 Yonggang Zhou
Yonggang Zhou Xiuxiu Xu
Xiuxiu Xu Haiming Wei
Haiming Wei
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 October 2021
Sec. Viral Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.716940
This article is part of the Research Topic ARDS in COVID-19 - Insights for Treatment View all 28 articles
At present, the global COVID-19 epidemic is still in a state of anxiety, and increasing the cure rate of critically ill patients is an important means to defeat the virus. From an immune perspective, ARDS driven by an inflammatory storm is still the direct cause of death in severe COVID-19 patients. Although some experience has been gained in the treatment of COVID-19, and intensive COVID-19 vaccination has been carried out recently, it is still effective to save lives to develop more effective programs to alleviate the inflammatory storm and ARDS in patients with SARS-CoV-2 or emerging variants of SARS-CoV-2. In reorganizing the ARDS-related inflammatory storm formation program in COVID-19 patients, we highlighted the importance of the vicious circle of inflammatory cytokines and inflammatory cell death, which is aggravated by blood circulation to form multi-system inflammation. Summarizes the interlocking and crisscrossing of inflammatory response and inflammatory cell death mechanisms including NETs, pyrolysis, apoptosis and PANoptosis in severe COVID-19. More importantly, in response to the inflammatory storm formation program we described, and on the premise of following ethical and clinical experimental norms, we propose a three-dimensional integrated program for future research based on boosting antiviral immune response at the initial stage, inhibiting inflammatory cytokine signaling at the exacerbation stage and inhibiting cell death before it’s worse to prevent and alleviate ARDS.
In the past 20 years, various coronaviruses including severe acute respiratory syndrome coronavirus (SARS-CoV) in 2003 (1), middle east respiratory syndrome coronavirus (MERS-CoV) in 2012 (2) and SARS-CoV-2 in 2020 (3, 4) have caused many health crises in different countries and regions around the world. The genome sequence of SARS-CoV-2 has nearly 80% homology with SARS-CoV and about 50% with MERS-CoV (5). In the latest and more extensive screening, samples of pneumonia patients collected from Sarawak Regional Hospital in Malaysia in the past few years were found to be canine-derived coronavirus infections (6) and in Haitian children, pig-derived delta coronaviruses were also found (7). The global pandemic of acute infectious pneumonia named “Coronavirus disease 2019 (COVID-19)”caused by SARS-CoV-2, is by far the most widespread, longest lasting and worst example among them.
From the beginning of 2020 to the present, the COVID-19 epidemic has risen and fallen throughout the world in just over a year, and the latest round of the epidemic caused by the rapid and continuous mutation of SARS-CoV-2 has also broken out (8). Globally, as of 16 September 2021, there have been exceeded 226 million confirmed cases of COVID-19, including >4.65 million deaths, reported to the World Health Organization (WHO). As a result, the overall prevalence of mortality in COVID-19 patients was ~2% (4.65/226). Although billions doses of vaccination that have been completed worldwide have brought hope to control the epidemic, the speed of vaccination is still a few days away from the universal immune barrier. The recent report of the centers for disease control and prevention in the United States also showed cases of COVID-19 vaccine breakthrough infections. And recent studies have also shown that existing vaccines, including mRNA vaccines, adenovirus vaccines, inactivated vaccines and RBD-subunit vaccines, have reduced the neutralizing activity against the SARS-CoV-2 mutant strains (9, 10). In addition, we still lack specific anti-SARS-CoV-2 drugs. Therefore, while developing a broader-spectrum vaccine and wonder drugs, we should further study the mechanism of death caused by COVID-19 and develop more effective treatment options.
The spectrum of COVID-19 presentations ranges from the asymptomatic infection, to a mild self-limiting influenza-like illness, to life-threatening multiorgan failure (11–13). Most COVID-19 patients present mild or moderate symptoms, about 15% of patients develop severe pneumonia and 5% progress to critically ill (14–16). So, reducing the incidence of multiorgan failure is the key to improve the cure rate and reduce the mortality of COVID-19 (17, 18). A large number of inflammatory macrophage infiltration and the distribution of inflammatory cytokines such as interleukin (IL)-1β, IL-6, and IL-18, were found in the pulmonary pathology of patients with severe COVID-19 (19–21). The inflammatory storm instigated by pathogenic T cells and inflammatory monocytes was considered to be the key to the severity of COVID-19 (22). These cells release proinflammatory factors represented by granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-6, which recruit more inflammatory cells into the lungs and other organs to form a “cytokine release syndrome”, and the further aggravated inflammatory storm will eventually lead to multiorgan failure and death in patients with severe COVID-19 (22–24). Based on these basic findings, the COVID-19 immunotherapy strategy that targets these inflammatory cytokines or their receptors to relieve the inflammatory storm have benefited patients with COVID-19 in the past year. The results of a Chinese study exploring the treatment of tocilizumab for COVID-19 including 21 patients in the intensive care unit (ICU) were the first to encourage this treatment strategy (25). Clinical trials focusing on blocking IL-6 signaling to treat COVID-19 benefit patients, including IL-6 receptor antagonists [tocilizumab (26, 27) and sarilumab (28)] and IL-6 inhibitors [siltuximab (29)], and tocilizumab needs to be combined with standard antiviral care to be highlighted in the comparison of international multi-center clinical trials (26, 30). Subsequent clinical trials of monoclonal antibody drugs showed that treatments targeting the GM-CSF receptor (mavrilimumab) and the IL-1 receptor (anakinra) were also related to clinical improvement of patients with severe COVID-19 (31, 32). Although these monoclonal antibody drugs that target inflammatory cytokine signals have shown some benefits, they were limited by many complex factors such as drug targets, the treatment time, the dosage, and differences in patient immune responses, and their performance in reducing patient mortality was unsatisfactory (24, 33, 34).
To further improve the COVID-19 immunotherapy strategy to better reduce the risk of patient death, it is necessary to re-analyze the process of COVID-19 inflammatory storm based on recent research findings. Here, we discuss the progression of acute respiratory distress syndrome (ARDS), a typical evolution of severe COVID-19, as a starting point, reorganize the process of severe inflammatory storms, and try to propose a targeted graded treatment plan for a future research based on the combination of antiviral immune response, inflammatory immune response and inflammatory cell death. Although targeting each individual aspect of this three-dimensional program has been shown to be effective, it has to be said that the overall treatment plan is still an idealized strategy. Therefore, to test its superiority and earlier application, we call for the three-dimensional graded treatment plan to be considered for clinical trials under the premise of ethical requirements.
ARDS is a common cause of respiratory failure in critically ill patients and is a severe pulmonary condition that leads to refractory hypoxemia (35, 36). Alveolar surfactant is a foamy substance that can keep the full expansion of the alveoli, which is essential for breathing. In ARDS, lung injury causes fluid to leak into the space between the alveoli and capillaries, and as the pressure increases, fluid builds up inside the alveoli to accumulate and degrade surfactants, forming a typical ARDS characteristic—accumulation of fluid in the lungs, causing the alveoli to collapse (36). These changes prevent the lungs from filling properly with air, disrupting the gas exchange in the lungs, and causing a series of serious cascade reactions that impact the oxygen supply of tissues and organs. Because of this, ARDS usually occurs in life-threatening conditions such as severe pneumonia, sepsis and severe trauma. The incidence of ICU patients worldwide is about 10%, and the mortality rate is as high as 30-40% (37, 38).
Viral infections, especially coronavirus and avian influenza virus (H5N1), cause pneumonia to be one of the main factors leading to ARDS (39). In the coronavirus epidemic caused by the SARS-CoV for the first time in 2003, reports showed that the incidence of ARDS was about 25% (40). ARDS had also occurred in some severe cases and animal models with MERS infection (41). A study from the early days of the COVID-19 epidemic showed that nearly 40% of severe and critical hospitalized patients developed ARDS, and more than half of those diagnosed died from the disease (42). So, ARDS is closely related to death caused by coronavirus infection. In those patients with ARDS who recovered, although the lung function gradually improved within a year or so, it was difficult to recover as before, lung volume was below normal, and scarring was present (14, 42).
In patients with symptomatic coronavirus infection, pulmonary inflammation was activated, and pneumonia develops into ARDS as the inflammation worsens (22, 40–42). More and more evidences suggest that the occurrence of ARDS seems to be less directly due to the infected virus itself and more related to excessive rather than effective inflammation in the body (15, 43, 44). This excessive inflammation is usually manifested as the continuous release of inflammatory factors, which is aggravated by blood circulation to form multi-system inflammation, which is called cytokine storm or inflammatory storm (22–24, 45).
Similar to the common influenza virus, the SARS-CoV-2 enters the respiratory tract of most people, it will also activate the antiviral immune response that causes inflammation, leading to mild symptoms such as sore throat, cough, fever. In some cases, the virus is difficult to control and escapes into the alveoli to stimulate excessive release of inflammatory factors, triggering an inflammatory storm and developing ARDS (Figure 1).
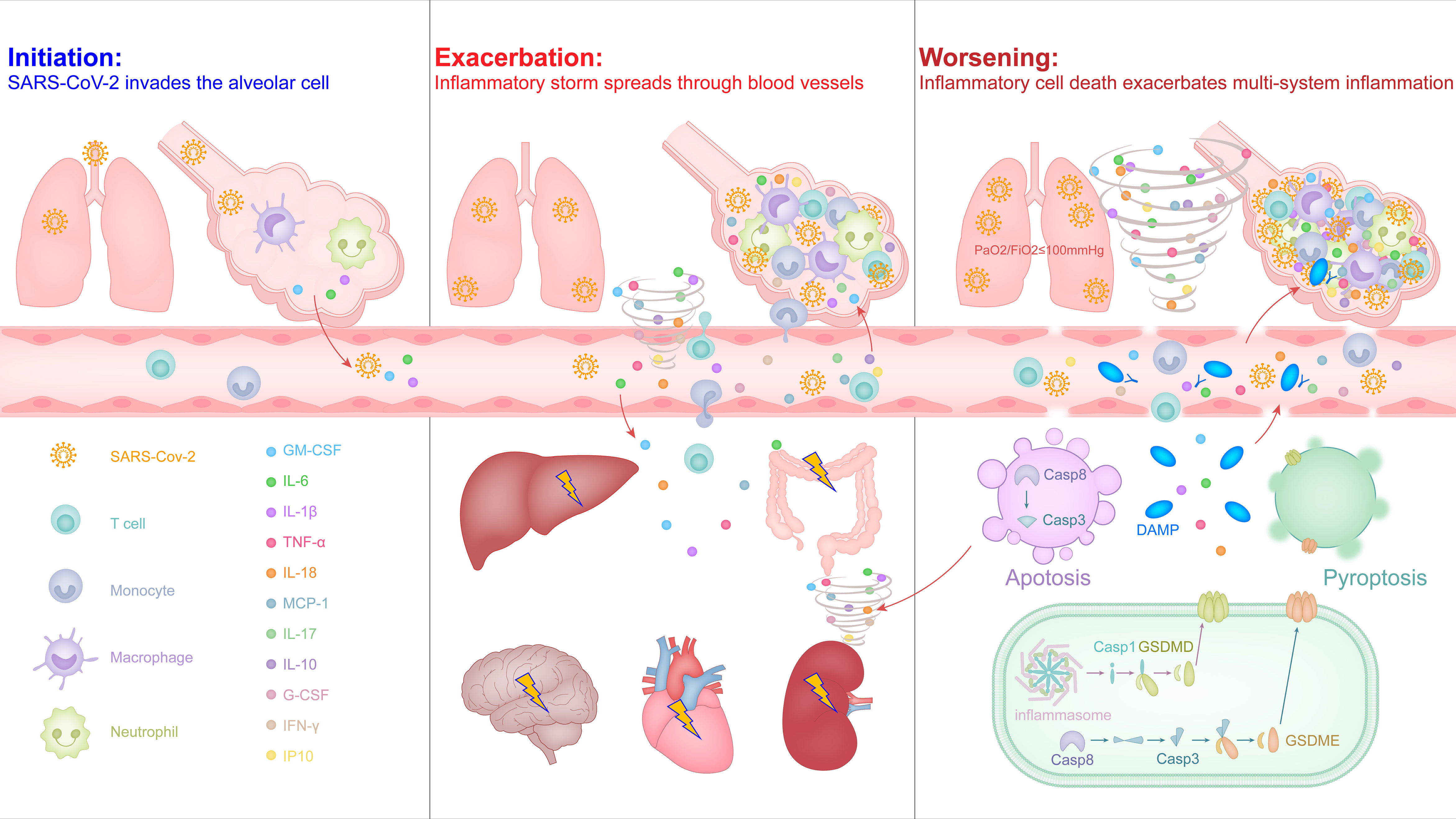
Figure 1 The malignant cycle of inflammatory factors and inflammatory cell death exacerbates the inflammatory storm to trigger ARDS and multiorgan failure in severe COVID-19.
Respiratory droplets are the main carrier of the SARS-CoV-2, and its journey begins in the nose, mouth and eyes, and travels down the alveoli in the lungs (46, 47). SARS-CoV-2 is an enveloped, positive-sense single-stranded RNA virus, which belongs to Betacoronavirus genus and is highly pathogenic (48, 49). SARS-CoV-2 encodes four structural proteins, among which the nucleocapsid (N) protein combines with RNA to form a helical capsid, spike (S), envelope (E), and membrane (M) constitute the viral membrane proteins, of which spike mediates virus entry into host cells (49–51). In the study of SARS-CoV, it has been confirmed that the main receptor of S protein is angiotensin-converting enzyme 2 (ACE2) expressed in type II alveolar epithelial cells (52). The S protein coding gene of SARS-CoV-2 is highly variable with SARS-CoV, and the nucleotide homology is less than 75% (5, 51). The S protein is trimeric-like clover-shaped, with three S1 heads and one trimeric S2 stem, and the receptor-binding domain (RBD) is located at the tip of each S1 head (49, 52, 53). After the RBD in the S protein mediates direct contact with ACE2 on the target cell surface, the transmembrane serine protease (TMPRSS2) cleaves the C-terminal peptide of ACE2 to enhance the virus invasion driven by the S protein (54). In addition, recent studies have shown that the CD147 molecule that can be expressed on most leukocytes, platelets and endothelial cells is also the host receptor for the RBD in S protein of SARS-CoV-2, participating in the interaction between the virus and the target cell and helping the virus invade (55). When the virus successfully infects a type II alveolar epithelial cell, it will inject its own RNA into the cell and achieve a large amount of replication, releasing more virus to infect other target cells nearby.
Due to the need to defend against pathogenic microorganisms brought in by breathing air, the liquid layer on the alveolar surface resides with immune cells, especially macrophages with phagocytic function, which account for more than 95% and are called alveolar macrophages (56). SARS-CoV-2 may directly infect these myeloid cells by binding to the C-type lectin receptor on the surface of cells via the glycosylation sites in the non-RBD region of the S protein, and this recognition mode did not induce the antiviral immune response of interferon, but instead led to the release of a large number of inflammatory factors (57). Macrophages can be polarized into pro-inflammatory M1 macrophages or M2 macrophages that inhibit inflammation, depending on the stimulus conditions they receive. Under physiological conditions, alveolar macrophages exhibit an anti-inflammatory M2 phenotype (56, 58). Recent studies have shown that the endosomal vesicles of M2 type macrophages are slightly alkaline, which can inhibit the separation of SARS-CoV-2 nucleic acid from viral particle components and help lysosomes to degrade the virus (59). This may be one of the reasons why most infected people have mild symptoms and can effectively control the virus in the early stage. In some severe cases, the out-of-control virus induces alveolar cells to release cytokines and higher proportions of M1 macrophages and neutrophils in the bronchoalveolar lavage fluid (60), intended to activate a stronger antiviral immune response, but it also produces a strong inflammatory response and alveolar injury (Figure 1, left). M1 type macrophages are softer and have better phagocytic effects, but the endosomal vesicles of M1 macrophages is acidic, which helps the SARS-CoV-2 nucleic acid to be separated from the viral particle components, thereby helping the virus amplification (20, 59). In addition, it also increases the risk of the virus spreading from M1 macrophages into the blood throughout the body.
The increased inflammation in COVID-19 patients leads to a further increase in body temperature and inflammation-related clinical indicators, such as C-reactive protein, serum ferritin, and IL-6 (13, 16). As inflammation and viruses spread to the blood, T cells are rapidly activated, and over-activated T cells develop into pathogenic T cells, producing factors such as GM-CSF and IL-6 (22). GM-CSF further activates CD14+CD16+ inflammatory monocytes to produce a larger amount of IL-6 and other inflammatory factors (e.g., IL-1β, IL-8, IL-18, and TNF-α), thereby forming an inflammatory storm, leading to severe immune damage in the lungs and other organs (15, 22, 23). Most patients with severe COVID-19 are diagnosed with lymphopenia based on blood routine reports, especially T cells (13, 16). This is not only related to apoptosis or death caused by syncytia after excessive activation of T cells (61), but may also be related to inflammatory infiltration of lungs and other organs. In the histopathological examination of the lungs, heart, intestines, etc. of critically ill patients, significant inflammatory cell infiltration was observed, including inflammatory macrophages, neutrophils, and pathological T cells (19, 20, 62).
Contrary to lymphopenia, the increase of neutrophils in the capillaries or inflammatory tissues of infected patients is also a sign of the severity of COVID-19 (13, 14), and most inflammatory factors can promote the activation of neutrophils (63). Activated neutrophils release cytokines and chemokines, and the networked DNA-protein complex structure forms neutrophil extracellular traps (NETs) to trap and kill pathogenic microorganisms (63, 64). During the formation of NETs, a variety of intracellular damage-associated molecular patterns (DAMPs) are released, activating pattern recognition receptors, causing the surrounding immune or non-immune cells to produce excessive pro-inflammatory cytokines and chemokines; and those released together include histone, DNA, myeloperoxidase (MPO), neutrophil elastase, cathepsin and proteinase-3 and other granular proteins, which cause increasing tissue necrosis (64). In severe COVID-19 patients, NETs-related signaling pathways in lung tissue are up-regulated, and the level of MPO-DNA complex in plasma is higher, suggesting lung tissue damage and platelet-triggered NETs formation is related (65, 66). Activated neutrophils can also activate complement by releasing NETs to cause endothelial damage and necrotizing inflammation, and further promote venous thrombosis (64). In addition, NETs can activate platelets through extracellular DNA and provide a scaffold for the combination of red blood cells and activated platelets, thereby promoting a wider connective network and amplifying the formation of immune thrombi (65, 67). Indeed, a recent study showed that SARS-CoV-2 can also directly infect vascular endothelial cells with the accumulation of inflammatory monocytes (e.g., neutrophils) in multiple organs of patients with severe COVID-19, such as lungs, heart, kidney, small intestine and liver (68). Patients with severe COVID-19 also have clinical symptoms of disseminated intravascular coagulation with elevated serum D-dimer and prolonged prothrombin times (14, 16, 69). Together, it is reasonable to believe that the direct attack of the virus and the infiltration of inflammatory immune cells caused by the infiltration of vascular endothelial cells will loosen the tight junctions of vascular endothelial cells, thereby promoting the spread of vascular leakage and inflammatory storms to multiple organs throughout the body through the circulatory system, and further aggravating lung damage (Figure 1, middle).
Although cell death (e.g., pyroptosis, apoptosis, and necroptosis) is an important mechanism for controlling pathogenic microbial infections, inflammatory cell death also leads to the release of inflammatory factors and cell contents, including alarmins and DAMPs, which causes severe inflammatory responses (64, 70).
Pyroptosis is a form of inflammatory cell death that is mediated by the caspases-inflammasome or -gasdermin cascade, which manifests as the continuous expansion of cells until the cell membrane ruptures, resulting in the release of cell contents and activating a strong inflammatory response (71). Pyrolysis is also the main mechanism for the release of non-signal peptide inflammatory factors, such as the release of IL-1β or IL-18 depends on the caspase-1-dependent gasdermin D cascade (72). In the lung pathology and peripheral blood from patients with severe COVID-19, it was also observed that the pyrolysis of macrophages led to the release of the IL-1β and IL-18 by NLRP3 inflammasome activation and cleavage of caspase-1 (20, 73, 74).
Apoptosis was originally thought to be a non-inflammatory form of cell death, which breaks down cells through membrane vesicles to avoid direct release of cell contents. However, more and more recent evidence shows that due to the crosstalk between the caspase family of apoptotic proteins and the gasdermin family of lysing cell executors, apoptosis is not always inflammatory silent (75–77). For example, in the caspase cascade that drives the onset of apoptosis, caspase 3 can cleave gasdermin E and caspase 8 can cleave gasdermin D to lyse cells under special conditions (75, 77), such as the ORF3a protein stimulation of SARS-CoV-2 (78). SARS-CoV-2 can also induce airway epithelial cells to show apoptosis and cytopathic characteristics (79).
Compared with the release of NETs by neutrophils, more DAMPs are released due to the inflammatory death of cells induced by thrombus and tissue damage (80). High levels of endogenous DAMP molecule S100A8/A9, HMGB1 and lactate dehydrogenase can be detected in the serum of severe COVID-19 patients (81, 82). The latest reports show that patients with severe COVID-19 produce a large number of autoantibodies against autoantigens including intracellular molecules, which indirectly supports the theory that inflammatory cell death promotes the formation of a hyperinflammatory state (83).
In a study on the effects of inflammatory factors released by COVID-19 on cell death, it was confirmed that tumor necrosis factor α (TNF-α) and interferon γ (IFN-γ), two inflammatory factors that were significantly elevated at the end of the inflammatory response, can induce PANoptosis, a regulated and extensive inflammatory cell death mode, and provide a molecular scaffold for the interaction and activation of mechanisms necessary for pyrolysis, apoptosis and necrosis (76, 84). Together, although more research is needed to fully clarify the inflammatory cell death pathway in the process of SARS-CoV-2 infection and the functional consequences of these processes, more and more evidence is pointing towards this. Due to the spread of the blood circulatory system, a large number of inflammatory factors, DAMPs and alarmins produced by inflammatory cell death completely amplify the inflammatory storm from the lungs into the multi-system of body, which not only makes the lungs worse, but also induces multiple organ failure and causes death that is difficult to save (Figure 1, right).
COVID-19 is a new infectious disease caused by the spread of SARS-CoV-2 mediated through respiratory particles, with complex clinical manifestations, ranging from no symptoms to critical illness associated with respiratory failure, septic shock, and multiorgan failure (14, 85). In the face of an increasing number of severe cases caused by the global spread of SARS-CoV-2, there is an urgent need for experimental therapies and drug repurposing to alleviate the COVID-19. Since the COVID-19 pandemic, global research institutes and hospitals have carried out intensive research work and clinical trials, and developed new treatment methods and multiple vaccines targeting SARS-CoV-2 at an unprecedented speed, making the management of COVID-19 significant progress. Therefore, in addition to symptomatic treatment, there are currently some treatments of proven benefit in antiviral and anti-inflammatory aspects, which are recommended for use under the emergency use authorization (EUA) or further evaluated in licensed clinical trials (86–88).
Antiviral medications are regarded as the essential requirement to control the outbreak of COVID-19, just like oseltamivir plays an important role in fighting the influenza virus (89). Multiple antiviral agents with anti-SARS-CoV-2 activity identified by in vitro screening during the early onset of the pandemic, including remdesivir, hydroxychloroquine and lopinavir/ritonavir, but subsequent randomized controlled clinical trials have shown little or no benefit (90–92). Ivermectin, as a cheap drug approved for antiparasitic use, has recently been reported to have a strong ability to inhibit SARS-CoV-2 replication in vitro (93), but unfortunately, the published clinical trial results do not support the conclusion of in vitro testing (94). In the latest living guideline of COVID-19 treatments by WHO issued on July 6, 2021, it is clearly recommended to against remdesivir for hospitalized patients with COVID-19 and against hydroxychloroquine, lopinavir/ritonavir or ivermectin for patients with COVID-19 of any severity (88).
Compared with the above-mentioned dilemma of small-molecule antiviral agents, the anti-SARS-CoV-2 neutralizing antibody cocktails have appeared promising in current clinical trials. Neutralizing antibodies, as an important antiviral weapon produced by the immune system, remain in the plasma of individuals recovering from the viral infection. As a traditional antiviral immunotherapy, convalescent plasma therapy was evaluated by clinical trials in China during the early onset of the pandemic (95), and subsequently authorized to be used for critically ill patients with COVID-19 under EUA in the United States (96, 97). This is only a stopgap measure due to the uncertain effects of the other composition from the plasma on therapeutic efficacy and safety. At present, the neutralizing antibody targeting SARS-CoV-2 obtained through recombinant expression technology has entered the stage of clinical trials. REGN-COV2, consisting of two monoclonal antibodies casirivimab and imdevimab, a neutralizing antibody cocktail drug to target the SARS-CoV-2 RBD domain, has been proven to reduce the viral load in the body compared with placebo (98), and it can effectively reduce hospitalization or mortality when administered to non-hospitalized patients with COVID-19 based on public clinical trial data (99). Bamlanivimab/Etesevimab, consisting of a cocktail of neutralizing antibodies targeting Skipe protein of the SARS-CoV-2, also benefits non-hospital patients in clinical trials, reducing hospitalization and mortality (100). Based on these clinical trials, REGN-COV2 and Bamlanivimab/Etesevimab have been licensed to treat non-hospitalized patients with COVID-19 under the EUA in the United States, but as the SARS-CoV-2 variants continue to update, their effectiveness needs further evaluation.
Interferon is a cytokine with antiviral and immunomodulatory activities produced by host cells when a virus infects the body, and is seen as the body’s first line of antiviral defence (101). Population studies have found that the COVID-19 severity is related to patients carrying autosomal genetic locus mutations associated with type I IFN genes (102) or the presence of neutralizing autoantibodies against type I IFN in patients (103). Moreover, the lack of type I IFN in the blood may be a potential predictor of the COVID-19 severity (104). These studies have highlighted the important role of type I IFN in the control of SARS-CoV-2 infection, so it is speculated that at least in the early stages of SARS-CoV-2 infection, the use of type I IFN may have therapeutic benefits for some patients. Some preliminary clinical trial data show that compared with the placebo group, inhaled interferon-α or interferon-β can achieve greater clinical improvement, reduce hospital stay and increase the chance of recovery (105, 106).
Contrary to the low antiviral immune response caused by the lack of type I interferon, the excessive immune response triggers a surge of inflammatory cytokines and the formation of an inflammatory storm that leads to a sudden turn of the disease. IL-1β is the pro-inflammatory cytokine produced by immune cells after recognizing viruses to activate inflammasomes, and it is also increased in COVID-19 patients (20). Anakinra, as an IL-1 receptor antagonist, is a drug approved for the treatment of rheumatoid arthritis and has the potential to reduce the need for invasive mechanical ventilation and mortality in severe COVID-19 patients based on a small case-control study (31). Pathological T cells that produce GM-CSF have been identified in COVID-19 patients (22), and the monoclonal antibody mavrilimumab that blocks the GM-CSF receptor has also shown promising prospects in preliminary clinical trials (32). But overall, the clinical research data targeting the early pro-inflammatory factor IL-1β or GM-CSF is still insufficient, and the efficacy of alleviating the inflammatory storm of COVID-19 still needs further confirmation.
In contrast, IL-6, as the core pro-inflammatory cytokine, has received extensive attention in the research on the inflammatory storm of COVID-19 (22, 33, 45). Three IL-6 signaling antagonists are used to try to alleviate the inflammatory storm of COVID-19, including the monoclonal antibody (Tocilizumab, Sarilumab) that blocks IL-6 receptors authorized for various rheumatological conditions and the monoclonal antibody (Siltuximab) that targets IL-6 authorized for Castleman’s syndrome. Tocilizumab’s confidence in alleviating the inflammatory storm of COVID-19 first began with the preliminary results of an clinical trial of Tocilizumab combined with conventional antiviral drugs in 21 patients with severe COVID-19 (25). Subsequently, the results of a large international multi-center randomized controlled trial (EMPACTA, NCT04372186) showed that for hospitalized patients with COVID-19 who were not mechanically ventilated, adding tocilizumab on the basis of standard care can reduce the risk of mechanical ventilation or death in patients (26). The results of another large randomized controlled trial (REMAP-CAP, NCT02735707) are consistent, and it also showed that treatment with tocilizumab or sarilumab to critically ill COVID-19 patients in the ICU can improve the outcomes including survival (107). Antagonists of IL-6 receptors (Tocilizumab, Sarilumab) have been authorized by the governments of China, the United Kingdom, and the United States to treat COVID-19 patients under EUA. In the latest living guideline of COVID-19 treatments issued by the WHO, tocilizumab or sarilumab is strongly recommended for use in severe and critical COVID-19 (88).
As mentioned above, we have accumulated some experience in the therapeuqcs of COVID-19, but there are still hundreds of thousands of new confirmed cases of COVID-19 and nearly 10,000 deaths every day in the world according to the data released by WHO. Stress from the frequent occurrence of SARS-CoV-2 variants is a well-known cause, and on the other hand, we also need to face up to the fact that we still haven’t found specific antiviral medicines, especially for SARS-CoV-2 variants including the highly contagious delta variant (B.1.617.2) (108) and the highly pathogenic lambda variant (C.37) (109).
From mild pneumonia symptoms to ARDS, to multiple organ failure, it is still the main cause of death in severe COVID-19 patients (14, 23, 45, 87). Reorganizing the process of inflammatory storms is not only important for understanding the progress of the disease, but also helps us to form a more complete treatment plan. Aiming at the mechanism that drives patients to progress from pneumonia to ARDS and multiple organ failure, the inflammatory storm that is gradually aggravated, we propose a new three-dimensional integrated treatment strategy for future research under ethical precursors: 1. Initiation phase: Block SARS-CoV-2 from entering cells and boost anti-viral immune response; 2. Exacerbation stage: Early and sufficient monoclonal drugs targeting inflammatory cytokines; 3. Before it’s worse: A three-dimensional unity based on anti-virus, anti-inflammatory, and anti-cell death (Figure 2). The drafting of this strategy is inspired by the results of published clinical trials. The single neutralizing antibody of anti-SARS-CoV-2 is not ideal (110), and the combination as a cocktail is recommended for mild patients with COVID-19 that plays a good role in blocking the infection of host cells in the early stage of viral infection (100). Tocilizumab, which is strongly recommended by the WHO, also requires a combination of antiviral agents because tocilizumab alone cannot be more effective than the placebo group (COVACTA, NCT04320615) (30), and adding tocilizumab on the basis of standard care benefit severe and critically ill patients with COVID-19 (25, 26, 107). This three-dimensional integrated treatment strategy not only highlights the combination of different drugs such as antiviral and anti-inflammatory, but also highlights the need for targeted addition of drugs at different stages of COVID-19, and calls for adding drugs to alleviate cell death before COVID-19 becomes life-threatening. At present, most of the screening inhibitors of targeted cell death are still in the cutting-edge basic research. At present, most inhibitors that target cell death is still in the cutting-edge basic research. Disulfiram, as a drug that has been approved for the treatment of alcohol addiction, has recently been reported to target gasdermin D to prevent it from making holes in the cell membrane, which can effectively alleviate the death of a mouse model of sepsis (111). Given that COVID-19 can produce an inflammatory syndrome that is similar to sepsis, whether disulfiram can be used to treat severe COVID-19 patients should be considered, and it can inhibit the Lpro protease of SARS-CoV-2P, which has the potential to inhibit virus replication (112). In addition, the combined treatment of neutralizing antibodies against TNF-α and IFN-γ in mice infected with SARS-CoV-2 can alleviate PANoptosis and protect mice from death (84). Although the existing evidence only comes from mouse models, anti-cell death is a potentially promising therapeutic idea in life-threatening infectious diseases caused by inflammatory storms including COVID-19.
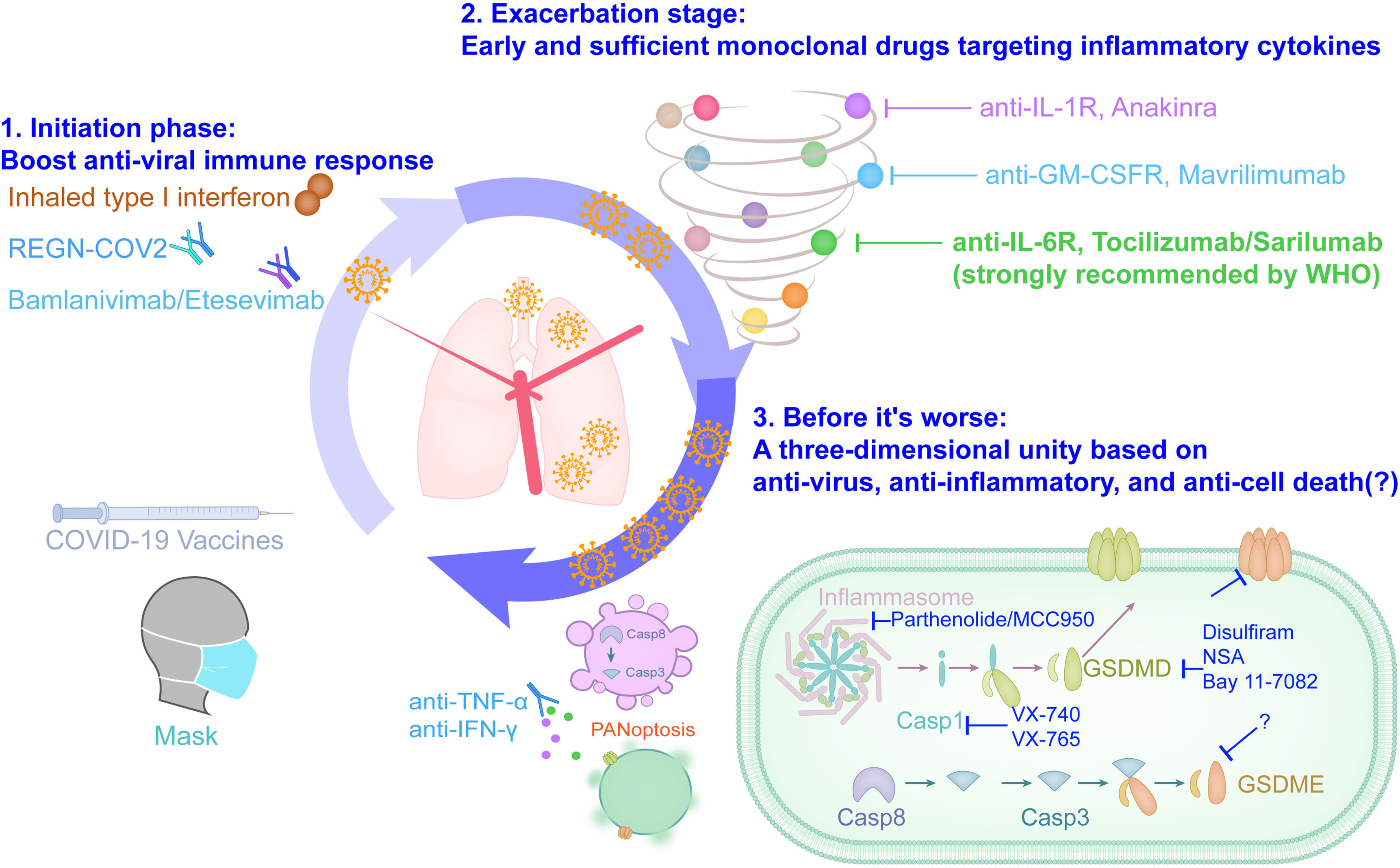
Figure 2 The three-dimensional integrated solution based on anti-viral, anti-inflammatory and anti-cell death slows down the ARDS clock of severe COVID-19.
The three-dimensional integrated treatment strategy including anti-viral, anti-inflammatory and anti-cell death is an ideal combination of saving the lives of COVID-19 patients, and its effectiveness needs to be repeatedly tested under the precursors of ethical and clinical research guidelines. Clinically, the definition of mild, severe and critical COVID-19 mainly refers to the imaging characteristics of pneumonia and blood oxygen related indexes, such as blood oxygen saturation and arterial oxygen partial pressure (13, 14). However, the clinical manifestations of the patient are delayed relative to the body damage. The blood biochemical test report of the patient showed a significant decrease in lymphocyte count, a significant increase in inflammation indicators (IL-6, C-reactive protein, ferritin, etc.) and an increase in blood coagulation function indicators (D-dimer, procalcitonin, thrombin time, etc.), which are potential early warning indicators for severe and critically ill patients with COVID-19 (14, 87). In addition, attempts to propose faster and more accurate COVID-19 prediction models from the aspects of clinical symptoms (113), transcriptome (114), serum protein (115), and metabolome (116) based on artificial intelligence algorithms have also been established, but they still need to be further confirmed in the future. And the limitations of these parameters and data models that are indicative of the progression of COVID-19 may also be discovered in the future exploration of the three-dimensional schedule, so that they can be further improved in a targeted manner, so as to indicate the medication window more timely and accurately.
All authors contributed to the article and approved the submitted version. YZ conceptualized and drafted the manuscript. XX drafted figures. HW edited/reviewed the manuscript.
This work was supported by China National Center for Biotechnology Development (2020YFC0843800 to HW), Jack Ma Foundation (ZB9100000001 to HW) and the project funded by USTC Research Funds of the Double First-Class Initiative (no. YD9100002008 to YZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We apologize to our colleagues in the field whose work could not be cited due to space limitations.
1. Drosten C, Günther S, Preiser W, van der Werf S, Brodt HR, Becker S, et al. Identification of a Novel Coronavirus in Patients With Severe Acute Respiratory Syndrome. N Engl J Med (2003) 348:1967–76. doi: 10.1056/NEJMoa030747
2. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a Novel Coronavirus From a Man With Pneumonia in Saudi Arabia. N Engl J Med (2012) 367:1814–20. doi: 10.1056/NEJMoa1211721
3. Wang C, Horby PW, Hayden FG, Gao GF. A Novel Coronavirus Outbreak of Global Health Concern. Lancet (2020) 395:470–3. doi: 10.1016/S0140-6736(20)30185-9
4. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A New Coronavirus Associated With Human Respiratory Disease in China. Nature (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
5. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
6. Vlasova AN, Diaz A, Damtie D, Xiu L, Toh TH, Lee JS, et al. Novel Canine Coronavirus Isolated From a Hospitalized Pneumonia Patient, East Malaysia. Clin Infect Dis (2021) ciab456. doi: 10.1093/cid/ciab456
7. Lednicky JA, Tagliamonte MS, White SK, Elbadry MA, Alam MM, Stephenson CJ, et al. Emergence of Porcine Delta-Coronavirus Pathogenic Infections Among Children in Haiti Through Independent Zoonoses and Convergent Evolution. medRxiv (2021) 2021.03.19.21253391. doi: 10.1101/2021.03.19.21253391
8. Kupferschmidt K. Viral Evolution may Herald New Pandemic Phase. Science (2021) 371:108–9. doi: 10.1126/science.371.6525.108
9. Abdool Karim SS, de Oliveira T. New SARS-CoV-2 Variants - Clinical, Public Health, and Vaccine Implications. N Engl J Med (2021) 384:1866–8. doi: 10.1056/NEJMc2100362
10. Cao Y, Yisimayi A, Bai Y, Huang W, Li X, Zhang Z, et al. Humoral Immune Response to Circulating SARS-CoV-2 Variants Elicited by Inactivated and RBD-Subunit Vaccines. Cell Res (2021) 31(7):732–41. doi: 10.1038/s41422-021-00514-9
11. Long QX, Tang XJ, Shi QL, Li Q, Deng HJ, Yuan J, et al. Clinical and Immunological Assessment of Asymptomatic SARS-CoV-2 Infections. Nat Med (2020) 26:1200–4. doi: 10.1038/s41591-020-0965-6
12. Gao Z, Xu Y, Sun C, Wang X, Guo Y, Qiu S, et al. A Systematic Review of Asymptomatic Infections With COVID-19. J Microbiol Immunol Infect (2021) 54:12–6. doi: 10.1016/j.jmii.2020.05.001
13. Vetter P, Vu DL, L’Huillier AG, Schibler M, Kaiser L, Jacquerioz F. Clinical Features of Covid-19. BMJ (2020) 369:m1470. doi: 10.1136/bmj.m1470
14. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA (2020) 324(8):782–93. doi: 10.1001/jama.2020.12839
15. Cao X. COVID-19: Immunopathology and Its Implications for Therapy. Nat Rev Immunol (2020) 20:269–70. doi: 10.1038/s41577-020-0308-3
16. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
17. Wang T, Du Z, Zhu F, Cao Z, An Y, Gao Y, et al. Comorbidities and Multi-Organ Injuries in the Treatment of COVID-19. Lancet (2020) 395:e52. doi: 10.1016/S0140-6736(20)30558-4
18. Zaim S, Chong JH, Sankaranarayanan V, Harky A. COVID-19 and Multiorgan Response. Curr Probl Cardiol (2020) 45:100618. doi: 10.1016/j.cpcardiol.2020.100618
19. Bian X-W, t.C.-P. Team. Autopsy of COVID-19 Victims in China. Natl Sci Rev (2020) 7(9):1414–8. doi: 10.1093/nsr/nwaa123
20. Zhang J, Wu H, Yao X, Zhang D, Zhou Y, Fu B, et al. Pyroptotic Macrophages Stimulate the SARS-CoV-2-Associated Cytokine Storm. Cell Mol Immunol (2021) 18:1305–7. doi: 10.1038/s41423-021-00665-0
21. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 Tissue Atlases Reveal SARS-CoV-2 Pathology and Cellular Targets. Nature (2021) 595(7865):107–13. doi: 10.1038/s41586-021-03570-8
22. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T-Cells and Inflammatory Monocytes Incite Inflammatory Storms in Severe COVID-19 Patients. Natl Sci Rev (2020) 7:998–1002. doi: 10.1093/nsr/nwaa041
23. Moore JB, June CH. Cytokine Release Syndrome in Severe COVID-19. Science (2020) 368:473–4. doi: 10.1126/science.abb8925
24. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
25. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective Treatment of Severe COVID-19 Patients With Tocilizumab. Proc Natl Acad Sci USA (2020) 117:10970–5. doi: 10.1073/pnas.2005615117
26. Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, et al. Tocilizumab in Patients Hospitalized With Covid-19 Pneumonia. N Engl J Med (2021) 384:20–30. doi: 10.1056/NEJMoa2030340
27. Soin AS, Kumar K, Choudhary NS, Sharma P, Mehta Y, Kataria S, et al. Tocilizumab Plus Standard Care Versus Standard Care in Patients in India With Moderate to Severe COVID-19-Associated Cytokine Release Syndrome (COVINTOC): An Open-Label, Multicentre, Randomised, Controlled, Phase 3 Trial. Lancet Respir Med (2021) 9:511–21. doi: 10.1016/S2213-2600(21)00081-3
28. Della-Torre E, Campochiaro C, Cavalli G, De Luca G, Napolitano A, La Marca S, et al. Interleukin-6 Blockade With Sarilumab in Severe COVID-19 Pneumonia With Systemic Hyperinflammation: An Open-Label Cohort Study. Ann Rheum Dis (2020) 79:1277–85. doi: 10.1136/annrheumdis-2020-218122
29. Gritti G, Raimondi F, Bottazzi B, Ripamonti D, Riva I, Landi F, et al. Siltuximab Downregulates Interleukin-8 and Pentraxin 3 to Improve Ventilatory Status and Survival in Severe COVID-19. Leukemia (2021) 35(9):2710–14. doi: 10.1038/s41375-021-01299-x
30. Rosas IO, Bräu N, Waters M, Go RC, Hunter BD, Bhagani S, et al. Tocilizumab in Hospitalized Patients With Severe Covid-19 Pneumonia. N Engl J Med (2021) 384:1503–16. doi: 10.1056/NEJMoa2028700
31. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 Blockade With High-Dose Anakinra in Patients With COVID-19, Acute Respiratory Distress Syndrome, and Hyperinflammation: A Retrospective Cohort Study. Lancet Rheumatol (2020) 2:e325–31. doi: 10.1016/S2665-9913(20)30127-2
32. De Luca G, Cavalli G, Campochiaro C, Della-Torre E, Angelillo P, Tomelleri A, et al. GM-CSF Blockade With Mavrilimumab in Severe COVID-19 Pneumonia and Systemic Hyperinflammation: A Single-Centre, Prospective Cohort Study. Lancet Rheumatol (2020) 2:e465–73. doi: 10.1016/S2665-9913(20)30170-3
33. Jones SA, Hunter CA. Is IL-6 a Key Cytokine Target for Therapy in COVID-19? Nat Rev Immunol (2021) 21(6):337–9. doi: 10.1038/s41577-021-00553-8
34. Parr JB. Time to Reassess Tocilizumab’s Role in COVID-19 Pneumonia. JAMA Intern Med (2021) 181:12–5. doi: 10.1001/jamainternmed.2020.6557
35. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute Respiratory Distress in Adults. Lancet (1967) 2:319–23. doi: 10.1016/S0140-6736(67)90168-7
36. Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA (2012) 307:2526–33. doi: 10.1001/jama.2012.5669
37. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA (2016) 315:788–800. doi: 10.1001/jama.2016.0291
38. Fan E, Brodie D, Slutsky AS. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA (2018) 319:698–710. doi: 10.1001/jama.2017.21907
39. Bauer TT, Ewig S, Rodloff AC, Müller EE. Acute Respiratory Distress Syndrome and Pneumonia: A Comprehensive Review of Clinical Data. Clin Infect Dis (2006) 43:748–56. doi: 10.1086/506430
40. Lew TW, Kwek TK, Tai D, Earnest A, Loo S, Singh K, et al. Acute Respiratory Distress Syndrome in Critically Ill Patients With Severe Acute Respiratory Syndrome. JAMA (2003) 290:374–80. doi: 10.1001/jama.290.3.374
41. Cockrell AS, Yount BL, Scobey T, Jensen K, Douglas M, Beall A, et al. A Mouse Model for MERS Coronavirus-Induced Acute Respiratory Distress Syndrome. Nat Microbiol (2016) 2:16226. doi: 10.1038/nmicrobiol.2016.226
42. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
43. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute Respiratory Distress Syndrome. Nat Rev Dis Primers (2019) 5:18. doi: 10.1038/s41572-019-0069-0
44. Harrison AG, Lin T, Wang P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol (2020) 41:1100–15. doi: 10.1016/j.it.2020.10.004
45. Turnquist C, Ryan BM, Horikawa I, Harris BT, Harris CC. Cytokine Storms in Cancer and COVID-19. Cancer Cell (2020) 38:598–601. doi: 10.1016/j.ccell.2020.09.019
46. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N Engl J Med (2020) 382:1199–207. doi: 10.1056/NEJMoa2001316
47. Lu CW, Liu XF, Jia ZF. 2019-Ncov Transmission Through the Ocular Surface Must Not be Ignored. Lancet (2020) 395:e39. doi: 10.1016/S0140-6736(20)30313-5
48. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-Ncov and Naming it SARS-CoV-2. Nat Microbiol (2020) 5:536–44. doi: 10.1038/s41564-020-0695-z
49. Yao H, Song Y, Chen Y, Wu N, Xu J, Sun C, et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell (2020) 183:730–8.e13. doi: 10.1016/j.cell.2020.09.018
50. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM Structure of the 2019-Ncov Spike in the Prefusion Conformation. Science (2020) 367:1260–3. doi: 10.1126/science.abb2507
51. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet (2020) 395:565–74. doi: 10.1016/S0140-6736(20)30251-8
52. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-Converting Enzyme 2 is a Functional Receptor for the SARS Coronavirus. Nature (2003) 426:450–4. doi: 10.1038/nature02145
53. Yi C, Sun X, Ye J, Ding L, Liu M, Yang Z, et al. Key Residues of the Receptor Binding Motif in the Spike Protein of SARS-CoV-2 That Interact With ACE2 and Neutralizing Antibodies. Cell Mol Immunol (2020) 17:621–30. doi: 10.1038/s41423-020-0458-z
54. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
55. Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, et al. CD147-Spike Protein Is a Novel Route for SARS-CoV-2 Infection to Host Cells. Signal Transduct Target Ther (2020) 5:283. doi: 10.1038/s41392-020-00426-x
56. Hussell T, Bell TJ. Alveolar Macrophages: Plasticity in a Tissue-Specific Context. Nat Rev Immunol (2014) 14:81–93. doi: 10.1038/nri3600
57. Lu Q, Liu J, Zhao S, Gomez Castro MF, Laurent-Rolle M, Dong J, et al. SARS-CoV-2 Exacerbates Proinflammatory Responses in Myeloid Cells Through C-Type Lectin Receptors and Tweety Family Member 2. Immunity (2021) 54:1304–19.e9. doi: 10.1016/j.immuni.2021.05.006
58. Hu G, Christman JW. Editorial: Alveolar Macrophages in Lung Inflammation and Resolution. Front Immunol (2019) 10:2275. doi: 10.3389/fimmu.2019.02275
59. Lv J, Wang Z, Qu Y, Zhu H, Zhu Q, Tong W, et al. Distinct Uptake, Amplification, and Release of SARS-CoV-2 by M1 and M2 Alveolar Macrophages. Cell Discovery (2021) 7:24. doi: 10.1038/s41421-021-00258-1
60. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
61. Zhang Z, Zheng Y, Niu Z, Zhang B, Wang C, Yao X, et al. SARS-CoV-2 Spike Protein Dictates Syncytium-Mediated Lymphocyte Elimination. Cell Death Differ (2021) 28(9):2765–77. doi: 10.1038/s41418-021-00782-3
62. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological Findings of COVID-19 Associated With Acute Respiratory Distress Syndrome. Lancet Respir Med (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
63. Borregaard N. Neutrophils, From Marrow to Microbes. Immunity (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
64. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18:134–47. doi: 10.1038/nri.2017.105
65. Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells (2020) 9(6):1383. doi: 10.3390/cells9061383
66. Wu M, Chen Y, Xia H, Wang C, Tan CY, Cai X, et al. Transcriptional and Proteomic Insights Into the Host Response in Fatal COVID-19 Cases. Proc Natl Acad Sci U S A (2020) 117:28336–43. doi: 10.1073/pnas.2018030117
67. Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial Dysfunction and Immunothrombosis as Key Pathogenic Mechanisms in COVID-19. Nat Rev Immunol (2021) 21:319–29. doi: 10.1038/s41577-021-00536-9
68. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
69. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-Associated Coagulopathy: Evidence From a Single-Centre, Cross-Sectional Study. Lancet Haematol (2020) 7:e575–82. doi: 10.1016/S2352-3026(20)30216-7
70. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host Cell Death and Inflammation. Nat Rev Microbiol (2009) 7:99–109. doi: 10.1038/nrmicro2070
71. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
72. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion From Living Macrophages. Immunity (2018) 48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013
73. van den Berg DF, Te Velde AA. Severe COVID-19: NLRP3 Inflammasome Dysregulated. Front Immunol (2020) 11:1580. doi: 10.3389/fimmu.2020.01580
74. Rodrigues TS, de Sá KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasome Activation in COVID-19 Patients. medRxiv (2020) 2020.08.05.20168872. 2020.08.05.20168872. doi: 10.1101/2020.08.05.20168872
75. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin Pores Permeabilize Mitochondria to Augment Caspase-3 Activation During Apoptosis and Inflammasome Activation. Nat Commun (2019) 10:1689. doi: 10.1038/s41467-019-09397-2
76. Lee S, Channappanavar R, Kanneganti TD. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol (2020) 41:1083–99. doi: 10.1016/j.it.2020.10.005
77. Place DE, Lee S, Kanneganti TD. PANoptosis in Microbial Infection. Curr Opin Microbiol (2021) 59:42–9. doi: 10.1016/j.mib.2020.07.012
78. Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a Protein of SARS-CoV-2 Induces Apoptosis in Cells. Cell Mol Immunol (2020) 17:881–3. doi: 10.1038/s41423-020-0485-9
79. Zhu N, Wang W, Liu Z, Liang C, Wang W, Ye F, et al. Morphogenesis and Cytopathic Effect of SARS-CoV-2 Infection in Human Airway Epithelial Cells. Nat Commun (2020) 11:3910. doi: 10.1038/s41467-020-17796-z
80. Rock KL, Kono H. The Inflammatory Response to Cell Death. Annu Rev Pathol (2008) 3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456
81. Gowda P, Patrick S, Joshi SD, Kumawat RK, Sen E. Glycyrrhizin Prevents SARS-CoV-2 S1 and Orf3a Induced High Mobility Group Box 1 (HMGB1) Release and Inhibits Viral Replication. Cytokine (2021) 142:155496. doi: 10.1016/j.cyto.2021.155496
82. Chen L, Long X, Xu Q, Tan J, Wang G, Cao Y, et al. Elevated Serum Levels of S100A8/A9 and HMGB1 at Hospital Admission Are Correlated With Inferior Clinical Outcomes in COVID-19 Patients. Cell Mol Immunol (2020) 17:992–4. doi: 10.1038/s41423-020-0492-x
83. Wang EY, Mao T, Klein J, Dai Y, Huck JD, Jaycox JR, et al. Diverse Functional Autoantibodies in Patients With COVID-19. Nature (2021) 595(7866):283–8. doi: 10.1101/2020.12.10.20247205
84. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell (2021) 184:149–68.e17. doi: 10.1016/j.cell.2020.11.025
85. Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA (2020) 323(20):2052–9. doi: 10.1001/jama.2020.6775
86. Coopersmith CM, Antonelli M, Bauer SR, Deutschman CS, Evans LE, Ferrer R, et al. The Surviving Sepsis Campaign: Research Priorities for Coronavirus Disease 2019 in Critical Illness. Crit Care Med (2021) 49:598–622. doi: 10.1097/CCM.0000000000004895
87. Aleem A, Akbar Samad AB, Slenker AK. Emerging Variants of SARS-CoV-2 And Novel Therapeutics Against Coronavirus (COVID-19). In: StatPearls. Treasure Island (FL: StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC. (2021).
88. W.H.O. (WHO), Update to Living WHO Guideline on Drugs for Covid-19. BMJ (2021) 374:n1703. doi: 10.1136/bmj.n1703
89. von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, et al. Rational Design of Potent Sialidase-Based Inhibitors of Influenza Virus Replication. Nature (1993) 363:418–23. doi: 10.1038/363418a0
90. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med (2020) 383:1813–26. doi: 10.1056/NEJMoa2007764
91. Horby P, Mafham M, Linsell L, Bell JL, Staplin N, Emberson JR, et al. Effect of Hydroxychloroquine in Hospitalized Patients With Covid-19. N Engl J Med (2020) 383:2030–40. doi: 10.1056/NEJMoa2022926
92. Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized With Severe Covid-19. N Engl J Med (2020) 382:1787–99. doi: 10.1056/NEJMoa2001282
93. Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM. The FDA-Approved Drug Ivermectin Inhibits the Replication of SARS-CoV-2 In Vitro. Antiviral Res (2020) 178:104787. doi: 10.1016/j.antiviral.2020.104787
94. López-Medina E, López P, Hurtado IC, Dávalos DM, Ramirez O, Martínez E, et al. Effect of Ivermectin on Time to Resolution of Symptoms Among Adults With Mild COVID-19: A Randomized Clinical Trial. JAMA (2021) 325:1426–35. doi: 10.1001/jama.2021.3071
95. Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 Critically Ill Patients With COVID-19 With Convalescent Plasma. JAMA (2020) 323:1582–9. doi: 10.1001/jama.2020.4783
96. Liu STH, Lin HM, Baine I, Wajnberg A, Gumprecht JP, Rahman F, et al. Convalescent Plasma Treatment of Severe COVID-19: A Propensity Score-Matched Control Study. Nat Med (2020) 26:1708–13. doi: 10.1038/s41591-020-1088-9
97. Joyner MJ, Carter RE, Senefeld JW, Klassen SA, Mills JR, Johnson PW, et al. Convalescent Plasma Antibody Levels and the Risk of Death From Covid-19. N Engl J Med (2021) 384:1015–27. doi: 10.1056/NEJMoa2031893
98. Baum A, Ajithdoss D, Copin R, Zhou A, Lanza K, Negron N, et al. REGN-COV2 Antibodies Prevent and Treat SARS-CoV-2 Infection in Rhesus Macaques and Hamsters. Science (2020) 370:1110–5. doi: 10.1126/science.abe2402
99. Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients With Covid-19. N Engl J Med (2021) 384:238–51. doi: 10.1056/NEJMoa2035002
100. Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA (2021) 325:632–44. doi: 10.1001/jama.2021.0202
101. Katze MG, He Y, Gale M Jr. Viruses and Interferon: A Fight for Supremacy. Nat Rev Immunol (2002) 2:675–87. doi: 10.1038/nri888
102. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn Errors of Type I IFN Immunity in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
103. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies Against Type I IFNs in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4585
104. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science (2020) 369:718–24. doi: 10.1126/science.abc6027
105. Zhou Q, Chen V, Shannon CP, Wei XS, Xiang X, Wang X, et al. Interferon-α2b Treatment for COVID-19. Front Immunol (2020) 11:1061. doi: 10.3389/fimmu.2020.01061
106. Monk PD, Marsden RJ, Tear VJ, Brookes J, Batten TN, Mankowski M, et al. Safety and Efficacy of Inhaled Nebulised Interferon Beta-1a (SNG001) for Treatment of SARS-CoV-2 Infection: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Respir Med (2021) 9:196–206. doi: 10.1016/S2213-2600(20)30511-7
107. Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, Arabi YM, et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients With Covid-19. N Engl J Med (2021) 384:1491–502. doi: 10.1056/NEJMoa2100433
108. Scudellari M. How the Coronavirus Infects Cells - and Why Delta Is So Dangerous. Nature (2021) 595:640–4. doi: 10.1038/d41586-021-02039-y
109. Kimura I, Kosugi Y, Wu J, Yamasoba D, Butlertanaka EP, Tanaka YL, et al. SARS-CoV-2 Lambda Variant Exhibits Higher Infectivity and Immune Resistance. bioRxiv (2021) 2020.08.05.20168872. doi: 10.1101/2021.07.28.454085
110. Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients With Covid-19. N Engl J Med (2021) 384:229–37. doi: 10.1056/NEJMoa2029849
111. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat Immunol (2020) 21:736–45. doi: 10.1038/s41590-020-0669-6
112. Sargsyan K, Lin CC, Chen T, Grauffel C, Chen YP, Yang WZ, et al. Multi-Targeting of Functional Cysteines in Multiple Conserved SARS-CoV-2 Domains by Clinically Safe Zn-Ejectors. Chem Sci (2020) 11:9904–9. doi: 10.1039/D0SC02646H
113. Liang W, Yao J, Chen A, Lv Q, Zanin M, Liu J, et al. Early Triage of Critically Ill COVID-19 Patients Using Deep Learning. Nat Commun (2020) 11:3543. doi: 10.1038/s41467-020-17280-8
114. Zhou Y, Zhang J, Wang D, Wang D, Guan W, Qin J, et al. Profiling of the Immune Repertoire in COVID-19 Patients With Mild, Severe, Convalescent, or Retesting-Positive Status. J Autoimmun (2021) 118:102596. doi: 10.1016/j.jaut.2021.102596
115. Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell (2020) 182:59–72.e15. doi: 10.1016/j.cell.2020.05.032
Keywords: COVID-19, ARDS, Inflammatory storm program, Inflammatory cytokine, Inflammatory cell death
Citation: Zhou Y, Xu X and Wei H (2021) Complex Pathophysiological Mechanisms and the Propose of the Three-Dimensional Schedule For Future COVID-19 Treatment. Front. Immunol. 12:716940. doi: 10.3389/fimmu.2021.716940
Received: 29 May 2021; Accepted: 05 October 2021;
Published: 20 October 2021.
Edited by:
Koichi Ikuta, Kyoto University, JapanReviewed by:
Luminița-Smaranda Iancu, Grigore T. Popa University of Medicine and Pharmacy, RomaniaCopyright © 2021 Zhou, Xu and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiming Wei, dXN0Y3dobUB1c3RjLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.