 Marta Vuerich1†
Marta Vuerich1† Na Wang
Na Wang Maria Serena Longhi
Maria Serena Longhi- 1Department of Anesthesia, Critical Care & Pain Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
- 2Department of Hematology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, China
- 3School of Medicine, Shandong University, Jinan, China
Autoimmune hepatitis (AIH) is a chronic inflammatory disorder characterized by hypergammaglobulinemia, presence of serum autoantibodies and histological features of interface hepatitis. AIH therapeutic management still relies on the administration of corticosteroids, azathioprine and other immunosuppressants like calcineurin inhibitors and mycophenolate mofetil. Withdrawal of immunosuppression often results in disease relapse, and, in some cases, therapy is ineffective or associated with serious side effects. Understanding the mechanisms underlying AIH pathogenesis is therefore of paramount importance to develop more effective and well tolerated agents capable of restoring immunotolerance to liver autoantigens. Imbalance between effector and regulatory cells permits liver damage perpetuation and progression in AIH. Impaired expression and regulation of CD39, an ectoenzyme key to immunotolerance maintenance, have been reported in Tregs and effector Th17-cells derived from AIH patients. Interference with these altered immunoregulatory pathways may open new therapeutic avenues that, in addition to limiting aberrant inflammatory responses, would also reconstitute immune homeostasis. In this review, we highlight the most recent findings in AIH immunopathogenesis and discuss how these could inform and direct the development of novel therapeutic tools.
Introduction
Autoimmune hepatitis (AIH) was initially described as a severe form of fluctuating persistent hepatitis, associated with acneiform rashes, spider angiomas, amenorrhea and marked elevation of serum immunoglobulins (1). Subsequent studies showed that AIH occurs in individuals of all ages and, although present in both sexes, is prevalent in females (2).
AIH is diagnosed based on the presence of hypergammaglobulinemia, serum autoantibodies and histological features of interface hepatitis (3). While positivity for anti-nuclear antibody (ANA) and/or anti-smooth muscle antibody (SMA) identifies type-1 AIH (AIH-1), liver-kidney-microsomal-type-1 (LKM-1) antibodies, which specifically target the cytochrome P4502D6 (CYP2D6) liver enzyme (4), are the serological hallmarks of type-2 AIH (AIH-2) (5). Additional autoantibodies might aid in AIH diagnosis, especially in patients negative for ANA, SMA and LKM-1 autoantibodies. Antibodies to soluble liver antigen (SLA) have been reported in 58% of AIH patients where they are often associated with severe disease (6). Anti-liver cytosol type-1 (LC1) antibodies, which target formiminotransferase cyclodeaminase (FTCD), identify AIH-2 (7), and their titer positively correlates with disease activity (8). Presence of anti-neutrophil cytoplasmic antibody (ANCA), particularly atypical p-ANCA (pANNA), might also help in AIH diagnosis, in the absence of positivity for the above-mentioned autoantibodies (9).
Interface hepatitis is present at disease presentation in 84-98% of cases (10) and is characterized by a dense mononuclear cell infiltrate eroding the limiting plate and invading the liver parenchyma (11).
In 50% of cases, AIH manifests with an insidious onset that is often associated with lethargy, malaise, arthralgia, and myalgia; 30-40% of patients present with clinical features of acute hepatitis, whereas the remaining 10-20% of cases are incidentally discovered as having elevated transaminase levels on biochemical screening (12). AIH clinical manifestations may differ, depending on the geographical location and ethnicity of the affected patients. As an example, cirrhosis is more frequent in African Americans (56-85%) than Europeans (38%) (13, 14); and when considering subjects of non-European Caucasoid ethnicity, AIH has earlier onset in African, Arabian and Asian individuals, who also show lower response to immunosuppressive treatment (13, 14). Japanese patients are reported having a later onset of the disease that in most cases improves upon treatment with immunosuppressive drugs (15).
AIH can be associated with autoimmune disorders, which can be also found in first-degree relatives in 40% of cases (16).
The aim of AIH current treatment is to control inflammation. The management of the acute phase includes the administration of prednisone or prednisolone, which is gradually decreased to lower doses, based on the decline of transaminase levels. The addition of the anti-metabolite immunosuppressant azathioprine can be considered when the transaminase levels stop decreasing on steroid treatment alone or in the presence of steroid-related side effects (17). Relapse may occur in up to 40% of cases due to low compliance or when attempting treatment withdrawal. Additional drugs have been used as steroid-saving agents with the aim of reducing steroid-related side effects, these including cyclosporine and tacrolimus (18–20) and, in difficult-to-treat-cases, mycophenolate mofetil in association with prednisone or prednisolone (21, 22). None of these treatments, however, restores immunotolerance by boosting the impaired regulatory cell compartment.
Several studies have been conducted to identify the mechanisms involved in AIH pathogenesis. It has been proposed that genetically predisposed individuals upon exposure to certain environmental conditions (23) can develop cell-mediated immune responses against liver autoantigens. The derived inflammation, permitted by defective immune regulation, progressively results in fibrosis and cirrhosis with aberrant liver re-generation. Genetic predisposition to the disease is conferred by the presence of specific genes located within the human-leukocyte-antigen (HLA) region on the short arm of chromosome 6, especially those encoding allelic variants of DQB1 and DRB1 (24, 25).
Studies on AIH pathogenesis have been furthered by the generation of animal models, developed using different strategies. In this regard, immunization of C57BL/6 female mice with a pCMV plasmid containing the N-terminal region of mouse CTLA-4 and human CYP2D6 and FTCD resulted in transaminase elevations peaking 4 and 7 months after injection (26). Additional models, obtained upon infection of mice with adenovirus Ad5 expressing human CYP2D6, supported a role for viral infection as a possible mechanism leading to tolerance breakdown and consequent development of autoimmunity (27, 28). In an additional model by Hardtke-Wolenski et al, self-limiting adenoviral infection triggered immune responses against the human FTCD in mice of NOD background and resulted in chronic AIH that was characterized by portal and lobular fibrosis (29). Other AIH models have been obtained in mice with medullary thymic epithelial cell depletion due to a conditional deletion of Traf6 expression in murine thymic epithelial cells (30); and in PD-1-/- mice upon neonatal thymectomy, which resulted in aberrant generation of follicular helper T-cells in the spleen (31). Overall, these studies indicate that experimental AIH may derive from different immunopathogenic routes that can be facilitated by viral infections and/or genetic predisposition, this recapitulating the human scenario.
From an immunological perspective, AIH liver damage is driven by overwhelming effector immune responses (32–35) that are not adequately controlled by suppressor/regulatory T-cells (Tregs) (36–39). Extracellular nucleotides and nucleosides are strong modulators of T-cell immunity (40). Mounting evidence links alterations of purinergic signaling with the immunological dysfunction present in autoimmune conditions (41). Both Treg and Th17-cells obtained from AIH patients express low levels and impaired activity of the immunoregulatory ectoenzyme CD39 (42, 43). We have recently reported that such dysfunction derives, at least in part, from alterations of aryl-hydrocarbon-receptor (AhR) signaling (44), suggesting that modulation of this pathway might be deployed to correct immunoregulatory defects while boosting Treg immunity in AIH.
Understanding the mechanisms underlying AIH immune dysregulation is of critical importance for developing more effective treatments. In this review, we will present and discuss the most recent experimental evidence of disrupted AhR/purinergic interactions as one of the prominent factors leading to immunotolerance breakdown in AIH. We will also review how these alterations might inform and direct towards novel therapeutic approaches that represent promising candidates for the treatment of AIH.
Mechanisms of Liver Damage
The mechanisms underlying the liver attack in AIH are still unclear. The autoimmune reaction is believed to be initiated by the presentation of a liver autoantigen by antigen presenting cells (APCs) to Th0 lymphocytes that upon antigen recognition become activated. There is evidence that, in AIH, HLA-class-II molecules are expressed not only on professional APCs but also on hepatocytes (45), this favoring the amplification of cellular immune responses. Following activation, Th0 lymphocytes can differentiate into Th1, Th2 or Th17-cells, this depending on the cytokines in the milieu. All these subsets are present in the hepatic inflammatory infiltrates of AIH patients (35, 46). The cytokines released by each cell subset lead to a cascade of events culminating with the maturation of B-lymphocytes into plasma-cells and consequent production of autoantibodies, which are involved in antibody-mediated-cell-cytotoxicity (47). Derived immune reactions include activation of cytotoxic T-cells with subsequent release of IL-2 and IFNγ, activation of macrophages and, importantly, upregulation of HLA-class-I and class-II molecules by hepatocytes (45). Th17-cells are also involved in AIH liver damage, by perpetrating inflammation and through induction of pro-fibrotic events (35). If this cascade of events is not opposed by effective immune regulation, perpetration of immune attack occurs with consequent progression of tissue damage.
There is evidence that the frequency of liver autoantigen-specific T-cells present in the portal infiltrate is low (33, 48); these cells could orchestrate the liver damage by favoring the recruitment of non-antigen specific lymphocytes that, in turn, carry on hepatocyte damage by producing IFNγ and other cytotoxic factors.
Following the identification of CYP2D6 as the target autoantigen of LKM-1 autoantibodies in AIH-2 (4), several investigations have been conducted to identify CYP2D6 epitopes recognized by B and T-cells. By performing a conformational epitope mapping of the CYP2D6 molecule, Ma and colleagues identified CYP2D6316-327 as key target for autoantibodies (49). Subsequent studies performed by the same group showed that CD4 T-cell immunity in AIH-2 was polyclonal and involved multiple subsets of effectors with IFNγ, IL-4 and IL-10 producing lymphocytes targeting specific epitope regions within CYP2D6 (32). CD4-mediated immune responses were associated with liver damage, a finding also confirmed when analyzing CYP2D6-specific CD8 T-cell immunity (33). Future investigations should elucidate where autoantigen specific CD8 T-cell immune responses are initiated and perpetuated, given previous animal studies showing poor cytotoxic function and pro-inflammatory cytokine secretion in CD8 T-cells activated within the liver microenvironment (50); and additional studies showing that the spleen could modulate immune regulation as well as the intensity of hepatic inflammation (51, 52).
There is evidence that overwhelming effector cell immunity in AIH results from failure of immune regulatory mechanisms permitting the autoimmune damage to unfold and perpetuate.
Defects in regulatory cells have been studied extensively over the years, although the reasons leading to these impairments and the factors contributing to it remain still unclear.
Treg Impairment in AIH: the Role of CD39
A wealth of studies has provided evidence that impairment of regulatory cells plays a permissive role in the initiation and progression of autoimmune tissue damage.
Among lymphocytes with immunoregulatory/suppressive properties, CD4+CD25highFOXP3+ Tregs have been those most extensively studied in the last decades. These cells play a key role in promoting and maintaining immunotolerance, by controlling effector immune responses. Tregs can be classified based on their developmental pathway. Generation of thymic Tregs (tTregs) is facilitated by intermediate affinity self-peptides/MHC interactions resulting in high intensity TCR signals (53). Peripheral Tregs (pTregs) can differentiate from T-cells in certain peripheral sites like the gut mucosa and acquire stable FOXP3 expression (54); whereas induced Tregs (iTregs) can be derived in vitro following Tconv exposure to suboptimal antigen stimulation in the presence of anti-inflammatory mediators (55, 56).
Tregs can suppress immune responses by different mechanisms that involve the downregulation of costimulatory molecules on APCs (57), release of cytokines like TGF-β (58), IL-10 (59) and IL-35 (60); activation of apoptosis (61), including the Galectin-9/T-cell-immunoglobulin-and-mucin-domain-3 (Tim-3) pathway (62); release of Granzymes and perforin (63); metabolic disruption via IL-2 deprivation from the environment (64); transfer of cyclic-adenosine-monophosphate (cAMP) to effector cells via gap junctions and subsequent upregulation of the inducible cAMP early repressor (65); or via CD39, an ectoenzyme that hydrolyzes pro-inflammatory ATP and ADP to ultimately generate immunosuppressive adenosine (40).
In vivo and in vitro studies have indicated that Treg impairment plays a key role in the pathogenesis of autoimmune diseases, permitting overwhelming effector cell immunity to perpetrate and perpetuate tissue damage (66–70).
Defects in suppressor lymphocytes/Tregs have been reported also in AIH and found to play an important role in permitting effectors like CD4 and CD8-cells to inflict hepatocyte damage (36–39, 71). Treg defects in AIH are multifactorial and include numerical impairment (37, 38, 72), functional defects (71, 73), high rate of immune exhaustion (74) as well as plasticity with tendency to acquire effector cell features, when exposed to or challenged with a proinflammatory stimulus (42). Subsequent investigations did not support these findings, likely as a result of differences in methodologies, patients’ demographics and clinical characteristics (75, 76).
Treg numerical impairment has been shown also in mice (77, 78). In a humanized mouse model of AIH, obtained upon injection of human CYP2D6/FTCD fusion protein into HLA-DR3- or HLA-DR3+ transgenic NOD recipients, Yuksel and colleagues showed that defective Tregs were associated with heightened Th1-cell immunity with HLA-DR3+ mice undergoing the most severe form of the disease (78). In another study by Lapierre et al, adoptive transfer of ex vivo expanded CXCR3+ Tregs restored peripheral tolerance to FTCD and induced AIH remission (77).
Different mechanisms might account for Treg impairment in AIH (Figure 1). We have shown that CD4+CD25high Tregs from AIH patients display low levels of Galectin-9, contain higher frequencies of IFNγ+ and IL-17+-cells, while displaying lower proportions of FOXP3+, IL-10+ and TGF-β+-cells. This impairment is associated with low expression of Tim-3, the receptor of Galectin-9 on effector CD4-cells (39); this implicating that regulatory cell defects are linked with low responsiveness of T-effectors to Treg control. In a subsequent study we have reported that AIH Tregs display reduced ability to produce IL-10, this resulting in low responsiveness to low dose IL-2, as demonstrated by inability to upregulate the phosphor-signal-transducer-and-activator-of-transcription-5 (pSTAT5) (73).
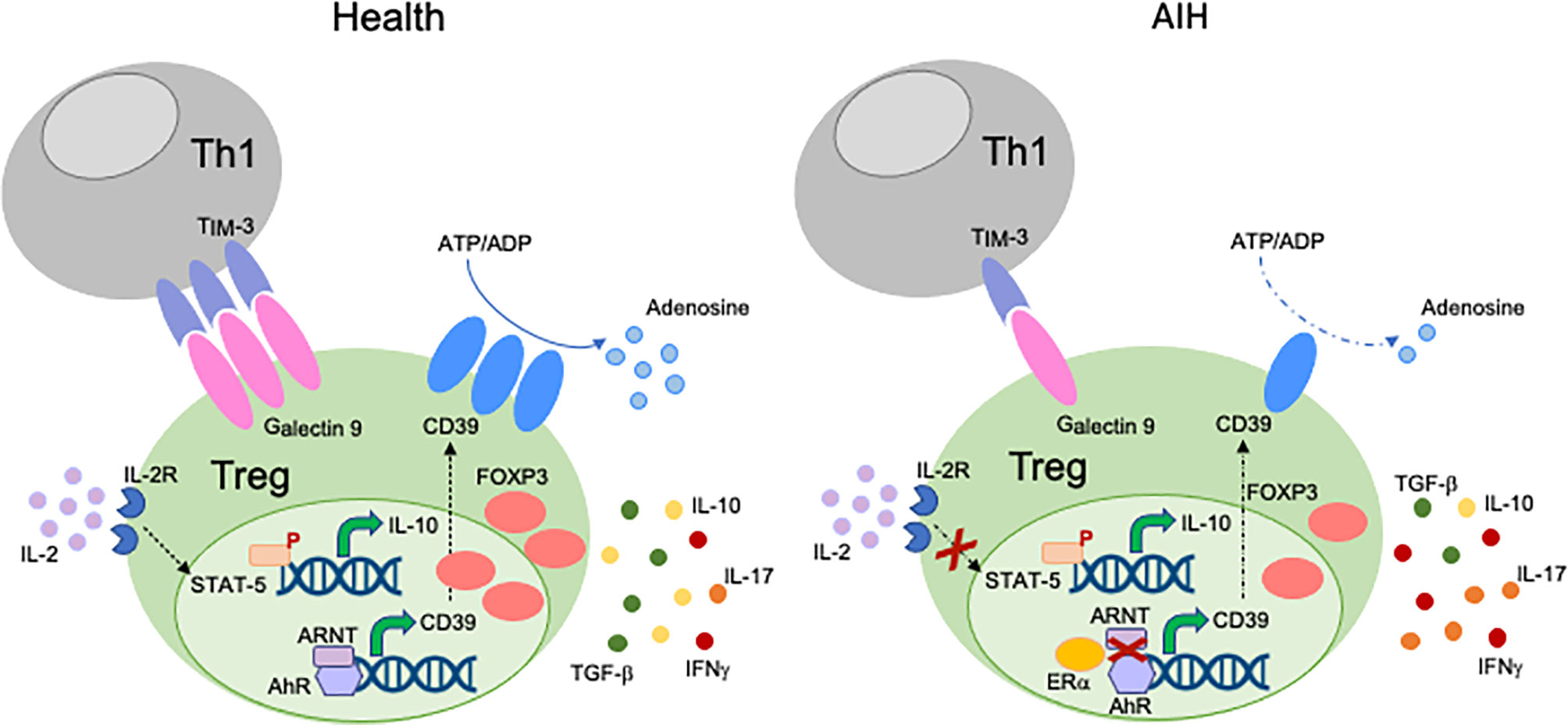
Figure 1 Mechanisms of Treg impairment in AIH. Different mechanisms account for Treg impairment in AIH. Low Galectin-9 in Tregs is associated with decreased TIM-3 levels on Th1-cells. This possibly results in reduced control of the effector potential of the Th1 subset through induction of apoptosis. AIH Tregs are also defective in their response to low dose IL-2, as reflected by impaired upregulation of pSTAT-5. AIH Tregs produce low amounts of IL-10 and TGF-β, while secreting effector cytokines like IFNγ and IL-17. This indicates that, in AIH, Tregs have high tendency to acquire effector cell features. AIH Tregs also display low levels and activity of CD39, an ectoenzyme that initiates a hydrolysis cascade culminating with the generation of immunosuppressive adenosine. In health, levels of CD39 are regulated by the aryl-hydrocarbon-receptor (AhR)/aryl-hydrocarbon-receptor-nuclear-translocator (ARNT) complex. In AIH, preferential AhR binding to estrogen-receptor-alpha (Erα), an AhR non-canonical partner results in less effective upregulation of CD39 by Tregs.
In de novo AIH occurring after liver transplant Treg impairment was proposed to derive from high secretion of IL-12 and IL-6 by monocytes/macrophages that induced aberrant IFNγ production by Tregs impacting their function (79, 80). Blockade of TLR2 and TLR4 on monocytes resulted in reduction of IFNγ production by Tregs, further supporting the role of monocytes/macrophages in conferring Tregs proinflammatory features (79, 80).
In a mouse model of AIH, characterized by hepatocellular expression of a MHC-class II restricted immunodominant epitope of the lymphocyte choriomeningitis virus and by accumulation of CD4 T-cells specifically recognizing this epitope, Preti et al. proposed that liver damage was fostered by selective failure of peripherally induced autoreactive Tregs (81). Notably these autoreactive Tregs not only were reduced in frequency but also displayed heightened IL-17 production and reduced epigenetic demethylation (81), postulating a role for altered epigenetic regulation in Treg impairment in this model. In human AIH, FOXP3 demethylation - a typical feature of bona fide Tregs - was retained in some studies (75, 79) and altered in others, where AIH derived Tregs were highly methylated (82); this indicates that further studies are needed to clearly establish the role of FOXP3 epigenetic regulation in AIH Tregs.
In subsequent studies, we have shown that Tregs obtained from the peripheral blood of AIH patients display low levels of CD39 ectoenzyme (42). In addition to displaying low CD39 levels, AIH Tregs are defective in their ectoenzymatic activity, this likely impacting their suppressive function due to lower adenosine generation (42). Impaired suppressive function of CD39+ Tregs is associated with increased tendency of these cells to upregulate CD127 and producing higher levels of IL-17 and IFNγ, when exposed to anti-CD3/CD28 (42).
CD39 can be regulated at genetic, transcriptional and post-transcriptional levels. Among the factors that regulate CD39 expression at the transcriptional level, the AhR signaling plays a pivotal role. AhR is a modulator of toxin responses that also regulates effector and Treg immunity (83). AhR is activated by endogenous ligands including products of heme catabolism like unconjugated bilirubin (UCB), tryptophan metabolites, 2-(1’ H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester, dietary compounds like quercetin, or environmental pollutants, i.e., dioxin and benzo-a-pyrene. Upon binding to its ligands, AhR translocates to the cell nucleus where it forms a complex with the aryl-hydrocarbon-receptor-nuclear-translocator (ARNT), the AhR canonical partner, to regulate various genes, including cytochrome P450 enzymes, cytokines (IL-22, IL-17, IL-10), drug transporters (ABCB1, ABCC4) and CD39.
Notably, AhR activation has been demonstrated playing a role in the pathogenesis of other liver diseases, like hepatitis C, where the AhR-cytochrome P4501A1 pathway was found to favor lipid accumulation along with virus replication and assembly (84); and primary biliary cholangitis (PBC) where dioxin activated dendritic cells promoted differentiation of naïve CD4-cells, derived from PBC patients, into effector Th1 and Th17-cells (85). Importance of xenobiotics as triggers for PBC was also reported in earlier investigations (86, 87). Conversely, in the setting of experimental primary sclerosing cholangitis, administration of indole-3-carboxaldehyde could alleviate hepatic inflammation and fibrosis upon activation of the AhR-IL-22 axis (88). Further, kynurenine, a tryptophane metabolite that serves as AhR endogenous ligand, was decreased in the serum of children with AIH, when compared to controls (89).
Given the important links between AhR and CD39, we tested whether defective CD39 levels could derive from decreased AhR or ARNT expression. We found that AIH Tregs display AhR and ARNT levels comparable to Tregs derived from healthy controls; notably AIH Tregs express high levels of estrogen-receptor-alpha (Erα) (44), an AhR non-canonical partner. Further, in AIH Tregs, AhR preferentially binds to Erα rather than ARNT, this potentially resulting in less effective upregulation of CD39 upon exposure to ligands like UCB, L-kynurenine and quercetin (44). As increased Erα levels are present in patients under immunosuppression, we postulate that immunosuppressive drugs, while enabling control over inflammation, might also promote liver damage perpetuation by upregulating Erα and leading to a less effective AhR activation cascade. As silencing of Erα results in increased frequency of CD39+-cells among Tregs (44), strategies potentially interfering with AhR non-canonical binding might have a role in boosting CD39 levels, by restoring AhR signaling.
There is evidence that AhR transcriptional efficiency could be regulated by factors that impact the chromatin structure (90), suggesting that AhR can undergo epigenetic regulation. Previous studies have documented global hypomethylation of CD4 T-cells in systemic lupus erythematosus, and systemic sclerosis (90, 91); it remains to be established whether these mechanisms are operative also in CD4 T cell subsets in AIH, possibly impacting AhR function and its ability to regulate downstream genes like CD39.
Aberrant CD39 Regulation in AIH Th17-Cells
Th17-cells are an effector subset that derives from CD4 lymphocytes upon exposure to IL-6 and TGF-β in mice and IL-6, IL-1β and TGF-β in humans (92). IL-23 plays an important role in the maintenance and stabilization of already differentiated Th17-cells (92). Transcription factors involved in the development of Th17-cells include RORγt, RORα and STAT-3 (92). Th17-cells can be regulated by additional factors, like AhR, which modulates CD39 and drug transporter levels (93, 94). Th17-cells have been involved in the pathogenesis of various autoimmune disorders including AIH (35).
Expression of CD39 by Th17-cells has been associated with attenuation of their pathogenic potential and acquisition of regulatory properties, as reflected by upregulation of FOXP3 and IL-10 production (93). Levels of CD39 in Th17-cells are regulated, at least in part, upon engagement of AhR by exogenous and endogenous ligands (93). Akin to Tregs, Th17-cells obtained from the peripheral blood of AIH patients express low CD39 levels and impaired ectoenzymatic activity (Figure 2). These defects are associated with reduced expression of A2A adenosine receptor, further confirming alterations in purinergic signaling mediators and, consequently, decreased response to adenosine. Due to lower generation of adenosine, AIH Th17-cells display impaired acquisition of regulatory properties that result in defective ability to control CD4+ CD25–cell proliferation and IL-17 production. As for Tregs, Th17-cells obtained from AIH patients cannot effectively upregulate CYP1A1 and CD39 when exposed to AhR ligands like UCB, L-kynurenine and quercetin, postulating alterations of AhR signaling as possible determinants of impaired CD39 levels. Differently from Tregs, however, no differences have been found in the expression of Erα. Instead, increase in the expression of hypoxia-inducible-factor-1alpha (HIF-1α), which is known to negatively regulate AhR expression (95) and signaling (94), is noted.
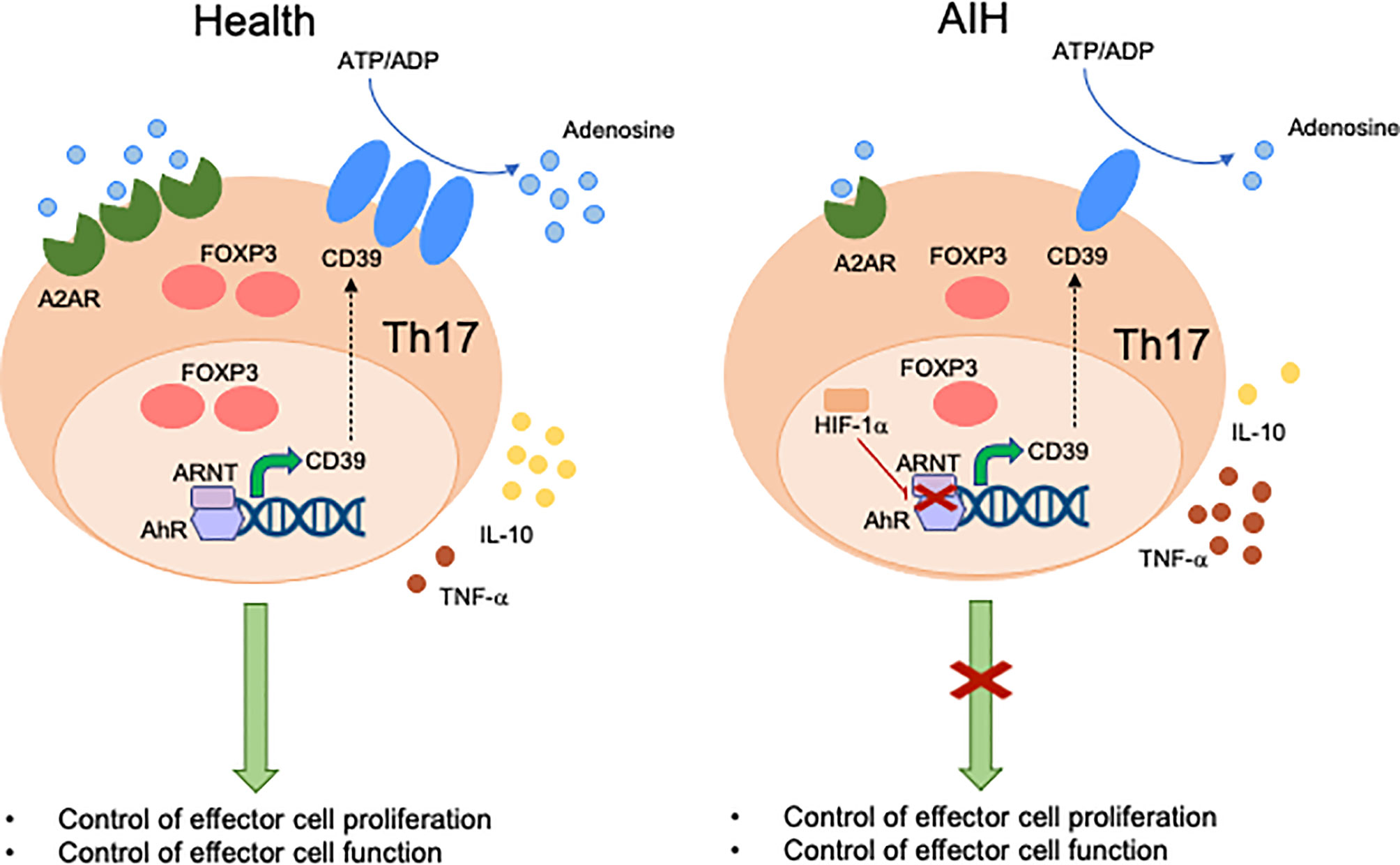
Figure 2 Mechanisms of aberrant Th17-cell immunity in AIH. Expression of CD39 by Th17-cells is associated with acquisition of regulatory properties. These include increased FOXP3 expression levels, IL-10 production and control of effector cell proliferation and function. In healthy subjects, levels of CD39 in Th17-cells are regulated by the aryl-hydrocarbon-receptor (AhR)/aryl-hydrocarbon-receptor-nuclear-translocator (ARNT) complex. In AIH, Th17-cells display decreased CD39 levels and activity. This is linked, at least in part, with high levels of hypoxia-inducible-factor-1alpha (HIF-1α) that inhibits AhR levels and signaling. Th17-cells from AIH patients also display reduced levels of A2A adenosine receptor, this implicating defective response of these cells to the immunosuppressive effects of adenosine.
As discussed, the AhR signaling alterations noted in AIH impact both Treg and Th17 cell ability to upregulate CD39, this accounting for their impaired suppressive function (Tregs) and inability to acquire regulatory properties (Th17-cells). This indicates that defective immune regulation involves not only the regulatory but also the effector cell compartment. Strategies aimed at restoring alterations of AhR signaling both in Tregs and Th17-cells should be developed to guarantee a broader control over effector cell immunity in this autoimmune setting.
New Immunotherapies
Given the multiple defects underlying immunotolerance breakdown in AIH, therapeutic strategies should be developed with the aim of restoring the pool of Tregs while dampening the effector potential of these cells (Figure 3). Reconstitution of the Treg pool could be achieved upon adoptive transfer. In previous studies we assessed Treg ability to expand under polyclonal conditions (96). Tregs expanded from the already existing regulatory cell pool express higher FOXP3 levels and suppress effector cells more effectively (96). This strategy also resulted in de novo generation of Tregs from CD4+CD25- effector lymphocytes, when using healthy controls and AIH-derived cells (96). In line with this strategy, inhibition of IL-17 was found to favor the differentiation of newly generated Tregs from CD4+CD25- effectors (97), supporting the postulate that immune regulation could be enhanced even further when effector cells are conditioned to become suppressive. Exposure of CD127+CD25+ activated cells to TGF-β and IL-2 boosts IL-10 production and suppressive function of these cells (98). As CD39 is strictly linked with Treg ability to suppress, adoptive transfer of CD39high Tregs (99) would be preferable; however, since this subset is substantially impaired in AIH patients (42), strategies that enable boosting CD39 in Tregs, prior to their transfer, should be implemented. A study by Oo and colleagues showed that polyclonal Tregs, isolated from leukapheresis products, labelled with indium tropolonate and re-infused in AIH patients, homed to the liver where they were detected for up to 72 hours (100). In a murine model of AIH in the context of autoimmune-polyendocrine-syndrome-type-1, a multiorgan autoimmune condition caused by mutations of the autoimmune regulator (AIRE) gene, adoptive transfer of polyspecific Tregs was effective in treating AIH (101).
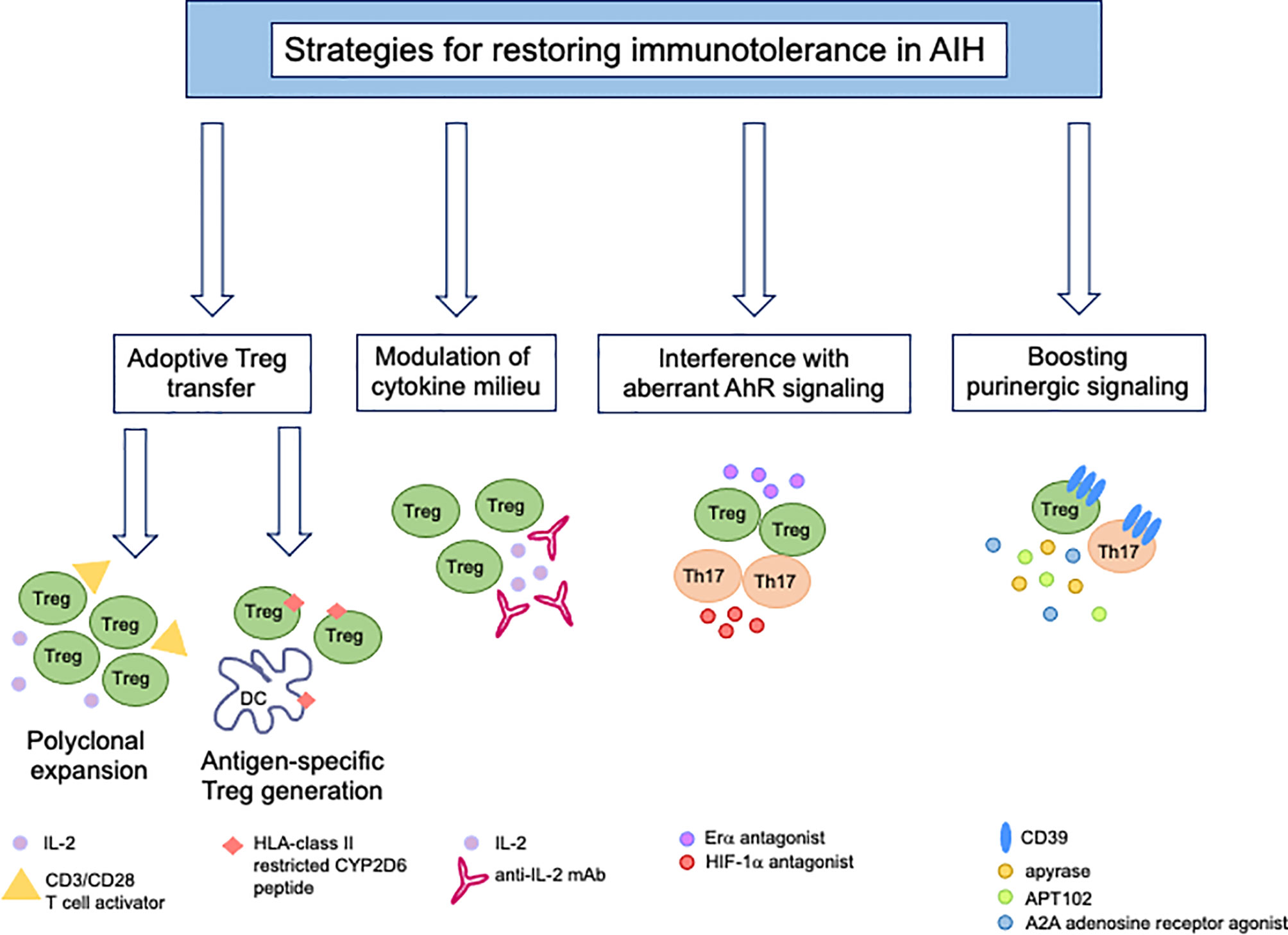
Figure 3 New immunotherapies for tolerance reconstitution in AIH. Strategies that might be considered as tools to re-establish immunotolerance in AIH include: adoptive transfer of Tregs, polyclonally expanded in vitro in the presence of high dose IL-2 and CD3/CD28 T-cell activator or generated under antigen-specific conditions upon co-culture with semi-mature dendritic cells; modulation of the cytokine milieu by administration of low dose IL-2 or monoclonal antibody/IL-2 complex; use of Erα or HIF-1α antagonists to interfere with aberrant AhR signaling in Tregs (Erα) and Th17-cells (HIF-1α); boosting of purinergic signaling through the use of exogenous apyrase, APT102, an ADPase that was found to enhance the beneficial effects of AhR activation in vivo and in vitro, and A2A adenosine receptor agonists to amplify the immunosuppressive properties of adenosine.
In the context of AIH-2 where the autoantigen and the CD4 and CD8 T-cell epitopes within it are known, adoptive transfer of autoantigen-specific Tregs would enable achieving a tailored and effective form of treatment to re-establish immunotolerance. In this regard, we could generate Tregs specific for HLA-DR3 and HLA-DR7 restricted CYP2D6 epitopes upon co-culture with semi-mature DCs that enable antigen presentation and promote immunotolerance induction (102). CYP2D6-Tregs suppress the proliferation and pro-inflammatory cytokine production by CD4 effectors of the same antigen specificity (102). As CYP2D6-specific Tregs showed the tendency to lose their suppressor properties when exposed to proinflammatory stimuli, treatment with retinoic acid could stabilize their phenotype and functional properties even in the presence of an inflammatory milieu (103). Effort has been made in subsequent studies to control autoantigen-specific effectors upon in vivo injections of peptide-major histocompatibility class-II (pMHC-II) nanomedicines with specificity for the immunodominant epitope CYP2D6398-412 in a mouse model of AIH, obtained by infecting NOD mice with replication defective adenovirus encoding for human FTCD (104). This approach resulted in the expansion of Tr1-cells (104). Preclinical studies have provided evidence that antigen-specific Tregs deliver targeted and superior immunosuppression, when compared to other approaches, including polyclonal Tregs (105). Since generation of antigen-specific Tregs is, however, hampered by their limited ability to expand, recent studies have shown that these cells could be also obtained from polyclonal Tregs through transduction of Treg T-cell receptor or chimeric antigen receptor (CAR) (105); or, through strategies based on the CRISPR Cas9 system where an endogenous TCR can be replaced by a recombinant one (105).
In addition to cell-based therapies, the implementation of which may be limited by substantial costs, Tregs could be expanded by modifying the cytokine environment. A study by Diestelhorst et al. proposed that the marked decrease in AIH Tregs during immunosuppressive therapy might derive from decreased levels of IL-2, a cytokine key for Treg survival (106). Exposure of PBMCs from patients with autoimmune liver diseases, including AIH, to very-low-dose Proleukin, used at less than 5 IU/ml, resulted in phosphorylation of STAT-5 and increased levels of CTLA-4 and FOXP3 in the Treg subset (107). Administration of low dose IL-2 would enable in vivo cell reconstitution, even when the autoantigen is unknown. In a study on two AIH patients with persistent disease activity, administration of low dose IL-2 resulted in Treg expansion in both. This was evident on day 9 but returned to baseline levels on day 28 (108). Clinical response was observed in one case, suggesting that the effects of IL-2-induced Treg expansion is transient and the clinical benefit limited, at least in the small cohort studied. Additional efforts should be made to optimize and successfully implement this approach.
To prolong the half-life of IL-2 in the bloodstream, mutants have been developed to promote Treg expansion in vivo. Another strategy has been attempted and consists of the administration of monoclonal antibody/IL-2 complex to expand Tregs in a mouse model of colitis (109) and AIH (110). A new complex of IL-2 and anti-IL-2 monoclonal antibodies was found to stimulate Treg expansion among human T-cells ex vivo and in rhesus macaques in vivo (111).
Transient increase in the proportion of splenic Tregs was achieved in a mouse model of AIH, resulting from xenoimmunization of C57BL/6 mice with DNA coding for human liver autoantigens, after treatment with low-dose anti-CD3 antibody (112). No differences in the proportion of splenic and liver derived Tregs were noted in xenoimmunized mice subjected to anti-CD20 antibody treatment, this approach inducing AIH remission by decreasing antigen presentation (through B-cells) and help to T lymphocytes (113). Whether belimumab, an inhibitor of serum B-cell activating factor that has been recently proposed as a promising treatment option for patients with refractory AIH and advanced liver-related fibrosis, induces disease remission having also a beneficial impact on Treg frequencies, is unknow (114).
As immunoregulatory defects in AIH involve both Tregs and effector Th17-cells, future approaches targeting both cell types should be considered. Our research has shown impaired CD39 levels in both Tregs and Th17-cells obtained from the peripheral blood of AIH patients. Reduced CD39 expression is present in AIH patients during active disease and remission, suggesting an intrinsic defect of this ectoenzyme in AIH Treg and Th17 lymphocytes (42, 43). This defect derives, at least in part, from aberrant increase in the levels of Erα, an alternative AhR partner, which is upregulated in AIH Tregs; and from high expression of HIF-1α, an AhR inhibitor, which is upregulated in AIH Th17-cells. In this regard, strategies inhibiting Erα and HIF-1α may help reconstituting the altered AhR signaling in AIH, and along with this, CD39 and immunotolerance.
Alternatively, interventions that boost CD39 levels might represent an additional option to bypass the altered AhR signaling/pathway and restore immunotolerance by enhancing the levels and/or activity of this ectoenzyme.
These approaches could be based on exogenous apyrase that has ectoenzymatic activity comparable to CD39; and APT102, the extracellular domain with improved ADPase activity of human nucleoside-triphosphate-diphosphohydrolase-3, a member of the CD39 family (115). APT102 was found beneficial in enhancing the immunoregulatory properties of the AhR ligand UCB in an experimental model of colitis in mice and in promoting increased levels of immunoregulatory molecules on Tregs and Tr1-cells (115). Notably, there were no safety or toxicity concerns when APT102 treatment was protracted after resolution of colitis in vivo (115). Additional studies reported on the safety profile of this molecule in the vascular setting (116, 117). However, since CD39 also plays an important role in tumor development and progression (99, 118–121) and inhibition of effector immune responses against pathogens (99, 122, 123), the safety of long-term treatments aimed at restoring the levels of this ectoenzyme in the context of autoimmune diseases should be considered and carefully evaluated. A2A receptor agonists that enhance the adenosinergic signal might also have a role in restoring immunotolerance by reconstituting the purinergic milieu (41).
Strategies targeting CD39 antisense RNA, a long noncoding RNA regulating CD39 at both mRNA and protein levels, should be taken into consideration. Silencing of CD39 antisense RNA has been effective in containing disease activity in an experimental mouse model of colitis in humanized NOD/scid/gamma mice, reconstituted with human CD4-cells (124).
Concluding Remarks
This review has discussed the mechanisms mediating liver damage perpetuation and progression in AIH with a specific focus on the imbalance between Tregs and Th17-cells.
Numerous pathways might be involved in AIH disordered immunity. We have discussed how defects in CD39 are linked with aberrant AhR signaling in AIH-derived Tregs and Th17-cells. Standard treatment of AIH is still based on corticosteroids and azathioprine, immunosuppressive drugs that currently enable control of inflammation without, however, restoring immunotolerance and preventing progression to end-stage liver disease. Strategies targeting factors that interfere with AhR canonical pathway or directly boosting CD39 expression and activity might represent new therapeutic avenues in the treatment of AIH.
Author Contributions
MV, NW, and AK wrote the manuscript. JG performed literature search. ML reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by the National Institutes of Health (R01 DK108894 and R01 DK124408 to ML) and the Department of Anesthesia Seed Grant Award (to ML).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Waldenstrom J. Liver, Blood Proteins and Food Proteins. Dtsch Z Verdau Stoffwechselkr (1952) 12(Spec No):113–21.
2. Al-Chalabi T, Underhill JA, Portmann BC, McFarlane IG, Heneghan MA. Impact of Gender on the Long-Term Outcome and Survival of Patients With Autoimmune Hepatitis. J Hepatol (2008) 48(1):140–7. doi: 10.1016/j.jhep.2007.08.013
3. Johnson PJ, McFarlane IG. Meeting Report: International Autoimmune Hepatitis Group. Hepatology (1993) 18(4):998–1005. doi: 10.1002/hep.1840180435
4. Manns MP, Johnson EF, Griffin KJ, Tan EM, Sullivan KF. Major Antigen of Liver Kidney Microsomal Autoantibodies in Idiopathic Autoimmune Hepatitis is Cytochrome P450db1. J Clin Invest (1989) 83(3):1066–72. doi: 10.1172/JCI113949
5. Vergani D, Alvarez F, Bianchi FB, Cancado EL, Mackay IR, Manns MP, et al. Liver Autoimmune Serology: A Consensus Statement From the Committee for Autoimmune Serology of the International Autoimmune Hepatitis Group. J Hepatol (2004) 41(4):677–83. doi: 10.1016/j.jhep.2004.08.002
6. Ma Y, Okamoto M, Thomas MG, Bogdanos DP, Lopes AR, Portmann B, et al. Antibodies to Conformational Epitopes of Soluble Liver Antigen Define a Severe Form of Autoimmune Liver Disease. Hepatology (2002) 35(3):658–64. doi: 10.1053/jhep.2002.32092
7. Muratori L, Sztul E, Muratori P, Gao Y, Ripalti A, Ponti C, et al. Distinct Epitopes on Formiminotransferase Cyclodeaminase Induce Autoimmune Liver Cytosol Antibody Type 1. Hepatology (2001) 34(3):494–501. doi: 10.1053/jhep.2001.27179
8. Muratori L, Cataleta M, Muratori P, Lenzi M, Bianchi FB. Liver/kidney Microsomal Antibody Type 1 and Liver Cytosol Antibody Type 1 Concentrations in Type 2 Autoimmune Hepatitis. Gut (1998) 42(5):721–6. doi: 10.1136/gut.42.5.721
9. Terziroli Beretta-Piccoli B, Mieli-Vergani G, Vergani D. Serology in Autoimmune Hepatitis: A Clinical-Practice Approach. Eur J Intern Med (2018) 48:35–43. doi: 10.1016/j.ejim.2017.10.006
10. Gleeson D, Heneghan MA, British Society of G. British Society of Gastroenterology (BSG) Guidelines for Management of Autoimmune Hepatitis. Gut (2011) 60(12):1611–29. doi: 10.1136/gut.2010.235259
11. Desmet VJ, Gerber M, Hoofnagle JH, Manns M, Scheuer PJ. Classification of Chronic Hepatitis: Diagnosis, Grading and Staging. Hepatology (1994) 19(6):1513–20. doi: 10.1002/hep.1840190629
12. McFarlane IG. Definition and Classification of Autoimmune Hepatitis. Semin Liver Dis (2002) 22(4):317–24. doi: 10.1055/s-2002-35702
13. Zolfino T, Heneghan MA, Norris S, Harrison PM, Portmann BC, McFarlane IG. Characteristics of Autoimmune Hepatitis in Patients Who are Not of European Caucasoid Ethnic Origin. Gut (2002) 50(5):713–7. doi: 10.1136/gut.50.5.713
14. Lim KN, Casanova RL, Boyer TD, Bruno CJ. Autoimmune Hepatitis in African Americans: Presenting Features and Response to Therapy. Am J Gastroenterol (2001) 96(12):3390–4. doi: 10.1111/j.1572-0241.2001.05272.x
15. Takahashi A, Arinaga-Hino T, Ohira H, Torimura T, Zeniya M, Abe M, et al. Autoimmune Hepatitis in Japan: Trends in a Nationwide Survey. J Gastroenterol (2017) 52(5):631–40. doi: 10.1007/s00535-016-1267-0
16. Gregorio GV, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, et al. Autoimmune Hepatitis in Childhood: A 20-Year Experience. Hepatology (1997) 25(3):541–7. doi: 10.1002/hep.510250308
17. Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli-Vergani G, Vergani D, et al. Diagnosis and Management of Autoimmune Hepatitis. Hepatology (2010) 51(6):2193–213. doi: 10.1002/hep.23584
18. Alvarez F, Ciocca M, Canero-Velasco C, Ramonet M, de Davila MT, Cuarterolo M, et al. Short-Term Cyclosporine Induces a Remission of Autoimmune Hepatitis in Children. J Hepatol (1999) 30(2):222–7. doi: 10.1016/S0168-8278(99)80065-8
19. Cuarterolo M, Ciocca M, Velasco CC, Ramonet M, Gonzalez T, Lopez S, et al. Follow-Up of Children With Autoimmune Hepatitis Treated With Cyclosporine. J Pediatr Gastroenterol Nutr (2006) 43(5):635–9. doi: 10.1097/01.mpg.0000235975.75120.38
20. Van Thiel DH, Carroll P, Abu-Elmagd K, Rodriguez-Rilo H, Irish W, McMichael J, et al. Tacrolimus (FK 506), a Treatment for Primary Sclerosing Cholangitis: Results of an Open-Label Preliminary Trial. Am J Gastroenterol (1995) 90(3):455–9.
21. Richardson PD, James PD, Ryder SD. Mycophenolate Mofetil for Maintenance of Remission in Autoimmune Hepatitis in Patients Resistant to or Intolerant of Azathioprine. J Hepatol (2000) 33(3):371–5. doi: 10.1016/S0168-8278(00)80271-8
22. Aw MM, Dhawan A, Samyn M, Bargiota A, Mieli-Vergani G. Mycophenolate Mofetil as Rescue Treatment for Autoimmune Liver Disease in Children: A 5-Year Follow-Up. J Hepatol (2009) 51(1):156–60. doi: 10.1016/j.jhep.2009.02.024
23. Lammert C, Chalasani SN, Atkinson EJ, McCauley BM, Lazaridis KN. Environmental Risk Factors are Associated With Autoimmune Hepatitis. Liver Int (2021) 41(5):1162–3. doi: 10.1111/liv.14944
24. Ma Y, Su H, Yuksel M, Longhi MS, McPhail MJ, Wang P, et al. Human Leukocyte Antigen Profile Predicts Severity of Autoimmune Liver Disease in Children of European Ancestry. Hepatology (2021). doi: 10.1002/hep.31893
25. Djilali-Saiah I, Fakhfakh A, Louafi H, Caillat-Zucman S, Debray D, Alvarez F. HLA Class II Influences Humoral Autoimmunity in Patients With Type 2 Autoimmune Hepatitis. J Hepatol (2006) 45(6):844–50. doi: 10.1016/j.jhep.2006.07.034
26. Lapierre P, Djilali-Saiah I, Vitozzi S, Alvarez F. A Murine Model of Type 2 Autoimmune Hepatitis: Xenoimmunization With Human Antigens. Hepatology (2004) 39(4):1066–74. doi: 10.1002/hep.20109
27. Hintermann E, Ehser J, Bayer M, Pfeilschifter JM, Christen U. Mechanism of Autoimmune Hepatic Fibrogenesis Induced by an Adenovirus Encoding the Human Liver Autoantigen Cytochrome P450 2d6. J Autoimmun (2013) 44:49–60. doi: 10.1016/j.jaut.2013.05.001
28. Holdener M, Hintermann E, Bayer M, Rhode A, Rodrigo E, Hintereder G, et al. Breaking Tolerance to the Natural Human Liver Autoantigen Cytochrome P450 2D6 by Virus Infection. J Exp Med (2008) 205(6):1409–22. doi: 10.1084/jem.20071859
29. Hardtke-Wolenski M, Fischer K, Noyan F, Schlue J, Falk CS, Stahlhut M, et al. Genetic Predisposition and Environmental Danger Signals Initiate Chronic Autoimmune Hepatitis Driven by CD4+ T Cells. Hepatology (2013) 58(2):718–28. doi: 10.1002/hep.26380
30. Bonito AJ, Aloman C, Fiel MI, Danzl NM, Cha S, Weinstein EG, et al. Medullary Thymic Epithelial Cell Depletion Leads to Autoimmune Hepatitis. J Clin Invest (2013) 123(8):3510–24. doi: 10.1172/JCI65414
31. Aoki N, Kido M, Iwamoto S, Nishiura H, Maruoka R, Tanaka J, et al. Dysregulated Generation of Follicular Helper T Cells in the Spleen Triggers Fatal Autoimmune Hepatitis in Mice. Gastroenterology (2011) 140(4):1322–33.e1-5. doi: 10.1053/j.gastro.2011.01.002
32. Ma Y, Bogdanos DP, Hussain MJ, Underhill J, Bansal S, Longhi MS, et al. Polyclonal T-Cell Responses to Cytochrome P450IID6 are Associated With Disease Activity in Autoimmune Hepatitis Type 2. Gastroenterology (2006) 130(3):868–82. doi: 10.1053/j.gastro.2005.12.020
33. Longhi MS, Hussain MJ, Bogdanos DP, Quaglia A, Mieli-Vergani G, Ma Y, et al. Cytochrome P450IID6-Specific CD8 T Cell Immune Responses Mirror Disease Activity in Autoimmune Hepatitis Type 2. Hepatology (2007) 46(2):472–84. doi: 10.1002/hep.21658
34. Longhi MS, Mitry RR, Samyn M, Scalori A, Hussain MJ, Quaglia A, et al. Vigorous Activation of Monocytes in Juvenile Autoimmune Liver Disease Escapes the Control of Regulatory T-Cells. Hepatology (2009) 50(1):130–42. doi: 10.1002/hep.22914
35. Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, et al. Interleukin-17 Contributes to the Pathogenesis of Autoimmune Hepatitis Through Inducing Hepatic Interleukin-6 Expression. PloS One (2011) 6(4):e18909. doi: 10.1371/journal.pone.0018909
36. Hodgson HJ, Wands JR, Isselbacher KJ. Alteration in Suppressor Cell Activity in Chronic Active Hepatitis. Proc Natl Acad Sci USA (1978) 75(3):1549–53. doi: 10.1073/pnas.75.3.1549
37. Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli-Vergani G, Vergani D. Impairment of CD4(+)CD25(+) Regulatory T-Cells in Autoimmune Liver Disease. J Hepatol (2004) 41(1):31–7. doi: 10.1016/j.jhep.2004.03.008
38. Longhi MS, Ma Y, Mitry RR, Bogdanos DP, Heneghan M, Cheeseman P, et al. Effect of CD4+ CD25+ Regulatory T-Cells on CD8 T-Cell Function in Patients With Autoimmune Hepatitis. J Autoimmun (2005) 25(1):63–71. doi: 10.1016/j.jaut.2005.05.001
39. Liberal R, Grant CR, Holder BS, Ma Y, Mieli-Vergani G, Vergani D, et al. The Impaired Immune Regulation of Autoimmune Hepatitis is Linked to a Defective Galectin-9/Tim-3 Pathway. Hepatology (2012) 56(2):677–86. doi: 10.1002/hep.25682
40. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic Signaling During Inflammation. N Engl J Med (2012) 367(24):2322–33. doi: 10.1056/NEJMra1205750
41. Vuerich M, Mukherjee S, Robson SC, Longhi MS. Control of Gut Inflammation by Modulation of Purinergic Signaling. Front Immunol (2020) 11:1882. doi: 10.3389/fimmu.2020.01882
42. Grant CR, Liberal R, Holder BS, Cardone J, Ma Y, Robson SC, et al. Dysfunctional CD39(POS) Regulatory T Cells and Aberrant Control of T-Helper Type 17 Cells in Autoimmune Hepatitis. Hepatology (2014) 59(3):1007–15. doi: 10.1002/hep.26583
43. Liberal R, Grant CR, Ma Y, Csizmadia E, Jiang ZG, Heneghan MA, et al. CD39 Mediated Regulation of Th17-Cell Effector Function is Impaired in Juvenile Autoimmune Liver Disease. J Autoimmun (2016) 72:102–12. doi: 10.1016/j.jaut.2016.05.005
44. Vuerich M, Harshe R, Frank LA, Mukherjee S, Gromova B, Csizmadia E, et al. Altered Aryl-Hydrocarbon-Receptor Signalling Affects Regulatory and Effector Cell Immunity in Autoimmune Hepatitis. J Hepatol (2021) 74(1):48–57. doi: 10.1016/j.jhep.2020.06.044
45. Lobo-Yeo A, Senaldi G, Portmann B, Mowat AP, Mieli-Vergani G, Vergani D. Class I and Class II Major Histocompatibility Complex Antigen Expression on Hepatocytes: A Study in Children With Liver Disease. Hepatology (1990) 12(2):224–32. doi: 10.1002/hep.1840120208
46. Senaldi G, Portmann B, Mowat AP, Mieli-Vergani G, Vergani D. Immunohistochemical Features of the Portal Tract Mononuclear Cell Infiltrate in Chronic Aggressive Hepatitis. Arch Dis Child (1992) 67(12):1447–53. doi: 10.1136/adc.67.12.1447
47. Cochrane AM, Moussouros A, Thomsom AD, Eddleston AL, Wiiliams R. Antibody-Dependent Cell-Mediated (K Cell) Cytotoxicity Against Isolated Hepatocytes in Chronic Active Hepatitis. Lancet (1976) 1(7957):441–4. doi: 10.1016/S0140-6736(76)91472-0
48. Mix H, Weiler-Normann C, Thimme R, Ahlenstiel G, Shin EC, Herkel J, et al. Identification of CD4 T-Cell Epitopes in Soluble Liver Antigen/Liver Pancreas Autoantigen in Autoimmune Hepatitis. Gastroenterology (2008) 135(6):2107–18. doi: 10.1053/j.gastro.2008.07.029
49. Ma Y, Thomas MG, Okamoto M, Bogdanos DP, Nagl S, Kerkar N, et al. Key Residues of a Major Cytochrome P4502D6 Epitope are Located on the Surface of the Molecule. J Immunol (2002) 169(1):277–85. doi: 10.4049/jimmunol.169.1.277
50. Holz LE, Benseler V, Vo M, McGuffog C, Van Rooijen N, McCaughan GW, et al. Naive CD8 T Cell Activation by Liver Bone Marrow-Derived Cells Leads to a “Neglected” IL-2low Bimhigh Phenotype, Poor CTL Function and Cell Death. J Hepatol (2012) 57(4):830–6. doi: 10.1016/j.jhep.2012.05.015
51. Kim MT, Harty JT. Splenectomy Alters Distribution and Turnover But Not Numbers or Protective Capacity of De Novo Generated Memory CD8 T-Cells. Front Immunol (2014) 5:568. doi: 10.3389/fimmu.2014.00568
52. Buitrago-Molina LE, Dywicki J, Noyan F, Trippler M, Pietrek J, Schlue J, et al. Splenectomy Prior to Experimental Induction of Autoimmune Hepatitis Promotes More Severe Hepatic Inflammation, Production of IL-17 and Apoptosis. Biomedicines (2021) 9(1):58. doi: 10.3390/biomedicines9010058
53. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic Selection of CD4+CD25+ Regulatory T Cells Induced by an Agonist Self-Peptide. Nat Immunol (2001) 2(4):301–6. doi: 10.1038/86302
54. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T Cells: Recommendations to Simplify the Nomenclature. Nat Immunol (2013) 14(4):307–8. doi: 10.1038/ni.2554
55. Apostolou I, von Boehmer H. In Vivo Instruction of Suppressor Commitment in Naive T Cells. J Exp Med (2004) 199(10):1401–8. doi: 10.1084/jem.20040249
56. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and Expanding Regulatory T Cell Populations by Foreign Antigen. Nat Immunol (2005) 6(12):1219–27. doi: 10.1038/ni1265
57. Cederbom L, Hall H, Ivars F. CD4+CD25+ Regulatory T Cells Down-Regulate Co-Stimulatory Molecules on Antigen-Presenting Cells. Eur J Immunol (2000) 30(6):1538–43. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X
58. Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A Critical Role for Transforming Growth Factor-Beta But Not Interleukin 4 in the Suppression of T Helper Type 1-Mediated Colitis by CD45RB(low) CD4+ T Cells. J Exp Med (1996) 183(6):2669–74. doi: 10.1084/jem.183.6.2669
59. Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An Essential Role for Interleukin 10 in the Function of Regulatory T Cells That Inhibit Intestinal Inflammation. J Exp Med (1999) 190(7):995–1004. doi: 10.1084/jem.190.7.995
60. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The Inhibitory Cytokine IL-35 Contributes to Regulatory T-Cell Function. Nature (2007) 450(7169):566–9. doi: 10.1038/nature06306
61. Ren X, Ye F, Jiang Z, Chu Y, Xiong S, Wang Y. Involvement of Cellular Death in TRAIL/DR5-Dependent Suppression Induced by CD4(+)CD25(+) Regulatory T Cells. Cell Death Differ (2007) 14(12):2076–84. doi: 10.1038/sj.cdd.4402220
62. Kashio Y, Nakamura K, Abedin MJ, Seki M, Nishi N, Yoshida N, et al. Galectin-9 Induces Apoptosis Through the Calcium-Calpain-Caspase-1 Pathway. J Immunol (2003) 170(7):3631–6. doi: 10.4049/jimmunol.170.7.3631
63. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and Perforin are Important for Regulatory T Cell-Mediated Suppression of Tumor Clearance. Immunity (2007) 27(4):635–46. doi: 10.1016/j.immuni.2007.08.014
64. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ Regulatory T Cells Induce Cytokine Deprivation-Mediated Apoptosis of Effector CD4+ T Cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
65. Vaeth M, Gogishvili T, Bopp T, Klein M, Berberich-Siebelt F, Gattenloehner S, et al. Regulatory T Cells Facilitate the Nuclear Accumulation of Inducible cAMP Early Repressor (ICER) and Suppress Nuclear Factor of Activated T Cell C1 (Nfatc1). Proc Natl Acad Sci USA (2011) 108(6):2480–5. doi: 10.1073/pnas.1009463108
66. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective Suppressor Function in CD4(+)CD25(+) T-Cells From Patients With Type 1 Diabetes. Diabetes (2005) 54(1):92–9. doi: 10.2337/diabetes.54.1.92
67. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised Function of Regulatory T Cells in Rheumatoid Arthritis and Reversal by Anti-TNFalpha Therapy. J Exp Med (2004) 200(3):277–85. doi: 10.1084/jem.20040165
68. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of Functional Suppression by CD4+CD25+ Regulatory T Cells in Patients With Multiple Sclerosis. J Exp Med (2004) 199(7):971–9. doi: 10.1084/jem.20031579
69. Mao C, Wang S, Xiao Y, Xu J, Jiang Q, Jin M, et al. Impairment of Regulatory Capacity of CD4+CD25+ Regulatory T Cells Mediated by Dendritic Cell Polarization and Hyperthyroidism in Graves’ Disease. J Immunol (2011) 186(8):4734–43. doi: 10.4049/jimmunol.0904135
70. Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, et al. Dysfunctional Blood and Target Tissue CD4+CD25high Regulatory T Cells in Psoriasis: Mechanism Underlying Unrestrained Pathogenic Effector T Cell Proliferation. J Immunol (2005) 174(1):164–73. doi: 10.4049/jimmunol.174.1.164
71. Longhi MS, Hussain MJ, Mitry RR, Arora SK, Mieli-Vergani G, Vergani D, et al. Functional Study of CD4+CD25+ Regulatory T Cells in Health and Autoimmune Hepatitis. J Immunol (2006) 176(7):4484–91. doi: 10.4049/jimmunol.176.7.4484
72. Ferri S, Longhi MS, De Molo C, Lalanne C, Muratori P, Granito A, et al. A Multifaceted Imbalance of T Cells With Regulatory Function Characterizes Type 1 Autoimmune Hepatitis. Hepatology (2010) 52(3):999–1007. doi: 10.1002/hep.23792
73. Liberal R, Grant CR, Holder BS, Cardone J, Martinez-Llordella M, Ma Y, et al. In Autoimmune Hepatitis Type 1 or the Autoimmune Hepatitis-Sclerosing Cholangitis Variant Defective Regulatory T-Cell Responsiveness to IL-2 Results in Low IL-10 Production and Impaired Suppression. Hepatology (2015) 62(3):863–75. doi: 10.1002/hep.27884
74. Jeffery HC, Braitch MK, Bagnall C, Hodson J, Jeffery LE, Wawman RE, et al. Changes in Natural Killer Cells and Exhausted Memory Regulatory T Cells With Corticosteroid Therapy in Acute Autoimmune Hepatitis. Hepatol Commun (2018) 2(4):421–36. doi: 10.1002/hep4.1163
75. Peiseler M, Sebode M, Franke B, Wortmann F, Schwinge D, Quaas A, et al. FOXP3+ Regulatory T Cells in Autoimmune Hepatitis are Fully Functional and Not Reduced in Frequency. J Hepatol (2012) 57(1):125–32. doi: 10.1016/j.jhep.2012.02.029
76. Taubert R, Hardtke-Wolenski M, Noyan F, Wilms A, Baumann AK, Schlue J, et al. Intrahepatic Regulatory T Cells in Autoimmune Hepatitis are Associated With Treatment Response and Depleted With Current Therapies. J Hepatol (2014) 61(5):1106–14. doi: 10.1016/j.jhep.2014.05.034
77. Lapierre P, Beland K, Yang R, Alvarez F. Adoptive Transfer of Ex Vivo Expanded Regulatory T Cells in an Autoimmune Hepatitis Murine Model Restores Peripheral Tolerance. Hepatology (2013) 57(1):217–27. doi: 10.1002/hep.26023
78. Yuksel M, Wang Y, Tai N, Peng J, Guo J, Beland K, et al. A Novel “Humanized Mouse” Model for Autoimmune Hepatitis and the Association of Gut Microbiota With Liver Inflammation. Hepatology (2015) 62(5):1536–50. doi: 10.1002/hep.27998
79. Arterbery AS, Osafo-Addo A, Avitzur Y, Ciarleglio M, Deng Y, Lobritto SJ, et al. Production of Proinflammatory Cytokines by Monocytes in Liver-Transplanted Recipients With De Novo Autoimmune Hepatitis Is Enhanced and Induces TH1-Like Regulatory T Cells. J Immunol (2016) 196(10):4040–51. doi: 10.4049/jimmunol.1502276
80. Arterbery AS, Yao J, Ling A, Avitzur Y, Martinez M, Lobritto S, et al. Inflammasome Priming Mediated via Toll-Like Receptors 2 and 4, Induces Th1-Like Regulatory T Cells in De Novo Autoimmune Hepatitis. Front Immunol (2018) 9:1612. doi: 10.3389/fimmu.2018.01612
81. Preti M, Schlott L, Lubbering D, Krzikalla D, Muller AL, Schuran FA, et al. Failure of Thymic Deletion and Instability of Autoreactive Tregs Drive Autoimmunity in Immune-Privileged Liver. JCI Insight (2021) 6(6):e141462. doi: 10.1172/jci.insight.141462
82. Chen J, Liu W, Zhu W. Foxp3(+) Treg Cells Are Associated With Pathological Process of Autoimmune Hepatitis by Activating Methylation Modification in Autoimmune Hepatitis Patients. Med Sci Monit (2019) 25:6204–12. doi: 10.12659/MSM.915408
83. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 Cell Differentiation by the Aryl Hydrocarbon Receptor. Nature (2008) 453(7191):65–71. doi: 10.1038/nature06880
84. Ohashi H, Nishioka K, Nakajima S, Kim S, Suzuki R, Aizaki H, et al. The Aryl Hydrocarbon Receptor-Cytochrome P450 1A1 Pathway Controls Lipid Accumulation and Enhances the Permissiveness for Hepatitis C Virus Assembly. J Biol Chem (2018) 293(51):19559–71. doi: 10.1074/jbc.RA118.005033
85. She C, Wang J, Tang N, Liu Z, Xu L, Liu B. Effects of 2,3,7,8-Tetrachlorodibenzo-P-Dioxin on T Cell Differentiation in Primary Biliary Cholangitis. BioMed Res Int (2020) 2020:1754975. doi: 10.1155/2020/1754975
86. Probert PM, Leitch AC, Dunn MP, Meyer SK, Palmer JM, Abdelghany TM, et al. Identification of a Xenobiotic as a Potential Environmental Trigger in Primary Biliary Cholangitis. J Hepatol (2018) 69(5):1123–35. doi: 10.1016/j.jhep.2018.06.027
87. Selmi C, Balkwill DL, Invernizzi P, Ansari AA, Coppel RL, Podda M, et al. Patients With Primary Biliary Cirrhosis React Against a Ubiquitous Xenobiotic-Metabolizing Bacterium. Hepatology (2003) 38(5):1250–7. doi: 10.1053/jhep.2003.50446
88. D’Onofrio F, Renga G, Puccetti M, Pariano M, Bellet MM, Santarelli I, et al. Indole-3-Carboxaldehyde Restores Gut Mucosal Integrity and Protects From Liver Fibrosis in Murine Sclerosing Cholangitis. Cells (2021) 10(7):1622. doi: 10.3390/cells10071622
89. Lytton SD, Osiecki M, Malgorzata W, Cukrowska B, Wierzbicka A, Goliszek M, et al. Tryptophan-Kynurenine Profile in Pediatric Autoimmune Hepatitis. Immunol Res (2019) 67(1):39–47. doi: 10.1007/s12026-019-9068-1
90. Mohammadi-Bardbori A, Akbarizadeh AR, Delju F, Rannug A. Chromatin Remodeling by Curcumin Alters Endogenous Aryl Hydrocarbon Receptor Signaling. Chem Biol Interact (2016) 252:19–27. doi: 10.1016/j.cbi.2016.03.037
91. Blobe GC, Schiemann WP, Lodish HF. Role of Transforming Growth Factor Beta in Human Disease. N Engl J Med (2000) 342(18):1350–8. doi: 10.1056/NEJM200005043421807
92. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
93. Longhi MS, Vuerich M, Kalbasi A, Kenison JE, Yeste A, Csizmadia E, et al. Bilirubin Suppresses Th17 Immunity in Colitis by Upregulating CD39. JCI Insight (2017) 2(9):e92791. doi: 10.1172/jci.insight.92791
94. Xie A, Robles RJ, Mukherjee S, Zhang H, Feldbrugge L, Csizmadia E, et al. HIF-1alpha-Induced Xenobiotic Transporters Promote Th17 Responses in Crohn’s Disease. J Autoimmun (2018) 94:122–33. doi: 10.1016/j.jaut.2018.07.022
95. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic Control of Type 1 Regulatory T Cell Differentiation by AHR and HIF1-Alpha. Nat Med (2015) 21(6):638–46. doi: 10.1038/nm.3868
96. Longhi MS, Meda F, Wang P, Samyn M, Mieli-Vergani G, Vergani D, et al. Expansion and De Novo Generation of Potentially Therapeutic Regulatory T Cells in Patients With Autoimmune Hepatitis. Hepatology (2008) 47(2):581–91. doi: 10.1002/hep.22071
97. Longhi MS, Liberal R, Holder B, Robson SC, Ma Y, Mieli-Vergani G, et al. Inhibition of Interleukin-17 Promotes Differentiation of CD25(-) Cells Into Stable T Regulatory Cells in Patients With Autoimmune Hepatitis. Gastroenterology (2012) 142(7):1526–35 e6. doi: 10.1053/j.gastro.2012.02.041
98. Liberal R, Grant CR, Yuksel M, Graham J, Kalbasi A, Ma Y, et al. Regulatory T-Cell Conditioning Endows Activated Effector T Cells With Suppressor Function in Autoimmune Hepatitis/Autoimmune Sclerosing Cholangitis. Hepatology (2017) 66(5):1570–84. doi: 10.1002/hep.29307
99. Zhao H, Bo C, Kang Y, Li H. What Else Can CD39 Tell Us? Front Immunol (2017) 8:727. doi: 10.3389/fimmu.2017.00727
100. Oo YH, Ackrill S, Cole R, Jenkins L, Anderson P, Jeffery HC, et al. Liver Homing of Clinical Grade Tregs After Therapeutic Infusion in Patients With Autoimmune Hepatitis. JHEP Rep (2019) 1(4):286–96. doi: 10.1016/j.jhepr.2019.08.001
101. Hardtke-Wolenski M, Taubert R, Noyan F, Sievers M, Dywicki J, Schlue J, et al. Autoimmune Hepatitis in a Murine Autoimmune Polyendocrine Syndrome Type 1 Model is Directed Against Multiple Autoantigens. Hepatology (2015) 61(4):1295–305. doi: 10.1002/hep.27639
102. Longhi MS, Hussain MJ, Kwok WW, Mieli-Vergani G, Ma Y, Vergani D. Autoantigen-Specific Regulatory T Cells, a Potential Tool for Immune-Tolerance Reconstitution in Type-2 Autoimmune Hepatitis. Hepatology (2011) 53(2):536–47. doi: 10.1002/hep.24039
103. Holder BS, Grant CR, Liberal R, Ma Y, Heneghan MA, Mieli-Vergani G, et al. Retinoic Acid Stabilizes Antigen-Specific Regulatory T-Cell Function in Autoimmune Hepatitis Type 2. J Autoimmun (2014) 53:26–32. doi: 10.1016/j.jaut.2014.02.001
104. Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J, et al. Suppression of a Broad Spectrum of Liver Autoimmune Pathologies by Single Peptide-MHC-Based Nanomedicines. Nat Commun (2019) 10(1):2150. doi: 10.1038/s41467-019-09893-5
105. Raffin C, Vo LT, Bluestone JA. Treg Cell-Based Therapies: Challenges and Perspectives. Nat Rev Immunol (2020) 20(3):158–72. doi: 10.1038/s41577-019-0232-6
106. Diestelhorst J, Junge N, Schlue J, Falk CS, Manns MP, Baumann U, et al. Pediatric Autoimmune Hepatitis Shows a Disproportionate Decline of Regulatory T Cells in the Liver and of IL-2 in the Blood of Patients Undergoing Therapy. PloS One (2017) 12(7):e0181107. doi: 10.1371/journal.pone.0181107
107. Jeffery HC, Jeffery LE, Lutz P, Corrigan M, Webb GJ, Hirschfield GM, et al. Low-Dose Interleukin-2 Promotes STAT-5 Phosphorylation, Treg Survival and CTLA-4-Dependent Function in Autoimmune Liver Diseases. Clin Exp Immunol (2017) 188(3):394–411. doi: 10.1111/cei.12940
108. Lim TY, Martinez-Llordella M, Kodela E, Gray E, Heneghan MA, Sanchez-Fueyo A. Low-Dose Interleukin-2 for Refractory Autoimmune Hepatitis. Hepatology (2018) 68(4):1649–52. doi: 10.1002/hep.30059
109. Spangler JB, Trotta E, Tomala J, Peck A, Young TA, Savvides CS, et al. Engineering a Single-Agent Cytokine/Antibody Fusion That Selectively Expands Regulatory T Cells for Autoimmune Disease Therapy. J Immunol (2018) 201(7):2094–106. doi: 10.4049/jimmunol.1800578
110. Buitrago-Molina LE, Pietrek J, Noyan F, Schlue J, Manns MP, Wedemeyer H, et al. Treg-Specific IL-2 Therapy can Reestablish Intrahepatic Immune Regulation in Autoimmune Hepatitis. J Autoimmun (2021) 117:102591. doi: 10.1016/j.jaut.2020.102591
111. Karakus U, Sahin D, Mittl PRE, Mooij P, Koopman G, Boyman O. Receptor-Gated IL-2 Delivery by an Anti-Human IL-2 Antibody Activates Regulatory T Cells in Three Different Species. Sci Transl Med (2020) 12(574):eabb9283. doi: 10.1126/scitranslmed.abb9283
112. Marceau G, Yang R, Lapierre P, Beland K, Alvarez F. Low-Dose Anti-CD3 Antibody Induces Remission of Active Autoimmune Hepatitis in Xenoimmunized Mice. Liver Int (2015) 35(1):275–84. doi: 10.1111/liv.12498
113. Beland K, Marceau G, Labardy A, Bourbonnais S, Alvarez F. Depletion of B Cells Induces Remission of Autoimmune Hepatitis in Mice Through Reduced Antigen Presentation and Help to T Cells. Hepatology (2015) 62(5):1511–23. doi: 10.1002/hep.27991
114. Arvaniti P, Giannoulis G, Gabeta S, Zachou K, Koukoulis GK, Dalekos GN. Belimumab is a Promising Third-Line Treatment Option for Refractory Autoimmune Hepatitis. JHEP Rep (2020) 2(4):100123. doi: 10.1016/j.jhepr.2020.100123
115. Robles RJ, Mukherjee S, Vuerich M, Xie A, Harshe R, Cowan PJ, et al. Modulation of CD39 and Exogenous APT102 Correct Immune Dysfunction in Experimental Colitis and Crohn’s Disease. J Crohns Colitis (2020) 14(6):818–30. doi: 10.1093/ecco-jcc/jjz182
116. Moeckel D, Jeong SS, Sun X, Broekman MJ, Nguyen A, Drosopoulos JH, et al. Optimizing Human Apyrase to Treat Arterial Thrombosis and Limit Reperfusion Injury Without Increasing Bleeding Risk. Sci Transl Med (2014) 6(248):248ra105. doi: 10.1126/scitranslmed.3009246
117. Ji Y, Adeola O, Strawn TL, Jeong SS, Chen R, Fay WP. Recombinant Soluble Apyrase APT102 Inhibits Thrombosis and Intimal Hyperplasia in Vein Grafts Without Adversely Affecting Hemostasis or Re-Endothelialization. J Thromb Haemost (2017) 15(4):814–25. doi: 10.1111/jth.13621
118. Sun X, Han L, Seth P, Bian S, Li L, Csizmadia E, et al. Disordered Purinergic Signaling and Abnormal Cellular Metabolism are Associated With Development of Liver Cancer in Cd39/ENTPD1 Null Mice. Hepatology (2013) 57(1):205–16. doi: 10.1002/hep.25989
119. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 Expression by CD4+Foxp3+ Regulatory T Cells Promotes Hepatic Metastatic Tumor Growth in Mice. Gastroenterology (2010) 139(3):1030–40. doi: 10.1053/j.gastro.2010.05.007
120. Allard D, Allard B, Stagg J. On the Mechanism of Anti-CD39 Immune Checkpoint Therapy. J Immunother Cancer (2020) 8(1):e000186. doi: 10.1136/jitc-2019-000186
121. Moesta AK, Li XY, Smyth MJ. Targeting CD39 in Cancer. Nat Rev Immunol (2020) 20(12):739–55. doi: 10.1038/s41577-020-0376-4
122. Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PloS Pathog (2015) 11(10):e1005177. doi: 10.1371/journal.ppat.1005177
123. Lee JS, Yilmaz O. Unfolding Role of a Danger Molecule Adenosine Signaling in Modulation of Microbial Infection and Host Cell Response. Int J Mol Sci (2018) 19(1):199. doi: 10.3390/ijms19010199
Keywords: autoimmune liver disease, Treg, Th17, ectonucleotidase, aryl hydrocarbon receptor
Citation: Vuerich M, Wang N, Kalbasi A, Graham JJ and Longhi MS (2021) Dysfunctional Immune Regulation in Autoimmune Hepatitis: From Pathogenesis to Novel Therapies. Front. Immunol. 12:746436. doi: 10.3389/fimmu.2021.746436
Received: 23 July 2021; Accepted: 10 September 2021;
Published: 28 September 2021.
Edited by:
Urs Christen, Goethe University Frankfurt, GermanyReviewed by:
Matthias Hardtke-Wolenski, Hannover Medical School, GermanyPatrick Leung, University of California, Davis, United States
Copyright © 2021 Vuerich, Wang, Kalbasi, Graham and Longhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Serena Longhi, bWxvbmdoaUBiaWRtYy5oYXJ2YXJkLmVkdQ==
†These authors have contributed equally to this work and share first authorship