock (141). Priming of the mice with TLR3 ligand to upregulate caspase-11 expression can bypass the requirement for TLR4 (84). Thus, the role of TLR4 in mouse endotoxic shock is largely limited to stimulation of caspase-11 expression. The re-defined role of TLR4 provides a plausible explanation for the failure of using the TLR4 antagonist Eritoran to treat patients with sepsis.
An important study conducted by Kayagaki et al. (25) revealed the crucial role of pro-inflammatory caspase-11 in caspase-1 activation and IL1b production in macrophages infected with E. coli, Citrobacter rodentium, or Vibrio cholerae. Following caspase 11 and caspase 1 gene manipulation in C57BL/6 mice, they provided evidence that caspase-11 (also known as human caspase-4) was critical for downstream caspase-1 activation. Their in vivo data also indicated that caspase-11 rather than caspase-1 is the critical effector of deleterious inflammatory responses in LPS-induced endothemia. Therefore, targeting human caspase-4 and caspase-5 (caspase 11 in mouse) may be more effective than inhibiting caspase-1 in patients with sepsis (25). In vivo, Gsdmd–/– and Casp11–/– mice show an increased susceptibility to infections with Salmonella enterica subsp. enterica serovar, Typhimurium, SifA, or Brucella abortus (142). In contrast, Gsdmd–/– mice were less susceptible to infection with Francisella novicida than Casp1–/– or Aim2–/– mice (143). Blood assays revealed that when primed with the TLR3 agonist poly(I:C), LPS-induced coagulation factor III (F3) release and lethal coagulation in mice suffering from sepsis required CASP11 but not CASP1-mediated GSDMD activation (69). Animal death caused by sepsis in response to intravenous injection of PAMP from E. coli was attenuated in mice with Casp1, Casp1/11, and Gsdmd depletion but not in Tlr4–/–, Casp11–/– mice or mice lacking receptors for IL-1B (Il1r–/–) and IL-18 (Il18r–/–). There is also evidence that intraperitoneal injection of necrosulfonamide, an inhibitor of the necroptosis effector mixed lineage kinase domain like pseudokinase, acts as a chemical inhibitor of GSDMD, thereby protecting mice from LPS-induced sepsis compared to controls (144). Thus, the critical role of intracellular LPS-caspase11/4/5-GSDMD axis-induced proptosis in sepsis provides a new avenue for the development of anti-septic therapeutics and merits further investigation (Table 1).
Other Diseases
Additionally, NLRP3/caspase-1/GSDMD activation and subsequent pyroptosis are associated with the pathogenesis of brain injury in a middle cerebral artery occlusion/reperfusion rat model (145). GSDMD activation and pytoptosis are induced in the brain tissues of mice after ischemia/reperfusion damage. Furthermore, GSDMD-NT can bind to membrane lipids and exhibit membrane-disrupting cytotoxicity in primary-cultured microglia, depending on the Glu15 and Leu156 amino acid sites. Inhibition of GSDMD and its upstream modulators, such as MCC950, LDC7559, and necrosulfonamide, with chemical compounds can attenuate the progression of diseases such as hepatic ischemia-reperfusion injury in rat models (146) and LPS-induced septic death (48, 144). Serum-derived exosomes from hepatic ischemia-reperfusion injury can not only cross the blood-brain barrier but also contribute to hippocampal and cortical neuronal pyroptosis via NLRP3-caspase-1-GSDMD activation and IL-1BIL-1B and IL-18 release in rat models (146). The small molecular specific inhibitor of NLRP3 MCC950 inhibits caspase-1 and GSDMD activation and attenuates subsequent hippocampal and cortical neuronal injury in a hepatic ischemia-reperfusion model of rats.
Concluding Remarks and Future Perspectives
Reports on GSDMD as a key effector of pyroptosis have improved the understanding of the GSDM family as an important executor of cell death. Many studies have described the role of gasdermins in regulating various physiological and pathological processes, including cell differentiation, cell death, coagulation, inflammation, and tumorigenesis. Studies of the structure and mutagenesis of full-length gasdermins revealed the mechanism of self-inhibition and membrane insertion. The GSDM-NT domains released following cleavage of GSDMA, GSDMB (or full-length GSDMB), GSDMC, GSDMD, or GSDME can form membrane pores, thereby acting as executors of pyroptosis. Specific gasdermin inhibitors have only recently been identified and require further analysis. Targeting the activity and function of GSMDs provides a potential strategy for treating diseases, particularly infections and inflammatory conditions. Therefore, studies are needed to evaluate the distinct resources of gasdermins in different cellular processes and their biological and pathological effects, identify shear molecules responsible for PJVK, discovering other functions besides cell lysis and pyroptosis, recognizing other regulators of GSDMs in cell death, and developing a GSDM-dependent strategy for disease treatment.
Author Contributions
Conceptualization, RC and JZ. Writing—original draft, RC. Writing—review and editing, DT, RK, YZ, YH, and JZ. All authors contributed to the article and approved the submitted version.
Funding
The manuscript was supported by grants from the National Natural Sciences Foundation of Hunan province (#2019JJ30041), National Natural Sciences Foundation of China (#82070613), and Innovation-Driven Project of Central South University (#2020CX044).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Editage for revising the manuscript.
Abbreviations
GSDM, gasdermin; GSDMA, gasdermin A; GSDMB, gasdermin B; GSDMC, gasdermin C; GSDMD, gasdermin D; DFNA5, deafness autosomal dominant 5; Rim3, recombination-induced mutation 3; CASP1, chloroquine down-regulated caspase-1; PJVK, pejvakin; GSDM-NT, GSDM N-terminal; GSDM-CT, GSDM C-terminal; LPS, lipopolysaccharide; NET, neutrophil extracellular trap; HL, hearing loss; FMF, familial Mediterranean fever; TLR4, Toll-like receptor 4.
References
1. Panganiban RA, Sun M, Dahlin A, Park HR, Kan M, Himes BE, et al. A Functional Splice Variant Associated With Decreased Asthma Risk Abolishes the Ability of Gasdermin B to Induce Epithelial Cell Pyroptosis. J Allergy Clin Immunol (2018) 142(5):1469–78.e2. doi: 10.1016/j.jaci.2017.11.040
2. Das S, Miller M, Broide DH. Chromosome 17q21 Genes ORMDL3 and GSDMB in Asthma and Immune Diseases. Adv Immunol (2017) 135:1–52. doi: 10.1016/bs.ai.2017.06.001
3. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
4. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy Drugs Induce Pyroptosis Through Caspase-3 Cleavage of a Gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
5. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
6. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
7. Xia SY. Biological Mechanisms and Therapeutic Relevance of the Gasdermin Family. Mol Aspects Med (2020) 76:100890. doi: 10.1016/j.mam.2020.100890
8. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and Intrinsic Apoptosis Activate Pannexin-1 to Drive NLRP3 Inflammasome Assembly. EMBO J (2019) 38(10):e101638. doi: 10.15252/embj.2019101638
9. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E Suppresses Tumour Growth by Activating Anti-Tumour Immunity. Nature (2020) 579(7799):415–20. doi: 10.1038/s41586-020-2071-9
10. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
11. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation During Apoptosis and Inflammasome Activation. Nat Commun (2019) 10(1):1689. doi: 10.1038/s41467-019-09397-2
12. Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep (2018) 25(6):1525–36.e7. doi: 10.1016/j.celrep.2018.09.071
13. Aglietti RA, Dueber EC. Recent Insights Into the Molecular Mechanisms Underlying Pyroptosis and Gasdermin Family Functions. Trends Immunol (2017) 38(4):261–71. doi: 10.1016/j.it.2017.01.003
14. Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. BioGPS: An Extensible and Customizable Portal for Querying and Organizing Gene Annotation Resources. Genome Biol (2009) 10(11):R130. doi: 10.1186/gb-2009-10-11-r130
15. Tanaka S, Mizushina Y, Kato Y, Tamura M, Shiroishi T. Functional Conservation of Gsdma Cluster Genes Specifically Duplicated in the Mouse Genome. G3 (Bethesda) (2013) 3(10):1843–50. doi: 10.1534/g3.113.007393
16. Rieckmann JC, Geiger R, Hornburg D, Wolf T, Kveler K, Jarrossay D, et al. Social Network Architecture of Human Immune Cells Unveiled by Quantitative Proteomics. Nat Immunol (2017) 18(5):583–93. doi: 10.1038/ni.3693
17. de Beeck KO, Laer LVP, Camp JV. DFNA5, a Gene Involved in Hearing Loss and Cancer: A Review. Genome Biol (2009) 10(11):R130. doi: 10.1177/000348941212100310
18. Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol Cell Proteomics (2014) 13(2):397–406. doi: 10.1074/mcp.M113.035600
19. Vrijens K, Van Camp G, Van Laer L. Characterization of the Murine Dfna5 Promoter and Regulatory Regions. Gene (2009) 432(1-2):82–90. doi: 10.1016/j.gene.2008.11.017
20. Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, et al. Mutations in the Gene Encoding Pejvakin, a Newly Identified Protein of the Afferent Auditory Pathway, Cause DFNB59 Auditory Neuropathy. Nat Genet (2006) 38(7):770–8. doi: 10.1038/ng1829
21. Maeda YFK, Kasai N, Maeta M, Nishizaki K. Quantification of TECTA and DFNA5 Expression in the Developing Mouse Cochlea. Neuroreport (2001) 12(15):3223–6. doi: 10.1097/00001756-200110290-00016
22. Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Megias D, Mota A, Rojo-Sebastian A, et al. Gasdermin-B Promotes Invasion and Metastasis in Breast Cancer Cells. PloS One (2014) 9(3):e90099. doi: 10.1371/journal.pone.0090099
23. Lin PH, Lin HY, Kuo CC, Yang LT. N-Terminal Functional Domain of Gasdermin A3 Regulates Mitochondrial Homeostasis via Mitochondrial Targeting. J BioMed Sci (2015) 22(1):44. doi: 10.1186/s12929-015-0152-0
24. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
25. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
26. Zhou Y, Jiang X, Gu P, Chen W, Zeng X, Gao X. Gsdma3 Mutation Causes Bulge Stem Cell Depletion and Alopecia Mediated by Skin Inflammation. Am J Pathol (2012) 180(2):763–74. doi: 10.1016/j.ajpath.2011.10.034
27. Yu J, Kang MJ, Kim BJ, Kwon JW, Song YH, Choi WA, et al. Polymorphisms in GSDMA and GSDMB Are Associated With Asthma Susceptibility, Atopy and BHR. Pediatr Pulmonol (2011) 46(7):701–8. doi: 10.1002/ppul.21424
28. Madore AM, Pain L, Boucher-Lafleur AM, Morin A, Meloche J, Simon MM, et al. Asthma-Associated Polymorphisms in 17q12-21 Locus Modulate Methylation and Gene Expression of GSDMA in Naive CD4(+) T Cells. J Genet Genomics (2020) 47(3):171–74. doi: 10.1016/j.jgg.2020.03.002
29. Al Tuwaijri A, Gagne-Ouellet V, Madore AM, Laprise C, Naumova AK. Local Genotype Influences DNA Methylation at Two Asthma-Associated Regions, 5q31 and 17q21, in a Founder Effect Population. J Med Genet (2016) 53(4):232–41. doi: 10.1136/jmedgenet-2015-103313
30. Das S, Miller M, Beppu AK, Mueller J, McGeough MD, Vuong C, et al. GSDMB Induces an Asthma Phenotype Characterized by Increased Airway Responsiveness and Remodeling Without Lung Inflammation. Proc Natl Acad Sci USA (2016) 113(46):13132–37. doi: 10.1073/pnas.1610433113
31. Park HJ, Cho HJ, Baek JI, Ben-Yosef T, Kwon TJ, Griffith AJ, et al. Evidence for a Founder Mutation Causing DFNA5 Hearing Loss in East Asians. J Hum Genet (2010) 55(1):59–62. doi: 10.1038/jhg.2009.114
32. Mujtaba G, Bukhari I, Fatima A, Naz S. A P.C343S Missense Mutation in PJVK Causes Progressive Hearing Loss. Gene (2012) 504(1):98–101. doi: 10.1016/j.gene.2012.05.013
33. Terao C, Kawaguchi T, Dieude P, Varga J, Kuwana M, Hudson M, et al. Transethnic Meta-Analysis Identifies GSDMA and PRDM1 as Susceptibility Genes to Systemic Sclerosis. Ann Rheum Dis (2017) 76(6):1150–58. doi: 10.1136/annrheumdis-2016-210645
34. Orvain C, Assassi S, Avouac J, Allanore Y. Systemic Sclerosis Pathogenesis: Contribution of Recent Advances in Genetics. Curr Opin Rheumatol (2020) 32(6):505–14. doi: 10.1097/BOR.0000000000000735
35. Soderman J, Berglind L, Almer S. Gene Expression-Genotype Analysis Implicates GSDMA GSDMB, and LRRC3C as Contributors to Inflammatory Bowel Disease Susceptibility. BioMed Res Int (2015) 2015:834805. doi: 10.1155/2015/834805
36. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 Inflammasome Inhibition is Disrupted in a Group of Auto-Inflammatory Disease CAPS Mutations. Nat Immunol (2016) 17(10):1176–86. doi: 10.1038/ni.3538
37. Xiao J, Wang C, Yao JC, Alippe Y, Xu C, Kress D, et al. Gasdermin D Mediates the Pathogenesis of Neonatal-Onset Multisystem Inflammatory Disease in Mice. PloS Biol (2018) 16(11):e3000047. doi: 10.1371/journal.pbio.3000047
38. Le Jan S, Muller C, Plee J, Durlach A, Bernard P, Antonicelli F. IL-23/IL-17 Axis Activates IL-1beta-Associated Inflammasome in Macrophages and Generates an Auto-Inflammatory Response in a Subgroup of Patients With Bullous Pemphigoid. Front Immunol (2019) 10:1972. doi: 10.3389/fimmu.2019.01972
39. Wan Z, Fan Y, Liu X, Xue J, Han Z, Zhu C, et al. NLRP3 Inflammasome Promotes Diabetes-Induced Endothelial Inflammation and Atherosclerosis. Diabetes Metab Syndr Obes (2019) 12:1931–42. doi: 10.2147/DMSO.S222053
40. Kumar S, Rathkolb B, Budde BS, Nürnberg P, de Angelis MH, Aigner B, et al. Gsdma3(I359N) Is a Novel ENU-Induced Mutant MouseLline for Studying the Function of Gasdermin A3 in the Hair Follicle and Epidermis. J Dermatol Sci (2012) 67(3):190–2. doi: 10.1016/j.jdermsci.2012.05.001
41. Watabe K, Ito A, Asada H, Endo Y, Kobayashi T, Nakamoto K’i, et al. Structure, Expression and Chromosome Mapping of MLZE, a Novel Gene Which Is Preferentially Expressed in Metastatic Melanoma Cells. Jpn J Cancer Res (2001) 92(2):140–51. doi: 10.1111/j.1349-7006.2001.tb01076.x
42. Laer LV, Huizing EH, Verstreken M, Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic Hearing Impairment Is Associated With a Mutation in DFNA5. Nat Genet (1998) 20(2):194–7. doi: 10.1038/2503
43. Sun Q, Zhang LQ, He FC. Progress of Researches on Gene Function of GSDMDC Family. Yi Chuan (2006) 28(5):596–600.
44. Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T. Gasdermin (Gsdm) Localizing to Mouse Chromosome 11 Is Predominantly Expressed in Upper Gastrointestinal Tract But Significantly Suppressed in Human Gastric Cancer Cells. Mamm Genome (2000) 11(9):718–24. doi: 10.1007/s003350010138
45. Cui H, Zhang L. Key Components of Inflammasome and Pyroptosis Pathways Are Deficient in Canines and Felines, Possibly Affecting Their Response to SARS-CoV-2 Infection. Front Immunol (2020) 11:592622. doi: 10.3389/fimmu.2020.592622
46. Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, et al. Members of a Novel Gene Family, Gsdm, Are Expressed Exclusively in the Epithelium of the Skin and Gastrointestinal Tract in a Highly Tissue-Specific Manner. Genomics (2007) 89(5):618–29. doi: 10.1016/j.ygeno.2007.01.003
47. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by Caspase-3 During Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
48. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat Chem Biol (2019) 15(6):556–59. doi: 10.1038/s41589-019-0277-7
49. Ruan J. Structural Insight of Gasdermin Family Driving Pyroptotic Cell Death. Adv Exp Med Biol (2019) 1172(1172):189–205. doi: 10.1007/978-981-13-9367-9_9
50. Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity (2019) 51(1):43–49.e4. doi: 10.1016/j.immuni.2019.04.017
51. Shi P, Tang A, Xian L, Hou S, Zou DY, Lv YS, et al. Loss of Conserved Gsdma3 Self-Regulation Causes Autophagy and Cell Death. Biochem J (2015) 468(2):325–36. doi: 10.1042/BJ20150204
52. Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM Structure of the Gasdermin A3 Membrane Pore. Nature (2018) 557(7703):62–7. doi: 10.1038/s41586-018-0058-6
53. Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Muller DJ. Mechanism of Membrane Pore Formation by Human Gasdermin-D. EMBO J (2018) 37(14):e98321. doi: 10.15252/embj.201798321
54. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD P30 Elicited by Caspase-11 During Pyroptosis Forms Pores in Membranes. Proc Natl Acad Sci USA (2016) 113(28):7858–63. doi: 10.1073/pnas.1607769113
55. Mangan DF, Welch GR, Wahl SM. Lipopolysaccharide, Tumor Necrosis Factor-Alpha, and IL-1 Beta Prevent Programmed Cell Death (Apoptosis) in Human Peripheral Blood Monocytes. J Immunol (1991) 146(5):1541–6.
56. Broz P, Pelegrin P, Shao F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat Rev Immunol (2020) 20(3):143–57. doi: 10.1038/s41577-019-0228-2
57. Saeki N, Kim DH, Usui T, Aoyagi K, Tatsuta T, Aoki K, et al. GASDERMIN, Suppressed Frequently in Gastric Cancer, Is a Target of LMO1 in TGF-Beta-Dependent Apoptotic Signalling. Oncogene (2007) 26(45):6488–98. doi: 10.1038/sj.onc.1210475
58. Tanaka S, Tamura M, Aoki A, Fujii T, Komiyama H, Sagai T, et al. A New Gsdma3 Mutation Affecting Anagen Phase of First Hair Cycle. Biochem Biophys Res Commun (2007) 359(4):902–7. doi: 10.1016/j.bbrc.2007.05.209
59. Lunny DP, Weed E, Nolan PM, Marquardt A, Augustin M, Porter RM. Mutations in Gasdermin 3 Cause Aberrant Differentiation of the Hair Follicle and Sebaceous Gland. J Invest Dermatol (2005) 124(3):615–21. doi: 10.1111/j.0022-202X.2005.23623.x
60. Watabe K, Ito A, Asada H, Endo Y, Kobayashi T, Nakamoto K, et al. Structure, Expression and Chromosome Mapping of MLZE, a Novel Gene Which Is Preferentially Expressed in Metastatic Melanoma Cells. Jpn J Cancer Res (2001) 92(2):140–51. doi: 10.1111/j.1349-7006.2001.tb01076.x
61. Katoh M, Katoh M. Identification and Characterization of Human DFNA5L, Mouse Dfna5l, and Rat Dfna5l Genes In Silico. Int J Oncol (2004) 25(3):765–70. doi: 10.3892/ijo.25.3.765
62. Leimeister C, Bach A, Woolf AS, Gessler M. Screen for Genes Regulated During Early Kidney Morphogenesis. Dev Genet (1999) 24(3-4):273–83. doi: 10.1002/(SICI)1520-6408(1999)24:3/4<273::AID-DVG10>3.0.CO;2-3
63. Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, et al. A High-Resolution Anatomical Atlas of the Transcriptome in the Mouse Embryo. PloS Biol (2011) 9(1):e1000582. doi: 10.1371/journal.pbio.1000582
64. Maeda Y, Fukushima K, Kasai N, Maeta M, Nishizaki K. Quantification of TECTA and DFNA5 Expression in the Developing Mouse Cochlea. Neuroreport (2001) 12(15):3223–6. doi: 10.1097/00001756-200110290-00016
65. Collin RW, Kalay E, Oostrik J, Oostrik J, Caylan R, Wollnik B, et al. Involvement of DFNB59 Mutations in Autosomal Recessive Nonsyndromic Hearing Impairment. Hum Mutat (2007) 28(7):718–23. doi: 10.1002/humu.20510
66. Liu W, Kinnefors A, Bostrom M, Edin F, Rask-Andersen H. Distribution of Pejvakin in Human Spiral Ganglion: An Immunohistochemical Study. Cochlear Implants Int (2013) 14(4):225–31. doi: 10.1179/1754762812Y.0000000027
67. Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive Expression and Function of Four GSDM Family Genes (GSDMA-D) in Normal and Malignant Upper Gastrointestinal Epithelium. Genes Chromosomes Cancer (2009) 48(3):261–71. doi: 10.1002/gcc.20636
68. Li J, Zhou Y, Yang T, Wang N, Lian X, Yang L. Gsdma3 Is Required for Hair Follicle Differentiation in Mice. Biochem Biophys Res Commun (2010) 403(1):18–23. doi: 10.1016/j.bbrc.2010.10.094
69. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome Activation Triggers Blood Clotting and Host Death Through Pyroptosis. Immunity (2019) 50(6):1401–11.e4. doi: 10.1016/j.immuni.2019.04.003
70. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial Endotoxin Activates the Coagulation Cascade Through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity (2019) 51(6):983–96.e6. doi: 10.1016/j.immuni.2019.11.005
71. Zhang H, Zeng L, Xie M, Liu J, Zhou B, Wu R, et al. TMEM173 Drives Lethal Coagulation in Sepsis. Cell Host Microbe (2020) 27(4):556–70.e6. doi: 10.1016/j.chom.2020.02.004
72. Sun Q, Yang J, Xing G, Sun Q, Zhang L, He F. Expression of GSDML Associates With Tumor Progression in Uterine Cervix Cancer. Transl Oncol (2008) 1(2):73–83. doi: 10.1593/tlo.08112
73. Miguchi M, Hinoi T, Shimomura M, Adachi T, Saito Y, Niitsu H, et al. Gasdermin C Is Upregulated by Inactivation of Transforming Growth Factor Beta Receptor Type II in the Presence of Mutated Apc, Promoting Colorectal Cancer Proliferation. PloS One (2016) 11(11):e0166422. doi: 10.1371/journal.pone.0166422
74. Xi G, Gao J, Wan B, Zhan P, Xu W, Lv T, et al. GSDMD is Required for Effector CD8(+) T Cell Responses to Lung Cancer Cells. Int Immunopharmacol (2019) 74:105713. doi: 10.1016/j.intimp.2019.105713
75. Gao J, Qiu X, Xi G, Liu HB, Zhang F, Lv TF, et al. Downregulation of GSDMD Attenuates Tumor Proliferation via the Intrinsic Mitochondrial Apoptotic Pathway and Inhibition of EGFR/Akt Signaling and Predicts a Good Prognosis in non−Small Cell Lung Cancer. Oncol Rep (2018) 40(4):1971–84. doi: 10.3892/or.2018.6634
76. Ibrahim J, Op de Beeck K, Fransen E, Peeters M, Van Camp G. The Gasdermin E Gene Has Potential as a Pan-Cancer Biomarker, While Discriminating Between Different Tumor Types. Cancers (Basel) (2019) 11(11):1810. doi: 10.3390/cancers11111810
77. De Schutter E, Croes L, Ibrahim J, Pauwels P, Op de Beeck K, Vandenabeele P, et al. GSDME and its Role in Cancer: From Behind the Scenes to the Front of the Stage. Int J Cancer (2021) 148(12):2872–83. doi: 10.1002/ijc.33390
78. Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, et al. Identification of DFNA5 as a Target of Epigenetic Inactivation in Gastric Cancer. Cancer Sci (2007) 98(1):88–95. doi: 10.1111/j.1349-7006.2006.00351.x
79. Kim MS, Lebron C, Nagpal JK, Chae YK, Chang X, Huang Y, et al. Methylation of the DFNA5 Increases Risk of Lymph Node Metastasis in Human Breast Cancer. Biochem Biophys Res Commun (2008) 370(1):38–43. doi: 10.1016/j.bbrc.2008.03.026
80. Kim MS, Chang X, Yamashita K, Nagpal JK, Baek JH, Wu J, et al. Aberrant Promoter Methylation and Tumor Suppressive Activity of the DFNA5 Gene in Colorectal Carcinoma. Oncogene (2008) 27(25):3624–34. doi: 10.1038/sj.onc.1211021
81. Jorgensen I, Miao EA. Pyroptotic Cell Death Defends Against Intracellular Pathogens. Immunol Rev (2015) 265(1):130–42. doi: 10.1111/imr.12287
82. Man SM, Kanneganti TD. Converging Roles of Caspases in Inflammasome Activation, Cell Death and Innate Immunity. Nat Rev Immunol (2016) 16(1):7–21. doi: 10.1038/nri.2015.7
83. Zambetti LP, Laudisi F, Licandro G, Ricciardi-Castagnoli P, Mortellaro A. The Rhapsody of NLRPs: Master Players of Inflammation...and a Lot More. Immunol Res (2012) 53(1-3):78–90. doi: 10.1007/s12026-012-8272-z
84. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS Activates Caspase-11 Implications in TLR4-Independent Endotoxic Shock. Science (2013) 341(6151):1250–3. doi: 10.1126/science.1240988
85. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Takamura SA, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science (2013) 341(6151):1246–9. doi: 10.5061/dryad.bt51g. 10.5061/dryad.bt51g).Supplementary.
86. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory Caspases are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
87. Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human Caspase-4 and Caspase-5 Regulate the One-Step Non-Canonical Inflammasome Activation in Monocytes. Nat Commun (2015) 6:8761. doi: 10.1038/ncomms9761
88. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1beta Secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
89. Agard NJ, Maltby D, Wells JA. Inflammatory Stimuli Regulate Caspase Substrate Profiles. Mol Cell Proteomics (2010) 9(5):880–93. doi: 10.1074/mcp.M900528-MCP200
90. Benaoudia S, Martin A, Puig Gamez M, Gay G, Lagrange B, Cornut M, et al. A Genome-Wide Screen Identifies IRF2 as a Key Regulator of Caspase-4 in Human Cells. EMBO Rep (2019) 20(9):e48235. doi: 10.15252/embr.201948235
91. Kayagaki N, Lee BL, Stowe IB, Kornfeld OS, O'Rourke K, Mirrashidi KM, et al. IRF2 Transcriptionally Induces GSDMD Expression for Pyroptosis. Sci Signal (2019) 12(582):eaax4917. doi: 10.1126/scisignal.aax4917
92. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J (2016) 35(16):1766–78. doi: 10.15252/embj.201694696
93. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion From Living Macrophages. Immunity (2018) 48(1):35–44.e6. doi: 10.1016/j.immuni.2017.11.013
94. Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin-D Pore Acts as a Conduit for IL-1beta Secretion in Mice. Eur J Immunol (2018) 48(4):584–92. doi: 10.1002/eji.201747404
95. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen Blockade of TAK1 Triggers Caspase-8-Dependent Cleavage of Gasdermin D and Cell Death. Science (2018) 362(6418):1064–69. doi: 10.1126/science.aau2818
96. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-Dependent Membrane Repairnegatively Regulates Pyroptosis Downstream of GSDMD Activation. Science (2018) 362(6417):956–60. doi: 10.1126/science.aar7607
97. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat Immunol (2020) 21(7):736–45. doi: 10.1038/s41590-020-0669-6
98. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang BW, Nemmara VV, et al. Succination Inactivates Gasdermin D and Blocks Pyroptosis. Science (2020) 369(6511):1633–37. doi: 10.1126/science.abb9818
99. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6676. doi: 10.1126/sciimmunol.aar6676
100. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D Exerts Anti-Inflammatory Effects by Promoting Neutrophil Death. Cell Rep (2018) 22(11):2924–36. doi: 10.1016/j.celrep.2018.02.067
101. Sollberger G, Choidas A, Burn GL, Habenberger P, Lucrezia RD, Kordes S, et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
102. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis Triggers Pore-Induced Intracellular Traps (PITs) That Capture Bacteria and Lead to Their Clearance by Efferocytosis. J Exp Med (2016) 213(10):2113–28. doi: 10.1084/jem.20151613
103. Lei M, Bai X, Yang T, Lai X, Qiu W, Yang L, et al. Gsdma3 is a New Factor Needed for TNF-Alpha-Mediated Apoptosis Signal Pathway in Mouse Skin Keratinocytes. Histochem Cell Biol (2012) 138(3):385–96. doi: 10.1007/s00418-012-0960-1
104. Chao KL, Kulakova L, Herzberg O. Gene Polymorphism Linked to Increased Asthma and IBD Risk Alters Gasdermin-B Structure, a Sulfatide and Phosphoinositide Binding Protein. Proc Natl Acad Sci USA (2017) 114(7):E1128–37. doi: 10.1073/pnas.1616783114
105. Binia A, Khorasani N, Bhavsar PK, Adcock I, Brightling CE, Chung KF, et al. Chromosome 17q21 SNP and Severe Asthma. J Hum Genet (2011) 56(1):97–8. doi: 10.1038/jhg.2010.134
106. Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, et al. GSDMB Promotes Non-Canonical Pyroptosis by Enhancing Caspase-4 Activity. J Mol Cell Biol (2019) 11(6):496–508. doi: 10.1093/jmcb/mjy056
107. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-Mediated Gasdermin C Expression Switches Apoptosis to Pyroptosis in Cancer Cells and Facilitates Tumour Necrosis. Nat Cell Biol (2020) 22(10):1264–75. doi: 10.1038/s41556-020-0575-z
108. Li YQ, Peng JJ, Peng J, Luo XJ. The Deafness Gene GSDME: Its Involvement in Cell Apoptosis, Secondary Necrosis, and Cancers. Naunyn Schmiedebergs Arch Pharmacol (2019) 392(9):1043–48. doi: 10.1007/s00210-019-01674-7
109. Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ, et al. Chemotherapeutic Paclitaxel and Cisplatin Differentially Induce Pyroptosis in A549 Lung Cancer Cells via Caspase-3/GSDME Activation. Apoptosis (2019) 24(3-4):312–25. doi: 10.1007/s10495-019-01515-1
110. Jiang M, Qi L, Li L, Li Y. The Caspase-3/GSDME Signal Pathway as a Switch Between Apoptosis and Pyroptosis in Cancer. Cell Death Discovery (2020) 6:112. doi: 10.1038/s41420-020-00349-0
111. Op de Beeck K, Van Camp G, Thys S, Cools N, Callebaut I, Vrijens K, et al. The DFNA5 Gene, Responsible for Hearing Loss and Involved in Cancer, Encodes a Novel Apoptosis-Inducing Protein. Eur J Hum Genet (2011) 19(9):965–73. doi: 10.1038/ejhg.2011.63
112. Van Rossom S, Op de Beeck K, Franssens V, Swinnen E, Schepers A, Ghillebert R, et al. The Splicing Mutant of the Human Tumor Suppressor Protein DFNA5 Induces Programmed Cell Death When Expressed in the Yeast Saccharomyces Cerevisiae. Front Oncol (2012) 2:77. doi: 10.3389/fonc.2012.00077
113. Bischoff AM, Luijendijk MW, Huygen PL, van Duijnhoven G, De Leenheer EM, Oudesluijs GG, et al. A Novel Mutation Identified in the DFNA5 Gene in a Dutch Family: A Clinical and Genetic Evaluation. Audiol Neurootol (2004) 9(1):34–46. doi: 10.1159/000074185
114. Masuda Y, Futamura M, Kamino H, Nakamura Y, Kitamura N, Ohnishi S, et al. The Potential Role of DFNA5, a Hearing Impairment Gene, in P53-Mediated Cellular Response to DNA Damage. J Hum Genet (2006) 51(8):652–64. doi: 10.1007/s10038-006-0004-6
115. Delmaghani S, Defourny J, Aghaie A, Beurg M, Dulon D, Thelen N, et al. Hypervulnerability to Sound Exposure Through Impaired Adaptive Proliferation of Peroxisomes. Cell (2015) 163(4):894–906. doi: 10.1016/j.cell.2015.10.023
116. Defourny J, Aghaie A, Perfettini I, Avan P, Delmaghani S, Petit C. Pejvakin-Mediated Pexophagy Protects Auditory Hair Cells Against Noise-Induced Damage. Proc Natl Acad Sci USA (2019) 116(16):8010–17. doi: 10.1073/pnas.1821844116
117. Harris SL, Kazmierczak M, Pangrsic T, Shah P, Chuchvara N, Barrantes-Freer A, et al. Conditional Deletion of Pejvakin in Adult Outer Hair Cells Causes Progressive Hearing Loss in Mice. Neuroscience (2017) 344:380–93. doi: 10.1016/j.neuroscience.2016.12.055
118. Kumar S, Rathkolb B, Budde BS, Nürnberg P, de Angelis MH, Aigner B, et al. Gsdma3(I359N) Is a Novel ENU-Induced Mutant Mouse Line for Studying the Function of Gasdermin A3 in the Hair Follicle and Epidermis. J Dermatol Sci (2012) 67(3):190–2. doi: 10.1016/j.jdermsci.2012.05.001
119. Kusumaningrum N, Lee DH, Yoon HS, Kim YK, Park CH, Chung JH. Gasdermin C Is Induced by Ultraviolet Light and Contributes to MMP-1 Expression via Activation of ERK and JNK Pathways. J Dermatol Sci (2018) 90(2):180–89. doi: 10.1016/j.jdermsci.2018.01.015
120. Hergueta-Redondo M, Sarrio D, Molina-Crespo Á, Vicario R, Bernadó-Morales C, Martínez L, et al. Gasdermin B Expression Predicts Poor Clinical Outcome in HER2-Positive Breast Cancer. Oncotarget (2016) 7(35):56295–308. doi: 10.18632/oncotarget.10787
121. de Beeck KO, Van Laer L, Camp GV. DFNA5 a Gene Involved in Hearing Loss and Cancer a Review. Ann Otol Rhinol Laryngol (2012) 121(3):197–207. doi: 10.1177/000348941212100310
122. Koppelman GH, Los H, Postma DS. Genetic and Environment in Asthma the Answer of Twin Studies. Eur Respir J (1999) 13(1):2–4. doi: 10.1183/09031936.99.13100299
123. Karunas AS, Iunusbaev BB, IuIu F, Gimalova GF, Ramazanova NN, Gur'eva LL, et al. Genome-Wide Association Study of Bronchial Asthma in the Volga-Ural Region of Russia. Mol Biol (Mosk) (2011) 45(6):992–1003. doi: 10.1134/S0026893311060057
124. Li FX, Tan JY, Yang XX, Wu YS, Wu D, Li M. Genetic Variants on 17q21 Are Associated With Asthma in a Han Chinese Population. Genet Mol Res (2012) 11(1):340–7. doi: 10.4238/2012.February.10.5
125. Yu X, Yu C, Ren Z, Deng Y, Song J, Zhang H, et al. Genetic Variants of 17q21 Are Associated With Childhood-Onset Asthma and Related Phenotypes in a Northeastern Han Chinese Population: A Case-Control Study. Tissue Antigens (2014) 83(5):330–6. doi: 10.1111/tan.12342
126. Stein MM, Thompson EE, Schoettler N, Helling BA, Magnaye KM, Stanhope C, et al. A Decade of Research on the 17q12-21 Asthma Locus: Piecing Together the Puzzle. J Allergy Clin Immunol (2018) 142(3):749–64.e3. doi: 10.1016/j.jaci.2017.12.974
127. Zhao CN, Fan Y, Huang JJ, Zhang HX, Gao T, Wang C, et al. The Association of GSDMB and ORMDL3 Gene Polymorphisms With Asthma: A Meta-Analysis. Allergy Asthma Immunol Res (2015) 7(2):175–85. doi: 10.4168/aair.2015.7.2.175
128. Bouzigon E, Corda E, Aschard H, Dizier MH, Boland A, Bousquet J, et al. Effect of 17q21 Variants and Smoking Exposure in Early-Onset Asthma. N Engl J Med (2008) 359(19):1985–94. doi: 10.1056/NEJMoa0806604
129. Cantero-Recasens G, Fandos C, Rubio-Moscardo F, Valverde MA, Vicente R. The Asthma-Associated ORMDL3 Gene Product Regulates Endoplasmic Reticulum-Mediated Calcium Signaling and Cellular Stress. Hum Mol Genet (2010) 19(1):111–21. doi: 10.1093/hmg/ddp471
130. Verlaan DJ, Berlivet S, Hunninghake GM, Madore AM, Lariviere M, Moussette S, et al. Allele-Specific Chromatin Remodeling in the ZPBP2/GSDMB/ORMDL3 Locus Associated With the Risk of Asthma and Autoimmune Disease. Am J Hum Genet (2009) 85(3):377–93. doi: 10.1016/j.ajhg.2009.08.007
131. Yu C, Meng XM, Zhang SF, Zhao GP, Hu LD, Kong X. A 3-Nucleotide Deletion in the Polypyrimidine Tract of Intron 7 of the DFNA5 Gene Causes Nonsyndromic Hearing Impairment in a Chinese Family. Genomics (2003) 82(5):575–79. doi: 10.1016/s0888-7543(03)00175-7
132. Cheng J, Han DY, Dai P, Sun HJ, Tao R, Sun Q, et al. A Novel DFNA5 Mutation, IVS8+4 A>G, in the Splice Donor Site of Intron 8 Causes Late-Onset Non-Syndromic Hearing Loss in a Chinese Family. Clin Genet (2007) 72(5):471–7. doi: 10.1111/j.1399-0004.2007.00889.x
133. Van LL, Pfister M, Thys S, Vrijens K, Mueller M, Umans L, et al. Mice Lacking Dfna5 Show a Diverging Number of Cochlear Fourth Row Outer Hair Cells. Neurobiol Dis (2005) 19(3):386–99. doi: 10.1016/j.nbd.2005.01.019
134. Zhang QJ, Lan L, Li N, Qi Y, Zong L, Shi W, et al. Identification of a Novel Mutation of PJVK in the Chinese Non-Syndromic Hearing Loss Population With Low Prevalence of the PJVK Mutations. Acta Otolaryngol (2015) 135(3):211–6. doi: 10.3109/00016489.2014.985799
135. Salime S, Charif M, Bousfiha A, Elrharchi S, Bakhchane A, Charoute H, et al. Homozygous Mutations in PJVK and MYO15A Genes Associated With Non-Syndromic Hearing Loss in Moroccan Families. Int J Pediatr Otorhinolaryngol (2017) 101:25–9. doi: 10.1016/j.ijporl.2017.07.024
136. Alimardani M, Hosseini SM, Khaniani MS, Haghi MR, Eslahi A, Farjami M, et al. Targeted Mutation Analysis of the SLC26A4, MYO6, PJVK and CDH23 Genes in Iranian Patients With AR Nonsyndromic Hearing Loss. Fetal Pediatr Pathol (2019) 38(2):93–102. doi: 10.1080/15513815.2018.1547336
137. Moreno-Moral A, Bagnati M, Koturan S, Ko JH, Fonseca C, Harmston N, et al. Changes in Macrophage Transcriptome Associate With Systemic Sclerosis and Mediate GSDMA Contribution to Disease Risk. Ann Rheum Dis (2018) 77(4):596–601. doi: 10.1136/annrheumdis-2017-212454
138. Kanneganti A, Malireddi RKS, Saavedra PHV, Vande Walle L, Van Gorp H, Kambara H, et al. GSDMD Is Critical for Autoinflammatory Pathology in a Mouse Model of Familial Mediterranean Fever. J Exp Med (2018) 215(6):1519–29. doi: 10.1084/jem.20172060
139. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
140. Yang J, Zhao Y, Shao F. Non-Canonical Activation of Inflammatory Caspases by Cytosolic LPS in Innate Immunity. Curr Opin Immunol (2015) 32:78–83. doi: 10.1016/j.coi.2015.01.007
141. Song DH, Lee JO. Sensing of Microbial Molecular Patterns by Toll-Like Receptors. Immunol Rev (2012) 250(1):216–29. doi: 10.1111/j.1600-065X.2012.01167.x
142. Cerqueira DM, Gomes MTR, Silva ALN, Rungue M, Assis NRG, Guimaraes ES, et al. Guanylate-Binding Protein 5 Licenses Caspase-11 for Gasdermin-D Mediated Host Resistance to Brucella Abortus Infection. PloS Pathog (2018) 14(12):e1007519. doi: 10.1371/journal.ppat.1007519
143. Schneider KS, Gross CJ, Dreier RF, Saller BS, Mishra R, Gorka O, et al. The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep (2017) 21(13):3846–59. doi: 10.1016/j.celrep.2017.12.018
144. Rathkey JK, Zhao J, Liu Z, Chen YH, Yang J, Kondolf HC, et al. Chemical Disruption of the Pyroptotic Pore-Forming Protein Gasdermin D Inhibits Inflammatory Cell Death and Sepsis. Sci Immunol (2018) 3(26):eaat2738. doi: 10.1126/sciimmunol.aat2738
145. Zhang D, Qian J, Zhang P, Li H, Shen H, Li X, et al. Gasdermin D Serves as a Key Executioner of Pyroptosis in Experimental Cerebral Ischemia and Reperfusion Model Both In Vivo and In Vitro. J Neurosci Res (2019) 97(6):645–60. doi: 10.1002/jnr.24385
Keywords: gasdermin, family gene, function, structure, GSDMD
Citation: Zou J, Zheng Y, Huang Y, Tang D, Kang R and Chen R (2021) The Versatile Gasdermin Family: Their Function and Roles in Diseases. Front. Immunol. 12:751533. doi: 10.3389/fimmu.2021.751533
Received: 01 August 2021; Accepted: 25 October 2021;
Published: 11 November 2021.
Edited by:
Cleni Mara Marzocchi-Machado, University of São Paulo, BrazilReviewed by:
Ambika MV Murthy, Australian National University, AustraliaYandan Yao, Sun Yat-sen University, China
Guy S Salvesen, Sanford Burnham Institute for Medical Research, United States
Copyright © 2021 Zou, Zheng, Huang, Tang, Kang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruochan Chen, ODQxNzIzMzJAcXEuY29t
†These authors have contributed equally to this work