 David W. Scott
David W. Scott- Department of Medicine (MED), Uniformed Services University of the Health Sciences, Bethesda, MD, United States
Chimeric antigen receptor (CAR) transduced T cells have significantly improved cancer immunotherapy. Similarly, engineering regulatory T cells (Treg) with specific receptors to endow specificity and increase efficacy of Tregs holds great promise for therapy of a variety of adverse immune responses. In this review, we focus on our approaches using retroviral transduction of specific T-cell receptors, single chain variable fragments (scFv) or antigen in models of monogenic diseases, autoimmunity and allergy. The advantages of each of these for different targets diseases are discussed as well as their potential for clinical translation.
Introduction
Nearly five decades ago, Gershon and colleagues at Yale proposed that immune responses can be controlled by a subset of T cells called “suppressor cells” that downregulate other lymphoid cells (1). At that time, immunologists lacked the reagents to fully characterize these suppressors and their mode of action other than cell mixing experiments. Ironically, the latter approach still remains a sine qua non to demonstrate their efficacy. Armed with flow cytometry and molecular biology approaches, including the discovery of FOXP3 and its association with human immunodeficiency and autoimmune diseases (2–5), suppressor cells were replaced by a well-defined entity, “regulatory T cells” (Tregs) (2, 6). Similarly, these cells have the ability to suppress a variety of immune responses in vitro and in vivo.
Clinical trials with expanded Tregs were initiated over a decade ago in transplantation and autoimmunity, as summarized in Romano et al. (7). While these cells have been used safely in multiple clinical trials, they are polyclonal and the frequency of specific Tregs is very low. Expanded Tregs express a broad repertoire of specificities, and have the potential to be non-specifically immunosuppressive (8). To overcome the latter issue and the rare frequency of specific T cells, our lab has focused the use of specifically engineered Tregs to suppress adverse immune responses in monogenic diseases, autoimmunity and allergy. Our studies are based on the seminal studies of Eshhar and colleagues, who first demonstrated expression of specific receptors in T cells (9). These pioneering studies have been successfully applied using single chain antibody fragments (scFv) in cancer immunotherapy worldwide (10, 11). A major example is the use of anti-CD20 engineered human T cells in the successful treatment of leukemia (10). The first successfully use of specific Tregs used expanded FoxP3-expressing transgenic T cells in an autoimmune model of multiple sclerosis (12). Since that time, multiple laboratories have made significant contributions by engineering specificity into murine and/or human Tregs (13–25). The purpose of this manuscript is to highlight our approaches in the context of this rapidly developing field focusing on targeting specific adverse responses.
Specificity in our lab has been achieved by engineering Tregs to express receptors that can recognize the targets of adverse immune responses. Thus, we have applied this protocol in autoimmunity, hemophilia A and allergy. To achieve this goal, we have used retroviral transduction of cloned T-cell receptors (TCR’s), scFv’s or antigen domains in thymic-derived human natural regulatory T cells (see Figure 1). In this review, we describe the basic principles and progress in each of these efforts by ourselves in three disease models to achieve the ultimate goal of modulating adverse human diseases (26–31). We also summarize the advantages and disadvantages of these approaches below and in Table 1. We have used retroviral transduction of Tregs with CD3 and CD28 signaling domains as the basic version; efforts to modify the signaling process has been reviewed by others (16, 32).
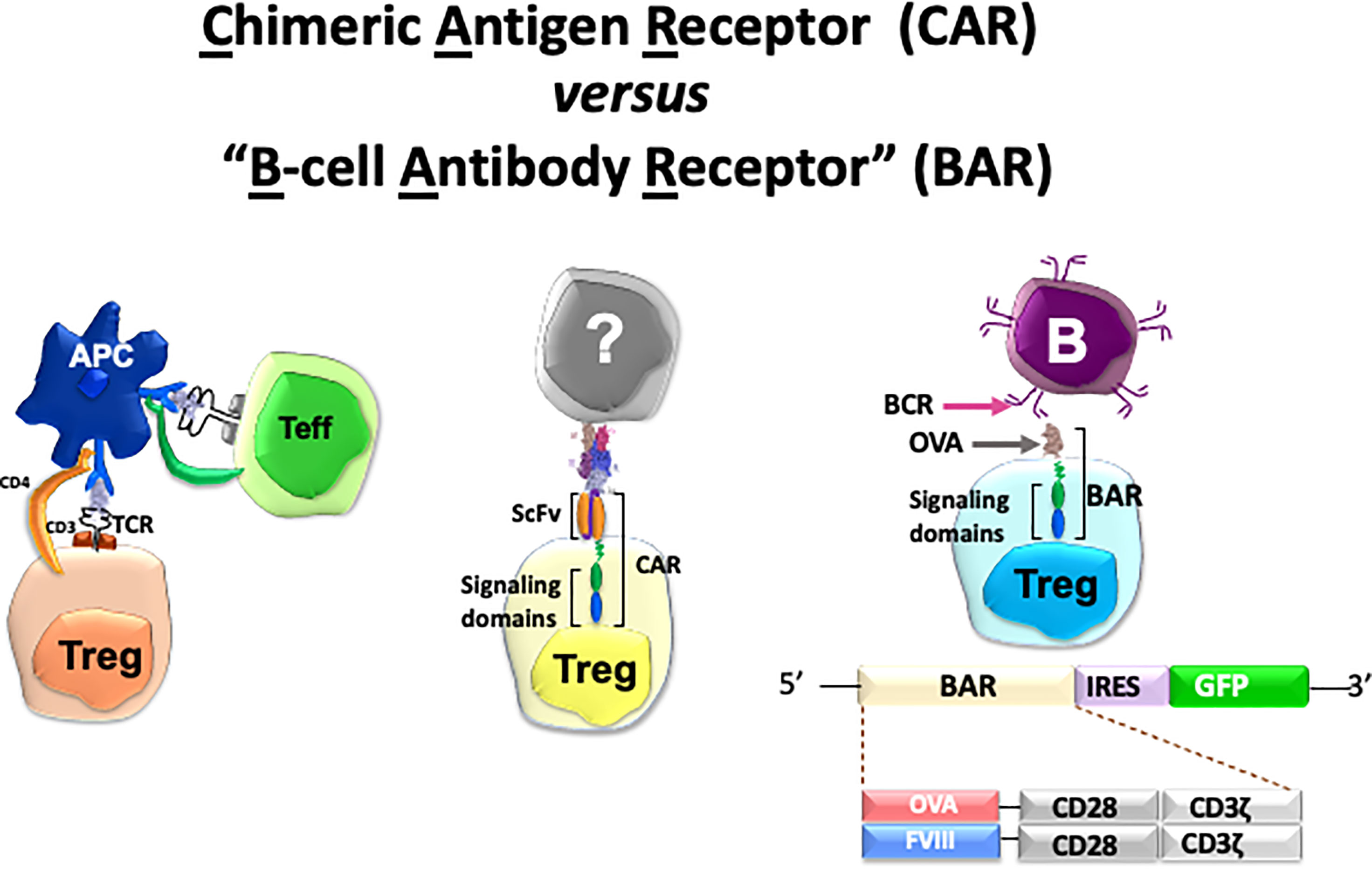
Figure 1 Cartoon of three types of specific Tregs and potential targets used in our lab. See Table 1 for details.
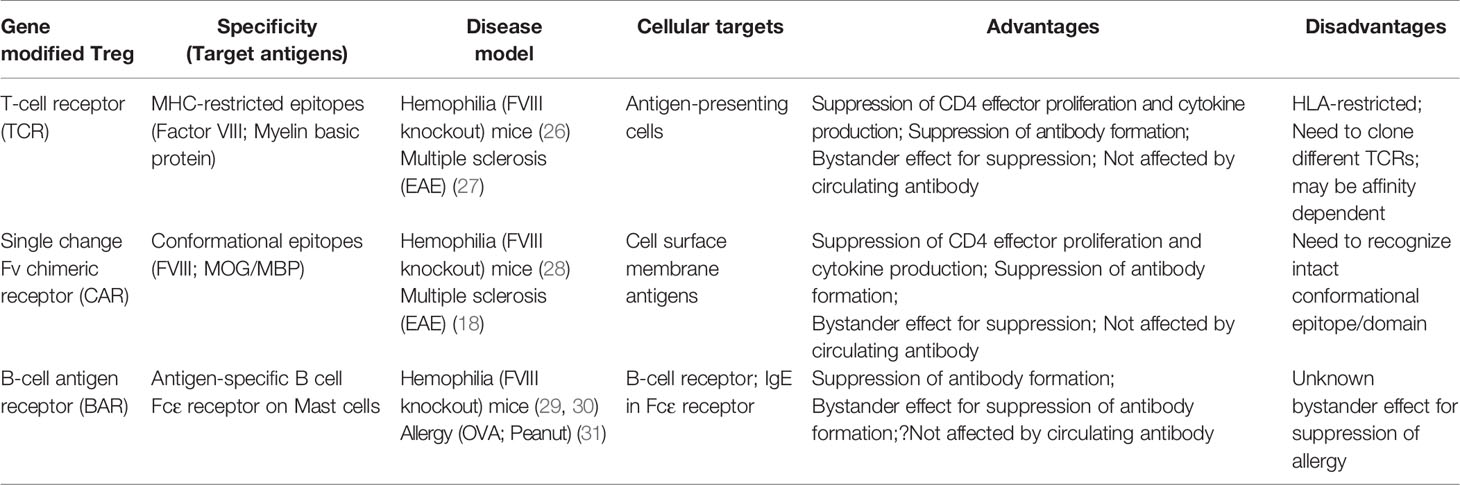
Table 1 Advantages and disadvantages of engineered Treg approaches.
Choosing Targets, Choosing Receptors, Establishing Collaboration
Typical pharmacologic therapies for adverse immune responses are broadly immunosuppressive. An issue to achieve any specific tolerogenic therapy is the choice of targets. Indeed, many adverse responses have multiple targets, with no unique or specific antigen being attacked. Moreover, any targeted antigen may have a variety of T-cell and B-cell epitopes. Thus, the challenge in rendering Treg cell-based therapy specific depends on multiple factors: knowing the target antigen and having the appropriate receptors. This is easier in monogenic diseases wherein the target antigen is known as patients lacking this protein often produce antibodies to the missing antigen when treated therapeutically. Patients are not tolerant to the missing protein and treat the therapeutic protein as a foreign antigen. Such is the case in hemophilia A (HA), an X-linked disease with a frequency of 1 in 5000 males in which patients have mutations in the F8 gene encoding pro-coagulant Factor VIII (FVIII). Approximately 30% of HA patients develop high-titer neutralizing antibodies against therapeutic FVIII following repeated infusions of this needed protein that inhibit the function of this life saving therapy (33, 34). Most of these inhibitors block FVIII activity by binding to two immunodominant domains, called C2 and A2, which are important for FVIII’s pro-coagulant activity.
In 2012, Yongchan Kim joined my lab after a successful post-doctoral fellowship with Ethan Shevach, a Treg expert at NIH. At the same time, Kathleen Pratt joined the faculty in our department. Dr. Pratt had cloned several T-cell lines from HA patients (35, 36), and one of these T cell clones, called 17195, recognized an HLA-restricted peptide in the C2 domain of FVIII, residues 2194-2210 (35, 37). With a determination of the TCR V regions, Yongchan then inserted them into a retroviral vector and used the vector to transduce FACS-purified human Tregs (see ref. (27) for detailed methods). Purified Tregs were CD25high and CD127low and typically expressed FOXP3 and Helios transcription factors. Although less than 20% of the initially transduced Tregs expressed the 17195 T-cell receptor (TCR), this population expanded upon stimulation with the FVIII 2194-2210 peptide on HLA DR1 antigen-presenting cells (26). This stimulation also led to an increased expression in FOXP3 and Helios transcription factor, markers that confirmed the expansion of Tregs in culture. Importantly, these expanded 17195-expressing Tregs suppressed the proliferation and cytokine production by FVIII-specific T effector cells even more effectively than polyclonal Tregs in vitro. Moreover, these expanded human 17195-expressing Tregs also suppressed the secondary antibody response of murine HLA-DR1 transgenic spleen cells stimulated with FVIII in vitro. Overall, human 17195-expressing Tregs were able to effectively suppress the antibody response to FVIII in vivo, even across a xenogeneic barrier, although they were rejected within two to three weeks (26). Notably, while human 17195-expressing Tregs were specific for a single peptide in the C2 domain of FVIII, they suppressed antibody formation against the multiple epitopes in the entire FVIII protein. This suggested that suppression, while “specific” for a single FVIII peptide, may have bystander effects on the response to other epitopes in FVIII locally, presumably at the level of the antigen-presenting cells.
TCR engineered Tregs are MHC-restricted, which limits their utility to HLA-matched donor recipient combinations and limited repertoires. To generate antigen-specific Tregs that were not MHC-restricted, Jeong-Heon Yoon collaborated with Christoph Königs and Anja (ne’ Naumann) Schmidt in Frankfurt. Anja had isolated several single chain (sc)Fv’s that recognized FVIII domains (38) and cloned them into our standard retroviral vector for expression in Tregs. The human Tregs transduced with retroviral vector encoding scFv recognized a conformational epitope in the A2 region of FVIII (38). When these scFv-expressing human Tregs were mixed with spleen cells from mice immunized with FVIII, they suppressed the secondary response in vitro, and blocked antibody formation in vivo in hemophilic mice. Compared to TCR-engineered Tregs, the scFv-transduced Tregs suppressed the anti-FVIII immune response to the same degree at certain ratios (28). It is worth noting, then, that engineered Tregs specific for the MHC-restricted peptide in the C2 domain of FVIII and Tregs engineered to express the scFv directed at the A2 domain of FVIII both suppressed the immune response to other domains in FVIII. Thus suggesting that both types of antigen-specific Tregs exerted bystander suppression to other epitopes in the same target protein locally, but they did not non-specifically suppress the response to an unrelated antigen, e.g., immunization with TNP hapten-conjugated red blood cells (28). Application of scFv-transduced Tregs in a transplant model was elegantly demonstrated in the Levings’ lab (15), thus demonstrating further application of engineered Tregs for tolerance.
Treg Effects in Autoimmunity
T-cell responses against self-proteins can result in a spectrum of autoimmune diseases. Unlike monogenic diseases in which the target antigens are known, multiple potential antigens and epitopes may be recognized in autoimmunity. For instance, several antigens have been identified in Type 1 diabetes including islet antigens such as glutamic acid decarboxylase or insulin. In the central nervous system (CNS), myelin basic protein (MBP) or myelin oligodendrocyte glycoprotein (MOG) can be targeted in multiple sclerosis (MS). Based on our success and the bystander effect in hemophilia with an engineered TCR recognizing FVIII, we hypothesized that transduction of an MBP-specific TCR into expanded human Tregs might be an effective therapy in MS. In collaboration with Kai Wucherpfennig at Harvard, we engineered human Tregs to express the Ob2F3 TCR V regions that targeted an immunodominant MBP peptide, p85-99 (39). Ob2F3-expressing Tregs from either healthy adults or MS patients could suppress MBP-specific T effector cells, but were also able to suppress T cells with other specificities after Tregs had been activated through the TCR (27). Surprisingly, these Ob2F3-expressing Tregs which were MBP-specific ameliorated EAE in MOG-immunized DR15 transgenic mice (22). This suggested that bystander suppression in vivo might be associated with soluble factors, enhanced by cell contact between Tregs and effectors (27). These results indicated that engineered MBP-specific Tregs were able to suppress autoimmune pathology in EAE.
An important question was whether Tregs engineered with scFv’s will work in autoimmunity. To answer this question, Anja Schmidt provided two single chain Fv’s reactive with MBP and MOG, and we showed that these cells recognized murine CNS tissue. Alessandra Pohl in my lab then cloned these into constructs in our retroviral vector for Treg transduction. Results showed that a mixture of human Tregs expressing these CNS “specific” scFv’s were able to suppress EAE in vivo similar to the TCR-expressing Tregs (18); however, direct comparison of the TCR- versus scFv-transduced Tregs has not yet been performed. Nevertheless, these important results indicate that engineered CNS targeting CAR-Tregs have the potential to be used as a cellular therapy for MS patients.
Possible Mechanisms of Suppression by Engineered Tregs
Using an ingenious version of a transwell system in which neighboring microtiter well liquid contents were connected via an opening above the cell layer (Figure 2), Kim showed that suppression of effector T cells in one well only occurred when the neighboring well contained both Tregs and effector T cells. Thus, contact between Tregs and effector cells in one well led to suppression of effector T cells not in direct contact in a neighboring microtiter well. Based on the observation that IL-2 signaling via interaction with CD25 can lead to STAT5 phosphorylation, we were able to show in kinetic studies that phospho-STAT5 decreased in effector CD4 T cells at the same time it was increasing in engineered Tregs (27). These data led us to propose that, similar to classical Tregs, engineered Tregs were able to capture IL-2 produced by effector T cells upon antigen presentation in the local milieu. Overall, these results indicate that the induction of engineered Treg growth led to the activation of Tregs and release of inhibitory cytokines which mediated the bystander suppressive effect observed in vivo.
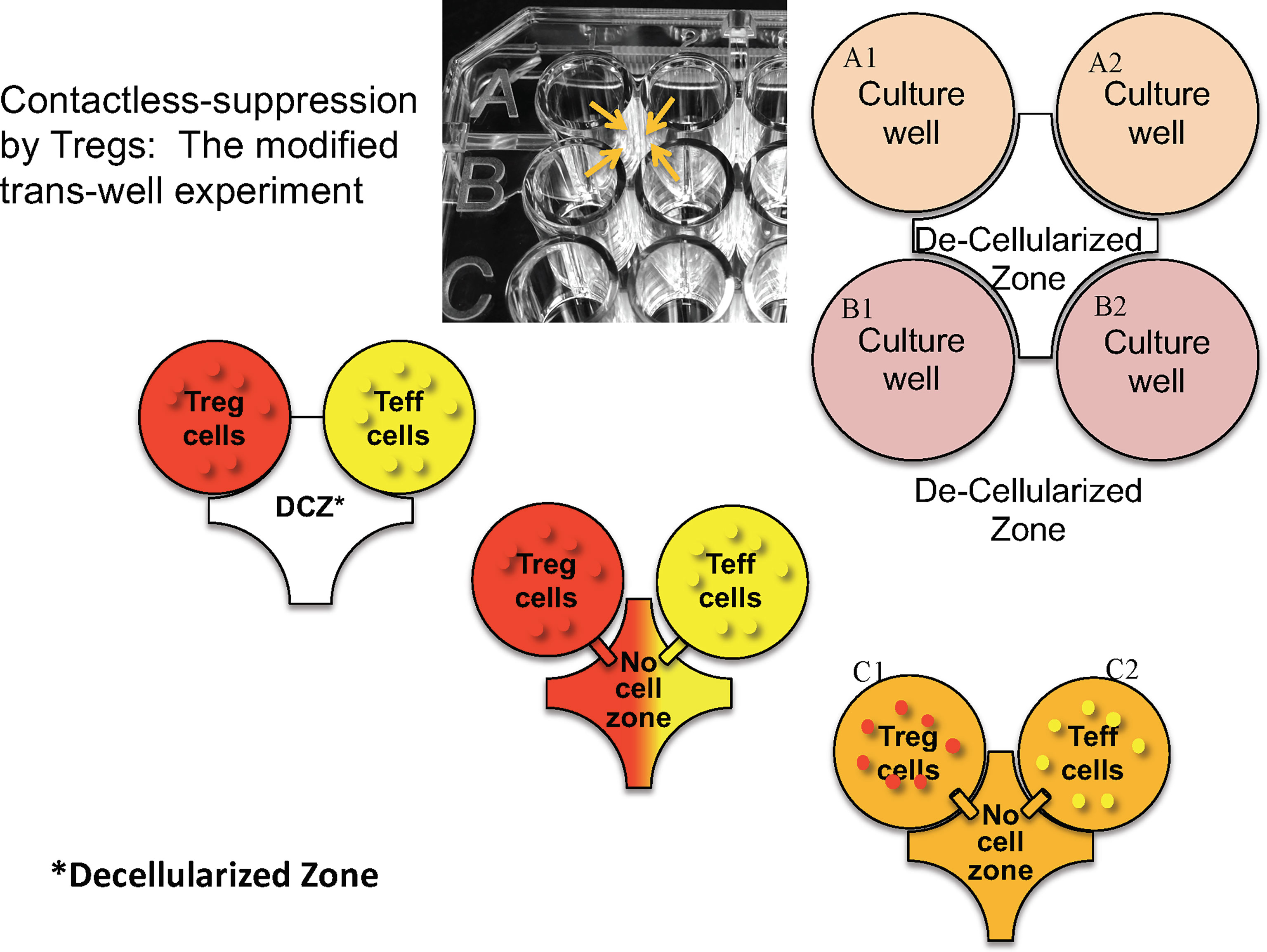
Figure 2 Design of microtiter plate to test whether direct cell contact is necessary for the bystander effect. Tregs and T effectors (Teff) and placed in the same or contiguous wells with a hole created above the bottom of the wells (arrows) so that fluid but no cells can migrate. “Decellularized” zone refers to the space between the four wells into which soluble products can diffuse to neighboring wells.
BAR Tregs to Target B Cells in Adverse Immune Responses
Our results suggested that specific Tregs may block adverse immune responses at the level of the antigen-presenting cells. While we knew that adverse inhibitory antibody responses that can occur in monogenic diseases were highly T-helper cell dependent, the culprits were alloreactive B cells. We reasoned that generating specific T cells expressing antigen should be recognized by (or recognize) B cells via their immunoglobulin receptors. Moreover, cytotoxic CAR CD8 T cells could kill specific B cells recognizing the expressed antigen as shown in pemphigus by the Payne group at Penn (20). Kalpana Parvathaneni in my lab, engineered human and murine cytotoxic T cells with FVIII antigen to target the B-cell antibody receptor (BAR) and showed that these BAR-engineered T cells were capable of killing FVIII-reactive B-cell hybridomas in vitro and in vivo (40). In addition, adoptive transfer of FVIII A2- and C2-BAR CD8 T cells significantly reduced the anti-FVIII antibody formation in hemophilic mice. These data suggest that BAR-engineered T cells are a promising approach for future prophylactic treatment for patients with severe hemophilia A who are at high risk of developing inhibitors.
Aihong (Allan) Zhang in the lab also demonstrated human BAR Tregs, which express only the C2 domain of FVIII, suppressed the secondary response of spleen cells from FVIII-immunized mice in vitro, thus confirming the bystander effect mediated by engineered Tregs (29). Moreover, FVIII-BAR Tregs also suppressed the primary response in vivo, and reduced priming to FVIII as evidenced by lower secondary antibody titers upon boosting. It is important to note that FVIII antigen domains on the surface of Tregs (or CD8 T cells) might be blocked by circulating antibody to immunodominant domains in immunized mice, thus competing with B cell targeting. However, this did not appear to be a major problem at least with low titered antibodies, as was shown in collaboration with Shiva Venkatesha (19), as well as by the Payne group in their model (20). Moreover, crosslinking the expressed domains on human T cells may even lead to expansion of these T cells.
Can BAR Tregs Work in Allergy Models?
Based on the success of BAR Tregs in hemophilia, we sought to test them in an allergy model, the IgE response to ovalbumin (OVA). We hypothesized that expression of OVA in Tregs would target the OVA-specific B cells as was done for FVIII-specific B cells in hemophilia. Maha Abdeladhim immunized mice with OVA in alum, eliciting both IgE and IgG responses, and treated OVA-primed mice with OVA-BAR Tregs. Forty-eight hours later, mice were challenged with a high dose of OVA intravenously to induce anaphylaxis as measured by an acute drop in body temperature of 2-6°C. Results showed that OVA-BAR Tregs, human or mouse, blunted this temperature drop, an effect that persisted for at least one month (31). Surprisingly, no significant drop in IgE titer in primed and Treg-treated mice was observed within two weeks. However, we predict that pretreatment with BAR Tregs would prevent sensitization as it does in the hemophilia model.
We then investigated whether OVA-BAR Treg treatment targeted mast cells loaded with IgE (see Figure 3, right side). To this end, Maha passively sensitized naive Balb/c mice with anti-OVA IgE and then injected mice with OVA-BAR Tregs. To our surprise and pleasure, these OVA-BAR Tregs were very effective at blocking passive anaphylaxis, thus implicating mast cells as an important target in this system (31). Notably, BAR Tregs alone did not cause detectable release of allergic mediators despite expressing the target antigen on their surface. While the mechanism for BAR Tregs blocking anaphylaxis but not eliciting a reaction per se is unknown, we currently attribute this to stoichiometric differences in antigen doses. Because mast cells in patients are loaded with IgE of diverse specificities, we are currently investigating whether specific BAR Treg activity can “desensitize” to other antigens for which the mast cells are sensitized, using a peanut allergy model.
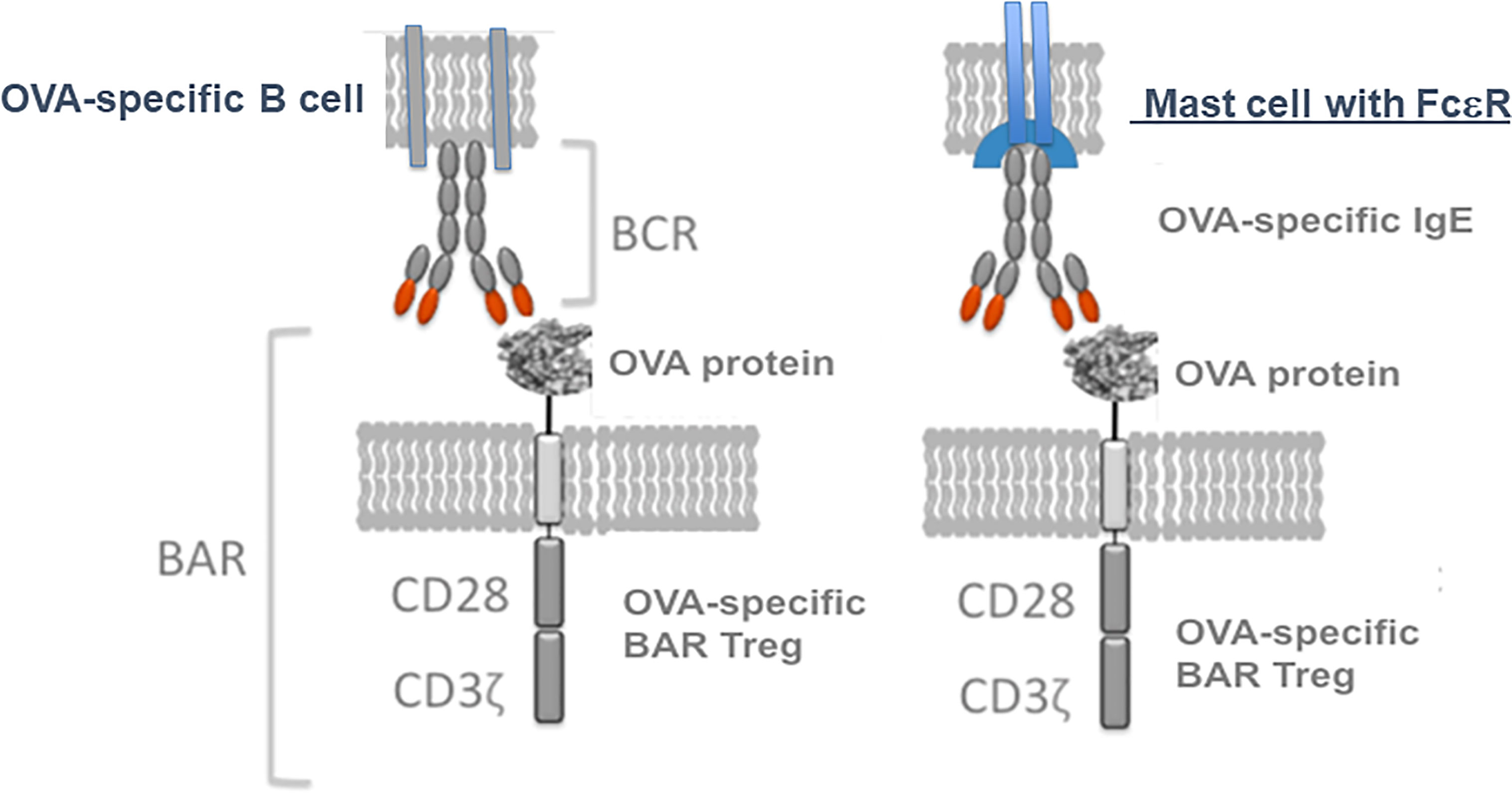
Figure 3 Model for BAR Tregs interaction not only with the immunoglobulin receptor on B cells, but also Ig(E) bound via FcεR on mast cells. Tregs expressing an antigen like ovalbumin (OVA) can interact with either specific B cell IgM (left, the BCR) or IgE bound to mast cells (right, in sensitized individuals).
Summary and Conclusions
The studies in my lab and others (13, 15, 16) have demonstrated that Tregs can be made specific to treat a variety of adverse immune responses. The choice of which specific receptor to employ will depend on the targeted disease. For example, TCR-engineered Tregs, while highly specific, are limited by MHC diversity and by the knowledge of the variable regions of the receptor. On the other hand, scFv (CAR)-Tregs require identification of a conformational epitope in a disease. The BAR Tregs are a choice in antibody-mediated diseases as they provide a clear target, the Ig receptors recognizing antigen. Our approaches have focused on the extracellular receptor, whereas other laboratories have attempted to increase Treg signaling, with variable success (16, 41). We hope our data will be followed up by multiple efforts to increase efficacy, as well as with clinical trials to prove their efficacy and safety in the future as depicted in Figure 4. Efforts to create “off the shelf” generic Tregs using CRISPR engineering should also prove fruitful. The future is bright and we hope that our approaches provide the framework for Treg treatments for adverse immune responses.
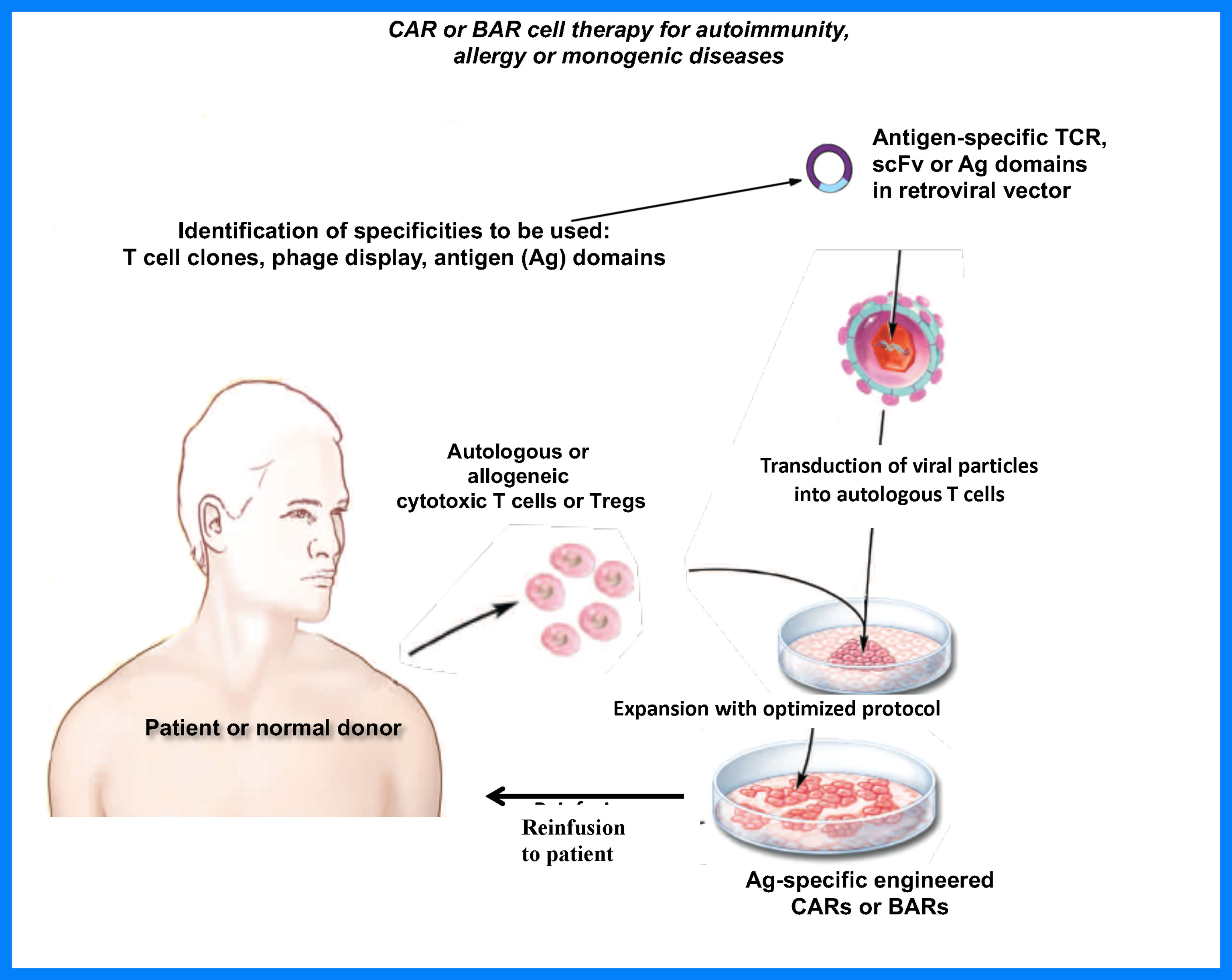
Figure 4 Proposed engineered Treg therapy in patients.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Author Disclaimer
The opinions expressed within are those solely of the author and do not necessarily reflect the official policy or position of the Uniformed Services University or the Department of Defense of the US government.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The studies reported here were supported by grants from the NIH (R01 HL126727, R01 HL061883, R21 HL127495 and R21 AI146687), the National Multiple Sclerosis Society and Pfizer. I am grateful for all of my colleagues, whose work is cited above, who performed these experiments or provided valuable reagents, suggestions for slides and (well-needed) advice, and to Kathleen Pratt for a critical review of the manuscript.
References
2. Sakaguchi S, Takahashi T, Nishizuka Y. Study on Cellular Events in Postthymectomy Autoimmune Oophoritis in Mice. I. Requirement of Lyt-1 Effector Cells for Oocytes Damage After Adoptive Transfer. J Exp Med (1982) 156(6):1565–76.
3. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome (IPEX) Is Caused by Mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
4. Ramsdell F, Ziegler SF. FOXP3 and Scurfy: How It All Began. Nat Rev Immunol (2014) 14(5):343–9. doi: 10.1038/nri3650
5. Steinmetz M, Minard K, Horvath S, McNicholas J, Srelinger J, Wake C, et al. A Molecular Map of the Immune Response Region From the Major Histocompatibility Complex of the Mouse. Nature (1982) 300(5887):35–42. doi: 10.1038/300035a0
6. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155(3):1151–64.
7. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front Immunol (2019) 10:43. doi: 10.3389/fimmu.2019.00043
8. Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, McKenna DH, et al. Adoptive Transfer of Umbilical Cord Blood-Derived Regulatory T Cells and Early Viral Reactivation. Biol Blood Marrow Transplant (2013) 19(8):1271–3. doi: 10.1016/j.bbmt.2013.06.004
9. Eshhar Z, Waks T, Bendavid A, Schindler DG. Functional Expression of Chimeric Receptor Genes in Human T Cells. J Immunol Methods (2001) 248(1-2):67–76. doi: 10.1016/S0022-1759(00)00343-4
10. Maus MV, Grupp SA, Porter DL, June CH. Antibody-Modified T Cells: CARs Take the Front Seat for Hematologic Malignancies. Blood (2014) 123(17):2625–35. doi: 10.1182/blood-2013-11-492231
11. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med (2018) 379(1):64–73. doi: 10.1056/NEJMra1706169
12. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-Engineered T Regulatory Cells Target the CNS and Suppress EAE Upon Intranasal Delivery. J Neuroinflamm (2012) 9:112. doi: 10.1186/1742-2094-9-112
13. Scott DW, Levings MK. Novel T Regulatory Cells Come of Age: The Curious Incident of a Mouse in Tennessee, Delayed Thymectomy and Chimeric Receptors! Cell Immunol (2021) 359:104253. doi: 10.1016/j.cellimm.2020.104253
14. Sicard A, Levings MK, Scott DW. Engineering Therapeutic T Cells to Suppress Alloimmune Responses Using TCRs, CARs, or BARs. Am J Transplant (2018) 18(6):1305–11. doi: 10.1111/ajt.14747
15. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-Specific Regulatory T Cells Generated With a Chimeric Antigen Receptor. J Clin Invest (2016) 126(4):1413–24. doi: 10.1172/JCI82771
16. Rana J, Biswas M. Regulatory T Cell Therapy: Current and Future Design Perspectives. Cell Immunol (2020) 356:104193. doi: 10.1016/j.cellimm.2020.104193
17. Fu RY, Chen AC, Lyle MJ, Chen CY, Liu CL, Miao CH. CD4(+) T Cells Engineered With FVIII-CAR and Murine Foxp3 Suppress Anti-Factor VIII Immune Responses in Hemophilia a Mice. Cell Immunol (2020) 358:104216. doi: 10.1016/j.cellimm.2020.104216
18. De Paula Pohl A, Schmidt A, Zhang AH, Maldonado T, Konigs C, Scott DW. Engineered Regulatory T Cells Expressing Myelin-Specific Chimeric Antigen Receptors Suppress EAE Progression. Cell Immunol (2020) 358:104222. doi: 10.1016/j.cellimm.2020.104222
19. Pohl AP, Venkatesha SH, Zhang AH, Scott DW. Suppression of FVIII-Specific Memory B Cells by Chimeric BAR Receptor-Engineered Natural Regulatory T Cells. Front Immunol (2020) 11:693. doi: 10.3389/fimmu.2020.00693
20. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering Chimeric Antigen Receptor T Cells for Targeted Therapy of Autoimmune Disease. Science (2016) 353(6295):179–84. doi: 10.1126/science.aaf6756
21. Janssens I, Cools N. Regulating the Regulators: Is Introduction of an Antigen-Specific Approach in Regulatory T Cells the Next Step to Treat Autoimmunity? Cell Immunol (2020) 358:104236. doi: 10.1016/j.cellimm.2020.104236
22. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-Generation Regulatory T Cell Therapy. Nat Rev Drug Discovery (2019) 18(10):749–69. doi: 10.1038/s41573-019-0041-4
23. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am J Transplant (2017) 17(4):917–30. doi: 10.1111/ajt.14175
24. Bézie S, Charreau B, Vimond N, Lasselin J, Gérard N, Nerrière-Daguin V, et al. Human CD8+ Tregs Expressing a MHC-Specific CAR Display Enhanced Suppression of Human Skin Rejection and GVHD in NSG Mice. Blood Adv (2019) 3(22):3522–38. doi: 10.1182/bloodadvances.2019000411
25. Mukhatayev Z, Dellacecca ER, Cosgrove C, Shivde R, Jaishankar D, Pontarolo-Maag K, et al. Antigen Specificity Enhances Disease Control by Tregs in Vitiligo. Front Immunol (2020) 11:581433. doi: 10.3389/fimmu.2020.581433
26. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered Antigen-Specific Human Regulatory T Cells: Immunosuppression of FVIII-Specific T- and B-Cell Responses. Blood (2015) 125(7):1107–15. doi: 10.1182/blood-2014-04-566786
27. Kim YC, Zhang AH, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-Specific Human Tregs Ameliorate MOG-Induced EAE Through IL-2-Triggered Inhibition of Effector T Cells. J Autoimmun (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
28. Yoon J, Schmidt A, Zhang AH, Königs C, Kim YC, Scott DW. FVIII-Specific Human Chimeric Antigen Receptor (CAR) T-Regulatory Cells Suppress T-And B-Cell Responses to FVIII. Blood (2016) 129(2):238–45. doi: 10.1182/blood-2016-07-727834
29. Zhang AH, Yoon J, Kim YC, Scott DW. Targeting Antigen-Specific B Cells Using Antigen-Expressing Transduced Regulatory T Cells. J Immunol (2018) 201(5):1434–41. doi: 10.4049/jimmunol.1701800
30. Parvathaneni K, Abdeladhim M, Pratt KP, Scott DW. Hemophilia A Inhibitor Treatment: The Promise of Engineered T-Cell Therapy. Transl Res (2017) 187:44–52. doi: 10.1016/j.trsl.2017.06.002
31. Abdeladhim M, Zhang AH, Kropp LE, Lindrose AR, Venkatesha SH, Mitre E, et al. Engineered Ovalbumin-Expressing Regulatory T Cells Protect Against Anaphylaxis in Ovalbumin-Sensitized Mice. Clin Immunol (2019) 207:49–54. doi: 10.1016/j.clim.2019.07.009
32. Dawson NAJ, Rosado-Sanchez I, Novakovsky GE, Fung VCW, Huang Q, McIver E, et al. Functional Effects of Chimeric Antigen Receptor Co-Receptor Signaling Domains in Human Regulatory T Cells. Sci Transl Med (2020) 12(557):eaaz3866. doi: 10.1126/scitranslmed.aaz3866
33. Scott DW, Pratt KP, Miao CH. Progress Toward Inducing Immunologic Tolerance to Factor VIII. Blood (2013) 121(22):4449–56. doi: 10.1182/blood-2013-01-478669
34. Pratt KP, Arruda VR, Lacroix-Desmazes S. Inhibitors-Recent Insights. Haemophilia (2020). doi: 10.1111/hae.14077
35. James EA, Kwok WW, Ettinger RA, Thompson AR, Pratt KP. T-Cell Responses Over Time in a Mild Hemophilia A Inhibitor Subject: Epitope Identification and Transient Immunogenicity of the Corresponding Self-Peptide. J Thromb Haemost (2007) 5(12):2399–407. doi: 10.1111/j.1538-7836.2007.02762.x
36. Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. Lineages of Human T-Cell Clones, Including T Helper 17/T Helper 1 Cells, Isolated at Different Stages of Anti-Factor VIII Immune Responses. Blood (2009) 114(7):1423–8. doi: 10.1182/blood-2009-01-200725
37. James EA, van Haren SD, Ettinger RA, Fijnvandraat K, Liberman JA, Kwok WW, et al. T-Cell Responses in Two Unrelated Hemophilia A Inhibitor Subjects Include an Epitope at the Factor VIII R593C Missense Site. J Thromb Haemost (2011) 9(4):689–99. doi: 10.1111/j.1538-7836.2011.04202.x
38. Naumann A, Scherger AK, Neuwirth J, Orlowski A, Kahle J, Schwabe D, et al. Selection and Characterisation of FVIII-Specific Single Chain Variable Fragments. Hamostaseologie (2013) 33 Suppl 1:S39–45. doi: 10.1055/s-0037-1619801
39. Wucherpfennig KW, Ota K, Endo N, Seidman JG, Rosenzweig A, Weiner HL, et al. Shared Human T Cell Receptor V Beta Usage to Immunodominant Regions of Myelin Basic Protein. Science (1990) 248(4958):1016–9. doi: 10.1126/science.1693015
40. Parvathaneni K, Scott DW. Engineered FVIII-Expressing Cytotoxic T Cells Target and Kill FVIII-Specific B Cells In Vitro and In Vivo. Blood Adv (2018) 2(18):2332–40. doi: 10.1182/bloodadvances.2018018556
Keywords: chimeric antigen receptor, tolerance, autoimmunity, hemophilia, allergy, EAE (experimental autoimmune encephalitis), regulatory T cell (Treg)
Citation: Scott DW (2021) Driving CARs to BARs: The Winding Road to Specific Regulatory T Cells for Tolerance. Front. Immunol. 12:742719. doi: 10.3389/fimmu.2021.742719
Received: 16 July 2021; Accepted: 16 August 2021;
Published: 06 September 2021.
Edited by:
Amy Rosenberg, United States Food and Drug Administration, United StatesReviewed by:
Xinhua Yu, Research Center Borstel (LG), GermanyRoland W. Herzog, Indiana University, United States
Copyright © 2021 Scott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David W. Scott, c2NvdHRkNDNAZ21haWwuY29t