
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 August 2021
Sec. Viral Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.730088
 Linda Francistiová1,2†
Linda Francistiová1,2† Adrián Klepe1,3,4†
Adrián Klepe1,3,4† Géza Curley1,3,4
Géza Curley1,3,4 Károly Gulya3
Károly Gulya3 András Dinnyés1,3,4*
András Dinnyés1,3,4* Kata Filkor1,3,4
Kata Filkor1,3,4In December 2019, a new viral disease emerged and quickly spread all around the world. In March 2020, the COVID-19 outbreak was classified as a global pandemic and by June 2021, the number of infected people grew to over 170 million. Along with the patients’ mild-to-severe respiratory symptoms, reports on probable central nervous system (CNS) effects appeared shortly, raising concerns about the possible long-term detrimental effects on human cognition. It remains unresolved whether the neurological symptoms are caused directly by the SARS-CoV-2 infiltration in the brain, indirectly by secondary immune effects of a cytokine storm and antibody overproduction, or as a consequence of systemic hypoxia-mediated microglia activation. In severe COVID-19 cases with impaired lung capacity, hypoxia is an anticipated subsidiary event that can cause progressive and irreversible damage to neurons. To resolve this problem, intensive research is currently ongoing, which seeks to evaluate the SARS-CoV-2 virus’ neuroinvasive potential and the examination of the antibody and autoantibody generation upon infection, as well as the effects of prolonged systemic hypoxia on the CNS. In this review, we summarize the current research on the possible interplay of the SARS-CoV-2 effects on the lung, especially on alveolar macrophages and direct and indirect effects on the brain, with special emphasis on microglia, as a possible culprit of neurological manifestation during COVID-19.
Over the last twenty years, there have been two major new human Coronavirus outbreaks, namely the SARS-CoV in 2002 in China and the MERS-CoV in 2012 in Saudi Arabia (1, 2). The third novel Coronavirus outbreak occurred at the end of 2019 caused by SARS-CoV-2 in Wuhan, China (3, 4).
Based on their serological properties, members of the Coronaviridae genus can be grouped into α, β, γ and δ coronaviruses. The most pathogenic, pandemic-related human coronaviruses belong to the β group (5). Coronaviruses have a long, positive-sense, single-stranded RNA genome of 26 to 32 kilobases in size. The genome of SARS-CoV-2 contains 14 open reading frames that encode 27 proteins. At the 5’ region, 15 non-structural proteins required for viral replication are encoded. Open reading frames at the 3’ region encode those structural proteins - namely spike (S), nucleocapsid (N), an envelope protein (E) and membrane protein (M) - that are required for infection and induce host immune response (6, 7). In a genome comparison between SARS-CoV and SARS-CoV-2, it was found that 79% of both were identical, and the structural organization of their genomes was the same. In contrast to SARS-CoV, SARS-CoV-2 and MERS displayed less similarity, as only 50% of their genome was identical (8).
Although SARS-CoV, MERS-CoV, and SARS-CoV-2 have strong structural similarities and mainly cause lower respiratory tract infections and shortness of breath, these viruses have some unique features. A comparison of these three human pathogenic coronaviruses is listed in Table 1.
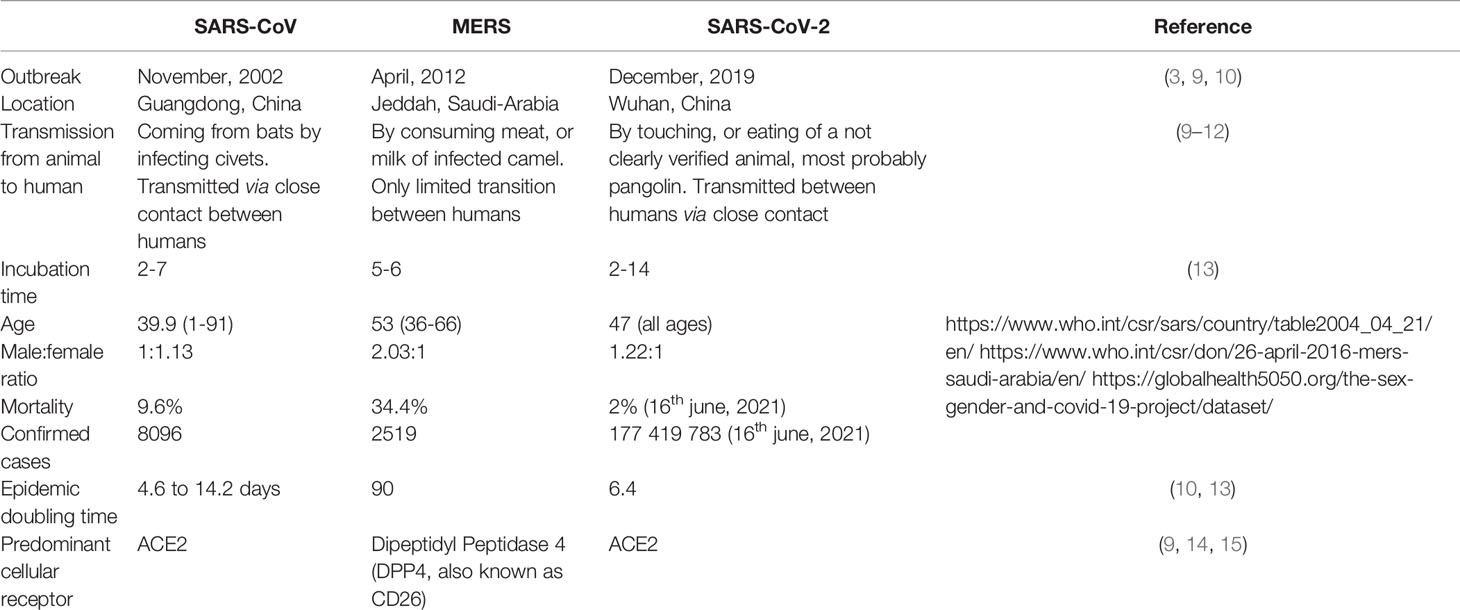
Table 1 Key comparison of geographical location and epidemiology data of SARS-CoV, MERS and SARS-CoV-2 caused infections.
The mildly pathogenic α Coronaviruses cause upper respiratory tract infections, while the highly pathogenic β Coronaviruses, including SARS-CoV, MERS-CoV, and SARS-CoV-2 cause serious lower respiratory tract symptoms (i.e., pneumonia), resulting in patients requiring respiratory support (16). Hence SARS-CoV-2 patients have a high risk of experiencing severe systemic hypoxia. Along with these, neurological symptoms may also develop, and the neuroinvasive tendencies of coronaviruses have been documented for almost all of the βCoVs, including SARS-CoV, MERS‐CoV (17), HCoV‐229E (18), HCoV‐OC43 (19) and the mouse hepatitis virus (20). SARS-CoV-2 also holds the potential for invading the nervous system. From the documented neurological symptoms, the mildest ones are anosmia and ageusia (sudden loss of smell taste) (21), but otolaryngeal symptoms, i.e., tinnitus, vertigo combined with a loss of hearing may appear (22). In severe cases, headache, seizures, delirium and even coma can develop (23). The presence of SARS-CoV-2 was confirmed in cerebrospinal fluid (CSF) taken from an encephalitis patient by next-generation sequencing (24) and by qRT-PCR indicating the presence of viral RNA in CSF (25, 26). In autopsy brain samples, Puelles and colleagues detected viral particles (27). Since the brain is one of the so-called immune-privileged sites of the human body, the investigation of anti-SARS-CoV-2 immunoglobulin G (IgG) production was also in focus, as the presence of antibodies in the CSF indicates intrathecal IgG production (28). However, in some cases, patients with the presence of SARS-CoV-2 IgG had normal CSF results, like ICP, cell counts, protein and glucose levels (29). In another, smaller trial, Barreras and her colleagues detected SARS-CoV-2 IgG in the CSF of patients with neurological symptoms. However, IgG levels did not correlate with the time between symptom development to sampling or disease severity (30).
There are several possible direct and indirect ways SARS-CoV-2 could interact with the CNS (31). In this review, we will discuss these possible interactions together with the impact of SARS-CoV-2 on alveolar macrophages and focus on the implications of hypoxia and hypoxia-induced factors on microglial cells (Figure 1).
SARS-CoV-2 uses a transmembrane protein angiotensin-converting enzyme 2 (ACE2), a metallo-peptidase expressed not only in the respiratory epithelial cells but in almost every organ of the body. ACE2 is on the membrane of the target cells to establish infection (14). Based on its structure, the S-protein belongs to the class I fusion proteins. It is formed by two subunits, namely S1 on the N-terminal surface mediates receptor binding; and the S2 subunit, a transmembrane peptide on the C-terminal, is responsible for the internalization of the virion in the host cell (32).The binding affinity between the S protein of SARS-CoV-2 and the ACE2 is nearly ten-fold higher than in the case of SARS-CoV. Besides the attachment to the host ACE2 receptor, the priming of S protein by transmembrane protease serine 2 (TMPRSS2), a host membrane serine protease on the cell membrane, is also needed to permit the entry of the SARS-CoV-2 (33). Upon the formation of the bond, conformation changes take place in the S protein. It is cleaved by TMPRSS2, thereby allowing the release of the S2 subunit, and facilitating the entry of viral RNA into the cytoplasm of the host cell (32). As potential strategies for the treatment of COVID-19, the pharmacological inhibition of TMPRSS2 or the bond between human recombinant soluble ACE2 and the receptor-binding domain of S-protein could significantly reduce infection by SARS-CoV-2 (34) (Figure 2A).
Figure 1 An overview of the topics covered in this review. Our review discusses how SARS-CoV-2 interacts with immune cells with a special focus on alveolar macrophages. Further, we will discuss how viral particles enter the brain and interacts with microglia cells.
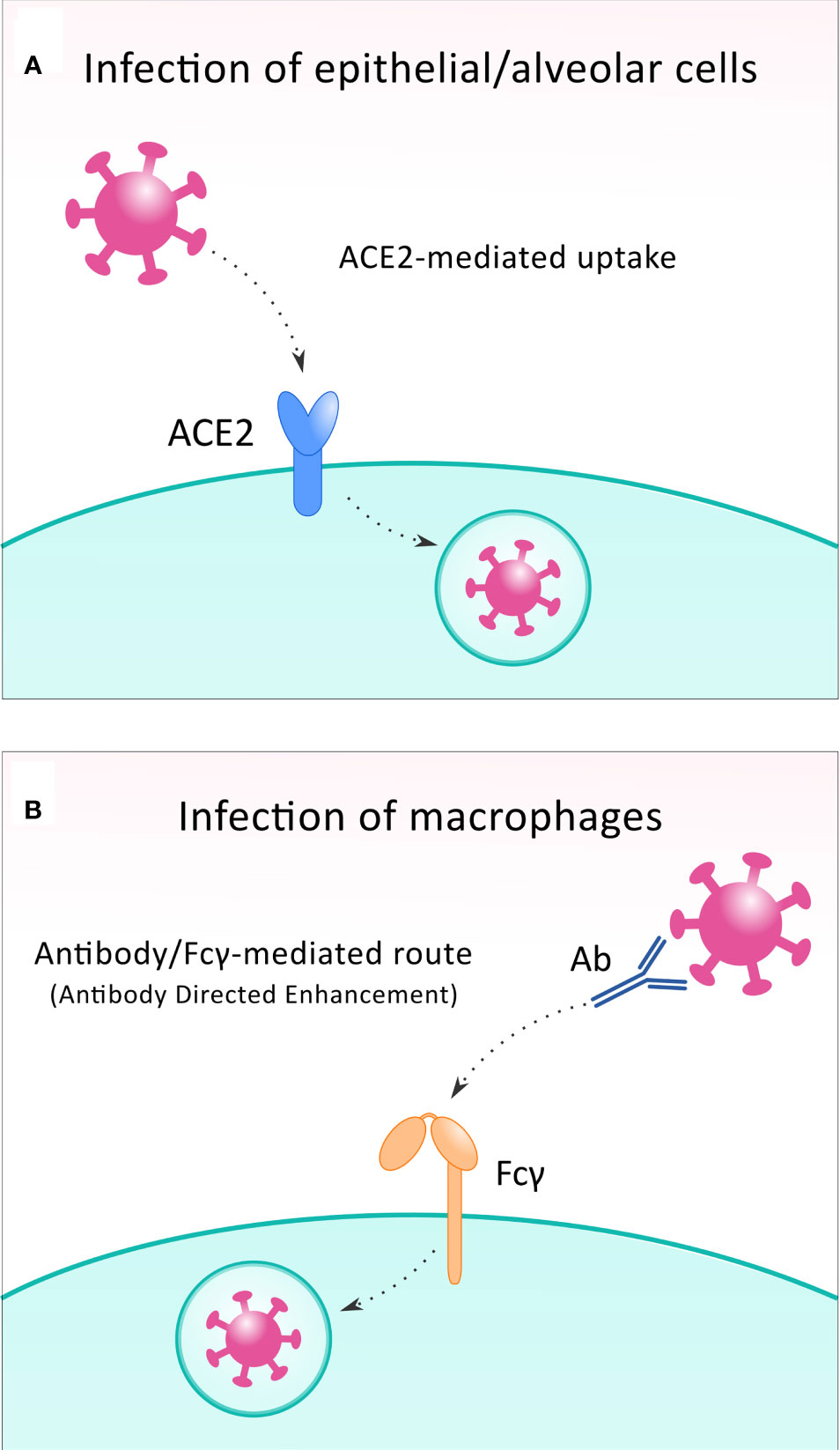
Figure 2 To infect the host cell, SARS-CoV-2 can exploit different receptors. In most infected cells, including epithelial and alveolar cells, the virus binds to ACE2 receptor (A), while during infection of macrophages, Fcγ receptors are utilized in the antibody-directed enhancement mechanism (B).
The ACE2 receptor plays a physiological role in the renin-angiotensin system, and it indirectly affects the signaling pathway. ACE2 converts Angiotensin II (Ang II) into Angiotensin 1-7 [Ang-(1-7)], which binds to the Mas receptor that has a protective role in the lungs, as well as in other organs. However, in the case of Ang II binding to its receptor, vascular permeability can be facilitated by the JAK/STAT signaling pathway (35). During acute lung injury fibrosis, and when patients are exposed to the SARS-CoV-2 infection, ACE inhibitors and Ang II receptor blockers (ARBs) could moderate the lung injury by shifting the system towards the protective pathway [ACE2/Ang-(1-7)] (36). ACE inhibitors reduce hypertension which is common among patients with SARS-CoV-2. The concerns related to high blood pressure and COVID-19 are justified here because the co-occurrence of the two diseases is relatively high and this has been reported in several studies (37, 38). Furthermore, ACE2 is cleaved by increased activity of the transmembrane proteinase (ADAM17), indicated by the activation of the receptor of the tumor necrosis factor-α (TNF-α). Upon the release of cytokines such as IL-6, IL-1β and IFN-γ along with TNF-α due to SARS-CoV-2 infection, inhibition of the expression of ACE2 has been reported (35), leading to an imbalance in homeostasis and to inflammation and a cytokine storm (34).
The primary infected organ of SARS-CoV-2 is the lung causing acute respiratory distress syndrome (ARDS) and respiratory failure (39). Initially the virus infects the respiratory epithelial cells, which highly express ACE2 that provides an excellent entry for the virus (14). After having formed the bond between the S protein and ACE2 receptor, the virus can evade the immune system by effectively inhibiting the activation of TNF receptor-associated factors (TRAF) 3/6, which are key molecules in activating downstream signaling such as in interferon regulatory factor (IRF) 3/7 and nuclear factor kappa B (NF-κB) signaling pathways (14, 40). Deactivation of further transcription factors and the suppression of early pro-inflammatory responses through type I interferon (IFN-I) signaling, the coronavirus can start limiting antiviral response mechanisms (14, 41). The evasion of IFN-I-mediated innate immunity is likely orchestrated by the viral protein N which acts as an antagonist of IFN signaling. In this scenario, novel coronaviruses can counteract IFN expression by inhibiting the phosphorylation and nuclear translocation of transcription factors of the JAK/STAT signaling pathway (14, 39, 42). The suppressed expression of IFN-I can lead to an insufficient response of the host cells and inadequate clearance of viral infections (43). Thus, the activation of early antiviral programs appears to be prohibited contributing to the evasion of innate antiviral immunity by the coronavirus (14, 39). Through the suppressed expression of IFN-I, virions can regulate IFN-I signaling in infected macrophages, in which an increased level of pro-inflammatory cytokine expression (especially TNF-α and IL-6) via the NF-κB cascade is auto-amplified through positive feedback loops. These can contribute to triggering hyperinflammation and to developing a cytokine storm (14, 41).
On the other hand, at a later stage, the activation of antiviral programs and the recruitment of non-infected immune cells occur. Virus particles released from the infected dead cells are recognized by endosomal RNA pattern recognition receptors (PRRs) which leads to the activation of the innate immune system and its cellular machinery (39, 44). Recent studies reported that not only the main RNA sensors, including cytoplasmic retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), have a key role in the detection and identification of coronavirus derived PAMPs (45, 46), but also pattern recognition toll-like receptors 3 and 7 (TLR-3 and TLR-7) are activated. TLRs induce IFN-I response and further increase the expression of IL-1β, IL-6 and TNF-α through MAVS, IRF3 and NF-κB cascades in immune cells (14, 44, 47). Therefore, the innate immune system is activated and macrophages are recruited to the lungs where hyperinflammation is triggered (14, 41) (Figure 3).
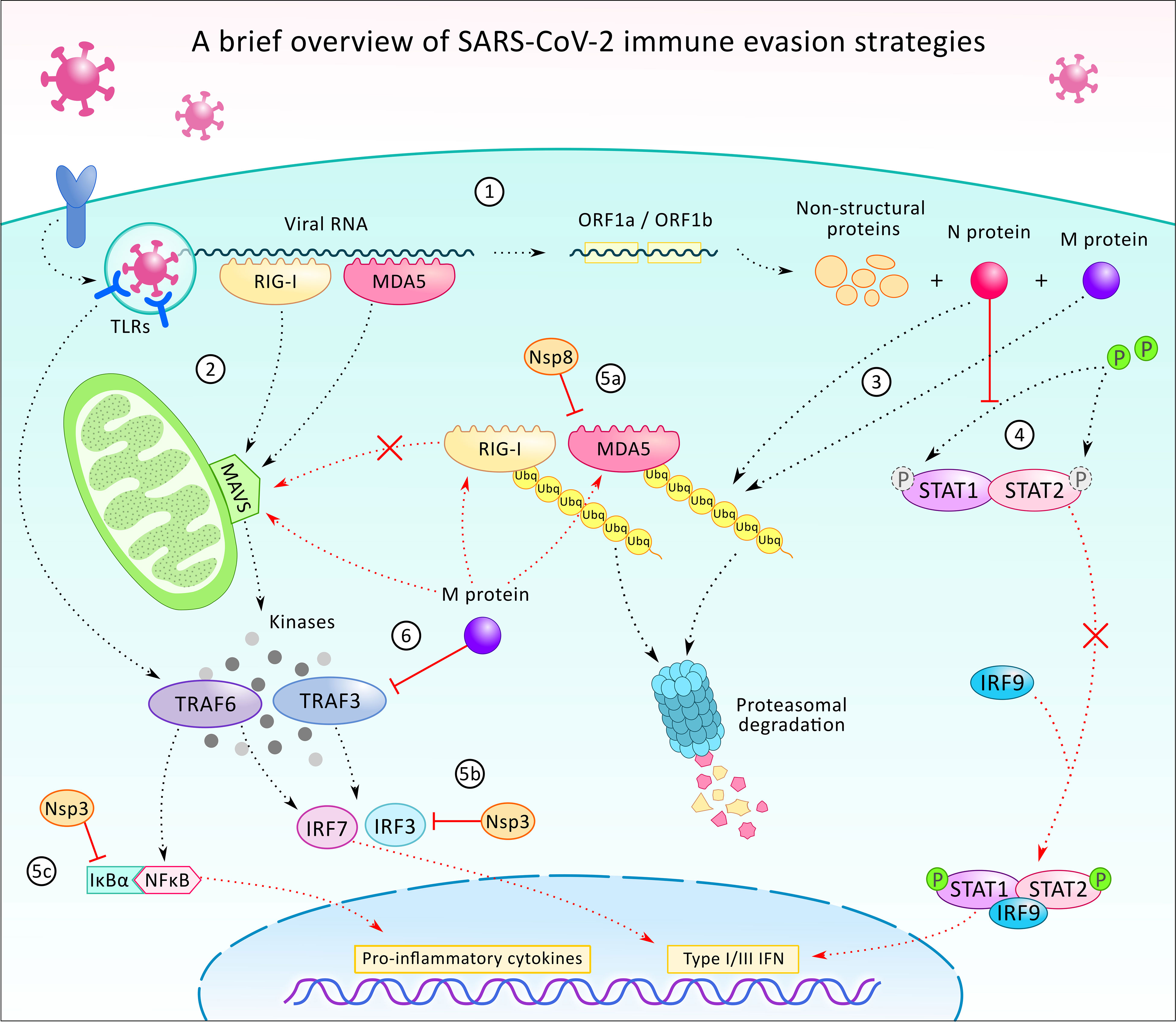
Figure 3 A brief overview of the SARS-CoV-2 virus’ immune evasion strategies. After infecting the host cell, the internalization of the viral particle, the transcription of the viral RNA starts with the translation of ORF1a and ORF2b, which generates proteins participating in the further transcription, host modulation, and immune evasion (1). In the cell, the presence of viral particles or viral RNA is being recognized by pattern recognition receptors such as toll-like receptors (TLR), retinoic acid-inducible gene I (RIG-1), and melanoma differentiation-associated protein 5 (MDA5) (2). TLRs (TLR 3, 7, and 8) are localized on endosomes and together with the cytoplasmatic viral sensors RIG-1 and MDA5, which orchestrate the translocation of NF-κB and IRF3 to the nucleus, leading to the expression of pro-inflammatory cytokines and type 1 and 3 interferons (INF-1/3). While TLRs activate TNF-associated factor 6 (TRAF6), RIG-1 and MDA5 act through mitochondrial antiviral protein (MAVS), to TRAF3/6, to IRF3/7. Viral protein N and M can alter the ubiquitination and degradation of RIG-1 and MDA5 sensors (3), thus restrains the activation of MAVS, TRAF3/6, and prevents IRF-3/7 and NF-κB signaling. N protein can also counteract IFN-1 production by inhibition of signal transducer and activator of transcription protein 1 and 2 (STAT1/2) phosphorylation and activation for further signaling (4). Additionally, some of the non-structural proteins (Nsp3, 8 and others - not shown) can also execute immunomodulatory functions, such as contributing to the inhibition of MDA5 (5a), IRF3 (5b), or stabilizing the NF-κB inhibitor IκBα (5c). Viral protein M also participates in immune evasion by blocking TRAF3 and binding to RIG-1, MDA5, and MAVS and preventing their interaction (6).
A less prevalent route for the immune evasion of SARS-CoV-2 is the infection of alveolar macrophages. While the “conventional” way of the entry of viral particles to the host cells is ACE2-mediated, in some cases, macrophages can be infected, and this happens via an antibody/Fcγ-mediated route. In this situation, SARS−CoV−2 virions are recognized by cross reactive neutralizing antibodies against seasonal coronaviruses (48); and then are taken up by the macrophage cells via Fcγ-receptors in a mechanism termed antibody directed enhancement (ADE) (49) (Figure 2B). Once taken up via Fcγ receptors, the virus particles inhibit the signaling of IFN-I in infected macrophages (14), leading to an increased expression of pro-inflammatory factors (IL-1β, IL-6 and TNF-α), which might result in hyperinflammation (14, 41). Besides this, the number of alveolar macrophages present during SARS-CoV-2 infection correlates well with disease severity (50). In the case of SARS-CoV, the delayed release of cytokines and chemokines was found in bronchial epithelial cells and macrophages at the early stage of infection in vitro cell experiments (51). Breathing difficulties caused by disrupted respiratory epithelial cells and the reduced alveolar partial pressure of oxygen (PO2) induce hypoxia, which causes inflammation and a hypoxic state in alveolar macrophages (52). The increased pulmonary expression of significant amounts of pro-inflammatory cytokines, NF-κB and hypoxia-inducible factor-1 α (HIF-1α) help uninfected, activated macrophages to invade into the alveoli, which recruit other immune cells such as CD8+ T effectors, and this produces more clones and causes tissue damage (50, 52, 53).
In hypoxic alveolar macrophages, the expression of neurokinin-1 receptors is upregulated (52), and this hypoxic event induces the production of multiple factors such as reactive oxygen species (ROS) enzymes, transcriptional factors, and MAPKs (54). In the hypoxic state, when the O2 levels are at 10% instead of the normoxic 21%, the activated alveolar macrophages contribute to the release of H2O2, leading to the inflammatory response (55). Likewise, in SARS-CoV-2, when alveolar macrophages are infected in the early phase of the disease, the secretion of pro-inflammatory cytokines (IL-1β, IL-6, IL-18 and TNF-α) and chemokines (CCL2, CCL3, CCL5) contributes to maintaining the emerged inflammation (14). Additionally, the autophagy of macrophages and activation via HIF-1α might result from hypoxia as well (56). HIF-1α, as a major transcription factor involved in response to reduced cellular oxygen levels, is significantly increased in the hypoxic state, while under physiological conditions, the levels of HIF-1α remain low (56). In response to hypoxia, increased mRNA, and protein levels of IL-8 and TNF-α have been observed, and these typical pro-inflammatory cytokines are released by macrophages and especially by microglia in the CNS (56).
Furthermore, the elevated expression of IFN-α/β and IL-6 can be detected (57), and secreted IFN-λ can disrupt the lung epithelial barrier by inhibiting the lung epithelial proliferation and its repair directly (58). While SARS-CoV-2 infects the macrophages, a viral cascade limits the antiviral response mechanisms by suppressing the activation of transcription factors (such as NF-κB and IRF3/7), thereby limiting the release of IFN-Is and allowing the secretion of the above-mentioned pro-inflammatory cytokines (14, 53). The positive feedback loops of inflammatory cascade by infected cells may contribute to hyperinflammation and the cytokine storm syndrome, the latter of which is also a key factor in extrapulmonary multiple-organ failure (51).
In the blood serum, high levels of C-reactive protein (CRP), ferritin, and D-dimer emerge during the course of COVID-19 infection (59, 60), suggesting an ongoing strong inflammation with self-perpetuating tendencies (61). Moreover, the increased serum levels of CCL7, CXCL10, and IL-1RA, and the presence of CCL2 and CCL7 in the bronchoalveolar fluid were found, pointing towards the activation of signaling towards macrophage recruitment. All of these changes might be associated with disease severity, lung disruption and have a potentially fatal outcome (62). To explain the elevated levels of these molecules, the high production of IL-6 and the activated macrophages should be taken into account. IL-6 is a cytokine of pleiotropic activity, but during viral infections, IL-6 is considered one of the most important cytokines for its regulation of T-cell response, prevention of viral-induced apoptosis of lung epithelial cells, regulation of IgG isotype switching, and other functions (63, 64). Moreover, an increased IL-6 level in the serum is associated with a negative prognosis in patients with SARS-CoV-2 (65). In COVID-19 patients, the gene expression of subsets of CD4+ and CD8+ T cells were observed in the bronchoalveolar fluid, and these subsets were activated differently; moreover, the B cell populations were altered, while the monocyte infiltration was minimal (53, 60). Furthermore, IgG levels declined about two months after symptoms of the early onset of the disease, but the recovered patients maintained high spike-specific IgG titers (60). The findings show that many factors and cells can be present in the blood and in the tissue fluid after a lung infection.
A key component of any immune response, including the response to SARS-CoV-2, is the activation of CD4+ and CD8+ T cells and the subsequent production of neutralizing antibodies (66). It is very interesting that the profile of anti-SARS-CoV-2 antibodies in the plasma and CSF differs within the same COVID-19 patient, suggesting a compartmentalized immune response within the brain (67). Such divergent humoral response indirectly supports neuroinvasion of SARS-CoV-2 during acute infection. Longitudinal profiling studies of neutralizing antibodies against SARS-CoV in 2005 revealed that recovered SARS patients had developed high levels of neutralizing antibodies, while patients with a shorter illness duration displayed higher neutralizing antibody activity compared to patients with a longer illness duration (68, 69). This suggests that antibody responses do indeed play a role in determining the disease outcome. Based on the similarities between SARS-CoV and SARS CoV -2, it is likely that neutralizing processes against SARS-CoV-2 antibodies also develop; and this could contribute to the course, duration, and possible termination of acute infection in the lung and other organs, including the CNS.
As is well known, the CNS contains CD4+ and CD8+ T cells, which patrol and protect the borders of CNS, while CD8+ T cells provide a cytotoxic defense against viral infections in the brain parenchyma (70–72). Aside from the direct cytotoxic reactions in the presence of viral particles in the brain, local cytokine and chemokine milieu is important for T cell generation, retention and infiltration to the brain. Thus, T cells might contribute to cytokine production and further exacerbate inflammatory conditions in the brain by paracrine signaling to microglia (73). For this reason, it might be worthwhile examining the T cell involvement during the course of SARS-CoV-2 infection in the brain-resident T cells. During CNS infection with neurotropic viruses, T cells infiltration is followed by B cells infiltration to ensure local secretion of antibodies since a passage of antibodies from serum into the brain is prevented by BBB (74).
Recently, several reports emerged describing the presence of autoantibodies in high proportions of patients with the most severe cases. These reports show that about 10% of the tested patients with severe COVID-19 had antibodies against the type I interferon molecules and other autoantibodies against proteins of blood vessels, heart, and brain (75, 76). These findings could explain the delay in the onset of the severe COVID-19 symptoms, how the “long COVID” develops, and why sometimes lung damage continues to progress long after the virus is no longer detectable in the body. Intensive research on this issue is currently ongoing. It could provide clues on the possibility that some people might be predisposed to producing autoantibodies and hence potentially be more likely to develop severe COVID-19 disease.
COVID-19 patients may be at a higher risk of infection of the nervous system, encephalitis and developing cognitive decline after displaying symptoms such as olfactory and gustatory dysfunctions, among others (72). Although CNS is not the primary organ affected by SARS-CoV-2, viruses can directly invade the CNS and damage nerves and neurons that were identified as targets of this viral infection (31). SARS-CoV-2 can migrate by retrograde or anterograde neuronal transport through motor proteins by infecting motor or sensory nerve endings (31). Moreover, SARS-CoV-2 can gain access to the brain either through the olfactory tract or via the lung epithelium (77) (Figure 4). In several COVID−19 cases, neurological complications were reported, including mild symptoms such as confusion, fatigue, anxiety, reversal of sleep-wake cycle, and headaches (78). But also severe neurological syndromes such as encephalopathies (79) (often with delirium or psychosis), meningo-encephalitis (25), ischemic stroke (80), acute necrotizing encephalopathy (81), and Guillain-Barré syndrome (82). An impressive cohort study by Peterson et al. provides a comparative analysis of neurological data from patients with COVID-19-related neurological disorders. Interestingly, many of the patients had only mild respiratory symptoms, showing that these complications were not related to the severity of the respiratory COVID−19, and thus suggesting that the primary affected organ was the brain. However, none of the patients tested for the presence of SARS−CoV−2 protein in CSF were positive (83). Another cohort study focused on neurological complications of COVID−19 additionally showed that the primary neurological impairments in younger patients comprise psychiatric diagnoses with alterations in mental status and encephalopathy or encephalitis, while in older patients (>60 years), the leading neurological complications were of cerebrovascular origin (84). Although extremely valuable, these studies present data sets collected from a small number of patients, and as authors remarked, it could show a bias towards severe disease. At the same time, they highlight the importance of future studies of the neurological complications accompanying COVID-19 and follow-up studies of the affected patients that will help uncover the long-term effects of the disease. In this section, we will explore the possibilities of the SARS−CoV−2 to directly infect the brain, while in the later sections, we will discuss the indirect, possibly immune-overactivation-based routes of COVID−19-related CNS damage.
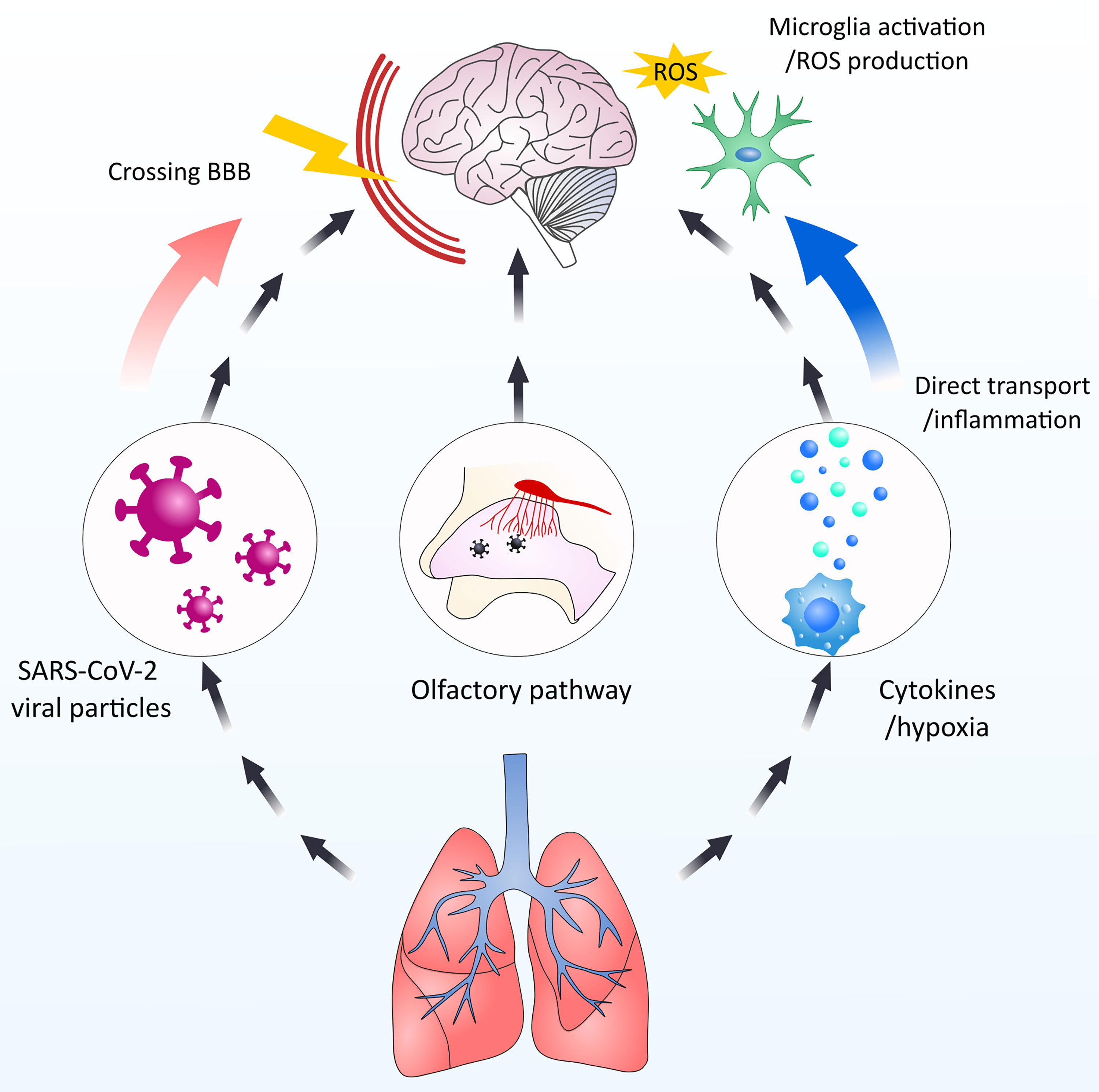
Figure 4 Schematic overview of the possible interactions of SARS-CoV-2 and CNS. SARS-CoV-2 particles could potentially and negatively influence the CNS by multiple means. 1. Directly entering the CNS via crossing the BBB. 2. By-passing the BBB by entering via the olfactory nerve. 3. Indirect influence via microglia activation by cytokines infiltration from the periphery and hypoxia-induced activation.
The CNS, sometimes described as “immune privileged”, is under normal conditions carefully sheltered against the invasion of pathogens from the outside environments as well as from the periphery. At the forefront of the brain’s immune privilege is the neurovascular unit, which consists of endothelial cells, associated blood-brain barrier (BBB) tight junctions (TJ), basal lamina, pericytes, parenchymal cells, astrocytes, neurons, and interneurons. The neurovascular unit ensures the maintenance of the BBB, cerebral homeostasis, and the cerebral blood flow (85). To further ensure protection against unwanted inflammation, along with the BBB, which separates the brain parenchyma from the peripheral blood (86), other biological barriers are present in the brain. The blood-retina barrier (BRB) guards the interface with retinae (87), and the blood-cerebrospinal fluid barrier (BCSFB) isolates the brain from contact with the CSF (88). Despite such sophisticated defense mechanisms, the invasion of the CNS by pathogens can occur. The entry of various pathogens, including viruses, into the CNS can be facilitated via multiple different routes: such as transcellular (e.g. by receptor-mediated transport of the viral particle or by pinocytosis uptake without disruption of the cellular barriers), paracellular (e.g. by disrupting the tight-junctions of the BBB and increasing the permeability of the BBB), as intracellular cargo (the “Trojan horse” method when the viral particle present inside of a cell is allowed to enter the CNS by circumventing the BBB) (89). Transcellular entry occurs as a receptor-mediated uptake or pinocytosis, but the cellular barriers are not disrupted. A potential target of SARS-CoV-2 infection is the endothelium, especially brain microvascular endothelial cells (BMVECs), which form the BBB, among other cell types. After crossing the basement membrane of the lung epithelium, the virus reaches the blood vessel and then the BMVECs, to be transported via the cellular pathway by binding to ACE2 receptor or via an intercellular pathway across the BBB into the brain (90). Potential direct viral infection can occur by the biding of the SARS-CoV-2 to ACE2 receptor on the surface of BMVECs. Moreover, internalization can lead to the production of ROS and the increased secretion of proinflammatory cytokines as well as chemokines (i.e., CXCL10) that can be detected in serum (91). And a recent study suggested that the coronavirus S1 protein might be cleaved from the virus by the host cell’s proteases and subsequently be available as a single infectious unit for crossing the BBB. The authors mention that this process could be executed via a mechanism similar to adsorptive transcytosis and uptake by peripheral tissue and be ACE2-independent. Once the protein enters the brain parenchyma, it might remain biologically highly active and toxic, and thus induce detrimental responses in the brain without the whole virions being involved (92). It should be added that this research was performed on mouse models and this mechanism could not be reproduced on a human induced pluripotent stem cell (iPSC)-based BBB in vitro model. Nevertheless, this result suggests an interesting avenue that deserves investigation.
Both inflammation and a potential cytokine storm induce the loosening of the TJ complex with alterations in the expression of TJ proteins such as in ZO-1, occludin, claudin-5 and VE-cadherin (93). Furthermore, cytoskeletal remodeling without a tight BBB leads to both vascular leakage and coagulation (57). The viral infection itself and inflammatory molecules bring about an enhanced permeability of the BBB (91). Through the compromised BBB, blood monocytes can infiltrate the CNS and produce a direct infection followed by the rapid activation of microglia among other glial cells (94). The balance- and type of cytokines and their cumulative effects at the BBB are complex and regulated by multiple signaling pathways and cell types. Next, the whole cascade results in a leaky and impaired BBB. Perrin and colleagues reported COVID-19 cases in which an elevated astroglia marker, S100B level was detected as a sign of increased BBB permeability (95). When an intense systemic inflammatory response occurs, more virus and peripheral cytokines such as IL-1β, IL-6, IL-17 and TNF-α, among others, can enter the brain via the damaged BBB. Hence, exacerbated, or triggered neuroinflammation by activating microglia, as the key cellular mediators of this process, may be developed and intensified by Th17 cells transmigration to the brain parenchyma and IL-17 upregulation, both caused by the augmentation of TNF-α (96). Together with the SARS-CoV-2 infection, the cascade promotes a cytokine storm that results in the increased secretion of pro-inflammatory factors (such as MIP1-α, IP-10, G-CSF, CRP and ferritin, among others) (97). Furthermore, the bond between cytokines/chemokines and their specific receptors on the cerebral microvascular endothelium provokes in addition to neuroinflammation, BBB breakdown and encephalitis (97).
The choroid plexus is a single layer of epithelial cells located in the brain’s ventricular system, and it produces cerebrospinal fluid. Neighboring cells of the choroid plexus are interconnected via TJs and form the BCSFB and contribute to the homeostatic regulation of the microenvironment in the brain (98). Recently, it has been found that certain cells of the choroid plexus express the ACE2 and other SARS-CoV-2 entry factors such as the TMPRSS2 (99). The same study using iPSC-derived brain organoids demonstrated that the infection of the choroid plexus cells by the SARS-CoV-2 causes substantial damage to the epithelium and a subsequent leakage across this important barrier. At the same time, the group did not observe infection of neuronal cells. These results overall suggest that the neurological symptoms observed in COVID-19 patients might be a secondary consequence of the infection of supportive CNS cells rather than the direct infection of neuronal cells (99). This hypothesis has yet to be confirmed experimentally, although it seems to be in line with the findings of a post-mortem study of the brains of 43 COVID-19 patients. The authors found no evidence of CNS damage caused directly by SARS-CoV-2 but found strong microglia and astrocyte activation and infiltration of cytotoxic T-lymphocytes (100), which might be a consequence of the disrupted brain barrier. On the other hand, a new study revealed the selective susceptibility of dopaminergic cells to SARS-CoV-2 infection and observed inflammatory and senescence on the transcriptional level (101), indicating that this issue among COVID-19 patients might require special attention.
Although the main route for the most viral infections of the lower airways is considered to be the oral-lung aspiration axis (102, 103), research of the novel coronavirus SARS-CoV-2 suggests that in this case, the more prevalent entry is via the aspiration of oropharyngeal mucus originating from the nasal cavity and containing the viral inoculum, into the deep lung. Evidence supporting this hypothesis comes from the knowledge that the nasal lining has a higher concentration of the ACE2 receptor compared to the lower airways, creating a gradient of high susceptibility of infection in the nasal cavity to lower parts in the deep areas of the lung (104). Moreover, the autopsies of patients who died as a consequence of COVID-19 showed that the macroscopic appearance of the infected areas of the lungs was described as “patchy, segmental and peripheral” (104). If the nasal surface indeed serves as the dominant initial site for SARS-CoV-2 infection, the threat of spreading into the olfactory nerves should be considered. The olfactory nerve projects from the nasal cavity directly into the olfactory bulb of the brain, while in the mucosal part of the nasal cavity, ciliated dendrites of the nerve extend to the mucus-lined airway space. There they gather odorant information and transmit it via olfactory sensory neurons’ axons through the cribriform plate directly to the brain (105). In this way, the olfactory sensory neurons are constantly exposed to the environment – including the potentially present pathogens (106); and hence serve as a direct single-cell route for neuroinvasion. Interestingly, the same study showed that despite the fact, that viruses can sometimes enter the CNS via the olfactory nerves and successfully enter the olfactory bulb of the brain while by-passing the protective barriers, another safety check is probably in operation in the olfactory bulb. In fact, after the infection of the olfactory bulb via the olfactory nerve, the infection was halted before it had the chance to spread to different CNS areas (106). To achieve such an inhibition of the spread of viral infection, the immune system needs to be ready to react in a way that neutralizes the viral particle yet does not damage the neurons. Such noncytolytic clearance is commonly facilitated by IFNs. INFs are cytokines primarily produced by lymphocytes (CD8+, CD4+ and natural killer cells (NK)) and serve as the primary protective measure against viruses (107). In the study of Moseman et al. (106), after the neuroinvasion of the viral particles to the brain, the first line of defense was represented by microglia, which shortly after the infection showed evidence of activation. The activation of cells characterized by the up-regulation of antigen-presenting molecules MHC I, CD80 and CD86, enabled microglia to present viral antigens to infiltrating CD8+ T cells. CD8+ T cells subsequently exert antiviral pressure and stop the viral spread throughout the CNS. It is interesting that in this study, microglia were not infected by the virus in question (vesicular stomatitis virus), yet it was able to present the viral antigens to T cells. This means that microglia acquired the antigen from nearby neurons and cross-presented them to the T cells (106). The scenario of the olfactory nerve infection by SARS-CoV-2 seems to be plausible as it has been reported that in many cases, the patients developed progressive ageusia and anosmia (108). Interestingly, another recent study presented evidence of the olfactory transmucosal invasion of SARS-CoV-2 infected individuals’ brains. These findings are supported by the detection of SARS-CoV-2 RNA as well as viral protein in the brain regions. Moreover, the authors described morphological changes of the tissue associated with such an infection, collectively supporting the SARS-CoV-2 neurotropism and the viral spread along the neuroanatomical structures receiving projections from the olfactory tract (109).
To determine how exactly the SARS-CoV-2 virus enters the olfactory nerve and propagates through it into the CNS, the ACE2 expression levels in the olfactory nerve and the CNS need to be investigated. Regardless of the manner of entry, once the virus successfully infects the CNS via the olfactory nerve, further regions of the brain are involved, such as the piriform and infralimbic cortices, basal ganglia, and dorsal raphae nuclei, while other areas such as the thalamus and hypothalamus are less frequently reported as positive (110). Such a distribution pattern would suggest a transneuronal spread (111). Despite these findings, the clear-cut neuro-invasive potential of SARS-CoV-2 is still yet to be determined, as some research also indicates that only epithelial (sustentacular) cells of the nasal mucosa can be infected, not olfactory neurons (112).
The family of HIF proteins is transcription regulators that respond to the prevalent oxygen levels and modify the gene transcription rates of specific DNA sequences. HIF-1 is a dimer that consists of HIF-1α and HIF-1β subunits. While HIF-1α is present at very low levels during normoxia, HIF-1β is continuously transcribed. In normoxic conditions, HIF-1α is hydroxylated in the presence of iron, oxygen, and 2-oxoglutarate, then HIF-1α undergoes ubiquitination and is destroyed (Figure 5) (113). However, in hypoxemic conditions, the oxygen required for HIF-1α ubiquitination is lacking. Therefore, HIF-1α does not degrade, but translocates to the nucleus, where it binds with HIF-1β, then recruits coactivator proteins at the hypoxia response element. Numerous target genes that assist in hypoxic adaptation are upregulated, such as VEGF, which induces angiogenesis, or erythropoietin, which generates more erythrocytes (114). However, some genes are downregulated (e.g., PDK1), and this reduces the oxygen consumption of the mitochondria. The regulation of genes that boost glucose flux to pyruvate causes a switch from oxidative to glycolytic cellular metabolism (115). Furthermore, through endothelial nitric oxide synthase modulation, HIF-1α can also affect pulmonary circulation (116).
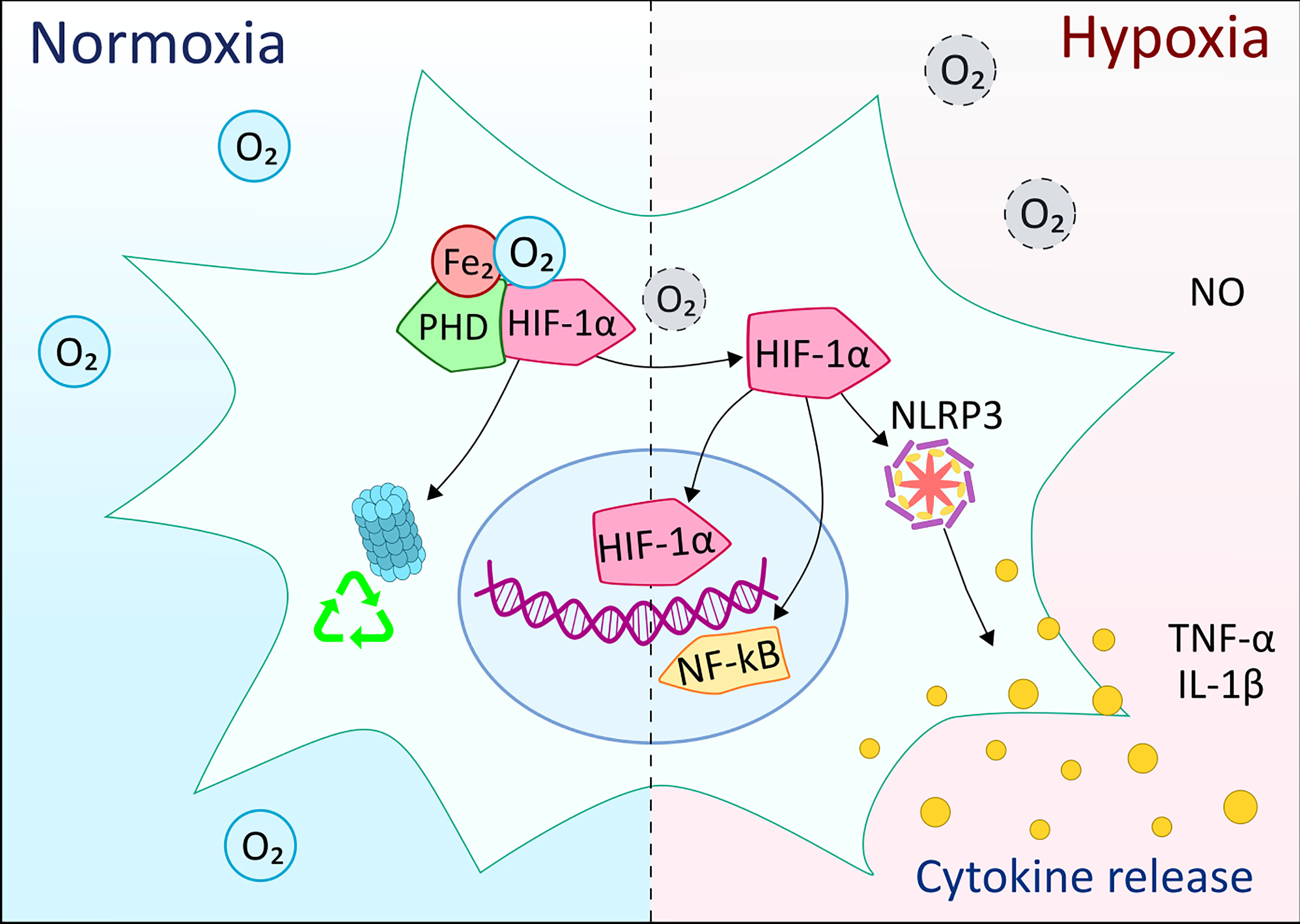
Figure 5 Overview of the effects of hypoxia on microglia cells compared to normal oxygen levels. HIF-1α is one of the main components of the response to hypoxia in myeloid cells. Under normoxic conditions, the oxygen molecules participate in the HIF-1α hydroxylation, ubiquitination and subsequent degradation by the proteasome complex. In hypoxia, the lack of oxygen molecules prevents ubiquitination and degradation of HIF-1α, which can then translocate to the nucleus and, together with other co-activating elements, initiate the expression of pro-inflammatory molecules. At the same time, HIF-1α activation might involve crosstalk with NF-κB, NLRP3 inflammasome, as well as nitric oxide (NO) production along with pro-inflammatory cytokines.
The interaction between HIF-1α and glucose levels is particularly important, as glycolytic flux is required for SARS-CoV-2 replication since high glucose levels promote viral replication and pro-inflammatory cytokine expression (117). Moreover, ROS are potent inducers of HIF-1α (118). The mitochondrial ROS (mtROS) production caused by SARS-CoV-2 stabilizes HIF-1α, which then upregulates IL-1β expression as well as glycolytic genes. These findings explain why uncontrolled diabetes can lead to lung dysfunction and a maladaptive immune response in patients with severe COVID-19 symptoms. They also suggest that the mtROS/HIF-1α/glycolysis axis could be a target for treating this disease (117).
Hypoxia plays a central role in endothelial activation and inflammation, and it creates a positive feedback loop via the reduction of the expression of the complement regulator CD55 by HIF-1α, IL-2, and TNF-α (119). In hypoxic and inflammatory microenvironments, HIF-1α is a key driver of myeloid cell response: (I) It modifies cellular energetics, (II) it upregulates glycolytic enzymes and glucose transporters to allow ATP generation under hypoxic conditions, and (III) it prevents the apoptosis of innate immune cells. In chronic infections, however, HIF-1α prevented excessive lymphocyte recruitment into lung interstitium and pathological immune consequences of the host (120). During the infection of SARS-CoV-2, the infected monocytes express higher levels of pro-inflammatory cytokines and multiple forms of IFN, such as IFN-α, β and γ (121). The presence of TNF-α, IL-1β, IL-6, and HIF-1α are associated with the COVID-19 cytokine storm. Dysregulated blood glucose levels in diabetic patients are a substantial risk factor for the severity of the disease. At the same time, elevated glucose levels might further augment TNF-α, IL-6, and IFN-α/β expression and initiate a vicious cycle of immune hyperactivity (121).
In patients with COVID-19, pneumonia and vascular permeability are related to the elevated thrombosis (119), which is stimulated by a hypoxic state in which the hypercoagulability is increased along with HIF-1α expression (122). Cells adapt to it by activating HIF-1 and HIF-2, which promote the expression of a wide array of genes involved in cell survival and specifically endothelial cell adaptation and energy metabolism. A transition from HIF-1 to HIF-2 suggests an adaptation from acute to prolonged hypoxia, although most genes can be regulated by both (123). HIF-1α has a short cytosolic half-life in normoxic conditions, low basal levels, and a high turnover, while in hypoxemic conditions, it is conserved (113). The cytosolic accumulation of succinate also prevents HIF-1α breakdown (124).
Besides being a coactivator of HIF-1α, pyruvate kinase isozyme 2 (PKM2) plays a key role in glycolysis and mediates acute inflammation (125). PKM2 dimers can directly interact with HIF-1α via nuclear translocation (125, 126). In the nucleus, HIF-1α regulates the adaptor response to hypoxia (125), transcribes pro-inflammatory cytokines and glycolytic machinery (124), especially IL- 1β, a key factor involved in acute and chronic inflammation and generation of fever response, and IL-6. This pro-inflammatory shift is accompanied by the downregulation of anti-inflammatory cytokines such as IL-10 (126). This condition is distinguished by excessive neutrophil-predominant inflammation and by disrupting the alveolar-capillary barrier that causes acute hypoxemic respiratory failure (127, 128).
The HIF-1 signaling pathway can interact with ErbB, PI3K-Akt, mTOR pathways and their connected proteins (129). Via a cascade: PI3K activation phosphorylates Akt, which activates mTOR. It is followed by the activation of 4E-BP1 and the eIF4 complex by Akt and mTORC1, which causes a translation of HIF-1α, an effector protein that initiates transcription and translation of host-specific genes (129, 130). The TNF signaling pathway is interlinked with HIF-1 signaling; hence, it can also increase HIF-1α by activating PI3K-Akt, MAPK, and NF-κB pathways and lead to the overexpression of HIF-1α mRNA translation and protein synthesis, but not affect its stability (131).
Microglia are the resident innate immune system cells in the brain and the first responders to damage of the CNS (132–135). Besides the maintenance of neuronal homeostasis and participating in the formation of the BBB (136), microglia perform critical physiological functions such as synaptic pruning (137), trophic support of the neurons, the removal of apoptotic debris (138), and continuous immune surveillance (139–141). Whenever the brain’s homeostasis is compromised, microglia can rapidly adapt their phenotype and functions in response to the signals from their environment, such as cellular damage or the infiltration of foreign entities (142). Depending on the nature of the activating signal, microglia change their phenotype into the phagocytic or cytokine-producing state. Often, this duality of activation states is referred to as M1 (IFN-γ-dependent classical activation; pro-inflammatory activation) and M2 (IL-4-dependent alternative activation; repair-inducing/phagocytic) activation states (143), during which microglia undergo morphological and metabolic changes in order to efficiently remove the detected threat (143). Although the long-standing notion of microglial cells having M1/M2 activation states is widely accepted (144), a growing body of research indicates that this model might be oversimplistic considering the complexity of microglial effector roles in both health and disease (145). However, as the M1/M2 model is in use within the context of microglial responses to the presence of pathogens, we will adhere to this nomenclature in the review.
At the time of homeostasis, the quiescent state of microglia is maintained mainly via the absence of activating factors, and it is supported by pacifying signals originating from neuronal and astroglial cells (e.g. CX3CL1 and CD200) (146). However, in the presence of an activating signal (e.g. a recognized bacterial or viral particle), microglia rapidly switch from the homeostatic stage with ramified morphology to the classically activated (M1) form. An outcome of an M1 polarizing event is the production of M1-associated factors such as pro-inflammatory cytokines: TNF-α, IL-1α, IL-1β, IL-6, IL-12, IL-23; chemokines; REDOX molecules and other co-stimulatory proteins while phagocytosis is inhibited during M1 activation (144). However, in the case of detection of cellular damage, sterile injury or neurodegenerative disease, M2 activation occurs, and the activation pattern includes events that lead to inflammation resolution through anti-inflammatory factors (TGF-β, IL-10, IL-13, VEGF, EGF and Arg1) in an attempt to re-establish homeostasis (147). One of the results of M2 activation is the transformation of the cells into the amoeboid type and the activation of phagocytosis, which attempts to remove cellular debris and recover tissue equilibrium (143).
As an innate immune response for viral infection in the CNS, microglial cells transit towards a proinflammatory M1 state and rapidly proliferate, leading to enhanced phagocytic activity of microglia (148). During the rapid division of microglial cells, the so-called Warburg effect occurs, in which the ATP is rapidly generated by the enhanced glycolytic energy pathway. This metabolic adaptation is an essential component for the polarization of microglia besides the increased production of cytokines such as IL-6 and TNF-α (148). When microglia polarizing to the M1 state during viral infection, the cells are likely to be exposed to hypoxic environments, which also activates HIF-1α expression (148). IFN-α/β genes play a pivotal role in the potent immune response against viral infections (149). Due to the interaction between the INF-α/β signaling and the IFN-γ pathway, microglia may become more responsive to the virus by its INF-γ antiviral program (150). Furthermore, the results of Zhou et al. clearly show that IFN priming prior to tissue cell infection with SARS-CoV results in the augmented expression of several molecules involved in the induction and upregulation of signaling pathways of IFN-β, among other factors (151).
Although astrocytes are the main source of IFN-α/β in mice (152), in the case of direct viral encephalitis, infected microglia also contribute to IFN-I production, which is dependent on signaling via the IFN-α/β receptor in an MDA-5 dependent pathway (153). Besides the IFN family, the critical role of IL-10 was found during an emerging inflammation caused by a viral infection. CD4+ cells are the pivotal source of IL-10, which cytokine blocks the production of IFN-γ of CD4+ lymphocytes. In the absence of IL-10, higher levels of INF-γ mRNA and protein were produced by CD4+ cells without alterations in the functions of CD8+ lymphocytes. In coronavirus infections, not only high numbers of microglia expressed iNOS and MHC-II, but the viral clearance was also more rapid in lesions of IL-10 deficient mice (154). Overall, in viral infections, the absence of IL-10 results in demyelination and cell death, while the presence of IL-10 protects against tissue damage.
Along with the above-mentioned processes, the inflammatory stimulus in COVID-19 can also be initiated by the cascade of a viral infection that can, in turn, activate the lung macrophages and affect the neutrophil influx; and this indirectly leads to NLRP3 inflammasome activation (155). NLRP3 inflammasome is crucial for the induction of acute lung injury when IL-1β signaling is induced by hypoxia. Similar to hypoxic alveolar macrophages in the lungs, microglia in the brain are the primary source of IL-1β (152). A higher level of another cytokine, IL-6, was observed in parallel with the downregulation of microglial functions in the case of hypoxia. Here, the pathological driver is the existent IL-6-mediated cytokine storm, which appears within two days, leading to neurotoxicity in less than a week (156). While astrocytes are the source of IL-6, an increased hypothalamic IL-6 level was observed along with microglia activation caused by lung infection (157). In contrast, a blockade of microglia in a hypoxic state led to induced astrocytic IL-6 production. A specific therapeutic blockade of circulating IL-6 can be achieved using tocilizumab or siltuximab, but the efficiency of such treatment is relatively low (158).
In general, when cells are injured, the normally intracellular molecules, such as ATP, appear in the extracellular space and serve as a danger-associated molecule pattern (DAMP), which is recognized by the PRRs of the macrophages (157). Therefore, this inflammatory cascade can be initiated by the activation of macrophages by upregulated cytosolic PRR signaling pathways in response to the tissue damage caused by SARS-CoV-2. Through the blood circulation, having released into the brain parenchyma, DAMPs and SARS-CoV-2-derived PAMPs can reactivate microglia. The activation of PRRs on microglia initiates the antiviral cascade via e.g.: the NF-κB (158). The production of IFN-I and -III promotes intracellular antiviral defense and the production and release of microglia and macrophage-dependent IL-1β and IL-6, are the primary response to viral infection (157). However, like M protein or N protein in the SARS-CoV, in SARS-CoV-2 these viral proteins can block the formation of the TRAF3, TANK and TBK1/IKK complex (159); and thereby inhibit the production of IFN-I, which happens in the case of dendritic cells (160). Additionally, elevated levels of IL-12, p40, TNF-α, IL-15, IL-6, and IL-1β, on both mRNA and protein levels, were observed in a neuron culture infected by a murine neurotrophic β Coronavirus then both in non-neurotrophic virus, MHV-2 infected cells and in the uninfected control cultures (161). Overall, microglia may contribute to viral control and nervous tissue damage in response to SARS-CoV-2 CNS infection (158, 159, 162); and thereby inhibit the production of IFN-I, which happens in the case of dendritic cells (160). Additionally, elevated levels of IL-12, p40, TNF-α, IL-15, IL-6, and IL-1β, on both mRNA and protein levels, were observed in a neuron culture infected by a murine neurotrophic β Coronavirus then both in non-neurotrophic virus, MHV-2 infected cells and in the uninfected control cultures (161). Supporting the idea, the quantities of cytokines are positively correlated with the viral titer, and the cytokine levels are high in the absence of microglia (153).
In this review, we have systematically described and analyzed the existing information on possible connections between infection-induced pulmonary events and neurological symptoms. Our analyses might encourage researchers to find early biomarkers and targeted therapeutic approaches to inhibit and/or block the development of neurologic symptoms.
The reported neurological problems in COVID-19 patients might come from the overstimulation of the immune system, and it is presumed that there is a strong involvement of microglia and possibly astroglia in response to peripheral inflammation rather than direct viral infection of the CNS (163). The described events could lead to an interplay between the lung and the brain in the case of SARS-CoV-2 infection, which is based on the fact that the hypoxia and systemic oxidative stress couple the disrupted respiratory epithelium to brain damage. It is known that during COVID-19 infection, there is an increased production of cytokines and chemokines in the periphery, especially in the lung macrophages and in bronchial epithelial cells. When hypoxia occurs owing to difficulties in breathing, a significant amount of proinflammatory factors is released due to the activation of HIF-1α. Certainly, in cases of prolonged and substantial elevation of cytokines in the blood, these can cross the BBB and activate microglia. Similarly, in the case of hypoxia, the lack of oxygen is inevitably sensed by the brain microglia, and this leads to its activation. In a healthy brain, this kind of activation might induce neuroinflammation and potentially lead to neuropsychiatric or neurodegenerative disorders. More severe effects of hypoxia are expected in those patients with pre-existing neurodegenerative diseases, including those which are pre-symptomatic and thus have not yet been diagnosed. In a relevant scenario for Alzheimer’s disease, it has been shown that hypoxia is a major risk factor. Specifically, HIF-1α upregulation in hippocampal microglia leads to reduced Aβ elimination by microglia and hence to increased Aβ-associated neuropathology (164). Knowing that both hypoxia and increased levels of systemic cytokine levels are very likely to cause some degree of damage to the brain, this possibility should not be overlooked and need to be investigated in the future.
Although the predominant clinical symptoms of COVID-19 are pulmonary issues with respiratory manifestations, neurological symptoms are increasingly noted. The loss of smell and taste was reported among the first non-respiratory symptoms, and many people reported otolaryngologic symptoms, headaches, fatigue, and a state called “brain fog” – a condition where patients had trouble thinking clearly (165). All these symptoms suggest possible effects of SARS-CoV-2 on the CNS. Several research groups examined the potential of viral particles of SARS-CoV-2 to invade the CNS tissue, and also the presence of the virus was confirmed in cerebrospinal fluid taken from an encephalitis patient by next-generation sequencing (24) as well as the presence of CSF-specific anti−SARS−CoV−2 antibodies (67). Furthermore, recently in human 3D organoids, it was shown that SARS-CoV-2 is indeed able to infect and kill neurons (166). A preprint publication by Chen et al. (101), claims that human pluripotent stem cell-derived midbrain dopamine neurons are selectively permissive to SARS-CoV-2 infection, with potential long-term implications for the risk of developing Parkinson’s disease-related symptoms. Nevertheless, despite finding further proofs of SARS-CoV-2 protein expression in the brains of the deceased COVID-19 patients, so far, it was not successfully proven that the presence of the virus is directly associated with the neuropathological changes (100). It is important to mention that the study of Song et al. (67) show evidence of the existence of antineuronal autoantibodies that were detectable specifically in COVID-19 patients, while the exact pathogenic relevance of these is yet to be discovered. In the meantime, these autoantibodies could be considered as one of the possible indirect source/contributors to COVID−19 related neuropathology. Presence of such autoantibodies is quite similar to prothrombotic autoantibodies causing occlusion of blood vessels identified recently in Covid-19 patients (75). Moreover, the same study hypothesizes about the similarities between severe COVID−19 phenotypes and those present in patients with lupus and antiphospholipid syndrome, both of which are typical for the presence of anti-phospholipid autoantibodies, and in extremely severe cases, can cause multiple organ failure (75). Additionally, a growing number of case reports describing a connection between SARS−CoV−2 and the Guillain-Barré syndrome (GBS) has emerged recently (167–171). GBS is a state of acute limb paralysis caused typically by an antecedent infectious disease (often a gastrointestinal or respiratory infection) or other immune stimulation that induces an aberrant autoimmune response that targets peripheral nerves and leads to weakness and loss of sensation (172, 173). The reported cases of COVID−19-associated GBS highlight the similarities with cytomegalovirus, Zika virus, and HIV-associated GBS, and thus also suggest a neuroinvasive potential of SARS−CoV−2 (170). At the same time, these findings further underline the importance of immune modulation and autoimmune processes in SARS−CoV−2 infected patients. Therefore, it is very likely that together with other pathological events occurring during COVID−19 illness, detrimental autoimmune effects could contribute to the overall disease progress.
A small Swedish clinical trial demonstrated that CSF of COVID-19 patients with neurological symptoms showed an elevated neurofilament light chain protein (NfL) level that resembles axon injury. Furthermore, the elevated NfL level in the CSF correlated with the seriousness of the neurological symptoms (Glasgow Coma Scale) and was higher in patients between serious and critical disease as compared with mild and moderate disease (174). Based on this, wider clinical trials are much needed to find reliable biomarkers for CNS injury to support the early rehabilitation of COVID-19 patients and to avoid the development of long-COVID. It is currently too early to tell what exactly the long-term effects of COVID-19 on patients’ mental health will be. With so many infected people worldwide, the overall number of patients with neurological complications might grow, and this could induce substantial social and economic costs (175). Moreover, even though the proportion of acute infections with neurological symptoms remains relatively small, neurological complications can lead to lifelong problems. Furthermore, in the cases of patients from the intensive care unit, it had been pointed out that several cases of brain injury could have escaped the attention due to patients being in a state of induced coma, and specific neurological examinations such as imaging were often undertaken only in those people that were slow to wake (83). At the same time, it should be kept in mind that many cases of COVID-related chronic neurological symptoms might not have been reported yet. Considering the global healthcare crisis caused by prioritizing COVID-19 patients with acute and life-threatening conditions, perhaps only after this crisis other COVID-related chronic complications will emerge. Therefore, it is of major importance to provide follow-up studies to determine the long-term neurological effects of this disease. Additionally, it has been observed that the course and the pathological consequences of COVID−19 might differ from patient to patient, future studies on the pathobiological mechanisms and contributing factors will be essential. For example, it would be important to describe the associated risk factors (including those of genetic origin), that can possibly contribute to the different disease outcomes in individual patients. Next, identification of biomarkers that help distinguish between direct viral infection of the brain and immune overactivation would be essential for the selection of proper treatment. Altogether, such knowledge would provide leverage in early recognition and short and long-term management of the disease and perhaps open new opportunities for personalized medicine based on the individual background and risk factors of each patient.
LF and AK wrote the original draft. GC contributed to writing of the paper. LF prepared the figures. KF supervised the conceptualization and writing. KG and AD reviewed and edited the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Grant Agreement No. 766124 (PurinesDx) and grant agreements No. 739593 (HCEMM) and No. 814978 (TUBE). Further financial support was granted by the New National Excellence Programme (20391-3/2018/FEKUSTRAT) and the TKP2020 Tématerületi Kiválósági Program (NKFIH-1279-2/2020) by the National Research, Development and Innovation Office, Hungary and the Doctoral Student Scholarship Program of the Co-operative Doctoral Program of the Ministry of Innovation and Technology financed from the National Research, Development and Innovation Fund (Grants No. 994735 and 973877 for AK and GC, respectively).
LF, AK, GC, AD, and KF are employed by BioTalentum Ltd. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Dr. David P. Curley for the linguistic correction of the manuscript.

1. Cui J, Li F, Shi ZL. Origin and Evolution of Pathogenic Coronaviruses. Nat Rev Microbiol (2019) 17(3):181–92. doi: 10.1038/s41579-018-0118-9
2. de Wit E, van Doremalen N, Falzarano D, Munster VJ, et al. SARS and MERS: Recent Insights Into Emerging Coronaviruses. Nat Rev Microbiol (2016) 14(8):523–34. doi: 10.1038/nrmicro.2016.81
3. Wang C, Horby PW, Hayden FG, Gao GF, et al. A Novel Coronavirus Outbreak of Global Health Concern. Lancet (2020) 395(10223):470–3. doi: 10.1016/S0140-6736(20)30185-9
4. Perlman S. Another Decade, Another Coronavirus. N Engl J Med (2020) 382(8):760–2. doi: 10.1056/NEJMe2001126
5. Chen Y, Liu Q, Guo D. Emerging Coronaviruses: Genome Structure, Replication, and Pathogenesis. J Med Virol (2020) 92(4):418–23. doi: 10.1002/jmv.25681
6. Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, et al. Coronavirus Infections and Immune Responses. J Med Virol (2020) 92(4):424–32. doi: 10.1002/jmv.25685
7. Malik YS, Sircar S, Bhat S, Sharun K, Dhama K, Dadar M, et al. Emerging Novel Coronavirus (2019-nCoV)-Current Scenario, Evolutionary Perspective Based on Genome Analysis and Recent Developments. Vet Q (2020) 40(1):68–76. doi: 10.1080/01652176.2020.1727993
8. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet (2020) 395(10224):565–74. doi: 10.1016/S0140-6736(20)30251-8
9. Anderson RM, Fraser C, Ghani AC, Donnelly CA, Riley S, Ferguson NM, et al. Epidemiology, Transmission Dynamics and Control of SARS: The 2002-2003 Epidemic. Philos Trans R Soc Lond B Biol Sci (2004) 359(1447):1091–105. doi: 10.1098/rstb.2004.1490
10. Chowell G, Abdirizak F, Lee S, Lee J, Jung E, Nishiura H, et al. Transmission Characteristics of MERS and SARS in the Healthcare Setting: A Comparative Study. BMC Med (2015) 13:210. doi: 10.1186/s12916-015-0450-0
11. Wang LF, Shi Z, Zhang S, Field H, Daszak P, Eaton BT, et al. Review of Bats and SARS. Emerg Infect Dis (2006) 12(12):1834–40. doi: 10.3201/eid1212.060401
12. Bhattacharya S, Basu P, Poddar S. Changing Epidemiology of SARS-CoV in the Context of COVID-19 Pandemic. J Prev Med Hyg (2020) 61(2):E130–6. doi: 10.2139/ssrn.3722801
13. Linton NM, Kobayashi T, Yang Y, Hayashi K, Akhmetzhanov AR, Jung SM, et al. Incubation Period and Other Epidemiological Characteristics of 2019 Novel Coronavirus Infections With Right Truncation: A Statistical Analysis of Publicly Available Case Data. J Clin Med (2020) 9(2):538. doi: 10.3390/jcm9020538
14. Felsenstein S, Herbert JA, McNamara PS, Hedrich CM, et al. COVID-19: Immunology and Treatment Options. Clin Immunol (2020) 215:108448. doi: 10.1016/j.clim.2020.108448
15. Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, et al. Dipeptidyl Peptidase 4 Is a Functional Receptor for the Emerging Human Coronavirus-EMC. Nature (2013) 495(7440):251–4. doi: 10.1038/nature12005
16. Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, et al. The Origin, Transmission and Clinical Therapies on Coronavirus Disease 2019 (COVID-19) Outbreak - An Update on the Status. Mil Med Res (2020) 7(1):11. doi: 10.1186/s40779-020-00240-0
17. Li K, Wohlford-Lenane C, Perlman S, Zhao J, Jewell AK, Reznikov LR, et al. Middle East Respiratory Syndrome Coronavirus Causes Multiple Organ Damage and Lethal Disease in Mice Transgenic for Human Dipeptidyl Peptidase 4. J Infect Dis (2016) 213(5):712–22. doi: 10.1093/infdis/jiv499
18. Talbot PJ, Ekande S, Cashman NR, Mounir S, Stewart JN, et al. Neurotropism of Human Coronavirus 229E. Adv Exp Med Biol (1993) 342:339–46. doi: 10.1007/978-1-4615-2996-5_52
19. Dube M, Le Coupanec A, Wong AHM, Rini JM, Desforges M, Talbot PJ, et al. Axonal Transport Enables Neuron-To-Neuron Propagation of Human Coronavirus Oc43. J Virol (2018) 92(17):e00404–18. doi: 10.1128/JVI.00404-18
20. Zhou X, Huang F, Xu L, Lin Z, de Vrij FMS, Ayo-Martin AC, et al. Hepatitis E Virus Infects Neurons and Brains. J Infect Dis (2017) 215(8):1197–206. doi: 10.1093/infdis/jix079
21. Giacomelli A, Pezzati L, Conti F, Bernacchia D, Siano M, Oreni L, et al. Self-Reported Olfactory and Taste Disorders in SARS-CoV-2 Patients: A Cross-Sectional Study. Clin Infect Dis (2020) 71(15):889–90. doi: 10.1093/cid/ciaa330
22. Ozcelik Korkmaz M, Korkmaz M, Egilmez OK, Ozcelik MA, Guven M, et al. Otolaryngological Manifestations of Hospitalised Patients With Confirmed COVID-19 Infection. Eur Arch Otorhinolaryngol (2021) 278(5):1675–85. doi: 10.1007/s00405-020-06396-8
23. Pun BT, Badenes R, Heras La Calle G, Orun OM, Chen W, Raman R, et al. Prevalence and Risk Factors for Delirium in Critically Ill Patients With COVID-19 (COVID-D): A Multicentre Cohort Study. Lancet Respir Med (2021). doi: 10.1016/S2213-2600(20)30552-X
24. Al Saiegh F, Ghosh R, Leibold A, Avery MB, Schmidt RF, Theofanis T, et al. Status of SARS-CoV-2 in Cerebrospinal Fluid of Patients With COVID-19 and Stroke. J Neurol Neurosurg Psychiatry (2020) 91(8):846–8. doi: 10.1136/jnnp-2020-323522
25. Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, et al. A First Case of Meningitis/Encephalitis Associated With SARS-Coronavirus-2. Int J Infect Dis (2020) 94:55–8. doi: 10.1016/j.ijid.2020.03.062
26. Huang YH, Jiang D, Huang JT. SARS-CoV-2 Detected in Cerebrospinal Fluid by PCR in a Case of COVID-19 Encephalitis. Brain Behav Immun (2020) 87:149. doi: 10.1016/j.bbi.2020.05.012
27. Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and Renal Tropism of SARS-CoV-2. N Engl J Med (2020) 383(6):590–2. doi: 10.1056/NEJMc2011400
28. Alexopoulos H, Magira E, Bitzogli K, Kafasi N, Vlachoyiannopoulos P, Tzioufas A, et al. Anti-SARS-CoV-2 Antibodies in the CSF, Blood-Brain Barrier Dysfunction, and Neurological Outcome: Studies in 8 Stuporous and Comatose Patients. Neurol Neuroimmunol Neuroinflamm (2020) 7(6). doi: 10.1212/NXI.0000000000000893
29. Espindola OM, Brandao CO, Gomes YCP, Siqueira M, Soares CN, Lima M, et al. Cerebrospinal Fluid Findings in Neurological Diseases Associated With COVID-19 and Insights Into Mechanisms of Disease Development. Int J Infect Dis (2021) 102:155–62. doi: 10.1016/j.ijid.2020.10.044
30. Garcia MA, Barreras PV, Lewis A, Pinilla G, Sokoll LJ, Kickler T, et al. Cerebrospinal Fluid in COVID-19 Neurological Complications: No Cytokine Storm or Neuroinflammation. medRxiv (2021). doi: 10.1101/2021.01.10.20249014
31. Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, et al. Nervous System Involvement After Infection With COVID-19 and Other Coronaviruses. Brain Behav Immun (2020) 87:18–22. doi: 10.1016/j.bbi.2020.03.031
32. Cataldi M, Pignataro G, Taglialatela M. Neurobiology of Coronaviruses: Potential Relevance for COVID-19. Neurobiol Dis (2020) 143:105007. doi: 10.1016/j.nbd.2020.105007
33. Ong WY, Go ML, Wang DY, Cheah IK, Halliwell B, et al. Effects of Antimalarial Drugs on Neuroinflammation-Potential Use for Treatment of COVID-19-Related Neurologic Complications. Mol Neurobiol (2020). doi: 10.1007/s12035-020-02093-z
34. Abd El-Aziz TM, Al-Sabi A, Stockand JD. Human Recombinant Soluble ACE2 (Hrsace2) Shows Promise for Treating Severe COVID-19. Signal Transduct Target Ther (2020) 5(1):258. doi: 10.1038/s41392-020-00374-6
35. Perrotta F, Matera MG, Cazzola M, Bianco A, et al. Severe Respiratory SARS-CoV2 Infection: Does ACE2 Receptor Matter? Respir Med (2020) 168:105996. doi: 10.1016/j.rmed.2020.105996
36. South AM, Brady TM, Flynn JT. ACE2 (Angiotensin-Converting Enzyme 2), COVID-19, and ACE Inhibitor and Ang II (Angiotensin II) Receptor Blocker Use During the Pandemic: The Pediatric Perspective. Hypertension (2020) 76(1):16–22. doi: 10.1161/HYPERTENSIONAHA.120.15291
37. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/S0140-6736(20)30566-3
38. Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA (2020) 323(16):1574–81. doi: 10.1001/jama.2020.4031
39. Chen DY, Khan N, Close BJ, Goel RK, Blum B, Tavares AH, et al. SARS-CoV-2 Disrupts Proximal Elements in the JAK-STAT Pathway. J Virol (2021), JVI0086221. doi: 10.1128/JVI.00862-21
40. Shah M, Woo HG. Molecular Perspectives of SARS-CoV-2: Pathology, Immune Evasion, and Therapeutic Interventions. Mol Cells (2021) 44(6):408–21. doi: 10.14348/molcells.2021.0026
41. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science (2020) 369(6504):718–24. doi: 10.1126/science.abc6027
42. Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, et al. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep (2020) 33(1):108234. doi: 10.1016/j.celrep.2020.108234
43. Lazear HM, Schoggins JW, Diamond MS. Shared and Distinct Functions of Type I and Type III Interferons. Immunity (2019) 50(4):907–23. doi: 10.1016/j.immuni.2019.03.025
44. Dixon DCK, Ratan C, Nair B, Mangalath S, Abraham R, Nath LR, et al. RNA Sensors as a Mechanism of Innate Immune Evasion Among SARS-CoV2, HIV and Nipah Viruses. Curr Protein Pept Sci (2021). doi: 10.2174/1389203722666210322142725
45. Chen K, Xiao F, Hu D, Ge W, Tian M, Wang W, et al. SARS-CoV-2 Nucleocapsid Protein Interacts With RIG-I and Represses RIG-Mediated IFN-Beta Production. Viruses (2020) 13(1):47. doi: 10.3390/v13010047
46. Sampaio NG, Chauveau L, Hertzog J, Bridgeman A, Fowler G, Moonen JP, et al. The RNA Sensor MDA5 Detects SARS-CoV-2 Infection. Sci Rep (2021) 11(1):13638. doi: 10.1038/s41598-021-92940-3
47. Zotta A, Hooftman A, O’Neill LAJ. SARS-CoV-2 Targets MAVS for Immune Evasion. Nat Cell Biol (2021) 23(7):682–3. doi: 10.1038/s41556-021-00712-y
48. Che XY, Qiu LW, Liao ZY, Wang YD, Wen K, Pan YX, et al. Antigenic Cross-Reactivity Between Severe Acute Respiratory Syndrome-Associated Coronavirus and Human Coronaviruses 229E and OC43. J Infect Dis (2005) 191(12):2033–7. doi: 10.1086/430355
49. Lee WS, Wheatley AK, Kent SJ, DeKosky BJ, et al. Antibody-Dependent Enhancement and SARS-CoV-2 Vaccines and Therapies. Nat Microbiol (2020) 5(10):1185–91. doi: 10.1038/s41564-020-00789-5
50. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
51. Ye Q, Wang B, Mao J. The Pathogenesis and Treatment of the `Cytokine Storm’ in COVID-19. J Infect (2020) 80(6):607–13. doi: 10.1016/j.jinf.2020.03.037
52. Chao J, Wood JG, Gonzalez NC. Alveolar Hypoxia, Alveolar Macrophages, and Systemic Inflammation. Respir Res (2009) 10:54. doi: 10.1186/1465-9921-10-54
53. Merad M, Martin JC. Pathological Inflammation in Patients With COVID-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol (2020) 20(6):355–62. doi: 10.1038/s41577-020-0331-4
54. Kiernan EA, Smith SM, Mitchell GS, Watters JJ, et al. Mechanisms of Microglial Activation in Models of Inflammation and Hypoxia: Implications for Chronic Intermittent Hypoxia. J Physiol (2016) 594(6):1563–77. doi: 10.1113/JP271502
55. Chao J, Wood JG, Blanco VG, Gonzalez NC, et al. The Systemic Inflammation of Alveolar Hypoxia Is Initiated by Alveolar Macrophage-Borne Mediator(s). Am J Respir Cell Mol Biol (2009) 41(5):573–82. doi: 10.1165/rcmb.2008-0417OC
56. Yang Z, Zhao TZ, Zou YJ, Zhang JH, Feng H, et al. Hypoxia Induces Autophagic Cell Death Through Hypoxia-Inducible Factor 1alpha in Microglia. PloS One (2014) 9(5):e96509. doi: 10.1371/journal.pone.0096509
57. Wei Y, Shah R. Substance Use Disorder in the COVID-19 Pandemic: A Systematic Review of Vulnerabilities and Complications. Pharm (Basel) (2020) 13(7):155–84. doi: 10.3390/ph13070155
58. Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A, et al. Type III Interferons Disrupt the Lung Epithelial Barrier Upon Viral Recognition. Science (2020) 369(6504):706–12. doi: 10.1126/science.abc3545
59. Soy M, Keser G, Atagunduz P, Tabak F, Atagunduz I, Kayhan S, et al. Cytokine Storm in COVID-19: Pathogenesis and Overview of Anti-Inflammatory Agents Used in Treatment. Clin Rheumatol (2020) 39(7):2085–94. doi: 10.1007/s10067-020-05190-5
60. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Zhang JE, et al. Deep Immune Profiling of COVID-19 Patients Reveals Distinct Immunotypes With Therapeutic Implications. Science (2020) 369(6508). doi: 10.1126/science.369.6508.1203-l
61. Wannamethee SG, Whincup PH, Lennon L, Papacosta O, Lowe GD, et al. Associations Between Fibrin D-Dimer, Markers of Inflammation, Incident Self-Reported Mobility Limitation, and All-Cause Mortality in Older Men. J Am Geriatr Soc (2014) 62(12):2357–62. doi: 10.1111/jgs.13133
62. Vaninov N. In the Eye of the COVID-19 Cytokine Storm. Nat Rev Immunol (2020) 20(5):277. doi: 10.1038/s41577-020-0305-6
63. Yang ML, Wang CT, Yang SJ, Leu CH, Chen SH, Wu CL, et al. IL-6 Ameliorates Acute Lung Injury in Influenza Virus Infection. Sci Rep (2017) 7:43829. doi: 10.1038/srep43829
64. Lauder SN, Jones E, Smart K, Bloom A, Williams AS, Hindley JP, et al. Interleukin-6 Limits Influenza-Induced Inflammation and Protects Against Fatal Lung Pathology. Eur J Immunol (2013) 43(10):2613–25. doi: 10.1002/eji.201243018
65. McGonagle D, Sharif K K, O'Regan A, Bridgewood C, et al. The Role of Cytokines Including Interleukin-6 in COVID-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev (2020) 19(6):102537. doi: 10.1016/j.autrev.2020.102537
66. Janice Oh HL, Ken-En Gan S, Bertoletti A, Tan YJ, et al. Understanding the T Cell Immune Response in SARS Coronavirus Infection. Emerg Microbes Infect (2012) 1(9):e23. doi: 10.1038/emi.2012.26
67. Song E, Bartley CM, Chow RD, Ngo TT, Jiang R, Zamecnik CR, et al. Divergent and Self-Reactive Immune Responses in the CNS of COVID-19 Patients With Neurological Symptoms. Cell Rep Med (2021) 2(5):100288. doi: 10.1016/j.xcrm.2021.100288
68. Liu STH, Lin HM, Baine I, Wajnberg A, Gumprecht JP, Rahman F, et al. Convalescent Plasma Treatment of Severe COVID-19: A Propensity Score-Matched Control Study. Nat Med (2020) 26:1708–13. doi: 10.1038/s41591-020-1088-9
69. Ni L, Ye F, Cheng ML, Feng Y, Deng YQ, Zhao H, et al. Detection of SARS-CoV-2-Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals. Immunity (2020) 52(6):971–7.e3. doi: 10.1016/j.immuni.2020.04.023
70. Smolders J, Heutinck KM, Fransen NL, Remmerswaal EBM, Hombrink P, Ten Berge IJM, et al. Tissue-Resident Memory T Cells Populate the Human Brain. Nat Commun (2018) 9(1):4593. doi: 10.1038/s41467-018-07053-9
71. Prasad S, Lokensgard JR. Brain-Resident T Cells Following Viral Infection. Viral Immunol (2019) 32(1):48–54. doi: 10.1089/vim.2018.0084
72. Heneka MT, Golenbock D, Latz E, Morgan D, Brown R, et al. Immediate and Long-Term Consequences of COVID-19 Infections for the Development of Neurological Disease. Alzheimers Res Ther (2020) 12(1):69. doi: 10.1186/s13195-020-00640-3
73. Zhan M, Qin Y, Xue X, Zhu S, et al. Death From Covid-19 of 23 Health Care Workers in China. N Engl J Med (2020) 382(23):2267–8. doi: 10.1056/NEJMc2005696
74. Lokensgard JR, Mutnal MB, Prasad S, Sheng W, Hu S, et al. Glial Cell Activation, Recruitment, and Survival of B-Lineage Cells Following MCMV Brain Infection. J Neuroinflamm (2016) 13(1):114. doi: 10.1186/s12974-016-0582-y
75. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies Against Type I IFNs in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
76. Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H, et al. Prothrombotic Autoantibodies in Serum From Patients Hospitalized With COVID-19. Sci Transl Med (2020) 12(570):eabd3876. doi: 10.1126/scitranslmed.abd3876
77. Iroegbu JD, Ifenatuoha CW, Ijomone OM. Potential Neurological Impact of Coronaviruses: Implications for the Novel SARS-CoV-2. Neurol Sci (2020) 41(6):1329–37. doi: 10.1007/s10072-020-04469-4
78. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol (2020) 77(6):683–90. doi: 10.1001/jamaneurol.2020.1127
79. Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic Features in Severe SARS-CoV-2 Infection. N Engl J Med (2020) 382(23):2268–70. doi: 10.1056/NEJMc2008597
80. Beyrouti R, Adams ME, Benjamin L, Cohen H, Farmer SF, Goh YY, et al. Characteristics of Ischaemic Stroke Associated With COVID-19. J Neurol Neurosurg Psychiatry (2020) 91(8):889–91. doi: 10.1136/jnnp-2020-323586
81. Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B, et al. COVID-19-Associated Acute Hemorrhagic Necrotizing Encephalopathy: Imaging Features. Radiology (2020) 296(2):E119–20. doi: 10.1148/radiol.2020201187
82. Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, et al. Guillain-Barre Syndrome Associated With SARS-CoV-2. N Engl J Med (2020) 382(26):2574–6. doi: 10.1056/NEJMc2009191
83. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The Emerging Spectrum of COVID-19 Neurology: Clinical, Radiological and Laboratory Findings. Brain (2020) 143(10):3104–20. doi: 10.1093/brain/awaa240
84. Varatharaj A, Thomas N, Ellul MA, Davies NWS, Pollak TA, Tenorio EL, et al. Neurological and Neuropsychiatric Complications of COVID-19 in 153 Patients: A UK-Wide Surveillance Study. Lancet Psychiatry (2020) 7(10):875–82. doi: 10.2139/ssrn.3601761
85. Abbott NJ, Ronnback L, Hansson E. Astrocyte-Endothelial Interactions at the Blood-Brain Barrier. Nat Rev Neurosci (2006) 7(1):41–53. doi: 10.1038/nrn1824
86. Obermeier B, Verma A, Ransohoff RM. The Blood-Brain Barrier. Handb Clin Neurol (2016) 133:39–59. doi: 10.1016/B978-0-444-63432-0.00003-7
87. Diaz-Coranguez M, Ramos C, Antonetti DA. The Inner Blood-Retinal Barrier: Cellular Basis and Development. Vision Res (2017) 139:123–37. doi: 10.1016/j.visres.2017.05.009
88. Ghersi-Egea JF, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, Engelhardt B, et al. Molecular Anatomy and Functions of the Choroidal Blood-Cerebrospinal Fluid Barrier in Health and Disease. Acta Neuropathol (2018) 135(3):337–61. doi: 10.1007/s00401-018-1807-1
89. Forrester JV, McMenamin PG, Dando SJ. CNS Infection and Immune Privilege. Nat Rev Neurosci (2018) 19(11):655–71. doi: 10.1038/s41583-018-0070-8
90. Li H, Xue Q, Xu X. Involvement of the Nervous System in SARS-CoV-2 Infection. Neurotox Res (2020) 38(1):1–7. doi: 10.1007/s12640-020-00219-8
91. Li F, Wang Y, Yu L, Cao S, Wang K, Yuan J, et al. Viral Infection of the Central Nervous System and Neuroinflammation Precede Blood-Brain Barrier Disruption During Japanese Encephalitis Virus Infection. J Virol (2015) 89(10):5602–14. doi: 10.1128/JVI.00143-15
92. Rhea EM, Logsdon AF, Hansen KM, Williams LM, Reed MJ, Baumann KK, et al. The S1 Protein of SARS-CoV-2 Crosses the Blood-Brain Barrier in Mice. Nat Neurosci (2020). doi: 10.1038/s41593-020-00771-8
93. Bleau C, Filliol A, Samson M, Lamontagne L, et al. Brain Invasion by Mouse Hepatitis Virus Depends on Impairment of Tight Junctions and Beta Interferon Production in Brain Microvascular Endothelial Cells. J Virol (2015) 89(19):9896–908. doi: 10.1128/JVI.01501-15
94. Das Sarma J. Microglia-Mediated Neuroinflammation Is an Amplifier of Virus-Induced Neuropathology. J Neurovirol (2014) 20(2):122–36. doi: 10.1007/s13365-013-0188-4
95. Perrin P, Collongues N, Baloglu S, Bedo D, Bassand X, Lavaux T, et al. Cytokine Release Syndrome-Associated Encephalopathy in Patients With COVID-19. Eur J Neurol (2020) 728:134952. doi: 10.20944/preprints202006.0103.v1
96. Najjar S, Najjar A, Chong DJ, Pramanik BK, Kirsch C, Kuzniecky RI, et al. Central Nervous System Complications Associated With SARS-CoV-2 Infection: Integrative Concepts of Pathophysiology and Case Reports. J Neuroinflamm (2020) 17(1):231. doi: 10.1186/s12974-020-01896-0
97. Alam SB, Willows S, Kulka M, Sandhu JK, et al. Severe Acute Respiratory Syndrome Coronavirus 2 May Be an Underappreciated Pathogen of the Central Nervous System. Eur J Neurol (2020) 27(11):2348–60. doi: 10.1111/ene.14442
98. Kaur C, Rathnasamy G, Ling EA. The Choroid Plexus in Healthy and Diseased Brain. J Neuropathol Exp Neurol (2016) 75(3):198–213. doi: 10.1093/jnen/nlv030
99. Jacob F, Pather SR, Huang WK, Zhang F, Wong SZH, Zhou H, et al. Human Pluripotent Stem Cell-Derived Neural Cells and Brain Organoids Reveal SARS-CoV-2 Neurotropism Predominates in Choroid Plexus Epithelium. Cell Stem Cell (2020) 27(6):937–50.e9. doi: 10.1016/j.stem.2020.09.016
100. Matschke J, Lutgehetmann M, Hagel C, Sperhake JP, Schroder AS, Edler C, et al. Neuropathology of Patients With COVID-19 in Germany: A Post-Mortem Case Series. Lancet Neurol (2020) 19(11):919–29. doi: 10.1016/S1474-4422(20)30308-2
101. Chen S, Han Y, Yang L, Kim T, Nair M, Harschnitz O, et al. SARS-CoV-2 Infection Causes Dopaminergic Neuron Senescence. Res Sq (2021). doi: 10.21203/rs.3.rs-513461/v1
102. Gaeckle NT, Pragman AA, Pendleton KM, Baldomero AK, Criner GJ, et al. The Oral-Lung Axis: The Impact of Oral Health on Lung Health. Respir Care (2020) 65(8):1211–20. doi: 10.4187/respcare.07332
103. Odani K, Tachibana M, Tamashima R, Tsutsumi Y, et al. Herpes Simplex Virus Pneumonia: Importance of Aspiration Etiology. Case Rep Pathol (2019) 2019:7623576. doi: 10.1155/2019/7623576
104. Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell (2020) 182(2):429–46.e14. doi: 10.1016/j.cell.2020.05.042
105. Barrios AW, Nunez G, Sanchez Quinteiro P, Salazar I, et al. Anatomy, Histochemistry, and Immunohistochemistry of the Olfactory Subsystems in Mice. Front Neuroanat (2014) 8:63. doi: 10.3389/fnana.2014.00063
106. Moseman EA, Blanchard AC, Nayak D, McGavern DB, et al. T Cell Engagement of Cross-Presenting Microglia Protects the Brain From a Nasal Virus Infection. Sci Immunol (2020) 5(48):eabb1817. doi: 10.1126/sciimmunol.abb1817
107. Broggi A, Granucci F, Zanoni I. Type III Interferons: Balancing Tissue Tolerance and Resistance to Pathogen Invasion. J Exp Med (2020) 217(1):e20190295. doi: 10.1084/jem.20190295
108. Vaira LA, Salzano G, Deiana G, De Riu G, et al. Anosmia and Ageusia: Common Findings in COVID-19 Patients. Laryngoscope (2020) 130(7):1787. doi: 10.1002/lary.28692
109. Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory Transmucosal SARS-CoV-2 Invasion as a Port of Central Nervous System Entry in Individuals With COVID-19. Nat Neurosci (2021) 24(2):168–75. doi: 10.1038/s41593-020-00758-5
110. Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S, et al. Severe Acute Respiratory Syndrome Coronavirus Infection Causes Neuronal Death in the Absence of Encephalitis in Mice Transgenic for Human ACE2. J Virol (2008) 82(15):7264–75. doi: 10.1128/JVI.00737-08
111. Al-Sarraj S, Troakes C, Hanley B, Osborn M, Richardson MP, Hotopf M, et al. Invited Review: The Spectrum of Neuropathology in COVID-19. Neuropathol Appl Neurobiol (2020) 47(1):3–16. doi: 10.1111/nan.12667
112. Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, et al. Non-Neuronal Expression of SARS-CoV-2 Entry Genes in the Olfactory System Suggests Mechanisms Underlying COVID-19-Associated Anosmia. Sci Adv (2020) 6(31):eabc5801. doi: 10.1126/sciadv.abc5801
113. West JB. Physiological Effects of Chronic Hypoxia. N Engl J Med (2017) 376(20):1965–71. doi: 10.1056/NEJMra1612008
114. Semenza GL, Wang GL. A Nuclear Factor Induced by Hypoxia via De Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Mol Cell Biol (1992) 12(12):5447–54. doi: 10.1128/mcb.12.12.5447-5454.1992
115. Kim JW, Tchernyshyov I, Semenza GL, Dang CV, et al. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab (2006) 3(3):177–85. doi: 10.1016/j.cmet.2006.02.002
116. Shimoda LA, Semenza GL. HIF and the Lung: Role of Hypoxia-Inducible Factors in Pulmonary Development and Disease. Am J Respir Crit Care Med (2011) 183(2):152–6. doi: 10.1164/rccm.201009-1393PP
117. Peter E. Evaluating the Impact of Nurse Practitioner Involvement in a Pleural Procedures Clinic. Br J Nurs (2020) 29(14):816–20. doi: 10.12968/bjon.2020.29.14.816
118. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell (2016) 167(2):457–70.e13. doi: 10.1016/j.cell.2016.08.064
119. Gupta N, Zhao YY, Evans CE. The Stimulation of Thrombosis by Hypoxia. Thromb Res (2019) 181:77–83. doi: 10.1016/j.thromres.2019.07.013
120. Marchetti M. COVID-19-Driven Endothelial Damage: Complement, HIF-1, and ABL2 Are Potential Pathways of Damage and Targets for Cure. Ann Hematol (2020) 99(8):1701–7. doi: 10.1007/s00277-020-04138-8
121. Zhu L, She ZG, Cheng X, Qin JJ, Zhang XJ, Cai J, et al. Association of Blood Glucose Control and Outcomes in Patients With COVID-19 and Pre-Existing Type 2 Diabetes. Cell Metab (2020) 31(6):1068–77.e3. doi: 10.1126/sciadv.abc5801
122. Pinno J, Bongartz H, Klepsch O, Wundrack N, Poli V, Schaper F, et al. Interleukin-6 Influences Stress-Signalling by Reducing the Expression of the mTOR-Inhibitor REDD1 in a STAT3-Dependent Manner. Cell Signal (2016) 28(8):907–16. doi: 10.1016/j.cellsig.2016.04.004
123. Mendez R, Menendez R, Amara-Elori I, Feced L, Piro A, Ramirez P, et al. Lymphopenic Community-Acquired Pneumonia Is Associated With a Dysregulated Immune Response and Increased Severity and Mortality. J Infect (2019) 78(6):423–31. doi: 10.1016/j.jinf.2019.04.006
124. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate Is an Inflammatory Signal That Induces IL-1beta Through HIF-1alpha. Nature (2013) 496(7444):238–42. doi: 10.1038/nature11986
125. Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell (2011) 145(5):732–44. doi: 10.1016/j.cell.2011.03.054
126. Corcoran SE, O’Neill LA. HIF1alpha and Metabolic Reprogramming in Inflammation. J Clin Invest (2016) 126(10):3699–707. doi: 10.1172/JCI84431
127. Donnelly SC, Strieter RM, Reid PT, Kunkel SL, Burdick MD, Armstrong I, et al. The Association Between Mortality Rates and Decreased Concentrations of Interleukin-10 and Interleukin-1 Receptor Antagonist in the Lung Fluids of Patients With the Adult Respiratory Distress Syndrome. Ann Intern Med (1996) 125(3):191–6. doi: 10.7326/0003-4819-125-3-199608010-00005
128. Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, et al. Inflammatory Cytokines in Patients With Persistence of the Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (1996) 154(3 Pt 1):602–11. doi: 10.1164/ajrccm.154.3.8810593
129. Appelberg S, Gupta S, Svensson Akusjarvi S, Ambikan AT, Mikaeloff F, Saccon E, et al. Dysregulation in Akt/mTOR/HIF-1 Signaling Identified by Proteo-Transcriptomics of SARS-CoV-2 Infected Cells. Emerg Microbes Infect (2020) 9(1):1748–60. doi: 10.1080/22221751.2020.1799723
130. Karar J, Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front Mol Neurosci (2011) 4:51. doi: 10.3389/fnmol.2011.00051
131. Zhou J, Callapina M, Goodall GJ, Brune B, et al. Functional Integrity of Nuclear Factor Kappab, Phosphatidylinositol 3’-Kinase, and Mitogen-Activated Protein Kinase Signaling Allows Tumor Necrosis Factor Alpha-Evoked Bcl-2 Expression to Provoke Internal Ribosome Entry Site-Dependent Translation of Hypoxia-Inducible Factor 1alpha. Cancer Res (2004) 64(24):9041–8. doi: 10.1158/0008-5472.CAN-04-1437
132. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate Mapping Analysis Reveals That Adult Microglia Derive From Primitive Macrophages. Science (2010) 330(6005):841–5. doi: 10.1126/science.1194637
133. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM, et al. Local Self-Renewal can Sustain CNS Microglia Maintenance and Function Throughout Adult Life. Nat Neurosci (2007) 10(12):1538–43. doi: 10.1038/nn2014
134. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell (2014) 159(6):1327–40. doi: 10.1016/j.cell.2014.11.023
135. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
136. Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, et al. Dual Microglia Effects on Blood Brain Barrier Permeability Induced by Systemic Inflammation. Nat Commun (2019) 10(1):5816. doi: 10.1038/s41467-019-13812-z
137. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science (2011) 333(6048):1456–8. doi: 10.1126/science.1202529
138. Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A, et al. The Role of Microglia in the Healthy Brain. J Neurosci (2011) 31(45):16064–9. doi: 10.1523/JNEUROSCI.4158-11.2011
139. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a Unique TGF-Beta-Dependent Molecular and Functional Signature in Microglia. Nat Neurosci (2014) 17(1):131–43. doi: 10.1038/nn.3599
140. Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, et al. A Milieu Molecule for TGF-Beta Required for Microglia Function in the Nervous System. Cell (2018) 174(1):156–71.e16. doi: 10.1016/j.cell.2018.05.027
141. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science (2005) 308(5726):1314–8. doi: 10.1126/science.1110647
142. Norris GT, Smirnov I, Filiano AJ, Shadowen HM, Cody KR, Thompson JA, et al. Neuronal Integrity and Complement Control Synaptic Material Clearance by Microglia After CNS Injury. J Exp Med (2018) 215(7):1789–801. doi: 10.1084/jem.20172244
143. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 Polarization and Metabolic States. Br J Pharmacol (2016) 173(4):649–65. doi: 10.1111/bph.13139
144. Boche D, Perry VH, Nicoll JA. Review: Activation Patterns of Microglia and Their Identification in the Human Brain. Neuropathol Appl Neurobiol (2013) 39(1):3–18. doi: 10.1111/nan.12011
145. Ransohoff RM. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat Neurosci (2016) 19(8):987–91. doi: 10.1038/nn.4338
146. Kierdorf K, Prinz M. Factors Regulating Microglia Activation. Front Cell Neurosci (2013) 7:44. doi: 10.3389/fncel.2013.00044
147. Martinez FO, Gordon S, Locati M, Mantovani A, et al. Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. J Immunol (2006) 177(10):7303–11. doi: 10.4049/jimmunol.177.10.7303
148. Ferraro E, Germano M, Mollace R, Mollace V, Malara N, et al. HIF-1, the Warburg Effect, and Macrophage/Microglia Polarization Potential Role in COVID-19 Pathogenesis. Oxid Med Cell Longev (2021) 2021:8841911. doi: 10.1155/2021/8841911
149. Chatterjee D, Biswas K, Nag S, Ramachandra SG, Das J, et al. Microglia Play a Major Role in Direct Viral-Induced Demyelination. Clin Dev Immunol (2013) 2013:510396. doi: 10.1155/2013/510396
150. Hwang M, Bergmann CC. Neuronal Ablation of Alpha/Beta Interferon (IFN-Alpha/Beta) Signaling Exacerbates Central Nervous System Viral Dissemination and Impairs IFN-Gamma Responsiveness in Microglia/Macrophages. J Virol (2020) 94(20):e00422–20. doi: 10.1128/JVI.00422-20
151. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature (2013) 493(7434):674–8. doi: 10.1038/nature11729
152. Facci L, Barbierato M, Marinelli C, Argentini C, Skaper SP, Giusti P, et al. Toll-Like Receptors 2, -3 and -4 Prime Microglia But Not Astrocytes Across Central Nervous System Regions for ATP-Dependent Interleukin-1beta Release. Sci Rep (2014) 4:6824. doi: 10.1038/srep06824
153. Wheeler DL, Sariol A, Meyerholz DK, Perlman S, et al. Microglia Are Required for Protection Against Lethal Coronavirus Encephalitis in Mice. J Clin Invest (2018) 128(3):931–43. doi: 10.1172/JCI97229
154. Savarin C, Bergmann CC. Fine Tuning the Cytokine Storm by IFN and IL-10 Following Neurotropic Coronavirus Encephalomyelitis. Front Immunol (2018) 9:3022. doi: 10.3389/fimmu.2018.03022
155. Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, et al. Critical Role for the NLRP3 Inflammasome During Acute Lung Injury. J Immunol (2014) 192(12):5974–83. doi: 10.4049/jimmunol.1400368
156. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric Antigen Receptor T-Cell Therapy - Assessment and Management of Toxicities. Nat Rev Clin Oncol (2018) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
157. Bobbo VCD, Jara CP, Mendes NF, Morari J, Velloso LA, Araujo EP, et al. Interleukin-6 Expression by Hypothalamic Microglia in Multiple Inflammatory Contexts: A Systematic Review. BioMed Res Int (2019) 2019:1365210. doi: 10.1155/2019/1365210
158. Vardhana SA, Wolchok JD. The Many Faces of the Anti-COVID Immune Response. J Exp Med (2020) 217(6):e20200678. doi: 10.1084/jem.20200678
159. Paces J, Strizova Z, Smrz D, Cerny J, et al. COVID-19 and the Immune System. Physiol Res (2020) 69(3):379–88. doi: 10.33549/physiolres.934492
160. Zhou H, Zhao J, Perlman S. Autocrine Interferon Priming in Macrophages But Not Dendritic Cells Results in Enhanced Cytokine and Chemokine Production After Coronavirus Infection. mBio (2010) 1(4):e00219–10. doi: 10.1128/mBio.00219-10
161. Li Y, Fu L, Gonzales DM, Lavi E, et al. Coronavirus Neurovirulence Correlates With the Ability of the Virus to Induce Proinflammatory Cytokine Signals From Astrocytes and Microglia. J Virol (2004) 78(7):3398–406. doi: 10.1128/JVI.78.7.3398-3406.2004
162. Carty M, Guy C, Bowie AG. Detection of Viral Infections by Innate Immunity. Biochem Pharmacol (2021) 183:114316. doi: 10.1016/j.bcp.2020.114316
163. Vargas G, Medeiros Geraldo LH, Gedeao Salomao N, Viana Paes M, Regina Souza Lima F, Carvalho Alcantara Gomes F, et al. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and Glial Cells: Insights and Perspectives. Brain Behav Immun Health (2020) 7:100127. doi: 10.1016/j.bbih.2020.100127
164. March-Diaz R, Lara-Ureña N, Romero-Molina C, Heras-Garvin A, Ortega-de San Luis C, Alvarez-Vergara MI, et al. Hypoxia Compromises the Mitochondrial Metabloism of Alzheimer’s Disease Microglia via HIF1. Nat Aging (2021) 1:385–99. doi: 10.1038/s43587-021-00054-2
165. Anu KR, Das S, Joseph A. Crosstalk Between Covid-19 And Associated Neurological Disorders: A Review. Curr Neuropharmacol (2021). doi: 10.2174/1570159X19666210113154342
166. Ramani A, Muller L, Ostermann PN, Gabriel E, Abida-Islam P, Muller-Schiffmann A, et al. SARS-CoV-2 Targets Neurons of 3D Human Brain Organoids. EMBO J (2020) 39(20):e106230. doi: 10.15252/embj.2020106230
167. Abu-Rumeileh S, Abdelhak A, Foschi M, Tumani H, Otto M, et al. Guillain-Barre Syndrome Spectrum Associated With COVID-19: An Up-to-Date Systematic Review of 73 Cases. J Neurol (2021) 268(4):1133–70. doi: 10.1007/s00415-020-10124-x
168. Nanda S, Handa R, Prasad A, Anand R, Zutshi D, Dass SK, et al. Covid-19 Associated Guillain-Barre Syndrome: Contrasting Tale of Four Patients From a Tertiary Care Centre in India. Am J Emerg Med (2021) 39:125–8. doi: 10.1016/j.ajem.2020.09.029
169. Caress JB, Castoro RJ, Simmons Z, Scelsa SN, Lewis RA, Ahlawat A, et al. COVID-19-Associated Guillain-Barre Syndrome: The Early Pandemic Experience. Muscle Nerve (2020) 62(4):485–91. doi: 10.1002/mus.27024
170. Khan F, Sharma P, Pandey S, Sharma D, Kumar N, et al. COVID-19-Associated Guillain-Barre Syndrome: Postinfectious Alone or Neuroinvasive Too? J Med Virol (2021). doi: 10.1002/jmv.27159
171. Finsterer J, Scorza FA. Guillain-Barre Syndrome in 220 Patients With COVID-19. Egypt J Neurol Psychiatr Neurosurg (2021) 57(1):55. doi: 10.1186/s41983-021-00310-7
172. Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barre Syndrome. Lancet (2016) 388(10045):717–27. doi: 10.1016/S0140-6736(16)00339-1
173. Shahrizaila N, Lehmann HC, Kuwabara S. Guillain-Barre Syndrome. Lancet (2021) 397(10280):1214–28. doi: 10.1016/S0140-6736(21)00517-1
174. Virhammar J, Naas A, Fallmar D, Cunningham JL, Klang A, Ashton NJ, et al. Biomarkers for Central Nervous System Injury in Cerebrospinal Fluid Are Elevated in COVID-19 and Associated With Neurological Symptoms and Disease Severity. Eur J Neurol (2020). doi: 10.1111/ene.14703
Keywords: COVID-19, SARS-CoV-2, neuroinvasion, microglia, hypoxia
Citation: Francistiová L, Klepe A, Curley G, Gulya K, Dinnyés A and Filkor K (2021) Cellular and Molecular Effects of SARS-CoV-2 Linking Lung Infection to the Brain. Front. Immunol. 12:730088. doi: 10.3389/fimmu.2021.730088
Received: 24 June 2021; Accepted: 27 July 2021;
Published: 13 August 2021.
Edited by:
Pei-Hui Wang, Shandong University, ChinaReviewed by:
Yunyun Jin, Sun Yat-Sen university, ChinaCopyright © 2021 Francistiová, Klepe, Curley, Gulya, Dinnyés and Filkor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: András Dinnyés, bWFudXNjcmlwdC5kaW5ueWVzQGJpb3RhbGVudHVtLmh1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.