
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 11 August 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.718719
 Vanitha Mariappan1*
Vanitha Mariappan1* Kumutha Malar Vellasamy2
Kumutha Malar Vellasamy2 Muttiah Barathan2A. S. Smiline Girija3
Muttiah Barathan2A. S. Smiline Girija3 Esaki M. Shankar4Jamuna Vadivelu2*
Esaki M. Shankar4Jamuna Vadivelu2*Burkholderia pseudomallei (B. pseudomallei) causes melioidosis, a potentially fatal disease for which no licensed vaccine is available thus far. The host-pathogen interactions in B. pseudomallei infection largely remain the tip of the iceberg. The pathological manifestations are protean ranging from acute to chronic involving one or more visceral organs leading to septic shock, especially in individuals with underlying conditions similar to COVID-19. Pathogenesis is attributed to the intracellular ability of the bacterium to ‘step into’ the host cell’s cytoplasm from the endocytotic vacuole, where it appears to polymerize actin filaments to spread across cells in the closer vicinity. B. pseudomallei effectively evades the host’s surveillance armory to remain latent for prolonged duration also causing relapses despite antimicrobial therapy. Therefore, eradication of intracellular B. pseudomallei is highly dependent on robust cellular immune responses. However, it remains ambiguous why certain individuals in endemic areas experience asymptomatic seroconversion, whereas others succumb to sepsis-associated sequelae. Here, we propose key insights on how the host’s surveillance radars get commandeered by B. pseudomallei.
Melioidosis is a systemic infectious disease caused by Burkholderia pseudomallei (B. pseudomallei), a Gram-negative environmental saprophyte commonly found in wet soils and contaminated waters of endemic areas (1). Melioidosis is sporadic in several countries but endemic in Southeast Asia, India and northern Australia, and some parts of the tropical and sub-tropical world (2, 3). The major route of entry is via inhalation or inoculation through skin breaks exposed to contaminated soil or water (4). However, laboratory-acquired cases, person-to-person spread (5), sexual transmission (6), breast milk (7) and mother-to-child transmission (8) have also been reported. The incubation period varies from less than a day to 21 days and can extend to several months or years (9, 10).
Melioidosis can occur either as primary syndrome or as a component of sepsis. The infectious melioidosis disease manifestation appears as abscess formation in visceral organs such as lungs, liver, spleen and soft tissue (11). Occasionally, infections can remain sub-clinical, while others can develop acute or chronic disease or can even progress to fatal sepsis (12). Symptoms extending for ≥2 months is defined as chronic melioidosis that usually occurs in ~10% of infected individuals (10, 13). Melioidosis relapse is relatively common due to failure by the host to eradicate B. pseudomallei during the primary stages of infection, especially in the immunocompromised, and even after prolonged antimicrobial therapy. The overall relapse rate can range between 15% and 30% in severe melioidosis (14, 15). Dormant sub-clinical infections are also recognized in several cases, whereby it could stimulate to cause disease, typically in association with an inter-current illness, classically with lung disease, surgical procedure or trauma. Additionally, late-onset diabetes, renal failure and immunosuppressive therapy could also contribute to reactivation (16, 17). The aspects that affect the disease exposition and presentation are still unknown. Nevertheless, alterations in the virulence of infecting strains together with host immune-competence could likely contribute to disease prognosis (18).
B. pseudomallei is an extra-ordinarily adapted facultative intracellular parasite that can adhere, invade, survive and replicate within pulmonary (airway) epithelial and phagocytic cells. For most pathogenic bacteria, the first and foremost critical step involves the establishment of infection predominantly via cell adhesion. For B. pseudomallei, cell adhesion is mediated by specific membrane proteins such as extracellular adherence protein, flagellin and adhesin that are regulated by temperature (19). On the other hand, type IV pili, flagella and motility also play a role in adhesion (20). Following adhesion B. pseudomallei tend to invade the epithelial cells via specific virulence systems viz., type III (T3SS) and VI (T6SS) secretion systems (21). Miscellaneous virulence factors that appear to play a role in virulence include a type II secretion system (T2SS) (22), quorum sensing (23), flagellin (24), type IV pili (20), lipopolysaccharide (LPS) and capsule (25), phospholipase C (26), lactonase family protein A (27). Following invasion, B. pseudomallei intracellular survival in the host cell explains the ability for latency. The bacterium can evade from digestion within endocytic vacuoles by entering into the cytoplasm, causing membrane protrusions by induction of actin polymerization at one of the bacterial poles (28). On the other hand, the bacterium also displays colony morphology variations leading to formation of small colony (SCVs) or mucoid variants, that can also produce biofilm attributing to high level resistance to antimicrobial agents (29). Besides, B. pseudomallei can survive hostile environmental conditions including differences in pH, lack of nutrients, low or high temperatures and disinfectant detergents and antiseptic solutions. Likewise, B. pseudomallei can outgrow several lines of cellular as well as humoral defenses. Further, B. pseudomallei produces several enzymes and toxins, including hemolysins, proteases, lipases, lechithinase, cytotoxic exolipids, catalase, perioxidase, and superoxide dismutase (SOD) (3). It has become evident that B. pseudomallei can resist several anti-microbial peptides, complement proteins, lysosomal defensins and cationic peptidases providing itself an edge to survive adverse conditions (30).
Infection of the host tissue by a pathogenic bacterium involves a complex interplay between a wide array of factors that determine disease prognosis. A pathogen must acquire necessary nutrients from the host, and must almost always produce virulence factors to inflict cell damage, also evading the host’s immune armamentaria in parallel. Understanding the communication of a bacterium with the host environment and the host’s response during infection has been explored at the whole-genome level. High-density host DNA microarray analysis in response to pathogenic infection offers a resourceful tactic to closely study the pathogen-host interactions. In the past few years, numerous studies have been carried out using DNA microarray analysis to identify and determine the regulation of host responses to bacterial infection (31, 32). The aptitude to review the responses of a great subsection of the host genome and to explore the patterns and trends between the profiles across diverse subsets of hosts and pathogens, permits numerous essential and ultimate questions to be addressed in regard to the foundation of pathogen recognition, the structures of host-pathogen interaction, and the host defense and microbial virulence mechanisms (33, 34).
Largely, host cells exhibit a transcriptional program that produces an intracellular anti-infective state, in response to bacterial infection, specifically relating type I interferons, rendering neighboring cells refractory to infection. Using a cDNA microarray, a powerful approach to understand host-pathogen interactions, the gene expression profiling is monitored. Studies suggest that genome-wide expression analyses, especially at the molecular level, required to comprehend the pathology instigated by B. pseudomallei (35). Host gene expression and regulation profiling of cells infected with B. pseudomallei would likely unveil the mechanisms adopted by the host in reply to bacterial infection and provide key clues to evasion of host defenses thereby necessitating pathogenic survival and growth within cells. This would also aid to identify newer therapeutic targets and strategies (35).
Chin et al. (2010) (36) established an experimental mice model for acute phase melioidosis and employed microarray approach to explore the global host-pathogen interactions. The study revealed that genes associated with immune responses, stress responses, cell cycle regulation, proteasomal degradation, cellular metabolism and signal transduction pathways were markedly altered. Of note, genes associated with ‘core host immune responses’ and acute inflammatory responses were upregulated signifying the onset of host tissue injury.
Vellasamy and colleagues (2016) (37) explored the transcriptional responses of human lung epithelial cells upon exposure to early infection of live B. pseudomallei and its secreted proteins using a microarray platform. The investigation showed that the host carbohydrate metabolism and apoptosis were activated, and simultaneously, amino acid metabolism and innate immune responses were curbed in the host cells exposed to both live bacteria as well as its secreted proteins. The authors concluded that early exposure prior to establishment of infection could be associated with primary activation of host genes engaged with bacterial spread from lungs to other target organs or to evade from likely sensing by macrophages.
Similarly, Rao et al. (2020) (38) carried out transcriptomic profiling of B. pseudomallei-infected human lung epithelial cells with an aim to describe the cellular responses during early stages of infection. Their findings showed that the PERK-mediated unfolded protein response (UPR) was enhanced in the signaling pathway of protein processing in the endoplasmic reticulum. Other genes related to inflammatory responses, cell migration, and apoptosis were also altered. Overall, findings from both Rao et al. (2020) (37) and Vellasamy et al. (2016) (38) provided significant primary insights into the close interaction between B. pseudomallei and lung epithelial cells, which can be further explored to elucidate the cellular mechanisms of B. pseudomallei infection.
However, recently, Heacock-Kang et al. (2021) (39) established B. pseudomallei mono-cells transcriptomic profiling via host cell infection at distinct stages (vacuole entry; cytoplasmic escape and replication; and membrane protrusion, promoting cell-to-cell spread) in order to yield pathophysiological insights through a ‘TRANSITomic’ approach. The B. pseudomallei ‘TRANSITome’ divulged dynamic gene expression alteration via the various stages of host cell infection. Several genes that are essential for pathogenesis and virulence were identified, including the secretion systems. Overall, the B. pseudomallei ‘TRANSITome’ offered mono-cell transcriptomics data permitting high-resolution understanding of host-pathogen interactions.
Innate immunity is described to fight almost against any infection and deploys immune cells at the front-line in respond to microbial pathogens that endeavor to breach into the host boundaries (40). As a general description, the innate immune system comprises all the host defense aspects including physical, biochemical and microbial components. The physical barriers include epithelial cell layers in addition to the respiratory, gastrointestinal, and the genitourinary tracts. Chemical and biological barriers comprise of complement proteins and antimicrobial peptides, for instance defensins that are continuously existing in body fluids, as well as indigenous microbiota. On the whole, the innate immunity recognizes pathogens using several pattern recognition receptors (PRRs) situated on or within the host cells. These PRRs bind to pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS), lipid-A, peptidoglycan, flagellin, and TTSS expressed by B. pseudomallei (41) eventually eliciting pro-inflammatory signaling pathways.
The innate immune responses to B. pseudomallei infection have been investigated in greater detail employing several noticeable cell types (e.g., macrophages and neutrophils) that are triggered following B. pseudomallei infection, together with their cell surface signaling molecules, as well as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and caspases via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) signaling pathway. Other important cytokines or interleukins include monocyte chemoattractant protein-1 (MCP-1), tumor necrosis factor alpha (TNF-α), interferon gamma (INF-γ), IL-12 and IL-8 (CXCL8).
The main forces in innate immune responses are the phagocytes, namely the macrophages and polymorphonuclear neutrophils (PMNs). With patrolling macrophages and neutrophils deployed at strategic tissue locations in the human body, B. pseudomallei employs versatile strategies to overcome the rivals and is equipped with multiple virulence factors in order for it to establish a successful infection.
The key strategy to evade potential elimination of B. pseudomallei by the host immune system is to circumvent their detection and phagocytosis. Different pathogens employ distinct strategies to avoid their recognition in the host. B. pseudomallei appears to escape from phagocytosis via production of a capsular polysaccharide (CPS), which abridges the deposition of C3b on their surfaces (42). CPS of B. pseudomallei has previously been characterized and identified as the major determinant of bacterial virulence (43). C3b is a vital component of the C3 complement system, which is pivotal to pathogenic opsonization (43). It has been demonstrated that encapsulated prototrophic B. pseudomallei allows poor C3b deposition rationalizing their high rates of persistence in the peripheral circulation as compared to its auxotrophic capsular-mutant counterpart. It has also been established that polymorphonuclear leukocytes (PMNLs) significantly engulfed more capsular mutants relative to capsule-producing wild types (WTs) (42). The above findings point to the role of CPS aiding in the successful evasion of the bacterium from phagocytosis.
Apart from phagocytosis, neutrophils also release neutrophilic extracellular traps (NET) (a.k.a. NETosis) upon their activation following their recruitment to the site of infection. NET, formed by neutrophilic DNA, targets bacterial virulence factors and irreversibly kills the bacteria trapped within (44). NETs are known to entrap both Gram-positive and Gram-negative bacteria. It has been demonstrated that activated neutrophils release NETS upon challenging by B. pseudomallei and the number of extracellular bacteria were shown to be reduced indicating the bactericidal activity of NETs. When DNase I treatment was applied to dissociate NETs, the killing of the bacteria was reduced (45). The results suggest that NETs do possess antibacterial activity against B. pseudomallei. The question as to whether B. pseudomallei is equipped with a surface endonuclease (similar to Streptococcus pneumoniae) (46) remain dubious as B. pseudomallei excretes various exoenzymes and its genome prediction was also found to contain a nuclease (47). The nuclease (if expressed) would degrade NETs to enable the bacteria to escape from killing by antimicrobial molecules present in NETs.
Fibrinolysis plays a paramount role in host immunity against invading bacteria. The fibrin clot ensures the closure of injured blood vessels preventing access to more pathogens, and concentrates and entraps intruders at a focused site preventing their systemic dissemination. During the process of fibrin clot formation, inflammatory responses appear to occur entailing phagocytic influx (48). Although it also appears that many pathogens likely interfere with the plasminogen-plasmin system for systemic dissemination and escape from phagocytes (49), there is no proof on such ability of B. pseudomallei. Indeed, it has been reported that urokinase-type plasminogen activator receptor (uPAR) that regulates fibrinolysis and inflammation is up-regulated upon challenging by B. pseudomallei in septic melioidosis and experimental melioidosis in mice (50). Even though phagocytosis of B. pseudomallei was impaired in uPAR knockout mice there was no difference in the fibrinolytic responses in both uPAR KO as well as WT mice. This suggests that unlike several other successful invading pathogens, B. pseudomallei may not interfere with the plasminogen-plasmin system for evasion from phagocytic activity.
One of the smartest ways of overcoming extracellular killing is to invade and adopt an intracellular lifestyle. The ability to adopt an intracellular lifestyle is one of the hallmarks of pathogenesis of B. pseudomallei (51). The various strategies employed by the bacterium for intracellular survival have been critically reviewed previously (52). As expected, T3SS cluster together with a few other virulence factors, namely adhesins and pilins, enhance their adhesion, invasion and intracellular survival within phagocytic and non-phagocytic cells (53).
For efficient survival and persistence, the bacteria must escape from the endocytic vesicles into the cytoplasm, which is rich in nutrients than the endocytic vesicles on gaining access into the intracellular milieu. B. pseudomallei will escape from endocytic vesicles within 15 minutes after internalization, according to an electron microscopy analysis (54). Research has also revealed that T3SS proteins like BopA, BsaZ, and BipD are most likely involved in the escape mechanisms (55) although the other possibilities remain unclear. Allwoods et al. (2011) (27) proposed two potential mechanisms based on the projected functional domains found in BopA: i) BopA has a carboxyl-terminal Rho GTPase inactivation domain that could purpose as a protease or acyltransferase on the vesicle. ii) BopA has a cholesterol-binding domain, which can prevent cholesterol depletion, resulting in phagolysosome or autophagolysosome formation and bacterial degradation (56).
Despite its aptitude to evade from endocytic vesicles, B. pseudomallei can suppress macrophage killing processes. The action of inducible nitric oxide synthase (iNOS) results in the development of the free radical species nitric oxide (NO) in macrophages necessary for the removal of intracellular pathogens (57). It was discovered that LPS of B. pseudomallei were responsible for lowering NO expression in macrophages as compared to LPS of E. coli or Salmonella enterica (58). This is believed to be caused by B. pseudomallei LPS, which prevents macrophages from secreting IFN-γ required to stimulate iNOS expression (59). Furthermore, intracellular B. pseudomallei can modulate the expression of a few proteins, including SOCS3 (suppressor of cytokine signaling), CIS (cytokine-inducible Src homology), and SIRP (signal regulatory protein), which together inhibit the expression of iNOS (58). These are the few efficient ways by which B. pseudomallei’s progress with intracellular survival within macrophages.
Intracellular bacteria can still succumb to autophagic clearance after escaping into the phagocyte cytosol, while others alter or evade autophagy to survive intracellularly (60). The survival of intracellular B. pseudomallei was found to be significantly reduced when infected RAW 264.7 cells treated with rapamycin (inducer of autophagy). In the control model, only a subgroup of bacteria co-localized with the autophagy marker protein, LC3, resulting in a higher number of intracellular B. pseudomallei that survived (56). Since the survival of BipD and BopA mutants in RAW 263.7, T3SS proteins are expected to be involved in avoiding autophagy, the cells were affected, and there was a complex degree of co-localization with LC3 and the lysosomal marker LAMP1. B. pseudomallei encodes a T6SS cluster-associated gene, bpss0180, which can regulate autophagy in both phagocytic and non-phagocytic host cells (61). This indicate that B. pseudomallei could alter autophagic attack in the host, but the underlying mechanism remain ambiguous. According to a unique theory, B. pseudomallei can also influence the ubiquitination pathway in host cells, where the bacteria can identify a target protein, ubiquitin. In this relation, TssM, a T6SS cluster 5 protein encoded by the bpss1512 gene was recognized in B. pseudomallei, which can obstruct the ubiquitination of essential signalling intermediates (62). B. pseudomallei also produces CHBP (Cif homolog in B. pseudomallei), a cyclomodulin that inhibits the eukaryotic ubiquitination pathway (27). Despite the lack of direct evidence, these findings indicate that B. pseudomallei may have the ability to modulate ubiquitination to avoid recognition and bacterial clearance through autophagy.
Some pathogenic bacteria may also use ubiquitination to target and suppress immune system components in the host (63). The capability of B. pseudomallei to degrade a transcriptional factor, ELT-2, with targeted ubiquitination targeting has been demonstrated (64). In C. elegans, the GATA transcriptional factor ELT-2 regulates the early immune response to an invading pathogen (65). The targeted suppression of ELT-2 appears to be restricted to infection with B. pseudomallei, as the protein level of ELT-2 in C. elegans decreased over time while remaining relatively constant when infected with other bacteria. P. aeruginosa is known to use T3SS and T4SS effector proteins to promote the ubiquitination of host target proteins to inactivate host defenses (66). T3SS of B. pseudomallei was also thought to be responsible for inducing unique host E3 ligases to target ELT-2 for ubiquitin-proteasome system (UPS) destruction (65). More research is in progress to determine the effectors and components of T3SS, as well as the mechanism involved in subverting UPS to degrade ELT-2 protein.
Several clinical studies have shown that chronic melioidosis can present a variety of clinical symptoms that differs from acute melioidosis. The acute disease is often linked to risk factors including diabetes, heavy alcohol use, and renal disease (9). Chronic B. pseudomallei infections, on the other hand, are considered less severe; however, they are more localized in specific organs or locations, and the bacteria are tough to treat in general (67, 68). Relapses are common even after antibiotic therapy, and chronic melioidosis may develop after acute symptoms have subsided, with symptoms lasting for months to years. The treatment strategies also vary for chronic infection in comparison to acute infection. Experimental evidences are documented on a higher end for acute infections rather than chronic conditions using in vitro melioidosis pulmonary infection models. As a result, very minor is known about the virulence mechanisms and pathology of B. pseudomallei chronic infections (69). However, these chronic model experiments have revealed relative resistance to B. pseudomallei infections, not expressing much relapses (68).
TLRs belong to IL-1 receptor superfamily host transmembrane receptors which identify the conserved B. pseudomallei molecular trends inducing a pro-inflammatory response in concert with nuclear transcription factor (NF-B) translocation. Furthermore, TLRs have been elucidated as key PRRs in B. pseudomallei infection (70). Previous studies have shown the critical roles of TLR 2 and TLR4 as receptors in the host pulmonary defense (71). The host is exposed to B. pseudomallei lipopeptides and peptidoglycan, which activates TLR2, followed by TLR1 or TLR6. TLR4 being a significant LPS receptor, it is considered as a key molecule in defense against B. pseudomallei (70). However, some studies have shown that TLR-independent activation involves a functional Bsa T3SS followed by B. pseudomallei internalization (72). They discovered that live B. pseudomallei can activate TLR2 even in the absence of the co-receptor CD14 substantiating the necessity of TLR4 stimulation in the occurrence of MD2 and CD14 (73). TLR2 and TLR4 expression were activated and increased during B. pseudomallei infection in vivo and in vitro, according to Wiersinga and colleagues (2007) (71). Additionally, patients with septic melioidosis have higher TLR2 and TLR4 expression and activation (74) by B. pseudomallei. They also discovered a defensive phenotype in TLR2-deficient mice infected with B. pseudomallei through intranasal infection. Surprisingly, they discovered TLR2 activation but not TLR4 stimulation in transfected HEK293 cells during infection with B. pseudomallei LPS.
In another in vivo melioidosis acute mice model, Chin et al. (2010) (36) found that the expression of several TLRs (TLR2, 3, 4, 5, 6, and 7) were curbed; TLR2 was strongly expressed, while TLR4, which perceives LPS, was weakly induced. Whiteley et al., 2018 (75) found a similar expression profile in B. pseudomallei-infected RAW264.7 macrophages. This indicates that TLR2 is primarily responsible for the processing of signals that activate pro-inflammatory cytokines in response to B. pseudomallei infection and that TLR4 is not included in host defense against melioidosis in vivo. On the other hand, the mechanism where TLR2 signaling contributes to B. pseudomallei pathogenesis is still unknown. According to Chin et al. (2010) (36), up-regulation of the TLR2 facilitated signaling pathway is liable for the identification and commencement of an inflammatory response in an acute B. pseudomallei infection. This supports a previous finding of enhanced survival in TLR2 with reduced bacterial burden in the KO mice lung. When the main TLR adaptor signalling protein (MyD88) lacks in the host, B. pseudomallei infection seem to be extremely susceptible due to a decrease in the neutrophil recruitment and activation (70). TLR-mediated MyD88-dependent cell signalling appears to have a protective function, but dysregulation of TLR-mediated immune response could lead to pathogenesis and further allow to the production of septic melioidosis. TLR polymorphisms are also known to influence the host resistance to melioidosis, but the mechanisms underlying this effect remain unknown.
At large, TNF-α, IL-1, IL-6, IL-8, and IL-12 are some of the interleukins involved in the inflammatory response, and NF-kB is a key transcription factor that modulates their expression (76). When TLRs detect B. pseudomallei infections, they activate the NF-kB pathway, lowering the host immune response. B. pseudomallei activates NF-kB in a MyD88-independent but partially NOD1 intracellular receptor-dependent manner that necessitates the activities of Cdc42 and Rac1. The role of NF-kB in infection resistance was demonstrated in a knockout mouse trial, in which mice lacking various mechanisms of the NF-kB pathway were inclined to different infections (77). Tan et al. (2010) (62) recently established that B. pseudomallei prevents the initiation of the NF-kB and type I IFN pathways, thus reducing host inflammatory responses. Since NF-kB is involved in the bacterial clearance via host’s immune response it is a fair play by many pathogenic bacteria by developing mechanisms to control the pathway to suppress host immune responses.
Altogether, NOD1, NOD2, NLRP2, NLRP3, and class II trans-activator (CIITA) are members of the NLR gene family, which functions as an intracellular sensor of cytosolic microbial products and “hazard signals” during infection. Finally, this process stimulates caspase (CASP) cascades, leading to apoptosis with an increase in inflammatory responses, both of which are critical in regulating the intracellular B. pseudomallei. The NLRs, NOD1 and NOD2 were found to control the development of IL-18 after B. pseudomallei infection (72). IL-18 has been demonstrated to protect against B. pseudomallei infections in response to inflammasome signalling (71). However, in vivo studies demonstrated that the mice lacking inflammasome mechanisms such as ASC, caspase-1, NLRC4, and NLRP3 were reportedly more vulnerable to B. pseudomallei infection than the wild-type mice (78).
Different caspases were shown to be activated in response to B. pseudomallei infections, including the inflammatory mediator subfamily (CASP1, CASP4), the apoptotic activator (CASP2, CASP8), and the apoptotic executioner (CASP7) (36, 79). B. pseudomallei infections have been shown to activate CASP1, resulting in the cell death of macrophages and the development of IL-1 and IL-18 (80). Subsequent to B. pseudomallei infection, the CASP1 pathway influenced the IFN-production. The CASP1/mice model was found to be extremely vulnerable to B. pseudomallei infection with elevated bacterial loads, and their IFN-release was significantly reduced (80).
MCP-1 is a mononuclear cell attractant and an activator that may be a key modulator of mononuclear cell migration (81). The MCP-1 gene deletion caused in a moderately impaired monocyte conscription, decreased bacterial elimination in the spleen, and increased infection susceptibility (82). This inconsistency in the expression is due to the role played by the NK cells as the primary producers of IFNc in the lungs, while epithelial cells are unable to produce IFNc. In B. pseudomallei-infected murine lung epithelial cells (LA-4), MCP-1 levels were found to be significantly higher than in uninfected control cells (83). In addition, Sim et al. (2009) (83) had earlier proposed that lung epithelial cells may produce pro-inflammatory cytokines and chemokines, as essential mechanisms of the innate immune response, during the early B. pseudomallei infection. MCP-1 upregulation in murine lung epithelial cells, on the other hand, was dependable with the earlier in vivo studies (84).
TNF- is a member of a set of cytokines that activate the acute phase reaction and is involved in systemic inflammation. These are generated by activated macrophages generate it, but can also be released by varies cell types such as CD4+ lymphocytes, NK cells, and neurons. Barnes et al. (2008) (85) demonstrated that TNF- is needed to regulate B. pseudomallei infection in a mouse model, in which the mice were more inclined to infection with increased number of bacterial in the spleen and liver, with elevated mortality rates in TNF-/, TNFR1, and TNFR2/mice. Increased TNF- levels in human melioidosis sepsis patients, on the other hand, have been linked to higher mortality rates (3). Furthermore, Bearss et al. (2017) (86) discovered that TNF- levels were significantly higher in the livers of BALB/c mice upon infection with B. pseudomallei, while levels were lesser in the liver of C57Bl/6 mice (86). This substantiates the dual role played by TNF- in B. pseudomallei infection associated defence and susceptibility control.
During B. pseudomallei infection, IFN- is an effective regulator for generating protective immunity (87). Previous studies indicate that IFN- could regulate the proliferation of B. pseudomallei in macrophages, as a preliminary target after its entry via inhalation. Activation of IFN-γ by macrophages helps host defence by phagocyting and destroying B. pseudomallei via release of reactive oxygen and nitrogen intermediates (ROIs and RNIs), cytokine and chemokine, and antigen presentation to T cells (88) (Figure 1).
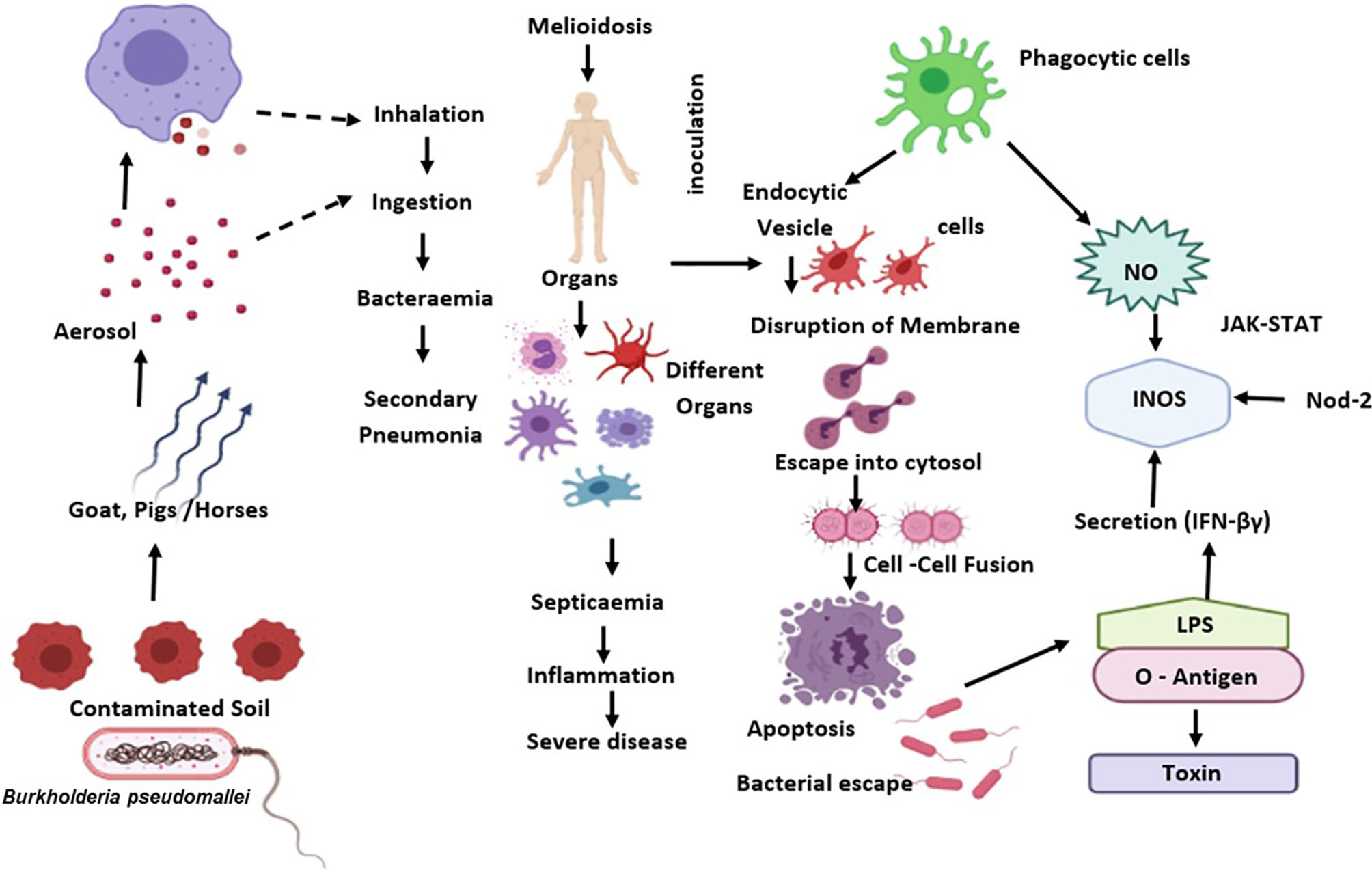
Figure 1 B. pseudomallei is diffused from the external reservoir to respiratory epithelial cells where the initial attachment initiates possibly through capsule and type IV pili. Upon invasion of epithelial cells, the T3SS-3 effectors promote in vacuolar evade and intracellular motility via BimA-mediated actin polymerization. Subsequent to phagocytosis, B. pseudomallei exits the phagosome and enters the host cytoplasm to replicates. This eventually causes the host cell death through initiation of apoptosis. The B. pseudomallei LPS regulates IFN secretion, which down-regulates iNOS expression and NO production. Also, B. pseudomallei influence the regulation of suppressor of cytokine signalling and cytokine-inducible Src homology 2-containing proteins that obstruct the JAK–STAT signalling pathway and iNOS activation.
Several other immune evasion strategies exist that includes inhibition of iNOS and development of TNF-α, up-regulation of the suppressor of cytokine signalling 3 (SOCS3) and cytokine-inducible Src homology 2-containing protein (CIS) that resulted in IFN-signaling reduction (53, 89). IFN- γ is primarily generated by NK cells and stimulated CD8+ T cells (90), but these cells are not actually involved in early defence, implying that the source of IFN- is highly redundant (91). The IFN- γ response is required for resistance in mice, particularly during early infection with B. pseudomallei, as established by the swift death of IFN- γ KO mice and mice treated with neutralizing monoclonal antibodies (mAb) against IFN- γ (71, 91, 92). They have revealed that the IFN-γ-inducing cytokines IL-12 and IL-18, are necessary for B. pseudomallei infection resistance (92).
Koo and Gan (2006) (93) observed overproduction of IFN-γ in mice during unrestrained bacterial loads. Additionally, Breitbach et al. (2009) (80) also found that macrophages from mice were not as susceptible to IFN-γ stimulated killing of B. pseudomallei. Recently, some researchers discovered that IFN-γ development controls B. pseudomallei intracellular killing, which is consistently higher in IFN-γ/- mice compared to WT infected with B. pseudomallei. They also suggested the initiation of IFN-γ production in vivo has not been completely illuminated; however, higher IFN-γ production by NK cells in the lungs have been shown (94).
B. pseudomallei evades humoral and cell-mediated immunity by rapidly spreading from cell to cell. In a human melioidosis infection, B. pseudomallei and its main microbial virulence factors not only promotes tissue invasion and necrosis but also enable evasion of normal humoral and cell-mediated immunity by surviving within both phagocytic and non-phagocytic host cells (37, 69). B. pseudomallei could colonize the host cell by performing phagosomal escape into the cytoplasm, resulting in multinucleated giant cells (MNGC) (54, 61). Although the exact function of MNGC in melioidosis relapse is unknown, we believe that the bacteria are encapsulated by MNGCs, preventing exposure to circulating antibodies and B cells that surround the host cell and further failure in neutralization. MNGC can fuse with surrounding cells under suitable conditions, releasing the dormant bacterial cells with further dissemination (95).
B. pseudomallei also uses an immune evasion technique called actin tail formation to replicate and spreads from cell to cell (96). This process prevents bacterial cells from coming into direct contact with the immune effector cells that are present around the host cells. This process is carried out by B. pseudomallei using actin-based mobility, which allows the pathogen to travel very rapidly across the cytoplasm. The filaments of actin that drive the bacteria in its movement are extremely stable (97). Rickettsia rickettsii, which propagates by a similar mechanism as B. pseudomallei and can travel at 4.8 m/min in the cytoplasm, is an example of bacteria that has successfully evaded the immune system (98). Based on the other results, it can be elucidated that B. pseudomallei could propagate much faster inside the cytoplasm, allowing them to spread systemically and outpace the human immune system’s surveillance rate (99).
During infection, the adaptive immune response is known as the third line of defense, and it has the potential to provide specific, long-term, and highly efficient protection against vital pathogens. Adaptive immune response gets triggered upon the breach of specific external barriers and B. pseudomallei is likely to compromise the adaptive immune system (37). Both T- and B-cells are involved in the cellular immune response, and they work together to suppress microbial infection (100). The cytotoxic T-cell, memory B-cell, and antibodies confers the third line of protection in the human body (101) (Figure 2).
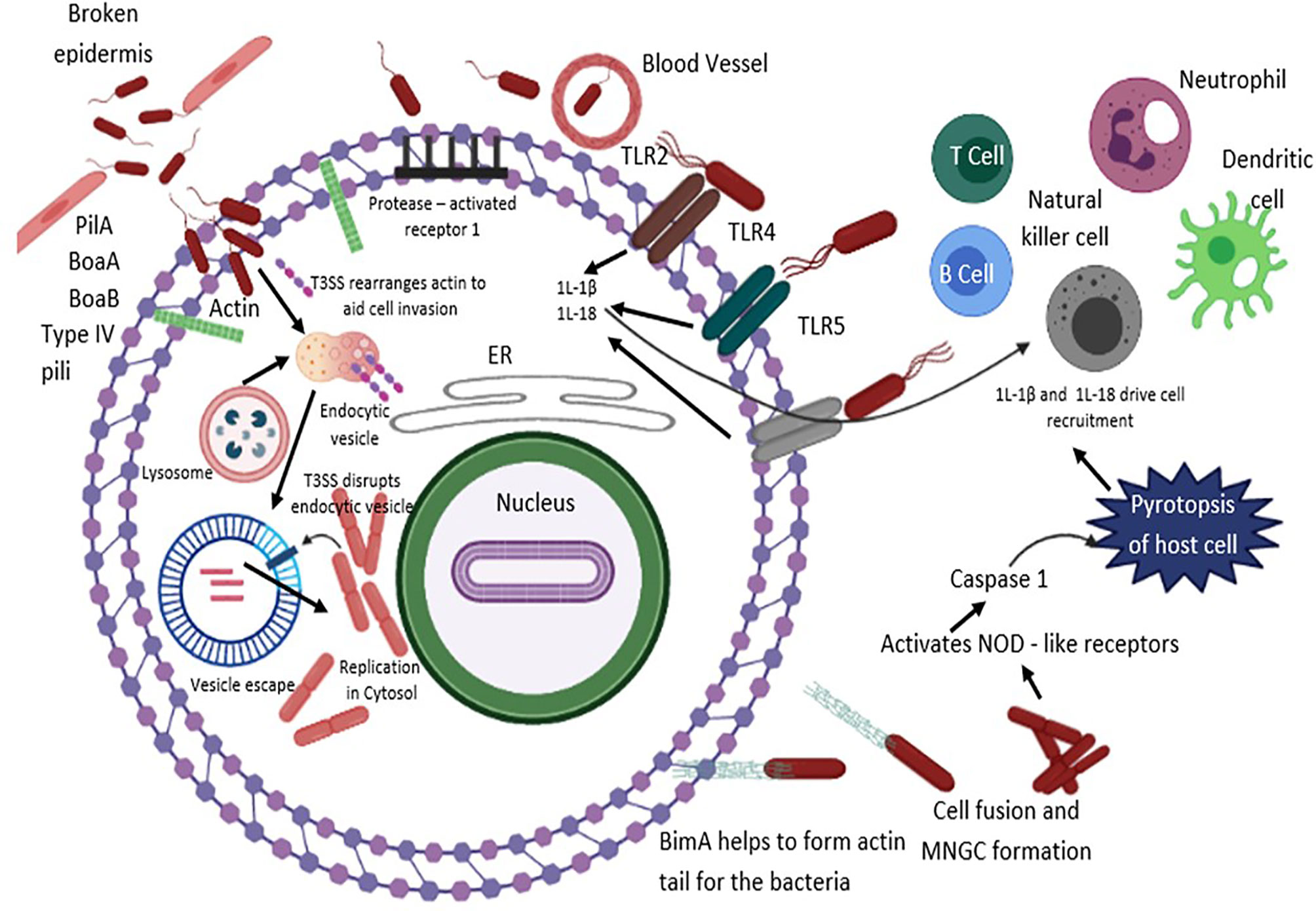
Figure 2 B. pseudomallei secretes N-acyl-homoserine lactone (AHL) signalling molecules that are implicated in coordinating attacks in contradiction of the host environment and biofilm formation. The T3SS effector proteins are vital for invasion and escape from host endosomal vesicle. Host cell entry is assisted by flagella, lipopolysaccharide (LPS), type IV pili and adhesion molecules. B. pseudomallei evades the vesicle and lyses the endosomal membrane using T3SS, T6SS and T2SS. Production of cationic peptides and ecotins facilitate B. pseudomallei to survive within an acidic endosomal environment. BopA (an effector protein) and BipD (translocator protein) obstruct sequestration in endosomal vesicles and avert microtubule-associated protein mediated autophagy. Upon entry into the cytoplasm compartment, B. pseudomallei replicates, and initiates the development of actin-based membrane protrusions and can pass through constant polymerization of host cell actin, thus accelerating dissemination to neighboring cells causing cell fusion and multinuclear giant cell (MNGC) formation. TLRs positioned on cell surfaces engage with the PAMPs and mediate NF-κB-induced initiation of immune responses, releasing pro-inflammatory cytokines IL-1β and IL-18 and facilitate caspase-1-mediated pyroptosis. Additionally, IL-18 warrants defensive IFNγ production, which allows recruitment of neutrophils, dendritic cells, B cells and T cells to the infection site, eventually activating the complement and coagulation cascades.
It is a well-known fact that cell-mediated immune responses mounted by lymphocytes activate leukocytes via cell-to-cell interaction or secretion of cytokines (100). The importance of CMI responses in melioidosis has become critical, as little is known about their role in the disease. Furthermore, since B. pseudomallei is an intracellular pathogen specific immune responses are key to antibacterial defence (16).
An avid understanding on the cellular immune response is essential for vaccine and drug delivery systems to induce the appropriate immune effectors for stimulating a defensive response and generation of specific antibodies against any pathogen (85, 102, 103). In view with this, Healey et al. (2005) (104) used dendritic cells (DC) to induce CMI responses to B. pseudomallei and are essential for the development of adaptive immune responses. There has also been clinical (90, 105) and experimental evidence (90, 106) to indicate that CMI may be vital for the existence of B. pseudomallei after infection. Individuals not affected by B. pseudomallei can undergo seroconversion developing a robust CMI, which may defend them from disease progression.
T-cell possess specific antigen receptors that can significantly bind to the target antigens leading to cytotoxicity. The foreign antigen must typically be recognized in combination with the self-major histocompatibility complexes (MHC) molecules to evoke T-cell immune responses (107) via expression of unique membrane molecules. The main receptors include CD4+ or CD8+ at the same time, and both help the T-cells to communicate with antigen-presenting cells (APC) by interacting with the antigen-presenting molecules. Though there are numerous reports on the role of CD4+ and CD8+ T cells in immunity to many pathogenic species (108), it still remains a ‘tip of the iceberg’ in the context of melioidosis.
Tippayawatet al. (2009) (90) found that individuals who are healthy with serological manifest of B. pseudomallei exposure and melioidosis patients produced CD4+ and CD8+ T cells that recognized the B. pseudomallei ATP-binding cassette (ABC) transporter family. A robust T-cell response might be needed for the determination of clinical melioidosis. In patients who improved from clinical melioidosis, B. pseudomallei reactive T-cells were stimulated and able of generating antigenic-specific gamma interferon (IFN) with the ability to thrive in-vitro in response to B. pseudomallei (3, 105).
Experimental evidences also have documented that mice infected with B. pseudomallei develop CMI, detected by delayed-type hypersensitivity (DTH) response with lymphocyte proliferation (88). The RAG-mice (absence of T- and B-cells) acquired infection more quickly than the C57BL/6 wild-type mice, according to a report by Haque and colleagues (2006) (109). The findings showed that primary infection with B. pseudomallei peaks the memory CD4+ and CD8+ T cell populations. In vitro studies demonstrated that the T cells isolated from B. pseudomallei-infected C57BL/6 mice developed Ag-specific IFN in response to re-stimulation. Further research into CD4+ or CD8+ T cell depletion revealed that CD4+ T-cell deficient mice died significantly sooner than the control mice. The CD8+ T cell depleted mice, on the other hand, had a brief survival time than the controls, but the alteration was not statistically substantial and indicated that CD4+ T cells, not CD8+ T cells, are involved in the elicited defence (86).
Not many studies have reported on T cell exhaustion in B. pseudomallei infection, which occurs due to scarce intracellular expression of IFN-γ (110). Programmed death 1 (PD-1) is a known T cell activation regulator purportedly expressed on various cell types, including T cells and myeloid cells and abundantly up-regulated during inflammation (111). Kleijn et al. (2013) (112) have stated that following exposure to a low dose of bacterial LPS, PD-1 was regulated on certain neutrophils and obstructs T cell function via the PD-1 pathway.
In B. pseudomallei infection, PD-1 mRNA was found to be elevated in the peripheral blood and also during the onset of inflammation in PMNs in vitro. In in vitro B. pseudomallei–infected PMNs, blocked inhibitory activity and reestablished proliferation of CD4+ T cells and IFN-γ proliferation were observed upon inclusion of anti–PD-1, suggesting that PD-L1 is a monitoring molecule of T cell functions and could play a major role in pathogenesis (113). Using an experimental in vivo mice model with persistent B. pseudomallei infection, See et al. (2016) (114) found a significant reduction in CD4+ T-cell functions. In addition, up-regulation of PD-1 on both CD4+ and CD8+ T cells was observed conceivably signifying that the T cells were enduring exhaustion and postulated that B. pseudomallei 'engineers' the exhaustion of CD4+ T cells and up-regulation of PD-1 that hypothetically enables bacterial persistence in the host.
In addition, using a mice model, See et al. (2017) (115) characterized the regulation of PD-1 and CTLA-4 levels on B cells, NK cells, and monocytes and confirmed that persistent infection with B. pseudomallei small colony variants (SVCs) can concomitantly cause PD-1 upregulation on B cells, NK cells and monocytes signifying host immune exhaustion. The SCVs were found to up-regulate the PD-1 on NK cells and monocytes in contrast to its WT counterpart. Based on the combined in vivo experiments, it was concluded that in B. pseudomallei SCVs infection, PD-1 was up-regulated on both adaptive and innate immune cells, whereas PD-1 up-regulation only was observed in B. pseudomallei WT infection (114, 115). They also speculated that SCVs initiate greater PD-1 expression to overwhelm the host immune responses and enable persistence, causing elevated bacterial burden. Recently, Menon et al. (2020) (116) demonstrated up-regulation of PD-1 on T cells in vitro, upon exposure to crude culture filtrate antigens of B. pseudomallei and suggested that this possibly contributes to deprived immune surveillance and pathogenesis.
Antibodies resulting from constant exposure to B. pseudomallei are normal among people living in melioidosis endemic areas. This likely could be attributed to the presence of high levels of B. pseudomallei-specific IgG, IgM, and IgA antibodies in melioidosis patients’ sera, which persistently raised for the length of infection (106, 117). According to a study by Khakhum et al. (2019) (116), IgG and their subclasses are inevitably significant in the detection of B. pseudomallei infection. Besides, a few scientific findings revealed that a discerning absence of one or more IgG sub-classes likely increases the susceptibility to recurrent infections or extend the course of an established infection. Furthermore, in melioidosis positive cases, IgG antibody exhibited a sturdier and more stable response than IgM antibody, which showed a more variable response. According to Patel et al. (2011) (88), the existence of B. pseudomallei in lymphoid tissues continues to induce recently maturing B cells to express IgM that can subsequently be substituted to more potent isotypes to combat the bacteria effectively. Vasu et al. (2003) (119) suggested that the levels of IgG antibodies (IgG1 or IgG2) generated against culture filtrate antigen in patients under antibiotic therapy could be indicative of the grade of infection and may be used as a standard guideline to regulate the extent of maintenance of antimicrobial therapy.
Since B. pseudomallei is known to be air-borne (74), the immune defence in the respiratory tract is paramount to prevention of infection. The lower respiratory tracthas a preponderance in IgG, whereas the upper respiratory fluids are enriched with secretory IgA, and with low levels of systemic IgG. Also, pathogens bound to IgA are picked up by airway macrophages via phagocytosis. As a result, we believe that B. pseudomallei uses macrophages as a vehicle for systemic dissemination in the host (120). Yi et al. (2019) (121) demonstrated that IgG and IgM defines the versatility of immunogenic proteins generated from the host challenged with B. pseudomallei via aerosol. The immune response was noticeable from the early stage of the infection and antigens elicited robust concentration for either or both of IgG and IgM.
Inactivation of reactive oxygen and nitrogen species, inhibition of phagolysosome development, and phagosomal escape are among the critical mechanisms employed by intracellular bacteria to evade host immune responses (122). Similarly, B. pseudomallei has unique characteristics that allow it to overcome its host and to meet its metabolic needs. This bacterium is currently being examined extensively from an immunological perspective to establish a void in its evasive strategies and, hopefully, find a perpetual solution to its intracellular persistence. B. pseudomallei causes a complement system deficiency, which dampens the humoral immunity. The capacity of B. pseudomallei to live in the human host is an intriguing phenomenon. B. pseudomallei does not lyse in human serum, according to Canadian scientists, because of its unusual CPS production, which aids in survival by inhibiting the deposition of C3b, adding to it's virulence. Further, B. pseudomallei decreased C3b deposition on the bacterium’s surface, rendering it resistant to phagocytosis leading to bacterial persistence in the host (25).
On the other hand, Carter and Fearon (1992) (123) showed that the B cell activation can be lowered with the co-ligation of the co-receptor and the B-cell antigen receptor (BCR), thus proving the role of complement influencing humoral responses (124). This fact was further supported by the presence of fusion proteins of multimers of C3d and lysozymes that lowered the in-vitro activation threshold of B cells (125). Animal models proved the relationship between the complement systems and humoral immunity control (126) substantiating the fact that if the complement-mediated immunity is decreased, the stimulus for humoral immunity will be reduced as well, leading to the elimination of B. pseudomallei from host tissues.
This review confers a better understanding on the immunological perspective behind B. pseudomallei infections. The organism secretes a slew of virulence proteins that amend host cell functions and further evading from the host immune response. Understanding the bacterium’s basic virulence factors and pathogenic mechanisms are essential for delving into the adaption of B. pseudomallei inducing a wide array of clinical manifestations. The host-parasite interactions are often influenced by both humoral and cellular immune response together with complement mediated pathways. The bacterium has also been known to evade the host’s front-line immune defenses and exploit the host’s responses to survive in the host while receiving adequate and long-term antibiotic treatment. Further elaborative elucidations on the immune mechanism of melioidosis is the need of the hour in the development and progression of novel therapeutic strategies to curb the menace of B. pseudomallei infections.
VM, KMV, and ES conceived the concept. VM and KMV wrote the manuscript. ES, MB, and AG co-wrote the manuscript. JV edited the manuscript. All authors contributed to the article and approved the submitted version.
We thank University of Malaya Impact-Oriented Interdisciplinary Research Grant (IIRG019-2019) by Ministry of Higher Education - University of Malaya and Frontiers Research Grant (FG012-17AFR) for the funding. ES is supported by the Department of Science and Technology-Science and Engineering Research Board, Government of India (Grant Number CRG/2019/006096). The funders had no role in the study design, data collection and analysis or the decision to publish or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ashdown LR, Clarke SG. Evaluation of Culture Techniques for Isolation of Pseudomonas Pseudomallei From Soil. Appl Environ Microbiol (1992) 58(12):4011–5. doi: 10.1128/aem.58.12.4011-4015.1992
2. Dance DA. Ecology of Burkholderia Pseudomallei and the Interactions Between Environmental Burkholderia Spp. And Human-Animal Hosts. Acta Tropica (2000) 74(2-3):159–68. doi: 10.1016/S0001-706X(99)00066-2
3. Cheng AC, Currie BJ. Melioidosis: Epidemiology, Pathophysiology, and Management. Clin Microbiol Rev (2005) 18(2):383–416. doi: 10.1128/CMR.18.2.383-416.2005
4. Baker A, Tahani D, Gardiner C, Bristow KL, Greenhill AR, Warner J. Groundwater Seeps Facilitate Exposure to Burkholderia Pseudomallei. Appl Environ Microbiol (2011) 77(20):7243–6. doi: 10.1128/AEM.05048-11
5. Holland DJ, Wesley A, Drinkovic D, Currie BJ. Cystic Fibrosis and Burkholderia Pseudomallei Infection: An Emerging Problem? Clin Infect Dis (2002) 35(12):138–40. doi: 10.1086/344447
6. Webling DD. Genito-Urinary Infections With Pseudomonas Pseudomallei in Australian Aboriginals. Trans R Soc Trop Med Hygiene (1980) 74(1):138–9. doi: 10.1016/0035-9203(80)90036-X
7. Ralph A, McBride J, Currie BJ. Transmission of Burkholderia Pseudomallei via Breast Milk in Northern Australia. Pediatr Infect Dis J (2004) 23(12):1169–71. doi: 10.1056/NEJM200104123441516
8. Abbink FC, Orendi JM, de Beaufort AJ. Mother-To-Child Transmission of Burkholderia Pseudomallei. New Engl J Med (2011) 344(15):1171–2. doi: 10.1056/NEJM200104123441516
9. Currie BJ, Fisher DA, Anstey NM, Jacups SP. Melioidosis: Acute and Chronic Disease, Relapse and Re-Activation. Trans R Soc Trop Med Hygiene (2000) 94(3):301–4. doi: 10.1016/S0035-9203(00)90333-X
10. Ngauy V, Lemeshev Y, Sadkowski L, Crawford G. Cutaneous Melioidosis in a Man Who was Taken as a Prisoner of War by the Japanese During World War II. J Clin Microbiol (2005) 43(2):970–2. doi: 10.1128/JCM.43.2.970-972.2005
11. Alsaif HS, Venkatesh SK. Melioidosis: Spectrum of Radiological Manifestations. Saudi J Med Med Sci (2016) 4(2):74–8. doi: 10.4103/1658-631X.178286
12. Limmathurotsakul D, Wongratanacheewin S, Teerawattanasook N, Wongsuvan G, Chaisuksant S, Chetchotisakd P, et al. Increasing Incidence of Human Melioidosis in Northeast Thailand. Am J Trop Med Hygiene (2010) 82(6):1113–7. doi: 10.4269/ajtmh.2010.10-0038
13. Gibney KB, Cheng AC, Currie BJ. Cutaneous Melioidosis in the Tropical Top End of Australia: A Prospective Study and Review of the Literature. Clin Infect Dis (2008) 47(5):603–9. doi: 10.1086/590931
14. Limmathuratsakul D, Chaowagul W, Chantratita N, Wuthienkanun V, Biaklang M, Tupama S, et al. A Simple Scoring System to Differentiate Between Relapse and Re-Infection in Patiens With Recurrent Melioidosis. PloS Neglected Trop Dis (2008) 2(10):327. doi: 10.1371/journal.pntd.0000327
15. Dance D. Treatment and Prophylaxis of Melioidosis. Int J Antimicrobial Agents (2014) 43(4):310–8. doi: 10.1016/j.ijantimicag.2014.01.005
16. Ramar PS, Stiles BG, Sethi G, Lim L. Melioidosis: Clinical Impact and Public Health Threat in the Tropics. PloS Neglected Trop Dis (2017) 11(5):e0004738. doi: 10.1371/journal.pntd.0004738
17. Nathan S, Chieng S, Kingsley PV, Mohan A, Podin Y, Ooi MH, et al. Melioidosis in Malaysia: Incidence, Clinical Challenges, and Advances in Understanding Pathogenesis. Trop Med Infect Dis (2019) 3(1):25. doi: 10.3390/tropicalmed3010025
18. Sarkar-Tyson M, Thwaite JE, Harding SV, Smither SJ, Oyston PCF, Atkins TP, et al. Polysaccharides and Virulence of Burkholderia pseudomallei. J Med Microbiol (2007) 56(Pt 8):1005–10. doi: 10.1099/jmm.0.47043-0
19. Haiko J, Westerlund-Wikström B. The Role of the Bacterial Flagellum in Adhesion and Virulence. Biology (2013) 2(4):1242–67. doi: 10.3390/biology2041242
20. Yang P, Zhang M, van Elsas JD. Role of Flagella and Type Four Pili in the Co-Migration of Burkholderia Terrae BS001 With Fungal Hyphae Through Soil. Sci Rep (2017) 7:2997. doi: 10.1038/s41598-017-02959-8
21. Burtnick MN, Brett PJ, Harding SV, Ngugi SA, Ribot WJ, Chantratita N, et al. The Cluster 1 Type VI Secretion System Is a Major Virulence Determinant in Burkholderia Pseudomallei. Infection Immun (2011) 79(4):1512–25. doi: 10.1128/IAI.01218-10
22. Burtnick MN, Brett PJ, DeShazer D. Proteomic Analysis of the Burkholderia Pseudomallei Type II Secretome Reveals Hydrolytic Enzymes, Novel Proteins, and the Deubiquitinase TssM. Infection Immun (2014) 82(8):3214–26. doi: 10.1128/IAI.01739-14
23. Ulrich RL, Deshazer D, Hines HB, Jeddeloh JA. Quorum Sensing: A Transcriptional Regulatory System Involved in the Pathogenicity of Burkholderia Mallei. Infection Immun (2004) 72(11):6589–96. doi: 10.1128/IAI.72.11.6589-6596.2004
24. Chen YS, Shiuan D, Chen SC, Chye SM, Chen YL. Recombinant Truncated Flagellin of Burkholderia Pseudomallei as a Molecular Probe for Diagnosis of Melioidosis. Clin Diagn Lab Immunol (2003) 10(3):423–5. doi: 10.1128/CDLI.10.3.423-425.2003
25. Reckseidler-Zenteno SL, DeVinney R, Woods DE. The Capsular Polysaccharide of Burkholderia Pseudomallei Contributes to Survival in Serum by Reducing Complement Factor C3b Deposition. Infection Immun (2005) 73(2):1106–15. doi: 10.1128/IAI.73.2.1106-1115.2005
26. Korbsrisate S, Tomaras AP, Damnin S, Ckumdee J, Srinon V, Lengwehasatit I, et al. Characterization of Two Distinct Phospholipase C Enzymes From Burkholderia Pseudomallei. Microbiology (2007) 153(Pt 6):1907–15. doi: 10.1099/mic.0.2006/003004-0
27. Allwood EM, Devenish RJ, Prescott M, Adler B, Boyce JD. Strategies for Intracellular Survival of Burkholderia Pseudomallei. Front Microbiol (2011) 2:170. doi: 10.3389/fmicb.2011.00170
28. Srinon V, Chaiwattanarungruengpaisan S, Korbsrisate S, Stevens JM. Burkholderia Pseudomallei BimC Is Required for Actin-Based Motility, Intracellular Survival, and Virulence. Front Cell Infection Microbiol (2019) 9:63. doi: 10.3389/fcimb.2019.00063
29. Ramli NS, Eng Guan C, Nathan S, Vadivelu J. The Effect of Environmental Conditions on Biofilm Formation of Burkholderia Pseudomallei Clinical Isolates. PloS One (2012) 7(9):e44104. doi: 10.1371/journal.pone.0044104
30. Cheng AC, Currie BJ, Dance D, Funnell S, Limmathurotsakul D, Simpson A, et al. Clinical Definitions of Melioidosis. Am J Trop Med Hygiene (2013) 88(3):411–3. doi: 10.4269/ajtmh.12-0555
31. Sakai T, Kohzaki K, Watanabe A, Tsuneoka H, Shimadzu M. Use of DNA Microarray Analysis in Diagnosis of Bacterial and Fungal Endophthalmitis. Clin Ophthalmol (Auckland NZ) (2012) 6:321–6. doi: 10.2147/OPTH.S29230
32. Ranjbar R, Behzadi P, Najafi A, Roudi R. DNA Microarray for Rapid Detection and Identification of Food and Water Borne Bacteria: From Dry to Wet Lab. Open Microbiol J (2017) 11:330–8. doi: 10.2174/1874285801711010330
33. Chieng S, Carreto L, Nathan S. Burkholderia Pseudomallei Transcriptional Adaptation in Macrophages (2012). BMC Genomics (2012) 13:328. doi: 10.1186/1471-2164-13-328
34. Birnie E, Claushuis T, Koh G, Limmathurotsakul D, Day N, Roelofs J, et al. Thrombocytopenia Impairs Host Defense Against Burkholderia Pseudomallei (Melioidosis). J Infect Dis (2019) 219(4):648–59. doi: 10.1093/infdis/jiy541
35. Krishnananthasivam S, Jayathilaka N, Sathkumara HD, Corea E, Natesan M, De Silva AD. Host Gene Expression Analysis in Sri Lankan Melioidosis Patients. PloS Neglected Trop Dis (2017) 11(6):e0005643. doi: 10.1371/journal.pntd.0005643
36. Chin CY, Monack DM, Nathan S. Genome Wide Transcriptome Profiling of a Murine Acute Melioidosis Model Reveals New Insights Into How Burkholderia Pseudomallei Overcomes Host Innate Immunity. BMC Genomics (2010) 11:672. doi: 10.1186/1471-2164-11-672
37. Vellasamy KM, Mariappan V, Shankar EM, Vadivelu J. Burkholderia Pseudomallei Differentially Regulates Host Innate Immune Response Genes for Intracellular Survival in Lung Epithelial Cells. PloS Neglected Trop Dis (2016) 10(7):e0004730. doi: 10.1371/journal.pntd.0004730
38. Rao C, Chan M, Xia Y, Zhang M, Hu Z, Yuan S, et al. Transcriptome Analysis Reveals Unfolded Protein Response was Induced During the Early Stage of Burkholderia Pseudomallei Infection in A549 Cells. J Front Genet (2020) 11:1522. doi: 10.3389/fgene.2020.585203
39. Heacock-Kang Y, McMillan IA, Norris MH, Sun Z, Zarzycki-Siek J, Bluhm AP, et al. The Burkholderia Pseudomallei Intracellular ‘Transitome’. Nat Commun (2021) 12:1907. doi: 10.1038/s41467-021-22169-1
40. Clarke TB. Microbial Programming of Systemic Innate Immunity and Resistance to Infection. PloS Pathog (2014) 10(12):e1004506. doi: 10.1371/journal.ppat.1004506
41. Vance RE, Isberg RR, Portnoy DA. Patterns of Pathogenesis: Discrimination of Pathogenic and Nonpathogenic Microbes by the Innate Immune System. Cell Host Microbe (2009) 6(1):10–21. doi: 10.1016/j.chom.2009.06.007
42. Scott AE, Burtnick MN, Stokes MG, Whelan AO, Williamson ED, Atkins TP, et al. Burkholderia Pseudomallei Capsular Polysaccharide Conjugates Provide Protection Against Acute Melioidosis. Infection Immun (2014) 82(8):3206–13. doi: 10.1128/IAI.01847-14
43. Estes DM, Dow SW, Schweizer HP, Torres AG. Present and Future Therapeutic Strategies for Melioidosis and Glanders. Expert Rev Anti-Infect Ther (2011) 8(3):325–38. doi: 10.1586/eri.10.4
44. de Jong HK, Koh GC, Achouiti A, van der Meer AJ, Bulder I, Stephan, et al. Neutrophil Extracellular Traps in the Host Defense Against Sepsis Induced by Burkholderia Pseudomallei (Melioidosis). Intensive Care Med Exp (2014) 2(1):21. doi: 10.1186/s40635-014-0021-2
45. Mohanty T, Fisher J, Bakochi A, Neumann A, Cardoso J, Karlsson C, et al. Neutrophil Extracellular Traps in the Central Nervous System Hinder Bacterial Clearance During Pneumococcal Meningitis. Nat Commun (2019) 10(1):1667. doi: 10.1038/s41467-019-09040-0
46. Riyapa D, Buddhisa S, Korbsrisate S, Cuccui J, Wren BW, Stevens M, et al. Neutrophil Extracellular Traps Exhibit Antibacterial Activity Against Burkholderia Pseudomallei and Are Influenced by Bacterial and Host Factors. Infection Immun (2012) 80(11):3921–9. doi: 10.1128/IAI.00806-12
47. Hayden HS, Lim R, Brittnacher MJ, Sims EH, Ramage ER, Fong, et al. Evolution of Burkholderia Pseudomallei in Recurrent Melioidosis. PloS One (2012) 7(5):e36507. doi: 10.1371/journal.pone.0036507
48. Motwani MP, Newson J, Kwong S, Richard-Loendt A, Colas R, Dalli J, et al. Prolonged Immune Alteration Following Resolution of Acute Inflammation in Humans. PloS One (2017) 12(10):e0186964. doi: 10.1371/journal.pone.0186964
49. Bhattacharya S, Ploplis VA, Castellino FJ. Bacterial Plasminogen Receptors Utilize Host Plasminogen System for Effective Invasion and Dissemination. J Biomed Biotechnol (2012) 2012:482096. doi: 10.1155/2012/482096
50. Birnie E, Weehuizen T, Lankelma JM, de Jong HK, Koh G, van Lieshout M, et al. Role of Toll-Like Receptor 5 (TLR5) in Experimental Melioidosis. Infection Immun (2019) 87(8):e00409–18. doi: 10.1128/IAI.00409-18
51. Wiersinga WJ, Virk HS, Torres AG, Currie BJ, Peacock SJ, Dance D, et al. Melioidosis. Nat Rev Dis Primers (2018) 4:17107. doi: 10.1038/nrdp.2017.107
52. Jones AL, Beveridge TJ, Woods DE. Intracellular Survival of Burkholderia Pseudomallei. Infection Immun (1996) 64(3):782–90. doi: 10.1128/iai.64.3.782-790.1996
53. Notti RQ, Stebbins CE. The Structure and Function of Type III Secretion Systems. Microbiol Spectr (2016) 4(1):10.1128. doi: 10.1128/microbiolspec.VMBF-0004-2015
54. Vadivelu J, Vellasamy KM, Thimma J, Mariappan V, Kang WT, Choh LC, et al. Survival and Cellular Trafficking of Persistent Bacteria. PloS Neglected Trop Dis (2017) 11(1):e0005241. doi: 10.1371/journal.pntd.0005241
55. Vander Broek CW, Chalmers KJ, Stevens MP, Stevens JM. Quantitative Proteomic Analysis of Burkholderia Pseudomallei Bsa Type III Secretion System Effectors Using Hypersecreting Mutants. Mol Cell Proteomics: MCP (2015) 14(4):905–16. doi: 10.1074/mcp.M114.044875
56. Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, et al. The Burkholderia Pseudomallei Type III Secretion System and BopA Are Required for Evasion of LC3-Associated Phagocytosis. PloS One (2011) 6(3):e17852. doi: 10.1371/journal.pone.0017852
57. Jamaati H, Mortaz E, Pajouhi Z, Folkerts G, Movassaghi M, Moloudizargari M, et al. Nitric Oxide in the Pathogenesis and Treatment of Tuberculosis. Front Microbiol (2017) 8:2008. doi: 10.3389/fmicb.2017.02008
58. Ekchariyawat P, Pudla S, Limposuwan K, Arjcharoen S, Sirisinha S, Utaisincharoen P. Burkholderia Pseudomallei-Induced Expression of Suppressor of Cytokine Signaling 3 and Cytokine-Inducible Src Homology 2-Containing Protein in Mouse Macrophages: A Possible Mechanism for Suppression of the Response to Gamma Interferon Stimulation. Infection Immun (2005) 73(11):7332–9. doi: 10.1128/IAI.73.11.7332-7339.2005
59. Arjcharoen S, Wikraiphat C, Pudla M, Limposuwan K, Woods DE, Sirisinha S, et al. Fate of a Burkholderia Pseudomallei Lipopolysaccharide Mutant in the Mouse Macrophage Cell Line RAW 264.7: Possible Role for the O-Antigenic Polysaccharide Moiety of Lipopolysaccharide in Internalization and Intracellular Survival. Infection Immun (2007) 75(9):4298–304. doi: 10.1128/IAI.00285-07
60. Pareja ME, Colombo MI. Autophagic Clearance of Bacterial Pathogens: Molecular Recognition of Intracellular Microorganisms. Front Cell Infection Microbiol (2013) 3:54. doi: 10.3389/fcimb.2013.00054
61. Singh AP, Lai SC, Nandi T, Chua HH, Ooi WF, Ong C, et al. Evolutionary Analysis of Burkholderia Pseudomallei Identifies Putative Novel Virulence Genes, Including a Microbial Regulator of Host Cell Autophagy. J Bacteriol (2013) 195(24):5487–98. doi: 10.1128/JB.00718-13
62. Tan KS, Chen Y, Lim YC, Tan GY, Liu Y, Lim YT, et al. Suppression of Host Innate Immune Response by Burkholderia Pseudomallei Through the Virulence Factor TssM. J Immunol (2010) 184(9):5160–71. doi: 10.4049/jimmunol.0902663
63. Alomairi J, Bonacci T, Ghigo E, Soubeyran P. Alterations of Host Cell Ubiquitination Machinery by Pathogenic Bacteria. Front Cell Infection Microbiol (2015) 5:17. doi: 10.3389/fcimb.2015.00017
64. Lee SH, Wong RR, Chin CY, Lim TY, Eng SA, Kong C, et al. Burkholderia Pseudomallei Suppresses Caenorhabditis Elegans Immunity by Specific Degradation of a GATA Transcription Factor. Proc Natl Acad Sci USA (2013) 110(37):15067–72. doi: 10.1073/pnas.1311725110
65. Block DH, Twumasi-Boateng K, Kang HS, Carlisle JA, Hanganu A, Lai TY, et al. The Developmental Intestinal Regulator ELT-2 Controls P38-Dependent Immune Responses in Adult. Caenorhabditis Elegans PloS Genet (2015) 11(5):e1005265. doi: 10.1371/journal.pgen.1005265
66. Galle M, Carpentier I, Beyaert R. Structure and Function of the Type III Secretion System of Pseudomonas Aeruginosa. Curr Protein Pept Sci (2012) 13(8):831–42. doi: 10.2174/138920312804871210
67. Wiersinga WJ, Poll TVD, White NJ, Day NP, Peacock SJ. Melioidosis: Insight Into the Pathogenesis of Burkholderia Pseudomallei. Nat Rev Microbiol (2006) 4:272–82. doi: 10.1038/nrmicro1385
68. Conejero L, Patel N, de Reynal M, Oberdorf S, Prior J, Felgner PL, et al. Low-Dose Exposure of C57BL/6 Mice to Burkholderia Pseudomallei Mimics Chronic Human Melioidosis. Am J Pathol (2011) 179(1):270–80. doi: 10.1016/j.ajpath.2011.03.031
69. Stone JK, DeShazer D, Brett PJ, Burtnick MN. Melioidosis: Molecular Aspects of Pathogenesis. Expert Rev Anti-Infect Ther (2004) 12(12):1487–99. doi: 10.1586/14787210.2014.970634
70. Wiersinga WJ, Wieland CW, Roelofs JJ, van der Poll T. MyD88 Dependent Signaling Contributes to Protective Host Defense Against Burkholderia Pseudomallei. PloS One (2008) 3(10):e3494. doi: 10.1371/journal.pone.0003494
71. Wiersinga WJ, Wieland CW, Dessing MC, Chantratita N, Cheng AC, Limmathurotsakul D, et al. Toll-Like Receptor 2 Impairs Host Defense in Gram-Negative Sepsis Caused by Burkholderia Pseudomallei (Melioidosis). PloS Med (2007) 4(7):e248. doi: 10.1371/journal.pmed.0040248
72. Kunyanee C, Kamjumphol W, Taweechaisupapong S, Kanthawong S, Wongwajana S, Wongratanacheewin S, et al. Burkholderia Pseudomallei Biofilm Promotes Adhesion, Internalization and Stimulates Proinflammatory Cytokines in Human Epithelial A549 Cells. PloS One (2016) 11(8):e0160741. doi: 10.1371/journal.pone.0160741
73. Weehuizen TA, Prior JL, van der Vaart TW, Ngugi SA, Nepogodiev SA, Field RA, et al. Differential Toll-Like Receptor-Signalling of Burkholderia Pseudomallei Lipopolysaccharide in Murine and Human Models. PloS One (2015) 10(12):e0145397. doi: 10.1371/journal.pone.0145397
74. West TE, Ernst RK, Jansson-Hutson MJ, Skerrett SJ. Activation of Toll-Like Receptors by Burkholderia Pseudomallei. BMC Immunol (2008) 9:46. doi: 10.1186/1471-2172-9-46
75. Whiteley L, Meffert T, Haug M, Weidenmaier C, Hopf V, Bitschar K, et al. Entry, Intracellular Survival, and Multinucleated-Giant-Cell-Forming Activity of Burkholderia Pseudomallei in Human Primary Phagocytic and Nonphagocytic Cells. Infection Immun (2017) 85(10):e00468–17. doi: 10.1128/IAI.00468-17
76. Liu T, Zhang L, Joo D, Sun SC. NF-κb Signaling in Inflammation. Signal Transduct Targeted Ther (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
77. Baker RG, Hayden MS, Ghosh S. NF-κb, Inflammation, and Metabolic Disease. Cell Metab (2011) 13(1):11–22. doi: 10.1016/j.cmet.2010.12.008
78. West TE, Myers ND, Chantratita N, Chierakul W, Limmathurotsakul D, Wuthiekanun V, et al. NLRC4 and TLR5 Each Contribute to Host Defense in Respiratory Melioidosis. PloS Neglected Trop Dis (2014) 8(9):e3178. doi: 10.1371/journal.pntd.0003178
79. Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, et al. Caspase-11-Dependent Pyroptosis of Lung Epithelial Cells Protects From Melioidosis While Caspase-1 Mediates Macrophage Pyroptosis and Production of IL-18. PloS Pathog (2018) 14(5):e1007105. doi: 10.1371/journal.ppat.1007105
80. Breitbach K, Sun GW, Köhler J, Eske K, Wongprompitak P, Tan G, et al. Caspase-1 Mediates Resistance in Murine Melioidosis. Infection Immun (2009) 77(4):1589–95. doi: 10.1128/IAI.01257-08
81. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J Interferon Cytokine Res: Off J Int Soc Interferon Cytokine Res (2009) 29(6):313–26. doi: 10.1089/jir.2008.0027
82. Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, et al. Additive Roles for MCP-1 and MCP-3 in CCR2-Mediated Recruitment of Inflammatory Monocytes During Listeria Monocytogenes Infection. J Immunol (Baltimore Md: 1950) (2008) 180(10):6846–53. doi: 10.4049/jimmunol.180.10.6846
83. Sim SH, Liu Y, Wang D, Novem V, Sivalingam SP, Thong TW, et al. Innate Immune Responses of Pulmonary Epithelial Cells to Burkholderia Pseudomallei Infection. PloS One (2009) 4(10):e7308. doi: 10.1371/journal.pone.0007308
84. Schneider D, Hong JY, Bowman ER, Chung Y, Nagarkar DR, McHenry CL, et al. Macrophage/epithelial Cell CCL2 Contributes to Rhinovirus-Induced Hyperresponsiveness and Inflammation in a Mouse Model of Allergic Airways Disease. Am J Physiol Lung Cell Mol Physiol (2013) 304(3):L162–9. doi: 10.1152/ajplung.00182.2012
85. Barnes JL, Williams NL, Ketheesan N. Susceptibility to Burkholderia Pseudomallei Is Associated With Host Immune Responses Involving Tumor Necrosis Factor Receptor-1 (TNFR1) and TNF Receptor-2 (TNFR2). FEMS Immunology and. Med Microbiol (2008) 52(3):379–88. doi: 10.1111/j.1574-695X.2008.00389.x
86. Bearss JJ, Hunter M, Dankmeyer JL, Fritts KA, Klimko CP, Weaver, et al. Characterization of Pathogenesis of and Immune Response to Burkholderia Pseudomallei K96243 Using Both Inhalational and Intraperitoneal Infection Models in BALB/c and C57BL/6 Mice. PloS One (2017) 12(2):e0172627. doi: 10.1371/journal.pone.0172627
87. Silva EB, Dow SW. Development of Burkholderia Mallei and Pseudomallei Vaccines. Front Cell Infection Microbiol (2013) 3:10. doi: 10.3389/fcimb.2013.00010
88. Patel N, Conejero L, De Reynal M, Easton A, Bancroft GJ, Titball RW. Development of Vaccines Against Burkholderia Pseudomallei. Front Microbiol (2011) 2:198. doi: 10.3389/fmicb.2011.00198
89. White CA, Nicola NA. SOCS3: An Essential Physiological Inhibitor of Signaling by Interleukin-6 and G-CSF Family Cytokines. JAK-STAT (2013) 2(4):e25045. doi: 10.4161/jkst.25045
90. Tippayawat P, Saenwongsa W, Mahawantung J, Suwannasaen D, Chetchotisakd P, Limmathurotsakul D, et al. Phenotypic and Functional Characterization of Human Memory T Cell Responses to Burkholderia Pseudomallei. PloS Neglected Trop Dis (2009) 3(4):e407. doi: 10.1371/journal.pntd.0000407
91. Wongratanacheewin S, Kespichayawattana W, Intachote P, Pichyangkul S, Sermswan RW, Krieg AM, et al. Immunostimulatory CpG Oligodeoxynucleotide Confers Protection in a Murine Model of Infection With Burkholderia Pseudomallei. Infection Immun (2004) 72(8):4494–502. doi: 10.1128/IAI.72.8.4494-4502.2004
92. Santanirand P, Harley VS, Dance DA, Drasar BS, Bancroft GJ. Obligatory Role of Gamma Interferon for Host Survival in a Murine Model of Infection With Burkholderia Pseudomallei. Infection Immun (1999) 67(7):3593–600. doi: 10.1128/IAI.67.7.3593-3600.1999
93. Koo GC, Gan YH. The Innate Interferon Gamma Response of BALB/c and C57BL/6 Mice to In Vitro Burkholderia Pseudomallei Infection. BMC Immunol (2006) 7:19. doi: 10.1186/1471-2172-7-19
94. Kessler B, Rinchai D, Kewcharoenwong C, Nithichanon A, Biggart R, Hawrylowicz CM, et al. Interleukin 10 Inhibits Pro-Inflammatory Cytokine Responses and Killing of Burkholderia Pseudomallei. Sci Rep (2017) 7:42791. doi: 10.1038/srep42791
95. Suparak S, Kespichayawattana W, Haque A, Easton A, Damnin S, Lertmemongkolchai G, et al. Multinucleated Giant Cell Formation and Apoptosis in Infected Host Cells Is Mediated by Burkholderia Pseudomallei Type III Secretion Protein BipB. J Bacteriol (2005) 187(18):6556–60. doi: 10.1128/JB.187.18.6556-6560.2005
96. Kespichayawattana W, Rattanachetkul S, Wanun T, Utaisincharoen P, Sirisinha S. Burkholderia Pseudomallei Induces Cell Fusion and Actin-Associated Membrane Protrusion: A Possible Mechanism for Cell-to-Cell Spreading. Infection Immun (2000) 68(9):5377–84. doi: 10.1128/IAI.68.9.5377-5384.2000
97. Benanti EL, Nguyen CM, Welch MD. Virulent Burkholderia Species Mimic Host Actin Polymerases to Drive Actin-Based Motility. Cell (2015) 161(2):348–60. doi: 10.1016/j.cell.2015.02.044
98. Sahni SK, Rydkina E. Host-Cell Interactions With Pathogenic Rickettsia Species. Future Microbiol (2009) 4(3):323–39. doi: 10.2217/fmb.09.6
99. Inglis TJ, Rigby P, Robertson TA, Dutton NS, Henderson M, Chang BJ. Interaction Between Burkholderia Pseudomallei and Acanthamoeba Species Results in Coiling Phagocytosis, Endamebic Bacterial Survival, and Escape. Infection Immun (2000) 68(3):1681–6. doi: 10.1128/IAI.68.3.1681-1686.2000
100. Bahjat KS, Meyer-Morse N, Lemmens EE, Shugart JA, Dubensky TW, Brockstedt DG, et al. Suppression of Cell-Mediated Immunity Following Recognition of Phagosome-Confined Bacteria. PloS Pathog (2009) 5(9):e1000568. doi: 10.1371/journal.ppat.1000568
101. Ratajczak W, Niedźwiedzka-Rystwej P, Tokarz-Deptuła B, Deptuła W. Immunological Memory Cells. Central-European J Immunol (2018) 43(2):194–203. doi: 10.5114/ceji.2018.77390
102. Clem AS. Fundamentals of Vaccine Immunology. J Global Infect Dis (2011) 3(1):73–8. doi: 10.4103/0974-777X.77299
103. Choh LC, Ong GH, Vellasamy KM, Kalaiselvam K, Kang WT, Al-Maleki AR, et al. Burkholderia Vaccines: Are We Moving Forward? Front Cell Infect Microbiol (2013) 3:5. doi: 10.3389/fcimb.2013.00005
104. Healey GD, Elvin SJ, Morton M, Williamson ED. Humoral and Cell-Mediated Adaptive Immune Responses Are Required for Protection Against Burkholderia Pseudomallei Challenge and Bacterial Clearance Postinfection. Infection Immun (2005) 73(9):5945–51. doi: 10.1128/IAI.73.9.5945-5951.2005
105. Dunachie SJ, Jenjaroen K, Reynolds CJ, Quigley KJ, Sergeant R, Sumonwiriya M, et al. Infection With Burkholderia Pseudomallei - Immune Correlates of Survival in Acute Melioidosis. Sci Rep (2017) 7(1):12143. doi: 10.1038/s41598-017-12331-5
106. Suwannasaen D, Mahawantung J, Chaowagul W, Limmathurotsakul D, Felgner PL, Davies H, et al. Human Immune Responses to Burkholderia Pseudomallei Characterized by Protein Microarray Analysis. J Infect Dis (2011) 203(7):1002–11. doi: 10.1093/infdis/jiq142
107. Wang M, Tang ST, Stryhn A, Justesen S, Larsen MV, Dziegiel MH, et al. Identification of MHC Class II Restricted T-Cell-Mediated Reactivity Against MHC Class I Binding Mycobacterium Tuberculosis Peptides. Immunology (2011) 132(4):482–91. doi: 10.1111/j.1365-2567.2010.03383.x
108. Cowley SC, Hamilton E, Frelinger JA, Su J, Forman J, Elkins KL. CD4-CD8- T Cells Control Intracellular Bacterial Infections Both In Vitro and In Vivo. J Exp Med (2005) 202(2):309–19. doi: 10.1084/jem.20050569
109. Haque A, Easton A, Smith D, O’Garra A, Rooijen NV, Lertmemongkolchai, et al. Role of T Cells in Innate and Adaptive Immunity Against Murine Burkholderia Pseudomallei Infection. J Infect Dis (2006) 193(3):370–9. doi: 10.1086/498983
110. Jenjaroen K, Chumseng S, Sumonwiriya M, Ariyaprasert P, Chantratita N, Sunyakumthorn P, et al. T-Cell Responses Are Associated With Survival in Acute Melioidosis Patients. PloS Negl Trop Dis (2015) 9(10):e0004152. doi: 10.1371/journal.pntd.0004152
111. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its Ligands in Tolerance and Immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
112. Kleijn S, Langereis JD, Leentjens J, Kox M, Netea MG, Koenderman L, et al. IFN-γ-Stimulated Neutrophils Suppress Lymphocyte Proliferation Through Expression of PD-L1. PloS One (2013) 8:e72249. doi: 10.1371/journal.pone.0072249
113. Buddhisa S, Rinchai D, Ato M, Bancroft GJ, Lertmemongkolchai G. Programmed Death Ligand 1 on Burkholderia Pseudomallei-Infected Human Polymorphonuclear Neutrophils Impairs T Cell Functions. J Immunol (2015) 194:4413–21. doi: 10.4049/jimmunol.1402417
114. See JX, Samudi C, Saeidi A, Menon N, Choh LC, Vadivelu J, et al. Experimental Persistent Infection of BALB/c Mice With Small-Colony Variants of Burkholderia Pseudomallei Leads to Concurrent Upregulation of PD-1 on T Cells and Skewed Th1 and Th17 Responses. PloS Negl Trop Dis (2016) 10:e0004503. doi: 10.1371/journal.pntd.0004503
115. See J-X, Chandramathi S, Abdulla MA, Vadivelu J, Shankar EM. Persistent Infection Due to a Small-Colony Variant of Burkholderia Pseudomallei Leads to PD-1 Upregulation on Circulating Immune Cells and Mononuclear Infiltration in Viscera of Experimental BALB/c Mice. PloS Negl Trop Dis (2017) 11(8):e0005702. doi: 10.1371/journal.pntd.0005702
116. Menon N, Mariappan V, Vellasamy KM, Samudi C, See JX, Ganesh PS, et al. Experimental Exposure of Burkholderia Pseudomallei Crude Culture Filtrate Upregulates PD-1 on T Lymphocytes. Access Microbiol (2020) 2(5):acmi000110. doi: 10.1099/acmi.0.000110
117. Rolim DB, Vilar DC, de Góes Cavalcanti LP, Freitas LB, Inglis TJ, Nobre Rodrigues JL, et al. Burkholderia Pseudomallei Antibodies in Individuals Living in Endemic Regions in Northeastern Brazil. Am J Trop Med Hygiene (2011) 84(2):302–5. doi: 10.4269/ajtmh.2011.10-0220
118. Khakhum N, Bharaj P, Myers JN, Tapia D, Kilgore PB, Ross BN, et al. Burkholderia Pseudomallei Δtonb Δhcp1 Live Attenuated Vaccine Strain Elicits Full Protective Immunity Against Aerosolized Melioidosis Infection. mSphere (2019) 4(1):e00570–18. doi: 10.1128/mSphere.00570-18
119. Vasu C, Vadivelu J, Puthucheary SD. The Humoral Response in Melioidosis Patients During Therapy. Infection (2003) 31:24–30. doi: 10.1007/s15010-002-3020-2
120. Mestecky J, Alexander RC, Wei Q, Moldoveanu Z. Methods for Evaluation of Humoral Immune Responses in Human Genital Tract Secretions. Am J Reprod Immunol (N Y NY Menkeir: 1989) (2011) 65(3):361–7. doi: 10.1111/j.1600-0897.2010.00923.x
121. Yi J, Simpanya MF, Settles EW, Shannon AB, Hernandez K, Pristo L, et al. Caprine Humoral Response to Burkholderia Pseudomallei Antigens During Acute Melioidosis From Aerosol Exposure. PloS Neglected Trop Dis (2019) 13(2):e0006851. doi: 10.1371/journal.pntd.0006851
122. Cruz-Adalia A, Veiga E. Close Encounters of Lymphoid Cells and Bacteria. Front Immunol (2016) 7:405. doi: 10.3389/fimmu.2016.00405
123. Carter RH, Fearon DT. CD19: Lowering the Threshold for Antigen Receptor Stimulation of B Lymphocytes. Science (1992) 256:5053. doi: 10.1126/science.1373518
124. Rossbacher J, Shlomchik MJ. The B Cell Receptor Itself can Activate Complement to Provide the Complement Receptor 1/2 Ligand Required to Enhance B Cell Immune Responses In Vivo. J Exp Med (2003) 198(4):591–602. doi: 10.1084/jem.20022042
125. Heesterbeek D, Angelier ML, Harrison RA, Rooijakkers S. Complement and Bacterial Infections: From Molecular Mechanisms to Therapeutic Applications. J Innate Immun (2018) 10(5-6):455–64. doi: 10.1159/000491439
Keywords: Burkholderia pseudomallei, immunology, melioidosis, pathogenesis, virulence
Citation: Mariappan V, Vellasamy KM, Barathan M, Girija ASS, Shankar EM and Vadivelu J (2021) Hijacking of the Host’s Immune Surveillance Radars by Burkholderia pseudomallei. Front. Immunol. 12:718719. doi: 10.3389/fimmu.2021.718719
Received: 01 June 2021; Accepted: 12 July 2021;
Published: 11 August 2021.
Edited by:
Elsa Anes, Universidade de Lisboa, PortugalReviewed by:
Jorge Leitāo, Universidade de Lisboa, PortugalCopyright © 2021 Mariappan, Vellasamy, Barathan, Girija, Shankar and Vadivelu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vanitha Mariappan, dmFuaXRoYS5tYUBnbWFpbC5jb20=; Jamuna Vadivelu, amFtdW5hQHVtLmVkdS5teQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.