 Marisierra Espinar-Buitrago1,2
Marisierra Espinar-Buitrago1,2 Ma Angeles Muñoz-Fernández1,2,3,4*
Ma Angeles Muñoz-Fernández1,2,3,4*- 1Section Head Immunology, Laboratorio InmunoBiología Molecular, Hospital General Universitario Gregorio Marañón (HGUGM), Madrid, Spain
- 2Instituto de Investigación Sanitaria Gregorio Marañón (IiSGM), Madrid, Spain
- 3Spanish Human Immunodeficiency Virus- Hospital Gregorio Marañón (HIV-HGM) BioBank, Madrid, Spain
- 4Networking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Madrid, Spain
Due to the success of combined antiretroviral therapy (cART) in recent years, the pathological outcome of Human Immunodeficiency Virus type 1 (HIV-1) infection has improved substantially, achieving undetectable viral loads in most cases. Nevertheless, the presence of a viral reservoir formed by latently infected cells results in patients having to maintain treatment for life. In the absence of effective eradication strategies against HIV-1, research efforts are focused on obtaining a cure. One of these approaches is the creation of therapeutic vaccines. In this sense, the most promising one up to now is based on the establishing of the immunological synapse between dendritic cells (DCs) and T lymphocytes (TL). DCs are one of the first cells of the immune system to encounter HIV-1 by acting as antigen presenting cells, bringing about the interaction between innate and adaptive immune responses mediated by TL. Furthermore, TL are the end effector, and their response capacity is essential in the adaptive elimination of cells infected by pathogens. In this review, we summarize the knowledge of the interaction between DCs with TL, as well as the characterization of the specific T-cell response against HIV-1 infection. The use of nanotechnology in the design and improvement of vaccines based on DCs has been researched and presented here with a special emphasis.
Introduction
Human Immunodeficiency Virus type 1 (HIV-1) continues to be a health problem worldwide, particularly in developing countries. Although the proportion of the population infected with the virus has stabilized since 2000, the total number of infected subjects continues to increase. According to UNAIDS data from 2019, 38 million people are living with HIV-1, increasing the number of new infections to 1.7 million people in that year.
The discovery of antiretroviral therapies, especially the cATR regimen, has helped the progression of the infection from a fatal to a chronic disease. Access and adherence to treatment are determining factors for the good medical prognosis of patients with this infection (1). However, both factors can be easily lost due to economic problems, social stigmas, and psychological side effects, especially in less developed countries. Despite deaths related to AIDS having drastically decreased in developed countries, many associated co-morbidities, commonly known as non-AIDS-events, decrease the quality of life of the infected and are the first cause of death in HIV populations, according to the CROI 2020 data. Therefore, a new long-term therapeutic/preventive approach against HIV-1 infection must be found (2).
In the absence of a complete cure for HIV-1 infection, a functional cure appears to be the most promising option (3). The definition of a functional cure states that, although the virus is still present in the host organism and remains latent in the genome of many cells as a reservoir, the immune system keeps the infection under control in the absence of cART (4).
The viral latency, the high mutability, and the variability of this virus make the search for a therapeutic cure extremely difficult. In this sense, the scientific community has spent many years working intensively to achieve this aim through different strategies, including the creation of vaccines against HIV-1. Therapeutic vaccines could contribute to HIV-1 therapy as their application immediately after the infection could limit the size of the virus reservoir and prevent future viral spread (5).
A significant number of vaccine candidates have been tested and failed. The main mechanism of all these vaccines is a delivery of HIV-1 antigenic peptides to antigen presenting cells (APC) to obtain a therapeutic response (6, 7).
Spread of Virus in the Immune System
HIV-1 Entry and Processing Inside Dendritic Cells
HIV-1 is capable of reaching a wide variety of immune cells. In this sense, the contact of the virus with the target TLs is an important mechanism for establishing a persistent infection. However, the first step is the arrival of the virus to APCs, in this case DCs, which will transmit HIV-1 to TLs (8). HIV-1 can use different receptors/binding pathways to infect DCs, including the CCR5 receptor, the CXC4 chemokine receptor (CXCR4), or C lectin type receptors, especially DC-SIGN (dendritic cell-specific ICAM-3 grabbing nonintegrin) that bind to the HIV-1 envelope, specifically, to gp120 (9). Then, the virus can transmit to the target cell or degrade in endosomes by binding to langergin before transmission occurs (10). In addition to this receptor dependent on the binding of gp120, Singlec-1 or CD169 is another receptor independent of binding to this glycoprotein. Singlec-1 is expressed in DCs; specifically it is found within the uropodia of migratory DCs, facilitating the capture and retention of HIV-1 by binding to ganglioside GM3 (11) (Figure 1). Furthermore, Singlec-1 has the quality of being within the compartments of DCs, and TLs can access to them in order to improve the interaction between DCs with TLs. In addition to this glycoprotein-dependent viral capture by DCs, HIV-1 can also be taken up by a lipid-dependent mechanism (12, 13). Moreover, HIV-1 has the ability to incorporate other virus envelope glycoproteins during assembly, a phenomenon known as pseudotyping. This allows HIV-1 to expand its cellular tropism and enables it to infect not only T cells (14).
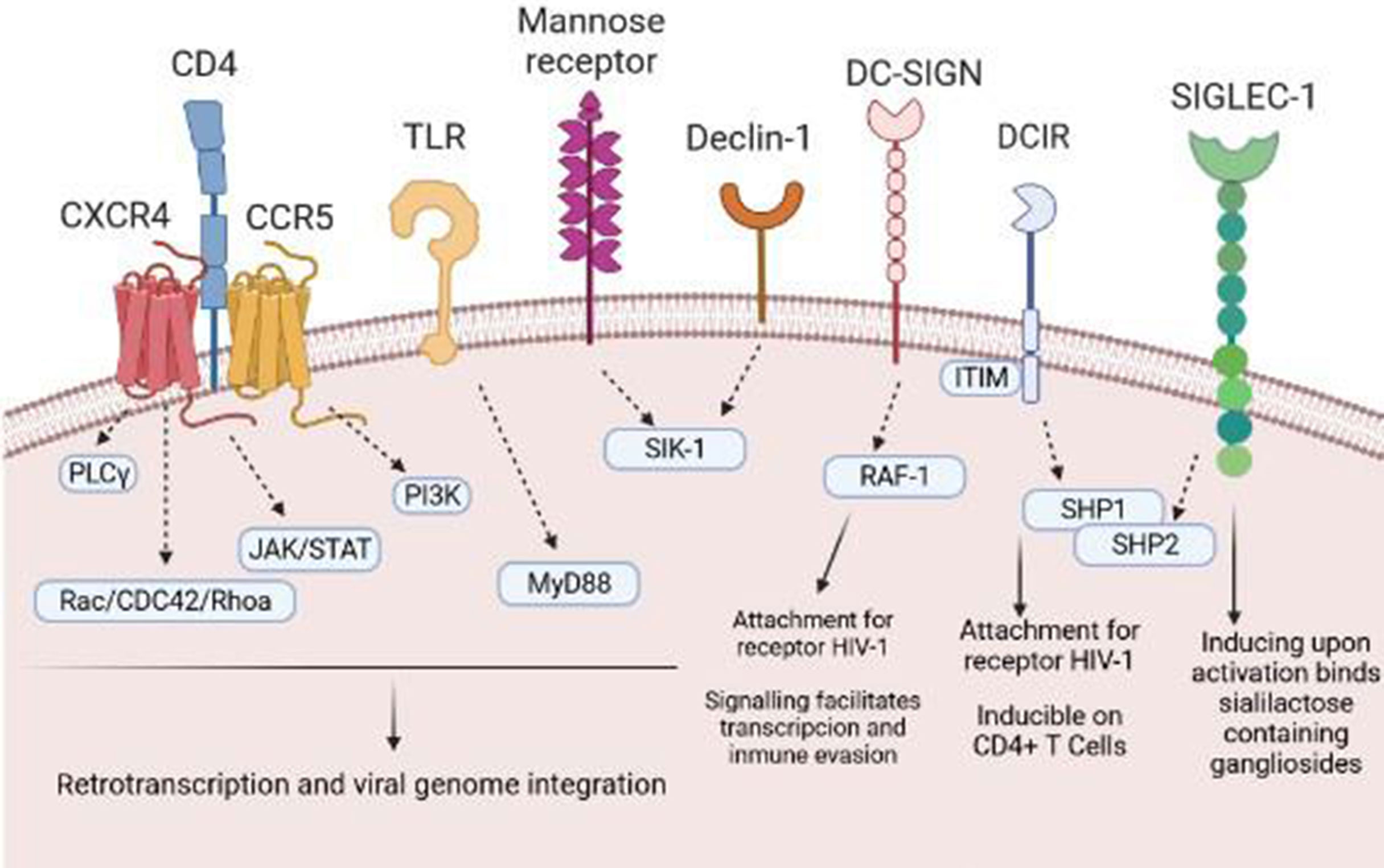
Figure 1 Receptors and pathways involved in the entry of HIV-1 into DCs. HIV-1 binds to several different DC surface receptors, determining the fate of the virus. Generally, HIV-1 is introduced to DCs through endocytosis after binding to DC-SIGN or other receptors, such as Declin-1 or Siglec-1. Binding to these receptors generates a series of intracellular signals that allow transinfection or immunological recognition and the consequent activation of T cells.
The process of interaction between viral particles and DCs generates a series of responses that lead to the maturation and regulation of chemokine receptors, which will help in the migration process to secondary lymphoid organs to initiate the adaptive immune response. DCs have been shown to undergo activation and maturation through different virus-induced mechanisms (15). These pathways of activation and maturation include those mediated by toll like receptor (TLR), the stimulator of the interferon gene (STING) (16), the cyclic pathway of guanosine monophosphate-adenosine monophosphate synthase (cGAS) (17), and the mitochondrial pathway (MAVs) (18). Despite HIV-1 infection, DCs can maintain their ability to migrate to the lymph nodes under the influence of environmental chemotactic factors such as sphingosine 1-phosphate (S1P) and CCL19/21, so DCs expression remains unaltered (19, 20).
Once the DCs reach the lymphoid organs, contact with the T cells begins. Although the molecular mechanisms are still not entirely clear, it is known that DCs capture the antigen by endocytosis, degrade it to peptides after endosome-lysosome fusion, and present it to T cells in the context of MHC (21). The union established between the antigen and MHC, with the collaboration of costimulatory signals on the surface of the DCs, forms an environment known as “immunological synapse”, capable of inducing the specific T response and, therefore, the adaptive immune response (22).
For optimal antigen presentation to T cells, DCs must execute good intracellular processing. However, cells infected with HIV-1 present the antigen through MHC-I, through cross-presentation (23). It is well known that HIV-1 exploits multiple mechanisms to evade immune recognition, including a high mutation rate, glycosylation of the envelope protein gp120, or in this case the virus has the ability to manipulate host antigen presentation and processing mechanisms (24).
Different HIV-1 proteins are involved in modifying these antigen presentation pathways so virus seeks the endosomal pathway, avoiding the lysosomal pathway, becoming a proliferating way for the virus to spread to other places such as the lymph nodes (24, 25). Endosomal processing may provide another opportunity to escape immunological recognition by promoting the destruction of key antigens by endosomal proteases. During cross-presentation, antigens are exposed to endosomal proteases. In this sense, the HIV-1 Nef protein interrupts the presentation of antigens on the cell surface by interfering with the normal trafficking pathway of MHC-I, through the AP-1-mediated signaling. This mechanism reduces the recognition of cytotoxic T lymphocytes and the lysis of infected cells, using an immune escape route (26, 27).
Surface DCs-T Cells Interactions in the Presence of HIV-1
During the immune synapse, T cells are able to probe the surface of the protrusions generated by DCs, inside which there is a large viral load. Due to exploratory contacts, the virus particles produced from these DCs can be transferred to T cells with high efficiency. In this context, a relatively small amount of DCs containing viral particles can generate an increase in T-cell infection, making it exponential (28, 29).The number of interactions established between DCs and TLs, as well as how long they remain in contact, cause T cells differentiation and acquisition of the ability to carry out effector and memory responses. Memory T cells can establish themselves as a reservoir during the chronic phase of viral infection, so that these target cells can cause virus reactivation favoring dissemination (30). A recent study in people with chronic HIV-1 infection has shown that CD8+ T cells not only responded to mutated HIV-1 epitopes which cause death of CD4+ T cells, but also led to increased maturation of cells producing higher transinfection of CD4+ T cells (31). The persistence of mutated viral strains gives HIV-1 an advantage over the immune system.
When T cells establish contact with viral particles embedded within membrane invaginations, a signaling cascade occurs that ends when T cells stop binding DCs. Knowledge of cell dynamics that orchestrate viral shedding from DCs to T cells is still lacking. HIV-1 binds to accessible surface adhesion and signaling receptors, forming a crucial point of contact with T cells. This leads to activation mediated by gp120 and lymphocyte function-associated antigen 1 (LFA-1) that initiates signal transduction by inducing T-cell entrapment and activation (32, 33). The interaction between DCs and T cells allows trapped HIV-1 particles to reach target T cells in a manner similar to that established in DCs, wherein DCs can capture ganglioside-rich exosomes (34). It has been shown that increasing numbers of viruses can bind directly to integrins (35). Particular importance is the adhesive interaction between LFA-1 and ICAM-1 that facilitate virologic synapse and cell-to-cell transmission (36). Blocking the binding between LFA1 and ICAM-1 prevents prolonged contact between DCs and T cells. Therefore, the established binding between gp120 and T cells is necessary for activation of LFA-1 (37). LFA-1 is activated when the binding of the gp120 protein to α4β7 occurs in primary T cells. The virological synapse is closely related to the activation of LFA-1, as well as to the signals that are established in the interaction of gp120 through α4β7 (38, 39). Therefore, the binding between the virus glycoprotein and T cells represents a limiting step in facilitating prolonged contacts of DCs and T cells (40) (Figure 2).
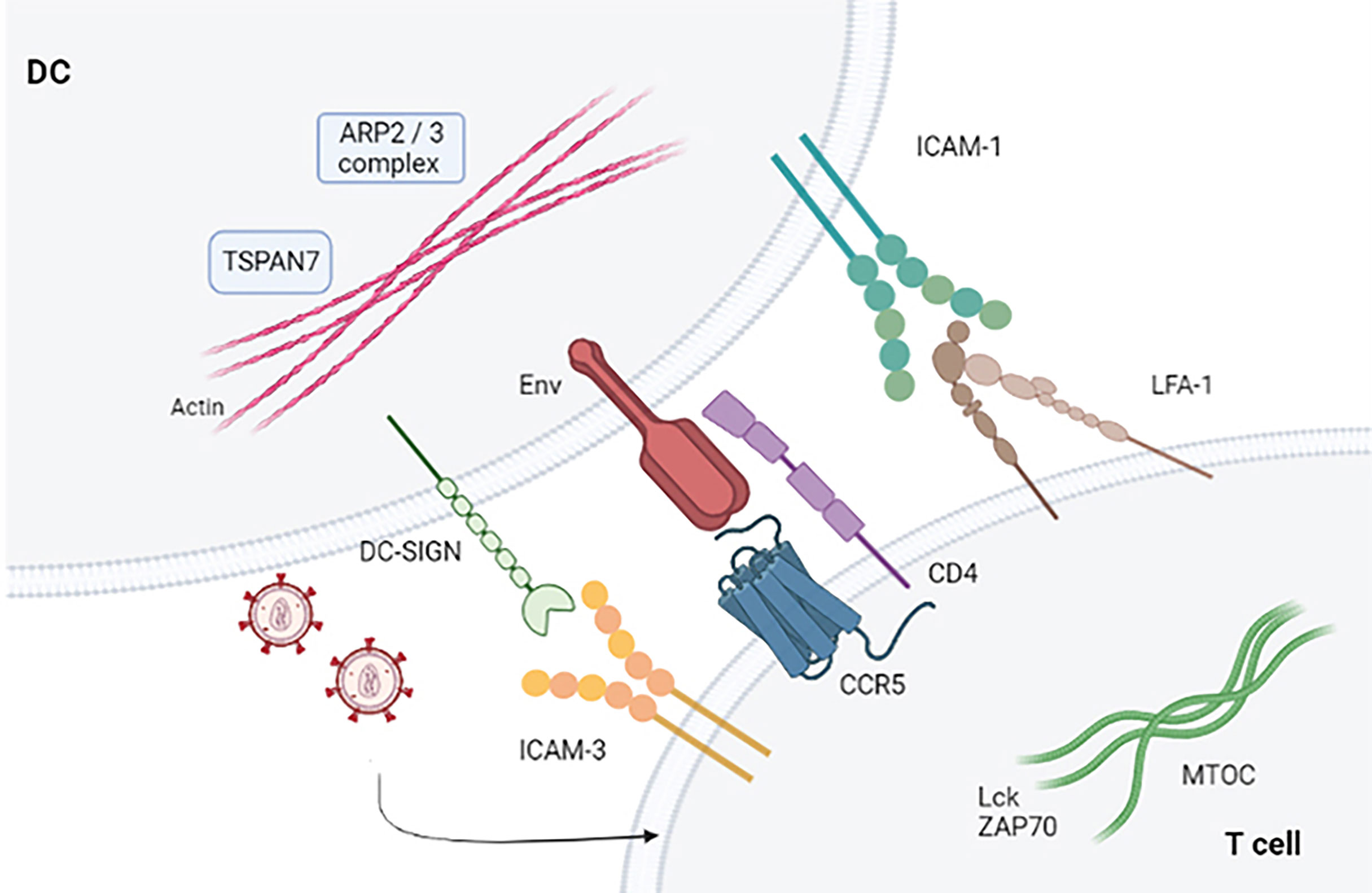
Figure 2 Virological synapse in the presence of HIV-1. Virological synapse in the presence of HIV. The interaction between the different receptors present in both DCs and T cells allows entry into the target cells (CD4+ T and CD8+ T lymphocytes), thus allowing the dissemination of HIV-1 that escapes the immune response mechanism produced by these cells.
The ability to capture and transmit viral particles possessed by DCs and their presentation to T cells at the site of cell-to-cell contact, known as transinfection, promotes systemic release after viral exposure (41, 42).
In addition to the interaction of the virus with the integrins, there is another family of proteins with a very important role in the transinfection process. Tetraspanins family is responsible for regulating intracellular traffic and modulating the function of other helper molecules, controlling their expression in the plasma membrane or classifying themselves into intracellular vesicles. Tetraspanins interact with other members of the superfamily, other transmembrane receptors, lipids, signaling molecules, and cytoskeletal components (43, 44). These proteins attend to the organization of tetraspanin-enriched microdomains (TEM) containing CD9, CD63, CD81, and CD82 in macrophages, DCs, and T cells (45). TEMs regulate the recognition and presentation of Ag and the activation and proliferation of T cells, as well as the extravasation of leukocytes associating with MHC-I and MHC-II on the surface of DCs (46). HIV-1 uses the mechanisms provided by the host cells to its advantage. Individually, CD63 has a dual function (26, 27, 47). On the one hand, it is essential for the replication of HIV-1 together with the CXCR4 and CCR5 receptor on T cells participating in virus entry or reverse transcription, and on the other hand, the expression of CD63 supports mediated fusion by Env during transduction, so it regulates the replication step. However, its activity and function have not yet been fully understood (48). In addition, CD9, CD63, and CD81 are involved in the immunological synapse at virion-enriched budding sites (49). CD81 is important in cross-presentation of antigen by DCs to cytotoxic T cells. In addition, it is capable of increasing the availability of dNTPs and therefore favoring HIV-1 infection thanks to the blocking SAMHD1, a restriction factor that allows DCs to escape productive HIV-1 infection (50). CD9 and CD81 are found in extracellular vesicles as exosomes. Blocking TEMs has previously been shown to result in a significant decrease in exosomal uptake efficiency in DCs (51). Both CD9 and CD81 bind to the cytoskeleton through proteins of the ezrin-radixin-moeisin family (ERM) which bind to actin, a key element in the interaction between DCs and TLs and in propagation of HIV-1 (52, 53). There is a membrane protein from tetraspanin family that, associated with the ARP2/3 complex, is capable of promoting nucleation and stabilizing actin. This protein is TSPAN7. TSPAN7 prevents the viral particles from being internalized, remaining close to the membrane of the DCs, sites rich in actin, favoring the dissemination to T cells (54).
Intracellular Signaling Pathways
The different interactions that are established between protein families during the virological synapse generate an intracellular signaling cascade (55). This activation induces the reorganization of MOTC and T cells polarization in cell-cell contact sites (56). HIV-1 infection of T cells occurs during the acute phase. One question is if virologic synapse favors the transmission of HIV-1 particles to T cells. Recent studies have shown that clustering of the viral Env protein with TLs initiated T cell receptor signaling, enhancing the transmission of HIV-1 between cells (57). Furthermore, signal amplification occurred in these T cells, suggesting that de novo expression of Env in infected T cells was able to initiate further activation signals during cell-cell contacts (58). The Env protein of HIV-1 induces a calcium flux which initiates an intracellular signaling cascade that produces MEK/ERK activation which induces phosphorylation of Lck and partial activation of ZAP-70, a tyrosine kinase protein from SyK family which plays a critical factor in T cell signaling (59).
These tyrosine kinases accomplish an essential function for T cells since they intervene in their maturation and differentiation. The Tec proteins trigger a signaling cascade in which LTK is phosphorylated and as a consequence the mobilization of Ca2 + occurs. Ca2+ involves the mobilization of actin responsible for cytoskeletal reorganization (60). This phosphorylation of LTK occurs after contact of ICAM-1 integrin with the gp120 protein in the presence of HIV-1 (61, 62). However, low levels of LTK activation occur, so the virus prevents actin from performing and reduces intracellular p24 levels. This suggests that the signaling pathway would be related to the transcription of the virus (63). Also, the interaction between the activation of the glycoprotein and the CD4+ T cells activates the PI3K pathway responsible for regulating the migration of T cells and promoting the entry of viral particles after HIV-1 infection (64). Furthermore, ERK phosphorylation is needed for reverse transcription, regulating virus infectivity (65, 66). Signaling is dependent on chemokine receptors, but these signals converge to regulate the behavior and function of T cell migration.
Another very important point at the intracellular signaling level is related to the ability of HIV-1 to spread to other cells. In this context, a little-studied family of proteins, the ESCRT, has great relevance (67). These will form cytosolic complexes establishing themselves as a machinery of remodeling both the cytoplasm and the cell membrane, allowing the viral budding process (68, 69). HIV-1 is able to recruit this machinery upon activation of ALIX and Tsg101 to mediate the budding process (70, 71). Furthermore, it seems that the ESCRT machinery is closely involved with the antigen presenting function of DCs by MHC-II (72). On the one hand, in non-activated DCs, ESCRT is capable of transporting antigens from MHC-II to phagolysosomes. On the other, when DCs have undergone activation, it supports the antigenic presentation process (73, 74).
There is a wide variety of signaling pathways connected and formed by mediators that intervene in multiples cellular processes, and are involved in viral dynamics. The study of these secondary effectors expand diverse treatment design strategies (75). One of the new discoveries that has been recently studied is lymphocyte activation pathway that does not require the recognition and presentation of antigens by the different APCs. This opens the possibility of a new pathway that could act in parallel or not from a canonical one (76).
Thereby, the antigenic presentation process in DCs and the consequent activation of T cells are a true platform to design new therapeutic vaccines. Knowledge of both the signaling pathways and all the participating intermediate molecules is essential to achieve the objective of an effective therapeutic vaccine.
Vaccines Based on DCs
Most DCs vaccines use viral peptides to stimulate autologous DCs and generate a specific cytotoxic T response (77). Accordingly, the protocol to be followed in most cases is based on the extraction and isolation of monocytes from PBMCs treated with GM-CSF and IL-4, deriving these monocytes to immature DCs (iDCs) (78, 79). During the maturation process of these iDCs, antigenic processing occurs, giving rise to monocyte-derived dendritic cells (MoDCs) (80). After that, the adaptive immune response begins with the objective of generating the T cells proliferation. CD4+ T cells will be able to interact with B lymphocytes for the generation of antibodies, while CD8+ T cells will give a cytotoxic response in order to eliminate the infected cells and reduce the viral load, preventing latency. To promote this process, expanding the immune response, MoDCs are capable of secreting a series of inflammatory cytokines such as IL-12p70 and TNFα (15) (Figure 3).
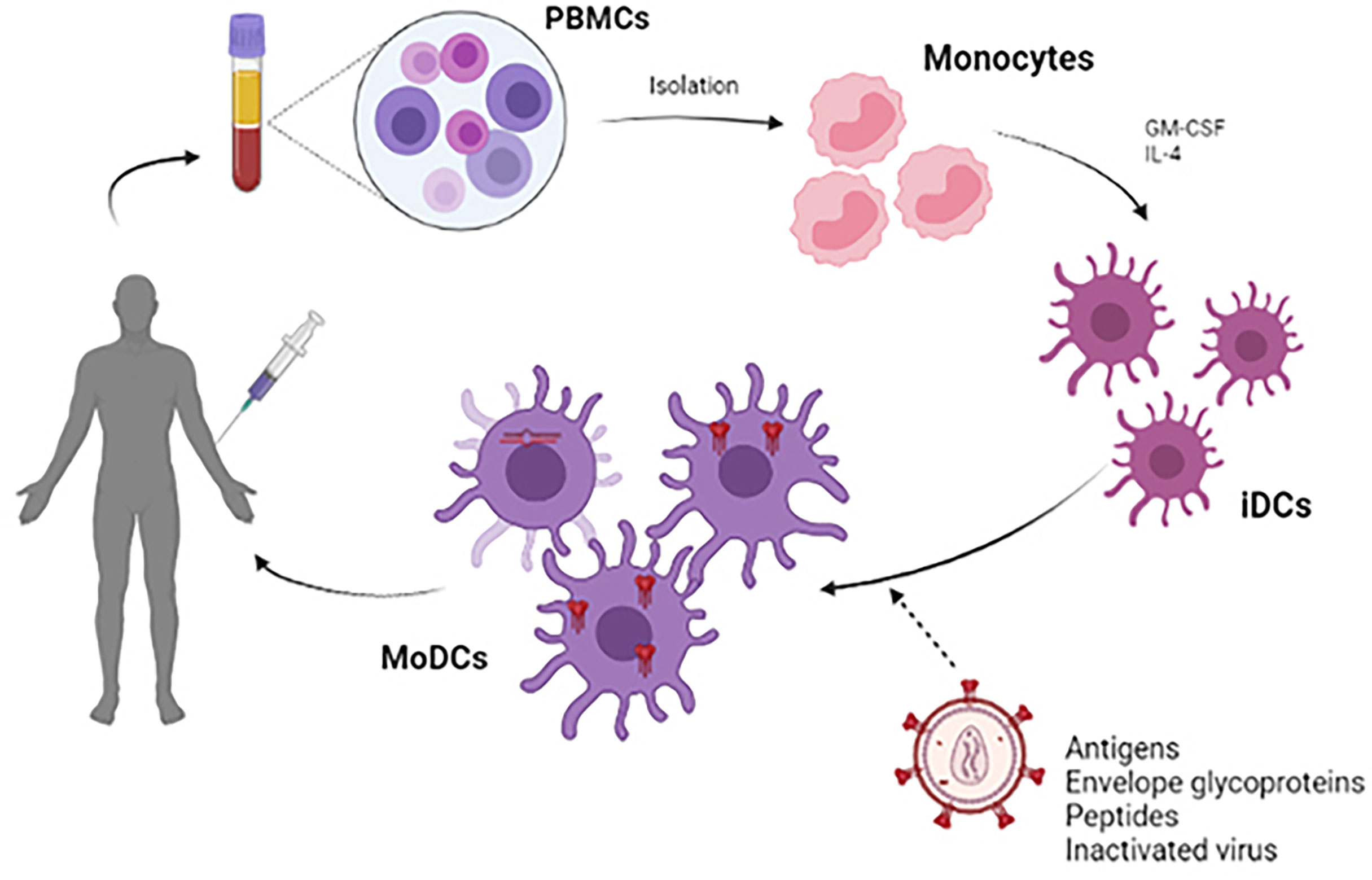
Figure 3 Scheme of the development of a therapeutic vaccine based on DCs. For the development of autologous dendritic cell vaccines, monocytes were extracted from PBMC obtained by leukapheresis from the patient himself. Monocytes are stimulated in vitro with GM-CSF and IL-4 to induce differentiation into immature dendritic cells (iDCs). These iDCs are loaded with HIV-1 derived antigen (antigenic peptides, inactivated whole virus, envelope glycoproteins) and will subsequently become mature antigen-presenting dendritic cells. These MoDCs can be used to formulate a vaccine that is administered to the patient to elicit a specific T-cell response to the HIV-1 antigen.
DCs vaccines have been developed using different antigens such as DNA vectors, recombinant proteins, and even the attenuated whole virus itself (81, 82).
Some of the first attempts at developing DCs vaccines were based on cancer immunotherapy studies. In these studies, nanotechnology has also been used to load proteins or lysates being used for numerous treatments (NCT02334735) (83) (NCT00045968) (84). Thus, some of these studies show the combination of DCs vaccines with treatments such as chemotherapy (NCT03688178, NCT03657966) (85, 86), radiotherapy (NCT03226236) (87), or agents or immunotherapeutic regimens (NCT03546426, NCT03450044) (88–90). All these studies seem to be a breakthrough and have led to the use of these vaccines for other purposes.
Immune Checkpoints in DCs Vaccines
There is a number of molecules at the immune level that act as immune checkpoints so that their function is to negatively regulate immune responses to maintain homeostasis. Nowadays, blocking immune checkpoint molecules is considered a strategy to enhance immune responses in patients. Recently, immunotherapy against cancer has attracted much attention due to the successful clinical application of inhibitors targeting the cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death protein (PD-1)/PD-L1 pathways (91). Studies on cancer vaccines, especially those based on DCs, have made significant progress in recent years. In fact, as mentioned above, the first DCs vaccines were developed as a cancer treatment. In particular, the identification and characterization of the cross-presenting allows effector T cells to mediate immunity against viruses, bacteria, and tumors. In this context, immune checkpoints are often improved during cancer and chronic infections as a mechanism of immune subversion and, therefore, have become a therapeutic target for cancer, but also for the fight against infectious diseases (92).
One of the most important factors that have been studied in recent years is programmed cell death ligand 1 (PDL-1) in DCs and its interaction with PD1 in T lymphocytes (93, 94). This interaction that is established between PDL-1 and PD1 assumes a control of activated T cells. It should be noted that this interaction takes place at two different time points in the life cycle of T cells: First, during the presentation of antigens to naïve T cells for their activation and differentiation, influencing the differentiation pathway of activated T cells leading to cytotoxic, antibody, or regulatory responses. Second, during antigen recognition (95). The PD-1/PD-L1 axis includes the action of effector TL and promotes the depletion of TL, thereby negatively affecting tumor infiltrating lymphocytes in the tumor microenvironment. PD-1 primarily exerts its inhibitory effect on T cells in the periphery where T cells meet PD-1 ligands (96). In cancer, tumor cells and myeloid cells are believed to be the main cell types that mediate T-cell suppression through this junction. In addition, PD-L1 utilizes the STAT3/caspase 7-dependent pathway and directly blocks interferon-γ-mediated cytotoxicity (97). Therefore, blocking the PD-1/PD-L1 axis will have a synergistic therapeutic action when co-administered with DCs vaccine therapy, thus affecting the tumor microenvironment, decreasing IL-10, interferon-γ, and enhancing the function of cytotoxic T lymphocytes (98).
In the case of HIV-1, this interaction between PDL-1 and PD1 has two phases. The first occurs in the early phase of infection where this PD-L1/PD-1 immune checkpoint is low and the CD8 + T cells perform their function by eliminating infected cells. However, in the later, even latent phases of the infection, the expression of PD-L1 is increased by the secretion of different cytokines and acts as a negative feedback system on CD8+ T cells. This circumstance is used by the virus to escape the immune response (99).
The other checkpoint that has been studied is CTLA-4. It was one of the first inhibitory receptors shown to play a role in suppressing T cell responses (100). CTLA-4 is structurally similar to CD28 and binds CD80 and CD86 with a higher affinity than it. In fact, CTLA-4 prevents CD28 binding to CD80/CD86 on DCs, inhibiting the activation of naïve T cells (101). On one hand, CTLA-4 expression by T reg cells serves as a mechanism to suppress excessive T cell responses, while intracellular reservoirs of CTLA-4 prevent tissue damage by T cells spontaneously activated pathogens. In a retrospective study of patients with advanced melanoma whose case progressed after DC vaccine therapy, the addition of ipilimumab (anti-CTLA-4) promoted tumor-specific cytotoxic T-lymphocytic action (102).
Both control points are also fundamental in the case of HIV-1, the use of anti PDL-1, anti PD1, and anti CTLA-4 treatments supposed to improve the immune response that occurs in the T cells and that are fundamental for the good performance of the vaccine based on DCs. However, not all HIV-1 infected patients respond favorably to these inhibitors, possibly due to the lack of immune cells. In addition, therapeutic vaccines have not achieved an adequate CD4+ T response with low antibody levels and poor viral control (103). Perhaps the combination of both, anti-checkpoints and DCs vaccines, can lead to the success of cancer and HIV-1 therapy.
Homing DCs Vaccine
During activation and differentiation into secondary lymphoid organs, T cells integrate TCR and are exposed to different costimulatory and cytokine signals, inducing the generation of memory and effector cells. The local lymphoid microenvironment is in charge of inducing this differentiation, in addition to selecting the specific properties of tissue localization expression on TLs (104). Localization and chemoattractant receptors can be up-regulated in secondary lymphoid organs and undergo selection during recirculation of cells moving through antigen-rich tissues. The ability to target effector lymphocytes to specific tissues helps increase the efficiency of pathogen clearance and prevents pathological inflammation (105).
Responding T cells program into secondary lymphoid organs and acquire these tissue localization phenotypes within two days of activation, before they leave their initial antigen-encounter site (106). Therefore, the possibility that these phenotypes are induced and randomized during successive rounds of recirculation in peripheral antigen-rich tissues is ruled out. The return of lymphocytes from the blood to the tissues is mediated by a series of sequential interactions between the lymphocyte and the vascular endothelium in specialized postcapillary venules (107).
This entire process is influenced by the pathway of entry of the pathogen into the body, which implies a variation in the availability of molecules that can be processed by dendritic cells at different sites in the lymphoid tissue (108). Some of the most widely studied site-specific phenotypes are those found in the cutaneous versus intestinal secondary lymphoid organs during initial T-cell activation. These would include vitamin A and D derivatives produced by DCs that drain the dermis or intestine positively regulate skin and gut localization markers on T cells (109). DCs can metabolize vitamin D3, a compound that is abundantly present in the skin, transforming it into its active form, and this metabolite suppresses the intestinal localization program in T cells at the same time that it induces the expression of the chemokine receptor, CCR10 (110), which allows localization in the skin. In contrast, dendritic cells located in gut-associated lymphoid tissue can convert vitamin A to retinoic acid. The production of retinoic acid by DCs, particularly those that express CD103, imprints intestinal tropism in T cells by inducing the expression of the integrin α4β7 and the chemokine receptor CCR9 (111–113).
Knowing the homing mechanisms that are established between DCs and TLs are essential when administering a possible therapeutic vaccine. The route of administration of antigen-loaded DCs affects the migration of DCs to lymphoid tissues and the magnitude of the antigen-specific TLs response.
Approaches in DCs Vaccines
The design of a therapeutic vaccine has two important points in its development. The first is how viral peptides can be introduced into DCs to be recognized as antigens and processed (114). The second is if the administered peptide is capable of stimulating the specific T response after contact with antigen-presenting DCs (115). Some of the most used strategies with the objective of introducing peptides to be recognized as antigens are mentioned below.
One of the ways that has been used to develop a delivery towards DCs has been to conjugate monoclonal antibodies specific for endocytic receptors of DCs, such as DEC205 and Clec9A, with the antigenic particle (116). DEC-205 is a C-type lectin receptor that acts as a recognition receptor for apoptotic and necrotic cells (117). These molecules have been used as a possible target molecule to selectively deliver antigens to DCs because DEC-205 and Clec9A are known to be highly expressed in DCs. Although DEC-205 and Clec9A are promising surface molecules for the targeted delivery of antigens by DCs, vaccines based on these molecules require adjuvants such as anti-CD40 (118) for efficient induction of CD8+ T cell responses. Some of the conjugates that have been used by this technique are those formed by the HIV-1 protein p24 or gag and the monoclonal antibody DEC-205, generating in both cases a B and T response (119). The conjugate with the Gag HIV-1 protein had better results both in the efficacy for the presentation of the antigen, and in achieving a better humoral response, with higher levels of antibodies, as well as cellular (120).
Another strategy that has been used is the formation of a trimer complex formed by the HIV-1 gp140 protein bound to the CD40 ligand (CD40L), incorporating itself directly into the DCs (121). An example of this chimeric strategy is that which was developed from a DNA vaccine encoding the Env protein of HIV-1 together with plasmids encoding the macrophage inflammatory protein alpha1 (MIP-1α) and the tyrosine ligand kinase 3 (Flt3L) (122, 123). This strategy resulted in the recruitment of DCs at the immunization site and induced the expression and maturation of CD11b, CD80, CD83, and MHC class II markers on DCs. One example is found in a DNA vaccine that encodes HIV-1 gp120 bound to the extracellular domain of Flt3L (124). In this case, the expansion of the DCs was induced, but the CD8+ T response was improved and the production of anti-gp120 antibodies increased, remaining up to 16 weeks after amplification of immunization. In addition, CTLA4 or PD-1 have also been incorporated as an alternative for the formation of these complexes as both are important checkpoints as mentioned before (125).
Another approach in recent years is the use of mRNA that codes for TAT, NEF, and REV, incorporating them into the MoDCs of HIV-1 patients. This technique achieves an improvement in the response of TL, both CD4+T and CD8+ T (126). Through the regulation of costimulatory molecules and cytokines, proteins such as Nef and Vpr can affect certain DCs maturation processes or the activation of T cells. The use of specific peptides in this case improves the immunogenic properties in contrast to the use of complete proteins (127).
There are also other types based on the use of the HIV-1 Gag and Env genes incorporating them into an adenovirus that codes for li (invariant chain), ces, rev, vif, and vpr (128). The invariant chain (Ii/CD74) has been identified as a surface receptor for migration inhibitory factor (MIFα). Most cells expressing Ii also synthesize major histocompatibility complex class II (MHC II) molecules, which depend on Ii as a companion and targeting factor. The membrane association of Ii-MHC II complexes allows MIF to target Ii-MHC II to antigen-clustered B-cell receptors (BCRs) and promote BCR-driven signaling and intracellular trafficking. The peptides that will bind to MHC-II are mainly of exogenous origin and are captured by endocytosis to be targeted to lysosomes (129).
In addition, there are also other types of vaccines like DNA vaccines (130, 131) or VLV vaccines (virus like vaccines) (132). Clinical trials have also shown that the current generation of DNA vaccines cannot induce a strong antibody response. Therefore, targeting antigen presentation in the MHC-II pathway to activate CD4 + Т cells seems especially advantageous (133).
However, these approaches are based on the use of the immune mechanisms offered by DCs in prophylactic therapy. DCs are used as intermediaries to generate an immune response, and recent improvements in this area consist in delivering immunogens towards these cells, facilitating their antigen capture and presentation functions through different strategies. Nevertheless, if we focus on the therapeutic use of vaccines based on DCs, in recent years, the use of therapeutic vaccines has been studied with the objective of inducing broad immune responses instead of specific responses of a single antigen to combat viral escape mutants and suppress viral rebound. In this sense, the studies that have been carried out on therapeutic vaccines based on DCs are mentioned below.
AGS-004, was one of the phase IIb trials based on autologous DCs co-electroporated with patient-derived HIV-1 RNA encoding three or four HIV-1 antigens and also CD40L. Although it seemed successful as there was an induction of the CD8+ T response, it was observed that there was no detectable antiviral effect compared to placebo (134). The next trial reached phase I and II using an autologous DCs HIV-1 ApB vaccine. For this, delivery was made with autologous apoptotic cells infected with HIV-1. Although it maintained a good safety profile and produced activation and lysis of infected cells, it did not prevent viral rebound during treatment interruption (135).
A vaccine (DCV-2) composed of autologous myeloid dendritic cells with high doses of heat-inactivated autologous HIV-1 was studied. In those patients in an initial phase of the infection, a reduction in plasma viral load was achieved along with a good T-cell response (136). This suggests that vaccines based on DCs could be used in newly infected patients with a greater chance of success. There is another study completed to phase I in which DCs were used for the delivery of HIV-1 lipopeptides. Despite the polyfunctional response, a virus rebound was observed after 14 days (137).
These studies have not achieved a very successful result, so other alternatives therapeutic vaccines continue to be developed. One of the latest advances is the ALVAC-HIV vaccine that combines the Nef, Pol, Env, and Gag genes of HIV-1 with LIPO-6T (tetanus toxoid class II-restricted universal CD4 epitope combined with 2 Gag, 2 Nef, and 1 Pol peptide) (138, 139). The novelty included in this vaccine is the incorporation of a lipid tail in the C-terminal region that favors the antigenic presentation function (140). In fact, there is a trial of immunized cART-treated HIV+ patients with autologous DCs enriched by ex vivo culture with GM-CSF and IFN-α and loaded with LIPO5 (2 Nef, 2 Gag, and 1 Pol lipopeptides) (141).
Despite all efforts, effective treatment is still not available with this type of vaccine. This is where nanotechnology plays a very important and advantageous role, both from a prophylactic and therapeutic treatment.
Nanoparticles in DCs Vaccines
One of the most used approaches in recent years is based on the use of nanoparticles as delivery agents. The size of these nanosystems is an advantage in drug delivery due to their ability to cross physiological barriers, reaching specific cells or intracellular compartments (142). The unique nanodimensional size, symmetrical shape, uniformity, and stable structure of the assembled nanoparticles, which closely resemble native viruses, are all advantages. Notably, smaller nanoparticles (25–40 nm in size) penetrate tissue barriers and traffic to the draining lymph nodes more rapidly than larger nanoparticles (greater than 100 nm in size), which are typically retained by cells at the site of injection and which need to be taken up and trafficked by dendritic cells (DCs) to facilitate their transport to the lymph nodes (143). From this perspective, numerous studies have been carried out in which nanoparticles have been used for the release of small molecules, proteins, or DNA, targeting them specifically to target cells, in this case DCs. Nanoparticles (NPs) are an extraordinary tool to provide different compounds such as DNA, siRNA, and peptides for different cells types (144). Several types of nanoparticles (NPs) such as gold, carbon, dendrimers, polymers, and liposomes nanoparticles have the ability to generate the production of cytokines and antibodies (145). Some of these particles that have been used for these purposes are inorganic NPs (iron and silica) (146), polymeric NPs (chitosan, PLGA, PVPONAlk, γ-PGA) (147), liposomes (cholesterol and lipids) (148, 149), virus-like particles (VLP) (150), and dendrimers (151, 152) (Figure 4).
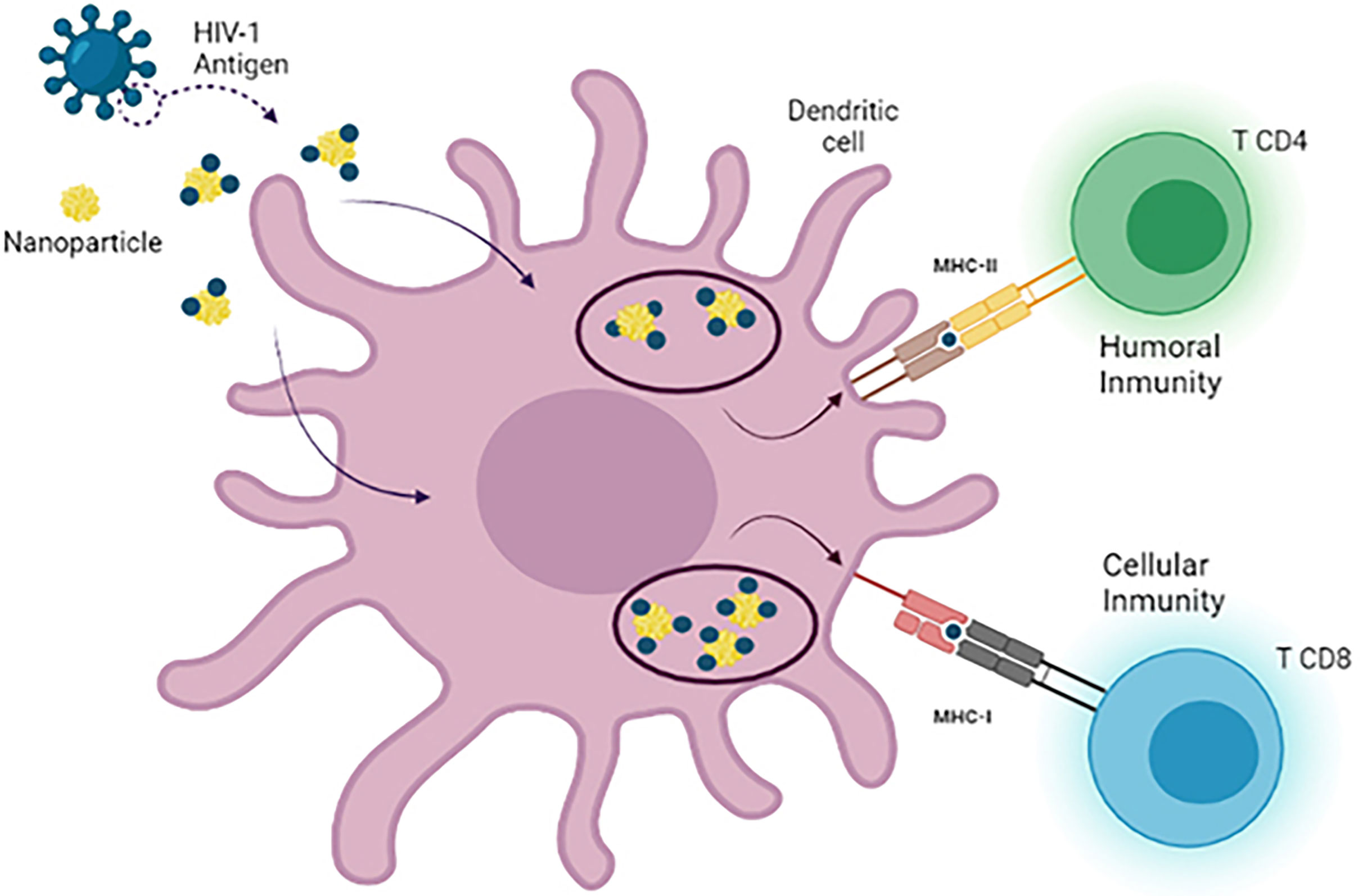
Figure 4 Use of nanotechnology as a delivery system in dendritic cell vaccines. There are different types of nanoparticles that can be used as nanocarriers for antigen delivery in DC-based immunotherapies against HIV-1. During the development of these vaccines, MoDCs are loaded with antigen-nanoparticle complexes that enhance antigen uptake by DCs, leading to the generation of a more specific and potent CD4+ T cell and CD8+ T cell response after reinjection of DCs.
DCs need the presence of an adjuvant for their correct activation and maturation. One of the advantages of NPs is that they can encapsulate both the antigen and the adjuvant, thus ensuring the release of both and favoring the establishment of an effective immune response (153). Fortunately, nanotechnology allows new forms of delivery to DCs. For example, studies using liposome-encapsulated HIV-1 gp160 or gp41 proteins or HIV-1 gag p24 protein coated on colloidal biodegradable polylactic acid (poly-d, llactide) could cause activation of MoDCs in patients of HIV-1 and induce specific cytotoxic CD8+ T responses, CD4+ T cell proliferation, and cytokine secretion (154). Polymeric NPs have been highlighted for their use in the development of therapeutic vaccines due to their excellent physico-chemical properties. These particles, in addition to having good safety and biocompatibility profiles, are biodegradable and capable of adjusting on their surface according to requests (155). The most commonly used polymeric NPs for vaccine administration are poly (lactic-co-glycolic acid; PLGA) or poly (lactic acid; PLA) (156). Other polymers of natural origin, such as inulins, alginate, and chitosan, have been used as adjuvants (157, 158).
There is a recent study related to the use of gold nanoparticles in vivo that are capable of transporting mannose oligosaccharides that are linked to HIV-1 peptides (159, 160). In this study, the use of these nanostructures used as peptide delivery was demonstrated; in comparison with antigenic peptides alone, they improved the CD4+ T cells and CD8+ T cells response and increased cytokine secretion (161). It seems that the adjuvant mechanism of these mannose ligands is through their binding to type C lectin receptors, such as DC-SIGN, present on DCs (162). As a result, a topical vaccine was developed in the form of a transdermal patch (DermaVir) based on polyethyleneimine mannose (Man-PEI), glucose and a DNA plasmid that encodes the HIV-1 antigen formulated in 100 nms nanoparticles and has reached phase II of clinical trials (163).
Regulating the processing of intracellular antigens is important for antigen presentation and subsequent priming of T cells. DCs have limited lysosomal capacity due to decreased proteolytic degradation, being slower than other APCs (164). The moderate lysosomal capacity of DCs favors the presentation of antigens and gives the opportunity to study another modulation pathway to enhance the degradation and presentation of antigens. Thanks to this mechanism of autophagy, intracellular degradation, and elimination of unneeded or dysfunctional components, the adequate presentation of antigens by DCs occurs, leading to an effective T response. An example is found in the covalent bond between Beclin1, autophagy-inducing peptide, OVA257-264 antigen peptide, and a polymer (165). This nanoconjugate represented an improvement in the induction of autophagy, increasing the efficiency of antigen presentation and the activation of T cells.
One of the nanoparticles that have been used for the design of vaccines are those based on lipids. This can enhance the stimulation of the host’s immune response. Until now, the lipid formulations have been formulated as a unilamellar or multilamellar structure composed by biodegradable phospholipids (phosphatidylserine, phosphatidylcholine, and cholesterol) (166). The mechanism by which these liposomes deliver vaccines is through membrane binding causing internalization of lipid vesicles by DCs (167). The studies that have been carried had lipid particles have been used with those in which phosphatidylserine is conjugated with virus peptides (168).
Other methods, such as the use of dendrimers, have facilitated delivery to dendritic cells. Dendrimers are branched structures that have multiple functional groups at the ends of the terminal branches (169). Thanks to this type of structure, the dendrimer is capable of generating multiple vaccines. A single dendrimer is capable of delivering several components or antigens to the target cell (170). The stimulation capacity of these nanoparticles must be verified to avoid inducing tolerance of the cells (171). Dendrimers, like other nanoparticles, must be capable of inducing the absorption of antigenic peptides by MoDCs and stimulating the immune response.
One of the dendrimeric structures that has been used in order to generate a therapeutic vaccine is the one formed by the glycodendrimers. In a recent study it was shown that these nanostructures can be used for the incorporation of HIV-1 peptides, and although there did not seem to be evidence of cytotoxicity, the glycodendrimers did not modify both the phenotype and some functions of the MoDCs such as the migration capacity and the cytokine profile (172). This is the first study that used a structure formed by the peptide-dendrimer junction forming complexes that are functionalized with maltose as a possible candidate for a DC-based vaccine capable of stimulating the immune system (173). In fact, studies have shown that the presence of maltose and maltosylated macromolecules are capable of binding to langerin, so that cells could potentially be activated by maltose when these nanoparticles are transported by langerin or DC-SIGN due to high structural similarity between them (174).
Another recent study uses G4-70/30 PAMAM polycationic dendrimer and AMC6 nanoparticle derived from β-cyclodextrin as peptide delivery agents (175). In this work, an antigenic peptide derived from the HIV-1 gag p24 protein was used in combination with these polycationic structures, binding to specific receptors. In the case of the G4-70/30 PAMAM dendrimer through mannose (MMR, DC-SIGN), it was linked to clathrin vehicleization, while the cyclodextrin structure could be clathrin-dependent or non-dependent (176). In case of this study, the efficacy of both structures as antigenic carriers was demonstrated, improving the presenting function of DCs and producing the release of pro-inflammatory cytokines IL-2 and TNF-α related to the activation of T cells.
Summary
HIV-1 infection involves a loss of immune system cells as a consequence of intense immune stimulation that leads to a prolonged and sustained inflammatory state that involves chronic immune activation (177). Controlling this disease state caused by the virus through the use of immunotherapy is a challenge. Therefore, DCs play a major role in the search for a functional vaccine or cure against HIV-1. Their role in the adaptive response, being responsible for the preparation of naïve T cells is a key point, and their manipulation opens the way to new forms of immunotherapy (28). In this sense, the first phase I and II trials in cancer therapy have generated encouraging results (178). Therefore, DCs are also promising candidates in the search for a functional HIV-1 cure or vaccine (69). The objective of the development of a therapeutic vaccine is to induce strong and broad specific immune responses against HIV-1 antigens and that these are capable of resisting viral escape due to mutation in the virus (179). The arsenals available to elicit such responses include DCs as critical mediators of innate and adaptive immunity, as well as proteins, nucleic acids, and viral vector vaccines (180).
Despite all the knowledge about the molecular and physiological mechanisms of both DCs and HIV-1 infection, it is still not known exactly how the response occurs, so more research is needed (181). DCs play a key role due to their ability to serve as a link between innate and adaptive immunity, including in the presence of HIV-1 infection. Knowing in depth the behavior of these cells would allow the design of modulating immune systems that function at different levels, either with a prophylactic or therapeutic purpose (69). To benefit from advantages offered by the field of nanomedicine, different mechanisms have been developed to improve activation by increasing the therapeutic efficacy of DC-based vaccines. Nanotechnology allows DCs activation in vivo and simulate natural antigen presentation, favoring lysosomal proteolysis for antigen presentation (182). Additionally, nanotechnology can further intervene in the presentation of antigens, acting directly on the APCs or serving as an adjuvant, improving the efficacy of immunotherapy based on DCs (183). One of the problems with conventional treatments based on DCs is that during the migration of these to the lymph nodes, these therapies can lose activity, being unable to interact with T cells to elicit a better specific response. However, nanoparticles can accumulate in the lymph nodes, which contributes to a high efficiency of lymph node drainage (184). Therefore, nanotechnology offers a promising therapeutic alternative that intervenes in the development of multiple disciplines, signifying great progress in clinical treatment (144).
Despite the fact that an increasing variety of nanoparticles are being directed towards the objective of providing a mechanism to achieve a potent immune response based on the use of DCs, additional studies in the field of modulation of these cells, antigen presentation, and DC-HIV-1 interaction are essential to determinate the foundations for the much desired functional cure of HIV-1 (185).
Author Contributions
ME-B wrote the initial draft. MM-F edited and corrected the draft, and provided funding. All authors contributed to the article and approved the submitted version.
Funding
This work has been (partially) supported by the RD16/0025/0019 projects as part of Acción Estratégica en Salud, Plan Nacional de Investigación Científica, and Desarrollo e Innovación Tecnológica (2020-2022) and co-financed by Instituto de Salud Carlos III (Subdirección General de Evaluación), Fondo Europeo de Desarrollo Regional (FEDER), RETIC PT17/0015/0042, and Fondo de Investigación Sanitaria (FIS) 2020-2022 (grant number P119/01638) and EPIICAL Project. This work has been supported partially by a EUROPARTNER: Strengthening and spreading international partnership activities of the Faculty of Biology and Environmental Protection for interdisciplinary research and innovation of the University of Lodz Programme: NAWA International Academic Partnership Programme. This article/publication is based upon work from COST Action CA 17140 “Cancer Nanomedicine from the Bench to the Bedside” supported by COST (European Cooperation in Science and Technology) 2018-2022
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge Dra. Laura Tarancón Díez and Dra. Elena Vázquez Alejo for their help and support in the implementation of this review.
Abbreviations
Ce, conserved element; Vif, viral infectivity factor; Vpr, viral protein R; li, MHC class II-associated invariant chain; Rev, regulator of expression of virion proteins; Nef, negative factor; Tat, regulatory protein HIV-1; PLGA, poly (lactic-co-glycolic acid); PVPONAlk, N-vinyl pyrrolidone-ran-propargyl acrylate; γ-PGA, Poly-γ-glutamic acid; Man-PEI, polyethyleneimine mannose; PAMAN, Poly-amidoamine; MMR, manose receptor.
References
1. Iacob SA, Iacob DG, Jugulete G. Improving the Adherence to Antiretroviral Therapy, a Difficult But Essential Task for a Successful HIV Treatment-Clinical Points of View and Practical Considerations. Front Pharmacol (2017) 8:831. doi: 10.3389/fphar.2017.00831
2. Tseng A, Seet J, Phillips EJ. The Evolution of Three Decades of Antiretroviral Therapy: Challenges, Triumphs and the Promise of the Future. Br J Clin Pharmacol (2015) 79(2):182–94. doi: 10.1111/bcp.12403
3. Gunthard HF, Saag MS, Benson CA, del Rio C, Eron JJ, Gallant JE, et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2016 Recommendations of the International Antiviral Society-USA Panel. JAMA (2016) 316(2):191–210. doi: 10.1001/jama.2016.8900
4. Bailon L, Mothe B, Berman L, Brander C. Novel Approaches Towards a Functional Cure of HIV/AIDS. Drugs (2020) 80(9):859–68. doi: 10.1007/s40265-020-01322-y
5. Schwarzer R, Gramatica A, Greene WC. Reduce and Control: A Combinatorial Strategy for Achieving Sustained HIV Remissions in the Absence of Antiretroviral Therapy. Viruses (2020) 12(2):188. doi: 10.3390/v12020188
6. Mohan T, Verma P, Rao DN. Novel Adjuvants & Delivery Vehicles for Vaccines Development: A Road Ahead. Indian J Med Res (2013) 138(5):779–95.
7. Moyer TJ, Zmolek AC, Irvine DJ. Beyond Antigens and Adjuvants: Formulating Future Vaccines. J Clin Invest (2016) 126(3):799–808. doi: 10.1172/JCI81083
8. Iwasaki A. Innate Immune Recognition of HIV-1. Immunity (2012) 37(3):389–98. doi: 10.1016/j.immuni.2012.08.011
9. Lin CL, Sewell AK, Gao GF, Whelan KT, Phillips RE, Austyn JM. Macrophage-Tropic HIV Induces and Exploits Dendritic Cell Chemotaxis. J Exp Med (2000) 192(4):587–94. doi: 10.1084/jem.192.4.587
10. Wilen CB, Tilton JC, Doms RW. HIV: Cell Binding and Entry. Cold Spring Harb Perspect Med (2012) 2(8):a006866. doi: 10.1101/cshperspect.a006866
11. Izquierdo-Useros N, Lorizate M, McLaren PJ, Telenti A, Krausslich HG, Martinez-Picado J. HIV-1 Capture and Transmission by Dendritic Cells: The Role of Viral Glycolipids and the Cellular Receptor Siglec-1. PloS Pathog (2014) 10(7):e1004146. doi: 10.1371/journal.ppat.1004146
12. Bergantz L, Subra F, Deprez E, Delelis O, Richetta C. Interplay Between Intrinsic and Innate Immunity During HIV Infection. Cells (2019) 8(8):922. doi: 10.3390/cells8080922
13. Wan D, Jiang W, Hao J. Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front Immunol (2020) 11:615. doi: 10.3389/fimmu.2020.00615
14. Tang Y, George AM, Petrechko O, Nouvet FJ, Sweet SD, Tanaka Y, et al. Pseudotyping of HIV-1 With Human T-Lymphotropic Virus 1 (HTLV-1) Envelope Glycoprotein During HIV-1-HTLV-1 Coinfection Facilitates Direct HIV-1 Infection of Female Genital Epithelial Cells: Implications for Sexual Transmission of HIV-1. mSphere (2018) 3(2):e00038–18. doi: 10.1128/mSphere.00038-18
15. Soto JA, Galvez NMS, Andrade CA, Pacheco GA, Bohmwald K, Berrios RV, et al. The Role of Dendritic Cells During Infections Caused by Highly Prevalent Viruses. Front Immunol (2020) 11:1513. doi: 10.3389/fimmu.2020.01513
16. Corrales L, Gajewski TF. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer. Clin Cancer Res (2015) 21(21):4774–9. doi: 10.1158/1078-0432.CCR-15-1362
17. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
18. Kumar S, Sunagar R, Gosselin E. Bacterial Protein Toll-Like-Receptor Agonists: A Novel Perspective on Vaccine Adjuvants. Front Immunol (2019) 10:1144. doi: 10.3389/fimmu.2019.01144
19. Lee C, Lee M, Rhee I. Distinct Features of Dendritic Cell-Based Immunotherapy as Cancer Vaccines. Clin Exp Vaccine Res (2018) 7(1):16–23. doi: 10.7774/cevr.2018.7.1.16
20. Ricart BG, John B, Lee D, Hunter CA, Hammer DA. Dendritic Cells Distinguish Individual Chemokine Signals Through CCR7 and CXCR4. J Immunol (2011) 186(1):53–61. doi: 10.4049/jimmunol.1002358
21. Gutierrez-Martinez E, Planes R, Anselmi G, Reynolds M, Menezes S, Adiko AC, et al. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Front Immunol (2015) 6:363. doi: 10.3389/fimmu.2015.00363
22. Embgenbroich M, Burgdorf S. Current Concepts of Antigen Cross-Presentation. Front Immunol (2018) 9:1643. doi: 10.3389/fimmu.2018.01643
23. Boucau J, Le Gall S. Antigen Processing and Presentation in HIV Infection. Mol Immunol (2019) 113:67–74. doi: 10.1016/j.molimm.2018.03.027
24. Frey BF, Jiang J, Sui Y, Boyd LF, Yu B, Tatsuno G, et al. Effects of Cross-Presentation, Antigen Processing, and Peptide Binding in HIV Evasion of T Cell Immunity. J Immunol (2018) 200(5):1853–64. doi: 10.4049/jimmunol.1701523
25. Cruz FM, Colbert JD, Merino E, Kriegsman BA, Rock KL. The Biology and Underlying Mechanisms of Cross-Presentation of Exogenous Antigens on MHC-I Molecules. Annu Rev Immunol (2017) 35:149–76. doi: 10.1146/annurev-immunol-041015-055254
26. Yang Y, Liu W, Hu D, Su R, Ji M, Huang Y, et al. HIV-1 Nef Interacts With LMP7 To Attenuate Immunoproteasome Formation and Major Histocompatibility Complex Class I Antigen Presentation. mBio (2020) 11(5):e02221–19. doi: 10.1128/mBio.02221-19
27. Roeth JF, Williams M, Kasper MR, Filzen TM, Collins KL. HIV-1 Nef Disrupts MHC-I Trafficking by Recruiting AP-1 to the MHC-I Cytoplasmic Tail. J Cell Biol (2004) 167(5):903–13. doi: 10.1083/jcb.200407031
28. Garcia F, Plana M, Climent N, Leon A, Gatell JM, Gallart T. Dendritic Cell Based Vaccines for HIV Infection: The Way Ahead. Hum Vaccin Immunother (2013) 9(11):2445–52. doi: 10.4161/hv.25876
29. Koh WH, Lopez P, Ajibola O, Parvarchian R, Mohammad U, Hnatiuk R, et al. HIV-Captured DCs Regulate T Cell Migration and Cell-Cell Contact Dynamics to Enhance Viral Spread. iScience (2020) 23(8):101427. doi: 10.1016/j.isci.2020.101427
30. Jin JH, Huang HH, Zhou MJ, Li J, Hu W, Huang L, et al. Virtual Memory CD8+ T Cells Restrain the Viral Reservoir in HIV-1-Infected Patients With Antiretroviral Therapy Through Derepressing KIR-Mediated Inhibition. Cell Mol Immunol (2020) 17(12):1257–65. doi: 10.1038/s41423-020-0408-9
31. Vajpayee M, Negi N, Kurapati S. The Enduring Tale of T Cells in HIV Immunopathogenesis. Indian J Med Res (2013) 138(5):682–99.
32. Capece T, Walling BL, Lim K, Kim KD, Bae S, Chung HL, et al. A Novel Intracellular Pool of LFA-1 Is Critical for Asymmetric CD8(+) T Cell Activation and Differentiation. J Cell Biol (2017) 216(11):3817–29. doi: 10.1083/jcb.201609072
33. Verma NK, Fazil MH, Ong ST, Chalasani ML, Low JH, Kottaiswamy A, et al. LFA-1/ICAM-1 Ligation in Human T Cells Promotes Th1 Polarization Through a GSK3beta Signaling-Dependent Notch Pathway. J Immunol (2016) 197(1):108–18. doi: 10.4049/jimmunol.1501264
34. Chistiakov DA, Grechko AV, Orekhov AN, Bobryshev YV. An Immunoregulatory Role of Dendritic Cell-Derived Exosomes Versus HIV-1 Infection: Take It Easy But Be Warned. Ann Transl Med (2017) 5(17):362. doi: 10.21037/atm.2017.06.34
35. Finke J, Mikulicic S, Loster AL, Gawlitza A, Florin L, Lang T. Anatomy of a Viral Entry Platform Differentially Functionalized by Integrins Alpha3 and Alpha6. Sci Rep (2020) 10(1):5356. doi: 10.1038/s41598-020-62202-9
36. Starling S, Jolly C. LFA-1 Engagement Triggers T Cell Polarization at the HIV-1 Virological Synapse. J Virol (2016) 90(21):9841–54. doi: 10.1128/JVI.01152-16
37. Verma NK, Kelleher D. Not Just an Adhesion Molecule: LFA-1 Contact Tunes the T Lymphocyte Program. J Immunol (2017) 199(4):1213–21. doi: 10.4049/jimmunol.1700495
38. Cicala C, Arthos J, Fauci AS. HIV-1 Envelope, Integrins and Co-Receptor Use in Mucosal Transmission of HIV. J Transl Med (2011) 9 Suppl 1:S2. doi: 10.1186/1479-5876-9-S1-S2
39. Hioe CE, Tuen M, Vasiliver-Shamis G, Alvarez Y, Prins KC, Banerjee S, et al. HIV Envelope Gp120 Activates LFA-1 on CD4 T-Lymphocytes and Increases Cell Susceptibility to LFA-1-Targeting Leukotoxin (LtxA). PloS One (2011) 6(8):e23202. doi: 10.1371/journal.pone.0023202
40. Jelicic K, Cimbro R, Nawaz F, Huang da W, Zheng X, Yang J, et al. The HIV-1 Envelope Protein Gp120 Impairs B Cell Proliferation by Inducing TGF-Beta1 Production and FcRL4 Expression. Nat Immunol (2013) 14(12):1256–65. doi: 10.1038/ni.2746
41. Manches O, Frleta D, Bhardwaj N. Dendritic Cells in Progression and Pathology of HIV Infection. Trends Immunol (2014) 35(3):114–22. doi: 10.1016/j.it.2013.10.003
42. Martin-Gayo E, Buzon MJ, Ouyang Z, Hickman T, Cronin J, Pimenova D, et al. Potent Cell-Intrinsic Immune Responses in Dendritic Cells Facilitate HIV-1-Specific T Cell Immunity in HIV-1 Elite Controllers. PloS Pathog (2015) 11(6):e1004930. doi: 10.1371/journal.ppat.1004930
43. Jankovicova J, Secova P, Michalkova K, Antalikova J. Tetraspanins, More Than Markers of Extracellular Vesicles in Reproduction. Int J Mol Sci (2020) 21(20):7568. doi: 10.3390/ijms21207568
44. Termini CM, Gillette JM. Tetraspanins Function as Regulators of Cellular Signaling. Front Cell Dev Biol (2017) 5:34. doi: 10.3389/fcell.2017.00034
45. Rocha-Perugini V, Sanchez-Madrid F, Martinez Del Hoyo G. Function and Dynamics of Tetraspanins During Antigen Recognition and Immunological Synapse Formation. Front Immunol (2015) 6:653. doi: 10.3389/fimmu.2015.00653
46. Saiz ML, Rocha-Perugini V, Sanchez-Madrid F. Tetraspanins as Organizers of Antigen-Presenting Cell Function. Front Immunol (2018) 9:1074. doi: 10.3389/fimmu.2018.01074
47. Thali M. The Roles of Tetraspanins in HIV-1 Replication. Curr Top Microbiol Immunol (2009) 339:85–102. doi: 10.1007/978-3-642-02175-6_5
48. Nydegger S, Khurana S, Krementsov DN, Foti M, Thali M. Mapping of Tetraspanin-Enriched Microdomains That Can Function as Gateways for HIV-1. J Cell Biol (2006) 173(5):795–807. doi: 10.1083/jcb.200508165
49. Suarez H, Rocha-Perugini V, Alvarez S, Yanez-Mo M. Tetraspanins, Another Piece in the HIV-1 Replication Puzzle. Front Immunol (2018) 9:1811. doi: 10.3389/fimmu.2018.01811
50. Su B, Biedma ME, Lederle A, Peressin M, Lambotin M, Proust A, et al. Dendritic Cell-Lymphocyte Cross Talk Downregulates Host Restriction Factor SAMHD1 and Stimulates HIV-1 Replication in Dendritic Cells. J Virol (2014) 88(9):5109–21. doi: 10.1128/JVI.03057-13
51. Sims B, Farrow AL, Williams SD, Bansal A, Krendelchtchikov A, Matthews QL. Tetraspanin Blockage Reduces Exosome-Mediated HIV-1 Entry. Arch Virol (2018) 163(6):1683–9. doi: 10.1007/s00705-018-3737-6
52. Rocha-Perugini V, Martinez Del Hoyo G, Gonzalez-Granado JM, Ramirez-Huesca M, Zorita V, Rubinstein E, et al. CD9 Regulates Major Histocompatibility Complex Class II Trafficking in Monocyte-Derived Dendritic Cells. Mol Cell Biol (2017) 37(15):e00202–17. doi: 10.1128/MCB.00202-17
53. Rocha-Perugini V, Suarez H, Alvarez S, Lopez-Martin S, Lenzi GM, Vences-Catalan F, et al. CD81 Association With SAMHD1 Enhances HIV-1 Reverse Transcription by Increasing dNTP Levels. Nat Microbiol (2017) 2(11):1513–22. doi: 10.1038/s41564-017-0019-0
54. Menager MM. TSPAN7, Effector of Actin Nucleation Required for Dendritic Cell-Mediated Transfer of HIV-1 to T Cells. Biochem Soc Trans (2017) 45(3):703–8. doi: 10.1042/BST20160439
55. Vasiliver-Shamis G, Dustin ML, Hioe CE. HIV-1 Virological Synapse Is Not Simply a Copycat of the Immunological Synapse. Viruses (2010) 2(5):1239–60. doi: 10.3390/v2051239
56. Lowin-Kropf B, Shapiro VS, Weiss A. Cytoskeletal Polarization of T Cells Is Regulated by an Immunoreceptor Tyrosine-Based Activation Motif-Dependent Mechanism. J Cell Biol (1998) 140(4):861–71. doi: 10.1083/jcb.140.4.861
57. Warren JA, Zhou S, Xu Y, Moeser MJ, MacMillan DR, Council O, et al. The HIV-1 Latent Reservoir Is Largely Sensitive to Circulating T Cells. Elife (2020) 9:e57246. doi: 10.7554/eLife.57246
58. Fan J, Liang H, Shen T, Wang S, Ji X, Yee C, et al. Early Env-Specific CTLs Effectively Suppress Viral Replication in SHIV Controller Macaques. Cell Immunol (2018) 331:30–7. doi: 10.1016/j.cellimm.2018.05.001
59. Sengupta S, Siliciano RF. Targeting the Latent Reservoir for HIV-1. Immunity (2018) 48(5):872–95. doi: 10.1016/j.immuni.2018.04.030
60. Freitas CMT, Johnson DK, Weber KS. T Cell Calcium Signaling Regulation by the Co-Receptor Cd5. Int J Mol Sci (2018) 19(5):1295. doi: 10.3390/ijms19051295
61. Kondo N, Melikyan GB. Intercellular Adhesion Molecule 1 Promotes HIV-1 Attachment But Not Fusion to Target Cells. PloS One (2012) 7(9):e44827. doi: 10.1371/journal.pone.0044827
62. Tardif MR, Tremblay MJ. Presence of Host ICAM-1 in Human Immunodeficiency Virus Type 1 Virions Increases Productive Infection of CD4+ T Lymphocytes by Favoring Cytosolic Delivery of Viral Material. J Virol (2003) 77(22):12299–309. doi: 10.1128/JVI.77.22.12299-12309.2003
63. Olowe OA, Mabayoje VO, Akanbi O, Adefioye OJ, Olowe RA, Fadeni EK, et al. HIV P24 Antigen Among HIV Antibody Seronegative Blood Donors in Osogbo Osun State, South Western Nigeria. Pathog Glob Health (2016) 110(4-5):205–8. doi: 10.1080/20477724.2016.1205311
64. Khanal S, Schank M, El Gazzar M, Moorman JP, Yao ZQ. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells (2021) 10(2):475. doi: 10.3390/cells10020475
65. Leng J, Ho HP, Buzon MJ, Pereyra F, Walker BD, Yu XG, et al. A Cell-Intrinsic Inhibitor of HIV-1 Reverse Transcription in CD4(+) T Cells From Elite Controllers. Cell Host Microbe (2014) 15(6):717–28. doi: 10.1016/j.chom.2014.05.011
66. Wei F, Yan J, Tang D, Lin X, He L, Xie Y, et al. Inhibition of ERK Activation Enhances the Repair of Double-Stranded Breaks via Non-Homologous End Joining by Increasing DNA-PKcs Activation. Biochim Biophys Acta (2013) 1833(1):90–100. doi: 10.1016/j.bbamcr.2012.10.016
67. Calistri A, Reale A, Palu G, Parolin C. Why Cells and Viruses Cannot Survive Without an ESCRT. Cells (2021) 10(3):483. doi: 10.3390/cells10030483
68. Foster TL, Pickering S, Neil SJD. Inhibiting the Ins and Outs of HIV Replication: Cell-Intrinsic Antiretroviral Restrictions at the Plasma Membrane. Front Immunol (2017) 8:1853. doi: 10.3389/fimmu.2017.01853
69. Martin-Moreno A, Munoz-Fernandez MA. Dendritic Cells, the Double Agent in the War Against HIV-1. Front Immunol (2019) 10:2485. doi: 10.3389/fimmu.2019.02485
70. Prescher J, Baumgartel V, Ivanchenko S, Torrano AA, Brauchle C, Muller B, et al. Super-Resolution Imaging of ESCRT-Proteins at HIV-1 Assembly Sites. PloS Pathog (2015) 11(2):e1004677. doi: 10.1371/journal.ppat.1004677
71. Votteler J, Sundquist WI. Virus Budding and the ESCRT Pathway. Cell Host Microbe (2013) 14(3):232–41. doi: 10.1016/j.chom.2013.08.012
72. van Niel G, Wubbolts R, Stoorvogel W. Endosomal Sorting of MHC Class II Determines Antigen Presentation by Dendritic Cells. Curr Opin Cell Biol (2008) 20(4):437–44. doi: 10.1016/j.ceb.2008.05.011
73. Roche PA, Furuta K. The Ins and Outs of MHC Class II-Mediated Antigen Processing and Presentation. Nat Rev Immunol (2015) 15(4):203–16. doi: 10.1038/nri3818
74. Watanabe N, Suzuki Y, Yonezu T, Nakagawa Y, Shiina T, Hirayama N, et al. A Cell-Based High-Throughput Screening Assay System for Inhibitor Compounds of Antigen Presentation by HLA Class II Molecule. Sci Rep (2017) 7(1):6798. doi: 10.1038/s41598-017-07080-4
75. Prokofjeva MM, Kochetkov SN, Prassolov VS. Therapy of HIV Infection: Current Approaches and Prospects. Acta Naturae (2016) 8(4):23–32. doi: 10.32607/20758251-2016-8-4-23-32
76. Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol (2018) 9:2379. doi: 10.3389/fimmu.2018.02379
77. Sabado RL, Balan S, Bhardwaj N. Dendritic Cell-Based Immunotherapy. Cell Res (2017) 27(1):74–95. doi: 10.1038/cr.2016.157
78. Chometon TQ, Siqueira MDS, Sant Anna JC, Almeida MR, Gandini M, Martins de Almeida Nogueira AC, et al. A Protocol for Rapid Monocyte Isolation and Generation of Singular Human Monocyte-Derived Dendritic Cells. PloS One (2020) 15(4):e0231132. doi: 10.1371/journal.pone.0231132
79. Posch W, Lass-Florl C, Wilflingseder D. Generation of Human Monocyte-Derived Dendritic Cells From Whole Blood. J Vis Exp (2016) 118):54968. doi: 10.3791/54968
80. Sabado RL, Bhardwaj N. Directing Dendritic Cell Immunotherapy Towards Successful Cancer Treatment. Immunotherapy (2010) 2(1):37–56. doi: 10.2217/imt.09.43
81. Liu MA. Immunologic Basis of Vaccine Vectors. Immunity (2010) 33(4):504–15. doi: 10.1016/j.immuni.2010.10.004
82. Nascimento IP, Leite LC. Recombinant Vaccines and the Development of New Vaccine Strategies. Braz J Med Biol Res (2012) 45(12):1102–11. doi: 10.1590/S0100-879X2012007500142
83. Pavlick AC, Blazquez A, Meseck M, Donovan MJ, Castillo-Martin M, Thin TH, et al. A Phase II Open Labeled, Randomized Study of Poly-ICLC Matured Dendritic Cells for NY-ESO-1 and Mean-A Peptide Vaccination Compared to Montanide, in Melanoma Patients in Complete Clinical Remission. J Clin Oncol (2019) 37: (15_suppl):9538–. doi: 10.1200/JCO.2019.37.15_suppl.9538
84. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First Results on Survival From a Large Phase 3 Clinical Trial of an Autologous Dendritic Cell Vaccine in Newly Diagnosed Glioblastoma. J Transl Med (2018) 16(1):142. doi: 10.1186/s12967-018-1507-6
85. McGranahan T, Therkelsen KE, Ahmad S, Nagpal S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr Treat Options Oncol (2019) 20(3):24. doi: 10.1007/s11864-019-0619-4
86. Sprooten J, Ceusters J, Coosemans A, Agostinis P, De Vleeschouwer S, Zitvogel L, et al. Trial Watch: Dendritic Cell Vaccination for Cancer Immunotherapy. Oncoimmunology (2019) 8(11):e1638212. doi: 10.1080/2162402X.2019.1638212
87. Tselis N, Chatzikonstantinou G. Treating the Chameleon: Radiotherapy in the Management of Renal Cell Cancer. Clin Transl Radiat Oncol (2019) 16:7–14. doi: 10.1016/j.ctro.2019.01.007
88. Li Z, Qiu Y, Lu W, Jiang Y, Wang J. Immunotherapeutic Interventions of Triple Negative Breast Cancer. J Transl Med (2018) 16(1):147. doi: 10.1186/s12967-018-1514-7
89. Nicolini F, Bocchini M, Bronte G, Delmonte A, Guidoboni M, Crino L, et al. Malignant Pleural Mesothelioma: State-Of-the-Art on Current Therapies and Promises for the Future. Front Oncol (2019) 9:1519. doi: 10.3389/fonc.2019.01519
90. Qian C, Yang LJ, Cui H. Recent Advances in Nanotechnology for Dendritic Cell-Based Immunotherapy. Front Pharmacol (2020) 11:960. doi: 10.3389/fphar.2020.00960
91. Intlekofer AM, Thompson CB. At the Bench: Preclinical Rationale for CTLA-4 and PD-1 Blockade as Cancer Immunotherapy. J Leukoc Biol (2013) 94(1):25–39. doi: 10.1189/jlb.1212621
92. Gu YZ, Zhao X, Song XR. Ex Vivo Pulsed Dendritic Cell Vaccination Against Cancer. Acta Pharmacol Sin (2020) 41(7):959–69. doi: 10.1038/s41401-020-0415-5
93. Dong Y, Sun Q, Zhang X. PD-1 and Its Ligands Are Important Immune Checkpoints in Cancer. Oncotarget (2017) 8(2):2171–86. doi: 10.18632/oncotarget.13895
94. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front Immunol (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
95. Francisco LM, Sage PT, Sharpe AH. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
96. Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The PD-1/PD-L1-Checkpoint Restrains T Cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell (2020) 38(5):685–700.e8. doi: 10.1016/j.ccell.2020.09.001
97. Hudson K, Cross N, Jordan-Mahy N, Leyland R. The Extrinsic and Intrinsic Roles of PD-L1 and Its Receptor PD-1: Implications for Immunotherapy Treatment. Front Immunol (2020) 11:568931. doi: 10.3389/fimmu.2020.568931
98. Correa-Rocha R, Lopez-Abente J, Gutierrez C, Perez-Fernandez VA, Prieto-Sanchez A, Moreno-Guillen S, et al. CD72/CD100 and PD-1/PD-L1 Markers Are Increased on T and B Cells in HIV-1+ Viremic Individuals, and CD72/CD100 Axis Is Correlated With T-Cell Exhaustion. PloS One (2018) 13(8):e0203419. doi: 10.1371/journal.pone.0203419
99. Schonrich G, Raftery MJ. The PD-1/PD-L1 Axis and Virus Infections: A Delicate Balance. Front Cell Infect Microbiol (2019) 9:207. doi: 10.3389/fcimb.2019.00207
100. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
101. Sansom DM. CD28, CTLA-4 and Their Ligands: Who Does What and to Whom? Immunology (2000) 101(2):169–77. doi: 10.1046/j.1365-2567.2000.00121.x
102. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
103. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:86. doi: 10.3389/fonc.2018.00086
104. Li MO, Rudensky AY. T Cell Receptor Signalling in the Control of Regulatory T Cell Differentiation and Function. Nat Rev Immunol (2016) 16(4):220–33. doi: 10.1038/nri.2016.26
105. Campbell DJ, Butcher EC. Rapid Acquisition of Tissue-Specific Homing Phenotypes by CD4(+) T Cells Activated in Cutaneous or Mucosal Lymphoid Tissues. J Exp Med (2002) 195(1):135–41. doi: 10.1084/jem.20011502
106. Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function Throughout Life. Immunity (2018) 48(2):202–13. doi: 10.1016/j.immuni.2018.01.007
107. Ager A. High Endothelial Venules and Other Blood Vessels: Critical Regulators of Lymphoid Organ Development and Function. Front Immunol (2017) 8:45. doi: 10.3389/fimmu.2017.00045
108. Kok L, Masopust D, Schumacher TN. The Precursors of CD8(+) Tissue Resident Memory T Cells: From Lymphoid Organs to Infected Tissues. Nat Rev Immunol (2021) 21:1–11. doi: 10.1038/s41577-021-00590-3
109. Pejoski D, Ballester M, Auderset F, Vono M, Christensen D, Andersen P, et al. Site-Specific DC Surface Signatures Influence CD4(+) T Cell Co-Stimulation and Lung-Homing. Front Immunol (2019) 10:1650. doi: 10.3389/fimmu.2019.01650
110. Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs Metabolize Sunlight-Induced Vitamin D3 to ‘Program’ T Cell Attraction to the Epidermal Chemokine CCL27. Nat Immunol (2007) 8(3):285–93. doi: 10.1038/ni1433
111. Cassani B, Villablanca EJ, Quintana FJ, Love PE, Lacy-Hulbert A, Blaner WS, et al. Gut-Tropic T Cells That Express Integrin Alpha4beta7 and CCR9 Are Required for Induction of Oral Immune Tolerance in Mice. Gastroenterology (2011) 141(6):2109–18. doi: 10.1053/j.gastro.2011.09.015
112. Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML, de Jong EC. Retinoic Acid Primes Human Dendritic Cells to Induce Gut-Homing, IL-10-Producing Regulatory T Cells. Mucosal Immunol (2015) 8(2):265–78. doi: 10.1038/mi.2014.64
113. Stagg AJ. Intestinal Dendritic Cells in Health and Gut Inflammation. Front Immunol (2018) 9:2883. doi: 10.3389/fimmu.2018.02883
114. Palucka K, Banchereau J, Mellman I. Designing Vaccines Based on Biology of Human Dendritic Cell Subsets. Immunity (2010) 33(4):464–78. doi: 10.1016/j.immuni.2010.10.007
115. Baldin AV, Savvateeva LV, Bazhin AV, Zamyatnin AA Jr. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers (Basel) (2020) 12(3):590. doi: 10.3390/cancers12030590
116. Hossain MK, Wall KA. Use of Dendritic Cell Receptors as Targets for Enhancing Anti-Cancer Immune Responses. Cancers (Basel) (2019) 11(3):418. doi: 10.3390/cancers11030418
117. Matsuo K, Yoshie O, Kitahata K, Kamei M, Hara Y, Nakayama T. Recent Progress in Dendritic Cell-Based Cancer Immunotherapy. Cancers (Basel) (2021) 13(10):2495. doi: 10.3390/cancers13102495
118. Macri C, Dumont C, Johnston AP, Mintern JD. Targeting Dendritic Cells: A Promising Strategy to Improve Vaccine Effectiveness. Clin Transl Immunol (2016) 5(3):e66. doi: 10.1038/cti.2016.6
119. Park CG, Rodriguez A, Ueta H, Lee H, Pack M, Matsuno K, et al. Generation of Anti-Human DEC205/CD205 Monoclonal Antibodies That Recognize Epitopes Conserved in Different Mammals. J Immunol Methods (2012) 377(1-2):15–22. doi: 10.1016/j.jim.2011.12.009
120. Baker K, Rath T, Lencer WI, Fiebiger E, Blumberg RS. Cross-Presentation of IgG-Containing Immune Complexes. Cell Mol Life Sci (2013) 70(8):1319–34. doi: 10.1007/s00018-012-1100-8
121. Melchers M, Matthews K, de Vries RP, Eggink D, van Montfort T, Bontjer I, et al. A Stabilized HIV-1 Envelope Glycoprotein Trimer Fused to CD40 Ligand Targets and Activates Dendritic Cells. Retrovirology (2011) 8:48. doi: 10.1186/1742-4690-8-48
122. Kutzler MA, Weiner DB. Developing DNA Vaccines That Call to Dendritic Cells. J Clin Invest (2004) 114(9):1241–4. doi: 10.1172/JCI23467
123. Lu Y, Xin KQ, Hamajima K, Tsuji T, Aoki I, Yang J, et al. Macrophage Inflammatory Protein-1alpha (MIP-1alpha) Expression Plasmid Enhances DNA Vaccine-Induced Immune Response Against HIV-1. Clin Exp Immunol (1999) 115(2):335–41. doi: 10.1046/j.1365-2249.1999.00793.x
124. Sailaja G, Husain S, Nayak BP, Jabbar AM. Long-Term Maintenance of Gp120-Specific Immune Responses by Genetic Vaccination With the HIV-1 Envelope Genes Linked to the Gene Encoding Flt-3 Ligand. J Immunol (2003) 170(5):2496–507. doi: 10.4049/jimmunol.170.5.2496
125. Nayak BP, Sailaja G, Jabbar AM. Enhancement of Gp120-Specific Immune Responses by Genetic Vaccination With the Human Immunodeficiency Virus Type 1 Envelope Gene Fused to the Gene Coding for Soluble CTLA4. J Virol (2003) 77(20):10850–61. doi: 10.1128/JVI.77.20.10850-10861.2003
126. Gandhi RT, Kwon DS, Macklin EA, Shopis JR, McLean AP, McBrine N, et al. Immunization of HIV-1-Infected Persons With Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J Acquir Immune Defic Syndr (2016) 71(3):246–53. doi: 10.1097/QAI.0000000000000852
127. Pawlak EN, Dirk BS, Jacob RA, Johnson AL, Dikeakos JD. The HIV-1 Accessory Proteins Nef and Vpu Downregulate Total and Cell Surface CD28 in CD4(+) T Cells. Retrovirology (2018) 15(1):6. doi: 10.1186/s12977-018-0388-3
128. Hussain A, Wesley C, Khalid M, Chaudhry A, Jameel S. Human Immunodeficiency Virus Type 1 Vpu Protein Interacts With CD74 and Modulates Major Histocompatibility Complex Class II Presentation. J Virol (2008) 82(2):893–902. doi: 10.1128/JVI.01373-07
129. Lindner R. Invariant Chain Complexes and Clusters as Platforms for MIF Signaling. Cells (2017) 6(1):6. doi: 10.3390/cells6010006
130. Rezaei T, Khalili S, Baradaran B, Mosafer J, Rezaei S, Mokhtarzadeh A, et al. Recent Advances on HIV DNA Vaccines Development: Stepwise Improvements to Clinical Trials. J Control Release (2019) 316:116–37. doi: 10.1016/j.jconrel.2019.10.045
131. Hokello J, Sharma AL, Tyagi M. An Update on the HIV DNA Vaccine Strategy. Vaccines (Basel) (2021) 9(6):605. doi: 10.3390/vaccines9060605
132. Schwerdtfeger M, Andersson AC, Neukirch L, Holst PJ. Virus-Like Vaccines Against HIV/SIV Synergize With a Subdominant Antigen T Cell Vaccine. J Transl Med (2019) 17(1):175. doi: 10.1186/s12967-019-1924-1
133. Starodubova ES, Isaguliants MG, Karpov VL. Regulation of Immunogen Processing: Signal Sequences and Their Application for the New Generation of DNA-Vaccines. Acta Naturae (2010) 2(1):53–60. doi: 10.32607/20758251-2010-2-1-53-59
134. Gay CL, DeBenedette MA, Tcherepanova IY, Gamble A, Lewis WE, Cope AB, et al. Immunogenicity of AGS-004 Dendritic Cell Therapy in Patients Treated During Acute HIV Infection. AIDS Res Hum Retroviruses (2018) 34(1):111–22. doi: 10.1089/aid.2017.0071
135. Macatangay BJ, Riddler SA, Wheeler ND, Spindler J, Lawani M, Hong F, et al. Therapeutic Vaccination With Dendritic Cells Loaded With Autologous HIV Type 1-Infected Apoptotic Cells. J Infect Dis (2016) 213(9):1400–9. doi: 10.1093/infdis/jiv582
136. Garcia F, Climent N, Guardo AC, Gil C, Leon A, Autran B, et al. A Dendritic Cell-Based Vaccine Elicits T Cell Responses Associated With Control of HIV-1 Replication. Sci Transl Med (2013) 5(166):166ra2. doi: 10.1126/scitranslmed.3004682
137. Leal L, Feher C, Richart V, Torres B, Garcia F. Antiretroviral Therapy Interruption (ATI) in HIV-1 Infected Patients Participating in Therapeutic Vaccine Trials: Surrogate Markers of Virological Response. Vaccines (Basel) (2020) 8(3):442. doi: 10.3390/vaccines8030442
138. Hsu DC, O’Connell RJ. Progress in HIV Vaccine Development. Hum Vaccin Immunother (2017) 13(5):1018–30. doi: 10.1080/21645515.2016.1276138
139. Ng’uni T, Chasara C, Ndhlovu ZM. Major Scientific Hurdles in HIV Vaccine Development: Historical Perspective and Future Directions. Front Immunol (2020) 11:590780. doi: 10.3389/fimmu.2020.590780
140. Levitz L, Koita OA, Sangare K, Ardito MT, Boyle CM, Rozehnal J, et al. Conservation of HIV-1 T Cell Epitopes Across Time and Clades: Validation of Immunogenic HLA-A2 Epitopes Selected for the GAIA HIV Vaccine. Vaccine (2012) 30(52):7547–60. doi: 10.1016/j.vaccine.2012.10.042
141. Surenaud M, Montes M, Lindestam Arlehamn CS, Sette A, Banchereau J, Palucka K, et al. Anti-HIV Potency of T-Cell Responses Elicited by Dendritic Cell Therapeutic Vaccination. PloS Pathog (2019) 15(9):e1008011. doi: 10.1371/journal.ppat.1008011
142. Thomas OS, Weber W. Overcoming Physiological Barriers to Nanoparticle Delivery-Are We There Yet? Front Bioeng Biotechnol (2019) 7:415. doi: 10.3389/fbioe.2019.00415
143. Smith DM, Simon JK, Baker JR Jr. Applications of Nanotechnology for Immunology. Nat Rev Immunol (2013) 13(8):592–605. doi: 10.1038/nri3488
144. Jia J, Zhang Y, Xin Y, Jiang C, Yan B, Zhai S. Interactions Between Nanoparticles and Dendritic Cells: From the Perspective of Cancer Immunotherapy. Front Oncol (2018) 8:404. doi: 10.3389/fonc.2018.00404
145. Pati R, Shevtsov M, Sonawane A. Nanoparticle Vaccines Against Infectious Diseases. Front Immunol (2018) 9:2224. doi: 10.3389/fimmu.2018.02224
146. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering Precision Nanoparticles for Drug Delivery. Nat Rev Drug Discovery (2021) 20(2):101–24. doi: 10.1038/s41573-020-0090-8
147. Bose RJ, Kim M, Chang JH, Paulmurugan R, Moon JJ, Koh WG, et al. Biodegradable Polymers for Modern Vaccine Development. J Ind Eng Chem (2019) 77:12–24. doi: 10.1016/j.jiec.2019.04.044
148. Beck Z, Matyas GR, Jalah R, Rao M, Polonis VR, Alving CR. Differential Immune Responses to HIV-1 Envelope Protein Induced by Liposomal Adjuvant Formulations Containing Monophosphoryl Lipid A With or Without QS21. Vaccine (2015) 33(42):5578–87. doi: 10.1016/j.vaccine.2015.09.001
149. Om K, Paquin-Proulx D, Montero M, Peachman K, Shen X, Wieczorek L, et al. Adjuvanted HIV-1 Vaccine Promotes Antibody-Dependent Phagocytic Responses and Protects Against Heterologous SHIV Challenge. PloS Pathog (2020) 16(9):e1008764. doi: 10.1371/journal.ppat.1008764
150. Chen CW, Saubi N, Joseph-Munne J. Design Concepts of Virus-Like Particle-Based HIV-1 Vaccines. Front Immunol (2020) 11:573157. doi: 10.3389/fimmu.2020.573157
151. Abdoli A, Radmehr N, Bolhassani A, Eidi A, Mehrbod P, Motevalli F, et al. Conjugated Anionic PEG-Citrate G2 Dendrimer With Multi-Epitopic HIV-1 Vaccine Candidate Enhance the Cellular Immune Responses in Mice. Artif Cells Nanomed Biotechnol (2017) 45(8):1762–8. doi: 10.1080/21691401.2017.1290642
152. Vacas-Cordoba E, Climent N, de la Mata FJ, Plana M, Gomez R, Pion M, et al. Dendrimers as Nonviral Vectors in Dendritic Cell-Based Immunotherapies Against Human Immunodeficiency Virus: Steps Toward Their Clinical Evaluation. Nanomed (Lond) (2014) 9(17):2683–702. doi: 10.2217/nnm.14.172
153. Kristoff J, Rinaldo CR, Mailliard RB. Role of Dendritic Cells in Exposing Latent HIV-1 for the Kill. Viruses (2019) 12(1):37. doi: 10.3390/v12010037
154. Martins C, Araujo F, Gomes MJ, Fernandes C, Nunes R, Li W, et al. Using Microfluidic Platforms to Develop CNS-Targeted Polymeric Nanoparticles for HIV Therapy. Eur J Pharm Biopharm (2019) 138:111–24. doi: 10.1016/j.ejpb.2018.01.014
155. Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted Polymeric Therapeutic Nanoparticles: Design, Development and Clinical Translation. Chem Soc Rev (2012) 41(7):2971–3010. doi: 10.1039/c2cs15344k
156. Pavot V, Berthet M, Resseguier J, Legaz S, Handke N, Gilbert SC, et al. Poly(lactic Acid) and Poly(Lactic-Co-Glycolic Acid) Particles as Versatile Carrier Platforms for Vaccine Delivery. Nanomed (Lond) (2014) 9(17):2703–18. doi: 10.2217/nnm.14.156
157. AbdelAllah NH, Gaber Y, Rashed ME, Azmy AF, Abou-Taleb HA, AbdelGhani S. Alginate-Coated Chitosan Nanoparticles Act as Effective Adjuvant for Hepatitis A Vaccine in Mice. Int J Biol Macromol (2020) 152:904–12. doi: 10.1016/j.ijbiomac.2020.02.287
158. Bayon E, Morlieras J, Dereuddre-Bosquet N, Gonon A, Gosse L, Courant T, et al. Overcoming Immunogenicity Issues of HIV P24 Antigen by the Use of Innovative Nanostructured Lipid Carriers as Delivery Systems: Evidences in Mice and Non-Human Primates. NPJ Vaccines (2018) 3:46. doi: 10.1038/s41541-018-0086-0
159. Carabineiro SAC. Applications of Gold Nanoparticles in Nanomedicine: Recent Advances in Vaccines. Molecules (2017) 22(5):857. doi: 10.3390/molecules22050857
160. Pudlarz A, Szemraj J. Nanoparticles as Carriers of Proteins, Peptides and Other Therapeutic Molecules. Open Life Sci (2018) 13:285–98. doi: 10.1515/biol-2018-0035
161. Dykman LA. Gold Nanoparticles for Preparation of Antibodies and Vaccines Against Infectious Diseases. Expert Rev Vaccines (2020) 19(5):465–77. doi: 10.1080/14760584.2020.1758070
162. van Dinther D, Stolk DA, van de Ven R, van Kooyk Y, de Gruijl TD, den Haan JMM. Targeting C-Type Lectin Receptors: A High-Carbohydrate Diet for Dendritic Cells to Improve Cancer Vaccines. J Leukoc Biol (2017) 102(4):1017–34. doi: 10.1189/jlb.5MR0217-059RR
163. Roy U, Rodriguez J, Barber P, das Neves J, Sarmento B, Nair M. The Potential of HIV-1 Nanotherapeutics: From In Vitro Studies to Clinical Trials. Nanomed (Lond) (2015) 10(24):3597–609. doi: 10.2217/nnm.15.160
164. Blum JS, Wearsch PA, Cresswell P. Pathways of Antigen Processing. Annu Rev Immunol (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
165. Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, et al. Identification of a Candidate Therapeutic Autophagy-Inducing Peptide. Nature (2013) 494(7436):201–6. doi: 10.1038/nature11866
166. Watson DS, Endsley AN, Huang L. Design Considerations for Liposomal Vaccines: Influence of Formulation Parameters on Antibody and Cell-Mediated Immune Responses to Liposome Associated Antigens. Vaccine (2012) 30(13):2256–72. doi: 10.1016/j.vaccine.2012.01.070
167. Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics (2017) 9(2):12. doi: 10.3390/pharmaceutics9020012
168. Wang N, Chen M, Wang T. Liposomes Used as a Vaccine Adjuvant-Delivery System: From Basics to Clinical Immunization. J Control Release (2019) 303:130–50. doi: 10.1016/j.jconrel.2019.04.025
169. Mittal P, Saharan A, Verma R, Altalbawy FMA, Alfaidi MA, Batiha GE, et al. Dendrimers: A New Race of Pharmaceutical Nanocarriers. BioMed Res Int (2021) 2021:8844030. doi: 10.1155/2021/8844030
170. Patra JK, Das G, Fraceto LF, Campos EVR, Rodriguez-Torres MDP, Acosta-Torres LS, et al. Nano Based Drug Delivery Systems: Recent Developments and Future Prospects. J Nanobiotechnol (2018) 16(1):71. doi: 10.1186/s12951-018-0392-8
171. Cifuentes-Rius A, Desai A, Yuen D, Johnston APR, Voelcker NH. Inducing Immune Tolerance With Dendritic Cell-Targeting Nanomedicines. Nat Nanotechnol (2021) 16(1):37–46. doi: 10.1038/s41565-020-00810-2
172. Fulcher JA, Hashimi ST, Levroney EL, Pang M, Gurney KB, Baum LG, et al. Galectin-1-Matured Human Monocyte-Derived Dendritic Cells Have Enhanced Migration Through Extracellular Matrix. J Immunol (2006) 177(1):216–26. doi: 10.4049/jimmunol.177.1.216
173. Cordoba EV, Pion M, Rasines B, Filippini D, Komber H, Ionov M, et al. Glycodendrimers as New Tools in the Search for Effective Anti-HIV DC-Based Immunotherapies. Nanomedicine (2013) 9(7):972–84. doi: 10.1016/j.nano.2013.03.004
174. Song CK, Jung SH, Kim DD, Jeong KS, Shin BC, Seong H. Disaccharide-Modified Liposomes and Their In Vitro Intracellular Uptake. Int J Pharm (2009) 380(1-2):161–9. doi: 10.1016/j.ijpharm.2009.07.014
175. Martin-Moreno A, Jimenez Blanco JL, Mosher J, Swanson DR, Garcia Fernandez JM, Sharma A, et al. Nanoparticle-Delivered HIV Peptides to Dendritic Cells a Promising Approach to Generate a Therapeutic Vaccine. Pharmaceutics (2020) 12(7):656. doi: 10.3390/pharmaceutics12070656
176. Sharma A, Porterfield JE, Smith E, Sharma R, Kannan S, Kannan RM. Effect of Mannose Targeting of Hydroxyl PAMAM Dendrimers on Cellular and Organ Biodistribution in a Neonatal Brain Injury Model. J Control Release (2018) 283:175–89. doi: 10.1016/j.jconrel.2018.06.003
177. Paiardini M, Muller-Trutwin M. HIV-Associated Chronic Immune Activation. Immunol Rev (2013) 254(1):78–101. doi: 10.1111/imr.12079
178. Calmeiro J, Carrascal MA, Tavares AR, Ferreira DA, Gomes C, Falcao A, et al. Dendritic Cell Vaccines for Cancer Immunotherapy: The Role of Human Conventional Type 1 Dendritic Cells. Pharmaceutics (2020) 12(2):158. doi: 10.3390/pharmaceutics12020158
179. Mylvaganam GH, Silvestri G, Amara RR. HIV Therapeutic Vaccines: Moving Towards a Functional Cure. Curr Opin Immunol (2015) 35:1–8. doi: 10.1016/j.coi.2015.05.001
180. Ko EJ, Robert-Guroff M. Dendritic Cells in HIV/SIV Prophylactic and Therapeutic Vaccination. Viruses (2019) 12(1):24. doi: 10.3390/v12010024
181. Wu L, KewalRamani VN. Dendritic-Cell Interactions With HIV: Infection and Viral Dissemination. Nat Rev Immunol (2006) 6(11):859–68. doi: 10.1038/nri1960
182. Pautler M, Brenner S. Nanomedicine: Promises and Challenges for the Future of Public Health. Int J Nanomed (2010) 5:803–9. doi: 10.2147/IJN.S13816
183. Figueiro Longo JP, Muehlmann LA. How has Nanomedical Innovation Contributed to the COVID-19 Vaccine Development? Nanomed (Lond) (2021) 16:1179–81. doi: 10.2217/nnm-2021-0035
184. Schudel A, Francis DM, Thomas SN. Material Design for Lymph Node Drug Delivery. Nat Rev Mater (2019) 4(6):415–28. doi: 10.1038/s41578-019-0110-7
Keywords: therapeutic vaccines, nanotechnology, T-cell response, dendritic cell, HIV-1
Citation: Espinar-Buitrago M and Muñoz-Fernández MA (2022) New Approaches to Dendritic Cell-Based Therapeutic Vaccines Against HIV-1 Infection. Front. Immunol. 12:719664. doi: 10.3389/fimmu.2021.719664
Received: 02 June 2021; Accepted: 07 December 2021;
Published: 04 January 2022.
Edited by:
Bernard J. C. Macatangay, University of Pittsburgh, United StatesReviewed by:
Maria Issagouliantis, Riga Stradiņš University, LatviaThorsten Demberg, Baylor College of Medicine, United States
Copyright © 2022 Espinar-Buitrago and Muñoz-Fernández. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ma Angeles Muñoz-Fernández, bW11bm96LmhndWdtQGdtYWlsLmNvbQ==; bW11bm96LmhndWdtQHNhbHVkLm1hZHJpZC5vcmc=