 Zicong He
Zicong He Shuixing Zhang
Shuixing Zhang- Department of Radiology, First Affiliated Hospital of Jinan University, Guangzhou, China
Tumor-associated macrophages (TAMs) are some of the most abundant immune cells within tumors and perform a broad repertoire of functions via diverse phenotypes. On the basis of their functional differences in tumor growth, TAMs are usually categorized into two subsets of M1 and M2. It is well established that the tumor microenvironment (TME) is characterized by hypoxia along with tumor progression. TAMs adopt an M1-like pro-inflammatory phenotype at the early phases of oncogenesis and mediate immune response that inhibits tumor growth. As tumors progress, anabatic hypoxia of the TME gradually induces the M2-like functional transformation of TAMs by means of direct effects, metabolic influence, lactic acidosis, angiogenesis, remodeled stroma, and then urges them to participate in immunosuppression, angiogenesis and other tumor-supporting procedure. Therefore, thorough comprehension of internal mechanism of this TAM functional transformation in the hypoxic TME is of the essence, and might provide some novel insights in hypoxic tumor immunotherapeutic strategies.
Introduction
The tumor microenvironment (TME) is now recognized as a major contributor to cancer progression. Hypoxia, resulting from an imbalance between oxygen supply and consumption (1), is an intrinsic property of the TME. The rapid proliferation of cells in the tumor mass necessitates extensive vascularization to sustain an adequate oxygen supply; however, tumor vessels are usually immature, disorganized, and hyperpermeable (2), leading to intratumoral oxygen deprivation. Cancer cells adapt to the resultant hypoxic microenvironment mainly via the hypoxia-inducible factor (HIF) signaling pathway, which regulates the expression of genes that contribute to immune evasion and malignant progression (3, 4). However, such inhospitable conditions are not favorable for infiltrating immune cells and promote their immunosuppressive functions (5).
Macrophages, which originate from circulating bone marrow-derived monocytic precursors, are among the most abundant immune cells within tumors and can be polarized into different phenotypes, each of which is associated with different and diverse functions (6, 7). According to their functional differences, these tumor-associated macrophages (TAMs) can be broadly categorized into two subsets, namely, M1 (pro-inflammatory and anti-tumor) and M2 (anti-inflammatory and pro-tumor) (8). M1-like TAMs are activated by IFN-γ, lipopolysaccharide, IL-1β, TNF, and/or GM-CSF and can recognize and destroy malignant cells via phagocytosis and cytotoxicity, in addition to producing pro-inflammatory cytokines that stimulate anti-tumor immunity (9–11). In contrast, M2-like TAMs are induced by Th2 cytokines such as IL-4, IL-10, IL-13, and/or M-CSF, and can favor tumor growth and promote TME remodeling by producing growth factors, immunosuppressive factors, pro-angiogenic molecules, and proteases (9, 12–14). However, this simplified distinction of M1/M2 polarization cannot strictly delineate the phenotypic and functional boundaries of TAMs as these cells are both highly dynamic and heterogeneous within and across tumors (15). TAMs have an extraordinary degree of plasticity, which enables them to finely modulate themselves in response to microenvironmental changes and thereby orchestrate various aspects of the TME (7, 15). Hypoxia is a microenvironmental cue that induces the tumor-supporting transformation of TAMs, an effect that is associated with disease progression and resistance to therapy (16). This highlights the need to integrate TAM-related hypoxic stress into tumor immunotherapy.
Here, we review the known mechanistic effects of a hypoxic TME on TAM functional transformation (Figure 1) and provide insights into immunotherapeutic strategies targeting hypoxic macrophages.
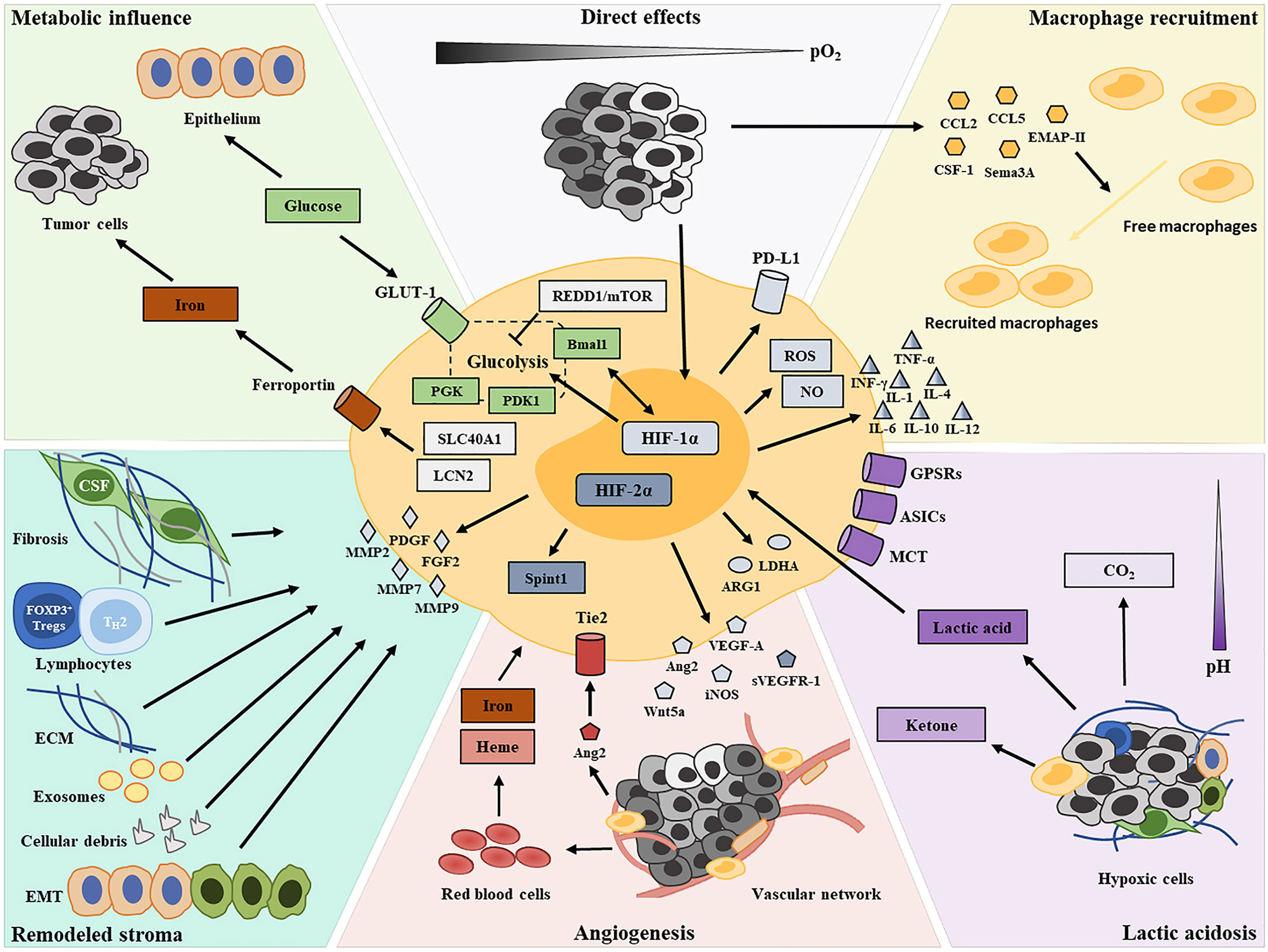
Figure 1 Graphical summary depicting the contributors of TAM functional transformation in the hypoxic TME.
Pro-Tumor Transformation of TAMs in the Hypoxic TME
Hypoxia-Driven TAM Recruitment
Due to unbalanced growth and a disorganized microvasculature, there is significant heterogeneity in oxygen content in a tumor mass. The hypoxic condition induces the production of a broad array of migratory stimulating factors, such as VEGF, CCL2, CCL5, CSF-1, EMAP-II, endothelin-2, SEMA3A, oncostatin M, and eotaxin, in tumor cells and the stroma within oxygen-deprived regions (17–24), resulting in macrophage recruitment and entrapment (25). When macrophages are recruited in hypoxic tumor areas, their polarization can be altered to an M2-like pro-tumor phenotype via the activity of the above-mentioned hypoxic tumor cell-derived cytokines (20, 24). A recent study revealed that neuropilin-1 (NRP-1) expression is significantly upregulated in hypoxic areas and induces pro-tumor phenotypes in recruited macrophages (26). Consequently, there is a greater abundance of M2-like TAMs at the invasive margin of tumors, where the hypoxic status is more severe, compared with that at the tumor center (27).
Direct Effects of Hypoxia
Hypoxia may also direct TAM polarization by affecting gene expression profiles. HIFs are key hypoxia-responsive transcription factors, the expression of which is upregulated in macrophages (28). Two isoforms of HIF—HIF-1 and HIF-2—elicit overlapping but sometimes opposing effects on macrophage transcriptional profiles, which endow macrophages with plasticity and shape their versatile phenotypes (29, 30). HIF activity in macrophages is dependent on the type of cytokine stimulus (31), with HIF-1α reported to be activated by Th1 cytokines and HIF-2α by Th2 cytokines. Additionally, HIF-1α and HIF-2α, via the regulation of respectively the inducible nitric oxide (NO) synthase and the arginase 1 genes, coordinately regulate NO availability to guide macrophage functional phenotypes (31). HIF-1α and HIF-2α are known to participate in the inflammatory function of macrophages. Macrophages sense changes in oxygen concentrations and then mediate IFN-γ production via HIF-1α, thereby enhancing their phagocytic functions and antigen presentation abilities (32). Meanwhile, HIF-1α promotes the production of inflammatory molecules in a TLR4-dependent fashion, including granule proteases, antimicrobial peptides, TNF-α, IL-1, IL-4, IL-6, and IL-12, thereby regulating the killing capacity of macrophages (33, 34). In vitro findings indicated that the absence of HIF-1α in macrophages leads to reduced ARG1 expression and the consequent suppression of T-cell activation (35). Additionally, there is evidence to indicate that HIF-1α affects the inflammatory function of macrophages by regulating their glycolytic capacity under hypoxic conditions (36). The contributions of HIF-2α to pro-inflammatory cytokine expression in hypoxic macrophages have also been documented (37). However, unlike HIF-1α, the regulation of inflammation by HIF-2α involves neither the production of NO nor the expression of costimulatory molecules (33, 37). Furthermore, HIF-1α and HIF-2α were found to exert antagonistic functions in angiogenesis. The role of HIF-1α as a positive regulator of macrophage-derived VEGF is well established (38). The knockout of HIF-1α in TAMs can attenuate their pro-angiogenic responses (39). In contrast, HIF-2α upregulates the production of soluble VEGF receptor 1 (sVERFR-1) by macrophages (40, 41). sVERFR-1 is an alternatively spliced variant of the membrane-bound VEGFR-1 expressed on endothelial cells and acts as a negative regulator of VEGF in tumor angiogenesis (40). Furthermore, HIF-1α was recently reported to upregulate the expression of PD-L1 in tumor-infiltrating macrophages, thereby promoting the establishment of an immunosuppressive TME (42). A recent study found that macrophage-derived HIF-2α regulates the expression of the serine protease inhibitor Kunitz type 1 (SPINT1), which contributes to the tumor-suppressive functions of TAMs in breast cancer development (43). Nonetheless, the latest evidence from single-cell RNA sequencing revealed that macrophages within both tumors and normal tissues do not show defined M1 or M2 polarization signature gene expression (44). The multifarious functional phenotypes of TAMs in hypoxic tumors might not be entirely dependent on gene expression profiles, but may also be influenced by the local environment.
At the early stages of oncogenesis, infiltrating macrophages adopt an M1-like phenotype that promotes the destruction of tumor cells and the inhibition of angiogenesis, concomitant with the activation of the inflammatory response (45). However, chronic inflammation resulting from M1-like TAM activity can accelerate genomic instability in malignant cells and serve as a driver of tumor progression (46, 47). As tumors progress, increasing levels of hypoxia lead to reduced secretion of pro-inflammatory mediators (e.g., IL-1β, TNF-α, and CCL17) by M1-polarized macrophages and facilitates macrophage differentiation toward the M2-like phenotype (48). Although hypoxia does not directly alter the relative abundance of macrophage subsets, it induces a pro-tumor gene expression profile in the M2-like macrophage subset (49), including the expression of growth factors (e.g., FGF2, PDGF, and VEGF) (50, 51), angiogenic molecules (e.g., VEGF, FGF2, CXCL8, and IL-8) (52), angiogenic modulators (e.g., COX2 and iNOS) (52), and matrix metalloproteinases (e.g., MMP2, MMP7, and MMP9) (53, 54). Furthermore, hypoxia can reportedly promote an increase in CCL20 expression in TAMs through the ERK/NF-κβ pathway, leading to the accumulation of CCR6+ Foxp3+ T regulatory cells (Tregs) (55). Although TAMs show no differences in M1 and M2 polarization capacity, they tend to exert M2-like pro-tumor functions in the hypoxic TME (35).
Metabolic Influence of Hypoxia
Hypoxia is known as a metabolic cue that shapes macrophage functional phenotypes within the TME. M1-like macrophages usually employ glycolytic metabolism for their energy supply and have a robust capacity for reactive oxygen species (ROS) production; in contrast, M2-like macrophages generally utilize oxidative phosphorylation to fuel their longer-term tissue repair functions (56). The crucial role of HIF-1α in regulating the glycolytic capacity of macrophages, as well as their survival and function, in the hypoxic TME has been documented (36). The expression of the glycolytic enzyme phosphoglycerate kinase (PGK) and glucose transporter 1 (GLUT-1) is markedly reduced in macrophages with deletion of myeloid HIF-1α, as is the cellular ATP pool, which leads to an impaired inflammatory response (33, 36). There is some evidence to suggest that pro-inflammatory macrophages redirect pyruvate away from pyruvate dehydrogenase (PDH) in a NO-dependent and HIF-1α-independent manner, thereby promoting their metabolic reprogramming (57). Pyruvate dehydrogenase kinase, isozyme 1 (PDK1), induced by HIF-1α in mildly hypoxic condition, has been found to regulate glycolytic reprograming of macrophages through the redirection of pyruvate flux into lactate, while leaving cytochrome c oxidase activity unaffected (58). Such active glycolysis promotes the redistribution of intracellular ATP, and plays an essential role in macrophage migratory capacity (58). However, long-term hypoxia in tumors still exerts a negative influence on TAM metabolism. Mammalian target of rapamycin (mTOR) functions as an integrative rheostat that couples cellular activation to nutrient sensing and metabolic status (59, 60). Hypoxia drives the upregulation of regulated in development and DNA damage response 1 (REDD1), an inhibitor of mTOR, which strongly hinders glycolysis in TAMs and reduces their metabolic competition with endothelial cells (61, 62). Such a REDD1/mTOR metabolic shift in TAMs culminates in endothelial cell hyperactivation, with the consequent formation of an abnormal vascular network (61, 62). A significant reduction in microRNA-30c levels is also observed in hypoxic TAMs, which impairs both mTOR activity and glycolysis, thereby inhibiting TAM M1-like polarization (63). BMAL1 is known as a molecular clock that regulates mitochondrial metabolism under metabolic stress in macrophages. A recent study found that BMAL1/HIF-1α crosstalk regulates macrophage energy metabolism, while metabolic dysregulation due to aberrant HIF-1α activation in TAMs contributes to an immunosuppressive TME (64).
Iron is an essential nutrient for malignant cell growth and proliferation and also contributes to both tumor progression and metastasis (65). Most iron is recycled and released to tissues by macrophages via erythrophagocytosis (66). M2-like TAMs exhibit a gene expression profile associated with iron efflux (increased ferroportin levels and reduced ferritin levels), whereas M1-like TAMs favor iron retention (67, 68). Tumor hypoxia supports such an iron-donor phenotype by upregulating solute carrier family 40, member 1 (SLC40A1) and lipocalin 2 (LCN2) expression in TAMs, resulting in increased iron availability in the TME and improved iron uptake by malignant cells (69–71).
Lactic Acidosis After Hypoxia
It is well established that the hypoxic TME is characterized by acidosis. Hypoxic tumor cells mainly obtain energy via anaerobic glycolysis, leading to increased concentrations of lactic acid (72). Meanwhile, such fermentative metabolism occurs in highly proliferating cells even in the presence of oxygen, known as the “Warburg Effect” (72). This byproduct of aerobic or anaerobic glycolysis (together with M-CSF) downregulates the NF-κB pathway, reduces the secretion of both NO and inflammation-related cytokines (such as TNF-α and IL-1), while simultaneously inducing the expression of VEGFA, ARG1, and other M2-associated genes (73–75). Besides, M2−like TAMs altered by lactic acid were found to promote T−cell apoptosis through the PD−L1/PD−1 pathway (76). Recent findings have shown that a pH of 6.1 without stimulation or a pH of 6.8 with IL-4 stimulation could promote the expression of ARG1 and VEGFA by macrophages in vitro (77, 78). These effects of tumor-cell-derived lactic acid are mediated by HIF-1α and promote TAM polarization toward an M2-like phenotype (73–75). A different study reported that lactic acid could inhibit ATP6V0d2 expression in TAMs, thereby promoting their HIF-2α-mediated pro-tumor functions (79). This suggests that lactic acid promotes the tumor-supporting phenotype of TAMs via the activation of HIF-1α3 and HIF-2α, albeit through distinct mechanisms. Under normoxic conditions, lactic acid normally exerts only a weak effect on TAMs. Under hypoxia, however, lactic acid greatly facilitates M2-like polarization via the HIF-1, Hedgehog, and mTOR pathways (80). Furthermore, G protein-coupled receptors (GPCRs) have been reported to function as key sensors of the acidification of the TME, inducing the expression of inducible cyclic AMP early repressor (ICER; transcriptional repressor), which enhances the pro-tumor transition of TAMs via NF-κB signaling inhibition (77, 81). Moreover, the activation of acid-sensing ion channels (ASICs) was identified as an important mediator of the endocytic functions of macrophages as well as their maturation (82). Recently, lactic acid was shown to be capable of skewing the macrophage phenotype toward the M2-like state via monocarboxylate channel transporter (MCT)/HIF-1α signaling (83). Lactate-derived histone lysine lactylation, a recently identified epigenetic modification, was demonstrated to induce the expression of M2-associated genes, including ARG1 (84). Moreover, the most recent evidence has indicated that tumor-released succinate can activate succinate receptor 1 (SUCNR1) signaling to polarize TAMs toward tumor-supporting phenotypes through a SUCNR1-activated PI3K/HIF-1α axis (85).
Angiogenesis in Hypoxic Areas
Hypoxia in the TME induces angiogenesis to meet the oxygen and nutrient needs of proliferating tumor cells. TAMs accumulate and transition into proangiogenic phenotypes in perivascular areas (86), especially those that are poorly vascularized (87). TIE2, an angiopoietin (ANG) receptor expressed by TAMs, is upregulated under hypoxic conditions and, together with ANG-2, enhances the pro-tumor functions of TAMs (88, 89). Compared with TIE2− TAMs, TIE2+ TAMs within the same tumor express higher levels of pro-angiogenic genes, including MMP9, VEGFA, COX2, WNT5A, and PDGFB (90, 91). ANG-2 expression is known to be increased in hypoxic regions and serves as a chemoattractant for macrophages (89). ANG-2, secreted from tumor and vasculature cells, can enhance IL-10 and mannose receptor expression, while decreasing that of TNF-α and IL-12, thereby weakening TAM anti-tumor activity under hypoxic conditions (88, 89).
The secretion of macrophage-derived VEGF-A is also markedly increased by HIF-1α at hypoxic sites, thereby enhancing tumor angiogenesis (92, 93). In contrast, under the regulation of HIF-2α, hypoxic TAMs generate high levels of sVEGFR-1, which selectively neutralizes VEGF activity and diminishes tumor angiogenesis (40, 41). This antagonistic effect of HIF-1α and HIF-2α on angiogenesis was suggested to facilitate the redistribution of the vascular network in hypoxic tumors to meet their growth and metabolic requirements. Of note, HIF-2α is also highly expressed in normoxic macrophages, leading to enhanced transcription of proangiogenic genes (52).
Neoangiogenesis can provide oxygen and nutrients to hypoxic areas, but can also result in erythrocyte extravasation and hemolysis. The release of heme and iron from hemolytic red blood cells can help convert M2-like TAMs into pro-inflammatory M1-like TAMs that display tumor-killing activity (94).
Hypoxia-Remodeled Stromal Components
Stromal fibrosis is a commonly occurring event in the hypoxic TME. Cancer-associated fibroblasts (CAFs) are considered to be the dominant component of fibrotic stroma and can be activated by tumor hypoxia through several mechanisms (95). These activated fibroblasts have been found to overexpress numerous pro-inflammatory cytokines (e.g., CCL2, CCL5, IL-4, IL-6, IL-8, GM-CSF, CXCL8, and CXCL14) that regulate TAM recruitment, differentiation, and activation (96). CAF-derived CXCL14 has been demonstrated to affect macrophage recruitment in tumors via NOS1-derived NO signaling. CAFs have also been reported to impair the maturation and differentiation of recruited macrophages, locking them in a suppressive state, through the induction of STAT3 phosphorylation (97, 98). In vitro observations have indicated that CAF might drive myeloid cells toward immunosuppressive differentiation via the production of IL-4, IL-6, and IL-8 (99).
Extensive lymphocyte subpopulations also constitute a major fraction of tumor stroma. These lymphocytes in the hypoxic TME engage the tumor-supporting activities of TAMs via a large array of cytokines. For instance, Th2 lymphocyte-derived IL-4 and IL-13 can enhance epidermal growth factor expression in TAMs, which promotes tumor cell metastasis, as well as the suppressive activity of TAMs, which blunts CD8+ T-cell responses to therapy (100, 101). Moreover, there is evidence showing that hypoxia can upregulate the expression of forkhead box P3 (FOXP3), a transcriptional activator of Tregs, through an HIF-1α-dependent mechanism (102), while FOXP3+ Tregs drive TAMs toward an immunosuppressive phenotype (103, 104).
Extracellular matrix (ECM), which serves as a structural scaffold for immune cell infiltration in the TME, is extensively remodeled under tumor hypoxia (105). Hyaluronic acid (HA), a primary ECM component, is associated with macrophage trafficking and tumor neovascularization (106). Hypoxia enhances the endogenous production of HA by tumor cells (107). Pro-angiogenic M2-like TAMs preferentially traffic to HA-rich areas in the TME (106). Tumor-derived HA has also been identified to trigger the transient, early activation of monocytes, thereby promoting M2-like immunosuppressive phenotypes among TAMs (108). Another study reported that periostin and collagen, both fibrosis-associated ECM components, respectively facilitated TAM recruitment via integrin binding (109) and promoted their M2-like polarization (110).
Cellular debris resulting from cell death is prevalent within hypoxic regions of tumors. The release of high mobility group protein B1 (HMGB1) was demonstrated to drive IL-10 production in TAMs selectively through the receptor for advanced glycation end products (RAGE), leading to an IL-10-rich milieu within the tumor (111). The recognition of apoptotic cells is also thought to suppress macrophage activation potential (112). TAMs can recognize dying tumor cells through the MER tyrosine-protein kinase (MERTK) receptor and upregulate the expression of wound-healing factors such as TGF-β, IL-10, and ARG1 that suppress anti-tumor immunity (113).
Research attention has increasingly focused on exosomes released by hypoxic tumor cells. Hypoxia can stimulate tumor cells to produce higher numbers of exosomes (114). Exosomes in hypoxic tumor areas contain large amounts of chemokines and immunomodulatory proteins, including CSF-1, CCL2, FTH, FTL, and TGF-β, which promote the differentiation of infiltrating myeloid cells toward an M2-like macrophage lineage (115). Exosomal miR-301a-3p derived from hypoxic pancreatic cancer cells was reported to promote M2-like macrophage polarization by activating the PTEN/PI3Kγ pathway (116). MiR-7a, another exosomal miRNA derived from hypoxic tumor cells, was shown to suppress several target genes of the insulin pathway, such as INS-1 and IGF1R, and thus trigger M2-like TAM polarization (117), similar to that seen for miR940 from exosomes derived from ovarian epithelial carcinoma cells (118). Recently, exosomal lncRNA BCRT1 was demonstrated to promote M2-like phenotype polarization and enhance macrophage-induced tumor progression (119). Additionally, miR-1246 in hypoxic glioma-derived exosomes was shown to mediate H-GDE-induced M2-like macrophage polarization by targeting TERF2IP via activating and inhibiting the STAT3 and NF-κB signaling pathways, respectively (120). Hypoxic stress was also demonstrated to suppress miR101 expression, which resulted in an increase in TAM-derived IL-1α and IL-6, which, in turn, promoted lung tumor cell growth (121).
Epithelial to mesenchymal transition (EMT) is also a common phenomenon associated with stroma remodeling in hypoxic tumors, helping to foster an immunosuppressive TME and facilitating tumor progression and metastasis (122, 123). A significant correlation has been confirmed to exist between EMT and TAM infiltration in hypoxic tumor tissues (124). Zinc finger E-box binding homeobox 1 (ZEB1) plays a critical role in the EMT program by restraining epithelial differentiation via the inhibition of members of the microRNA-200 family (125). The high expression of ZEB1 in hypoxic regions has a positive relationship with M2-like TAM abundance, i.e., it recruits M2-like TAMs by activating CCL8 transcription (126). Moreover, high HIF-1α expression under hypoxic conditions leads to increased secretion of the cytokine IL-1β by M2 TAMs, which, in turn, enhances EMT progression (127).
Immunotherapeutic Strategies Targeting Hypoxic TAMs
Substantial evidence supports that the hypoxia-induced immunosuppressive TME elicits a more aggressive tumor phenotype and promotes resistance to treatment (128). Several studies have reported that TAM polarization might counterproductively be skewed towards an M2-like pro-tumor phenotype after chemotherapy and radiotherapy, which contributes to tumor revascularization and relapse, while increasing levels of hypoxia after therapy could further enhance the tumor-supporting functions of TAMs (129, 130). This highlights the potential of TAMs as immunotherapeutic targets for hypoxic tumors. Macrophage-centered therapeutic strategies for treating hypoxic tumors should focus on improving the hypoxic status of the TME, inhibiting the tumor-promoting functions of M2-like TAMs, or reactivating the anti-tumor activity of M1-like TAMs.
Improving the Hypoxic Status of the TME
As described above, the hypoxic TME is responsible for the pro-tumor transformation of TAMs. Redressing hypoxia in the TME may be beneficial for reversing the malignant TAM phenotypes and improving responses to immunotherapy. Oxygen delivery to hypoxic areas via nanomaterials may be an attractive means for achieving this. Various strategies for delivering O2 to the hypoxic TME have been reported, such as using certain oxygen carriers for transporting O2 to tumor sites or generating O2 from endogenous hydrogen peroxide in situ using nanocatalysts (131–134). Recently, a TAM-targeted biomimetic nano red blood cell system was designed for precise O2 delivery and M2-like TAM depletion within the TME (135). This nanosystem alleviated tumor hypoxia and markedly enhanced chemoimmunotherapeutic effects. Normalization of the tumor vasculature represents another possible approach for directly alleviating tumor hypoxia. Vessel normalization is now thought to be beneficial for tumor immune reprogramming (136). As is generally acknowledged, a wide spectrum of highly expressed pro-angiogenic proteins are responsible for the abnormal vasculature networks found in hypoxic tumors. Scheduling a proper dose of anti-angiogenic drugs that block these pro-angiogenic proteins or their receptors, such as VEGF/VEGFR, could help restore functional vessels, thus alleviating tumor hypoxia (137). Low-dose anti-VEGFR2 therapy has been reported to improve the perfusion of hypoxic tumors and promote an immunosuppressive-to-immunostimulatory TAM phenotype conversion (138). Counterintuitively, monotherapy with anti-angiogenic drugs at high doses might be counterproductive owing to the associated excessive pruning of tumor vessels (137). Modification of the HIF signaling pathway might be another way of alleviating hypoxia in the TME. Vorinostat (suberoylanilide hydroxamic acid, SAHA) is a histone deacetylase inhibitor that has been approved by the United States Food and Drug Administration (FDA) and has been demonstrated to negatively regulate the expression and function of HIF-1α through the inhibition of an eIF3G-dependent translation mechanism (139). Meanwhile, topotecan, a FDA-approved topoisomerase I inhibitor, has been shown to inhibit HIF-1α protein accumulation through a DNA damage-independent mechanism and thus delay both angiogenesis and tumor growth (140).
Inhibiting the Tumor-Promoting Functions of M2-like TAMs
The depletion of M2-like TAMs represents a possible therapeutic approach for lessening pro-tumor functions. Liposomal clodronate treatment was shown to attenuate lung cancer progression through depleting TAMs (141). Additionally, trabectedin (ET-743), originally developed as an anti-proliferative agent for soft tissue sarcoma and relapsed ovarian cancer, was reported to activate the extrinsic apoptotic pathway via TRAIL receptors, followed by TAM depletion in tumors (142). However, anti-cancer therapy with trabectedin might elicit undesirable effects on monocyte/macrophage-mediated host defenses because of the indiscriminate depletion of macrophages (142). As a consequence, molecular-targeting has emerged as a promising direction for M2-like TAM depletion. Cieslewicz and colleagues constructed an M2-targeting fusion peptide to selectively exhaust M2-like TAMs, thereby reducing systemic damage (143).
Because macrophages are recruited and entrapped in hypoxic areas of tumors by tumor- and stroma-derived chemoattractants, preventing macrophage recruitment via pharmacological modulation may be another effective treatment method for inhibiting the pro-tumor functions of TAMs. Several antibodies selectively targeting chemoattractant receptors, including CCL2R, VEGFR2, and CSF-1R, have been shown to reduce macrophage infiltration and suppress tumor growth (144–146). Accordingly, interfering pharmacologically with other macrophage chemoattractants, such as CXCL12 and CCL5, as a means of inhibiting tumor growth merits further investigation (147, 148).
Reactivating the Anti-Tumor Activity of M1-like TAMs
As mentioned above, M1-like TAMs possess anti-tumor activity, such as the ability to inhibit tumor angiogenesis as well as the activation of inflammatory responses. This suggests that repolarizing TAMs to an M1-like phenotype may be an additional supplement to the arsenal of anti-cancer therapies. One study found that zoledronic acid, a nitrogen-containing bisphosphonate used for the treatment of cancer patients with bone metastases, could convert the TAM phenotype from M2-like to M1-like by targeting the mevalonate pathway (149). Additionally, M2-like TAMs activated using CD40 agonists can reportedly reacquire antigen-presenting capabilities and become tumoricidal, resulting in the reestablishment of tumor immune surveillance and the short-term reduction of tumor volume (150). Meanwhile, it has been shown that Toll-like receptor 3 (TLR3) signaling can transform tumor-supporting TAMs into tumor suppressors by rapidly inducing the production of pro-inflammatory cytokines (151). Furthermore, there is evidence to support that the structural and functional restoration of the tumor vasculature might restore the anti-tumor functions of TAMs. It has been demonstrated that histidine-rich glycoprotein (HRG) can downregulate placental growth factor (PlGF) levels, leading to the restoration of tumor vessel functionality and TAM repolarization (152). CSF-1R inhibition has also been reported to alter TAM polarization in combination with glioma-secreted factors, including GM-CSF and IFN-γ (20). Anti-CD47-elicited antibody-dependent cellular phagocytosis might also lead to the skewing of TAM polarization toward an M1-like phenotype (153). Recent studies have found that PI3Kγ signaling represents a crucial mediator of the switching between immunostimulatory and immunosuppressive macrophage phenotypes. The selective inactivation of PI3Kγ can stimulate and prolong NF-κB activation while inhibiting that of C/EBPβ, thereby restoring the pro-inflammatory functions of macrophages (154). However, whether the anti-tumor functions of repolarized TAMs will be overridden by the hypoxic TME remains unclear and warrants further investigation.
Concluding Remarks
Hypoxia is a critical modulator of tumor immunity. TAMs, an important component of tumor immunity, are recruited into the hypoxic regions of tumors, where they acquire a pro-tumor phenotype following direct or indirect stimulation by the hypoxic TME. TAMs subsequently become important contributors to tumor immune escape, angiogenesis, matrix remodeling, metabolic changes, and treatment resistance through a vast array of pathophysiological processes. Although hypoxia-modified gene expression profiles endow TAMs with plasticity and versatility, the interaction with the hypoxic TME finally defines their specific functions. Consequently, a close characterization of the cross-talk between the TAM functional state and other components of the TME might offer significant insight into the development of new treatment regimens. Alleviating hypoxia in the TME and the phenotypic conversion of TAMs might be the focus of future efforts for cancer immunotherapy.
Author Contributions
Conceptualization, ZH. Investigation and Resources, ZH. Writing - Original Draft Preparation, ZH. Writing - Review and Editing, SZ. Visualization, ZH. Graphics, ZH. Supervision, SZ. and Project Administration, SZ. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (81871323, 81801665, 81901709) and the Natural Science Foundation of Guangdong Province (2018B030311024, 2019A1515011918).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The Hypoxic Tumour Microenvironment. Oncogenesis (2018) 7:10. doi: 10.1038/s41389-017-0011-9
2. Siemann DW. The Unique Characteristics of Tumor Vasculature and Preclinical Evidence for Its Selective Disruption by Tumor-Vascular Disrupting Agents. Cancer Treat Rev (2011) 37:63–74. doi: 10.1016/j.ctrv.2010.05.001
3. Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer (2016) 2:758–70. doi: 10.1016/j.trecan.2016.10.016
4. Vaupel P, Multhoff G. Hypoxia-/HIF-1alpha-Driven Factors of the Tumor Microenvironment Impeding Antitumor Immune Responses and Promoting Malignant Progression. Adv Exp Med Biol (2018) 1072:171–5. doi: 10.1007/978-3-319-91287-5_27
5. Vito A, El-Sayes N, Mossman K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells (2020) 9:992. doi: 10.3390/cells9040992
6. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
7. Poh AR, Ernst M. Targeting Macrophages in Cancer: From Bench to Bedside. Front Oncol (2018) 8:49. doi: 10.3389/fonc.2018.00049
8. Rhee I. Diverse Macrophages Polarization in Tumor Microenvironment. Arch Pharm Res (2016) 39:1588–96. doi: 10.1007/s12272-016-0820-y
9. Martinez FO, Gordon S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
10. Crusz SM, Balkwill FR. Inflammation and Cancer: Advances and New Agents. Nat Rev Clin Oncol (2015) 12:584–96. doi: 10.1038/nrclinonc.2015.105
11. Mantovani A, Allavena P. The Interaction of Anticancer Therapies With Tumor-Associated Macrophages. J Exp Med (2015) 212:435–45. doi: 10.1084/jem.20150295
12. Coussens LM, Zitvogel L, Palucka AK. Neutralizing Tumor-Promoting Chronic Inflammation: A Magic Bullet? Sci (N Y NY) (2013) 339:286–91. doi: 10.1126/science.1232227
13. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
14. Cassetta L, Pollard JW. Targeting Macrophages: Therapeutic Approaches in Cancer. Nat Rev Drug Discov (2018) 17:887–904. doi: 10.1038/nrd.2018.169
15. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
16. Henze AT, Mazzone M. The Impact of Hypoxia on Tumor-Associated Macrophages. J Clin Invest (2016) 126:3672–9. doi: 10.1172/JCI84427
17. Leek RD, Hunt NC, Landers RJ, Lewis CE, Royds JA, Harris AL. Macrophage Infiltration is Associated With VEGF and EGFR Expression in Breast Cancer. J Pathol (2000) 190:430–6. doi: 10.1002/(sici)1096-9896(200003)190:4<430::Aid-path538>3.0.Co;2-6
18. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature (2011) 475:222–5. doi: 10.1038/nature10138
19. Wang SW, Liu SC, Sun HL, Huang TY, Chan CH, Yang CY, et al. CCL5/CCR5 Axis Induces Vascular Endothelial Growth Factor-Mediated Tumor Angiogenesis in Human Osteosarcoma Microenvironment. Carcinogenesis (2015) 36:104–14. doi: 10.1093/carcin/bgu218
20. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat Med (2013) 19:1264–72. doi: 10.1038/nm.3337
21. Matschurat S, Knies UE, Person V, Fink L, Stoelcker B, Ebenebe C, et al. Regulation of EMAP II by Hypoxia. Am J Pathol (2003) 162:93–103. doi: 10.1016/S0002-9440(10)63801-1
22. Grimshaw MJ, Wilson JL, Balkwill FR. Endothelin-2 is a Macrophage Chemoattractant: Implications for Macrophage Distribution in Tumors. Eur J Immunol (2002) 32:2393–400. doi: 10.1002/1521-4141(200209)32:9<2393::Aid-immu2393>3.0.Co;2-4
23. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding Macrophage Entry Into Hypoxic Tumor Areas by Sema3A/Nrp1 Signaling Blockade Inhibits Angiogenesis and Restores Antitumor Immunity. Cancer Cell (2013) 24:695–709. doi: 10.1016/j.ccr.2013.11.007
24. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages Are Recruited to Hypoxic Tumor Areas and Acquire a Pro-Angiogenic M2-Polarized Phenotype via Hypoxic Cancer Cell Derived Cytokines Oncostatin M and Eotaxin. Oncotarget (2014) 5:5350–68. doi: 10.18632/oncotarget.2110
25. Murdoch C, Giannoudis A, Lewis CE. Mechanisms Regulating the Recruitment of Macrophages Into Hypoxic Areas of Tumors and Other Ischemic Tissues. Blood (2004) 104:2224–34. doi: 10.1182/blood-2004-03-1109
26. Chen XJ, Wu S, Yan RM, Fan LS, Yu L, Zhang YM, et al. The Role of the Hypoxia-Nrp-1 Axis in the Activation of M2-Like Tumor-Associated Macrophages in the Tumor Microenvironment of Cervical Cancer. Mol Carcinog (2019) 58:388–97. doi: 10.1002/mc.22936
27. Zheng X, Weigert A, Reu S, Guenther S, Mansouri S, Bassaly B, et al. Spatial Density and Distribution of Tumor-Associated Macrophages Predict Survival in Non-Small-Cell Lung Carcinoma. Cancer Res (2020) 80:4414–25. doi: 10.1158/0008-5472.CAN-20-0069
28. Elbarghati L, Murdoch C, Lewis CE. Effects of Hypoxia on Transcription Factor Expression in Human Monocytes and Macrophages. Immunobiology (2008) 213:899–908. doi: 10.1016/j.imbio.2008.07.016
29. Fang HY, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia-Inducible Factors 1 and 2 Are Important Transcriptional Effectors in Primary Macrophages Experiencing Hypoxia. Blood (2009) 114:844–59. doi: 10.1182/blood-2008-12-195941
30. Tausendschon M, Rehli M, Dehne N, Schmidl C, Doring C, Hansmann ML, et al. Genome-Wide Identification of Hypoxia-Inducible Factor-1 and -2 Binding Sites in Hypoxic Human Macrophages Alternatively Activated by IL-10. Biochim Biophys Acta (2015) 1849:10–22. doi: 10.1016/j.bbagrm.2014.10.006
31. Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, et al. Differential Activation and Antagonistic Function of HIF-{Alpha} Isoforms in Macrophages Are Essential for NO Homeostasis. Genes Dev (2010) 24:491–501. doi: 10.1101/gad.1881410
32. Acosta-Iborra B, Elorza A, Olazabal IM, Martin-Cofreces NB, Martin-Puig S, Miro M, et al. Macrophage Oxygen Sensing Modulates Antigen Presentation and Phagocytic Functions Involving IFN-Gamma Production Through the HIF-1 Alpha Transcription Factor. J Immunol (2009) 182:3155–64. doi: 10.4049/jimmunol.0801710
33. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1alpha Expression Regulates the Bactericidal Capacity of Phagocytes. J Clin Invest (2005) 115:1806–15. doi: 10.1172/JCI23865
34. Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting Edge: Essential Role of Hypoxia Inducible Factor-1alpha in Development of Lipopolysaccharide-Induced Sepsis. J Immunol (2007) 178:7516–9. doi: 10.4049/jimmunol.178.12.7516
35. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage Expression of Hypoxia-Inducible Factor-1 Alpha Suppresses T-Cell Function and Promotes Tumor Progression. Cancer Res (2010) 70:7465–75. doi: 10.1158/0008-5472.CAN-10-1439
36. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1alpha is Essential for Myeloid Cell-Mediated Inflammation. Cell (2003) 112:645–57. doi: 10.1016/s0092-8674(03)00154-5
37. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, et al. Hypoxia-Inducible Factor 2alpha Regulates Macrophage Function in Mouse Models of Acute and Tumor Inflammation. J Clin Invest (2010) 120:2699–714. doi: 10.1172/jci39506
38. Choi SM, Oh H, Park H. Microarray Analyses of Hypoxia-Regulated Genes in an Aryl Hydrocarbon Receptor Nuclear Translocator (Arnt)-Dependent Manner. FEBS J (2008) 275:5618–34. doi: 10.1111/j.1742-4658.2008.06686.x
39. Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K, et al. Knockout of HIF-1α in Tumor-Associated Macrophages Enhances M2 Polarization and Attenuates Their Pro-Angiogenic Responses. Carcinogenesis (2010) 31:1863–72. doi: 10.1093/carcin/bgq088
40. Eubank TD, Roda JM, Liu H, O’Neil T, Marsh CB. Opposing Roles for HIF-1alpha and HIF-2alpha in the Regulation of Angiogenesis by Mononuclear Phagocytes. Blood (2011) 117:323–32. doi: 10.1182/blood-2010-01-261792
41. Roda JM, Sumner LA, Evans R, Phillips GS, Marsh CB, Eubank TD. Hypoxia-Inducible Factor-2alpha Regulates GM-CSF-Derived Soluble Vascular Endothelial Growth Factor Receptor 1 Production From Macrophages and Inhibits Tumor Growth and Angiogenesis. J Immunol (2011) 187:1970–6. doi: 10.4049/jimmunol.1100841
42. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 Is a Novel Direct Target of HIF-1α, and its Blockade Under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J Exp Med (2014) 211:781–90. doi: 10.1084/jem.20131916
43. Susen RM, Bauer R, Olesch C, Fuhrmann DC, Fink AF, Dehne N, et al. Macrophage HIF-2alpha Regulates Tumor-Suppressive Spint1 in the Tumor Microenvironment. Mol Carcinog (2019) 58:2127–38. doi: 10.1002/mc.23103
44. He D, Wang D, Lu P, Yang N, Xue Z, Zhu X, et al. Single-Cell RNA Sequencing Reveals Heterogeneous Tumor and Immune Cell Populations in Early-Stage Lung Adenocarcinomas Harboring EGFR Mutations. Oncogene (2021) 40:355–68. doi: 10.1038/s41388-020-01528-0
45. Zaynagetdinov R, Sherrill TP, Polosukhin VV, Han W, Ausborn JA, McLoed AG, et al. A Critical Role for Macrophages in Promotion of Urethane-Induced Lung Carcinogenesis. J Immunol (2011) 187:5703–11. doi: 10.4049/jimmunol.1100558
46. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-Induced Cancer: Crosstalk Between Tumours, Immune Cells and Microorganisms. Nat Rev Cancer (2013) 13:759–71. doi: 10.1038/nrc3611
47. Canli Ö, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M, et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell (2017) 32:869–83.e5. doi: 10.1016/j.ccell.2017.11.004
48. Ke X, Chen C, Song Y, Cai Q, Li J, Tang Y, et al. Hypoxia Modifies the Polarization of Macrophages and Their Inflammatory Microenvironment, and Inhibits Malignant Behavior in Cancer Cells. Oncol Lett (2019) 18:5871–8. doi: 10.3892/ol.2019.10956
49. Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, et al. Tumor Hypoxia Does Not Drive Differentiation of Tumor-Associated Macrophages But Rather Fine-Tunes the M2-Like Macrophage Population. Cancer Res (2014) 74:24–30. doi: 10.1158/0008-5472.Can-13-1196
50. Kuwabara K, Ogawa S, Matsumoto M, Koga S, Clauss M, Pinsky DJ, et al. Hypoxia-Mediated Induction of Acidic/Basic Fibroblast Growth Factor and Platelet-Derived Growth Factor in Mononuclear Phagocytes Stimulates Growth of Hypoxic Endothelial Cells. Proc Natl Acad Sci USA (1995) 92:4606–10. doi: 10.1073/pnas.92.10.4606
51. Harmey JH, Dimitriadis E, Kay E, Redmond HP, Bouchier-Hayes D. Regulation of Macrophage Production of Vascular Endothelial Growth Factor (VEGF) by Hypoxia and Transforming Growth Factor Beta-1. Ann Surg Oncol (1998) 5:271–8. doi: 10.1007/BF02303785
52. White JR, Harris RA, Lee SR, Craigon MH, Binley K, Price T, et al. Genetic Amplification of the Transcriptional Response to Hypoxia as a Novel Means of Identifying Regulators of Angiogenesis. Genomics (2004) 83:1–8. doi: 10.1016/s0888-7543(03)00215-5
53. Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, et al. Hypoxia-Induced Gene Expression in Human Macrophages: Implications for Ischemic Tissues and Hypoxia-Regulated Gene Therapy. Am J Pathol (2003) 163:1233–43. doi: 10.1016/S0002-9440(10)63483-9
54. Grimshaw MJ, Hagemann T, Ayhan A, Gillett CE, Binder C, Balkwill FR. A Role for Endothelin-2 and Its Receptors in Breast Tumor Cell Invasion. Cancer Res (2004) 64:2461–8. doi: 10.1158/0008-5472.can-03-1069
55. Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology (2019) 70:198–214. doi: 10.1002/hep.30593
56. Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab (2017) 26:49–70. doi: 10.1016/j.cmet.2017.06.004
57. Palmieri EM, Gonzalez-Cotto M, Baseler WA, Davies LC, Ghesquiere B, Maio N, et al. Nitric Oxide Orchestrates Metabolic Rewiring in M1 Macrophages by Targeting Aconitase 2 and Pyruvate Dehydrogenase. Nat Commun (2020) 11:698. doi: 10.1038/s41467-020-14433-7
58. Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. HIF-1α-PDK1 Axis-Induced Active Glycolysis Plays an Essential Role in Macrophage Migratory Capacity. Nat Commun (2016) 7:11635. doi: 10.1038/ncomms11635
59. Weichhart T, Hengstschlager M, Linke M. Regulation of Innate Immune Cell Function by mTOR. Nat Rev Immunol (2015) 15:599–614. doi: 10.1038/nri3901
60. Mazzone M, Menga A, Castegna A. Metabolism and TAM Functions-it Takes Two to Tango. FEBS J (2018) 285:700–16. doi: 10.1111/febs.14295
61. Wenes M, Shang M, Di Matteo M, Goveia J, Martin-Perez R, Serneels J, et al. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab (2016) 24:701–15. doi: 10.1016/j.cmet.2016.09.008
62. Mantovani A, Locati M. Macrophage Metabolism Shapes Angiogenesis in Tumors. Cell Metab (2016) 24:887–8. doi: 10.1016/j.cmet.2016.11.007
63. Zhihua Y, Yulin T, Yibo W, Wei D, Yin C, Jiahao X, et al. Hypoxia Decreases Macrophage Glycolysis and M1 Percentage by Targeting microRNA-30c and mTOR in Human Gastric Cancer. Cancer Sci (2019) 110:2368–77. doi: 10.1111/cas.14110
64. Alexander RK, Liou YH, Knudsen NH, Starost KA, Xu C, Hyde AL, et al. Bmal1 Integrates Mitochondrial Metabolism and Macrophage Activation. Elife (2020) 9:e54090. doi: 10.7554/eLife.54090
65. Torti SV, Torti FM. Iron and Cancer: More Ore to be Mined. Nat Rev Cancer (2013) 13:342–55. doi: 10.1038/nrc3495
66. Beaumont C, Delaby C. Recycling Iron in Normal and Pathological States. Semin Hematol (2009) 46:328–38. doi: 10.1053/j.seminhematol.2009.06.004
67. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential Regulation of Iron Homeostasis During Human Macrophage Polarized Activation. Eur J Immunol (2010) 40:824–35. doi: 10.1002/eji.200939889
68. Corna G, Campana L, Pignatti E, Castiglioni A, Tagliafico E, Bosurgi L, et al. Polarization Dictates Iron Handling by Inflammatory and Alternatively Activated Macrophages. Haematologica (2010) 95:1814–22. doi: 10.3324/haematol.2010.023879
69. Mertens C, Akam EA, Rehwald C, Brune B, Tomat E, Jung M. Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype With Consequences on Tumor Progression. PloS One (2016) 11:e0166164. doi: 10.1371/journal.pone.0166164
70. Oren B, Urosevic J, Mertens C, Mora J, Guiu M, Gomis RR, et al. Tumour Stroma-Derived Lipocalin-2 Promotes Breast Cancer Metastasis. J Pathol (2016) 239:274–85. doi: 10.1002/path.4724
71. Mertens C, Mora J, Oren B, Grein S, Winslow S, Scholich K, et al. Macrophage-Derived Lipocalin-2 Transports Iron in the Tumor Microenvironment. Oncoimmunology (2018) 7:e1408751. doi: 10.1080/2162402X.2017.1408751
72. Pillai SR, Damaghi M, Marunaka Y, Spugnini EP, Fais S, Gillies RJ. Causes, Consequences, and Therapy of Tumors Acidosis. Cancer Metastasis Rev (2019) 38:205–22. doi: 10.1007/s10555-019-09792-7
73. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature (2014) 513:559–63. doi: 10.1038/nature13490
74. Zhao Y, Wang DG, Xu T, Liu PF, Cao YW, Wang YH, et al. Bladder Cancer Cells Re-Educate TAMs Through Lactate Shuttling in the Microfluidic Cancer Microenvironment. Oncotarget (2015) 6:39196–210. doi: 10.18632/oncotarget.5538
75. Paolini L, Adam C, Beauvillain C, Preisser L, Blanchard S, Pignon P, et al. Lactic Acidosis Together With GM-CSF and M-CSF Induces Human Macrophages Toward an Inflammatory Protumor Phenotype. Cancer Immunol Res (2020) 8:383–95. doi: 10.1158/2326-6066.Cir-18-0749
76. Shan T, Chen S, Chen X, Wu T, Yang Y, Li S, et al. M2−TAM Subsets Altered by Lactic Acid Promote T−cell Apoptosis Through the PD−L1/PD−1 Pathway. Oncol Rep (2020) 44:1885–94. doi: 10.3892/or.2020.7767
77. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, et al. Tumor Immunoevasion via Acidosis-Dependent Induction of Regulatory Tumor-Associated Macrophages. Nat Immunol (2018) 19:1319–29. doi: 10.1038/s41590-018-0226-8
78. El-Kenawi A, Gatenbee C, Robertson-Tessi M, Bravo R, Dhillon J, Balagurunathan Y, et al. Acidity Promotes Tumour Progression by Altering Macrophage Phenotype in Prostate Cancer. Br J Cancer (2019) 121:556–66. doi: 10.1038/s41416-019-0542-2
79. Liu N, Luo J, Kuang D, Xu S, Duan Y, Xia Y, et al. Lactate Inhibits ATP6V0d2 Expression in Tumor-Associated Macrophages to Promote HIF-2alpha-Mediated Tumor Progression. J Clin Invest (2019) 129:631–46. doi: 10.1172/JCI123027
80. Zhao Y, Zhao B, Wang X, Guan G, Xin Y, Sun YD, et al. Macrophage Transcriptome Modification Induced by Hypoxia and Lactate. Exp Ther Med (2019) 18:4811–9. doi: 10.3892/etm.2019.8164
81. Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, et al. Gpr132 Sensing of Lactate Mediates Tumor-Macrophage Interplay to Promote Breast Cancer Metastasis. Proc Natl Acad Sci USA (2017) 114:580–5. doi: 10.1073/pnas.1614035114
82. Ni L, Fang P, Hu ZL, Zhou HY, Chen JG, Wang F, et al. Identification and Function of Acid-Sensing Ion Channels in RAW 264.7 Macrophage Cells. Curr Med Sci (2018) 38:436–42. doi: 10.1007/s11596-018-1897-y
83. Zhang L, Li S. Lactic Acid Promotes Macrophage Polarization Through MCT-Hif1α Signaling in Gastric Cancer. Exp Cell Res (2020) 388:111846. doi: 10.1016/j.yexcr.2020.111846
84. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
85. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol Cell (2020) 77:213–27 e5. doi: 10.1016/j.molcel.2019.10.023
86. Lewis CE, Harney AS, Pollard JW. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell (2016) 30:365. doi: 10.1016/j.ccell.2016.07.009
87. Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of Vascular Endothelial Growth Factor by Macrophages is Up-Regulated in Poorly Vascularized Areas of Breast Carcinomas. J Pathol (2000) 192:150–8. doi: 10.1002/1096-9896(2000)9999:9999<::Aid-path687>3.0.Co;2-g
88. Lewis CE, De Palma M, Naldini L. Tie2-Expressing Monocytes and Tumor Angiogenesis: Regulation by Hypoxia and Angiopoietin-2. Cancer Res (2007) 67:8429–32. doi: 10.1158/0008-5472.CAN-07-1684
89. Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by Human Monocytes and Their Responses to Angiopoietin-2. J Immunol (2007) 178:7405–11. doi: 10.4049/jimmunol.178.11.7405
90. Pucci F, Venneri MA, Biziato D, Nonis A, Moi D, Sica A, et al. A Distinguishing Gene Signature Shared by Tumor-Infiltrating Tie2-Expressing Monocytes, Blood “Resident” Monocytes, and Embryonic Macrophages Suggests Common Functions and Developmental Relationships. Blood (2009) 114:901–14. doi: 10.1182/blood-2009-01-200931
91. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 Regulates Gene Expression in TIE2-Expressing Monocytes and Augments Their Inherent Proangiogenic Functions. Cancer Res (2010) 70:5270–80. doi: 10.1158/0008-5472.Can-10-0012
92. Bingle L, Lewis CE, Corke KP, Reed MW, Brown NJ. Macrophages Promote Angiogenesis in Human Breast Tumour Spheroids. Vivo Br J Cancer (2006) 94:101–7. doi: 10.1038/sj.bjc.6602901
93. Guruvayoorappan C. Tumor Versus Tumor-Associated Macrophages: How Hot is the Link? Integr Cancer Ther (2008) 7:90–5. doi: 10.1177/1534735408319060
94. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron Induces Anti-Tumor Activity in Tumor-Associated Macrophages. Front Immunol (2017) 8:1479. doi: 10.3389/fimmu.2017.01479
95. Giaccia AJ, Schipani E. Role of Carcinoma-Associated Fibroblasts and Hypoxia in Tumor Progression. Curr Top Microbiol Immunol (2010) 345:31–45. doi: 10.1007/82_2010_73
96. Kalluri R. The Biology and Function of Fibroblasts in Cancer. Nat Rev Cancer (2016) 16:582–98. doi: 10.1038/nrc.2016.73
97. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res (2013) 73:3007–18. doi: 10.1158/0008-5472.CAN-12-4601
98. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity (2016) 44:303–15. doi: 10.1016/j.immuni.2016.01.014
99. Kim JH, Oh SH, Kim EJ, Park SJ, Hong SP, Cheon JH, et al. The Role of Myofibroblasts in Upregulation of S100A8 and S100A9 and the Differentiation of Myeloid Cells in the Colorectal Cancer Microenvironment. Biochem Biophys Res Commun (2012) 423:60–6. doi: 10.1016/j.bbrc.2012.05.081
100. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell (2009) 16:91–102. doi: 10.1016/j.ccr.2009.06.018
101. Shiao SL, Ruffell B, DeNardo DG, Faddegon BA, Park CC, Coussens LM. TH2-Polarized CD4(+) T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol Res (2015) 3:518–25. doi: 10.1158/2326-6066.CIR-14-0232
102. Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia Controls CD4+CD25+ Regulatory T-Cell Homeostasis. Via Hypoxia-Inducible Factor-1alpha Eur J Immunol (2008) 38:2412–8. doi: 10.1002/eji.200838318
103. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 Expression Identifies a Novel Suppressive Macrophage Population in Human Ovarian Carcinoma. J Exp Med (2006) 203:871–81. doi: 10.1084/jem.20050930
104. Kryczek I, Wei S, Zhu G, Myers L, Mottram P, Cheng P, et al. Relationship Between B7-H4, Regulatory T Cells, and Patient Outcome in Human Ovarian Carcinoma. Cancer Res (2007) 67:8900–5. doi: 10.1158/0008-5472.Can-07-1866
105. Xu S, Xu H, Wang W, Li S, Li H, Li T, et al. The Role of Collagen in Cancer: From Bench to Bedside. J Transl Med (2019) 17:309. doi: 10.1186/s12967-019-2058-1
106. Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, et al. Hyaluronan Deficiency in Tumor Stroma Impairs Macrophage Trafficking and Tumor Neovascularization. Cancer Res (2010) 70:7073–83. doi: 10.1158/0008-5472.CAN-09-4687
107. Chen JE, Lumibao J, Blazek A, Gaskins HR, Harley B. Hypoxia Activates Enhanced Invasive Potential and Endogenous Hyaluronic Acid Production by Glioblastoma Cells. Biomater Sci (2018) 6:854–62. doi: 10.1039/c7bm01195d
108. Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L. Tumor-Derived Hyaluronan Induces Formation of Immunosuppressive Macrophages Through Transient Early Activation of Monocytes. Blood (2007) 110:587–95. doi: 10.1182/blood-2007-01-068031
109. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour-Associated Macrophages and Promotes Malignant Growth. Nat Cell Biol (2015) 17:170–82. doi: 10.1038/ncb3090
110. Stahl M, Schupp J, Jager B, Schmid M, Zissel G, Muller-Quernheim J, et al. Lung Collagens Perpetuate Pulmonary Fibrosis via CD204 and M2 Macrophage Activation. PloS One (2013) 8:e81382. doi: 10.1371/journal.pone.0081382
111. Huber R, Meier B, Otsuka A, Fenini G, Satoh T, Gehrke S, et al. Tumour Hypoxia Promotes Melanoma Growth and Metastasis via High Mobility Group Box-1 and M2-Like Macrophages. Sci Rep (2016) 6:29914. doi: 10.1038/srep29914
112. Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity (2017) 47:913–27.e6. doi: 10.1016/j.immuni.2017.10.006
113. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK Inhibition in Tumor Leukocytes Decreases Tumor Growth and Metastasis. J Clin Invest (2013) 123:3231–42. doi: 10.1172/JCI67655
114. King HW, Michael MZ, Gleadle JM. Hypoxic Enhancement of Exosome Release by Breast Cancer Cells. BMC Cancer (2012) 12:421. doi: 10.1186/1471-2407-12-421
115. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-Induced Tumor Exosomes Promote M2-Like Macrophage Polarization of Infiltrating Myeloid Cells and microRNA-Mediated Metabolic Shift. Oncogene (2019) 38:5158–73. doi: 10.1038/s41388-019-0782-x
116. Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, et al. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kgamma to Promote Pancreatic Cancer Metastasis. Cancer Res (2018) 78:4586–98. doi: 10.1158/0008-5472.CAN-17-3841
117. Wu Q, Li J, Li Z, Sun S, Zhu S, Wang L, et al. Exosomes From the Tumour-Adipocyte Interplay Stimulate Beige/Brown Differentiation and Reprogram Metabolism in Stromal Adipocytes to Promote Tumour Progression. J Exp Clin Cancer Res (2019) 38:223. doi: 10.1186/s13046-019-1210-3
118. Chen X, Ying X, Wang X, Wu X, Zhu Q, Wang X. Exosomes Derived From Hypoxic Epithelial Ovarian Cancer Deliver microRNA-940 to Induce Macrophage M2 Polarization. Oncol Rep (2017) 38:522–8. doi: 10.3892/or.2017.5697
119. Liang Y, Song X, Li Y, Chen B, Zhao W, Wang L, et al. LncRNA BCRT1 Promotes Breast Cancer Progression by Targeting miR-1303/PTBP3 Axis. Mol Cancer (2020) 19:85. doi: 10.1186/s12943-020-01206-5
120. Qian M, Wang S, Guo X, Wang J, Zhang Z, Qiu W, et al. Hypoxic Glioma-Derived Exosomes Deliver microRNA-1246 to Induce M2 Macrophage Polarization by Targeting TERF2IP via the STAT3 and NF-kappaB Pathways. Oncogene (2020) 39:428–42. doi: 10.1038/s41388-019-0996-y
121. Li J, Xu P, Wu D, Guan M, Weng X, Lu Y, et al. Hypoxic Stress Suppresses Lung Tumor-Secreted Exosomal Mir101 to Activate Macrophages and Induce Inflammation. Cell Death Dis (2021) 12:776. doi: 10.1038/s41419-021-04030-x
122. Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia Induced EMT: A Review on the Mechanism of Tumor Progression and Metastasis in OSCC. Oral Oncol (2018) 80:23–32. doi: 10.1016/j.oraloncology.2018.03.004
123. Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, et al. Hypoxia-Induced Epithelial-To-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res (2016) 76:818–30. doi: 10.1158/0008-5472.CAN-15-0977
124. Zhang WJ, Chen C, Zhou ZH, Gao ST, Tee TJ, Yang LQ, et al. Hypoxia-Inducible Factor-1 Alpha Correlates With Tumor-Associated Macrophages Infiltration, Influences Survival of Gastric Cancer Patients. J Cancer (2017) 8:1818–25. doi: 10.7150/jca.19057
125. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-Activator ZEB1 Promotes Tumorigenicity by Repressing Stemness-Inhibiting microRNAs. Nat Cell Biol (2009) 11:1487–95. doi: 10.1038/ncb1998
126. Chen XJ, Deng YR, Wang ZC, Wei WF, Zhou CF, Zhang YM, et al. Hypoxia-Induced ZEB1 Promotes Cervical Cancer Progression via CCL8-Dependent Tumour-Associated Macrophage Recruitment. Cell Death Dis (2019) 10:508. doi: 10.1038/s41419-019-1748-1
127. Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T, et al. Hypoxia-Inducible Factor-1α/Interleukin-1β Signaling Enhances Hepatoma Epithelial-Mesenchymal Transition Through Macrophages in a Hypoxic-Inflammatory Microenvironment. Hepatology (2018) 67:1872–89. doi: 10.1002/hep.29681
128. Wilson WR, Hay MP. Targeting Hypoxia in Cancer Therapy. Nat Rev Cancer (2011) 11:393–410. doi: 10.1038/nrc3064
129. Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, et al. Perivascular M2 Macrophages Stimulate Tumor Relapse After Chemotherapy. Cancer Res (2015) 75:3479–91. doi: 10.1158/0008-5472.CAN-14-3587
130. Chiang CS, Fu SY, Wang SC, Yu CF, Chen FH, Lin CM, et al. Irradiation Promotes an M2 Macrophage Phenotype in Tumor Hypoxia. Front Oncol (2012) 2:89. doi: 10.3389/fonc.2012.00089
131. Wang L, Niu M, Zheng C, Zhao H, Niu X, Li L, et al. A Core-Shell Nanoplatform for Synergistic Enhanced Sonodynamic Therapy of Hypoxic Tumor via Cascaded Strategy. Adv Healthc Mater (2018) 7:e1800819. doi: 10.1002/adhm.201800819
132. Ji C, Si J, Xu Y, Zhang W, Yang Y, He X, et al. Mitochondria-Targeted and Ultrasound-Responsive Nanoparticles for Oxygen and Nitric Oxide Codelivery to Reverse Immunosuppression and Enhance Sonodynamic Therapy for Immune Activation. Theranostics (2021) 11:8587–604. doi: 10.7150/thno.62572
133. Yang B, Chen Y, Shi J. Nanocatalytic Medicine. Adv Mater (2019) 31:e1901778. doi: 10.1002/adma.201901778
134. Jiang W, Zhang Z, Wang Q, Dou J, Zhao Y, Ma Y, et al. Tumor Reoxygenation and Blood Perfusion Enhanced Photodynamic Therapy Using Ultrathin Graphdiyne Oxide Nanosheets. Nano Lett (2019) 19:4060–7. doi: 10.1021/acs.nanolett.9b01458
135. Wang Y, Yu J, Luo Z, Shi Q, Liu G, Wu F, et al. Engineering Endogenous Tumor-Associated Macrophage-Targeted Biomimetic Nano-RBC to Reprogram Tumor Immunosuppressive Microenvironment for Enhanced Chemo-Immunotherapy. Adv Mater (2021) e2103497. doi: 10.1002/adma.202103497
136. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual Regulation of Tumour Vessel Normalization and Immunostimulatory Reprogramming. Nature (2017) 544:250–4. doi: 10.1038/nature21724
137. Jain RK. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
138. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular Normalizing Doses of Antiangiogenic Treatment Reprogram the Immunosuppressive Tumor Microenvironment and Enhance Immunotherapy. Proc Natl Acad Sci USA (2012) 109:17561–6. doi: 10.1073/pnas.1215397109
139. Hutt DM, Roth DM, Vignaud H, Cullin C, Bouchecareilh M. The Histone Deacetylase Inhibitor, Vorinostat, Represses Hypoxia Inducible Factor 1 Alpha Expression Through Translational Inhibition. PloS One (2014) 9:e106224. doi: 10.1371/journal.pone.0106224
140. Rapisarda A, Zalek J, Hollingshead M, Braunschweig T, Uranchimeg B, Bonomi CA, et al. Schedule-Dependent Inhibition of Hypoxia-Inducible Factor-1alpha Protein Accumulation, Angiogenesis, and Tumor Growth by Topotecan in U251-HRE Glioblastoma Xenografts. Cancer Res (2004) 64:6845–8. doi: 10.1158/0008-5472.Can-04-2116
141. Fritz JM, Tennis MA, Orlicky DJ, Lin H, Ju C, Redente EF, et al. Depletion of Tumor-Associated Macrophages Slows the Growth of Chemically Induced Mouse Lung Adenocarcinomas. Front Immunol (2014) 5:587. doi: 10.3389/fimmu.2014.00587
142. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell (2013) 23:249–62. doi: 10.1016/j.ccr.2013.01.008
143. Cieslewicz M, Tang J, Yu JL, Cao H, Zavaljevski M, Motoyama K, et al. Targeted Delivery of Proapoptotic Peptides to Tumor-Associated Macrophages Improves Survival. Proc Natl Acad Sci USA (2013) 110:15919–24. doi: 10.1073/pnas.1312197110
144. Gazzaniga S, Bravo AI, Guglielmotti A, van Rooijen N, Maschi F, Vecchi A, et al. Targeting Tumor-Associated Macrophages and Inhibition of MCP-1 Reduce Angiogenesis and Tumor Growth in a Human Melanoma Xenograft. J Invest Dermatol (2007) 127:2031–41. doi: 10.1038/sj.jid.5700827
145. Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, et al. Vascular Endothelial Growth Factor Receptor 2 Mediates Macrophage Infiltration Into Orthotopic Pancreatic Tumors in Mice. Cancer Res (2008) 68:4340–6. doi: 10.1158/0008-5472.Can-07-6705
146. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 Receptor Improves the Antitumor Efficacy of Adoptive Cell Transfer Immunotherapy. Cancer Res (2014) 74:153–61. doi: 10.1158/0008-5472.Can-13-1816
147. Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell (2016) 29:587–601. doi: 10.1016/j.ccell.2016.03.005
148. Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, et al. TIE2-Expressing Macrophages Limit the Therapeutic Efficacy of the Vascular-Disrupting Agent Combretastatin A4 Phosphate in Mice. J Clin Invest (2011) 121:1969–73. doi: 10.1172/jci44562
149. Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, et al. Zoledronic Acid Repolarizes Tumour-Associated Macrophages and Inhibits Mammary Carcinogenesis by Targeting the Mevalonate Pathway. J Cell Mol Med (2010) 14:2803–15. doi: 10.1111/j.1582-4934.2009.00926.x
150. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Sci (N Y NY) (2011) 331:1612–6. doi: 10.1126/science.1198443
151. Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, et al. Toll-Like Receptor 3 Signaling Converts Tumor-Supporting Myeloid Cells to Tumoricidal Effectors. Proc Natl Acad Sci USA (2012) 109:2066–71. doi: 10.1073/pnas.1113099109
152. Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization Through Downregulation of PlGF. Cancer Cell (2011) 19:31–44. doi: 10.1016/j.ccr.2010.11.009
153. Sockolosky JT, Dougan M, Ingram JR, Ho CC, Kauke MJ, Almo SC, et al. Durable Antitumor Responses to CD47 Blockade Require Adaptive Immune Stimulation. Proc Natl Acad Sci USA (2016) 113:E2646–54. doi: 10.1073/pnas.1604268113
Keywords: tumor hypoxia, hypoxic tumor microenvironment, tumor-associated macrophages, macrophage polarization, macrophage functional transformation
Citation: He Z and Zhang S (2021) Tumor-Associated Macrophages and Their Functional Transformation in the Hypoxic Tumor Microenvironment. Front. Immunol. 12:741305. doi: 10.3389/fimmu.2021.741305
Received: 14 July 2021; Accepted: 01 September 2021;
Published: 16 September 2021.
Edited by:
Sara Labiano, University of Navarra Clinic, SpainReviewed by:
Julián Aragones Lopez, Autonomous University of Madrid, SpainJamshid Hadjati, Tehran University of Medical Sciences, Iran
Copyright © 2021 He and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuixing Zhang, c2h1aTc1MTVAMTI2LmNvbQ==