 Yunxin Lai
Yunxin Lai Xinru Wei
Xinru Wei Ting Ye
Ting Ye Lilin Hang
Lilin Hang Ling Mou
Ling Mou Jin Su
Jin Su
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 November 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.747335
This article is part of the Research Topic Immunomodulatory Mechanism in Inflammatory Interstitial Lung Disease View all 12 articles
Interstitial lung diseases (ILDs) are a heterogeneous group of diseases characterized by varying degrees of inflammation and fibrosis of the pulmonary interstitium. The interrelations between multiple immune cells and stromal cells participate in the pathogenesis of ILDs. While fibroblasts contribute to the development of ILDs through secreting extracellular matrix and proinflammatory cytokines upon activation, T cells are major mediators of adaptive immunity, as well as inflammation and autoimmune tissue destruction in the lung of ILDs patients. Fibroblasts play important roles in modulating T cell recruitment, differentiation and function and conversely, T cells can balance fibrotic sequelae with protective immunity in the lung. A more precise understanding of the interrelation between fibroblasts and T cells will enable a better future therapeutic design by targeting this interrelationship. Here we highlight recent work on the interactions between fibroblasts and T cells in ILDs, and consider the implications of these interactions in the future development of therapies for ILDs.
The interstitial lung diseases (ILDs) are a large, heterogeneous group of several hundred generally rare pulmonary pathologies, involving injury, inflammation and/or scarring in the lung. Fibrosing ILDs, especially idiopathic pulmonary fibrosis (IPF) with unknown aetiology, manifest a progressive phenotype characterized by decline in lung function, life quality and early mortality. To date, only two approved drugs (Nintedanib and Pirfenidone) are available for IPF, which can only delay disease progression. Connective tissue diseases (CTDs) are a heterogeneous group of autoimmune disorders that can affect any of the body’s connective tissues, which frequently evolve to ILDs (so called CTD-ILDs) (1). Rheumatoid arthritis-associated ILDs (RA-ILDs)and systemic sclerosis-associated ILDs (SSc-ILDs) are two major CTD-ILDs. As many as 10% patients with RA have been diagnosed with ILDs over the course of the disease (2). Both genetic and environmental factors are involved in the development of ILDs. The mutations of genes critical in telomere maintenance are well-known for their involvement in IPF. Exposure to air pollutants such as ozone (O3), nitrogen dioxide (NO2) and particulate matter may trigger oxidative stress and chronic inflammation (3), which accelerate telomere shortening and dysfunction, and critically short telomeres trigger genomic instability and cellular senescence (4), contributing to the development of ILDs. In addition, respiratory infections are increasingly deemed as critical causes in ILDs pathogenesis.
The lung contains more than 40 different cell types and cellular interactions between them are extremely complex which take part in the pathogenesis of ILDs (5). Fibroblasts have been acknowledged as a ‘non-classical’ branch of the innate immunity (6). It is suggested that chronic inflammation occurs because of disordered fibroblast behavior in which failure to switch off their inflammatory program leads to the inappropriate survival and retention of leukocytes within inflamed tissue (7). Accordingly, fibroblasts are important sentinel cells that play a critical role in the switch from acute inflammation to adaptive immunity and tissue repair. T cells are the most important inducers of adaptive immunity which clear infections and also cause autoimmunity under specific conditions. Whether T cells are necessary for the development of ILDs remains unknown. Although bleomycin causes lung fibrosis in immunodeficient mice without T cells (8), bleomycin based murine models cannot truly recapitulate the characteristics of IPF in humans. T cell anomaly may cause repetitive alveolar epithelial injury and repair which lead to gradual destruction of functional lung parenchyma and its replacement by extracellular matrix (ECM). Furthermore, failure to remove excessive numbers of apoptotic cells may induce a persistent inflammatory state. Global Treg impairment has been found in IPF patients which strongly correlates with disease severity (9). While the fibroblasts-macrophages reciprocal interactions in ILDs have gained much attention (10), the interactions between fibroblasts and T cells in ILDs are underrated. In this review, we will summarize recent research advances on the roles of fibroblasts and T cells and the interrelation between them in infections and ILDs, in hope to ignite new interests in this area.
Fibroblasts are mesenchymal cells residing in all tissue types. The primary function of fibroblasts is the maintenance of the structural integrity of the connective tissues, through the secretion of ECM proteins such as collagens, matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). However, aberrant accumulation of activated fibroblasts (myofibroblasts) and ECM may lead to organ fibrosis, a hallmark of ILDs. Lung myofibroblasts in injury and fibrosis are heterogenous in terms of their origins. The predominant sources are pericytes (11) and resident fibroblasts, along with minor sources such as hematopoietic CXCR4+ fibrocytes (12), alveolar epithelial cells (AECs) (13), endothelial cells and MSCs (14) (Figure 1A). Foxd1 progenitor–derived lung pericytes, localized within basal membranes or perivascular linings, are a major source of myofibroblasts after lung injury (11), as well as in kidney fibrosis (15). Increased number of circulating fibrocytes was detected in subjects with autoimmune ILDs compared with healthy controls (16). AECs trans-differentiate into fibroblasts through epithelial-mesenchymal transition (EMT), a process requiring prolonged exposure to TGFβ for nearly 2 weeks, and AECs overlying fibroblastic foci in IPF/UIP appear histologically similar to fibroblasts, suggesting ongoing EMT (13). Recently, a unique HAS1hi ECM-producing fibroblast subset was identified markedly enriched in lungs from patients with IPF, and a population of epithelial cells (KRT5-/KRT17+) expressing collagen and other ECM components was found conserved across a subset of histopathologic patterns of pulmonary fibrosis (17). Interestingly, human fibroblasts can transdifferentiate to endothelial cells (ECs) through TLR3 (toll-like receptor 3) agonist Poly I:C induced innate immune signaling and ECs growth factors (18). In addition, fibroblasts can be reprogrammed into functional antigen-presenting DCs when overexpressing three transcription factors PU.1, IRF8 and BATF3 (19) (Figure 1B). Therefore, fibroblasts are not only effectors of fibrosis, but also hold the potential to reorganize tissue infrastructures through ECM and transdifferentiation.
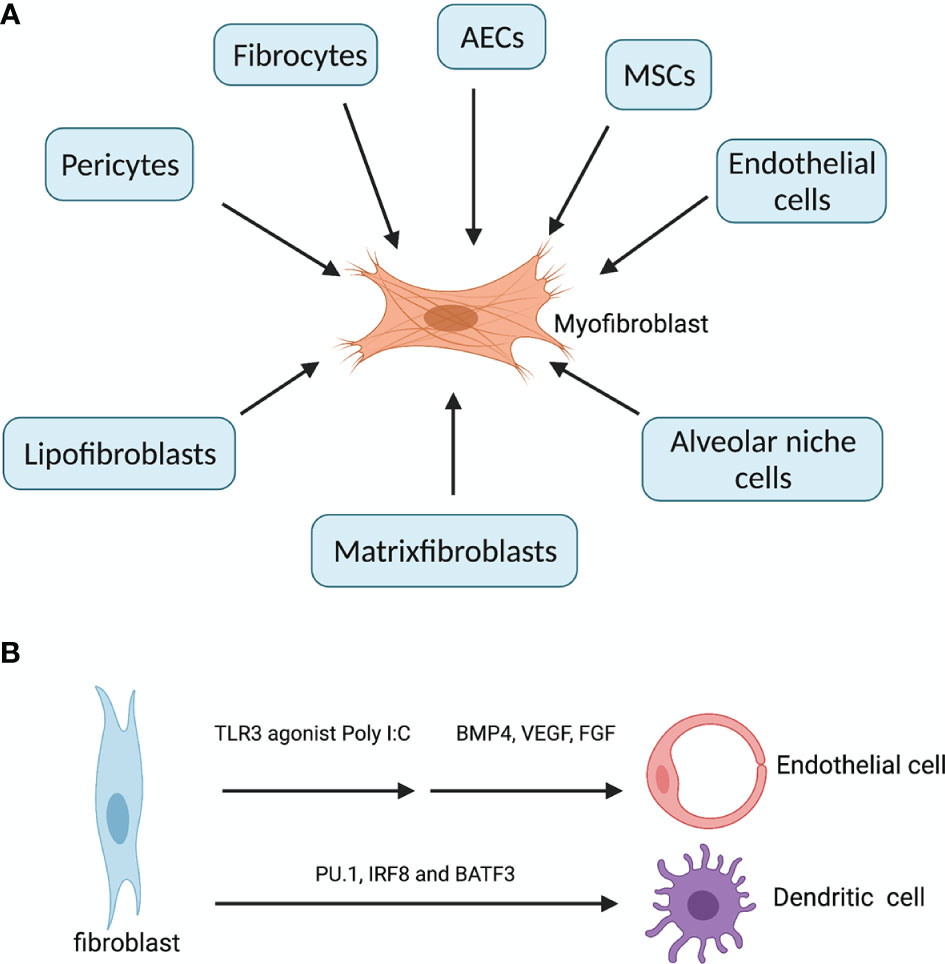
Figure 1 Diverse origins of myofibroblasts and transdifferentation of fibroblast. (A) In fibrosing ILDs, the effector myofibroblasts can be derived from three other resident fibroblasts (lipofibroblasts, matrixfibroblasts and alveolar niche cells), pericytes (residing within basal membranes or perivascular linings), hematopoietic CXCR4+ fibrocytes, alveolar epithelial cells (AECs), endothelial cells (ECs) and mesenchymal stem cells (MSCs) (14). (B) fibroblasts can transdifferentiate into endothelial cells when treated with innate immune agonist Poly I:C and growth factors BMP4, VEGF and FGF. Fibroblasts overexpressing PU.1, IRF8 and BATF3 simultaneously can be reprogrammed into functional dendritic cells. Created with BioRender.com.
Alveolar resident fibroblasts comprise four functionally distinct populations: myofibroblasts, lipofibroblasts, matrixfibroblasts and alveolar niche cells. Lipofibroblasts, matrixfibroblasts and alveolar niche cells can transdifferentiate into myofibroblasts, and the imbalance between these populations are associated with various fibrotic ILDs (20). While myofibroblasts are driver of fibrosis, matrixfibroblasts, lipofibroblasts and alveolar niche cells are regulators of alveolar epithelial cell growth and differentiation from type II to type I cells. In IPF, the proliferating myofibroblasts within the fibroblastic foci are Thy-1-, whereas normal lung fibroblasts are predominantly Thy-1+, and Thy-1- myofibroblasts are more resistant to apoptosis than Thy-1+ ones (21). The resistance to apoptosis by lung fibroblasts from IPF patients has been well established. IPF lung fibroblasts are resistant to Fas-mediated apoptosis, probably through anti-apoptotic proteins ILP and FLIPL (22). Aged lung fibroblasts from IPF patients show persistent activation of mTOR and reduced autophagy activity, which contributes to apoptosis resistance (23). Moreover, mitochondrial dysfunctions such as inhibited opening of mitochondrial permeability transition pore (mPTP) and reduced release of cytochrome c contribute to apoptosis resistance by IPF fibroblasts (24). FasL+ myofibroblasts from fibrotic lungs can induce lung epithelial cell apoptosis through Fas ligation in recipient mice (25). Normally fibroblasts are early players in initiating irritation in response to invading microorganisms and tissue damage; they respond to wound healing through proliferating and migrating to the sites of tissue injury to restructure the ECMs; and they also monitor any deviation from tissue homeostasis by sensing changes in mechanical stress through integrin connectors which substantially link ECM with their cytoskeleton, enabling transmission of force in both directions. Therefore, the plastic and versatile fibroblasts are heterogenous in terms of both origins and functions. Blocking the profibrotic programs in fibroblasts is key to the treatment of various fibrosing ILDs. It has been shown that simply eliminating the activated fibroblasts through FAP-specific chimeric antigen receptor T cells showed therapeutic efficacy against fibrosis (26).
Lung microbiome is deemed increasingly as a notable player in the initiation and exacerbation of ILDs. Humans are in constant combat with respiratory viruses such as influenza virus, coronavirus, Epstein-Barr virus (27) and adenovirus (28), and bacteria such as mycobacterium tuberculosis and Haemophilus influenzae, all of which may play key roles in the pathogenesis of ILDs (29). Defects in the immune system, when dealing with lung microbiome, may be key drivers for ILDs development. Mutation of the innate sensor TLR3, which recognizes viral dsRNA, is associated with IPF and pulmonary sarcoidosis (30, 31). Staphylococcus nepalensis released corisin, a peptide conserved in diverse staphylococci, induces apoptosis of lung epithelial cells and is upregulated in IPF patients with acute exacerbation compared to patients without disease exacerbation (32). Microbial DNA with hypomethylated CpG motifs, ligand for innate immune receptor TLR9, promotes profibrotic cytokine and chemokine synthesis in IPF fibroblasts which is associated with the rapidly progressive IPF phenotype (33). Innate immune receptor TLR4 deficiency was shown to exacerbate pulmonary fibrosis through promoting formation of an immunosuppressive tissue microenvironment and attenuating autophagy-associated degradation of collagen (34). A substantial proportion of patients with acute respiratory distress syndrome (ARDS) following viral infection developed or even died from progressive pulmonary fibrosis (35), and the rate of fibrosis was positively correlated with disease duration (36). Influenza infection in aged mice leads to non-resolving inflammation and persistent chronic lung pathology, and age-associated lung CD8 TRM accumulation is not protective but rather drives inflammatory and fibrotic sequelae after primary respiratory viral infection (37). These studies indicate that failure to eradicating infectious agents from the lung can be a primary cause to ILDs development.
Resident memory T (TRM) cells take up residence in the lung after respiratory infections to facilitate rapid and localized immune responses during reinfection. Lung TRM cell pool comprises two distinct subsets, airway TRM cells and interstitial TRM cells, the homeostasis of which are regulated by integrated stress response (ISR) induced by airway cues such as viral infection and amino acid starvation (38). CD8 TRM cells typically localize in the epithelial layers of barrier tissues where they are optimally positioned to act as sentinels to trigger antigen-specific protection, while CD4 TRM cells typically localize below the epithelial layers and cluster in lymphoid structures designed to optimize interactions with antigen-presenting cells (39). However, in the lung, CD4 TRM cells in the lung interstitium are maintained predominantly within inducible bronchus-associated lymphoid tissue (iBALT), and interstitial CD8 TRM cells are predominantly maintained within the repair-associated memory depots (RAMD) that are temporarily created at the site of tissue injury which is crucial for protection against secondary infections (40, 41). The structural characteristics of RAMD differ from iBALT as most CD8 TRM cells in the RAMD do not form organized lymphoid structures. The size of the RAMD shrinks over time as tissue repair proceeds and tends to disappear several months post-infection. TGFβ signaling is required for TRM cell development in peripheral tissues, and IL-15 signaling is required for the maintenance of CD8 TRM cells. Coordinated downregulation of both T-bet and Eomes in CD8+ TRM cells was shown to be required for optimal TGFβ signaling and residual level of T-bet was necessary for IL-15R (CD122) expression (42). Moreover, long-term TRM cell maintenance depends on local persistence of cognate antigens (43), but it is still unresolved which type of cells these antigens persist in (Figure 2A). It is interesting to speculate that lung resident fibroblasts are ideal hosts for these antigens since they are resistant to external stress and persist long.
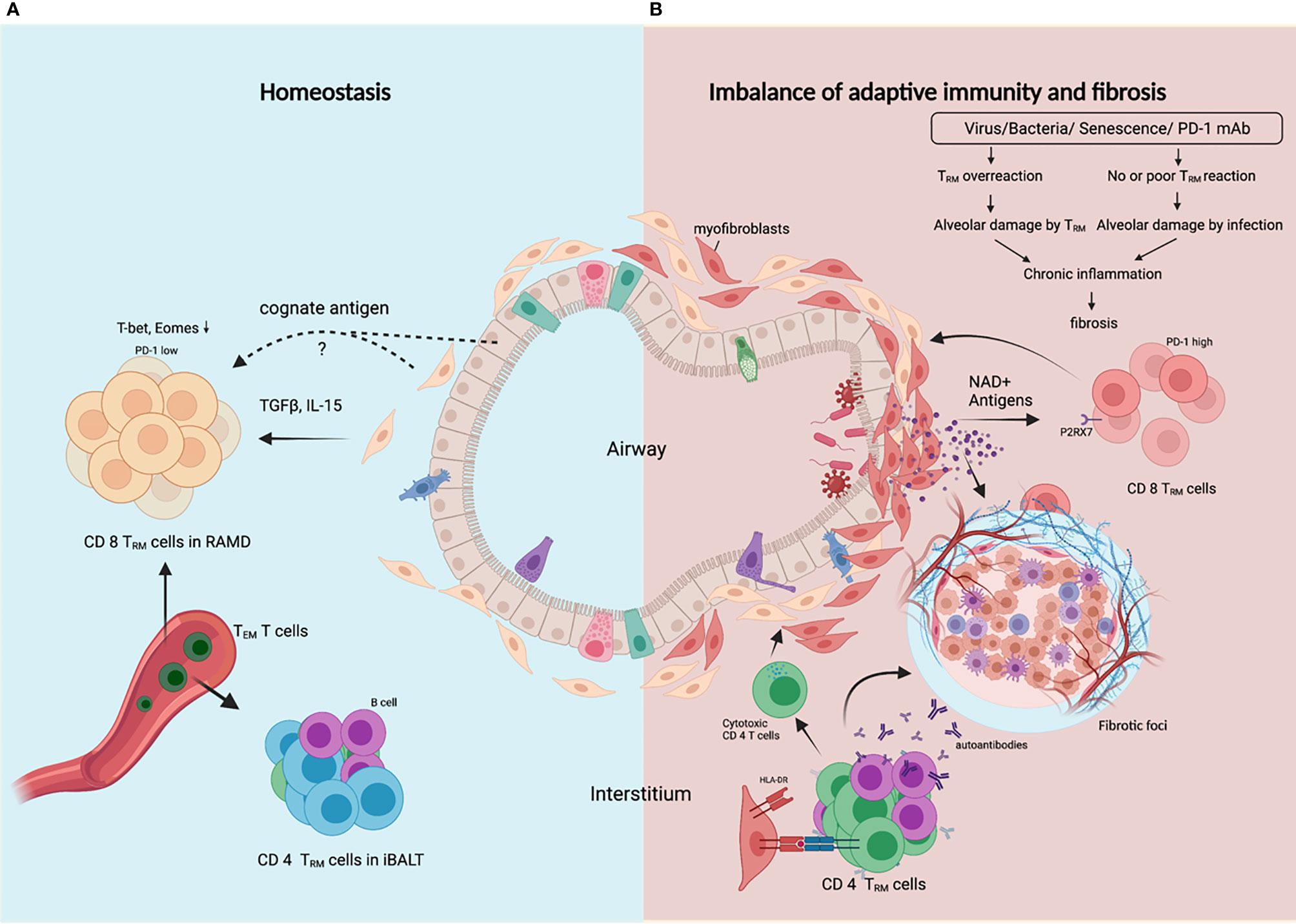
Figure 2 TRM cells control the balance between protective adaptive immunity and progressive fibrosis. (A) In homeostasis following resolution of lung infection or injury, CD8 TRM cells and CD4 TRM cells are generated and transiently maintained in RAMD and iBALT respectively, and can be replenished by TEM cells from the bloodstream. TRM cells are poised to render robust protection against secondary infection or injury, and ensure subsequent rapid tissue repair. (B) In fibrosing lung, dysregulated CD8 TRM cells in response to certain external insults lead to constant alveolar damage, subsequent chronic inflammation and progressive fibrosis. Alveolar damage release high level of NAD+ which selectively delete P2RX7+ TRM cells. CD4 TRM cells in iBALT may help B cells generate autoantibodies, or become cytotoxic or regulatory CD4 T cells that worsen tissue injury and fibrosis. Created with BioRender.com.
Lung TRM cells express high level of PD-1, a marker reflecting tissue residency rather than exhaustion (44). Peripheral T cells in age-related IPF and sarcoidosis also showed upregulated PD-1, reduced proliferative capacity and increased TGFβ and IL-17A production (45). TRM cells have been demonstrated to promote chronic parenchymal inflammation and fibrosis in aged mice following viral infection, and aged TRM cells are insufficient to provide protective immunity due to defects in genes involved in TCR signaling and effector function (37). For PD-1+ Th17 cells, PD-1 blockade is antifibrotic for reducing IL-17A and TGFβ expression, but for PD-1hi TRM CD8 T cells, PD-1 blockade is profibrotic for disrupting the balance between protective immunity and fibrotic sequelae controlled by TRM cell ‘exhaustion’ (46). In light of this, lung fibrosis caused by PD-1 blockade therapies in lung cancer patients can be attributed to activation of unrelated viral TRM cells. Unlike T cells in lymphoid organs, lung TRM cells can be reactivated by not only CD11c+ DCs, but also non-hematopoietic cells (47), and excessive bystander activation of lung TRM cells may result in amplified inflammatory and fibrotic signals contributing to the development and/or exacerbation of preexisting fibrotic respiratory diseases.
Alveolar damage is a hallmark of progressive ILDs, which induces senescence of fibroblasts and depletion of TRM cells. External insults like irradiation cause DNA damage and DNA damage response (DDR) in fibroblasts. GMP-AMP synthase (cGAS) is crucial in perpetuating IPF lung fibroblast senescence by binding damaged DNA released into the cytosol (12). Lung fibroblasts upregulate type I interferon in response to microbial or self DNA (48). Local concentration of extracellular ATP and NAD+ is strongly increased during tissue damage and inflammation, as both nucleotides are released into the extracellular space. Their receptor, P2RX7, is expressed on TRM cells and P2RX7 activation by NAD+ resulted in selective cell death of TRM cells (49) (Figure 2B). However, P2RX7 sensing of ATP promotes CD8 TRM cell generation by enhancing their sensitivity to TGFβ (50), perhaps a dominant effect in healthy lungs without alveolar damage. The gradual shrinkage of lung TRM cell pool caused by destruction of alveoli and fibrosis may hamper immunity against recurrent infections and allow for further inflammation and fibrosis.
Cellular autoreactivities are integral players in idiopathic pulmonary fibrosis (51). Certain autoantigens have been identified, such as nuclear factor (52) annexin 1 (53), alanyl-tRNA synthetase (54) and HSP-70 (55), which also generate antibody responses presumably with the help ofCD4 T cells with the same antigen-specificity. Epstein–Barr protein has homology to alanyl-tRNA synthetase, which suggests a link between viral infection and autoimmunity in ILDs. HLA-DR alleles are involved in the pathogenesis of ILDs. DRB1*1501 is over-represented in IPF patients (56, 57). A self-epitope has been identified from the α3 chain of Type IV collagen (α3135-145). HLA-DR15-α3135-145 recognizing CD4 T cells exhibit a conventional T cell phenotype that secretes pro-inflammatory cytokines; however, HLA-DR1-α3135-145 recognizing T cells are predominantly Tregs expressing tolerogenic cytokines (58). Only recently, widespread HLA-DR expression on lung epithelial and endothelial cells was found in COVID-19 patients, accompanied by increased cytotoxic CD4 T cells in lung infiltrate, contributing to increased apoptosis of epithelial cells, lung inflammation and eventually to fibrosis in severe COVID-19 (59). Moreover, abnormal CD4 T cell clonal expansions were found in all IPF patients, with 82% of these subjects also generating IgG autoantibodies against cellular antigens. These CD4 T cells have characteristics typical of cell-mediated pathologic responses, including augmented effector functions, help for autoantibody production (51). Alveolar CD103+ resident CD4 T cells from the bronchoalveolar lavage fluid of ILDs patients were shown to exert a Th1-like inflammatory phenotype (60), and these cells may play a notorious role in the aggressive injury of alveoli. Human lung fibroblasts have been proved to engulf live nontypeable Haemophilus influenzae (NTHi), bacteria highly prevalent in human respiratory tract, and present antigens to CD4 T cells through HLA-DR, inducing IFNγ and IL-17A production (61). These studies suggest that lung fibroblasts might play a crucial role in regulating the balance between inflammatory, cytotoxic and regulatory CD4 T cells and autoreactive and/or cytotoxic CD4 T cells may be critical contributors or even drivers in the pathogenesis of ILDs (Figure 2B).
Tertiary lymphoid structures (TLS) are present in tumors, infected and inflamed tissues including the lung with ILDs. TLS formation begin with an initial phase of stromal cell priming that occurs independently of lymphotoxin and precedes tissue infiltration by adaptive immune cells, followed by the maturation of fibroblasts to a full lymphoid tissue organizer cell phenotype which appears to be dependent in most settings on lymphotoxin and TNFα (62, 63). Transient activation of stromal cells that often occurs in acute phases of inflammation is not sufficient to support complete lymphoid-like fibroblast maturation. Upon resolution of inflammation, the “primed state” of fibroblasts is likely to be lost, but antigen persistence or chronic inflammation may drive the development of lymphoid tissue-like mesenchyme.
In respiratory infections, iBALT as one type of TLS, serves as a general priming site for T cells (64), and dissolve upon antigen clearance (64). The role of fibroblasts in TLS formation and function has been extensively elaborated elsewhere (62, 63). Viral infection induced type I IFN upregulates CXCL13 in pulmonary PDGFRα+ fibroblasts which recruit CXCR5+ B cells to support ectopic germinal center formation (65). Resident CD4+ T cells tightly colocalize with B cells in iBALT and promote humoral responses against viral infection in the lung (66). IL-17 produced by γδ T cells has been demonstrated to provide the trigger for priming of lung fibroblasts in iBALT formation (67, 68). And Th2-derived IL-13 and Th17-derived IL-17A synergistically stimulate pulmonary fibroblasts to produce CXCL13 which is required for iBALT formation (69). Autoantibodies generated in iBALT may further worsen fibrosis, which is exemplified by antifibroblast autoantibodies capable of binding to the surface of fibroblasts and induce profibrotic chemokines (70).
Fibroblasts and T cells interact with each other in cell contact-dependent manners in all tissues, healthy, inflamed, fibrotic, or tumors. This interaction regulates tissue remodeling and immune responses. Activated T cells inhibit collagen I and III production by dermal and synovial fibroblasts (71, 72), but stimulate FLS cells to produce proinflammatory cytokines in a contact-dependent manner which required membrane-bound TNFα, but not LFA-1/ICAM-1 (73). Coculture of T cells with bronchial fibroblasts, which involves CD40L/α5β1 interaction, increases the production of IL-6 by fibroblasts (74). T-cells overexpressing the integrins αvβ3 and αvβ5 are profibrotic on cultured primary human pulmonary fibroblasts, probably through a TGFβ-dependent mechanism (75). The contact between synovial fibroblasts and T cells induces ICAM-1 and VCAM-I expression in fibroblasts, and TNFα, IFNγ and IL-6 secretion from T cells; however, contact between dermal fibroblasts and T cells only induces ICAM-1 on fibroblasts, suggesting tissue-dependent outcome (76). Another study showed that coculture of either Th1 or Th17 cells with synovial fibroblasts promoted CD40、CD54 and MHC-II expression and production of IL-6 and IL-8 by synovial fibroblasts (77). Normal fibrocytes contact with naive CD4 T cells in vitro induced release of Th2 cytokines IL-4 and IL-10, but trans-differentiation of fibrocytes reversed Th2 cytokine production (78). Th17 cells promote inflammatory and antigen-presenting functions of fibrocytes in autoimmunity (79). Fibrocytes were also shown to be potent stimulators of anti-virus cytotoxic T cells (80).
Fibroblasts express MHC-I and MHC-II and can cross-present antigens to T cells; however, fibroblasts lack expression of typical costimulatory molecules CD80/86 and IPF T cells exhibit much lower expression of CD28 (81), making IPF fibroblasts unable to activate T cells but rather able to induce T cell anergy or Treg cells (82). IPF fibroblasts acquire an invasive phenotype that is essential for progressive fibrosis, and upregulated PD-L1 on them was required for their invasiveness (8). Furthermore, CAFs in tumors were shown able to kill CD8 T cells in an antigen-specific and antigen-dependent manner via PD-L2 and FASL (83). A recent study revealed that senescent fibroblasts expressing HLA-E can evade immune attack by inhibiting cytotoxicity of CD8 T cells and NK cells through HLA-E -NKG2A axis (84) (Figure 3). Unlike resident fibroblasts, circulating fibrocytes express CD80low and CD86high as costimulatory molecules, and express PD-L1high, but not PD-L2, as a coinhibitory molecule; therefore they strongly enhance the proliferation of CD8 T cells, an effect enhanced by PD-L1 blockade, and have the ability of antigen cross-presentation to CD8 T cells (85). And fibrocytes were also shown able to enhance Th17 response and inflammation (79).
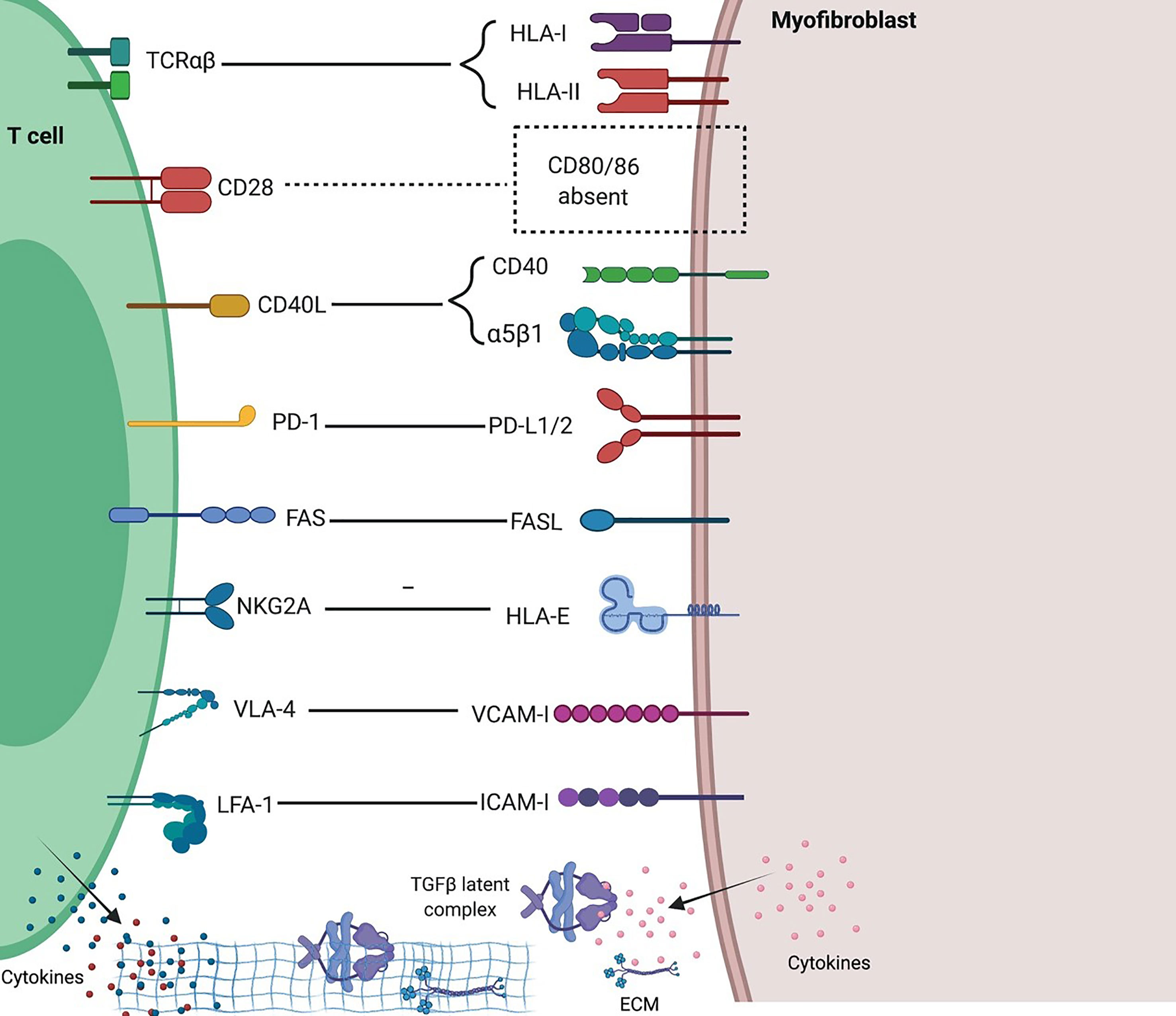
Figure 3 Interactions between T cells and fibroblasts and their implications in fibrosing ILDs. Distinct molecular interactions and reciprocal intracellular signaling exist between heterogenous T cells and fibroblasts in different tissue and immune contexts. Generally, fibroblasts inhibit T cell activation and proliferation due to a lack of potent costimulation and presence of multiple inhibitory signals, and in inflammatory settings such as ILDs, mediate Th1 to Th2 and iTreg to Th17 conversions, thus favoring fibrosis and inflammation. Created with BioRender.com.
In response to different T cells-derived stimuli, fibroblasts can be immune-activating or suppressing, tissue-destructive or reparative. The roles of different T cell subsets in fibrosis has been reviewed before (86). Th1 cytokines enhance antigen presentation and inflammatory cytokine production, but reduce ECM synthesis in fibroblasts (87). Th2 cytokines IL-4 and IL-13 stimulated upregulation of procollagen and TIMP-1 but downregulation of MMPs, effects partially opposed by Th1 cytokines IFNγ and TNFα (88–91). Likewise, IL-4 is an active player in stimulating collagen synthesis and hyperproliferation of Thy-1+ lung fibroblasts, creating a fibrotic environment in the lung (92). IL-4 also enhances expression of adhesion molecules such as beta1 integrin, ICAM-1 and VCAM-1 (93, 94). However, IL-4 secreting Th2 cells from Ssc patients inhibit collagen production by dermal fibroblasts via the dominance of membrane-associated TNFα over IL-4 (95). Trans-presentation of cytokines, similar to IL-6 trans-presentation between DCs and T cells (96), occur between fibroblasts and T cells. Fibroblasts produce inflammatory cytokine IL-6 after stimulation with IL-1 or the CD40 pathway (97). Environmental stimulations like irradiation increase expression of IL-6R and gp130 in fibroblasts (98), and IL-6 trans-signaling-STAT3 pathway pe se is able to promote expression of ECM and proliferation markers c-Myc, Bcl-xl and cycline D1 in fibroblasts (99). IL-6 trans-signaling in fibroblasts also induces Tfh (follicular helper T) and B cell differentiation factors responsible for GC formation and fibrosis in the development of IgG4-related disease (100). Treg cells promote lung epithelial proliferation (101) and repair of acute lung injury through production of TGFβ (102) and inhibition of fibrocyte recruitment to the lung along the CXCL12-CXCR4 axis (103).
Fibroblasts are able to orchestrate T cell function and plasticity. Senescent fibroblasts in IPF show abnormal activation, apoptosis resistance, telomere shortening, mitochondrial dysfunction, autophagy deficiency, and senescence-associated secretory phenotypes (SASP) (104). Lung myofibroblasts in bleomycin-induced murine model are sensitive for the activation of SHP2, STAT-3 and SOCS3, but are resistant to IFNγ-STAT-1 activation (105), thus favoring a shift from Th1 to Th2 immune responses. Synovial fibroblasts mediate conversion of iTreg cells to inflammatory Foxp3- Th17 cells by secreting IL-6 (33, 106, 107). Fibroblasts inhibit activation and proliferation of CD4 T cells, inducing a significant reduction of transcription and protein expression of TNFα, CD69, LFA-1 and CD28 in activated CD4 T cells (108). But FLS cells from RA patients induced proliferation of autologous T cells through secreting thrombospondin-1, which recognized CD47 on T cells and induced T cell adhesion, aggregation and costimulation (109). Progressive pulmonary fibrosis is associated with elevated TGFβ production from mechano-stressed AT2 cells during impaired alveolar regeneration (110). Under mechanical stress, integrin-mediated myofibroblast contraction releases active TGFβ from the ECM (111). Constitutive activation of TGFβ2 (but not TGFβ1 or TGFβ3) caused by epigenetic modulation of a discrete TGFβ2 enhancer, which is enforced by BRD4 and NF-κB, drives profibrotic programs in SSc fibroblasts (112). TGFβ1 enhances Fas-mediated lung epithelial cell apoptosis (113), but activates the FAK and the PI3K/Akt anti-apoptotic pathways in lung fibroblasts (114), favoring fibroblast senescence. TGFβ stimulated-human lung fibroblasts secret PD-L1 into extracellular vesicles which are capable of inhibiting T cell proliferation in response to T cell receptor stimulation (115). However, MMPs can cleave membrane-bound PD-L1 on Thy-1+ fibroblasts to ablate its suppressive effect on Th1 and Th17 responses (116). PGE2 produced by lung fibroblasts also suppresses apoptosis and activation-induced cell death (AICD) of T cells (117). Fibroblasts also support Th17 expansion through PGE2-mediated upregulation of IL-23 production by DCs (118). IL-11 can be induced by TGFβ (119), and it has been shown to be critical in TGFβ-mediated pulmonary fibrosis (120), which is an ideal target for treatment of ILDs (121, 122). IL-11-expressing fibroblasts and IL-11-ERK signaling in fibroblasts have been demonstrated to play pivotal roles in cancer (123) and Ssc (124). IL-11 polarizes Th17 differentiation through STAT3-RORγt axis in autoimmune diseases (125). However, earlier studies showed that IL-11 polarized Th2 differentiation and inhibited Th1 responses (126), thus preventing acute GVHD. Production of IL-15 by RA synovial fibroblasts induces the proinflammatory cytokines in cocultured T cells which involves IL-15 trans-signaling (127).
Fibroblasts also regulate T cell migration and retention through chemokine gradient and ECM-dependent sequestration of T cells. CD90+ adventitial fibroblasts expand and transition to VCAM1+ perivascular adventitial fibroblasts in spongiotic dermatitis and lupus which are associated with the retention of dense perivascular T cells (128). Fibroblasts can highly secrete βig-h3, a TGFβ-induced ECM protein, which interacts with CD61 on T cells resulting in phosphorylation of Lck at Y505 and the subsequent inhibition of this early kinase of the TCR signaling pathway (129). In summary, the reciprocal regulation between fibroblasts and T cells is extremely complex in both health and diseases due to the heterogeneity of fibroblasts, T cells and tissues they reside.
In progressive ILDs, continuously damaged lung tissues are replaced by ECM and myofibroblasts which impede tissue regeneration and lead to gradual decline in lung function. TRM cells and circulating T cells exert adaptive immunity against respiratory infections, as well as inflammation regulation and autoimmunity against self-tissues. The interactions between fibroblasts and T cells in the lung regulate immune responses and tissue remodeling. Understanding thoroughly the interactions between fibroblasts and T cells in the initiation and exacerbation of distinct ILDs, especially IPF, is crucial for developing novel therapies for patients with IPF or other types of ILDs.
Fibroblasts are able to orchestrate T cell function and plasticity. Senescent fibroblasts in IPF are resistant to apoptosis and can also evade immune attack by T cells, favoring fibrosis. Targeting fibroblasts directly has been the mainstay of ILDs therapies, however, specific modulations of T cell function may also indirectly inhibit the profibrotic events of fibroblasts. Harnessing cytotoxicity of T cells against fibroblasts through chimeric antigen receptors has been shown plausible in treating cardiac fibrosis in mice, but remains untested in humans with ILDs. Abrogating TGFβ signaling in TGFBR2-deleted CD4 T cells inhibited tumor growth by reorganizing the reticular vessel system in tumors (130). These CD4 T cells may orchestrate the reshaping of vessel system through interacting with fibroblasts. Therefore, targeting TGFβ signaling in CD4 T cells also holds the potential to treat fibrosing ILDs.
YL, XW, and TY contributed equally to this review. All authors contributed to the article and approved the submitted version.
This work was supported by Project of The State Key Laboratory of Respiratory Disease (Grant No. SKLRD-Z-202017), grants of the State Key Laboratory of Respiratory Disease, Guangdong-Hong Kong-Macao Joint Laboratory of Respiratory Infectious Disease (Grant No. GHMJLRID-Z-202116 and GHMJLRID-Z-202117), and National Natural Science Foundation of China (Grant No. 82003265).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Spagnolo P, Distler O, Ryerson CJ, Tzouvelekis A, Lee JS, Bonella F, et al. Mechanisms of Progressive Fibrosis in Connective Tissue Disease (CTD)-Associated Interstitial Lung Diseases (ILDs). Ann Rheum Dis (2021) 80:143–50. doi: 10.1136/annrheumdis-2020-217230
2. Bongartz T, Nannini C, Medina-Velasquez YF, Achenbach SJ, Crowson CS, Ryu JH, et al. Incidence and Mortality of Interstitial Lung Disease in Rheumatoid Arthritis: A Population-Based Study. Arthritis Rheum (2010) 62:1583–91. doi: 10.1002/art.27405
3. Harari S, Raghu G, Caminati A, Cruciani M, Franchini M, Mannucci P. Fibrotic Interstitial Lung Diseases and Air Pollution: A Systematic Literature Review. Eur Respir Rev (2020) 29(157). doi: 10.1183/16000617.0093-2020
4. Barnes RP, Fouquerel E, Opresko PL. The Impact of Oxidative DNA Damage and Stress on Telomere Homeostasis. Mech Ageing Dev (2019) 177:37–45. doi: 10.1016/j.mad.2018.03.013
5. Bagnato G, Harari S. Cellular Interactions in the Pathogenesis of Interstitial Lung Diseases. Eur Respir Rev (2015) 24:102–14. doi: 10.1183/09059180.00003214
6. Davidson S, Coles M, Thomas T, Kollias G, Ludewig B, Turley S, et al. Fibroblasts as Immune Regulators in Infection, Inflammation and Cancer. Nat Rev Immunol (2021) 21(11):704–17. doi: 10.1038/s41577-021-00540-z
7. Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts Regulate the Switch From Acute Resolving to Chronic Persistent Inflammation. Trends Immunol (2001) 22:199–204. doi: 10.1016/S1471-4906(01)01863-4
8. Geng Y, Liu X, Liang J, Habiel DM, Kulur V, Coelho AL, et al. PD-L1 on Invasive Fibroblasts Drives Fibrosis in a Humanized Model of Idiopathic Pulmonary Fibrosis. JCI Insight (2019) 4(6). doi: 10.1172/jci.insight.125326
9. Kotsianidis I, Nakou E, Bouchliou I, Tzouvelekis A, Spanoudakis E, Steiropoulos P, et al. Global Impairment of CD4+CD25+FOXP3+ Regulatory T Cells in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2009) 179:1121–30. doi: 10.1164/rccm.200812-1936OC
10. Buechler MB, Fu W, Turley SJ. Fibroblast-Macrophage Reciprocal Interactions in Health, Fibrosis, and Cancer. Immunity (2021) 54:903–15. doi: 10.1016/j.immuni.2021.04.021
11. Hung C, Linn G, Chow YH, Kobayashi A, Mittelsteadt K, Altemeier WA, et al. Role of Lung Pericytes and Resident Fibroblasts in the Pathogenesis of Pulmonary Fibrosis. Am J Respir Crit Care Med (2013) 188:820–30. doi: 10.1164/rccm.201212-2297OC
12. Schuliga M, Read J, Blokland KEC, Waters DW, Burgess J, Prele C, et al. Self DNA Perpetuates IPF Lung Fibroblast Senescence in a cGAS-Dependent Manner. Clin Sci (Lond) (2020) 134:889–905. doi: 10.1042/CS20191160
13. Willis BC, duBois RM, Borok Z. Epithelial Origin of Myofibroblasts During Fibrosis in the Lung. Proc Am Thorac Soc (2006) 3:377–82. doi: 10.1513/pats.200601-004TK
14. Barron L, Gharib SA, Duffield JS. Lung Pericytes and Resident Fibroblasts: Busy Multitaskers. Am J Pathol (2016) 186:2519–31. doi: 10.1016/j.ajpath.2016.07.004
15. Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate Tracing Reveals the Pericyte and Not Epithelial Origin of Myofibroblasts in Kidney Fibrosis. Am J Pathol (2010) 176:85–97. doi: 10.2353/ajpath.2010.090517
16. Odackal J, Yu V, Gomez-Manjerres D, Field JJ, Burdick MD, Mehrad B. Circulating Fibrocytes as Prognostic Biomarkers of Autoimmune Interstitial Lung Disease. ERJ Open Res (2020) 6(4). doi: 10.1183/23120541.00481-2020
17. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-Cell RNA Sequencing Reveals Profibrotic Roles of Distinct Epithelial and Mesenchymal Lineages in Pulmonary Fibrosis. Sci Adv (2020) 6:eaba1972. doi: 10.1126/sciadv.aba1972
18. Sayed N, Wong WT, Ospino F, Meng S, Lee J, Jha A, et al. Transdifferentiation of Human Fibroblasts to Endothelial Cells: Role of Innate Immunity. Circulation (2015) 131:300–9. doi: 10.1161/CIRCULATIONAHA.113.007394
19. Rosa FF, Pires CF, Kurochkin I, Ferreira AG, Gomes AM, Palma LG, et al. Direct Reprogramming of Fibroblasts Into Antigen-Presenting Dendritic Cells. Sci Immunol (2018) 3(30). doi: 10.1126/sciimmunol.aau4292
20. Ushakumary MG, Riccetti M, Perl AT. Resident Interstitial Lung Fibroblasts and Their Role in Alveolar Stem Cell Niche Development, Homeostasis, Injury, and Regeneration. Stem Cells Transl Med (2021) 10(7):1021–32. doi: 10.1002/sctm.20-0526
21. Sanders YY, Kumbla P, Hagood JS. Enhanced Myofibroblastic Differentiation and Survival in Thy-1(-) Lung Fibroblasts. Am J Respir Cell Mol Biol (2007) 36:226–35. doi: 10.1165/rcmb.2006-0178OC
22. Tanaka T, Yoshimi M, Maeyama T, Hagimoto N, Kuwano K, Hara N. Resistance to Fas-Mediated Apoptosis in Human Lung Fibroblast. Eur Respir J (2002) 20:359–68. doi: 10.1183/09031936.02.00252602
23. Romero Y, Bueno M, Ramirez R, Alvarez D, Sembrat JC, Goncharova EA, et al. mTORC1 Activation Decreases Autophagy in Aging and Idiopathic Pulmonary Fibrosis and Contributes to Apoptosis Resistance in IPF Fibroblasts. Aging Cell (2016) 15:1103–12. doi: 10.1111/acel.12514
24. Luis-Garcia ER, Becerril C, Salgado-Aguayo A, Aparicio-Trejo OE, Romero Y, Flores-Soto E, et al. Mitochondrial Dysfunction and Alterations in Mitochondrial Permeability Transition Pore (mPTP) Contribute to Apoptosis Resistance in Idiopathic Pulmonary Fibrosis Fibroblasts. Int J Mol Sci (2021) 22(15). doi: 10.3390/ijms22157870
25. Golan-Gerstl R, Wallach-Dayan SB, Amir G, Breuer R. Epithelial Cell Apoptosis by Fas Ligand-Positive Myofibroblasts in Lung Fibrosis. Am J Respir Cell Mol Biol (2007) 36:270–5. doi: 10.1165/rcmb.2006-0133OC
26. Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, et al. Targeting Cardiac Fibrosis With Engineered T Cells. Nature (2019) 573:430–3. doi: 10.1038/s41586-019-1546-z
27. Egan JJ, Stewart JP, Hasleton PS, Arrand JR, Carroll KB, Woodcock AA. Epstein-Barr Virus Replication Within Pulmonary Epithelial Cells in Cryptogenic Fibrosing Alveolitis. Thorax (1995) 50:1234–9. doi: 10.1136/thx.50.12.1234
28. Kuwano K, Nomoto Y, Kunitake R, Hagimoto N, Matsuba T, Nakanishi Y, et al. Detection of Adenovirus E1A DNA in Pulmonary Fibrosis Using Nested Polymerase Chain Reaction. Eur Respir J (1997) 10:1445–9. doi: 10.1183/09031936.97.10071445
29. Atabati E, Dehghani-Samani A, Mortazavimoghaddam SG. Association of COVID-19 and Other Viral Infections With Interstitial Lung Diseases, Pulmonary Fibrosis, and Pulmonary Hypertension: A Narrative Review. Can J Respir Ther (2020) 56:1–9. doi: 10.29390/cjrt-2020-021
30. Cooke G, Kamal I, Strengert M, Hams E, Mawhinney L, Tynan A, et al. Toll-Like Receptor 3 L412F Polymorphism Promotes a Persistent Clinical Phenotype in Pulmonary Sarcoidosis. QJM (2018) 111:217–24. doi: 10.1093/qjmed/hcx243
31. O'Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, et al. The Toll-Like Receptor 3 L412F Polymorphism and Disease Progression in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2013) 188:1442–50. doi: 10.1164/rccm.201304-0760OC
32. D'Alessandro-Gabazza CN, Kobayashi T, Yasuma T, Toda M, Kim H, Fujimoto H, et al. A Staphylococcus Pro-Apoptotic Peptide Induces Acute Exacerbation of Pulmonary Fibrosis. Nat Commun (2020) 11:1539. doi: 10.1038/s41467-020-15344-3
33. Hogaboam CM, Trujillo G, Martinez FJ. Aberrant Innate Immune Sensing Leads to the Rapid Progression of Idiopathic Pulmonary Fibrosis. Fibrogenesis Tissue Repair (2012) 5:S3. doi: 10.1186/1755-1536-5-S1-S3
34. Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, et al. TLR4 Activity Is Required in the Resolution of Pulmonary Inflammation and Fibrosis After Acute and Chronic Lung Injury. Am J Pathol (2012) 180:275–92. doi: 10.1016/j.ajpath.2011.09.019
35. George PM, Wells AU, Jenkins RG. Pulmonary Fibrosis and COVID-19: The Potential Role for Antifibrotic Therapy. Lancet Respir Med (2020) 8:807–15. doi: 10.1016/S2213-2600(20)30225-3
36. Thille AW, Esteban A, Fernandez-Segoviano P, Rodriguez JM, Aramburu JA, Vargas-Errazuriz P, et al. Chronology of Histological Lesions in Acute Respiratory Distress Syndrome With Diffuse Alveolar Damage: A Prospective Cohort Study of Clinical Autopsies. Lancet Respir Med (2013) 1:395–401. doi: 10.1016/S2213-2600(13)70053-5
37. Goplen NP, Wu Y, Son YM, Li C, Wang Z, Cheon IS, et al. Tissue-Resident CD8(+) T Cells Drive Age-Associated Chronic Lung Sequelae After Viral Pneumonia. Sci Immunol (2020) 5(53). doi: 10.1101/2020.04.13.040196
38. Hayward SL, Scharer CD, Cartwright EK, Takamura S, Li ZT, Boss JM, et al. Environmental Cues Regulate Epigenetic Reprogramming of Airway-Resident Memory CD8(+) T Cells. Nat Immunol (2020) 21:309–20. doi: 10.1038/s41590-019-0584-x
39. Takamura S. Niches for the Long-Term Maintenance of Tissue-Resident Memory T Cells. Front Immunol (2018) 9:1214. doi: 10.3389/fimmu.2018.01214
40. Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, et al. Specific Niches for Lung-Resident Memory CD8+ T Cells at the Site of Tissue Regeneration Enable CD69-Independent Maintenance. J Exp Med (2016) 213:3057–73. doi: 10.1084/jem.20160938
41. Takamura S, Kohlmeier JE. Establishment and Maintenance of Conventional and Circulation-Driven Lung-Resident Memory CD8(+) T Cells Following Respiratory Virus Infections. Front Immunol (2019) 10:733. doi: 10.3389/fimmu.2019.00733
42. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-Box Transcription Factors Combine With the Cytokines TGF-Beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity (2015) 43:1101–11. doi: 10.1016/j.immuni.2015.11.008
43. Uddback I, Cartwright EK, Scholler AS, Wein AN, Hayward SL, Lobby J, et al. Long-Term Maintenance of Lung Resident Memory T Cells is Mediated by Persistent Antigen. Mucosal Immunol (2021) 14:92–9. doi: 10.1038/s41385-020-0309-3
44. Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, et al. Single-Cell Transcriptomic Analysis of Tissue-Resident Memory T Cells in Human Lung Cancer. J Exp Med (2019) 216:2128–49. doi: 10.1084/jem.20190249
45. Shimada Y, Hayashi M, Nagasaka Y, Ohno-Iwashita Y, Inomata M. Age-Associated Up-Regulation of a Negative Co-Stimulatory Receptor PD-1 in Mouse CD4+ T Cells. Exp Gerontol (2009) 44:517–22. doi: 10.1016/j.exger.2009.05.003
46. Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, et al. PD-1(Hi) CD8(+) Resident Memory T Cells Balance Immunity and Fibrotic Sequelae. Sci Immunol (2019) 4(36). doi: 10.1126/sciimmunol.aaw1217
47. Low JS, Farsakoglu Y, Amezcua Vesely MC, Sefik E, Kelly JB, Harman CCD, et al. Tissue-Resident Memory T Cell Reactivation by Diverse Antigen-Presenting Cells Imparts Distinct Functional Responses. J Exp Med (2020) 217(8). doi: 10.1084/jem.20192291
48. Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-Dependent Matrix Remodelling is Required for the Generation and Maintenance of Cancer-Associated Fibroblasts. Nat Cell Biol (2013) 15:637–46. doi: 10.1038/ncb2756
49. Stark R, Wesselink TH, Behr FM, Kragten NAM, Arens R, Koch-Nolte F, et al. T RM Maintenance is Regulated by Tissue Damage via P2RX7. Sci Immunol (2018) 3(30). doi: 10.1126/sciimmunol.aau1022
50. Borges da Silva H, Peng C, Wang H, Wanhainen KM, Ma C, Lopez S, et al. Sensing of ATP via the Purinergic Receptor P2RX7 Promotes CD8(+) Trm Cell Generation by Enhancing Their Sensitivity to the Cytokine TGF-Beta. Immunity (2020) 53:158–71.e156. doi: 10.1016/j.immuni.2020.06.010
51. Feghali-Bostwick CA, Tsai CG, Valentine VG, Kantrow S, Stoner MW, Pilewski JM, et al. Cellular and Humoral Autoreactivity in Idiopathic Pulmonary Fibrosis. J Immunol (2007) 179:2592–9. doi: 10.4049/jimmunol.179.4.2592
52. Nagaya H, Sieker HO. Pathogenetic Mechanisms of Interstitial Pulmonary Fibrosis in Patients With Serum Antinuclear Factor. A Histologic and Clinical Correlation. Am J Med (1972) 52:51–62. doi: 10.1016/0002-9343(72)90007-1
53. Kurosu K, Takiguchi Y, Okada O, Yumoto N, Sakao S, Tada Y, et al. Identification of Annexin 1 as a Novel Autoantigen in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. J Immunol (2008) 181:756–67. doi: 10.4049/jimmunol.181.1.756
54. Takahashi T, Wada I, Ohtsuka Y, Munakata M, Homma Y, Kuroki Y. Autoantibody to alanyl-tRNA Synthetase in Patients With Idiopathic Pulmonary Fibrosis. Respirology (2007) 12:642–53. doi: 10.1111/j.1440-1843.2007.01140.x
55. Kahloon RA, Xue J, Bhargava A, Csizmadia E, Otterbein L, Kass DJ, et al. Patients With Idiopathic Pulmonary Fibrosis With Antibodies to Heat Shock Protein 70 Have Poor Prognoses. Am J Respir Crit Care Med (2013) 187:768–75. doi: 10.1164/rccm.201203-0506OC
56. Xue J, Gochuico BR, Alawad AS, Feghali-Bostwick CA, Noth I, Nathan SD, et al. The HLA Class II Allele DRB1*1501 Is Over-Represented in Patients With Idiopathic Pulmonary Fibrosis. PloS One (2011) 6:e14715. doi: 10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A4272
57. Fingerlin TE, Zhang W, Yang IV, Ainsworth HC, Russell PH, Blumhagen RZ, et al. Genome-Wide Imputation Study Identifies Novel HLA Locus for Pulmonary Fibrosis and Potential Role for Auto-Immunity in Fibrotic Idiopathic Interstitial Pneumonia. BMC Genet (2016) 17:74. doi: 10.1186/s12863-016-0377-2
58. Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH, et al. Dominant Protection From HLA-Linked Autoimmunity by Antigen-Specific Regulatory T Cells. Nature (2017) 545:243–7. doi: 10.1038/nature22329
59. Kaneko N, Boucau J, Kuo HH, Perugino C, Mahajan VS, Farmer JR, et al. Expansion of Cytotoxic CD4+ T Cells in the Lungs in Severe COVID-19. medRxiv (2021). doi: 10.2139/ssrn.3813278
60. Braun RK, Foerster M, Grahmann PR, Haefner D, Workalemahu G, Kroegel C. Phenotypic and Molecular Characterization of CD103+ CD4+ T Cells in Bronchoalveolar Lavage From Patients With Interstitial Lung Diseases. Cytometry B Clin Cytom (2003) 54:19–27. doi: 10.1002/cyto.b.10021
61. Hutton AJ, Polak ME, Spalluto CM, Wallington JC, Pickard C, Staples KJ, et al. Human Lung Fibroblasts Present Bacterial Antigens to Autologous Lung Th Cells. J Immunol (2017) 198:110–8. doi: 10.4049/jimmunol.1600602
62. Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA, et al. Stromal Fibroblasts in Tertiary Lymphoid Structures: A Novel Target in Chronic Inflammation. Front Immunol (2016) 7:477. doi: 10.3389/fimmu.2016.00477
63. Buckley CD, Barone F, Nayar S, Benezech C, Caamano J. Stromal Cells in Chronic Inflammation and Tertiary Lymphoid Organ Formation. Annu Rev Immunol (2015) 33:715–45. doi: 10.1146/annurev-immunol-032713-120252
64. Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S, et al. Induced Bronchus-Associated Lymphoid Tissue Serves as a General Priming Site for T Cells and is Maintained by Dendritic Cells. J Exp Med (2009) 206:2593–601. doi: 10.1084/jem.20091472
65. Denton AE, Innocentin S, Carr EJ, Bradford BM, Lafouresse F, Mabbott NA, et al. Type I Interferon Induces CXCL13 to Support Ectopic Germinal Center Formation. J Exp Med (2019) 216:621–37. doi: 10.1084/jem.20181216
66. Swarnalekha N, Schreiner D, Litzler LC, Iftikhar S, Kirchmeier D, Kunzli M, et al. T Resident Helper Cells Promote Humoral Responses in the Lung. Sci Immunol (2021) 6(55). doi: 10.1126/sciimmunol.abb6808
67. Fleige H, Ravens S, Moschovakis GL, Bolter J, Willenzon S, Sutter G, et al. IL-17-Induced CXCL12 Recruits B Cells and Induces Follicle Formation in BALT in the Absence of Differentiated FDCs. J Exp Med (2014) 211:643–51. doi: 10.1084/jem.20131737
68. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The Development of Inducible Bronchus-Associated Lymphoid Tissue Depends on IL-17. Nat Immunol (2011) 12:639–46. doi: 10.1038/ni.2053
69. Eddens T, Elsegeiny W, Garcia-Hernadez ML, Castillo P, Trevejo-Nunez G, Serody K, et al. Pneumocystis-Driven Inducible Bronchus-Associated Lymphoid Tissue Formation Requires Th2 and Th17 Immunity. Cell Rep (2017) 18:3078–90. doi: 10.1016/j.celrep.2017.03.016
70. Fineschi S, Goffin L, Rezzonico R, Cozzi F, Dayer JM, Meroni PL, et al. Antifibroblast Antibodies in Systemic Sclerosis Induce Fibroblasts to Produce Profibrotic Chemokines, With Partial Exploitation of Toll-Like Receptor 4. Arthritis Rheum (2008) 58:3913–23. doi: 10.1002/art.24049
71. Chizzolini C, Rezzonico R, Ribbens C, Burger D, Wollheim FA, Dayer JM. Inhibition of Type I Collagen Production by Dermal Fibroblasts Upon Contact With Activated T Cells: Different Sensitivity to Inhibition Between Systemic Sclerosis and Control Fibroblasts. Arthritis Rheum (1998) 41:2039–47. doi: 10.1002/1529-0131(199811)41:11<2039::AID-ART20>3.0.CO;2-1
72. Rezzonico R, Burger D, Dayer JM. Direct Contact Between T Lymphocytes and Human Dermal Fibroblasts or Synoviocytes Down-Regulates Types I and III Collagen Production via cell-associated cytokines. J Biol Chem (1998) 273:18720–8. doi: 10.1074/jbc.273.30.18720
73. Tran CN, Lundy SK, White PT, Endres JL, Motyl CD, Gupta R, et al. Molecular Interactions Between T Cells and Fibroblast-Like Synoviocytes: Role of Membrane Tumor Necrosis Factor-Alpha on Cytokine-Activated T Cells. Am J Pathol (2007) 171:1588–98. doi: 10.2353/ajpath.2007.070004
74. Loubaki L, Semlali A, Boisvert M, Jacques E, Plante S, Aoudjit F, et al. Crosstalk Between T Cells and Bronchial Fibroblasts Obtained From Asthmatic Subjects Involves CD40L/alpha 5 Beta 1 Interaction. Mol Immunol (2010) 47:2112–8. doi: 10.1016/j.molimm.2010.03.011
75. Luzina IG, Todd NW, Nacu N, Lockatell V, Choi J, Hummers LK, et al. Regulation of Pulmonary Inflammation and Fibrosis Through Expression of Integrins Alphavbeta3 and Alphavbeta5 on Pulmonary T Lymphocytes. Arthritis Rheum (2009) 60:1530–9. doi: 10.1002/art.24435
76. Bombara MP, Webb DL, Conrad P, Marlor CW, Sarr T, Ranges GE, et al. Cell Contact Between T Cells and Synovial Fibroblasts Causes Induction of Adhesion Molecules and Cytokines. J Leukoc Biol (1993) 54:399–406. doi: 10.1002/jlb.54.5.399
77. Kato H, Endres J, Fox DA. The Roles of IFN-Gamma Versus IL-17 in Pathogenic Effects of Human Th17 Cells on Synovial Fibroblasts. Mod Rheumatol (2013) 23:1140–50. doi: 10.3109/s10165-012-0811-x
78. Medina A, Ghahary A. Reprogrammed Fibrocytes Induce a Mixed Th1/Th2 Cytokine Response of Naive CD4(+) T Cells. Mol Cell Biochem (2011) 346:89–94. doi: 10.1007/s11010-010-0595-2
79. Fang, Huang Y, Liu X, Zhong S, Wang N, Zhao B, et al. Interaction Between CCR6+ Th17 Cells and CD34+ Fibrocytes Promotes Inflammation: Implications in Graves' Orbitopathy in Chinese Population. Invest Ophthalmol Vis Sci (2018) 59:2604–14. doi: 10.1167/iovs.18-24008
80. Balmelli C, Ruggli N, McCullough K, Summerfield A. Fibrocytes are Potent Stimulators of Anti-Virus Cytotoxic T Cells. J Leukoc Biol (2005) 77:923–33. doi: 10.1189/jlb.1204701
81. Gilani SR, Vuga LJ, Lindell KO, Gibson KF, Xue J, Kaminski N, et al. CD28 Down-Regulation on Circulating CD4 T-Cells is Associated With Poor Prognoses of Patients With Idiopathic Pulmonary Fibrosis. PloS One (2010) 5:e8959. doi: 10.1371/journal.pone.0008959
82. De Jaeghere EA, Denys HG, De Wever O. Fibroblasts Fuel Immune Escape in the Tumor Microenvironment. Trends Cancer (2019) 5:704–23. doi: 10.1016/j.trecan.2019.09.009
83. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-Associated Fibroblasts Induce Antigen-Specific Deletion of CD8 (+) T Cells to Protect Tumour Cells. Nat Commun (2018) 9:948. doi: 10.1038/s41467-018-03347-0
84. Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, et al. Senescent Cells Evade Immune Clearance via HLA-E-Mediated NK and CD8(+) T Cell Inhibition. Nat Commun (2019) 10:2387. doi: 10.1038/s41467-019-10335-5
85. Afroj T, Mitsuhashi A, Ogino H, Saijo A, Otsuka K, Yoneda H, et al. Blockade of PD-1/PD-L1 Pathway Enhances the Antigen-Presenting Capacity of Fibrocytes. J Immunol (2021) 206:1204–14. doi: 10.4049/jimmunol.2000909
86. Wynn TA, Ramalingam TR. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat Med (2012) 18:1028–40. doi: 10.1038/nm.2807
87. Jang JY, Choi GH, Ji S. IFN-Gamma or IL-4 Polarization Impacts the Response of Gingival Fibroblasts to Oral Bacteria. J Periodontal Res (2021) 56:462–70. doi: 10.1111/jre.12837
88. Leonardi A, Cortivo R, Fregona I, Plebani M, Secchi AG, Abatangelo G. Effects of Th2 Cytokines on Expression of Collagen, MMP-1, and TIMP-1 in Conjunctival Fibroblasts. Invest Ophthalmol Vis Sci (2003) 44:183–9. doi: 10.1167/iovs.02-0420
89. Fujitsu Y, Fukuda K, Kumagai N, Nishida T. IL-4-Induced Cell Proliferation and Production of Extracellular Matrix Proteins in Human Conjunctival Fibroblasts. Exp Eye Res (2003) 76:107–14. doi: 10.1016/S0014-4835(02)00248-8
90. Chambers M, Kirkpatrick G, Evans M, Gorski G, Foster S, Borghaei RC. IL-4 Inhibition of IL-1 Induced Matrix Metalloproteinase-3 (MMP-3) Expression in Human Fibroblasts Involves Decreased AP-1 Activation via Negative Crosstalk Involving of Jun N-Terminal Kinase (JNK). Exp Cell Res (2013) 319:1398–408. doi: 10.1016/j.yexcr.2013.04.010
91. Ihn H, Yamane K, Asano Y, Kubo M, Tamaki K. IL-4 Up-Regulates the Expression of Tissue Inhibitor of Metalloproteinase-2 in Dermal Fibroblasts via the P38 Mitogen-Activated Protein Kinase Dependent Pathway. J Immunol (2002) 168:1895–902. doi: 10.4049/jimmunol.168.4.1895
92. Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of Murine Lung Fibroblasts Express Membrane-Bound and Soluble IL-4 Receptors. Role of IL-4 in Enhancing Fibroblast Proliferation and Collagen Synthesis. J Immunol (1994) 152:3606–14.
93. Doucet C, Brouty-Boye D, Pottin-Clemenceau C, Jasmin C, Canonica GW, Azzarone B. IL-4 and IL-13 Specifically Increase Adhesion Molecule and Inflammatory Cytokine Expression in Human Lung Fibroblasts. Int Immunol (1998) 10:1421–33. doi: 10.1093/intimm/10.10.1421
94. Piela-Smith TH, Broketa G, Hand A, Korn JH. Regulation of ICAM-1 Expression and Function in Human Dermal Fibroblasts by IL-4. J Immunol (1992) 148:1375–81.
95. Chizzolini C, Parel Y, De Luca C, Tyndall A, Akesson A, Scheja A, et al. Systemic Sclerosis Th2 Cells Inhibit Collagen Production by Dermal Fibroblasts via Membrane-Associated Tumor Necrosis Factor Alpha. Arthritis Rheum (2003) 48:2593–604. doi: 10.1002/art.11129
96. Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, et al. Trans-Presentation of IL-6 by Dendritic Cells Is Required for the Priming of Pathogenic TH17 Cells. Nat Immunol (2017) 18:74–85. doi: 10.1038/ni.3632
97. Epstein Shochet G, Brook E, Bardenstein-Wald B, Shitrit D. TGF-Beta Pathway Activation by Idiopathic Pulmonary Fibrosis (IPF) Fibroblast Derived Soluble Factors is Mediated by IL-6 Trans-Signaling. Respir Res (2020) 21:56. doi: 10.1186/s12931-020-1319-0
98. Tabata C, Kubo H, Tabata R, Wada M, Sakuma K, Ichikawa M, et al. All-Trans Retinoic Acid Modulates Radiation-Induced Proliferation of Lung Fibroblasts via IL-6/IL-6R System. Am J Physiol Lung Cell Mol Physiol (2006) 290:L597–606. doi: 10.1152/ajplung.00282.2005
99. Ray S, Ju X, Sun H, Finnerty CC, Herndon DN, Brasier AR. The IL-6 Trans-Signaling-STAT3 Pathway Mediates ECM and Cellular Proliferation in Fibroblasts From Hypertrophic Scar. J Invest Dermatol (2013) 133:1212–20. doi: 10.1038/jid.2012.499
100. Zongfei J, Rongyi C, Xiaomeng C, Lili M, Lingying M, Xiufang K, et al. In Vitro IL-6/IL-6r Trans-Signaling in Fibroblasts Releases Cytokines That May Be Linked to the Pathogenesis of IgG4-Related Disease. Front Immunol (2020) 11:1272. doi: 10.3389/fimmu.2020.01272
101. Mock JR, Garibaldi BT, Aggarwal NR, Jenkins J, Limjunyawong N, Singer BD, et al. Foxp3+ Regulatory T Cells Promote Lung Epithelial Proliferation. Mucosal Immunol (2014) 7:1440–51. doi: 10.1038/mi.2014.33
102. D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4+CD25+Foxp3+ Tregs Resolve Experimental Lung Injury in Mice and are Present in Humans With Acute Lung Injury. J Clin Invest (2009) 119:2898–913. doi: 10.1172/JCI36498
103. Garibaldi BT, D'Alessio FR, Mock JR, Files DC, Chau E, Eto Y, et al. Regulatory T Cells Reduce Acute Lung Injury Fibroproliferation by Decreasing Fibrocyte Recruitment. Am J Respir Cell Mol Biol (2013) 48:35–43. doi: 10.1165/rcmb.2012-0198OC
104. Waters DW, Blokland KEC, Pathinayake PS, Burgess JK, Mutsaers SE, Prele CM, et al. Fibroblast Senescence in the Pathology of Idiopathic Pulmonary Fibrosis. Am J Physiol Lung Cell Mol Physiol (2018) 315:L162–72. doi: 10.1152/ajplung.00037.2018
105. Chang CJ, Lin CF, Lee CH, Chuang HC, Shih FC, Wan SW, et al. Overcoming Interferon (IFN)-Gamma Resistance Ameliorates Transforming Growth Factor (TGF)-Beta-Mediated Lung Fibroblast-to-Myofibroblast Transition and Bleomycin-Induced Pulmonary Fibrosis. Biochem Pharmacol (2021) 183:114356. doi: 10.1016/j.bcp.2020.114356
106. Yang S, Zhang X, Chen J, Dang J, Liang R, Zeng D, et al. Induced, But Not Natural, Regulatory T Cells Retain Phenotype and Function Following Exposure to Inflamed Synovial Fibroblasts. Sci Adv (2020) 6(44). doi: 10.1126/sciadv.abb0606
107. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic Conversion of Foxp3+ T Cells Into TH17 Cells in Autoimmune Arthritis. Nat Med (2014) 20:62–8. doi: 10.1038/nm.3432
108. Vancheri C, Mastruzzo C, Trovato-Salinaro E, Gili E, Lo Furno D, Pistorio MP, et al. Interaction Between Human Lung Fibroblasts and T-Lymphocytes Prevents Activation of CD4+ Cells. Respir Res (2005) 6:103. doi: 10.1186/1465-9921-6-103
109. Vallejo AN, Yang H, Klimiuk PA, Weyand CM, Goronzy JJ. Synoviocyte-Mediated Expansion of Inflammatory T Cells in Rheumatoid Synovitis Is Dependent on CD47-Thrombospondin 1 Interaction. J Immunol (2003) 171:1732–40. doi: 10.4049/jimmunol.171.4.1732
110. Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell (2021) 184:845–6. doi: 10.1016/j.cell.2021.01.020
111. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast Contraction Activates Latent TGF-Beta1 From the Extracellular Matrix. J Cell Biol (2007) 179:1311–23. doi: 10.1083/jcb.200704042
112. Shin JY, Beckett JD, Bagirzadeh R, Creamer TJ, Shah AA, McMahan Z, et al. Epigenetic Activation and Memory at a TGFB2 Enhancer in Systemic Sclerosis. Sci Transl Med (2019) 11(497). doi: 10.1126/scitranslmed.aaw0790
113. Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, Fujita M, et al. TGF-Beta 1 as an Enhancer of Fas-Mediated Apoptosis of Lung Epithelial Cells. J Immunol (2002) 168:6470–8. doi: 10.4049/jimmunol.168.12.6470
114. Horowitz JC, Rogers DS, Sharma V, Vittal R, White ES, Cui Z, et al. Combinatorial Activation of FAK and AKT by Transforming Growth Factor-Beta1 Confers an Anoikis-Resistant Phenotype to Myofibroblasts. Cell Signal (2007) 19:761–71. doi: 10.1016/j.cellsig.2006.10.001
115. Kang JH, Jung MY, Choudhury M, Leof EB. Transforming Growth Factor Beta Induces Fibroblasts to Express and Release the Immunomodulatory Protein PD-L1 Into Extracellular Vesicles. FASEB J (2020) 34:2213–26. doi: 10.1096/fj.201902354R
116. Aguirre JE, Beswick EJ, Grim C, Uribe G, Tafoya M, Chacon Palma G, et al. Matrix Metalloproteinases Cleave Membrane-Bound PD-L1 on CD90+ (Myo-)Fibroblasts in Crohn's Disease and Regulate Th1/Th17 Cell Responses. Int Immunol (2020) 32:57–68. doi: 10.1093/intimm/dxz060
117. Yarovinsky TO, Hunninghake GW. Lung Fibroblasts Inhibit Activation-Induced Death of T Cells Through PGE(2)-Dependent Mechanisms. Am J Physiol Lung Cell Mol Physiol (2001) 281:L1248–56. doi: 10.1152/ajplung.2001.281.5.L1248
118. Schirmer C, Klein C, von Bergen M, Simon JC, Saalbach A. Human Fibroblasts Support the Expansion of IL-17-Producing T Cells via Up-Regulation of IL-23 Production by Dendritic Cells. Blood (2010) 116:1715–25. doi: 10.1182/blood-2010-01-263509
119. Tang W, Yang L, Yang YC, Leng SX, Elias JA. Transforming Growth Factor-Beta Stimulates Interleukin-11 Transcription via Complex Activating Protein-1-Dependent Pathways. J Biol Chem (1998) 273:5506–13. doi: 10.1074/jbc.273.10.5506
120. Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, et al. IL-11 Is a Crucial Determinant of Cardiovascular Fibrosis. Nature (2017) 552:110–5. doi: 10.1038/nature24676
121. Targeting IL-11 in Idiopathic Pulmonary Fibrosis. Nat Rev Drug Discov (2019) 18:904. doi: 10.1038/d41573-019-00183-9
122. Kortekaas RK, Burgess JK, van Orsoy R, Lamb D, Webster M, Gosens R. Therapeutic Targeting of IL-11 for Chronic Lung Disease. Trends Pharmacol Sci (2021) 42:354–66. doi: 10.1016/j.tips.2021.01.007
123. Nishina T, Deguchi Y, Ohshima D, Takeda W, Ohtsuka M, Shichino S, et al. Interleukin-11-Expressing Fibroblasts Have a Unique Gene Signature Correlated With Poor Prognosis of Colorectal Cancer. Nat Commun (2021) 12:2281. doi: 10.1038/s41467-021-22450-3
124. Adami E, Viswanathan S, Widjaja AA, Ng B, Chothani S, Zhihao N, et al. IL11 Is Elevated in Systemic Sclerosis and IL11-Dependent ERK Signaling Underlies TGFbeta-Mediated Activation of Dermal Fibroblasts. Rheumatol (Oxford) (2021). doi: 10.1093/rheumatology/keab168
125. Zhang X, Kiapour N, Kapoor S, Khan T, Thamilarasan M, Tao Y, et al. IL-11 Induces Encephalitogenic Th17 Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. J Immunol (2019) 203:1142–50. doi: 10.4049/jimmunol.1900311
126. Curti A, Ratta M, Corinti S, Girolomoni G, Ricci F, Tazzari P, et al. Interleukin-11 Induces Th2 Polarization of Human CD4(+) T Cells. Blood (2001) 97:2758–63. doi: 10.1182/blood.V97.9.2758
127. Miranda-Carus ME, Balsa A, Benito-Miguel M, Perez de Ayala C, Martin-Mola E. IL-15 and the Initiation of Cell Contact-Dependent Synovial Fibroblast-T Lymphocyte Cross-Talk in Rheumatoid Arthritis: Effect of Methotrexate. J Immunol (2004) 173:1463–76. doi: 10.4049/jimmunol.173.2.1463
128. Barron AMS, Mantero JC, Ho JD, Nazari B, Horback KL, Bhawan J, et al. Perivascular Adventitial Fibroblast Specialization Accompanies T Cell Retention in the Inflamed Human Dermis. J Immunol (2019) 202:56–68. doi: 10.4049/jimmunol.1801209
129. Goehrig D, Nigri J, Samain R, Wu Z, Cappello P, Gabiane G, et al. Stromal Protein Betaig-H3 Reprogrammes Tumour Microenvironment in Pancreatic Cancer. Gut (2019) 68:693–707. doi: 10.1136/gutjnl-2018-317570
Keywords: fibroblasts, T cells, interrelation, ILDs, fibrosis
Citation: Lai Y, Wei X, Ye T, Hang L, Mou L and Su J (2021) Interrelation Between Fibroblasts and T Cells in Fibrosing Interstitial Lung Diseases. Front. Immunol. 12:747335. doi: 10.3389/fimmu.2021.747335
Received: 26 July 2021; Accepted: 19 October 2021;
Published: 05 November 2021.
Edited by:
Dong Weng, Tongji University, ChinaReviewed by:
Leslie Chavez-Galan, Instituto Nacional de Enfermedades Respiratorias-México (INER), MexicoCopyright © 2021 Lai, Wei, Ye, Hang, Mou and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Su, c3VqaW5AZ2lyZC5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.