
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 30 September 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.734229
This article is part of the Research TopicThe Role of Reactive Oxygen Species in Protective ImmunityView all 13 articles
 Marcella Canton1,2*
Marcella Canton1,2* Ricardo Sánchez-Rodríguez1,2
Ricardo Sánchez-Rodríguez1,2 Iolanda Spera3Francisca C. Venegas1,2Maria Favia3
Iolanda Spera3Francisca C. Venegas1,2Maria Favia3 Antonella Viola1,2
Antonella Viola1,2 Alessandra Castegna2,3*
Alessandra Castegna2,3*Reactive oxygen species (ROS) are fundamental for macrophages to eliminate invasive microorganisms. However, as observed in nonphagocytic cells, ROS play essential roles in processes that are different from pathogen killing, as signal transduction, differentiation, and gene expression. The different outcomes of these events are likely to depend on the specific subcellular site of ROS formation, as well as the duration and extent of ROS production. While excessive accumulation of ROS has long been appreciated for its detrimental effects, there is now a deeper understanding of their roles as signaling molecules. This could explain the failure of the “all or none” pharmacologic approach with global antioxidants to treat several diseases. NADPH oxidase is the first source of ROS that has been identified in macrophages. However, growing evidence highlights mitochondria as a crucial site of ROS formation in these cells, mainly due to electron leakage of the respiratory chain or to enzymes, such as monoamine oxidases. Their role in redox signaling, together with their exact site of formation is only partially elucidated. Hence, it is essential to identify the specific intracellular sources of ROS and how they influence cellular processes in both physiological and pathological conditions to develop therapies targeting oxidative signaling networks. In this review, we will focus on the different sites of ROS formation in macrophages and how they impact on metabolic processes and inflammatory signaling, highlighting the role of mitochondrial as compared to non-mitochondrial ROS sources.
The immune system orchestrates a complex defensive strategy against pathogens or tissue injury. In vertebrates, two types of immunity are used to protect the host from infections: innate and adaptive. The innate system, which constitutes the first line of defense, is genetically programmed to recognize structures that are broadly shared by invading microbes (named PAMPs, pathogen-associated molecular patterns) and by cell damage (named DAMPs, damage-associated molecular patterns). Cells of the innate immune system include macrophages, dendritic cells, neutrophils, eosinophils, basophils, mast cells and Natural Killer cells. In contrast, the adaptive system, also referred as the acquired immune system, employs antigen-specific receptors that are specifically developed (“acquired”) by lymphocytes during the lifetime of the organism.
Macrophages are large, specialized cells that rapidly recognize, engulf, and destroy pathogens or apoptotic cells. Indeed, the term macrophage is formed by the combination of the Greek terms “makro” meaning big and “phagein” meaning eat. One of the fundamental features of macrophages is their high plasticity, which allows them to respond to stimuli from the complex tissue microenvironment, by changing rapidly their functional profile through a process named “polarization”. In fact, they initially adopt a proinflammatory phenotype and then later they acquire an anti-inflammatory profile to repair the tissue damage (1, 2). Due to the complex stimulating network, the process of macrophage polarization in an in vivo setting cannot be recapitulated by the static vision of M1-M2 polarization adopted in in vitro experiments, reached by stimulation with lipopolysaccharide (LPS)/interferon-γ (IFN-γ) or Interleukin (IL)-4/IL-13 or IL-10, respectively (2, 3). However, macrophages with predominantly proinflammatory properties are commonly referred as M1, whereas those with a pro-fibrotic and anti-inflammatory signature as M2. They are found ubiquitously in tissues as resident cells patrolling their surroundings, thus maintaining tissue integrity. Resident macrophages have different names according to where they function in the body. For instance, macrophages in brain are termed microglia, while in liver they are called Kupffer cells (4). Moreover, in case of tissue damage or infection, monocytes leave the bloodstream to enter the affected tissues and undergo a series of changes to become macrophages.
Reactive oxygen species (ROS) have been known for many years as fundamental for macrophages to kill invasive microorganisms through the oxidative burst mediated by NADPH oxidase (5). However, more recent studies have shown that mitochondrial ROS play essential roles in several innate immune functions, through subtle changes in the intracellular redox state (6).
Oxygen is a highly electronegative element that readily accepts electrons generated by normal oxidative metabolism within cells, thereby producing ROS. The term “ROS” includes superoxide anion (O2-.), hydrogen peroxide (H2O2), hydroxyl radical and singlet oxygen, which are produced as described in (6). Besides ROS, other endogenous small, reactive signaling molecules include reactive nitrogen species (RNS), such as nitric oxide, as well as hydrogen sulfide and carbon monoxide. Several reviews discuss their roles in macrophage function (7–9), so they will not be considered in the present review.
While ROS have been considered for a long time as dangerous by-products of mitochondrial metabolism, it is now widely accepted that they play crucial roles as signaling molecules, regulating cell growth, differentiation, and apoptosis (10). As discussed extensively in other reviews (5, 6), ROS appear to play different -sometime opposing- roles depending on their subcellular origin and levels. Among ROS, H2O2 is by far the most prevalent and best studied cellular oxidant and plays a major role in redox regulation of biological activities (11). Similar to other signaling molecules, the intracellular concentrations of H2O2 are maintained very low (in the range of 1-100 nM) and are tightly regulated (6). They are produced in such a low level to be confined to a restricted subcellular location and to induce signaling pathways, supporting normal physiological processes. This range of concentration must be considered just as an order of magnitude, because it depends on many factors, as cell type, local concentrations, etc. Different stimuli, such as growth factors or chemokines, trigger a physiological increase of H2O2, which targets specific proteins leading to their reversible oxidation, thereby altering their activity, localization and interactions. These protein modifications contribute to orchestration of various processes in cells and organs, including cell proliferation, differentiation, migration and angiogenesis. On the other hand, high levels of ROS (roughly reaching micromolar concentrations) are likely to become involved in non-specific oxidation of targets, causing damage to macromolecules, impairing their function and triggering stress response mechanisms, as inflammation, fibrogenesis, tumor growth, metastasis and, at higher levels, cell death (6, 12). In this review, we will focus on the different sites of ROS formation in macrophages, the major molecular redox targets, and their related cellular response.
NADPH oxidases (NOXs) are a family of transmembrane enzymes specifically dedicated to produce cytosolic ROS (cytROS) (13, 14), mainly located in the plasma membrane (Figure 1). NOXs catalyze superoxide formation by transferring one electron from NADPH to oxygen (15). Superoxide can be further converted to H2O2 either by spontaneous dismutation or catalyzed by superoxide dismutase. So far, seven members of the family have been described (NOX1-5 and Duox1-2) (16), and three of them (NOX1, NOX2 and NOX4) have been identified in phagocytes (17–27). NOX2 is the most well-characterized isoform for its role in phagocytic function. During phagocytosis, the plasma membrane is internalized and becomes the interior wall of the phagocytic vesicle. Next, the produced by NOX2 (named “oxidative burst”) is released into the vesicle to kill the internalized target. The relevance of cytROS in host immunity has been demonstrated in chronic granulomatous disease (CGD), a genetic disorder characterized by mutations in genes encoding components of the NOX2 complex (28, 29). Patients with CGD are hypersensitive to both bacterial and fungal infections, as their phagocytic cells fail in killing pathogens due to the extremely low oxidative burst during phagocytosis (30, 31). More recently, it has been shown that also NOX4-mediated ROS production is selectively required for the host defense against Toxoplasma gondii infection (26).
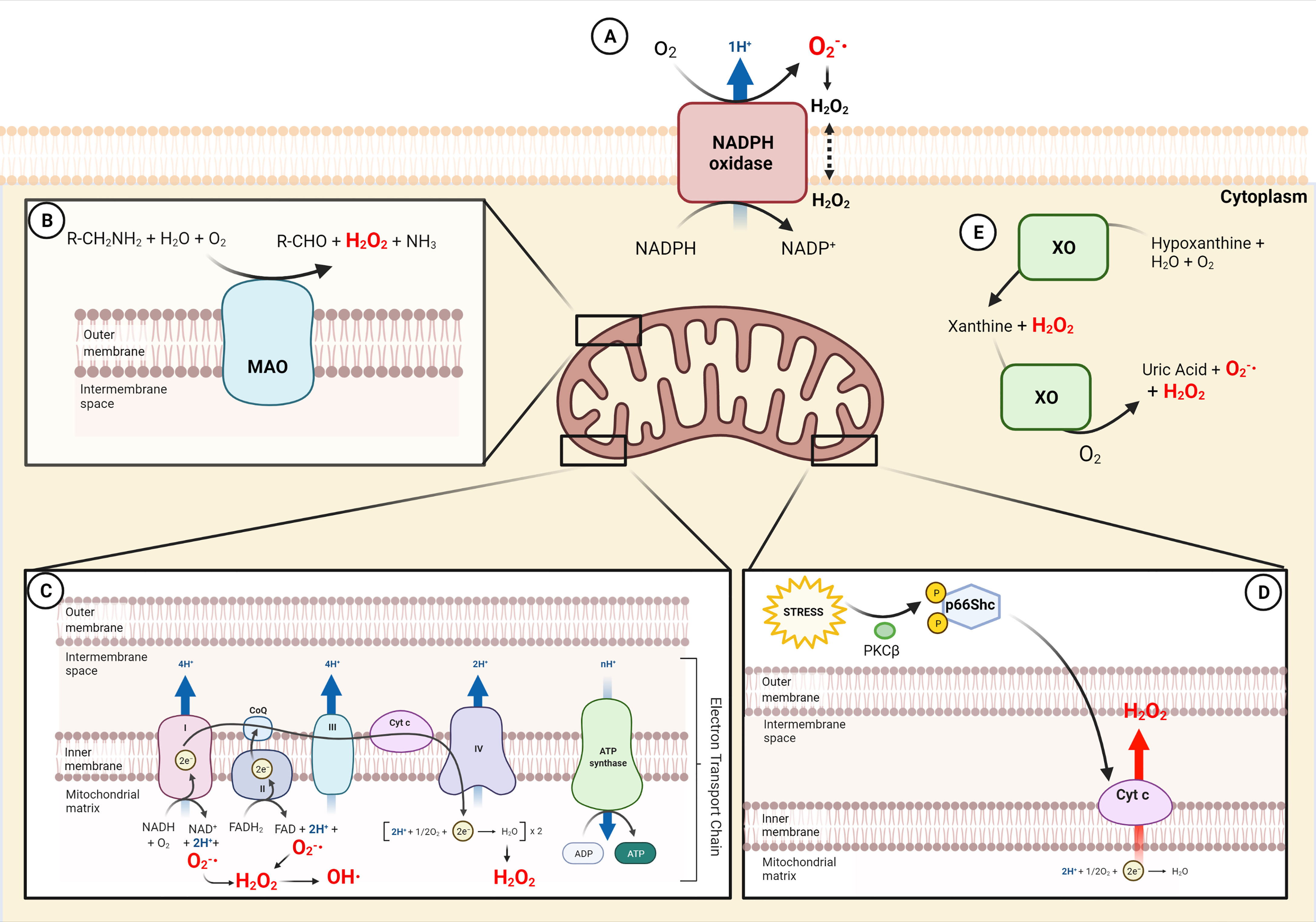
Figure 1 Sources of ROS in macrophages. (A) Generation of and consecutive formation of H2O2 by NADPH oxidase (NOX) in the external cell membrane. (B) Production of H2O2 in the outer mitochondria membrane by oxidative deamination of biogenic and xenobiotic amines by MAO. (C) Electron Transport chain in the inner mitochondrial membrane generates , H2O2 and OH· in the mitochondrial matrix. (D) Cytochrome c in the inner mitochondrial membrane produces H2O2 following p66Shc activation by stress. (E) The xanthine metabolism produces H2O2 and by XO in the cytoplasm. NADP+, Nicotinamide adenine dinucleotide phosphate; MAO, monoamine oxidases; CoQ, coenzyme Q; FAD, flavin adenine dinucleotide; Cyt c, cytochrome c; ADP, adenosine diphosphate; ATP, adenosine triphosphate; PKCβ, protein kinase C β; XO, xanthine oxidase.
Besides killing invasive microorganisms, NOX-dependent ROS production influences many metabolic processes and disease states. For instance, NOX1 and NOX2 are critical for the differentiation of monocytes to macrophages and the M2-type polarization, as assessed in macrophages from NOX1/NOX2 double knockout mice (19). Interestingly, ROS generated by NOX2 were found to contribute to fatty liver disease (27). Indeed, Nox2-deficient mice were protected against hepatic steatosis induced by high-fat diet and insulin resistance (27). Mechanistically, palmitate triggers endocytosis of the Toll-like receptor 4 (TLR4)-MD2 complex, leading to NOX2 activation, ROS generation and proinflammatory cytokine production in hepatic infiltrating macrophages (27). By the way, although palmitate has been considered a TLR4 ligand, a subsequent study demonstrated that it does not directly bind this receptor (32). More recently, NOX1-dependent ROS production has been found to be neurotoxic for microglia located in the retina (33). Its overactivation is mediated by the translocator protein TSPO. Using different NOX-deficient mice, the study shows that the TSPO-NOX1 axis controls the phagocyte-triggered angiogenesis in the eye of a mouse model of age-related macular degeneration, a major cause of blindness in the elderly (33). NOX4 has also been shown to be an inducible source of ROS, driving cell death when monocytes and macrophages were exposed to oxidized low density lipoproteins (oxLDL) (17).
Finally, different studies investigated the impact of NOX in the activation of the NLRP3 inflammasome in macrophages. The NLRP3 inflammasome is a molecular platform activated upon signs of cellular danger (PAMP or DAMP) to trigger innate immune defenses through the maturation of pro-inflammatory cytokines (34, 35). For its activation it requires the adapter protein apoptosis associated speck-like protein containing a CARD (ASC) to activate caspase-1, which cleaves pro-interleukin (IL)-1β and pro-IL-18 in IL-1β and IL-18 (34). The results linking NOX to the NLRP3 inflammasome are controversial, and this could be ascribed to species differences, differential regulation of monocytes and macrophages or redundant NOX enzymes, as discussed (23, 36, 37). Also the localization of NOX4 is quite arguable/disputed, as some studies found this isoform even in mitochondria, others in the plasma membrane and in the endoplasmic reticulum (6).
Xanthine dehydrogenase is an enzyme that can be converted to its oxygenase form xantine oxidase (XO) upon oxidative stress. Both forms generate uric acid from hypoxanthine or xanthine, but XO also produces ROS (38) (Figure 1). The conversion of xanthine dehydrogenase to XO can be due to irreversible proteolytic cleavage, or to reversible sulfhydryl modification (39, 40). Its role has been widely studied for many years, but scarce information is available with respect to the innate immune system. Interestingly, convincing evidence reports that XO is a source of ROS that mediates NLRP3 inflammasome activation in macrophages (41) and that represents a key factor to trigger inflammation against parasitic infection (42).
Mitochondria are important sources of ROS, as they are the main oxygen consumers in the cell (43). These organelles produce ROS through various mechanisms, including electron leak from the electron transport chain to oxygen, or as by-products of the catalytic activity of several oxidases (44–46). Interestingly, it has been suggested that also mitochondrial ROS (mtROS) represent an important component of the antibacterial responses, thereby revealing a novel pathway linking innate immune signaling to mitochondria (47). Specifically, the engagement of a subset of Toll-like receptors (TLR1, TLR2 and TLR4) was found to cause the recruitment of mitochondria to macrophage phagosomes and augments mtROS, although the mechanism is still partially unclear (47). Another study highlighted the relevance of mtROS as compared to cytROS. Bulua and Coworkers showed that mtROS are responsible for excessive LPS-driven production of proinflammatory cytokines in cells from patients with an autoinflammatory disorder caused by missense mutations in the type-1 TNF receptor (TNFR1), named TNF receptor-associated periodic syndrome (TRAPS) (48). On the other hand, NOXs are not the source of proinflammatory ROS, as NOX subunits were found to be dispensable for inflammatory cytokine production (48). The authors hypothesized several mechanisms by which TNFR1 mutations enhance mitochondrial respiration, although further research should more specifically address this issue. The endoplasmic reticulum, where mutant TNFR1 resides, can provide signals to activate mitochondrial respiration (49). Moreover, mutant TNFR1 may increase activation of the riboflavin kinase, which can associate with TNFR1 (50), possibly leading to enhanced charging of FAD-dependent enzymes in the mitochondria.
The electron transport chain (ETC) is a series of electron-carrier proteins located in the mitochondrial inner membrane (Figure 1). It transfers electrons from the reduced coenzymes NADH and FADH2, generated by catabolic processes, to molecular oxygen. Thanks to this process, three of the four protein complexes pump protons across the mitochondrial inner membrane to maintain the protonmotive force driving ATP synthesis. A physiological consequence of the electron transfer is the generation of mitochondrial ROS (mtROS). In fact, the electron leak from complexes I, II, and III mediates the one-electron reduction of oxygen to superoxide (), which can then be rapidly converted to H2O2 by manganese superoxide dismutase (MnSOD) within the mitochondrial matrix. H2O2 can then freely diffuse in the cell and trigger thiol oxidation of proteins.
mtROS can be also produced at complex I through reverse electron transfer (RET). This process, observed in vitro in the sixties of the last century (51, 52), has been considered of uncertain physiological relevance for many years. More recently, several studies highlighted that RET at complex I is a process underlying mitochondrial redox signaling in physiological and pathological conditions (45, 53–56). It occurs when electrons flow back through complex I, in contrast to conventional forward transport, because of elevated mitochondrial membrane potential coupled to highly reduced coenzyme Q. Thus, the electrons can reduce NAD+ to NADH and drive superoxide formation. Indeed, in macrophages stimulated with LPS, Mills et al. showed that the accumulation of succinate, that is oxidized by complex II (succinate dehydrogenase), results in mtROS production, seemingly from RET at complex I (45).
A relevant mitochondrial-specific source of ROS is monoamine oxidase (MAO), although its impact on inflammation has been quite overlooked. MAO is located in the outer mitochondrial membrane and catalyzes the oxidative deamination of neurotransmitters (i.e. catecholamines) and dietary amines, generating aldehydes, ammonia and H2O2 (57) (Figure 1). The two isoforms MAO-A and MAO-B differ for substrate specificity and inhibitor sensitivity. MAO-A has greater affinity for hydroxylated amines, i.e. serotonin and noradrenaline, whereas MAO-B has greater affinity for non-hydroxylated ones, i.e. β-phenylethylamine. Notwithstanding, they show similar affinity for dopamine and tyramine. MAO physiologic role is well established in the central nervous system: it terminates neurotransmitter signaling and, by doing this, it generates H2O2 that is constantly removed by endogenous scavengers (57). On the contrary, in pathological conditions, the increased activity of the enzyme overcomes the cellular antioxidant defenses, altering the redox homeostasis and eliciting deleterious effects, as in muscular dystrophy and cardiac injury (58–62). Only few studies have characterized the role of MAO in innate immunity. It has been shown that MAO-A is upregulated by LPS or by IL-4/IL-13 in phagocytic cells (63–66). These studies also suggest that IL-13 and IL-4 induced MAO-A-mediated ROS generation through Jak signaling pathways (65).
Few years ago, a study from Tschopp’s group highlighted the crucial role of mtROS in NLRP3 inflammasome activation, although the source of ROS production was unclear (65, 67). Recently, it has been shown that H2O2 produced by MAO-B plays a non-redundant role in sustaining NLRP3 inflammasome activation (68) in human and murine macrophages. Mechanistically, MAO-B-dependent ROS formation causes mitochondrial dysfunction and NF-κB activation, resulting in NLRP3 and pro-IL-1β overexpression. Both in vitro and in vivo, MAO-B inhibition by the clinical grade drug rasagiline prevents IL-1β secretion, and MAO-B deficient mice display an impaired response to LPS-mediated endotoxemia (68).
Remarkably, two interesting studies highlight the critical role of MAO in macrophages, considering the enzyme as a catecholamine consumer and not as a ROS producer. Briefly, macrophages from adipose tissue of aged mice displayed upregulation of MAO-A in a NLRP3 inflammasome-dependent manner. The enhanced activity of MAO-A increases catecholamine catabolism, thereby dampening the activation of lipolytic signaling in adipocytes as this process depends on noradrenaline levels (69). In the same context, MAO-A was identified in a subset of cells called macrophages associated with sympathetic neurons (SAMs) that work in the clearance of noradrenaline, acting as a sink (70). Moreover, SAMs were increased in two models of obesity and contributed to the disease by excessive import and metabolism of noradrenaline (70, 71).
p66Shc is a cytosolic adaptor protein that regulates the cellular redox state and apoptosis (72, 73). Oxidative stress activates protein kinase C-β (PKCβ), which phosphorylates p66Shc. As a result, p66Shc translocates to mitochondria, where it generates H2O2. Indeed, this redox enzyme utilizes reducing equivalents of the mitochondrial ETC through the oxidation of cytochrome c to catalyze the partial reduction of molecular oxygen (72). Mice lacking p66Shc are long-lived, and their cells are both resistant to oxidative stress and produce less ROS. A few studies characterized p66Shc in the immune system and were mainly focused on its role in the macrophages of the atherosclerotic lesions induced by high-fat diet or diabetes (74, 75). Chronic high-fat diet was reported to increase the atherosclerotic lesion area more in wild-type than p66Shc knockout mice. Early lesions from p66Shc knockout mice had fewer macrophage-derived foam cells as compared to those from wild-type mice (74). A cross-talk between p66Shc and NOX has been observed in murine macrophages, as NOX activation is defective in p66Shc-deficient mice, leading to decreased superoxide production (76).
It is widely accepted that physiological levels of ROS can cause reversible post-translational modifications in proteins to regulate signaling pathways. More in detail, H2O2 can oxidize thiol groups (-SH) on cysteine residues to form sulfenic acid (-SOH), which can react with GSH to become glutathionylated (-SSG), or with adjacent thiols to form a disulfide bond (-SS-) (6, 77). Cysteine thiols can be considered as redox sensors, as they are particularly sensitive to oxidants. The reactivity of thiols for ROS is limited to the cysteinyl residues placed in sites that enable the formation of the thiolate form (-S-), which is more nucleophilic and susceptible to oxidation (6). These modifications can change the activity of the target proteins, thus altering their function and their downstream signaling pathway and metabolism (78). Supraphysiological concentration of ROS leads to oxidative stress, a state of imbalance between ROS production and ROS removal, which can be either due to increased ROS formation or to reduced antioxidant defenses.
In this review, we summarize several examples of signaling targets for ROS that have been identified in macrophages, as the list of targets is too extensive to be covered exhaustively (6). Importantly, cysteine oxidation networks have been recently become accessible (79) and provide a quantitative tissue-specific overview of the redox-regulated proteome, called the Oximouse dataset (79).
Nrf2 (nuclear factor-erythroid 2 p45-related factor 2) is the master regulator of the antioxidant response: it is responsible for maintaining the redox homeostasis under oxidative stress by regulating the expression of detoxifying enzymes involved in glutathione, NADPH and thioredoxin systems (including GCLC, GCLM, NQO1, G6DH, TRX and HO-1) (12, 80, 81). Under basal conditions, Nrf2 is constitutively degraded by the proteasome, as its cytosolic repressor Keap1 (Kelch-like ECH-associated protein 1) enables the Cul3-Ring-box 1 E3 ubiquitin ligase to ubiquitinate it. Oxidative stress induces the oxidation of Cys151, Cys273 and Cys288 in two domains of the negative regulator Keap1, inducing its conformational change and disruption of the Nrf2/Keap1 interaction (82). This results in the stabilization of Nrf2, which translocates to the nucleus and binds specific DNA sequences, the Antioxidants Response Elements (ARE) (83–85). The role of Nrf2 in macrophages is still controversial, as both anti-inflammatory and pro-inflammatory mechanisms have been described, as summarized here below. Several studies demonstrate that the Keap1/Nrf2/ARE signaling pathway attenuates inflammation. Nfr2 deletion causes exacerbation of inflammation in different experimental murine models, such as septic shock and emphysema (86–88), and antioxidant treatments reduce these effects. A recent study reports that murine macrophages achieve self-protection against oxidative stress through the Mst-Nrf2 axis (89). The kinases Mst1/2 sense ROS and maintain the cellular redox balance by modulating Nrf2 stability. Mechanistically, both phagosomal and mtROS activate Mst1/2, which phosphorylate Keap1, thereby blocking Nrf2 ubiquitination and degradation to protect cells against oxidative damage and to maintain the phagocytosis properties (89). Nrf2 was found to inhibit the NLRP3 inflammasome assembly by buffering ROS and controlling the expression of thioredoxin, which inhibits a protein necessary for NLRP3 complex stabilization, the thioredoxin-interacting protein (TXNIP) (90) (see 3.6 Thioredoxins and ASK1 for further details). On the other hand, other studies provide evidence for a proinflammatory role. Indeed, Nrf2 plays an essential role in ROS-mediated inflammasome activation, as Nrf2-deficient macrophages show a reduced formation of ASC specks and IL-1β release in response to NLRP3 and AIM2 inflammasome stimuli (91). ASC is an adaptor protein which assembles into a large protein complex, termed “speck”, upon inflammasome activation. Hence, ASC speck formation is commonly used as a simple upstream readout for inflammasome activation (92).
The paradoxical role of Nrf2 might be related to the kind of inflammatory stimuli and/or the timing of activation. In fact, the early activation of Nrf2 triggered by antioxidant treatments can enhance the antioxidant response, reduce ROS levels and induce an anti-inflammatory signature, whereas the late activation can mainly contribute to the NLRP3 assembling, such as the one triggered by cholesterol crystals.
Despite its strong association with redox biology, the activation of Nrf2 by mtROS induces also the overexpression of macrophage-specific genes that are not classified as anti-oxidative stress-response genes, such as the gene encoding MARCO, a scavenger receptor required for bacteria phagocytosis (93). Furthermore, Nrf2 can inhibit the expression of proinflammatory cytokine genes in a redox-independent manner. Indeed, Nrf2 was found to inhibit the recruitment of RNA polymerase II onto the proinflammatory cytokine gene loci for Il-1β and Il-6 (94).
Hypoxia Inducible factors (HIFs) are transcription factors, consisting of an oxygen-labile subunit (HIF-α) and a constitutively stable subunit (HIF-β), which play pivotal roles in inducing cellular responses to hypoxia and in regulating immune cell effector functions (95, 96). They form a heterodimeric complex, which binds to hypoxia-responsive elements (HREs), thus activating the transcription of their target genes and promoting a metabolic and functional cell reprogramming (97). In homeostatic conditions, HIF is hydroxylated by prolyl hydroxylase (PHD), an enzyme belonging to the 2-oxoglutarate (2-OG)-dependent dioxygenase (2-OGDD) family, which catalyzes the conversion of 2-OG to succinate and employs molecular oxygen for HIF hydroxylation. The hydroxylation of HIF drives its ubiquitylation by the E3 ligase von Hippel–Lindau tumour-suppressor protein (VHL) and its consequent degradation by the proteasome (5). Upon LPS stimulation, macrophages reprogram their metabolism towards glycolysis, thereby driving ROS generation which can sustain hypoxic adaptation by HIF stabilization (3, 45, 98) and concomitantly support the expression of cytokines, such as IL-1β via HIF-1α. The crucial role of ROS is documented by a strong body of evidence. Limiting ROS production by uncoupling mitochondria or by expressing the alternative oxidase (AOX) inhibits the inflammatory phenotype of macrophages (45). Moreover, ROS contribute to HIF-1α stabilization by (I) diverting the PHD substrate 2-OG toward a non-enzymatic decarboxylation (99), and (II) oxidizing the PHD cofactor Fe2+ to Fe3+ (100). Finally, ROS act as inhibitor of Factor Inhibiting HIF (FIH), an asparaginyl hydroxylase belonging to the 2-OGDD family. FIH hydroxylation, occurring in a different site from PHD hydroxylation (101), impairs HIF-1α function by reducing its C-terminal transactivation domain activity (101). It is worth noting that HIF asparaginyl hydroxylation (OH) differs from the prolyl OH in the fact that is more sensitive to low concentrations of H2O2 than prolyl OH, whereas in moderate hypoxia asparaginyl OH is less effectively inhibited than prolyl OH. This suggests that hypoxia and ROS can provide different levels of regulation of HIF transcriptional output (101). In support of this finding, although in cancer cell lines, mtDNA depletion is sufficient to prevent HIF-1α stabilization under hypoxia (102). In bone-derived murine macrophages mtROS generated by spermidine activate AMP-activated protein kinase (AMPK), which in turn enhances mitochondrial function, and upregulates HIF-1α (103). Moreover, HIF-1α reduces mitochondrial mass through mitophagy, thus limiting oxygen consumption. MtROS target SDH subunit A (SDHA), leading to the inhibition of its enzymatic activity, which in turn stabilizes HIF-1α and causes the subsequent, sustained expression of IL-1β together with TCA intermediates accumulation (104).
HIF-1α mediates the expression of genes encoding for glycolytic enzymes, for the glucose transporter GLUT1 (98), and the pyruvate dehydrogenase kinase isoform 1 (PDK1) in macrophages (105, 106). When HIF1 is stabilized, PDK activity inhibits the pyruvate dehydrogenase complex, limiting the flux of pyruvate into the tricarboxylic acid (TCA) cycle. This attenuates mitochondrial respiration through ETC flux, preventing mtROS production from overpowering the antioxidant endogenous defense in hypoxic conditions (107, 108). In this scenario, PDK1 activation might also represent a strategy to divert glucose from glycolysis to the pentose phosphate pathway, leading to a higher NADPH/NADP+ ratio that might prevent or compensate for uncontrolled oxidative stress, which could be harmful. Indeed, LPS lethality in mice can be prevented by limiting ROS production (45). PDK activity can also be inhibited by mtROS, providing a regulatory flexibility that is functionally important in the migration setting of macrophages (109).
Besides PHD, ROS can also inhibit the activity of Jumonji domain-containing histone demethylase (JMJD), a family of Fe2+-dependent 2-OG oxygenases essential for epigenetic reprogramming in macrophages through histone demethylation. Remarkably, the increase in the succinate/2-OG ratio inhibits PHD and JMJD function activating HIF-1α and sustaining the glycolytic switch and proinflammatory phenotype (110). In this way the HIF-ROS axis represents a central functional cycle of mutual support where ROS production is a mediator of hypoxia adaptation, by translating oxygen limitation into transcriptional regulation for a metabolic reprogramming.
More recently, it has been described that SARS-CoV2 infection triggers mtROS production in monocytes, and this induces HIF-1α stabilization and consequently promotes glycolytic reprogramming, thereby enhancing the viral replication (111). SARS-CoV2 infection induced downregulation of several proteins of the ETC, such as NDUFV1 (complex I), SDHA (complex II), UQCRC2 (complex III), limiting oxygen consumption rate (111). Defects of the mitochondrial respiratory chain have been related to increased mtROS levels (45), although the precise mechanisms linking infection and mtROS production are still to be defined.
NF-κB is a transcription factor that plays a crucial role in inflammatory and immune responses (112). It displays a plethora of modulatory mechanisms due to the different DNA binding affinities of their homo- and hetero-dimeric complexes emanating from the five monomers (RelA/p65, RelB, cRel, NF-κB1 p50, and NF-κB2 p52). This heterogeneity is further increased by interactions of the NF-κB dimers with other transcription factors (112). The heterodimeric complex p50/p65 is one of the better characterized during the inflammatory response (113). NF-κB in the cytosol is inactivated by binding to the regulatory protein IkBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha). Inflammatory stimuli drive the phosphorylation of the IKK complex, consisting of two catalytic (IKKα and IKKβ) and one regulatory subunit (NEMO or IKKϒ), the latter acting as scaffold protein for IKKβ activity. The activated complex phosphorylates IκBα, marking the protein for proteasome-linked degradation (114). NF-κB is then free to translocate to the nucleus and start the transcription of several genes, including inflammatory and antiapoptotic genes. The mechanisms linking ROS formation to NF-κB activation are multiple. In macrophages, mtROS are known to mediate IKK complex activation by forming a disulfide bridge between Cys54 and Cys347 on NEMO, which is crucial for IKK complex activation (115). Under proinflammatory stimulus, the IKK complex can be activated by ROS produced by the GTP-binding protein Rac1 (116), leading to signal transduction pathways that contribute to TNF-α secretion. In line with this finding, ROS effects can be suppressed by SOD, reducing the pro-inflammatory immune responses by blocking the p38-MAPK/NF-κB signaling activation (117).
NF-κB is also involved for different transduction outcomes related with anti-inflammatory response. This is particularly evident in the context of the tumor microenvironment, in which ROS formation activates NF-κB signaling, which binds the Pdl1 promoter in a transcriptional specific manner, leading to PD-L1 expression and release of immunosuppressive chemokines. Indeed, the described ROS-mediated NF-κB activation does not induce expression of the classical NF-κB target IL-6 (118). Similarly, in the context of colitis, mtROS lead to induction of NF-κB signaling responsible of a protective effect associated to the recruitment and polarization of intestinal macrophages to the M2 anti-inflammatory phenotype (119).
Notably, NF-κB is involved also in hypoxic conditions, and evidence of a crosstalk between NF-κB and HIF-1α is growing. PHD inhibition regulates the activity of IKKβ, inducing the nuclear translocation of NF-κB (120). Conversely, hypoxia-mediated NF-κB induction controls HIF-1α activity in macrophages, enhancing the production of pro-inflammatory cytokines and chemokines to sustain the host defense response (121). The link between NF-κB and HIF-1α is further underlined by the finding that HIF-1α promoter contains a NF-κB binding site that can trigger HIF-1α upregulation under conditions of NADPH oxidase-mediated ROS formation (122).
The crosstalk between HIF and NF-κB pathways is extensive, intensive (123) and bi-directional (124). In fact, they are reciprocally regulated (124). An additional level of functional crosstalk between HIF and NF-κB includes common activating stimuli, shared regulators and target genes (123). The overlap of common regulators between HIF and NF-κB consequently finds functional involvement of HIF in processes in which NF-κB is involved, such as infection and inflammation. Many important genes are regulated by HIF and NF-κB (123), including cytokines and chemokines, such as TNF-α, IL- 1β and IL-8. In addition, cell death related proteins, such as Noxa and BNIP3, and other important cellular proteins such as PKM2, Tert, Cyclin D1, and Cox-2 are also shared HIF and NF-κB targets. However, it is not known whether these genes are targeted by these transcription factors at the same time or independently of each other (123).
Moreover, HIF and NF-κB synergistically respond against pathogens. In fact, it has been demonstrated that macrophages, infected by Gram-negative and Gram-positive bacteria, are characterized by a defective HIF-1α expression following the ablation of IKKβ, essential regulator of NF-κB activity (121, 125).
Signal transducer and activator of transcription (STAT) proteins are a family of transcription factors that are essential for the cellular response to cytokines and growth factors. STATs are latent in the cytoplasm under resting conditions. When extracellular stimuli, such as cytokines, bind to specific cell-surface receptors, they activate the tyrosine kinases Jak (‘Janus kinase’), that phosphorylates STAT proteins, thereby allowing translocation to the nucleus to drive transcription of several chemokines and cytokines. The JAK-STAT signaling pathway is fundamental for the immune system (126–129). So far, seven members of the family (STAT1-4, STAT5A, STAT5B, and STAT6) have been identified. STATs are fundamental in the inflammatory/anti-inflammatory response, like the antiviral response through interferon production, and in wound healing (130, 131). The JAK-STAT pathway plays an essential role in macrophage polarization: STAT1 shifts macrophages towards a pro-inflammatory M1 profile activated by interferon gamma (IFN-γ), whereas STAT6 is associated to an M2 anti-inflammatory profile through IL-4. The role of ROS during STAT activation in macrophages is still under scrutiny. A positive feedback in the ROS-p38MAPK-STAT1 axis has been described, as STAT1-/- mice showed impairment in p38MAPK activation in a ROS-dependent manner (132). As a further suggestion of a cooperative role between ROS and STAT1 activation, in NOX-deficient diabetic mice the reduced ROS levels were found to impair STAT1 activation, and to increase STAT6 activation, thereby promoting a M2 signature during diabetes progression (133). On the other hand, the H2O2 produced by Cu-Zn SOD activity in alveolar murine macrophages was found to activate STAT6 by redox regulation of a critical cysteine during the polarization toward M2-like macrophages (134). Taken together, these findings suggest that further studies will be warranted to understand the tight crosstalk between ROS and STAT, considering the sources and the kind of ROS which are involved.
The Stimulator of Interferon Genes (STING) pathway senses cytosolic double-strand (ds) DNA, that is a sign of microbial infection, cell injury or nuclear DNA damage (135). Cytosolic dsDNA triggers the activation of the cyclic-GMP-AMP-synthase (cGAS), leading to endogenous generation of cyclic GMP-AMP (cGAMP), a unique second messenger. cGAMP binds to and activates the endoplasmic reticulum transmembrane receptor STING, finally resulting in the production and release of type I interferons (IFN), which are potent anti-viral and anti-cancer cytokines (136). On the other hand, the hyperactivity of STING pathway has been implicated in several debilitating autoimmune syndromes (137–139) (i.e. systemic lupus erythematosus) and in acute and chronic inflammation (140).
STING forms a domain-swapped homodimer in the absence of ligands, whereas, upon cGAMP binding, it undergoes extensive conformational rearrangements, leading to oligomerization (141). Recently, a structural analysis by Ergun et al. demonstrated that STING polymerization is necessary for its activation through the formation of intermolecular disulfide bonds via Cys148 (142). Moreover, the increase of mtROS led to cGAS-STING induction of type I IFN (143, 144). Controversially, during herpesvirus infection ROS were found to dampen the type I IFN production in a STING-dependent manner (145). To try to reconcile these findings, the authors speculated that the differences could be ascribed to the amount of ROS, as cysteines can be susceptible to different post-translation modifications, which can either activate or inhibit protein function. More in detail, high ROS levels could oxidize STING thus preventing its polymerization and interferon production, whereas lower levels can promote its assembly. Thus, further studies will be warranted to elucidate how the post-translational modifications of STING by redox regulation affect the innate immune responses against DNA viruses, especially to identify novel immunotherapy targeting IFN production.
Thioredoxins (TRXs) are small proteins that represent a key protection system against oxidative stress through their disulfide reductase activity. TRXs contain two redox-active cysteines in a Cys-X-X-Cys motif, that can be reversibly oxidized to keep intracellular redox balance (146). Besides the reducing activity, TRXs are also important components of redox signaling pathways. Indeed, TRXs control the activity of several proteins by direct physical interaction (146). For instance, TRX binds the apoptosis signal-regulating kinase 1 (ASK1), thus forming an inactive TRX-ASK1 complex. An increase of intracellular ROS induces disulfide bond formation in TRX. Such a conformational change allows ASK1 release, which activates p38MAPK and NF-κB pathways, and triggers a cell death program (147–149). This axis is supported in macrophages, as LPS stimulation, in a Myd88- dependent manner, induces the production of H2O2 to activate NF-κB (148). Remarkably, during tuberculosis infection (150) the recognition of tuberculin protein by TLR2 triggered an increase in ROS production that enhanced a positive feedback burst of ROS by TRX-ASK1-p38 activation (150). Another mechanism, described in microglia, indicates that ASK1 activation occurs by sensing extracellular ATP through the P2X7 receptor, which triggers ROS generation leading to TRX-ASK1 release (151). Furthermore, TRX binds to the thioredoxin-interacting protein (TXNIP). TXNIP activation appears to be essential for NLRP3 assembling by different inflammasome agonists, such as monosodium urate crystals (MSU), ATP or high concentration of glucose (152). These stimuli increase ROS production that allows TRX cysteine oxidation, inducing the breakdown of TRX-TXNIP interaction, and allowing TXNIP to bind NLRP3 to stabilize the inflammasome assembly (152). Moreover, endoplasmic reticulum stress induces ROS-mediated IRE1α activation, that increases the TXNIP mRNA stability, enhancing the NLRP3 activation in sterile inflammation (153).
The studies summarized in this review highlight the crucial and versatile functions of cytosolic and mitochondrial ROS in macrophages. The signaling and damaging properties of ROS can drive the inflammatory response, as well as the diseases resulting from chronic or overwhelming inflammation. The redox balance depends on many parameters, including the levels and the compartmentalization of ROS, their specific sources and subspecies and the specificity/selectivity for their targets.
The great impact of oxidative stress in many diseases explains the enormous number of studies and clinical trials targeting ROS for therapeutic purposes. However, non-selective antioxidants at high doses did not prove effective in either preventing or treating disease processes. This is probably to be ascribed to the disruption of crucial intracellular redox signaling. Even worse, clinical trials have showed harmful effects of antioxidants (154). Indeed, the physiological relevance of H2O2 signaling was still unclear when most of these trials were performed (6, 155). It is now widely accepted that ROS are part of a signaling network with different sources and targets. Their different subcellular compartmentalization and expression suggest the presence of multiple hot spots of ROS with different roles, rather than a homogenous intracellular redox level. The expanding knowledge about the pleiotropy of ROS signaling requires that therapeutic interventions use strategies aimed at addressing the specific disease-relevant mechanisms without disrupting other crucial signaling pathways.
With this respect the development of novel therapeutic approaches targeting ROS will require to identify and dampen the main sources of deleterious ROS relevant for a specific disease, without altering the vital physiological sources of ROS and their downstream signaling pathways. The clinical status of these mechanism-based redox therapies is summarized in (154). An example is the inhibition of specific NOX isoforms or the use of Nrf2 agonists enhancing the expression of endogenous antioxidant enzymes at their physiological sites. Our studies support the relevance of targeting the mitochondrial enzyme MAO to counteract inflammatory diseases for several reasons. Indeed, MAO inhibitors, for which the mechanism of action, pharmacodynamics and pharmacokinetics are well established (they are approved drugs from Parkinson’s disease), block the formation of a specific subset of mitochondrial ROS relevant in pathological conditions, thus preventing mitochondrial dysfunction. More research targeting specific ROS sources and defined mechanisms are strongly awaited.
MC, RS-R, and AC: conceptualization, writing original draft, reviewing, and editing. AV: supervision, reviewing. IS and MF: supervision, literature searching, reviewing, and editing. FV: supervision, reviewing, and editing the figures. All authors contributed to the article and approved the submitted version.
The study was supported by ERC-2019-PoC n. 899770 to AV and IRP-PENTA Grant 18/07-1 to MC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We gratefully acknowledge Dr. Martinvalet, Dr. Bindoli and Dr. Rigobello for helpful discussion.
1. Arnold L, Henry A, Poron F, Baba-Amer Y, Van Rooijen N, Plonquet A, et al. Inflammatory Monocytes Recruited After Skeletal Muscle Injury Switch Into Antiinflammatory Macrophages to Support Myogenesis. J Exp Med (2007) 204(5):1057–69. doi: 10.1084/jem.20070075
2. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
3. Castegna A, Gissi R, Menga A, Montopoli M, Favia M, Viola A, et al. Pharmacological Targets of Metabolism in Disease: Opportunities From Macrophages. Pharmacol Ther (2020) 210:107521. doi: 10.1016/j.pharmthera.2020.107521
4. Lavin Y, Mortha A, Rahman A, Merad M. Regulation of Macrophage Development and Function in Peripheral Tissues. Nat Rev Immunol (2015) 15:731–44. doi: 10.1038/nri3920
5. Nathan C, Cunningham-Bussel A. Beyond Oxidative Stress: An Immunologist’s Guide to Reactive Oxygen Species. Nat Rev Immunol (2013) 13:349–61. doi: 10.1038/nri3423
6. Sies H, Jones DP. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat Rev Mol Cell Biol (2020) 21:363–83. doi: 10.1038/s41580-020-0230-3
7. Martínez MC, Andriantsitohaina R. Reactive Nitrogen Species: Molecular Mechanisms and Potential Significance in Health and Disease. Antioxid Redox Signal (2009) 11(3):669–702. doi: 10.1089/ars.2007.1993
8. Bhatia M, Gaddam RR. Hydrogen Sulfide in Inflammation: A Novel Mediator and Therapeutic Target. Antioxid Redox Signal (2021) 34:1368–77. doi: 10.1089/ars.2020.8211
9. Brüne B, Dehne N, Grossmann N, Jung M, Namgaladze D, Schmid T, et al. Redox Control of Inflammation in Macrophages. Antioxid Redox Signal (2013) 19(6):595–637. doi: 10.1089/ars.2012.4785
10. Martínez-Reyes I, Chandel NS. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
11. Winterbourn CC. Biological Production, Detection, and Fate of Hydrogen Peroxide. Antioxid Redox Signal (2018) 29:541–51. doi: 10.1089/ars.2017.7425
12. Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative Stress in Cancer. Cancer Cell (2020) 38(2):167–97. doi: 10.1016/j.ccell.2020.06.001
13. Lambeth JD. NOX Enzymes and the Biology of Reactive Oxygen. Nat Rev Immunol (2004) 4(3):181–9. doi: 10.1038/nri1312
14. Brandes RP, Weissmann N, Schröder K. Nox Family NADPH Oxidases: Molecular Mechanisms of Activation. Free Radic Biol Med (2014) 76:208–26. doi: 10.1016/j.freeradbiomed.2014.07.046
15. Panday A, Sahoo MK, Osorio D, Batra S. NADPH Oxidases: An Overview From Structure to Innate Immunity-Associated Pathologies. Cell Mol Immunol (2015) 12(1):5–23. doi: 10.1038/cmi.2014.89
16. Augsburger F, Filippova A, Rasti D, Seredenina T, Lam M, Maghzal G, et al. Pharmacological Characterization of the Seven Human NOX Isoforms and Their Inhibitors. Redox Biol (2019) 26:101272. doi: 10.1016/j.redox.2019.101272
17. Lee CF, Qiao M, Schröder K, Zhao Q, Asmis R. Nox4 is a Novel Inducible Source of Reactive Oxygen Species in Monocytes and Macrophages and Mediates Oxidized Low Density Lipoprotein-Induced Macrophage Death. Circ Res (2010) 106(9):1489–97. doi: 10.1161/CIRCRESAHA.109.215392
18. Liang S, Ma HY, Zhong Z, Dhar D, Liu X, Xu J, et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology (2019) 156(4):1156–72. doi: 10.1053/j.gastro.2018.11.019
19. Xu Q, Choksi S, Qu J, Jang J, Choe M, Banfi B, et al. NADPH Oxidases are Essential for Macrophage Differentiation. J Biol Chem (2016) 291(38):20030–41. doi: 10.1074/jbc.M116.731216
20. Mongue-Din H, Patel AS, Looi YH, Grieve DJ, Anilkumar N, Sirker A, et al. NADPH Oxidase-4 Driven Cardiac Macrophage Polarization Protects Against Myocardial Infarction–Induced Remodeling. JACC Basic Transl Sci (2017) 2(6):688–98. doi: 10.1016/j.jacbts.2017.06.006
21. Ullevig S, Zhao Q, Lee CF, Kim HS, Zamora D, Asmis R. NADPH Oxidase 4 Mediates Monocyte Priming and Accelerated Chemotaxis Induced by Metabolic Stress. Arter Thromb Vasc Biol (2012) 32(2):415–26. doi: 10.1161/ATVBAHA.111.238899
22. He C, Larson-Casey JL, Davis D, Hanumanthu VS, Longhini ALF, Thannickal VJ, et al. NOX4 Modulates Macrophage Phenotype and Mitochondrial Biogenesis in Asbestosis. JCI Insight (2019) 4(16):e126551. doi: 10.1172/jci.insight.126551
23. Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabón MA, et al. NOX4-Dependent Fatty Acid Oxidation Promotes NLRP3 Inflammasome Activation in Macrophages. Nat Med (2016) 22(9):1002–12. doi: 10.1038/nm.4153
24. Short JD, Downs K, Tavakoli S, Asmis R. Protein Thiol Redox Signaling in Monocytes and Macrophages. Antioxid Redox Signal (2016) 25(15):816–35. doi: 10.1089/ars.2016.6697
25. Helfinger V, Palfi K, Weigert A, Schröder K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an Nfκb-Dependent Manner. Oxid Med Cell Longev (2019) 2019:3264858. doi: 10.1155/2019/3264858
26. Kim JH, Lee J, Bae SJ, Kim Y, Park BJ, Choi JW, et al. NADPH Oxidase 4 is Required for the Generation of Macrophage Migration Inhibitory Factor and Host Defense Against Toxoplasma Gondii Infection. Sci Rep (2017) 7(1):6361. doi: 10.1038/s41598-017-06610-4
27. Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, et al. Pro-Inflammatory Hepatic Macrophages Generate ROS Through NADPH Oxidase 2 via Endocytosis of Monomeric TLR4-MD2 Complex. Nat Commun (2017) 8(1):2247. doi: 10.1038/s41467-017-02325-2
28. Lekstrom-Himes JA, Gallin JI. Immunodeficiency Diseases Caused by Defects in Phagocytes. N Engl J Med (2000) 343(23):1703–14. doi: 10.1056/nejm200012073432307
29. Pollock JD, Williams DA, Gifford MAC, Li LL, Du X, Fisherman J, et al. Mouse Model of X–linked Chronic Granulomatous Disease, an Inherited Defect in Phagocyte Superoxide Production. Nat Genet (1995) 9(2):202–9. doi: 10.1038/ng0295-202
30. Holland SM. Chronic Granulomatous Disease. Clinic Rev Allerg Immunol (2010) 38:3–10. doi: 10.1007/s12016-009-8136-z
31. Arnold DE. & Heimall, J. R. A Review of Chronic Granulomatous Disease. Adv Ther (2017) 34:2543–57. doi: 10.1007/s12325-017-0636-2
32. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence That TLR4 Is Not a Receptor for Saturated Fatty Acids But Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab (2018) 27(5):1096–1110.e5. doi: 10.1016/j.cmet.2018.03.014
33. Wolf A, Herb M, Schramm M, Langmann T. The TSPO-NOX1 Axis Controls Phagocyte-Triggered Pathological Angiogenesis in the Eye. Nat Commun (2020) 11(1):2709. doi: 10.1038/s41467-020-16400-8
34. Broz P, Dixit VM. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat Rev Immunol (2016) 16(7):407–20. doi: 10.1038/nri.2016.58
35. Swanson KV, Deng M, Ting JPY. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
36. Latz E. NOX-Free Inflammasome Activation. Blood (2010) 116(9):1393–4. doi: 10.1182/blood-2010-06-287342
37. Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome Activation in NADPH Oxidase Defective Mononuclear Phagocytes From Patients With Chronic Granulomatous Disease. Blood (2010) 116(9):1570–3. doi: 10.1182/blood-2010-01-264218
38. Bortolotti M, Polito L, Battelli MG, Bolognesi A. Xanthine Oxidoreductase: One Enzyme for Multiple Physiological Tasks. Redox Biol (2021) 41:101882. doi: 10.1016/j.redox.2021.101882
39. Nishino T, Okamoto K, Kawaguchi Y, Hori H, Matsumura T, Eger BT, et al. Mechanism of the Conversion of Xanthine Dehydrogenase to Xanthine Oxidase. J Biol Chem (2005) 280(26):24888–94. doi: 10.1074/jbc.m501830200
40. Saksela M, Lapatto R, Raivio KO. Irreversible Conversion of Xanthine Dehydrogenase Into Xanthine Oxidase by a Mitochondrial Protease. FEBS Lett (1999) 443(2):117–20. doi: 10.1016/S0014-5793(98)01686-X
41. Ives A, Nomura J, Martinon F, Roger T, LeRoy D, Miner JN, et al. Xanthine Oxidoreductase Regulates Macrophage IL1β Secretion Upon NLRP3 Inflammasome Activation. Nat Commun (2015) 6:6555. doi: 10.1038/ncomms7555
42. Ty MC, Zuniga M, Götz A, Kayal S, Sahu PK, Mohanty A, et al. Malaria Inflammation by Xanthine Oxidase-Produced Reactive Oxygen Species. EMBO Mol Med (2019) 11(8):e9903. doi: 10.15252/emmm.201809903
43. Turrens JF. Mitochondrial Formation of Reactive Oxygen Species. J Physiol (2003) 552(Pt 2):335–44. doi: 10.1113/jphysiol.2003.049478
44. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol Rev (2014) 94(3):909–50. doi: 10.1152/physrev.00026.2013
45. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell (2016) 167(2):457–70 e13. doi: 10.1016/j.cell.2016.08.064
46. Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of Superoxide and Hydrogen Peroxide From Specific Mitochondrial Sites Under Different Bioenergetic Conditions. J Biol Chem (2017) 292(41):16804–9. doi: 10.1074/jbc.R117.789271
47. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR Signalling Augments Macrophage Bactericidal Activity Through Mitochondrial ROS. Nature (2011) 472(7344):476–80. doi: 10.1038/nature09973
48. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, et al. Mitochondrial Reactive Oxygen Species Promote Production of Proinflammatory Cytokines and are Elevated in TNFR1-Associated Periodic Syndrome (TRAPS). J Exp Med (2011) 208(3):519–33. doi: 10.1084/jem.20102049
49. Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell (2010) 142(2):270–83. doi: 10.1016/j.cell.2010.06.007
50. Yazdanpanah B, Wiegmann K, Tchikov V, Krut O, Pongratz C, Schramm M, et al. Riboflavin Kinase Couples TNF Receptor 1 to NADPH Oxidase. Nature (2009) 460(7259):1159–63. doi: 10.1038/nature08206
51. Hinkle PC, Butow RA, Racker E. & Chance, B. Partial Resolution of the Enzymes Catalyzing Oxidative Phosphorylation. XV. Reverse Electron Transfer in the Flavin-Cytochrome Beta Region of the Respiratory Chain of Beef Heart Submitochondrial Particles. J Biol Chem (1967) 242(22):5169–73.
52. Chance B, Hollunger G. The Interaction of Energy and Electron Transfer Reactions in Mitochondria. I. General Properties and Nature of the Products of Succinate-Linked Reduction of Pyridine Nucleotide. J Biol Chem (1961) 236:1562–8. doi: 10.1016/S0021-9258(18)64210-3
53. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury Through Mitochondrial ROS. Nature (2014) 515(7527):431–5. doi: 10.1038/nature13909
54. Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, et al. A Unifying Mechanism for Mitochondrial Superoxide Production During Ischemia-Reperfusion Injury. Cell Metab (2016) 23(2):254–63. doi: 10.1016/j.cmet.2015.12.009
55. Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, et al. Control of Mitochondrial Superoxide Production by Reverse Electron Transport at Complex I. J Biol Chem (2018) 293(25):9869–79. doi: 10.1074/jbc.RA118.003647
56. Scialò F, Sriram A, Fernández-Ayala D, Gubina N, Lõhmus M, Nelson G, et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab (2016) 23(4):725–34. doi: 10.1016/j.cmet.2016.03.009
57. Youdim MBH, Edmondson D, Tipton KF. The Therapeutic Potential of Monoamine Oxidase Inhibitors. Nat Rev Neurosci (2006) 7:295–309. doi: 10.1038/nrn1883
58. Menazza S, Blaauw B, Tiepolo T, Toniolo L, Braghetta P, Spolaore B, et al. Oxidative Stress by Monoamine Oxidases is Causally Involved in Myofiber Damage in Muscular Dystrophy. Hum Mol Genet (2010) 19:4207–15. doi: 10.1093/hmg/ddq339
59. Vitiello L, Marabita M, Sorato E, Nogara L, Forestan G, Mouly V, et al. Drug Repurposing for Duchenne Muscular Dystrophy: The Monoamine Oxidase B Inhibitor Safinamide Ameliorates the Pathological Phenotype in Mdx Mice and in Myogenic Cultures From DMD Patients. Front Physiol (2018) 9:1087. doi: 10.3389/fphys.2018.01087
60. Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, et al. Oxidative Stress by Monoamine Oxidase Mediates Receptor-Independent Cardiomyocyte Apoptosis by Serotonin and Postischemic Myocardial Injury. Circulation (2005) 112(21):3297–305. doi: 10.1161/CIRCULATIONAHA.104.528133
61. Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, et al. Monoamine Oxidase A-Mediated Enhanced Catabolism of Norepinephrine Contributes to Adverse Remodeling and Pump Failure in Hearts With Pressure Overload. Circ Res (2010) 106:193–202. doi: 10.1161/CIRCRESAHA.109.198366
62. Kaludercic N, Carpi A, Nagayama T, Sivakumaran V, Zhu G, Lai EW, et al. Monoamine Oxidase B Prompts Mitochondrial and Cardiac Dysfunction in Pressure Overloaded Hearts. Antioxid Redox Signal (2014) 20(2):267–80. doi: 10.1089/ars.2012.4616
63. Flierl MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, et al. Phagocyte-Derived Catecholamines Enhance Acute Inflammatory Injury. Nature (2007) 449(7163):721–5. doi: 10.1038/nature06185
64. Chaitidis P, Billett EE, O’Donnell VB, Fajardo AB, Fitzgerald J, Kuban RJ, et al. Th2 Response of Human Peripheral Monocytes Involves Isoform-Specific Induction of Monoamine Oxidase-A. J Immunol (2004) 173(8):4821–7. doi: 10.4049/jimmunol.173.8.4821
65. Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, Cathcart MK. IL-4 and IL-13 Employ Discrete Signaling Pathways for Target Gene Expression in Alternatively Activated Monocytes/Macrophages. Free Radic Biol Med (2013) 54:1–16. doi: 10.1016/j.freeradbiomed.2012.10.553
66. Dhabal S, Das P, Biswas P, Kumari P, Yakubenko VP, Kundu S, et al. Regulation of Monoamine Oxidase A (MAO-A) Expression, Activity, and Function in IL-13–Stimulated Monocytes and A549 Lung Carcinoma Cells. J Biol Chem (2018) 293(36):14040–64. doi: 10.1074/jbc.RA118.002321
67. Zhou R, Yazdi AS, Menu P, Tschopp J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
68. Sánchez-Rodríguez R, Munari F, Angioni R, Venegas F, Agnellini A, Castro-Gil MP, et al. Targeting Monoamine Oxidase to Dampen NLRP3 Inflammasome Activation in Inflammation. Cell Mol Immunol (2021) 18:1311–3. doi: 10.1038/s41423-020-0441-8
69. Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, et al. Inflammasome-Driven Catecholamine Catabolism in Macrophages Blunts Lipolysis During Ageing. Nature (2017) 550(7674):119–23. doi: 10.1038/nature24022
70. Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, et al. Sympathetic Neuron-Associated Macrophages Contribute to Obesity by Importing and Metabolizing Norepinephrine. Nat Med (2017) 23(11):1309–18. doi: 10.1038/nm.4422
71. Larabee CM, Neely OC, Domingos AI. Obesity: A Neuroimmunometabolic Perspective. Nat Rev Endocrinol (2020) 16(1):30–43. doi: 10.1038/s41574-019-0283-6
72. Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, et al. Electron Transfer Between Cytochrome C and p66Shc Generates Reactive Oxygen Species That Trigger Mitochondrial Apoptosis. Cell (2005) 122:221–33. doi: 10.1016/j.cell.2005.05.011
73. Gertz M, Steegborn C. The Mitochondrial Apoptosis Pathway and p66Shc—a Regulatory Redox Enzyme or an Adapter Protein Snuggling Around? Cell Cycle (2010) 9:4425–6. doi: 10.4161/cc.9.22.14053
74. Napoli C, Martin-Padura I, De Nigris F, Giorgio M, Mansueto G, Somma P, et al. Deletion of the p66Shc Longevity Gene Reduces Systemic and Tissue Oxidative Stress, Vascular Cell Apoptosis, and Early Atherogenesis in Mice Fed a High-Fat Diet. Proc Natl Acad Sci U S A (2003) 100(4):2112–6. doi: 10.1073/pnas.0336359100
75. Shahzad K, Gadi I, Nazir S, Al-Dabet MM, Kohli S, Bock F, et al. Activated Protein C Reverses Epigenetically Sustained p66Shc Expression in Plaque-Associated Macrophages in Diabetes. Commun Biol (2018) 1:104. doi: 10.1038/s42003-018-0108-5
76. Tomilov A, Bicocca V, Schoenfeld Ra, Giorgio M, Migliaccio E, Ramsey JJ, et al. Decreased Superoxide Production in Macrophages of Long-Lived p66Shc Knock-Out Mice. J Biol Chem (2010) 285:1153–65. doi: 10.1074/jbc.M109.017491
77. Finkel T. From Sulfenylation to Sulfhydration: What a Thiolate Needs to Tolerate. Sci Signal (2012) 5(215):pe10. doi: 10.1126/scisignal.2002943
78. Muri J, Kopf M. Redox Regulation of Immunometabolism. Nat Rev Immunol (2021) 21(6):363–81. doi: 10.1038/s41577-020-00478-8
79. Xiao H, Jedrychowski MP, Schweppe DK, Huttlin EL, Yu Q, Heppner DE, et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation During Aging. Cell (2020) 180(5):968–83.e24. doi: 10.1016/j.cell.2020.02.012
80. Cuadrado A, Manda G, Hassan A, Alcaraz MJ, Barbas C, Daiber A, et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol Rev (2018) 70(2):348–83. doi: 10.1124/pr.117.014753
81. Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, et al. Therapeutic Targeting of the NRF2 and KEAP1 Partnership in Chronic Diseases. Nat Rev Drug Discov (2019) 18(4):295–317. doi: 10.1038/s41573-018-0008-x
82. Saito R, Suzuki T, Hiramoto K, Asami S, Naganuma E, Suda H, et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol Cell Biol (2015) 36(2):271–84. doi: 10.1128/mcb.00868-15
83. Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, et al. Mechanisms of Activation of the Transcription Factor Nrf2 by Redox Stressors, Nutrient Cues, and Energy Status and the Pathways Through Which it Attenuates Degenerative Disease. Free Radic Biol Med (2015) 88(Pt B):108–46. doi: 10.1016/j.freeradbiomed.2015.06.021
84. Suzuki T, Muramatsu A, Saito R, Iso T, Shibata T, Kuwata K, et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep (2019) 28(3):746–758.e4. doi: 10.1016/j.celrep.2019.06.047
85. Zhang DD, Hannink M. Distinct Cysteine Residues in Keap1 Are Required for Keap1-Dependent Ubiquitination of Nrf2 and for Stabilization of Nrf2 by Chemopreventive Agents and Oxidative Stress. Mol Cell Biol (2003) 23(22):8137–51. doi: 10.1128/mcb.23.22.8137-8151.2003
86. Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, et al. Nrf2 is a Critical Regulator of the Innate Immune Response and Survival During Experimental Sepsis. J Clin Invest (2006) 116(4):984–95. doi: 10.1172/JCI25790
87. Ma Q, Battelli L, Hubbs AF. Multiorgan Autoimmune Inflammation, Enhanced Lymphoproliferation, and Impaired Homeostasis of Reactive Oxygen Species in Mice Lacking the Antioxidant-Activated Transcription Factor Nrf2. Am J Pathol (2006) 168(6):1960–74. doi: 10.2353/ajpath.2006.051113
88. Ishii Y, Itoh K, Morishima Y, Kimura T, Kiwamoto T, IIzuka T, et al. Transcription Factor Nrf2 Plays a Pivotal Role in Protection Against Elastase-Induced Pulmonary Inflammation and Emphysema. J Immunol (2005) 175(10):6968–75. doi: 10.4049/jimmunol.175.10.6968
89. Wang P, Geng J, Gao J, Zhao H, Li J, Shi Y, et al. Macrophage Achieves Self-Protection Against Oxidative Stress-Induced Ageing Through the Mst-Nrf2 Axis. Nat Commun (2019) 10(1):755. doi: 10.1038/s41467-019-08680-6
90. Hou Y, Wang Y, He Q, Li L, Xie H, Zhao Y, et al. Nrf2 Inhibits NLRP3 Inflammasome Activation Through Regulating Trx1/TXNIP Complex in Cerebral Ischemia Reperfusion Injury. Behav Brain Res (2018) 336:32–9. doi: 10.1016/j.bbr.2017.06.027
91. Zhao C, Gillette DD, Li X, Zhang Z, Wen H. Nuclear Factor E2-Related Factor-2 (Nrf2) is Required for NLRP3 and AIM2 Inflammasome Activation. J Biol Chem (2014) 289(24):17020–9. doi: 10.1074/jbc.M114.563114
92. Stutz A, Horvath GL, Monks BG, Latz E. ASC Speck Formation as a Readout for Inflammasome Activation. Methods Mol Biol (2013) 1040:91–101. doi: 10.1007/978-1-62703-523-1-8
93. Dehn S, DeBerge M, Yeap X-Y, Yvan-Charvet L, Fang D, Eltzschig HK, et al. HIF-2α in Resting Macrophages Tempers Mitochondrial Reactive Oxygen Species To Selectively Repress MARCO-Dependent Phagocytosis. J Immunol (2016) 197(9):3639–49. doi: 10.4049/jimmunol.1600402
94. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat Commun (2016) 7:11624. doi: 10.1038/ncomms11624
95. Ryan DG, O’Neill LAJ. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu Rev Immunol (2020) 38:289–313. doi: 10.1146/annurev-immunol-081619-104850
96. Corcoran SE, O’Neill LAJ. Hif1α and Metabolic Reprogramming in Inflammation. J Clin Invest (2016) 126(10):3699–707. doi: 10.1172/JCI84431
97. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF Transcription Factors, Inflammation, and Immunity. Immunity (2014) 41(4):518–28. doi: 10.1016/j.immuni.2014.09.008
98. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic Reprogramming of Macrophages: Glucose Transporter 1 (GLUT1)-Mediated Glucose Metabolism Drives a Proinflammatory Phenotype. J Biol Chem (2014) 289(11):7884–96. doi: 10.1074/jbc.M113.522037
99. Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, et al. Coupling Krebs Cycle Metabolites to Signalling in Immunity and Cancer. Nat Metab (2019) 1:16–33. doi: 10.1038/s42255-018-0014-7
100. Kaelin WG, Ratcliffe PJ. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol Cell (2008) 30(4):393–402. doi: 10.1016/j.molcel.2008.04.009
101. Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, et al. The FIH Hydroxylase is a Cellular Peroxide Sensor That Modulates HIF Transcriptional Activity. EMBO Rep (2012) 13(3):251–7. doi: 10.1038/embor.2012.9
102. Sena LA, Chandel NS. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol Cell (2012) 48(2):158–67. doi: 10.1016/j.molcel.2012.09.025
103. Liu R, Li X, Ma H, Yang Q, Shang Q, Song L, et al. Spermidine Endows Macrophages Anti-Inflammatory Properties by Inducing Mitochondrial Superoxide-Dependent AMPK Activation, Hif-1α Upregulation and Autophagy. Free Radic Biol Med (2020) 161:339–50. doi: 10.1016/j.freeradbiomed.2020.10.029
104. Fuhrmann DC, Wittig I, Brüne B. TMEM126B Deficiency Reduces Mitochondrial SDH Oxidation by LPS, Attenuating HIF-1α Stabilization and IL-1β Expression. Redox Biol (2019) 20:204–16. doi: 10.1016/j.redox.2018.10.007
105. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab (2006) 3(3):177–85. doi: 10.1016/j.cmet.2006.02.002
106. Finkel T. Signal Transduction by Mitochondrial Oxidants. J Biol Chem (2012) 287:4434–40. doi: 10.1074/jbc.R111.271999
107. Kirito K, Hu Y, Komatsu N. HIF-1 Prevents the Overproduction of Mitochondrial ROS After Cytokine Stimulation Through Induction of PDK-1. Cell Cycle (2009) 8(17):2844–9. doi: 10.4161/cc.8.17.9544
108. Deng X, Wang Q, Cheng M, Chen Y, Yan X, Guo R, et al. Pyruvate Dehydrogenase Kinase 1 Interferes With Glucose Metabolism Reprogramming and Mitochondrial Quality Control to Aggravate Stress Damage in Cancer. J Cancer (2020) 11(4):962–73. doi: 10.7150/jca.34330
109. Strehl C, Fangradt M, Fearon U, Gaber T, Buttgereit F, Veale DJ. Hypoxia: How Does the Monocyte-Macrophage System Respond to Changes in Oxygen Availability? J Leukoc Biol (2014) 95(2):233–41. doi: 10.1189/jlb.1212627
110. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunity (2015) 42(3):406–17. doi: 10.1016/j.immuni.2015.02.002
111. Codo AC, Davanzo GG, Monteiro L de B, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response Through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab (2020) 32(3):437–446.e5. doi: 10.1016/j.cmet.2020.07.007
112. Dorrington MG, Fraser IDC. NF-κb Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front Immunol (2019) 10:705. doi: 10.3389/fimmu.2019.00705
113. Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective Requirement for C-Rel During IL-12 P40 Gene Induction Macrophages. Proc Natl Acad Sci USA (2000) 97(23):12705–10. doi: 10.1073/pnas.230436397
114. Schröfelbauer B, Polley S, Behar M, Ghosh G, Hoffmann A. NEMO Ensures Signaling Specificity of the Pleiotropic Ikkβ by Directing Its Kinase Activity Toward Iκbα. Mol Cell (2012) 47(1):111–21. doi: 10.1016/j.molcel.2012.04.020
115. Herb M, Gluschko A, Wiegmann K, Farid A, Wolf A, Utermöhlen O, et al. Mitochondrial Reactive Oxygen Species Enable Proinflammatory Signaling Through Disulfide Linkage of NEMO. Sci Signal (2019) 12(568):eaar5926. doi: 10.1126/scisignal.aar5926
116. Sanlioglu S, Williams CM, Samavati L, Butler NS, Wang G, McCray PB, et al. Lipopolysaccharide Induces Rac1-Dependent Reactive Oxygen Species Formation and Coordinates Tumor Necrosis Factor-α Secretion Through IKK Regulation of NF-κb. J Biol Chem (2001) 276(32):30188–98. doi: 10.1074/jbc.M102061200
117. Hwang J, Jin J, Jeon S, Moon SH, Park MY, Yum DY, et al. SOD1 Suppresses Pro-Inflammatory Immune Responses by Protecting Against Oxidative Stress in Colitis. Redox Biol (2020) 37:101760. doi: 10.1016/j.redox.2020.101760
118. Roux C, Jafari SM, Shinde R, Duncan G, Cescon DW, Silvester J, et al. Reactive Oxygen Species Modulate Macrophage Immunosuppressive Phenotype Through the Up-Regulation of PD-L1. Proc Natl Acad Sci USA (2019) 116(10):4326–35. doi: 10.1073/pnas.1819473116
119. Formentini L, Santacatterina F, Núñez de Arenas C, Stamatakis K, López-Martínez D, Logan A, et al. Mitochondrial ROS Production Protects the Intestine From Inflammation Through Functional M2 Macrophage Polarization. Cell Rep (2017) 19(6):1202–13. doi: 10.1016/j.celrep.2017.04.036
120. Corrado C, Fontana S. Hypoxia and HIF Signaling: One Axis With Divergent Effects. Int J Mol Sci (2020) 21(16):5611. doi: 10.3390/ijms21165611
121. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-κb Links Innate Immunity to the Hypoxic Response Through Transcriptional Regulation of HIF-1α. Nature (2008) 453(7196):807–11. doi: 10.1038/nature06905
122. Bonello S, Zähringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive Oxygen Species Activate the HIF-1α Promoter via a Functional Nfκb Site. Arter Thromb Vasc Biol (2007) 27(4):755–61. doi: 10.1161/01.ATV.0000258979.92828.bc
123. Bandarra D, Rocha S. A Tale of Two Transcription Factors: NF-kB and HIF Crosstalk. OA Mol Cell Biol (2013) 1(1):1–7. doi: 10.13172/2054-7331-1-1-924
124. D’Ignazio L, Batie M, Rocha S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-κb. Biomedicines (2017) 5(2):21. doi: 10.3390/biomedicines5020021
125. D’Ignazio L, Bandarra D, Rocha S. NF-κb and HIF Crosstalk in Immune Responses. FEBS J (2016) 283(3):413–24. doi: 10.1111/febs.13578
126. Levy DE, Darnell JE. STATs: Transcriptional Control and Biological Impact. Nat Rev Mol Cell Biol (2002) 3(9):651–62. doi: 10.1038/nrm909
127. Stark GR, Darnell JE. The JAK-STAT Pathway at Twenty. Immunity (2012) 36(4):503–14. doi: 10.1016/j.immuni.2012.03.013
128. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and Consequences of Jak-STAT Signaling in the Immune System. Nat Immunol (2017) 18(4):374–84. doi: 10.1038/ni.3691
129. Salas A, Hernandez-Rocha C, Duijvestein M, Faubion W, McGovern D, Vermeire S, et al. JAK–STAT Pathway Targeting for the Treatment of Inflammatory Bowel Disease. Nat Rev Gastroenterol Hepatol (2020) 17(6):323–37. doi: 10.1038/s41575-020-0273-0
130. Shuai K, Liu B. Regulation of JAK-STAT Signalling in the Immune System. Nat Rev Immunol (2003) 3(11):900–11. doi: 10.1038/nri1226
131. Darnell JE, Kerr IM, Stark GR. Jak-STAT Pathways and Transcriptional Activation in Response to IFNs and Other Extracellular Signaling Proteins. Sci (80-) (1994) 264(5164):1415–21. doi: 10.1126/science.8197455
132. Kim HS, Lee M-S. Essential Role of STAT1 in Caspase-Independent Cell Death of Activated Macrophages Through the P38 Mitogen-Activated Protein Kinase/STAT1/Reactive Oxygen Species Pathway. Mol Cell Biol (2005) 25(15):6821–33. doi: 10.1128/mcb.25.15.6821-6833.2005
133. Padgett LE, Burg AR, Lei W, Tse HM. Loss of NADPH Oxidase-Derived Superoxide Skews Macrophage Phenotypes to Delay Type 1 Diabetes. Diabetes (2015) 64(3):937–46. doi: 10.2337/db14-0929
134. He C, Ryan AJ, Murthy S, Carter AB. Accelerated Development of Pulmonary Fibrosis via Cu,Zn-Superoxide Dismutase-Induced Alternative Activation of Macrophages. J Biol Chem (2013) 288(28):20745–57. doi: 10.1074/jbc.M112.410720
135. Li T, Chen ZJ. The cGAS-cGAMP-STI NG Pathway Connects DNA Damage to Inflammation, Senescence, and Cancer. J Exp Med (2018) 215(5):1287–99. doi: 10.1084/jem.20180139
136. Burdette DL, Vance RE. STING and the Innate Immune Response to Nucleic Acids in the Cytosol. Nat Immunol (2013) 14(1):19–26. doi: 10.1038/ni.2491
137. Ahn J, Gutman D, Saijo S, Barber GN. STING Manifests Self DNA-Dependent Inflammatory Disease. Proc Natl Acad Sci U S A (2012) 109(47):19386–91. doi: 10.1073/pnas.1215006109
138. Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING Activation by Translocation From the ER is Associated With Infection and Autoinflammatory Disease. Cell Host Microbe (2015) 18(2):157–68. doi: 10.1016/j.chom.2015.07.001
139. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-Activating Mutation Underlies a Familial Inflammatory Syndrome With Lupus-Like Manifestations. J Clin Invest (2014) 124(12):5516–20. doi: 10.1172/JCI79100
140. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP, et al. IRF3 and Type I Interferons Fuel a Fatal Response to Myocardial Infarction. Nat Med (2017) 23(12):1481–7. doi: 10.1038/nm.4428
141. Shang G, Zhang C, Chen ZJ, Bai X, Chen, Zhang X. Cryo-EM Structures of STING Reveal its Mechanism of Activation by Cyclic GMP–AMP. Nature (2019) 567(7748):389–93. doi: 10.1038/s41586-019-0998-5
142. Ergun SL, Fernandez D, Weiss TM, Li L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell (2019) 178(2):290–301.e10. doi: 10.1016/j.cell.2019.05.036
143. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature (2015) 520:553–7. doi: 10.1038/nature14156
144. Moran HBT, Turley JL, Andersson M, Lavelle EC. Immunomodulatory Properties of Chitosan Polymers. Biomaterials (2018) 184:1–9. doi: 10.1016/j.biomaterials.2018.08.054
145. Tao L, Lemoff A, Wang G, Zarek C, Lowe A, Yan N, et al. Reactive Oxygen Species Oxidize Sting and Suppress Interferon Production. Elife (2020) 9:e57837. doi: 10.7554/ELIFE.57837
146. Lee S, Kim SM, Lee RT. Thioredoxin and Thioredoxin Target Proteins: From Molecular Mechanisms to Functional Significance. Antioxid Redox Signal (2013) 18(10):1165–20. doi: 10.1089/ars.2011.4322
147. Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, et al. ROS-Dependent Activation of the TRAF6-ASK1-P38 Pathway is Selectively Required for TLR4-Mediated Innate Immunity. Nat Immunol (2005) 6(6):587–92. doi: 10.1038/ni1200
148. Kohchi C, Inagawa H, Nishizawa T, Soma GI. ROS and Innate Immunity. Anticancer Res (2009) 29(3):817–21.
149. Shu N, Hägglund P, Cai H, Hawkins CL, Davies MJ. Modification of Cys Residues in Human Thioredoxin-1 by P-Benzoquinone Causes Inhibition of its Catalytic Activity and Activation of the ASK1/p38-MAPK Signalling Pathway. Redox Biol (2020) 29:101400. doi: 10.1016/j.redox.2019.101400
150. Yang CS, Shin DM, Lee HM, Son JW, Lee SJ, Akira S, et al. ASK1-P38 MAPK-P47phox Activation is Essential for Inflammatory Responses During Tuberculosis via TLR2-ROS Signalling. Cell Microbiol (2008) 10(3):741–54. doi: 10.1111/j.1462-5822.2007.01081.x
151. Hirata Y, Nada Y, Yamada Y, Toyama T, Fukunaga K, Hwang GW, et al. Elaidic Acid Potentiates Extracellular ATP-Induced Apoptosis via the P2X7-ROS-ASK1-P38 Axis in Microglial Cell Lines. Biol Pharm Bull (2020) 43(10):1562–9. doi: 10.1248/bpb.b20-00409
152. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-Interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat Immunol (2010) 11(2):136–40. doi: 10.1038/ni.1831
153. Lerner AG, Upton JP, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, et al. Ire1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death Under Irremediable ER Stress. Cell Metab (2012) 16(2):250–64. doi: 10.1016/j.cmet.2012.07.007
154. Schmidt HHHW, Stocker R, Vollbracht C, Paulsen G, Riley D, Daiber A, et al. Antioxidants in Translational Medicine. Antioxid Redox Signal (2015) 23(14):1130–43. doi: 10.1089/ars.2015.6393
Keywords: macrophages, reactive oxygen species (ROS), mitochondria, innate immunity, redox signaling, inflammasome, monoamine oxidase, protein oxidation
Citation: Canton M, Sánchez-Rodríguez R, Spera I, Venegas FC, Favia M, Viola A and Castegna A (2021) Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 12:734229. doi: 10.3389/fimmu.2021.734229
Received: 30 June 2021; Accepted: 15 September 2021;
Published: 30 September 2021.
Edited by:
Michael Walch, Université de Fribourg, SwitzerlandReviewed by:
Cristina Lopez-Rodriguez, Pompeu Fabra University, SpainCopyright © 2021 Canton, Sánchez-Rodríguez, Spera, Venegas, Favia, Viola and Castegna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcella Canton, bWFyY2VsbGEuY2FudG9uQHVuaXBkLml0; Alessandra Castegna, YWxlc3NhbmRyYS5jYXN0ZWduYUB1bmliYS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.