
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 August 2021
Sec. T Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.718621
This article is part of the Research Topic Impact of Disease and Prior Therapies on T Cell Function in Cancer View all 8 articles
 Sanjay Chandrasekaran1*
Sanjay Chandrasekaran1* Christopher Ronald Funk1Troy Kleber1
Christopher Ronald Funk1Troy Kleber1 Chrystal M. Paulos2
Chrystal M. Paulos2 Mala Shanmugam1
Mala Shanmugam1 Edmund K. Waller1
Edmund K. Waller1PI3K-δ and PI3K-γ are critical regulators of T-cell differentiation, senescence, and metabolism. PI3K-δ and PI3K-γ signaling can contribute to T-cell inhibition via intrinsic mechanisms and regulation of suppressor cell populations, including regulatory T-cells and myeloid derived suppressor cells in the tumor. We examine an exciting new role for using selective inhibitors of the PI3K δ- and γ-isoforms as modulators of T-cell phenotype and function in immunotherapy. Herein we review the current literature on the implications of PI3K-δ and -γ inhibition in T-cell biology, discuss existing challenges in adoptive T-cell therapies and checkpoint blockade inhibitors, and highlight ongoing efforts and future directions to incorporate PI3K-δ and PI3K-γ as synergistic T-cell modulators in immunotherapy.
T-cell based immunotherapies aim to reinvigorate immunity against malignant cells either via infusion of effector T-cells or activation of existing T-cells in the body. Here, we provide an overview of the mechanism of PI3K signaling in T-cells, particularly PI3K-δ and -γ, down-stream of T-cell receptor activation. Particular attention is given to how inhibition of PI3K-δ and -γ signaling with drug inhibitors regulates and activates pathways related to T-cell proliferation, differentiation, senescence, exhaustion, and metabolism. In this context, we consider the current state of therapies targeting T-cell immune checkpoint pathways and the effects of synergizing PI3K inhibition with immune checkpoint inhibitors (ICIs) to re-model the activity of T-cells and other immunosuppressive cells in the tumor microenvironment (TME). We also consider PI3K-δ and -γ signaling and inhibition in the context of Adoptive T-cell Transfer (ACT) therapies. ACT therapies entail harvesting a patient’s immune cells, culturing and potentially modifying them ex vivo, and reinfusing them back into the patient. Within ACT, three commonly utilized approaches used for patients are chimeric antigen receptor T-cell (CART), TCR (T-cell Receptor) therapy, and tumor infiltrating lymphocyte (TIL) therapies. Manufacturing ACT products are costly, time consuming, technically challenging, and clinical responses are promising but not consistently achieved (1, 2). We subsequently review the current preclinical and clinical progress with ACT therapies, highlight notable differences between their manufacturing processes, and discuss specific areas for process improvement in the context of surrogate measures of clinical outcomes. Finally, while reviewing current limitations in ACT, we discuss how PI3K-δ and -γ are promising pharmacological targets for improving T-cell response in ACT given their ubiquitous expression in T-cells.
PI3K proteins are divided into class IA, IB, II, and III and are named by order of discovery. Class IA subunits include -α, -β and -δ, and are generally phosphorylated by receptor tyrosine kinases (RTKs). Class IB is comprised of the PI3K-γ isoform and is expressed and co-localized with G-protein coupled receptors (GPCRs). In healthy tissue, the PI3K-α and -β isoforms are ubiquitously expressed whereas the PI3K-δ and -γ isoforms are mainly expressed in hematopoietic cells (3, 4). PI3K-δ and/or -γ inhibition and knockout in lymphocytes reduces cytokine-mediated chemotaxis. In particular, knockout of PI3K-γ reduced T-cell migration, while PI3K-δ knockout generated deficiencies in B cell chemotaxis (5).
When over-expressed in cancers, PI3K-α and -β drive tumor growth and metastasis while expression of PI3K-δ and -γ in hematopoietic cells regulates immune cell activity, especially lymphocyte and myeloid cell differentiation and activation (6–8). In many solid tumor malignancies, including breast cancer, lung, head and neck cancer, and melanoma, increased activity within the PI3K pathway occurs through activating mutations and gene amplifications in PIK3CA (PI3K-α), or loss of expression of the PI3K tumor suppressor, PTEN (8–15). Efforts to develop pan-PI3K inhibitors into successful anti-cancer therapy have been stymied by low response rates to PI3K inhibitors and significant toxicities (16–23). Unlike the use of PI3K inhibitors in solid tumor malignancies, the anti-tumor effects of PI3K inhibition in lymphoid malignancies are not dependent on gene mutations, amplifications, or deletions within the PI3K pathway. Instead, PI3K-δ and -γ inhibitors exert direct inhibitory effects on lymphoid cancer cell survival, and indirect effects by targeting survival and homing of normal lymphoid and myeloid cells into the TME (24–26). These bi-functional properties are a result of PI3K-δ and -γ signaling regulating activation and differentiation of normal B- and T-cells while simultaneously limiting the proliferation of the cancer counterparts of normal lymphoid cells following their neoplastic transformation.
FDA approved therapies now exist for relapsed B-cell malignancies like CLL and Follicular Lymphoma (FL) with idelalisib (PI3K-δ), duvelisib (PI3K-δ/γ), and copanlisib (PI3K-α/δ) (16, 27–31). Clinical trials are ongoing in Richter Syndrome or transformed FL, Relapsed/Refractory T-cell Lymphomas, Relapsed or Refractory Peripheral T-cell Lymphoma (PTCL), ALL, maintenance post autologous transplant for T-cell and indolent B cell lymphomas, DLBCL, Mantle Cell lymphoma, and Marginal Zone Lymphoma (NCT03892044, NCT02783625, NCT03372057, NCT04331119, NCT03742323, NCT03133221, NCT04233697, NCT04263584, NCT03877055, and NCT03474744) (32).
The direct anti-cancer activity of PI3K-δ and -γ inhibitors in hematologic malignancies occurs by inhibiting the proliferation of malignant cells with a simultaneous indirect effect on normal hematopoietic cells that is related to limiting the immune suppressive properties of the TME (8, 33, 34). Studies in healthy T-cell biology have unequivocally demonstrated that PI3K signaling drives T-cell differentiation and senescence and supports immune homeostasis by Tregs (3, 6, 35, 36).
The TCR/CD3 complex and related CAR constructs work in concert with co-stimulatory signals like CD28 to induce T-cell activation via PI3K pathway signaling (37, 38). In brief, CD3-mediated signaling through Lck, LAT, and PLC protein leads to phosphorylation of the p110δ and p110γ subunits of the PI3K-δ and PI3K-γ isoforms (Figure 1). The p110δ and p110γ subunits work by phosphorylating phosphatidylinositol 4,5 bisphosphate (PIP2) to yield phosphatidylinositol 3, 4,5 triphosphate (PIP3), which permits anchorage and association of cytosolic proteins near the lipid bilayer to facilitate downstream signal transduction (39). PIP3 thus functions as a second messenger initiating multiple signaling cascades, most notably facilitating the phosphorylation of AKT by PDK1 which subsequently leads to downstream survival and differentiation signals secondary to activation of the mechanistic target of rapamycin 1 (mTORC1) (40, 41) (Figure 1). We have shown that inhibition of PI3K-δ and -γ in T-cells with duvelisib simultaneously activates signaling through the RAS/RAF/MEK pathway (42).
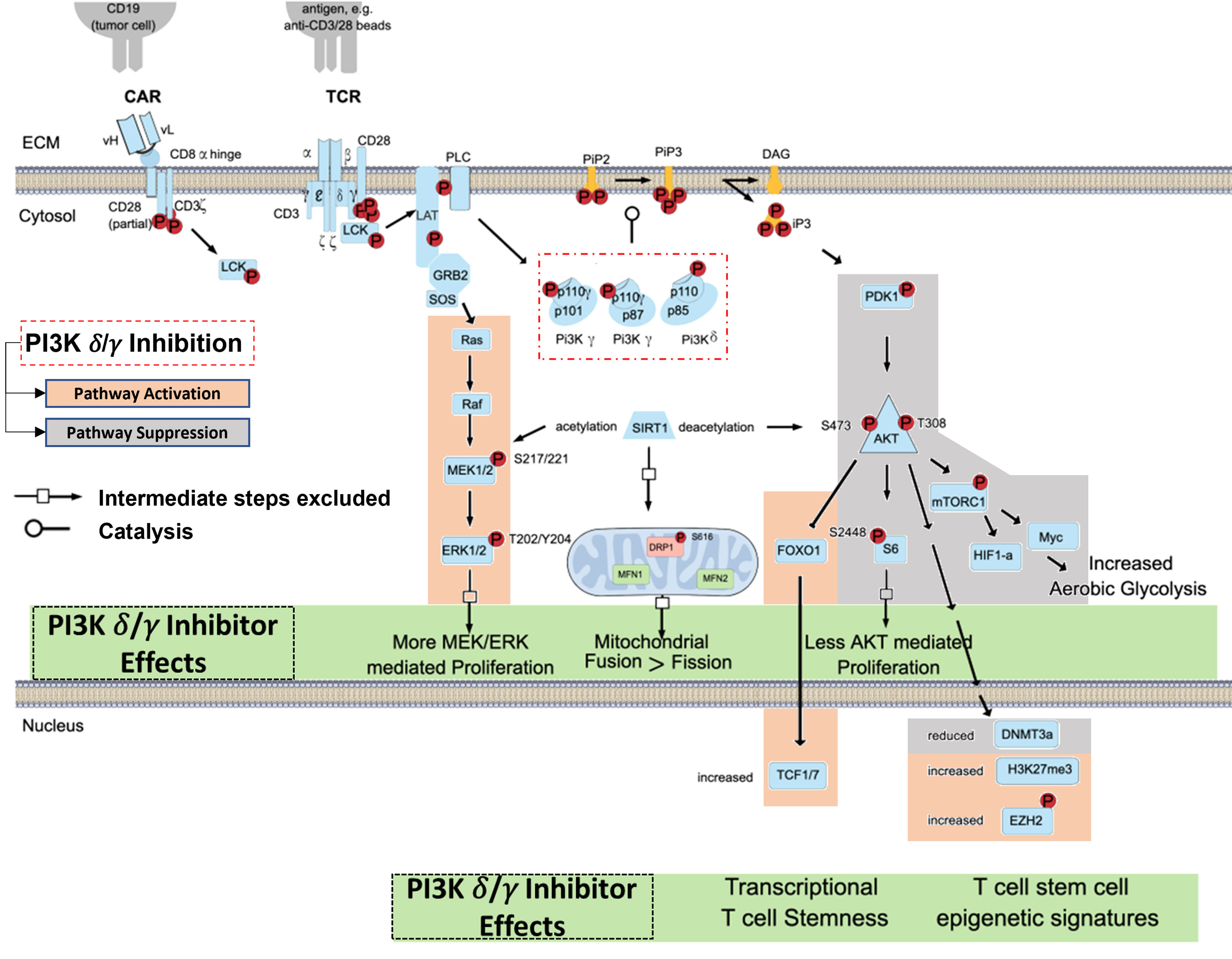
Figure 1 Blocking TCR/CAR Mediated Activation of PI3K Signaling in T cells. TCR/CAR binding by antigen results in downstream signaling through PI3K δ/γ, AKT, and mTORC1. This signal cascade promotes AKT mediated proliferation, aerobic glycolysis, and FOXO1 inhibition, and loss of TCF1/7 and the stem-cell like epigenetic markers phosphorylated EZH2 and H3K27me3, leading effector T-cell generation. PI3K δ/γ reverse these effects and in turn increasing proliferative signaling through MEK and ERK, increase mitochondrial fusion, and promote epigenetic changes associated with T cell stemness.
Upon T-cell activation, there is an increase in glucose and amino acid uptake and activation of mTORC1, which is required to maintain T-cell effector functions (43–46). Activation of mTORC1 also promotes induction and maintenance of aerobic glycolysis, which leads to increased differentiation and effector functions (46). In the context of adoptive T-cell therapies, mTORC1 mediated differentiation into effector cells render ACT less efficacious, as the survival and expansion potential of differentiated T cells is limited. Inhibition of mTORC1 activity through direct inhibition, genetic modification, inhibition of AKT, or STAT3 activation promotes formation of a pool of T-cells that are smaller in size with a stem cell memory (Tscm) and naïve (Tn) cell phenotype by inhibiting aerobic glycolysis and preventing terminal differentiation (47–50) (Figure 2).
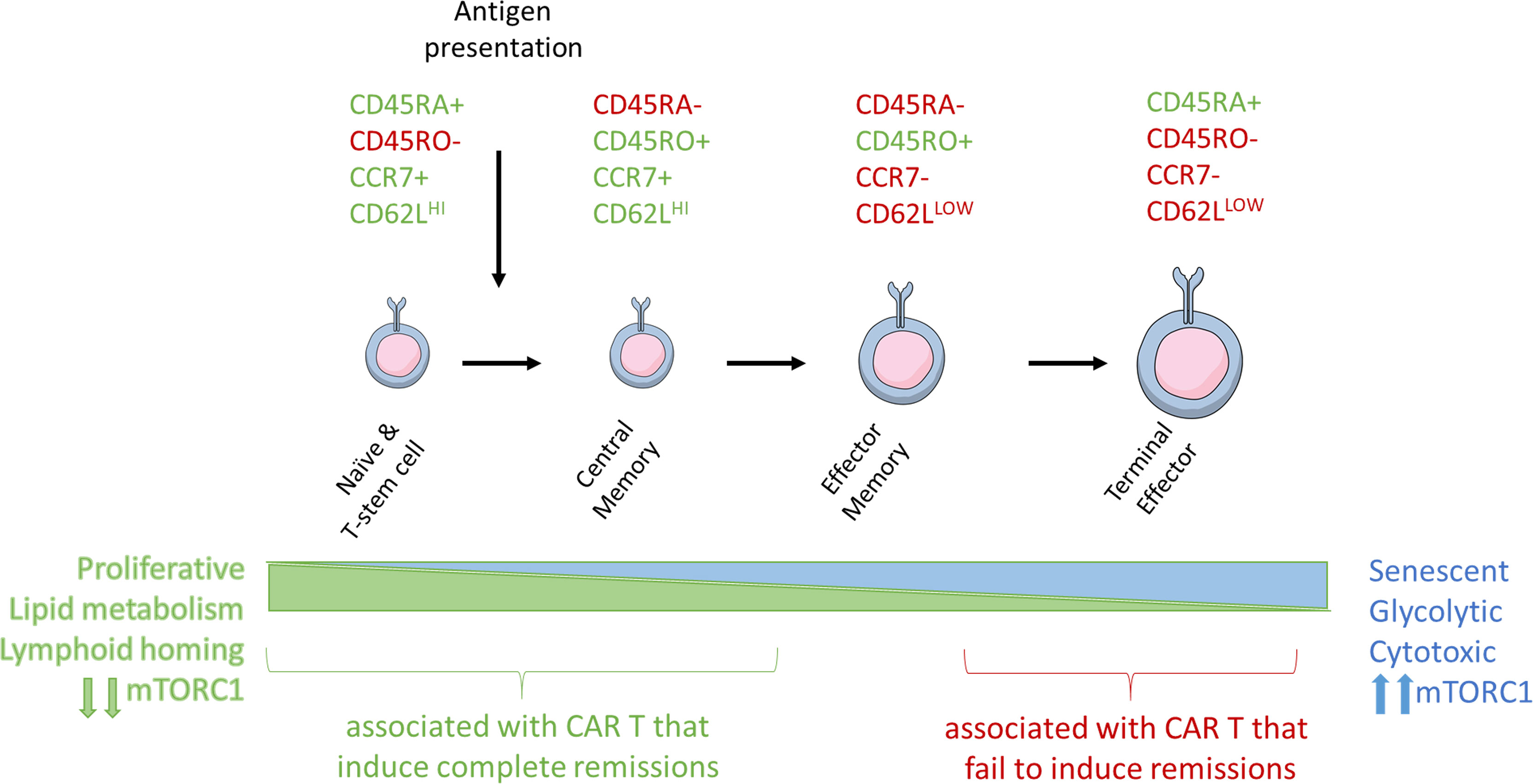
Figure 2 Antigen stimulation induces T-cell differentiation and associated changes in T-cell metabolism and size. Naïve, stem cell, and central memory phenotype T cells are smaller and associated with catabolic metabolism. Antigen stimulation of the TCR signals through PI3K and mTORC1 to induce metabolic reprogramming, increase in T cell size, and eventual senescence.
The effects of PI3K activation are recapitulated clinically in APDS (Activated phosphoinositide 3-kinase delta syndrome (APDS), an autosomal dominant primary human immunodeficiency caused by heterozygous gain-of-function mutations in PIK3CD which encodes the p110δ catalytic subunit of PI3K. Similar to TCR overstimulation, increased PI3K-δ and downstream mTORC1 activity in APDS alters T-cell glucose transport, leading to unregulated differentiation and increased senescence by shifting CD8+ T-cells towards short-lived effector cells that are unable to yield memory lymphocytes (51, 52). Altered T-cell differentiation due to activating mutations in the PI3K p110δ subunit thus leads to reduced numbers of circulating reduced CCR7+ Tn and central memory (Tcm) cells and increased numbers of CD45RA−CCR7− effector memory (Tem) and CD45RA+CCR7− (Te) CD8+ T-cells. The over representation of terminally differentiated and senescent T-cells leads to lymphadenopathy, functional immunodeficiency, and increased risk for sinopulmonary infections, nodular lymphoid hyperplasia and viremia from cytomegalovirus (CMV) and/or Epstein-Barr virus (EBV) and associated B-cell malignancies (51, 52).
PI3K-δ and -γ mediated activation is critical to the processes of antigen driven T-cell differentiation and induction of exhaustion, senescence, and metabolic reprogramming mechanisms (53). During ex vivo manufacturing of the ACT product or following chronic cancer-induced antigen presentation, constant engagement of the TCR alters T-cell phenotypes and functions, ultimately contributing to reduced cytotoxic responses.
The different T-cell subsets and their memory and effector functions have been previously reviewed in extensive detail (54). Clinically meaningful populations can be defined by patterns of expression of extracellular T cell markers including CD45RA, CD45RO, CD62L, and CCR7 (55) (Figure 2). As T-cells become further differentiated in response to antigen presentation, CD45RA isoform is switched to CD45RO, CCR7 is lost, and CD62L expression is reduced.
Tn and minimally differentiated Tscm cells can be phenotypically defined by their expression of the co-stimulatory molecules CD27 and CD28 in the contect of other T cell markers. Stimulation via CD3 and CD28 in response to a cognate antigen promotes the expansion and differentiation of Tn and Tscm T-cells into CD27+/CD28+/CD45RO+/CCR7+ Tcm cells. These cells, which have previously encountered antigen, are capable of significant proliferation and have increased activity upon antigen re-exposure, express CCR7 that mediates their homing into the peripheral tissues (56, 57). Effector-memory T-cells (Tem) retain CD45RO but lose CCR7, CD27, and CD28, which significantly reduces their homing and proliferative capacity (58). Terminal effector (Te) T-cells switch to the CD45RA+ CD45 isoform back and retain CCR7 negativity. Te cells are capable of engaging a robust cytotoxic response, but their persistence is transient, and they lack the proliferative and homing capabilities of less differentiated cells. As a consequence of the proliferative capacity of T cells at different stages of differentiation, ACT composed primarily of Te cells is predicted to have a limited persistence in vivo and shorter response duration.
Constant T-cell stimulation can also lead to onset of T-cell exhaustion or senescence, both dysfunctional T-cell states that share similar characteristics of reduced proliferation, cytotoxic activity, and metabolic capacity but have different underlying etiologies, cytokine profiles, and cell surface marker phenotypes (59). While both phenotypes often overlap in immuno-oncology, exhaustion typically occurs due to constant antigenic stimulation due to the inflammatory cancer-state, while senescence ensues when cells are forced to endure multiple, rapid signals to enterthe cell-cycle and undergo proliferative cell divisions in the face of repeated antigenic stimulation, exposure to DNA damaging agents, or other stress signals (59).
The T-cell exhaustion phenotype has been well characterized in models of chronic viral infection (60). A similar process of constant antigenic stimulation of the TCR is thought to occur within the cancer TME and contribute to establishing a population of exhausted T-cells phenotypically identified by increased expression of inhibitory receptors such as PD-1 (programmed cell death-1), CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), TIM-3, LAG-3 (anti-lymphocyte activation gene-3), and VISTA (52, 61). PD-1 itself has been shown to block cell cycle progression by inhibiting CD28-mediated activation of PI3K through its immunoreceptor tyrosine-based switch motif located in the cytoplasmic tail, and its increased expression in the setting of activating PI3K mutations may be compensatory (62–64). Antibody therapies called immune checkpoint inhibitors (ICIs) targeting molecules like CTLA-4, PD-1, and its ligand, PD-L1, are now commonly used in a wide variety of cancers and will be discussed in detail further on in this review.
As a result of repeated antigen-stimulated replication and differentiation, similar to the pathophysiology seen in APDS, T-cells can become senescent and lose their ability to proliferate, manifesting less cytotoxic activity and greater cell cycle arrest (59). T-cell senescence is either replicative (telomerase-dependent) or premature (telomerase independent) (65). In replicative senescence, frequent TCR engagement leads to inactivation of the telomerase promoter, decreased telomerase expression, and activation of DNA damage signals. In aggregate, this leads to cell cycle arrest, increased expression of CD57, and loss of co-stimulatory molecules CD27 and CD28 (51, 66). On the other hand, cancer mediated T-cell senescence may be telomerase independent, and impairs T-cell stimulation via the TCR and reduces response rates to CAR-T therapy in conditions like CLL (67, 68). We have shown that T-cells from patients with DLBCL sorted for co-expression of CD27 and CD28 proliferate whereas T-cells lacking CD27 and CD28 are senescent (69). Further implications and mechanisms of T-cell senescence in the setting of CAR-T therapy for malignant hematologic conditions has been reviewed prior in great detail (65).
T-cell differentiation is associated with the transition from reliance on catabolic metabolism to anabolic metabolism (70). Tn, Tscm, and Tcm cells rely upon fatty acid oxidation (FAO) and oxidative metabolism (OXPHOS) to meet metabolic needs. As T-cells encounter antigen and differentiate they increase their reliance upon glycolysis to rapidly meet bioenergetic and biosynthetic demands for rapidly dividing cells, such that Te cells almost entirely rely upon glycolytic metabolism (71) (Figure 3). Antigen binding at the TCR upregulates glucose and amino acid transporters at the T-cell surface, driving a process of metabolic reprogramming (47, 72). During metabolic re-programming, memory cells demonstrate increases in mitochondrial mass associated with an induction of PGC1-alpha and increased respiratory capacity (73, 74). The switch to glycolytic metabolism while supporting the Te cytotoxic phenotype is also thought to reduce their longevity (75). Thus, less differentiated and less glycolytic Tn, Tscm, and Tcm cells are the preferred cell populations for ACT and other cancer immunotherapies, and strategies to abrogate metabolic reprogramming are promising mechanisms by which to improve therapy efficacy. Increased activity in the AKT/mTOR pathway drives T-cell metabolic re-programming and glycolysis, and strategies to inhibit this pathway in ACT are being tested (47–50). Changes within the tumor microenvironment also influence non-cancer cell metabolism. PD-L1 expression on cancer cells can engage with infiltrating immune effector cells that express PD-1, limiting activation of downstream mTOR signaling (72). Interestingly, PD-1 blockade has been shown to restore oxidative phosphorylation (76). Both exhaustion and senescence alter T-cell mitochondrial biogenesis and respiratory capacity, but in mechanistically different ways. In exhaustion, both OXPHOS and glycolytic metabolic mechanisms are suppressed and associated with increased PD-1 expression, while senescence is associated with a shift to towards anaerobic glycolysis (59). The significance of these changes in signaling pathways remains under investigation.
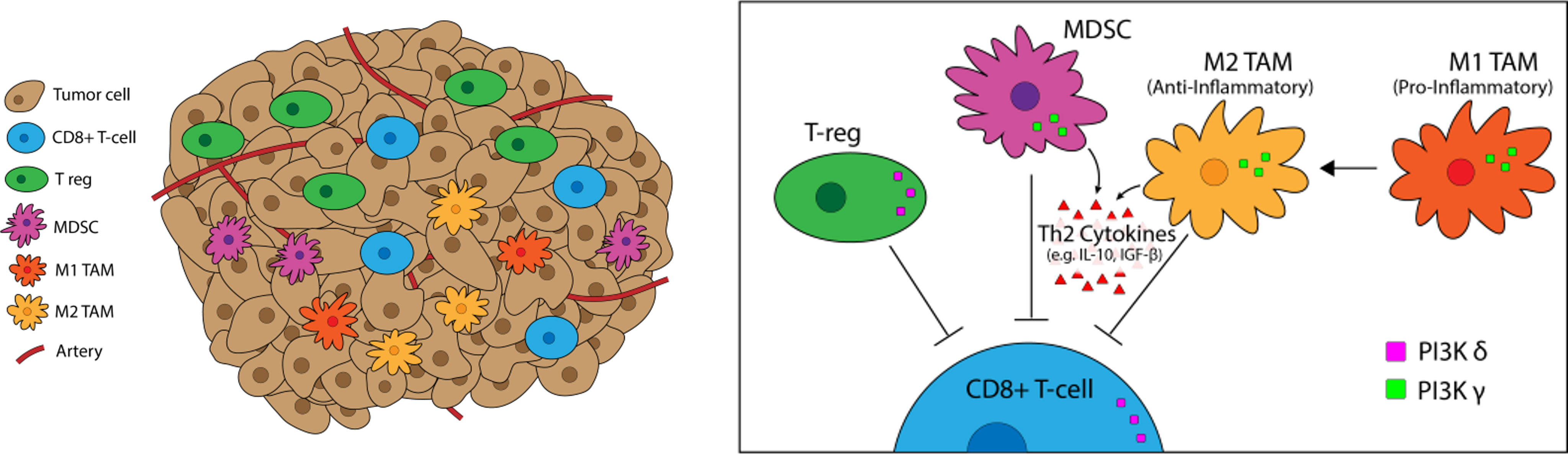
Figure 3 PI3K-δ and -γ in the Tumor Microenvironement. PI3K-δ and -γ signaling Tregs, TAMs, and MDSCs regulates suppression and trafficking of CD8+ tumor infiltrating lymphocytes in the TME. PI3K-δ signaling drives Treg suppression of CD8+ tumor-infiltrating T-cells while TAMs and MDSCs rely on PI3K-γ for their immunosuppressive function.
Immune Checkpoint Inhibitors (ICI) bind to and interfere with inhibitory surface receptors on T-cells, thus allowing T-cells to remain active, participate in antigen recognition, and induce anti-tumor immune responses. Current clinical ICI applications target CTLA-4, PD-1, or PD-L1 molecules, and the FDA has approved ICI therapies in multiple malignancies, including melanoma, Merkel cell, non-small cell lung, head and neck, gastroesophageal, renal, bladder, and hepatocellular cancers (77, 78). The CTLA-4 inhibitor ipilimumab was the first ICI to be FDA approved (79, 80), and subsequently nivolumab and pembrolizumab (anti-PD-1) received approvals. Now, multiple additional ICI agents have been approved including cemiplimab (anti-PD-1) and the anti-PD-L1 antibodies avelumab, atezolizumab, and durvalumab (77). Globally, many more anti-CTLA-4, anti-PD-1, and anti-PD-L1 antibodies are under clinical study.
ICI have demonstrated tremendous clinical benefit in multiple solid tumor indications. For example, in patients with unresectable advanced stage (III/IV) cutaneous melanoma, ICIs have achieved single-agent response rates up to 40% in the first-line setting, higher than the prior 10% seen with chemotherapy alone. While these improvements in clinical outcomes are exciting, a majority of patients will not experience a long-term clinical response and not all malignancies are sensitive to T-cell checkpoint inhibitors, in large part due to poor intratumoral T-cell trafficking. A common strategy currently being used is to use a multi-targeted approach to simultaneously inhibit additional co-inhibitory receptors other than PD-1 and CTLA-4 associated with primary or acquired ICI resistance (81, 82). Promising preclinical data with co-inhibition of TIM-3 or LAG-3 with anti-PD-1 therapy (83, 84) has led to multiple ongoing clinical trials testing such combinations in solid tumor malignancies, including with nvel dual-targeting bispecific antibodies (NCT03219268, NCT04080804, NCT04140500, NCT03250832, NCT01968109, NCT04370704, NCT03005782, NCT04139902, NCT03680508, NCT02817633, NCT03708328, NCT03744468, NCT02608268, NCT03630159) (32).
An alternate strategy, however, is to combine non-ICI pharmacologic inhibitors with ICI with the intent of enhancing T-cell anti-tumor activity in ICI-resistant cancers (85, 86). Mechanistically, CTLA-4 inhibition reduces CD4+ Treg proliferation and induces the expansion of a Th1-like CD4+ effector populations, while inhibiting PD-1 reduces T-cell exhaustion and increases CD8+ tumor infiltrating subsets (87–90). ICI failure stems from fundamental deficiencies in mechanisms of innate and acquired resistance (82, 91). Variability in cancer type, prior treatment history, tumor heterogeneity, and the immunosuppressive tumor microenvironment also influence poor therapy response (92, 93). Seven immune escape mechanisms for ineffective immune mediated anti-tumor response to anti-PD-1/PD-L1 therapy have been previously described (94, 95), and can be broadly summarized into three categories, namely 1) T-cell priming and activation, 2) T-cell trafficking and infiltration, and 3) tumor cell recognition and killing.
Tumor microenvironments can be classified as immunogenic or immune-restricted, which refers to the infiltration and presence of T-cells and other antigen presenting immune cell populations (96). Immunogenic or ICI-responsive tumor types include cutaneous melanoma, lung cancers, renal cancers, and bladder cancers, while colorectal, pancreatic, prostatic, breast, or cancers of central nervous system origin are regarded as immune-restricted and ICI-resistant malignancies (97–100).
Lack of ICI response in immune-restricted cancers occurs from deficiencies in T-cell priming, activation, trafficking, and infiltration into the TME. Research has focused on increasing tumor antigen release, improving the presence and efficiency of antigen presenting cells, and augmenting T-cell intra-tumoral homing and co-stimulation (100, 101). In immunogenic tumors, ICI resistance and/or failure is more complicated and follows three distinct phenotypes: a) patients that do not respond (innate resistance); b) those that respond initially but fail to respond in later stages (acquired resistance); and c) those that respond initially and continue to respond (92, 93). Despite intratumoral T-cell presence, deficiencies in tumor cell recognition and killing driven by alterations in MHC or other co-inhibitory signals can lead to immune evasion and therapy failure (102–104). Even if adequate antigen presentation exists, T-cell exhaustion, senescence, and inactivation due to concomitant checkpoint pathways, or direct suppression by other immune cells including regulatory T-cells (Tregs) and M2-like tumor associated macrophages (TAMs) or MDSCs limit the efficacy of ICI. Therefore, much need exists for therapies that synergize with ICI to elicit activation, proliferation, and long-term persistence of antigen experienced tumor-reactive T-cells (105).
PI3K signaling plays a multifactorial role in shaping the TME, particularly the milieu of other T-cell suppressive immune cells (Figure 5). We have previously linked PI3K pathway signaling to tumor antigen presentation mechanisms in head and neck cancers. PTEN loss and PI3K activation downregulated major histocompatibility complex (MHC) Class I and Class II induction by IFN-γ, and clinical tumor samples demonstrated inverse staining associations of MHC and Phospho-S6, a serine/threonine kinase downstream of PI3K (106). Downregulation of MHC expression in HNSCC and melanoma has a clinical correlation with treatment resistance and poorer clinical outcomes (102–104). Furthermore, PTEN loss has been shown to promote resistance to T-cell targeted immunotherapy in melanoma (107). The effects of PI3K inhibition on T-cell infiltration and activation in tumors, thus leading to improved T-cell mediated cytotoxicity with ICI, has been well characterized in multiple pre-clinical models of immunogenic and immune-restricted cancers, however the mechanism of such effects depends which PI3K isoforms are targeted (108, 109). Importantly, PI3K-δ and -γ isoforms regulate lymphocyte trafficking, intratumoral lymphocyte recruitment, T-cell differentiation, activation, and proliferation, myeloid cell and macrophage differentiation and function, and immune cell metabolism (5, 109–113). Chemoattractants produced by cancers also activate GPCRs and RTKs involved in PI3K phosphorylation and activation, like PI3K-δ in immature and immunosuppressive myeloid cells, further driving and sustaining tumor inflammation (114).
In cancer, PI3K-δ inhibition has been shown to play a role in augmenting intra-tumoral T-cell activation. In preclinical models of lung, breast, and colon cancer, in vivo treatment with the PI3K-α/δ inhibitor AZD8835 and the PI3K-δ inhibitor idelalisib favorably increased CD8+ TIL/Treg ratios by ~2-fold (36). Furthermore, ex vivo cultures of conventional CD8+ T-cells with AZD8835 and idelalisib demonstrated a dose-dependent enhancement in T-cell survival, cell size, increased CD69+ activation marker expression, and increased expression of the IL-2 receptor CD25 without negatively impacting proliferation (36).
In a triple negative breast cancer-like transgenic MMTV-PyMT murine model of breast cancer, an increase of more than two-fold in the percentage of intra-tumoral CD4+ and CD8+ T-cells were found in the mammary fat-pad tumors and lung metastases of PI3K-γ knockout mice versus PI3K-γ competent mice (115), along with a 50% reduction in primary tumor growth volume at 5-weeks, reduced metastases formation, increased TNF-alpha secretion by CD4+ and CD8+ TILs, and increased TIL expression of the T-cell activation marker CD69+. In PI3K-γ competent mice, inhibition with the pan-PI3K inhibitor buparlisib recapitulated the above findings. The most compelling finding is that while single-agent anti-PD-1 antibody therapy is minimally effective in this TNBC model, combination therapy of anti-PD-1 antibody with buparlisib inhibited tumor growth in 100% of the mice. Similar findings were also seen in syngeneic mouse models of pancreatic ductal cancer, with tumors from PI3K-γ knockout mice exhibiting significantly more CD4+ and CD8+ T-cell content than tumors from wild-type mice (116). The most likely explanation for these observations is that PI3K-δ blockade inhibits Tregs and PI3K-γ inhibition suppresses MDSCs and TAMs, thereby indirectly and favorably altering the T-cell immune infiltrate (3, 6, 109, 117).
Tregs represent a diverse and functionally distinct population of T-cells involved in immunological self-tolerance. Deficiencies in Treg function can lead to clinical manifestations of autoimmune disease while increased Treg presence in cancer enhances immune escape by inducing poor CD8+ T-cell tumor infiltration and cytotoxic activity (118). A commonly accepted Treg phenotype is CD4+CD25+Foxp3+CD127lo, and the critical role of PI3K-δ in maintaining Treg proliferation and immunosuppressive function was initially demonstrated in mouse models of colitis (119). Specific to cancer immunology, work in murine models has shown PI3K-δ knockout impairs Treg proliferation and redundancy in the PI3K-α and -β pathways in conventional T-cells spares their function in the face of pharmacologic PI3K-δ inhibition. Similar results have been seen with pharmacologic inhibition in human T-cell populations as well (3, 35). PI3K-δ inhibition with AZD8835 (PI3K-α/δ) and PI-3065 (PI3K-δ specific) decreased tumor Treg infiltration by over 50% as early as 3 days after treatment in the CT26 colorectal mouse model (36). Similar results of increased tumor reduction and reduced numbers of Tregs, M2-TAMs, and MDSCs were seen in A20 lymphoma and CT26 colorectal mouse models with combination duvelisib and anti-PD-1 antibody therapy versus anti-PD-1 therapy alone (120).
Characterizing Treg in blood samples from patients with CLL prior to and on treatment with idelalisib suggest that idelalisib therapy diminished Treg immunosuppressive activity (121). In patients with advanced melanoma, increased numbers of Tregs have been seen in the peripheral blood and tumors (primary, lymph nodes, and metastatic sites) of patients (15). Overall, large meta-analysis from clinical studies have shown that increased Treg infiltration is associated with reduced OS in a majority of solid tumors, including cervical, renal, melanomas, and breast cancers (122).
TAMCs encompass a wide phenotype of myeloid-derived cells that include MDSCs and TAMs. Our current understanding of myeloid cell biology suggests that cancers generate pro-inflammatory factors that disrupt normal bone marrow myelopoiesis, increase the expression of immunosuppressive factors like arginase and inducible nitric oxide synthase, and lead to the expansion of a heterogenous population of immunosuppressive immature myeloid cells (IMCs), also known as MDSCs (123, 124). While variability in the literature exists, MDSCs are phenotypically accepted to be CD33+CD11b+HLA-DR-/low in humans and CD11b+Gr1+ phenotype in mice (123, 125), and can be further divided according to either a granulocytic or monocytic (mMDSC) phenotype (126). In humans, CD14+HLA-DR-/low and CCR2+ mMDSCs are regarded as the most immunosuppressive subtype (127–129). TAMs comprise a heterogenous population of CD45+ cells residing within the TME that originate from normal tissue precursors or circulating myelomonocytic progenitors (130). TAMs are often characterized based on the classically accepted pro-inflammatory M1- or anti-inflammatory M2- macrophage phenotypes, named after the respective Th1 and Th2 cytokines with which their responses are associated. While MDSCs and TAMs are often simplified as M2-like (M2*) and characterized by high levels of IL-10 secretion (131, 132), TAM diversity is complex and unlikely yet fully elucidated, but we surmise that the majority of TAMs functionally fall on a spectrum somewhere in-between (133–136). The complex myelomonocytic checkpoints governing the differentiation of TAMs from myelomonocytic progenitors can broadly be considered as 3 steps: 1) IMC expansion and differentiation into MDSCs, 2) MDSC migration and differentiation into TAMs, and 3) TAM polarization, with each step being mediated by growth factors (GM-CSF (granulocyte macrophage colony stimulating factor); G-CSF), cytokines (IL-10, IL-6) and chemokines (CCL2, CCR5) released either by the tumor cells or the surrounding stroma (137).
Within the TME, analysis of the myeloid infiltrate with PI3K-γ knockout or inhibition demonstrates an increase in M1/M2 TAM ratios (109, 116). Findings are similar with the dual PI3K-δ/γ inhibitor duvelisib in PDX models of T-cell lymphoma, in which duvelisib reduces the percentage of M2-phenotype macrophages in the mouse spleens with a concomitant 50% increase in M1-phenotype macrophages (138). In PI3K-γ murine knockout models, intratumoral migration of TAMs and their immunosuppressive activity (as measured by cytokine production) is severely impaired in the knockout versus wild-type mice, demonstrating the critical, non-redundant role PI3K-γ plays in myeloid cell activity (6, 115, 116). PI3K-γ expression in myeloid cells in murine pancreatic cancer models is associated with transcription of genes associated with the M2-macrophage phenotype in pancreatic cancer, including immunosuppressive factors like Arg1, TGF-beta, and IL-10. Inhibiting PI3K-γ induced expression of these genes permits for CD8+ T-cell activation and reduced cancer survival (116).
De Henau et al. demonstrated that combination anti-PD-1 and anti-CTLA-4 therapy had minimal tumor growth impact in vivo in mouse models of 4T1 (breast) and B16-GMCSF (melanoma), but that adding the PI3K-γ selective inhibitor eganelisib to the combination therapy resulted in 30% and 80% complete remissions in each model, respectively (109). Dual PI3K-δ/γ inhibition, in combination with PD-1 pathway inhibition, resulted in MDSC inhibition and increased CD8+ T-cell infiltration in preclinical models of HNSCC and osteosarcoma (108, 139). These findings are further supported by the fact that selective PI3K-γ inhibition had minimal impact in a B16F10 melanoma model, presumably due to lower numbers of baseline suppressive TAMCs, in contrast to results seen in the GMCSF expressing B16-GMCSF model. Clinically, ICI resistance has been well characterized to correlate with the presence of TAMCs, as increased circulating levels of MDSCs have been shown to correspond with poor response to anti-CTLA-4 therapy in melanoma patients (140).
CART and TIL therapies use similar fundamental steps of harvesting T-cells, ex vivo manufacturing, lymphodepletion therapy, and infusion of final T-cell product. T-cells can be harvested from peripheral blood via apheresis for CART and TCR or isolated from tumor for TIL therapy. For CART therapy, the T harvested cells are transduced or transfected with genetic material encoding a new synthetic TCR (T-cell receptor), called a chimeric antigen receptor (CAR), which is able to bind to a specific antigen expressed on the surface of tumor cells. In contrast to ICIs, no ACT therapies are currently approved to be used in the first-line treatment setting.
The majority of efforts in CART therapy have focused on targeting CD19 in B-cell mediated hematologic malignancies. Clinically, CART is reserved for patients who have disease refractory to multiple prior lines of therapy with limited third-line options and poor prognosis. In DLBCL, for example, approximately 50% of patients with relapsed or refractory disease will progress following stem cell transplant, and median survival is ~6 months without any additional treatment, like anti-CD19-CART (141).
Four commercially available FDA approved anti-CD19-CART therapies currently exist. Kymriah (tisagenlecleucel) is the only CART approved for two distinct indications: adults with relapsed or refractory DLBCL, high-grade B cell lymphoma, and DLBCL arising from follicular lymphoma (142) and young adults up to age 25 years of age with relapsed or refractory B cell acute lymphoblastic leukemia (B-ALL) (143). Axicabtagene lisoleucel (Axi-cel; Yescarta) is also approved for adult patients with relapsed or refractory large B cell lymphomas, including DLBCL, primary mediastinal B cell lymphoma, high grade B cell lymphoma, and DLBCL from transformed follicular lymphoma based on results from the ZUMA-1 trial (144). The similarly approved lisocabtagene maraleucel (liso-cell; Breyanzi) is the only CART product in which CD4+ and CD8+ CART are separately manufactured and administered as sequential components in equal doses (145). Approved for the treatment of adult patients with relapsed/refractory mantle cell lymphoma (MCL) is brexucabtagene autoleucel (brex-cel; Tecartus; KTE-X19), a CD19 CART construct similar to axi-cel (146). KTE-X19 differs in that manufacturing incorporates a process of T-cell selection and lymphocyte enrichment to prevent contamination of the CART product with circulating mantle cell lymphoma cells that could contribute to disease relapse. The most recently approved CART is idecabtagene vicleucel (ide-cell; Abecma), which targets BCMA (B cell maturation antigen), a TNF family cell surface receptor commonly expressed in multiple myeloma, for the treatment of patients with multiple myeloma refractory to or relapsed after at least three prior treatments (147, 148). Another anti-BCMA CART therapy, JNJ-4528, has received breakthrough designation by the FDA based on results from the CARTITUDE-1 trial for patients with relapsed/refractory multiple myeloma (149, 150).
In contrast to CART, in TCR therapy, the T-cells are transduced with an engineered-HLA specific TCR that binds to MHC on cancer cells, can target a wider variety of cancer antigens, and is more sensitive than engineered CART (151). Unlike CART, however, no TCR therapies are currently approved for clinical use, but multiple agents are in clinical trials with a particular interest in targeting the cancer-testis family antigen NY-ESO-1 (New York esophageal squamous cell carcinoma 1). The clinical development of TCR therapies, including common indications and antigen targets, have been excellently reviewed (152). For the purposes of this review, both technologies face similar challenges with respect T-cell harvesting, ex vivo expansion, final product infusion, and clinical toxicity, and in this context, we will primarily focus on CART in the context of clinical challenges and limitations.
In contrast to T-cell harvesting from peripheral blood, TIL therapy harnesses the power of a patient’s own tumor fighting T-cells by isolating them from autologous tumor tissue (isolation step), growing and activating those cells ex vivo (expansion step), treatment with lymphodepleting chemotherapy prior to TIL infusion (pre-conditioning step), infusing expanded TILs, and supporting in vivo T-cell activation and proliferation post TIL infusion with high-dose interleukin-2 therapy (HD-IL-2) (153). While no FDA approved TIL therapies currently exist, promising options are on the horizon (154). The history of research work on TIL therapy over the past 30 years and recent advancements in treating melanoma and other cancers with TIL has been excellently reviewed previously (155–157). Initial studies were performed in patients with metastatic melanoma in the age prior to ICI when therapy options were limited (153), and since then TIL therapy has been further refined and clinically explored in other epithelial cancers, including ovarian, cervical, gastrointestinal and renal cell (156, 158–161). TIL therapy requires patients must have (1) a resectable tumor from which (2) TILs can be isolated that must (3) exhibit tumor-specific reactivity. Ex vivo TIL expansion is a resource intensive process that traditionally takes 5-8 weeks (162).
All ACT therapies require engaging the antigen receptors to promote ex vivo expansion to produce enough manufactured product and in vivo expansion and persistence to generate and maintain therapeutic response. Many pitfalls to the manufacturing process exist, including the challenges of sustained antigen stimulation that promotes T-cell differentiation, exhaustion, and eventually senescence (65, 163). Cancer and its inherent chronic inflammatory state can further impair T-cell mitochondrial function and metabolic fitness, rendering ACT less effective (67). Current strategies designed to promote formation of less differentiated cells and improve metabolic and cytotoxic capacity involve synergistically combining ACT with other treatments, like anti-PD-1 therapy and anti-OX40 antibody therapy, or directly modifying CART to express costimulatory receptors (CD27), stimulatory cytokines, or ICIs (“Armored CAR”) (164). Adding additional agents to the ex vivo T-cell expansion process to improve T-cell selection and expansion are also under consideration, including Bruton’s tyrosine kinase (BTK) inhibitors and homeostatic cytokines like IL-7, IL-15, and IL-21 to promote the memory stem cell phenotype (165, 166).
Both the T-cell intrinsic properties of the ACT product and extrinsic factors related to the patient clinical history and tumor microenvironment (TME) may promote or limit the clinical efficacy of ACT (Figure 4). Intrinsic characteristics pertain to either the CART construct or the phenotypes of T-cell subsets in the ACT product, while extrinsic characteristics relate to cancer biology or prior therapy induced negative effects on T-cells. The impact of these factors directly augment or inhibit the success of the manufacturing process and long-term efficacy of the transferred cells.

Figure 4 Addressing Challenges Associated with Intrinsic and Extrinsic Product Characteristics in ACT. Challenges with ACT pertain to intrinsic and extrinsic characteristics that limit success of the manufactured product. Intrinsic characteristics are T cell specific and extrinsic include cancer-induced pressures or prior therapy effects. Measured outcomes can be considered as clinical or surrogate outcomes.
Functional and phenotypic differences between the manufactured products are a consequence of heterogeneity between CART constructs, patient demographics, and the manufacturing process. These factors, taken together with the heterogeneity of patients across difference clinical trial designs, make direct comparisons of the efficacy of competitor products and trial outcomes challenging. All FDA-approved therapies are second generation CARTs and contain both a TCR stimulatory domain (from the T-cell surface glyco-protein CD3 ζ-chain (CD3ζ) and a co-stimulatory domain (167). Co-stimulatory domains of currently approved CART are either CD28 or 41BB (CD137). Axicabtagene ciloleucel and Brexucabtagene autoleucel are CD28-based, while tisagenlecleucel (CD19), lisocabtagene maraleucel (CD19), and idecabtagene vicleucel (BCMA) utilize 41BB. Data comparing the effect of these co-stimulatory domains, while predominantly preclinical or from small-scale clinical investigations, suggest that the type of co-stimulatory molecule expressed may contribute to differences in expansion kinetics, persistence, and efficacy amongst approved CART products (168–171).
41BB co-stimulation appears to induce growth of CD8+ Tcm cells with increased respiratory capacity, fatty acid oxidation, and mitochondrial biogenesis, while CD28 co-stimulation induces Tem with augmented glycolytic activity (171). The reason for these differences appears to lie within the distinct signaling pathways each co-stimulatory domain activates (172). In pre-clinical models, 41BB-CD19-CART demonstrated increased gene transcription of transcription factors associated with memory function (KLF6, JUN, JUNB), while similar CD28-based CART showed increased surface markers of exhaustion (TIM-3, LAG-3, CTLA-4). These differences in gene expression and enrichment pathways suggest 41BB signaling may prevent T-cell exhaustion, improving the persistence of T-cells in vivo following treatment of leukemic mice. That said, a limitation of 41BB-CART is the inability to rapidly proliferate to control tumor burden (173). In contrast, CD28-CART proliferate more rapidly than 41BB-CART but fail to persist long-term. Taken together, the use of each co-stimulatory molecule in the CART construct contributes distinct beneficial properties, with CD28-CART possessing a greater proportion of Tem cells capable of eliciting a rapid short-term effector memory response and 41BB-CART having more Tcm cells that induce long-term effector T-cell (Te) function with increased persistence (167, 173). Not surprisingly, development of third generation CART constructs that encode both CD28 and 41BB is underway (174).
T-cell differentiation has broad implications on proliferative capacity, effector function, and metabolic reprogramming. The intrinsic phenotypic differences, existing either pre- or post-manufacturing, thereby significantly influence quality of the manufactured product and subsequent therapeutic success.
Recent work by Fraietta et al. in a study of 41 patients with relapsed and refractory CLL having received anti-CD19-CART retrospectively associated complete remissions following CART therapy with an increased frequency of a class of memory-like CD8+CD27+CD45RO- T-cells in the leukapheresis sample prior to CART manufacture based on cluster analysis (68). Similarly, complete remissions following CART therapy were more likely in patients receiving products containing reduced frequencies of senescent CD8+PD-1+ CART cells (20% vs 50% or higher in PR/non-responders) and increased frequencies of Tcm-like CD8+CD27+PD-1- CART cells. The improved disease remission rate seen with the CD27+ memory like T-cells is anticipated to have a direct effect on in vivo expansion and subsequently long-term CART persistence. Clinical trial data with axi-cel (axicabtagene ciloleucel) demonstrated that higher CART levels in peripheral blood in the first 4 weeks following treatment were associated with increased treatment response and that detection of CART in the peripheral blood up to 2 years correlated with long-term ORRs (objective response rates) (144). Conversely, we have reported that oligoclonal expansion of CD27/CD28 double-negative T-cells in response to pancytopenic aplasia, led to rapid disease relapse in a patient following CART therapy (175).
Taken together, optimization of CAR constructs and a greater understanding of favorable CART T-cell phenotypes on intrinsic function are areas for further research. Unfortunately, standard methods for CART manufacturing at this time do not automatically enrich the product for favorable T-cell phenotypes and inadvertently may also promote antigen-driven terminal differentiation (41, 176–178).
TIL products rely on the intra-tumoral presence and isolation of tumor antigen specific T-cells. In less immunogenic malignancies or malignancies that lack high acquired/somatic mutational load, expanding tumor reactive T-cells has been challenging. In recent years, this process has benefited from newer high-throughput genetic sequencing technologies that can identify tumor-specific mutations and neoantigens that can be targeted by TILs. Advances in whole exome sequencing have enabled investigators to selectively isolate and expand tumor and peripheral blood T-cells reactive against those tumor epitopes for TIL therapy (179). Such advancements allow researchers to identify patient specific, targetable, and somatic mutations in epithelial cancers like breast (BC), esophageal, and ovarian cancers and employ them in TIL therapy (180). A recent study of a patient with chemo-refractory metastatic HR+ (Hormone Receptor) BC with a complete response following treatment with autologous TILs demonstrated the infused product was reactive against mutant versions of 4 different proteins (SLC3A2, KIAA0368, CADPS2, CTSB) (181). Other approaches include selecting TILs from the expanded product that demonstrate a specific phenotype, such as with LN-145-S1, a PD-1 selected TIL therapy under investigation in head and neck cancers (Clinicaltrials.gov Identifier: NCT03645928) (32).
Patients with hematologic malignancies, especially those having had multiple lines of cytotoxic chemotherapy, have more senescent T-cells (67, 68) and fewer of the memory-like CD8+ cells that are associated with durable remissions after CART therapy. The level of cancer induced T-cell senescence also varies by cancer-type, especially among chronic versus acute CD19+ B cell malignancies. In CLL, there is a T-cell-intrinsic disadvantage that exists prior to initiating CART manufacturing that is likely related to CLL-induced immune dysfunction (67, 68). These T-cell intrinsic defects may potentially explain the finding that meta-analysis of clinical trials shows lower rates of CD19-CART-mediated remissions in CLL compared to other hematologic malignancies (2, 182).
Disease relapse after CD19-CART therapy can be antigen positive (CD19+) or antigen negative (CD19-). 10-20% of patients with CD19+ malignancies can develop native-antigen negative disease (CD19 mutation or down regulation) that is challenging to treat or re-treat with CD19 CART (183). Approaches to overcome antigen loss include designing CAR constructs that target multiple antigens, such as CD19 and CD22 for ALL (Acute Lymphoblastic Leukemia) (NCT03241940 and NCT03289455), ALL and diffuse large B cell lymphoma (DLBCL) (NCT03233854), ALL and non-Hodgkin lymphoma (NHL) (NCT03330691 and NCT03448393), and NHL and CLL (NCT03019055). With dual-recognition, CARTs can engage either antigen and tumor cells must lose expression of both antigens concomitantly for escape.
Clinically, disease relapse due to antigen loss or lack of antigen presence is less common than antigen positive relapse, which more often is related to lack of CART persistence, low CART potency, or B cell aplasia (183). In the Phase II JULIET trial with tisagenlecleucel in patients with DLBCL, no differences in response between groups stratified by tumor expression of CD19 were seen (142). With axi-cel, further analysis of CD19 expression at the time of progression showed that only 3 (27%) out of 11 patients with CD19+ baseline disease had CD19- disease at progression (144).
The sensitivity of methods used to assess epitope presence also matters. In the axi-cel trial, 8 patients with assay-tested CD19- disease were included in the trial, and response rates in these 8 patients were similar to trial participants with assay-confirmed CD19+ disease. While not powered to specifically examine this, the positive responses to anti-CD19 CART even with epitope negative disease suggests that current antigen detection assays may not entirely or accurately assess target antigen presence.
Most FDA approved indications for CART include patients with relapsed or refractory aggressive B cell lymphomas, and therapy for such patients often involve high dose chemotherapy with stem cell transplantation. An unintended consequence of cytotoxic chemotherapy is the depletion of Tn, Tscm, and Tcm subsets that possess the greatest expansion potential and anti-cancer activity (184, 185). DLBCL patients with a history of extensive prior chemotherapy have fewer naïve and minimally differentiated T-cells, including increased populations of CD27 and CD28 double- negative senescent cells, when compared to healthy controls and newly diagnosed DLBCL patients (69).
Lymphodepletion with chemotherapy prior to ACT infusion also impacts clinical efficacy. In patients with NHL undergoing CD19 CART, patients who received combination cyclophosphamide with fludarabine versus cyclophosphamide alone had increased CART expansion, persistence, and objective response rates, especially CRs (50% vs 8%) (186). Lymphodepletion has broad benefits, including killing of immunosuppressive regulatory T-cells (Tregs) and myeloid derived suppressor cells (MDSCs), elimination of T-cells with cytokine receptors that function as homeostatic cytokine sinks resulting in increased levels of cytokines like IL-7, IL-15, and IL-21 necessary for in vivo ACT expansion (187), and direct modulation of tumor IDO (indoleamine 2,3-dioxygenase) (188), all of which together beneficially support T-cell persistence and cytotoxic activity (189). In preclinical models, TBI (Total Body Irradiation) has been shown to increase microbial Toll-Like Receptors (TLRs) that activate antigen presenting cells (190). The impact of prior systemic therapies on CART success suggests there is potential for treatment with additional immunomodulatory therapies that favorably alter in vivo frequencies of Tn and Tcm cells prior to T-cell isolation or suppress Tregs and other immunosuppressive cell populations and alter circulating cytokine levels prior to CART infusion.
Clinical outcomes with CD19 CART vary based on the B-cell malignancy histology, patient demographics, and intrinsic features of the manufactured product, including co-stimulatory domains engaged and CD4/CD8 T-cell ratios. Meta-analysis of 42 trials of anti-CD19 CAR T-cells in various B cell hematologic malignancies showed higher complete response (CR) rates of 77% in acute lymphoblastic leukemia patients (ALL) versus 25% in chronic lymphocytic leukemia (CLL) and 54% in non-Hodgkin lymphoma (NHL) (2). Across all four commercially available CD19 CART therapy approval trials, except for B-cell ALL, the primary end points were ORR (CR or PR (partial response)), with additional metrics of time to response, duration of response (DOR), relapse free survival (RFS), overall survival, and CART kinetics also investigated. Clinical response rates (CR and PR) from pertinent clinical trials of the above CART therapies are reviewed in detail in Table 1.
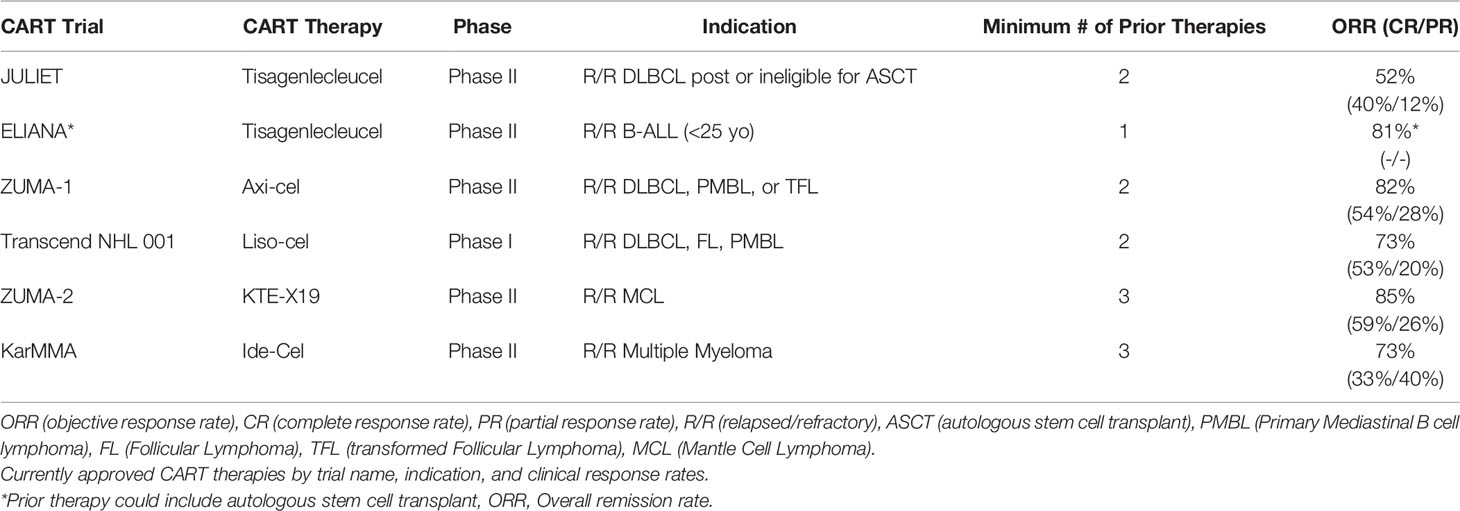
Table 1 Clinical Outcomes from CART FDA Approval Trials.
Taking these findings together, CART response rates in high grade B-cell lymphomas are generally >50% regardless of the therapy given. Of note, the median time to clinical response appears to vary according to clinical trial-specified assessments (1 vs 3 months), and in some cases, delayed CRs have been observed. In Zuma-1, the first tumor assessment was at 1-month, which corresponded with the median time to response, however 23 patients (11 of 35 with a partial response and 12 of 25 with stable disease) developed CRs in the absence of additional therapies up to 15 months after CART. The heterogeneity in patient populations and prior therapy received between trials, variability in observed CR and PR rates with similarly designed CART products, and observation of delayed ORRs beyond pre-determined clinical timepoints suggest we still have much learn about how we assess clinical outcomes with CART.
Toxicity following CART infusion is common, with the most worrisome being CRS (cytokine release syndrome) and neurotoxicity (2). In CRS, infusion of cytotoxic T-cells leads to a rampant release of cytokines like IFN-γ, GM-CSF, TNF, IL-10, and IL-6, triggering an inflammatory response characterized by tachycardia, risk for acute respiratory distress syndrome (ARDS) acute hypoxic respiratory failure, and multiorgan failure (191). CRS is treated by administering the IL-6 inhibitor tocilizumab or steroids, both of which can reduce the morbidity and mortality associated with CRS. Tocilizumab does not appear to suppress the cytotoxic activity of the infused T-cells, and is generally preferred to steroids as initial therapy (192). Neurotoxicity from CART is biologically less well understood, but symptomatically recognizable. Neurotoxicity can occur with CRS, after CRS, or in delayed form multiple weeks after infusion, has been observed occurring in all grades as high as 64% (axi-cel). Across the aforementioned approval trials, Grade 3 & 4 CRS rates were highest with tisagenlecleucel (DLBCL: 22% and ALL: 46%) and lower for axi-cel (13%), liso-cel (2%), KTE-X19 (15%), and ide-cel (5%). The most common other Grade 3 or higher adverse event with CART is cytopenia, with neutropenic predominance. In the KTE-X19 and ide-cel trials, more than 90% of patients experienced Grade 3 or higher cytopenias. Direct comparisons between individual CART product toxicities are challenging due to differences in the grading criteria used between trials.
TIL therapy has shown promise as an effective cancer therapy. Results from 93 patients with metastatic melanoma receiving lymphodepletion and TIL infusion demonstrated an ORR of >50% with 22% experiencing (n=20) complete tumor regression. At 3 years, 19/20 were still in complete response (193). In comparison, single agent HD-IL-2 therapy, which is FDA approved in this therapy refractory setting, has a 5-10% ORR (194). In a smaller study of 9 patients with recurrent metastatic cervical cancer, 3 patients had an ORR with 2/3 being CRs (159). Studies in ovarian cancer, kidney, gastrointestinal, and head and neck cancers have also been attempted, with mixed results (156, 158, 195). A critical challenge to assessing the clinical value of these data in today’s clinical context is that many past studies with TIL therapy occurred prior to the development of ICI therapies like anti-PD-1/PD-L1 and anti-CTLA-4.
Recent advancements in commercial TIL production have improved upon the initial limitations of TIL therapy, and recent clinical trial data strongly suggests this modality will become part of the clinical paradigm as an option after current standard of care in the near future. LN-144 (lifileucel) is a commercially produced TIL therapy that has a TIL manufacturing time of 22 days (Gen-2 TIL) and has demonstrated clinical benefit in cancers like melanoma, cervical cancer, and head and neck cancers. In a Phase 2 clinical trial (NCT02360579), LN-144 demonstrated a 38% (n=18) ORR (1 CR, 17 PR) in 47 metastatic melanoma patients previously treated with anti- PD-1 antibody and/or BRAF inhibitor (DOR 1.3-14.0 months) (154). Similar results were seen in 13 recurrent metastatic squamous cell carcinomas of the head and neck cancer patients (31% ORR) (NCT03083873) (196) and in 27 recurrent, metastatic or persistent cervical carcinoma patients (44% ORR, 11% CR, 33% PR) treated with lifiluecel LN-145 TIL therapy (NCT03108495) (197). LN-145 trials are ongoing in patients with metastatic triple negative BC (NCT04111510) and other bone and soft tissue sarcomas (NCT03449108) (32).
TIL therapy toxicity lacks the CRS and neurotoxicity seen with CAR-T therapy, is most often related to the consequences of the pre-conditioning non-myeloablative therapy or post-infusion HD-IL-2 therapy, and is generally short lived (<2 weeks).
In trials with commercially produced TILs (NCT02360579, cohort 2, n=47), the most frequently reported toxicities of any grade (>50% of patients) were thrombocytopenia, chills, neutropenia, febrile neutropenia, anemia, and pyrexia. Approximately 95% of patients reported Grade 3 & 4 toxicities, with the most common being thrombocytopenia (81%), neutropenia (53%), febrile neutropenia (53%), anemia (47%), and leukopenia (43%) (154). Similar results were seen in cervical cancer (NCT03108495) (n=27), with Grade 3 & 4 toxicities observed in 96% of patients, the most frequent being anemia (56%), thrombocytopenia (44%), neutropenia (30%) and febrile neutropenia (30%) (197). Myeloablative regimens, typically combining chemotherapy with TBI, may improve ORRs and CRs, albeit with increased clinical toxicity. NCT01319565 is a clinical trial evaluating the addition of TBI to pre-conditioning regimens prior to TIL therapy (32).
While there is much interest in developing surrogate endpoints to determine ACT success, no standardized metrics currently exist. However, surrogate outcomes like manufacturing success and ACT cell product persistence in vivo may become valuable metrics in future clinical trials.
Failure to manufacture or complications while awaiting CART therapy are measurable outcomes that indirectly affect planned CART therapy for patients with aggressive disease. While most patients that undergo leukapheresis are able to receive CART therapy, manufacturing failure remains a limitation in the success of this novel therapy. Reasons contributing to manufacturing failure include the collection of insufficient numbers of T-cells from pre-treated patients or contamination of the apheresis product with granulocytes and monocytes (198). Other quality control issues related to post-manufacturing viability, product purity, or product potency, as determined by quantitative or qualitative cytotoxic or cytokine release assays, may also render the final product unsuitable for infusion (198). Early non-commercial CART products had higher manufacturing failure rates from 2% as high as 14%. In the post-marketing-approval use of CART, rates of manufacturing failure have continually decreased (Table 2).
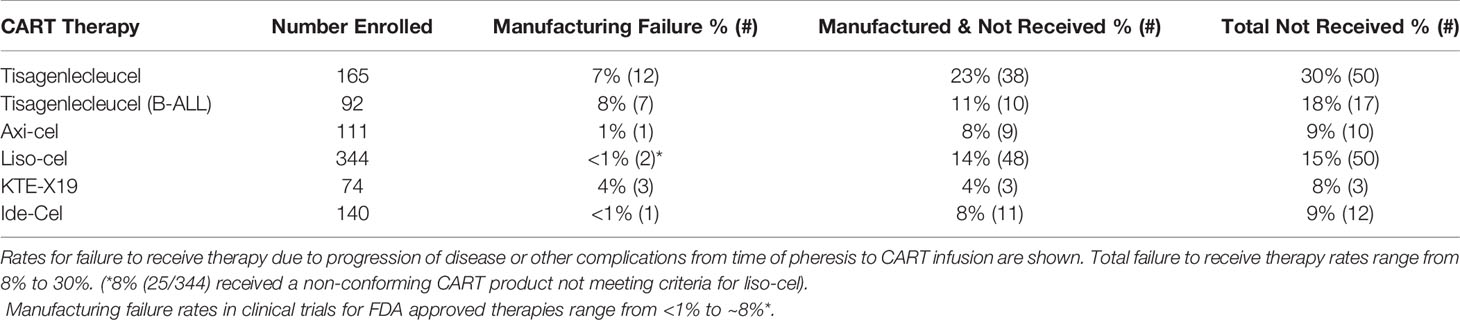
Table 2 Clinical Experience With CART Manufacturing.
Long-term CART persistence correlates with response rates. In the Zuma-1 Phase II trial, CART cells peaked in the blood within 14 days post infusion and were detectable up to 180 days for most patients. At 24 months, 3 patients with CRs still had detectable peripheral blood CART transgene levels (144). Similarly, with tisagenlecleucel, CAR transgene levels were measurable in the peripheral blood up to 2 years in patients with durable responses (142). Kinetic studies with BCMA CART showed 29 of 49 patients (59%) had cells present at 6 months and 4 of 11 patients (36%) at 12 months post infusion (148). Delayed CRs, especially those occurring 3 months or more after therapy infusion, raise important questions about CART pharmacometrics and time to optimal response. In Zuma-2, the median time to CR was 3.0 months but ranged from 0.9 up to 9.3 months (146). Even more impressive, delayed CRs reported in ZUMA-1 were seen up to 15 months in the absence of additional therapies (144).
Multiple strategies to improve CART persistence have been considered. Controlling the ratio of CD4 and CD8 T-cells in the infused product and using lymphodepleting fludarabine chemotherapy prior to infusion are some promising methods (199, 200). Additional strategies being considered include building newer generation CARs with multiple costimulatory domains in addition to CD28 or 41BB, like ICOS, or modifications in CD28 amino acid residues (201, 202). Reduction in target CAR antigen levels, either in direct response to CART therapy or through cancer-mediated downregulation results in loss of CAR stimulation and CART persistence. Strategies involving maintenance therapy that bolsters target antigen levels have demonstrated promise in preclinical models (203).
Manufacturing time indirectly affects CART efficacy. Patients awaiting CART therapy have aggressive disease and are at high risk of complications by cancer, cancer progression, or other co-morbidities. Adverse events while awaiting CART inevitably renders a portion of patients ineligible to receive the manufactured product (Table 2). Typical manufacturing times range from 2-4 weeks, with the median times from leukapheresis to product delivery for axi-cel and KTE-X19 reported as 17 and 16 days, respectively (144, 146). For JULIET, 38 (23%) of 165 enrolled patients discontinued study participation for reasons unrelated to manufacturing, of which 34 of 38 were related to disease progression (142). In the tisagenlecleucel ALL trial, ~11% (10/92) of patients died prior to receiving CART due to disease progression (n=4), complications from sepsis, respiratory failure and fungemia (n=3), and other complications (n=3) (143). In the ZUMA-1 axi-cel trial, 8% (9/111) enrolled patients did not receive CART due to progressive disease or serious adverse reactions following leukapheresis (144). 14% (48/344) of the enrolled patients in the liso-cell trial had lymphoma complications or died before receiving CART, even with allowance for bridging therapy between leukapheresis and CART infusion (145). Similarly, 4% (3/74) of patients for whom KTE-X19 CART were successfully manufactured did not receive therapy (146), and ~8% (11/140) did not receive the manufactured ide-cel product (148). Combining the manufacturing failure and pre-infusion drop-off rates means ~9-30% of patients in CART clinical trials fail to receive the manufactured product (Table 1). Comprehensive data showing the real-world success of manufacturing commercial CART products in patients treated outside of a clinical trial is still pending, but the challenges of patient selection outside of the clinical trial setting, increasing adoption of commercial CART therapies at more cancer treatment centers, and additional challenges related to increased production and demand raises concerns whether or not these rates will increase.
TIL therapies face similar challenges in efficient manufacturing. Using the selected TIL manufacturing protocol with the traditional pre-REP, selection, and REP process typically takes 5-8 weeks for production, and the final expansion products are given immediately so as to avoid the risk of product loss due to cryopreservation (162). Combining the inability to grow TILs in ~20-25% of patients with the risk of disease progression during the long expansion times, drop-out rates in clinical trials initially ranged from 25-70% (156, 204–206). Use of a newer expansion method called the young TIL protocol has reduced manufacturing times by up to 3 weeks, but left the issue of concern with cryopreservation, limiting the ability to streamline the process with centralized expansion and direct distribution to clinical sites. Newer Gen-2 and Gen-3 versions of the commercially produced lifileucel LN-144/LN-145 TIL therapy seek to address these limitations. The Gen-1 TIL manufacturing time, which included phenotype selection, was 38 days in duration, and yielded a fresh, hypothermic product. The Gen-2 TIL process omits TIL selection and allows for production of a final, shippable, cryopreserved product within 22 days. Newer Gen-3 versions of LN-145 now seek to reduce the manufacturing time to just 16 days (207).
Taking into consideration the signaling cascade linking the CAR and TCR with PI3K signaling, the role of PI3K on T-cell differentiation and metabolic reprogramming, and the standard methods of expanding CARTs and TILs ex vivo using anti-CD3/CD28 stimulation, the evidence for utilizing PI3K-δ and -γ inhibition as a means to improve the ACT manufacturing process is compelling.
We identify five points of intervention within the ACT therapy manufacturing process wherein strategic changes can be made to modify intrinsic and extrinsic T-cell product characteristics and improve clinical and surrogate outcomes, in particular those related to prior therapy and cancer mediated T-cell differentiation, CD4/CD8 expansion ratios, Treg activity, T-cell exhaustion, and post treatment toxicity (Figure 5). These points of intervention include: 1) In vivo treatment with a PI3K inhibitor (PI3Ki) therapy prior to leukapheresis or T-cell isolation, 2) Ex vivo culture conditions, 3) Lymphodepleting therapy, 4) Maintenance therapy, and 5) Toxicity prevention strategies. We discuss the exciting current preclinical and clinical data supporting the role of PI3K-δ and -γ inhibition in each of these situations while giving consideration to other non-PI3K mediated strategies currently being studied.
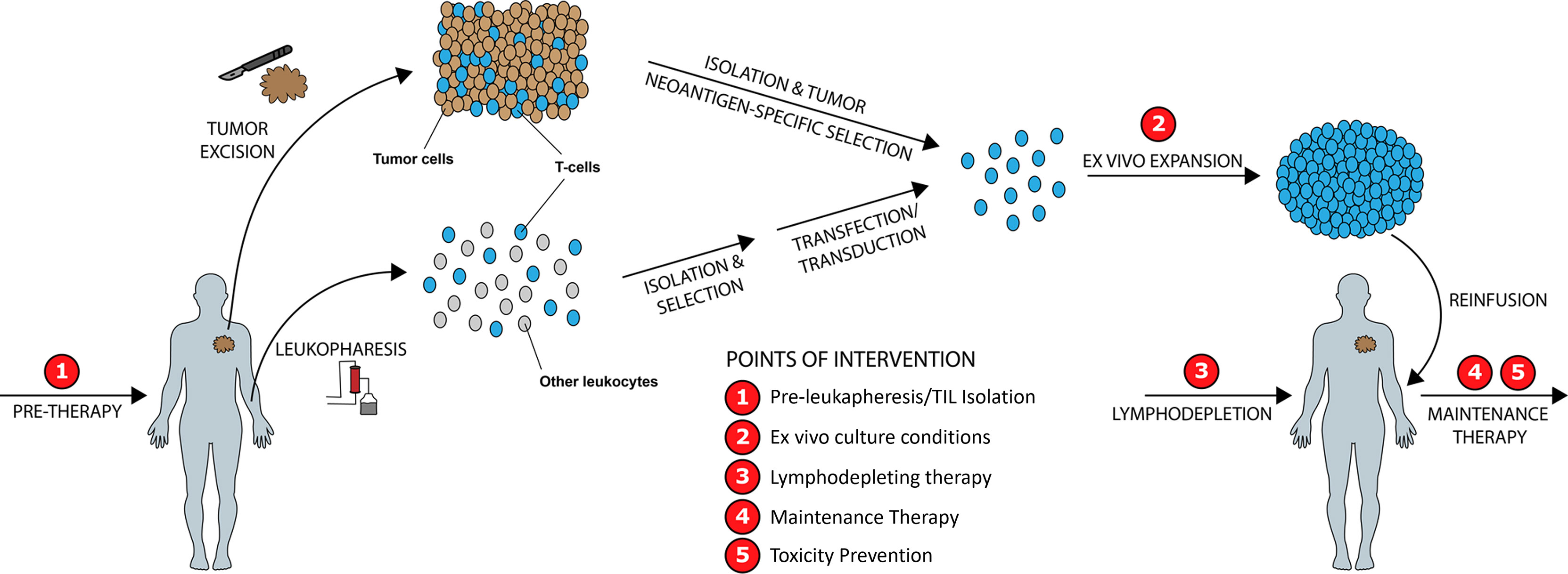
Figure 5 Improving ACT Therapy with PI3K-δ and PI3K-γ Inhibition. Five distinct timepoints during ACT manufacturing are identified wherein intervention with PI3K-δ/-γ inhibitors may improve manufactured product quality and clinical outcomes.
The potential benefits to in vivo PI3K therapy prior to leukapheresis or T-cell isolation may have direct impacts on having a more favorable baseline T-cell phenotype for CART or increased T-cell yield from tumor for TIL therapy.
As previously discussed, the effects of prior systemic therapy and malignancy-driven T-cell terminal differentiation together reduce frequencies of memory and naïve cells in the leukaphereses T-cell product, thus reducing expansion capability and subsequent manufactured product response rates. As previously discussed, data from Fraietta et al. in CLL patients associated increased frequencies of CD27+CD45RO-CD8+ memory-like T-cells prior to CART manufacturing with improved disease remission rates (68). This finding supports exploring strategies to enhance memory-like T-cell expansion in vivo prior to pheresis with PI3Ki therapy. Such studies are planned but have yet to be undertaken. The majority of evidence to test this hypothesis derives from ex vivo pre-clinical and clinical studies and will be further discussed in the following sections.
In TIL therapy, early studies noted high patient dropout rates of ~33% due to lack of T-cells in the tumor for successful expansion. These studies employed 2-phase TIL expansion protocols comprised of an initial pre-rapid expansion phase wherein tumor tissues are cultured with IL-2 to support T-cell division and survival, and a subsequent process of T-cell selection based on tumor reactivity followed by 14-days of rapid expansion (REP). During REP, T-cells were activated via anti-CD3 binding and co-cultured with irradiated feeder cells (either autologous or allogeneic) (208, 209). Challenges with this process include poor expansion during the pre-REP phase if no T-cells are present, lack of tumor-reactive T-cells to undergo rapid expansion, and reduction in T-cell health associated with rapid expansion and prolonged ex vivo culture (210). Therefore, methods to augment T-cell infiltration into the tumor microenvironment prior to T-cell isolation could therefore have positive effects on improving TIL manufacturing success. As prior noted, in vivo preclinical studies have demonstrated that PI3K-δ inhibition and knockout favorably increase the CD8+ TIL/Treg ratios in mouse models of lung, breast, and colon cancer (36). Not surprisingly, clinical studies are now underway to further assess such findings and characterize a role for PI3K inhibition prior to TIL isolation (NCT04142554 & NCT02646748).
During ex vivo culture, preferential expansion of either the CD4+ cells vs CD8+ cells can skew CD4/CD8 ratios in infused product and influences the CART product persistence and efficacy (211). Concomitant stimulation of CD8+ TCRs and CARs promotes CD8+ CART-cell exhaustion with increased PD-1 and LAG-3 surface expression and decreased long-term persistence (212). Such divergences in maturation may also persistent following infusion, as T-cell phenotypes from the bone marrow of multiple myeloma patients post anti-BMCA-CART demonstrated increased frequencies of CD8+ Tscm and Tcm populations vs CD4+ Tcm and Te populations, suggesting CD4 and CD8 cells may undergo divergent courses of maturation in vivo following infusion (213). One approach, explored to counter this effect is to engineer paired CD4/CD8 CART products and infuse a final 1:1 therapy. Use of this strategy in a non-commercial anti-CD-19 CART product in a phase 1 trial of children and young adults with relapsed or refractory B-ALL reported a ~90% remission rate with 93% manufacturing success (NCT02028455) (214). These results are not uniform, however, as 25 patients in the Transcend NHL 00 Liso-cel trial received non-conforming CART products that did not contain equivalent CD4+/CD8+ product ratios (145). An alternate thought is that the presence of CD4+ cells beneficially influence and support CD8+ cell expansion. Ex vivo studies using pheresis samples from healthy donors and lymphoma patients demonstrated that initial CD4:CD8 ratios of at least 40%:60% more than doubled CD8+ CART expansion yields. A final 1:1 product was most closely achieved with starting ratios of 70%:30%, and this product also demonstrated the greatest anti-tumor effect against Raji (human B-lymphocyte cell line) lymphoma cells in an immunodeficient mouse model (215).
Growing separate CD4 and CD8 cultures or manipulating pre-expansion CD4/CD8 ratios introduces additional technical challenges. As an alternative, pharmacological manipulation of culture conditions may offer a simpler approach. Several groups, including our own, however, have shown that adding PI3K inhibitors during CART manufacturing may be an alternative to separately manufacturing CD4+ and CD8+ CART products. The Paulos lab has shown that PI3K-δ activity alters T-cell differentiation in murine and human CD8+ adoptively transferred T-cells using both murine transgenic TCR pmel-1 CD8+ T-cell models and a human peripheral blood T-cell model wherein T-cells are transduced with a tumor antigen specific CAR that recognizes the mesothelin, 4-1BB, and CD3ζ signaling domains (mesoCAR) (216). Inhibition of PI3K-δ during ex vivo manufacturing for 7-days with idelalisib resulted in a less differentiated T-cell products possessing a Tcm phenotype (increased CD62L/CCR7, CD127, and Tcf7) (216). These T-cell products also demonstrated increased anti-tumor activity against melanoma and mesothelioma in mice.
Phenotypically, altering PI3K signaling during ex vivo expansion appears to reduce T-cell exhaustion. We and others have shown PI3K inhibitors promote dose-responsive decreases in the expression of immune checkpoint molecules and exhaustion markers like TIM-3, LAG-3, and PD-1, thus restoring the Tcm phenotype (42, 113, 217). In the mesoCAR-T model, treatment with eganelisib (PI3K-γ) reduced surface TIM-3 expression (113). Altering the exhaustion phenotype of the manufactured CART product may have significant clinical implications, since in JULIET 11 patients with the highest percentages of LAG-3+ T-cells did not respond to tisagenlecleucel or had early relapse (142). Similarly, TILs from patients with HNSCC (head and neck squamous cell cancers) refractory to anti-PD-1 therapy demonstrated an enhanced exhaustion phenotype with TIM-3 upregulation that appears to be mediated by PI3K-Akt pathway activity (218). Further supporting these findings, the Paulos lab demonstrated that TIL cultures expanded from patients with lung carcinoma demonstrated reduced TIM-3 expression and higher CD62L when the cells were subsequently cultured with idelalisib for 2 weeks (216).
Functionally, PI3Ki-expanded CART cells demonstrate increased cytotoxicity, superior persistence and in vivo expansion, and greater anti-leukemia activity against human CLL cells engrafted in an immunodeficient NOG (NOD/Shi-scid/IL-2Rγnull) mouse model (42). In these experiments, short-term exposure to PI3Ki during CART cell manufacturing led to persistent alterations in CART cell function, even weeks after in vitro exposure to duvelisib, suggesting critical transcriptional and epigenetic changes occur in PI3Ki-expanded T-cells. Such changes may have lasting implications on cytotoxic function. In vivo mouse model studies with idelalisib demonstrated increased mesoCART-cell persistence up to 55 days post T-cell transfer, with the percentage of CD45+ peripheral blood lymphocytes almost 3-fold higher in the idelalisib cultured group vs control cultures (216). Pmel-1 transgenic CD8+ T-cells expanded ex vivo with either idelalisib (PI3K-δ) or eganelisib (PI3K-γ) showed enhanced in vivo tumor control against a B16F10 melanoma (median survival 70 days and ~60 days, respectively) vs control CD8+ cultures (30 days) (113).
Mechanistically, as previously reviewed, TCR stimulation with anti-CD3/CD28 beads during CART-cell manufacturing can induce PI3K/AKT signaling and T-cell terminal differentiation (68), similar to how PI3K/mTOR signaling is differentially regulated during antigen-driven expansion of CD4+ and CD8+ T-cells. Western blot analysis of murine CD8+ T-cells transduced with a TCR against the cancer-testis PLAC1 prior to in vivo infusion demonstrated that the transduced cells expressed increased PI3K-γ and phospo-AKT (219). Furthermore, lymphocyte differentiation studies have shown that daughter T-cells with increased PI3K/mTOR signaling differentiate into an effector cells, while those with reduced PI3K/mTOR signaling retain self-renewal capacity (220, 221). We also believe that minimal negative effects seen on ex vivo T-cell expansion in the presence of PI3K inhibition is mediated by concomitant signaling through the MEK/ERK pathway. We have previously shown that CART products cultured and expanded in the presence of duvelisib simultaneously demonstrated reduced phosphorylation of downstream PI3K pathway proteins with increased MEK and ERK phosphorylation (42).
A plausible explanation for the increased cytotoxicity seen in PI3Ki-expanded CART cells is secondary to downstream increases in CD27 and CD28 expression, a feature of T-cells noted to have increased in vivo persistence and cytotoxic activity (222). It has been well characterized that rapid expansion of TILs with IL-2 reduces T-cell surface CD27 and CD28 expression and results in an increased fraction of terminally differentiated T-cells in the final product (223, 224). In more recent years, cells expanded via the young TIL method has mitigated the adverse effects of rapid expansion, with young TIL products demonstrating longer telomeres and higher expression of CD27 and CD28 (210, 225). Pharmacologically achieving similar results with PI3Ki, however, may be less technically challenging. Already Dwyer et al. has shown that mesoCART cultured with eganelisib demonstrate reduced T-cell differentiation and increased CD27/CD28+ surface expression (113). Inhibition of PI3K-δ, either alone or simultaneously with PI3K-γ, also increased CD28 expression in these models.
Taken together, PI3K inhibition during ex vivo T-cell culture has the ability to inhibit differentiation and exhaustion mechanisms without affecting proliferation. Further supporting these preclinical and clinical correlate studies is data from a recent phase I study of bb21217, an anti-BCMA CART therapy based on ide-cel that adds the PI3K inhibitor bb007 during ex vivo culture to enrich the drug product for memory-like T-cells (226). Comparison of T-cell populations from peripheral blood versus the manufactured drug product showed bb21217 had increased enrichment for CD27+/CCR7+ Tm cells, depletion of CD57+ senescent cells, increased CD127 expression (a marker of persistent Tm formation), and higher peak in vivo CART expansion.
As prior discussed, prolonged manufacturing time and manufacturing failure remain as major clinical hurdles. While data is lacking, early studies promising suggest the addition of PI3Ki to ACT expansion cultures can shorten manufacturing times or reduce the manufacturing failure rate.
As previously discussed, lymphodepletion with chemotherapy prior to CART infusion improves T-cell persistence and cytotoxic activity (186) by killing immunosuppressive Tregs and MDSCs, eliminating homeostatic cytokine sinks (187), and modulating tumor IDO (indoleamine 2,3-dioxygenase) (188). Interestingly, PI3K-δ inhibition with idelalisib in patients with CLL has been shown to inhibit CD4+CD25+CD127- Treg proliferation and Treg-induced suppression of CD4+ and CD8+ T-cells, suggesting pre-infusion pharmacologic PI3Ki therapy may favorably augment the effects of lymphodepletion (121). Multiple studies in TIL therapy have already been performed to add additional therapy, like radiation, to the to the pre-conditioning regimen (227–229), but none with PI3K inhibitors.
Another consideration is to continue PI3Ki therapy following ACT product infusion. One reason for this is to mitigate the ill effects of rapid in vivo expansion. Similar to ex vivo anti-CD3/CD28 stimulation, rapid in vivo CART expansion induces increased expression of the T-cell exhaustion marker PD-1. Adding anti-PD-1/PD-L1 therapy after ACT is one approach being considered to rescue T-cells from exhaustion, but responses from CART trials have not been uniformly positive (230). Multiple clinical trials are now studying post-TIL anti-CTLA-4 or anti-PD-1 therapies in advanced or metastatic cutaneous melanoma, HNSCC, NSCLC, or cervical cancers (NCT02278887, NCT03645928, NCT03108495). Post-TIL HD-IL-2 therapy, which is given to support TIL expansion and persistence, may also unfavorably stimulate in vivo Treg expansion. In one study, melanoma patients with the highest fold expansions of these ICOS+ Treg-like cells following TIL therapy and HD-IL-2 were noted to have worse clinical outcomes than patients with fewer ICOS+ Tregs (231). Taken together, post ACT PI3Ki therapy, with or without checkpoint blockade, may favorably modulate cytotoxic T-cell and suppress Treg expansion post ACT.
Reducing toxicities like CRS would also improve outcomes with CART. While tocilizumab and steroids are useful treatments for CRS, tocilizumab requires IV infusion and steroids can inhibit the CART effect. There remains much need for safe and tolerable therapies to use in the post-infusion period to reduce risk of CRS without affecting CART efficacy. Recent preclinical evidence suggests that in ex vivo CRS assays and in vivo murine models of CD19 CART, the dual PI3K-δ and -γ inhibitor duvelisib, antagonizes and reduces IL-6 secretion better than single agent isoform selective PI3K-δ and -γ inhibitors without negatively inhibiting CART function (232). More data from pre-clinical humanized mouse models and from the clinical setting are necessary to further validate and test these promising approaches.
Despite the recent success of T-cell immunotherapies, much room for improvement remains. With ACT, the timing of cell manufacturing, availability of normal lymphocytes and tumor antigen for ACT manufacturing, and the complexity of the manufacturing process must be intimately integrated with the clinical treatment timeline to achieve success. This poses unique challenges, and we have highlighted herein five points in the ACT manufacturing process where the addition of PI3K inhibitors might result in improvement in manufactured product or the clinical outcome of ACT. T-cell targeted immunotherapies face challenges related to immune cell infiltration and T-cell activity in the TME. Multiple therapeutic synergies pairing anti-PD/PD-L1 agents with chemotherapy, radiation therapy, molecular targeted therapies like HER-2 targeted agents and inhibitors of VEGF, CDK4/6, PARP, HDAC, BRAF, MEK, and other checkpoint inhibitors like TIM-3 and LAG-3 (NCT04370704) have been considered in order to overcome the well-characterized challenges of tumor immune escape mechanisms (95, 233–236). Furthermore, targeting metabolic pathways of cancer and related cells in the TME contributing to nutrient and metabolic stress has become an area of growing interest with multiple therapeutic targets under consideration, including IDO, MCT1/MCT4, mitochondrial complex I, and the mitochondrial tricarboxylic acid (TCA) cycle (237–240).
While the PI3K pathway is critical to cancer development and regulates activity of multiple immune cell populations in the TME, clinical development of pan-PI3K inhibitors for the treatment of solid tumor malignancies in melanoma, breast, lung, colorectal, and head and neck cancers has faced challenges of inefficacy and clinical toxicity (16–23).
The toxicities of PI3K inhibitors must be considered during clinical applications. Clinically, they can manifest with autoimmune or autoinflammatory signatures, including liver toxicity, hyperglycemia, rash, and colitis. Notably, these symptoms overlap during treatment of both solid and hematologic malignancies, vary by route of administration, and appear to improve with isoform-selectivity (241, 242). Furthermore, while the mechanisms of immunomodulation governing these toxicities are still yet to be fully characterized, many of them toxicities overlap with those seen with ICIs, which must be further considered during synergy trials (78).
Isoform selectivity and drug formulation have a direct effect on clinical side-effects and toxicities (241, 242). Hyperglycemia is most commonly seen with PI3K-α inhibition (selective and pan-inhibitors), due to therapy induced disruptions in insulin signaling and glucose hemostasis leading to clinically evident metabolic changes (243). Comparison of idelalisib (oral, PI3K-δ) with copanlisib (IV, PI3K-α/δ) shows that Grade 3 diarrhea or colitis is rarely seen with copanlisib, but common (>15%) with idelalisib, likely due to the IV formulation (241). Furthermore, severe colitis and pneumonitis risk is higher with PI3K-δ and -γ inhibition (idelalisib, copanlisib, and duvelisib) (31, 242, 244).
Clinical trials have shown PI3K-δ and -γ inhibition are associated with increased immune suppression and opportunistic infection risk (111, 244). In DYNAMO, a Phase II study of duvelisib in patients with refractory indolent non-Hodgkin lymphoma, dual -δ/-γ inhibition led to frequent AEs of any-grade of diarrhea (48.8%), nausea (29.5%), neutropenia (28.7%), fatigue (27.9%), and cough (27.1%) (244). The most frequent grade 3/4 AEs were neutropenia (24.8%), diarrhea (14.7%), anemia (14.7%), and thrombocytopenia (11.6%) and similar rates of grade 3 neutropenia (27%) were observed in a Phase I trial of the PI3K-δ selective idelalisib, also in patients with relapsed non-Hodgkin lymphoma (29).
The combination of a greater understanding of the redundant and non-redundant functions of PI3K signaling in T-cells, the systemic toxicities of targeting different isoforms, and the broad availability of isoform selective agents, has initiated a new wave of clinical testing, particularly with using PI3K-α inhibitors for PIK3CA mutated cancers (245, 246), PI3K-β in cancers with PTEN loss (247, 248), and PI3K-δ and -γ inhibitors as cancer immunotherapies and T-cell immunomodulators.
Clinical evaluation of PI3K-δ and -γ inhibitors as immunomodulators of T-cell activity, metabolism, and the TME is still in the early stages, but multiple efforts are underway. A search of actively recruiting or pending trials in ClinicalTrials.Gov related to “PI3K” and “Cancer” was conducted. 177 studies were identified that were either recruiting, not yet recruiting, or active/not recruiting. Attention was given to trials incorporating inhibitors of PI3K-δ and -γ with T-cell targeted immunotherapies. We excluded trials for cancer specific mutations in PIK3CA, PIK3CB, AKT, or PTEN and trials for inhibitors of PI3K-α and -β (alpelisib, taselisib, serabelisib, HS-10352, GSK2636771), AKT (ipatasertib, AZD5363), mTOR (everolimus, TAK-228), or dual PI3K/mTOR (gedatolisib, samotolisib, HEC68498, paxalisib).
We identified 18 clinical trials combining copanlisib, duvelisib, eganelisib, idelalisib, parsaclisib, SF1126, and TGR-1202 with checkpoint inhibitors of PD-1 or PD-L1 in solid and hematologic malignancies (Table 3). Particularly interesting are trials incorporating expression of biomarker to clinically assess immune cell changes following PI3K-δ and -γ inhibition. Of note, 7 trials utilize PI3K-α or -α/-β targeting inhibitors. Of these trials, 4 are in solid tumor indications, and in the absence testing for mutations, amplifications or gene loss in PI3K or PTEN, the primary motive for these is to investigate the -δ and -γ effects.
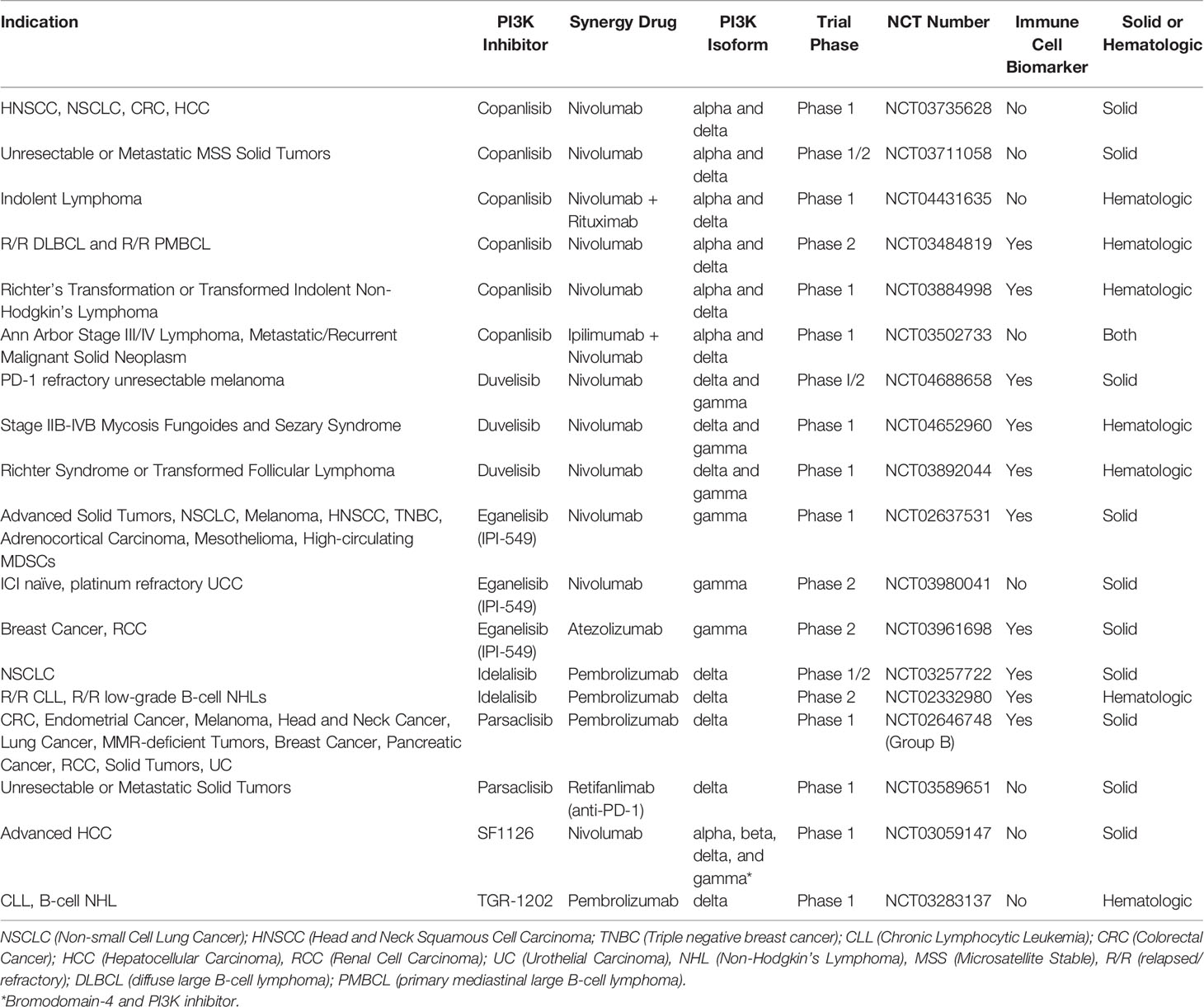
Table 3 Current clinical trials combining PI3K-δ and -γ inhibitors with T-cell targeted therapies.
NCT04688658 is a phase I/II trial in melanoma studying changes in immune cell function from tumor and peripheral blood prior to and while on duvelisib therapy. MARIO-3 (Macrophage Reprogramming in Immuno-Oncology) (NCT03961698) incorporates pre- and on-treatment tissue biopsies to correlate the effects of eganelisib on PD-L1 expression in tumor-infiltrating immune cells. In hematologic malignancies, NCT03484819 will characterize the effects of the copanlisib and nivolumab combination regimen on tumor cells, tumor microenvironment and the immune response in relapsed/refractory DLBCL and Primary Mediastinal Large B-cell Lymphoma. NCT03502733 will report changes in PD-1, PD-L1, PD-L2, and immune cell profiles and markers of immune-modulation in CLL and low-grade B cell NHL patients with copanlisib and combination ICI.
Two trials (NCT02646748 and NCT03257722) are combining the PI3K-δ inhibitors parsaclisib and idelalisib, respectively, with pembrolizumab to assess the effects on Tregs. In NCT02646748, pre- and post- therapy changes in the total number of tumor infiltrating lymphocytes and ratio of CD8+/FoxP3+ Tregs will be measured. NCT03257722 will use Treg suppression (target of 80% suppression in 80% of patients) to calculate the optimal idelalisib immunomodulatory dose. The latter trial design raises an important consideration of incorporating meaningful biomarker driven correlates into clinical trials to facilitate identification of the optimal immunomodulatory dose versus maximally tolerated dose.
Macrophage remodeling with PI3K-γ inhibitors is also of interest. NCT03795610 is a window of opportunity study in HNSCC to determine whether 3 weeks of the PI3K-γ inhibitor eganelisib prior to surgery is sufficient to drive macrophage phenotype switching in tumors. NCT02637531 is a multi-group study combining eganelisib with nivolumab, in which one group is stratified by the presence of high-circulating peripheral blood MDSCs. Similarly, Mario-275 (NCT03980041) evaluates eganelisib with nivolumab in patients with advanced urothelial carcinoma and incorporates MDSC levels as a part of its randomization process.
Taken together, our current knowledge combined with the results of these trials and future studies has the potential to drastically change how we re-purpose PI3K inhibitors, in particular PI3K-δ and -γ inhibitors, as immunomodulatory agents. While current focus heavily remains on T-cell activation, differentiation, and anti-tumor activity persistence and memory in developing immunotherapies, we anticipate future investigations focusing on immune cell metabolism and other immune cell suppressive functions in the TME will yield biologically meaningful and clinically significant changes in the field of immuno-oncology.
SC conceived, drafted, wrote, and revised the manuscript and accompanying figures and tables. CF assisted with writing and developing figures. TK developed and revised the figures and tables. CP critically reviewed and revised the manuscript regarding TIL therapy and immune checkpoint blockade. MS critically reviewed and revised the manuscript regarding T-cell metabolism and differentiation. EW was involved with drafting and critically reviewing and revising the manuscript. All authors contributed to the article and approved the submitted version.
SC: NIH T32CA160040, ECOG-ACRIN Paul Carbone, MD Fellowship Award; EW: NIH 5R01AI145231-03; MS: NIH 1R01CA208328-01A1, LLS (Leukemia/Lymphoma Society) Award # 6573-19.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber-Fiebert JD. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Multiply Relapsed or Refractory Adult Large B-Cell Lymphoma. J Clin Oncol (2019) 37:2105–19. doi: 10.1200/JCO.18.02079
2. Grigor EJM, Fergusson D, Kekre N, Montroy J, Atkins H, Seftel MD, et al. Risks and Benefits of Chimeric Antigen Receptor T-Cell (CAR-T) Therapy in Cancer: A Systematic Review and Meta-Analysis. Transfusion Med Rev (2019) 33:98–110. doi: 10.1016/j.tmrv.2019.01.005
3. Ahmad S, Abu-eid R, Shrimali R, Webb M, Verma V, Doroodchi A, et al. Differential PI3K D Signaling in CD4 Þ T-Cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res (2017) 77:1892–904. doi: 10.1158/0008-5472.CAN-16-1839
4. Deane JA, Fruman DA. Phosphoinositide-3-Kinase: Diverse Roles in Immune Cell Activation. Annu Rev Immunol (2004) 22:563–98. doi: 10.1146/annurev.immunol.22.012703.104721
5. Reif K, Okkenhaug K, Sasaki T, Penninger JM, Vanhaesebroeck B, Cyster JG. Cutting Edge : Differential Roles for Phosphoinositide. J Immunol (2004) 173(4):2236–40. doi: 10.4049/jimmunol.173.4.2236
6. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. Pi3kγ is a Molecular Switch That Controls Immune Suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
7. Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov (2016) 6:1090–105. doi: 10.1158/2159-8290.CD-16-0716
8. Fruman DA, Rommel C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat Rev Drug Discov (2014) 13:140–56. doi: 10.1038/nrd4204
9. Lee Y-R, Chen M, Pandolfi PP. The Functions and Regulation of the PTEN Tumour Suppressor: New Modes and Prospects. Nat Rev Mol Cell Biol (2018) 19:547–62. doi: 10.1038/s41580-018-0015-0
10. Song MS, Salmena L, Pandolfi PP. The Functions and Regulation of the PTEN Tumour Suppressor. Nat Rev Mol Cell Biol (2012) 13:283–96. doi: 10.1038/nrm3330
11. Miller TW, Hennessy BT, González-Angulo AM, Fox EM, Mills GB, Chen H, et al. Hyperactivation of Phosphatidylinositol-3 Kinase Promotes Escape From Hormone Dependence in Estrogen Receptor–Positive Human Breast Cancer. J Clin Invest (2010) 120:2406–13. doi: 10.1172/JCI41680
12. Abramson VG, Cooper Lloyd M, Ballinger T, Sanders ME, Du L, Lai D, et al. Characterization of Breast Cancers With PI3K Mutations in an Academic Practice Setting Using SNaPshot Profiling. Breast Cancer Res Treat (2014) 145:389–99. doi: 10.1007/s10549-014-2945-3
13. Marquard FE, Jücker M. PI3K/AKT/mTOR Signaling as a Molecular Target in Head and Neck Cancer. Biochem Pharmacol (2020) 172:113729. doi: 10.1016/j.bcp.2019.113729
14. Samuels Y. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science (2004) 304:554–4. doi: 10.1126/science.1096502
15. Karachaliou N, Pilotto S, Teixidó C, Viteri S, González-Cao M, Riso A, et al. Melanoma: Oncogenic Drivers and the Immune System. Ann Trans Med (2015) 3:265. doi: 10.3978/j.issn.2305-5839.2015.08.06
16. Di Leo A, Johnston S, Lee KS, Ciruelos E, Lønning PE, Janni W, et al. Buparlisib Plus Fulvestrant in Postmenopausal Women With Hormone-Receptor-Positive, HER2-Negative, Advanced Breast Cancer Progressing on or After mTOR Inhibition (BELLE-3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2018) 19:87–100. doi: 10.1016/S1470-2045(17)30688-5
17. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR Pathways: Cross-Talk and Compensation. Trends Biochem Sci (2011) 36:320–8. doi: 10.1016/j.tibs.2011.03.006
18. Li X, Dai D, Chen B, Tang H, Xie X, Wei W. Efficacy of PI3K/AKT/mTOR Pathway Inhibitors for the Treatment of Advanced Solid Cancers: A Literature-Based Meta-Analysis of 46 Randomised Control Trials. PLoS One (2018) 13:1–18. doi: 10.1371/journal.pone.0192464
19. Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, et al. PI3K Inhibitors as New Cancer Therapeutics: Implications for Clinical Trial Design. OncoTargets Ther (2016) 10:203. doi: 10.2147/OTT.S89967
20. Ando Y, Iwasa S, Takahashi S, Saka H, Kakizume T, Natsume K, et al. A Phase 1 Study of Alpelisib (BYL719), an Alpha- Specific PI3K Inhibitor. In: Japanese Patients With Advanced Solid Tumors. John Wiley & Sons Australia: Cancer Science (2018). p. 1–11. doi: 10.1111/cas.13923
21. Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, et al. Pictilisib for Oestrogen Receptor-Positive, Aromatase Inhibitor-Resistant, Advanced or Metastatic Breast Cancer (FERGI): A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Oncol (2016) 17:811–21. doi: 10.1016/S1470-2045(16)00106-6
22. Vuylsteke P, Huizing M, Petrakova K, Roylance R, Laing R, Chan S, et al. Pictilisib PI3Kinase Inhibitor (a Phosphatidylinositol 3-Kinase [PI3K] Inhibitor) Plus Paclitaxel for the Treatment of Hormone Receptor-Positive, HER2-Negative, Locally Recurrent, or Metastatic Breast Cancer: Interim Analysis of the Multicentre, Placebo-C. Ann Oncol (2016) 27:2059–66. doi: 10.1093/annonc/mdw320
23. Baselga J, Im S-A, Iwata H, Cortés J, De Laurentiis M, Jiang Z, et al. Buparlisib Plus Fulvestrant Versus Placebo Plus Fulvestrant in Postmenopausal, Hormone Receptor-Positive, HER2-Negative, Advanced Breast Cancer (BELLE-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2017) 18:904–16. doi: 10.1016/S1470-2045(17)30376-5
24. Hirsch E, Ciraolo E, Franco I, Ghigo A, Martini M. PI3K in Cancer-Stroma Interactions: Bad in Seed and Ugly in Soil. Oncogene (2014) 33:3083–90. doi: 10.1038/onc.2013.265
25. Macias-Perez IM, Flinn IW. GS-1101: A Delta-Specific PI3K Inhibitor in Chronic Lymphocytic Leukemia. Curr Hematol Malignancy Rep (2013) 8:22–7. doi: 10.1007/s11899-012-0142-1
26. So L, Fruman DA. PI3K Signalling in B- and T-Lymphocytes: New Developments and Therapeutic Advances. Biochem J (2012) 442:465–81. doi: 10.1042/BJ20112092
27. Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-In-Human Phase I Study of Pictilisib (GDC-0941), a Potent Pan–Class I Phosphatidylinositol-3-Kinase (PI3K) Inhibitor, in Patients With Advanced Solid Tumors. Clin Cancer Res (2015) 21:77–86. doi: 10.1158/1078-0432.CCR-14-0947
28. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med (2014) 370:997–1007. doi: 10.1056/NEJMoa1315226
29. Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. Pi3kδ Inhibition by Idelalisib in Patients With Relapsed Indolent Lymphoma. N Engl J Med (2014) 370:1008–18. doi: 10.1056/NEJMoa1314583
30. Dreyling M, Morschhauser F, Bouabdallah K, Bron D, Cunningham D, Assouline SE, et al. Phase II Study of Copanlisib, a PI3K Inhibitor, in Relapsed or Refractory, Indolent or Aggressive Lymphoma. Ann Oncol (2017) 28:2169–78. doi: 10.1093/annonc/mdx289
31. Flinn IW, Hillmen P, Montillo M, Nagy Z, Illés Á, Etienne G, et al. The Phase 3 DUO Trial: Duvelisib vs Ofatumumab in Relapsed and Refractory CLL/SLL. Blood (2018) 132:2446–55. doi: 10.1182/blood-2018-05-850461
33. Okkenhaug K. Two Birds With One Stone: Dual P110δ and P110γ Inhibition. Chem Biol (2013) 20:1309–10. doi: 10.1016/j.chembiol.2013.11.002
34. Okkenhaug K, Burger JA. PI3K Signaling in Normal B Cells and Chronic Lymphocytic Leukemia (CLL). Curr topics Microbiol Immunol (2016) 393:123–42. doi: 10.1007/82_2015_484
35. Abu-Eid R, Samara RN, Ozbun L, Abdalla MY, Berzofsky JA, Friedman KM, et al. Selective Inhibition of Regulatory T Cells by Targeting the PI3K-Akt Pathway. Cancer Immunol Res (2014) 2:1080–9. doi: 10.1158/2326-6066.CIR-14-0095
36. Carnevalli LS, Sinclair C, Taylor MA, Gutierrez PM, Langdon S, Coenen-Stass AML, et al. PI3Kalpha/delta Inhibition Promotes Anti-Tumor Immunity Through Direct Enhancement of Effector CD8(+) T-Cell Activity. J Immunother Cancer (2018) 6:158. doi: 10.1186/s40425-018-0457-0
37. Appleman LJ, van Puijenbroek AAFL, Shu KM, Nadler LM, Boussiotis VA. CD28 Costimulation Mediates Down-Regulation of P27 Kip1 and Cell Cycle Progression by Activation of the PI3K/PKB Signaling Pathway in Primary Human T Cells. J Immunol (2002) 168:2729–36. doi: 10.4049/jimmunol.168.6.2729
38. Garçon F, Patton DT, Emery JL, Hirsch E, Rottapel R, Sasaki T, et al. CD28 Provides T-Cell Costimulation and Enhances PI3K Activity at the Immune Synapse Independently of Its Capacity to Interact With the P85/P110 Heterodimer. Blood (2008) 111:1464–71. doi: 10.1182/blood-2007-08-108050
39. Lewis C. Cantley. The Phosphoinositide 3-Kinase Pathway. Science (2002) 296:1655–8. doi: 10.1126/science.296.5573.1655
40. Vangapandu HV, Jain N, Gandhi V. Duvelisib: A Phosphoinositide-3 Kinase δ/γ Inhibitor for Chronic Lymphocytic Leukemia. Expert Opin Investigational Drugs (2017) 26:625–32. doi: 10.1080/13543784.2017.1312338
41. Petersen CT, Hassan M, Morris AB, Lee K, Jagirdar N, Staton AD, et al. Improving Ex Vivo T Cell Expansion From DLBCL Patients for T Cell Therapies via Antagonism of PI3K δ and VIP. Blood (2017) 130:3195. doi: 10.1182/bloodadvances.2017011254
42. Funk CRF, Wang S, Waller A, Edgar C, Sharma A, Chen K, et al. Dual Inhibition of PI3KDelta/Gamma During Manufacturing Reprograms Metabolism of CAR T Cells to Enhance Expansion and Cytotoxicity Against CLL. Blood (2020) 136.
43. Finlay DK, Rosenzweig E, Sinclair LV, Carmen FC, Hukelmann JL, Rolf J, et al. PDK1 Regulation of mTOR and Hypoxia-Inducible Factor 1 Integrate Metabolism and Migration of CD8+ T Cells. J Exp Med (2012) 209:2441–53. doi: 10.1084/jem.20112607
44. Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. Ampkα1: A Glucose Sensor That Controls CD8 T-Cell Memory. Eur J Immunol (2013) 43:889–96. doi: 10.1002/eji.201243008
45. Hukelmann JL, Anderson KE, Sinclair LV, Grzes KM, Murillo AB, Hawkins PT, et al. The Cytotoxic T Cell Proteome and Its Shaping by Mammalian Target of Rapamycin. Nat Immunol (2016) 17:104–12. doi: 10.1038/ni.3314.The
46. Salmond RJ. mTOR Regulation of Glycolytic Metabolism in T Cells. Front Cell Dev Biol (2018) 6:122. doi: 10.3389/fcell.2018.00122
47. Sukumar M, Kishton RJ, Restifo NP. Metabolic Reprograming of Anti-Tumor Immunity. Curr Opin Immunol (2017) 46:14–22. doi: 10.1016/j.coi.2017.03.011
48. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt Inhibition Enhances Expansion of Potent Tumor-Specific Lymphocytes With Memory Cell Characteristics. Cancer Res (2015) 75:296–305. doi: 10.1158/0008-5472.CAN-14-2277
49. Scholz G, Jandus C, Zhang L, Grandclément C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR Signalling Triggers the Formation of Stem Cell-Like Memory T Cells. EBioMedicine (2016) 4:50–61. doi: 10.1016/j.ebiom.2016.01.019
50. Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An Interleukin-21- Interleukin-10-STAT3 Pathway Is Critical for Functional Maturation of Memory CD8+ T Cells. Immunity (2011) 35:792–805. doi: 10.1016/j.immuni.2011.09.017
51. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-Activating Germline Mutations in the Gene Encoding the PI(3)K Catalytic Subunit P110δ Result in T Cell Senescence and Human Immunodeficiency. Nat Immunol (2014) 15:88–97. doi: 10.1038/ni.2771
52. Cannons JL, Preite S, Kapnick SM, Uzel G, Schwartzberg PL. Genetic Defects in Phosphoinositide 3-Kinase δ Influence CD8+T Cell Survival, Differentiation, and Function. Front Immunol (2018) 9:1758. doi: 10.3389/fimmu.2018.01758
53. Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, et al. Construction and Preclinical Evaluation of an Anti-CD19 Chimeric Antigen Receptor. J Immunother (2009) 32:689–702. doi: 10.1097/CJI.0b013e3181ac6138
54. Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (2016) 8:36. doi: 10.3390/cancers8030036
55. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The Who’s Who of T-Cell Differentiation: Human Memory T-Cell Subsets. Eur J Immunol (2013) 43:2797–809. doi: 10.1002/eji.201343751
56. Rosenblum MD, Way SS, Abbas AK. Regulatory T Cell Memory. Nat Rev Immunol (2016) 16:90–101. doi: 10.1038/nri.2015.1
57. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central Memory Self/Tumor-Reactive CD8+ T Cells Confer Superior Antitumor Immunity Compared With Effector Memory T Cells. Proc Natl Acad Sci (2005) 102:9571–6. doi: 10.1073/pnas.0503726102
58. Jensen MC, Riddell SR. Design and Implementation of Adoptive Therapy With Chimeric Antigen Receptor-Modified T Cells. Immunol Rev (2014) 257:127–44. doi: 10.1111/imr.12139
59. Zhao Y, Shao Q, Peng G. Exhaustion and Senescence: Two Crucial Dysfunctional States of T Cells in the Tumor Microenvironment. Cell Mol Immunol (2020) 17:27–35. doi: 10.1038/s41423-019-0344-8
60. Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular Signature of CD8+ T Cell Exhaustion During Chronic Viral Infection. Immunity (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
61. Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a Cancer Immunotherapy Target. Immunol Rev (2017) 276:80–96. doi: 10.1111/imr.12519
62. Patsoukis N, Weaver JD, Strauss L, Herbel C, Seth P, Boussiotis VA. Immunometabolic Regulations Mediated by Coinhibitory Receptors and Their Impact on T Cell Immune Responses. Front Immunol (2017) 8:330. doi: 10.3389/fimmu.2017.00330
63. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol Cell Biol (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
64. Wherry EJ, Kurachi M. Molecular and Cellular Insights Into T Cell Exhaustion. Nat Rev Immunol (2015) 15:486–99. doi: 10.1038/nri3862
65. Kasakovski D, Xu L, Li Y. T Cell Senescence and CAR-T Cell Exhaustion in Hematological Malignancies. J Hematol Oncol (2018) 11:1–9. doi: 10.1186/s13045-018-0629-x
66. Bellon M, Nicot C. Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses (2017) 9:289. doi: 10.3390/v9100289
67. van Bruggen JAC, Martens AWJ, Fraietta JA, Hofland T, Tonino SH, Eldering E, et al. Chronic Lymphocytic Leukemia Cells Impair Mitochondrial Fitness in CD8+ T Cells and Impede CAR T-Cell Efficacy. Blood (2019) 134:44–58. doi: 10.1182/blood.2018885863
68. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
69. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N, et al. Improving T-Cell Expansion and Function for Adoptive T-Cell Therapy Using Ex Vivo Treatment With PI3Kδ Inhibitors and VIP Antagonists. Blood Adv (2018) 2:210–23. doi: 10.1182/bloodadvances.2017011254
70. Windt GJW, Pearce EL. Metabolic Switching and Fuel Choice During T-Cell Differentiation and Memory Development. Immunol Rev (2012) 249:27–42. doi: 10.1111/j.1600-065X.2012.01150.x
71. Xu K, Yin N, Peng M, Stamatiades EG, Shyu A, Li P, et al. Glycolysis Fuels Phosphoinositide 3-Kinase Signaling to Bolster T Cell Immunity. Science (2021) 371:405–10. doi: 10.1126/science.abb2683
72. Herbel C, Patsoukis N, Bardhan K, Seth P, Weaver JD, Boussiotis VA. Clinical Significance of T Cell Metabolic Reprogramming in Cancer. Clin Trans Med (2016) 5. doi: 10.1186/s40169-016-0110-9
73. Verma V, Jafarzadeh N, Boi S, Kundu S, Jiang Z, Fan Y, et al. MEK Inhibition Reprograms CD8+ T Lymphocytes Into Memory Stem Cells With Potent Antitumor Effects. Nat Immunol (2021) 22:53–66. doi: 10.1038/s41590-020-00818-9
74. van der Windt GJW, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8 + T Cell Memory Development. Immunity (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
75. Kishton RJ, Sukumar M, Restifo NP. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab (2017) 26:94–109. doi: 10.1016/j.cmet.2017.06.016
76. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
77. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors Per NCCN Guidelines With the Level of Evidence. Cancers (2020) 12:1–19. doi: 10.3390/cancers12030738
78. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060
79. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. New Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
80. Robert C, Thomas L, Bondarenko I, O’Day S, JW MD, Garbe C, et al. Ipilimumab Plus Dacarbazine for Previously Untreated Metastatic Melanoma. New Engl J Med (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
81. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive Resistance to Therapeutic PD-1 Blockade is Associated With Upregulation of Alternative Immune Checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
82. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br J Cancer (2018) 118:9–16. doi: 10.1038/bjc.2017.434
83. Woo S-R, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
84. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J Exp Med (2011) 208:1331–1. doi: 10.1084/jem.201006432011512c
85. Sanlorenzo M, Vujic I, Moy A, Quaglino P, Fierro MT, Gammaitoni L, et al. Synergy of Molecular Targeted Approaches and Immunotherapy in Melanoma: Preclinical Basis and Clinical Perspectives. Expert Opin Biol Ther (2015) 15:1491–500. doi: 10.1517/14712598.2015.1069272
86. Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab Alone in Advanced Melanoma (CheckMate 067): 4-Year Outcomes of a Multicentre, Randomised, Phase 3 Trial. Lancet Oncol (2018) 19:1480–92. doi: 10.1016/S1470-2045(18)30700-9
87. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol: Cancer Clin Trials (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
88. Peggs KS, Quezada SA, Allison JP. Cell Intrinsic Mechanisms of T-Cell Inhibition and Application to Cancer Therapy. Immunol Rev (2008) 224:141–65. doi: 10.1111/j.1600-065X.2008.00649.x
89. Okazaki T, Honjo T. PD-1 and PD-1 Ligands: From Discovery to Clinical Application. Int Immunol (2007) 19:813–24. doi: 10.1093/intimm/dxm057
90. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-AAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170:1120–33.e17. doi: 10.1016/j.cell.2017.07.024
91. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
92. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity (2016) 44:1255–69. doi: 10.1016/j.immuni.2016.06.001
93. O’Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 Checkpoint Inhibition. Cancer Treat Rev (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
94. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
95. Kim JM, Chen DS. Immune Escape to PD-L1/PD-1 Blockade: Seven Steps to Success (or Failure). Ann Oncol (2016) 27:1492–504. doi: 10.1093/annonc/mdw217
96. Hegde PS, Karanikas V, Evers S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res (2016) 22:1865–74. doi: 10.1158/1078-0432.CCR-15-1507
97. Gajewski TF, Woo S-R, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer Immunotherapy Strategies Based on Overcoming Barriers Within the Tumor Microenvironment. Curr Opin Immunol (2013) 25:268–76. doi: 10.1016/j.coi.2013.02.009
98. Joyce JA, Fearon DT. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science (2015) 348:74–80. doi: 10.1126/science.aaa6204
99. Maleki Vareki S. High and Low Mutational Burden Tumors Versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J ImmunoTher Cancer (2018) 6:4–8. doi: 10.1186/s40425-018-0479-7
100. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol (2019) 10:168. doi: 10.3389/fimmu.2019.00168
101. Zemek RM, Chin WL, Nowak AK, Millward MJ, Lake RA, Lesterhuis WJ. Sensitizing the Tumor Microenvironment to Immune Checkpoint Therapy. Front Immunol (2020) 11:223. doi: 10.3389/fimmu.2020.00223
102. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated With Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
103. Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-Specific MHC-II Expression Represents a Tumour-Autonomous Phenotype and Predicts Response to Anti-PD-1/PD-L1 Therapy. Nat Commun (2016) 7:10582. doi: 10.1038/ncomms10582
104. Yoo SH, Keam B, Ock C-Y, Kim S, Han B, Kim J-W, et al. Prognostic Value of the Association Between MHC Class I Downregulation and PD-L1 Upregulation in Head and Neck Squamous Cell Carcinoma Patients. Sci Rep (2019) 9:7680. doi: 10.1038/s41598-019-44206-2
105. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of Human Solid Tumors From T-Cell Recognition: Molecular Mechanisms and Functional Significance. Adv Immunol (2000) 74:181–273. doi: 10.1016/s0065-2776(08)60911-6
106. Chandrasekaran S, Sasaki M, Scharer CD, Kissick HT, Patterson DG, Magliocca KR, et al. Phosphoinositide 3-Kinase Signaling can Modulate MHC Class I and II Expression. Mol Cancer Res (2019) 17:2395–409. doi: 10.1158/1541-7786.MCR-19-0545. molcanres.0545.2019.
107. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov (2016) 6:202–16. doi: 10.1158/2159-8290.CD-15-0283
108. Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 Efficacy can be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells With a Selective Inhibitor of PI3Kd/G. Cancer Res (2017) 77:2607–19. doi: 10.1158/0008-5472.CAN-16-2534
109. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming Resistance to Checkpoint Blockade Therapy by Targeting PI3Kγ in Myeloid Cells. Nature (2016) 539:443–7. doi: 10.1038/nature20554
110. Uehara M, McGrath MM, Ohori S, Solhjou Z, Banouni N, Routray S, et al. Regulation of T Cell Alloimmunity by PI3Kγ and PI3Kδ. Nat Commun (2017) 8:951. doi: 10.1038/s41467-017-00982-x
111. Winkler DG, Faia KL, Dinitto JP, Ali JA, White KF, Brophy EE, et al. PI3K-δ and PI3K-γ Inhibition by IPI-145 Abrogates Immune Responses and Suppresses Activity in Autoimmune and Inflammatory Disease Models. Chem Biol (2013) 20:1364–74. doi: 10.1016/j.chembiol.2013.09.017
112. Joshi S, Singh AR, Zulcic M. Durden DL. A Macrophage-Dominant PI3K Isoform Controls Hypoxia-Induced Hif1α and HIF2α Stability and Tumor Growth, Angiogenesis, and Metastasis. Mol Cancer Res (2014) 12:1520–31. doi: 10.1158/1541-7786.MCR-13-0682
113. Dwyer CJ, Arhontoulis DC, Rangel Rivera GO, Knochelmann HM, Smith AS, Wyatt MM, et al. Ex Vivo Blockade of PI3K Gamma or Delta Signaling Enhances the Antitumor Potency of Adoptively Transferred CD8+ T Cells. Eur J Immunol (2020) 50:1386–99. doi: 10.1002/eji.201948455
114. Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, et al. Receptor Tyrosine Kinases and TLR/IL1Rs Unexpectedly Activate Myeloid Cell Pi3kγ, A Single Convergent Point Promoting Tumor Inflammation and Progression. Cancer Cell (2011) 19:715–27. doi: 10.1016/j.ccr.2011.04.016
115. Sai J, Owens P, Novitskiy SV, Hawkins OE, Vilgelm AE, Yang J, et al. PI3K Inhibition Reduces Mammary Tumor Growth and Facilitates Antitumor Immunity and Anti-PD1 Responses. Clin Cancer Res (2017) 23:3371–84. doi: 10.1158/1078-0432.CCR-16-2142
116. Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3K Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov (2016) 6:870–85. doi: 10.1158/2159-8290.CD-15-1346
117. Ali K, Soond DR, Piñeiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K P110δ Breaks Regulatory T-Cell-Mediated Immune Tolerance to Cancer. Nature (2014) 510:407–11. doi: 10.1038/nature13444
118. Tanaka A, Sakaguchi S. Targeting Treg Cells in Cancer Immunotherapy. Eur J Immunol (2019) 49:1140–6. doi: 10.1002/eji.201847659
119. Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, et al. Cutting Edge: The Phosphoinositide 3-Kinase P110 Is Critical for the Function of CD4+CD25+Foxp3+ Regulatory T Cells. J Immunol (2006) 177:6598–602. doi: 10.4049/jimmunol.177.10.6598
120. Coma S, Funk CR, Wang S, Waller EK, Weaver DT, Pachter JA. “Abstract CT045: Synergistic Anti-Tumor Efficacy of the Dual PI3K-δ/PI3K-γ Inhibitor Duvelisib With PD-1 Blockade in Solid Tumor and Lymphoma Models”. In: Proceedings of the Annual Meeting of the American Association for Cancer Research. Philadelphia (PA): AACR (2020) 80(16 Suppl):Abstract nr CT045. doi: 10.1158/1538-7445.AM2020-CT045
121. Chellappa S, Kushekhar K, Munthe LA, Tjønnfjord GE, Aandahl EM, Okkenhaug K, et al. The PI3K P110δ Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J Immunol (2019) 202:ji1701703. doi: 10.4049/jimmunol.1701703
122. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci Rep (2015) 5:1–9. doi: 10.1038/srep15179
123. Umansky V, Sevko A, Gebhardt C, Utikal J. Myeloid-Derived Suppressor Cells in Malignant Melanoma. JDDG: J der Deutschen Dermatologischen Gesellschaft (2014) 12:1021–7. doi: 10.1111/ddg.12411
124. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
125. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
126. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The Immunosuppressive Tumour Network: Myeloid-Derived Suppressor Cells, Regulatory T Cells and Natural Killer T Cells. Immunology (2013) 138:105–15. doi: 10.1111/imm.12036
127. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. Korangy F. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology (2008) 135:234–43. doi: 10.1053/j.gastro.2008.03.020
128. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2+ Myeloid-Derived Suppressor Cells Promote Immune Escape by Limiting Activated CD8 T-Cell Infiltration Into the Tumor Microenvironment. Cancer Res (2012) 72:876–86. doi: 10.1158/0008-5472.CAN-11-1792
129. Serbina NV, Pamer EG. Monocyte Emigration From Bone Marrow During Bacterial Infection Requires Signals Mediated by Chemokine Receptor CCR2. Nat Immunol (2006) 7:311–7. doi: 10.1038/ni1309
130. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-Induced Myeloid Deviation: When Myeloid-Derived Suppressor Cells Meet Tumor-Associated Macrophages. J Clin Invest (2015) 125:3365–76. doi: 10.1172/JCI80006
131. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-Talk Between Myeloid-Derived Suppressor Cells (MDSC), Macrophages, and Dendritic Cells Enhances Tumor-Induced Immune Suppression. Semin Cancer Biol (2012) 22:275–81. doi: 10.1016/j.semcancer.2012.01.011
132. Sierra-Filardi E, Nieto C, Domínguez-Soto Á, Barroso R, Sánchez-Mateos P, Puig-Kroger A, et al. CCL2 Shapes Macrophage Polarization by GM-CSF and M-CSF: Identification of CCL2/CCR2-Dependent Gene Expression Profile. J Immunol (2014) 192:3858–67. doi: 10.4049/jimmunol.1302821
133. Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-Associated Macrophages and the Related Myeloid-Derived Suppressor Cells as a Paradigm of the Diversity of Macrophage Activation. Hum Immunol (2009) 70:325–30. doi: 10.1016/j.humimm.2009.02.008
134. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
135. Lee KY. M1 and M2 Polarization of Macrophages: A Mini-Review. Med Biol Sci Eng (2019) 2:1–5. doi: 10.30579/mbse.2019.2.1.1
136. Elliott LA, Doherty GA, Sheahan K, Ryan EJ. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front Immunol (2017) 8:86. doi: 10.3389/fimmu.2017.00086
137. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
138. Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, et al. Activity of the PI3K-δ,G Inhibitor Duvelisib in a Phase 1 Trial and Preclinical Models of T-Cell Lymphoma. Blood (2018) 131:888–98. doi: 10.1182/blood-2017-08-802470
139. Shi X, Li X, Wang H, Yu Z, Zhu Y, Gao Y. Specific Inhibition of PI3Kδ/γ Enhances the Efficacy of Anti-PD1 Against Osteosarcoma Cancer. J Bone Oncol (2018) 16:100206. doi: 10.1016/j.jbo.2018.11.001
140. Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, et al. Clinical Significance of Circulating CD33+ CD11bHLA-DR Myeloid Cells in Patients With Stage IV Melanoma Treated With Ipilimumab. Clin Cancer Res (2016) 22:5661–72. doi: 10.1158/1078-0432.CCR-15-3104
141. Crump M, Neelapu SS, Farooq U, van den Neste E, Kuruvilla J, Westin J, et al. Outcomes in Refractory Diffuse Large B-Cell Lymphoma: Results From the International SCHOLAR-1 Study. Blood (2017) 130:1800–8. doi: 10.1182/blood-2017-03-769620
142. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
143. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
144. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
145. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene Maraleucel for Patients With Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
146. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
147. Lee L, Bounds D, Paterson J, Herledan G, Sully K, Seestaller-Wehr LM, et al. Evaluation of B Cell Maturation Antigen as a Target for Antibody Drug Conjugate Mediated Cytotoxicity in Multiple Myeloma. Br J Haematology (2016) 174:911–22. doi: 10.1111/bjh.14145
148. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
149. Madduri D, Usmani SZ, Jagannath S, Singh I, Zudaire E, Yeh T-M, et al. Results From CARTITUDE-1: A Phase 1b/2 Study of JNJ-4528, a CAR-T Cell Therapy Directed Against B-Cell Maturation Antigen (BCMA), in Patients With Relapsed and/or Refractory Multiple Myeloma (R/R Mm). Blood (2019) 134:577–7. doi: 10.1182/blood-2019-121731
150. Berdeja JG, Madduri D, Usmani SZ, Singh I, Zudaire E, Yeh T-M, et al. Update of CARTITUDE-1: A Phase Ib/II Study of JNJ-4528, a B-Cell Maturation Antigen (BCMA)-Directed CAR-T-Cell Therapy, in Relapsed/Refractory Multiple Myeloma. J Clin Oncol (2020) 38:8505–5. doi: 10.1200/JCO.2020.38.15_suppl.8505
151. Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J Immunol (2018) 200:1088–100. doi: 10.4049/jimmunol.1700236
152. Zhang J, Wang L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol Cancer Res Treat (2019) 18:153303381983106. doi: 10.1177/1533033819831068
153. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients With Metastatic Melanoma. N Engl J Med (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
154. Sarnaik A, Thomas SS, Davar D, Kirkwood JM, Kluger HM, Lutzky J, et al. Safety and Efficacy of Cryopreserved Autologous Tumor Infiltrating Lymphocyte Therapy (LN-144, Lifileucel) in Advanced Metastatic Melanoma Patients Previously Treated With at Least One Prior Systemic Therapy. J Clin Oncol (2019) 37:136–6. doi: 10.1200/JCO.2019.37.8_suppl.136
155. Lee S, Margolin K. Tumor-Infiltrating Lymphocytes in Melanoma. Curr Oncol Rep (2012) 14:468–74. doi: 10.1007/s11912-012-0257-5
156. Geukes Foppen MH, Donia M, Svane IM, Haanen JBAG. Tumor-Infiltrating Lymphocytes for the Treatment of Metastatic Cancer. Mol Oncol (2015) 9:1918–35. doi: 10.1016/j.molonc.2015.10.018
157. Rohaan MW, Van Den Berg JH, Kvistborg P, Haanen JBAG. Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Melanoma: A Viable Treatment Option 11 Medical and Health Sciences 1107 Immunology 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. J ImmunoTher Cancer (2018) 6:1–16. doi: 10.1186/s40425-018-0391-1
158. Andersen R, Donia M, Westergaard MCW, Pedersen M, Hansen M, Svane IM. Tumor Infiltrating Lymphocyte Therapy for Ovarian Cancer and Renal Cell Carcinoma. Hum Vaccines Immunotherapeutics (2015) 11:2790–5. doi: 10.1080/21645515.2015.1075106
159. Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete Regression of Metastatic Cervical Cancer After Treatment With Human Papillomavirus-Targeted Tumor-Infiltrating T Cells. J Clin Oncol (2015) 33:1543–50. doi: 10.1200/JCO.2014.58.9093
160. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient With Epithelial Cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
161. Dafni U, Michielin O, Lluesma SM, Tsourti Z, Polydoropoulou V, Karlis D, et al. Efficacy of Adoptive Therapy With Tumor-Infiltrating Lymphocytes and Recombinant Interleukin-2 in Advanced Cutaneous Melanoma: A Systematic Review and Meta-Analysis. Ann Oncol (2019) 30:1902–13. doi: 10.1093/annonc/mdz398
162. Sadeghi A, Ullenhag G, Wagenius G, Tötterman TH, Eriksson F. Rapid Expansion of T Cells: Effects of Culture and Cryopreservation and Importance of Short-Term Cell Recovery. Acta Oncol (2013) 52:978–86. doi: 10.3109/0284186X.2012.737020
163. Tang L, Zhang Y, Hu Y. Mei H. T Cell Exhaustion and CAR-T Immunotherapy in Hematological Malignancies. BioMed Res Int (2021) 2021:1–8. doi: 10.1155/2021/6616391
164. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
165. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib Enhances Chimeric Antigen Receptor T-Cell Engraftment and Efficacy in Leukemia. Blood (2016) 127:1117–27. doi: 10.1182/blood-2015-11-679134
166. Mardiana S, Solomon BJ, Darcy PK, Beavis PA. Supercharging Adoptive T Cell Therapy to Overcome Solid Tumor–Induced Immunosuppression. Sci Trans Med (2019) 11:eaaw2293. doi: 10.1126/scitranslmed.aaw2293
167. Shah NN, Fry TJ. Mechanisms of Resistance to CAR T Cell Therapy. Nat Rev Clin Oncol (2019) 16:372–85. doi: 10.1038/s41571-019-0184-6
168. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell (2015) 28:415–28. doi: 10.1016/j.ccell.2015.09.004
169. Zhao X, Yang J, Zhang X, Lu X-A, Xiong M, Zhang J, et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol Ther - Oncolytics (2020) 18:272–81. doi: 10.1016/j.omto.2020.06.016
170. Ying Z, He T, Wang X, Zheng W, Lin N, Tu M, et al. Parallel Comparison of 4-1BB or CD28 Co-Stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol Ther - Oncolytics (2019) 15:60–8. doi: 10.1016/j.omto.2019.08.002
171. Kawalekar OU, O’ Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity (2016) 44:712. doi: 10.1016/j.immuni.2016.02.023
172. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat Med (2015) 21:581–90. doi: 10.1038/nm.3838
173. van der Stegen SJC, Hamieh M, Sadelain M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat Rev Drug Discov (2015) 14:499–509. doi: 10.1038/nrd4597
174. Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In Vivo Fate and Activity of Second- Versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol Ther (2018) 26:2727–37. doi: 10.1016/j.ymthe.2018.09.009
175. Funk C, Petersen C, Jagirdar N, Ravindranathan S, Jaye D, Flowers C, et al. Oligoclonal T Cells Transiently Expand and Express Tim-3 and PD-1 Following Anti-CD19 CAR T Cell Therapy: A Case Report. Int J Mol Sci (2018) 19:4118. doi: 10.3390/ijms19124118
176. Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, et al. Idelalisib Given Front-Line for Treatment of Chronic Lymphocytic Leukemia Causes Frequent Immune-Mediated Hepatotoxicity. Blood (2016) 128:195–203. doi: 10.1182/blood-2016-03-707133
177. Balagoni H, Chaudhari D, Reddy C, Young M. Idelalisib: A Rare Cause of Enterocolitis. Ann Gastroenterology (2016) 29:233–5. doi: 10.20524/aog.2016.0022
178. Louie CY, DiMaio MA, Matsukuma KE, Coutre SE, Berry GJ, Longacre TA. Idelalisib-Associated Enterocolitis: Clinicopathologic Features and Distinction From Other Enterocolitides. Am J Surg Pathol (2015) 39:1653–60. doi: 10.1097/PAS.0000000000000525
179. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of Neoantigen-Specific T Cells From Tumor and Peripheral Lymphocytes. J Clin Invest (2015) 125:3981–91. doi: 10.1172/JCI82416
180. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med (2017) 9:1–14. doi: 10.1186/s13073-017-0424-2
181. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, et al. Immune Recognition of Somatic Mutations Leading to Complete Durable Regression in Metastatic Breast Cancer. Nat Med (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
182. Lemal R, Tournilhac O. State-Of-the-Art for CAR T-Cell Therapy for Chronic Lymphocytic Leukemia in 2019. J ImmunoTher Cancer (2019) 7:1–6. doi: 10.1186/s40425-019-0686-x
183. Xu X, Sun Q, Liang X, Chen Z, Zhang X, Zhou X, et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front Immunol (2019) 10:2664. doi: 10.3389/fimmu.2019.02664
184. Mackall C, Fleisher T, Brown M, Magrath I, Shad A, Horowitz M, et al. Lymphocyte Depletion During Treatment With Intensive Chemotherapy for Cancer. Blood (1994) 84:2221–8. doi: 10.1182/blood.V84.7.2221.bloodjournal8472221
185. Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, Ouyang C, et al. Memory T Cell–Driven Differentiation of Naive Cells Impairs Adoptive Immunotherapy. J Clin Invest (2015) 126:318–34. doi: 10.1172/JCI81217
186. Turtle CJ, Hanafi L-A, Berger C, Sommermeyer D, Pender B, Robinson EM, et al. Addition of Fludarabine to Cyclophosphamide Lymphodepletion Improves In Vivo Expansion of CD19 Chimeric Antigen Receptor-Modified T Cells and Clinical Outcome in Adults With B Cell Acute Lymphoblastic Leukemia. Blood (2015) 126:3773–3. doi: 10.1182/blood.V126.23.3773.3773
187. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of Homeostatic Cytokine Sinks by Lymphodepletion Enhances the Efficacy of Adoptively Transferred Tumor-Specific CD8+ T Cells. J Exp Med (2005) 202:907–12. doi: 10.1084/jem.20050732
188. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor Indoleamine 2,3-Dioxygenase (IDO) Inhibits CD19-CAR T Cells and is Downregulated by Lymphodepleting Drugs. Blood (2015) 125:3905–16. doi: 10.1182/blood-2015-01-621474
189. Neelapu SS. CAR-T Efficacy: Is Conditioning the Key? Blood (2019) 133:1799–800. doi: 10.1182/blood-2019-03-900928
190. Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial Translocation Augments the Function of Adoptively Transferred Self/Tumor-Specific CD8+ T Cells via TLR4 Signaling. J Clin Invest (2007) 117:2197–204. doi: 10.1172/JCI32205
191. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome After CD19 Chimeric Antigen Receptor–Modified T-Cell Therapy. Blood (2017) 130:2295–306. doi: 10.1182/blood-2017-06-793141
192. Kotch C, Barrett D, Teachey DT. Tocilizumab for the Treatment of Chimeric Antigen Receptor T Cell-Induced Cytokine Release Syndrome. Expert Rev Clin Immunol (2019) 15:813–22. doi: 10.1080/1744666X.2019.1629904
193. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable Complete Responses in Heavily Pretreated Patients With Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
194. Rosenberg SA, Yang JC, White DE, Steinberg SM. Durability of Complete Responses in Patients With Metastatic Cancer Treated With High-Dose Interleukin-2. Ann Surg (1998) 228:307–19. doi: 10.1097/00000658-199809000-00004
195. Zhang GQ, Zhao H, Wu JY, Li JY, Yan X, Wang G, et al. Prolonged Overall Survival in Gastric Cancer Patients After Adoptive Immunotherapy. World J Gastroenterology (2015) 21:2777–85. doi: 10.3748/wjg.v21.i9.2777
196. Leidner RS, Sukari A, Chung CH, Ohr J, Haigentz M, Cohen EEW, et al. A Phase 2, Multicenter Study to Evaluate the Efficacy and Safety of Autologous Tumor Infiltrating Lymphocytes (LN-145) for the Treatment of Patients With Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck (HNSCC). J Clin Oncol (2018) 36(15_suppl):TPS6096–TPS6096. doi: 10.1200/JCO.2018.36.15_suppl.TPS6096
197. Jazaeri AA, Zsiros E, Amaria RN, Artz AS, Edwards RP, Wenham RM, et al. Safety and Efficacy of Adoptive Cell Transfer Using Autologous Tumor Infiltrating Lymphocytes (LN-145) for Treatment of Recurrent, Metastatic, or Persistent Cervical Carcinoma. J Clin Oncol (2019) 37:2538–8. doi: 10.1200/JCO.2019.37.15_suppl.2538
198. Gee AP. GMP CAR-T Cell Production. Best Pract Research: Clin Haematology (2018) 31:126–34. doi: 10.1016/j.beha.2018.01.002
199. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
200. Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s Lymphoma With a Defined Ratio of CD8 + and CD4 + CD19-Specific Chimeric Antigen Receptor–Modified T Cells. Sci Trans Med (2016) 8:355ra116–355ra116. doi: 10.1126/scitranslmed.aaf8621
201. Guedan S, Posey AD, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T Cell Persistence Through ICOS and 4-1BB Costimulation. JCI Insight (2018) 3. doi: 10.1172/jci.insight.96976
202. Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, et al. Single Residue in CD28-Costimulated CAR-T Cells Limits Long-Term Persistence and Antitumor Durability. J Clin Invest (2020) 130:3087–97. doi: 10.1172/JCI133215
203. Ramakrishna S, Highfill SL, Walsh Z, Nguyen SM, Lei H, Shern JF, et al. Modulation of Target Antigen Density Improves CAR T-Cell Functionality and Persistence. Clin Cancer Res (2019) 25:5329–41. doi: 10.1158/1078-0432.CCR-18-3784
204. Borch TH, Andersen R, Ellebaek E, Met Ö, Donia M, Marie Svane I. Future Role for Adoptive T-Cell Therapy in Checkpoint Inhibitor-Resistant Metastatic Melanoma. J Immunother Cancer (2020) 8:1–7. doi: 10.1136/jitc-2020-000668
205. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical Responses in a Phase II Study Using Adoptive Transfer of Short-Term Cultured Tumor Infiltration Lymphocytes in Metastatic Melanoma Patients. Clin Cancer Res (2010) 16:2646–55. doi: 10.1158/1078-0432.CCR-10-0041
206. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Patients With Metastatic Melanoma: Intent-To-Treat Analysis and Efficacy After Failure to Prior Immunotherapies. Clin Cancer Res (2013) 19:4792–800. doi: 10.1158/1078-0432.CCR-13-0380
207. Iovance_Biotherapeutics. Available at: https://www.biospace.com/article/releases/iovance-biotherapeutics-reports-fourth-quarter-and-full-year-2019-financial-results-and-provides-corporate-update/.
208. Wu R, Forget M-A, Chacon J, Bernatchez C, Haymaker C, Chen JQ, et al. Adoptive T-Cell Therapy Using Autologous Tumor-Infiltrating Lymphocytes for Metastatic Melanoma. Cancer J (2012) 18:160–75. doi: 10.1097/PPO.0b013e31824d4465
209. Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of Tumor-Infiltrating Lymphocyte Cultures for Use in Adoptive Transfer Therapy for Melanoma Patients. J Immunother (2003) 26:332–42. doi: 10.1097/00002371-200307000-00005
210. Donia M, Junker N, Ellebaek E, Andersen MH, Straten PT, Svane IM. Characterization and Comparison of ‘Standard’ and ‘Young’ Tumour-Infiltrating Lymphocytes for Adoptive Cell Therapy at a Danish Translational Research Institution. Scandinavian J Immunol (2012) 75:157–67. doi: 10.1111/j.1365-3083.2011.02640.x
211. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric Antigen Receptor-Modified T Cells Derived From Defined CD8+ and CD4+ Subsets Confer Superior Antitumor Reactivity In Vivo. Leukemia (2016) 30:492–500. doi: 10.1038/leu.2015.247
212. Yang Y, Kohler ME, Chien CD, Sauter CT, Jacoby E, Yan C, et al. TCR Engagement Negatively Affects CD8 But Not CD4 CAR T Cell Expansion and Leukemic Clearance. Sci Trans Med (2017) 9:1–13. doi: 10.1126/scitranslmed.aag1209
213. Zudaire E, Madduri D, Usmani SZ, Jakubowiak A, Berdeja JG, Geng D, et al. Translational Analysis From CARTITUDE-1, an Ongoing Phase 1b/2 Study of JNJ-4528 BCMA-Targeted CAR-T Cell Therapy in Relapsed and/or Refractory Multiple Myeloma (R/R MM), Indicates Preferential Expansion of CD8+ T Cell Central Memory Cell Subset. Blood (2019) 134:928–8. doi: 10.1182/blood-2019-127309
214. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-To-Treat Leukemia Remission by CD19 CAR T Cells of Defined Formulation and Dose in Children and Young Adults. Blood (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
215. Lee DH, Cervantes-Contreras F, Lee SY, Green DJ, Till BG. Improved Expansion and Function of CAR T Cell Products From Cultures Initiated at Defined CD4:CD8 Ratios. Blood (2018) 132:3334–4. doi: 10.1182/blood-2018-99-111576
216. Bowers JS, Majchrzak K, Nelson MH, Aksoy BA, Wyatt MM, Smith AS, et al. Pi3kδ Inhibition Enhances the Antitumor Fitness of Adoptively Transferred CD8+T Cells. Front Immunol (2017) 8:1221. doi: 10.3389/fimmu.2017.01221
217. Stock S, Übelhart R, Schubert MM-L, Fan F, He B, Hoffmann JJ-M, et al. Idelalisib for Optimized CD19-Specific Chimeric Antigen Receptor T Cells in Chronic Lymphocytic Leukemia Patients. Int J Cancer (2019) 145:1312–24. doi: 10.1002/ijc.32201
218. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive Resistance to Anti-PD1 Therapy by Tim-3 Upregulation is Mediated by the PI3k-Akt Pathway in Head and Neck Cancer. OncoImmunology (2017) 6:1–11. doi: 10.1080/2162402X.2016.1261779
219. Li Q, Liu M, Wu M, Zhou X, Wang S, Hu Y, et al. PLAC1-Specific TCR-Engineered T Cells Mediate Antigen-Specific Antitumor Effects in Breast Cancer. Oncol Lett (2018) 15:5924–32. doi: 10.3892/ol.2018.8075
220. Lin W-HW, Adams WC, Nish SA, Chen Y-H, Yen B, Rothman NJ, et al. Asymmetric PI3K Signaling Driving Developmental and Regenerative Cell Fate Bifurcation. Cell Rep (2015) 13:2203–18. doi: 10.1016/j.physbeh.2017.03.040
221. Nish SA, Zens KD, Kratchmarov R, Lin WHW, Adams WC, Chen YH, et al. CD4+ T Cell Effector Commitment Coupled to Self-Renewal by Asymmetric Cell Divisions. J Exp Med (2017) 214:39–47. doi: 10.1084/jem.20161046
222. Hendriks J, Xiao Y, Borst J. CD27 Promotes Survival of Activated T Cells and Complements CD28 in Generation and Establishment of the Effector T Cell Pool. J Exp Med (2003) 198:1369–80. doi: 10.1084/jem.20030916
223. Powell DJ Jr, Dudley ME, Robbins PF. Rosenberg S a. Transition of Late-Stage Effector T Cells to CD27. Blood (2005) 105:241–50. doi: 10.1182/blood-2004-06-2482.An
224. Huang J, Kerstann KW, Ahmadzadeh M, Li YF, El-Gamil M, Rosenberg SA, et al. Modulation by IL-2 of CD70 and CD27 Expression on CD8 + T Cells: Importance for the Therapeutic Effectiveness of Cell Transfer Immunotherapy. J Immunol (2006) 176:7726–35. doi: 10.4049/jimmunol.176.12.7726
225. Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere Length of Transferred Lymphocytes Correlates With In Vivo Persistence and Tumor Regression in Melanoma Patients Receiving Cell Transfer Therapy. J Immunol (2005) 175:7046–52. doi: 10.4049/jimmunol.175.10.7046
226. Alsina M, Shah N, Raje NS, Jagannath S, Madduri D, Kaufman JL, et al. Updated Results From the Phase I CRB-402 Study of Anti-Bcma CAR-T Cell Therapy Bb21217 in Patients With Relapsed and Refractory Multiple Myeloma: Correlation of Expansion and Duration of Response With T Cell Phenotypes. Blood (2020) 136:25–6. doi: 10.1182/blood-2020-140410
227. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive Cell Therapy for Patients With Metastatic Melanoma: Evaluation of Intensive Myeloablative Chemoradiation Preparative Regimens. J Clin Oncol (2008) 26:5233–9. doi: 10.1200/JCO.2008.16.5449
228. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive Cell Transfer Therapy Following non-Myeloablative But Lymphodepleting Chemotherapy for the Treatment of Patients With Refractory Metastatic Melanoma. J Clin Oncol (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
229. Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, et al. CD8+ Enriched “Young” Tumor Infiltrating Lymphocytes can Mediate Regression of Metastatic Melanoma. Clin Cancer Res (2010) 16:6122–31. doi: 10.1158/1078-0432.CCR-10-1297
230. News In Brief. Augmenting CAR T Cells With PD-1 Blockade. Cancer Discov (2019) 9:158.1–158. doi: 10.1158/2159-8290.CD-NB2018-165
231. Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, et al. IL-2 Therapy Promotes Suppressive ICOS+ Treg Expansion in Melanoma Patients. J Clin Invest (2014) 124:99–110. doi: 10.1172/JCI46266
232. Amatya PN, Carter AJ, Ritchey JK, Niswonger J, Cooper ML, Pachter JA, et al. The Dual Pi3kδγ Inhibitor Duvelisib Potently Inhibits IL-6 Production and Cytokine Release Syndrome (CRS) While Maintaining CAR-T Function in Vitro and In Vivo. Blood (2020) 136:1–2. doi: 10.1182/blood-2020-139904
233. Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, et al. Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy. Front Immunol (2018) 9:1739. doi: 10.3389/fimmu.2018.01739
234. Page DB, Bear H, Prabhakaran S, Gatti-Mays ME, Thomas A, Cobain E, et al. Two may be Better Than One: PD-1/PD-L1 Blockade Combination Approaches in Metastatic Breast Cancer. NPJ Breast Cancer (2019) 5:34. doi: 10.1038/s41523-019-0130-x
235. Wang D, Lin J, Yang X, Long J, Bai Y, Yang X, et al. Combination Regimens With PD-1/PD-L1 Immune Checkpoint Inhibitors for Gastrointestinal Malignancies. J Hematol Oncol (2019) 12:42. doi: 10.1186/s13045-019-0730-9
236. Ascierto PA, Ferrucci PF, Fisher R, del Vecchio M, Atkinson V, Schmidt H, et al. Dabrafenib, Trametinib and Pembrolizumab or Placebo in BRAF-Mutant Melanoma. Nat Med (2019) 25:941–6. doi: 10.1038/s41591-019-0448-9
237. Yap TA, Rodon Ahnert J, Piha-Paul SA, Fu S, Janku F, Karp DD, et al. Phase I Trial of IACS-010759 (IACS), a Potent, Selective Inhibitor of Complex I of the Mitochondrial Electron Transport Chain, in Patients (Pts) With Advanced Solid Tumors. J Clin Oncol (2019) 37:3014–4. doi: 10.1200/JCO.2019.37.15_suppl.3014
238. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat Plus Pembrolizumab Versus Placebo Plus Pembrolizumab in Patients With Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
239. Alistar AT, Morris B, Harrison L, Bickenbach K, Starker L, Ginder N, et al. Alpert J. A Single-Arm, Open-Label, Phase I Study of CPI-613 (Devimistat) in Combination With Gemcitabine and Nab-Paclitaxel for Patients With Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J Clin Oncol (2020) 38:4635–5. doi: 10.1200/JCO.2020.38.15_suppl.4635
240. Halford SER, Jones P, Wedge S, Hirschberg S, Katugampola S, Veal G, et al. A First-in-Human First-in-Class (FIC) Trial of the Monocarboxylate Transporter 1 (MCT1) Inhibitor AZD3965 in Patients With Advanced Solid Tumours. J Clin Oncol (2017) 35:2516–6. doi: 10.1200/JCO.2017.35.15_suppl.2516
241. Esposito A, Viale G, Curigliano G. Safety, Tolerability, and Management of Toxic Effects of Phosphatidylinositol 3-Kinase Inhibitor Treatment in Patients With Cancer: A Review. JAMA Oncol (2019) 5:1347–54. doi: 10.1001/jamaoncol.2019.0034
242. Curigliano G, Shah RR. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf (2019) 42:247–62. doi: 10.1007/s40264-018-0778-4
243. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. Leading Edge The PI3K Pathway in Human Disease. Cell (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
244. Flinn IW, Miller CB, Ardeshna KM, Tetreault S, Assouline SE, Mayer J, et al. DYNAMO: A Phase II Study of Duvelisib (IPI-145) in Patients With Refractory Indolent non-Hodgkin Lymphoma. J Clin Oncol (2019) 37:912–22. doi: 10.1200/JCO.18.00915
245. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA -Mutated, Hormone Receptor–Positive Advanced Breast Cancer. New Engl J Med (2019) 380:1929–40. doi: 10.1056/NEJMoa1813904
246. Mayer IA, Prat A, Egle D, Blau S, Alejandro Perez Fidalgo J, Gnant M, et al. A Phase II Randomized Study of Neoadjuvant Letrozole Plus Alpelisib for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer (NeO-ORB). Clin Cancer Res (2019) 25:2975–87. doi: 10.1158/1078-0432.CCR-18-3160
247. Bonnevaux H, Lemaitre O, Vincent L, Levit MN, Windenberger F, Halley F, et al. Concomitant Inhibition of PI3Kβ and BRAF or MEK in PTEN-Deficient/BRAF-Mutant Melanoma Treatment: Preclinical Assessment of SAR260301 Oral Pi3kβ-Selective Inhibitor. Am Assoc Cancer Res (2016) 15:1460–71. doi: 10.1158/1535-7163.MCT-15-0496
Keywords: adoptive cell immunotherapy, TIL (tumor infiltrating lymphocytes), CAR T cancer therapy, immune checkpoint inhibition (ICI), PI3K delta, PI3K gamma, T cell differentiation
Citation: Chandrasekaran S, Funk CR, Kleber T, Paulos CM, Shanmugam M and Waller EK (2021) Strategies to Overcome Failures in T-Cell Immunotherapies by Targeting PI3K-δ and –γ. Front. Immunol. 12:718621. doi: 10.3389/fimmu.2021.718621
Received: 01 June 2021; Accepted: 06 August 2021;
Published: 26 August 2021.
Edited by:
Robert Weinkove, Victoria University of Wellington, New ZealandReviewed by:
Rachel Perret, Victoria University of Wellington, New ZealandCopyright © 2021 Chandrasekaran, Funk, Kleber, Paulos, Shanmugam and Waller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanjay Chandrasekaran, c2FuamF5LmNoYW5kcmFzZWthcmFuQHV0c291dGh3ZXN0ZXJuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.