 Chenchen Zhao
Chenchen Zhao Yi Zhang
Yi Zhang Hong Zheng
Hong Zheng- 1Penn State Cancer Institute, Penn State University College of Medicine, Hershey, PA, United States
- 2Fels Institute for Cancer Research and Molecular Biology, Temple University, Philadelphia, PA, United States
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a potentially curative therapy for hematological malignancies. This beneficial effect is derived mainly from graft-versus-leukemia (GVL) effects mediated by alloreactive T cells. However, these alloreactive T cells can also induce graft-versus-host disease (GVHD), a life-threatening complication after allo-HSCT. Significant progress has been made in the dissociation of GVL effects from GVHD by modulating alloreactive T cell immunity. However, many factors may influence alloreactive T cell responses in the host undergoing allo-HSCT, including the interaction of alloreactive T cells with both donor and recipient hematopoietic cells and host non-hematopoietic tissues, cytokines, chemokines and inflammatory mediators. Interferons (IFNs), including type I IFNs and IFN-γ, primarily produced by monocytes, dendritic cells and T cells, play essential roles in regulating alloreactive T cell differentiation and function. Many studies have shown pleiotropic effects of IFNs on allogeneic T cell responses during GVH reaction. Epigenetic mechanisms, such as DNA methylation and histone modifications, are important to regulate IFNs’ production and function during GVHD. In this review, we discuss recent findings from preclinical models and clinical studies that characterize T cell responses regulated by IFNs and epigenetic mechanisms, and further discuss pharmacological approaches that modulate epigenetic effects in the setting of allo-HSCT.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) provides the long-term effective and curative treatment for patients with hematological malignancies. The therapeutic benefit of allo-HSCT is primarily attributed to the graft-versus-leukemia (GVL) effect, which is mainly mediated by infused donor T cells (1). However, these allogeneic T cells can also cause harmful graft-versus-host disease (GVHD) (2–4). Acute GVHD is a major risk for non-relapse mortality in the first 200 days after allo-HSCT (5). Therefore, maintaining the beneficial GVL effect while reducing GVHD is the holy grail of allo-HSCT.
Upon stimulation by host antigen-presenting cells (APC), infused donor T cells are activated to undergo robust proliferation and effector differentiation (2, 6). These APCs express high levels of antigen-presenting molecule MHC class II and costimulatory molecules (e.g., CD80, CD86), which are required to activate allogeneic T cells and promote expansion of activated T cells, respectively. Many cytokines, such as IL-2 and IL-12, are important for instructing these activated T cells to differentiate into effector cells mediating host tissue injury (7, 8). Notably, interferons (IFN) have an essential role in regulating T-cell activities during GVHD (9, 10). Type I (mainly IFN-α/β) and type II (IFN-γ) are two major IFNs that mediate pathophysiologic changes during infection, cancer and autoimmune diseases (11–15). IFN-γ is primarily derived from T helper 1 (Th1) CD4+ T cells and cytotoxic CD8+ T cells once adaptive immunity develops, whereas IFN-α can be produced by plasmacytoid dendritic cells (pDCs) (16, 17). Both IFN-γ and IFN-α are pivotal regulators of alloreactive T cell responses that mediate GVHD (18–20). However, optimal control of GVHD by modulating IFN signaling remains challenging. IFN signaling is complex and frequently context-dependent: it can lead to distinct effects at different times or stages of a disease course. IFNs regulate T cell functions by regulating a group of intracellular transcription programs. Epigenetic regulations of molecules in the IFN signaling pathway and the interferon-stimulated genes (ISG) are crucial for T cell activity (21, 22). This review focuses on how IFNs regulate alloreactive T cell responses and what role epigenetic regulation plays in this process.
Effects of IFNs on T Cell Differentiation and Function During GVHD
Type I IFNs
Type I IFNs contain a subgroup of highly related polypeptides that have proven essential in regulating innate and adaptive immunity (23). Approximately 12-14 types of IFN-α and one type of IFN-β, IFN-ϵ, IFN-κ, and IFN-ω have been identified (24). Intriguingly, although type I IFNs are structurally divergent, only one form of heterodimer receptor, IFNAR, has been found. Thus, all type I IFNs activate the same receptor and many subsequent cell-signaling activities are shared. IFN-α and IFN-β are well defined and are the main subtypes from the immunological perspective. Virtually all cell types reserve the ability to produce variable level of IFN-β, whereas pDCs are the main source of IFN-α (23). Host tissue injuries triggered by conditioning regimens, such as preparative irradiation and chemotherapy, induce damage-associated molecular patterns (DAMP) and foreign pathogen-associated molecular patterns (PAMP). Type I IFNs are among the early cytokines whose production is triggered by the host and donor APCs after the detection of these danger signals by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and nucleic acid sensors that located on or within the cytosol of cells (25) (Figure 1). IFN-α/β can exert antiviral and antitumor activity by up-regulating MHC-I and subsequently promoting antigen presentations.
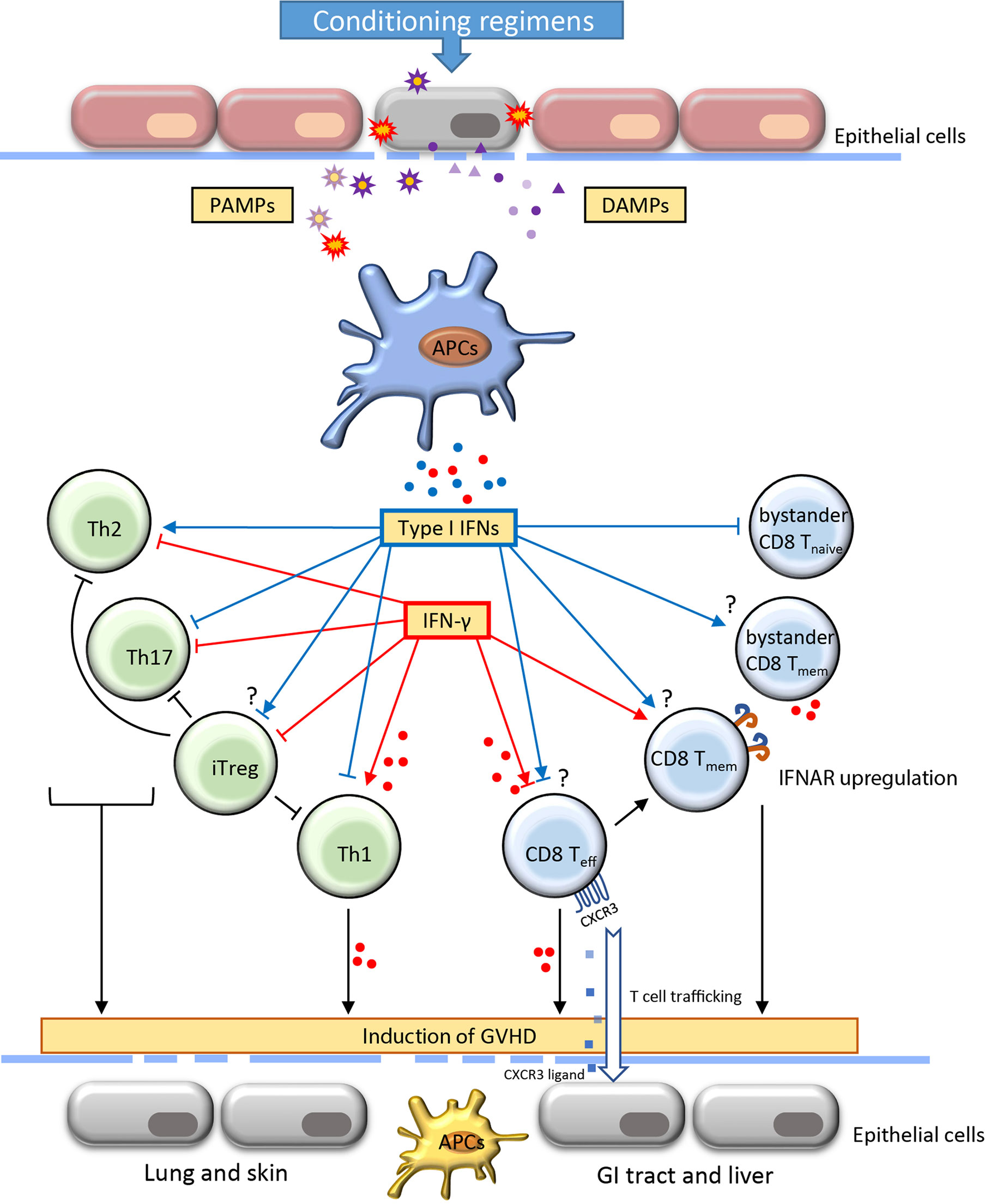
Figure 1 Role of type I IFNs and IFN-γ in the development of acute graft-versus-host-disease. Acute graft-versus-host disease (GVHD) is often initiated with the destruction of the epithelial barrier through the conditioning regimens including irradiation and chemotherapy. The signal of pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP) released from damaged cells and microbiota induce the activation of antigen-presenting cells (APCs). Consequently, the production of IFNs by APCs interact with the alloreactive T cells and regulate their activation, differentiation, function and contraction. The proliferation of Th1 cells and effector CD8 T cells result in increased secretion of IFN-γ. The induction of activated alloreactive T cells and cytokines further affect the resident APCs and host tissues contributing to extensive functional incapability and damages of different organs. The IFNs play a critical role in orchestrating T cell activities throughout the induction and effector phase of GVHD. The blue and red dots indicate the IFNs (type I IFNs and IFN-γ, respectively) secreted by adjacent cells. CXCR, CXC chemokine receptors; GI, gastrointestinal; Th, T helper cells; iTreg, induced regulatory T cells; Teff, effector T cell; Teff, memory T cell.
IFN-α/β has context-dependent roles in CD4+ T cell activation, differentiation and survival (26). IFN-α is associated with CD4+ T cell activation and contributes to the IFN-γ-mediated Th1 response (27). In contrast, IFN-α/β may suppress Th2 differentiation of human CD4+ T cells. Importantly, IFN-α regulation of T cell differentiation appears to be context-dependent. Dichotomous T cell polarization towards either Th1 or TFH was recently observed depending on the IFN-α exposure at different times (28, 29). In a colon-targeted GVHD murine model, IFN-α signaling prevented donor CD4+ T cell proliferation and differentiation, resulting in alleviating colon tissue damage (30, 31). Regulatory T cells (Tregs) play essential roles in controlling immune tolerance after allo-HSCT (32). Adoptive transfer of Treg ameliorates GVHD and improves survival in a murine model (33). However, IFN-α/β has shown some controversial impacts on Tregs. Some studies suggested that overexpression of IFN-α significantly reduced the frequency of Tregs in the tolerogenic tumor environment (34). Other studies suggested that IFN-α stimulation may increase differentiation of CD4+CD25+FOXP3+ Tregs (iTregs) (35) (Figure 1).
IFN-α/β signaling is essential for antigen-driven CD8+ T cell responses. First, differentiation of effector CD8+ T cell was associated with decreased IFNAR but increased IL-12 receptor, whereas augmented IFNAR favors the development of central memory T (TCM) cell (36, 37). In IFNAR deficient mice, CD8+ T cells lose the ability to become memory T cells during lymphocytic choriomeningitis virus (LCMV) infection (37, 38). Paradoxically, withdrawal of IFN-α monotherapy in clinical chronic myeloid leukemia resulted in elevated frequency of peripheral CD8+ TCM cells (39). Given that many different types of cells express IFNAR, the difference in regulating memory formation between IFNAR deficiency and IFN-α monotherapy may be attributable to both direct and indirect mechanisms. Second, the activation of IFN-α/β signaling in T cells could benefit cytokine secretion and cytolytic activity. In mice, injection of IFN-α incited substantial primary CD8+ T responses through cross-priming by DCs that were independent of CD4+ T-cell help (40, 41). IFN-α/β signaling plays a co-stimulatory role in CD8+ T activation and slows the death of activated T cells (42, 43). Moreover, direct activation of granzyme B transcription through IFN-α/β in effector CD8+ T cells contributes to tumor suppression as well as autoimmunity (44, 45). Consistent results were found in the context of GVHD that both CD8-dependent GVHD and GVL effects were enhanced through IFN-α/β signaling (30). In addition, despite IFN-α/β signaling induces transient attrition of bystander naïve T cells in the wake of T-cell response, it can rapidly activate nonspecific bystander memory CD8+ T cells. Activation of memory T cells contributes to rapid production of proinflammatory cytokines including IFN-γ (Figure 1) (46–48).
Clinically, recombinant IFN-α has been used alone or in combination with donor lymphocytes infusions or other cytokines such as granulocyte-macrophage colony-stimulating factor to establish GVL effects in patients with minimal residual disease or relapse after allo-HSCT (49–53). Studies using animal models demonstrate the effectiveness of IFN-α treatment against leukemia cells. A most recent trial showed that the proportion of granzyme positive CD8+ effector and effector memory subsets is positively correlated with GVHD incidence (53). This suggests that IFN-α-induced CD8+ T cells may be a double-edged sword against both malignant and normal cells.
In response to IFN-α/β signaling, the trimolecular interferon-stimulated gene factor 3 (ISGF3), which comprises signal transducer and activator of transcription (STAT) 1, STAT2 and interferon regulatory factor (IRF) 9, leads to most of the cellular effects of IFN-α/β (23). The dysregulation of ISGF3 results in aberrant T cell functions. For example, the absence of IRF2, a negative regulator of ISGF3, induces hyperresponsiveness of CD8+ T cells and promotes spontaneous inflammatory skin lesion in mice (54). Furthermore, in a LCMV infection mouse model, the STAT1 deficiency leads to a CD4+ T cell-mediated lethal disease. This effect is independent of IFN-γ, but it coincides with exaggerated proinflammatory cytokine production as well as increased frequency of LCMV-specific CD4+ and CD8+ T cells (55). These observations suggest that the effects of IFN-α/β signaling are not only divergent on CD4+ or CD8+ T cells, but also highly dependent on the pathophysiological backgrounds. Of note, several molecules in the downstream of the IFN-α/β pathway, including Janus Kinase (JAK) 1, STAT1 and STAT3, are shared with IFN-γ signaling, which indicates the possibility of crosstalk between the two signals in the transcriptional level.
IFN-γ
IFN-γ promotes CD4+ T cell differentiation towards Th1 lymphocytes and drives CD8+ T cell expansion and differentiation towards both effector and memory cells (Figure 1). Early studies in GVHD models suggest that IFN-γ contributes as a pathogenic factor to alloreactive responses. For instance, high serum levels of IFN-γ correlated with increased severity of GVHD after allo-HSCT (56). IFN-γ induced apoptosis of intestinal epithelial crypt cells, leading to extensive erosion of intestinal epithelium and GVHD propagation (57, 58). Genetic deletion of IFNGR in T cells prevents lethal GVHD while preserving the robust GVL effect (59). Furthermore, evidence from live-cell imaging reveals that both motility and cytotoxicity of CD8+ T cells are enhanced in alloreactive tissue due to autocrine/paracrine IFN-γ (60). The expression of CXCR3 induced by IFN-γ signaling is one of the mechanisms that drive the T cells to the sites of GVHD target organs (59).
Intriguingly, some studies suggested that IFN-γ may negatively regulate alloreactive T cells and prevent tissue damages. Evidence from IFN-γ knockout mice shows that IFN-γ could be protective against GVHD depending on the extent of conditioning in mouse models (18, 61). Infusion of IFN-γ-null donor T cells increased mortality of GVHD compared to that of wild-type T cells (62). One possible reason might be that IFN-γ is required for normal T cell contraction since IFN-γ deficiency would lead to delayed apoptosis of CD8+ T cell population, leading to prolonged inflammation (63–65). In addition, PD-L1, which is considered as an inhibitory checkpoint molecule in infections and tumors, was identified as a positive contributor to T cell-mediated GVHD in the murine model, as decreased inflammatory cytokines and increased apoptosis were observed in both Pdl1-/- allogeneic CD4+ and CD8+ T cells. Of note, both Ifngr-/- CD4+ and CD8+ donor T cells showed impaired PD-L1 expression, suggesting that loss of IFN-γ signaling mitigates tissue damages in GVHD via the PD-L1 pathway (66).
Interestingly, manipulation of IFN-γ signaling in alloreactive T cells results in variable lesions in GVHD target organs. IFN-γ produced by alloreactive T cells is the primary mediator contributing to the apoptosis of intestinal stem cells and intestinal damage (57). In addition, both clinical and preclinical studies suggest that IFN-γ-producing Th1 cells mediate damages in the gastrointestinal (GI) tract (67), whereas IFN-γ KO model results in exacerbated skin and lung injury (68). In the absence of IFN-γ signaling, alloantigen-primed CD4+ T cells showed decreased capacity to produce IFN-γ-secreting Th1 cells while skewing toward both Th2 and Th17 cells (68). Further studies are needed to define the correlation between the effects of IFN-γ on alloreactive T cells and the consequence of GVHD.
The binding of IFN-γ to the receptor, IFNGR1 and IFNGR2 complex, induces recruitment and phosphorylation of receptor-associated JAK1/2, which triggers subsequent signaling pathways predominantly through STAT1 (Figure 2). Interestingly, TCR stimuli initiate the translocation of STAT to IFNGR1-rich regions of the membrane similar to IFN-γ ligation (69). Blocking the JAK1/2 molecule significantly abrogates the polarization and proliferation of activated T cells as well as downregulates activation markers, such as CD69 and CD25, and reduces the production of proinflammatory cytokines (70). In light of the suppressive effect on T cell responses, the JAK inhibitors were reported to control GVHD in both mice and humans. Recently, ruxolitinib was approved for the treatment of steroid-refractory acute GVHD. JAK inhibitors mitigate GVHD via pleiotropic effects on T cells. For example, ruxolitinib mitigates acute GVHD by reducing CXCR3 expression, which results in less T-cell infiltrates in target organs (59, 71, 72), and by decreasing IFN-γ and IL17A production in CD4+ T cells (73). Similarly, another JAK1/2 selective inhibitor, baricitinib, can abrogate IFN-γ and IL-6 signaling in CD4+ T cells and significantly decrease Th1 and Th2 cell differentiation while augmenting the frequency of Tregs (74). In addition to the reduction of GVHD, baricitinib could also improve GVL (74). Despite these promising observations, the transcriptional regulations of the downstream genes in T cells are yet to be found.
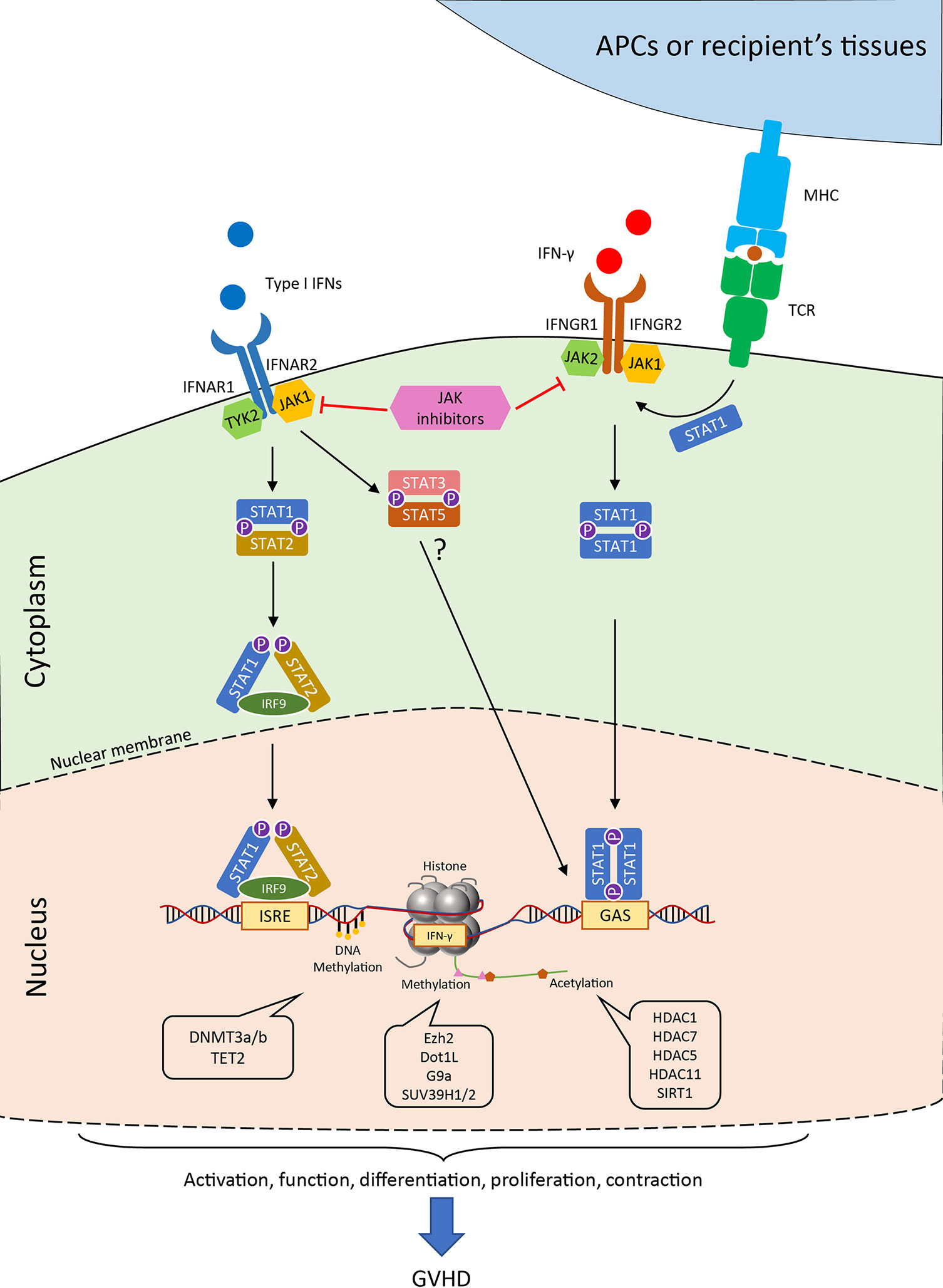
Figure 2 Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathways and the epigenetic regulations of IFN signaling in T cell. Generally, the binding of type I IFN to the receptor initiates the engagement of IFNAR1 associated Tyk2 protein tyrosine kinase and the IFNAR2 associated JAK1 protein tyrosine kinase. The signal further passes to the phosphorylation and the heterodimerize of STAT1 and STAT2, which together with IRF9 form the ISGF3 complex in the cytoplasm. ISGF3 translocates into the nucleus and binds to IFN-stimulated response elements (ISREs) found in most of IFN-stimulated genes (ISGs). Alternatively, STAT3 and STAT5 heterodimer are also observed after IFNAR activation in the absence of STAT1. Canonical IFN-γ signaling occurs through IFNGR and activates the JAK1/2 kinases, which further induce the phosphorylation of STAT1. The STAT1 homodimer can directly move into the nucleus and binds to gamma-activated sequence (GAS) sites. The activation of T-cell receptor could also help the co-localization of STAT1 to IFNGR-rich regions of the membrane. The transcription of the downstream genes as well as the production of IFN-γ are tightly regulated by numerous epigenetic enzymes, which control the modification of the DNA and histones. These regulators critically control the T cell activities in the process of GVHD, which allows for possible therapeutic interventions. JAK, Janus kinase; STAT, signal transducer and activator of transcription; IFNAR, interferon alpha receptor; IFNGR, interferon gamma receptor; ISREs, IFN-stimulated response elements; GAS, gamma-activated sequence; TCR, T cell receptor; MHC, major histocompatibility complex; IRF, interferon regulatory factor; HDAC, histone deacetylase; SIRT, sirtuin.
Notably, as the IFNGR can be expressed on almost all cell types, the generated IFN-γ from activated allogeneic T cells could have a remarkable influence on the innate immunity and break the homeostasis of the surrounding tissues. For instance, IFN-γ signaling acts as a ‘super-activator’ of macrophages, inducing transcriptional activation of proinflammatory genes (e.g., IL-6 and TNF-α) and enhancing antigen presentation. Furthermore, recent evidence suggests that IFN-γ directly inhibits the proliferation of hematopoietic stem/progenitor cells (HSPCs) and their generation of pDCs that can induce immune tolerance against GVHD (20). On the other hand, exposure to IFN-γ reduces the proliferation of intestinal epithelial cells and further induces apoptosis by modulation of the AKT/β-catenin and Wnt/β-catenin pathway (75). Thus, although IFN-γ plays dichotomous roles in the regulation of T cells, its proinflammatory damage to the tissues in GVHD is generally acknowledged (Figure 1).
Type III IFNs
Recent studies have discovered other members of type III IFNs, including IFN-λ1 (IL-29), IFN-λ2 (IL28A), IFN-λ3 (IL-28B) and IFN-λ4 (76–78). They participate in the antiviral activities similar to the type I IFNs, but primarily in barrier tissues such as mucosal epithelial cells (79, 80). Although IFN-α/β and IFN-λ engage different receptors, they induce similar downstream signaling pathways through the phosphorylation of STAT1/2 and the subsequent transcription factor ISGF3 (81). However, our knowledge of IFN-λ in GVHD is limited. An initial study found that IFN-λ2 did not significantly modulate GVHD mortality in a murine model upon deleting its receptor (Ifnlr1-/-) or administration of recombinant IFN-λ2 (82). Intriguingly, a most recent study revealed that Ifnlr1 deletion led to exaggerated damages in the GI tract and recombinant IFN-λ treatment reduced GVHD lethality (83). Deletion of Ifnlr1 led to increases of donor T-cell expansion and serum IFN-γ levels, however, it did not affect the proliferation and apoptosis of alloreactive T cells. Interestingly, the effect of IFN-λ on T cells seemed to be indirect since the T-cell expansion was influenced by early engraftment, which was related to IFN-λ signaling in NK cells (83). Further studies of IFN-λ in the modulation of GVHD the underlying mechanisms are warranted.
Epigenetic Regulation of IFN Expression and Function in T Cells
The epigenomic signatures, including DNA methylation on cytosine nucleotides, histone modifications and chromatin accessibility, reflect previous and present gene expression, and can positively or negatively regulate future transcription according to environmental stimuli. The labeled or ‘bookmarked’ chromatin organized as ‘epigenetic code’ that can be recognized by protein complexes called ‘readers’. It is also closely controlled by enzymes, called ‘writers and erasers’, that are able to manipulate different modifications mounted on specific residues (84). The major contributors comprise DNA methyltransferases (DNMTs) or DNA demethylases on DNA level; histone acetyltransferase (HATs), histone deacetylase (HDACs), histone methyltransferases (HMTs) and lysine demethylases (KDM) on histone level. Other epigenetic mechanism includes the microRNAs (miRNAs), which negatively control target gene expression post-transcriptionally via interaction with the complementary sequences. It has been well established that IFN-signaling can generate ‘interferon epigenomic signatures’ and reprogram cell response (21).
During antigen-driven immune responses, such as GVHD, T cells are located at the downstream of type I IFN signaling as they receive this signal from innate immune cells (20, 85). Although many regulators and pathways of type I IFN signaling from innate cells may be interchangeable in T cells, the cell type and context-dependent mechanisms have yet to be characterized. As discussed earlier, the canonical signal of type I IFN depends on the activation of STAT1 and STAT2, which leads to the activation of ISGs by transcription factor IRF9 (also known as ISGF3) (23). Notably, multiple pathways co-exist in the downstream of IFNAR1/2 and control T cell immune responses (Figure 2). For example, IFN-α/β induced STAT1 is responsible for the suppression of CD8+ T cell expansion, whereas STAT3 and STAT5 mediate antiapoptotic and mitogenic effects in T cells in the absence of STAT1 (86).
The epigenetic regulation of ISGs in T cells is far less documented compared with studies in innate immune cells. Evidence has been widely found on innate immune cells that ISG promoters are associated with increased level of histone acetylation, which in part mediated by STAT1 and STAT2 (87), STAT1/2 promotes histone acetylation after IFN-α/β stimulation in T cells as well. Early study has linked IFN-α signaling with histone hyperacetylation at the granzyme B and eomesodermin (Eomes) loci during CD8+ T cell differentiation (88). Similarly, IFN-α/β signaling enhances H3K4me3 and H3K9ac (transcription permissive) at the promoter region of Eomes and activates it in an IRF9-dependent manner (89). Interestingly, T-bet is found to counteract aberrant IFN-α/β signaling during Th1 cell development by repressing ISGs such as Isg15, Mx1, Oasl1a, etc. Deletion of T-bet results in accumulation of STAT2 and elevation of transcription active mark H3K27ac at ISGs activated by IFN-β (90), highlighting the complexity of interactions between the extrinsic cytokine influence and the intrinsic regulation of cell development. In addition, the epigenetic modulation of T cells in responding to IFN-α/β is likely to be context-dependent. For example, although STAT1 mRNA levels are both increased in lupus and normal CD8+ T cells with IFN-α stimulation, the signature of hypomethylated DNA sites in lupus CD8+ T cells facilitates the upregulation of HLA-DRB1 in a STAT1-signaling-dependent manner (91). In addition to histone and DNA modification, the miRNA-155 downregulates the T-cell responsiveness to IFN-α/β via IFNAR-STAT pathway. Despite the direct targets of miRNA-155 were not defined, the loss of microRNA-155 results in impaired antiviral CD8 T cell response (92). To reconcile these paradoxical and the highly context-dependent effects of IFN-α/β on T cells, it will be important to map how the ISGs are regulated on the epigenetic level in alloreactive T cells during GVHD.
Compared to IFN-α/β, the epigenetic regulation of IFN-γ is much more complicated. IFN-γ not only promotes antigen-driven T cell differentiation, but also is the major mediator for tissue injury. Much work has investigated the epigenetic regulation of Ifng locus in T cells. The CpG dinucleotide at Ifng promoter in naïve CD8+ T cells is substantially methylated and undergoes demethylation when these CD8+ T cells are activated. Memory CD8+ T cells retain relative hypomethylated status to enable a rapid gene expression for the re-activation in the future (93, 94). Similar regulation can be found in Th1 CD4+ T cells. The CpG for Ifng promoter in naïve CD4+ T cells is mostly methylated, but only approximately 50% of CpG regions are methylated in resting memory cells (95). Conversely, in the lineages other than Th1 development, their suppression of IFN-γ can also be achieved in part with DNA methylation (96). The ten-eleven translocation (TET) 2 enzyme, which mediates DNA demethylation, positively regulates Ifng transcription by promoting 5-hydroxymethylcytosine level in CD4+ T cells (97). From another view, histone modifications act in accordance with the gene regulation of DNA methylation. For example, the Ifng promoter region in Th1 cells is associated with hyperacetylation of histones H3 and H4, but not in Th2 cells (98). Additionally, the Ifng suppression in Th2 cells is accompanied by repressive H3K9me3, which is governed by enzyme SUV39H1. Loss of this enzyme results in skewed lineage stability (99).
Epigenetic Programs and Pharmacological Modulations That Control IFNs in Allogeneic T Cells During GVHD
The function and differentiation of T cells are closely intertwined with IFN expression. Epigenetic processes, including DNA methylation, histone modification and chromatin remodeling, are the key mechanisms that control T cell differentiation and function (100, 101). Multiple epigenetic enzymes have been identified to regulate the production and subsequent effect of IFNs in allogeneic T cells (102–104). A number of chemical compounds that selectively inhibit these enzymes are made available, and their effect on IFN signaling and therapeutic potentials are under active investigation (104–106). Since there are very limited reports studying the epigenetic regulation of type I IFNs in the GVHD context, our continual discussion will focus on the epigenetic effects on IFN-γ.
DNA Methylation
DNA methyltransferase (DNMT) 3a or DNMT3b contributes to de novo DNA methylation, resulting in genetic silencing. In T cells, DNMT3a expression is regulated by TCR signaling (107). The promoter of Ifng locus remains hypomethylated during Th1 differentiation from naïve CD4+ T cells, albeit de novo DNA methylation at Ifng promoter is observed in other commitments, such as Th2, Th17 and iTreg cells (108). DNMT3a is responsible for maintaining the silence of Ifng gene in non-Th1 lineages (109). Accordingly, deletion of DNMT3a after T cell activation selectively reduces the level of Ifng methylation (107), and allows significant IFN-γ production from non-IFN-γ producing CD4+ T cells (109). During secondary contact with antigen, DNA demethylation at the IFN-γ promoter takes place in memory T cells in order to facilitate rapid effector responses (94). Furthermore, the functional exhaustion of the CD8+ T cells couples with persistent DNA hypermethylation at Ifng loci, even if the cells are treated with anti-PD-1 blockade. Inhibition of DNA methylation by hypomethylating agent together with anti-PD-L1 significantly promotes IFN-γ secretion by exhausted CD8+ T cells (110). The above studies suggest that inhibition of DNMT may promote alloreactive T cell activities in GVHD by suppressing DNA methylation and subsequently enhance IFN-γ production.
Although hypomethylating agents globally alter DNA methylation levels, their influence on gene expression in T cells shows preference. Compelling evidence indicates that both IFN-γ and FOXP3 locus are demethylated by Azacytidine (Aza) (94, 111, 112). In vitro studies revealed that Aza and decitabine could directly induce FOXP3 expression in T cells, whereas IFN-γ gene expression along with other cell-cycle related genes were significantly down-regulated by Aza (104). Consistently, decitabine significantly suppressed differentiation of naïve CD4+ T cells into Th1 subsets but not Tregs (113). These findings applied to the alloreactive T cells in GVHD. It has been observed that Aza mitigates GVHD in murine models by converting alloreactive CD4+CD25+FOXP3- cells to suppressive CD4+CD25+FOXP3+ Tregs and directly increase Treg proliferation. In addition, the frequency of IFN-γ-producing CD4+ T cells was significantly decreased (114–116). The inhibition of naïve CD4+ T cell proliferation by decitabine is also accompanied by the elevation of the TET2, an enzyme that acts opposite to DNMTs, which promote DNA demethylation (113).
Despite the above in vitro data supporting that hypomethylating agents up-regulate Treg and suppress conventional CD4+ T cells (117, 118), post-transplantation Aza treatment in patients with high risk of AML and MDS shows no significant differences in terms of overall survival and GVHD incidence in patients compared to the control arm (119). It is possible that additional epigenetic mechanisms are involved in the IFN-γ regulation of alloreactive T cells. For example, in DNMT3a-null Th2 or Th17 cells, decreased level of DNA methylation at the Ifng loci correlated with low level of H3K4 and high H3K27 methylation, which permits and inhibits DNA transcription, respectively (109).
Histone Methylation
Histone methylation is predominantly restricted to the N-terminal tails of H3 and H4 histones and is usually presented by one, two, or three lysine residues (120). The effects of histone methylation on gene expression are loci-specific. Genes that bound by H3K4, H3K36 and H4K20 are more likely to be actively transcribed, whereas H3K9, H3K27 and H3K79 are usually associated with gene suppression (121–124). The histone methylation level at each site is controlled by one or a set of HMTs and KDMs (120, 124). Thus, the activities of these enzymes are the key factors that determine gene transcription.
CD4+ and CD8+ T cells display unique patterns of histone methylation landscapes at Ifng locus based on the stages of cell differentiation. Once activated by TCR signaling or specific cytokines, the histone methylation markers of T cells are dynamically catalyzed by their dedicated enzymes. During the quiescent stage of naïve T cells, the Ifng promoter of both CD4+ and CD8+ T cells are occupied with repressive H3K27me3 but low level of permissive H3K4me3 (125, 126). Upon activation, Ifng region of both CD4+ and CD8+ T cells loses H3K27me3 markers (125, 127, 128). However, effector CD8+ T cells gain H3K4me3 at Ifng locus (127, 129). CD4+ Th1 cell differentiation increases both H3K4me3 and H3K9me2 (permissive and repressive, respectively), whereas CD4+ Th2-cells rapidly extinguish H3K9 methylation by STAT6 and GATA-3 dependent mechanisms (130).
Ezh2 is a crucial enzyme that catalyzes H3K27 methylation and remarkably silences target genes to facilitate T cell differentiation and function. During Th2 cell development, Ezh2 is recruited with STAT6 and GATA3 to the Ifng locus and is responsible for the silencing of Ifng locus through H3K27 methylation (130). In vitro studies revealed that Ezh2 affected CD4+ T cell differentiation depending on the context of the extracellular environment. For instance, Ezh2-deficiency could enhance the CD4+ T cell production of either IFN-γ or IL-4, depending on the cell-inducing cytokines in vitro, such as IL-12 or IL-4, respectively (131–133). Further, both T-bet and Eomes are required for the regulation of IFN-γ production by Ezh2 (131). The role of Ezh2 in GVHD is complex. In an MHC-mismatched B6 anti-BALB/c GVHD murine model, loss of Ezh2 in donor T cells resulted in impaired IFN-γ production and reduced GVHD. Specifically, Ezh2 promoted Th1 development by stimulating Ifng, Tbx21 and Stat4 expression (102). Similar results could also be found from Th1 cells in aplastic anemia, in which Ezh2 directly activated Tbx21 transcription by direct binding to its promoter (134). Contradictive results are also found with CD8+ T cells. Ezh2 inhibition resulted in increased frequency of IFN-γ producing tumor-infiltrating CD8+ T cells (135), whereas Ezh2-deficient CD8+ T cells exhibit an impaired ability to produce IFN-γ in a virus infection model (136). How to explain the discrepancy observed in these studies remains elusive. In addition, Ezh2 could co-localize with FOXP3 and assist in silencing the IFN-γ expression (137). Consistently, the absence of Ezh2 resulted in defective Treg differentiation, which could further contribute to autoimmune colitis (132).
Dot1L, a solo H3K79 methyltransferase, has been recently identified to regulate T cell activation and polarization. In general, H3K79 methylation strongly correlates with active gene transcription (138, 139), but exceptions are also reported (140, 141). When T cells were cultured in Th1 cell-polarizing conditions, IFN-γ production was enhanced by Dot1L inhibition with a small molecule inhibitor (SGC0946) at the beginning of polarization and was associated with the reduction of H3K79me2. Interestingly, the proliferative capacity was not affected (142). These observations indicate that Dot1L may play a negative role in regulating Th1 cell differentiation and IFN-γ production. Another group recently used a T-cell-specific Dot1L-deficient infection mouse model and observed that the repressive effect of IFN-γ production by Dot1L was T-bet dependent. In this study, the enhanced IFN-γ secreting ability via Dot1L inhibition (with chemical probe SGC0946) in Th2 cells was abrogated by T-bet deletion (143). However, the opposite phenomenon was observed in GVHD setting. Inhibition of Dot1L with the same chemical probe attenuated xenogeneic GVHD by globally suppressing T cell activation-induced genes, in which IFN-γ production was significantly reduced (103). Of note, this effect was only observed in T cells with low-avidity TCR interaction. Therefore, Dot1L inhibition increased the TCR stimulation threshold and was controlled in an ERK phosphorylation-dependent manner (103). The inconsistent findings among different studies are likely due to the different roles of Dot1L in the regulations of upstream and downstream of IFN-γ signaling. Similar to the data found in CD4+ T cells, Dot1L also remarkably controls the differentiation of CD8+ T cells. Dot1L-deficiency resulted in the induction of memory-like transcriptome feature in antigen inexperienced CD8+ T cells. Furthermore, these cells were functionally impaired as they were incompetent to produce IFN-γ upon stimulation with anti-CD3 and anti-CD28 antibodies (144). In addition, using the approach of genetic Dot1L deletion and a specific inhibitor, EPZ004777, the repression of Dot1L resulted in inhibition of H3K79me2 in CD8+ T cells that associated with increased CD8+ T cell apoptosis and suppressed IFN-γ and TNF-α secretion. Besides, the methionine metabolism in the microenvironment also affects the methylation status of H3K79 in CD8+ T cells, further promoting the dysregulation of the immune response (145). However, genetic approaches are required to define the precise role of Dot1l in T cells.
Both G9a and SUV39H1/2 contribute to the methylation of the H3K9 site. However, G9a catalyzes H3K9 residue to mono- or dimethylation (H3K9me/me2), whereas SUV39H1/2 is responsible for di- to tri-methylation (H3K9me3) (146, 147). These enzymes could be found in multiple repressive complexes that promote transcription inhibition. Importantly, the heterochromatin protein 1α (HP1α) directly recognizes and binds to H3K9me3 and initiates the chromatin remodeling by forming heterochromatin (148). Despite their similarity in histone modification, G9a and SUV39H1/2 are remarkably divergent in epigenetic regulation of IFN and its subsequent effects on T cell functions. During Th2 development, G9a facilitates the transcriptional silence of Ifng locus since increased IFN-γ production was observed in G9a deficiency CD4+ T cells along with a decreased level of H3K9me2 (149). However, given that G9a deficiency and inhibition do not affect the development of Th1 cells as well as their capacity in secreting IFN-γ both in vitro and in vivo, G9a is currently considered dispensable for Th1 cell response (149, 150). Similarly, the ability to produce IFN-γ in CD8+ T cells is not affected in G9a knockout cells, but G9a is crucial to repress helper T lineage genes after the activation of CD8+ T cells (151). These studies indicate a moderate role of G9a in epigenetic control of IFN-γ. In addition, although not verified in T cells, evidence suggests that the downstream of IFN-α/β signaling and ISGs are negatively regulated by G9a (152). On the other hand, SUV39H1-H3K9me3-HP1α pathway also contributes to Th2 stability by decorating H3K9me3 at Ifng promoter (99). Less is known whether this signaling redundancy may mutually compensate for both G9a and SUV39H1 when activated via different upstream pathways.
In addition, SETDB1, which belongs to the SUV39H family, is responsible for H3K9me3 deposition at specific promoters. Adoue et al. demonstrated that SETDB1 was required to maintain IFN-γ silencing in Th2 cells. Instead of directly catalyzing H3K9me3 on the target gene, SETDB1 represses adjacent endogenous retrovirus location that affects the transcription of Th1 genes (153). This study reveals the spatial regulation of histone modification in the epigenetic control of Th cell differentiation. In contrast to the inhibitory regulation of IFN-γ in CD4+ T cells, wild-type CD8+ T cells exhibited a higher ability to produce IFN-γ compared to SUV39H1-conditional knockout mice that infected with L. monocytogenes. SUV39H1 was responsible for silencing stem/memory gene programs while enhancing the functions of effector cells in CD8+ T cells (154).
Histone Acetylation
Unlike methylation, histone acetylation uniformly assists gene transcription because the acetyl group neutralizes the positive charge on the histones, thereby reducing the electrostatic force between histone and the negatively charged DNA molecules (155). On the other hand, together with methylation, phosphorylation and other covalent modifications, histone acetylation also takes part in the formation of ‘epigenetic code’, which allows the recognition by the protein complexes that help amplify the gene transcription (156). HAT and HDAC regulate acetylation status on both H3 and H4 histones.
The anti-inflammatory properties of the HDAC inhibitors have long been recognized by numerous experimental and clinical studies, including GVHD. Early studies that first linked histone acetylation with GVHD revealed that the panoramic HDAC inhibitor (pan-HDACi) suberoylanilide hydroxamic acid (SAHA) ameliorate and delayed the development of GVHD and reduced the serum level of proinflammatory cytokines such as IFN-γ and TNF-α following allogeneic bone marrow transplantation (157, 158). Although the STAT1 phosphorylation was inhibited during this process, the T cell proliferation and cytotoxic responses in GVL activity remained intact. Several clinical trials using SAHA as prophylactic treatment after allo-HSCT also observed reduction of GVHD in patients (159, 160). These trials revealed higher Treg cell numbers in the peripheral blood after HDACi administration as well as lower GVHD-related biomarkers, such as ST2 and Reg3α, and the proinflammatory cytokine IL-6 in the plasma. Another clinical trial uses pan-HDACi panobinostat, combined with glucocorticoids, as primary treatment for acute GVHD demonstrates an enhanced H3 acetylation in both CD4+ and CD8+ T cells (161). However, in contrast to the mouse model, the level of plasma IFN-γ was not significantly changed in patients (160, 161). It will be interesting to determine whether functional changes of T cells are controlled by HDACi and correlate with clinical responses and outcomes of patients.
Accumulating evidence from recent studies discovered the regulation of IFN-γ by specific HDAC members in T cell responses with or without allogeneic antigens. HDAC1-deficiency does not affect the development and effector functions but increases the STAT1 activity in CD4+ T cells, which results in the elevated level of IFN-γ production in activated Th1 cells (162, 163). Similar effects were also detected in effector CD8+ T cells (164), indicating a negative role of HDAC1 in controlling IFN-γ transcription. Besides, HDAC7 and SIRT1 may synergize with HDAC1 in repressing T cell activation and IFN-γ production via separate pathways (165, 166). On the contrary, HDAC5 and HDAC11 positively regulate IFN-γ production. Both HDAC5 deletion in CD8+ T cells and HDAC11 knockout in CD4+ and CD8+ T cells induce increased IFN-γ production upon anti-CD3 and anti-CD28 activation in vitro (167, 168). In addition, potent GVHD in the murine model can be induced in HDAC11 KO mice. Both CD4+ and CD8+ T cells with HDAC11 deletion are hyperresponsive to alloantigen and associated with increased expression of Eomes and T-bet (167).
Of note, inconsistent results can be found among studies. These discrepancies may partially attribute to the fact that some HDACs can both have histone and non-histone targets, which increases the complexity of gene regulation by introducing indirect effects. For example, genetic deletion and inhibition of SIRT1 reduces T-cell alloreactivity and promotes the function of iTreg through the enhancement of p53 acetylation, leading to the attenuation of GVHD (169).
Compared to the extensive investigations exploring the features of HDACs, the regulatory role of HAT in IFN related T cell responses are not well understood. In Th2 cells, the HAT p300 is recruited by Gata3 and Chd4 complex to promote the transcription of Th2 cytokine, whereas HDAC2 is recruited in the Gata3-Chd4-NuRD complex to suppress Tbx21 and the subsequent IFN-γ expression (170). Moreover, the CREB-binding protein and p300 complex regulates the differentiation of human Treg via H3K27 acetylation. Although much evidence has been found in innate immune cells that p300 and other HATs essentially regulate STAT-ISG signaling and type I IFN production, our understanding of the epigenetic control of HATs remains low in regard to functional regulation of IFN in T cells. Especially, further studies of HATs should be conducted to validate the roles of both HATs and HDACs in GVHD models.
Conclusion and Perspectives
Although extensive efforts have been made in defining the roles of IFNs in alloreactive T cells, the mechanisms underlying the effects of IFNs in the setting of GVHD remain largely unknown. Much of our knowledge about the IFN-related regulations in T cells mostly come from infection, tumor and autoimmune diseases. However, considering the release of DAMP and PAMP, anti-leukemia effect and the exposure of alloreactive antigens in patients after allo-HSCT, the T cell response in GVHD and GVL scenarios reflect combined effects of these conditions. In addition, the effect of IFNs in different organs and tissues, which have distinct microenvironments, may also affect T cell response. Development of novel genetic approaches is important to further dissect the impact of IFNs on T cell alloimmunity and tumor immunity.
The advancement in epigenetics of T cell biology opens a unique way to understand the molecular mechanisms of IFN regulation. Much effort has been made to identify key epigenetic enzymes and pathways that affect IFN expression in T cells. However, most of the effects of the enzymes are still unknown in the context of GVHD. It will be interesting to determine the connection between the inhibitors of epigenetic enzymes and the outcomes of GVHD models and clinical patients. Future studies mapping epigenetic mechanisms of IFN regulations in allogeneic T cells may be beneficial to elucidate how IFN modulates GVHD and GVL.
Author Contributions
CZ conceived the manuscript and drew the figures. YZ and HZ discussed the concepts and critically revised the article. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Blazar BR, Hill GR, Murphy WJ. Dissecting the Biology of Allogeneic HSCT to Enhance the GvT Effect Whilst Minimizing Gvhd. Nat Rev Clin Oncol (2020) 17(8):475–92. doi: 10.1038/s41571-020-0356-4
2. Zeiser R, Blazar BR. Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med (2017) 377(22):2167–79. doi: 10.1056/NEJMra1609337
3. Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hulsdunker J, Varelias A, et al. Mhc Class II Antigen Presentation by the Intestinal Epithelium Initiates Graft-Versus-Host Disease and Is Influenced by the Microbiota. Immunity (2019) 51(5):885–98.e7. doi: 10.1016/j.immuni.2019.08.011
4. Jones SC, Murphy GF, Friedman TM, Korngold R. Importance of Minor Histocompatibility Antigen Expression by Nonhematopoietic Tissues in a CD4+ T Cell-Mediated Graft-Versus-Host Disease Model. J Clin Invest (2003) 112(12):1880–6. doi: 10.1172/JCI19427
5. McDonald GB, Sandmaier BM, Mielcarek M, Sorror M, Pergam SA, Cheng GS, et al. Survival, Nonrelapse Mortality, and Relapse-Related Mortality After Allogeneic Hematopoietic Cell Transplantation: Comparing 2003-2007 Versus 2013-2017 Cohorts. Ann Intern Med (2020) 172(4):229–39. doi: 10.7326/M19-2936
6. Zhang Y, Louboutin JP, Zhu J, Rivera AJ, Emerson SG. Preterminal Host Dendritic Cells in Irradiated Mice Prime CD8+ T Cell-Mediated Acute Graft-Versus-Host Disease. J Clin Invest (2002) 109(10):1335–44. doi: 10.1172/JCI14989
7. Kalia V, Sarkar S. Regulation of Effector and Memory Cd8 T Cell Differentiation by IL-2-A Balancing Act. Front Immunol (2018) 9:2987. doi: 10.3389/fimmu.2018.02987
8. Tait Wojno ED, Hunter CA, Stumhofer JS. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity (2019) 50(4):851–70. doi: 10.1016/j.immuni.2019.03.011
9. Kumar S, Mohammadpour H, Cao X. Targeting Cytokines in GVHD Therapy. J Immunol Res Ther (2017) 2(1):90–9.
10. Henden AS, Hill GR. Cytokines in Graft-versus-Host Disease. J Immunol (2015) 194(10):4604–12. doi: 10.4049/jimmunol.1500117
11. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
12. Crow MK, Olferiev M, Kirou KA. Type I Interferons in Autoimmune Disease. Annu Rev Pathol (2019) 14:369–93. doi: 10.1146/annurev-pathol-020117-043952
13. Arico E, Castiello L, Capone I, Gabriele L, Belardelli F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers (Basel) (2019) 11(12):1943. doi: 10.3390/cancers11121943
14. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-Gamma in Tumor Progression and Regression: A Review. Biomark Res (2020) 8:49. doi: 10.1186/s40364-020-00228-x
15. Pollard KM, Cauvi DM, Toomey CB, Morris KV, Kono DH. Interferon-Gamma and Systemic Autoimmunity. Discovery Med (2013) 16(87):123–31.
16. Ali S, Mann-Nuttel R, Schulze A, Richter L, Alferink J, Scheu S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front Immunol (2019) 10:778. doi: 10.3389/fimmu.2019.00778
17. Schoenborn JR, Wilson CB. Regulation of Interferon-Gamma During Innate and Adaptive Immune Responses. Adv Immunol (2007) 96:41–101. doi: 10.1016/S0065-2776(07)96002-2
18. Wang H, Yang YG. The Complex and Central Role of Interferon-Gamma in Graft-Versus-Host Disease and Graft-Versus-Tumor Activity. Immunol Rev (2014) 258(1):30–44. doi: 10.1111/imr.12151
19. Robb RJ, Hill GR. The Interferon-Dependent Orchestration of Innate and Adaptive Immunity After Transplantation. Blood (2012) 119(23):5351–8. doi: 10.1182/blood-2012-02-368076
20. Tian Y, Meng L, Wang Y, Li B, Yu H, Zhou Y, et al. Graft-Versus-Host Disease Depletes Plasmacytoid Dendritic Cell Progenitors to Impair Tolerance Induction. J Clin Invest (2021) 131(1):e136774. doi: 10.1172/JCI136774
21. Barrat FJ, Crow MK, Ivashkiv LB. Interferon Target-Gene Expression and Epigenomic Signatures in Health and Disease. Nat Immunol (2019) 20(12):1574–83. doi: 10.1038/s41590-019-0466-2
22. Ivashkiv LB. Ifngamma: Signalling, Epigenetics and Roles in Immunity, Metabolism, Disease and Cancer Immunotherapy. Nat Rev Immunol (2018) 18(9):545–58. doi: 10.1038/s41577-018-0029-z
23. Ivashkiv LB, Donlin LT. Regulation of Type I Interferon Responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
24. Pestka S. Purification and Cloning of Interferon Alpha. Curr Top Microbiol Immunol (2007) 316:23–37. doi: 10.1007/978-3-540-71329-6_3
25. Porritt RA, Hertzog PJ. Dynamic Control of Type I IFN Signalling by an Integrated Network of Negative Regulators. Trends Immunol (2015) 36(3):150–60. doi: 10.1016/j.it.2015.02.002
26. Kawashima K, Isogawa M, Onishi M, Baudi I, Saito S, Nakajima A, et al. Restoration of Type I Interferon Signaling in Intrahepatically Primed CD8+ T Cells Promotes Functional Differentiation. JCI Insight (2021) 6(3):e145761. doi: 10.1172/jci.insight.145761
27. Jarry A, Malard F, Bou-Hanna C, Meurette G, Mohty M, Mosnier JF, et al. Interferon-Alpha Promotes Th1 Response and Epithelial Apoptosis Via Inflammasome Activation in Human Intestinal Mucosa. Cell Mol Gastroenterol Hepatol (2017) 3(1):72–81. doi: 10.1016/j.jcmgh.2016.09.007
28. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription Factor STAT3 and Type I Interferons Are Corepressive Insulators for Differentiation of Follicular Helper and T Helper 1 Cells. Immunity (2014) 40(3):367–77. doi: 10.1016/j.immuni.2014.02.005
29. Barbet G, Sander LE, Geswell M, Leonardi I, Cerutti A, Iliev I, et al. Sensing Microbial Viability Through Bacterial RNA Augments T Follicular Helper Cell and Antibody Responses. Immunity (2018) 48(3):584–98.e5. doi: 10.1016/j.immuni.2018.02.015
30. Robb RJ, Kreijveld E, Kuns RD, Wilson YA, Olver SD, Don AL, et al. Type I-IFNs Control GVHD and GVL Responses After Transplantation. Blood (2011) 118(12):3399–409. doi: 10.1182/blood-2010-12-325746
31. Fischer JC, Bscheider M, Eisenkolb G, Lin CC, Wintges A, Otten V, et al. Rig-I/MAVS and STING Signaling Promote Gut Integrity During Irradiation- and Immune-Mediated Tissue Injury. Sci Transl Med (2017) 9(386):eaag2513. doi: 10.1126/scitranslmed.aag2513
32. Whangbo JS, Antin JH, Koreth J. The Role of Regulatory T Cells in Graft-Versus-Host Disease Management. Expert Rev Hematol (2020) 13(2):141–54. doi: 10.1080/17474086.2020.1709436
33. Riegel C, Boeld TJ, Doser K, Huber E, Hoffmann P, Edinger M. Efficient Treatment of Murine Acute GvHD by In Vitro Expanded Donor Regulatory T Cells. Leukemia (2020) 34(3):895–908. doi: 10.1038/s41375-019-0625-3
34. Hashimoto H, Ueda R, Narumi K, Heike Y, Yoshida T, Aoki K. Type I IFN Gene Delivery Suppresses Regulatory T Cells Within Tumors. Cancer Gene Ther (2014) 21(12):532–41. doi: 10.1038/cgt.2014.60
35. Daniel V, Sadeghi M, Wang H, Opelz G. Cd4+Cd25+Foxp3+IFN-gamma+ Human Induced T Regulatory Cells Are Induced by Interferon-Gamma and Suppress Alloresponses Nonspecifically. Hum Immunol (2011) 72(9):699–707. doi: 10.1016/j.humimm.2011.05.020
36. Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. Reciprocal Responsiveness to Interleukin-12 and Interferon-Alpha Specifies Human CD8+ Effector Versus Central Memory T-Cell Fates. Blood (2009) 113(22):5516–25. doi: 10.1182/blood-2008-11-188458
37. Gangaplara A, Martens C, Dahlstrom E, Metidji A, Gokhale AS, Glass DD, et al. Type I Interferon Signaling Attenuates Regulatory T Cell Function in Viral Infection and in the Tumor Microenvironment. PloS Pathog (2018) 14(4):e1006985. doi: 10.1371/journal.ppat.1006985
38. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I Interferons Act Directly on CD8 T Cells to Allow Clonal Expansion and Memory Formation in Response to Viral Infection. J Exp Med (2005) 202(5):637–50. doi: 10.1084/jem.20050821
39. Ilander M, Kreutzman A, Rohon P, Melo T, Faber E, Porkka K, et al. Enlarged Memory T-cell Pool and Enhanced Th1-type Responses in Chronic Myeloid Leukemia Patients Who Have Successfully Discontinued IFN-alpha Monotherapy. PloS One (2014) 9(1):e87794. doi: 10.1371/journal.pone.0087794
40. Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-Priming of CD8+ T Cells Stimulated by Virus-Induced Type I Interferon. Nat Immunol (2003) 4(10):1009–15. doi: 10.1038/ni978
41. Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, et al. Direct Stimulation of T Cells by Type I IFN Enhances the CD8+ T Cell Response During Cross-Priming. J Immunol (2006) 176(8):4682–9. doi: 10.4049/jimmunol.176.8.4682
42. Marrack P, Kappler J, Mitchell T. Type I Interferons Keep Activated T Cells Alive. J Exp Med (1999) 189(3):521–30. doi: 10.1084/jem.189.3.521
43. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I Ifns Provide a Third Signal to CD8 T Cells to Stimulate Clonal Expansion and Differentiation. J Immunol (2005) 174(8):4465–9. doi: 10.4049/jimmunol.174.8.4465
44. Lu C, Klement JD, Ibrahim ML, Xiao W, Redd PS, Nayak-Kapoor A, et al. Type I Interferon Suppresses Tumor Growth Through Activating the STAT3-Granzyme B Pathway in Tumor-Infiltrating Cytotoxic T Lymphocytes. J Immunother Cancer (2019) 7(1):157. doi: 10.1186/s40425-019-0635-8
45. Newby BN, Brusko TM, Zou B, Atkinson MA, Clare-Salzler M, Mathews CE. Type 1 Interferons Potentiate Human Cd8(+) T-Cell Cytotoxicity Through a STAT4- and Granzyme B-Dependent Pathway. Diabetes (2017) 66(12):3061–71. doi: 10.2337/db17-0106
46. Marshall HD, Urban SL, Welsh RM. Virus-Induced Transient Immune Suppression and the Inhibition of T Cell Proliferation by Type I Interferon. J Virol (2011) 85(12):5929–39. doi: 10.1128/JVI.02516-10
47. Freeman BE, Hammarlund E, Raue HP, Slifka MK. Regulation of Innate CD8+ T-Cell Activation Mediated by Cytokines. Proc Natl Acad Sci USA (2012) 109(25):9971–6. doi: 10.1073/pnas.1203543109
48. Lertmemongkolchai G, Cai G, Hunter CA, Bancroft GJ. Bystander Activation of CD8+ T Cells Contributes to the Rapid Production of IFN-Gamma in Response to Bacterial Pathogens. J Immunol (2001) 166(2):1097–105. doi: 10.4049/jimmunol.166.2.1097
49. Mo XD, Zhang XH, Xu LP, Wang Y, Yan CH, Chen H, et al. IFN-Alpha Is Effective for Treatment of Minimal Residual Disease in Patients With Acute Leukemia After Allogeneic Hematopoietic Stem Cell Transplantation: Results of a Registry Study. Biol Blood Marrow Transplant (2017) 23(8):1303–10. doi: 10.1016/j.bbmt.2017.04.023
50. Lin XJ, Dai HP, Wang AJ, Chen F, Ma X, Sun AN, et al. Effects of Preemptive Interferon-Alpha Monotherapy in Acute Leukemia Patients With Relapse Tendency After Allogeneic Hematopoietic Stem Cell Transplantation: A Case-Control Study. Ann Hematol (2018) 97(11):2195–204. doi: 10.1007/s00277-018-3429-z
51. Bezerra ED, Flowers ME, Onstad LE, Chielens D, Radich J, Higano CS. A Phase 2 Study of Alpha Interferon for Molecularly Measurable Residual Disease in Chronic Myeloid Leukemia After Allogeneic Hematopoietic Cell Transplantation. Leuk Lymphoma (2019) 60(11):2754–61. doi: 10.1080/10428194.2019.1605508
52. Cooper N, Rao K, Goulden N, Amrolia P, Veys P. Alpha Interferon Augments the Graft-Versus-Leukaemia Effect of Second Stem Cell Transplants and Donor Lymphocyte Infusions in High-Risk Paediatric Leukaemias. Br J Haematol (2012) 156(4):550–2. doi: 10.1111/j.1365-2141.2011.08889.x
53. Henden AS, Varelias A, Leach J, Sturgeon E, Avery J, Kelly J, et al. Pegylated Interferon-2alpha Invokes Graft-Versus-Leukemia Effects in Patients Relapsing After Allogeneic Stem Cell Transplantation. Blood Adv (2019) 3(20):3013–9. doi: 10.1182/bloodadvances.2019000453
54. Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T, et al. Cd8(+) T Cell-Mediated Skin Disease in Mice Lacking IRF-2, the Transcriptional Attenuator of Interferon-Alpha/Beta Signaling. Immunity (2000) 13(5):643–55. doi: 10.1016/s1074-7613(00)00064-9
55. Hofer MJ, Li W, Manders P, Terry R, Lim SL, King NJ, et al. Mice Deficient in STAT1 But Not STAT2 or IRF9 Develop a Lethal CD4+ T-Cell-Mediated Disease Following Infection With Lymphocytic Choriomeningitis Virus. J Virol (2012) 86(12):6932–46. doi: 10.1128/JVI.07147-11
56. Imamura M, Hashino S, Kobayashi H, Kubayashi S, Hirano S, Minagawa T, et al. Serum Cytokine Levels in Bone Marrow Transplantation: Synergistic Interaction of Interleukin-6, Interferon-Gamma, and Tumor Necrosis Factor-Alpha in Graft-Versus-Host Disease. Bone Marrow Transplant (1994) 13(6):745–51.
57. Takashima S, Martin ML, Jansen SA, Fu Y, Bos J, Chandra D, et al. T Cell-Derived Interferon-Gamma Programs Stem Cell Death in Immune-Mediated Intestinal Damage. Sci Immunol (2019) 4(42):eaay8556. doi: 10.1126/sciimmunol.aay8556
58. Eriguchi Y, Nakamura K, Yokoi Y, Sugimoto R, Takahashi S, Hashimoto D, et al. Essential Role of IFN-gamma in T Cell-Associated Intestinal Inflammation. JCI Insight (2018) 3(18):e121886. doi: 10.1172/jci.insight.121886
59. Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, et al. Ifngammar Signaling Mediates Alloreactive T-Cell Trafficking and GVHD. Blood (2012) 120(19):4093–103. doi: 10.1182/blood-2012-01-403196
60. Bhat P, Leggatt G, Waterhouse N, Frazer IH. Interferon-Gamma Derived From Cytotoxic Lymphocytes Directly Enhances Their Motility and Cytotoxicity. Cell Death Dis (2017) 8(6):e2836. doi: 10.1038/cddis.2017.67
61. Welniak LA, Blazar BR, Anver MR, Wiltrout RH, Murphy WJ. Opposing Roles of Interferon-Gamma on CD4+ T Cell-Mediated Graft-Versus-Host Disease: Effects of Conditioning. Biol Blood Marrow Transplant (2000) 6(6):604–12. doi: 10.1016/s1083-8791(00)70025-5
62. Sun K, Hsiao HH, Li M, Ames E, Bouchlaka M, Welniak LA, et al. IFN-Gamma Receptor-Deficient Donor T Cells Mediate Protection From Graft-Versus-Host Disease and Preserve Graft-Versus-Tumor Responses After Allogeneic Bone Marrow Transplantation. J Immunol (2012) 189(4):2033–42. doi: 10.4049/jimmunol.1102853
63. Badovinac VP, Tvinnereim AR, Harty JT. Regulation of Antigen-Specific CD8+ T Cell Homeostasis by Perforin and Interferon-Gamma. Science (2000) 290(5495):1354–8. doi: 10.1126/science.290.5495.1354
64. Refaeli Y, Van Parijs L, Alexander SI, Abbas AK. Interferon Gamma Is Required for Activation-Induced Death of T Lymphocytes. J Exp Med (2002) 196(7):999–1005. doi: 10.1084/jem.20020666
65. Dalton DK, Haynes L, Chu CQ, Swain SL, Wittmer S. Interferon Gamma Eliminates Responding CD4 T Cells During Mycobacterial Infection by Inducing Apoptosis of Activated CD4 T Cells. J Exp Med (2000) 192(1):117–22. doi: 10.1084/jem.192.1.117
66. Saha A, O’Connor RS, Thangavelu G, Lovitch SB, Dandamudi DB, Wilson CB, et al. Programmed Death Ligand-1 Expression on Donor T Cells Drives Graft-Versus-Host Disease Lethality. J Clin Invest (2016) 126(7):2642–60. doi: 10.1172/JCI85796
67. Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 Mediate Acute Graft-Versus-Host Disease, Each With Distinct End-Organ Targets. J Clin Invest (2000) 105(9):1289–98. doi: 10.1172/JCI7894
68. Yi T, Chen Y, Wang L, Du G, Huang D, Zhao D, et al. Reciprocal Differentiation and Tissue-Specific Pathogenesis of Th1, Th2, and Th17 Cells in Graft-Versus-Host Disease. Blood (2009) 114(14):3101–12. doi: 10.1182/blood-2009-05-219402
69. Maldonado RA, Soriano MA, Perdomo LC, Sigrist K, Irvine DJ, Decker T, et al. Control of T Helper Cell Differentiation Through Cytokine Receptor Inclusion in the Immunological Synapse. J Exp Med (2009) 206(4):877–92. doi: 10.1084/jem.20082900
70. Parampalli Yajnanarayana S, Stubig T, Cornez I, Alchalby H, Schonberg K, Rudolph J, et al. JAK1/2 Inhibition Impairs T Cell Function In Vitro and in Patients With Myeloproliferative Neoplasms. Br J Haematol (2015) 169(6):824–33. doi: 10.1111/bjh.13373
71. Choi J, Cooper ML, Alahmari B, Ritchey J, Collins L, Holt M, et al. Pharmacologic Blockade of JAK1/JAK2 Reduces GvHD and Preserves the Graft-Versus-Leukemia Effect. PloS One (2014) 9(10):e109799. doi: 10.1371/journal.pone.0109799
72. Carniti C, Gimondi S, Vendramin A, Recordati C, Confalonieri D, Bermema A, et al. Pharmacologic Inhibition of JAK1/JAK2 Signaling Reduces Experimental Murine Acute GVHD While Preserving Gvt Effects. Clin Cancer Res (2015) 21(16):3740–9. doi: 10.1158/1078-0432.CCR-14-2758
73. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of Therapeutic JAK 1/2 Blockade in Graft-Versus-Host Disease. Blood (2014) 123(24):3832–42. doi: 10.1182/blood-2013-12-543736
74. Choi J, Cooper ML, Staser K, Ashami K, Vij KR, Wang B, et al. Baricitinib-Induced Blockade of Interferon Gamma Receptor and Interleukin-6 Receptor for the Prevention and Treatment of Graft-Versus-Host Disease. Leukemia (2018) 32(11):2483–94. doi: 10.1038/s41375-018-0123-z
75. Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, et al. Interferon-Gamma Regulates Intestinal Epithelial Homeostasis Through Converging Beta-Catenin Signaling Pathways. Immunity (2010) 32(3):392–402. doi: 10.1016/j.immuni.2010.03.001
76. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-Lambdas Mediate Antiviral Protection Through a Distinct Class II Cytokine Receptor Complex. Nat Immunol (2003) 4(1):69–77. doi: 10.1038/ni875
77. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. Il-28, IL-29 and Their Class II Cytokine Receptor IL-28R. Nat Immunol (2003) 4(1):63–8. doi: 10.1038/ni873
78. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A Variant Upstream of IFNL3 (IL28B) Creating a New Interferon Gene IFNL4 Is Associated With Impaired Clearance of Hepatitis C Virus. Nat Genet (2013) 45(2):164–71. doi: 10.1038/ng.2521
79. Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, et al. IFN-Lambda Determines the Intestinal Epithelial Antiviral Host Defense. Proc Natl Acad Sci USA (2011) 108(19):7944–9. doi: 10.1073/pnas.1100552108
80. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-Lambda (IFN-Lambda) Is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PloS Pathog (2008) 4(3):e1000017. doi: 10.1371/journal.ppat.1000017
81. Lazear HM, Schoggins JW, Diamond MS. Shared and Distinct Functions of Type I and Type III Interferons. Immunity (2019) 50(4):907–23. doi: 10.1016/j.immuni.2019.03.025
82. Fischer JC, Lin CC, Heidegger S, Wintges A, Schlapschy M, Beudert M, et al. Regeneration After Radiation- and Immune-Mediated Tissue Injury Is Not Enhanced by Type Iii Interferon Signaling. Int J Radiat Oncol Biol Phys (2019) 103(4):970–6. doi: 10.1016/j.ijrobp.2018.11.038
83. Henden AS, Koyama M, Robb RJ, Forero A, Kuns RD, Chang K, et al. Ifnlambda Therapy Prevents Severe Gastrointestinal Graft-versus-Host Disease. Blood (2021). doi: 10.1182/blood.2020006375
84. Bhan A, Deb P, Mandal SS. Epigenetic Code. Gene Regul Epigenet Hormone Signaling (2017) . p:29–58. doi: 10.1002/9783527697274.ch2
85. Toubai T, Mathewson N, Reddy P. The Role of Dendritic Cells in Graft-Versus-Tumor Effect. Front Immunol (2014) 5:66. doi: 10.3389/fimmu.2014.00066
86. Tanabe Y, Nishibori T, Su L, Arduini RM, Baker DP, David M. Cutting Edge: Role of STAT1, STAT3, and STAT5 in IFN-Alpha Beta Responses in T Lymphocytes. J Immunol (2005) 174(2):609–13. doi: 10.4049/jimmunol.174.2.609
87. Stark GR, Darnell JE Jr. The JAK-STAT Pathway at Twenty. Immunity (2012) 36(4):503–14. doi: 10.1016/j.immuni.2012.03.013
88. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, et al. Gene Regulation and Chromatin Remodeling by IL-12 and Type I IFN in Programming for CD8 T Cell Effector Function and Memory. J Immunol (2009) 183(3):1695–704. doi: 10.4049/jimmunol.0900592
89. Martinet V, Tonon S, Torres D, Azouz A, Nguyen M, Kohler A, et al. Type I Interferons Regulate Eomesodermin Expression and the Development of Unconventional Memory CD8(+) T Cells. Nat Commun (2015) 6:7089. doi: 10.1038/ncomms8089
90. Iwata S, Mikami Y, Sun HW, Brooks SR, Jankovic D, Hirahara K, et al. The Transcription Factor T-Bet Limits Amplification of Type I Ifn Transcriptome and Circuitry in T Helper 1 Cells. Immunity (2017) 46(6):983–91.e4. doi: 10.1016/j.immuni.2017.05.005
91. Miller S, Tsou PS, Coit P, Gensterblum-Miller E, Renauer P, Rohraff DM, et al. Hypomethylation of STAT1 and HLA-DRB1 Is Associated With Type-I Interferon-Dependent HLA-DRB1 Expression in Lupus CD8+ T Cells. Ann Rheum Dis (2019) 78(4):519–28. doi: 10.1136/annrheumdis-2018-214323
92. Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, et al. The microRNA miR-155 Controls CD8(+) T Cell Responses by Regulating Interferon Signaling. Nat Immunol (2013) 14(6):593–602. doi: 10.1038/ni.2576
93. Yang R, Cheng S, Luo N, Gao R, Yu K, Kang B, et al. Distinct Epigenetic Features of Tumor-Reactive CD8+ T Cells in Colorectal Cancer Patients Revealed by Genome-Wide DNA Methylation Analysis. Genome Biol (2019) 21(1):2. doi: 10.1186/s13059-019-1921-y
94. Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM, et al. Rapid Demethylation of the IFN-Gamma Gene Occurs in Memory But Not Naive CD8 T Cells. J Immunol (2006) 176(7):4083–93. doi: 10.4049/jimmunol.176.7.4083
95. Komori HK, Hart T, LaMere SA, Chew PV, Salomon DR. Defining CD4 T Cell Memory by the Epigenetic Landscape of CpG DNA Methylation. J Immunol (2015) 194(4):1565–79. doi: 10.4049/jimmunol.1401162
96. Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, et al. Comprehensive Epigenetic Profiling Identifies Multiple Distal Regulatory Elements Directing Transcription of the Gene Encoding Interferon-Gamma. Nat Immunol (2007) 8(7):732–42. doi: 10.1038/ni1474
97. Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, et al. The Methylcytosine Dioxygenase Tet2 Promotes DNA Demethylation and Activation of Cytokine Gene Expression in T Cells. Immunity (2015) 42(4):613–26. doi: 10.1016/j.immuni.2015.03.005
98. Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. T(H) Cell Differentiation Is Accompanied by Dynamic Changes in Histone Acetylation of Cytokine Genes. Nat Immunol (2002) 3(7):643–51. doi: 10.1038/ni808
99. Allan RS, Zueva E, Cammas F, Schreiber HA, Masson V, Belz GT, et al. An Epigenetic Silencing Pathway Controlling T Helper 2 Cell Lineage Commitment. Nature (2012) 487(7406):249–53. doi: 10.1038/nature11173
100. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic Control of CD8(+) T Cell Differentiation. Nat Rev Immunol (2018) 18(5):340–56. doi: 10.1038/nri.2017.146
101. Yerinde C, Siegmund B, Glauben R, Weidinger C. Metabolic Control of Epigenetics and Its Role in CD8(+) T Cell Differentiation and Function. Front Immunol (2019) 10:2718. doi: 10.3389/fimmu.2019.02718
102. He S, Xie F, Liu Y, Tong Q, Mochizuki K, Lapinski PE, et al. The Histone Methyltransferase Ezh2 Is a Crucial Epigenetic Regulator of Allogeneic T-Cell Responses Mediating Graft-Versus-Host Disease. Blood (2013) 122(25):4119–28. doi: 10.1182/blood-2013-05-505180
103. Kagoya Y, Nakatsugawa M, Saso K, Guo T, Anczurowski M, Wang CH, et al. DOT1L Inhibition Attenuates Graft-Versus-Host Disease by Allogeneic T Cells in Adoptive Immunotherapy Models. Nat Commun (2018) 9(1):1915. doi: 10.1038/s41467-018-04262-0
104. Stubig T, Badbaran A, Luetkens T, Hildebrandt Y, Atanackovic D, Binder TM, et al. 5-Azacytidine Promotes an Inhibitory T-cell Phenotype and Impairs Immune Mediated Antileukemic Activity. Mediators Inflamm (2014) 2014:418292. doi: 10.1155/2014/418292
105. Nast R, Choepak T, Luder CGK. Epigenetic Control of IFN-Gamma Host Responses During Infection With Toxoplasma Gondii. Front Immunol (2020) 11:581241. doi: 10.3389/fimmu.2020.581241
106. Li A, Abraham C, Wang Y, Zhang Y. New Insights Into the Basic Biology of Acute Graft-Versus-Host-Disease. Haematologica (2020) 105(11):2540–9. doi: 10.3324/haematol.2019.240291
107. Gamper CJ, Agoston AT, Nelson WG, Powell JD. Identification of DNA Methyltransferase 3a as a T Cell Receptor-Induced Regulator of Th1 and Th2 Differentiation. J Immunol (2009) 183(4):2267–76. doi: 10.4049/jimmunol.0802960
108. Winders BR, Schwartz RH, Bruniquel D. A Distinct Region of the Murine IFN-Gamma Promoter Is Hypomethylated From Early T Cell Development Through Mature Naive and Th1 Cell Differentiation, But Is Hypermethylated in Th2 Cells. J Immunol (2004) 173(12):7377–84. doi: 10.4049/jimmunol.173.12.7377
109. Thomas RM, Gamper CJ, Ladle BH, Powell JD, Wells AD. De Novo DNA Methylation Is Required to Restrict T Helper Lineage Plasticity. J Biol Chem (2012) 287(27):22900–9. doi: 10.1074/jbc.M111.312785
110. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell (2017) 170(1):142–57 e19. doi: 10.1016/j.cell.2017.06.007
111. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
112. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA Methylation Controls Foxp3 Gene Expression. Eur J Immunol (2008) 38(6):1654–63. doi: 10.1002/eji.200838105
113. Wang X, Wang J, Yu Y, Ma T, Chen P, Zhou B, et al. Decitabine Inhibits T Cell Proliferation Via a Novel TET2-dependent Mechanism and Exerts Potent Protective Effect in Mouse Auto- and Allo-Immunity Models. Oncotarget (2017) 8(34):56802–15. doi: 10.18632/oncotarget.18063
114. Choi J, Ritchey J, Prior JL, Holt M, Shannon WD, Deych E, et al. In Vivo Administration of Hypomethylating Agents Mitigate Graft-Versus-Host Disease Without Sacrificing Graft-Versus-Leukemia. Blood (2010) 116(1):129–39. doi: 10.1182/blood-2009-12-257253
115. Cooper ML, Choi J, Karpova D, Vij K, Ritchey J, Schroeder MA, et al. Azacitidine Mitigates Graft-Versus-Host Disease Via Differential Effects on the Proliferation of T Effectors and Natural Regulatory T Cells In Vivo. J Immunol (2017) 198(9):3746–54. doi: 10.4049/jimmunol.1502399
116. Ehx G, Fransolet G, de Leval L, D’Hondt S, Lucas S, Hannon M, et al. Azacytidine Prevents Experimental Xenogeneic Graft-Versus-Host Disease Without Abrogating Graft-Versus-Leukemia Effects. Oncoimmunology (2017) 6(5):e1314425. doi: 10.1080/2162402X.2017.1314425
117. Field T, Perkins J, Huang Y, Kharfan-Dabaja MA, Alsina M, Ayala E, et al. 5-Azacitidine for Myelodysplasia Before Allogeneic Hematopoietic Cell Transplantation. Bone Marrow Transplant (2010) 45(2):255–60. doi: 10.1038/bmt.2009.134
118. Lubbert M, Bertz H, Ruter B, Marks R, Claus R, Wasch R, et al. Non-Intensive Treatment With Low-Dose 5-Aza-2’-Deoxycytidine (DAC) Prior to Allogeneic Blood SCT of Older MDS/AML Patients. Bone Marrow Transplant (2009) 44(9):585–8. doi: 10.1038/bmt.2009.64
119. Oran B, de Lima M, Garcia-Manero G, Thall PF, Lin R, Popat U, et al. A Phase 3 Randomized Study of 5-Azacitidine Maintenance vs Observation After Transplant in High-Risk AML and MDS Patients. Blood Adv (2020) 4(21):5580–8. doi: 10.1182/bloodadvances.2020002544
120. Greer EL, Shi Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat Rev Genet (2012) 13(5):343–57. doi: 10.1038/nrg3173
121. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell (2007) 129(4):823–37. doi: 10.1016/j.cell.2007.05.009
122. Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic Maps and Comparative Analysis of Histone Modifications in Human and Mouse. Cell (2005) 120(2):169–81. doi: 10.1016/j.cell.2005.01.001
123. Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Spatial Distribution of Di- and Tri-Methyl Lysine 36 of Histone H3 at Active Genes. J Biol Chem (2005) 280(18):17732–6. doi: 10.1074/jbc.M500796200
124. Jambhekar A, Dhall A, Shi Y. Roles and Regulation of Histone Methylation in Animal Development. Nat Rev Mol Cell Biol (2019) 20(10):625–41. doi: 10.1038/s41580-019-0151-1
125. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global Mapping of H3K4me3 and H3K27me3 Reveals Specificity and Plasticity in Lineage Fate Determination of Differentiating CD4+ T Cells. Immunity (2009) 30(1):155–67. doi: 10.1016/j.immuni.2008.12.009
126. Denton AE, Russ BE, Doherty PC, Rao S, Turner SJ. Differentiation-Dependent Functional and Epigenetic Landscapes for Cytokine Genes in Virus-Specific CD8+ T Cells. Proc Natl Acad Sci U.S.A. (2011) 108(37):15306–11. doi: 10.1073/pnas.1112520108
127. Zediak VP, Johnnidis JB, Wherry EJ, Berger SL. Cutting Edge: Persistently Open Chromatin at Effector Gene Loci in Resting Memory CD8+ T Cells Independent of Transcriptional Status. J Immunol (2011) 186(5):2705–9. doi: 10.4049/jimmunol.1003741
128. Li Q, Zou J, Wang M, Ding X, Chepelev I, Zhou X, et al. Critical Role of Histone Demethylase Jmjd3 in the Regulation of CD4+ T-Cell Differentiation. Nat Commun (2014) 5:5780. doi: 10.1038/ncomms6780
129. Araki Y, Wang Z, Zang C, Wood WH 3rd, Schones D, Cui K, et al. Genome-Wide Analysis of Histone Methylation Reveals Chromatin State-Based Regulation of Gene Transcription and Function of Memory CD8+ T Cells. Immunity (2009) 30(6):912–25. doi: 10.1016/j.immuni.2009.05.006
130. Chang S, Aune TM. Dynamic Changes in Histone-Methylation ‘Marks’ Across the Locus Encoding Interferon-Gamma During the Differentiation of T Helper Type 2 Cells. Nat Immunol (2007) 8(7):723–31. doi: 10.1038/ni1473
131. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The Polycomb Protein Ezh2 Regulates Differentiation and Plasticity of CD4(+) T Helper Type 1 and Type 2 Cells. Immunity (2013) 39(5):819–32. doi: 10.1016/j.immuni.2013.09.012
132. Yang XP, Jiang K, Hirahara K, Vahedi G, Afzali B, Sciume G, et al. EZH2 Is Crucial for Both Differentiation of Regulatory T Cells and T Effector Cell Expansion. Sci Rep (2015) 5:10643. doi: 10.1038/srep10643
133. Zhang Y, Kinkel S, Maksimovic J, Bandala-Sanchez E, Tanzer MC, Naselli G, et al. The Polycomb Repressive Complex 2 Governs Life and Death of Peripheral T Cells. Blood (2014) 124(5):737–49. doi: 10.1182/blood-2013-12-544106
134. Tong Q, He S, Xie F, Mochizuki K, Liu Y, Mochizuki I, et al. Ezh2 Regulates Transcriptional and Posttranslational Expression of T-Bet and Promotes Th1 Cell Responses Mediating Aplastic Anemia in Mice. J Immunol (2014) 192(11):5012–22. doi: 10.4049/jimmunol.1302943
135. He S, Liu Y, Meng L, Sun H, Wang Y, Ji Y, et al. Ezh2 Phosphorylation State Determines Its Capacity to Maintain CD8(+) T Memory Precursors for Antitumor Immunity. Nat Commun (2017) 8(1):2125. doi: 10.1038/s41467-017-02187-8
136. Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, et al. Early Transcriptional and Epigenetic Regulation of CD8(+) T Cell Differentiation Revealed by Single-Cell RNA Sequencing. Nat Immunol (2017) 18(4):422–32. doi: 10.1038/ni.3688
137. Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation-Induced Repression of Chromatin Bound by the Transcription Factor Foxp3 in Regulatory T Cells. Nat Immunol (2014) 15(6):580–7. doi: 10.1038/ni.2868
138. Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, et al. DOT1L/KMT4 Recruitment and H3K79 Methylation Are Ubiquitously Coupled With Gene Transcription in Mammalian Cells. Mol Cell Biol (2008) 28(8):2825–39. doi: 10.1128/MCB.02076-07
139. Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial Patterns of Histone Acetylations and Methylations in the Human Genome. Nat Genet (2008) 40(7):897–903. doi: 10.1038/ng.154
140. Wood K, Tellier M, Murphy S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules (2018) 8(1):11. doi: 10.3390/biom8010011
141. Cao K, Ugarenko M, Ozark PA, Wang J, Marshall SA, Rendleman EJ, et al. DOT1L-Controlled Cell-Fate Determination and Transcription Elongation Are Independent of H3K79 Methylation. Proc Natl Acad Sci USA (2020) 117(44):27365–73. doi: 10.1073/pnas.2001075117
142. Scheer S, Ackloo S, Medina TS, Schapira M, Li F, Ward JA, et al. A Chemical Biology Toolbox to Study Protein Methyltransferases and Epigenetic Signaling. Nat Commun (2019) 10(1):19. doi: 10.1038/s41467-018-07905-4
143. Scheer S, Runting J, Bramhall M, Russ B, Zaini A, Ellemor J, et al. The Methyltransferase Dot1l Controls Activation and Lineage Integrity in CD4(+) T Cells During Infection and Inflammation. Cell Rep (2020) 33(11):108505. doi: 10.1016/j.celrep.2020.108505
144. Kwesi-Maliepaard EM, Aslam MA, Alemdehy MF, van den Brand T, McLean C, Vlaming H, et al. The Histone Methyltransferase DOT1L Prevents Antigen-Independent Differentiation and Safeguards Epigenetic Identity of CD8(+) T Cells. Proc Natl Acad Sci USA (2020) 117(34):20706–16. doi: 10.1073/pnas.1920372117
145. Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer SLC43A2 Alters T Cell Methionine Metabolism and Histone Methylation. Nature (2020) 585(7824):277–82. doi: 10.1038/s41586-020-2682-1
146. Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of Histone H3 Lysine 9 Creates a Binding Site for HP1 Proteins. Nature (2001) 410(6824):116–20. doi: 10.1038/35065132
147. Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. G9a Histone Methyltransferase Plays a Dominant Role in Euchromatic Histone H3 Lysine 9 Methylation and Is Essential for Early Embryogenesis. Genes Dev (2002) 16(14):1779–91. doi: 10.1101/gad.989402
148. Zeng W, Ball AR Jr, Yokomori K. HP1: Heterochromatin Binding Proteins Working the Genome. Epigenetics (2010) 5(4):287–92. doi: 10.4161/epi.5.4.11683
149. Lehnertz B, Northrop JP, Antignano F, Burrows K, Hadidi S, Mullaly SC, et al. Activating and Inhibitory Functions for the Histone Lysine Methyltransferase G9a in T Helper Cell Differentiation and Function. J Exp Med (2010) 207(5):915–22. doi: 10.1084/jem.20100363
150. Scheer S, Zaph C. The Lysine Methyltransferase G9a in Immune Cell Differentiation and Function. Front Immunol (2017) 8:429. doi: 10.3389/fimmu.2017.00429
151. Verbaro DJ, Sakurai N, Kim B, Shinkai Y, Egawa T. Cutting Edge: The Histone Methyltransferase G9a Is Required for Silencing of Helper T Lineage-Associated Genes in Proliferating Cd8 T Cells. J Immunol (2018) 200(12):3891–6. doi: 10.4049/jimmunol.1701700
152. Fang TC, Schaefer U, Mecklenbrauker I, Stienen A, Dewell S, Chen MS, et al. Histone H3 Lysine 9 Di-Methylation as an Epigenetic Signature of the Interferon Response. J Exp Med (2012) 209(4):661–9. doi: 10.1084/jem.20112343
153. Adoue V, Binet B, Malbec A, Fourquet J, Romagnoli P, van Meerwijk JPM, et al. The Histone Methyltransferase SETDB1 Controls T Helper Cell Lineage Integrity by Repressing Endogenous Retroviruses. Immunity (2019) 50(3):629–44.e8. doi: 10.1016/j.immuni.2019.01.003
154. Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, et al. The Epigenetic Control of Stemness in CD8(+) T Cell Fate Commitment. Science (2018) 359(6372):177–86. doi: 10.1126/science.aah6499
155. Lee DY, Hayes JJ, Pruss D, Wolffe AP. A Positive Role for Histone Acetylation in Transcription Factor Access to Nucleosomal DNA. Cell (1993) 72(1):73–84. doi: 10.1016/0092-8674(93)90051-q
157. Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA, et al. Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Reduces Acute Graft-Versus-Host Disease and Preserves Graft-Versus-Leukemia Effect. Proc Natl Acad Sci USA (2004) 101(11):3921–6. doi: 10.1073/pnas.0400380101
158. Leng C, Gries M, Ziegler J, Lokshin A, Mascagni P, Lentzsch S, et al. Reduction of Graft-Versus-Host Disease by Histone Deacetylase Inhibitor Suberonylanilide Hydroxamic Acid Is Associated With Modulation of Inflammatory Cytokine Milieu and Involves Inhibition of STAT1. Exp Hematol (2006) 34(6):776–87. doi: 10.1016/j.exphem.2006.02.014
159. Choi SW, Braun T, Chang L, Ferrara JL, Pawarode A, Magenau JM, et al. Vorinostat Plus Tacrolimus and Mycophenolate to Prevent Graft-Versus-Host Disease After Related-Donor Reduced-Intensity Conditioning Allogeneic Haemopoietic Stem-Cell Transplantation: A Phase 1/2 Trial. Lancet Oncol (2014) 15(1):87–95. doi: 10.1016/S1470-2045(13)70512-6
160. Choi SW, Braun T, Henig I, Gatza E, Magenau J, Parkin B, et al. Vorinostat Plus Tacrolimus/Methotrexate to Prevent GVHD After Myeloablative Conditioning, Unrelated Donor HCT. Blood (2017) 130(15):1760–7. doi: 10.1182/blood-2017-06-790469
161. Perez L, Fernandez H, Horna P, Riches M, Locke F, Field T, et al. Phase I Trial of Histone Deacetylase Inhibitor Panobinostat in Addition to Glucocorticoids for Primary Therapy of Acute Graft-Versus-Host Disease. Bone Marrow Transplant (2018) 53(11):1434–44. doi: 10.1038/s41409-018-0163-z
162. Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, et al. Conditional Deletion of Histone Deacetylase 1 in T Cells Leads to Enhanced Airway Inflammation and Increased Th2 Cytokine Production. J Immunol (2010) 185(6):3489–97. doi: 10.4049/jimmunol.0903610
163. Goschl L, Preglej T, Hamminger P, Bonelli M, Andersen L, Boucheron N, et al. A T Cell-Specific Deletion of HDAC1 Protects Against Experimental Autoimmune Encephalomyelitis. J Autoimmun (2018) 86:51–61. doi: 10.1016/j.jaut.2017.09.008
164. Tschismarov R, Firner S, Gil-Cruz C, Goschl L, Boucheron N, Steiner G, et al. HDAC1 Controls CD8+ T Cell Homeostasis and Antiviral Response. PloS One (2014) 9(10):e110576. doi: 10.1371/journal.pone.0110576
165. Myers DR, Lau T, Markegard E, Lim HW, Kasler H, Zhu M, et al. Tonic LAT-HDAC7 Signals Sustain Nur77 and Irf4 Expression to Tune Naive Cd4 T Cells. Cell Rep (2017) 19(8):1558–71. doi: 10.1016/j.celrep.2017.04.076
166. Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, et al. The Type III Histone Deacetylase Sirt1 Is Essential for Maintenance of T Cell Tolerance in Mice. J Clin Invest (2009) 119(10):3048–58. doi: 10.1172/JCI38902
167. Woods DM, Woan KV, Cheng F, Sodre AL, Wang D, Wu Y, et al. T Cells Lacking HDAC11 Have Increased Effector Functions and Mediate Enhanced Alloreactivity in a Murine Model. Blood (2017) 130(2):146–55. doi: 10.1182/blood-2016-08-731505
168. Xiao H, Jiao J, Wang L, O’Brien S, Newick K, Wang LC, et al. HDAC5 Controls the Functions of Foxp3(+) T-Regulatory and CD8(+) T Cells. Int J Cancer (2016) 138(10):2477–86. doi: 10.1002/ijc.29979
169. Daenthanasanmak A, Iamsawat S, Chakraborty P, Nguyen HD, Bastian D, Liu C, et al. Targeting Sirt-1 Controls GVHD by Inhibiting T-Cell Allo-Response and Promoting Treg Stability in Mice. Blood (2019) 133(3):266–79. doi: 10.1182/blood-2018-07-863233
Keywords: type I interferon, IFN- γ, GVHD, epigenetic regulation, alloreactive T cells
Citation: Zhao C, Zhang Y and Zheng H (2021) The Effects of Interferons on Allogeneic T Cell Response in GVHD: The Multifaced Biology and Epigenetic Regulations. Front. Immunol. 12:717540. doi: 10.3389/fimmu.2021.717540
Received: 31 May 2021; Accepted: 25 June 2021;
Published: 08 July 2021.
Edited by:
Jaebok Choi, Washington University School of Medicine in St. Louis, United StatesReviewed by:
Cameron Scott Bader, Stanford University, United StatesDaniel Peltier, University of Michigan, United States
Copyright © 2021 Zhao, Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Zhang, eWkuemhhbmdAdGVtcGxlLmVkdQ==; Hong Zheng, aHpoZW5nQHBlbm5zdGF0ZWhlYWx0aC5wc3UuZWR1