 Xiaofeng Zhou
Xiaofeng Zhou Bethany B. Moore
Bethany B. Moore- 1Dept. of Microbiology and Immunology, University of Michigan Medical School, Ann Arbor, MI, United States
- 2Division of Pulmonary and Critical Care Medicine, Dept. of Internal Medicine, University of Michigan Medical School, Ann Arbor, MI, United States
Pulmonary infections remain a major cause of morbidity and mortality in hematopoietic cell transplantation (HCT) recipients. The prevalence and type of infection changes over time and is influenced by the course of immune reconstitution post-transplant. The interaction between pathogens and host immune responses is complex in HCT settings, since the conditioning regimens create periods of neutropenia and immunosuppressive drugs are often needed to prevent graft rejection and limit graft-versus-host disease (GVHD). Experimental murine models of transplantation are valuable tools for dissecting the procedure-related alterations to innate and adaptive immunity. Here we review mouse models of post-HCT infectious pulmonary complications, primarily focused on three groups of pathogens that frequently infect HCT recipients: bacteria (often P. aeruginosa), fungus (primarily Aspergillus fumigatus), and viruses (primarily herpesviruses). These mouse models have advanced our knowledge regarding how the conditioning and HCT process negatively impacts innate immunity and have provided new potential strategies of managing the infections. Studies using mouse models have also validated clinical observations suggesting that prior or occult infections are a potential etiology of noninfectious pulmonary complications post-HCT as well.
Introduction
Hematopoietic cell transplantation (HCT) is a potentially curative treatment for high-risk hematopoietic neoplastic disorders, metabolic, genetic and immune-mediated diseases. It involves eradication or suppression of the recipient’s hematopoietic cells using a conditioning regimen followed by infusion of stem cells collected from the bone marrow, placenta (cord blood) or peripheral blood (1). The source of hematopoietic cells can be either autologous (auto, recipient-derived) or allogeneic (allo, matched related or unrelated donor-derived) hematopoietic cells. HCT has been carried out increasingly over the years with 47,468 transplants in 50 European and associated countries (2), and 22,573 transplants in the United States in 2018 (3).
Unfortunately, the toxicity of conditioning regimens, alloimmune responses and immunosuppressive therapies cause severe post-transplant complications, in which the lung is one of the most common target organs. Pulmonary complications occur in up to 60% of allo-HCT recipients (4) and 25% of auto-HCT recipients (5). The frequent pulmonary complications and their significant contribution to post-transplant morbidity and mortality limit the success of HCT (6–8). These complications are heterogeneous, and include pathologies generated by infectious agents and noninfectious disorders. Although infectious pulmonary complications after HCT have been significantly reduced due to aggressive prophylaxis and the use of broad-spectrum antimicrobial medications, these infections still remain problematic, especially among the patients with graft-versus-host disease (GVHD). Major noninfectious pulmonary complications include early onset idiopathic pneumonia syndrome (IPS) (7), and late onset bronchiolitis obliterans syndrome (BOS) (9). Current experimental data support alloimmunity as an underlying mechanism of these idiopathic noninfectious lung injuries (7, 9). For a review on non-infectious pulmonary complications of stem cell transplantation, please see references (10, 11).
Animal models have been extensively used for the establishment and improvement of HCT therapy (12, 13). Animal models allow manipulation of single factors during the development of complications associated with HCT, and thus are crucial for successfully improving clinical applications. Most of the current knowledge regarding defects in immune responses during infectious pulmonary complications come from studies using mouse models of HCT. In this review, we will first briefly introduce infectious pulmonary complications post-HCT, and then describe relevant mouse models and current understanding of host immune responses to lung infections post-HCT that have been acquired from studying these models.
Clinical Phenotypes Of Infectious Pulmonary Complications Post-Hct
The immune system of HCT recipients is eradicated or weakened by either myeloablative or less intense nonmyeloablative conditioning regimens before transplant to eradicate/reduce tumor burdens and to prevent graft rejection. Thus, it is not surprising that infections are a major complication post-HCT. Infectious complications are more frequent and severe in patients with allo-HCT due to prolonged immunosuppressive therapy and GVHD (14). The timing of reconstitution of the immune system post-HCT varies considerably among patients, depending on the type of transplant (autologous vs allogeneic), the intensity of conditioning regimen, the source of hematopoietic cells, the presence of GVHD and the length of immunosuppressive therapies. Nevertheless, post-HCT reconstitution can be roughly divided into three phases: severe neutropenia or pre-engraftment phase (first 2-4 weeks), early engraftment phase (second and third month) and late engraftment phase (after second or third month) (14).
The prevalence and types of infection change over time and often follow the course of immune reconstitution post-transplant in patients (14). During the pre-engraftment phase, the depletion of neutrophils and damage to the mucosal barriers caused by conditioning regimens allow opportunistic pathogens to become infectious. The predominant pathogens during this phase are Pseudomonas, Candida and Aspergillus species (15–18). During the early engraftment phase, most innate immune cell subsets such as monocytes, neutrophils, and natural killer cells repopulate at normal levels (19), but lymphocyte counts are still low. This allows the reactivation of herpesviruses, such as cytomegalovirus (CMV), Epstein–Barr virus (EBV), human herpesvirus 6 (HHV-6), and new infections with respiratory viruses (20–22). A second peak of invasive Aspergillus infection occurs at the end of the early engraftment phase in allo-HCT recipients due to prolonged GVHD and its immunosuppressive therapy (18). During the late posttransplant phase (about three months after transplant), innate immunity is mostly reconstituted, but the recovery of T cells takes about a year and B cells may take even longer to completely repopulate (23). Bacterial pneumonia is less common during this late phase, but allo-HCT recipients are still at risk of late CMV reactivation and fungal infection. Current preemptive therapeutic strategies have significantly reduced early onset CMV infections after allo-HCT, but the incidence of late CMV infections have increased (24, 25). CMV reactivation remains a life-threatening infectious complication that is difficult to manage following allo-HCT (26–28). Allo-HCT recipients with chronic GVHD and immunosuppressive therapy continue to be susceptible to Aspergillus and Gram-positive bacteria as well (29). It is thus important to understand the interplay among host immunity, pathogens and GVHD in an allo-HCT setting.
Mouse Models Of Infectious Pulmonary Complications Following Bone Marrow Transplantation
There have been many functional studies on immune responses to pathogens in mouse models of HCT. These have included both syngeneic (syn) and allogeneic strain combinations to recapitulate autologous or allogeneic HCT in patients. The pneumonia pathogens studied span bacteria (mostly Pseudomonas aeruginosa), fungus (primarily Aspergillus fumigatus), and viruses (primarily herpesviruses). There are also reports of sepsis subsequent to gastrointestinal damage due to conditioning regimens in HCT mouse models (30).
Mouse Models of Post-HCT Bacterial Pneumonia
Bacterial pneumonia usually occurs early after HCT during the neutropenic period (31), but can also occur post-engraftment. P. aeruginosa is the most common pathogen isolated from the lower respiratory tract within 100 days post-transplant (15). P. aeruginosa is a ubiquitous environmental bacterium, and if inhaled into the lung airway by a immunocompetent individual, it is quickly cleared by alveolar macrophages (AMs) (32). Because P. aeruginosa has become increasingly resistant to multiple antibiotics over the years (33), it can be difficult to treat multidrug resistant Pseudomonal pneumonia (MDRPa) (16). About 40% of the hematologic malignancy patients infected with MDRPa will die in 30 days (34) and MDRPa outbreaks are associated with a death rate as high as 80% (35).
A syn-HCT mouse model was established in our laboratory to understand why HCT recipients are susceptible to P. aeruginosa (36). This model is clinically relevant, as both autologous and allogeneic transplant patients are susceptible to P. aeruginosa infection (37). Like auto-HCT recipients, mouse syn-HCT recipients have no risk of GVHD, and thus the model can be used to explore how the transplant procedure alone impacts pulmonary immunity. Recipient C57BL/6 mice are given a split dose of 13 Gy total body irradiation (TBI) from either a 137Cs or x-ray orthovoltage source with an interval of 3 hours between doses. Bone marrow is harvested from donor C57BL/6 mice, and 5x106 whole bone marrow cells are infused into the recipients via tail vein injection. Five weeks after transplant, the percentage of donor-derived cells is approximately 95% in the spleen and the percentage of donor-derived AMs in the lung is about 83% (38). At this time point, HCT or age-matched non-HCT control mice are infected with P. aeruginosa PAO1 via intratracheal (i.t.) inoculation (36). These experiments demonstrated increased bacterial burden in the lung and dissemination to the blood at 24 h post-infection in HCT mice compared to non-transplant controls (36). See Figure 1 for schematic illustration of the model system.
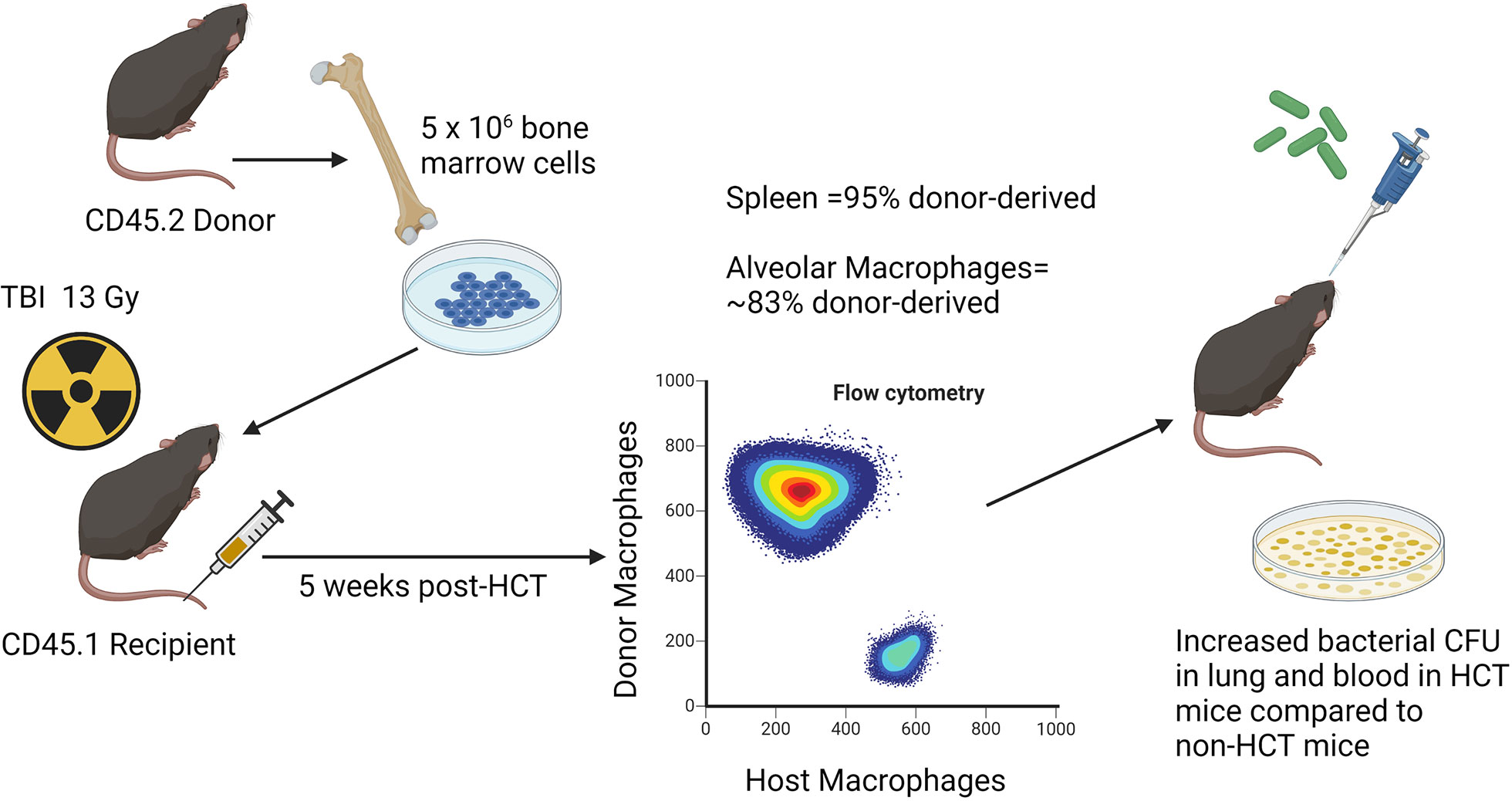
Figure 1 Schematic illustration of syn HCT mouse model. The figure shows the process of syn HCT used in experiments to test impaired host defense against Pseudomonas aeruginosa as described in the text. The figure is created with Biorender.com.
The defect in bacterial clearance in HCT mice is associated with reduced phagocytosis and killing of P. aeruginosa in lung AMs (39, 40) and impaired killing and defective formation of neutrophil extracellular traps (NETs) in neutrophils (41). Similar to HCT patients, the levels of immunosuppressive prostaglandin E2 (PGE2) are elevated in HCT mice (39, 42). Subsequent studies found that overproduction of PGE2 impairs the functions of both AMs and neutrophils, and pharmacologic inhibition of PGE2 production in vivo restores host defense of HCT mice (39, 41).
The syn-HCT model permits further dissection of the mechanisms explaining how the HCT procedure promotes AMs to overproduce PGE2. Conditioning-associated cellular stress stimulates alveolar epithelial cells to produce TGF-β (43). TGF-β signaling stimulates AMs to transcribe microRNA (miR)-29b which suppresses the expression of DNA methyltransferases (DNMTs) (44). Under homeostatic conditions, DNMT3a and DNMT3b methylate the promoter region of cyclooxygenase (COX)-2 gene which encodes a critical enzyme for the production of PGE2 (40). Methylation of the COX-2 promoter limits transcription and reduces COX-2 gene expression. Suppression of DNMTs by the TGF-β-miR-29b axis releases this endogenous break on COX-2 expression and thus increases the production of PGE2 in AMs (44). Interestingly, these epigenetic changes can be long lived with human HCT patients (even 6 years post-HCT) showing elevated levels of PGE2 in the lung. Thus, this mechanism likely accounts for long-lived innate immune impairment post-HCT.
The immunosuppressive function of PGE2 is mediated by its receptors E prostanoid receptor 2 (EP2) and EP4 (44). Signaling via these receptors can activate a cyclic adenosine monophosphate (cAMP)-mediated signaling cascade with multiple downstream effects. One effect is downregulation of the scavenger receptor MARCO which is critical for recognition and phagocytosis of P. aeruginosa (45). Another effect is upregulation of IL-1 receptor associated kinase M (IRAK-M), which is an inhibitor of TLR signaling (46). Ultimately, this alteration impairs the proinflammatory cytokine response (e.g. TNF-α and IFN-γ) that could help clear bacterial infection. At the same time, PGE2 promotes transcription of IL-1β which is a mediator of tissue damage in the lung (47). Furthermore, PGE2 stimulates the expression of phosphatase and tensin homolog deleted on chromosome 10 (PTEN), which negatively regulates phagocytosis and killing of P. aeruginosa (48). A summary figure describing some of the innate immune changes in AMs post-HCT is found in Figure 2.
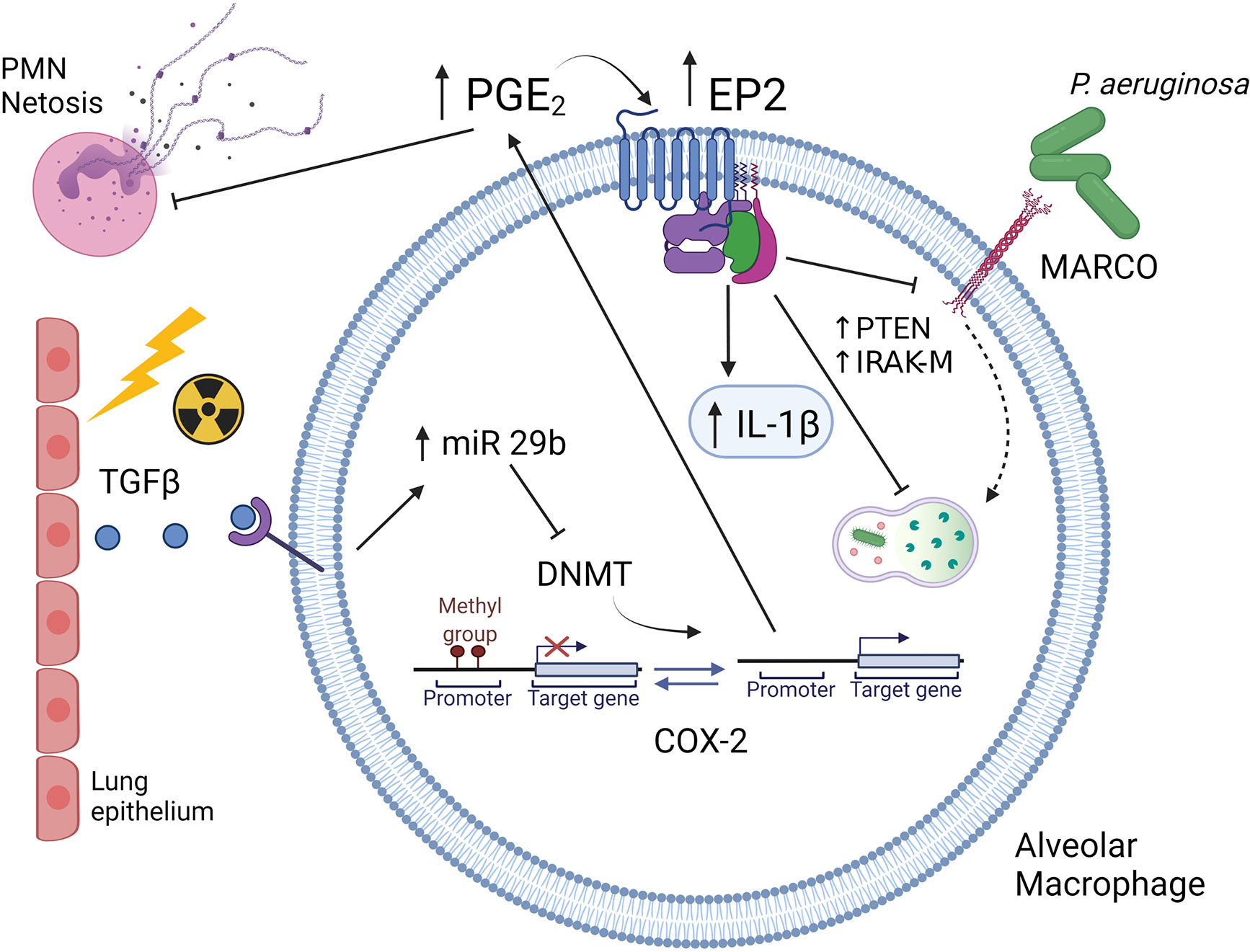
Figure 2 Schematic illustration of Syn HCT induced changes in innate immunity. Conditioning with TBI causes injury to lung epithelium resulting in production of TGF-β. Binding of TGF-β to alveolar macrophages results in increased miR29b expression which then limits expression of DNA methyltransferases (DNMT). This allows for the promoter of the cyclooxygenase 2 (COX-2) gene to be unmethylated resulting in increased production of prostaglandin E2 (PGE2). PGE2 then binds to the E prostanoid 2 (EP2) receptor which is also upregulated post-HCT. Downstream signaling by PGE2 results in upregulation of IL-1, elevations in phosphatase and tensin homolog on chromosome 10 (PTEN) and elevations in IL-1 receptor associated kinase (IRAK-M). These changes impair intracellular killing while downregulation of the MARCO scavenger receptor impairs phagocytosis of P. aeruginosa. Original references described in text. The figure is created with Biorender.com.
In contrast to the impaired phagocytosis of P. aeruginosa post-HCT by AMs, phagocytosis of Staphylococcus aureus is actually enhanced (45). This increased phagocytosis of S. aureus, is also regulated by PGE2, by stimulating the expression of miR-155 which upregulates scavenger receptor (SR-)AI/II (45). However, despite the enhanced uptake of S. aureus, AMs from HCT mice are unable to effectively kill the pathogen intracellularly as a result of the impacts on IRAK-M and PTEN described above. Additionally, the impaired innate immune function of neutrophils likely contributes to poor S. aureus clearance. Interestingly, a study by Zimecki et al. explored the use of bacteriophages as a therapeutic strategy for syn-HCT mice infected with S. aureus strain L (49). Similar to the findings reported above, HCT mice were highly susceptible to S. aureus infection (only 8.3% of infected mice survived whereas mice treated with phage showed 72% survival. It was also noted that the phage therapy increased the circulating leukocyte and neutrophil counts.
While the majority of the studies reviewed above show defective innate immune function, there is one study from 1989 showing that early after marrow transfer in allogeneic radiation chimeras that macrophages can be non-specifically activated by the conditioning milieu and show enhanced resistance to Listeria monocytogenes initially, but that this protection eventually declines with time (50).
Mouse Models of Post-HCT Fungal Pneumonia
Invasive fungi have become the leading infectious cause of morbidity and mortality in HCT recipients in the era of improved prophylaxis and treatment of bacterial and viral infections (51, 52). Invasive pulmonary aspergillosis (IPA) is the most common fungal infection in the lung of HCT recipients. IPA causes high mortality among HCT patients, ranging from 30% to 70% (53), accounting for 10% of all death among the recipients (54). IPA is highly associated with neutropenia, GVHD and its related immunosuppressive therapy, and thus the incidence of IPA peaks early in the pre-engraftment phase and then later post-engraftment in allo-HCT recipients (18). This bimodal distribution of IPA post-HCT reflects different etiologies of Aspergillus infection: early IPA is due to prolonged neutropenia, especially when using myeloablative conditioning regimens, while late IPA is secondary to receiving corticosteroid or other immunosuppressive therapies to treat GVHD (55). Interestingly, the pro-inflammatory status noted in allo-HCT mice without immunosuppressive treatment may enhance clearance of Aspergillus as noted in one study by Hildebrandt et al. (56). The significant shift to using lower intensity non-myeloablative regimens allows a shorter neutropenic period and the systematic use of antifungal prophylaxis has led to a decrease in the incidence of early IPA (57). The early and late IPA time periods present distinct immunopathology patterns in HCT recipients. Early IPA in the neutropenic phase is characterized by rapid fungal growth and low levels of inflammation, but late IPA in immunosuppressed patients usually presents with overabundant inflammation including excessive neutrophil infiltration with insufficient fungal clearance (58).
Accordingly, several animal models have been established to understand the pathogenesis of early or late aspergillosis in HCT patients (59). Neutropenic models include treating mice with chemotherapeutic agents such as cyclophosphamide (60), TBI (61, 62), and using antibody depletion of neutrophils (63). Immunomodulated models usually involve the use of corticosteroids (64). These two types of mouse models recapitulate the different pathologies of IPA that present in neutropenic and immunosuppressed patients respectively (64, 65). For the neutropenic mouse models, it is most common to administer cyclophosphamide at 150 mg/kg via the intraperitoneal route thrice weekly before infection. Some studies have used monoclonal antibodies to achieve neutrophil depletion. A dose of 100µg of anti-Ly6 (Gr1) rat IgG2b MAb57 (clone RB6-8C5) via intraperitoneal injection on the day before and 2 days after fungal intranasal inoculation dramatically reduces the number of neutrophils for up to 5 days (63). The lethal dose of irradiation varies depending on mouse strain receiving the treatment. For example, a single lethal dose of 9 Gy given to C3H/HeJ mice followed by transplants with 2x106 T-cell-depleted allogeneic bone marrow cells from DBA/2 mice shows profound neutropenia 3 days after transplant (63). Immunomodulated models commonly administer cortisone at 100 to 200 mg/kg via subcutaneous injection thrice weekly for 1 or 2 weeks before experimental infection. In a cortisone-treated immunosuppressive model, myeloid cells such as neutrophils and macrophages, are massively recruited to the lungs upon infection, but lymphocytes fail to be recruited to the lung, indicating the requirement of lymphocytes to efficiently clear the infection (64). The most common routes of inoculation of Aspergillus are intranasal and intratracheal administration. A conidial suspension of A. fumigatus inoculated into the nares is close to natural infection, but due to upper mucociliary clearance of mice, only about 10% of the inoculum actually enters into the lungs (66). As a result, the development of IPA is highly variable in intranasally inoculated mice. Delivery of spore suspension directly into the trachea, by either tracheotomy or oropharyngeal aspiration in anesthetized mice, can more tightly control the fungal inoculum and lead to reproducible IPA (59). The most commonly used A. fumigatus strains are the low virulent strain AF293 (ATCC MYA4609 or CBS101355) and the high virulent strain Dal/CEA10 (ATCC MYA1163 or CBS 144.89). Depending on the strain of A. fumigatus and the route, the dose ranges from 1.0×102 to 1.0×109 conidia for mice infected through the intranasal route, and 1.0 to 2.0x107 conidia through the intratracheal route (59).
Studies on mouse models and human patients have greatly increased our knowledge of the host immune responses to A. fumigatus and have aided in the development of novel therapeutic targets to treat IPA [reviewed in references (67, 68)]. Here we highlight a few advances in the field during recent years. Mouse models have confirmed or identified several important pattern recognition receptors (PRRs) on the cell surface of innate immune cells, such as AMs, which include dectin-1 (61), TLR2 (69, 70), TLR4 (70), TLR9 (71–73), NOD2 (74), soluble pentraxin-3 (75) and TREM1 (76) in recognizing Aspergillus components. Inflammatory cytokines such as IL-1α and IL-1β are critical for host defense against A. fumigatus in neutropenic mice (77, 78). The role of antigen presentation and development of antigen specific T cell subsets has been studied with respect to clearance of IPA, but with some conflicting results. For example, adoptive transfer of dendritic cells pulsed with conidia stimulates a T helper type 1 (Th1) responses and improves survival in a syn-HCT model suggesting an important role of Th responses to clear infection (79). This is consistent with several other studies in mouse HCT models that also demonstrated that Th1-mediated immunity is important in clearing A. fumigatus infection (80–82). Interestingly, it appears paradoxical that CCR7 deficient HCT mice whose dendritic cells cannot enter draining lymph nodes to prime T lymphocytes show improved survival in a monoclonal antibody (anti-Gr1) induced neutropenic model (83). This study suggests that retaining CD11b+ dendritic cells inside the neutropenic lung during initial infection with Aspergillus is beneficial, potentially by complimenting the loss of neutrophils. The role of the Th17 response in the pathogenesis of IPA is somewhat controversial, as it can be either protective (84, 85) or pathogenic (80). Th2 responses and the production of IL-4 are detrimental to control A. fumigatus infection, as IL-4-/- mice are protected from IPA (86). Finally, regulatory T cells are producers of IL-10 which is linked to disease progression in steroid immunosuppressive experimental IPA (87, 88). Figure 3 provides a summary of recent insights from mouse models of HCT or neutropenia with regards to Aspergillus infection.
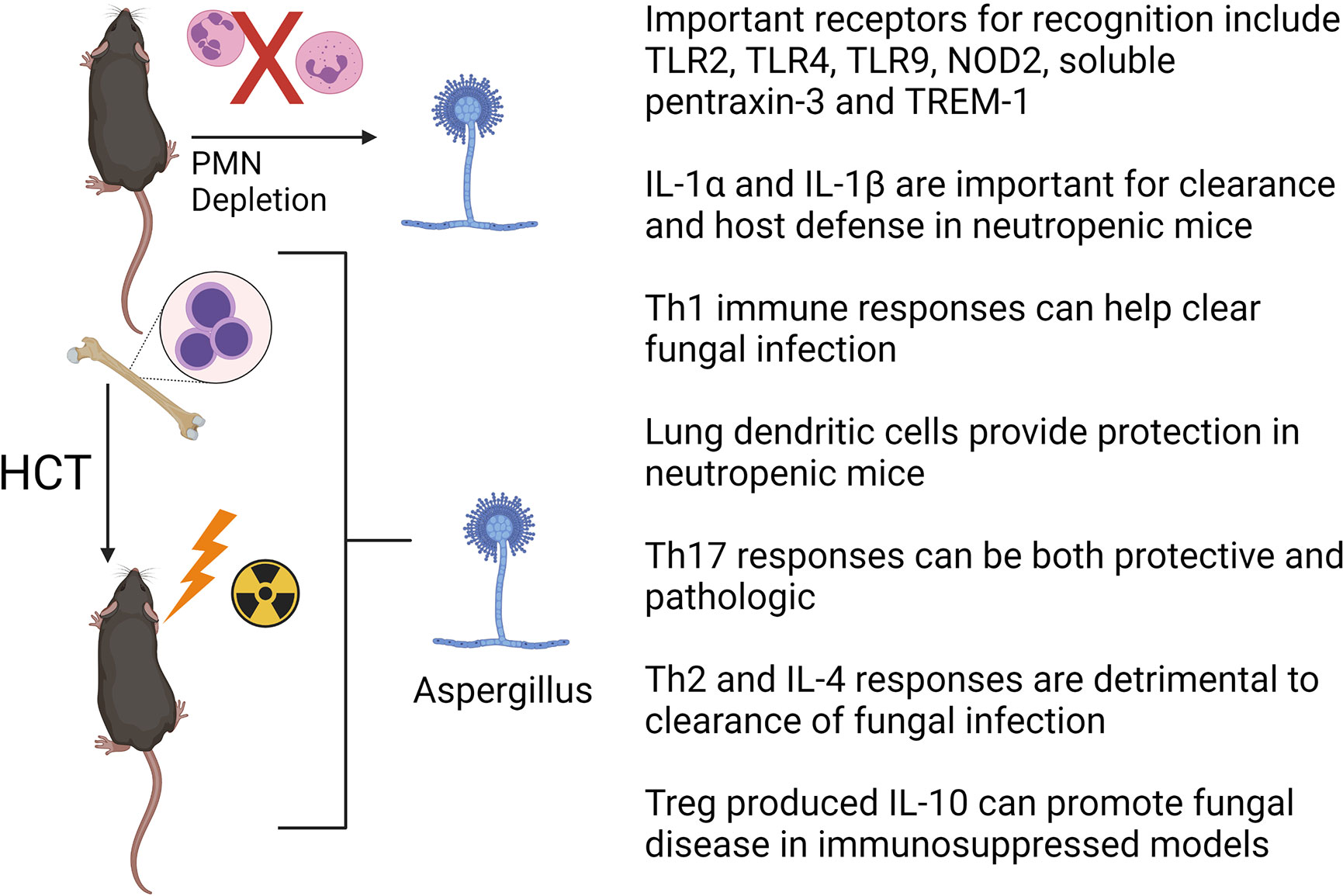
Figure 3 Factors important for clearance of Aspergillus fumigatus in HCT or neutrophil (PMN) depleted mouse models. See text for details. The figure is created with Biorender.com.
Mouse Models of Post-HCT Viral Pneumonia
Viral infections often occur after engraftment when the reconstitution of lymphocytes is not yet complete, or when immunosuppression due to prophylaxis or treatment of GVHD in allo-HCT recipients is needed. Nearly every human being is infected with one or more herpesviruses in the first two decades of life and the viruses can establish life-long latency to escape immune surveillance and detection (20). Viral pneumonia can be caused by reactivation of latent human herpesviruses or new infection with community acquired respiratory viruses. Additionally, primary herpesvirus infections can occur in seronegative patients receiving grafts from seropositive donors (89). Besides causing direct lung injuries, such as, cytomegalovirus (CMV) pneumonia (20), occult or prior herpesvirus infections appear to trigger the development of “noninfectious” pulmonary complications at later time points after allo-HCT (21, 22, 90).
Most human herpesviruses have very strict host-species specificity, and it is thus difficult to study human herpesviruses in mice. Herpes simplex virus type 1 (HSV-1) is an exception, as it can directly cause pneumonia in HCT mice (91). To bypass this hurdle, some researchers have generated transgenic mouse models that express receptors for human herpesvirus. For example, human CD46, an HHV-6A receptor, is expressed in the brain of a mouse line to study host innate immunity against HHV-6A (92). Other researchers engrafted human CD34+ hematopoietic progenitor cells into NOD-scid IL2Rγcnull (NSG) mice which can then be directly infected with human herpesvirus (93). More often, murine homologs of their corresponding human herpesviruses are used in mouse models to study the principles of virus-host interactions that are thought to be shared among human and mouse systems. Mouse CMV (mCMV), murine gammaherpesvirus 68 (MHV-68) and murine roseolovirus (MRV) are frequently used to study human CMV, Epstein-Barr virus (EBV) and human herpesvirus (HHV)-6A/B, respectively in HCT settings.
Most murine herpesvirus models fall into two categories: pre-HCT latent infection or post-HCT infection models. Both syngeneic and allogeneic HCT have been studied. Latent infection models usually involve a primary infection in neonates or adult mice to mimic the natural history of herpesviral infection, followed by various lengths of “waiting time” to let the virus enter latency (21, 94). After that, an allo-HCT is usually performed to stimulate viral reactivation. Latently infected mouse models are most suitable for studying the reactivation of herpesvirus, their subsequent effects and for testing novel therapeutic strategies. This model has facilitated the discovery of the critical role of humoral immunity in controlling the reactivation of mCMV (94). The half-life of preexisting antibodies in latently infected mice and the elimination of recipient plasma cells due to GVHD can lead to a loss of anti-mCMV antibodies, which eventually leads to mCMV reactivation in recipients. Importantly, the reactivation of mCMV in allo-HCT recipients can be prevented by the transfer of immune serum (94). In a similar latent infection model, two doses of leukotriene B4 administered via intravenous route effectively reduced the reactivation of mCMV in allo-HCT mice through yet unknown mechanisms (95). To determine the relationship between herpesviral reactivation and noninfectious pulmonary complications, MRV, a mouse homolog of HHV-6, was given to neonatal mice and then reactivated in response to a minor histocompatibility antigen mismatched allo-HCT 8 weeks later. Indeed, the reactivation of MRV not only caused IPS-like pathology but also exacerbated histologic signs of acute GVHD in the gut (21).
Due to variable waiting times for entering latency and the heterogenous nature of post-HCT viral reactivation among latently infected mice, some researchers infect mice post-HCT or concurrent with HCT to mimic the reactivation of herpesvirus. The HCT procedure creates an immunosuppressive lung microenvironment, characterized by increased levels of PGE2 (39), TGF-β (91, 96) and Kynurenine (97). Reduced influx or altered function of CD8+ T cells, which are critical for clearance of mCMV (98, 99), HSV-1 (91) and community acquired respiratory viruses (100, 101), were observed in both syn- and allo-HCT recipients. As a result, most HCT mice experienced delayed viral clearance and persistent pneumonitis. Interestingly, the impact of HCT on T cell immunity does not seem to be mediated by the elevated levels of PGE2 (101), but rather by TGF-β (91).
Recently, studies using MHV-68, a mouse herpesvirus genetically related to Kaposi’s sarcoma-associated herpesvirus (KSHV) and EBV, in a syn-HCT model in our laboratory have advanced our understanding of host immune response to herpesvirus infection in the HCT setting. A C57BL/6 to C57BL/6 syn-HCT mouse model as described above was adopted to study MHV-68 infection. The HCT procedure not only causes an immunosuppressive environment, but also changes the structure of the lung microbiome (102). Together, these alterations in the lung microenvironment have significant impacts on the biology of conventional dendritic cells (cDCs) in HCT lungs. After exposure to MHV-68, the cDCs in syn-HCT mice increased their expression of pro-Th17 cytokines such as IL-6 IL-23 and TGF-β relative to the responses noted in untransplanted mice (103). These HCT lung cDCs also become deficient for delta like ligand 4 (DLL4), a Notch ligand, on their cell surface which further permits Th17 polarization (104). In addition, the migration of cDCs into mediastinal draining lymph nodes is impaired, significantly reducing Th1 responses, but augmenting Th17 responses which appear to be primed locally in the lung (105). Thus, the functional changes of lung cDCs post-HCT tip the balance of Th responses against MHV-68 infection from protective Th1 responses to pathogenic Th17 responses (96, 103).
Excessive IL-17A due to Th17 responses eventually causes the development of pneumonitis and pulmonary fibrosis 3 weeks after infection, when lytic MHV-68 is no longer detectable (103, 106). Administration of anti-IL-17A antibodies or using bone marrow cells isolated from IL-17A-/- donor mice protects HCT recipients from pneumonitis and fibrosis after infection with MHV-68 (103). Figure 4 highlights some of the changes noted in HCT lungs post-infection with herpesvirus. Note that the pathology seen in this mouse model also resembles many histological features seen in noninfectious complications such as IPS and restrictive lung disease, suggesting a potential etiology of noninfectious pulmonary complications caused by prior or occult infections that trigger pathogenic immune responses leading to lung injury and improper repair. This hypothesis is supported by recent discoveries of occult infections in IPS patients (90), and the strong association between the infections with herpesviruses (21) or community acquired respiratory viruses (22) and noninfectious complications in HCT recipients. Furthermore, direct evidence comes from a study mentioned above showing that reactivation of MRV in allo-HCT mice causes IPS-like pathology and exacerbates acute GVHD (21).
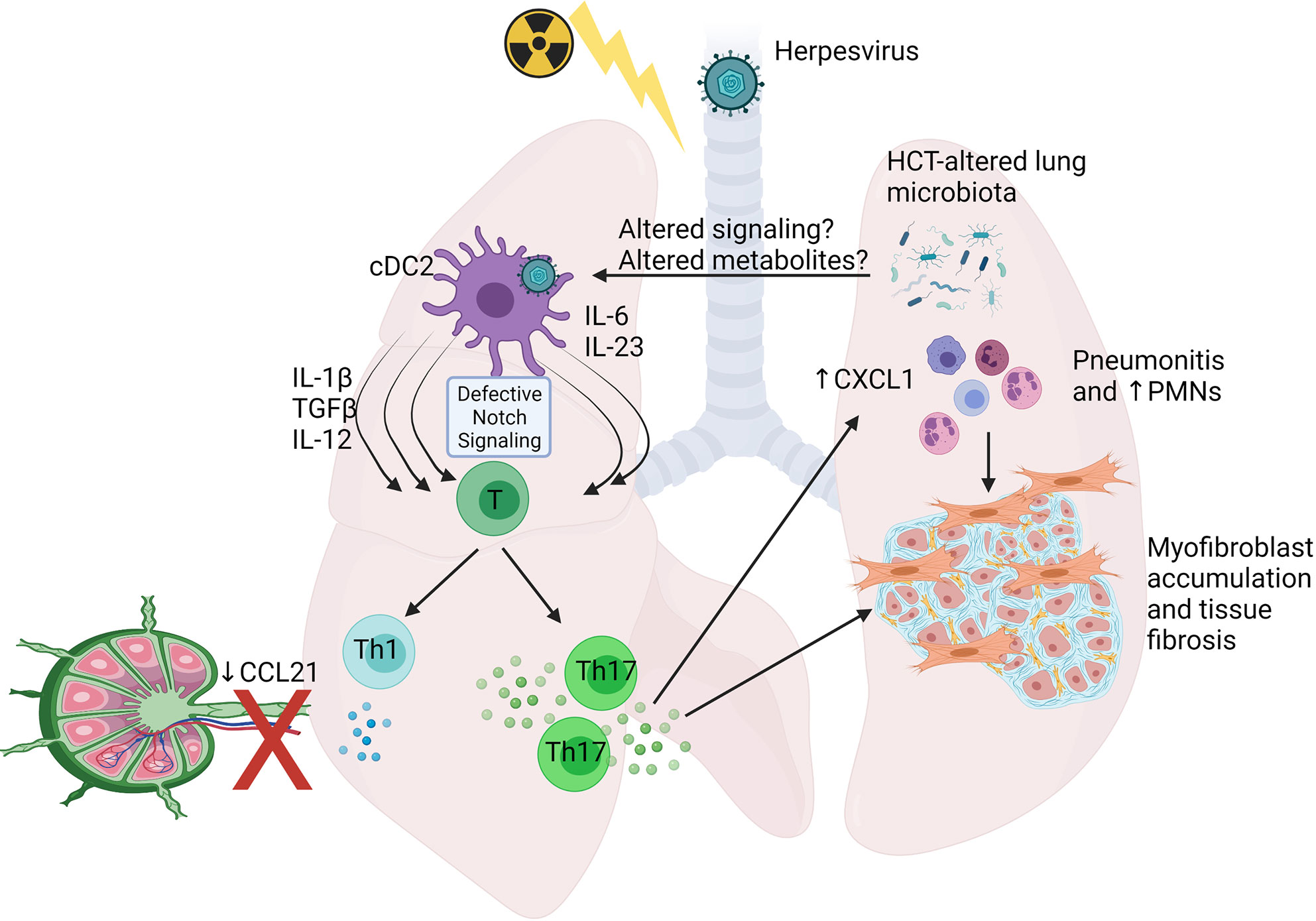
Figure 4 Schematic showing alterations shown to occur in Syn HCT mouse models of murine gammaherpesvirus infection. In responses to HCT, lung conventional dendritic cells 2 (cDC2) display defective delta like ligand 4 leading to impaired notch signaling when interacting with virus-specific T cells. This leads to production of cytokines able to drive both Th1 and Th17 cell differentiation, but because of poor ability of the cDCs to migrate to the draining lymph nodes secondary to reduced CCL21 levels, the Th1 response is impaired, while the Th17 response is primed efficiently in the lungs. Elevated IL-17 levels stimulate lung epithelium to produce CXCL1 and recruit PMNs prominently, although other cell types also accumulate. The IL-17 can also directly activate myofibroblast proliferation and extracellular matrix production leading to fibrosis. Alterations to the lung and gut associated microbiome are also prominent post-HCT and may contribute to altered immune cell functions. See text for details. The figure is created with Biorender.com.
Discussion
Pulmonary infections remain a major cause of morbidity and mortality in HCT patients. This is not surprising given that the conditioning regimens create periods of cytopenia and immunosuppressive drugs are often needed to limit GVHD. The rise of antibiotic resistant strains of bacteria, challenges of vaccinating immunocompromised patients, and limited availability of vaccines for many of the common pathogens in this patient population all help explain the infectious challenges facing HCT patients. Additionally, mucosal tissue damage as a result of GVHD and bacterial colonization can significantly increase the likelihood of opportunistic infection (107–109). As discussed above, non-myeloablative conditioning and fungal and viral prophylaxis have helped reduced infections during the cytopenic/neutropenic phases; however, there is a growing appreciation that even post-engraftment, innate and adaptive immune cells display altered immune function.
Even as early as 1982, it was noted that AMs from HCT patients were defective at phagocytosis, chemotaxis towards, and killing of fungal and bacterial pathogens when studied 4 months post-HCT (110). Importantly, while phagocytic and chemotactic defects normalized, the killing defects persisted even 12 months post-HCT (110). This suggests that alterations in the lung milieu, most likely caused by conditioning regimens may interfere with the function of innate immune cells. It also suggests that long-lived alterations may result from epigenetic alterations. A decade later, elevated levels of PGE2 were noted in both auto and allo-HCT patients; however, this finding was not linked to impaired innate immune function (42). It was not until 2006 that murine models were able to provide a mechanistic link between these two observations and demonstrate that overproduction of PGE2 post-HCT was responsible for impaired phagocytosis and killing by AMs (39). It took almost another decade to describe the epigenetic alterations in methylation of the COX-2 promoter that were caused by elevated levels of TGFβ1 caused by the conditioning regimen (44). Interestingly, this same mechanism that impairs innate immune function in HCT, has recently been shown to explain defective macrophage responses to wound healing and wound infection in diabetes as well (111). No doubt there are many other, yet to be discovered pathways that impair the function of innate immune cells in the transplant setting that are likely to be regulated via epigenetic alterations induced by the altered lung milieu.
We are also starting to learn more about how alterations of the normal microbiota in the lung and gut of HCT recipients may alter immune tone. The process of allo-HCT has been shown to reduce the diversity of the gut microbiota in humans (112). Furthermore, low diversity of gut flora at the time of neutrophil engraftment predicts mortality (113). Interestingly, when focused on the connection between gut microbiome and pulmonary complications, an observational study found that HCT patients that had low baseline gut microbiome diversity or proteobacteria domination early post-HCT had the highest incidence of pulmonary complications (114). This highlights the potential for a gut-lung axis when considering regulation of pulmonary immunity and such a concept has previously been suggested (115–118). More recently, the concept of alteration of the lung microbiome in the setting of HCT has demonstrated that dysbiosis is associated with alterations in inflammatory cytokines and poor outcomes (102, 119). Interestingly, in the murine studies, it was found that while the HCT procedure alone altered the microbiota, there was a more profound and prolonged alteration in the setting of herpesviral infection as well (102). Similarly, HCT patients with confirmed transcriptionally active pathogens in the bronchoalveolar lavage fluid display overall lower microbiome diversity in the lung, and many of these patients had viral infections (120). Whether the alterations caused by disturbances of the lung or gut microbiota are related to altered signaling via pathogen recognition receptors on immune cells or altered metabolites secreted by the microbiota or both remains to be determined. Interestingly, such findings may offer new diagnostic approaches. Zinter et al. recently performed metatranscriptomic analysis of pre-HCT bronchoalveolar lavage fluid in pediatric patients and found that children with evidence of viral enrichment and innate immune activation had the highest incidence of post-HCT lung injury while patients with diverse oropharyngeal taxa and lacking inflammatory signatures rarely developed post-HCT lung injury (119). While there are known differences in the composition of human and murine microbiota, murine models should still be useful for proof-of-concept studies regarding the role of potential prebiotics, fecal microbiome transplant and other potential therapies to improve outcomes post-HCT.
Our understanding of the host-pathogen interaction in HCT recipients has been accumulated over decades from studying animal models. Mice and humans have fairly similar organs and systems, immunity and pathology. Mouse models permit tightly controlled experimental conditions and unified genetics of host and pathogens. One of the most important advantages of mice is the availability of a huge collection of gene knockout or transgenic mouse strains, and it is now relatively easy to generate such mice if they are not readily available. In practical aspects, the cost of mice is inexpensive and experiments are reproducible. However, there are a few important limitations that need to be kept in consideration. First, the immune system in mice is considerably different from that in humans (121), and thus the knowledge acquired from mice may serve as “proof-of-concept”, but may not readily be translated to human clinical treatment. Second, mice are small and have short live spans. The small sizes of the body and lungs of mice may contribute to the different kinetics of immune reconstitution post-HCT and disease course in the lung compared with humans, as mouse lungs can be quickly overwhelmed by pathogens or immune cell infiltration. While there have been mouse models of BOS in mice that have provided important insights (122–128), it is not clear that the evolution of this disease in mice which have short lifespans fully recapitulate the features of disease that evolve over years in humans. Similarly, long term effects of chronic latency and reactivation of herpesviruses may be difficult to capture. Third, the host-pathogen interactions, especially with viral infections, is usually species-specific, which reduces clinical translatability. Other limitations of mouse models include a lack of parallel methodologies with the ones commonly used in clinical settings. Many of the studies have used syn-HCT models to avoid the complications of alloimmune responses, yet clinically, allo-HCT patients often have the most severe pulmonary complications and many of the current mouse models do not sufficiently explore the effects of GVHD or immunosuppressive therapy and how those factors impact immune function. Additionally, for convenience, mouse models often use TBI as the conditioning regimen, yet human HSCT is often accomplished with chemotherapy, reduced intensity irradiation or combinations. Despite these limitations however, the power of mouse genetics and the ability to genetically modify gene expression in a cell-type specific manner makes these murine models important tools that enable the dissection of fundamental mechanisms which underlie disease. Given that we do not have good anti-microbial strategies for many of the common pathogens that plague patients post-HCT, it is important to better understand how we can quickly repopulate the immune cells of the host and how we can manipulate the HCT regimens to improve the functionality of these immune cells. The power of HCT to cure inherited genetic diseases and malignancies will never be fully realized until the infectious complications, particularly in the lung can be better managed.
Author Contributions
XZ and BM reviewed the literature, drafted the manuscript, created figures and edited the final document. All authors contributed to the article and approved the submitted version.
Funding
Supported by R35HL144481 and DK124290.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Granot N, Storb R. History of Hematopoietic Cell Transplantation: Challenges and Progress. Haematologica (2020) 105(12):2716–29. doi: 10.3324/haematol.2019.245688
2. Passweg JR, Baldomero H, Chabannon C, Basak GW, Corbacioglu S, Duarte R, et al. The EBMT Activity Survey on Hematopoietic-Cell Transplantation and Cellular Therapy 2018: CAR-T's Come Into Focus. Bone Marrow Transplant (2020) 55(8):1604–13. doi: 10.1038/s41409-020-0826-4
3. D'Souza A, Fretham C, Lee SJ, Arora M, Brunner J, Chhabra S, et al. Current Use of and Trends in Hematopoietic Cell Transplantation in the United States. Biol Blood marrow Transplant J Am Soc Blood Marrow Transplant (2020) 26(8):e177–82. doi: 10.1016/j.bbmt.2020.04.013
4. Diab KJ, Yu Z, Wood KL, Shmalo JA, Sheski FD, Farber MO, et al. Comparison of Pulmonary Complications After Nonmyeloablative and Conventional Allogeneic Hematopoietic Cell Transplant. Biol Blood marrow Transplant J Am Soc Blood Marrow Transplant (2012) 18(12):1827–34. doi: 10.1016/j.bbmt.2012.06.013
5. Afessa B, Abdulai RM, Kremers WK, Hogan WJ, Litzow MR, Peters SG. Risk Factors and Outcome of Pulmonary Complications After Autologous Hematopoietic Stem Cell Transplant. Chest (2012) 141(2):442–50. doi: 10.1378/chest.10-2889
6. Tomblyn M, Chiller T, Einsele H, Gress R, Sepkowitz K, Storek J, et al. Guidelines for Preventing Infectious Complications Among Hematopoietic Cell Transplant Recipients: A Global Perspective. Preface Bone Marrow Transplant (2009) 44(8):453–5. doi: 10.1038/bmt.2009.254
7. Panoskaltsis-Mortari A, Griese M, Madtes DK, Belperio JA, Haddad IY, Folz RJ, et al. An Official American Thoracic Society Research Statement: Noninfectious Lung Injury After Hematopoietic Stem Cell Transplantation: Idiopathic Pneumonia Syndrome. Am J Respir Crit Care Med (2011) 183(9):1262–79. doi: 10.1164/rccm.2007-413ST
8. Tamburro RF, Cooke KR, Davies SM, Goldfarb S, Hagood JS, Srinivasan A, et al. Pulmonary Complications of Pediatric Hematopoietic Cell Transplantation. A National Institutes of Health Workshop Summary. Ann Am Thorac Soc (2021) 18(3):381–94. doi: 10.1513/AnnalsATS.202001-006OT
9. Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, Fowler DH, et al. The Biology of Chronic Graft-Versus-Host Disease: A Task Force Report From the National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-Versus-Host Disease. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant (2017) 23(2):211–34. doi: 10.1016/j.bbmt.2016.09.023
10. Carreras E, Cooke KR. Noninfectious Pulmonary Complications. In: Carreras E, Dufour C, Mohty M, Kröger N, editors. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. Cham (CH: Springer Copyright 2019, EBMT and the Author(s (2019). p. 393–401.
11. Rowan C, Baloglu O, McArthur J. Non-Infectious Pulmonary Complications of Hematopoietic Stem Cell Transplantation. J Pediatr Intensive Care (2014) 3(3):133–46. doi: 10.3233/PIC-14095
12. Boieri M, Shah P, Dressel R, Inngjerdingen M. The Role of Animal Models in the Study of Hematopoietic Stem Cell Transplantation and GvHD: A Historical Overview. Front Immunol (2016) 7:333. doi: 10.3389/fimmu.2016.00333
13. Graves SS, Parker MH, Storb R. Animal Models for Preclinical Development of Allogeneic Hematopoietic Cell Transplantation. ILAR J (2018) 59(3):263–75. doi: 10.1093/ilar/ily006
14. Wingard JR, Hsu J, Hiemenz JW. Hematopoietic Stem Cell Transplantation: An Overview of Infection Risks and Epidemiology. Infect Dis Clin North Am (2010) 24(2):257–72. doi: 10.1016/j.idc.2010.01.010
15. Lossos IS, Breuer R, Or R, Strauss N, Elishoov H, Naparstek E, et al. Bacterial Pneumonia in Recipients of Bone Marrow Transplantation. A Five-Year Prospective Study. Transplantation (1995) 60(7):672–8. doi: 10.1097/00007890-199510150-00010
16. Olearo F, Kronig I, Masouridi-Levrat S, Chalandon Y, Khanna N, Passweg J, et al. Optimal Treatment Duration of Pseudomonas Aeruginosa Infections in Allogeneic Hematopoietic Cell Transplant Recipients. Open Forum Infect Dis (2020) 7(7):ofaa246. doi: 10.1093/ofid/ofaa246
17. Cesaro S, Tridello G, Blijlevens N, Ljungman P, Craddock C, Michallet M, et al. Incidence, Risk Factors, and Long-Term Outcome of Acute Leukemia Patients With Early Candidemia After Allogeneic Stem Cell Transplantation: A Study by the Acute Leukemia and Infectious Diseases Working Parties of European Society for Blood and Marrow Transplantation. Clin Infect Dis (2018) 67(4):564–72. doi: 10.1093/cid/ciy150
18. Wald A, Leisenring W, van Burik JA, Bowden RA. Epidemiology of Aspergillus Infections in a Large Cohort of Patients Undergoing Bone Marrow Transplantation. J Infect Dis (1997) 175(6):1459–66. doi: 10.1086/516480
19. Storek J, Zhao Z, Lin E, Berger T, McSweeney PA, Nash RA, et al. Recovery From and Consequences of Severe Iatrogenic Lymphopenia (Induced to Treat Autoimmune Diseases). Clin Immunol (2004) 113(3):285–98. doi: 10.1016/j.clim.2004.07.006
20. Reid GE, Lynch JP 3rd, Weigt S, Sayah D, Belperio JA, Grim SA, et al. Herpesvirus Respiratory Infections in Immunocompromised Patients: Epidemiology, Management, and Outcomes. Semin Respir Crit Care Med (2016) 37(4):603–30. doi: 10.1055/s-0036-1584793
21. Zhou X, O'Dwyer DN, Xia M, Miller HK, Chan PR, Trulik K, et al. First-Onset Herpesviral Infection and Lung Injury in Allogeneic Hematopoietic Cell Transplantation. Am J Respir Crit Care Med (2019) 200(1):63–74. doi: 10.1164/rccm.201809-1635OC
22. Versluys AB, Rossen JW, van Ewijk B, Schuurman R, Bierings MB, Boelens JJ. Strong Association Between Respiratory Viral Infection Early After Hematopoietic Stem Cell Transplantation and the Development of Life-Threatening Acute and Chronic Alloimmune Lung Syndromes. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant (2010) 16(6):782–91. doi: 10.1016/j.bbmt.2009.12.534
23. Park BG, Park CJ, Jang S, Chi HS, Kim DY, Lee JH, et al. Reconstitution of Lymphocyte Subpopulations After Hematopoietic Stem Cell Transplantation: Comparison of Hematologic Malignancies and Donor Types in Event-Free Patients. Leuk Res (2015) 39(12):1334–41. doi: 10.1016/j.leukres.2015.09.010
24. Boeckh M, Nichols WG, Papanicolaou G, Rubin R, Wingard JR, Zaia J. Cytomegalovirus in Hematopoietic Stem Cell Transplant Recipients: Current Status, Known Challenges, and Future Strategies. Biol Blood marrow Transplant J Am Soc Blood Marrow Transplant (2003) 9(9):543–58. doi: 10.1016/S1083-8791(03)00287-8
25. Green ML, Leisenring W, Xie H, Mast TC, Cui Y, Sandmaier BM, et al. Cytomegalovirus Viral Load and Mortality After Haemopoietic Stem Cell Transplantation in the Era of Pre-Emptive Therapy: A Retrospective Cohort Study. Lancet Haematol (2016) 3(3):e119–27. doi: 10.1016/S2352-3026(15)00289-6
26. Erard V, Guthrie KA, Seo S, Smith J, Huang M, Chien J, et al. Reduced Mortality of Cytomegalovirus Pneumonia After Hematopoietic Cell Transplantation Due to Antiviral Therapy and Changes in Transplantation Practices. Clin Infect Dis (2015) 61(1):31–9. doi: 10.1093/cid/civ215
27. Einsele H, Ljungman P, Boeckh M. How I Treat CMV Reactivation After Allogeneic Hematopoietic Stem Cell Transplantation. Blood (2020) 135(19):1619–29. doi: 10.1182/blood.2019000956
28. Ljungman P, de la Camara R, Robin C, Crocchiolo R, Einsele H, Hill JA, et al. Guidelines for the Management of Cytomegalovirus Infection in Patients With Haematological Malignancies and After Stem Cell Transplantation From the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect Dis (2019) 19(8):e260–e72. doi: 10.1016/S1473-3099(19)30107-0
29. Diab M, ZazaDitYafawi J, Soubani AO. Major Pulmonary Complications After Hematopoietic Stem Cell Transplant. Exp Clin Transplant (2016) 14(3):259–70. doi: 10.6002/ect.2015.0275
30. Duran-Struuck R, Hartigan A, Clouthier SG, Dyson MC, Lowler K, Gatza E, et al. Differential Susceptibility of C57BL/6NCr and B6.Cg-Ptprca Mice to Commensal Bacteria After Whole Body Irradiation in Translational Bone Marrow Transplant Studies. J Transl Med (2008) 6:10. doi: 10.1186/1479-5876-6-10
31. Cheretakis C, Leung R, Sun CX, Dror Y, Glogauer M. Timing of Neutrophil Tissue Repopulation Predicts Restoration of Innate Immune Protection in a Murine Bone Marrow Transplantation Model. Blood (2006) 108(8):2821–6. doi: 10.1182/blood-2006-04-018184
32. Kobayashi H, Kobayashi O, Kawai S. Pathogenesis and Clinical Manifestations of Chronic Colonization by Pseudomonas Aeruginosa and its Biofilms in the Airway Tract. J Infect Chemother (2009) 15(3):125–42. doi: 10.1007/s10156-008-0691-3
33. Averbuch D, Tridello G, Hoek J, Mikulska M, Akan H, Yanez San Segundo L, et al. Antimicrobial Resistance in Gram-Negative Rods Causing Bacteremia in Hematopoietic Stem Cell Transplant Recipients: Intercontinental Prospective Study of the Infectious Diseases Working Party of the European Bone Marrow Transplantation Group. Clin Infect Dis (2017) 65(11):1819–28. doi: 10.1093/cid/cix646
34. Trecarichi EM, Tumbarello M, Caira M, Candoni A, Cattaneo C, Pastore D, et al. Multidrug Resistant Pseudomonas Aeruginosa Bloodstream Infection in Adult Patients With Hematologic Malignancies. Haematologica (2011) 96(1):e1–3; author reply e4. doi: 10.3324/haematol.2010.036640
35. Mudau M, Jacobson R, Minenza N, Kuonza L, Morris V, Engelbrecht H, et al. Outbreak of Multi-Drug Resistant Pseudomonas Aeruginosa Bloodstream Infection in the Haematology Unit of a South African Academic Hospital. PloS One (2013) 8(3):e55985. doi: 10.1371/journal.pone.0055985
36. Ojielo CI, Cooke K, Mancuso P, Standiford TJ, Olkiewicz KM, Clouthier S, et al. Defective Phagocytosis and Clearance of Pseudomonas Aeruginosa in the Lung Following Bone Marrow Transplantation. J Immunol (2003) 171(8):4416–24. doi: 10.4049/jimmunol.171.8.4416
37. Hakki M, Limaye AP, Kim HW, Kirby KA, Corey L, Boeckh M. Invasive Pseudomonas Aeruginosa Infections: High Rate of Recurrence and Mortality After Hematopoietic Cell Transplantation. Bone Marrow Transplant (2007) 39(11):687–93. doi: 10.1038/sj.bmt.1705653
38. Hubbard LL, Ballinger MN, Wilke CA, Moore BB. Comparison of Conditioning Regimens for Alveolar Macrophage Reconstitution and Innate Immune Function Post Bone Marrow Transplant. Exp Lung Res (2008) 34(5):263–75. doi: 10.1080/01902140802022518
39. Ballinger MN, Aronoff DM, McMillan TR, Cooke KR, Olkiewicz K, Toews GB, et al. Critical Role of Prostaglandin E2 Overproduction in Impaired Pulmonary Host Response Following Bone Marrow Transplantation. J Immunol (2006) 177(8):5499–508. doi: 10.4049/jimmunol.177.8.5499
40. Domingo-Gonzalez R, Huang SK, Laouar Y, Wilke CA, Moore BB. COX-2 Expression is Upregulated by DNA Hypomethylation After Hematopoietic Stem Cell Transplantation. J Immunol (2012) 189(9):4528–36. doi: 10.4049/jimmunol.1201116
41. Domingo-Gonzalez R, Martinez-Colon GJ, Smith AJ, Smith CK, Ballinger MN, Xia M, et al. Inhibition of Neutrophil Extracellular Trap Formation After Stem Cell Transplant by Prostaglandin E2. Am J Respir Crit Care Med (2016) 193(2):186–97. doi: 10.1164/rccm.201501-0161OC
42. Cayeux SJ, Beverley PC, Schulz R, Dorken B. Elevated Plasma Prostaglandin E2 Levels Found in 14 Patients Undergoing Autologous Bone Marrow or Stem Cell Transplantation. Bone Marrow Transplant (1993) 12(6):603–8.
43. Ballinger MN, McMillan TR, Moore BB. Eicosanoid Regulation of Pulmonary Innate Immunity Post-Hematopoietic Stem Cell Transplantation. Arch Immunol Ther Exp (Warsz) (2007) 55(1):1–12. doi: 10.1007/s00005-007-0001-2
44. Domingo-Gonzalez R, Wilke CA, Huang SK, Laouar Y, Brown JP, Freeman CM, et al. Transforming Growth Factor-Beta Induces microRNA-29b to Promote Murine Alveolar Macrophage Dysfunction After Bone Marrow Transplantation. Am J Physiol Lung Cell Mol Physiol (2015) 308(1):L86–95. doi: 10.1152/ajplung.00283.2014
45. Domingo-Gonzalez R, Katz S, Serezani CH, Moore TA, Levine AM, Moore BB. Prostaglandin E2-Induced Changes in Alveolar Macrophage Scavenger Receptor Profiles Differentially Alter Phagocytosis of Pseudomonas Aeruginosa and Staphylococcus Aureus Post-Bone Marrow Transplant. J Immunol (2013) 190(11):5809–17. doi: 10.4049/jimmunol.1203274
46. Hubbard LL, Ballinger MN, Thomas PE, Wilke CA, Standiford TJ, Kobayashi KS, et al. A Role for IL-1 Receptor-Associated Kinase-M in Prostaglandin E2-Induced Immunosuppression Post-Bone Marrow Transplantation. J Immunol (2010) 184(11):6299–308. doi: 10.4049/jimmunol.0902828
47. Martinez-Colon GJ, Taylor QM, Wilke CA, Podsiad AB, Moore BB. Elevated Prostaglandin E2 Post-Bone Marrow Transplant Mediates Interleukin-1beta-Related Lung Injury. Mucosal Immunol (2018) 11(2):319–32. doi: 10.1038/mi.2017.51
48. Hubbard LL, Wilke CA, White ES, Moore BB. PTEN Limits Alveolar Macrophage Function Against Pseudomonas Aeruginosa After Bone Marrow Transplantation. Am J Respir Cell Mol Biol (2011) 45(5):1050–8. doi: 10.1165/rcmb.2011-0079OC
49. Zimecki M, Artym J, Kocieba M, Weber-Dabrowska B, Borysowski J, Górski A. Prophylactic Effect of Bacteriophages on Mice Subjected to Chemotherapy-Induced Immunosuppression and Bone Marrow Transplant Upon Infection With Staphylococcus Aureus. Med Microbiol Immunol (2010) 199(2):71–9. doi: 10.1007/s00430-009-0135-4
50. Roesler J, Gröttrup E, Baccarini M, Lohmann-Mattes ML. Efficient Natural Defense Mechanisms Against Listeria Monocytogenes in T and B Cell-Deficient Allogeneic Bone Marrow Radiation Chimeras. Preactivated Macrophages are the Main Effector Cells in an Early Phase After Bone Marrow Transfer. J Immunol (1989) 143(5):1710–5.
51. Chen CS, Boeckh M, Seidel K, Clark JG, Kansu E, Madtes DK, et al. Incidence, Risk Factors, and Mortality From Pneumonia Developing Late After Hematopoietic Stem Cell Transplantation. Bone Marrow Transplant (2003) 32(5):515–22. doi: 10.1038/sj.bmt.1704162
52. Kontoyiannis DP, Marr KA, Park BJ, Alexander BD, Anaissie EJ, Walsh TJ, et al. Prospective Surveillance for Invasive Fungal Infections in Hematopoietic Stem Cell Transplant Recipients, 2001-2006: Overview of the Transplant-Associated Infection Surveillance Network (TRANSNET) Database. Clin Infect Dis (2010) 50(8):1091–100. doi: 10.1086/651263
53. Karthaus M. Prophylaxis and Treatment of Invasive Aspergillosis With Voriconazole, Posaconazole and Caspofungin: Review of the Literature. Eur J Med Res (2011) 16(4):145–52. doi: 10.1186/2047-783X-16-4-145
54. McNeil MM, Nash SL, Hajjeh RA, Phelan MA, Conn LA, Plikaytis BD, et al. Trends in Mortality Due to Invasive Mycotic Diseases in the United States, 1980-1997. Clin Infect Dis (2001) 33(5):641–7. doi: 10.1086/322606
55. Segal BH, Walsh TJ. Current Approaches to Diagnosis and Treatment of Invasive Aspergillosis. Am J Respir Crit Care Med (2006) 173(7):707–17. doi: 10.1164/rccm.200505-727SO
56. Hildebrandt GC, Olkiewicz K, Hogaboam CM, Ferrara JL, Cooke KR. Increased Clearance Ofaspergillus Fumigatus in Mice With Chronic Lung Injury After Allogeneic Stem Cell Transplantation. Biol Blood Marrow Transplant (2006) 12(2):6. doi: 10.1016/j.bbmt.2005.11.024
57. Lortholary O, Gangneux JP, Sitbon K, Lebeau B, de Monbrison F, Le Strat Y, et al. Epidemiological Trends in Invasive Aspergillosis in France: The SAIF Network (2005-2007). Clin Microbiol Infect (2011) 17(12):1882–9. doi: 10.1111/j.1469-0691.2011.03548.x
58. Dagenais TR, Keller NP. Pathogenesis of Aspergillus Fumigatus in Invasive Aspergillosis. Clin Microbiol Rev (2009) 22(3):447–65. doi: 10.1128/CMR.00055-08
60. Sheppard DC, Rieg G, Chiang LY, Filler SG, Edwards JE Jr., Ibrahim AS. Novel Inhalational Murine Model of Invasive Pulmonary Aspergillosis. Antimicrob Agents Chemother (2004) 48(5):1908–11. doi: 10.1128/AAC.48.5.1908-1911.2004
61. Cunha C, Di Ianni M, Bozza S, Giovannini G, Zagarella S, Zelante T, et al. Dectin-1 Y238X Polymorphism Associates With Susceptibility to Invasive Aspergillosis in Hematopoietic Transplantation Through Impairment of Both Recipient- and Donor-Dependent Mechanisms of Antifungal Immunity. Blood (2010) 116(24):5394–402. doi: 10.1182/blood-2010-04-279307
62. Arber C, Bitmansour A, Shashidhar S, Wang S, Tseng B, Brown JM. Protection Against Lethal Aspergillus Fumigatus Infection in Mice by Allogeneic Myeloid Progenitors is Not Major Histocompatibility Complex Restricted. J Infect Dis (2005) 192(9):1666–71. doi: 10.1086/491743
63. Cenci E, Mencacci A, Spreca A, Montagnoli C, Bacci A, Perruccio K, et al. Protection of Killer Antiidiotypic Antibodies Against Early Invasive Aspergillosis in a Murine Model of Allogeneic T-Cell-Depleted Bone Marrow Transplantation. Infect Immun (2002) 70(5):2375–82. doi: 10.1128/IAI.70.5.2375-2382.2002
64. Kalleda N, Amich J, Arslan B, Poreddy S, Mattenheimer K, Mokhtari Z, et al. Dynamic Immune Cell Recruitment After Murine Pulmonary Aspergillus Fumigatus Infection Under Different Immunosuppressive Regimens. Front Microbiol (2016) 7:1107. doi: 10.3389/fmicb.2016.01107
65. Balloy V, Huerre M, Latge JP, Chignard M. Differences in Patterns of Infection and Inflammation for Corticosteroid Treatment and Chemotherapy in Experimental Invasive Pulmonary Aspergillosis. Infect Immun (2005) 73(1):494–503. doi: 10.1128/IAI.73.1.494-503.2005
66. Markaryan A, Morozova I, Yu H, Kolattukudy PE. Purification and Characterization of an Elastinolytic Metalloprotease From Aspergillus Fumigatus and Immunoelectron Microscopic Evidence of Secretion of This Enzyme by the Fungus Invading the Murine Lung. Infect Immun (1994) 62(6):2149–57. doi: 10.1128/iai.62.6.2149-2157.1994
67. Al-Bader N, Sheppard DC. Aspergillosis and Stem Cell Transplantation: An Overview of Experimental Pathogenesis Studies. Virulence (2016) 7(8):950–66. doi: 10.1080/21505594.2016.1231278
68. Zinter MS, Hume JR. Effects of Hematopoietic Cell Transplantation on the Pulmonary Immune Response to Infection. Front Pediatr (2021) 9:634566. doi: 10.3389/fped.2021.634566
69. Balloy V, Si-Tahar M, Takeuchi O, Philippe B, Nahori MA, Tanguy M, et al. Involvement of Toll-Like Receptor 2 in Experimental Invasive Pulmonary Aspergillosis. Infect Immun (2005) 73(9):5420–5. doi: 10.1128/IAI.73.9.5420-5425.2005
70. Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, et al. The Contribution of the Toll-Like/IL-1 Receptor Superfamily to Innate and Adaptive Immunity to Fungal Pathogens In Vivo. J Immunol (2004) 172(5):3059–69. doi: 10.4049/jimmunol.172.5.3059
71. Kasperkovitz PV, Cardenas ML, Vyas JM. TLR9 is Actively Recruited to Aspergillus Fumigatus Phagosomes and Requires the N-Terminal Proteolytic Cleavage Domain for Proper Intracellular Trafficking. J Immunol (2010) 185(12):7614–22. doi: 10.4049/jimmunol.1002760
72. Khan NS, Kasperkovitz PV, Timmons AK, Mansour MK, Tam JM, Seward MW, et al. Dectin-1 Controls TLR9 Trafficking to Phagosomes Containing Beta-1,3 Glucan. J Immunol (2016) 196(5):2249–61. doi: 10.4049/jimmunol.1401545
73. Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-Dependent Activation of a TLR9-BTK-Calcineurin-NFAT Pathway Co-Ordinates Innate Immunity to Aspergillus Fumigatus. EMBO Mol Med (2015) 7(3):240–58. doi: 10.15252/emmm.201404556
74. Gresnigt MS, Cunha C, Jaeger M, Goncalves SM, Malireddi RKS, Ammerdorffer A, et al. Genetic Deficiency of NOD2 Confers Resistance to Invasive Aspergillosis. Nat Commun (2018) 9(1):2636. doi: 10.1038/s41467-018-04912-3
75. Bottazzi B, Garlanda C, Cotena A, Moalli F, Jaillon S, Deban L, et al. The Long Pentraxin PTX3 as a Prototypic Humoral Pattern Recognition Receptor: Interplay With Cellular Innate Immunity. Immunol Rev (2009) 227(1):9–18. doi: 10.1111/j.1600-065X.2008.00719.x
76. Bernal-Martinez L, Goncalves SM, de Andres B, Cunha C, Gonzalez Jimenez I, Lagrou K, et al. TREM1 Regulates Antifungal Immune Responses in Invasive Pulmonary Aspergillosis. Virulence (2021) 12(1):570–83. doi: 10.1080/21505594.2021.1879471
77. Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, Lamkanfi M, et al. Concerted Activation of the AIM2 and NLRP3 Inflammasomes Orchestrates Host Protection Against Aspergillus Infection. Cell Host Microbe (2015) 17(3):357–68. doi: 10.1016/j.chom.2015.01.006
78. Said-Sadier N, Padilla E, Langsley G, Ojcius DM. Aspergillus Fumigatus Stimulates the NLRP3 Inflammasome Through a Pathway Requiring ROS Production and the Syk Tyrosine Kinase. PloS One (2010) 5(4):e10008. doi: 10.1371/journal.pone.0010008
79. Bozza S, Perruccio K, Montagnoli C, Gaziano R, Bellocchio S, Burchielli E, et al. A Dendritic Cell Vaccine Against Invasive Aspergillosis in Allogeneic Hematopoietic Transplantation. Blood (2003) 102(10):3807–14. doi: 10.1182/blood-2003-03-0748
80. de Luca A, Bozza S, Zelante T, Zagarella S, D'Angelo C, Perruccio K, et al. Non-Hematopoietic Cells Contribute to Protective Tolerance to Aspergillus Fumigatus via a TRIF Pathway Converging on IDO. Cell Mol Immunol (2010) 7(6):459–70. doi: 10.1038/cmi.2010.43
81. Bozza S, Gaziano R, Lipford GB, Montagnoli C, Bacci A, Di Francesco P, et al. Vaccination of Mice Against Invasive Aspergillosis With Recombinant Aspergillus Proteins and CpG Oligodeoxynucleotides as Adjuvants. Microbes Infect (2002) 4(13):1281–90. doi: 10.1016/S1286-4579(02)00007-2
82. Cenci E, Mencacci A, Bacci A, Bistoni F, Kurup VP, Romani L. T Cell Vaccination in Mice With Invasive Pulmonary Aspergillosis. J Immunol (2000) 165(1):381–8. doi: 10.4049/jimmunol.165.1.381
83. Hartigan AJ, Westwick J, Jarai G, Hogaboam CM. CCR7 Deficiency on Dendritic Cells Enhances Fungal Clearance in a Murine Model of Pulmonary Invasive Aspergillosis. J Immunol (2009) 183(8):5171–9. doi: 10.4049/jimmunol.0901027
84. Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM, Schwiebert LM, et al. Requisite Role for the Dectin-1 Beta-Glucan Receptor in Pulmonary Defense Against Aspergillus Fumigatus. J Immunol (2009) 182(8):4938–46. doi: 10.4049/jimmunol.0804250
85. Urb M, Snarr BD, Wojewodka G, Lehoux M, Lee MJ, Ralph B, et al. Evolution of the Immune Response to Chronic Airway Colonization With Aspergillus Fumigatus Hyphae. Infect Immun (2015) 83(9):3590–600. doi: 10.1128/IAI.00359-15
86. Cenci E, Mencacci A, Del Sero G, Bacci A, Montagnoli C, d'Ostiani CF, et al. Interleukin-4 Causes Susceptibility to Invasive Pulmonary Aspergillosis Through Suppression of Protective Type I Responses. J Infect Dis (1999) 180(6):1957–68. doi: 10.1086/315142
87. Clemons KV, Grunig G, Sobel RA, Mirels LF, Rennick DM, Stevens DA. Role of IL-10 in Invasive Aspergillosis: Increased Resistance of IL-10 Gene Knockout Mice to Lethal Systemic Aspergillosis. Clin Exp Immunol (2000) 122(2):186–91. doi: 10.1046/j.1365-2249.2000.01382.x
88. Del Sero G, Mencacci A, Cenci E, d'Ostiani CF, Montagnoli C, Bacci A, et al. Antifungal Type 1 Responses Are Upregulated in IL-10-Deficient Mice. Microbes Infect (1999) 1(14):1169–80. doi: 10.1016/S1286-4579(99)00245-2
89. Nichols WG, Corey L, Gooley T, Davis C, Boeckh M. High Risk of Death Due to Bacterial and Fungal Infection Among Cytomegalovirus (CMV)-Seronegative Recipients of Stem Cell Transplants From Seropositive Donors: Evidence for Indirect Effects of Primary CMV Infection. J Infect Dis (2002) 185(3):273–82. doi: 10.1086/338624
90. Seo S, Renaud C, Kuypers JM, Chiu CY, Huang ML, Samayoa E, et al. Idiopathic Pneumonia Syndrome After Hematopoietic Cell Transplantation: Evidence of Occult Infectious Etiologies. Blood (2015) 125(24):3789–97. doi: 10.1182/blood-2014-12-617035
91. Adler H, Beland JL, Kozlow W, Del-Pan NC, Kobzik L, Rimm IJ. A Role for Transforming Growth Factor-Beta1 in the Increased Pneumonitis in Murine Allogeneic Bone Marrow Transplant Recipients With Graft-Versus-Host Disease After Pulmonary Herpes Simplex Virus Type 1 Infection. Blood (1998) 92(7):2581–9. doi: 10.1182/blood.V92.7.2581
92. Reynaud JM, Jegou JF, Welsch JC, Horvat B. Human Herpesvirus 6A Infection in CD46 Transgenic Mice: Viral Persistence in the Brain and Increased Production of Proinflammatory Chemokines via Toll-Like Receptor 9. J Virol (2014) 88(10):5421–36. doi: 10.1128/JVI.03763-13
93. Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB, et al. Granulocyte-Colony Stimulating Factor Reactivates Human Cytomegalovirus in a Latently Infected Humanized Mouse Model. Cell Host Microbe (2010) 8(3):284–91. doi: 10.1016/j.chom.2010.08.001
94. Martins JP, Andoniou CE, Fleming P, Kuns RD, Schuster IS, Voigt V, et al. Strain-Specific Antibody Therapy Prevents Cytomegalovirus Reactivation After Transplantation. Science (2019) 363(6424):288–93. doi: 10.1126/science.aat0066
95. Gosselin J, Borgeat P, Flamand L. Leukotriene B4 Protects Latently Infected Mice Against Murine Cytomegalovirus Reactivation Following Allogeneic Transplantation. J Immunol (2005) 174(3):1587–93. doi: 10.4049/jimmunol.174.3.1587
96. Coomes SM, Wilke CA, Moore TA, Moore BB. Induction of TGF-Beta 1, Not Regulatory T Cells, Impairs Antiviral Immunity in the Lung Following Bone Marrow Transplant. J Immunol (2010) 184(9):5130–40. doi: 10.4049/jimmunol.0901871
97. Gurczynski SJ, Pereira NL, Hrycaj SM, Wilke C, Zemans RL, Moore BB. Stem Cell Transplantation Uncovers TDO-AHR Regulation of Lung Dendritic Cells in Herpesvirus-Induced Pathology. JCI Insight (2021) 6(2):e139965. doi: 10.1172/jci.insight.139965
98. Holtappels R, Podlech J, Geginat G, Steffens HP, Thomas D, Reddehase MJ. Control of Murine Cytomegalovirus in the Lungs: Relative But Not Absolute Immunodominance of the Immediate-Early 1 Nonapeptide During the Antiviral Cytolytic T-Lymphocyte Response in Pulmonary Infiltrates. J Virol (1998) 72(9):7201–12. doi: 10.1128/JVI.72.9.7201-7212.1998
99. Podlech J, Holtappels R, Pahl-Seibert MF, Steffens HP, Reddehase MJ. Murine Model of Interstitial Cytomegalovirus Pneumonia in Syngeneic Bone Marrow Transplantation: Persistence of Protective Pulmonary CD8-T-Cell Infiltrates After Clearance of Acute Infection. J Virol (2000) 74(16):7496–507. doi: 10.1128/JVI.74.16.7496-7507.2000
100. Gowdy KM, Martinu T, Nugent JL, Manzo ND, Zhang HL, Kelly FL, et al. Impaired CD8(+) T Cell Immunity After Allogeneic Bone Marrow Transplantation Leads to Persistent and Severe Respiratory Viral Infection. Transpl Immunol (2015) 32(1):51–60. doi: 10.1016/j.trim.2014.10.005
101. McCarthy MK, Procario MC, Wilke CA, Moore BB, Weinberg JB. Prostaglandin E2 Production and T Cell Function in Mouse Adenovirus Type 1 Infection Following Allogeneic Bone Marrow Transplantation. PloS One (2015) 10(9):e0139235. doi: 10.1371/journal.pone.0139235
102. O'Dwyer DN, Zhou X, Wilke CA, Xia M, Falkowski NR, Norman KC, et al. Lung Dysbiosis, Inflammation, and Injury in Hematopoietic Cell Transplantation. Am J Respir Crit Care Med (2018) 198(10):1312–21. doi: 10.1164/rccm.201712-2456OC
103. Zhou X, Loomis-King H, Gurczynski SJ, Wilke CA, Konopka KE, Ptaschinski C, et al. Bone Marrow Transplantation Alters Lung Antigen-Presenting Cells to Promote TH17 Response and the Development of Pneumonitis and Fibrosis Following Gammaherpesvirus Infection. Mucosal Immunol (2016) 9(3):610–20. doi: 10.1038/mi.2015.85
104. Gurczynski SJ, Zhou X, Flaherty M, Wilke CA, Moore BB. Bone Marrow Transplant-Induced Alterations in Notch Signaling Promote Pathologic Th17 Responses to Gamma-Herpesvirus Infection. Mucosal Immunol (2018) 11(3):881–93. doi: 10.1038/mi.2017.85
105. Wilke CA, Chadwick MM, Chan PR, Moore BB, Zhou X. Stem Cell Transplantation Impairs Dendritic Cell Trafficking and Herpesvirus Immunity. JCI Insight (2019) 4(18):e130210. doi: 10.1172/jci.insight.130210
106. Coomes SM, Farmen S, Wilke CA, Laouar Y, Moore BB. Severe Gammaherpesvirus-Induced Pneumonitis and Fibrosis in Syngeneic Bone Marrow Transplant Mice is Related to Effects of Transforming Growth Factor-Beta. Am J Pathol (2011) 179(5):2382–96. doi: 10.1016/j.ajpath.2011.08.002
107. Ford CD, Gazdik MA, Lopansri BK, Webb B, Mitchell B, Coombs J, et al. Vancomycin-Resistant Enterococcus Colonization and Bacteremia and Hematopoietic Stem Cell Transplantation Outcomes. Biol Blood marrow Transplant J Am Soc Blood Marrow Transplant (2017) 23(2):340–6. doi: 10.1016/j.bbmt.2016.11.017
108. Sadowska-Klasa A, Piekarska A, Prejzner W, Bieniaszewska M, Hellmann A. Colonization With Multidrug-Resistant Bacteria Increases the Risk of Complications and a Fatal Outcome After Allogeneic Hematopoietic Cell Transplantation. Ann Hematol (2018) 97(3):509–17. doi: 10.1007/s00277-017-3205-5
109. Satwani P, Freedman JL, Chaudhury S, Jin Z, Levinson A, Foca MD, et al. A Multicenter Study of Bacterial Blood Stream Infections in Pediatric Allogeneic Hematopoietic Cell Transplantation Recipients: The Role of Acute Gastrointestinal Graft-Versus-Host Disease. Biol Blood marrow Transplant J Am Soc Blood Marrow Transplant (2017) 23(4):642–7. doi: 10.1016/j.bbmt.2017.01.073
110. Winston DJ, Territo MC, Ho WG, Miller MJ, Gale RP, Golde DW. Alveolar Macrophage Dysfunction in Human Bone Marrow Transplant Recipients. Am J Med (1982) 73(6):859–66. doi: 10.1016/0002-9343(82)90777-X
111. Davis FM, Gallagher KA. Epigenetic Mechanisms in Monocytes/Macrophages Regulate Inflammation in Cardiometabolic and Vascular Disease. Arteriosclerosis thrombosis Vasc Biol (2019) 39(4):623–34. doi: 10.1161/ATVBAHA.118.312135
112. Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, et al. Intestinal Domination and the Risk of Bacteremia in Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Clin Infect Dis (2012) 55(7):905–14. doi: 10.1093/cid/cis580
113. Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, et al. The Effects of Intestinal Tract Bacterial Diversity on Mortality Following Allogeneic Hematopoietic Stem Cell Transplantation. Blood (2014) 124(7):1174–82. doi: 10.1182/blood-2014-02-554725
114. Harris B, Morjaria SM, Littmann ER, Geyer AI, Stover DE, Barker JN, et al. Gut Microbiota Predict Pulmonary Infiltrates After Allogeneic Hematopoietic Cell Transplantation. Am J Respir Crit Care Med (2016) 194(4):450–63. doi: 10.1164/rccm.201507-1491OC
115. Marsland BJ, Trompette A, Gollwitzer ES. The Gut-Lung Axis in Respiratory Disease. Ann Am Thorac Soc (2015) 12(Suppl 2):S150–6. doi: 10.1513/AnnalsATS.201503-133AW
116. Samuelson DR, Welsh DA, Shellito JE. Regulation of Lung Immunity and Host Defense by the Intestinal Microbiota. Front Microbiol (2015) 6:1085. doi: 10.3389/fmicb.2015.01085
117. O'Dwyer DN, Dickson RP, Moore BB. The Lung Microbiome, Immunity, and the Pathogenesis of Chronic Lung Disease. J Immunol (2016) 196(12):4839–47. doi: 10.4049/jimmunol.1600279
118. Yanik G, Cooke KR. The Lung as a Target Organ of Graft-Versus-Host Disease. Semin Hematol (2006) 43(1):42–52. doi: 10.1053/j.seminhematol.2005.09.004
119. Zinter MS, Lindemans CA, Versluys BA, Mayday MY, Sunshine S, Reyes G, et al. The Pulmonary Metatranscriptome Prior to Pediatric HCT Identifies Post-HCT Lung Injury. Blood (2021) 137(12):1679–89. doi: 10.1182/blood.2020009246
120. Langelier C, Zinter MS, Kalantar K, Yanik GA, Christenson S, O'Donovan B, et al. Metagenomic Sequencing Detects Respiratory Pathogens in Hematopoietic Cellular Transplant Patients. Am J Respir Crit Care Med (2018) 197(4):524–8. doi: 10.1164/rccm.201706-1097LE
121. Shay T, Jojic V, Zuk O, Rothamel K, Puyraimond-Zemmour D, Feng T, et al. Conservation and Divergence in the Transcriptional Programs of the Human and Mouse Immune Systems. Proc Natl Acad Sci USA (2013) 110(8):2946–51. doi: 10.1073/pnas.1222738110
122. Flynn R, Du J, Veenstra RG, Reichenbach DK, Panoskaltsis-Mortari A, Taylor PA, et al. Increased T Follicular Helper Cells and Germinal Center B Cells Are Required for cGVHD and Bronchiolitis Obliterans. Blood (2014) 123(25):3988–98. doi: 10.1182/blood-2014-03-562231
123. Verghese DA, Chun N, Paz K, Fribourg M, Woodruff TM, Flynn R, et al. C5aR1 Regulates T Follicular Helper Differentiation and Chronic Graft-Versus-Host Disease Bronchiolitis Obliterans. JCI Insight (2018) 3(24):e124646. doi: 10.1172/jci.insight.124646
124. Panoskaltsis-Mortari A, Tram KV, Price AP, Wendt CH, Blazar BR. A New Murine Model for Bronchiolitis Obliterans Post-Bone Marrow Transplant. Am J Respir Crit Care Med (2007) 176(7):713–23. doi: 10.1164/rccm.200702-335OC
125. Srinivasan M, Flynn R, Price A, Ranger A, Browning JL, Taylor PA, et al. Donor B-Cell Alloantibody Deposition and Germinal Center Formation Are Required for the Development of Murine Chronic GVHD and Bronchiolitis Obliterans. Blood (2012) 119(6):1570–80. doi: 10.1182/blood-2011-07-364414
126. Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, et al. Ibrutinib Treatment Ameliorates Murine Chronic Graft-Versus-Host Disease. J Clin Invest (2014) 124(11):4867–76. doi: 10.1172/JCI75328
127. Flynn R, Allen JL, Luznik L, MacDonald KP, Paz K, Alexander KA, et al. Targeting Syk-Activated B Cells in Murine and Human Chronic Graft-Versus-Host Disease. Blood (2015) 125(26):4085–94. doi: 10.1182/blood-2014-08-595470
Keywords: hematopoietic cell transplantation, bone marrow transplantation, infectious pulmonary complications, herpesvirus, Pseudomonas aeruginosa, Aspergillus fumigatus
Citation: Zhou X and Moore BB (2021) Experimental Models of Infectious Pulmonary Complications Following Hematopoietic Cell Transplantation. Front. Immunol. 12:718603. doi: 10.3389/fimmu.2021.718603
Received: 01 June 2021; Accepted: 26 July 2021;
Published: 16 August 2021.
Edited by:
Antiopi Varelias, The University of Queensland, AustraliaReviewed by:
Kenneth Cooke, Johns Hopkins Medicine, United StatesMahzad Akbarpour, University of Chicago Medicine, United States
Copyright © 2021 Zhou and Moore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofeng Zhou, eGlhemhvdUB1bWljaC5lZHU=