 Lauren M. Quinn
Lauren M. Quinn F. Susan Wong
F. Susan Wong Parth Narendran
Parth Narendran
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 01 October 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.737964
This article is part of the Research TopicImmunopathology of Type 1 DiabetesView all 21 articles
The rising incidence of type 1 diabetes (T1D) cannot be ascribed to genetics alone, and causative environmental triggers and drivers must also be contributing. The prospective TEDDY study has provided the greatest contributions in modern time, by addressing misconceptions and refining the search strategy for the future. This review outlines the evidence to date to support the pathways from association to causality, across all stages of T1D (seroconversion to beta cell failure). We focus on infections and vaccinations; infant growth and childhood obesity; the gut microbiome and the lifestyle factors which cultivate it. Of these, the environmental determinants which have the most supporting evidence are enterovirus infection, rapid weight gain in early life, and the microbiome. We provide an infographic illustrating the key environmental determinants in T1D and their likelihood of effect. The next steps are to investigate these environmental triggers, ideally though gold-standard randomised controlled trials and further prospective studies, to help explore public health prevention strategies.
An estimated 1.1 million people under 20 years of age are affected by type 1 diabetes (T1D) worldwide (1, 2). T1D represents 5-10% of the global diabetes burden (3) and is not a disease of childhood alone, with almost half diagnosed in adulthood (4–6). Overall annual increase in T1D is estimated at 3% (2-5%) (1, 7), with rising trends observed across all age groups over the last three decades (1, 3). Some of the greatest increases are observed in historically low prevalence countries (4, 8).
T1D is a chronic autoimmune condition, characterised by hyperglycaemia and long-term insulin dependency (9). T1D pathophysiology is defined by three stages of disease progression (10). Stage one is seroconversion to one or more autoantibodies (10), including glutamic acid decarboxylase (GAD), anti-insulin, insulinoma-associated antigen 2 (IA2) and zinc-transporter 8 (Zn-T8). Presence of two or more antibodies will see 70% of children develop T1D in the next 10 years, whilst four autoantibodies invariably confer 100% risk (11). Stage 2 is damage to the beta-cells causing pre-symptomatic dysglycaemia and stage 3 is overt T1D due to beta-cell failure with requirement for exogenous insulin (10). This provides different targets for prevention at the stages of primary prevention (preventing seroconversion, in those genetically at risk), and secondary prevention (preventing loss of and damage to the beta-cells in individuals with autoimmunity/autoantibodies) (12).
The primary risk factor for T1D is genetic. It is strongly associated with HLA-DR3-DQ2 and/or HLA-DR4-DQ8 haplotypes (13). The significance of genetics is further evidenced by the increased risk observed where a sibling (8%), father (5%) or mother (3%) has T1D. The major histocompatibility complex (MHC) encoding the HLA region confers 50% of the genetic risk for T1D (13). Genome-wide association studies have identified an additional 50 loci that confer susceptibility (13).
However, genetics alone does not equate to causality and an array of environmental factors are implicated to trigger T1D seroconversion and disease progression (14). The threshold hypothesis describes a model in which genetic and environmental factors represent intersecting and reciprocal trend lines, defined by odds ratios in rank order, which cumulatively confer risk of progression to T1D until a critical threshold is met (15).
Evidence to support the nurture side of the argument for T1D continues to expand. Firstly, the rising incidence of T1D in recent decades is too rapid to be explained by genetic changes alone (8). Secondly, T1D concordance rates between monozygotic twins is <50% (16). Thirdly, T1D incidence also demonstrates significant geographic and latitudinal differences, with higher incidence in Nordic countries (17–20), and migration studies show that the incident risk of the new location is assumed (21). Fourthly, incidence is increasing across all age groups, and the incidence in younger children is rising (1), despite highest risk genotypes declining over the last 20-40 years (22, 23). The highest rates are observed in previously low incidence countries and countries with the highest economic growth (8).
It may be more than coincidence that T1D is therefore a heterogenous condition determined by a combination of genetic, immunological, and metabolic factors, and this complexity reflects the array of environmental triggers implicated in the pathogenesis.
Numerous other reviews have explored environmental contributions to T1D (8, 12, 14, 24, 25). These have largely outlined association studies between different environmental determinants and the development of T1D. Some of these reviews also predate the results of the seminal, ‘The Environmental Determinants of Diabetes in the Young’ (TEDDY) study, from which different aspects have been published over the last 6 years (26).
In this narrative review, we explore the putative environmental risk factors for T1D with an emphasis on testing causality. Since causality is best tested in the setting of a double-blind randomised controlled trial (RCT), we outline the different RCTs in this area. As part of the review, we outline potential underlying mechanisms to the different environmental determinants and the stage of pre-T1D at which they could exert an influence.
We undertook this review though searching PubMed and Medline. We used the following search terms ‘environment AND type 1 diabetes’; type 1 diabetes (T1D) OR islet autoimmunity (IA) combined with the following - enterovirus; rotavirus; influenza; COVID-19; vaccine OR vaccination; birth weight; weight gain; BMI; childhood obesity; gut microbiome OR gut microbiota; diet; breast milk OR breastfeeding; cow’s milk; formula milk; gluten; antibiotic; probiotic; vitamin D; and nicotinamide; omega-3. We included systematic reviews, meta-analyses, RCT, cohort studies (prospective and retrospective) and case-control studies. We have included the largest retrospective and prospective European cohort and case control studies, including ‘Type 1 Diabetes Prediction and Prevention’ (DIPP) (27), ‘Early Childhood Diabetes in Finland’ (DiMe) (28), ‘Danish National Birth Cohort’ (DNBC), ‘Norwegian Mother and Child cohort’ (MOBA) (29), ‘Diabetes Autoimmunity Study in the Young’ (DAISY) (30), DIABIMMUNE, Environmental Triggers of T1D (MIDIA - Norwegian acronym) (31), Finnish Dietary Intervention Trial for the Prevention of T1D (FINDIA) (32), TEDDY study (26), and trials including ‘Trial to Reduce IDDM in the Genetically at Risk’ (TRIGR) (33), the ‘European Nicotinamide Diabetes Intervention Trial’ (ENDIT) (34), and the Deutsche Nicotinamide Intervention Study (DENIS) (35). Reference lists were also screened for relevant articles.
Viruses are important contenders for environmental triggers of T1D (36, 37).
Acute fulminant diabetes, termed type 1b diabetes, is reported following infection with Mumps, Coxsackie B3 and B4, Rubella, and Influenza B infection (38). The hyperglycaemic ketosis is sudden, symptoms occur for one week, islet antibodies are negative, and C-peptide is extremely low. In this instance, beta-cell damage occurs secondary to direct lytic effects from viral invasion, causing widespread beta-cell destruction and absolute insulin deficiency, without autoimmunity (37, 38).
An alternative association, expanded below, appears to be a more chronic, repeated viral exposure. Implicated mechanisms here include molecular mimicry, where viral epitope sequences bear resemblance to beta-cell antigens and potentially trigger a cross-reactive autoimmune response (36). Viral infection of beta-cells will also result in over-expression of MHC class I, resulting in presentation of self-antigens and perpetuation of further autoimmunity (36, 37, 39). Viruses implicated in this more chronic-repeated infection model are outlined below.
The most robust evidence for a viral trigger exists for enteroviruses (EV) (40–42). T1D incidence correlates with enteroviral infection rates and the seasonal variation in T1D is preceded by enteroviral epidemics (41, 43). However, EV is a common childhood infection, hence HLA susceptibility to T1D, combined with genetically determined susceptibility and inflammatory response to EV are critical determinants of risk (41, 44–46). EV infection potentially initiates and accelerates all three stages of T1D pathophysiology (42, 43).
EV spreads and replicates via the upper respiratory and gastrointestinal tracts, and invades the beta islet cells via the coronavirus adeno receptor (CAR) (36). Inefficient viral clearage of EV (47) and induction of a chemokine response from the beta-cells triggers islet autoimmunity (IA) through molecular mimicry, inflammation, bystander effects, and T cell suppression (48). EV chronicity appears critical to sustain beta-cell autoimmunity; repeated infection with EV strains further increases risk (36).
Systematic reviews (41, 49) and cohort studies (27, 28, 50, 51) demonstrate positive associations between persistent EV infection, autoimmunity, and progression to stage 3. The DiMe study (28) and the DIPP (27) study showed that EV infection in pregnancy or early childhood respectively increased risk of T1D. Interestingly, studies show evidence of seroconversion to islet autoantibodies in both mother postnatally and child, following enteroviral infection during pregnancy (52, 53). A meta-analysis showed that maternal infections were also significantly associated with T1D progression, and most notably for maternal enterovirus infections, odds ratio (OR) 1.54 (confidence interval (CI) 1.05-2.27) (54). The postulated mechanism is transfer of epitopes/molecular mimicry that triggers autoimmunity in the offspring, but the supporting evidence is limited. Cross-reactivity between EV with GAD and IA2 epitopes could trigger seroconversion, but equally, secondary to chronic and cumulative viral infections, primed auto-reactive T cells could promote progression to stage 2, in auto-antibody positive individuals (36).
Most recently, the TEDDY study showed from a genetically predisposed cohort of children (n=8676) followed-up over 15 years, that chronicity of Coxsachie B (EVB), from persistent shedding in the stool, predicted development of IA, particularly anti-insulin antibodies (51, 55, 56). Conversely, acute EVB infection, without prolonged stool shedding, did not associate with autoimmunity or T1D in this cohort (51). A systematic review and meta-analysis by Yeung et al. (41) further showed that persistent EVB increases risk of IA and T1D with an OR of 3.7 (CI: 2.1-6.8) and 9.8 (5.5-17.4) respectively. Problems remain in detecting the specific strain(s) of EV that confer the highest risk.
Based on this strong association between chronic EV infection and T1D (51, 56), a RCT with a polyvalent vaccine to EV is currently planned. An EV vaccine would target primary and secondary prevention by reducing recurrent EV infection, limiting exposure to potential reactive epitopes and chronic beta-cell inflammation that could contribute to IA (48, 57).
Introduction of the childhood rotavirus (RV) vaccination in Australia, and the fall in T1D incidence that followed, was postulated to implicate a causal environmental trigger (58). The outer capsid protein of the human RV (VP7) shares 56% identity and 100% similarity with a dominant epitope in IA2, and both bind to (HLA-DR4) (*0401) (59), suggesting molecular mimicry and cross-reactive T cells as mediators of IA (59, 60). However, the DIPP longitudinal cohort study exploring RV in T1D pathophysiology failed to show an association between RV and auto-antibodies (61). A study demonstrated that RV infection prior to 6 months of age was significantly associated with human and bovine-insulin binding antibodies, but this was strongest in children receiving cow’s milk prior to 3 months (62), representing an important confounding factor.
An Australian interrupted time series analysis found a 15% T1D risk reduction in children aged 0-4 years receiving the RV vaccine compared to those not vaccinated (58). However, the Finnish population-based study with 11–14 year follow-up did not replicate these findings (63, 64). Although Rogers et al. (65) found a 33% reduced risk of T1D in vaccinated compared to unvaccinated children, Burke et al. found no association between RV vaccination and T1D incidence in a US cohort of children with commercial insurance (66). Similarly, Glanz et al. (67) found no association. Hence the evidence supporting RV vaccination as a protective environmental factor is inconclusive.
Studies investigating the impact of the influenza infection on stage 1 and 2 T1D risk, have also been inconclusive. Valdes et al. (68) and Nenna et al. (69) showed increased risk of T1D following influenza infection whereas Kondrashova et al. (70) showed no increased risk in children genetically susceptible to T1D.
With regard to vaccination, Ruiz et al. (71) and Bardage et al. (72) showed no association between T1D risk and influenza vaccination. The Pandemrix vaccine, which caused narcolepsy in genetically susceptible individuals, raised concerns for T1D cross-reactivity and was investigated in the TEDDY study in a Finnish and Swedish cohort (73). Here, the Pandemrix vaccine did not increase risk of seroconversion for one or two islet autoantibodies [HR 0.75 (0.55-1.03) and HR 0.85 (0.57-1.26)] respectively, after adjusting for confounders (73). Interestingly, 73% received a second dose in Sweden compared to 0.6% in Finland, and the Finnish cohort had a lower risk of IA; one antibody [HR (0.47 (0.29-0.75)], or more than one antibody [HR 0.50 (0.28-0.90)], and risk of T1D [HR 0.38 (0.20-0.72)] (73). The TEDDY study is ongoing to explore links between influenza vaccination and progression to stage 3 T1D, but no RCTs are planned to test the associates listed above.
Individuals with T1D are at greater risk of the Sars-CoV2 coronavirus (COVID-19) and more susceptible to severe infection (74). Global studies have suggested an increased incidence of T1D during the COVID-19 pandemic and higher frequency of presentation in severe diabetic ketoacidosis (DKA) (37, 75–78). However, important confounders include delay in seeking medical assistance and the resulting later presentations in DKA (37, 75, 78), as well as the high numbers of patients with type 2 diabetes presenting in DKA. The latter is evidenced by cases of COVID-19 associated DKA who were eventually weaned off insulin onto oral therapies (79, 80).
The coronavirus gains access to lung and gut epithelium via the angiotensin converting enzyme-2 (ACE2) functional receptor, which is also highly expressed in islet cells (81–83). The previous SARS-CoV1 2003 epidemic was associated with elevated fasting plasma glucose, with hyperglycaemia as an independent predictor of morbidity and mortality, even in mild pneumonitis cases with no steroid requirement (84–86). Similarly, in SARS-CoV2, hyperglycaemia in non-diabetic patients is attributed to the inflammatory response and cytokine activation, in addition to viral infection of beta-cells, which decreases insulin production (37, 86). It remains unclear if COVID-19 is simply triggering a fulminant diabetogenic state or presents the final trigger for T1D progression. However, in the former scenario, the beta-cell damage has not always persisted, evidenced by cases that were weaned off insulin and onto oral therapies (79, 80). Moving forward, the CoviDiab study (87) and roll-out of COVID-19 vaccination programmes (88) may help delineate causal relationships between COVID-19 and T1D risk.
The steady rise in autoimmune and allergic diseases in industrialised countries has been linked to a reduction in infectious diseases. Contributing factors include geography and climate (North-South gradient), childhood mixing and subsequent exposure to childhood infection (20), and vaccination programmes, and this relationship underlines the hygiene hypothesis. The incidence of T1D is lower in countries without nationwide vaccination programmes and contrasts starkly to countries with established (or newly implemented) vaccination programmes, where there is a rising T1D incidence (8). In vivo studies have shown in nonobese diabetic mice for T1D models, T1D incidence is higher in the mice bred in specific pathogen free (SPF) conditions compared to those bred in conventional facilities (20, 89, 90).
In terms of vaccinations being directly linked to T1D, cohort studies and meta-analysis performed to explore a causative association have failed to identify a link to date (91, 92). Childhood vaccinations for measles, rubella, mumps, pertussis, Bacillus Calmette-Guerin (BCG), Haemophilus influenza B (HiB), Tetanus Diptheria poliomyelitis, Measles/Mumps/Rubella and Diptheria/Tetanus/Pertussis showed no association with T1D, harmful or protective (91). Kühtreiber et al. performed a randomised 8 year study in T1D and showed that three years after receiving two doses of the BCG vaccine, HbA1c was lowered to near normal levels for five years (93). The mechanism was demonstrated in vitro and in vivo, and can be explained by a metabolism shift from oxidative phosphorylation to aerobic glycolysis, the latter of which is a high glucose usage state. The BCG vaccine also had a role in re-programming tolerance, through epigenetic demethylation of regulatory T cell signature genes, resulting in upregulation of mRNA expression and subsequent induction of T regulatory cells (93).
The evidence of viral infections as a risk factor for T1D is strong. The strongest evidence appears to be through a direct lytic effect on beta-cells (37), for example following infection with mumps. Alternatively, the more insidious classical autoimmune T1D appears to have the strongest association with EV infection (36) and RCT involving vaccination programmes to test a causative link are in development. Associations between T1D and RV or COVID-19 need further evidence - either way there are other major population benefits to vaccinating against these two diseases. There is currently no evidence that any of the childhood vaccination programmes associate with T1D risk.
Worldwide prevalence of obesity has risen to 5.6% in girls and 7.8% in boys (94), which is a 10-fold rise in four decades (8, 95–98). The roots lie in genetic, epigenetic, and environmental factors (99, 100). We have observed a rising incidence of childhood obesity along with that of T1D, depicting a double diabetes state which combines features of autoimmunity with insulin resistance (8, 99, 101). The obesity induced insulin resistance in children, increases the burden on the islets cells and potentially initiates, and accelerates the autoimmune processes in genetically predisposed individuals (102, 103). This has been termed the accelerator hypothesis (102, 103). Adiposity also contributes to systemic chronic inflammation, which together contributes to beta-cell damage and apoptosis, and progression to stage 2 and 3 of T1D (104).
It has been suggested that the stress on these beta-cells, either through inflammation or metabolic demand risks derailing stringently controlled events relating to protein transcription, translation or folding (26, 105–107). The aberrant proteins and peptides resulting from this “beta-cell stress”, in conjunction with local inflammation, are then capable of stimulating an autoimmune response (108–110).
Higher birth weight and infant growth rate contribute to T1D pathogenesis. A systematic review and meta-analysis by Harder et al., comprising 2.4 million children, found high birth weight (>4kg) increased risk of T1D by 17% (1.09-1.26) (111). The Danish National Birth Cohort (DNBC) and Norwegian Mother and Child cohort (MoBa) comprised 99,832 children and found an increased birth weight up to 12 months of age was significantly associated with T1D risk [HR 1.24 (1.09-1.41)] (29). Similarly, the Goldacre UK population study found that children with higher birth weight (3.5-<4kg and 4-5.49kg) compared to medium birth weight (3-3.49kg) had higher incidence of T1D, HR 1.13 (1.03-1.23) and HR 1.16 (1.02-1.31) respectively (112).
The TEDDY study showed that higher infantile weight gain was associated with increased risk of IA [HR 1.09 per 1 kg/year (1.02-1.17)] (113). In children with first autoantibody GAD, there was also an increased risk of progression from IA to overt T1D [HR 2.57 per 1 kg/year (1.34-4.91)] (113). Increased progression from IA to T1D was also observed when height-growth pattern was lower in infancy, but higher in early childhood (113). Interestingly, Yassouridis et al. (114) used pooled analyses to show that IA was only linked with rising BMI up to three years of age, in non-diabetic mothers (adjusted OR 2.02 (1.03-3.73) (114). Finally, in the TRIGR study, although annual growth did not associate with autoantibody status, being overweight at 2-10 years of age increased risk (HR 2.39 (1.46-3.92) of progression to T1D (stage 2-3) but not risk of seroconversion (stage 1) (33).
A meta-analysis showed a positive dose response relationship between childhood BMI and T1D risk, OR 1.25 (1.04–1.51) (115). A Mendelian randomisation study found an OR of 2.76 (1.40-5.44) for T1D risk (116). Further, a Danish study showed that higher BMIz score at 7 years [OR 1.23 (1.09-1.37)] and 13 years [OR 1.20 (1.04-1.40)] of age was associated with an increased risk of T1D (117). The TrialNet pathway to prevention study found no link between BMI, BMI percentile, insulin resistance of progression to T1D (118), although Ferrara et al. showed that cumulative excess BMI was associated (119).
Given birth weight (111), body weight (115) and weight gain (113) correlate with T1D risk at an early stage, and this excess weight is most amenable to intervention in early childhood (120), this justifies early recognition and treatment. However, to date there are limited studies exploring whether reducing obesity decreases T1D risk. A number of studies have explored whether exercise programmes that reduce the insulin resistance and weight associated with obesity also reduce T1D. Exercise in both the NOD mouse model of T1D (121–124) and in people newly diagnosed with T1D appear to preserve beta-cell function (125), with evidence of reduced immune cell inflammation and insulitis in the former model (126, 127).
There is now reasonable evidence that increased birth weight (111), early weight gain (113) and obesity in children (115) matters, and associates with risk of IA as well as overt T1D, i.e. that this environmental factor may act to progress people into stage 1, 2, and stage 3 pre-T1D (51, 112, 113, 115). Unfortunately, RCT evidence to test causality are lacking. Surrogate studies demonstrating that exercise interventions can preserve beta-cells at stage 3 T1D do however show promise (125).
The gut microbiota, established in early life, is influenced by perinatal factors and nutrition, and modulates the innate and adaptive immune systems (128, 129). Signature profiles of gut flora are observed in those genetically predisposed to, and incident cases of, T1D (130). The hallmark characteristics are decreased bacterial diversity, reduced microbiota stability, increased frequency of Bacterioides species and decreased frequency of Prevotella, Bifidobacteria, and Lactobacillus, the latter of which confer immunomodulatory properties through production of short chain fatty acids (SCFA) (130). The gut microbiome potentially contributes to beta-cell autoimmunity through enhanced intestinal inflammation, increased permeability, loss of barrier function, and subsequent exposure to dietary antigens (129).
The TEDDY study used 16S ribosomal ribonucleic acid (rRNA) and metagenomic sequencing to reveal gut taxonomy from stool samples of healthy controls compared to genetically at-risk children, aged 3-46 months (131). Weak associations were identified between the gut microbial taxonomies and progression from stages 1-3 of T1D (seroconversion or progression to overt T1D) (131). Conversely, Vatanen et al. (132) showed the gut microbiome in healthy controls expressed genes which stimulated fermentation and synthesis of SCFA, although taxonomy did not significantly differ from the case subjects. This reflects functionally protective properties among healthy gut flora which are lost in predisposed and seroconverted individuals (132). The DIABIMMUNE study, which included 1000 genetically predisposed newborns, showed reduction in gut microbial diversity when progressing from autoantibody positivity to T1D. In autoantibody positive subjects, gene expression was shifted to enhance sugar transport and reduce amino acid biosynthesis, compared to non-seroconverters (31). The ABIS study, included 17,000 babies from Sweden born between 1997 and 1999 followed-up for 12 years, and showed that HLA haplotype determines gut microbial composition (133). Zhao et al. compared serial faecal samples in 11 seroconverted cases (5 of whom developed T1D) compared to controls, and found a higher bacteriophage Shannon diversity index in the controls, and these differences increased with age (134). Random Forests analysis revealed T1D-associated viral bacteriophage contigs, separate from the age-associated bacteriophage contigs, which were linked to gut microbial composition. The best predictive contig had nucletoid sequence homolog consistent with B.dorei (134). Overall, the gut microbiome is a key window to IA.
In the TEDDY study, mode of birth delivery was an important determinant of gut taxonomy in the first year of life (131). Mode of delivery cultivates the neonatal gut microbiome, which determines microbial composition, succession and function, and contributes to risk of autoimmune disease, allergy and obesity in later life (130). Vaginal delivery leads to neonatal gut colonisation that reflects the vaginal flora, comprising Lactobacillus and Bifidobacterium (130). The TEDDY study suggests that vaginal delivery leads to Bacteroide colonisation, which supports gut maturation and enhances microbial diversity (130, 131). Alternatively, delivery by caesarean section (CS) is associated with gut microbiota seeded from the mothers skin commensals, namely Clostridium and Staphylococcal species (130). The lack of colonisation by Lactobacillus and Bifidobacterium species confers dysfunctional immunomodulation with resultant implications for autoimmunity (130, 131). Risk of T1D following CS vs vaginal delivery was evaluated in a meta-analysis, including 20 studies. After adjustment for confounders, CS increased risk of T1D by 23% (1.15-1.32) compared to vaginal delivery (135). Another systematic review, comprising 9 observational studies and including 5 million births found that elective CS increased childhood T1D risk by 12% (1.05-1.20) compared to vaginal delivery. However, following adjustment, risk differences did not remain due to large study heterogeneity (136). Separate analyses focussed on cohort studies, which reduced the heterogeneity and showed T1D risk was significantly higher in elective CS [OR 1.12 (1.01-1.24)]. In contrast, the DIPP (137) and DIABIMMUNE (132) studies both demonstrated higher levels of Bacteroide colonisation in genetically predisposed children who seroconverted and progressed to T1D, contrary to evidence that early colonisation with Bacteroidetes species comprised healthy gut flora (130). The gut microbiome is therefore complex, and we need large metagenomic studies to taxonomise the gut microbiota and identify the species which confer protective vs damaging effects, and how they relate to each other.
Indeed, multiple environmental factors shape the gut microbiome in the first years of life, including geography and household exposures such as pets and siblings (131, 138). The ABIS study showed that exposure during pregnancy to cats and dogs conferred no increased risk of T1D, but hamsters did (139).
Obesity also negatively influences the gut microbiome. The obese adult individuals’ gut microbiome lacks diversity and the resultant dysbiosis, triggers immune dysregulation, inflammation and promotes diet-sustained obesity (140). The lack of diversity and composition in the gut of an obese individual is therefore similar to the T1D gut milieu. Consequently, there is research interest in interventions which negate these effects and help restore healthy gut microbiota. In mouse studies, faecal transplant from obese humans to germ free mice triggered greater weight gain, and the opposite also remains true. Allogenic healthy donor faecal transplant to individuals with metabolic syndrome improved insulin sensitivity and restored healthy gut flora (140). Trials of donor faecal transplant for T1D prevention have yet to be attempted, but de Groot et al. performed an RCT in new-onset T1D (<6 weeks) and found preservation of C-peptide following faecal transplantation (141).
In all cases, further studies need to address the range of factors which cultivate the gut microbiome, but dysbiosis appears to be an important hallmark for T1D pathogensis (142, 143).
TEDDY showed that the most significant determinant of gut taxonomy in the first year of life is breastfeeding (131). The World Health Organisation (WHO) recommend exclusive breastfeeding until ≥6 months of age, to support growth, development, immunity and the developing gut microbiome (144). However, practices differ across societies and cultures regarding duration of breast feeding, and the type and timing of solid foods (145). Unique benefits of breast feeding include transference of biologically active substances, such as antibodies, cytokines and hormones that modulate the developing immune system (130, 146). It is postulated that breast milk also confers protection from T1D through reduced frequency of infantile respiratory and gastrointestinal infections (147), delayed exposure to dietary antigens (gluten and bovine insulin) (12), and promotion of a healthy gut flora, seeding Bifidobacterium species (131). The DNBC and MOBA (29) population-based cohort studies, included 155,392 children and showed a two-fold increased risk [HR 2.29 (1.14-4.61)] of T1D in children not breastfed at 6-12 months compared to any breastfeeding for ≥12 months (30). There was no difference in T1D incidence between those fully or partially breastfed, and no association with age of introduction of solid foods (30). The MIDIA study explored breast feeding and age at introduction of solid foods with T1D risk in genetically susceptible children (148). Similarly, they found breastfeeding for ≥12 months predicted decreased risk of progression to T1D (HR 0.35 (0.13-0.94), with no effect on IA (148). Duration of full breastfeeding, age at introduction of solid foods and combination with breastfeeding, did not associate with risk of IA or T1D (148). Importantly, the prospective TEDDY study (149, 150) and the TRIGR RCT (151) showed no effect with duration of exclusive breastfeeding on seroconversion or progression to T1D. Despite the mixed results, ability to extrapolate further insights is limited, as the general health benefits of breastfeeding outweigh risk.
Cow’s milk, which contains bovine insulin, could potentially induce autoimmune responses through molecular mimicry to human insulin, leading to T1D seroconversion in children (152). A Finnish cohort study found that children exposed to cow’s milk formula before 3 months of age, had higher rates of IgG binding to bovine insulin antigen and these antibodies cross-reacted with human insulin (153); however none of these children went onto develop IA. The bovine insulin binding antibodies also inversely correlated with age at introduction of formula feed. Bovine insulin autoantibodies declined at 12 and 18 months, except in the anti-insulin antibody seropositive children, where levels significantly increased (153). The FINDIA study investigated bovine-insulin free formula feed with randomisation to three treatment arms (cow’s milk, whey-based hydrolysed formula and bovine-insulin free formula) and showed a reduced incidence of seroconversion in the bovine-insulin free formula feed group (32).
In light of concerns around introduction of cow’s milk (standard/conventional formulas), protein hydrolysed formula alternatives were trialled to determine risk reduction in T1D. In the Finnish TRIGR study, genetically susceptible children were randomised to either cow’s milk (CM) or casein-hydrosylate formula (CHF) feed, during the first 6-8 months of life where breast feeding was not possible, and found a reduced incidence of IA in the CHF group compared to CM group, with one [HR 0.51 (0.28-0.91)] or ≥two autoantibody positivity [HR 0.47 (0.19-1.07)] (154). The TRIGR study was a double-blind RCT including 2159 genetically at-risk children from 15 countries, followed-up for at least 10 years (33). TRIGR showed that weaning to hydrolysed formula compared with conventional formula (casein hydrosylate or adapted cow’s milk formula) did not decrease the cumulative incident risk of T1D after 11.5 years follow-up (33). Similarly, the TEDDY study generally showed no significant association between IA and hydrolysed or conventional formula feed (155). However, extensively hydrolysed formula feed was associated with an increased risk of IA when introduced in the first 7 days of life [HR 1.57 (1.04-2.38)] (155).
Coeliac disease is triggered by an autoimmune reaction to gluten, leading to villous atrophy in the small intestine and subsequent malabsorption (156). Coeliac disease affects 2.5% to 16.4% (5.7% overall) of individuals with T1D (157). Gluten is thought to trigger progression to beta-cell autoimmunity through molecular mimicry (158). The Finnish DIPP study, in 5545 genetical predisposed children, showed that higher intake of oats and gluten-containing foods increased risk of IA (159). The DAISY study showed cumulative gluten intake in the first 12 months did not associate with IA or T1D; however, introduction of gluten prior to 4 months of age significantly increased risk of T1D (160). On the contrary, the prospective TEDDY study showed that delaying introduction of gluten increased the risk of IA. Risk of developing islet antibodies was lower with introduction of gluten at <4 months of age compared to 4-9 months [HR 0.68 (0.47-0.99)], but higher compared to >9 months [HR 1.57 (1.07-2.31)] (161). However, TEDDY also showed that higher gluten intake in the first 5 years of life was associated with an increased risk for celiac disease (156).
Risks of other solid foods included in a weaning regimen have been explored but evidence is limited. The DIPP study linked early introduction of fruits, berries, and root vegetables, between 3-4 months of age, with increased risk of IA in genetically predisposed infants (162). Moreover, the TEDDY study showed protection against IA with introduction of egg, but the association was weak and did not remain when examined in a dose-response relationship (161). Virtanen et al. showed that early introduction of egg, at <8 months of age increased risk of IA in the first three years of life, but the relationship did not remain beyond 3 years follow-up (163).
Overall, we can deduce that introduction of solid foods presents a critical window to the gut microbiota, which may be protected by continuation of breastfeeding during this period (164, 165).
Antibiotics carry potential to chronically disrupt the gut microbiome, particularly in immunosuppressed individuals (130). The same concern applies to antibiotic treatment in early life, where new environmental exposures can shift microbial colonisation, conferring risk to T1D. Mikelson et al. (166) showed in a population case-control study that broad-spectrum antibiotic use in the first two years of life increased risk of T1D. A Finnish case-control study found T1D risk was associated with maternal pre-natal phenoxymethyl penicillin [OR 1.70 (1.08–2.68)] or quinolone use [OR 2.43 (1.16–5.10)] (167). Importantly though, antenatal antibiotic use did not affect risk. The UK Health Improvement Network (THIN) database revealed increased antibiotic exposure was associated with T1D risk, observed when taking 2-5 courses of cephalosporins [OR 1.41 (1.11–1.78)] or >5 courses of penicillins [OR 1.63 (1.26–2.11)] (168). However, the TEDDY study showed cumulative antibiotic use within the first four years of life did not associate with seroconversion [HR 0.98 (0.95-1.10)] or autoantibody progression [HR 0.99 (0.95-1.02)] (169). Similarly, Tapia et al. (170) showed no link between acetaminophen use in the first 6-9 months of life and risk of T1D in a Norwegian cohort.
Agents which alter gut bacterial flora provide opportunities to restore a healthy microbiome for primary and secondary preventative purposes (171). However, evidence in support of their beneficial impact in reducing T1D risk is lacking. Probiotics consist of live micro-organisms and are engineered to restore healthy gut microbiota; protect gut membrane integrity; increase SCFA/butyrate production; reduce proinflammatory cytokines; and promote anti-inflammatory cytokines (171). In the TEDDY study, probiotic use in the first 27 days of life reduced risk of IA, compared to probiotic use after 27 days of life or no probiotic use, HR 0.66 (0.46-0.94), but this was only observed in genetically predisposed individuals (150). A double-blind placebo RCT compared maternal and infant probiotic supplementation in 1223 babies at risk of allergy and found no association with IA by 5 years, or overt T1D by 13 years, but this was a small sample size in a population not at risk of T1D (172). Prebiotics similarly aim to restore healthy gut flora and confer immunomodulatory benefits. Prebiotics comprise fructo-oligosaccharides, galacto-oligosaccharides, lactulose, or indigestible carbohydrates, are selectively up taken by gut microbiota and are associated with SCFA production, but have not been tested as a protective agent in T1D. Overall, evidence to support probiotics or prebiotics in the primary or secondary prevention of T1D is limited, but represent novel targets for therapeutic trials in genetically predisposed and seroconverted individuals (171).
Vitamin D is a candidate for protection against T1D due to its anti-inflammatory effects, role in regulation of the immune system and induction of T regulatory cells, which modulate autoimmune risk (8). Cathelicidin was recently proposed to link vitamin D with the gut microbiota and protective effects on beta-cell function (173). Further evidence stems from the higher incident cases of T1D observed at northern latitudes and in winter months compared to summer, where sunlight exposure inversely correlates with T1D cases on a monthly basis (174). However, studies exploring the relationship between vitamin D concentration and supplementation with T1D risk demonstrate mixed results (175–178).
A higher serum vitamin D reduces risk of IA, as demonstrated by a dose-response meta-analysis which found a U-shaped relationship with an OR 0.91 (0.90-0.93) for T1D per 10nmol/L increase in vitamin D (179). In contrast, the prospective DAISY study found no association between vitamin D concentration and seroconversion or T1D disease progression in IA positive individuals (175). This finding was corroborated by the prospective DIABIMMUNE study (180). Importantly however, the TEDDY study confirmed that higher plasma 25‐hydroxyvitamin D correlated with lower risk for IA in genetically predisposed children (181). More copies of the Vitamin D Receptor allele (VDR) due to a Single Nucleotide Polymorphism (SNP-86), conferred greater protection. Interestingly, dairy product vitamin D supplementation in Finland has since been associated with the stabilising incidence of T1D in this region (181).
Regarding supplementation, the Finnish birth cohort study found that in cases where the recommended dose was supplemented in the first year of life, >2000 units per day compared to <2000 units per day, relative risk (RR) for T1D was much reduced at 0.22 (0.05-0.89) (182). A Norwegian study showed that vitamin D and cod liver oil supplementation from 7-12 months of age reduced risk of T1D compared to supplementation from birth to 6 months of age (183). Further, the EURODIAB study showed that vitamin D supplementation in infancy was associated with reduced risk of T1D (184). A meta-analysis also showed a 29% (0.60-0.84) risk reduction for T1D with vitamin D supplementation (178). However, the DAISY prospective cohort did not identify an association between vitamin D and IA or risk of progression to T1D (175). In the ABIS study, infantile, intermediate vitamin D supplementation also did not associate with IA (176). Analysis of the TEDDY cohort for infantile vitamin D supplementation and T1D risk is awaited. However, the TEDDY study and a meta-analysis showed no association between maternal vitamin D supplementation and offspring’s T1D risk (177). The jury is out but further trials are warranted to further explore the value of vitamin D in the T1D risk story.
Nicotinamide delays beta-cell failure enhances resistance to beta-cell toxins and increased regenerative capacities observed in NOD mice (185, 186). The ENDIT RCT investigated islet cell antibody (ICA) positive, first degree relatives of people with T1D but found no significant association with T1D (34). The DENIS study similarly showed no benefit with high dose nicotinamide at 3 years follow-up in genetically predisposed first-degree relatives (35).
Omega-3 poly-unsaturated fatty acids (PUFA) reduce pro-inflammatory cytokines and may protect against T1D (187). Studies exploring benefit with omega-3 supplementation however have shown mixed results (188, 189). The TRIALNET Pathway to Prevention study compared omega-3 supplementation in the third trimester of pregnancy compared to infants aged 5 months and found no difference in pro-inflammatory cytokine profiles (188). In the DAISY study, Norris et al. identified a risk reduction in IA in infants supplemented with omega-3 PUFA from 12 months of age (189). This association was strongest in participants who were positive for more than 2 autoantibodies. The DAISY study further showed this increased risk was associated with reduced omega-3 PUFA in the red blood cell membranes. Reduced membrane concentration of docosapentaenoic acid predicted increased risk of IA and an individual’s genotype determined protective effects of α-linolenic acid supplementation (165, 190).
The role of the gut microbiome and diet has been an area of active interest and research. There is strong evidence for an association between the microbiome (and factors that affect it) and T1D, and this is worth further exploration (129–132). However, the association of T1D with the many dietary agents that have been postulated remain to be confirmed and tested in an RCT setting.
Despite over 40 years of investigation, with multiple, international case-control, cohort, and prospective studies, we are still in search of those critical environmental triggers for T1D. The TEDDY study has provided the largest evaluation of environmental triggers in genetically predisposed children to date (26). Lessons learned are that T1D is a highly heterogenous condition, influenced by both genetic (13) and environmental factors (14), which interact through the threshold hypothesis (15, 26), to initiate and promote T1D over time.
We would suggest that a way forward for this field is first to explore and establish those environmental factors that probably associate with risk for IA and/or T1D. Once identified, they can then be tested, ideally through a RCT.
This proposal comes with challenges. The challenges of recruiting, defining and measuring exposure to the environmental agent, and allowing a sufficient period of follow up for IA and T1D to develop should not be under-estimated and has been outlined by others (191). Bearing these issues in mind, our review suggests probable associations with enterovirus infections; birth weight; early growth; childhood obesity; and with changes in the gut microbiome (Table 1). Several other possible associations exist but these need further evaluation. Figure 1 summarises the likelihood of effect influenced by the environmental discussed in this review.
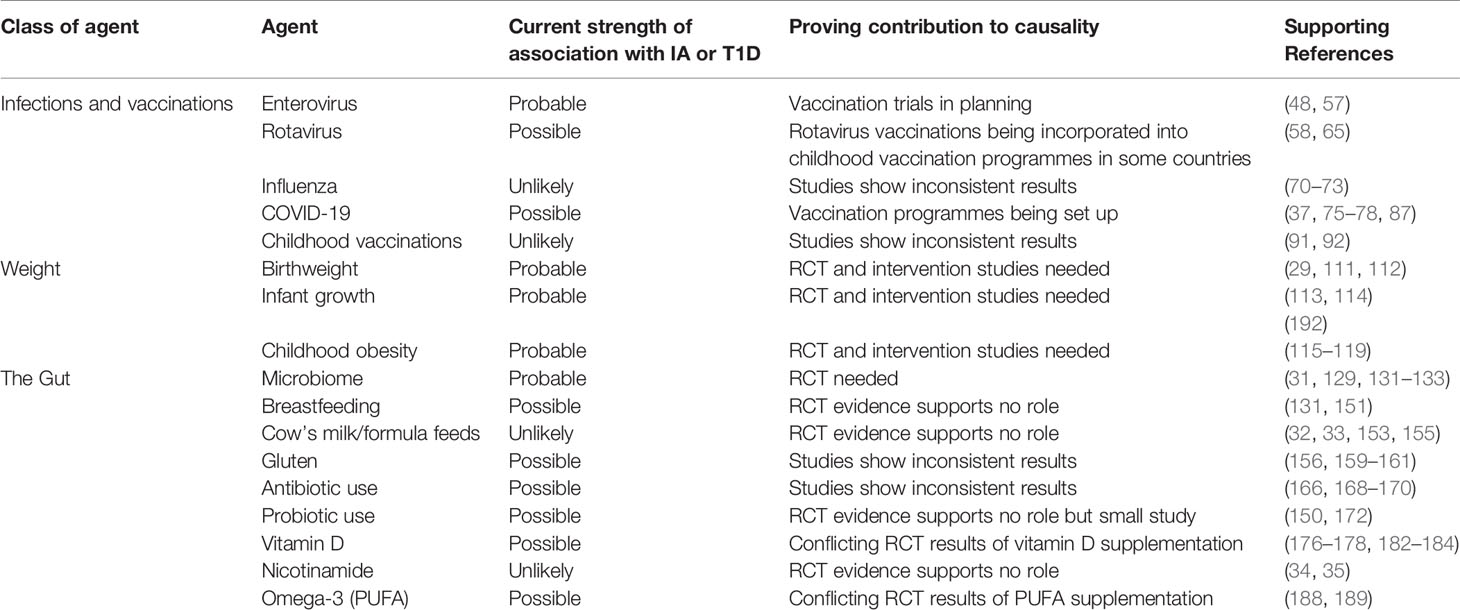
Table 1 List of the key environmental determinants outlined in this review and the evidence supporting a causal framework.
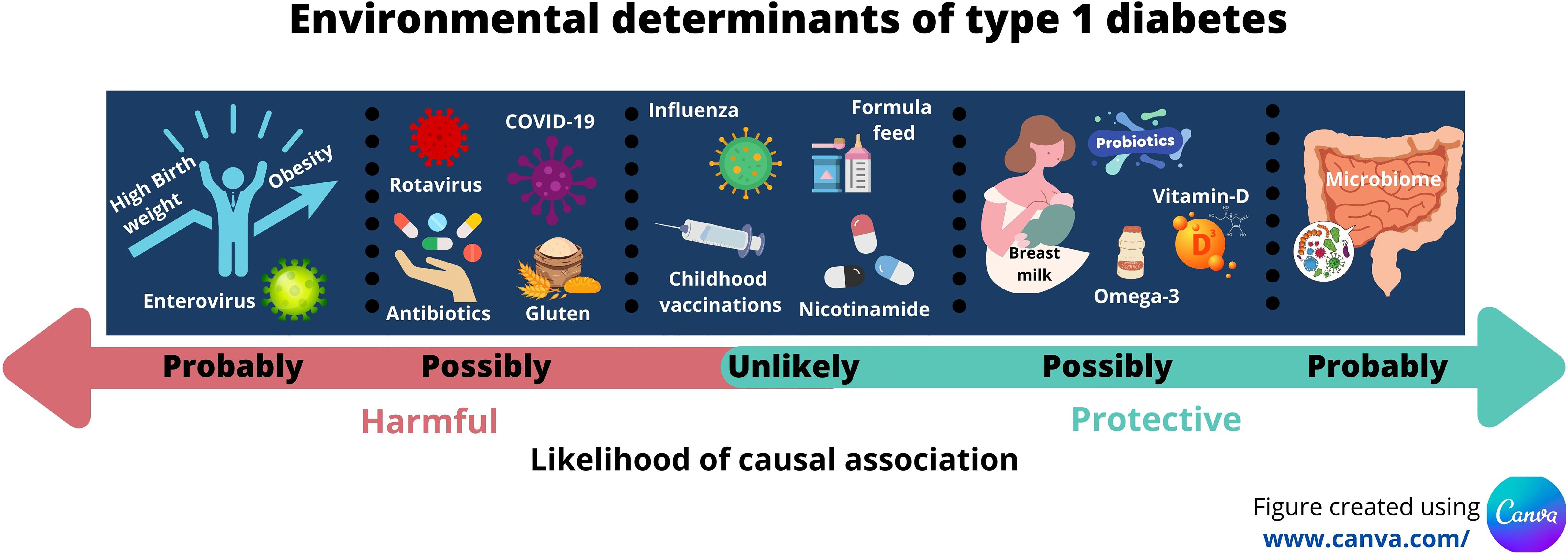
Figure 1 Infographic illustrating the key environmental determinants of type 1 diabetes and their likelihood of contributing to causality.
The subsequent testing of ‘probable association’ also brings challenges. Some agents do not lend themselves easily to testing with a gold-standard RCT (birth weight and rate of childhood growth), and others cannot be tested because programmes to control the putative agent have been, or are being, implemented for other public health reasons (rotavirus, COVID-19) (88, 193). Yet other environmental agents such as childhood obesity may be considered unethical to test because there are good arguments for establishing a national programme to address this major global health burden (8). Proving causality for these agents will require means of assessment other than RCTs. However, well conducted RCTs, as was undertaken for the TRIGR study comparing hydrolyzed infant formula compared to cow’s milk-based formula (33), can be effective at addressing long-standing concerns about the T1D risk of particular environmental agents.
In conclusion, we present a summary of the environmental determinants according to the leading hypotheses; infection and vaccinations, the accelerator hypothesis, and the gut microbiome, and we outline the necessary routes to transition from association to causality.
LQ, FW, and PN made substantial contributions to the following: conception or design of the work; drafting the work or revising it critically for important intellectual content; providing approval for publication of the content; and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Patterson CC, Karuranga S, Salpea P, Saeedi P, Dahlquist G, Soltesz G, et al. Worldwide Estimates of Incidence, Prevalence and Mortality of Type 1 Diabetes in Children and Adolescents: Results From the International Diabetes Federation Diabetes Atlas, 9th Edition. Diabetes Res Clin Pract (2019) 157:107842. doi: 10.1016/j.diabres.2019.107842
2. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and Regional Diabetes Prevalence Estimates for 2019 and Projections for 2030 and 2045: Results From the International Diabetes Federation Diabetes Atlas, 9(Th) Edition. Diabetes Res Clin Pract (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
3. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and Incidence of Type 1 Diabetes in the World: A Systematic Review and Meta-Analysis. Health Promot Perspect (2020) 10(2):98–115. doi: 10.34172/hpp.2020.18
4. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 Diabetes. Lancet (2014) 383(9911):69–82. doi: 10.1016/S0140-6736(13)60591-7
5. NHS Digital. National Diabetes Audit Diabetes Prevention Programme- Quarterly Report: 1 January to 31 December 2020. NHS Digital (2021). Available at: https://digital.nhs.uk/data-and-information/publications/statistical/national-diabetes-audit/diabetes-prevention-programme--quarterly-report-1-january-to-31-december-2020.
6. Thomas NJ, Jones SE, Weedon MN, Shields BM, Oram RA, Hattersley AT. Frequency and Phenotype of Type 1 Diabetes in the First Six Decades of Life: A Cross-Sectional, Genetically Stratified Survival Analysis From UK Biobank. Lancet Diabetes Endocrinol (2018) 6(2):122–9. doi: 10.1016/S2213-8587(17)30362-5
7. Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Epidemiology of Type 1 Diabetes. Endocrinol Metab Clin North Am (2010) 39(3):481–97. doi: 10.1016/j.ecl.2010.05.011
8. Xia Y, Xie Z, Huang G, Zhou Z. Incidence and Trend of Type 1 Diabetes and the Underlying Environmental Determinants. Diabetes Metab Res Rev (2019) 35(1):e3075. doi: 10.1002/dmrr.3075
9. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 Diabetes. Lancet (2018) 391(10138):2449–62. doi: 10.1016/S0140-6736(18)31320-5
10. Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, et al. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care (2015) 38(10):1964–74. doi: 10.2337/dc15-1419
11. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion to Multiple Islet Autoantibodies and Risk of Progression to Diabetes in Children. JAMA (2013) 309(23):2473–9. doi: 10.1001/jama.2013.6285
12. Skyler JS. Primary and Secondary Prevention of Type 1 Diabetes. Diabetes Med (2013) 30(2):161–9. doi: 10.1111/dme.12100
13. Pociot F, Lernmark A. Genetic Risk Factors for Type 1 Diabetes. Lancet (2016) 387(10035):2331–9. doi: 10.1016/S0140-6736(16)30582-7
14. Rewers M, Ludvigsson J. Environmental Risk Factors for Type 1 Diabetes. Lancet (2016) 387(10035):2340–8. doi: 10.1016/S0140-6736(16)30507-4
15. Wasserfall C, Nead K, Mathews C, Atkinson MA. The Threshold Hypothesis: Solving the Equation of Nurture vs Nature in Type 1 Diabetes. Diabetologia (2011) 54(9):2232–6. doi: 10.1007/s00125-011-2244-z
16. Nistico L, Iafusco D, Galderisi A, Fagnani C, Cotichini R, Toccaceli V, et al. Emerging Effects of Early Environmental Factors Over Genetic Background for Type 1 Diabetes Susceptibility: Evidence From a Nationwide Italian Twin Study. J Clin Endocrinol Metab (2012) 97(8):E1483–91. doi: 10.1210/jc.2011-3457
17. Levy-Marchal C, Patterson C, Green A. Variation by Age Group and Seasonality at Diagnosis of Childhood IDDM in Europe. EURODIAB ACE Study Group Diabetologia (1995) 38(7):823–30. doi: 10.1007/s001250050359
18. Berhan Y, Waernbaum I, Lind T, Mollsten A, Dahlquist G, Swedish Childhood Diabetes Study G. Thirty Years of Prospective Nationwide Incidence of Childhood Type 1 Diabetes: The Accelerating Increase by Time Tends to Level Off in Sweden. Diabetes (2011) 60(2):577–81. doi: 10.2337/db10-0813
19. Oilinki T, Otonkoski T, Ilonen J, Knip M, Miettinen PJ. Prevalence and Characteristics of Diabetes Among Somali Children and Adolescents Living in Helsinki, Finland. Pediatr Diabetes (2012) 13(2):176–80. doi: 10.1111/j.1399-5448.2011.00783.x
20. Bach JF. The Effect of Infections on Susceptibility to Autoimmune and Allergic Diseases. N Engl J Med (2002) 347(12):911–20. doi: 10.1056/NEJMra020100
21. Soderstrom U, Aman J, Hjern A. Being Born in Sweden Increases the Risk for Type 1 Diabetes - a Study of Migration of Children to Sweden as a Natural Experiment. Acta Paediatr (2012) 101(1):73–7. doi: 10.1111/j.1651-2227.2011.02410.x
22. Fourlanos S, Varney MD, Tait BD, Morahan G, Honeyman MC, Colman PG, et al. The Rising Incidence of Type 1 Diabetes Is Accounted for by Cases With Lower-Risk Human Leukocyte Antigen Genotypes. Diabetes Care (2008) 31(8):1546–9. doi: 10.2337/dc08-0239
23. Norris JM, Johnson RK, Stene LC. Type 1 Diabetes-Early Life Origins and Changing Epidemiology. Lancet Diabetes Endocrinol (2020) 8(3):226–38. doi: 10.1016/S2213-8587(19)30412-7
24. Butalia S, Kaplan GG, Khokhar B, Rabi DM. Environmental Risk Factors and Type 1 Diabetes: Past, Present, and Future. Can J Diabetes (2016) 40(6):586–93. doi: 10.1016/j.jcjd.2016.05.002
25. Kanta A, Lyka E, Koufakis T, Zebekakis P, Kotsa K. Prevention Strategies for Type 1 Diabetes: A Story of Promising Efforts and Unmet Expectations. Hormones (Athens) (2020) 19(4):453–65. doi: 10.1007/s42000-020-00207-9
26. Rewers M, Hyoty H, Lernmark A, Hagopian W, She JX, Schatz D, et al. The Environmental Determinants of Diabetes in the Young (TEDDY) Study: 2018 Update. Curr Diabetes Rep (2018) 18(12):136. doi: 10.1007/s11892-018-1113-2
27. Lonnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, et al. Enterovirus Infection as a Risk Factor for Beta-Cell Autoimmunity in a Prospectively Observed Birth Cohort: The Finnish Diabetes Prediction and Prevention Study. Diabetes (2000) 49(8):1314–8. doi: 10.2337/diabetes.49.8.1314
28. Hyoty H, Hiltunen M, Knip M, Laakkonen M, Vahasalo P, Karjalainen J, et al. A Prospective Study of the Role of Coxsackie B and Other Enterovirus Infections in the Pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes (1995) 44(6):652–7. doi: 10.2337/diab.44.6.652
29. Magnus MC, Olsen SF, Granstrom C, Joner G, Skrivarhaug T, Svensson J, et al. Infant Growth and Risk of Childhood-Onset Type 1 Diabetes in Children From 2 Scandinavian Birth Cohorts. JAMA Pediatr (2015) 169(12):e153759. doi: 10.1001/jamapediatrics.2015.3759
30. Lund-Blix NA, Dydensborg Sander S, Stordal K, Nybo Andersen AM, Ronningen KS, Joner G, et al. Infant Feeding and Risk of Type 1 Diabetes in Two Large Scandinavian Birth Cohorts. Diabetes Care (2017) 40(7):920–7. doi: 10.2337/dc17-0016
31. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, et al. The Dynamics of the Human Infant Gut Microbiome in Development and in Progression Toward Type 1 Diabetes. Cell Host Microbe (2015) 17(2):260–73. doi: 10.1016/j.chom.2015.01.001
32. Vaarala O, Ilonen J, Ruohtula T, Pesola J, Virtanen SM, Harkonen T, et al. Removal of Bovine Insulin From Cow's Milk Formula and Early Initiation of Beta-Cell Autoimmunity in the FINDIA Pilot Study. Arch Pediatr Adolesc Med (2012) 166(7):608–14. doi: 10.1001/archpediatrics.2011.1559
33. Writing Group for the TSG, Knip M, Akerblom HK, Al Taji E, Becker D, Bruining J, et al. Effect of Hydrolyzed Infant Formula vs Conventional Formula on Risk of Type 1 Diabetes: The TRIGR Randomized Clinical Trial. JAMA (2018) 319(1):38–48. doi: 10.1001/jama.2017.19826
34. Gale EA, Bingley PJ, Emmett CL, Collier T, European Nicotinamide Diabetes Intervention Trial G. European Nicotinamide Diabetes Intervention Trial (ENDIT): A Randomised Controlled Trial of Intervention Before the Onset of Type 1 Diabetes. Lancet (2004) 363(9413):925–31. doi: 10.1016/S0140-6736(04)15786-3
35. Lampeter EF, Klinghammer A, Scherbaum WA, Heinze E, Haastert B, Giani G, et al. The Deutsche Nicotinamide Intervention Study: An Attempt to Prevent Type 1 Diabetes. DENIS Group. Diabetes (1998) 47(6):980–4. doi: 10.2337/diabetes.47.6.980
36. Rodriguez-Calvo T. Enterovirus Infection and Type 1 Diabetes: Unraveling the Crime Scene. Clin Exp Immunol (2019) 195(1):15–24. doi: 10.1111/cei.13223
37. Boddu SK, Aurangabadkar G, Kuchay MS. New Onset Diabetes, Type 1 Diabetes and COVID-19. Diabetes Metab Syndr (2020) 14(6):2211–7. doi: 10.1016/j.dsx.2020.11.012
38. Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y. A Novel Subtype of Type 1 Diabetes Mellitus Characterized by a Rapid Onset and an Absence of Diabetes-Related Antibodies. Osaka IDDM Study Group. N Engl J Med (2000) 342(5):301–7. doi: 10.1056/NEJM200002033420501
39. Christen U, Edelmann KH, McGavern DB, Wolfe T, Coon B, Teague MK, et al. A Viral Epitope That Mimics a Self Antigen can Accelerate But Not Initiate Autoimmune Diabetes. J Clin Invest (2004) 114(9):1290–8. doi: 10.1172/JCI200422557
40. Richer MJ, Horwitz MS. Coxsackievirus Infection as an Environmental Factor in the Etiology of Type 1 Diabetes. Autoimmun Rev (2009) 8(7):611–5. doi: 10.1016/j.autrev.2009.02.006
41. Yeung WC, Rawlinson WD, Craig ME. Enterovirus Infection and Type 1 Diabetes Mellitus: Systematic Review and Meta-Analysis of Observational Molecular Studies. BMJ (2011) 342:d35. doi: 10.1136/bmj.d35
42. Geravandi S, Liu H, Maedler K. Enteroviruses and T1D: Is It the Virus, the Genes or Both Which Cause T1D. Microorganisms (2020) 8(7):1017. doi: 10.3390/microorganisms8071017
43. Filippi CM, von Herrath MG. Viral Trigger for Type 1 Diabetes: Pros and Cons. Diabetes (2008) 57(11):2863–71. doi: 10.2337/db07-1023
44. Stene LC, Oikarinen S, Hyoty H, Barriga KJ, Norris JM, Klingensmith G, et al. Enterovirus Infection and Progression From Islet Autoimmunity to Type 1 Diabetes: The Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes (2010) 59(12):3174–80. doi: 10.2337/db10-0866
45. Sin J, Mangale V, Thienphrapa W, Gottlieb RA, Feuer R. Recent Progress in Understanding Coxsackievirus Replication, Dissemination, and Pathogenesis. Virology (2015) 484:288–304. doi: 10.1016/j.virol.2015.06.006
46. Honkanen H, Oikarinen S, Nurminen N, Laitinen OH, Huhtala H, Lehtonen J, et al. Detection of Enteroviruses in Stools Precedes Islet Autoimmunity by Several Months: Possible Evidence for Slowly Operating Mechanisms in Virus-Induced Autoimmunity. Diabetologia (2017) 60(3):424–31. doi: 10.1007/s00125-016-4177-z
47. Richardson SJ, Morgan NG. Enteroviral Infections in the Pathogenesis of Type 1 Diabetes: New Insights for Therapeutic Intervention. Curr Opin Pharmacol (2018) 43:11–9. doi: 10.1016/j.coph.2018.07.006
48. Dunne JL, Richardson SJ, Atkinson MA, Craig ME, Dahl-Jorgensen K, Flodstrom-Tullberg M, et al. Rationale for Enteroviral Vaccination and Antiviral Therapies in Human Type 1 Diabetes. Diabetologia (2019) 62(5):744–53. doi: 10.1007/s00125-019-4811-7
49. Faulkner CL, Luo YX, Isaacs S, Rawlinson WD, Craig ME, Kim KW. The Virome in Early Life and Childhood and Development of Islet Autoimmunity and Type 1 Diabetes: A Systematic Review and Meta-Analysis of Observational Studies. Rev Med Virol (2020) e2209. doi: 10.1002/rmv.2209
50. Lonnrot M, Salminen K, Knip M, Savola K, Kulmala P, Leinikki P, et al. Enterovirus RNA in Serum Is a Risk Factor for Beta-Cell Autoimmunity and Clinical Type 1 Diabetes: A Prospective Study. Childhood Diabetes in Finland (DiMe) Study Group. J Med Virol (2000) 61(2):214–20.
51. Vehik K, Lynch KF, Wong MC, Tian X, Ross MC, Gibbs RA, et al. Prospective Virome Analyses in Young Children at Increased Genetic Risk for Type 1 Diabetes. Nat Med (2019) 25(12):1865–72. doi: 10.1038/s41591-019-0667-0
52. Dahlquist GG, Ivarsson S, Lindberg B, Forsgren M. Maternal Enteroviral Infection During Pregnancy as a Risk Factor for Childhood IDDM. A Population-Based Case-Control Study. Diabetes (1995) 44(4):408–13. doi: 10.2337/diab.44.4.408
53. Resic Lindehammer S, Honkanen H, Nix WA, Oikarinen M, Lynch KF, Jonsson I, et al. Seroconversion to Islet Autoantibodies After Enterovirus Infection in Early Pregnancy. Viral Immunol (2012) 25(4):254–61. doi: 10.1089/vim.2012.0022
54. Yue Y, Tang Y, Tang J, Shi J, Zhu T, Huang J, et al. Maternal Infection During Pregnancy and Type 1 Diabetes Mellitus in Offspring: A Systematic Review and Meta-Analysis. Epidemiol Infect (2018) 146(16):2131–8. doi: 10.1017/S0950268818002455
55. Krischer JP, Liu X, Vehik K, Akolkar B, Hagopian WA, Rewers MJ, et al. Predicting Islet Cell Autoimmunity and Type 1 Diabetes: An 8-Year TEDDY Study Progress Report. Diabetes Care (2019) 42(6):1051–60. doi: 10.2337/dc18-2282
56. Lindfors K, Lin J, Lee HS, Hyoty H, Nykter M, Kurppa K, et al. Metagenomics of the Faecal Virome Indicate a Cumulative Effect of Enterovirus and Gluten Amount on the Risk of Coeliac Disease Autoimmunity in Genetically at Risk Children: The TEDDY Study. Gut (2020) 69(8):1416–22. doi: 10.1136/gutjnl-2019-319809
57. Hyoty H, Leon F, Knip M. Developing a Vaccine for Type 1 Diabetes by Targeting Coxsackievirus B. Expert Rev Vaccines (2018) 17(12):1071–83. doi: 10.1080/14760584.2018.1548281
58. Perrett KP, Jachno K, Nolan TM, Harrison LC. Association of Rotavirus Vaccination With the Incidence of Type 1 Diabetes in Children. JAMA Pediatr (2019) 173(3):280–2. doi: 10.1001/jamapediatrics.2018.4578
59. Honeyman MC, Stone NL, Falk BA, Nepom G, Harrison LC. Evidence for Molecular Mimicry Between Human T Cell Epitopes in Rotavirus and Pancreatic Islet Autoantigens. J Immunol (2010) 184(4):2204–10. doi: 10.4049/jimmunol.0900709
60. Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE, et al. Association Between Rotavirus Infection and Pancreatic Islet Autoimmunity in Children at Risk of Developing Type 1 Diabetes. Diabetes (2000) 49(8):1319–24. doi: 10.2337/diabetes.49.8.1319
61. Blomqvist M, Juhela S, Erkkila S, Korhonen S, Simell T, Kupila A, et al. Rotavirus Infections and Development of Diabetes-Associated Autoantibodies During the First 2 Years of Life. Clin Exp Immunol (2002) 128(3):511–5. doi: 10.1046/j.1365-2249.2002.01842.x
62. Makela M, Vaarala O, Hermann R, Salminen K, Vahlberg T, Veijola R, et al. Enteral Virus Infections in Early Childhood and an Enhanced Type 1 Diabetes-Associated Antibody Response to Dietary Insulin. J Autoimmun (2006) 27(1):54–61. doi: 10.1016/j.jaut.2006.04.003
63. Vaarala O, Jokinen J, Lahdenkari M, Leino T. Rotavirus Vaccination and the Risk of Celiac Disease or Type 1 Diabetes in Finnish Children at Early Life. Pediatr Infect Dis J (2017) 36(7):674–5. doi: 10.1097/INF.0000000000001600
64. Hemming-Harlo M, Lahdeaho ML, Maki M, Vesikari T. Rotavirus Vaccination Does Not Increase Type 1 Diabetes and May Decrease Celiac Disease in Children and Adolescents. Pediatr Infect Dis J (2019) 38(5):539–41. doi: 10.1097/INF.0000000000002281
65. Rogers MAM, Basu T, Kim C. Lower Incidence Rate of Type 1 Diabetes After Receipt of the Rotavirus Vaccine in the United State-2017. Sci Rep (2019) 9(1):7727. doi: 10.1038/s41598-019-44193-4
66. Burke RM, Tate JE, Dahl RM, Saydah S, Imperatore G, Gregg EW, et al. Rotavirus Vaccination and Type 1 Diabetes Risk Among US Children With Commercial Insurance. JAMA Pediatr (2020) 174(4):383–5. doi: 10.1001/jamapediatrics.2019.5513
67. Glanz JM, Clarke CL, Xu S, Daley MF, Shoup JA, Schroeder EB, et al. Association Between Rotavirus Vaccination and Type 1 Diabetes in Children. JAMA Pediatr (2020) 174(5):455–62. doi: 10.1001/jamapediatrics.2019.6324
68. Valdes C, Unanue N, Hernandez M, Garcia R, Castro M, Vasquez L, et al. Is There a Link Between Influenza and Type I Diabetes? Increased Incidence of TID During the Pandemic H1N1 Influenza of 2009 in Chile. Pediatr Endocrinol Rev (2013) 11(2):161–6.
69. Nenna R, Papoff P, Moretti C, Pierangeli A, Sabatino G, Costantino F, et al. Detection of Respiratory Viruses in the 2009 Winter Season in Rome: 2009 Influenza A (H1N1) Complications in Children and Concomitant Type 1 Diabetes Onset. Int J Immunopathol Pharmacol (2011) 24(3):651–9. doi: 10.1177/039463201102400311
70. Kondrashova A, Nurminen N, Patrikainen M, Huhtala H, Lehtonen J, Toppari J, et al. Influenza A Virus Antibodies Show No Association With Pancreatic Islet Autoantibodies in Children Genetically Predisposed to Type 1 Diabetes. Diabetologia (2015) 58(11):2592–5. doi: 10.1007/s00125-015-3723-4
71. Ruiz PLD, Stene LC, Gulseth HL, Tapia G, Trogstad L, Bakken IJ, et al. Pandemic Influenza A H1N1 Vaccination and Subsequent Risk of Type 1 Diabetes in Norway. Epidemiology (2018) 29(1):e6–8. doi: 10.1097/EDE.0000000000000748
72. Bardage C, Persson I, Ortqvist A, Bergman U, Ludvigsson JF, Granath F. Neurological and Autoimmune Disorders After Vaccination Against Pandemic Influenza A (H1N1) With a Monovalent Adjuvanted Vaccine: Population Based Cohort Study in Stockholm, Sweden. BMJ (2011) 343:d5956. doi: 10.1136/bmj.d5956
73. Elding Larsson H, Lynch KF, Lonnrot M, Haller MJ, Lernmark A, Hagopian WA, et al. Pandemrix(R) Vaccination Is Not Associated With Increased Risk of Islet Autoimmunity or Type 1 Diabetes in the TEDDY Study Children. Diabetologia (2018) 61(1):193–202. doi: 10.1007/s00125-017-4448-3
74. Holman N, Knighton P, Kar P, O'Keefe J, Curley M, Weaver A, et al. Risk Factors for COVID-19-Related Mortality in People With Type 1 and Type 2 Diabetes in England: A Population-Based Cohort Study. Lancet Diabetes Endocrinol (2020) 8(10):823–33. doi: 10.1016/S2213-8587(20)30271-0
75. Lazzeroni P, Bernardi L, Pecora F, Motta M, Bianchi L, Ruozi MB, et al. Diabetic Ketoacidosis at Type 1 Diabetes Onset: Indirect Impact of COVID-19 Pandemic. Acta BioMed (2020) 91(4):e2020193. doi: 10.23750/abm.v91i4.10943
76. Rabbone I, Schiaffini R, Cherubini V, Maffeis C, Scaramuzza A, Diabetes Study Group of the Italian Society for Pediatric E, et al. Has COVID-19 Delayed the Diagnosis and Worsened the Presentation of Type 1 Diabetes in Children? Diabetes Care (2020) 43(11):2870–2. doi: 10.2337/dc20-1321
77. Tittel SR, Rosenbauer J, Kamrath C, Ziegler J, Reschke F, Hammersen J, et al. Did the COVID-19 Lockdown Affect the Incidence of Pediatric Type 1 Diabetes in Germany? Diabetes Care (2020) 43(11):e172–3. doi: 10.2337/dc20-1633
78. Unsworth R, Wallace S, Oliver NS, Yeung S, Kshirsagar A, Naidu H, et al. New-Onset Type 1 Diabetes in Children During COVID-19: Multicenter Regional Findings in the U.K. Diabetes Care (2020) 43(11):e170–e1. doi: 10.2337/dc20-1551
79. Kuchay MS, Reddy PK, Gagneja S, Mathew A, Mishra SK. Short Term Follow-Up of Patients Presenting With Acute Onset Diabetes and Diabetic Ketoacidosis During an Episode of COVID-19. Diabetes Metab Syndr (2020) 14(6):2039–41. doi: 10.1016/j.dsx.2020.10.015
80. Reddy PK, Kuchay MS, Mehta Y, Mishra SK. Diabetic Ketoacidosis Precipitated by COVID-19: A Report of Two Cases and Review of Literature. Diabetes Metab Syndr (2020) 14(5):1459–62. doi: 10.1016/j.dsx.2020.07.050
81. Harmer D, Gilbert M, Borman R, Clark KL. Quantitative mRNA Expression Profiling of ACE 2, a Novel Homologue of Angiotensin Converting Enzyme. FEBS Lett (2002) 532(1-2):107–10. doi: 10.1016/S0014-5793(02)03640-2
82. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J Pathol (2004) 203(2):631–7. doi: 10.1002/path.1570
83. Fignani D, Licata G, Brusco N, Nigi L, Grieco GE, Marselli L, et al. SARS-CoV-2 Receptor Angiotensin I-Converting Enzyme Type 2 (ACE2) Is Expressed in Human Pancreatic Beta-Cells and in the Human Pancreas Microvasculature. Front Endocrinol (Lausanne) (2020) 11:596898. doi: 10.3389/fendo.2020.596898
84. Yang JK, Lin SS, Ji XJ, Guo LM. Binding of SARS Coronavirus to Its Receptor Damages Islets and Causes Acute Diabetes. Acta Diabetol (2010) 47(3):193–9. doi: 10.1007/s00592-009-0109-4
85. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80 e8. doi: 10.1016/j.cell.2020.02.052
86. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science (2020) 367(6485):1444–8. doi: 10.1126/science.abb2762
87. CoviDiab-Registry. The Global Registry of New-Onset Covid-19 Related Diabetes. CoviDiab (Internet) (2020). Available at: https://www.diabetes.org.uk/research/take-part-in-research/covidiab.
88. Xing K, Tu XY, Liu M, Liang ZW, Chen JN, Li JJ, et al. Efficacy and Safety of COVID-19 Vaccines: A Systematic Review. Zhongguo Dang Dai Er Ke Za Zhi (2021) 23(3):221–8. doi: 10.7499/j.issn.1008-8830.2101133
89. Tao L, Reese TA. Making Mouse Models That Reflect Human Immune Responses. Trends Immunol (2017) 38(3):181–93. doi: 10.1016/j.it.2016.12.007
90. Okada H, Kuhn C, Feillet H, Bach JF. The 'Hygiene Hypothesis' for Autoimmune and Allergic Diseases: An Update. Clin Exp Immunol (2010) 160(1):1–9. doi: 10.1111/j.1365-2249.2010.04139.x
91. Morgan E, Halliday SR, Campbell GR, Cardwell CR, Patterson CC. Vaccinations and Childhood Type 1 Diabetes Mellitus: A Meta-Analysis of Observational Studies. Diabetologia (2016) 59(2):237–43. doi: 10.1007/s00125-015-3800-8
92. Hviid A, Stellfeld M, Wohlfahrt J, Melbye M. Childhood Vaccination and Type 1 Diabetes. N Engl J Med (2004) 350(14):1398–404. doi: 10.1056/NEJMoa032665
93. Kuhtreiber WM, Tran L, Kim T, Dybala M, Nguyen B, Plager S, et al. Long-Term Reduction in Hyperglycemia in Advanced Type 1 Diabetes: The Value of Induced Aerobic Glycolysis With BCG Vaccinations. NPJ Vaccines (2018) 3:23. doi: 10.1038/s41541-018-0062-8
94. Di Cesare M, Soric M, Bovet P, Miranda JJ, Bhutta Z, Stevens GA, et al. The Epidemiological Burden of Obesity in Childhood: A Worldwide Epidemic Requiring Urgent Action. BMC Med (2019) 17(1):212. doi: 10.1186/s12916-019-1449-8
95. de Onis M, Blossner M, Borghi E. Global Prevalence and Trends of Overweight and Obesity Among Preschool Children. Am J Clin Nutr (2010) 92(5):1257–64. doi: 10.3945/ajcn.2010.29786
96. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, Regional, and National Prevalence of Overweight and Obesity in Children and Adults During 1980-2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet (2014) 384(9945):766–81. doi: 10.1016/S0140-6736(14)60460-8
97. Collaboration NCDRF. Worldwide Trends in Blood Pressure From 1975 to 2015: A Pooled Analysis of 1479 Population-Based Measurement Studies With 19.1 Million Participants. Lancet (2017) 389(10064):37–55. doi: 10.1016/S0140-6736(16)31919-5
98. WHO. Tenfold Increase in Childhood and Adolescent Obesity in Four Decades. Saudi Med J (2017) 38(11):1162–3. doi: 10.1038/s42255-020-0183-z
99. Sahoo K, Sahoo B, Choudhury AK, Sofi NY, Kumar R, Bhadoria AS. Childhood Obesity: Causes and Consequences. J Family Med Prim Care (2015) 4(2):187–92. doi: 10.4103/2249-4863.154628
100. Kopp W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab Syndr Obes (2019) 12:2221–36. doi: 10.2147/DMSO.S216791
101. Islam ST, Srinivasan S, Craig ME. Environmental Determinants of Type 1 Diabetes: A Role for Overweight and Insulin Resistance. J Paediatr Child Health (2014) 50(11):874–9. doi: 10.1111/jpc.12616
102. Wilkin TJ. The Accelerator Hypothesis: A Unifying Explanation for Type-1 and Type-2 Diabetes. Nestle Nutr Workshop Ser Clin Perform Programme (2006) 11:139–53. doi: 10.1159/000094447
103. Wilkin TJ. The Convergence of Type 1 and Type 2 Diabetes in Childhood: The Accelerator Hypothesis. Pediatr Diabetes (2012) 13(4):334–9. doi: 10.1111/j.1399-5448.2011.00831.x
104. Odegaard JI, Chawla A. Connecting Type 1 and Type 2 Diabetes Through Innate Immunity. Cold Spring Harb Perspect Med (2012) 2(3):a007724. doi: 10.1101/cshperspect.a007724
105. Cnop M, Foufelle F, Velloso LA. Endoplasmic Reticulum Stress, Obesity and Diabetes. Trends Mol Med (2012) 18(1):59–68. doi: 10.1016/j.molmed.2011.07.010
106. Szabat M, Page MM, Panzhinskiy E, Skovso S, Mojibian M, Fernandez-Tajes J, et al. Reduced Insulin Production Relieves Endoplasmic Reticulum Stress and Induces Beta Cell Proliferation. Cell Metab (2016) 23(1):179–93. doi: 10.1016/j.cmet.2015.10.016
107. Marre ML, James EA, Piganelli JD. Beta Cell ER Stress and the Implications for Immunogenicity in Type 1 Diabetes. Front Cell Dev Biol (2015) 3:67. doi: 10.3389/fcell.2015.00067
108. O'Sullivan-Murphy B, Urano F. ER Stress as a Trigger for Beta-Cell Dysfunction and Autoimmunity in Type 1 Diabetes. Diabetes (2012) 61(4):780–1. doi: 10.2337/db12-0091
109. Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A. Type 1 Diabetes Mellitus as a Disease of the Beta-Cell (do Not Blame the Immune System? Nat Rev Endocrinol (2021) 17(3):150–61. doi: 10.1038/s41574-020-00443-4
110. Thomaidou S, Zaldumbide A, Roep BO. Islet Stress, Degradation and Autoimmunity. Diabetes Obes Metab (2018) 20(Suppl 2):88–94. doi: 10.1111/dom.13387
111. Harder T, Roepke K, Diller N, Stechling Y, Dudenhausen JW, Plagemann A. Birth Weight, Early Weight Gain, and Subsequent Risk of Type 1 Diabetes: Systematic Review and Meta-Analysis. Am J Epidemiol (2009) 169(12):1428–36. doi: 10.1093/aje/kwp065
112. Goldacre RR. Associations Between Birthweight, Gestational Age at Birth and Subsequent Type 1 Diabetes in Children Under 12: A Retrospective Cohort Study in England 1998-2012. Diabetologia (2018) 61(3):616–25. doi: 10.1007/s00125-017-4493-y
113. Liu X, Vehik K, Huang Y, Elding Larsson H, Toppari J, Ziegler AG, et al. Distinct Growth Phases in Early Life Associated With the Risk of Type 1 Diabetes: The TEDDY Study. Diabetes Care (2020) 43(3):556–62. doi: 10.2337/dc19-1670
114. Yassouridis C, Leisch F, Winkler C, Ziegler AG, Beyerlein A. Associations of Growth Patterns and Islet Autoimmunity in Children With Increased Risk for Type 1 Diabetes: A Functional Analysis Approach. Pediatr Diabetes (2017) 18(2):103–10. doi: 10.1111/pedi.12368
115. Verbeeten KC, Elks CE, Daneman D, Ong KK. Association Between Childhood Obesity and Subsequent Type 1 Diabetes: A Systematic Review and Meta-Analysis. Diabetes Med (2011) 28(1):10–8. doi: 10.1111/j.1464-5491.2010.03160.x
116. Censin JC, Nowak C, Cooper N, Bergsten P, Todd JA, Fall T. Childhood Adiposity and Risk of Type 1 Diabetes: A Mendelian Randomization Study. PloS Med (2017) 14(8):e1002362. doi: 10.1371/journal.pmed.1002362
117. Antvorskov JC, Aunsholt L, Buschard K, Gamborg M, Kristensen K, Johannesen J, et al. Childhood Body Mass Index in Relation to Subsequent Risk of Type 1 Diabetes-A Danish Cohort Study. Pediatr Diabetes (2018) 19(2):265–70. doi: 10.1111/pedi.12568
118. Meah FA, DiMeglio LA, Greenbaum CJ, Blum JS, Sosenko JM, Pugliese A, et al. The Relationship Between BMI and Insulin Resistance and Progression From Single to Multiple Autoantibody Positivity and Type 1 Diabetes Among TrialNet Pathway to Prevention Participants. Diabetologia (2016) 59(6):1186–95. doi: 10.1007/s00125-016-3924-5
119. Ferrara-Cook C, Geyer SM, Evans-Molina C, Libman IM, Becker DJ, Gitelman SE, et al. Excess BMI Accelerates Islet Autoimmunity in Older Children and Adolescents. Diabetes Care (2020) 43(3):580–7. doi: 10.2337/dc19-1167
120. Brown T MT, Hooper L, Gao Y, Zayegh A, Ijaz S, Elwenspoek M, et al. Do Diet and Physical Activity Strategies Help Prevent Obesity in Children (Aged 0 to 18 Years)? Cochrane Database Syst Rev (2019) CD001871. doi: 10.3310/signal-000817
121. Choi SB, Jang JS, Hong SM, Jun DW, Park S. Exercise and Dexamethasone Oppositely Modulate Beta-Cell Function and Survival via Independent Pathways in 90% Pancreatectomized Rats. J Endocrinol (2006) 190(2):471–82. doi: 10.1677/joe.1.06400
122. Choi SB, Jang JS, Park S. Estrogen and Exercise may Enhance Beta-Cell Function and Mass via Insulin Receptor Substrate 2 Induction in Ovariectomized Diabetic Rats. Endocrinology (2005) 146(11):4786–94. doi: 10.1210/en.2004-1653
123. Coskun O, Ocakci A, Bayraktaroglu T, Kanter M. Exercise Training Prevents and Protects Streptozotocin-Induced Oxidative Stress and Beta-Cell Damage in Rat Pancreas. Tohoku J Exp Med (2004) 203(3):145–54. doi: 10.1620/tjem.203.145
124. Oharomari LK, de Moraes C, Navarro AM. Exercise Training But Not Curcumin Supplementation Decreases Immune Cell Infiltration in the Pancreatic Islets of a Genetically Susceptible Model of Type 1 Diabetes. Sports Med Open (2017) 3(1):15. doi: 10.1186/s40798-017-0082-3
125. Narendran P, Jackson N, Daley A, Thompson D, Stokes K, Greenfield S, et al. Exercise to Preserve Beta-Cell Function in Recent-Onset Type 1 Diabetes Mellitus (EXTOD) - a Randomized Controlled Pilot Trial. Diabetes Med (2017) 34(11):1521–31. doi: 10.1111/dme.13439
126. Chimen M, Kennedy A, Nirantharakumar K, Pang TT, Andrews R, Narendran P. What Are the Health Benefits of Physical Activity in Type 1 Diabetes Mellitus? A lit review Diabetologia (2012) 55(3):542–51. doi: 10.1007/s00125-011-2403-2
127. Narendran P, Greenfield S, Troughton J, Doherty Y, Quann N, Thompson C, et al. Development of a Group Structured Education Programme to Support Safe Exercise in People With Type 1 Diabetes: The EXTOD Education Programme. Diabetes Med (2020) 37(6):945–52. doi: 10.1111/dme.14064
128. Thomas RM, Jobin C. Microbiota in Pancreatic Health and Disease: The Next Frontier in Microbiome Research. Nat Rev Gastroenterol Hepatol (2020) 17(1):53–64. doi: 10.1038/s41575-019-0242-7
129. Verduci E, Mameli C, Amatruda M, Petitti A, Vizzuso S, El Assadi F, et al. Early Nutrition and Risk of Type 1 Diabetes: The Role of Gut Microbiota. Front Nutr (2020) 7:612377. doi: 10.3389/fnut.2020.612377
130. Dedrick S, Sundaresh B, Huang Q, Brady C, Yoo T, Cronin C, et al. The Role of Gut Microbiota and Environmental Factors in Type 1 Diabetes Pathogenesis. Front Endocrinol (Lausanne) (2020) 11:78. doi: 10.3389/fendo.2020.00078
131. Stewart CJ, Ajami NJ, O'Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal Development of the Gut Microbiome in Early Childhood From the TEDDY Study. Nature (2018) 562(7728):583–8. doi: 10.1038/s41586-018-0617-x
132. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The Human Gut Microbiome in Early-Onset Type 1 Diabetes From the TEDDY Study. Nature (2018) 562(7728):589–94. doi: 10.1038/s41586-018-0620-2
133. Russell JT, Roesch LFW, Ordberg M, Ilonen J, Atkinson MA, Schatz DA, et al. Genetic Risk for Autoimmunity Is Associated With Distinct Changes in the Human Gut Microbiome. Nat Commun (2019) 10(1):3621. doi: 10.1038/s41467-019-11460-x
134. Zhao G, Vatanen T, Droit L, Park A, Kostic AD, Poon TW, et al. Intestinal Virome Changes Precede Autoimmunity in Type I Diabetes-Susceptible Children. Proc Natl Acad Sci USA (2017) 114(30):E6166–75. doi: 10.1073/pnas.1706359114
135. Cardwell CR, Stene LC, Joner G, Cinek O, Svensson J, Goldacre MJ, et al. Caesarean Section Is Associated With an Increased Risk of Childhood-Onset Type 1 Diabetes Mellitus: A Meta-Analysis of Observational Studies. Diabetologia (2008) 51(5):726–35. doi: 10.1007/s00125-008-0941-z
136. Tanoey J, Gulati A, Patterson C, Becher H. Risk of Type 1 Diabetes in the Offspring Born Through Elective or Non-Elective Caesarean Section in Comparison to Vaginal Delivery: A Meta-Analysis of Observational Studies. Curr Diabetes Rep (2019) 19(11):124. doi: 10.1007/s11892-019-1253-z
137. Davis-Richardson AG, Ardissone AN, Dias R, Simell V, Leonard MT, Kemppainen KM, et al. Bacteroides Dorei Dominates Gut Microbiome Prior to Autoimmunity in Finnish Children at High Risk for Type 1 Diabetes. Front Microbiol (2014) 5:678. doi: 10.3389/fmicb.2014.00678
138. Kemppainen KM, Ardissone AN, Davis-Richardson AG, Fagen JR, Gano KA, Leon-Novelo LG, et al. Early Childhood Gut Microbiomes Show Strong Geographic Differences Among Subjects at High Risk for Type 1 Diabetes. Diabetes Care (2015) 38(2):329–32. doi: 10.2337/dc14-0850
139. Faresjo AO, Ludvigsson J. Pet Exposure in the Family During Pregnancy and Risk for Type 1 Diabetes-The Prospective ABIS Study. Pediatr Diabetes (2018) 19(7):1206–10. doi: 10.1111/pedi.12721
140. Valdes AM, Walter J, Segal E, Spector TD. Role of the Gut Microbiota in Nutrition and Health. BMJ (2018) 361:k2179. doi: 10.1136/bmj.k2179
141. de Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, et al. Faecal Microbiota Transplantation Halts Progression of Human New-Onset Type 1 Diabetes in a Randomised Controlled Trial. Gut (2021) 70(1):92–105. doi: 10.1136/gutjnl-2020-322630
142. Han H, Li Y, Fang J, Liu G, Yin J, Li T, et al. Gut Microbiota and Type 1 Diabetes. Int J Mol Sci (2018) 19(4):995. doi: 10.3390/ijms19040995
143. Abdellatif AM, Sarvetnick NE. Current Understanding of the Role of Gut Dysbiosis in Type 1 Diabetes. J Diabetes (2019) 11(8):632–44. doi: 10.1111/1753-0407.12915
144. WHO. World Health Organisation - Breastfeeding. World Health Organisation (2021). Available at: https://www.who.int/health-topics/breastfeeding.
145. Riikonen A, Hadley D, Uusitalo U, Miller N, Koletzko S, Yang J, et al. Milk Feeding and First Complementary Foods During the First Year of Life in the TEDDY Study. Matern Child Nutr (2018) 14(4):e12611. doi: 10.1111/mcn.12611
146. Pereira PF, Alfenas Rde C, Araujo RM. Does Breastfeeding Influence the Risk of Developing Diabetes Mellitus in Children? A Review of Current Evidence. J Pediatr (Rio J) (2014) 90(1):7–15. doi: 10.1016/j.jped.2013.02.024
147. Frank NM, Lynch KF, Uusitalo U, Yang J, Lonnrot M, Virtanen SM, et al. The Relationship Between Breastfeeding and Reported Respiratory and Gastrointestinal Infection Rates in Young Children. BMC Pediatr (2019) 19(1):339. doi: 10.1186/s12887-019-1693-2
148. Lund-Blix NA, Stene LC, Rasmussen T, Torjesen PA, Andersen LF, Ronningen KS. Infant Feeding in Relation to Islet Autoimmunity and Type 1 Diabetes in Genetically Susceptible Children: The MIDIA Study. Diabetes Care (2015) 38(2):257–63. doi: 10.2337/dc14-1130
149. Hummel S, Weiss A, Bonifacio E, Agardh D, Akolkar B, Aronsson CA, et al. Associations of Breastfeeding With Childhood Autoimmunity, Allergies, and Overweight: The Environmental Determinants of Diabetes in the Young (TEDDY) Study. Am J Clin Nutr (2021) 114(1):134–42. doi: 10.1093/ajcn/nqab065
150. Uusitalo U, Liu X, Yang J, Aronsson CA, Hummel S, Butterworth M, et al. Association of Early Exposure of Probiotics and Islet Autoimmunity in the TEDDY Study. JAMA Pediatr (2016) 170(1):20–8. doi: 10.1001/jamapediatrics.2015.2757
151. Sorkio S, Cuthbertson D, Barlund S, Reunanen A, Nucci AM, Berseth CL, et al. Breastfeeding Patterns of Mothers With Type 1 Diabetes: Results From an Infant Feeding Trial. Diabetes Metab Res Rev (2010) 26(3):206–11. doi: 10.1002/dmrr.1074
152. Adler K, Mueller DB, Achenbach P, Krause S, Heninger AK, Ziegler AG, et al. Insulin Autoantibodies With High Affinity to the Bovine Milk Protein Alpha Casein. Clin Exp Immunol (2011) 164(1):42–9. doi: 10.1111/j.1365-2249.2011.04324.x
153. Vaarala O, Knip M, Paronen J, Hamalainen AM, Muona P, Vaatainen M, et al. Cow's Milk Formula Feeding Induces Primary Immunization to Insulin in Infants at Genetic Risk for Type 1 Diabetes. Diabetes (1999) 48(7):1389–94. doi: 10.2337/diabetes.48.7.1389
154. Knip M, Virtanen SM, Seppa K, Ilonen J, Savilahti E, Vaarala O, et al. Dietary Intervention in Infancy and Later Signs of Beta-Cell Autoimmunity. N Engl J Med (2010) 363(20):1900–8. doi: 10.1056/NEJMoa1004809
155. Hummel S, Beyerlein A, Tamura R, Uusitalo U, Andren Aronsson C, Yang J, et al. First Infant Formula Type and Risk of Islet Autoimmunity in The Environmental Determinants of Diabetes in the Young (TEDDY) Study. Diabetes Care (2017) 40(3):398–404. doi: 10.2337/dc16-1624
156. Andren Aronsson C, Lee HS, Hard Af Segerstad EM, Uusitalo U, Yang J, Koletzko S, et al. Association of Gluten Intake During the First 5 Years of Life With Incidence of Celiac Disease Autoimmunity and Celiac Disease Among Children at Increased Risk. JAMA (2019) 322(6):514–23. doi: 10.1001/jama.2019.10329
157. Hagopian W, Lee HS, Liu E, Rewers M, She JX, Ziegler AG, et al. Co-Occurrence of Type 1 Diabetes and Celiac Disease Autoimmunity. Pediatrics (2017) 140(5):e20171305. doi: 10.1542/peds.2017-1305
158. Hogg-Kollars S, Al Dulaimi D, Tait K, Rostami K. Type 1 Diabetes Mellitus and Gluten Induced Disorders. Gastroenterol Hepatol Bed Bench (2014) 7(4):189–97.
159. Hakola L, Miettinen ME, Syrjala E, Akerlund M, Takkinen HM, Korhonen TE, et al. Association of Cereal, Gluten, and Dietary Fiber Intake With Islet Autoimmunity and Type 1 Diabetes. JAMA Pediatr (2019) 173(10):953–60. doi: 10.1001/jamapediatrics.2019.2564
160. Lund-Blix NA, Dong F, Marild K, Seifert J, Baron AE, Waugh KC, et al. Gluten Intake and Risk of Islet Autoimmunity and Progression to Type 1 Diabetes in Children at Increased Risk of the Disease: The Diabetes Autoimmunity Study in the Young (DAISY). Diabetes Care (2019) 42(5):789–96. doi: 10.2337/dc18-2315
161. Uusitalo U, Lee HS, Andren Aronsson C, Vehik K, Yang J, Hummel S, et al. Early Infant Diet and Islet Autoimmunity in the TEDDY Study. Diabetes Care (2018) 41(3):522–30. doi: 10.2337/dc17-1983
162. Virtanen SM, Kenward MG, Erkkola M, Kautiainen S, Kronberg-Kippila C, Hakulinen T, et al. Age at Introduction of New Foods and Advanced Beta Cell Autoimmunity in Young Children With HLA-Conferred Susceptibility to Type 1 Diabetes. Diabetologia (2006) 49(7):1512–21. doi: 10.1007/s00125-006-0236-1
163. Virtanen SM, Takkinen HM, Nevalainen J, Kronberg-Kippila C, Salmenhaara M, Uusitalo L, et al. Early Introduction of Root Vegetables in Infancy Associated With Advanced Ss-Cell Autoimmunity in Young Children With Human Leukocyte Antigen-Conferred Susceptibility to Type 1 Diabetes. Diabetes Med (2011) 28(8):965–71. doi: 10.1111/j.1464-5491.2011.03294.x
164. Norris JM, Barriga K, Klingensmith G, Hoffman M, Eisenbarth GS, Erlich HA, et al. Timing of Initial Cereal Exposure in Infancy and Risk of Islet Autoimmunity. JAMA (2003) 290(13):1713–20. doi: 10.1001/jama.290.13.1713
165. Frederiksen B, Kroehl M, Lamb MM, Seifert J, Barriga K, Eisenbarth GS, et al. Infant Exposures and Development of Type 1 Diabetes Mellitus: The Diabetes Autoimmunity Study in the Young (DAISY). JAMA Pediatr (2013) 167(9):808–15. doi: 10.1001/jamapediatrics.2013.317
166. Mikkelsen KH, Knop FK, Vilsboll T, Frost M, Hallas J, Pottegard A. Use of Antibiotics in Childhood and Risk of Type 1 Diabetes: A Population-Based Case-Control Study. Diabetes Med (2017) 34(2):272–7. doi: 10.1111/dme.13262
167. Kilkkinen A, Virtanen SM, Klaukka T, Kenward MG, Salkinoja-Salonen M, Gissler M, et al. Use of Antimicrobials and Risk of Type 1 Diabetes in a Population-Based Mother-Child Cohort. Diabetologia (2006) 49(1):66–70. doi: 10.1007/s00125-005-0078-2
168. Boursi B, Mamtani R, Haynes K, Yang YX. The Effect of Past Antibiotic Exposure on Diabetes Risk. Eur J Endocrinol (2015) 172(6):639–48. doi: 10.1530/EJE-14-1163
169. Kemppainen KM, Vehik K, Lynch KF, Larsson HE, Canepa RJ, Simell V, et al. Association Between Early-Life Antibiotic Use and the Risk of Islet or Celiac Disease Autoimmunity. JAMA Pediatr (2017) 171(12):1217–25. doi: 10.1001/jamapediatrics.2017.2905
170. Tapia G, Stordal K, Marild K, Kahrs CR, Skrivarhaug T, Njolstad PR, et al. Antibiotics, Acetaminophen and Infections During Prenatal and Early Life in Relation to Type 1 Diabetes. Int J Epidemiol (2018) 47(5):1538–48. doi: 10.1093/ije/dyy092
171. Mishra SP, Wang S, Nagpal R, Miller B, Singh R, Taraphder S, et al. Probiotics and Prebiotics for the Amelioration of Type 1 Diabetes: Present and Future Perspectives. Microorganisms (2019) 7(3):67. doi: 10.3390/microorganisms7030067
172. Savilahti E, Harkonen T, Savilahti EM, Kukkonen K, Kuitunen M, Knip M. Probiotic Intervention in Infancy Is Not Associated With Development of Beta Cell Autoimmunity and Type 1 Diabetes. Diabetologia (2018) 61(12):2668–70. doi: 10.1007/s00125-018-4738-4
173. Cristelo C, Machado A, Sarmento B, Gama FM. The Roles of Vitamin D and Cathelicidin in Type 1 Diabetes Susceptibility. Endocr Connect (2021) 10(1):R1–R12. doi: 10.1530/EC-20-0484
174. Miller KM, Hart PH, de Klerk NH, Davis EA, Lucas RM. Are Low Sun Exposure and/or Vitamin D Risk Factors for Type 1 Diabetes? Photochem Photobiol Sci (2017) 16(3):381–98. doi: 10.1039/C6PP00294C
175. Simpson M, Brady H, Yin X, Seifert J, Barriga K, Hoffman M, et al. No Association of Vitamin D Intake or 25-Hydroxyvitamin D Levels in Childhood With Risk of Islet Autoimmunity and Type 1 Diabetes: The Diabetes Autoimmunity Study in the Young (DAISY). Diabetologia (2011) 54(11):2779–88. doi: 10.1007/s00125-011-2278-2
176. Brekke HK, Ludvigsson J. Vitamin D Supplementation and Diabetes-Related Autoimmunity in the ABIS Study. Pediatr Diabetes (2007) 8(1):11–4. doi: 10.1111/j.1399-5448.2006.00223.x
177. Silvis K, Aronsson CA, Liu X, Uusitalo U, Yang J, Tamura R, et al. Maternal Dietary Supplement Use and Development of Islet Autoimmunity in the Offspring: TEDDY Study. Pediatr Diabetes (2019) 20(1):86–92. doi: 10.1111/pedi.12794
178. Zipitis CS, Akobeng AK. Vitamin D Supplementation in Early Childhood and Risk of Type 1 Diabetes: A Systematic Review and Meta-Analysis. Arch Dis Child (2008) 93(6):512–7. doi: 10.1136/adc.2007.128579
179. Hou Y, Song A, Jin Y, Xia Q, Song G, Xing X. A Dose-Response Meta-Analysis Between Serum Concentration of 25-Hydroxy Vitamin D and Risk of Type 1 Diabetes Mellitus. Eur J Clin Nutr (2020) 75(7):1010–23. doi: 10.1038/s41430-020-00813-1
180. Reinert-Hartwall L, Honkanen J, Harkonen T, Ilonen J, Simell O, Peet A, et al. No Association Between Vitamin D and Beta-Cell Autoimmunity in Finnish and Estonian Children. Diabetes Metab Res Rev (2014) 30(8):749–60. doi: 10.1002/dmrr.2550
181. Norris JM, Lee HS, Frederiksen B, Erlund I, Uusitalo U, Yang J, et al. Plasma 25-Hydroxyvitamin D Concentration and Risk of Islet Autoimmunity. Diabetes (2018) 67(1):146–54. doi: 10.2337/db17-0802
182. Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM. Intake of Vitamin D and Risk of Type 1 Diabetes: A Birth-Cohort Study. Lancet (2001) 358(9292):1500–3. doi: 10.1016/S0140-6736(01)06580-1
183. Stene LC, Joner G, Norwegian Childhood Diabetes Study G. Use of Cod Liver Oil During the First Year of Life Is Associated With Lower Risk of Childhood-Onset Type 1 Diabetes: A Large, Population-Based, Case-Control Study. Am J Clin Nutr (2003) 78(6):1128–34. doi: 10.1093/ajcn/78.6.1128
184. EURODIAB. Vitamin D Supplement in Early Childhood and Risk for Type I (Insulin-Dependent) Diabetes Mellitus. The EURODIAB Substudy 2 Study Group. Diabetologia (1999) 42(1):51–4. doi: 10.1007/s001250051112
185. Kawasaki E, Abiru N, Eguchi K. Prevention of Type 1 Diabetes: From the View Point of Beta Cell Damage. Diabetes Res Clin Pract (2004) 66(Suppl 1):S27–32. doi: 10.1016/j.diabres.2003.09.015
186. Kolb H, Martin S. Environmental/lifestyle Factors in the Pathogenesis and Prevention of Type 2 Diabetes. BMC Med (2017) 15(1):131. doi: 10.1186/s12916-017-0901-x
187. Bi X, Li F, Liu S, Jin Y, Zhang X, Yang T, et al. Omega-3 Polyunsaturated Fatty Acids Ameliorate Type 1 Diabetes and Autoimmunity. J Clin Invest (2017) 127(5):1757–71. doi: 10.1172/JCI87388
188. Greenberg JA, Bell SJ, Ausdal WV. Omega-3 Fatty Acid Supplementation During Pregnancy. Rev Obstet Gynecol (2008) 1(4):162–9. doi: 10.1002/14651858.CD003402.pub3
189. Norris JM, Yin X, Lamb MM, Barriga K, Seifert J, Hoffman M, et al. Omega-3 Polyunsaturated Fatty Acid Intake and Islet Autoimmunity in Children at Increased Risk for Type 1 Diabetes. JAMA (2007) 298(12):1420–8. doi: 10.1001/jama.298.12.1420
190. Norris JM, Kroehl M, Fingerlin TE, Frederiksen BN, Seifert J, Wong R, et al. Erythrocyte Membrane Docosapentaenoic Acid Levels are Associated With Islet Autoimmunity: The Diabetes Autoimmunity Study in the Young. Diabetologia (2014) 57(2):295–304. doi: 10.1007/s00125-013-3106-7
191. Butalia S, Kaplan GG, Khokhar B, Haubrich S, Rabi DM. The Challenges of Identifying Environmental Determinants of Type 1 Diabetes: In Search of the Holy Grail. Diabetes Metab Syndr Obes (2020) 13:4885–95. doi: 10.2147/DMSO.S275080
192. Nucci AM, Virtanen SM, Cuthbertson D, Ludvigsson J, Einberg U, Huot C, et al. Growth and Development of Islet Autoimmunity and Type 1 Diabetes in Children Genetically at Risk. Diabetologia (2021) 64:826–35. doi: 10.1007/s00125-020-05358-3
Keywords: type 1 diabetes (T1D), seroconversion, auto-antibodies, autoimmunity, environmental factors, gut micro biome, obesity, infection - immunology
Citation: Quinn LM, Wong FS and Narendran P (2021) Environmental Determinants of Type 1 Diabetes: From Association to Proving Causality. Front. Immunol. 12:737964. doi: 10.3389/fimmu.2021.737964
Received: 08 July 2021; Accepted: 08 September 2021;
Published: 01 October 2021.
Edited by:
Antonio Toniolo, University of Insubria, ItalyReviewed by:
Magdalena Zoledziewska, Institute of Genetic and Biomedical Research (CNR), ItalyCopyright © 2021 Quinn, Wong and Narendran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lauren M. Quinn, bC5xdWlubi4xQGJoYW0uYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.