 Esther L. Jones
Esther L. Jones Stephen M. Laidlaw
Stephen M. Laidlaw Lynn B. Dustin
Lynn B. Dustin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 September 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.738473
This article is part of the Research Topic Regulators of Immune System Function in Autoimmunity and Aging - Molecular and Cellular Research View all 16 articles
TRIM21 (Ro52/SSA1) is an E3 ubiquitin ligase with key roles in immune host defence, signal transduction, and possibly cell cycle regulation. It is also an autoantibody target in Sjögren’s syndrome, systemic lupus erythematosus, and other rheumatic autoimmune diseases. Here, we summarise the structure and function of this enzyme, its roles in innate immunity, adaptive immunity and cellular homeostasis, the pathogenesis of autoimmunity against TRIM21, and the potential impacts of autoantibodies to this intracellular protein.
The immune system is a balanced network of interacting cells which distinguish between self and non-self to effectively respond to invading pathogens. Failures in these peripheral and central tolerance mechanisms can lead to immune cells reacting to self-antigens, causing the extensive inflammation and tissue damage observed in autoimmune diseases (1).
TRIM21 (also called Ro52) autoantibodies have been detected in at least thirteen autoimmune diseases, with frequencies of detection in patients ranging from 5% to 95% (2, 3). The most common TRIM21-associated autoimmune diseases are systemic lupus erythematosus (SLE) affecting the central nervous system, skin, kidneys and joints, and Sjögren’s syndrome (SS), which primarily affects the tear and salivary glands (4, 5). Beyond SS and SLE, anti-TRIM21 antibodies have also been identified in patients with primary biliary cirrhosis, idiopathic inflammatory myopathies (mainly polymyositis and dermatomyositis), and infants with congenital heart block (CHB) associated with maternal autoantibody transfer (6–9). In fact, for primary Sjögren’s syndrome (pSS), serological detection of anti-TRIM21 antibodies is a diagnostic criterion, with detection rates in patients ranging from 50-70% according to assay method (10, 11).
For TRIM21-associated SS, single nucleotide polymorphism (SNP) and genome-wide association (GWAS) studies have identified polymorphisms in HLA, interferon regulatory factor-5 (IRF5) (suggesting TRIM21 is an interferon-stimulated gene; see below), and B cell activating factor (BAFF) loci (12, 13). For example, it is postulated that specific gene polymorphisms, including a CGGGG indel repeat in the IRF5 gene promoter, can alter IRF5 mRNA expression. This specific repeat may act as a binding site for transcription factor SP1, driving chronic type I interferon (IFN-I) proinflammatory cytokine production (14, 15).
Despite the identification of genetic risk factors (e.g. the influence of SNPs in autoantibody induction) and some of the cellular interactions involved, antigen-specific mechanisms which initiate and drive autoimmune pathologies remain poorly understood. Analysis of peripheral blood mononuclear cells (PBMCs) isolated from SLE and SS patients provides evidence of elevated TRIM21 transcript expression; however, questions remain as to whether TRIM21 is a key autoantigen driving B-cell activation, autoantibody production and autoimmune pathogenesis in these diseases (16).
Comprising more than 80 members, the TRIM protein family is a group of E3 ubiquitin ligases with roles in multiple cellular processes including cell cycle regulation, autophagy and innate immunity (17, 18). They have a conserved multidomain architecture, exclusively found in metazoans (both vertebrate and invertebrate species), consisting of the tripartite motif (N-terminal Really Interesting New Gene (RING) domain, B-box domain and coiled-coil domain) that may be associated with a variable C-terminal domain.
Specific C-terminal domains mediate substrate recognition, localisation within the cell and are used to classify TRIM proteins into sub-families according to function (19). This is illustrated by the C-terminal subgroup One Signature domain which enables localisation of TRIM18 (MID1) to microtubules for polyubiquitination and proteasomal degradation of the protein phosphatase 2A catalytic subunit (PP2Ac) (20, 21). Involved in multiple signalling pathways, PP2Ac regulates the mammalian Target of Rapamycin Complex 1 (mTORC1) pathway which controls tumour cell growth and metabolism, intracellular transport, cell migration, autophagy and cell cycle dynamics. When active, TRIM18 polyubiquitinates PP2Ac leading to its degradation, thus inhibiting mTORC1 complex formation to prevent downstream signalling (22). Therefore, it is unsurprising that mutations that alter or inhibit TRIM protein functions have complex downstream effects.
Other family members contain single or combinations of the following C-terminal domains; fibronectin type 3, plant homeodomain, transmembrane, ADP ribosylation factor-like, MATH (meprin and tumour-necrosis factor receptor (TNFR)-associated factor (TRAF) homology), filamin-type immunoglobulin, NHL repeats, or most commonly, a PRY/SPRY domain (23). Given the number of C-terminal domains and proteins in this family, it is conceivable that there is little or no functional redundancy between family members, yet this remains to be confirmed.
TRIM21 consists of an N-terminal RING domain, B-box domain, central coiled-coil domain (anticipated based on TRIM family homology) and terminal PRY/SPRY domain (24). The full crystal structure of TRIM21 is yet to be solved. However, TRIM21’s structure can be predicted by combining confirmed structures, binding kinetics and homology. The coiled-coil domain is believed to mediate TRIM21 homodimerization. This enables the dimer’s two PRY/SPRY domains to form a high-affinity binding pocket for immunoglobulin Fc domains, Figure 1 (25, 27).
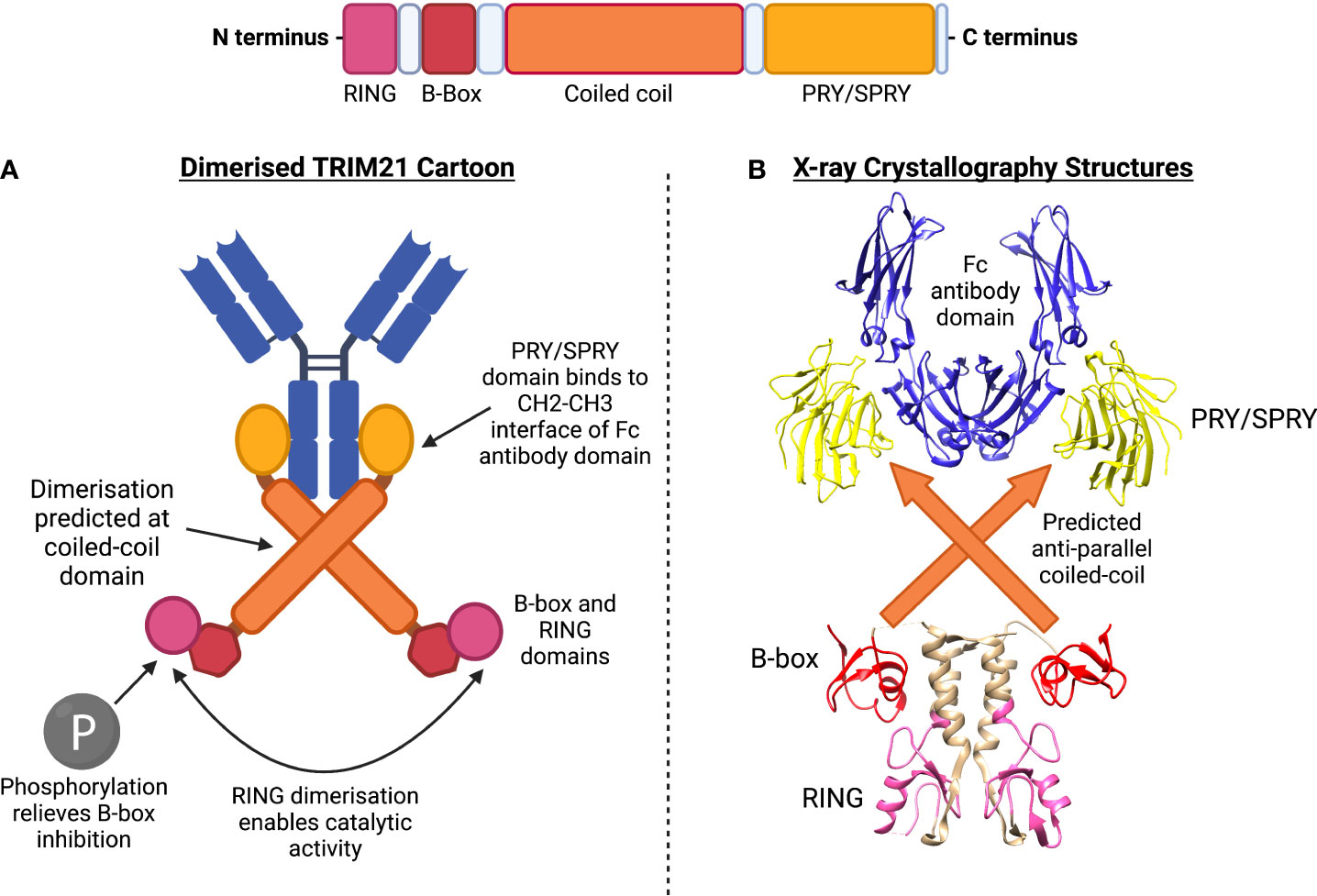
Figure 1 TRIM21 structure includes RING, B-box, coiled-coil and PRY/SPRY domains. (A) Dimerised TRIM21 cartoon shows binding of PRY/SPRY domain at constant region (Fc) of antibody. Dimerisation at the coiled-coil is predicted according to homology to other TRIM family members. Phosphorylation of the LxxIS motif of the RING domain relieves B-box inhibition, allowing RING dimerisation for catalytic activity. (B) X-ray crystallography structures have been obtained for the (1) PRY/SPRY domain, identified in complex with the Fc antibody domain (PDB:2IWG) (25). (2) The B-box and RING domains have been crystallised in a dimer confirmation (PDB:5OLM) (26). The central predicted coiled-coil structure has not been formally identified.
The RING domain is characterised by a series of conserved cysteine and histidine residues (28). These residues coordinate zinc atom binding, enabling the folding of an E3 ubiquitin ligase domain. The E3 zinc finger motifs mediate interactions with ubiquitin-bound E2 enzymes, catalysing the transfer of ubiquitin to target proteins (29, 30). Ubiquitin transfer involves the formation of lysine-specific, covalent polyubiquitin chains, which determine specific cellular activities. For example, proteins modified with Lysine 48 (K48)-specific ubiquitin chains are targeted for proteasomal degradation, whilst K63-specific chains regulate signalling pathways including DNA repair and inflammatory signalling (31).
A specific mechanism for self-anchored TRIM21 ubiquitin transfer has been recently uncovered, showing the RING domain acts as both substrate and catalyst for ubiquitination. Following mono-ubiquitination at the N-terminus (RING domain) by the E2 enzyme Ube2W, TRIM21 is activated. This ubiquitin-priming promotes recruitment of an E2 Ube2N/Ube2V2 heterodimer during K63-specific ubiquitination. Two dimerised RING domains hold the E2-Ub heterodimer in place, catalysing the transfer of K63-linked ubiquitin chains to a third acceptor mono-ubiquitinated RING, during self-anchored trans-ubiquitination. Such K63-linked ubiquitination is required for targeting the TRIM21-protein (e.g. antibody) complex for degradation (32).
The RING-domain of TRIM21 also mediates K48-linked ubiquitination. This was demonstrated by the ubiquitination and degradation of DEAD-box protein DDX41, an intracellular dsDNA sensor in myeloid dendritic cells (DCs) and monocytes (33). Binding to DNA via the DEAD domain, DDX41 activates the STING pathway for proinflammatory IFN-I stimulation (34). By targeting DDX41 for degradation, TRIM21-mediated DDX41 ubiquitination inhibits the IFN-I response to dsDNA. IFN-I responses to self-DNA are central in SLE pathogenesis (35). It is seemingly contradictory that such IFN-I responses are elevated in SLE patients when TRIM21 mRNA expression is also increased, as one would expect greater DDX41 ubiquitin-mediated degradation (16). This discrepancy may suggest inherent TRIM21 functions in downregulating DNA sensors such as DDX41 are impaired, by some yet unknown mechanism.
The B-box domain is less well-characterised but may have distinct functions in different TRIM family members. It may help coordinate TRIM self-association, may contain a zinc finger motif, in some TRIM proteins might confer E3 ubiquitin ligase activity, or in others has a regulatory role (26, 36, 37).
The B-box domain can mediate higher-order complex formation (38). This was demonstrated for TRIM5α which recognizes and binds to capsids of multiple retroviruses including HIV-1 (39). TRIM5α spontaneously assembles into a hexagonal lattice via hydrophobic interactions utilizing a key Arg residue located at the B-box2 domain (40). This hexagonal array mimics that of the target viral capsids and enables efficient binding of the SPRY domains to multiple capsid sites for subsequent ubiquitination and degradation during antiretroviral defense (39).
Despite also having a B-box2 domain, TRIM21 does not form higher-order assemblies. Instead, TRIM21’s B-box domain has a regulatory role via interactions with the RING domain (41). Whilst most TRIM proteins are constitutively active, X-ray crystallography has shown that the TRIM21 B-box domain represses its ubiquitin ligase activity by occupying the E2 binding site. Overexpression of kinases IKKβ or TBK1 led to phosphorylation of a LxxIS motif in the RING domain and was sufficient to relieve B-box inhibition (26). These findings may be of interest in an autoimmunity context, as disruption of this autoinhibition mechanism could contribute to excessive proinflammatory signalling. Polymorphisms located in regions encoding the LxxIS motif of TRIM21 have not yet been identified and in general, GWAS studies relating to TRIM21 in autoimmunity are limited. However, one SS patient study identified polymorphisms in multiple coding regions, with certain SNPs correlating with the presence of anti-TRIM21 antibodies (42). Therefore, GWAS studies should be expanded to identify additional polymorphisms associated with TRIM21, including those potentially encoding the LxxIS motif.
The central coiled-coil domain of TRIM21 is predicted to be an α-helical, supercoiled structure involved in dimerisation and TRIM self-association, Figure 1 (43). Except for TRIM19 which forms a torus-shaped homo-tetramer, TRIM proteins which associate via coiled-coil domains exist either as monomers, homodimers or in monomer: homodimer equilibria, with hetero-dimerisation being uncommon (44, 45). TRIM homodimers (and possibly higher-order oligomers) formed by anti-parallel coiled-coils also allow dimerisation at the RING domain (46, 47).
Studies have shown dimerisation of the catalytic RING domains to be necessary for ubiquitinating activity in most TRIM proteins studied so far (48). This holds true for TRIM21, with a recent study demonstrating that dimerisation at TRIM21’s coiled-coil directly enabled catalytic activity (49). Mutation of residues at the antiparallel coiled-coil dimer interface was sufficient to inhibit auto-ubiquitination of the RING domain, following infection of HEK293T cells with antibody-coated adenovirus. Furthermore, forced RING domain dimerisation (via RING-linker-RING constructs) increased ubiquitin discharge activity. A proposed model of “clustering-induced activation” suggests dimerisation of RING domains makes E2-ubiquitin engagement with TRIM21 and subsequent polyubiquitin discharge more energetically favourable (49).
The variable C-terminal region of TRIM family proteins is involved in protein-protein interaction and subcellular localisation, with TRIM21 containing a PRY/SPRY C-terminal domain, Figure 1 (25, 45).
The specificity and roles of PRY/SPRY domains have been demonstrated in a number of studies (50). Domain-swapping experiments show that TRIM5α’s PRY/SPRY domain is essential for viral restriction (51). Mutations in TRIM18, most often in the PRY/SPRY domain, alter TRIM18’s subcellular distribution and cause X-linked Opitz Syndrome (52, 53). Furthermore, PRY/SPRY mutations in TRIM20 cause Familial Mediterranean Fever (54), and in TRIM36 cause anencephaly (55).
For TRIM21, the PRY/SPRY domain contains a high-affinity immunoglobulin Fc binding site, the structure of which has been solved, Figure 1 (25). TRIM21’s PRY/SPRY domain binds to the CH2-CH3 interface of the Fc, and does not overlap with the Fc domain’s binding sites for FcγR and C1q. The CH2-CH3 interface is highly conserved, allowing TRIM21 binding to 98% of circulating immunoglobulins regardless of their antigen specificity (25).
Human TRIM21 displays promiscuous antibody interactions, binding to all four human IgG subclasses and even binding IgGs from different mammalian species in a 2:1 binding ratio (TRIM21: antibody Fc) (56). TRIM21 also binds both IgA (Kd: 54 μM) and IgM (Kd: 17 μM) (57, 58). These affinities are substantially lower than that of IgG binding, which has been measured at Kd: 37 nM for TRIM21-Fc fragment interactions (25). Importantly, except for IgG binding to its cognate high-affinity FcγRI (Kd: 4.2 nm), most FcRs bind IgG Fc with lower-affinity binding in the micromolar range (59, 60). Therefore, the nanomolar TRIM21-Fc binding highlights the strength and high affinity of IgG for this intracellular receptor. Whether TRIM21 can bind to IgE or IgD remains to be investigated (24, 25).
The TRIM21 PRY/SPRY domain is also subject to regulation by acetylation. Histone deacetylase 6 (HDAC6) interacts with TRIM21 via the PRY/SPRY domain, where it deacetylases TRIM21 at K385 and K387, promoting TRIM21 homodimerization. When HDAC6 is inhibited, TRIM21 remains hyperacetylated, preventing TRIM21 dimerisation, thus impairing its enzymatic activity (61).
Antibodies extracellularly neutralise antigens, preventing targeted pathogens from penetrating cell membranes. This occurs either directly or indirectly through opsonisation and/or complement system activation, whilst antibody-mediated neutralisation within intracellular compartments was thought unlikely due to membrane exclusion (62). Extracellular neutralisation is not always possible, as many neutralising epitopes are shielded, for example by glycans (63).
In vivo studies have demonstrated that even in the presence of saturating concentrations of neutralising antibody, TRIM21-deficient mice are highly susceptible to mouse adenovirus 1 infection. In contrast, wild-type mice upregulate TRIM21, thus controlling viraemia. Interestingly, Trim21+/- heterozygous mice display an intermediate phenotype, leading to an increased viral load but lower than that of Trim21-/- mice, suggesting that TRIM21 levels directly influence the efficiency of antiviral defence (64).
When bound to non-neutralising antibodies, intracellular pathogens including viruses, bacteria and parasites, may be rapidly sensed by cytosolic TRIM21 (64–67). The mechanisms by which antibody-bound pathogens access the cytoplasm vary and are pathogen-specific (68). Some enveloped viruses, such as the α-herpesvirus HSV, deliver their capsids directly to the cytosol by fusing their envelope with the plasma membrane (69). Others use endocytosis pathways such as clathrin-mediated endocytosis, micropinocytosis or lipid raft-mediated endocytosis (70). Multiple bacterial species use caveolin-mediated endocytosis to enter cells, including E. coli, C. jejuni, S. typhimurium, and P. aeruginosa, as have a number of viruses, such as SV40 (71). This mechanism might be preferred for pathogens, as unlike clathrin-coated pit entry, caveolae-internalized bacteria may avoid lysosomal degradation (72).
TRIM21 binds internalised antibody-coated pathogens within the cell via PRY/SPRY-Fc interactions. However, why and how antibody-coated pathogens are available for binding in the cytosol, remains largely unsolved. TRIM21 recruitment may be limited to non-enveloped DNA and RNA-viruses that enter the cytosol with attached immunoglobulins. McEwan et al. showed that the enveloped Respiratory Syncytial virus shed attached antibodies upon entry and did not activate TRIM21, whereas non-enveloped feline calicivirus did (73). Additionally, TRIM21 can detect picornavirus HRV14, which promotes the lysis of endosomal membranes for cytosolic virion release, whilst it cannot not detect HRV2, which delivers its genome into the cytoplasm through a pore (74).
TRIM21 facilitates antibody-dependent intracellular neutralisation of human adenovirus type 5 (Ad5). Ad5 bound to as few as 1.6 antibody molecules recruits TRIM21 in the cytosol and is degraded in a TRIM21-dependent manner. This involves TRIM21 autoubiquitination and possibly ubiquitin transfer also to the TRIM21-associated viral particle prior to proteasomal degradation, Figure 2 (58, 67). Adenoviral capsids can be released from early endosomes into the cytosol during “endosomal escape” (75). This stepwise endosomal rupture process is promoted after initial engagement of Ad5 with coxsackievirus adenovirus receptor (CAR) and coreceptor αv integrin at the host cell membrane (76). Interaction with these receptors may promote early capsid disassembly, occurring just as Ad5 enters the cell membrane (77, 78). Disassembly leads to the exposure of protein VI, a lytic factor which disrupts the endosomal membrane, releasing the capsid into the cytosol for subsequent TRIM21 engagement, Figure 2 (58, 79).
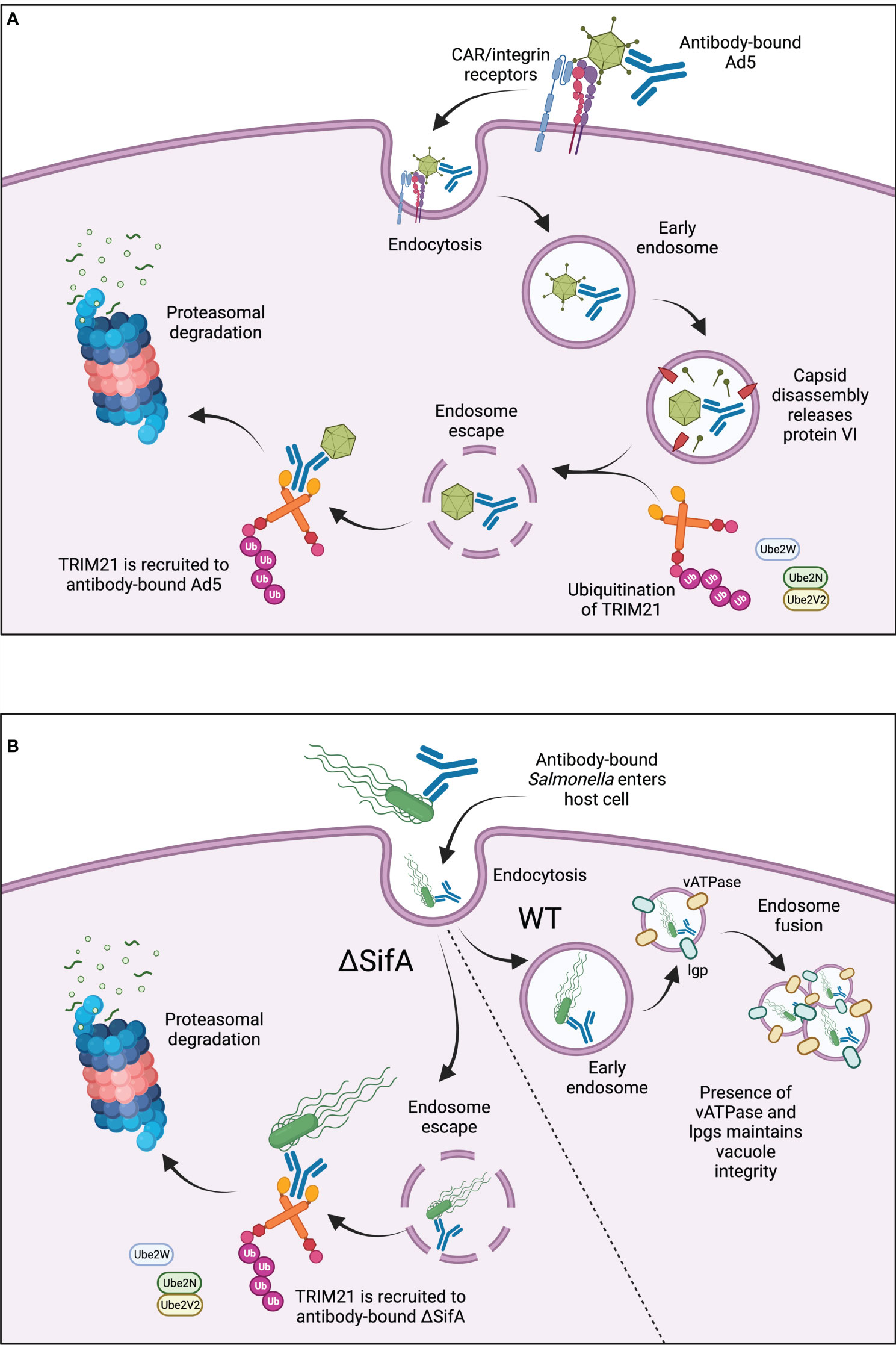
Figure 2 (A) TRIM21 in antiviral responses. TRIM21 is recruited to cytosolic antibody-bound-Ad5 after virus entry into the cell. Ad5 binds to CAR/αv integrin receptors at the cell surface, triggering capsid disassembly upon cell entry. Lytic protein VI is released leading to endosomal disassembly and “endosomal escape” of the antibody-bound-Ad5. Active, ubiquitinated TRIM21 binds to the Fc region of the antibody and promotes subsequent proteasomal degradation. (B) TRIM21 in responses to intracellular bacteria. TRIM21 is recruited to antibody-coated SifA-mutant S. typhimurium. Wild-type S. typhimurium are protected from TRIM21-mediated degradation due to the presence of vATPase and lgps, which maintain SCV integrity and enable endosome fusion for bacterial replication. ΔSifA bacteria cannot maintain SCV integrity, leading to endosomal escape into the cytosol for TRIM21-mediated degradation.
TRIM21 also localises with antibody-bound S. typhimurium in the cytosol, Figure 2 (73). Normally within host cells, S. typhimurium grows within Salmonella-containing vacuoles (SCVs) which protect the bacteria from antibacterial responses (80). However, mutant S. typhimurium defective in SifA, an effector translocated by type III secretion systems, have been identified. ΔSifA S. typhimurium cannot maintain SCV membrane integrity due to an absence of lysosomal membrane glycoproteins (lgps) and vacuolar ATPase (vATPase), necessary for membrane fusion during bacterial replication. Therefore, ΔSifA mutants are released into the cytosol, Figure 2 (81). Surprisingly, a follow-up study showed that ΔSifA S. typhimurium actually replicated more rapidly within the cytosol of epithelial cells compared to those residing within vacuoles, suggesting there may be some bacterial advantage for vacuole-escape (82). TRIM21 has been shown to recognise antibody-bound-ΔSifA mutants more readily than wild-type S. typhimurium, leading to greater activation of NF-κB proinflammatory signalling (73). Although the bactericidal effects of this recognition and enhanced signalling were not explored, it does suggest TRIM21 is important for rapid intracellular detection and binding of pathogens which escape into the cytosol, Figure 2.
By binding IgM, TRIM21 provides protection from primary infections, and by binding IgG it can protect against secondary infection. In vivo challenge protection has been demonstrated using antibodies targeting the viral nucleoprotein of various viruses including influenza, coronaviruses, and lymphocytic choriomeningitis virus (LCMV) (65, 83–89). Nucleoprotein-specific antibodies are non-neutralising, as viral nucleoprotein is an internal virus antigen, and protection does not require the FcγR (90, 91). TRIM21 and anti-nucleoprotein antibodies play a critical role in stimulating nucleoprotein-specific cytotoxic T cells during LCMV infection (65).
Once bound to cytosolic antibody-coated pathogens, TRIM21 induces coordinated effector and signalling responses, both dependent on TRIM21’s E3 ubiquitin ligase activity. Upon cellular infection with Ad5-antibody complexes, two independent, concurrent activities occur. Firstly, the effector response results in proteasomal degradation of the virus. Secondly, an intracellular antiviral response is induced via innate-immune signalling molecules such as IFN-I (67, 68). Additionally, TRIM21 initiates signalling cascades, activating the transcription factor NF-κB, upregulating more inflammatory cytokines and inducing an antiviral state (73).
Some viruses have hijacked TRIM21 to enhance their replication. In the case of severe fever with thrombocytopenia syndrome virus, its non-structural protein (NSs) interacts with and inhibits TRIM21. This prevents the activation of nuclear factor erythroid 2-related factor 2, which is responsible for the expression of a number of antioxidant effectors, thus promoting viral replication and pathogenesis (92). Enterovirus 71 (EV71) is restricted by the host restriction factor SAMHD1. Upon infection, EV71 upregulates TRIM21 in an IFN-dependent manner; TRIM21 interacts with and degrades SAMHD1 through K48-ubiquitination and proteasomal degradation, thus promoting EV71 replication (93). Human papillomavirus presents an alternative method for immune escape by utilising the oncoprotein E7. This recruits TRIM21 to promote the K33-linked ubiquitination and degradation of the host IFI16 inflammasome (94).
TRIM21 also has indirect effects on innate immunity by promoting pattern recognition receptor (PRR) detection of exposed immunostimulatory ligands that are otherwise shielded by viral capsids. This induces two waves of TRIM21-dependent transcription, the first caused by TRIM21 antigen recognition via Fc binding and a later wave following recognition of viral PAMPs by cGAS and STING (74). TRIM21 also interacts through its PRY/SPRY domain with another PRR, MAVS. TRIM21 is upregulated following early detection of host cell viral invasion from Hepatitis C virus, Newcastle disease virus or Sendai virus (SEV). Then its RING domain conjugates K27-linked polyubiquitin onto MAVS, promoting TBK1 binding for subsequent MAVS downstream signalling (95, 96).
Effects of TRIM21 on virus-encoded proteins are less well-understood than those of the IFN pathway. TRIM21 reportedly interacts with hepatitis B virus (HBV) to prevent HBV DNA replication, by promoting the K48-linked ubiquitination and degradation of HBV DNA Pol (97). Additionally, Porcine Epidemic Diarrhoea Virus (PEDV) is inhibited by TRIM21, which targets the nucleoprotein for proteasomal degradation. Conversely, following in vitro PEDV infection, the endogenous expression of TRIM21 was downregulated, increasing both PEDV viral titres and nucleoprotein levels (98).
Co-immunoprecipitation has shown TRIM21 to interact with intracellular proteins that regulate antiviral responses. This includes direct interactions with IFN-inducible protein 35 (IFI35) and indirectly, the N-Myc and STAT interactor (NMI). Subsequent K63-linked ubiquitination of NMI stabilises the TRIM21/IFI35/NMI complex and downregulates innate antiviral signalling pathways by inhibiting IFN-I production (99).
IFNs bind to cell surface receptors, triggering signalling cascades for downstream transcription of genes involved in innate and adaptive immunity. IFN-α and IFN-β are type I IFNs (IFN-I) and IFN-γ is type II (IFN-II) (100). Early indications that TRIM21 is regulated by IFN signalling were shown by Rhodes et al. In unstimulated HeLa cells, TRIM21 was detected at low concentrations, whilst TRIM21 mRNA was rapidly upregulated in response to IFN-II (101). Furthermore, TRIM21 was upregulated in cultured macrophages and DCs, in response to either influenza virus infection or CpG oligonucleotide TLR9 agonists. TRIM21 induction was dependent on IFN-I signalling, as it was diminished in IFNAR-/- cells (102). IFN-I and to a lesser extent IFN-II, induced TRIM21 mRNA in the mouse T cell line EL4. Promoter-mapping and luciferase-reporter experiments identified an IFN-stimulated response element (ISRE) in the TRIM21 promoter, Figure 3 (103).
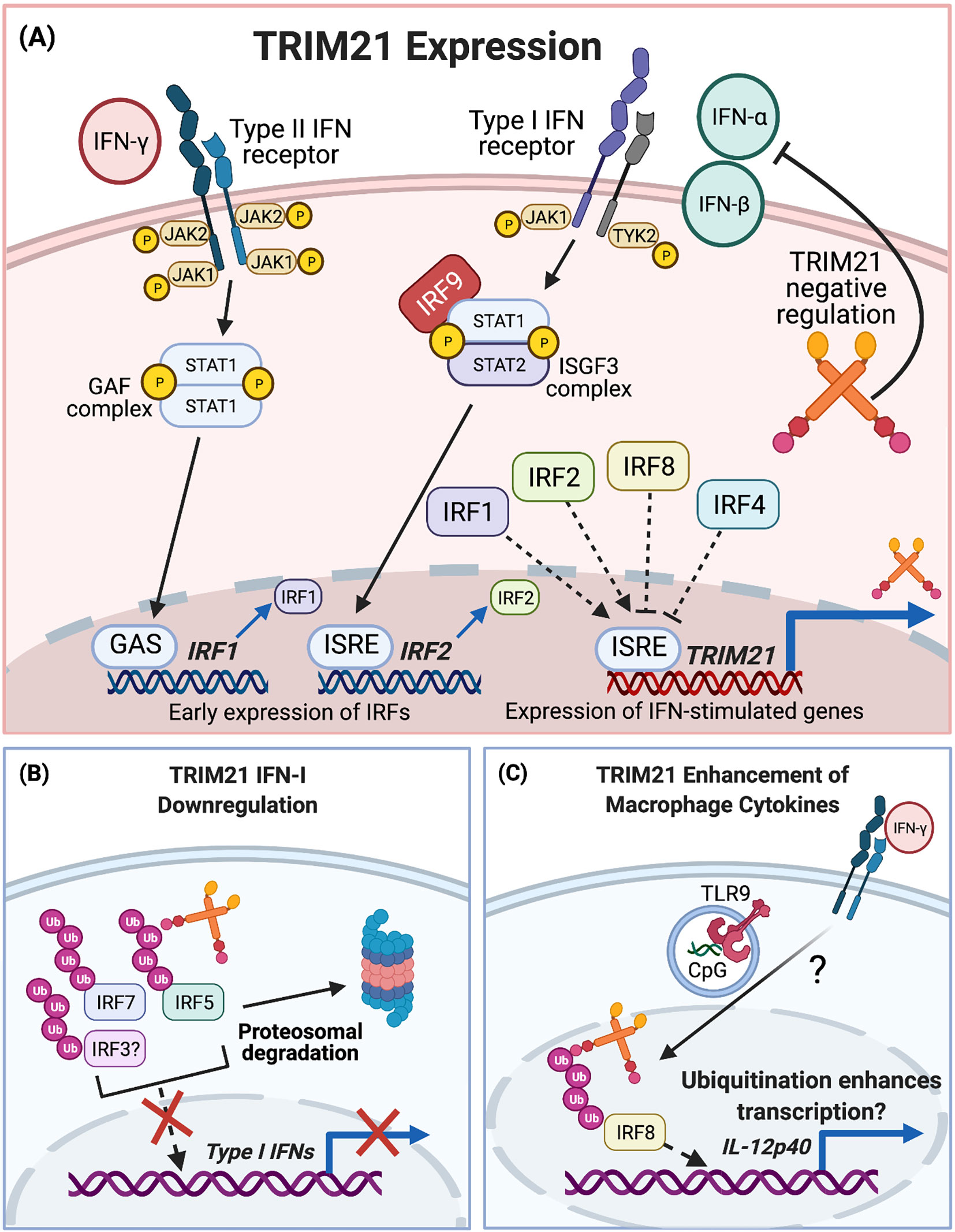
Figure 3 TRIM21 and IFN signalling. (A) TRIM21 expression is upregulated by IFN-I and IFN-II signalling. IFN-I signalling involves JAK/STAT phosphorylation and ISGF3 complex formation for early IRF expression. Nuclear translocation of IRF1/2 and binding to the ISRE leads to expression of IFN-stimulated genes including TRIM21. IFN-II signalling also occurs via JAK/STAT signalling, leading to formation of the GAF complex. This binds to GAS elements for IRF1 expression and subsequent TRIM21 expression. TRIM21 upregulation is inhibited by IRF4 and IRF8, although much of the upstream signalling pathway remains to be fully elucidated. (B) After upregulation, TRIM21 downregulates the IFN-I response via ubiquitination of IRF7, IRF5 and possibly IRF3. (C) TRIM21 enhances proinflammatory macrophage cytokine expression after IRF8 ubiquitination.
IFN-I stimulation induces phosphorylation of the JAK1 and TYK2 kinases, which in turn phosphorylate STAT1 and STAT2. pSTAT1 and pSTAT2 complex with IRF9, forming the IFN-stimulated gene factor 3 (ISGF3) complex which translocates to the nucleus. ISGF3 activates transcription by binding ISREs in promoters of IFN-stimulated genes, Figure 3 (104). IFN-II signalling induces JAK1 and JAK2 phosphorylation, leading to STAT1 phosphorylation and the formation of pSTAT1 homodimers, referred to as the gamma-activated factor (GAF) complex. GAF translocates to the nucleus, binding to gamma IFN activation sites (GAS) to activate transcription, Figure 3 (105).
Both IRF1 and IRF2 bind to a TRIM21 gene ISRE to upregulate TRIM21 expression, Figure 3 (103, 106). Despite the abrogation of IFN-II-stimulated TRIM21 induction in IRF1-/- macrophages, the TRIM21 locus does not contain a GAS site. This suggests IFN-II stimulates TRIM21 expression by an indirect mechanism involving IRF1. TRIM21 expression was inhibited by IRF4 and IRF8, which bound to the ISRE and blocked IRF1 and IRF2-mediated TRIM21 upregulation, Figure 3 (103). The roles of non-canonical IFN signal transduction pathways in TRIM21 gene regulation are not yet known (107).
TRIM21 participates in a negative feedback loop to downregulate IFN-I production, modulating the IFN response and inhibiting further TRIM21 expression, Figure 3 (108, 109). Downregulation of IFN-I results from TRIM21-mediated IRF7 ubiquitination and subsequent proteasomal degradation, Figure 3. In TRIM21-deficient cells, these inhibitory effects were abrogated and the IFN-α4 promoter was overstimulated by IRF7 (109).
Similarly, TRIM21 ubiquitinates IRF5 to mediate its degradation and prevent IRF5-driven IFN-α expression, Figure 3 (110, 111). Interestingly, one study showed that although TRIM21 interacted with and ubiquitinated all IRF5 isoforms studied, the subsequent effects on protein stability were isoform specific. For example, IRF5 isoforms lacking 48 nucleotides in a central Proline, Glutamic acid, Serine, Threonine-rich (PEST) domain were ubiquitinated by TRIM21 but resistant to subsequent degradation (110). Increased stability of certain isoforms may have relevance in SLE pathogenesis, whereby patients show enhanced expression and alternative splicing of IRF5, due to increased spliceosome activity (112). IRF5 polymorphisms affecting PEST domain expression are associated with elevated risk for SLE and may modulate IFN-I promoter activity. However, it remains unclear whether these isoforms contribute to SLE pathology through increased IFN-I expression (113).
Evidence for IFN-β downregulation following TRIM21-mediated IRF3 degradation, is more conflicting. In a human glial cell line (CHE3), Japanese encephalitis virus infection stimulated IFN-β upregulation via IRF3 phosphorylation. TRIM21 overexpression abrogated IFN-β induction, whilst TRIM21 silencing enhanced it (108). These findings agreed with a previous study in HEK293 cells, whereby IRF3-induced IFN-β expression was negatively regulated by TRIM21 following SEV infection, or stimulation by LPS or the TLR3 agonist polyI:C, Figure 3 (114). In contrast, after SEV infection of HEK293 cells, TRIM21 bound IRF3, blocking Pin1-mediated pIRF3 degradation and enhancing IRF3-stimulated IFN-β expression (115, 116). Reasons for these conflicting results remain unknown and the impact of TRIM21-mediated ubiquitination on the stability of IRF3 is uncertain.
Studies have also investigated IFN-mediated upregulation of TRIM21 and the downstream effects leading to apoptosis and macrophage cytokine expression (117, 118). Imaging using fluorescent anti-TRIM21 monoclonal antibodies showed that IFN-I stimulation of HeLa cells led to increased cytoplasmic TRIM21 protein levels. After 48 hours of stimulation TRIM21 had undergone nuclear translocation which preceded apoptosis, detected by TUNEL-assays in parallel-treated cultures (117). However, whether TRIM21 nuclear translocation actively induced apoptosis remains to be confirmed. The mechanisms of nuclear translocation, and the functions, if any, of intranuclear TRIM21 are yet to be explored.
Intranuclear roles of TRIM21 have been demonstrated in macrophages following IFN-II and TLR stimulation. IRF8-TRIM21 complexes were identified in nuclear extracts of macrophages stimulated with IFN-II plus TLR9 agonist CpG oligonucleotides (118, 119). This was verified in live macrophages whereby YFP fluorescent signals dependent on IRF8-TRIM21 interactions were detected in the nucleus, also corresponding to increased IRF8 ubiquitination. However, unlike previous reports, this led to enhanced proinflammatory IL-12p40 expression, suggesting that ubiquitination actually enhances IRF8-mediated transcription, Figure 3 (118). Much of the upstream signalling pathway and how IRF8-ubiquitination enhances IL-12p40 transcription remains unclear. It may be worth confirming whether similar examples of enhanced TRIM21-mediated transcription exist for other proinflammatory gene targets in activated macrophages. These could include promotors requiring coordinated regulation by both IRF8 and IRF1, including SOCS7 for NF-κB signalling, H28 for antigen presentation, c-Myc for cell growth and survival, and CXCL16, which is elevated in SLE patient sera and expressed by macrophages for adhesion, chemotaxis and inducing inflammatory cell infiltration (120, 121).
Inducing IFN-I signalling helps eliminate virus and prevents host morbidity and mortality (122–124). However, IFN levels must be balanced; too much may over-stimulate innate immunity and inflammation, leading to pathological outcomes including autoimmunity. Therefore, signal termination is necessary to prevent an excessive IFN response. TRIM21 controls innate antiviral responses by degrading the transcription factors IRF7 and IRF3 (108, 125, 126). For Influenza A virus, NMI is upregulated, promoting TRIM21 ubiquitination of IRF7 (125), whilst for SEV infection, FoxO1 destabilises IRF3 by promoting TRIM22 or TRIM21-mediated proteasomal degradation, following K-48 ubiquitination (126).
Autoimmunity as a result of aberrations in these fine-tuned signalling pathways has been demonstrated for SS using gene expression array analysis. Compared to healthy controls, PBMCs isolated from SS patients displayed elevated TRIM21 transcript expression, correlating with increased IRF1 and IRF2 mRNA (103). One might expect enhanced ubiquitination of TRIM21-regulated IRFs, leading to greater downregulation of the IFN response, yet this was not the case. Instead, IRF3 and IRF5 protein levels remained high even in the presence of increased TRIM21. This may suggest other signalling pathways contribute to impaired proteasomal degradation and altered IFN signalling homeostasis in autoimmune individuals (127). A clearer understanding of these signalling pathways is needed.
TRIM21 downregulation has been implicated in unfavourable outcomes for multiple cancer types including breast cancer, hepatocellular carcinoma, diffuse large B-cell lymphomas (DLBCL) and most recently, colitis-associated cancers (128–131). Lower TRIM21 mRNA expression was correlated with reduced patient survival in DLBCLs and in hepatocellular carcinomas; in vitro studies implicated enhanced cell proliferation as a possible cause (129, 130). In liver cancer cell lines, siRNA-mediated TRIM21 silencing abrogated apoptosis, whilst promoting cellular proliferation and migration in a transwell system (129). Conversely, TRIM21 overexpression in a B cell line reduced proliferation and increased apoptosis compared to control cells, and promoted apoptosis following anti-CD40 treatment (16). Together, these studies suggest TRIM21 has tumour suppressor capabilities, influencing cellular and tissue homeostasis.
Autoantibodies against nuclear and cytoplasmic autoantigens have been detected in cancer patients, with possible causal mechanisms including autoantigen overexpression from tumour cells and autoantigen release following chemotherapy-induced cell death (132). Recently, anti-TRIM21 antibodies have been detected in patients with ovarian cancer, the presence of which correlated with better patient survival. Analysis showed these antibodies differed in epitope profile to anti-TRIM21 antibodies detected in SLE and SS, although why this is the case remains to be discovered (133).
Dysregulation of B cell function has been identified in both SS and SLE patients. This is characterised by higher levels of circulating activated B cells which express co-stimulatory markers and secrete autoantibodies, driving inflammation and autoimmune pathology (2). Similarly to the cancer studies, TRIM21 deficiency has been explored as a potential driver of SLE, by altering B cell regulation. TRIM21-/- mice have been developed with differing phenotypes, most likely due to specific gene disruption strategies (134). One knockout showed no phenotypic abnormalities, likely due to compensatory upregulation of related TRIMs (135). However, other TRIM21-/- mice showed inflammatory phenotypes when subjected to minor injury by ear tagging. They developed site-specific severe dermatitis and displayed SLE-specific autoimmune characteristics, including elevated proinflammatory cytokine production (136). This discrepancy may occur due to production of a truncated TRIM21 protein (RING, B-box and partial coiled-coil domain) in the second strategy, which enabled and promoted autoantibody production.
Another study showed how TRIM21 gene disruption in a lupus-prone mouse model (TRIM21−/−MRL/lpr) altered SLE pathology. Compared to lupus-prone (MRL/lpr) mice retaining functional TRIM21 activity, the spleens of TRIM21−/−MRL/lpr mice had significantly increased mature B cell numbers. Once isolated, these cells differentiated into antibody-secreting plasma cells at higher rates in vitro, compared to cells from the MRL/lpr spleens (137). One model proposed TRIM21 may regulate plasma cell differentiation by ubiquitinating IRF5, thus blocking IRF5-induced upregulation of IRF4 and subsequent Blimp-1 mRNA expression, Figure 4 (110, 137–139). However, much of this pathway, especially how IRF5 alters IRF4 expression and whether TRIM21 has a definitive role in this IRF4/5 axis, requires clarification.
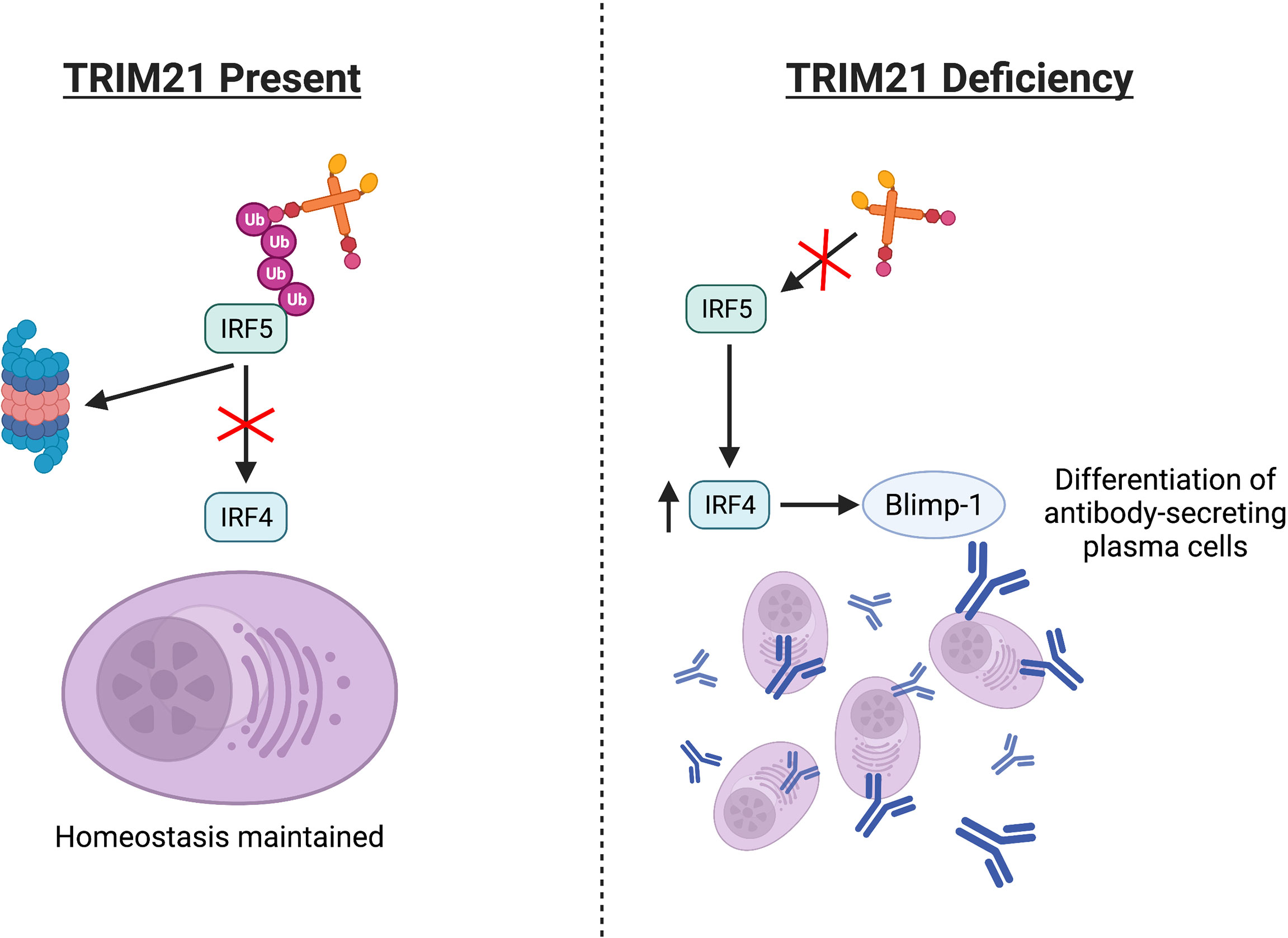
Figure 4 Absence or deficiencies in TRIM21 may alter the IRF4/5 axis. TRIM21 regulates IRF5 via ubiquitination, potentially affecting downstream IRF4 and Blimp-1 expression. Aberrant regulation may increase differentiation of antibody-secreting plasma cells.
Alternatively to expression deficiencies, impaired TRIM21 function may involve antibody-dependent mechanisms. For example, anti-TRIM21 seropositivity in SLE patients correlates with impaired TRIM21 ubiquitinylating activity. PBMCs isolated from anti-TRIM21 seropositive patients display enhanced IFN-I gene expression, suggesting an association between the presence of TRIM21 autoantibodies and attenuated negative regulation of IFN-pathway proteins by TRIM21 (127).
How autoantibodies may downregulate native TRIM21 function has been explored in vitro. In a cell-free system, anti-TRIM21 antibodies from SS patients sterically hindered TRIM21’s interaction with the E2 ubiquitin-conjugating enzyme, preventing E3 ligase autoubiquitination via the RING domain (140). This may impair the subsequent ubiquitination of target proteins such as IRF5, although whether anti-TRIM21 antibodies can indeed access and inhibit TRIM21 in vivo has not been directly tested.
Under normal conditions, TRIM21 is uniformly distributed throughout the cytoplasm, with small amounts detected within the nucleus (141). Thus, whether and how autoantibodies access this cytoplasmic protein remains a key question for TRIM21-associated autoimmune diseases.
Recently, TRIM21 was detected at the surface of antigen-presenting cells in pSS patient blood (142). Anti-TRIM21 antibodies are associated with pSS pathogenesis, inducing damage and driving inflammation. This could involve recognition of anti-TRIM21 Fc domains by lymphocytes, driving inflammatory pathways such as antibody-dependent cellular cytotoxicity (143, 144). Therefore, cell surface TRIM21 might exacerbate proinflammatory pathways in autoimmune diseases.
IFN-β, the most potent inducer of TRIM21 expression, induces changes in TRIM21 expression and cellular location in PBMCs (142, 145). TRIM21 was strongly upregulated both intracellularly and at monocyte cell surfaces, with a modest increase observed in plasmacytoid DCs (142). Although this evidence suggests IFN upregulates TRIM21 expression, it remains unclear how TRIM21 may be transported and expressed at the cell surface.
These recent novel findings contradict previous publications suggesting TRIM21 is only exposed at the cell surface during cell death, with anti-TRIM21 antibodies possibly driving apoptosis (141–143). TRIM21 exposure following apoptosis has been demonstrated in the context of foetal CHB which is strongly associated with maternal SLE and SS (146). High titres of cross-reactive maternal anti-TRIM21 antibodies induced apoptosis of foetal cardiomyocytes in vitro, Figure 5 (9). Although CHB pathogenesis remains unclear, one proposed mechanism involves cross-reactive, inhibitory binding of maternal anti-Ro52/60 and anti-La antibodies to α1-subunits of L-Type foetal cardiomyocyte calcium channels (147). Channel inhibition may then dysregulate calcium signalling, leading to abnormal atrioventricular conduction and eventual apoptosis, Figure 5 (148, 149). Apoptosis of cultured, non-permeabilised human foetal cardiomyocytes led to the redistribution of Ro52/60 and La antigens into apoptotic blebs and their emergence at the cell surface (150). Furthermore, in a co-culture system, apoptotic foetal cardiomyocytes treated with anti-Ro/La antibodies stimulated macrophages to secrete TNF-α (151). Apoptotic blebs rich in autoantigens such as TRIM21 may be insufficiently cleared, facilitating antigen-autoantibody interactions, or enabling DC ingestion and presentation to activate T cells and autoreactive B cells, Figure 5 (152–154).
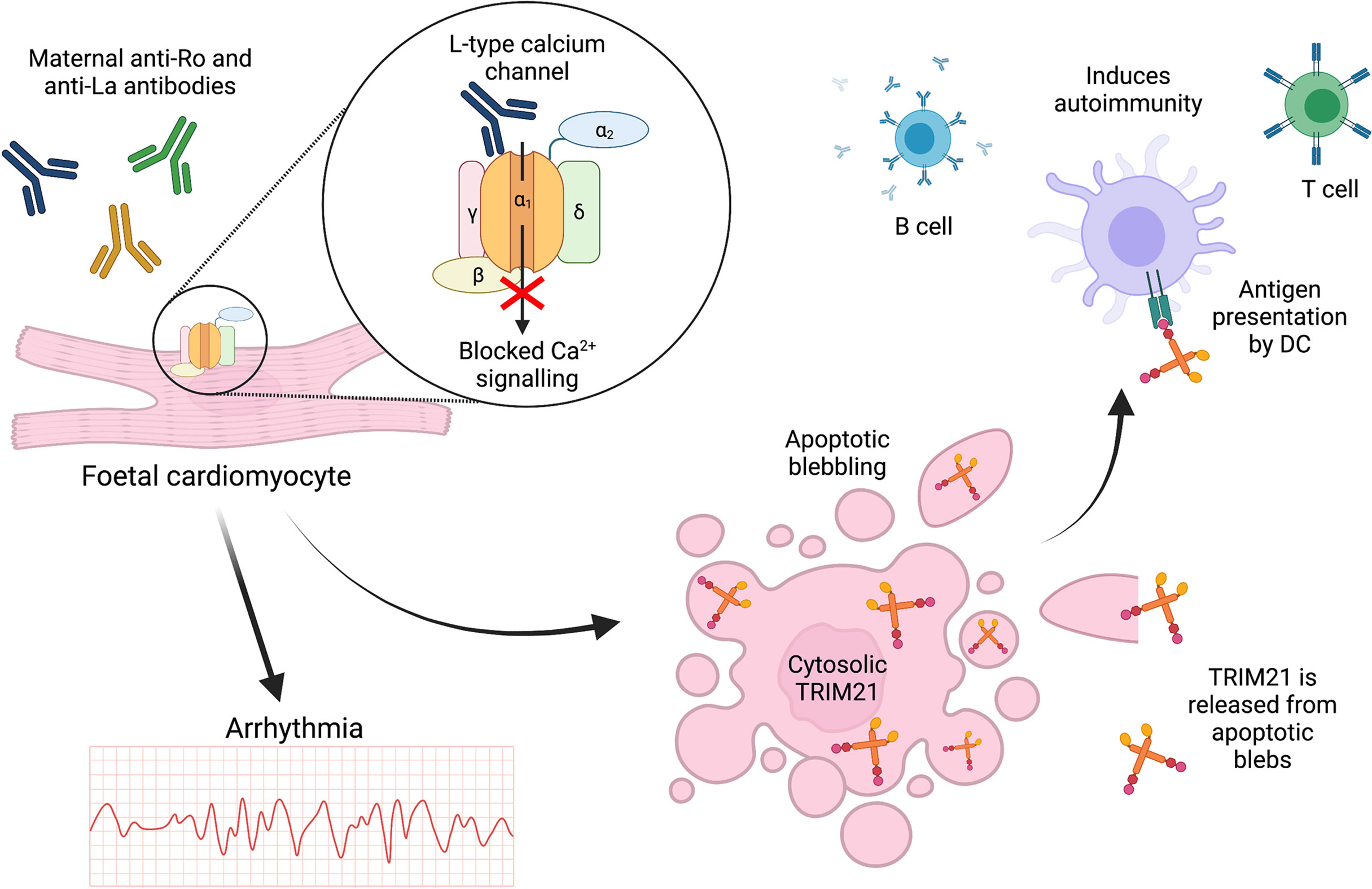
Figure 5 TRIM21 exposure at the cell surface may occur due to apoptosis. In foetal CHB, cross-reactive maternal autoantibodies may bind to and block L-type calcium channels leading to arrhythmia and cardiomyocyte apoptosis. This releases intracellular antigens such as TRIM21 for antigen presentation, driving subsequent autoimmunity.
TRIM21 may also be exposed in non-apoptotic cells following UV-B/C exposure or oxidative stress (155, 156). For example, UV-irradiated keratinocytes displayed TRIM21 at the cell surface, a phenomenon which was inhibited in a concentration-dependent manner by the reactive oxygen species (ROS) scavenger N-acetyl-L-cysteine (157, 158). Surface TRIM21 exposure was also induced in keratinocytes treated with diamide, a thiol-oxidising agent (157). Diamide disrupts intracellular glutathione redox reactions, inducing oxidative stress and promoting mitochondrial-induced apoptosis pathways (159). Together, these results indicate that oxidative stress and altered redox homeostasis may induce cell surface TRIM21 exposure. Increased expression of molecular chaperones may also be involved, aiding TRIM21 protein synthesis and translocation, although this has not been confirmed experimentally (157).
Oxidative damage has been identified in the salivary glands of SS patients, with increased saliva detection of the oxidative stress biomarker 8-Hydroxy-2-deoxyguanosine, generated following ROS-induced oxidation of DNA guanine residues (160, 161). The findings by Saegusa et al., suggest that ROS may drive increased exposure of TRIM21 during SS pathogenesis (157). Other studies have confirmed the translocation of SLE autoantigens to keratinocyte cell surfaces following stimulation by UV-B, heat-shock or certain inflammatory cytokines (144). However, the optimal irradiation dose (200mJ/cm2) used in one study far exceeds levels required to induce significant keratinocyte cell death (162, 163). Thus, the autoantigen redistribution observed in these studies might have occurred during early apoptotic events.
Whilst the mechanism by which TRIM21 is translocated to the surface remains uncertain, one study reported that TRIM21 cytoplasmic bodies localise along microtubule networks. Visualised using live cell fluorescence microscopy, these bodies were highly mobile, undergoing multidirectional movements across a range of distances within the cytosol. However, motility was only observed beneath the plasma membrane and not at the cell surface (164). Therefore, whether this motility contributes to TRIM21 membrane exposure in response to stimulation or oxidative stress remains unclear.
Together these reports suggest that TRIM21 translocation to the cell surface may occur following apoptosis or cellular stress. Coming back to the recent findings by Hillen et al., it is possible the observed TRIM21 surface expression actually occurred during early apoptotic events (142). Repeating this study using Annexin V as a discriminator between apoptotic and non-apoptotic cells may elucidate whether TRIM21 is indeed expressed at the cell surface under non-apoptotic conditions (165).
Defective tolerance mechanisms, elevated antigen presentation and inflammatory cytokine expression are thought to drive autoreactive B cell activation, leading to pathogenic autoantibody production (166). Therefore, it is noteworthy that TRIM21 protein levels are significantly elevated in the salivary gland ductal epithelium of SS patients, correlating with inflammation and lymphocyte infiltration (167). Studies have directly correlated serum anti-TRIM21 antibody levels with disease severity. Higher anti-TRIM21 autoantibody titres in pSS are associated with greater localised and systemic disease manifestations including parotid enlargement, haematological abnormalities and central nervous system involvement (168). Studies of patients undergoing hematopoietic stem cell transplantation suggest a link between elevated BAFF expression and anti-TRIM21 autoantibodies (169). Increased BAFF may support the survival of TRIM21-targeting autoreactive memory B cells. However, questions remain as to where this tolerance breakdown occurs, whether TRIM21 specifically drives autoreactivity and whether BAFF expression is necessary for anti-TRIM21 responses.
Another potential mechanism driving anti-TRIM21 autoantibody production is epitope spreading, whereby during chronic inflammation, the exposure of normally sequestered self-antigens induces a secondary immune response against endogenous epitopes (170, 171). Such epitope spreading was demonstrated in mice immunised with the Ro60 autoantigen plus Freund’s complete adjuvant. After repeated Ro60 booster immunizations, mice developed an antibody response against the unrelated TRIM21/Ro52 autoantigen (172). This corresponds with SS patient data whereby individuals may display anti-TRIM21 antibodies alone, or in combination with anti-Ro60 antibodies (173). Interestingly, the autoantibodies from anti-TRIM21/anti-Ro60 doubly-seropositive patients exhibit a distinct epitope profile with frequent reactivity to the TRIM21 RING domain, in contrast to patients reactive to TRIM21 alone (174). Thus, Ro60 autoimmune reactivity might influence subsequent reactivity against TRIM21.
Importantly, TRIM21 and Ro60 are structurally and functionally distinct proteins occupying different cellular compartments. Ro60 (TROVE2) a ring-shaped protein which forms ribonucleoprotein complexes with non-coding (Y) RNA. By binding misfolded Y-RNAs, Ro60 may assist in their degradation (175). Direct interactions and complex formation between Ro60 and TRIM21/Ro52 have not been established, although transient interactions may be possible (175, 176). One hypothesis for how epitope spreading could induce double-positive TRIM21/Ro60 autoreactivity, is that if an autoimmune patient displays reactivity against one of these antigens (Ro60 or TRIM21), transient antigen interactions may expose epitopes from the other antigen, driving a secondary autoimmune response (177). However, questions remain regarding the autoimmune contributions of reactivity against certain TRIM21 epitopes and why only some autoimmune patients are double-positive for anti-Ro60 and anti-TRIM21 antibodies.
Autoimmune induction of anti-TRIM21 antibodies has been explored in mouse models (178). In particular, the New Zealand Mixed 2758 (NZM2758) mouse strain, obtained by backcrossing and selectively breeding New Zealand Black and New Zealand White mice, has been used to model SS development (179, 180). These mice display classical SS manifestations including glandular histopathology and dysfunction, lymphocyte infiltration, inflammation and autoantibody production (180). TRIM21-immunized NZM2758 mice developed salivary gland dysfunction which correlated with levels of anti-TRIM21 produced (181). Passive serum transfer from TRIM21-immunized NZM2758 mice induced salivary gland dysfunction in adjuvant-primed (intraperitoneally), non-immunized recipient NZM2758 mice (181).
Of note, TRIM21 immunization induced IgG deposition in, and dysfunction of, the lacrimal glands in female but not male NZM2758 mice (182). This observation is interesting in light of the known sex differences in pSS presentation, with women accounting for approximately 90% of cases but men typically presenting with more severe disease (183). Male mice in the Trzeciak et al., study showed no significant clinical disease and therefore do not model the severe disease observed in men with pSS (182). Sex differences in the pathogenesis of rheumatic diseases such as SS and SLE may be related to hormonal effects (184) and to gene dosage effects of immune-related genes such as TLR7 on the X chromosome (185).
Immunisation of NZM2758 mice with MBP-fusion proteins corresponding to different murine TRIM21 domains, was used to determine anti-TRIM21 antibody epitope targets and how they relate to autoimmune pathologies. No differences were detected in the levels of autoantibodies generated against the different TRIM21 domains, however certain epitopes correlated with salivary gland dysfunction in these mice. Specifically, antibodies raised against the coiled-coil domain of TRIM21 reduced saliva production, suggesting a link between specific antibody targets and SS pathology (186). This corresponds with patient findings, confirming that the coiled-coil domain is the most antigenic epitope. For example, 97% of patients with autoimmune rheumatic diseases, and who are also anti-TRIM21 antibody positive, generate antibodies targeting the coiled-coil domain, Figure 1 (187).
Extensive SS-patient epitope analysis has demonstrated the presence of autoantibodies directed against other TRIM21 regions, including the immunoglobulin binding C-terminal (PRY/SPRY) domain, Figure 1. Using pull-down assays, it was demonstrated that antibodies from patient serum bound with high affinity to the C-terminal region of both wild-type TRIM21 and a mutant which cannot bind to immunoglobulin Fc. One model proposes that TRIM21’s role in targeting antibody-coated pathogens for proteasomal degradation may promote autoantibody production against these antigenic regions (24, 188). However, whether or not autoantibodies directed against the C-terminal domain are pathogenic and actively drive SS clinical manifestations (e.g. glandular dysfunction), remains to be elucidated.
TRIM21 is an intracellular receptor that binds with high affinity to immunoglobulin Fc regions via its PRY/SPRY domains. This binding is vital for intracellular detection of pathogens that escape extracellular antibody neutralisation, enter host cells and access the cytosol. Subsequently, ubiquitination targets the pathogens for proteasomal degradation and immune signalling is activated. In particular, TRIM21 is both enhanced by and regulates IFN signalling for effective immune responses to infection. This also includes the upregulation of other proinflammatory transcription factors such as NF-κB, inducing an antiviral state for pathogen destruction.
TRIM21 dysregulation has been implicated in cancers and autoimmune diseases which include SS and SLE. In particular, excessive IFN responses are correlated with elevated TRIM21 transcript expression. However, these findings are seemingly counter-intuitive as more TRIM21 should promote greater IRF ubiquitination, thus decreasing the IFN response. Therefore, clearer understanding of TRIM21’s roles in these signalling pathways is required to explain these contradictory findings.
The associations between anti-TRIM21 seropositivity and impaired intracellular TRIM21 ubiquitination activities are intriguing. However, the main question these findings raise is how autoantibodies are generated against an intracellular antigen. Whether TRIM21 is expressed at the cell surface remains unclear, whilst exposure after apoptosis, as shown by apoptosis of foetal cardiomyocytes in the presence of maternal anti-Ro52 antibodies, provides the most suitable explanation for the generation of anti-TRIM21 antibodies.
Why only some autoimmune patients generate antibodies against both Ro60 and TRIM21/Ro52, whilst others generate antibodies against just one, is another unanswered question. So far, NZM2758 mice have provided a model of female-specific SS. Further studies using these animal models may answer key questions related to anti-TRIM21 antibody generation, autoantibody epitope specificity, and how this relates to SS clinical manifestations. Certainly, answering these questions will go some way to enhancing our understanding of TRIM21 biology, its roles in innate and adaptive immunity, and its contributions to autoimmunity.
EJ, SL, and LD: Researched the topic, wrote, and edited the manuscript. EJ: Prepared the illustrations. All authors contributed to the article and approved the submitted version.
EJ is supported by a KTRR studentship from the Kennedy Trust for Rheumatology Research (KENN 19 20 01), and by a Henni Mester Fellowship (University College, University of Oxford). SL was supported by the Kennedy Trust for Rheumatology Research (research grant KENN 18 19 01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Figures were created with BioRender.com.
1. Yang SH, Gao CY, Li L, Chang C, Leung PSC, Gershwin ME, et al. The Molecular Basis of Immune Regulation in Autoimmunity. Clin Sci (Lond) (2018) 132:43–67. doi: 10.1042/CS20171154
2. Oke V, Wahren-Herlenius M. The Immunobiology of Ro52 (TRIM21) in Autoimmunity: A Critical Review. J Autoimmun (2012) 39:77–82. doi: 10.1016/j.jaut.2012.01.014
3. Cavazzana I, Franceschini F, Quinzanini M, Manera C, Papa ND, Maglione W, et al. Anti-Ro/SSA Antibodies in Rheumatoid Arthritis: Clinical and Immunologic Associations. Clin Exp Rheumatol (2006) 24:59–64.
4. Brito-Zeron P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, et al. Sjogren Syndrome. Nat Rev Dis Primers (2016) 2:16047. doi: 10.1038/nrdp.2016.47
5. Fugger L, Jensen LT, Rossjohn J. Challenges, Progress, and Prospects of Developing Therapies to Treat Autoimmune Diseases. Cell (2020) 181:63–80. doi: 10.1016/j.cell.2020.03.007
6. Granito A, Muratori P, Muratori L, Pappas G, Cassani F, Worthington J, et al. Antibodies to SS-A/Ro-52kd and Centromere in Autoimmune Liver Disease: A Clue to Diagnosis and Prognosis of Primary Biliary Cirrhosis. Aliment Pharmacol Ther (2007) 26:831–8. doi: 10.1111/j.1365-2036.2007.03433.x
7. Rutjes SA, Vree Egberts WTM, Jongen P, Van Den Hoogen F, Pruijn GJM, Venrooij WJV. Anti-Ro52 Antibodies Frequently Co-Occur With Anti-Jo-1 Antibodies in Sera From Patients With Idiopathic Inflammatory Myopathy. Clin Exp Immunol (2003) 109:32–40. doi: 10.1046/j.1365-2249.1997.4081308.x
8. Infantino M, Manfredi M, Grossi V, Benucci M, Morozzi G, Tonutti E, et al. An Effective Algorithm for the Serological Diagnosis of Idiopathic Inflammatory Myopathies: The Key Role of Anti-Ro52 Antibodies. Clin Chim Acta (2017) 475:15–9. doi: 10.1016/j.cca.2017.10.002
9. Salomonsson S, Sonesson SE, Ottosson L, Muhallab S, Olsson T, Sunnerhagen M, et al. Ro/SSA Autoantibodies Directly Bind Cardiomyocytes, Disturb Calcium Homeostasis, and Mediate Congenital Heart Block. J Exp Med (2005) 201:11–7. doi: 10.1084/jem.20041859
10. Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification Criteria for Sjögren’s Syndrome: A Revised Version of the European Criteria Proposed by the American-European Consensus Group. Ann Rheum Dis (2002) 61:554–8. doi: 10.1136/ard.61.6.554
11. Fayyaz A, Kurien BT, Scofield RH. Autoantibodies in Sjogren's Syndrome. Rheum Dis Clin North Am (2016) 42:419–34. doi: 10.1016/j.rdc.2016.03.002
12. Nossent JC, Lester S, Zahra D, Mackay CR, Rischmueller M. Polymorphism in the 5' Regulatory Region of the B-Lymphocyte Activating Factor Gene is Associated With the Ro/La Autoantibody Response and Serum BAFF Levels in Primary Sjogren's Syndrome. Rheumatol (Oxford) (2008) 47:1311–6. doi: 10.1093/rheumatology/ken246
13. Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at Multiple Loci Implicated in Both Innate and Adaptive Immune Responses Are Associated With Sjogren's Syndrome. Nat Genet (2013) 45:1284–92. doi: 10.1038/ng.2792
14. Reed JH, Verstappen GM, Rischmueller M, Bryant VL. When B Cells Break Bad: Development of Pathogenic B Cells in Sjögren's Syndrome. Clin Exp Rheumatol (2020) 38:271–82.
15. Sigurdsson S, Goring HH, Kristjansdottir G, Milani L, Nordmark G, Sandling JK, et al. Comprehensive Evaluation of the Genetic Variants of Interferon Regulatory Factor 5 (IRF5) Reveals a Novel 5 Bp Length Polymorphism as Strong Risk Factor for Systemic Lupus Erythematosus. Hum Mol Genet (2008) 17:872–81. doi: 10.1093/hmg/ddm359
16. Espinosa A, Zhou W, Ek M, Hedlund M, Brauner S, Popovic K, et al. The Sjogren's Syndrome-Associated Autoantigen Ro52 Is an E3 Ligase That Regulates Proliferation and Cell Death. J Immunol (2006) 176:6277–85. doi: 10.4049/jimmunol.176.10.6277
17. Di Rienzo M, Romagnoli A, Antonioli M, Piacentini M, Fimia GM. TRIM Proteins in Autophagy: Selective Sensors in Cell Damage and Innate Immune Responses. Cell Death Differ (2020) 27:887–902. doi: 10.1038/s41418-020-0495-2
18. Yang W, Gu Z, Zhang H, Hu H. To TRIM the Immunity: From Innate to Adaptive Immunity. Front Immunol (2020) 11:2157. doi: 10.3389/fimmu.2020.02157
19. Sardiello M, Cairo S, Fontanella B, Ballabio A, Meroni G. Genomic Analysis of the TRIM Family Reveals Two Groups of Genes With Distinct Evolutionary Properties. BMC Evol Biol (2008) 8:225. doi: 10.1186/1471-2148-8-225
20. Wright KM, Du H, Dagnachew M, Massiah MA. Solution Structure of the Microtubule-Targeting COS Domain of MID1. FEBS J (2016) 283:3089–102. doi: 10.1111/febs.13795
21. Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH, et al. MID1, Mutated in Opitz Syndrome, Encodes an Ubiquitin Ligase That Targets Phosphatase 2A for Degradation. Nat Genet (2001) 29:287–94. doi: 10.1038/ng762
22. Baldini R, Mascaro M, Meroni G. The MID1 Gene Product in Physiology and Disease. Gene (2020) 747:144655. doi: 10.1016/j.gene.2020.144655
23. Ozato K, Shin DM, Chang TH, Morse HC 3rd. TRIM Family Proteins and Their Emerging Roles in Innate Immunity. Nat Rev Immunol (2008) 8:849–60. doi: 10.1038/nri2413
24. Foss S, Bottermann M, Jonsson A, Sandlie I, James LC, Andersen JT. TRIM21-From Intracellular Immunity to Therapy. Front Immunol (2019) 10:2049. doi: 10.3389/fimmu.2019.02049
25. James LC, Keeble AH, Khan Z, Rhodes DA, Trowsdale J. Structural Basis for PRYSPRY-Mediated Tripartite Motif (TRIM) Protein Function. Proc Natl Acad Sci USA (2007) 104:6200–5. doi: 10.1073/pnas.0609174104
26. Dickson C, Fletcher AJ, Vaysburd M, Yang JC, Mallery DL, Zeng J, et al. Intracellular Antibody Signalling Is Regulated by Phosphorylation of the Fc Receptor TRIM21. Elife (2018) 7. doi: 10.7554/eLife.32660
27. D'Cruz AA, Babon JJ, Norton RS, Nicola NA, Nicholson SE. Structure and Function of the SPRY/B30.2 Domain Proteins Involved in Innate Immunity. Protein Sci (2013) 22:1–10. doi: 10.1002/pro.2185
28. Freemont PS, Hanson IM, Trowsdale J. A Novel Cysteine-Rich Sequence Motif. Cell (1991) 64:483–4. doi: 10.1016/0092-8674(91)90229-r
29. Freemont PS. Ubiquitination: RING for Destruction? Curr Biol (2000) 10:R84–87. doi: 10.1016/s0960-9822(00)00287-6
30. Wada K, Kamitani T. Autoantigen Ro52 Is an E3 Ubiquitin Ligase. Biochem Biophys Res Commun (2006) 339:415–21. doi: 10.1016/j.bbrc.2005.11.029
31. Ohtake F, Tsuchiya H. The Emerging Complexity of Ubiquitin Architecture. J Biochem (2017) 161:125–33. doi: 10.1093/jb/mvw088
32. Kiss L, Clift D, Renner N, Neuhaus D, James LC. RING Domains Act as Both Substrate and Enzyme in a Catalytic Arrangement to Drive Self-Anchored Ubiquitination. Nat Commun (2021) 12:1220. doi: 10.1038/s41467-021-21443-6
33. Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. The E3 Ubiquitin Ligase TRIM21 Negatively Regulates the Innate Immune Response to Intracellular Double-Stranded DNA. Nat Immunol (2013) 14:172–8. doi: 10.1038/ni.2492
34. Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, et al. The Helicase DDX41 Recognizes the Bacterial Secondary Messengers Cyclic Di-GMP and Cyclic Di-AMP to Activate a Type I Interferon Immune Response. Nat Immunol (2012) 13:1155–61. doi: 10.1038/ni.2460
35. Bai Y, Tong Y, Liu Y, Hu H. Self-dsDNA in the Pathogenesis of Systemic Lupus Erythematosus. Clin Exp Immunol (2018) 191:1–10. doi: 10.1111/cei.13041
36. Bell JL, Malyukova A, Holien JK, Koach J, Parker MW, Kavallaris M, et al. TRIM16 Acts as an E3 Ubiquitin Ligase and can Heterodimerize With Other TRIM Family Members. PloS One (2012) 7:e37470. doi: 10.1371/journal.pone.0037470
37. Massiah MA, Simmons BN, Short KM, Cox TC. Solution Structure of the RBCC/TRIM B-Box1 Domain of Human MID1: B-Box With a RING. J Mol Biol (2006) 358:532–45. doi: 10.1016/j.jmb.2006.02.009
38. Wagner JM, Roganowicz MD, Skorupka K, Alam SL, Christensen D, Doss G, et al. Mechanism of B-Box 2 Domain-Mediated Higher-Order Assembly of the Retroviral Restriction Factor TRIM5alpha. Elife (2016) 5. doi: 10.7554/eLife.16309
39. Ganser-Pornillos BK, Pornillos O. Restriction of HIV-1 and Other Retroviruses by TRIM5. Nat Rev Microbiol (2019) 17:546–56. doi: 10.1038/s41579-019-0225-2
40. Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M. Hexagonal Assembly of a Restricting TRIM5alpha Protein. Proc Natl Acad Sci USA (2011) 108:534–9. doi: 10.1073/pnas.1013426108
41. Wallenhammar A, Anandapadamanaban M, Lemak A, Mirabello C, Lundstrom P, Wallner B, et al. Solution NMR Structure of the TRIM21 B-Box2 and Identification of Residues Involved in Its Interaction With the RING Domain. PloS One (2017) 12:e0181551. doi: 10.1371/journal.pone.0181551
42. Nakken B, Jonsson R, Bolstad AI. Polymorphisms of the Ro52 Gene Associated With Anti–Ro 52-Kd Autoantibodies in Patients With Primary Sjögren’s Syndrome. Arthritis Rheum (2001) 44:638–46. doi: 10.1002/1529-0131(200103)44:3<638::AID-ANR112>3.0.CO;2-J
43. Sanchez JG, Okreglicka K, Chandrasekaran V, Welker JM, Sundquist WI, Pornillos O. The Tripartite Motif Coiled-Coil Is an Elongated Antiparallel Hairpin Dimer. Proc Natl Acad Sci USA (2014) 111:2494–9. doi: 10.1073/pnas.1318962111
44. Wang P, Benhenda S, Wu H, Lallemand-Breitenbach V, Zhen T, Jollivet F, et al. RING Tetramerization Is Required for Nuclear Body Biogenesis and PML Sumoylation. Nat Commun (2018) 9:1277. doi: 10.1038/s41467-018-03498-0
45. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The Tripartite Motif Family Identifies Cell Compartments. EMBO J (2001) 20:2140–5. doi: 10.1093/emboj/20.9.2140
46. Esposito D, Koliopoulos MG, Rittinger K. Structural Determinants of TRIM Protein Function. Biochem Soc Trans (2017) 45:183–91. doi: 10.1042/BST20160325
47. Dawidziak DM, Sanchez JG, Wagner JM, Ganser-Pornillos BK, Pornillos O. Structure and Catalytic Activation of the TRIM23 RING E3 Ubiquitin Ligase. Proteins (2017) 85:1957–61. doi: 10.1002/prot.25348
48. Fiorentini F, Esposito D, Rittinger K. Does It Take Two to Tango? RING Domain Self-Association and Activity in TRIM E3 Ubiquitin Ligases. Biochem Soc Trans (2020) 48:2615–24. doi: 10.1042/BST20200383
49. Zeng J, Santos AF, Mukadam AS, Osswald M, Jacques DA, Dickson CF, et al. Target-Induced Clustering Activates Trim-Away of Pathogens and Proteins. Nat Struct Mol Biol (2021) 28:278–89. doi: 10.1038/s41594-021-00560-2
50. Watanabe M, Hatakeyama S. TRIM Proteins and Diseases. J Biochem (2017) 161:135–44. doi: 10.1093/jb/mvw087
51. Li X, Li Y, Stremlau M, Yuan W, Song B, Perron M, et al. Functional Replacement of the RING, B-Box 2, and Coiled-Coil Domains of Tripartite Motif 5alpha (TRIM5alpha) by Heterologous TRIM Domains. J Virol (2006) 80:6198–206. doi: 10.1128/JVI.00283-06
52. Li B, Zhou T, Zou Y. Mid1/Mid2 Expression in Craniofacial Development and a Literature Review of X-Linked Opitz Syndrome. Mol Genet Genomic Med (2016) 4:95–105. doi: 10.1002/mgg3.183
53. Schweiger S, Schneider R. The MID1/PP2A Complex: A Key to the Pathogenesis of Opitz BBB/G Syndrome. Bioessays (2003) 25:356–66. doi: 10.1002/bies.10256
54. Consortium TIF. Ancient Missense Mutations in a New Member of the RoRet Gene Family Are Likely to Cause Familial Mediterranean Fever. Cell (1997) 90:797–807. doi: 10.1016/s0092-8674(00)80539-5
55. Singh N, Kumble Bhat V, Tiwari A, Kodaganur SG, Tontanahal SJ, Sarda A, et al. A Homozygous Mutation in TRIM36 Causes Autosomal Recessive Anencephaly in an Indian Family. Hum Mol Genet (2017) 26:1104–14. doi: 10.1093/hmg/ddx020
56. Keeble AH, Khan Z, Forster A, James LC. TRIM21 Is an IgG Receptor That Is Structurally, Thermodynamically, and Kinetically Conserved. Proc Natl Acad Sci USA (2008) 105:6045–50. doi: 10.1073/pnas.0800159105
57. Bidgood SR, Tam JC, McEwan WA, Mallery DL, James LC. Translocalized IgA Mediates Neutralization and Stimulates Innate Immunity Inside Infected Cells. Proc Natl Acad Sci USA (2014) 111:13463–8. doi: 10.1073/pnas.1410980111
58. Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. Antibodies Mediate Intracellular Immunity Through Tripartite Motif-Containing 21 (TRIM21). Proc Natl Acad Sci USA (2010) 107:19985–90. doi: 10.1073/pnas.1014074107
59. Kiyoshi M, Caaveiro JM, Kawai T, Tashiro S, Ide T, Asaoka Y, et al. Structural Basis for Binding of Human IgG1 to Its High-Affinity Human Receptor FcgammaRI. Nat Commun (2015) 6:6866. doi: 10.1038/ncomms7866
60. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and Affinity of Human Fcgamma Receptors and Their Polymorphic Variants for Human IgG Subclasses. Blood (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
61. Xie S, Zhang L, Dong D, Ge R, He Q, Fan C, et al. HDAC6 Regulates Antibody-Dependent Intracellular Neutralization of Viruses via Deacetylation of TRIM21. J Biol Chem (2020) 295:14343–51. doi: 10.1074/jbc.RA119.011006
62. Forthal DN. Functions of Antibodies. Microbiol Spectr (2014) 2:1–17. doi: 10.1128/microbiolspec.AID-0019-2014
63. Walls AC, Tortorici MA, Frenz B, Snijder J, Li W, Rey FA, et al. Glycan Shield and Epitope Masking of a Coronavirus Spike Protein Observed by Cryo-Electron Microscopy. Nat Struct Mol Biol (2016) 23:899–905. doi: 10.1038/nsmb.3293
64. Vaysburd M, Watkinson RE, Cooper H, Reed M, O'Connell K, Smith J, et al. Intracellular Antibody Receptor TRIM21 Prevents Fatal Viral Infection. Proc Natl Acad Sci USA (2013) 110:12397–401. doi: 10.1073/pnas.1301918110
65. Caddy SL, Vaysburd M, Papa G, Wing M, O'Connell K, Stoycheva D, et al. Viral Nucleoprotein Antibodies Activate TRIM21 and Induce T Cell Immunity. EMBO J (2021) 40:e106228. doi: 10.15252/embj.2020106228
66. Foltz C, Napolitano A, Khan R, Clough B, Hirst EM, Frickel EM. TRIM21 Is Critical for Survival of Toxoplasma Gondii Infection and Localises to GBP-Positive Parasite Vacuoles. Sci Rep (2017) 7:5209. doi: 10.1038/s41598-017-05487-7
67. McEwan WA, Hauler F, Williams CR, Bidgood SR, Mallery DL, Crowther RA, et al. Regulation of Virus Neutralization and the Persistent Fraction by TRIM21. J Virol (2012) 86:8482–91. doi: 10.1128/JVI.00728-12
68. Fletcher AJ, James LC. Coordinated Neutralization and Immune Activation by the Cytosolic Antibody Receptor Trim21. J Virol (2016) 90:4856–9. doi: 10.1128/JVI.00050-16
69. Maurer UE, Sodeik B, Grunewald K. Native 3D Intermediates of Membrane Fusion in Herpes Simplex Virus 1 Entry. Proc Natl Acad Sci USA (2008) 105:10559–64. doi: 10.1073/pnas.0801674105
70. Mercer J, Schelhaas M, Helenius A. Virus Entry by Endocytosis. Annu Rev Biochem (2010) 79:803–33. doi: 10.1146/annurev-biochem-060208-104626
71. Pelkmans L, Helenius A. Endocytosis Via Caveolae. Traffic (2002) 3:311–20. doi: 10.1034/j.1600-0854.2002.30501.x
72. O'Donoghue EJ, Krachler AM. Mechanisms of Outer Membrane Vesicle Entry Into Host Cells. Cell Microbiol (2016) 18:1508–17. doi: 10.1111/cmi.12655
73. McEwan WA, Tam JC, Watkinson RE, Bidgood SR, Mallery DL, James LC. Intracellular Antibody-Bound Pathogens Stimulate Immune Signaling via the Fc Receptor TRIM21. Nat Immunol (2013) 14:327–36. doi: 10.1038/ni.2548
74. Watkinson RE, McEwan WA, Tam JC, Vaysburd M, James LC. TRIM21 Promotes cGAS and RIG-I Sensing of Viral Genomes During Infection by Antibody-Opsonized Virus. PloS Pathog (2015) 11:e1005253. doi: 10.1371/journal.ppat.1005253
75. Gastaldelli M, Imelli N, Boucke K, Amstutz B, Meier O, Greber UF. Infectious Adenovirus Type 2 Transport Through Early But Not Late Endosomes. Traffic (2008) 9:2265–78. doi: 10.1111/j.1600-0854.2008.00835.x
76. Burckhardt CJ, Suomalainen M, Schoenenberger P, Boucke K, Hemmi S, Greber UF. Drifting Motions of the Adenovirus Receptor CAR and Immobile Integrins Initiate Virus Uncoating and Membrane Lytic Protein Exposure. Cell Host Microbe (2011) 10:105–17. doi: 10.1016/j.chom.2011.07.006
77. Maier O, Marvin SA, Wodrich H, Campbell EM, Wiethoff CM. Spatiotemporal Dynamics of Adenovirus Membrane Rupture and Endosomal Escape. J Virol (2012) 86:10821–8. doi: 10.1128/JVI.01428-12
78. Wiethoff CM, Nemerow GR. Adenovirus Membrane Penetration: Tickling the Tail of a Sleeping Dragon. Virology (2015) 479-480:591–9. doi: 10.1016/j.virol.2015.03.006
79. Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. Adenovirus Protein VI Mediates Membrane Disruption Following Capsid Disassembly. J Virol (2005) 79:1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005
80. Steele-Mortimer O. The Salmonella-Containing Vacuole: Moving With the Times. Curr Opin Microbiol (2008) 11:38–45. doi: 10.1016/j.mib.2008.01.002
81. Beuzón CR, Méresse S, Unsworth KE, Ruíz-Albert J, Garvis S, Waterman SR, et al. Salmonella Maintains the Integrity of Its Intracellular Vacuole Through the Action of SifA. EMBO J (2000) 19:3235–49. doi: 10.1093/emboj/19.13.3235
82. Beuzón CR, Salcedo SP, Holden DW. Growth and Killing of a Salmonella Enterica Serovar Typhimurium sifA Mutant Strain in the Cytosol of Different Host Cell Lines. Microbiology (2002) 148:2705–15. doi: 10.1099/00221287-148-9-2705
83. Nakanaga K, Yamanouchi K, Fujiwara K. Protective Effect of Monoclonal Antibodies on Lethal Mouse Hepatitis Virus Infection in Mice. J Virol (1986) 59:168–71. doi: 10.1128/JVI.59.1.168-171.1986
84. Fujimoto Y, Tomioka Y, Takakuwa H, Uechi G-I, Yabuta T, Ozaki K, et al. Cross-Protective Potential of Anti-Nucleoprotein Human Monoclonal Antibodies Against Lethal Influenza A Virus Infection. J Gen Virol (2016) 97:2104–16. doi: 10.1099/jgv.0.000518
85. Carragher DM, Kaminski DA, Moquin A, Hartson L, Randall TD. A Novel Role for Non-Neutralizing Antibodies Against Nucleoprotein in Facilitating Resistance to Influenza Virus. J Immunol (2008) 181:4168–76. doi: 10.4049/jimmunol.181.6.4168
86. Richter K, Oxenius A. Non-Neutralizing Antibodies Protect From Chronic LCMV Infection Independently of Activating FcgammaR or Complement. Eur J Immunol (2013) 43:2349–60. doi: 10.1002/eji.201343566
87. Sambhara S, Kurichh A, Miranda R, Tumpey T, Rowe T, Renshaw M, et al. Heterosubtypic Immunity Against Human Influenza A Viruses, Including Recently Emerged Avian H5 and H9 Viruses, Induced by FLU-ISCOM Vaccine in Mice Requires Both Cytotoxic T-Lymphocyte and Macrophage Function. Cell Immunol (2001) 211:143–53. doi: 10.1006/cimm.2001.1835
88. Straub T, Schweier O, Bruns M, Nimmerjahn F, Waisman A, Pircher H. Nucleoprotein-Specific Nonneutralizing Antibodies Speed Up LCMV Elimination Independently of Complement and FcgammaR. Eur J Immunol (2013) 43:2338–48. doi: 10.1002/eji.201343565
89. Lecomte J, Cainelli-Gebara V, Mercier G, Mansour S, Talbot PJ, Lussier G, et al. Protection From Mouse Hepatitis Virus Type 3-Induced Acute Disease by an Anti-Nucleoprotein Monoclonal Antibody. Arch Virol (1987) 97:123–30. doi: 10.1007/BF01310740
90. Bergthaler A, Flatz L, Verschoor A, Hegazy AN, Holdener M, Fink K, et al. Impaired Antibody Response Causes Persistence of Prototypic T Cell-Contained Virus. PloS Biol (2009) 7:e1000080. doi: 10.1371/journal.pbio.1000080
91. LaMere MW, Lam HT, Moquin A, Haynes L, Lund FE, Randall TD, et al. Contributions of Antinucleoprotein IgG to Heterosubtypic Immunity Against Influenza Virus. J Immunol (2011) 186:4331–9. doi: 10.4049/jimmunol.1003057
92. Choi Y, Jiang Z, Shin W-J, Jung JU. Severe Fever With Thrombocytopenia Syndrome Virus NSs Interacts With TRIM21 to Activate the P62-Keap1-Nrf2 Pathway. J Virol (2020) 94:e01684–01619. doi: 10.1128/JVI
93. Li Z, Huan C, Wang H, Liu Y, Liu X, Su X, et al. TRIM21-Mediated Proteasomal Degradation of SAMHD1 Regulates Its Antiviral Activity. EMBO Rep (2020) 21:e47528. doi: 10.15252/embr.201847528
94. Song Y, Wu X, Xu Y, Zhu J, Li J, Zou Z, et al. HPV E7 Inhibits Cell Pyroptosis by Promoting TRIM21-Mediated Degradation and Ubiquitination of the IFI16 Inflammasome. Int J Biol Sci (2020) 16:2924–37. doi: 10.7150/ijbs.50074
95. Liu H, Li M, Song Y, Xu W. TRIM21 Restricts Coxsackievirus B3 Replication, Cardiac and Pancreatic Injury via Interacting With MAVS and Positively Regulating IRF3-Mediated Type-I Interferon Production. Front Immunol (2018) 9:2479. doi: 10.3389/fimmu.2018.02479
96. Xue B, Li H, Guo M, Wang J, Xu Y, Zou X, et al. TRIM21 Promotes Innate Immune Response to RNA Viral Infection Through Lys27-Linked Polyubiquitination of MAVS. J Virol (2018) 86:8482–91. doi: 10.1128/JVI.00728-12
97. Mu T, Zhao X, Zhu Y, Fan H, Tang H. The E3 Ubiquitin Ligase TRIM21 Promotes HBV DNA Polymerase Degradation. Viruses (2020) 12:346. doi: 10.3390/v12030346
98. Wang H, Chen X, Kong N, Jiao Y, Sun D, Dong S, et al. TRIM21 Inhibits Porcine Epidemic Diarrhea Virus Proliferation by Proteasomal Degradation of the Nucleocapsid Protein. Arch Virol (2021) 166:1903–11. doi: 10.1007/s00705-021-05080-4
99. Das A, Dinh PX, Pattnaik AK. Trim21 Regulates Nmi-IFI35 Complex-Mediated Inhibition of Innate Antiviral Response. Virol (2015) 485:383–92. doi: 10.1016/j.virol.2015.08.013
100. Platanias LC. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat Rev Immunol (2005) 5:375–86. doi: 10.1038/nri1604
101. Rhodes DA, Ihrke G, Reinicke AT, Malcherek G, Towey M, Isenberg DA, et al. The 52 000 MW Ro/SS-A Autoantigen in Sjögren's Syndrome/Systemic Lupus Erythematosus (Ro52) Is an Interferon-γ Inducible Tripartite Motif Protein Associated With Membrane Proximal Structures. Immunol (2002) 106:246–56. doi: 10.1046/j.1365-2567.2002.01417.x
102. Rajsbaum R, Stoye JP, O'Garra A. Type I Interferon-Dependent and -Independent Expression of Tripartite Motif Proteins in Immune Cells. Eur J Immunol (2008) 38:619–30. doi: 10.1002/eji.200737916
103. Sjostrand M, Ambrosi A, Brauner S, Sullivan J, Malin S, Kuchroo VK, et al. Expression of the Immune Regulator Tripartite-Motif 21 Is Controlled by IFN Regulatory Factors. J Immunol (2013) 191:3753–63. doi: 10.4049/jimmunol.1202341
104. Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska M, et al. Direct Inhibition of IRF-Dependent Transcriptional Regulatory Mechanisms Associated With Disease. Front Immunol (2019) 10:1176. doi: 10.3389/fimmu.2019.01176
105. Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol (2018) 9:847. doi: 10.3389/fimmu.2018.00847
106. Michalska A, Blaszczyk K, Wesoly J, Bluyssen HAR. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front Immunol (2018) 9:1135. doi: 10.3389/fimmu.2018.01135
107. Mazewski C, Perez RE, Fish EN, Platanias LC. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front Immunol (2020) 11:606456. doi: 10.3389/fimmu.2020.606456
108. Manocha GD, Mishra R, Sharma N, Kumawat KL, Basu A, Singh SK. Regulatory Role of TRIM21 in the Type-I Interferon Pathway in Japanese Encephalitis Virus-Infected Human Microglial Cells. J Neuroinflamm (2014) 11:1–12. doi: 10.1186/1742-2094-11-24
109. Higgs R, Lazzari E, Wynne C, Ni Gabhann J, Espinosa A, Wahren-Herlenius M, et al. Self Protection From Anti-Viral Responses–Ro52 Promotes Degradation of the Transcription Factor IRF7 Downstream of the Viral Toll-Like Receptors. PloS One (2010) 5:e11776. doi: 10.1371/journal.pone.0011776
110. Lazzari E, Korczeniewska J, Ni Gabhann J, Smith S, Barnes BJ, Jefferies CA. TRIpartite Motif 21 (TRIM21) Differentially Regulates the Stability of Interferon Regulatory Factor 5 (IRF5) Isoforms. PloS One (2014) 9:e103609. doi: 10.1371/journal.pone.0103609
111. Jefferies CA. Regulating IRFs in IFN Driven Disease. Front Immunol (2019) 10:325. doi: 10.3389/fimmu.2019.00325
112. Feng D, Stone RC, Eloranta ML, Sangster-Guity N, Nordmark G, Sigurdsson S, et al. Genetic Variants and Disease-Associated Factors Contribute to Enhanced Interferon Regulatory Factor 5 Expression in Blood Cells of Patients With Systemic Lupus Erythematosus. Arthritis Rheum (2010) 62:562–73. doi: 10.1002/art.27223
113. Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, et al. Three Functional Variants of IFN Regulatory Factor 5 (IRF5) Define Risk and Protective Haplotypes for Human Lupus. Proc Natl Acad Sci USA (2007) 104:6758–63. doi: 10.1073/pnas.0701266104
114. Higgs R, Gabhann JN, Larbi NB, Breen EP, Fitzgerald KA, Jefferies CA. The E3 Ubiquitin Ligase Ro52 Negatively Regulates IFN-Beta Production Post-Pathogen Recognition by Polyubiquitin-Mediated Degradation of IRF3. J Immunol (2008) 181:1780–6. doi: 10.4049/jimmunol.181.3.1780
115. Yang K, Shi HX, Liu XY, Shan YF, Wei B, Chen S, et al. TRIM21 is Essential to Sustain IFN Regulatory Factor 3 Activation During Antiviral Response. J Immunol (2009) 182:3782–92. doi: 10.4049/jimmunol.0803126
116. Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, et al. Negative Regulation of Interferon-Regulatory Factor 3-Dependent Innate Antiviral Response by the Prolyl Isomerase Pin1. Nat Immunol (2006) 7:598–605. doi: 10.1038/ni1347
117. Strandberg L, Ambrosi A, Espinosa A, Ottosson L, Eloranta ML, Zhou W, et al. Interferon-Alpha Induces Up-Regulation and Nuclear Translocation of the Ro52 Autoantigen as Detected by a Panel of Novel Ro52-Specific Monoclonal Antibodies. J Clin Immunol (2008) 28:220–31. doi: 10.1007/s10875-007-9157-0
118. Kong HJ, Anderson DE, Lee CH, Jang MK, Tamura T, Tailor P, et al. Cutting Edge: Autoantigen Ro52 Is an Interferon Inducible E3 Ligase That Ubiquitinates IRF-8 and Enhances Cytokine Expression in Macrophages. J Immunol (2007) 179:26–30. doi: 10.4049/jimmunol.179.1.26
119. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
120. Dror N, Alter-Koltunoff M, Azriel A, Amariglio N, Jacob-Hirsch J, Zeligson S, et al. Identification of IRF-8 and IRF-1 Target Genes in Activated Macrophages. Mol Immunol (2007) 44:338–46. doi: 10.1016/j.molimm.2006.02.026
121. Qin M, Guo Y, Jiang L, Wang X. Elevated Levels of Serum Scxcl16 in Systemic Lupus Erythematosus; Potential Involvement in Cutaneous and Renal Manifestations. Clin Rheumatol (2014) 33:1595–601. doi: 10.1007/s10067-014-2741-9
122. Ellermann-Eriksen S, Justesen J, Mogensen SC. Genetically Determined Difference in the Antiviral Action of α/β Interferon in Cells From Mice Resistant or Susceptible to Herpes Simplex Virus Type 2. J Gen Virol (1986) 67:1859–66. doi: 10.1099/0022-1317-67-9-1859
123. Zawatzky R, Gresser I, DeMaeyer E, Kirchner H. The Role of Interferon in the Resistance of C57BL/6 Mice to Various Doses of Herpes Simplex Virus Type 1. J Infect Dis (1982) 146:405–10. doi: 10.1093/infdis/146.3.405
124. Gresser I, Maury C, Kress C, Blangy D, Maunoury MT. Role of Interferon in the Pathogenesis of Virus Diseases in Mice as Demonstrated by the Use of Anti-Interferon Serum. VI. Polyoma Virus Infection. Int J Cancer (1979) 24:178–83. doi: 10.1002/ijc.2910240209
125. Ouyang W, Cen M, Yang L, Zhang W, Xia J, Xu F. NMI Facilitates Influenza A Virus Infection by Promoting Degradation of IRF7 Through Trim21. Am J Respir Cell Mol Biol (2021) 65:30–40. doi: 10.1165/rcmb.2020-0391OC
126. Ma Z, Zhang W, Fan W, Wu Y, Zhang M, Xu J, et al. Forkhead Box O1-Mediated Ubiquitination Suppresses RIG-I-Mediated Antiviral Immune Responses. Int Immunopharmacol (2021) 90:107152. doi: 10.1016/j.intimp.2020.107152
127. Kamiyama R, Yoshimi R, Takeno M, Iribe Y, Tsukahara T, Kishimoto D, et al. Dysfunction of TRIM21 in Interferon Signature of Systemic Lupus Erythematosus. Mod Rheumatol (2018) 28:993–1003. doi: 10.1080/14397595.2018.1436028
128. Zhou W, Zhang Y, Zhong C, Hu J, Hu H, Zhou D, et al. Decreased Expression of TRIM21 Indicates Unfavorable Outcome and Promotes Cell Growth in Breast Cancer. Cancer Manag Res (2018) 10:3687–96. doi: 10.2147/CMAR.S175470
129. Ding Q, He D, He K, Zhang Q, Tang M, Dai J, et al. Downregulation of TRIM21 Contributes to Hepatocellular Carcinoma Carcinogenesis and Indicates Poor Prognosis of Cancers. Tumour Biol (2015) 36:8761–72. doi: 10.1007/s13277-015-3572-2
130. Brauner S, Zhou W, Backlin C, Green TM, Folkersen L, Ivanchenko M, et al. Reduced Expression of TRIM21/Ro52 Predicts Poor Prognosis in Diffuse Large B-Cell Lymphoma Patients With and Without Rheumatic Disease. J Intern Med (2015) 278:323–32. doi: 10.1111/joim.12375
131. Zhou G, Wu H, Lin J, Lin R, Feng B, Liu Z. TRIM21 Is Decreased in Colitis-Associated Cancer and Negatively Regulates Epithelial Carcinogenesis. Inflammation Bowel Dis (2021) 27:458–68. doi: 10.1093/ibd/izaa229
132. de Jonge H, Iamele L, Maggi M, Pessino G, Scotti C. Anti-Cancer Auto-Antibodies: Roles, Applications and Open Issues. Cancers (Basel) (2021) 13:1–33. doi: 10.3390/cancers13040813
133. Bogdanos DP, Gkoutzourelas A, Papadopoulos V, Liaskos C, Patrikiou E, Tsigalou C, et al. Anti-Ro52 Antibody Is Highly Prevalent and a Marker of Better Prognosis in Patients With Ovarian Cancer. Clin Chim Acta (2021) 521:199–205. doi: 10.1016/j.cca.2021.07.006
134. Ozato K, Yoshimi R, Chang TH, Wang H, Atsumi T, Morse HC 3rd, et al. Comment on "Gene Disruption Study Reveals a Nonredundant Role for TRIM21/Ro52 in NF-Kappa B-Dependent Cytokine Expression in Fibroblasts". J Immunol (2009) 183:7619; author reply 7720–7611. doi: 10.4049/jimmunol.0990103
135. Yoshimi R, Chang TH, Wang H, Atsumi T, Morse HC 3rd, Ozato K. Gene Disruption Study Reveals a Nonredundant Role for TRIM21/Ro52 in NF-kappaB-Dependent Cytokine Expression in Fibroblasts. J Immunol (2009) 182:7527–38. doi: 10.4049/jimmunol.0804121
136. Espinosa A, Dardalhon V, Brauner S, Ambrosi A, Higgs R, Quintana FJ, et al. Loss of the Lupus Autoantigen Ro52/Trim21 Induces Tissue Inflammation and Systemic Autoimmunity by Disregulating the IL-23-Th17 Pathway. J Exp Med (2009) 206:1661–71. doi: 10.1084/jem.20090585
137. Kunishita Y, Yoshimi R, Kamiyama R, Kishimoto D, Yoshida K, Hashimoto E, et al. TRIM21 Dysfunction Enhances Aberrant B-Cell Differentiation in Autoimmune Pathogenesis. Front Immunol (2020) 11:98. doi: 10.3389/fimmu.2020.00098
138. De S, Zhang B, Shih T, Singh S, Winkler A, Donnelly R, et al. B Cell-Intrinsic Role for IRF5 in TLR9/BCR-Induced Human B Cell Activation, Proliferation, and Plasmablast Differentiation. Front Immunol (2017) 8:1938. doi: 10.3389/fimmu.2017.01938
139. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded Expression of Interferon Regulatory Factor-4 Coordinates Isotype Switching With Plasma Cell Differentiation. Immunity (2006) 25:225–36. doi: 10.1016/j.immuni.2006.07.009
140. Espinosa A, Hennig J, Ambrosi A, Anandapadmanaban M, Abelius MS, Sheng Y, et al. Anti-Ro52 Autoantibodies From Patients With Sjogren's Syndrome Inhibit the Ro52 E3 Ligase Activity by Blocking the E3/E2 Interface. J Biol Chem (2011) 286:36478–91. doi: 10.1074/jbc.M111.241786
141. Ohlsson M, Jonsson R, Brokstad KA. Subcellular Redistribution and Surface Exposure of the Ro52, Ro60 and La48 Autoantigens During Apoptosis in Human Ductal Epithelial Cells: A Possible Mechanism in the Pathogenesis of Sjögren's Syndrome. Scand J Immunol (2002) 56:456–69. doi: 10.1046/j.1365-3083.2002.01072_79.x
142. Hillen MR, Urso K, Koppe E, Lopes AP, Blokland SLM, Pandit A, et al. Autoantigen TRIM21/Ro52 Is Expressed on the Surface of Antigen-Presenting Cells and Its Enhanced Expression in Sjogren's Syndrome Is Associated With B Cell Hyperactivity and Type I Interferon Activity. RMD Open (2020) 6:1–5. doi: 10.1136/rmdopen-2020-001184
143. Yoshimi R, Ishigatsubo Y, Ozato K. Autoantigen TRIM21/Ro52 as a Possible Target for Treatment of Systemic Lupus Erythematosus. Int J Rheumatol (2012) 2012:718237. doi: 10.1155/2012/718237
144. Norris DA. Pathomechanisms of Photosensitive Lupus Erythematosus. J Invest Dermatol (1993) 100:S58–68. doi: 10.1038/jid.1993.25
145. Lee AJ, Ashkar AA. The Dual Nature of Type I and Type II Interferons. Front Immunol (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
146. Hardy JD, Solomon S, Banwell GS, Beach R, Wright V, Howard FM. Congenital Complete Heart Block in the Newborn Associated With Maternal Systemic Lupus Erythematosus and Other Connective Tissue Disorders. Arch Dis Child (1979) 54:7–13. doi: 10.1136/adc.54.1.7
147. Xiao G-Q, Hu K, Boutjdir M. Direct Inhibition of Expressed Cardiac L- and T-Type Calcium Channels by IgG From Mothers Whose Children Have Congenital Heart Block. Circ Res (2001) 103:1599–604. doi: 10.1161/01.cir.103.11.1599
148. Boutjdir M, Chen L, Zhang ZH, Tseng CE, DiDonato F, Rashbaum W, et al. Arrhythmogenicity of IgG and Anti-52-kD SSA/Ro Affinity-Purified Antibodies From Mothers of Children With Congenital Heart Block. Circ Res (1997) 80:354–62. doi: 10.1161/01.res.80.3.354
149. Capone C, Buyon JP, Friedman DM, Frishman WH. Cardiac Manifestations of Neonatal Lupus: A Review of Autoantibody-Associated Congenital Heart Block and Its Impact in an Adult Population. Cardiol Rev (2012) 20:72–6. doi: 10.1097/CRD.0b013e31823c808b
150. Miranda ME, Tseng C-E, Rashbaum W, Ochs RL, Casiano CA, Donato FD, et al. Accessibility of SSA/Ro and SSB/La Antigens to Maternal Autoantibodies in Apoptotic Human Fetal Cardiac Myocytes. J Immunol (1998) 161:5061–9.
151. Miranda-Carus ME, Askanase AD, Clancy RM, Di Donato F, Chou TM, Libera MR, et al. Anti-SSA/Ro and Anti-SSB/La Autoantibodies Bind the Surface of Apoptotic Fetal Cardiocytes and Promote Secretion of TNF-Alpha by Macrophages. J Immunol (2000) 165:5345–51. doi: 10.4049/jimmunol.165.9.5345
152. Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of Immunological Tolerance by Apoptotic Cells Requires Caspase-Dependent Oxidation of High-Mobility Group Box-1 Protein. Immunity (2008) 29:21–32. doi: 10.1016/j.immuni.2008.05.013
153. Fransen JH, Hilbrands LB, Ruben J, Stoffels M, Adema GJ, van der Vlag J, et al. Mouse Dendritic Cells Matured by Ingestion of Apoptotic Blebs Induce T Cells to Produce Interleukin-17. Arthritis Rheum (2009) 60:2304–13. doi: 10.1002/art.24719
154. Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, et al. Apoptosis Facilitates Antigen Presentation to T Lymphocytes Through MHC-I and CD1 in Tuberculosis. Nat Med (2003) 9:1039–46. doi: 10.1038/nm906
155. LeFeber WP, Norris DA, Ryan SR, Huff JC, Lee LA, Kubo M, et al. Ultraviolet Light Induces Binding of Antibodies to Selected Nuclear Antigens on Cultured Human Keratinocytes. J Clin Invest (1984) 74:1545–51. doi: 10.1172/JCI111569
156. Lawley W, Doherty A, Denniss S, Chauhan D, Pruijn G, Venrooij WJ, et al. Rapid Lupus Autoantigen Relocalization and Reactive Oxygen Species Accumulation Following Ultraviolet Irradiation of Human Keratinocytes. Rheumatology (2000) 39:253–61. doi: 10.1093/rheumatology/39.3.253
157. Saegusa J, Kawano S, Koshiba M, Hayashi N, Kosaka H, Funasaka Y, et al. Oxidative Stress Mediates Cell Surface Expression of SS-A/Ro Antigen on Keratinocytes. Free Radic Biol Med (2002) 32:1006–16. doi: 10.1016/s0891-5849(02)00797-9
158. Zhitkovich A. N-Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem Res Toxicol (2019) 32:1318–9. doi: 10.1021/acs.chemrestox.9b00152
159. Aquilano K, Baldelli S, Ciriolo MR. Glutathione: New Roles in Redox Signaling for an Old Antioxidant. Front Pharmacol (2014) 5:196. doi: 10.3389/fphar.2014.00196
160. Ryo K, Yamada H, Nakagawa Y, Tai Y, Obara K, Inoue H, et al. Possible Involvement of Oxidative Stress in Salivary Gland of Patients With Sjogren's Syndrome. Pathobiology (2006) 73:252–60. doi: 10.1159/000098211
161. Kasai H, Crain PF, Kuchino Y, Nishimura S, Ootsuyama A, Tanooka H. Formation of 8-Hydroxyguanine Moiety in Cellular DNA by Agents Producing Oxygen Radicals and Evidence for Its Repair. Carcinogenesis (1986) 7:1849–51. doi: 10.1093/carcin/7.11.1849
162. Furukawa F, Kashihara-Sawami M, Lyons MB, Norris DA. Binding of Antibodies to the Extractable Nuclear Antigens SS-A/Ro and SS-B/La Is Induced on the Surface of Human Keratinocytes by Ultraviolet Light (UVL): Implications for the Pathogenesis of Photosensitive Cutaneous Lupus. J Invest Dermatol (1990) 94:77–85. doi: 10.1111/1523-1747.ep12873930
163. Paz ML, Maglio DHG, Weill FS, Bustamante J, Leoni J. Mitochondrial Dysfunction and Cellular Stress Progression After Ultraviolet B Irradiation in Human Keratinocytes. Photodermatol Photoimmunol Photomed (2008) 24:115–22. doi: 10.1111/j.1600-0781.2008.00348.x
164. Tanaka M, Tanji K, Niida M, Kamitani T. Dynamic Movements of Ro52 Cytoplasmic Bodies Along Microtubules. Histochem Cell Biol (2010) 133:273–84. doi: 10.1007/s00418-009-0669-y
165. Vermes I, Haanen C, Reutelingsperger C. Flow Cytometry of Apoptotic Cell Death. J Immunol Methods (2000) 243:167–90. doi: 10.1016/s0022-1759(00)00233-7
166. Theofilopoulos AN, Kono DH, Baccala R. The Multiple Pathways to Autoimmunity. Nat Immunol (2017) 18:716–24. doi: 10.1038/ni.3731
167. Aqrawi LA, Kvarnstrom M, Brokstad KA, Jonsson R, Skarstein K, Wahren-Herlenius M. Ductal Epithelial Expression of Ro52 Correlates With Inflammation in Salivary Glands of Patients With Primary Sjogren's Syndrome. Clin Exp Immunol (2014) 177:244–52. doi: 10.1111/cei.12341
168. Retamozo S, Akasbi M, Brito-Zerón P, Bosch X, Bove A, Perez-De-Lis M, et al. Anti-Ro52 Antibody Testing Influences the Classification and Clinical Characterisation of Primary Sjögren's Syndrome. Clin Exp Rheumatol (2012) 30:686–92.
169. Yang K, Chen Y, Qi H, Ye Y, Fan Z, Huang F, et al. Anti-Ro52 Autoantibodies Are Related to Chronic Graft-Vs.-Host Disease After Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol (2020) 11:1505. doi: 10.3389/fimmu.2020.01505
170. Tong L, Koh V, Thong BY. Review of Autoantigens in Sjogren's Syndrome: An Update. J Inflammation Res (2017) 10:97–105. doi: 10.2147/JIR.S137024
171. Vanderlugt CJ, Miller SD. Epitope Spreading. Curr Opin Immunol (1996) 8:831–6. doi: 10.1016/s0952-7915(96)80012-4
172. Tseng CE, Chan EK, Miranda E, Gross M, Donato FD, Buyon JP. The 52-Kd Protein as a Target of Intermolecular Spreading of the Immune Response to Components of the SS-A/Ro-SS-B/La Complex. Arthritis Rheum (1997) 40:936–44. doi: 10.1002/art.1780400523
173. Murng SHK, Thomas M. Clinical Associations of the Positive Anti Ro52 Without Ro60 Autoantibodies: Undifferentiated Connective Tissue Diseases. J Clin Pathol (2018) 71:12–9. doi: 10.1136/jclinpath-2015-203587
174. Gkoutzourelas A, Liaskos C, Mytilinaiou MG, Simopoulou T, Katsiari C, Tsirogianni A, et al. Anti-Ro60 Seropositivity Determines Anti-Ro52 Epitope Mapping in Patients With Systemic Sclerosis. Front Immunol (2018) 9:2835. doi: 10.3389/fimmu.2018.02835
175. Yoshimi R, Ueda A, Ozato K, Ishigatsubo Y. Clinical and Pathological Roles of Ro/SSA Autoantibody System. Clin Dev Immunol (2012) 2012:606195. doi: 10.1155/2012/606195
176. Schulte-Pelkum J, Fritzler M, Mahler M. Latest Update on the Ro/SS-A Autoantibody System. Autoimmun Rev (2009) 8:632–7. doi: 10.1016/j.autrev.2009.02.010
177. Didona D, Di Zenzo G. Humoral Epitope Spreading in Autoimmune Bullous Diseases. Front Immunol (2018) 9:779. doi: 10.3389/fimmu.2018.00779
178. Bagavant H, Michrowska A, Deshmukh US. The NZB/W F1 Mouse Model for Sjogren's Syndrome: A Historical Perspective and Lessons Learned. Autoimmun Rev (2020) 19:102686. doi: 10.1016/j.autrev.2020.102686
179. Rudofsky UH, Lawrence DA. New Zealand Mixed Mice: A Genetic Systemic Lupus Erythematosus Model for Assessing Environmental Effects. Environ Health Perspect (1999) 107:713–21. doi: 10.1289/ehp.99107s5713
180. Bagavant H, Nandula SR, Kaplonek P, Rybakowska PD, Umesh S. Deshmukh P. Alum, an Aluminum Based Adjuvant, Induces Sjögren’s Syndrome-Like Disorder in Mice. Clin Exp Rheumatol (2014) 32:251–5.
181. Szczerba BM, Kaplonek P, Wolska N, Podsiadlowska A, Rybakowska PD, Dey P, et al. Interaction Between Innate Immunity and Ro52-Induced Antibody Causes Sjogren's Syndrome-Like Disorder in Mice. Ann Rheum Dis (2016) 75:617–22. doi: 10.1136/annrheumdis-2014-206297
182. Trzeciak M, Bagavant H, Papinska J, Deshmukh US. Immune Response Targeting Sjogren's Syndrome Antigen Ro52 Suppresses Tear Production in Female Mice. Int J Mol Sci (2018) 19:1–10. doi: 10.3390/ijms19102935
183. Ramirez Sepulveda JI, Kvarnstrom M, Brauner S, Baldini C, Wahren-Herlenius M. Difference in Clinical Presentation Between Women and Men in Incident Primary Sjogren's Syndrome. Biol Sex Differ (2017) 8:16. doi: 10.1186/s13293-017-0137-7
184. Sequeira JF, Keser G, Greenstein B, Wheeler MJ, Duarte PC, Khamashta MA, et al. Systemic Lupus Erythematosus: Sex Hormones in Male Patients. Lupus (1993) 2:315–7. doi: 10.1177/096120339300200507
185. Souyris M, Cenac C, Azar P, Daviaud D. l., Canivet A, Grunenwald S, et al. TLR7 Escapes X Chromosome Inactivation in Immune Cells. Sci Immunol (2018) 3:eaap8855. doi: 10.1126/sciimmunol.aap8855
186. Sroka M, Bagavant H, Biswas I, Ballard A, Deshmukh US. Immune Response Against the Coiled Coil Domain of Sjögren’s Syndrome Associated Autoantigen Ro52 Induces Salivary Gland Dysfunction. Clin Exp Rheumatol (2018) 36:41–6.
187. Infantino M, Meacci F, Grossi V, Benucci M, Morozzi G, Tonutti E, et al. Serological Epitope Profile of Anti-Ro52-Positive Patients With Systemic Autoimmune Rheumatic Diseases. Arthritis Res Ther (2015) 17:365. doi: 10.1186/s13075-015-0871-3
Keywords: innate immunity, E3 ubiquitin ligase, autoimmune disease, Sjogren’s syndrome, Fc receptor, systemic lupus erythematosus, intracellular antibodies
Citation: Jones EL, Laidlaw SM and Dustin LB (2021) TRIM21/Ro52 - Roles in Innate Immunity and Autoimmune Disease. Front. Immunol. 12:738473. doi: 10.3389/fimmu.2021.738473
Received: 08 July 2021; Accepted: 16 August 2021;
Published: 06 September 2021.
Edited by:
Poornima Paramasivan, Concept Life Sciences, United KingdomReviewed by:
Jillian M. Richmond, University of Massachusetts Medical School, United StatesCopyright © 2021 Jones, Laidlaw and Dustin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lynn B. Dustin, bHlubi5kdXN0aW5Aa2VubmVkeS5veC5hYy51aw==
†Present address: Stephen M. Laidlaw, Wellcome Centre for Human Genetics, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.