 Lim Jack Lee1†
Lim Jack Lee1† Thamil Vaani Komarasamy1†
Thamil Vaani Komarasamy1† Nur Amelia Azreen Adnan1William James2
Nur Amelia Azreen Adnan1William James2 Vinod RMT Balasubramaniam1*
Vinod RMT Balasubramaniam1*- 1Infection and Immunity Research Strength, Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia, Bandar Sunway, Malaysia
- 2Sir William Dunn School of Pathology, University of Oxford, Oxford, United Kingdom
Zika virus (ZIKV) received worldwide attention over the past decade when outbreaks of the disease were found to be associated with severe neurological syndromes and congenital abnormalities. Unlike most other flaviviruses, ZIKV can spread through sexual and transplacental transmission, adding to the complexity of Zika pathogenesis and clinical outcomes. In addition, the spread of ZIKV in flavivirus-endemic regions, and the high degree of structural and sequence homology between Zika and its close cousin Dengue have raised questions on the interplay between ZIKV and the pre-existing immunity to other flaviviruses and the potential immunopathogenesis. The Zika epidemic peaked in 2016 and has affected over 80 countries worldwide. The re-emergence of large-scale outbreaks in the future is certainly a possibility. To date, there has been no approved antiviral or vaccine against the ZIKV. Therefore, continuing Zika research and developing an effective antiviral and vaccine is essential to prepare the world for a future Zika epidemic. For this purpose, an in-depth understanding of ZIKV interaction with many different pathways in the human host and how it exploits the host immune response is required. For successful infection, the virus has developed elaborate mechanisms to escape the host response, including blocking host interferon response and shutdown of certain host cell translation. This review provides a summary on the key host factors that facilitate ZIKV entry and replication and the mechanisms by which ZIKV antagonizes antiviral innate immune response and involvement of adaptive immune response leading to immunopathology. We also discuss how ZIKV modulates the host immune response during sexual transmission and pregnancy to induce infection, how the cross-reactive immunity from other flaviviruses impacts ZIKV infection, and provide an update on the current status of ZIKV vaccine development.
Introduction
Zika virus (ZIKV) is a mosquito-borne arbovirus that was brought to attention in the past decade due to its link to serious neuropathogenesis in new-borns and adults. ZIKV is an enveloped, positive sense single-stranded RNA virus of the flavivirus genus in the Flaviviridae family, with a 50 nm diameter and a genome of about 10.8 kilobases in length (1). It was incidentally discovered in 1947 in the Zika Forest of Uganda, during a study on the vectors of sylvatic yellow fever. Until recent years, ZIKV never garnered much attention as there were only sporadic infections occurring in Africa and Asia (2), and the disease was thought to be either asymptomatic or mild and self-limiting. Symptoms and signs of ZIKV infection typically include low-grade fever, arthalgia, pruritic rash, conjunctivitis, myalgia, retro-orbital pain, headache, dysesthesia, and asthenia. Abdominal pain, diarrhea, nausea and mucous membrane ulcerations are some of the less common symptoms and signs (3–5). Thrombocytopenia has been reported, which could be due to an immune-mediated mechanism (6).
A string of outbreaks occurred since 2007 which eventually led to the observation of an increased incidence of new-borns with microcephaly and adults with Guillain-Barre syndrome in ZIKV-endemic populations. This prompted the World Health Organization (WHO) to declare ZIKV as a Public Health Emergency of International Concern (PHEIC) on 1 February 2016 to investigate the associations (7). These unique characteristics were not seen in infections by other similar viruses and has thus prompted researchers to investigate the mechanisms of ZIKV infection and develop preventative and treatment strategies against ZIKV infection.
Extensive research on ZIKV found that the effects on a new-born is not just limited to microcephaly, but also include various other adverse pregnancy outcomes such as miscarriage, fetal growth restriction, a range of fetal brain anomalies (ventriculomegaly, intracranial calcification), ocular abnormalities and hearing loss (8–11). In the developing mammalian brain, ZIKV efficiently targets the neural progenitor cells (NPCs), astrocytes, microglia and oligodendrocyte precursor cells (12). Studies on tissue tropism identified ZIKV from various tissues such as the placenta, fetal brain, male and female reproductive tract and body fluids, consistent with cases reporting vertical and sexual transmission. ZIKV RNA can persist for weeks in body fluids, and up to 6 months post-infection in seminal fluid (13). In adults, other than Guillain-Barré syndrome (GBS), very few cases of CNS involvement have been reported. Some of the complications include encephalitis, meningitis, myelitis, meningoencephalitis, transverse myelitis and neuropsychiatric symptoms (14–18). To characterize the cellular responses and molecular mechanisms involved in Zika pathogenesis, extensive virus-host interactome analyses have been conducted. These studies identified key cellular proteins that are associated with neuronal development and neurological diseases (19–21).
ZIKV infection is diagnosed by molecular and serological testing. For symptomatic individuals (≤7 days after onset of symptoms), ZIKV infections is established by real-time reverse transcription polymerase chain reaction (rRT-PCR) for ZIKV RNA in serum, urine or whole blood. If the rt-PCR results are negative, serologic testing: IgM ELISA and plaque reduction neutralization test (PRNT) are prompted. As for symptomatic individuals (>7 days after onset of symptoms), the diagnostics testing for ZIKV includes IgM and PRNT (22). However, cross-reactivity with other flavivirus antibodies complicates interpretation of serologic results. Although PRNT is generally the most specific, it may not be sufficient to distinguish between ZIKV and DENV infection (23). ZIKV NS1-sepcific IgM antibody approach was found to be more specific and was not detected in patients with previous Dengue (24).
As of today, there are no approved vaccines or antivirals for ZIKV infection (25). With the low number of active cases, clinical trials on potential vaccines and antivirals were halted until they can be tested on more patients in a future outbreak (26). This review article aims to summarize the current knowledge on immune evasion strategies used by ZIKV leading to its pathogenesis to aid in the development of vaccine and therapeutics. There are various host defence mechanisms involved in the detection and elimination of ZIKV. These defences can be broadly categorized three categories, namely intrinsic, innate and adaptive immunity.
ZIKV Replication Cycle and Targeted Host Factors
ZIKV like other viruses’ hijacks host cell machineries for its replication. Moreover, being an RNA virus with a small genome size, successful replication of ZIKV requires utilization of many host factors. In addition, small RNA viruses are highly adaptive, with a high degree of genetic variation in every cycle of replication. This presents a challenge for the development of an effective anti-ZIKV antiviral that target epitopes of ZIKV. Instead, perhaps host factors can be a better target for ZIKV therapeutics and vaccine, as they mutate at a much slower rate.
Entry of ZIKV into host cells is facilitated by the binding of ZIKV surface glycoproteins with host cell surface receptors. Expression of some host receptors have been found to increase ZIKV infectivity. These include TIM, TAM (Tyro3, Axl, Mertk) and DC-SIGN receptors (27). TIM and TAM are families of transmembrane receptors that recognizes and binds to phosphatidylserine (PS), which is a molecule which signals for phagocytosis of cells expressing it. PS signalling is a characteristic feature of apoptotic cells, but viruses such as ZIKV and DENV also express PS on its envelope as a disguise to gain access to host cells. TIM directly binds to PS, whereas TAM receptors require a bridging ligand Gas6 to bind PS (28). DC-SIGN is a receptor present on innate immune cells such as dendritic cells and macrophages. It binds to glycans and facilitates uptake of antigens expressing these glycans (29). Binding of ZIKV envelope glycoprotein to these receptors initiate clathrin-mediated endocytosis, which brings in the virus in a clathrin-coated endosome. Other cofactors on host cell membranes such as heat shock protein 70 (Hsp70) may also play a role in mediating ZIKV entry (30).
As the endosome matures, hydrogen ions are pumped in resulting in an acidic internal environment. The low pH triggers a structural change in the envelope of ZIKV, causing it to fuse with the endosomal membrane, releasing the viral genome into the cytoplasm (31). The ZIKV genome comprises a 5’ and a 3’ non-coding region, with a single open reading frame which encodes for three structural proteins – envelope (E) proteins, capsid (C) proteins, premembrane/membrane (prM/M) proteins and seven non-structural proteins – NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5. The structural proteins form structural components of virions and assemble new virus particles, and mediate virus entry and encapsidation. The E protein contains a transmembrane domain, and three ectodomains (EDI, EDII and EDIII) located outside the membrane. The protein is involved viral attachment, membrane fusion and cellular receptor binding. It also represents the main target for neutralising antibodies (1, 32). The non-structural proteins have a variety of functions which includes evasion of host immune response, alteration of host cell signalling pathways, and replication of ZIKV RNA, all of which contributes to effective viral replication and pathogenesis (1). The NS1 protein is essential for viral RNA replication, and the immune evasion and pathogenesis by interacting with host immune factors such as RIG-I-like receptors (33–35). It is involved in virus maturation through interaction with viral prM and E protein (20). NS1 also elicits production of protective antibodies (36). The NS3 and NS5 proteins, possess all enzymatic functions required for RNA replication. The NS3 protein consists of an N-terminal containing a serine protease domain (NS3pro), which is essential for proteolytic processing of the viral polyprotein, and a C-terminal bearing RNA helicase, RNA triphosphatase and RNA-stimulated NTPase domain that are critical for RNA replication (37, 38). ZIKV NS5 consists of an RNA dependent RNA polymerase (RdRp) domain, which is responsible for viral RNA synthesis, and a methyltransferase (MTase) domain, that is involved in translation and evasion of host immune response (34).
The released positive-sense single-stranded RNA virus (+ssRNA) is then translated by the host cell’s translational machinery. Translation begins in the cytosol and is directed to the endoplasmic reticulum (ER) via ER-localizing signals on the nascent polypeptide chain. The polypeptide chain embeds and translocates into the ER with the help of Sec61 translocon, ER membrane complexes (EMCs), signal peptidases and oligotransferases (39). The completed polyprotein is subsequently cleaved by host signal peptidase and viral NS2B-NS3 protease complex into individual viral proteins, which then localizes to different components of the cell to carry out their respective functions (1).
At the ER, ZIKV enhances genome replication, virion assembly and transportation by remodelling the ER architecture, forming an assortment of virus-induced membrane structures, which includes vesicle packets, convoluted membranes, zippered ER and pancrystalline arrays (39). NS4A interacts with reticulon 3.1A, a host factor responsible for regulation of membrane structures, to induce curvature of the ER membrane, forming vesicles where ZIKV genome replication occurs. Knockdown of this host factor have been shown to reduce virus-induced structures and ZIKV replication (40).
For the maturation and eventual release of ZIKV virion, utilization of the host cell secretory machinery is required. Newly assembled virions go through a series of maturation processes in the golgi network. The acidic environment of the trans-golgi network once again induces a conformational change in the ZIKV E proteins from a spiky trimeric heterodimer to a flat homodimer. This exposes the furin cleavage site, enabling the cleavage of prM proteins into mature M proteins by furin (1), which is a host protease abundant in golgi bodies. Vesicles containing mature ZIKV then fuses with the plasma membrane to release the mature virions into the extracellular space.
Host Intrinsic Defenses Against ZIKV
Intrinsic immunity are host defences that are constantly present in host cells. These defences detect and restrict viral replication via host cellular mechanisms such as autophagy, apoptosis, RNA interference/decay and formation of stress granules (41). Several studies have identified intrinsic defences that restrict ZIKV replication. Stress granules (SG) are collections of ribonucleoproteins made up of mRNA complexes stalled in the initiation phase of translation. This can be due to the phosphorylation of eukaryotic initiation factor eIF2α by kinases such as protein kinase R (PKR), PKR-like endoplasmic reticulum kinase (PERK) and general control nonderepressible (GCN) that are activated at times of cellular stress (42). Stress granule proteins G3BP, TIA-1 and TIAR are often targeted by viruses to inhibit SG formation. Flaviviruses such as DENV and WNV have been known to sequester TIAR and TIA-1 to be used for their RNA replication (43). Studies by Hou et al. and Amorim et al. highlighted ZIKV’s ability to inhibit phosphorylation of eIF2α, thereby preventing formation of stress granules and ensuring the continuity of viral replication (44, 45). However, Hou et al. also demonstrated inhibition of SGs formed via eIF2α-independent mechanisms by ZIKV in HFA and A549 cells (45), while Amorim et al. demonstrated ZIKV’s inability to inhibit SGs formed via eIF2α-independent mechanisms in Vero cells (44). Although ZIKV has the ability to prevent SG formation, both studies found that ZIKV infection did not significantly induce formation of SGs.
Reticulophagy on the other hand is another intrinsic defence mechanism that is likely to be more important in restricting ZIKV replication. As ZIKV modulates the ER to aid in its replication and assembly, ER degradation by reticulophagy can be an important step for host cells to inhibit ZIKV replication. Lennemann and Coyne demonstrated that inhibition of the reticulophagy receptor FAM134B resulted in a significant increase in ZIKV and DENV RNA levels. They also identified the ability of flavivirus NS2B3 to cleave FAM134B, disrupting the reticulophagy process (46).
Host Innate Immune Response Against ZIKV
The innate immune system is activated by detection and subsequent phagocytosis of foreign antigens by innate immune cells such as dendritic cells and macrophages. Innate immunity plays an important role in restricting ZIKV pathogenesis, mainly through production of interferons (IFN) and interferon-stimulated genes (ISGs) which encodes various proteins that antagonize processes of ZIKV replication (47). For ISGs to be produced, a long, multistep immune signalling pathway is required. This process generally begins when ZIKV enters a host innate immune cell. Here, different types of pattern recognition receptors (PRRs) detect specific pathogen associated molecular patterns (PAMP). The RIG-I-like receptors (RLR) are PRRs present in the cytoplasm of these immune cells. RIG-I and MDA5 are RLRs that detect viral RNA molecules. Upon detection of RNA, the RLRs translocate to the mitochondria to activate the mitochondrial antiviral signalling protein (MAVS), which then sends a downstream signal to activate TANK-binding kinase 1 (TBK1), an enzyme required for the phosphorylation of transcription factors IRF3, IRF5, IRF7 and NF-κB. These activated transcription factors then translocate into the nucleus to aid in the production of interferons (48). Apart from the RLRs, toll-like receptors (TLR) are another type of PRR. The endosomal TLR3 and TLR7/8 function to detect viral dsRNA and ssRNA respectively. Once activated, TLR3 signals TRIF, which then activates TRAF6 and transcription factors IRF3, IRF 7 and NFkB. On the other hand, TLR7/8 signals MyD88, an adapter protein to activate transcription factors such as NF-κB and IRF7 (49). The third type of PRRs are NOD-like receptors (NLRs). Detection of PAMPs triggers oligomerization of NLRs and subsequent inflammasome formation. NLRP3 inflammasomes activate caspase 1, resulting in the activation of proinflammatory cytokines such as IL-1β and IL-18 (50). Another type of PRR involves the cGAS-STING pathway. This is a cytosolic DNA-sensing mechanism that detects damaged DNA and signals for downstream processes to mount a type I IFN response (51).
Type I interferons (IFN α and β) play an important role in the immune response towards flaviviruses. After the production of type I IFN via the multistep process initiated by detection of viral RNA by PRRs, the IFNs bind to IFN receptors consisting of two dimerized subunits - IFNAR1 and IFNAR2. Binding by type 1 IFN induces activation of Janus kinase (JAK), which phosphorylates signal transducers and activators of transcription 1 (STAT1) and 2 (STAT2). Phosphorylated STAT1 and STAT2 forms a heterodimer which associates with Interferon Regulatory Factor 9 (IRF9), forming the IFN-stimulated gene factor 3 (ISGF-3), which results in the increased expression of ISGs (47).
Evasion of the Innate Immune Response by ZIKV
To ensure successful replication, ZIKV employs several strategies to evade the host innate immune system. These evasion processes are carried out by ZIKV non-structural proteins, and they target various points of the IFN signalling pathway. At the RIG-I-like receptors, ZIKV NS3 binds to the 14-3-3 protein, preventing translocation of RIG-I and MDA5 to the mitochondria for the activation of MAVS (52). In addition, ZIKV NS4A was found to interact with MAVS, preventing its activation by RIG-I and MDA5 (53) (Figure 1). Co-immunoprecipitation studies by Wu et al. identified interactions between TBK1 and ZIKV NS1 and NS4B, which prevents TBK1 from phosphorylating transcription factors of IFN genes (54) (Figure 1). Another group reported suppression of TBK1 by NS1, NS2A, NS2B and NS4B (55) (Figure 1). As we move downstream, ZIKV NS5 has been shown to interact with IRF3, reducing the induction of IFN-β genes (55). Another mechanism is the ability of ZIKV NS1 to activate inflammasomes by preventing proteosomal degradation of caspase-1 (Figure 1). This seems contradictory to the function of inflammasomes but caspase-1 levels were found to be negatively correlated with type I IFN response, making high caspase-1 levels beneficial for ZIKV replication. The stabilization of caspase-1 as a result of inflammasome activation by NS1 was also found to facilitate cleavage of cGAS by caspase-1. This disrupts the cGAS-STING pathway, reducing the induction of type I IFN signalling downstream (56) (Figure 1).
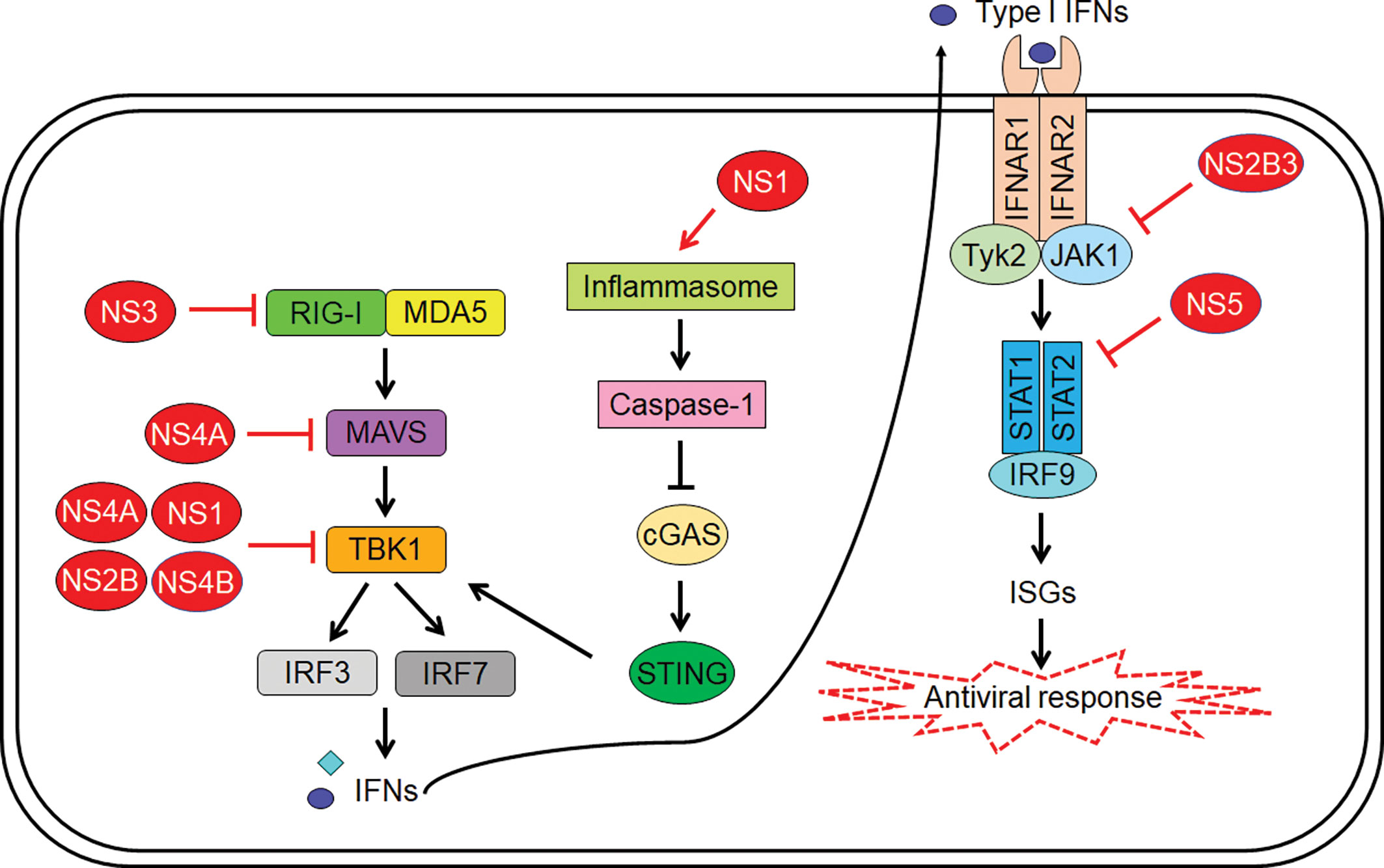
Figure 1 Zika virus (ZIKV)-mediated inhibition of host innate immunity. The viral proteins indicated in red color interfere with signalling pathways at multiple steps leading to suppression of type I interferon (IFN) induction as well as IFN-mediated expression of IFN-stimulated genes (ISGs).
At the JAK-STAT signalling pathway, ZIKV NS5 can bind to STAT2, facilitating its degradation by proteasomes (Figure 1). This is a species-specific process that occurs in human and nonhuman primate models but not in murine models (57), which is one of the reasons immunocompetent mice are resistant to ZIKV infection. Besides STAT2 degradation, NS5 was also found to inhibit phosphorylation of STAT1 and STAT2. Another mechanism is the ability of ZIKV NS2B3 to interact with and degrade JAK1, inhibiting phosphorylation of STAT and subsequent downstream processes (54) (Figure 1). Although ZIKV downregulates type I and type III IFN, it was found to upregulate type II IFN. Chaudhary et al. found that suppression of type II IFN signalling pathway led to decreased viral replication, and increased replication was observed in cells that were treated with IFN-γ before subjected to ZIKV (58). Selective degradation of STAT2 and sparing of STAT1 leads to the formation of STAT1 homodimers, which results in the activation of IFN-γ-stimulated genes, such as CXCL10. CXCL10 has shown to play an essential role in CD8 T cell recruitment during West Nile virus (WNV) infection in the central nervous system (59). Importantly it also has been associated to neuronal damage by causing apoptosis in fetal neurons (60). Therefore, the activation of type II IFN seems to play an active role in aiding ZIKV replication and may have implication on Zika neuropathogenesis.
Role of the Adaptive Immune Response in ZIKV Infection
Several studies have identified the role of host adaptive immunity in response to ZIKV infection. ZIKV infection has shown to induce T cell activation in both humans and mouse models. Winkler and colleagues identified proliferation of CD4+ and CD8+ T cells peaking at 7 days post-infection in adult wild-type mice despite undetectable ZIKV levels. In the same study, depletion of CD4+ and CD8+ T cells only resulted in transient weight loss in immunocompetent mice but causes significant disease in mice with anti-IFNAR antibodies. Also, mice treated with anti-IFNAR antibodies without T cell depletion did not suffer from disease or experience weight loss despite increased viral replication, suggesting that these T cells play an important role in the restriction of ZIKV infection only when type I IFN response is compromised (61).
CD4+ T cells were found to differentiate into Th1 cells during ZIKV infection as evidenced by the increased production of IFN-γ, IL-2 and TNF-α cytokines and transcription factor T-bet. Effector CD8+ T-cells are also found to produce IFN-γ and TNF-α, and CD8+ levels are increased in ZIKV infection, accompanied with increased expression of granzyme B on them (62). A study using IFNAR KO mice demonstrated the role of T Cells in ZIKV Pathogenesis. Depletion of CD8+ T cells resulted in an increase in viral load in the brain but exhibited improved survival and reduced paralysis. On the other hand, depletion of CD4+ T cells alone caused paralysis in all mice, while depletion of both CD4+ and CD8+ T cells resulted in an intermediate phenotype with an increase in survival and reduction in paralysis (63). Another study showed that depletion of CD4+ T cells caused high viral load in the CNS and decreased survival (64). These findings suggest the role of CD8 T cells in causing neuropathology and potential regulatory role of CD4 T cells through reduction of immunopathology caused by CD8 T cells (Figure 2). In addition, it was also found that depletion of CD8+ T cells led to an increase in ZIKV-positive neurons and it was shown that CD8 T cells mediate lysis of ZIKV-infected neurons. These observations suggest that CD8 T cells limit ZIKV replication within the neurons, but cause neuropathogenesis leading to paralysis (Figure 2).
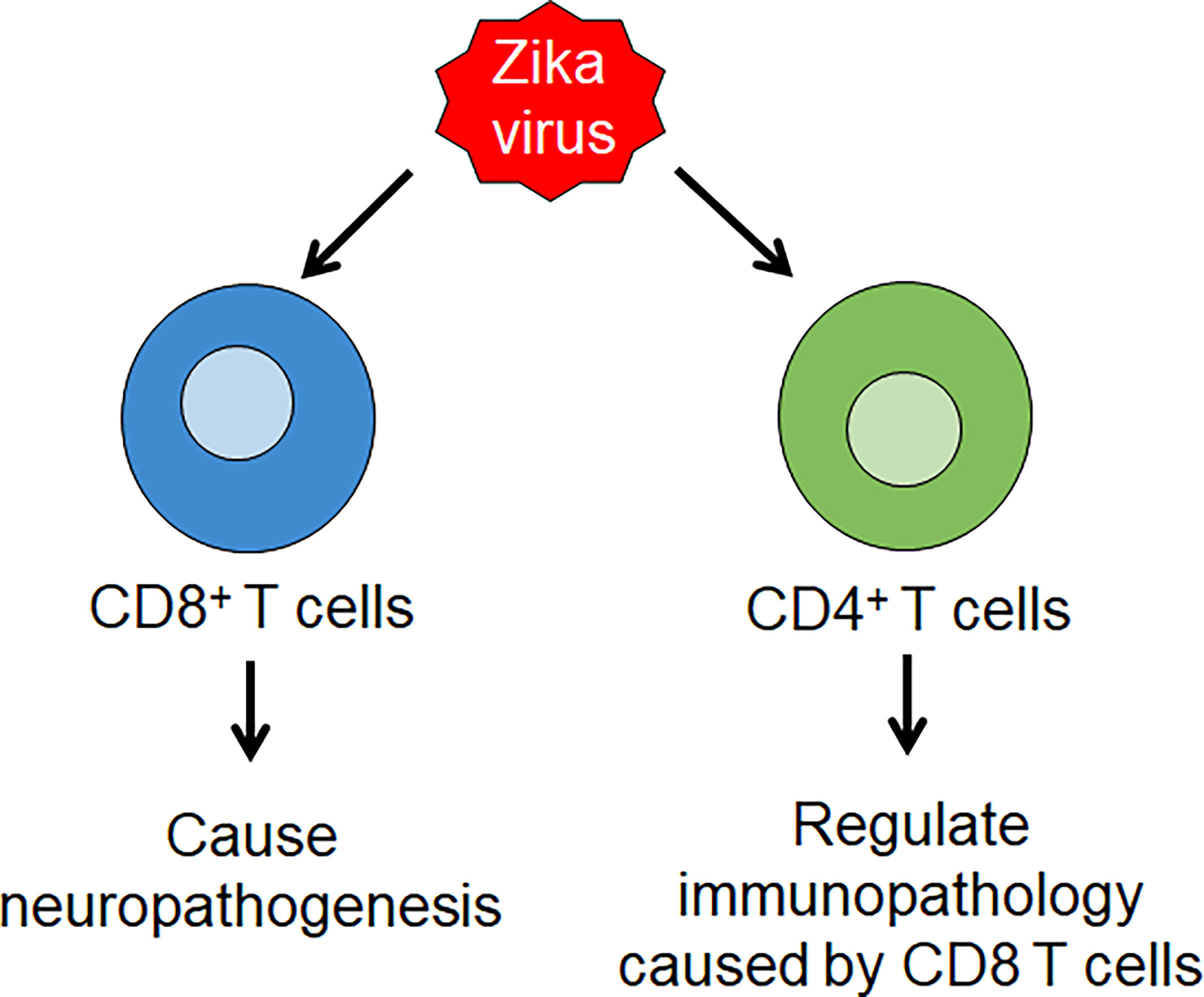
Figure 2 Zika virus (ZIKV) infection induces T cell immunity. ZIKV infection leads to the production of Th 1 CD4 T cell and effector CD8 T cell responses. CD8 T cells lead to ZIKV neuropathology, while CD4 T cells play a regulatory role through reduction of immunopathology caused by CD8 T cells.
In terms of humoral immunity, B cell response to ZIKV have been elicited by the detection of neutralizing antibodies in mice models (62). CD4 T cells have shown to be essential for production of ZIKV-specific humoral response by driving antibody production (65). In humans, Lai et al. detected anti-ZIKV IgM, ZIKV-specific plasmablasts and neutralizing antibodies in ZIKV-infected patients’ sera (66). The majority of the neutralizing mAbs derived from memory B cells of ZIKV-infected patients were found to primarily target EDIII or quaternary epitopes of whole ZIKV. In different mouse models, the neutralizing EDIII-specific antibodies have shown to provide protection against lethal infection of ZIKV (67, 68). Researchers have also explored the effects of cross-reactivity between ZIKV and DENV. Monoclonal antibodies isolated from plasmablasts of patients with past DENV infection were also found to cross-react with ZIKV, binding to components of ZIKV but are mostly ineffective in neutralizing it. Potent neutralizing ability was mostly limited to type-specific antibodies (69). This brings about the possibility of antibody-dependent enhancement, which was elicited in a few studies (67, 70), where poorly neutralizing antibodies bind to and bring viruses to infect immune cells expressing Fc receptors, increasing virion production and worsening disease outcomes.
Zika Autoimmunity and Guillain-Barré Syndrome
A recent systematic review and meta-analysis characterized ZIKV-associated GBS as a broad sensory demyelinating illness with frequent facial paralysis and a severe disease course (71). A study found that ZIKV patients with GBS had significantly higher levels of anti-ganglioside autoantibodies compared to Zika patients without GBS (72). As anti-gangliosides have been implicated in the pathogenesis of GBS (73), these autoantibodies could be a factor in ZIKV-linked GBS. In several ZIKV-infected patients with GBS, mononuclear lymphocytic infiltration and demyelination associated with inflammation have been observed (74).
Role of the Immune Response in Sexual Transmission of ZIKV
Zika virus (ZIKV) is transmitted to humans primarily by Aedes mosquitoes. However, several evidence supports that ZIKV can be transmitted sexually (75, 76). There is no evidence of sexual transmissibility in any other flaviviruses. Sexual transmission poses a serious threat to humans who are outside of the ZIKV epidemic-prone locations. Several studies found high titers of ZIKV RNA in human seminal fluid for months after clearance of viremia (77, 78). In addition, the presence of ZIKV in seminal fluid is more robust than in vaginal fluids (79). In males, the immunological responses facilitate the persistent infection of the MRT, while in females, a robust immune response controls the virus infection (80, 81). In line with this, male-to-female transmission is more common than female-to-male transmission (82, 83)
ZIKV has shown strong tropism for the male reproductive tract MRT. In animal models, ZIKV-infected Sertoli cells, spermatogonia, primary spermatocytes, Leydig cells, peritubular myoid cells, and epithelial cells of the lumen (84, 85). While in the human testis ex vivo model, ZIKV was found to infect somatic and germ cells, including Sertoli cells, testicular macrophages, Leydig cells and peritubular cells (86). The inflammatory response that occurs following ZIKV infection mediate the recruitment of innate cells (myeloid DCs, non-classic monocytes, and NK cells) as early targets of the virus and subsequently, the virus travel in these cells to the testis. To reach the testis, it is likely that ZIKV manipulates the antiviral mechanism to disrupt the blood testicular barrier (BTB) junctions, or the virus uses its proteins to destroy the junctions to cross the barrier (80, 87, 88).
The Sertoli cells, which consist of the blood-testis barrier (BTB) have shown to be highly susceptible to ZIKV infection. ZIKV has been shown to effectively infect Sertoli cells for the long-term without causing cell death (86, 89, 90). Infection of Sertoli cells induces pro-inflammatory mediators and genes linked to multiple innate immune response pathways. Among these, the IFN signaling was most significantly modulated. Down-regulation of adherent junction genes were also observed (90). These findings suggest that ZIKV can induce a strong antiviral response in Sertoli cells, which can alter the permeability of BTB.
Another study demonstrated that ZIKV infection of Sertoli cells induced cell adhesion molecules (VCAM-1 and ICAM-1), which mediate the adhesion of naive immune cells to Sertoli cells and increase BTB permeability. In addition, ZIKV infection in macrophages results in the production of inflammatory cytokines and chemokines that can degrade the tight junction protein ZO-1 and directly affect the integrity of the BTB (87). Proteomic analyses of ZIKV-infected human Sertoli cells revealed dysregulation of different proteins and cellular signaling pathways involved in the maintenance of Sertoli cells tight junction permeability and BTB, spermatogenesis as well as testicular development. Mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK), Metalloproteinase inhibitor 1 (TIMP1), IL-6, Stanniocalcin-1 (STC1), nuclear factor-κB (NF-κB), insulin-like growth factor 1 (IGF1) and fibroblast growth factor (FGF) were among them (91)
ZIKV infection elicited virus-specific IgG in the lumen of the vagina and recruited virus-specific T cells (CD4+ and CD8+ T cells) to the female reproductive tract (FRT). Transfer of virus-specific IgG and circulating memory T cells reduced viral replication, with the humoral response providing greater protection compared to cellular response. These findings suggest that the humoral and cellular responses confer protection against intravaginal ZIKV infection (81). Overall, these findings support the dual role of the immune response in the pathogenesis of ZIKV sexual transmission: a promoter of viral dissemination in males and as a protective factor in females.
Relationship Between Immunological Response During Pregnancy and ZIKV Infection
It is generally known that the immune system changes during pregnancy, whereby it must be finely balanced to avoid fetal and placental rejection and protect the fetus from infections. During pregnancy, hormones play a critical role in modulating the immune response by lowering the antigen-presenting potential of dendritic cells (DCs), reducing the number of monocytes and macrophages, as well as blocking natural killer cells, T and B cells (92). Evidence supports the production of both pro-inflammatory and anti-inflammatory factors at different stages of pregnancy (93). ZIKV can cross the placental barrier and has been shown to cause damage in the decidua and chorionic villi of the placenta. The virus has been shown to infect the placental cells such as cytotrophoblast (CTB), endothelial cells (En), syncytiotrophoblast (STB), mesenchymal cells (MS), fibroblasts (Fi), Hofbauer (Hf), macrophages and decidual cells (94–96). ZIKV can persist in the placenta for a long time after the onset of maternal symptoms and may serve as a latent viral source of fetal infection (94, 95, 97).
In a mouse model, infection with ZIKV at a gestational age (similar to the mid and late first trimester in humans) showed that type I IFN led to detrimental effects in pregnancy. IFNAR signaling in the conceptus reduced ZIKV replication in the placenta. However, IFNAR signaling mediated apoptosis of fetal endothelial cells and trophoblasts, suppressed the development of the placental labyrinth, disrupted the maternal-fetal blood barrier, fetal hypoxia and subsequently contributing to severe intrauterine growth restriction and fetal demise (98). Activation of type I IFN induced by ZIKV infection leads to significant neuroinflammation and tissue injury.
ZIKV infection during pregnancy has been shown to induce production of pro-inflammatory cytokines (IFN-γ, IFN-α, IL-6 and IL-17A) and chemokines (CXCL10, CCL2, CXCL9, and CXCL8). Expression of cytokines such as TNF-α and IFN-γ were observed in placental tissues (96). Increased levels of CXCL10, IL-22, MCP-1, and TNF-α were seen in pregnant women carrying babies with brain anomalies (99). Oher than that, higher levels of CCL2, CXCL10, IL-6, IL-8 and VEGF in amniotic fluid of ZIKV-infected pregnant women who gave birth to microcephaly babies (100). The increase in CCL2, CCL3, CCL4, CXCL9, and CXCL8 has been linked to neurological damage and fetal abnormalities (100, 101). The elevated level of CCL2 and its inverse correlation with the levels of CD163, TNFRSF1A, and CCL22 were linked to ZIKV-induced abnormal birth. In addition, distinct subsets of cytokines were detected at different trimesters. Notably, most of these cytokines are involved in the infiltration of leukocytes, in particular NK cells and monocytes, which are associated with pregnancy outcomes (101).
ZIKV-infected pregnant mice displayed increased levels of DCs associated with immunotolerance, reduced levels of cells expressing pro-inflammatory IL-12 and lower levels of Ag-experienced CD8+ T cells. These findings suggest that the immunotolerance during pregnancy may hinder efficient activation of the antiviral T cell response (102). In another study, ZIKV-infected pregnant mice caused a significant reduction of proliferating CD4+ and CD8+ T cells compared to ZIKV-infected non-pregnant mice. Furthermore, ZIKV-infected pregnant mice had considerably less granzyme B positive CD8+ T cells, indicating a loss in cytolytic effector activity (61). Hence, it is evident that pregnancy alters of the innate and adaptive immune responses to ZIKV infection. However, no significant differences were observed in the level of neutralizing antibodies between pregnant and non-pregnant mice, suggesting that pregnancy may not significantly alter the humoral immune response to ZIKV (61).
Cross-Reactive Immunity Between ZIKV and Other Flaviviruses
The structure of ZIKV E protein, which contains EDI, EDII, and EDIII, is similar to the E protein of other flaviviruses, including DENV, WNV and YFV. The amino acid sequence E proteins of ZIKV and DENV type 2 share an antigenic similarity of 53.9% (103). The high sequence similarity in the E protein between ZIKV and other flaviviruses, particularly DENV, raises a major concern of cross-reactivity as well as antibody-dependent enhancement (ADE). Various studies have demonstrated that monoclonal antibodies (mAb) against DENV and sera from DENV patients can both enhance and neutralize ZIKV infection in vitro and in vivo (104–107).
ZIKV infection in individuals previously exposed to DENV induced a rapid and strong plasmablast response, mostly originating from the memory B cell compartment. The acute B cell response to ZIKV exhibited preferential binding and neutralization of DENV, providing evidence of original antigenic sin (OAS). Although most of the antibodies were broadly cross-reactive, they were poorly neutralizing against ZIKV and enhanced its infection, supporting that pre-existing immunity to DENV may have a deleterious impact on early protective B cell responses to ZIKV (69). In pregnant mice, pre-existing DENV antibodies increased vertical transmission, enhanced ZIKV replication in placenta and resulted in adverse outcomes. The enhancement effect observed in these pregnant mice was dependent on Fc receptors (108, 109). In contrast, ZIKV infection of nonhuman primates immunized with DENV or YFV did not cause an increase in ZIKV titer or adverse clinical outcomes despite modulation of the immune response (110). A cohort of individuals from Salvador (epicenter of ZIKV epidemic in Brazil) showed that pre-existing antibodies against DENV contributed to decreased risk of ZIKV infection and symptoms (111). In another cohort study involving pregnant women infected with ZIKV, previous DENV infection did not cause disease severity and abnormal birth outcomes (112).
In a mouse model, administration of low concentrations of DENV-immune plasma, displayed enhancement of ZIKV infection and resulted in a higher mortality rate. However, at higher concentrations, mice displayed milder symptoms and 100% survival after ZIKV infection (70). Therefore, the cross-reactivity or cross-protection against ZIKV is likely dependent on the concentrations of the anti-DENV antibodies. Another important factor determining the antibodies’ protective effects on DENV is the time interval between ZIKV infection and previous dengue fever incidence. A study found that the protective effect could last up to 6 years (113). A study proposed that the cross-protection against congenital Zika syndrome (CZS) conferred by DENV neutralizing antibodies was not dependent on the antibody titer but likely mediated by the immune response (114). As observed in humans, DENV antibodies have been shown to increase CD4+ and CD8+ T-cell responses during ZIKV infections (115, 116). Also, in pregnant mice, DENV cross-reactive CD8+ T cells have been demonstrated to protect the fetus during ZIKV infection (117, 118).
The cross-reactivity effects induced by pre-existing antibodies against other flaviviruses, may represent a challenge for the development of vaccine for ZIKV. Pre-existing antibodies may produce a strong immune response against the ZIKV vaccine through cross-reactive T cells, thus enhancing the vaccine efficacy (65). However, vaccination against ZIKV could also lead to ADE of DENV infection and vice versa, enhancing virus entry and potentially resulting in severe disease. Increasing evidence suggests that previous ZIKV immunity leads to increased DENV infection and risk for severe dengue (119, 120). Hence, designing an ideal vaccine for ZIKV requires careful consideration of potential ADE.
Zika Virus Vaccine Development
Nearly half of the world’s population lives in regions at risk of Zika transmission, and the possibility of future Zika epidemics remains high (121). A mathematical model based in Nicaragua has predicted a possible ZIKV outbreak in 2047, affecting mainly the women of reproductive age. However, if protective immunity wanes, ZIKV recurrence may happen sooner (122). With the association between ZIKV and adverse pregnancy outcomes, congenital diseases, and severe developmental problems, the search for an effective vaccine has become even more urgent.
The WHO has proposed two strategies for ZIKV vaccine development roadmap: emergency outbreak response and endemic use. The emergency outbreak response entails mass vaccination of pregnant women and women of child-bearing age to prevent ZIKV-related adverse pregnancy outcomes and neurological complications. On the other hand, endemic use involves a broad vaccination of the general population and routine immunization to establish population immunity and prevent ZIKV transmission (123). Several ZIKV vaccine candidates under various platforms have been developed over the past 5 years. These include inactivated, live attenuated, viral-vectored, subunit, nucleic acid and messenger RNA (mRNA) vaccines. Some of these vaccines have demonstrated promising results in preclinical studies and have advanced into clinical trials. In animal models, a number of vaccine candidates were found to reduce ZIKV RNA levels in maternal, placental, testis and fetal tissues, and subsequently prevent their damage (124–126). Another study demonstrated the ability of live-attenuated ZIKV vaccine candidates to prevent viral transmission during pregnancy and protect against testis infection and injury in mice (125). As of September 2021, 12 ZIKV vaccine candidates are in clinical evaluation (Table 1) (ClinicalTrials.gov).
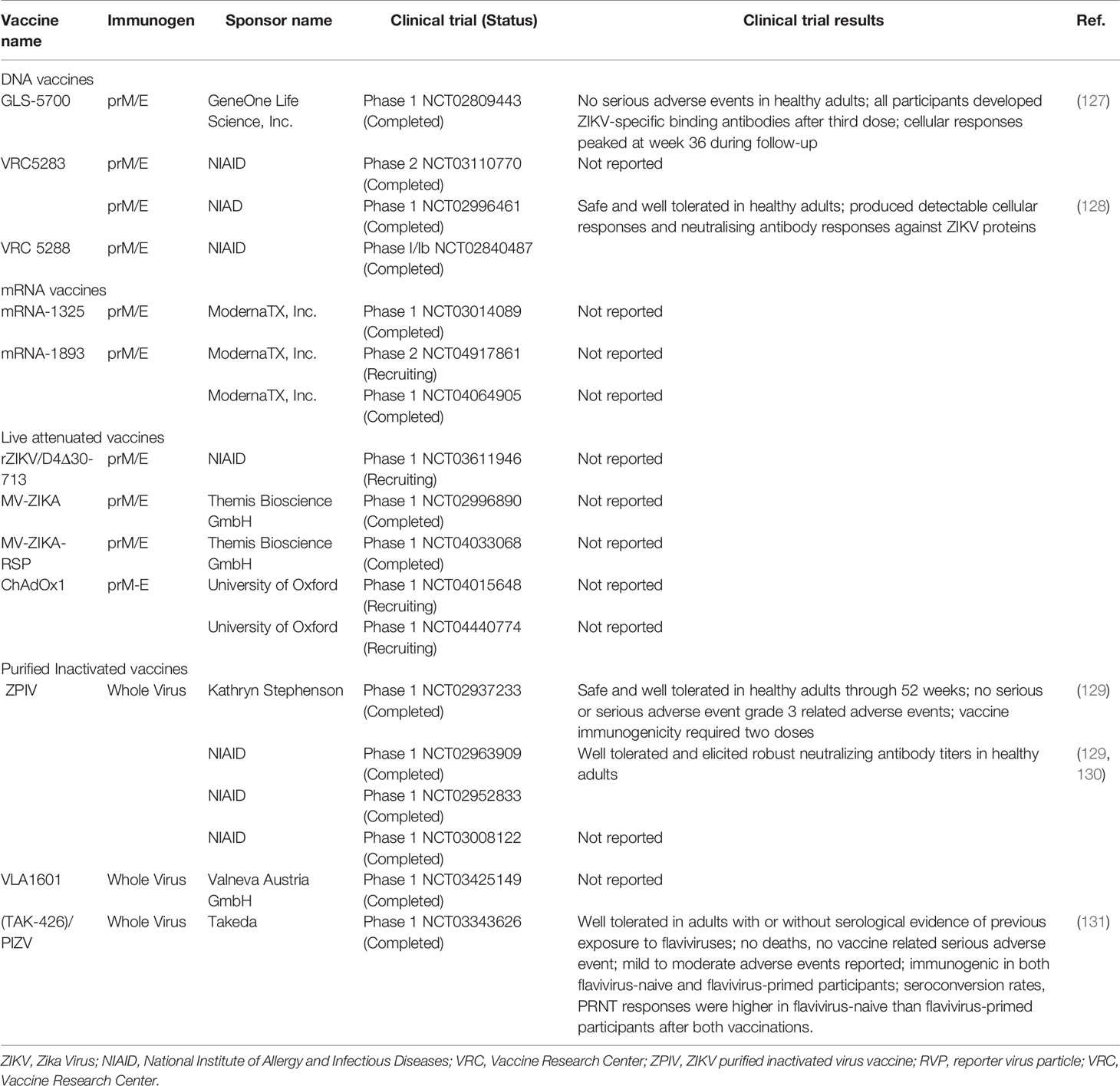
Table 1 Zika vaccine candidates in clinical trials.
NHPs with immune responses primed by infection with an African ZIKV strain were protected when re-challenged with an Asian ZIKV strain (132). Hence, a ZIKV vaccine based on a single strain could be adequate and protect against other strains. Since ZIKV causes severe birth outcomes and congenital malformations, the ability of the vaccines to adequately protect pregnant women, women of reproductive age and children need to be determined. To protect the fetus from ZIKV infection, protective immunity needs to be achieved during the first trimester or early second trimester. Importantly, the safety of the vaccines during pregnancy is required to be critically accessed. Another hurdle is the potential impact of the pre-existing flavivirus antibodies in people who have been previously infected or vaccinated against other flaviviruses. In light of this, a study conducted by Larocca et al. demonstrated that pre-existing DENV immunity did not reduce the immunogenicity or protective immunity of ZIKV candidate vaccines in animal models (rhesus macaques and mice) (133). However, the designing and administration of ZIKV vaccines require extreme caution, and their safety and immunogenicity need to be evaluated both in flavivirus-exposed and naïve populations.
Conclusion
Zika virus (ZIKV) has been shown to modulate both the innate and adaptive immune system of the host defence mechanism to buy time to effectively induce infection and cause neuropathogenesis. Identification and understanding of the role of effector molecules in antiviral response and signal transduction pathways are crucial to decipher the host-ZIKV interactions. ZIKV uses multiple mechanisms to evade the host immune response. The majority of these mechanisms are triggered by the non-structural proteins. It modulates the innate immune response primarily by inhibiting the expression of type I IFNs and ISGs. Suppression of type I IFN increases dependency on adaptive immune response in circumventing infection.
Importantly, the ZIKV-induced host immune response is dependent on several factors: gender, pregnancy and pre-existing immunity against other flaviviruses. In males, the immune response facilitates ZIKV persistence through inflammatory factors and alteration of BTB permeability. While in females, the immune response seems to control ZIKV virus infection. However, the ZIKV cellular targets in the reproductive organs and the underlying mechanisms need to be further investigated. Alterations of the innate and adaptive immune responses during pregnancy have demonstrated to have significant effects on ZIKV infection and pathogenesis. Activation of type I IFN along with the expression of proinflammatory cytokines and impaired T cell response in ZIKV-infected pregnant women could be the major factors contributing to adverse fetal and birth outcomes. Given the important effects of pregnancy-induced immune modulation on ZIKV infection and the possible adverse outcomes, this area of research requires thorough understanding. This is also critical for successful vaccine development and administration in women of reproductive age and pregnant women. In addition, follow-ups of children born with in utero ZIKV exposure may aid in assessing the effects caused by the immune response and providing support for early intervention to improve the neurodevelopment of the children.
The effects of pre-existing flavivirus antibodies on ZIKV immunopathogenesis and their potential impact on Zika vaccine development have been a major concern. Various studies have demonstrated that pre-existing flavivirus immunity, particularly against DENV can both enhance and neutralize ZIKV infection in vitro and in vivo. The detrimental or protective effects of the pre-existing immunity is likely determined by the concentration of antibodies, time interval between exposure with different flaviviruses, and immune response. The majority of the studies conducted in vitro and in mice showed enhanced ZIKV replication and disease severity due to pre-existing flavivirus immunity. However, most of the investigations in nonhuman primates and humans showed the opposite and suggest that high rates of immunity may present a barrier to future ZIKV outbreaks. Other factors such as the ZIKV strains, the type of animal models used, and Fc receptors may also contribute to the effects of pre-exiting flavivirus antibodies. Future studies in nonhuman primates and humans are necessary to fully understand the effects of pre-existing antibodies on ZIKV infection, particularly in the placenta and also their impact on Zika vaccines. Studies also need to address the effects of Zika vaccines on DENV infection and potential ADE. Dissecting the dynamics of pre-existing flavivirus immunity will aid in understanding flavivirus pathogenesis and vaccine development.
Author Contributions
Conceptualization by LL and VB. Methodology by LL and TK. Writing—original draft preparation by LL and TK. Writing, review and editing by LL, TK, NA, WJ, and VB. Supervision by VB and WJ. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the anonymous reviewer for critical comments and suggestions. We would also like to thank the school and administration staff of Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia for their continuous support to this project and lab members of Infectious Disease Laboratory for critically reading this review.
References
1. Sirohi D, Kuhn RJ. Zika Virus Structure, Maturation, and Receptors. J Infect Dis (2017) 216(suppl_10):S935–S44. doi: 10.1093/infdis/jix515
2. Gubler DJ, Vasilakis N, Musso D. History and Emergence of Zika Virus. J Infect Dis (2017) 216(suppl_10):S860–S7. doi: 10.1093/infdis/jix451
3. Brasil P, Calvet GA, Siqueira AM, Wakimoto M, de Sequeira PC, Nobre A, et al. Zika Virus Outbreak in Rio De Janeiro, Brazil: Clinical Characterization, Epidemiological and Virological Aspects. PloS Negl Trop Dis (2016) 10(4):e0004636. doi: 10.1371/journal.pntd.0004636
4. Chen LH. Zika Virus Infection in a Massachusetts Resident After Travel to Costa Rica: A Case Report. Ann Intern Med (2016) 164(8):574–6. doi: 10.7326/L16-0075
5. Lazear HM, Diamond MS. Zika Virus: New Clinical Syndromes and Its Emergence in the Western Hemisphere. J Virol (2016) 90(10):4864–75. doi: 10.1128/JVI.00252-16
6. Van Dyne EA, Neaterour P, Rivera A, Bello-Pagan M, Adams L, Munoz-Jordan J, et al. Incidence and Outcome of Severe and Nonsevere Thrombocytopenia Associated With Zika Virus Infection-Puerto Rico, 2016. Open Forum Infect Dis (2019) 6(1):ofy325. doi: 10.1093/ofid/ofy325
7. Maurice J. The Zika Virus Public Health Emergency: 6 Months on. Lancet (2016) 388(10043):449–50. doi: 10.1016/S0140-6736(16)31207-7
8. Brasil P, Pereira JP Jr., Moreira ME, Ribeiro Nogueira RM, Damasceno L, Wakimoto M, et al. Zika Virus Infection in Pregnant Women in Rio De Janeiro. N Engl J Med (2016) 375(24):2321–34. doi: 10.1056/NEJMoa1602412
9. de Paula Freitas B, de Oliveira Dias JR, Prazeres J, Sacramento GA, Ko AI, Maia M, et al. Ocular Findings in Infants With Microcephaly Associated With Presumed Zika Virus Congenital Infection in Salvador, Brazil. JAMA Ophthalmol (2016) 134(5):529–35. doi: 10.1001/jamaophthalmol.2016.0267
10. Vouga M, Baud D. Imaging of Congenital Zika Virus Infection: The Route to Identification of Prognostic Factors. Prenat Diagn (2016) 36(9):799–811. doi: 10.1002/pd.4880
11. Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika Virus and Birth Defects–Reviewing the Evidence for Causality. N Engl J Med (2016) 374(20):1981–7. doi: 10.1056/NEJMsr1604338
12. Wen Z, Song H, Ming GL. How Does Zika Virus Cause Microcephaly? Genes Dev (2017) 31(9):849–61. doi: 10.1101/gad.298216.117
13. Miner JJ, Diamond MS. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe (2017) 21(2):134–42. doi: 10.1016/j.chom.2017.01.004
14. Carteaux G, Maquart M, Bedet A, Contou D, Brugieres P, Fourati S, et al. Zika Virus Associated With Meningoencephalitis. N Engl J Med (2016) 374(16):1595–6. doi: 10.1056/NEJMc1602964
15. da Silva IRF, Frontera JA, Bispo de Filippis AM, Nascimento O, Group R-G-ZR. Neurologic Complications Associated With the Zika Virus in Brazilian Adults. JAMA Neurol (2017) 74(10):1190–8. doi: 10.1001/jamaneurol.2017.1703
16. Galliez RM, Spitz M, Rafful PP, Cagy M, Escosteguy C, Germano CS, et al. Zika Virus Causing Encephalomyelitis Associated With Immunoactivation. Open Forum Infect Dis (2016) 3(4):ofw203. doi: 10.1093/ofid/ofw203
17. Mecharles S, Herrmann C, Poullain P, Tran TH, Deschamps N, Mathon G, et al. Acute Myelitis Due to Zika Virus Infection. Lancet (2016) 387(10026):1481. doi: 10.1016/S0140-6736(16)00644-9
18. Zucker J, Neu N, Chiriboga CA, Hinton VJ, Leonardo M, Sheikh A, et al. Zika Virus-Associated Cognitive Impairment in Adolescent, 2016. Emerg Infect Dis (2017) 23(6):1047–8. doi: 10.3201/eid2306.162029
19. Golubeva VA, Nepomuceno TC, Gregoriis G, Mesquita RD, Li X, Dash S, et al. Network of Interactions Between ZIKA Virus Non-Structural Proteins and Human Host Proteins. Cells (2020) 9(1):153. doi: 10.3390/cells9010153
20. Scaturro P, Cortese M, Chatel-Chaix L, Fischl W, Bartenschlager R. Dengue Virus Non-Structural Protein 1 Modulates Infectious Particle Production via Interaction With the Structural Proteins. PloS Pathog (2015) 11(11):e1005277. doi: 10.1371/journal.ppat.1005277
21. Shah PS, Link N, Jang GM, Sharp PP, Zhu T, Swaney DL, et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell (2018) 175(7):1931–45.e18. doi: 10.1016/j.cell.2018.11.028
22. Sharp TM, Fischer M, Munoz-Jordan JL, Paz-Bailey G, Staples JE, Gregory CJ, et al. Dengue and Zika Virus Diagnostic Testing for Patients With a Clinically Compatible Illness and Risk for Infection With Both Viruses. MMWR Recomm Rep (2019) 68(1):1–10. doi: 10.15585/mmwr.rr6801a1
23. Announcement: Guidance for U.S. Laboratory Testing for Zika Virus Infection: Implications for Health Care Providers. MMWR Morb Mortal Wkly Rep (2016) 65(46):1304. doi: 10.15585/mmwr.mm6546a7
24. Barzon L, Percivalle E, Pacenti M, Rovida F, Zavattoni M, Del Bravo P, et al. Virus and Antibody Dynamics in Travelers With Acute Zika Virus Infection. Clin Infect Dis (2018) 66(8):1173–80. doi: 10.1093/cid/cix967
25. Baz M, Boivin G. Antiviral Agents in Development for Zika Virus Infections. Pharmaceuticals (Basel). (2019) 12(3):101. doi: 10.3390/ph12030101
26. Wilder-Smith A, Vannice K, Durbin A, Hombach J, Thomas SJ, Thevarjan I, et al. Zika Vaccines and Therapeutics: Landscape Analysis and Challenges Ahead. BMC Med (2018) 16(1):84. doi: 10.1186/s12916-018-1067-x
27. Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, et al. Biology of Zika Virus Infection in Human Skin Cells. J Virol (2015) 89(17):8880–96. doi: 10.1128/JVI.00354-15
28. Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, et al. The TIM and TAM Families of Phosphatidylserine Receptors Mediate Dengue Virus Entry. Cell Host Microbe (2012) 12(4):544–57. doi: 10.1016/j.chom.2012.08.009
29. Garcia-Vallejo JJ, van Kooyk Y. The Physiological Role of DC-SIGN: A Tale of Mice and Men. Trends Immunol (2013) 34(10):482–6. doi: 10.1016/j.it.2013.03.001
30. Pujhari S, Brustolin M, Macias VM, Nissly RH, Nomura M, Kuchipudi SV, et al. Heat Shock Protein 70 (Hsp70) Mediates Zika Virus Entry, Replication, and Egress From Host Cells. Emerg Microbes Infect (2019) 8(1):8–16. doi: 10.1080/22221751.2018.1557988
31. Persaud M, Martinez-Lopez A, Buffone C, Porcelli SA, Diaz-Griffero F. Infection by Zika Viruses Requires the Transmembrane Protein AXL, Endocytosis and Low Ph. Virology (2018) 518:301–12. doi: 10.1016/j.virol.2018.03.009
32. Dai L, Song J, Lu X, Deng YQ, Musyoki AM, Cheng H, et al. Structures of the Zika Virus Envelope Protein and Its Complex With a Flavivirus Broadly Protective Antibody. Cell Host Microbe (2016) 19(5):696–704. doi: 10.1016/j.chom.2016.04.013
33. Kim JA, Seong RK, Son SW, Shin OS. Insights Into ZIKV-Mediated Innate Immune Responses in Human Dermal Fibroblasts and Epidermal Keratinocytes. J Invest Dermatol (2019) 139(2):391–9. doi: 10.1016/j.jid.2018.07.038
34. Shi Y, Gao GF. Structural Biology of the Zika Virus. Trends Biochem Sci (2017) 42(6):443–56. doi: 10.1016/j.tibs.2017.02.009
35. Amorim JH, Alves RP, Boscardin SB, Ferreira LC. The Dengue Virus non-Structural 1 Protein: Risks and Benefits. Virus Res (2014) 181:53–60. doi: 10.1016/j.virusres.2014.01.001
36. Bailey MJ, Duehr J, Dulin H, Broecker F, Brown JA, Arumemi FO, et al. Human Antibodies Targeting Zika Virus NS1 Provide Protection Against Disease in a Mouse Model. Nat Commun (2018) 9(1):4560. doi: 10.1038/s41467-018-07008-0
37. Xu S, Ci Y, Wang L, Yang Y, Zhang L, Xu C, et al. Zika Virus NS3 is a Canonical RNA Helicase Stimulated by NS5 RNA Polymerase. Nucleic Acids Res (2019) 47(16):8693–707. doi: 10.1093/nar/gkz650
38. Li K, Phoo WW, Luo D. Functional Interplay Among the Flavivirus NS3 Protease, Helicase, and Cofactors. Virol Sin (2014) 29(2):74–85. doi: 10.1007/s12250-014-3438-6
39. Mohd Ropidi MI, Khazali AS, Nor Rashid N, Yusof R. Endoplasmic Reticulum: A Focal Point of Zika Virus Infection. J BioMed Sci (2020) 27(1):27. doi: 10.1186/s12929-020-0618-6
40. Aktepe TE, Liebscher S, Prier JE, Simmons CP, Mackenzie JM. The Host Protein Reticulon 3.1a Is Utilized by Flaviviruses to Facilitate Membrane Remodelling. Cell Rep (2017) 21(6):1639–54. doi: 10.1016/j.celrep.2017.10.055
41. Serman TM, Gack MU. Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus. Viruses (2019) 11(10):970. doi: 10.3390/v11100970
42. Protter DSW, Parker R. Principles and Properties of Stress Granules. Trends Cell Biol (2016) 26(9):668–79. doi: 10.1016/j.tcb.2016.05.004
43. Emara MM, Brinton MA. Interaction of TIA-1/TIAR With West Nile and Dengue Virus Products in Infected Cells Interferes With Stress Granule Formation and Processing Body Assembly. Proc Natl Acad Sci USA (2007) 104(21):9041–6. doi: 10.1073/pnas.0703348104
44. Amorim R, Temzi A, Griffin BD, Mouland AJ. Zika Virus Inhibits Eif2alpha-Dependent Stress Granule Assembly. PloS Negl Trop Dis (2017) 11(7):e0005775. doi: 10.1371/journal.pntd.0005775
45. Hou S, Kumar A, Xu Z, Airo AM, Stryapunina I, Wong CP, et al. Zika Virus Hijacks Stress Granule Proteins and Modulates the Host Stress Response. J Virol (2017) 91(16):e00474–17. doi: 10.1128/JVI.00474-17
46. Lennemann NJ, Coyne CB. Dengue and Zika Viruses Subvert Reticulophagy by NS2B3-Mediated Cleavage of FAM134B. Autophagy (2017) 13(2):322–32. doi: 10.1080/15548627.2016.1265192
47. Schneider WM, Chevillotte MD, Rice CM. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
48. Loo YM, Gale M Jr. Immune Signaling by RIG-I-Like Receptors. Immunity (2011) 34(5):680–92. doi: 10.1016/j.immuni.2011.05.003
49. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
50. Martinon F, Mayor A, Tschopp J. The Inflammasomes: Guardians of the Body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
51. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS-STING Pathway in Health and Disease. Nat Rev Genet (2019) 20(11):657–74. doi: 10.1038/s41576-019-0151-1
52. Riedl W, Acharya D, Lee JH, Liu G, Serman T, Chiang C, et al. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe (2019) 26(4):493–503 e6. doi: 10.1016/j.chom.2019.09.012
53. Ma J, Ketkar H, Geng T, Lo E, Wang L, Xi J, et al. Zika Virus Non-Structural Protein 4a Blocks the RLR-MAVS Signaling. Front Microbiol (2018) 9:1350. doi: 10.3389/fmicb.2018.01350
54. Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, et al. Zika Virus Evades Interferon-Mediated Antiviral Response Through the Co-Operation of Multiple Nonstructural Proteins In Vitro. Cell Discov (2017) 3:17006. doi: 10.1038/celldisc.2017.6
55. Xia H, Luo H, Shan C, Muruato AE, Nunes BTD, Medeiros DBA, et al. An Evolutionary NS1 Mutation Enhances Zika Virus Evasion of Host Interferon Induction. Nat Commun (2018) 9(1):414. doi: 10.1038/s41467-017-02816-2
56. Zheng Y, Liu Q, Wu Y, Ma L, Zhang Z, Liu T, et al. Zika Virus Elicits Inflammation to Evade Antiviral Response by Cleaving cGAS via NS1-Caspase-1 Axis. EMBO J (2018) 37(18):e99347. doi: 10.15252/embj.201899347
57. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe (2016) 19(6):882–90. doi: 10.1016/j.chom.2016.05.009
58. Chaudhary V, Yuen KS, Chan JF, Chan CP, Wang PH, Cai JP, et al. Selective Activation of Type II Interferon Signaling by Zika Virus NS5 Protein. J Virol (2017) 91(14):e00163–17. doi: 10.1128/JVI.00163-17
59. Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, et al. Neuronal CXCL10 Directs CD8+ T-Cell Recruitment and Control of West Nile Virus Encephalitis. J Virol (2005) 79(17):11457–66. doi: 10.1128/JVI.79.17.11457-11466.2005
60. Liu M, Guo S, Hibbert JM, Jain V, Singh N, Wilson NO, et al. CXCL10/IP-10 in Infectious Diseases Pathogenesis and Potential Therapeutic Implications. Cytokine Growth Factor Rev (2011) 22(3):121–30. doi: 10.1016/j.cytogfr.2011.06.001
61. Winkler CW, Myers LM, Woods TA, Messer RJ, Carmody AB, McNally KL, et al. Adaptive Immune Responses to Zika Virus Are Important for Controlling Virus Infection and Preventing Infection in Brain and Testes. J Immunol (2017) 198(9):3526–35. doi: 10.4049/jimmunol.1601949
62. Pardy RD, Rajah MM, Condotta SA, Taylor NG, Sagan SM, Richer MJ. Analysis of the T Cell Response to Zika Virus and Identification of a Novel CD8+ T Cell Epitope in Immunocompetent Mice. PloS Pathog (2017) 13(2):e1006184. doi: 10.1371/journal.ppat.1006184
63. Jurado KA, Yockey LJ, Wong PW, Lee S, Huttner AJ, Iwasaki A. Antiviral CD8 T Cells Induce Zika-Virus-Associated Paralysis in Mice. Nat Microbiol (2018) 3(2):141–7. doi: 10.1038/s41564-017-0060-z
64. Hassert M, Wolf KJ, Schwetye KE, DiPaolo RJ, Brien JD, Pinto AK. Cd4+T Cells Mediate Protection Against Zika Associated Severe Disease in a Mouse Model of Infection. PloS Pathog (2018) 14(9):e1007237. doi: 10.1371/journal.ppat.1007237
65. Elong Ngono A, Shresta S. Cross-Reactive T Cell Immunity to Dengue and Zika Viruses: New Insights Into Vaccine Development. Front Immunol (2019) 10:1316. doi: 10.3389/fimmu.2019.01316
66. Lai L, Rouphael N, Xu Y, Natrajan MS, Beck A, Hart M, et al. Innate, T-, and B-Cell Responses in Acute Human Zika Patients. Clin Infect Dis (2018) 66(1):1–10. doi: 10.1093/cid/cix732
67. Stettler K, Beltramello M, Espinosa DA, Graham V, Cassotta A, Bianchi S, et al. Specificity, Cross-Reactivity, and Function of Antibodies Elicited by Zika Virus Infection. Science (2016) 353(6301):823–6. doi: 10.1126/science.aaf8505
68. Wang Q, Yang H, Liu X, Dai L, Ma T, Qi J, et al. Molecular Determinants of Human Neutralizing Antibodies Isolated From a Patient Infected With Zika Virus. Sci Transl Med (2016) 8(369):369ra179. doi: 10.1126/scitranslmed.aai8336
69. Rogers TF, Goodwin EC, Briney B, Sok D, Beutler N, Strubel A, et al. Zika Virus Activates De Novo and Cross-Reactive Memory B Cell Responses in Dengue-Experienced Donors. Sci Immunol (2017) 2(14):eaan6809. doi: 10.1126/sciimmunol.aan6809
70. Bardina SV, Bunduc P, Tripathi S, Duehr J, Frere JJ, Brown JA, et al. Enhancement of Zika Virus Pathogenesis by Preexisting Antiflavivirus Immunity. Science (2017) 356(6334):175–80. doi: 10.1126/science.aal4365
71. Leonhard SE, Bresani-Salvi CC, Lyra Batista JD, Cunha S, Jacobs BC, Brito Ferreira ML, et al. Guillain-Barre Syndrome Related to Zika Virus Infection: A Systematic Review and Meta-Analysis of the Clinical and Electrophysiological Phenotype. PloS Negl Trop Dis (2020) 14(4):e0008264. doi: 10.1371/journal.pntd.0008264
72. Rivera-Correa J, de Siqueira IC, Mota S, do Rosario MS, Pereira de Jesus PA, Alcantara LCJ, et al. Anti-Ganglioside Antibodies in Patients With Zika Virus Infection-Associated Guillain-Barre Syndrome in Brazil. PloS Negl Trop Dis (2019) 13(9):e0007695. doi: 10.1371/journal.pntd.0007695
73. Willison HJ, Yuki N. Peripheral Neuropathies and Anti-Glycolipid Antibodies. Brain (2002) 125(Pt 12):2591–625. doi: 10.1093/brain/awf272
74. Dirlikov E, Torres JV, Martines RB, Reagan-Steiner S, Perez GV, Rivera A, et al. Postmortem Findings in Patient With Guillain-Barre Syndrome and Zika Virus Infection. Emerg Infect Dis (2018) 24(1):114–7. doi: 10.3201/eid2401.171331
75. Turmel JM, Abgueguen P, Hubert B, Vandamme YM, Maquart M, Le Guillou-Guillemette H, et al. Late Sexual Transmission of Zika Virus Related to Persistence in the Semen. Lancet (2016) 387(10037):2501. doi: 10.1016/S0140-6736(16)30775-9
76. Arsuaga M, Bujalance SG, Diaz-Menendez M, Vazquez A, Arribas JR. Probable Sexual Transmission of Zika Virus From a Vasectomised Man. Lancet Infect Dis (2016) 16(10):1107. doi: 10.1016/S1473-3099(16)30320-6
77. Mead PS, Duggal NK, Hook SA, Delorey M, Fischer M, Olzenak McGuire D, et al. Zika Virus Shedding in Semen of Symptomatic Infected Men. N Engl J Med (2018) 378(15):1377–85. doi: 10.1056/NEJMoa1711038
78. Epelboin S, Dulioust E, Epelboin L, Benachi A, Merlet F, Patrat C. Zika Virus and Reproduction: Facts, Questions and Current Management. Hum Reprod Update (2017) 23(6):629–45. doi: 10.1093/humupd/dmx024
79. Gornet ME, Bracero NJ, Segars JH. Zika Virus in Semen: What We Know and What We Need to Know. Semin Reprod Med (2016) 34(5):285–92. doi: 10.1055/s-0036-1592312
80. Arevalo Romero H, Vargas Pavia TA, Velazquez Cervantes MA, Flores Pliego A, Helguera Repetto AC, Leon Juarez M. The Dual Role of the Immune Response in Reproductive Organs During Zika Virus Infection. Front Immunol (2019) 10:1617. doi: 10.3389/fimmu.2019.01617
81. Scott JM, Lebratti TJ, Richner JM, Jiang X, Fernandez E, Zhao H, et al. Cellular and Humoral Immunity Protect Against Vaginal Zika Virus Infection in Mice. J Virol (2018) 92(7):e00038–18. doi: 10.1128/JVI.00038-18
82. Deckard DT, Chung WM, Brooks JT, Smith JC, Woldai S, Hennessey M, et al. Male-To-Male Sexual Transmission of Zika Virus–Texas, January 2016. MMWR Morb Mortal Wkly Rep (2016) 65(14):372–4. doi: 10.15585/mmwr.mm6514a3
83. Hamer DH, Wilson ME, Jean J, Chen LH. Epidemiology, Prevention, and Potential Future Treatments of Sexually Transmitted Zika Virus Infection. Curr Infect Dis Rep (2017) 19(4):16. doi: 10.1007/s11908-017-0571-z
84. Uraki R, Hwang J, Jurado KA, Householder S, Yockey LJ, Hastings AK, et al. Zika Virus Causes Testicular Atrophy. Sci Adv (2017) 3(2):e1602899. doi: 10.1126/sciadv.1602899
85. Govero J, Esakky P, Scheaffer SM, Fernandez E, Drury A, Platt DJ, et al. Zika Virus Infection Damages the Testes in Mice. Nature (2016) 540(7633):438–42. doi: 10.1038/nature20556
86. Matusali G, Houzet L, Satie AP, Mahe D, Aubry F, Couderc T, et al. Zika Virus Infects Human Testicular Tissue and Germ Cells. J Clin Invest (2018) 128(10):4697–710. doi: 10.1172/JCI121735
87. Siemann DN, Strange DP, Maharaj PN, Shi PY, Verma S. Zika Virus Infects Human Sertoli Cells and Modulates the Integrity of the In Vitro Blood-Testis Barrier Model. J Virol (2017) 91(22):e00623–17. doi: 10.1128/JVI.00623-17
88. O'Connor MA, Tisoncik-Go J, Lewis TB, Miller CJ, Bratt D, Moats CR, et al. Early Cellular Innate Immune Responses Drive Zika Viral Persistence and Tissue Tropism in Pigtail Macaques. Nat Commun (2018) 9(1):3371. doi: 10.1038/s41467-018-05826-w
89. Kumar A, Jovel J, Lopez-Orozco J, Limonta D, Airo AM, Hou S, et al. Human Sertoli Cells Support High Levels of Zika Virus Replication and Persistence. Sci Rep (2018) 8(1):5477. doi: 10.1038/s41598-018-23899-x
90. Strange DP, Green R, Siemann DN, Gale M Jr., Verma S. Immunoprofiles of Human Sertoli Cells Infected With Zika Virus Reveals Unique Insights Into Host-Pathogen Crosstalk. Sci Rep (2018) 8(1):8702. doi: 10.1038/s41598-018-27027-7
91. Rashid MU, Zahedi-Amiri A, Glover KKM, Gao A, Nickol ME, Kindrachuk J, et al. Zika Virus Dysregulates Human Sertoli Cell Proteins Involved in Spermatogenesis With Little Effect on Tight Junctions. PloS Negl Trop Dis (2020) 14(6):e0008335. doi: 10.1371/journal.pntd.0008335
92. Teixeira FME, Pietrobon AJ, Oliveira LM, Oliveira L, Sato MN. Maternal-Fetal Interplay in Zika Virus Infection and Adverse Perinatal Outcomes. Front Immunol (2020) 11:175. doi: 10.3389/fimmu.2020.00175
93. King NJC, Teixeira MM, Mahalingam S. Zika Virus: Mechanisms of Infection During Pregnancy. Trends Microbiol (2017) 25(9):701–2. doi: 10.1016/j.tim.2017.05.005
94. Jurado KA, Simoni MK, Tang Z, Uraki R, Hwang J, Householder S, et al. Zika Virus Productively Infects Primary Human Placenta-Specific Macrophages. JCI Insight (2016) 1(13):e88461. doi: 10.1172/jci.insight.88461
95. Simoni MK, Jurado KA, Abrahams VM, Fikrig E, Guller S. Zika Virus Infection of Hofbauer Cells. Am J Reprod Immunol (2017) 77(2):10.1111/aji.12613. doi: 10.1111/aji.12613
96. Rabelo K, de Souza LJ, Salomao NG, Machado LN, Pereira PG, Portari EA, et al. Zika Induces Human Placental Damage and Inflammation. Front Immunol (2020) 11:2146. doi: 10.3389/fimmu.2020.02146
97. Bhatnagar J, Rabeneck DB, Martines RB, Reagan-Steiner S, Ermias Y, Estetter LB, et al. Zika Virus RNA Replication and Persistence in Brain and Placental Tissue. Emerg Infect Dis (2017) 23(3):405–14. doi: 10.3201/eid2303.161499
98. Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, et al. Type I Interferons Instigate Fetal Demise After Zika Virus Infection. Sci Immunol (2018) 3(19):eaao1680. doi: 10.1126/sciimmunol.aao1680
99. Kam YW, Leite JA, Lum FM, Tan JJL, Lee B, Judice CC, et al. Specific Biomarkers Associated With Neurological Complications and Congenital Central Nervous System Abnormalities From Zika Virus-Infected Patients in Brazil. J Infect Dis (2017) 216(2):172–81. doi: 10.1093/infdis/jix261
100. Ornelas AM, Pezzuto P, Silveira PP, Melo FO, Ferreira TA, Oliveira-Szejnfeld PS, et al. Immune Activation in Amniotic Fluid From Zika Virus-Associated Microcephaly. Ann Neurol (2017) 81(1):152–6. doi: 10.1002/ana.24839
101. Foo SS, Chen W, Chan Y, Lee WS, Lee SA, Cheng G, et al. Biomarkers and Immunoprofiles Associated With Fetal Abnormalities of ZIKV-Positive Pregnancies. JCI Insight (2018) 3(21):e124152. doi: 10.1172/jci.insight.124152
102. Lesteberg KE, Fader DS, Beckham JD. Pregnancy Alters Innate and Adaptive Immune Responses to Zika Virus Infection in the Reproductive Tract. J Immunol (2020) 205(11):3107–21. doi: 10.4049/jimmunol.2000882
103. Priyamvada L, Quicke KM, Hudson WH, Onlamoon N, Sewatanon J, Edupuganti S, et al. Human Antibody Responses After Dengue Virus Infection are Highly Cross-Reactive to Zika Virus. Proc Natl Acad Sci USA (2016) 113(28):7852–7. doi: 10.1073/pnas.1607931113
104. Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, et al. Dengue Virus Sero-Cross-Reactivity Drives Antibody-Dependent Enhancement of Infection With Zika Virus. Nat Immunol (2016) 17(9):1102–8. doi: 10.1038/ni.3515
105. Keasey SL, Pugh CL, Jensen SMR, Smith JL, Hontz RD, Durbin AP, et al. Antibody Responses to Zika Virus Infections in Environments of Flavivirus Endemicity. Clin Vaccine Immunol (2017) 24(4):e00036–17. doi: 10.1128/CVI.00036-17
106. Lima NS, Rolland M, Modjarrad K, Trautmann L. T Cell Immunity and Zika Virus Vaccine Development. Trends Immunol (2017) 38(8):594–605. doi: 10.1016/j.it.2017.05.004
107. Montecillo-Aguado MR, Montes-Gomez AE, Garcia-Cordero J, Corzo-Gomez J, Vivanco-Cid H, Mellado-Sanchez G, et al. Cross-Reaction, Enhancement, and Neutralization Activity of Dengue Virus Antibodies Against Zika Virus: A Study in the Mexican Population. J Immunol Res (2019) 2019:7239347. doi: 10.1155/2019/7239347
108. Brown JA, Singh G, Acklin JA, Lee S, Duehr JE, Chokola AN, et al. Dengue Virus Immunity Increases Zika Virus-Induced Damage During Pregnancy. Immunity (2019) 50(3):751–62.e5. doi: 10.1016/j.immuni.2019.01.005
109. Rathore APS, Saron WAA, Lim T, Jahan N, St John AL. Maternal Immunity and Antibodies to Dengue Virus Promote Infection and Zika Virus-Induced Microcephaly in Fetuses. Sci Adv (2019) 5(2):eaav3208. doi: 10.1126/sciadv.aav3208
110. Pantoja P, Perez-Guzman EX, Rodriguez IV, White LJ, Gonzalez O, Serrano C, et al. Zika Virus Pathogenesis in Rhesus Macaques is Unaffected by Pre-Existing Immunity to Dengue Virus. Nat Commun (2017) 8:15674. doi: 10.1038/ncomms15674
111. Rodriguez-Barraquer I, Costa F, Nascimento EJM, Nery NJ, Castanha PMS, Sacramento GA, et al. Impact of Preexisting Dengue Immunity on Zika Virus Emergence in a Dengue Endemic Region. Science (2019) 363(6427):607–10. doi: 10.1126/science.aav6618
112. Gordon A, Gresh L, Ojeda S, Katzelnick LC, Sanchez N, Mercado JC, et al. Prior Dengue Virus Infection and Risk of Zika: A Pediatric Cohort in Nicaragua. PloS Med (2019) 16(1):e1002726. doi: 10.1371/journal.pmed.1002726
113. Carvalho MS, Freitas LP, Cruz OG, Brasil P, Bastos LS. Association of Past Dengue Fever Epidemics With the Risk of Zika Microcephaly at the Population Level in Brazil. Sci Rep (2020) 10(1):1752. doi: 10.1038/s41598-020-58407-7
114. Pedroso C, Fischer C, Feldmann M, Sarno M, Luz E, Moreira-Soto A, et al. Cross-Protection of Dengue Virus Infection Against Congenital Zika Syndrome, Northeastern Brazil. Emerg Infect Dis (2019) 25(8):1485–93. doi: 10.3201/eid2508.190113
115. Delgado FG, Torres KI, Castellanos JE, Romero-Sanchez C, Simon-Loriere E, Sakuntabhai A, et al. Improved Immune Responses Against Zika Virus After Sequential Dengue and Zika Virus Infection in Humans. Viruses (2018) 10(9):480. doi: 10.3390/v10090480
116. Grifoni A, Pham J, Sidney J, O'Rourke PH, Paul S, Peters B, et al. Prior Dengue Virus Exposure Shapes T Cell Immunity to Zika Virus in Humans. J Virol (2017) 91(24):e01469–17. doi: 10.1128/JVI.01469-17
117. Regla-Nava JA, Elong Ngono A, Viramontes KM, Huynh AT, Wang YT, Nguyen AT, et al. Cross-Reactive Dengue Virus-Specific CD8(+) T Cells Protect Against Zika Virus During Pregnancy. Nat Commun (2018) 9(1):3042. doi: 10.1038/s41467-018-05458-0
118. Wen J, Tang WW, Sheets N, Ellison J, Sette A, Kim K, et al. Identification of Zika Virus Epitopes Reveals Immunodominant and Protective Roles for Dengue Virus Cross-Reactive CD8(+) T Cells. Nat Microbiol (2017) 2:17036. doi: 10.1038/nmicrobiol.2017.36
119. Castanha PMS, Erdos G, Watkins SC, Falo LD Jr, Marques ETA, Barratt-Boyes SM. Reciprocal Immune Enhancement of Dengue and Zika Virus Infection in Human Skin. JCI Insight (2020) 5(3):e133653. doi: 10.1172/jci.insight.133653
120. Katzelnick LC, Narvaez C, Arguello S, Lopez Mercado B, Collado D, Ampie O, et al. Zika Virus Infection Enhances Future Risk of Severe Dengue Disease. Science (2020) 369(6507):1123–8. doi: 10.1126/science.abb6143
121. Messina JP, Brady OJ, Pigott DM, Golding N, Kraemer MU, Scott TW, et al. The Many Projected Futures of Dengue. Nat Rev Microbiol (2015) 13(4):230–9. doi: 10.1038/nrmicro3430
122. Counotte MJ, Althaus CL, Low N, Riou J. Impact of Age-Specific Immunity on the Timing and Burden of the Next Zika Virus Outbreak. PloS Negl Trop Dis (2019) 13(12):e0007978. doi: 10.1371/journal.pntd.0007978
123. WHO, UNICEF. WHO/UNICEF Zika Virus (ZIKV) Vaccine Target Product Profile (TPP): Vaccine to Protect Against Congenital Zika Syndrome for Use During an Emergency. (2017). Available at: https://www.unicef.org/supply/media/1346/file/Target%20product%20profile%20-%20Zika%20vaccine.pdf.
124. Li XF, Dong HL, Wang HJ, Huang XY, Qiu YF, Ji X, et al. Development of a Chimeric Zika Vaccine Using a Licensed Live-Attenuated Flavivirus Vaccine as Backbone. Nat Commun (2018) 9(1):673. doi: 10.1038/s41467-018-02975-w
125. Shan C, Muruato AE, Jagger BW, Richner J, Nunes BTD, Medeiros DBA, et al. A Single-Dose Live-Attenuated Vaccine Prevents Zika Virus Pregnancy Transmission and Testis Damage. Nat Commun (2017) 8(1):676. doi: 10.1038/s41467-017-00737-8
126. Richner JM, Jagger BW, Shan C, Fontes CR, Dowd KA, Cao B, et al. Vaccine Mediated Protection Against Zika Virus-Induced Congenital Disease. Cell (2017) 170(2):273–83.e12. doi: 10.1016/j.cell.2017.06.040
127. Tebas P, Roberts CC, Muthumani K, Reuschel EL, Kudchodkar SB, Zaidi FI, et al. Safety and Immunogenicity of an Anti-Zika Virus DNA Vaccine. N Engl J Med (2017) 385(12):e35. doi: 10.1056/NEJMoa1708120
128. Gaudinski MR, Houser KV, Morabito KM, Hu Z, Yamshchikov G, Rothwell RS, et al. Safety, Tolerability, and Immunogenicity of Two Zika Virus DNA Vaccine Candidates in Healthy Adults: Randomised, Open-Label, Phase 1 Clinical Trials. Lancet (2018) 391(10120):552–62. doi: 10.1016/S0140-6736(17)33105-7
129. Stephenson KE, Tan CS, Walsh SR, Hale A, Ansel JL, Kanjilal DG, et al. Safety and Immunogenicity of a Zika Purified Inactivated Virus Vaccine Given via Standard, Accelerated, or Shortened Schedules: A Single-Centre, Double-Blind, Sequential-Group, Randomised, Placebo-Controlled, Phase 1 Trial. Lancet Infect Dis (2020) 20(9):1061–70. doi: 10.1016/S1473-3099(20)30085-2
130. Modjarrad K, Lin L, George SL, Stephenson KE, Eckels KH, de la Barrera RA, et al. Preliminary Aggregate Safety and Immunogenicity Results From Three Trials of a Purified Inactivated Zika Virus Vaccine Candidate: Phase 1, Randomised, Double-Blind, Placebo-Controlled Clinical Trials. Lancet (2018) 391(10120):563–71. doi: 10.1016/S0140-6736(17)33106-9
131. Han HH, Diaz C, Acosta CJ, Liu M, Borkowski A. Safety and Immunogenicity of a Purified Inactivated Zika Virus Vaccine Candidate in Healthy Adults: An Observer-Blind, Randomised, Phase 1 Trial. Lancet Infect Dis (2021) 21(9):1282–92. doi: 10.1016/S1473-3099(20)30733-7
132. Aliota MT, Dudley DM, Newman CM, Mohr EL, Gellerup DD, Breitbach ME, et al. Heterologous Protection Against Asian Zika Virus Challenge in Rhesus Macaques. PloS Negl Trop Dis (2016) 10(12):e0005168. doi: 10.1371/journal.pntd.0005168
Keywords: Zika virus, congenital abnormalities, immunopathogenesis, innate and adaptive immunity, sexual transmission, pregnancy, cross-reactive immunity, vaccine
Citation: Lee LJ, Komarasamy TV, Adnan NAA, James W and RMT Balasubramaniam V (2021) Hide and Seek: The Interplay Between Zika Virus and the Host Immune Response . Front. Immunol. 12:750365. doi: 10.3389/fimmu.2021.750365
Received: 30 July 2021; Accepted: 04 October 2021;
Published: 21 October 2021.
Edited by:
Bikash Sahoo, Howard Hughes Medical Institute (HHMI), United StatesReviewed by:
Pinar Mesci, University of California, San Diego, United StatesKishan Kumar Nyati, Osaka University, Japan
Copyright © 2021 Lee, Komarasamy, Adnan, James and RMT Balasubramaniam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinod RMT Balasubramaniam, dmlub2QuYmFsYXN1YnJhbWFuaWFtQG1vbmFzaC5lZHU=
†These authors have contributed equally to this work and share first authorship