 Alba Llibre1*
Alba Llibre1* Martin Dedicoat2
Martin Dedicoat2 Julie G. Burel3
Julie G. Burel3 Caroline Demangel4†
Caroline Demangel4† Matthew K. O’Shea2,5†
Matthew K. O’Shea2,5† Claudio Mauro1*†
Claudio Mauro1*†- 1Institute of Inflammation and Ageing, College of Medical and Dental Sciences, University of Birmingham, Birmingham, United Kingdom
- 2Department of Infectious Diseases, Heartlands Hospital, University Hospitals Birmingham NHS Foundation Trust, Birmingham, United Kingdom
- 3Division of Vaccine Discovery, La Jolla Institute for Immunology, La Jolla, CA, United States
- 4Immunobiology of Infection Unit, Institut Pasteur, INSERM U1224, Paris, France
- 5Institute of Immunology and Immunotherapy, University of Birmingham, Birmingham, United Kingdom
Mycobacterial diseases are a major public health challenge. Their causative agents include, in order of impact, members of the Mycobacterium tuberculosis complex (causing tuberculosis), Mycobacterium leprae (causing leprosy), and non-tuberculous mycobacterial pathogens including Mycobacterium ulcerans. Macrophages are mycobacterial targets and they play an essential role in the host immune response to mycobacteria. This review aims to provide a comprehensive understanding of the immune-metabolic adaptations of the macrophage to mycobacterial infections. This metabolic rewiring involves changes in glycolysis and oxidative metabolism, as well as in the use of fatty acids and that of metals such as iron, zinc and copper. The macrophage metabolic adaptations result in changes in intracellular metabolites, which can post-translationally modify proteins including histones, with potential for shaping the epigenetic landscape. This review will also cover how critical tuberculosis co-morbidities such as smoking, diabetes and HIV infection shape host metabolic responses and impact disease outcome. Finally, we will explore how the immune-metabolic knowledge gained in the last decades can be harnessed towards the design of novel diagnostic and therapeutic tools, as well as vaccines.
Introduction
Mycobacteria have been a major cause of human disease for millennia, with the effects of Mycobacterium tuberculosis (M.tb) seen in the skeletons of mummified human remains from over 4000 years ago (1). The main mycobacteria of public health importance today are members of the mycobacterium tuberculosis complex (which includes M.tb, M.bovis, M.africanum, M.microti, M.pinnepedi, M.caprae), which cause tuberculosis (TB), Mycobacterium leprae (M.leprae) the cause of leprosy, and nontuberculous mycobacteria (NTM) leading to a wide variety of clinical presentations. NTM include Mycobacterium ulcerans (M.ulcerans), the cause of Buruli ulcer (BU).
Around 2 billion people worldwide are infected with M.tb. Approximately 10 million people fall ill with TB each year and there are around 1.5 million deaths (2). Great progress has been made with TB control over the past decade, but these gains have been undermined to some extent by the ongoing Covid-19 pandemic, especially in less well-resourced settings (3). Current treatment for drug sensitive TB is a minimum of 6 months and considerably longer for drug resistant disease. Shorter more effective treatments are needed, and host-directed therapies (HDT) arise as a promising strategy.
Leprosy caused by chronic infection with M.leprae is predominantly a disease of the skin and peripheral nerves. It is curable with a prolonged course of antibiotics. The incidence of leprosy has declined over the past century, but the rate of decline is currently sluggish. A large proportion of endemic countries (118/161) reported new cases in 2019 (202,256 or 26 per million population) (4). Although antibiotics can still cure the disease, permanent changes to nerves can occur leading to lifelong disabilities. There is still a great amount of stigma around the diagnosis of leprosy which can delay detection and effective treatment. Also, weak health systems can make early detection and treatment difficult. Current therapies although effective are prolonged and commonly associated with adverse effects (5).
Both TB and leprosy present a spectrum of clinical manifestations. TB can be described as a dynamic continuum from asymptomatic M.tb infection to active infectious disease, including latent infection as well as incipient, subclinical and active TB disease (6, 7). Leprosy also comprises ample clinical variability. The two extremes of the spectrum are tuberculoid and lepromatous forms of the disease, the first being paucibacillary and mild, while the latter is multibacillary and presents increased disease severity (8, 9).
There are over 200 species of NTM identified to date (10), of which the vast majority (over 95%) have not been associated with human disease. NTM are predominantly environmental organisms of low pathogenicity to humans that only cause disease in specific circumstances. An exception is M.ulcerans, which causes chronic skin ulcers in immunocompetent individuals, through production of a diffusible cytotoxin called mycolactone (11). BU is, after TB and leprosy, the third most common mycobacterial disease worldwide and together with leprosy one of the 20 Neglected Tropical Diseases prioritized by the WHO (12). West African countries are the worst impacted by BU, with prevalence rates reaching 26.9 cases per 10,000 in Benin. Other NTMs primarily affect immunocompromised people. Recently, NTM infections have been associated with health care procedures with infections due to M.chimerae occurring after cardiac surgery (13) and infections with rapid growing NTM’s such as M.abscessus being associated with cosmetic surgery procedures (14). M.abscessus is also associated with progressive lung infection in patients with cystic fibrosis (15). Treatment of NTM infections is complex requiring multiple prolonged antibiotics which may not be effective or well tolerated. Overall, novel treatments for mycobacterial diseases are needed. HTDs are a promising approach but we need a greater understanding of how the host responds to infection.
Macrophages as Targets of Mycobacterial Infection
Macrophages not only play an essential role in the host immune response to mycobacteria, but they also are mycobacterial targets. They fulfil a variety of essential functions in homeostasis and disease, including phagocytosis, uptake and killing of pathogens, tissue repair and inflammation resolution (16, 17). Mirroring their wide range of functions, macrophages constitute a highly diverse and heterogeneous population (18, 19). Traditionally, they have been classified into M1, i.e. classically-activated (LPS + IFNγ) and pro-inflammatory, and M2, i.e. alternatively-activated (IL-4) and anti-inflammatory. While the oversimplified M1/M2 dichotomy has been a useful tool when studying macrophage diversity, most macrophages exist as a spectrum and contain features of both (20, 21). Additionally, it is increasingly recognised that specific environmental cues can promote certain macrophage phenotypes and functions, demonstrating their plasticity. Metabolically, M1-like macrophages are predominantly glycolytic and present a broken TCA cycle (22). Their pentose phosphate pathway as well as fatty acid synthesis are highly active, ensuring availability of biosynthetic precursors. In contrast, M2-like macrophages mainly rely on oxidative metabolism, present an intact TCA cycle and fatty acid oxidation is upregulated. This review will explore macrophage metabolic diversity and flexibility and how these metabolic changes can drive specific phenotypes and functions, focusing on their relevance in the context of mycobacterial pathogenesis.
M.tb infection occurs when aerosols containing the bacilli are inhaled by a susceptible host. When M.tb reaches the lung, alveolar macrophages (AM), the first cellular target (23), are infected. Infected AMs can migrate to the lung interstitium, facilitating infection of other cell types, including newly-recruited monocyte-derived macrophages and neutrophils. Although abundant evidence points towards M.leprae being transmitted through the respiratory route (24, 25), the precise mechanisms for M.leprae spread remain to be fully elucidated (26). M.leprae primary cell targets are macrophages and Schwann cells, the latter contributing to build the myelin sheath that covers nerve fibres. Following introduction into the skin, M. ulcerans bacilli are phagocytosed by macrophages and multiply intracellularly until bacterial production of mycolactone causes host cell apoptosis (11, 27). Therefore, macrophages are a clear target for mycobacterial pathogens which have evolved successful strategies to survive and replicate within them (28), including metabolic manipulation.
In the context of mycobacterial disease, macrophages play an essential role in driving innate and adaptive immune responses and inflammation, while mediating both tissue destruction and repair (29). Two major macrophage populations cohabit in the lung: AMs and interstitial macrophages (IMs). They are distinct at the ontogenic, phenotypic, metabolic and functional levels (30). AMs display an M2-like phenotype (31) with predominant oxidative metabolism (30, 32), similar to IL-4 treated hMDM (33). In contrast, IMs are mainly derived from recruited monocytes (34, 35), and this has been particularly shown in the context of M.tb infection (30, 36). IMs are M1-like and operate at high levels of glycolysis (30). Other types of macrophages relevant in the context of mycobacterial infection include epithelioid cells, multinucleate giant cells (MGCs) and foamy, lipid-rich macrophages (37). MGCs are the result of macrophage fusion within the granuloma, organised cellular aggregates hallmark of TB and leprosy (38).
In leprosy, skin lesions from the milder, tuberculoid forms of the disease have been shown to have an M1-like macrophage predominant population, whereas in more severe skin lesions of multibacillary patients the balance shifts towards M2-like macrophages (39).
BU manifests as chronic ulcerative skin lesions with a distinctive lack of pain and inflammation, which if untreated enlarge over time (40). Tissue necrosis, local analgesia and defective inflammation are all attributed to bacterial production of mycolactone, a diffusible macrolide targeting the entry point of the secretory pathway in eukaryotic cells: the Sec61 translocon (11, 41, 42). By inhibiting Sec61, mycolactone prevents host cell’s production of secreted proteins, and most of its transmembrane proteins, leading to their cytosolic degradation by the proteasome (43–45). In the skin regions surrounding bacterial foci, complete and sustained inhibition of Sec61 in host cells triggers endoplasmic reticulum (ER) stress responses culminating in apoptosis (46). In immune cells recruited to the site of infection, including macrophages, exposure to non-cytotoxic concentrations of mycolactone prevents the production of cytokines, chemokines and the transduction of receptor-mediated signals, thereby the generation of protective immune responses (41). How M.tb, M.leprae and M.ulcerans impact the metabolic reprogramming of immune cells is explored in the following section, and is summarised in Table 1.

Table 1 M1-like vs M2-like macrophages in mycobacterial infection.
Immune-Metabolic Adaptations of the Macrophage to Mycobacterial Infection
This section provides an overview of the current knowledge regarding the effects of M.tb, M.leprae and M.ulcerans on host macrophage glycolysis, oxidative and lipid metabolism (Figure 1).
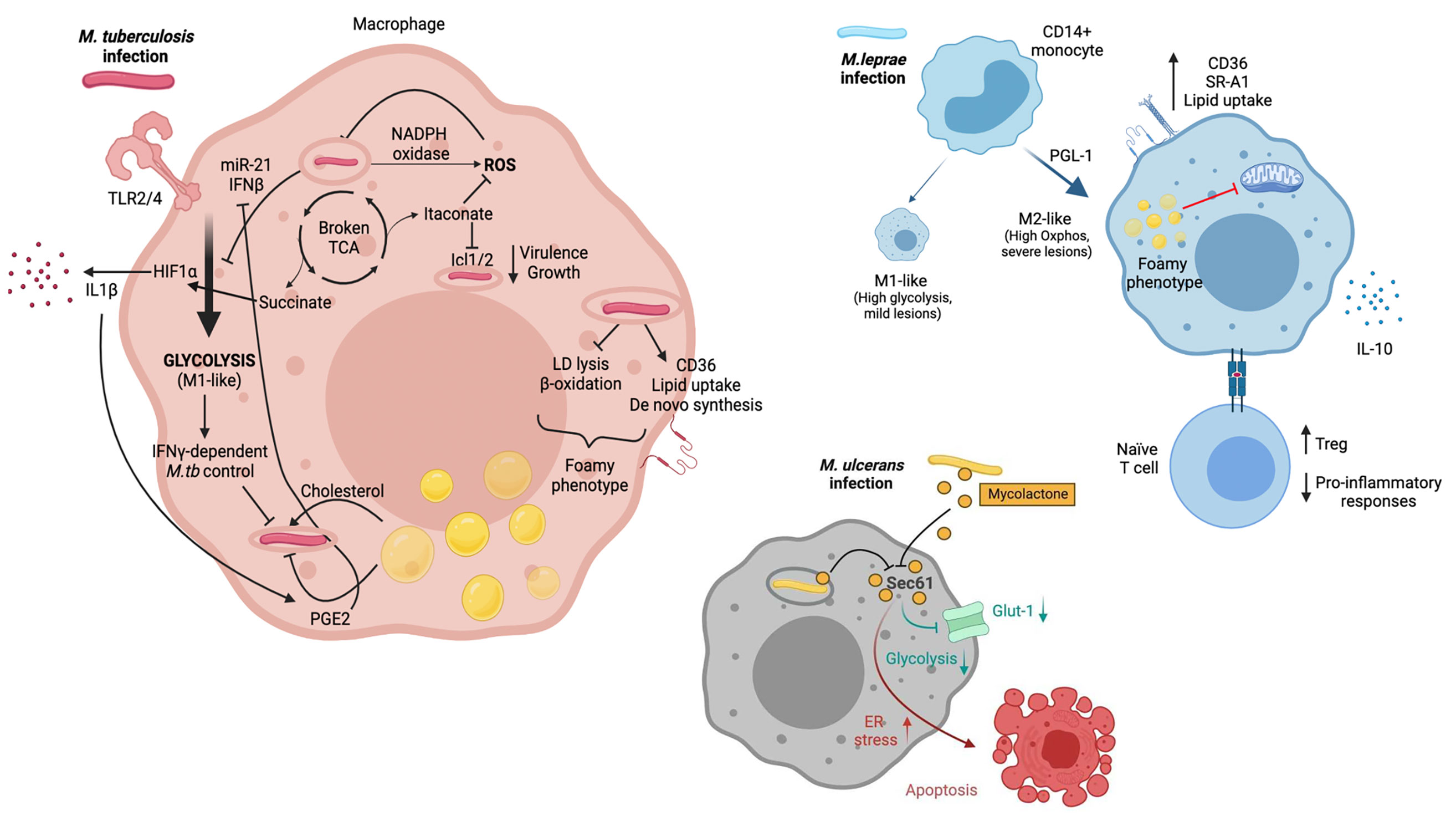
Figure 1 M.tb, M.leprae and M.ulcerans infect macrophages and alter their metabolism. M.tb is primarily sensed by host macrophages through TLR2 and TLR4. Macrophage metabolism shifts towards glycolysis (M1-like), which allows infection control. M.tb has developed strategies to counteract this metabolic switch though induction of miR-21 and IFNβ. M1-like macrophages present a broken TCA cycle, with elevated succinate concentrations that stabilise HIF1α, essential for induction of glycolysis and secretion of pro-inflammatory cytokines such as IL-1β. Itaconate also arises as a consequence of a broken TCA cycle and has multiple immune-regulatory functions, including inhibition of isocitrate lyases Icl1/2. M.tb infection triggers ROS production through NAPDH oxidase. It also promotes increased intracellular lipid content. This results in the development of lipid droplets and the foamy macrophage phenotype, which can be beneficial (i.e. providing cholesterol as nutrient) or detrimental (i.e. secretion of prostaglandins) for the pathogen. M.leprae infection of CD14+ monocytes drives macrophages towards an M2-like phenotype, with a key role for PGL-1. M2-like macrophages rely on oxidative metabolism and are associated with severe lesions. Upon M.leprae infection, macrophages upregulate CD36 and SR-A1 which translates in increased lipid uptake. Lipid droplets and the foamy phenotype have been linked to suppressed mitochondrial function. Furthermore, M.leprae-infected macrophages promote a Treg phenotype when interacting with naïve T cells, together with an impairment of pro-inflammatory cytokine release. Mycolactone released by phagocytozed and extracellular M. ulcerans bacilli diffuses into the cytoplasm of host macrophages and neighboring cells, respectively, gains access to the Sec61 translocon and blocks its activity. An immediate effect of Sec61 blockade by mycolactone is the downregulation of secretory and transmembrane proteins, among which the glucose importer Glut-1, likely resulting in decreased glycolysis. Sustained Sec61 blockade in mycolactone-exposed cells, including macrophages, triggers ER stress responses culminating in apoptosis. Created with BioRender.com.
Glycolysis Versus Oxidative Metabolism: Shaping Macrophage Polarisation
Activation of Toll-like receptors (TLRs) by mycobacterial components induces dynamic and coordinated changes in the energy metabolism of host macrophages similar to those occurring during macrophagic differentiation into the M1-like phenotype. They include a switch towards glycolysis, a disruption of the TCA cycle leading to the accumulation of succinate, and an impaired oxidative phosphorylation. Such alterations are promoted by the Hypoxia-Inducible Factor (HIF)-1α, an oxygen sensor and key glycolysis regulator that is activated by the TCA cycle intermediate succinate and mediates the production of Interleukin (IL)-1β (56). Induction of aerobic glycolysis and HIF-1α are beneficial for both innate and IFNγ-dependent control of intracellular M.tb infection by host macrophages (47, 57, 58). Importantly, in contrast to killed M.tb and the vaccine strain Bacille Calmette-Guérin (BCG), live M.tb was recently shown to specifically prevent the glycolytic switch in infected macrophages, pointing towards M.tb having evolved specific strategies to modulate host cell metabolism to its own benefit (48–50). Thus, there is an arms race to control macrophage metabolism as it is essential in determining infection outcome, and this topic has been extensively covered in recent excellent reviews (59–61). Validating this idea, it was shown in a mouse model of M.tb infection that ontologically and metabolically distinct lung macrophage populations (AMs and IMs) have differential capacities to control bacterial burden (30). Specifically, IMs which are predominantly monocyte-derived and glycolytic, present an increased ability to control M.tb growth compared to AMs, a subset of embryonic origin committed to fatty acid oxidation. Although these findings require further validation in humans, a recent single cell RNA-seq study demonstrated that the majority of lung macrophage populations are conserved between mouse and human (62).
Reprogramming of energy metabolism in M.tb-infected macrophages is also associated with increased levels of NADPH oxidase and inducible nitric oxide synthase (iNOS), promoting the production of reactive oxygen species (ROS) including nitric oxide (NO) with antimycobacterial activity (63, 64). In addition to yielding succinate, disruption of the TCA cycle upon M.tb infection promotes the generation of itaconate from cis-aconitate by aconitate decarboxylase, also known as immune-responsive-gene 1 (IRG1). Although itaconate suppresses the production of inflammatory cytokines and ROS by infected macrophages (65, 66), it directly inhibits M.tb enzymes isocitrate lyases Icl1/2, which are required for bacterial virulence and growth in vivo (67). IRG1 expression was shown to potentiate macrophage capacity to control intracellular M.tb (68) and prevent immunopathology in a mouse model of M.tb infection (69), demonstrating the key importance of this metabolic pathway in host defence against M.tb.
Studies in Schwann cells suggested that infection with M.leprae may also triggers major metabolic reprogramming in infected macrophages. Following infection by M.leprae, Schwann cells increase the expression of insulin-like growth factor (IGF), upregulating glucose transporter 1 (GLUT-1) and glucose uptake by Akt signalling (70). Glucose is redirected from glycolysis to the pentose phosphate pathway through the activation of G6PD, increasing the carbon flux to lipid biosynthesis, while both mitochondrial activity and lactate production are reduced (71, 72). Since M.leprae infection success depends on the pentose phosphate pathway, which generates reducing power for glutathione antioxidant system, it was proposed that M.leprae subverts host cell glucose metabolism to facilitate glutathione regeneration and thereby free-radical control (71).
In vitro experiments demonstrated that M1-like macrophages acquire M2-like phenotypes in the presence of M.leprae and apoptotic cells (which occur in skin lesions), contributing to mycobacterial persistence (52). Exposure of human CD14+ monocytes to M.leprae (MOI 5:1) inhibited M1 polarization, and this effect was likely mediated by the lipid component PGL-1 (73). IL-10 (anti-inflammatory) and IL-15 (pro-inflammatory) were shown to drive distinct macrophage responses which translated into progressive versus self-healing leprosy lesions, respectively (54). Live (but not killed) M.leprae promoted an M2-like phenotype in hMDM, based on IL-1β, IL-6, TNF, IL-10, CD163 and MHC-II expression (55). This M2-skewing was confirmed by measuring IL-10 and IL-12 transcript and protein levels in a different study (74). When M.leprae-infected macrophages were exposed to naïve T cells, diminished pro-inflammatory responses together with increased T regulatory phenotypes were reported (55, 74). These changes occurred using M.leprae harvested from both tuberculoid and lepromatous skin lesions (74). These findings suggest that the M2-like macrophages with predominant oxidative metabolism – same as in the context of M.tb infection - would favour pathogen persistence rather than infection resolution.
Production of mycolactone by intracellular M.ulcerans causes the apoptosis of host macrophages, and bacteria grow primarily extracellularly in infected skin during active BU (11). However, mycolactone released by bacteria diffuses broadly in infected organisms, interfering with the metabolism of both skin-resident and more distant cells (11, 75–77). Mycolactone-mediated Sec61 blockade is likely to impair host cell energy metabolism via the downregulation of nutrient tranporters, such as the glucose transporter Glut1 (SLC2A1). In support of this hypothesis, our metabolomic analysis of Jurkat T cells exposed to mycolactone revealed decreased intracellular levels of Glut1 substrates, glucose-1-phosphate and mannose-6-phosphate, suggesting impaired glycolytic activity (77). We speculate that during infection with M.ulcerans, inhibition of Sec61 impairs glycolysis reprogramming in infected macrophages, and more generally in all mycolactone-exposed immune cells.
Lipid Metabolism
In addition to reprogramming energetic metabolism, M.tb infection rewires the lipid metabolism of host macrophages through inhibition of catabolic pathways (lipid droplet [LD] lysis and β-oxidation of fatty acids) and concomitant activation of lipid uptake, mobilization and de novo synthesis (78). This leads M.tb-infected macrophages to acquire a foamy phenotype, due to the cytoplasmic accumulation of LDs mainly composed of triacylglycerol (TAGs), a storage form of fatty acids, and cholesteryl esters. Whether LD accumulation is beneficial to host macrophages or intracellular M.tb is a matter of debate. Since intracellular M.tb has the ability to import fatty acids deriving from host TAGs and foamy macrophages are a hallmark of chronic TB, accumulation of LDs in infected macrophages may provide the pathogen with essential nutrients (37, 79–81). However, recent data indicate that LD maintenance requires IFNγ-driven induction of HIF-1α, which inhibits lipolysis and prevents M.tb’s acquisition of host lipids (82). Besides, LDs are the major sites of eicosanoid production including the anti-mycobacterial prostaglandin E2 (PGE2) (83), and have direct antibacterial properties (84). PGE2 is required for M.tb control (85, 86) Leukotriene B4 (LTB4) is elevated in pulmonary TB compared to latent M.tb infected individuals (LTBI) (87) and contributes to M.tb immunopathogenesis (86). Beside eicosanoids, fatty acids released by LD lysis can be shuttled across the mitochondrial cell wall for β-oxidation, generating acetyl-CoA that enters the TCA cycle, and co-enzymes used in the respiratory chain to produce ATP. Interestingly, inhibiting fatty acid oxidation augmented macrophage ability to control M.tb infection (30, 53). Rather than starving intracellular M.tb from lipid nutrients, blocking β-oxidation of fatty acids may favour the generation of mitochondrial ROS that promote the phagosomal recruitment of NADPH oxidase and the xenophagic elimination of M.tb (53).
Similar to M.tb, M.leprae upregulates lipid uptake and biosynthetic pathways in infected macrophages, particularly cholesterol (88, 89). Mechanistically, infection with M.leprae increases macrophage expression of scavenger receptor (SR)A-I and CD36, promoting the foamy macrophage phenotype (90, 91). The mycobacteria seem to take shelter within lipid bodies, formed abundantly by host cells, possibly as a strategy to cover and hide surface antigens from innate immune receptors in the cytosol (92, 93). Contrary to M.tb (94), M.leprae cannot degrade or utilize cholesterol as a nutritional source (92), leaving the mechanism by which host cholesterol metabolism supports its in vivo persistence undefined. Notably, M.leprae infection reduces host cell mitochondrial activity in a distinctive manner (88). Oliveira et al. proposed that cytosolic accumulation of lipids in M.leprae-infected macrophages may contribute to mitochondrial shutdown and suppression of their innate immune functions (88).
Besides, macrophages infected with M.tb deprive intracellular bacteria from essential micronutrients like iron and manganese, while using copper and zinc to poison them (95). In turn, M.tb has developed sophisticated strategies to ensure micronutrient acquisition and resist metal toxicity. Interestingly, iron release prevails in AMs facilitating M.tb access to iron, whereas IMs have the capacity to sequester it, contributing to an iron starvation M.tb phenotype (96). Further information on this topic can be found in an excellent recent review by Neyrolles et al. (95). Proteins involved in iron uptake and metabolism are upregulated in lepromatous leprosy lesions, compared to tuberculoid forms, suggesting an association between iron storage in M.leprae-infected macrophages and intracellular bacterial persistence (97).
Together, these studies revealed aerobic glycolysis and fatty acid oxidation as key metabolic pathways enhancing or decreasing the anti-mycobacterial responses of macrophages, respectively. They highlighted species-specific mechanisms used by M.tb, M.leprae and M. ulcerans to subvert the glycolytic switch that is induced by TLR stimulation in infected macrophages, and take advantage of the increased lipid anabolism in host macrophages.
Metabolic-Epigenetic Crosstalk in Mycobacterial-Infected Macrophages
The macrophage metabolic adaptations to mycobacterial infection translate into changes of the intracellular metabolome. The concentration of particular metabolites therefore increases with the predominance of certain metabolic pathways. For example, lactate generation is enhanced with glycolysis, and so are citrate and acetyl-CoA with an active TCA cycle. These metabolic intermediates have the potential to post-translationally modify a wide range of proteins (e.g. histones), increasing proteome diversity and modulating function, including immune function, according to the cell needs (98–100). DNA and histones can thus be modified through metabolic substrates (e.g. methionine, acetyl-CoA, lactate), and metabolic genes can also be targets of such modifications (101). There is increasing recognition that epigenetics play an important role in shaping host-pathogen interactions and infection outcomes.
Zhang and colleagues showed that macrophage histones can be lactylated promoting an M2-like phenotype upon bacterial challenge (102), although this idea has been recently challenged (103). Since elevated glycolysis and lactate production occur in the lung of M.tb-infected hosts [reviewed in (104)], lactate could potentially drive macrophages towards an M2-phenotpye, favouring M.tb survival.
M.tb components switch host cellular metabolism toward aerobic glycolysis in human peripheral blood mononuclear cells (PBMC) through a TLR2-dependent but NOD2-independent mechanism which is partly mediated via the activation of the AKT/mTOR pathway (58). This seems to be of functional relevance as inhibition of the AKT/mTOR pathway inhibits cellular responses to M.tb both in human PBMC and a murine model of TB (58). Insights into the possible mechanisms underlying this relationship has come from studies of BCG vaccination, which has been shown to be a potent inducer of trained immunity due to cellular metabolism reprogramming arising from epigenetic changes (105). BCG-stimulated monocytes have been shown to undergo chromatin remodeling due to histone modification, namely increases in H3K4me3 and H3K9me3 at promoter sites of essential glycolytic genes. The resultant activation of the AKT/mTOR/HIF-1α pathway switches cellular metabolism from oxidative phosphorylation to aerobic glycolysis and as a consequence facilitates increased production of cytokines such as TNF and IL-6 that promote mycobacterial killing (106, 107). Conversely, these epigenetic changes are dependent on the induction of metabolic processes, as inhibition of glycolysis results in reversal of changes in H3K4me3 and H3K9me3 at promoter sites of TNF and IL-6 (107). In contrast to BCG, M.tb has been shown to impair macrophage trained immunity through activation of the type I interferon/iron axis in hematopoietic stem cells (108). This immune-metabolic reprogramming also resulted in suppressed myelopoiesis, overall enhancing host susceptibility to M.tb infection (108).
Another recently described epigenetic modification shown to cause immunomodulation in M.tb infection involves Alu repeat elements. Alu repeats are mobile interspersed repetitive DNA sequences that are transposable from one site in the genome to another, resulting in mutations, insertions and recombination events in protein-coding mRNAs (109). Analysis of genes adjacent to H3K4me1-associated Alu repeats linked to macrophage metabolic responses to M.tb infection has shown that Liver X Receptor-α signaling can be initiated at response elements present in Alu repeats and significantly reduces M.tb viability by altering cholesterol metabolism and enhancing macrophage apoptosis (110). Furthermore, levels of Alu methylation have been found to be significantly lower in paediatric TB patients and the detection of Alu DNA methylation may serve as a diagnostic and prognostic tool of TB disease in this population (111).
Co-Morbidities Impact on Metabolism and Immunity
Vulnerability to infection with M.tb and progression to active disease can be affected by several co-morbidities and social risk factors. The main co-morbidities associated with TB progression and poor outcomes are HIV infection, diabetes, renal disease and smoking. Social risk factors such as excess alcohol consumption, air pollution, incarceration and poor housing are also important. Diabetes, tobacco smoking and HIV infection have a profound impact on the host metabolic state, at both the macrophage (Figure 2) and systemic levels. Understanding how these metabolic changes occur will contribute to elucidating why these particular conditions worsen TB.
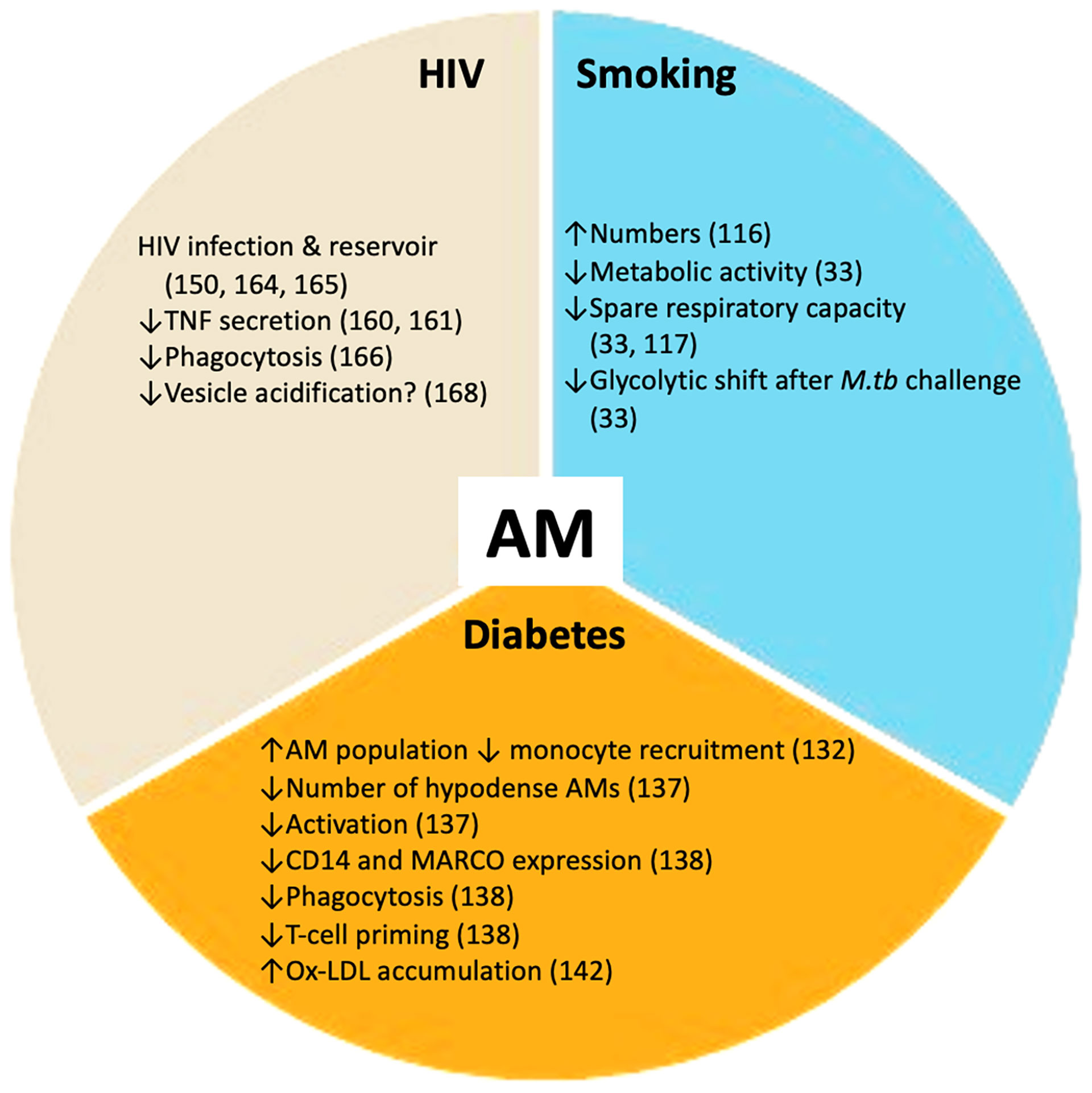
Figure 2 TB co-morbidities impact alveolar macrophages (AMs). Smoking, HIV infection and diabetes mellitus perturb the healthy alveolar macrophage phenotype. These co-morbidities promote changes in alveolar macrophage metabolism and essential functions including phagocytosis and cytokine secretion. These disturbances render the alveolar macrophage population more susceptible to M.tb infection. ↑ increased; ↓ decreased.
Smoking
The fact that tobacco smoking impacts the immune system and increases the prevalence of both respiratory and distal organ-associated diseases has been known for decades [reviewed in (112–116)]. Cigarette smoke (CS) contains abundant compounds (including toxins and carcinogens) which can directly modify immune function [reviewed in (115)].
Few studies have investigated the direct metabolic impact of smoking in M.tb infection. A macrophage shift to glycolysis is essential for effective control of M.tb (30, 47, 49). AMs from smokers presented decreased capacity to control H37Ra M.tb compared to non-smokers (117). This was probably due to impaired secretion of key cytokines for infection control, including IL-1β, TNF and IFNγ, and some of these altered immune responses remained in AMs from ex-smokers (117).
Both reduced metabolic activity (measured by oxygen consumption rate [OCR] and extracellular acidification rate [ECAR]) and metabolic reserves (glycolytic reserve and spare respiratory capacity [SRC]) have been reported at baseline in AM from smokers, measured by extracellular flux analysis (33). Interestingly, baseline metabolism of smokers’ AMs was skewed towards glycolysis, matching previous results (118). AMs from smokers showed diminished glycolysis than AMs from non-smokers when challenged with M.tb, measured by extracellular flux analysis and lactate secretion (33). No differences in oxidative metabolism were detected. Of note, the essential shift towards glycolysis (ECAR/OCR) for M.tb control was severely impaired in smokers’ AMs compared to their non-smokers counterparts, and was confirmed at the transcriptional level (2-fold change reduction in the glycolysis rate-limiting enzyme hexokinase 1). This translated in a trend towards attenuated pro-inflammatory responses (IL-1β and PGE2 secretion). The described deficient shift to glycolysis, decreased lactate and IL-1β secretion were confirmed in an in vitro model of hMDMs treated with CSE (33). The bioenergetic profiling experiments were performed using H37Rv γ-irradiated M.tb. This is important since live/dead M.tb have been shown to induce profoundly different metabolic adaptations in hMDMs measured by extracellular flux analysis (48). Other studies using hMDM and a THP-1 model and CSE have shown impaired responses to LPS, based on reduced basal and induced glycolysis, NLRP3 activation and subsequent IL-1β and IL-18 secretion (119).
Although further mechanistic studies are needed, there is enough evidence supporting the notion that CS causes dysfunctional AM metabolic profiles, which directly impact the orchestration of effective immune responses against respiratory pathogens, and might help explaining the worsening of TB reported in the smoking population.
Diabetes Mellitus
The Increasing Overlap of Two Epidemics
Up until a few years ago, the epidemics of TB and diabetes mellitus (DM) were, for the most part, geographically disconnected. Today, the geographic overlap between these two epidemics arises as a worldwide threat, as TB and diabetes have the potential to make each other worse (120, 121). It is estimated that about 15% of current TB cases are associated with T2D (122, 123). Here, we will discuss how diabetes detrimentally impacts TB.
Animal studies have further confirmed that DM worsens TB outcomes. In a mouse model of DM using streptozotocin (STZ) to destroy pancreatic islets, macrophages presented a 90% reduction in their phagocytic capacity, although their intracellular killing abilities were intact (124). When these mice were challenged with M.tb (Schacht strain) 90% of them died, compared to 10% in the non-diabetic group (124). Other mice, rat and guinea pig models of diabetes/hyperglycaemia have reported increased bacterial burden compared to control animals, together with diminished IFNγ responses (125–129).
Immune Alterations
Chemotaxis of monocytes has been shown to be impaired in diabetic patients (130, 131), potentially affecting recruitment into the lung. Furthermore, monocytes from DM patients presented diminished binding and phagocytic capacity towards H37Rv M.tb compared to healthy controls (132).
Within the lungs of diabetic C57BL/6 mice (STZ model) and using an aerosol challenge model of Erdman M.tb, it was shown that the increased TB susceptibility may arise from delayed innate immune responses (133). In particular, at 2 weeks post-infection (pi) there was abundant recruited monocytes at the site of infection in the control group. In contrast, in the lungs of diabetic mice, M.tb-infected AMs prevailed, and monocyte recruitment was limited (133). This translated in delayed delivery of M.tb-antigens to the lymph nodes, and delayed presence of IFNγ+ M.tb-specific T cells in the lymph nodes and in the lungs compared to the non-diabetic group, slowing appearance of effective immune responses and potentially contributing to bacterial persistence (133). The mechanisms underlying impaired chemotaxis/recruitment remain unknown. The reliance of AMs on oxidative metabolism which facilitates M.tb persistence, in comparison to the more glycolytic recruited monocytes, could further explain the observed differences in mycobacterial control.
PBMCs from healthy individuals treated with M.tb lysate in high concentrations of glucose (40mM, but not 25mM or lower) resulted in increased secretion of TNF, IL-1β, IL-6, but not IFNγ IL-17A and IL-22 (134). Differentiated macrophages at 25 mM glucose promoted enhanced cytokine production after stimulation with M.tb lysate and LPS. However, no differences were reported in phagocytosis and M.tb killing capacities (134). Macrophages differentiated from healthy individuals and diabetic patients were characterised at baseline and after M.tb infection (MOI 5:1 for 24h) with different strains (135). Expression of key molecules for antigen presentation (HLA-DR, CD80 and CD86), the inhibitory molecule PD-L1, and cytokines/chemokines secretion patterns differed between DM patients and controls. MDMs from T2D patients presented attenuated capacity to bind, internalise and clear M.tb, and worse outcomes were reported with more virulent strains (135). Similarly, MDMs from chronic diabetic patients presented impaired M.tb killing capacity (136). Monocytes from T2D patients and healthy controls infected with M.tb presented similar bacterial growth (137). However, monocytes cultured at 30mM glucose to mimic hyperglycaemic conditions, compared to monocytes cultured at 11mM glucose, presented decreased IL-8 production (a key neutrophil chemoattractant) as well as increased H37ra M.tb survival 3 days pi (137).
In the particular context of AM, TB-DM patients presented decreased numbers of a particular hypodense AM subset, compared to TB only patients (138). Within the active pulmonary TB patients, a negative correlation was observed between the percentage of hypodense AMs and sputum bacterial load, together with disease severity assessed by chest X-ray. Overall AMs from TB-DM patients were less activated (138). Although primarily reporting descriptive findings, this is, to our knowledge, the first study showing direct impact of DM in the AM lung-resident population. Another key study focusing on AMs used the STZ mice diabetes model and showed that AMs from diabetic mice presented reduced CD14 and MARCO expression, the latter being essential in the recognition of trehalose 6,6′-dimycolate (TDM) within the bacterial cell wall (139). This translated in reduced M.tb (Erdman strain) phagocytosis and was specific to the AM population (peritoneal or BMDM did not present this altered phenotype). These AMs defects resulted in impaired T-cell priming, which was observed when AMs from diabetic animals were transferred to control animals (139).
Other major immune cell types including dendritic cells, neutrophils, natural killer cells and T cells are also impacted in the context of diabetes and hyperglycaemia. It is beyond the scope of this review to explore their precise mechanisms, and they have been reviewed elsewhere (140, 141).
Immune-Metabolic Alterations Beyond Hyperglycaemia
DM not only comprises hyperglycaemia but also a wide range of further metabolic alterations [reviewed in (142)] which have the potential to impact immune responses to M.tb (i.e. dyslipidaemia, redox and hormone balance). DM is often associated with dyslipidaemia (e.g. increased oxidised-low density lipoproteins [ox-LDLs)] and although it is difficult to establish causal links, it is well described that M.tb thrives in lipid-rich environments. Ox-LDLs accumulate in AMs of guinea pigs infected with M.tb, increasing bacterial burden (143). Similarly, in vitro human studies showed that ox-LDLs promoted M.tb survival by impairing lysosomal function (144).
It is challenging to draw conclusive answers regarding the underlying mechanisms of TB enhanced susceptibility in DM patients due to the limited number of human studies and the variety of models used (ie. mouse and human monocytes/macrophages, from diabetic subjects or treated with varying concentrations of glucose). Nonetheless, there is ample evidence suggesting altered function of the myeloid compartment in the context of DM, including chemotaxis, bacterial recognition, phagocytosis, cytokine secretion and metabolism, which could facilitate TB disease. The described immune alterations have the potential to impact the outcome of other infectious diseases [reviewed in (145, 146)]. Although there is scarce literature exploring the link between diabetes and leprosy, early studies reported an increase in diabetes in lepromatous leprosy patients (147).
HIV
According to the WHO, people living with HIV have an increased risk (16-27 times) of developing active TB compared to non-infected individuals (2). One of the key mechanisms behind this enhanced risk is the fact that HIV causes CD4+ T cell depletion [reviewed in (148)], a cell subset essential for control of M.tb infection (149–151). There is increasing awareness of HIV-driven impairment in the innate immune compartment as a key contributor to increased TB risk in M.tb-infected individuals. Here, we explore the impact of HIV infection on macrophages and how that may facilitate TB progression.
Monocyte chemotaxis (152) and monocyte oxidative burst capacities have been shown to be impaired in HIV-infected individuals (153). HIV infection downregulated essential TLRs in the myeloid compartment which are essential for M.tb recognition (154, 155). In vitro, HIV infection of hMDMs promotes an M1-like phenotype (156) and impacts macrophage metabolism, inducing mitochondrial fusion, reduced oxidative phosphorylation and no changes in glycolysis (157, 158). Enhanced lipid accumulation together with increased uptake of ox-LDL was reported in hMDMs from HIV-infected individuals compared to controls, which could potentially contribute to the foamy macrophage phenotype and aid M.tb survival (159). HIV-infected hMDMs prevented GM-CSF-mediated activation of STAT5A, a signalling pathway essential for TB control (160). TNF secretion was reduced in HIV/M.tb co-infected macrophages compared to M.tb alone in a THP-1 cell model and human AMs (161, 162), as well as TNF-dependent apoptosis (161–163). There is evidence of HIV capacity to infect AMs (164, 165) which can act as viral reservoirs (166). HIV-infected AMs present impaired phagocytic function (167), and HIV inhibits phagocytosis in hMDMs in a Nef-dependent manner (168). Restricted vesicle acidification in AMs from HIV-TB patients has been reported (169). One study in HIV-TB coinfected individuals reported no differences in phagocytosis or acidification capacities due to HIV infection (170). The overall evidence suggests that HIV makes AMs more permissive to M.tb and promotes early bacterial growth, but further research is needed to precisely elucidate the role of macrophage metabolism in this co-infection setting.
In contrast to TB, an increased risk of leprosy or enhanced severe disease has not been described in HIV-infected individuals [reviewed in (171)]. However, leprosy patients receiving highly active antiretroviral therapy (HAART) are at increased risk of reversal reaction (RR), an inflammatory exacerbation (172). The monocyte/macrophage phenotype has been shown to be distinct in skin lesions from RR/HIV patients compared to RR alone (173) including increased expression of CD209, vascular endothelial growth factor (VEGF), arginase 2 (ARG2) and PPARγ in the former. The clinical implications of these findings remain unknown. HIV-BU co-infection is rare and there is very limited research on the topic. However, current studies point towards BU patients being more likely to be infected with HIV, and HIV infection increasing BU severity (174, 175).
Identifying Immune-Metabolic Host Therapeutic Targets
Given the significant disease burden of TB and leprosy, the rise in drug resistance and the challenges associated with the development of novel anti-mycobacterial agents, HDT strategies which augment host responses to mycobacterial infections, have become an increasing focus of interest (176–178) (Figure 3). Greater understanding of the role of immunometabolism in both protection against, and susceptibility to, infection provides potential targets to promote resistance and modulate tolerance to chronic infection.
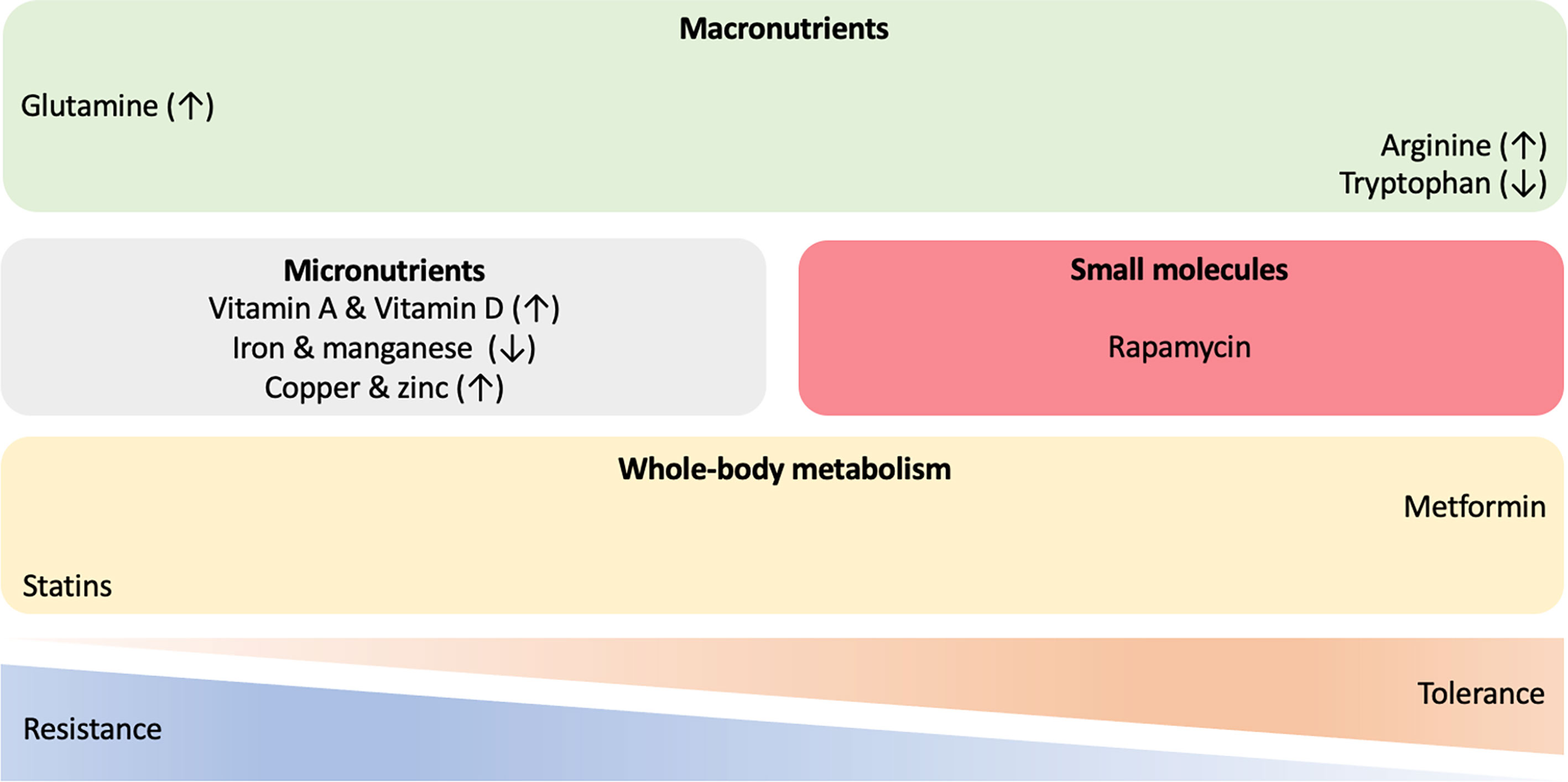
Figure 3 Host-directed therapies in mycobacterial infections. A wide range of approaches and molecules are being investigated to identify novel immune-metabolic targets for treatment of mycobacterial diseases. Therapeutic interventions with macronutrients, micronutrients and small chemical molecules, as well as compounds targeting whole-body metabolism have the potential to shift the resistance/tolerance balance of the host when challenged with mycobacteria. ↑ increased; ↓ decreased.
As discussed, it is well established epidemiologically that individuals with metabolic conditions such as T2D are at increased risk of developing active TB disease, suggesting not only the important role of host metabolic regulation in anti-M.tb responses but also pathways for possible therapeutic intervention (179).
In the context of T2D, the antihyperglycemic biguanide drug metformin appears to be a good candidate to modulate mycobacterial tolerance [reviewed in (180)]. Among patients with T2D metformin reduces the risk of M.tb infection, the progression to and the severity of TB disease, mortality, lung cavitation, relapse, accelerates sputum conversion and enhances the efficacy of anti-TB drugs (181–186). Several possible mechanisms for the advantageous effects of metformin have been suggested, including enhancing phagocytosis, phagolysosome fusion and autophagy to increase M.tb killing in macrophages; the upregulation of mitochondrial ROS production and intracellular antimycobacterial responses; an anti-inflammatory effect to reduce deleterious inflammation (182, 187, 188). Recent work suggests that metformin also educates CD8+ T-cells which results in increased mitochondrial mass, oxidative phosphorylation, and fatty acid oxidation. Such reprogramming of immune-metabolic circuits increases survival capacity and anti-mycobacterial properties of CD8+ T-cells, as seen by enhancement of BCG vaccine protective efficacy and improvements in the sterilizing ability of antibiotics (189). Since butyrate increases susceptibility to TB (134), increasing its concentration by altering gut microbiota might offer another HDT against M.tb. Eicosanoids have also been proposed as potential drug candidates for treating TB. In elegant mice experiments, IL-1 induced PGE2 production which promoted M.tb control by suppressing type I IFN (85). Furthermore, PGE2 administration resulted in decreased pulmonary M.tb load and associated pathology, as well as increased animal survival (85). These promising findings need to be validated in humans and recent evidence highlights the potential of PGE2 as an HDT candidate for treating TB (190). However, a comprehensive understanding of PGE2 kinetics and its effects on the overall immune system will be needed before eicosanoids can be clinically applied (191).
Similar to the action of metformin in targeting whole-body metabolism, statin intake has been associated with significantly reduced risk of developing TB in both T2D patients and non-diabetic general populations (192). Statins reduce inflammation, modulate the immune responses and have direct antimicrobial effects (193). Used as an adjunct, statins enhance the bactericidal activity of first-line TB drugs against intracellular M.tb and shortens TB treatment duration (194). Mechanistic studies indicate that statin-mediated reduction in cholesterol levels within phagosomal membranes counteract M.tb-induced inhibition of phagosomal maturation and promotes host-induced autophagy, therefore augmenting host responses against M.tb (195).
The role of vitamins in host immunity to M.tb are of increasing interest. The active form of vitamin A, all-trans retinoic acid (ATRA), has been shown to promote autophagy. This results in a reduced bacterial burden in human macrophages infected with M.tb, which is induced by cytosolic sensing of double-stranded DNA via the STING/TBK1/IRF3 axis (196). Furthermore, ATRA induces a reduction in total cellular cholesterol concentration and promotes lysosomal acidification in M.tb-infected monocytes via an NPC2-dependent mechanism which results in enhanced antimicrobial activity (197). The possible benefit derived from sunlight and vitamin D supplementation was first suggested in the pre-antibiotic era and has shown to modulate both innate and adaptive immune responses. The biologically active form of vitamin D, 1,25-dihydroxy- vitamin D3 (1,25(OH)2D3), enhances the expression of LL-37 in macrophages, the only cathelicidin-derived antimicrobial peptide found in humans, which promotes the destruction of M.tb and consequently autophagy (198, 199). While a series of clinical trials have failed to show vitamin D supplementation impacts on clinical outcomes in TB, there is some evidence that high-dose vitamin D improves the resolution of inflammatory responses during TB therapy and may be beneficial in a subset of TB patients who have a specific polymorphism in the vitamin D receptor (VDR) (200, 201).
The exploration of possible roles of immunomodulatory macronutrient has, to date, focused on glutamine, arginine and tryptophan. Glutamine pathway genes are differentially expressed in M.tb-infected macrophages and in the blood of individuals with LTBI or active TB. Glutamine has been identified as the main nitrogen M.tb source within infected macrophages (202). Inhibiting glutaminolysis or reducing the availability of glutamine impairs the production of key cytokines by T-cells (IFNγ, IL-1β, IL-17 and IL-22) in response to challenge with M.tb, as does genetic polymorphisms in glutamine metabolism genes (including GLS2, SLC1A5, and SLC7A5) (203). Similarly, L-citrulline and L-arginine are necessary for antimycobacterial responses, mediated by microbicidal NO production via inducible NO synthase–mediated L-arginine metabolism in infected macrophages. It has been suggested that targeting this pathway might provide novel approaches for enhancing immunity in mycobacterial disease (204). Interestingly, Arg1 conditional gene-deleted mice presented decreased M.tb burden compared to wild types, probably due to enhanced macrophage M.tb killing capacities (205). In contrast, L-Arg has been shown to contribute to macrophage M.tb clearance (206). Observed differences could be explained by different location of Arg1+ macrophages within the granuloma, as well as their role in different infection stages (207). Finally, tryptophan biosynthesis by mycobacteria under stress conditions has been shown to protect M.tb from CD4+ T-cell-mediated killing by IFNγ. Inhibition of M.tb tryptophan synthesis by the small-molecule 2-amino-6-fluorobenzoic acid (6-FABA) converts M.tb into a tryptophan auxotroph and restores the efficacy of failed CD4+ T-cell-mediated host defence (208). Interestingly, the indole propionic acid (IPA), produced by the gut microbiota, also blocks tryptophan biosynthesis in M.tb via inhibition of anthranilate synthase (TrpE) which catalyses the first committed step in the tryptophan biosynthesis pathway, by mimicking the physiological allosteric inhibitor of this enzyme (209). Therefore, in contrast to macronutrient supplementation by glutamine and arginine, tryptophan depletion may have a role in M.tb control. In the specific context of macrophages, M.tb infection causes indoleamine 2,3-dioxygenase (IDO) upregulation, the first rate-limiting enzyme of tryptophan catabolism (210, 211). This not only decreases tryptophan concentrations, but also produces metabolites which activate the aryl hydrocarbon receptor (AHR), which in turns modulate immune activity. For instance, Ahr-/- mice were unable to control M.tb H37Rv infection (212). Excellent recent reviews have further covered the potential intervention of metabolic pathways for HDT against TB (213–216).
Treating infected macrophages with statins reduces the viability of intracellular M. leprae, similar to that seen with M.tb, raising the possible use of statins as an adjuvant HDT for leprosy (217). Also similar to M.tb, autophagy is an exciting possible target for HDT leprosy as it promotes bacterial clearance and antigen presentation (218, 219). Not only does autophagy clear M. leprae from macrophages but recent evidence suggests it can modulate leprosy disease presentation, driving paucibacillary tuberculoid leprosy in individuals with more autophagic control with a predominance of IL-26, IFNγ, and TNF, autophagy-inducing cytokines (51, 220–223). Rapamycin, metformin and the antiprotozoal drug nitazoxanide have been proposed as HDTs against leprosy via modulating autophagic mechanisms to promote the antimicrobial response against M.leprae and decrease inflammation-mediated immunopathology (93).
Conclusion
Mycobacterial infections still represent a major public health issue and we need better treatments. A greater understanding of the host-induced metabolic and immune responses to mycobacterial challenge will aid the design of novel, and needed, host-directed therapeutic strategies. Macrophages are mycobacterial targets and constitute a heterogenous cell population; as reflected by their metabolic diversity and plasticity. Mycobacterial infection shapes host cell metabolism and there are clear metabolic and coupled functional phenotypes associated with particular infection outcomes (ie. M1-like favours infection control, while M2-like macrophages are more permissive to infection). Changes in macrophage polarisation upon infection directly impact the concentration of certain intracellular metabolites. They have the potential to post-translationally modify proteins, including histones. Therefore, changes in host cell metabolism can change the epigenetic landscape of the cell, with long term consequences regarding the host ability to control mycobacterial infection (107, 108). Mycobacterial diseases including TB and leprosy do not exist in isolation. Co-morbidities such as smoking, diabetes and HIV infection can worsen TB outcomes, and this can partially be explained by disease-induced metabolic changes. Gaining mechanistic understanding of this phenomenon brings us closer to the design of new and effective therapies, including expanding the use of current drugs such as metformin. There has been outstanding research in the last decades unravelling the links between cell metabolism and induced immune responses. This is a promising avenue that needs pursuing as it holds great potential to identify new targets for therapy that will aid the fight against ancient and devastating epidemics such as TB and leprosy.
Author Contributions
AL and CM conceptualised the manuscript. AL, MD, JB, CD, MO’S, and CM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
AL is supported by the European Commission (H2020-761 MSCA-IF-2018, 841729). CM is supported by the Medical Research Council (MR/T016736/1) and by a Professorial Fellowship and Translational Funds from the University of Birmingham.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zink AR, Sola C, Reischl U, Grabner W, Rastogi N, Wolf H, et al. Characterization of Mycobacterium Tuberculosis Complex Dnas From Egyptian Mummies by Spoligotyping. J Clin Microbiol (2003) 41(1):359–67. doi: 10.1128/JCM.41.1.359-367.2003
3. Thekkur P, Tweya H, Phiri S, Mpunga J, Kalua T, Kumar AMV, et al. Assessing the Impact of Covid-19 on Tb and Hiv Programme Services in Selected Health Facilities in Lilongwe, Malawi: Operational Research in Real Time. Trop Med Infect Dis (2021) 6(2):1–16. doi: 10.3390/tropicalmed6020081
4. World Health Organization. Towards Zero Leprosy Towards Zero Leprosy - Global Leprosy (Hansen’s Disease) Strategy 2021–2030 (2021). Available at: https://www.who.int/publications/i/item/9789290228509.
5. Deps P, Geurra P, Nasser S, Simon M. Hemolytic Anemia in Patients Receiving Daily Dapsone for the Treatment of Leprosy. Lepr Rev (2012) 83(3):305–7. doi: 10.47276/lr.83.3.305
6. Achkar JM, Jenny-Avital ER. Incipient and Subclinical Tuberculosis: Defining Early Disease States in the Context of Host Immune Response. J Infect Dis (2011) 204(SUPPL. 4):1179–86. doi: 10.1093/infdis/jir451
7. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Prim (2016) 2:1–23. doi: 10.1038/nrdp.2016.76
8. Gaschignard J, Grant AV, Van Thuc N, Orlova M, Cobat A, Huong NT, et al. Pauci- and Multibacillary Leprosy: Two Distinct, Genetically Neglected Diseases. PloS Negl Trop Dis (2016) 10(5):1–20. doi: 10.1371/journal.pntd.0004345
9. da Silva Prata RB, de mattos Barbosa MG, de Andrade Silva BJ, Paixao de Oliveira JA, Lameira Bittencourt T, Olmo Pinheiro R. Macrophages in the Pathogenesis of Leprosi. IntechOpen (2016) 1:1–19. doi: 10.5772/intechopen.88754
10. Tortoli E, Fedrizzi T, Meehan CJ, Trovato A, Grottola A, Giacobazzi E, et al. The New Phylogeny of the Genus Mycobacterium: The Old and the News. Infect Genet Evol (2017) 56(October):19–25. doi: 10.1016/j.meegid.2017.10.013
11. Demangel C. Immunity Against Mycobacterium Ulcerans: The Subversive Role of Mycolactone. Immunol Rev (2021) 301(1):209–21. doi: 10.1111/imr.12956
12. Omansen TF, Erbowor-Becksen A, Yotsu R, van der Werf TS, Tiendrebeogo A, Grout L, et al. Global Epidemiology of Buruli Ulcer, 2010–2017, and Analysis of 2014 WHO Programmatic Targets. Emerg Infect Dis (2019) 25(12)::2183–90. doi: 10.3201/eid2512.190427
13. Kasperbauer SH, Daley CL. Mycobacterium Chimaera Infections Related to the Heater-Cooler Unit Outbreak: A Guide to Diagnosis and Management. Clin Infect Dis (2019) 68(7):1244–50. doi: 10.1093/cid/ciy789
14. Leto Barone A, Grzelak M, Frost C, Ngagge L, Ge S, Kolegraff K, et al. Atypical Mycobacterial Infections After Plastic Surgery Procedures Abroad: A Multidisciplinary Algorithm for Diagnosis and Treatment. Ann Plast Surg (2020) 84(3):257–62. doi: 10.1097/SAP.0000000000002061
15. Martiniano SL, Esther CR, Haworth CS, Kasperbauer SH, Zemanick ET, Caverly LJ. Challenging Scenarios in Nontuberculous Mycobacterial Infection in Cystic Fibrosis. Pediatr Pulmonol (2020) 55(2):521–5. doi: 10.1002/ppul.24604
16. Tauber AI. Metchnikoff and the Phagocytosis Theory. Nat Rev Mol Cell Biol (2003) 4(11):897–901. doi: 10.1038/nrm1244
17. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The Role of Macrophages in the Resolution of Inflammation. J Clin Invest (2019) 129(7):2619–28. doi: 10.1172/JCI124615
18. Murray PJ, Wynn TA. Protective and Pathogenic Functions of Macrophage Subsets. Nat Rev Immunol (2011) 11(11):723–37. doi: 10.1038/nri3073
19. Lawrence T, Natoli G. Transcriptional Regulation of Macrophage Polarization: Enabling Diversity With Identity. Nat Rev Immunol (2011) 11(11):750–61. doi: 10.1038/nri3088
20. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immun (2014) 40(2):274–88. doi: 10.1016/j.immuni.2014.01.006
21. Martinez FO, Gordon S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep (2014) 6:1–13. doi: 10.12703/P6-13
22. O’Neill LAJ, Pearce EJ. Immunometabolism Governs Dendritic Cell and Macrophage Function. J Exp Med (2016) 213(1):15–23. doi: 10.1084/jem.20151570
23. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar Macrophages Provide an Early Mycobacterium Tuberculosis Niche and Initiate Dissemination. Cell Host Microbe (2018) 24(3):439–46 e4. doi: 10.1016/j.chom.2018.08.001
24. Davey T, Rees R. The Nasal Dicharge in Leprosy: Clinical and Bacteriological Aspects. Lepr Rev (1974) 45(2):121–34. doi: 10.5935/0305-7518.19740014
25. Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. The Continuing Challenges of Leprosy. Clin Microbiol Rev (2006) 19(2):338–81. doi: 10.1128/CMR.19.2.338-381.2006
26. Bratschi MW, Steinmann P, Wickenden A, Gillis TP. Current Knowledge on Mycobacterium Leprae Transmission: A Systematic Literature Review. Lepr Rev (2015) 86(2):142–55. doi: 10.47276/lr.86.2.142
27. Torrado E, Fraga AG, Castro AG, Stragier P, Meyers WM, Portaels F, et al. Evidence for an Intramacrophage Growth Phase of Mycobacterium Ulcerans. Infect Immun (2007) 75(2):977–87. doi: 10.1128/IAI.00889-06
28. Philips JA, Ernst JD. Tuberculosis Pathogenesis and Immunity. Annu Rev Pathol Mech Dis (2012) 7:353–84. doi: 10.1146/annurev-pathol-011811-132458
29. Marakalala MJ, Martinez FO, Plüddemann A, Gordon S. Macrophage Heterogeneity in the Immunopathogenesis of Tuberculosis. Front Microbiol (2018) 9:1–15. doi: 10.3389/fmicb.2018.01028
30. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium Tuberculosis In Vivo Segregates With Host Macrophage Metabolism and Ontogeny. J Exp Med (2018) 215(4):1135–52. doi: 10.1084/jem.20172020
31. Gordon S. Alternative Activation of Macrophages. Nat Rev Immunol (2003) 3(1):23–35. doi: 10.1038/nri978
32. Guirado E, Schlesinger LS, Kaplan G. Macrophages in Tuberculosis: Friend or Foe. Semin Immunopathol (2013) 35:563–83. doi: 10.1007/s00281-013-0388-2
33. Gleeson LE, O’Leary SM, Ryan D, McLaughlin AM, Sheedy FJ, Keane J. Cigarette Smoking Impairs the Bioenergetic Immune Response to Mycobacterium Tuberculosis Infection. Am J Respir Cell Mol Biol (2018) 59(5):572–9. doi: 10.1165/rcmb.2018-0162OC
34. Tan SYS, Krasnow MA. Developmental Origin of Lung Macrophage Diversity. Dev (2016) 143(8):1318–27. doi: 10.1242/dev.129122
35. Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am J Respir Cell Mol Biol (2017) 57(1):66–76. doi: 10.1165/rcmb.2016-0361OC
36. Srivastava S, Ernst JD, Desvignes L. Beyond Macrophages: The Diversity of Mononuclear Cells in Tuberculosis. Immunol Rev (2014) 262(1):179–92. doi: 10.1111/imr.12217
37. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy Macrophages and the Progression of the Human Tuberculosis Granuloma. Nat Immunol (2009) 10(9):943–8. doi: 10.1038/ni.1781
38. Chambers T, Spector W. Inflammatory Giant Cells. Immunobiology (1982) 161((3-4):283–9. doi: 10.1016/S0171-2985(82)80084-3
39. Moura DF, de Mattos KA, Amadeu TP, Andrade PR, Sales JS, Schmitz V, et al. CD163 Favors Mycobacterium Leprae Survival and Persistence by Promoting Anti-Inflammatory Pathways in Lepromatous Macrophages. Eur J Immunol (2012) 42(11):2925–36. doi: 10.1002/eji.201142198
40. Guarner J. Buruli Ulcer : Review of a Neglected Skin Mycobacterial. J Clin Microbiol (2018) 2):1–8. doi: 10.1128/JCM.01507-17
41. Baron L, Paatero AO, Morel JD, Impens F, Guenin-Macé L, Saint-Auret S, et al. Mycolactone Subverts Immunity by Selectively Blocking the Sec61 Translocon. J Exp Med (2016) 213(13):2885–96. doi: 10.1084/jem.20160662
42. Demangel C, High S. Sec61 Blockade by Mycolactone: A Central Mechanism in Buruli Ulcer Disease. Biol Cell (2018) 110(11):237–48. doi: 10.1111/boc.201800030
43. McKenna M, Simmonds RE, High S. Mechanistic Insights Into the Inhibition of Sec61-Dependent Co- and Post-Translational Translocation by Mycolactone. J Cell Sci (2016) 129(7):1404–15. doi: 10.1242/jcs.182352
44. McKenna M, Simmonds RE, High S. Mycolactone Reveals the Substrate-Driven Complexity of Sec61-Dependent Transmembrane Protein Biogenesis. J Cell Sci (2017) 130(7):1307–20. doi: 10.1242/jcs.198655
45. Hall BS, Hill K, McKenna M, Ogbechi J, High S, Willis AE, et al. The Pathogenic Mechanism of the Mycobacterium Ulcerans Virulence Factor, Mycolactone, Depends on Blockade of Protein Translocation Into the ER. PloS Pathog (2014) 10(4):15–7. doi: 10.1371/journal.ppat.1004061
46. Morel JD, Paatero AO, Wei J, Yewdell JW, Guenin-Macé L, Van Haver D, et al. Proteomics Reveals Scope of Mycolactone-Mediated Sec61 Blockade and Distinctive Stress Signature. Mol Cell Proteomics (2018) 17(9):1750–65. doi: 10.1074/mcp.RA118.000824
47. Gleeson LE, Sheedy FJ, Palsson-McDermott EM, Triglia D, O’Leary SM, O’Sullivan MP, et al. Cutting Edge: Mycobacterium Tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J Immunol (2016) 196(6):2444–9. doi: 10.4049/jimmunol.1501612
48. Cumming BM, Addicott KW, Adamson JH, Steyn AJ. Mycobacterium Tuberculosis Induces Decelerated Bioenergetic Metabolism in Human Macrophages. Elife (2018) 7:1–28. doi: 10.7554/eLife.39169
49. Hackett EE, Charles-Messance H, O’Leary SM, Gleeson LE, Munoz-Wolf N, Case S, et al. Mycobacterium Tuberculosis Limits Host Glycolysis and IL-1beta by Restriction of PFK-M via MicroRNA-21. Cell Rep (2020) 30(1):124–136 e4. doi: 10.1016/j.celrep.2019.12.015
50. Olson GS, Murray TA, Jahn AN, Mai D, Diercks AH, Gold ES, et al. Type I Interferon Decreases Macrophage Energy Metabolism During Mycobacterial Infection. Cell Rep (2021) 35(9):109195. doi: 10.1016/j.celrep.2021.109195
51. Yamamura M. Erratum: Defining Protective Responses to Pathogens: Cytokine Profiles in Leprosy Lesions (Science (277)). Science (1992) 255(5040):12. doi: 10.1126/science.1925582
52. Fulco T de O, Andrade PR, Barbosa MG de M, Pinto TGT, Ferreira PF, Ferreira H, et al. Effect of Apoptotic Cell Recognition on Macrophage Polarization and Mycobacterial Persistence. Infect Immun (2014) 82(9):3968–78. doi: 10.1128/IAI.02194-14
53. Chandra P, He L, Zimmerman M, Yang G, Köster S, Ouimet M, et al. Inhibition of Fatty Acid Oxidation Promotes Macrophage Control of Mycobacterium Tuberculosis. MBio (2020) 11(4):1–15. doi: 10.1128/mBio.01139-20
54. Montoya D, Cruz D, Teles RMB, Lee DJ, Ochoa MT, Krutzik SR, et al. Divergence of Macrophage Phagocytic and Antimicrobial Programs in Leprosy. Cell Host Microbe (2009) 6(4):343–53. doi: 10.1016/j.chom.2009.09.002
55. Yang D, Shui T, Miranda JW, Gilson DJ, Song Z, Chen J, et al. Mycobacterium Leprae-Infected Macrophages Preferentially Primed Regulatory T Cell Responses and Was Associated With Lepromatous Leprosy. PloS Negl Trop Dis (2016) 10(1):1–13. doi: 10.1371/journal.pntd.0004335
56. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, McGettrick AF, Goel G, et al. Succinate is an Inflammatory Signal That Induces IL-1β Through HIF-1α. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
57. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. Hif-1α is an Essential Mediator of IFN-γ–Dependent Immunity to Mycobacterium Tuberculosis. J Immunol (2016) 197(4):1287–97. doi: 10.4049/jimmunol.1600266
58. Lachmandas E, Beigier-Bompadre M, Cheng SC, Kumar V, van Laarhoven A, Wang X, et al. Rewiring Cellular Metabolism via the AKT/mTOR Pathway Contributes to Host Defence Against Mycobacterium Tuberculosis in Human and Murine Cells. Eur J Immunol (2016) 46(11):2574–86. doi: 10.1002/eji.201546259
59. Cumming BM, Pacl HT, Steyn AJC. Relevance of the Warburg Effect in Tuberculosis for Host-Directed Therapy. Front Cell Infect Microbiol (2020) 10(506):576596. doi: 10.3389/fcimb.2020.576596
60. Hackett EE, Sheedy FJ. An Army Marches on its Stomach: Metabolic Intermediates as Antimicrobial Mediators in Mycobacterium Tuberculosis Infection. Front Cell Infect Microbiol (2020) 10(446):446. doi: 10.3389/fcimb.2020.00446
61. Sheedy FJ, Divangahi M. Targeting Immunometabolism in Host Defence Against Mycobacterium Tuberculosis. Immunology (2021) 162(2):145–59. doi: 10.1111/imm.13276
62. Pisu D, Huang L, Narang V, Theriault M, Lê-Bury G, Lee B, et al. Single Cell Analysis of M. Tuberculosis Phenotype and Macrophage Lineages in the Infected Lung. J Exp Med (2021) 218(9):1–25. doi: 10.1084/jem.20210615
63. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell (2016) 167(2):457–470.e13. doi: 10.1016/j.cell.2016.08.064
64. Shi L, Jiang Q, Bushkin Y, Subbian S, Tyagi S. Biphasic Dynamics of Macrophage Immunometabolism During Mycobacterium Tuberculosis Infection. MBio (2019) 10(2):1–19. doi: 10.1128/mBio.02550-18
65. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate Is an Anti-Inflammatory Metabolite That Activates Nrf2 via Alkylation of KEAP1. Nature (2018) 556(7699):113–7. doi: 10.1038/nature25986
66. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase With Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24(1):158–66. doi: 10.1016/j.cmet.2016.06.004
67. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-Responsive Gene 1 Protein Links Metabolism to Immunity by Catalyzing Itaconic Acid Production. Proc Natl Acad Sci USA (2013) 110(19):7820–5. doi: 10.1073/pnas.1218599110
68. Hoffmann E, Machelart A, Belhaouane I, Deboosere N, Pauwels A-M, Saint-André J-P, et al. IRG1 Controls Immunometabolic Host Response and Restricts Intracellular Mycobacterium Tuberculosis Infection. bioRxiv (2019), 1–40. doi: 10.1101/761551
69. Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, et al. Irg1 Expression in Myeloid Cells Prevents Immunopathology During M. tuberculosis Infection. J Exp Med (2018) 215(4):1035–45. doi: 10.1084/jem.20180118
70. Batista-Silva LR, Rodrigues LS, Vivarini ADC, Costa FDMR, De Mattos KA, Costa MRSN, et al. Mycobacterium Leprae-Induced Insulin-Like Growth Factor I Attenuates Antimicrobial Mechanisms, Promoting Bacterial Survival in Macrophages. Sci Rep (2016) 6(May):1–13. doi: 10.1038/srep27632
71. Medeiros RCA, De Vasconcelos Girardi KDC, Cardoso FKL, De Siqueira Mietto B, De Toledo Pinto TG, Gomez LS, et al. Subversion of Schwann Cell Glucose Metabolism by Mycobacterium Leprae. J Biol Chem (2016) 291(41):21375–87. doi: 10.1074/jbc.M116.725283
72. Borah K, Girardi DV, Mendum TA, Santos M, Beste DJV, Lara A, et al. Intracellular Mycobacterium Leprae Utilizes Host Glucose as a Carbon Source in Schwann Cells. Am Soc Microbiol (2019) 10(6):1–9. doi: 10.1128/mBio.02351-19
73. Fallows D, Peixoto B, Kaplan G, Manca C. Mycobacterium Leprae Alters Classical Activation of Human Monocytes In Vitro. J Inflamm (United Kingdom) (2016) 13(1):4–8. doi: 10.1186/s12950-016-0117-4
74. Ma Y, Pei Q, Zhang L, Lu J, Shui T, Chen J, et al. Live Mycobacterium Leprae Inhibits Autophagy and Apoptosis of Infected Macrophages and Prevents Engulfment of Host Cell by Phagocytes. Am J Transl Res (2018) 10(9):2929–39.
75. Sarfo FS, Chevalier F, Aka N, Phillips RO, Amoako Y, Boneca IG, et al. Mycolactone Diffuses Into the Peripheral Blood of Buruli Ulcer Patients - Implications for Diagnosis and Disease Monitoring. PloS Negl Trop Dis (2011) 5(7):1–8. doi: 10.1371/journal.pntd.0001237
76. Hong H, Coutanceau E, Leclerc M, Caleechurn L, Leadlay PF, Demangel C. Mycolactone Diffuses From Mycobacterium Ulcerans-Infected Tissues and Targets Mononuclear Cells in Peripheral Blood and Lymphoid Organs. PloS Negl Trop Dis (2008) 2(10):1–8. doi: 10.1371/journal.pntd.0000325
77. Niang F, Sarfo FS, Frimpong M, Guenin-Macé L, Wansbrough-Jones M, Stinear T, et al. Metabolomic Profiles Delineate Mycolactone Signature in Buruli Ulcer Disease. Sci Rep (2015) 5:1–14. doi: 10.1038/srep17693
78. Laval T, Chaumont L, Demangel C. Not Too Fat to Fight: The Emerging Role of Macrophage Fatty Acid Metabolism in Immunity to Mycobacterium Tuberculosis. Immunol Rev (2021) 301(1):84–97.10.1111/imr.12952
79. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy Macrophages From Tuberculous Patients’ Granulomas Constitute a Nutrient-Rich Reservoir for M. Tuberculosis Persistence. PloS Pathog (2008) 4(11):1–14. doi: 10.1371/journal.ppat.1000204
80. Elamin AA, Stehr M, Singh M. Lipid Droplets and Mycobacterium Leprae Infection. J Pathog (2012) 2012:1–10. doi: 10.1155/2012/361374
81. Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium Tuberculosis Uses Host Triacylglycerol to Accumulate Lipid Droplets and Acquires a Dormancy-Like Phenotype in Lipid-Loaded Macrophages. PloS Pathog (2011) 7(6):1–16. doi: 10.1371/journal.ppat.1002093
82. Knight M, Braverman J, Asfaha K, Gronert K, Stanley S. Lipid Droplet Formation in Mycobacterium Tuberculosis Infected Macrophages Requires IFN-γ/Hif-1α Signaling and Supports Host Defense. PloS Pathog (2018) 14(1):e1006874. doi: 10.1371/journal.ppat.1006874
83. Mayer-Barber KD, Sher A. Cytokine and Lipid Mediator Networks in Tuberculosis. Immunol Rev (2015) 264(1):264–75. doi: 10.1111/imr.12249
84. Bosch M, Sánchez-Álvarez M, Fajardo A, Kapetanovic R, Steiner B, Dutra F, et al. Mammalian Lipid Droplets Are Innate Immune Hubs Integrating Cell Metabolism and Host Defense. Science (2020) 370(6514):1–12. doi: 10.1126/science.aay8085
85. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-Directed Therapy of Tuberculosis Based on Interleukin-1 and Type I Interferon Crosstalk. Nat (2014) 511(7507):99–103. doi: 10.1038/nature13489
86. Sorgi CA, Soares EM, Rosada RS, Bitencourt CS, Zoccal KF, Pereira PAT, et al. Eicosanoid Pathway on Host Resistance and Inflammation During Mycobacterium Tuberculosis Infection Is Comprised by LTB4 Reduction But Not PGE2 Increment. Biochim Biophys Acta - Mol Basis Dis (2020) 1866(3):165574. doi: 10.1016/j.bbadis.2019.165574
87. Nore KG, Jørgensen MJ, Dyrhol-Riise AM, Jenum S, Tonby K. Elevated Levels of Anti-Inflammatory Eicosanoids and Monocyte Heterogeneity in Mycobacterium Tuberculosis Infection and Disease. Front Immunol (2020) 11(November):1–12. doi: 10.3389/fimmu.2020.579849
88. Oliveira MF, Medeiros RCA, Mietto BS, Calvo TL, Mendonça APM, Rosa TLSA, et al. Reduction of Host Cell Mitochondrial Activity as Mycobacterium Leprae’s Strategy to Evade Host Innate Immunity. Immunol Rev (2021) 301(1):193–208. doi: 10.1111/imr.12962
89. Mattos KA, Oliveira VCG, Berrêdo-Pinho M, Amaral JJ, Antunes LCM, Melo RCN, et al. Mycobacterium Leprae Intracellular Survival Relies on Cholesterol Accumulation in Infected Macrophages: A Potential Target for New Drugs for Leprosy Treatment. Cell Microbiol (2014) 16(6):797–815. doi: 10.1111/cmi.12279
90. Cruz D, Watson AD, Miller CS, Montoya D, Ochoa MT, Sieling PA, et al. Host-Derived Oxidized Phospholipids and HDL Regulate Innate Immunity in Human Leprosy. J Clin Invest (2008) 118(8):2917–28. doi: 10.1172/JCI34189
91. Kaur G, Kaur J. Multifaceted Role of Lipids in Mycobacterium Leprae. Future Microbiol (2017) 12(4):315–35. doi: 10.2217/fmb-2016-0173
92. Marques MAM, Berrêdo-Pinho M, Rosa TLSA, Pujari V, Lemes RMR, Lery LMS, et al. The Essential Role of Cholesterol Metabolism in the Intracellular Survival of Mycobacterium Leprae Is Not Coupled to Central Carbon Metabolism and Energy Production. J Bacteriol (2015) 197(23):3698–707. doi: 10.1128/JB.00625-15
93. Toledo Pinto TG, Batista-Silva LR, Medeiros R, Lara FA, Moraes M. Type I Interferons, Autophagy and Host Metabolism in Leprosy. Front Immunol (2018) 9(APR):1–11. doi: 10.3389/fimmu.2018.00806
94. Wilburn KM, Fieweger RA, VanderVen BC. Cholesterol and Fatty Acids Grease the Wheels of Mycobacterium Tuberculosis Pathogenesis. Pathog Dis (2018) 76(2):1–14. doi: 10.1093/femspd/fty021
95. Neyrolles O, Wolschendorf F, Mitra A, Niederweis M. Mycobacteria, Metals, and the Macrophage. Immunol Rev (2015) 264(1):249–63. doi: 10.1111/imr.12265
96. Pisu D, Huang L, Grenier JK, Russell DG. Dual RNA-Seq of Mtb-Infected Macrophages In Vivo Reveals Ontologically Distinct Host-Pathogen Interactions. Cell Rep (2020) 30(2):335–350.e4. doi: 10.1016/j.celrep.2019.12.033
97. de Mattos Barbosa MG, da Silva Prata RB, Andrade PR, Ferreira H, de Andrade Silva BJ, da Paixão de Oliveira JA, et al. Indoleamine 2,3-Dioxygenase and Iron Are Required for Mycobacterium Leprae Survival. Microbes Infect (2017) 19(11):505–14. doi: 10.1016/j.micinf.2017.06.006
98. Prabakaran S, Lippens G, Steen H, Gunawardena J. Post-Translational Modification: Nature’s Escape From Genetic Imprisonment and the Basis for Dynamic Information Encoding. Wiley Interdiscip Rev Syst Biol Med (2012) 4(6):565–83. doi: 10.1002/wsbm.1185
99. Karve TM, Cheema AK. Small Changes Huge Impact: The Role of Protein Posttranslational Modifications in Cellular Homeostasis and Disease. J Amino Acids (2011) 2011:1–13. doi: 10.4061/2011/207691
100. Liu J, Qian C, Cao X. Post-Translational Modification Control of Innate Immunity. Immun (2016) 45(1):15–30. doi: 10.1016/j.immuni.2016.06.020
101. Allis CD, Jenuwein T. The Molecular Hallmarks of Epigenetic Control. Nat Rev Genet (2016) 17(8):487–500. doi: 10.1038/nrg.2016.59
102. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
103. Dichtl S, Lindenthal L, Zeitler L, Behnke K, Schlösser D, Strobl B, et al. Lactate and IL6 Define Separable Paths of Inflammatory Metabolic Adaptation. Scie Adv (2021) 7(26):1–11. doi: 10.1126/sciadv.abg3505
104. Llibre A, Grudzinska FS, O’Shea MK, Duffy D, Thickett DR, Mauro C, et al. Lactate Crosstalk in Host-Pathogen Interactions. Biochem J (2021) 478(17):3157–78. doi: 10.1042/BCJ20210263
105. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining Trained Immunity and its Role in Health and Disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
106. Chao WC, Yen CL, Wu YH, Chen SY, Hsieh CY, Chang TC, et al. Increased Resistin may Suppress Reactive Oxygen Species Production and Inflammasome Activation in Type 2 Diabetic Patients With Pulmonary Tuberculosis Infection. Microbes Infect (2015) 17(3):195–204. doi: 10.1016/j.micinf.2014.11.009
107. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep (2016) 17(10):2562–71. doi: 10.1016/j.celrep.2016.11.011
108. Khan N, Downey J, Sanz J, Kaufmann E, Blankenhaus B, Pacis A, et al. M. Tuberculosis Reprograms Hematopoietic Stem Cells to Limit Myelopoiesis and Impair Trained Immunity. Cell (2020) 183(3):752–770.e22. doi: 10.1016/j.cell.2020.09.062
109. Batzer MA, Deininger PL. Alu Repeats and Human Genomic Diversity. Nat Rev Genet (2002) 3(5):370–9. doi: 10.1038/nrg798
110. Bouttier M, Laperriere D, Memari B, Mangiapane J, Fiore A, Mitchell E, et al. Alu Repeats as Transcriptional Regulatory Platforms in Macrophage Responses to M.Tuberculosis Infection. Nucleic Acids Res (2016) 44(22):10571–87. doi: 10.1093/nar/gkw782
111. Maruthai K, Subramanian M. Methylation Status of Alu Repetitive Elekents in Children With Tuberculosis Disease. Int J Mycobacteriol (2017) 6(3):239–45. doi: 10.4103/ijmy.ijmy_86_18
112. Holt PG, Keast D. Environmentally Induced Changes in Immunological Function: Acute and Chronic Effects of Inhalation of Tobacco Smoke and Other Atmospheric Contaminants in Man and Experimental Animals. Bacteriol Rev (1977) 41(1):205–16. doi: 10.1128/br.41.1.205-216.1977
113. Holt PG. Immune and Inflammatory Function in Cigarette Smokers. Thorax (1987) 42(4):241–9. doi: 10.1136/thx.42.4.241
114. Sopori M. Effects of Cigarette Smoke on the Immune System. Nat Rev Immunol (2002) 2:372–7. doi: 10.1038/nri803
115. Stämpfli MR, Anderson GP. How Cigarette Smoke Skews Immune Responses to Promote Infection, Lung Disease and Cancer. Nat Rev Immunol (2009) 9(5):377–84. doi: 10.1038/nri2530
116. Qiu F, Liang CL, Liu H, Zeng YQ, Hou S, Huang S, et al. Impacts of Cigarette Smoking on Immune Responsiveness: Up and Down or Upside Down? Oncotarget (2017) 8(1):268–84. doi: 10.18632/oncotarget.13613
117. O’Leary SM, Coleman MM, Chew WM, Morrow C, McLaughlin AM, Gleeson LE, et al. Cigarette Smoking Impairs Human Pulmonary Immunity to Mycobacterium Tuberculosis. Am J Respir Crit Care Med (2014) 190(12):1430–6. doi: 10.1164/rccm.201407-1385OC
118. Aridgides DS, Mellinger DL, Armstrong DA, Hazlett HF, Dessaint JA, Hampton TH, et al. Functional and Metabolic Impairment in Cigarette Smoke-Exposed Macrophages is Tied to Oxidative Stress. Sci Rep (2019) 9(1):1–11. doi: 10.1038/s41598-019-46045-7
119. Buscetta M, Di Vincenzo S, Miele M, Badami E, Pace E, Cipollina C. Cigarette Smoke Inhibits the NLRP3 Inflammasome and Leads to Caspase-1 Activation via the TLR4-TRIF-Caspase-8 Axis in Human Macrophages. FASEB J (2020) 34(1):1819–32. doi: 10.1096/fj.201901239R
120. Stevenson CR, Critchley JA, Forouhi NG, Roglic G, Williams BG, Dye C, et al. Diabetes and the Risk of Tuberculosis: A Neglected Threat to Public Health? Chronic Illn (2007) 3(3):228–45. doi: 10.1177/1742395307081502
121. Dooley KE, Chaisson RE. Tuberculosis and Diabetes Mellitus: Convergence of Two Epidemics. Lancet Infect Dis (2009) 9(12):737–46. doi: 10.1016/S1473-3099(09)70282-8
122. Lönnroth K, Roglic G, Harries AD. Improving Tuberculosis Preven- Tion and Care Through Addressing the Global Diabetes Epidemic: From Evidence to Policy and Practice. Lancet Diabetes Endocrinol (2014) 2(9):730–9. doi: 10.1016/S2213-8587(14)70109-3
123. Workneh MH, Bjune GA, Yimer SA. Prevalence and Associated Factors of Tuberculosis and Diabetes Mellitus Comorbidity: A Systematic Review1. PloS One (2017) 12(4):1–25. doi: 10.1371/journal.pone.0175925
124. Saiki O, Negoro S, Tsuyuguchi I, Yamamura Y. Depressed Immunological Defense Mechanisms in Mice With Experimentally Induced Diabetes. Infect Immun (1980) 28(1):127–31. doi: 10.1128/iai.28.1.127-131.1980
125. Yamashiro S, Kawakami K, Uezu K, Kinjo T, Miyagi K, Nakamura K, et al. Lower Expression of Th1-Related Cytokines and Inducible Nitric Oxide Synthase in Mice With Streptozotocin-Induced Diabetes Mellitus Infected With Mycobacterium Tuberculosis. Clin Exp Immunol (2005) 139(1):57–64. doi: 10.1111/j.1365-2249.2005.02677.x
126. Martens GW, Arikan MC, Lee J, Ren F, Greiner D, Kornfeld H. Tuberculosis Susceptibility of Diabetic Mice. Am J Respir Cell Mol Biol (2007) 37(5):518–24. doi: 10.1165/rcmb.2006-0478OC
127. Sugawara I, Mizuno S. Higher Susceptibility of Type 1 Diabetic Rats to Mycobacterium Tuberculosis Infection. Tohoku J Exp Med (2008) 216(4):363–70. doi: 10.1620/tjem.216.363
128. Sugawara I, Yamada H, Mizuno S. Pulmonary Tuberculosis in Spontaneously Diabetic Goto Kakizaki Rats. Tohoku J Exp Med (2004) 204(2):135–45. doi: 10.1620/tjem.204.135
129. Podell BK, Ackart DF, Kirk NM, Eck SP, Bell C, Basaraba RJ. Non-Diabetic Hyperglycemia Exacerbates Disease Severity in Mycobacterium Tuberculosis Infected Guinea Pigs. PloS One (2012) 7(10):1–10. doi: 10.1371/journal.pone.0046824
130. Moutschen M, Scheen A, Lefebvre P. Impaired Immune Responses in Diabetes Mellitus: Analysis of the Factors and Mechanisms Involved. Relevance to the Increased Susceptibility of Diabetic Patients to Specific Infections. Diabete Metab (1992) 18(3):187–201.
131. Stew SS, Martinez PJ, Schlesinger LS, Restrepo BI. Differential Expression of Monocyte Surface Markers Among TB Patients With Diabetes Co-Morbidity. Tuberculosis (2013) 93(SUPPL.):578–82. doi: 10.1016/S1472-9792(13)70015-5
132. Gomez DI, Twahirwa M, Schlesinger LS, Restrepo BI. Reduced Mycobacterium Tuberculosis Association With Monocytes From Diabetes Patients That Have Poor Glucose Control. Tuberculosis (2013) 93(2):192–7. doi: 10.1016/j.tube.2012.10.003
133. Vallerskog T, Martens GW, Kornfeld H. Diabetic Mice Display a Delayed Adaptive Immune Response to Mycobacterium Tuberculosis. J Immunol (2010) 184(11):6275–82. doi: 10.4049/jimmunol.1000304
134. Lachmandas E, Van Den Heuvel CNAM, Damen MSMA, Cleophas MCP, Netea MG, Van Crevel R. Diabetes Mellitus and Increased Tuberculosis Susceptibility: The Role of Short-Chain Fatty Acids. J Diabetes Res (2016) 2016:36–8. doi: 10.1155/2016/6014631
135. Lopez-Lopez N, Martinez AGR, Garcia-Hernandez MH, Hernandez-Pando R, Castañeda-Delgado JE, Lugo-Villarino G, et al. Type-2 Diabetes Alters the Basal Phenotype of Human Macrophages and Diminishes Their Capacity to Respond, Internalise, and Control Mycobacterium Tuberculosis. Mem Inst Oswaldo Cruz (2018) 113(4):1–11. doi: 10.1590/0074-02760170326
136. Restrepo BI, Khan A, Singh VK, de-Leon E, Aguillón-Durán GP, Ledezma-Campos E, et al. Human Monocyte-Derived Macrophage Responses to M. Tuberculosis Differ by the Host’s Tuberculosis, Diabetes or Obesity Status, and are Enhanced by Rapamycin. Tuberculosis (2021) 126:1–8. doi: 10.1016/j.tube.2020.102047
137. Torres M, Herrera MT, Fabián-San-Miguel G, Gonzalez Y. The Intracellular Growth of M. Tuberculosis is More Associated With High Glucose Levels Than With Impaired Responses of Monocytes From T2D Patients. J Immunol Res (2019) 2019:1–10. doi: 10.1155/2019/1462098
138. Wang CH, Yu CT, Lin HC, Liu CY, Kuo HP. Hypodense Alveolar Macrophages in Patients With Diabetes Mellitus and Active Pulmonary Tuberculosis. Tuber Lung Dis (1999) 79(4):235–42. doi: 10.1054/tuld.1998.0167
139. Martinez N, Ketheesan N, West K, Vallerskog T, Kornfeld H. Impaired Recognition of Mycobacterium Tuberculosis by Alveolar Macrophages From Diabetic Mice. J Infect Dis (2016) 214(11):1629–37. doi: 10.1093/infdis/jiw436
140. Hodgson K, Morris J, Bridson T, Govan B, Rush C, Ketheesan N. Immunological Mechanisms Contributing to the Double Burden of Diabetes and Intracellular Bacterial Infections. Immunology (2015) 144(2):171–85. doi: 10.1111/imm.12394
141. Ferlita S, Yegiazaryan A, Noori N, Lal G, Nguyen T, To K, et al. Type 2 Diabetes Mellitus and Altered Immune System Leading to Susceptibility to Pathogens, Especially Mycobacterium Tuberculosis. J Clin Med (2019) 8(12):2219. doi: 10.3390/jcm8122219
142. Segura-Cerda CA, López-Romero W, Flores-Valdez MA. Changes in Host Response to Mycobacterium Tuberculosis Infection Associated With Type 2 Diabetes: Beyond Hyperglycemia. Front Cell Infect Microbiol (2019) 9:1–10. doi: 10.3389/fcimb.2019.00342
143. Palanisamy GS, Kirk NM, Ackart DF, Obregón-Henao A, Shanley CA, Orme IM, et al. Uptake and Accumulation of Oxidized Low-Density Lipoprotein During Mycobacterium Tuberculosis Infection in Guinea Pigs. PloS One (2012) 7(3):1–10. doi: 10.1371/journal.pone.0034148
144. Vrieling F, Wilson L, Rensen PCN, Walzl G, Ottenhoff THM, Joosten SA. Oxidized Low-Density Lipoprotein (Oxldl) Supports Mycobacterium Tuberculosis Survival in Macrophages by Inducing Lysosomal Dysfunction. PloS Pathog (2019) 15(4):1–27. doi: 10.1371/journal.ppat.1007724
145. Alves C, Casqueiro J, Casqueiro J. Infections in Patients With Diabetes Mellitus: A Review of Pathogenesis. Indian J Endocrinol Metab (2012) 16(7):27. doi: 10.4103/2230-8210.94253
146. van Crevel R, van de Vijver S, Moore DAJ. The Global Diabetes Epidemic: What Does It Mean for Infectious Diseases in Tropical Countries? Lancet Diabetes Endocrinol (2017) 5(6):457–68. doi: 10.1016/S2213-8587(16)30081-X
147. Nigam P, Dayal SG, Srivastava P, Joshi LD, Goyal BM, Dutt B, et al. Diabetic Status in Leprosy. Hansenol Int (1979) 4(1):7–14.
148. Okoye AA, Picker LJ. Cd4+ T-Cell Depletion in Hiv Infection: Mechanisms of Immunological Failure. Immunol Rev (2013) 254(1):54–64. doi: 10.1111/imr.12066
149. Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, Flynn JL. Mice Deficient in CD4 T Cells Have Only Transiently Diminished Levels of IFN-Gamma, Yet Succumb to Tuberculosis. J Immunol (1999) 162(9):5407–16.
150. Scanga CA, Mohan VP, Yu K, Joseph H, Tanaka K, Chan J, et al. Depletion of CD4+ T Cells Causes Reactivation of Murine Persistent Tuberculosis Despite Continued Expression of Interferon γ and Nitric Oxide Synthase 2. J Exp Med (2000) 192(3):347–58. doi: 10.1084/jem.192.3.347
151. Lawn SD, Myer L, Edwards D, Bekker LG, Wood R. Short-Term and Long-Term Risk of Tuberculosis Associated With CD4 Cell Recovery During Antiretroviral Therapy in South Africa. Aids (2009) 23(13):1717–25. doi: 10.1097/QAD.0b013e32832d3b6d
152. Wahl SM, Allen JB, Gartner S, Orenstein JM, Popovic M, Chenoweth DE, et al. HIV-1 and its Envelope Glycoprotein Down-Regulate Chemotactic Ligand Receptors and Chemotactic Function of Peripheral Blood Monocytes. J Immunol (1989) 142(10):3553–9.
153. Spear GT, Kessler HA, Rothberg L, Phair J, Landay AL. Decreased Oxidative Burst Activity of Monocytes From Asymptomatic HIV-Infected Individuals. Clin Immunol Immunopathol (1990) 54(2):184–91. doi: 10.1016/0090-1229(90)90080-A
154. Nicol MQ, Mathys J-M, Pereira A, Ollington K, Ieong MH, Skolnik PR. Human Immunodeficiency Virus Infection Alters Tumor Necrosis Factor Alpha Production via Toll-Like Receptor-Dependent Pathways in Alveolar Macrophages and U1 Cells. J Virol (2008) 82(16):7790–8. doi: 10.1128/JVI.00362-08
155. Richardson ET, Shukla S, Sweet DR, Wearsch PA, Tsichlis PN, Henry Boom W, et al. Toll-Like Receptor 2-Dependent Extracellular Signal-Regulated Kinase Signaling in Mycobacterium Tuberculosis-Infected Macrophages Drives Anti-Inflammatory Responses and Inhibits Th1 Polarization of Responding T Cells. Infect Immun (2015) 83(6):2242–54. doi: 10.1128/IAI.00135-15
156. Porcheray F, Léone C, Samah B, Rimaniol AC, Dereuddre-Bosquet N, Gras G. Glutamate Metabolism in HIV-Infected Macrophages: Implications for the CNS. Am J Physiol - Cell Physiol (2006) 291(4):618–26. doi: 10.1152/ajpcell.00021.2006
157. Castellano P, Prevedel L, Valdebenito S, Eugenin EA. HIV Infection and Latency Induce a Unique Metabolic Signature in Human Macrophages. Sci Rep (2019) 9(1):1–14. doi: 10.1038/s41598-019-39898-5
158. Hollenbaugh JA, Munger J, Kim B. Metabolite Profiles of Human Immunodeficiency Virus Infected CD4+ T Cells and Macrophages Using LC-MS/MS Analysis. Virology (2011) 415(2):153–9. doi: 10.1016/j.virol.2011.04.007
159. Bowman ER, Cameron CM, Richardson B, Kulkarni M, Gabriel J, Cichon MJ, et al. Macrophage Maturation From Blood Monocytes is Altered in People With HIV, and is Linked to Serum Lipid Profiles and Activation Indices: A Model for Studying Atherogenic Mechanisms. PloS Pathog (2020) 16(10):1–24. doi: 10.1371/journal.ppat.1008869
160. Bryson BD, Rosebrock TR, Tafesse FG, Itoh CY, Nibasumba A, Babunovic GH, et al. Heterogeneous GM-CSF Signaling in Macrophages is Associated With Control of Mycobacterium Tuberculosis. Nat Commun (2019) 10(1):1–11. doi: 10.1038/s41467-019-10065-8
161. Kumawat K, Pathak SK, Spetz AL, Kundu M, Basu J. Exogenous Nef is an Inhibitor of Mycobacterium Tuberculosis-Induced Tumor Necrosis Factor-α Production and Macrophage Apoptosis. J Biol Chem (2010) 285(17):12629–37. doi: 10.1074/jbc.M109.073320
162. Patel NR, Zhu J, Tachado SD, Zhang J, Wan Z, Saukkonen J, et al. Hiv Impairs Tnf-α Mediated Macrophage Apoptotic Response to Mycobacterium Tuberculosis. J Immunol (2007) 179(10):6973–80. doi: 10.4049/jimmunol.179.10.6973
163. Patel NR, Swan K, Li X, Tachado SD, Koziel H. Impaired M. Tuberculosis -Mediated Apoptosis in Alveolar Macrophages From HIV+ Persons: Potential Role of IL-10 and BCL-3. J Leukoc Biol (2009) 86(1):53–60. doi: 10.1189/jlb.0908574
164. Lawn SD, Pisell TL, Hirsch CS, Wu M, Butera ST, Toossi Z. Anatomically Compartmentalized Human Immunodeficiency Virus Replication in HLA-DR+ Cells and CD14+ Macrophages at the Site of Pleural Tuberculosis Coinfection. J Infect Dis (2001) 184(9):1127–33. doi: 10.1086/323649
165. Schiff AE, Linder AH, Luhembo SN, Banning S, Deymier MJ, Diefenbach TJ, et al. T Cell-Tropic HIV Efficiently Infects Alveolar Macrophages Through Contact With Infected CD4+ T Cells. Sci Rep (2021) 11(1):1–14. doi: 10.1038/s41598-021-82066-x
166. Cribbs SK, Lennox J, Caliendo AM, Brown LA, Guidot DM. Healthy HIV-1-Infected Individuals on Highly Active Antiretroviral Therapy Harbor HIV-1 in Their Alveolar Macrophages. AIDS Res Hum Retroviruses (2015) 31(1):64–70. doi: 10.1089/aid.2014.0133
167. Jambo KC, Banda DH, Kankwatira AM, Sukumar N, Allain TJ, Heyderman RS, et al. Small Alveolar Macrophages are Infected Preferentially by HIV and Exhibit Impaired Phagocytic Function. Mucosal Immunol (2014) 7(5):1116–26. doi: 10.1038/mi.2013.127
168. Mazzolini J, Herit F, Bouchet J, Benmerah A, Benichou S, Niedergang F. Inhibition of Phagocytosis in HIV-1-Infected Macrophages Relies on Nef-Dependent Alteration of Focal Delivery of Recycling Compartments. Blood (2010) 115(21):4226–36. doi: 10.1182/blood-2009-12-259473
169. Deretic V, Vergne I, Chua J, Master S, Singh SB, Fazio JA, et al. Endosomal Membrane Traffic: Convergence Point Targeted by Mycobacterium Tuberculosis and HIV. Cell Microbiol (2004) 6(11):999–1009. doi: 10.1111/j.1462-5822.2004.00449.x
170. Mwandumba HC, Russell DG, Nyirenda MH, Anderson J, White SA, Molyneux ME, et al. Mycobacterium Tuberculosis Resides in Nonacidified Vacuoles in Endocytically Competent Alveolar Macrophages From Patients With Tuberculosis and HIV Infection. J Immunol (2004) 172(7):4592–8. doi: 10.4049/jimmunol.172.7.4592
171. Ustianowski AP, Lawn SD, Lockwood DN. Interactions Between HIV Infection and Leprosy: A Paradox. Lancet Infect Dis (2006) 6(6):350–60. doi: 10.1016/S1473-3099(06)70493-5
172. Menezes VM, Nery JAC, Sales AM, Miranda A, Galhardo MCG, Bastos FI, et al. Epidemiological and Clinical Patterns of 92 Patients Co-Infected With HIV and Mycobacterium Leprae From Rio De Janeiro State, Brazil. Trans R Soc Trop Med Hyg (2014) 108(2):63–70. doi: 10.1093/trstmh/trt113
173. da Silva TP, Bittencourt TL, de Oliveira AL, Prata RB da S, Menezes V, Ferreira H, et al. Macrophage Polarization in Leprosy–HIV Co-Infected Patients. Front Immunol (2020) 11:1–11. doi: 10.3389/fimmu.2020.01493
174. O’Brien DP, Comte E, Serafini M, Ehounou G, Antierens A, Vuagnat H, et al. The Urgent Need for Clinical, Diagnostic, and Operational Research for Management of Buruli Ulcer in Africa. Lancet Infect Dis (2014) 14(5):435–40. doi: 10.1016/S1473-3099(13)70201-9
175. Vincent QB, Ardant MF, Marsollier L, Chauty A, Alcaïs A. HIV Infection and Buruli Ulcer in Africa. Lancet Infect Dis (2014) 14(9):796–7. doi: 10.1016/S1473-3099(14)70882-5
176. Kiran D, Podell BK, Chambers M, Basaraba RJ. Host-Directed Therapy Targeting the Mycobacterium Tuberculosis Granuloma: A Review. Semin Immunopathol (2016) 38(2):167–83. doi: 10.1007/s00281-015-0537-x
177. Hawn TR, Shah JA, Kalman D. New Tricks for Old Dogs: Countering Antibiotic Resistance in Tuberculosis With Host-Directed Therapeutics. Immunol Rev (2015) 264(1):344–62. doi: 10.1111/imr.12255
178. Wallis RS, Hafner R. Advancing Host-Directed Therapy for Tuberculosis. Nat Rev Immunol (2015) 15(4):255–63. doi: 10.1038/nri3813
179. Chai Q, Zhang Y, Liu CH. Mycobacterium Tuberculosis: An Adaptable Pathogen Associated With Multiple Human Diseases. Front Cell Infect Microbiol (2018) 8:1–15. doi: 10.3389/fcimb.2018.00158
180. Naicker N, Sigal A, Naidoo K. Metformin as Host-Directed Therapy for TB Treatment: Scoping Review. Front Microbiol (2020) 11:1–11. doi: 10.3389/fmicb.2020.00435
181. Magee MJ, Salindri AD, Kornfeld H, Singhal A. Reduced Prevalence of Latent Tuberculosis Infection in Diabetes Patients Using Metformin and Statins. Eur Respir J (2019) 53(3):1–4. doi: 10.1183/13993003.01695-2018
182. Singhal A, Jie L, Kumar P, Hong GS, Leow MKS, Paleja B, et al. Metformin as Adjunct Antituberculosis Therapy. Sci Transl Med (2014) 6(263):1–10. doi: 10.1126/scitranslmed.3009885
183. Pan SW, Yen YF, Kou YR, Chuang PH, Su VYF, Feng JY, et al. The Risk of TB in Patients With Type 2 Diabetes Initiating Metformin vs Sulfonylurea Treatment. Chest (2018) 153(6):1347–57. doi: 10.1016/j.chest.2017.11.040
184. Lin SY, Tu HP, Lu PL, Chen TC, Wang WH, Chong IW, et al. Metformin is Associated With a Lower Risk of Active Tuberculosis in Patients With Type 2 Diabetes. Respirology (2018) 23(11):1063–73. doi: 10.1111/resp.13338
185. Degner NR, Wang JY, Golub JE, Karakousis PC. Metformin Use Reverses the Increased Mortality Associated With Diabetes Mellitus During Tuberculosis Treatment. Clin Infect Dis (2018) 66(2):198–205. doi: 10.1093/cid/cix819
186. Lee YJ, Han SK, Park JH, Lee JK, Kim DK, Chung HS, et al. The Effect of Metformin on Culture Conversion in Tuberculosis Patients With Diabetes Mellitus. Korean J Intern Med (2018) 33(5):933–40. doi: 10.3904/kjim.2017.249
187. Łabuzek K, Liber S, Gabryel B, Adamczyk J, Okopień B. Metformin Increases Phagocytosis and Acidifies Lysosomal/Endosomal Compartments in AMPK-Dependent Manner in Rat Primary Microglia. Naunyn Schmiedebergs Arch Pharmacol (2010) 381(2):171–86. doi: 10.1007/s00210-009-0477-x
188. Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF. Targeting Mitochondrial Reactive Oxygen Species as Novel Therapy for Inflammatory Diseases and Cancers. J Hematol Oncol (2013) 6(1):1–19. doi: 10.1186/1756-8722-6-19
189. Böhme J, Martinez N, Li S, Lee A, Marzuki M, Tizazu AM, et al. Metformin Enhances Anti-Mycobacterial Responses by Educating CD8+ T-Cell Immunometabolic Circuits. Nat Commun (2020) 11(1):1–15. doi: 10.1038/s41467-020-19095-z
190. Pellegrini JM, Tateosian NL, Morelli MP, Rollandelli A, Amiano NO, Palmero D, et al. Immunosuppressive Role of PGE2 During Human Tuberculosis Instituto De Química Biológica De La Facultad De Ciencias Exactas Y Naturales ( IQUIBICEN ). Universidad de Buenos Instituto de Histología y Embriología de: Facultad de Ciencias Exactas y Naturales (2020). Cp 5500.
191. Moreno JR, García IE, de la Luz García Hernández M, Leon DA, Marquez R, Pando RH. The Role of Prostaglandin E2 in the Immunopathogenesis of Experimental Pulmonary Tuberculosis. Immunology (2002) 106(2):257–66. doi: 10.1046/j.1365-2567.2002.01403.x
192. Duan H, Liu T, Zhang X, Yu A, Cao Y. Statin Use and Risk of Tuberculosis: A Systemic Review of Observational Studies. Int J Infect Dis (2020) 93:168–74. doi: 10.1016/j.ijid.2020.01.036
193. Hennessy E, Adams C, Reen FJ, O’Gara F. Is There Potential for Repurposing Statins as Novel Antimicrobials? Antimicrob Agents Chemother (2016) 60(9):5111–21. doi: 10.1128/AAC.00192-16
194. Dutta NK, Bruiners N, Pinn ML, Zimmerman MD, Prideaux B, Dartois V, et al. Statin Adjunctive Therapy Shortens the Duration of TB Treatment in Mice. J Antimicrob Chemother (2016) 71(6):1570–7. doi: 10.1093/jac/dkw014
195. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin Therapy Reduces the Mycobacterium Tuberculosis Burden in Human Macrophages and in Mice by Enhancing Autophagy and Phagosome Maturation. J Infect Dis (2014) 209(5):754–63. doi: 10.1093/infdis/jit550
196. Coleman MM, Basdeo SA, Coleman AM, Cheallaigh CN, De Castro CP, McLaughlin AM, et al. All-Trans Retinoic Acid Augments Autophagy During Intracellular Bacterial Infection. Am J Respir Cell Mol Biol (2018) 59(5):548–56. doi: 10.1165/rcmb.2017-0382OC
197. Wheelwright M, Kim EW, Inkeles MS, De Leon A, Pellegrini M, Krutzik SR, et al. All- Trans Retinoic Acid–Triggered Antimicrobial Activity Against Mycobacterium Tuberculosis is Dependent on NPC2. J Immunol (2014) 192(5):2280–90. doi: 10.4049/jimmunol.1301686
198. Dürr UHN, Sudheendra US, Ramamoorthy A. LL-37, the Only Human Member of the Cathelicidin Family of Antimicrobial Peptides. Biochim Biophys Acta - Biomembr (2006) 1758(9):1408–25. doi: 10.1016/j.bbamem.2006.03.030
199. Liu PT, Stenger S, Tang DH, Modlin RL. Cutting Edge: Vitamin D-Mediated Human Antimicrobial Activity Against Mycobacterium Tuberculosis Is Dependent on the Induction of Cathelicidin. J Immunol (2007) 179(4):2060–3. doi: 10.4049/jimmunol.179.4.2060
200. Martineau AR, Timms PM, Bothamley GH, Hanifa Y, Islam K, Claxton AP, et al. High-Dose Vitamin D3 During Intensive-Phase Antimicrobial Treatment of Pulmonary Tuberculosis: A Double-Blind Randomised Controlled Trial. Lancet (2011) 377(9761):242–50. doi: 10.1016/S0140-6736(10)61889-2
201. Coussens AK, Wilkinson RJ, Hanifa Y, Nikolayevskyy V, Elkington PT, Islam K, et al. Vitamin D Accelerates Resolution of Inflammatory Responses During Tuberculosis Treatment. Proc Natl Acad Sci USA (2012) 109(38):15449–54. doi: 10.1073/pnas.1200072109
202. Borah K, Beyß M, Theorell A, Wu H, Basu P, Mendum TA, et al. Intracellular Mycobacterium Tuberculosis Exploits Multiple Host Nitrogen Sources During Growth in Human Macrophages. Cell Rep (2019) 29(11):3580–3591.e4. doi: 10.1016/j.celrep.2019.11.037
203. Koeken VACM, Lachmandas E, Riza A, Matzaraki V, Li Y, Kumar V, et al. Role of Glutamine Metabolism in Host Defense Against Mycobacterium Tuberculosis Infection. J Infect Dis (2019) 219(10):1662–70. doi: 10.1093/infdis/jiy709
204. Rapovy SM, Zhao J, Bricker RL, Schmidt SM, Setchell KDR, Qualls JE. Differential Requirements for L-Citrulline and L-Arginine During Antimycobacterial Macrophage Activity. J Immunol (2015) 195(7):3293–300. doi: 10.4049/jimmunol.1500800
205. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-Like Receptor-Induced Arginase 1 in Macrophages Thwarts Effective Immunity Against Intracellular Pathogens. Nat Immunol (2008) 9(12):1399–406. doi: 10.1038/ni.1671
206. Lange SM, McKell MC, Schmidt SM, Zhao J, Crowther RR, Green LC, et al. L-Arginine Synthesis From L-Citrulline in Myeloid Cells Drives Host Defense Against Mycobacteria In Vivo. J Immunol (2019) 202(6):1747–54. doi: 10.4049/jimmunol.1801569
207. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in Tuberculous Granulomas are Delineated by Distinct Populations of Macrophage Subsets and Expression of Nitric Oxide Synthase and Arginase Isoforms. J Immunol (2013) 191(2):773–84. doi: 10.4049/jimmunol.1300113
208. Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, et al. Tryptophan Biosynthesis Protects Mycobacteria From CD4 T-Cell-Mediated Killing. Cell (2013) 155(6):1296–308. doi: 10.1016/j.cell.2013.10.045
209. Abate Negatu D, Yamada Y, Xi Y, Lin Go M, Zimmerman M, Ganapathy U, et al. Gut Microbiota Metabolite Indole Propionic Acid Targets Tryptophan Biosynthesis in Mycobacterium Tuberculosis. MBio (2019) 10(2):1–15. doi: 10.1128/mBio.02781-18
210. Blumenthal A, Nagalingam G, Huch JH, Walker L, Guillemin GJ, Smythe GA, et al. M. Tuberculosis Induces Potent Activation of Ido-1, But This is Not Essential for the Immunological Control of Infection. PloS One (2012) 7(5):1–11. doi: 10.1371/journal.pone.0037314
211. Mehra S, Pahar B, Dutta NK, Conerly CN, Philippi-Falkenstein K, Alvarez X, et al. Transcriptional Reprogramming in Nonhuman Primate (Rhesus Macaque) Tuberculosis Granulomas. PloS One (2010) 5(8):1–12. doi: 10.1371/journal.pone.0012266
212. Moura-Alves P, Faé K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, et al. Ahr Sensing of Bacterial Pigments Regulates Antibacterial Defence. Nature (2014) 512(7515):387–92. doi: 10.1038/nature13684
213. Kim JS, Kim YR, Yang CS. Host-Directed Therapy in Tuberculosis: Targeting Host Metabolism. Front Immunol (2020) 11:1–12. doi: 10.3389/fimmu.2020.01790
214. Park JH, Shim D, Kim KES, Lee W, Shin SJ. Understanding Metabolic Regulation Between Host and Pathogens: New Opportunities for the Development of Improved Therapeutic Strategies Against Mycobacterium Tuberculosis Infection. Front Cell Infect Microbiol (2021) 11:1–21. doi: 10.3389/fcimb.2021.635335
215. Crowther RR, Qualls JE. Metabolic Regulation of Immune Responses to Mycobacterium Tuberculosis: A Spotlight on L-Arginine and L-Tryptophan Metabolism. Front Immunol (2021) 11:1–16. doi: 10.3389/fimmu.2020.628432
216. Kiran D, Basaraba RJ. Lactate Metabolism and Signaling in Tuberculosis and Cancer: A Comparative Review. Front Cell Infect Microbiol (2021) 11(37):624607. doi: 10.3389/fcimb.2021.624607
217. Lobato LS, Rosa PS, Da Silva Ferreira J, Da Silva Neumann A, Da Silva MG, Nascimento DC, et al. Statins Increase Rifampin Mycobactericidal Effect. Antimicrob Agents Chemother (2014) 58(10):5766–74. doi: 10.1128/AAC.01826-13
218. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy Protects Against Active Tuberculosis by Suppressing Bacterial Burden and Inflammation. Proc Natl Acad Sci USA (2012) 109(46):1–9. doi: 10.1073/pnas.1210500109
219. Saini NK, Baena A, Ng TW, Venkataswamy MM, Kennedy SC, Kunnath-Velayudhan S, et al. Suppression of Autophagy and Antigen Presentation by Mycobacterium Tuberculosis PE-PGRS47. Nat Microbiol (2016) 1(52):1–12. doi: 10.1038/nmicrobiol.2016.133
220. Evans MJ, Levy L. Ultrastructural Changes in Cells of the Mouse Footpad Infected With Mycobacterium Leprae. Infect Immun (1972) 5(2):238–47. doi: 10.1128/iai.5.2.238-247.1972
221. Silva BJ de A, Barbosa MG de M, Andrade PR, Ferreira H, Nery JA da C, Côrte-Real S, et al. Autophagy Is an Innate Mechanism Associated With Leprosy Polarization. PloS Pathog (2017) 13(1):1–29. doi: 10.1371/journal.ppat.1006103
222. Sieling PA, Modlin RL. Cytokine Patterns at the Site of Mycobacterial Infection. Immunobiology (1994) 4–5:378–87. doi: 10.1016/S0171-2985(11)80443-2
Keywords: mycobacteria, macrophage, immunometabolism, host-directed therapies, tuberculosis
Citation: Llibre A, Dedicoat M, Burel JG, Demangel C, O’Shea MK and Mauro C (2021) Host Immune-Metabolic Adaptations Upon Mycobacterial Infections and Associated Co-Morbidities. Front. Immunol. 12:747387. doi: 10.3389/fimmu.2021.747387
Received: 26 July 2021; Accepted: 08 September 2021;
Published: 23 September 2021.
Edited by:
Suzie Hingley-Wilson, University of Surrey, United KingdomReviewed by:
Ioannis Mitroulis, Democritus University of Thrace, GreeceDavid George Russell, Cornell University, United States
Copyright © 2021 Llibre, Dedicoat, Burel, Demangel, O’Shea and Mauro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alba Llibre, YS5sbGlicmVAYmhhbS5hYy51aw==; Claudio Mauro, Yy5tYXVyb0BiaGFtLmFjLnVr
†These authors have contributed equally to this work