 Jean-Yves Dubé
Jean-Yves Dubé Vinicius M. Fava
Vinicius M. Fava Erwin Schurr
Erwin Schurr Marcel A. Behr
Marcel A. Behr
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 30 June 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.714808
This article is part of the Research Topic Exploring Immune Variability in Susceptibility to Tuberculosis Infection in Humans View all 12 articles
Human genetic control is thought to affect a considerable part of the outcome of infection with Mycobacterium tuberculosis (Mtb). Most of us deal with the pathogen by containment (associated with clinical “latency”) or sterilization, but tragically millions each year do not. After decades of studies on host genetic susceptibility to Mtb infection, genetic variation has been discovered to play a role in tuberculous immunoreactivity and tuberculosis (TB) disease. Genes encoding pattern recognition receptors (PRRs) enable a consistent, molecularly direct interaction between humans and Mtb which suggests the potential for co-evolution. In this review, we explore the roles ascribed to PRRs during Mtb infection and ask whether such a longstanding and intimate interface between our immune system and this pathogen plays a critical role in determining the outcome of Mtb infection. The scientific evidence to date suggests that PRR variation is clearly implicated in altered immunity to Mtb but has a more subtle role in limiting the pathogen and pathogenesis. In contrast to ‘effectors’ like IFN-γ, IL-12, Nitric Oxide and TNF that are critical for Mtb control, ‘sensors’ like PRRs are less critical for the outcome of Mtb infection. This is potentially due to redundancy of the numerous PRRs in the innate arsenal, such that Mtb rarely goes unnoticed. Genetic association studies investigating PRRs during Mtb infection should therefore be designed to investigate endophenotypes of infection – such as immunological or clinical variation – rather than just TB disease, if we hope to understand the molecular interface between innate immunity and Mtb.
Tuberculosis (TB) was the number one cause of death due to a single infectious agent, Mycobacterium tuberculosis (Mtb), in the year 2019 according to the WHO. SARS-CoV-2 has surpassed Mtb in the last year; however, deployment of vaccines and experience with containment measures should blunt the death rate from COVID-19 in the years to come, such that TB may reprise its role as the most important cause of infectious mortality. Near 40 million people have died from TB in the last 20 years while treatment has saved 60 million (WHO). Yet, in the same interval, an estimated 10- to 20-fold more people were infected but did not progress to disease (1, 2). Together, this suggests broad host control or tolerance of this pathogen, despite the important minority who progress to disease each year.
Our time together with Mtb has potentially spurred human adaptation to allow us as a population to subsist with this obligate pathogen. Mtb has been evolving to parasitize humans for millennia and within that time the relationship has possibly changed us too, when and where Mtb was endemic (3–5). Current and past abundance of human genetic diversity allows researchers to test the importance of genetic variation in Mtb infection outcomes and infer an evolutionary response by our species to survive the Mtb pandemic. One example where Mtb has potentially exerted a purifying selection on humans is that of the TYK2 P1104A variant, which was calculated to have decreased in western Europeans concomitant with endemic TB over the last two millennia (6, 7). The TYK2 P1104A variant is known to disrupt IL-23-dependent IFN-γ production (6) and was associated with a 5-fold increased risk for developing TB in the contemporary UK biobank (8). We are not aware of any evidence of positive selection of a TB resistance gene to date.
Is every case of TB a situation where the host genetic combination is vulnerable to Mtb? We can hypothesize a genetic combination impervious to Mtb. We may not have to extend our imagination very far, as there are documented cases of people who remain TST negative in high-burden settings, such that it is statistically unlikely that they have never inhaled Mtb [recently reviewed in (9)]. Therefore, developing TB is, in part, a result of genetics, and not just being a human exposed to Mtb, a postulate supported by the 21% heritability estimate for household contacts in Peru progressing from TST positivity to TB (10). Environmental parameters can also have an effect (e.g. level of exposure, lung damage, HIV co-infection) and thus in theory identical twins could have different outcomes with Mtb infection. Mtb also has variation which might contribute to a different outcome for the bacterium and the host: there are 9 lineages described to date (11–13) with some being deemed more virulent in experimental models (14).
Genetic variation creates differences that can fine tune a host-pathogen interaction, or abrogate it completely, resulting in altered immunity. One modality where there is a direct opportunity for co-evolution is in physical interactions between host molecules and Mtb molecules. These interactions can be placed into a few camps including: 1) between classical T-cell receptors and MHC molecules presenting microbial epitopes (15); 2) between antibodies and cognate microbial ligands (16); 3) between donor-unrestricted T cells and their respective mycobacterial epitopes presented on invariant host molecules operating analogously to MHC (17, 18); 4) between inborn sensors of microbial products, otherwise known as pattern-recognition receptors (PRRs), and their cognate microbe-associated molecular patterns (MAMPs). By their nature as structural molecules, MAMPs are subjected to a stronger purifying selection than many proteins. Unlike T-cell receptors, PRRs cannot generate diversity within an individual, yet there is variability amongst human population PRR gene pools as discussed further below. Most of all, should we even expect strong selective pressure on host PRRs to recognize Mtb MAMPs? In this paper, we sought to review what is known about the relative importance of the MAMP-PRR interaction for the mammalian host during Mtb infection primarily through two sources of data: 1) controlled animal experiments using engineered genetic knockouts (KOs) of PRRs; 2) natural experiments in humans where genetic diversity permits us to seek associations between polymorphisms and the course of Mtb infection. We later place this in perspective with genes known to have strong effects on animal outcomes and lastly discuss how to approach human genetic studies of PRRs in the years to come.
The interactions between many PRRs, Mtb and Mtb MAMPs have been described over the last few decades and are summarized in Figure 1. Various mechanisms have been uncovered by which PRR recognition of Mtb leads to a cellular effect. Immediately below, we briefly review the molecular functionality of the PRRs which have been demonstrated to mediate an immune response to mycobacteria. Whether these molecular and/or cellular effects translate to protection or pathology in the whole animal is examined in the subsequent section.
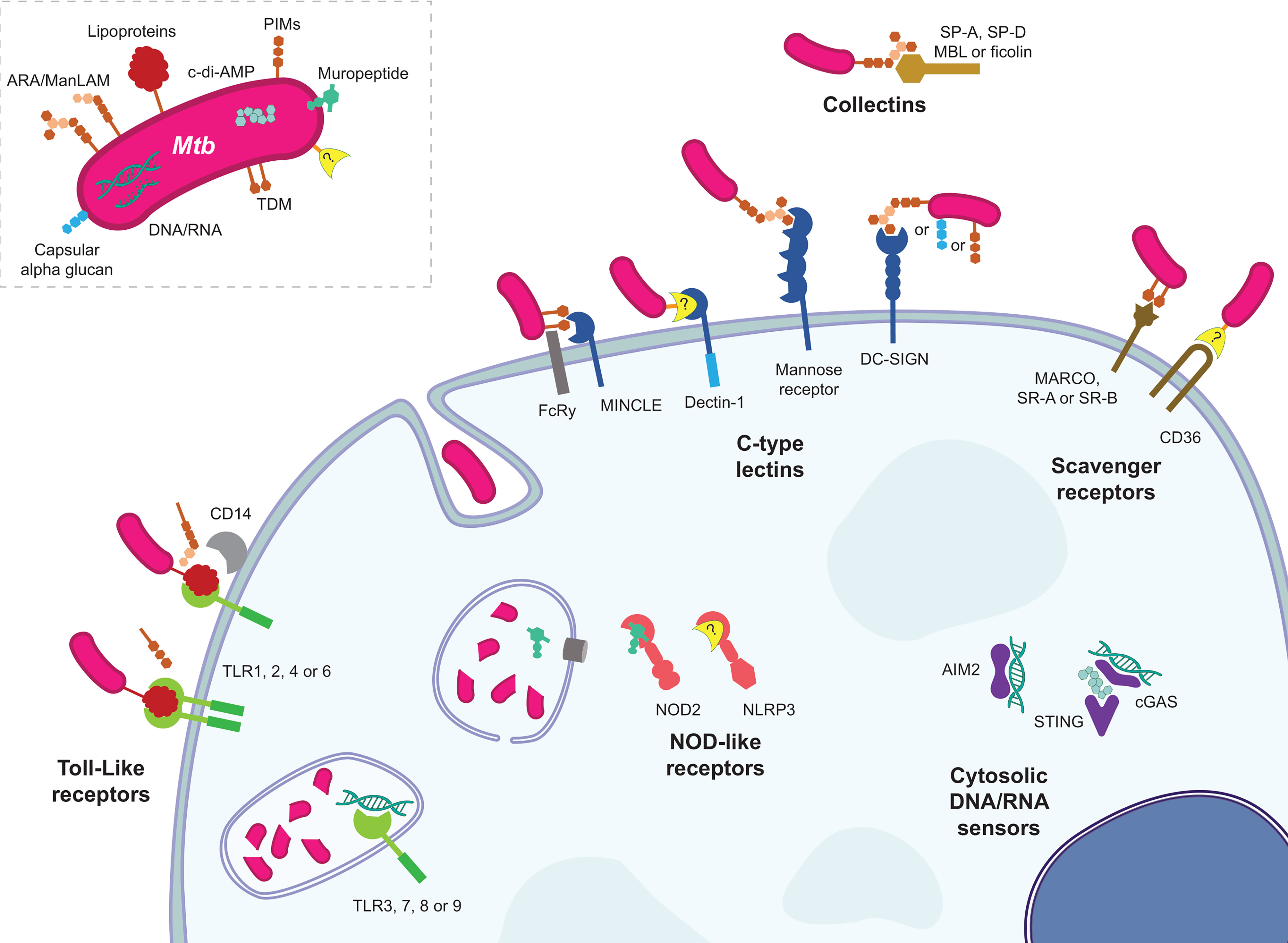
Figure 1 The various host PRR and Mtb MAMP interactions. Representative MAMP-PRR interaction are depicted in their approximate cellular locations highlighting the numerous ways in which Mtb announces its arrival to a phagocyte.
Toll-like receptors (TLRs) were the prototypical PRR fitting the hypothesis proposed earlier by Janeway Jr (19) that there existed inborn sensors in animals for products common among groups of microbes but absent from the host, allowing host recognition of non-self invading microbes – a form of antibody or T-cell receptor for innate immunity. The discoveries in the 1990s on the Toll gene of Drosophila, followed by work exploiting mutant forms of TLR4 in human cells and mice demonstrated that mammalian TLR4 was a sensor of Gram-negative endotoxin (a.k.a. lipopolysaccharide, LPS) (20–23). In total there are 10 TLRs in humans (13 in mice), each with different microbial ligands and slightly varying effects. TLR2 cooperates with TLR1 or TLR6, as well as other PRRs like CD14, to sense mycobacterial lipoproteins and lipoglycans. Identified mycobacterial TLR2 ligands include LAM (non-capped araLAM and not ManLAM) (24, 25), 19 kDa lipoprotein (LpqH), 38 kDa lipoprotein (PstS1) (26), PIMs (with differing activities) (27, 28), 27 kDa lipoprotein (LprG) (29), and LprA (30) to name a few (31). Mycobacteria including Mtb shed membrane vesicles containing TLR2 ligands that are sufficient to generate a TLR2-dependent immune response (32). More recently Mtb sulfoglycolipids have been shown to be competitive TLR2 antagonists (33). TLR4’s mycobacterial ligands are less clear, but Mtb extracts have TLR4-dependent stimulatory activity; many proteins have been proposed as TLR4 agonists, with GroEL1 and 2 being examples (34). TLR5, which recognizes flagellin, does not have a known mycobacterial ligand (mycobacteria do not swim – they float). TLR3, TLR7 and TLR8 recognize RNA, and recent reports revealed that they may respond to host and/or mycobacterial RNA during infection (35–37). TLR9 recognizes CG-rich DNA (i.e. CpG motifs) and has been shown to contribute to the cellular response to Mtb’s CG-rich genomic DNA (38, 39). TLR10 has no known ligands, mycobacterial or otherwise. Murine TLR11, 12, and 13 are not reviewed here because they have no direct relevance to human health.
A complete review of TLR signalling, not specific to mycobacteria, has been recently published elsewhere (40). Briefly, when TLRs are engaged and oligomerize on the membrane, adaptor proteins MyD88 or TRIF are recruited to the cytoplasmic side to form ‘myddosomes’ or ‘triffosomes’, respectively. These supramolecular platforms direct signalling events that lead to activation of MAPK and NF-κB pathways, for example. Such signalling begins an inflammatory response by the cell which includes upregulation of costimulatory molecules and antigen presentation by MHC molecules, plus secretion of soluble factors like cytokines, in the cases of macrophages and dendritic cells (DCs).
C-type lectin receptors (CLRs) are a large, diverse category of receptors of which some members function as PRRs by binding to MAMPs; other CLRs bind endogenous ligands or non-microbial exogenous ligands. The etymology of the name originates from some members requiring calcium (Ca++, hence “C”) to bind their respective carbohydrate ligands (hence “lectin”). There are both membrane-bound and soluble forms of CLRs. A full review of this complex category of PRRs has recently been published and provides more mechanistic detail than is presented here (41).
The mannose receptor (CD206) expressed on macrophages was shown to assist these phagocytes in uptake of the tubercule bacillus (42) with ManLAM being the mycobacterial ligand for CD206 (43). The ManLAM-CD206 interaction was later demonstrated to uniquely induce IL-8 and cyclooxengenase expression via PPARγ, while PPARγ knockdown was associated with reduced bacterial growth and increased TNF production during monocyte-derived macrophage infection (44). Pparg-KO mice had about half the pulmonary bacterial burden and reduced lung pathology than WT counterparts when aerosol infected with Mtb (45).
The CLR called DC-SIGN (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin, a.k.a. CD209) is a main receptor on DCs for binding to Mtb (46). DC-SIGN expressed on dendritic cells has been show to interact with ManLAM (47), PIM6 (48) and capsular alpha-glucan (49). The DC-SIGN homologues L-SIGN (human) and SIGNR1 (mouse, one of five homologues), have been shown to interact with ManLAM too (50). DC-SIGN ManLAM ligation modulated TLR-induced signalling (e.g. NF-κB pathway) via Raf-1 (51, 52).
MINCLE (Macrophage inducible Ca++-dependent lectin receptor, encoded by CLEC4E) associates with FcRγ to bind mycobacterial cord factor trehalose-6,6’-dimycolate (TDM) and TDM is sufficient to induce granuloma formation in murine lungs if functional MINCLE and FcRγ are present (53). MINCLE signals via the SYK-CARD9 pathway to lead to the production of proinflammatory cytokines (54). Card9-KO mice succumb more quickly to Mtb than WT, associated with defective anti-inflammatory signalling presumably leading to lethal immunopathology (55). MINCLE expression is low in resting macrophages, and first requires induction via signalling through MCL (encoded by Clec4d and not to be confused with MCL-1). MCL is also a FcRγ-coupled and TDM CLR but cannot mediate the same pro-inflammatory response on its own (56). MINCLE and MCL expression are co-dependent (57, 58).
Dectin-1 was shown to mediate part of the immune response of splenic DCs to Mtb (59). Mif-KO mice have impaired survival and immunity compared to WT during aerosol infection with Mtb HN878 strain, while bacterial killing and cytokine production were nearly restored when Mif-KO cells were complemented with Dectin-1 (overexpressed). These results suggest the MIF defect mostly manifests in defective Dectin-1 signalling (60). The mycobacterial ligand for Dectin-1 remains unknown. Dectin-2 was reported to recognize ManLAM (61, 62), but pathogenesis studies have yet to be done with this CLR.
Recently, DCAR (dendritic cell immunoactivating receptor; encoded by Clec4b1), also a FcRγ-coupled CLR, was demonstrated to be a receptor of PIMs. DCAR is expressed on monocytes and macrophages. Clec4b1-KO mice had partially defective immune responses and bacterial control during BCG infection (63).
Collectins are soluble, non-cell-bound proteins; they are CLRs in that they specifically associate with sugars on the surface of microbes to mediate an effect. Surfactant proteins (SP) are collectins that exist in pulmonary surfactant. SP-A promoted attachment and phagocytosis of Mtb by alveolar macrophages by a mechanism that required mannose receptor but not SP-A contacting Mtb (64, 65). SP-A supressed nitrite production from AMs preventing Mtb killing and controlling bacterial growth (66). SP-A was shown to bind to ManLAM (67) and APA, the alanine- and proline-rich antigenic glycoprotein (68). SP-D binds to ManLAM and agglutinates Mtb, but in contrast to SP-A, SP-D reduced Mtb binding to macrophages (69). Reduced uptake occurred without agglutination using a modified SP-D (70). However, SP-D increased phagosome-lysosome fusion, but did not alter the respiratory burst (71).
The collectin mannan-binding lectin (MBL) and ficolins are serum-borne receptors that bind to microbes to initiate the complement cascade. MBL was first demonstrated to interact with Mtb and M. leprae sonicate (72). Ficolins, of which there are at least three in humans and two in mice, are also part of the lectin-complement system. Ficolin-2 was shown to bind to Mtb to play a protective role involving opsonization and inflammatory signalling in macrophages (73). Another group suggested ficolin-3 was important for agglutination and phagocytosis of Mtb (74). MBL and ficolins were suggested to bind to ManLAM and/or Ag85 (75, 76).
Nucleotide-binding Oligomerization Domain-containing (NOD)-like receptors (NLRs) are a group of cytoplasmic sensors. Reviews with more detail on their mechanisms of action than presented here have been published, for example (77). Its members NOD1 and NOD2 are essential in the detection of peptidoglycan fragments D-glutamyl-meso-diaminopimelic acid (iE-DAP) and muramyl dipeptide (MDP), respectively (78–81). Both NOD1 and NOD2 signal through the adaptor protein RIPK2 to reach NF-κB and MAPK pathways. Few reports have been published on an important role for NOD1 during Mtb infection, one showing NOD1 plays a role in cytokine production only in the absence of NOD2 or after LPS-pretreatment of BMDMs (82). Mycobacteria do possess the iE-DAP moiety in their peptidoglycan (83). In contrast, NOD2 has been well-studied in Mtb infection [Pubmed searches of “Mycobacterium tuberculosis AND NOD1” or “…NOD2” yielded 7 and 57 hits, respectively, at the time of writing (15-2-2021)].
Mycobacteria produce a distinct NOD2 ligand, N-glycolyl MDP, while most other bacteria produce N-acetyl MDP (84, 85). N-glycolylated peptidoglycan and MDP were shown to be better inducers of immune responses compared to the N-acetylated forms by comparing with mycobacterial KOs and synthetic MDPs (86–88). The absence of NOD2 during Mtb infection was accompanied by reduced nitric oxide and cytokine production from mouse macrophages (89, 90) and reduced iNOS from human macrophages (91). NOD2 signalling has been called “non-redundant” in that although there are shared pathways with TLR and CLR signalling (e.g. NF-κB), NOD2 signalling appears to work synergistically with other MAMPs and little immune response is produced with MDP stimulation alone (86, 88, 92).
Only one other NLR has been significantly studied in the context of Mtb infection: NLRP3 (NLR family pyrin domain containing 3). There is no known mycobacterial ligand for this NLR – it has been suggested that NLRP3 can sense specific host products in the context of infection (77). Recently, ESX-1-mediated membrane damage has been tied to caspase-1 activation, NLRP3 oligomerization, inflammasome formation and finally IL-1β release (93).
AIM2 (absent in melanoma 2) appears to play a role in Mtb infection. This cytosolic DNA receptor was necessary for full caspase-1 cleavage/activation and IL-1β release during Mtb infection and Mtb DNA transfection (94). STING (Stimulator of interferon genes), part of a cytosolic DNA sensing system, was essential for autophagy targeting of ESAT-6-producing mycobacteria in mouse bone marrow-derived macrophages (95) and zebrafish embryos (96). cGAS (cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) synthase), a DNA sensor that works with STING, was required for Mtb autophagy in addition to STING (97). Type I IFN production during Mtb infection elicited by cGAS-produced, STING-sensed cGAMP was dependent on RD-1 (98, 99). Mtb-produced c-di-AMP was also shown to contribute to type I IFN production through STING (100). It has been suggested that Mtb DNA engagement of the AIM2 inflammasome leads to the inhibition of host cell-protective STING functions (101).
Scavenger receptors (SR) are a diverse and poorly defined group of cell surface receptors that interact with endogenous and microbial ligands. Details of these receptors have been recently reviewed elsewhere (102, 103). Inhibitors of scavenger receptors reduced Mtb binding to macrophages (104).
MARCO is a scavenger receptor that was suggested to bind and “tether” Mtb to a macrophage’s surface by interacting with TDM (105), and zebrafish lacking MARCO expression had reduced macrophage uptake of M. marinum (106). Similarly, blocking MARCO on human mesenchymal stems cells reduced Mtb uptake (107).
KO of the gene encoding scavenger receptor A (SR-A) did not affect inflammatory gene transcription during Mtb infection (108). The KO increased TNF and MIP-1α production from AMs treated with TDM (109). Overexpression of Scavenger receptor B1 (SR-B1) in immortal cells was associated with increased BCG and Mtb binding to the cells, but the corresponding KO in murine macrophages had no effect on BCG binding (110). SR-B1 was essential for EsxA-mediated transcytosis of Mtb across M cells (111). A related SR-B family member, CD36, was identified with a Drosophila RNAi screen to be essential for uptake of M. fortuitum (112). CD36 knockdown in human monocyte-derived macrophages reduced surfactant lipid uptake as well as intracellular growth of Mtb, suggesting CD36 normally promotes intracellular Mtb growth or survival (113). Another group showed Cd36 KO macrophages control Mtb, BCG and M. marinum infections better, independent of phagocytosis rate, nitric oxide and ROS production. Similarly, mice receiving BCG i.p. had lower bacterial loads with Cd36 KO vs WT (114). Homologues of SR-BII and CD36 in Dictyostelium discoideum (a social amoeba) are similarly involved in phagocytosis of M. marinum (115).
The overall importance of individual genes during Mtb infection is best addressed in two ways: observing what happens to individuals with diverse expression or functionality of the gene of interest who perchance become infected with Mtb (the natural experiment); alternatively, individuals of known or controlled gene status can be intentionally infected with Mtb – unethical in humans and therefore animal models are necessary. As mentioned above, many PRRs are important for specific cellular processes relevant to Mtb infection. It is therefore hypothesized that in the absence of a PRR, certain aspects of the host-Mtb interaction are lost, which should result in a phenotype in the whole animal. We have also assumed that the animal would suffer most from the aberrant immune response. This assumption is perhaps too simplistic: Mtb is a professional, obligate pathogen, and although it is perceived as hard-to-kill, it might also suffer from a host environment that does not behave as Mtb has evolved to ‘expect’. Additionally, we have suggested that the potential coevolution of humans and Mtb has shaped the PRR-MAMP interaction, which clearly would not apply in infections of animals like mice, which are not natural hosts for Mtb. However, one can still use mice to generate testable hypothesis for human studies and validate genetic effects observed first in humans. Numerous KO mouse studies of Mtb infection have been performed over the years with hypotheses of defective immunity in the animal that should manifest as decreased survival, increased bacterial burden and/or detectable differences in the immune response (e.g. bronchoalveolar lavage cytokines or T-cell defects).
To non-biasedly form a conclusion as to the importance of PRRs during Mtb infection in animal models, we used the Medline database via Pubmed to repeatedly search every known PRR and its role in Mtb infection in a living animal with the term below (where “[PRR]” was changed in each search):
“Mycobacterium tuberculosis AND [PRR]”
Where PRR names were ambiguous, we searched multiple times using the different names. After removing duplicates, this search produced over 1100 papers, which were screened for data using KO animals during Mtb infection. The results of this in silico exercise are summarized in Table 1. We have added a few studies of which we were aware but that were missed by the screen (noted in Table 1). It is possible that other appropriate data are absent; however the non-biased approach reinforces the validity of our subsequent conclusions.
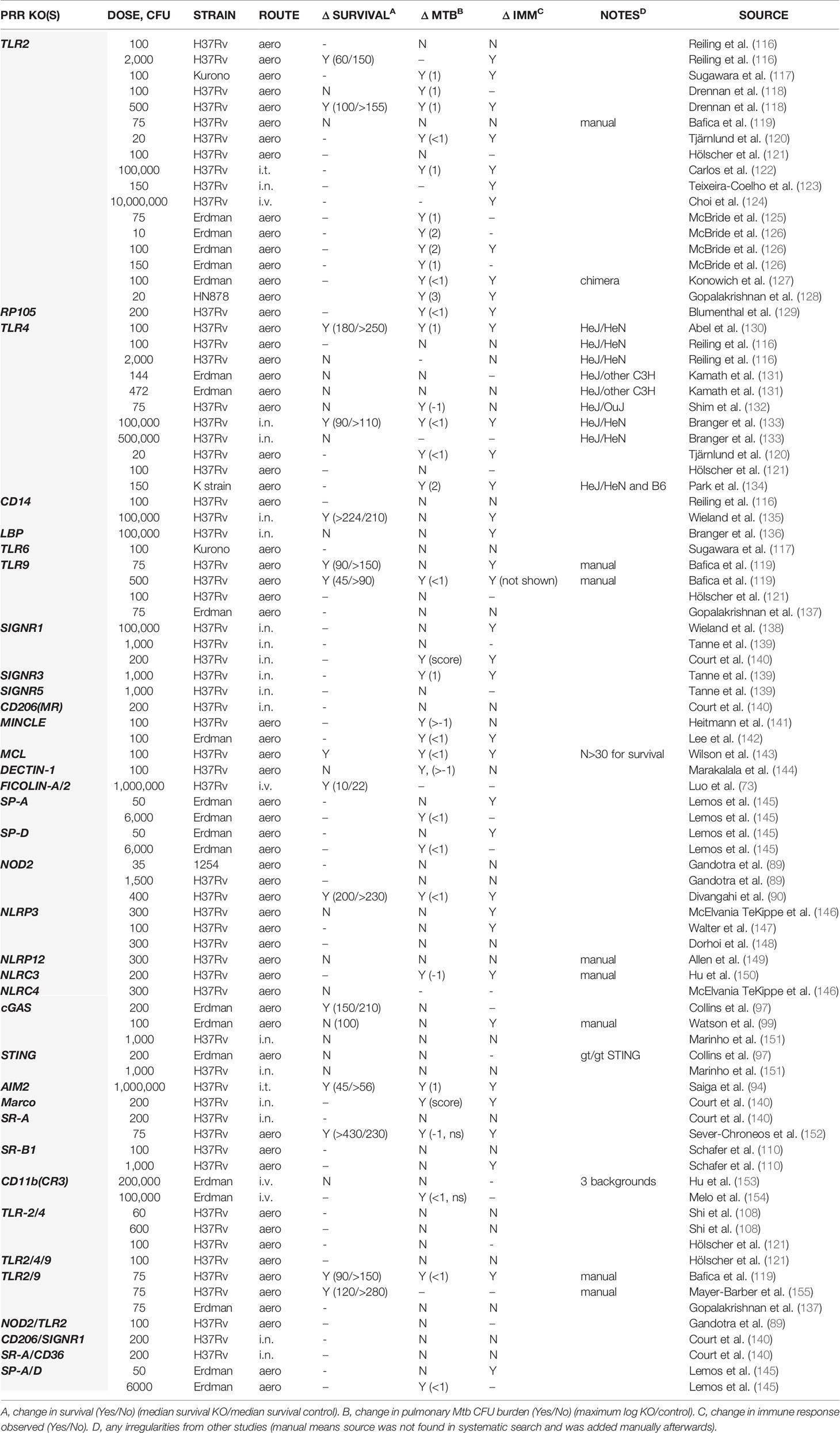
Table 1 Results of KO mouse studies in Mtb infection.
In Table 1, we summarized the results of individual experiments presented in the literature on murine Mtb infections comparing a PRR KO to the ‘WT’ control animal. All data found were exclusive to the mouse. We have included the dose, Mtb strain and route of infection per experiment. Mtb can establish an infection via the lungs with just a few bacilli (156, 157), and therefore models using large doses and atypical routes may represent different aspects of Mtb disease but not necessarily follow the normal mode of infection. The magnitude of disease in mice also changes with the strain of Mtb, where for example the H37Rv strain is expected to be less virulent than the related Erdman strain and the HN878 strain. It is possible that some of the different outcomes across different studies addressing the same PRR knockout were due to differences in the infection model. However, our review of the data did not reveal an obvious effect of dose, strain nor route (Table 1).
Where survival data were present, it was clear that PRRs can have an effect on survival, although in most cases there was either no significant difference in survival from WT to KO, or it was quantitatively small. There were two instances where KO mice survived longer than WT [CD14 (135) and SR-A (152)], demonstrating that some host systems are detrimental to Mtb tolerance.
For TLR2, two of four experiments showed reduced survival in KOs. A single Tlr6-KO study did not show a difference in bacterial burden nor immune response (117). For TLR4, two of seven experiments showed reduced survival in KOs. Note that many TLR4 studies took advantage of the C3H/HeJ mouse (having a spontaneous Tlr4 loss-of-function mutation) employing other only somewhat related C3H strains as wildtype control. The maximum difference in pulmonary bacterial burden observed in most of these papers was approximately one log more in Tlr2 or Tlr4 KOs vs WT. Defects in immune responses were observed in a majority of Tlr2 KO experiments and a minority of Tlr4 KO experiments. Two experiments with Tlr9 KO from one study showed more rapid death with high dose infection compared to low dose, and only the high-dose resulted in a statistically significant increase in pulmonary bacterial burden (158). No survival data for other TLRs have been published. Interestingly, most experiments with combination KOs of Tlr2, 4 and/or 9 resulted in no differences in bacterial burden nor immunologic responses. Two of seven experiments (Tlr2/9 double KOs) resulted in shortened survival times, but with small or unreported differences in bacterial burden. Together, mutations in TLRs, even multiple, had only modest or negligible effects on the host’s survival and bacterial control but were frequently associated with altered immune responses. In particular, TLR2 and 9 stood out.
Of note, Myd88-KO mice succumbed rapidly (all dead within 1-2 months) to Mtb infection, despite TLRs seeming to be largely dispensable. This was attributed to the necessity of MyD88 for IL-1R signalling (Il1r1 KO mice are equally susceptible) and intrinsic macrophage function requiring MyD88 (121, 159, 160). An earlier report with Myd88-KO mice showed a nearly 2-log increase in pulmonary colony-forming units (CFU) compared to WT but mice survived at least 12 weeks with limited immunological changes; no survival was presented (161).
For CLRs, only MCL and Dectin-1 were found by us to have been disrupted in Mtb survival challenges. In one report, MCL (Clec4d)-KO caused a significant difference in survival, but specifically this was 20% mortality by 6 weeks, after which no Clec4d-KO mice died to week 10 (when the experiment was ended) (143). However, in the same study, pulmonary bacterial burden was less than half a log higher in the KO at four months (no significant difference at 2 months). Proinflammatory immunologic responses were elevated in the KO. Thus, MCL might play a role early in infection to control the immune response, but not so much for bacterial control.
Another lone report showed Dectin-1 (Clec7a)-KO mice did not have changed mortality after infection with Mtb, and in fact had slightly lower bacterial burdens compared to WT at 2 and 4 months post infection (144). Therefore, Dectin-1 is likely not required by the host during Mtb infection. No survival data was found for murine DC-SIGN homologues, mannose receptor, nor MINCLE. Only one of three studies reported a difference in bacterial burden with SIGNR1 (Cd209b)-KO at one- and nine-months post infection, but by scoring Ziehl-Neelsen-stained lung sections rather than directly counting CFUs (140). When assessed, altered immunity was consistently observed with this KO. A single study found a difference in bacterial burden and immunological response with a SIGNR3 (CD209d)-KO but not a SIGNR5 (Cd209a)-KO (139). Another lone study addressing the mannose receptor showed no bacterial or immunologic effect with KO (140). Two studies on MINCLE presented opposing data on pulmonary bacterial burden (more or less a half log compared to WT) and only one identified significant immunological changes with KO. Double KO of the genes encoding mannose receptor plus SIGNR1 showed no bacterial or immunologic differences. KOs of other membrane-bound CLRs have not been tested during in vivo Mtb infection.
KOs of genes encoding collectins SP-A and SP-D had no long-term effect on bacterial burden during Mtb infection – survival was not tested/presented. Immunologic responses were similar to WT but with decreased neutrophil numbers in the lung. Double KO for SP-A and SP-D genes was similar to the SP-A single KO (145). KO of the gene encoding ficolin-A (homologue of human ficolin-2 and/or 3) decreased the survival of mice given one million CFU H37Rv strain i.v. compared to WT, but survival was enhanced compared to WT when KO mice were given a plasmid containing ficolin-A or ficolin-2 by i.m. electroporation on the day of infection (73). This suggests ficolins might help control systemic or bloodborne Mtb. Thus, as with TLRs, CLRs are generally dispensable for Mtb immunity. The few exceptions seem to suggest an early, minor role in Mtb infection for CLRs like MCL and SP-A/D.
NOD2 disruption produced a late survival phenotype: KO mice died faster than WT near 6 months post Mtb infection. Bacterial burden was slightly higher and immunological responses were also reduced in KO mice in this study (90). A separate study did not find significant differences in bacterial burden nor immunological responses with Nod2-KO nor Nod2-Tlr2-double KO, but survival was not evaluated (89). In contrast, Nlrp3-KO had no effect on bacterial burden in three studies. Immunological responses with Nlrp3-KO can be altered, but survival did not change.
Aim2-KO mice succumbed rapidly to infection with one million CFU H37Rv strain delivered i.t. compared to WT. The KO had greater bacterial burden and pathology and altered immunity (94). However, no other independent studies were found besides this one, and the high dose delivery makes the result difficult to compare to other PRR-KO survival studies with the more physiological low-dose aerosol infection. The importance of AIM2 during mycobacterial infection is supported by data with BCG, where repeated infection of WT and Aim2-KO mice vial the tail vein showed KO mice were defective in controlling bacterial burden which was associated with altered immunity (enhance type I IFN, reduced type II IFN) (101). Additionally, the adaptor protein ASC (a.k.a. PYCARD) was also shown to be important for survival in at least two separate studies (146, 155). This supports the importance of AIM2 and/or another inflammasome sensor for Mtb infection, with NLRP3 seemingly dispensable.
Pulmonary burden of Mtb in Cgas-KO and Sting1gt/gt mice was unchanged from WT at 3 and 6 weeks post aerosol infection of 200 CFU of the Erdman strain, although Cgas-KO mice had late reduced survival (deaths between 100 and 200 days p.i.) while the STING mutant did not differ from WT (97). In another study, an Erdman-strain aerosol experiment running 100 days did not reveal a difference between WT and Cgas-KO mice in terms of survival and bacterial burden, but less type I IFN was present in the lungs and serum (99). In a third study with i.n. infection with 1000 CFU H37Rv strain, cGAS and STING mutations did not affect survival past 250 days (no mice died as with WT), although Cgas-KO mice did not maintain weight as well. Bacterial burden and immunology were the same as WT too (151). Together, these studies suggest cGAS plays a minor role during Mtb infection (that emerged as a death phenotype late in one study), while STING is dispensable. These findings are difficult to reconcile with the proposed model where cGAS functions upstream of STING as the mycobacterial DNA sensor; accordingly a STING mutant should be defective for cGAS functions. Furthermore, Mtb CDC1551 mutants that either lack their own c-di-AMP production, or overexpress it, significantly decreased and increased survival relative to WT, respectively (100), contributing further confusion regarding the importance of STING. It is possible that the importance of cGAS during mouse survival of Mtb infection is related to a STING-independent function of cGAS, and that the STING phenotype with mutant Mtb is more valuable as a mechanistic lesson than a biologically relevant one.
No data was found showing scavenger receptor KOs were detrimental to Mtb control. A difference in bacterial burden during Mtb infection of Marco-KO mice was only detected by scoring Ziehl-Neelsen-stained lungs, not by CFU enumeration, at 6 and 9 months post infection (140). KOs for SR-A and SR-B genes did not result in increased bacterial burdens nor were they consistently associated with immunological changes. Two independent studies examining the complement receptor CR3/Cd11b KO during Mtb infection found no evidence that they play a role in Mtb control nor survival (153, 154).
In summary, most PRR KO experiments presented did not show reduced survival compared to WT. Control of bacterial burden was either unaffected or just slightly increased by PRR KOs in most experiments. We suspect that publication bias against negative data would also mean that PRR KO effects are, if anything, over-represented in the literature. In contrast, altered immunity was found often in PRR KOs during Mtb infection. It is possible that the effects on immunity with some PRR KOs are not large or relevant enough to result in changes in survival and bacterial burden that are sufficiently robust to be statistically detectable with a practical number of animals.
Selective pressure caused by human-microbe interactions coupled with population admixture has helped shape the response of modern humans to pathogens (162). A recent example is a locus controlling COVID-19 severity in modern humans that can be traced to Neanderthal introgression (163). Mtb and humans have coexisted for an estimated 2,000 – 6,000 years (3, 4) and purifying selection of human genes by Mtb was traced to the bronze age for the TYK2 P1104A mutation. Similarly, as members of the first line of host innate immune defense PRRs have been subjected to purifying selection (164). PRR diversity in humans may explain, at least in part, the variable susceptibility to Mtb across populations.
For example, humans express 10 functional TLRs which are subdivided in two categories: cell surface (TLR1, 2, 4 – 6 and 10) and intracellular endolysosomal (TLR3, 7 – 9). The intracellular TLRs underwent strong purifying selection and have poor tolerance to loss of function mutations (165). Conversely, cell surface TLRs are more permissive to genetic variation across human populations (166). This difference may be attributed to the nature of ligands. Bacterial antigens detected by cell surface TLRs are clearly distinct from host molecules while nucleic acids detected by intracellular TLRs (RNA or DNA with CpGs) can resemble host endogenous factors. It has been proposed that mutations in intracellular TLRs are less tolerated to prevent “autoimmunity” (167–169). Mtb is detected by heterodimers of TLR1, 2 and 6, therefore presenting redundancy in the host response. Interestingly, mutations in the TLR1 (S248N, I602S), TLR6 (P249S) and TLR10 (I775V) genes clustered on chromosome 4p14 have shown signs of recent positive selection in Europeans (165). It has been suggested, although not confirmed, that tuberculosis and leprosy epidemics in Europe have played a role in this selective pressure (170). Of particular interest is the TLR1 I602S mutation which has been associated with both TB and leprosy (171–175). The TLR1 602S amino acid was shown to impair NF-κB activity in response to Mtb and decrease IL-6 production (174). Studies evaluating TLR2 mutations in TB have provided inconsistent results, which limited the interpretation of its role in TB pathogenesis (176, 177). Moreover, TLR4 and TLR9 have also been suggested to contribute to TB susceptibility (178–180).
NLR is another group of PRRs that shows signs of diversity between populations. NLRs encompass three families of cytosolic PRRs (NOD receptors, NLRPs and IPAFs) involved in viral and intracellular bacterial pathogen recognition. An excess of rare NOD1 non-synonymous variants segregating in the human population provided evidence for weak negative selection against these variants (181). In contrast, there was evidence among Asians and Europeans of positive selection for rare variants in NOD2 (181). In a meta-analysis, the NOD2 R702W amino acid change was associated with protection from TB (182, 183). Curiously, the same NOD2 R702W mutation is one of the strongest known genetic risk factors for Crohn’s disease, suggesting a pivotal role for NOD2 in balancing host inflammatory responses (184). Most NLRPs shows signs of strong selective constrains emphasizing their essential function in the human innate immune response (181). Macrophages challenged with Mtb or M. marinum in vitro showed a NLRP3-dependent increase in IL1β production (176). In a small population of cases with HIV/Mtb co-infection a non-coding variant in NLRP3 had a weak association with early mortality (185).
DC-SIGN (CD209), a member of CLR family, is a major dendritic cell receptor of Mtb (46). In ancient humans a duplication of CD209 gave origin to the CD209L gene. Interestingly, natural selection has prevented accumulation of amino acid changes in CD209 while the closely related CD209L gene was permissive (186). This discrepancy in selective pressures highlights the importance of function for CD209 while diversity in CD209L might have benefitted human adaptation to pathogens. Two promoter variants in CD209 are associated with TB in multiple African populations (187–189), South Asians (190) and Brazilians (191). Other PRRs, such as ficolins, have been evaluated for association with TB (192, 193), while studies with genes encoding proteins of the complement and PRRs for the RIG-1 family have not yet been reported.
Through animal experimentation, certain genes and associated pathways have been shown to be major determinants of the host outcome upon Mtb infection. Here, we define how some of these pathways compare to PRR pathways at the molecular level, and the level of importance to Mtb infection, as internal positive controls to our review.
For the sake of controls, similar systems to the PRR-MAMP interaction would include endogenous receptor-ligand systems. Receptor-cytokine interactions are an example which includes mechanisms that are even functionally related to PRR signalling pathways (e.g. the IL-1R/IL-1 system, which uses MyD88 like the TLRs as mentioned above).
IL-1R deficient mice were more susceptible to Mtb after intranasal infection with 105 CFU H37Rv strain, with a median survival of around 110 days, while no WT had died by 140 days; the remaining KO mice had 4 logs more pulmonary CFU than WT at 140 days post infection (194). Another study by a different group showed that with 100 CFU Kurono strain aerosol infection Il1r1 KO mice had died after 45 days (KO mice had 3 logs more pulmonary CFU than WT at 35 days) (195). During another H37Rv strain infection (200 CFU i.n.), Il1r1 KO mice phenocopied Myd88 KO mice (died around 4 weeks post infection) (159). There have been variable phenotypes with IL-1α and IL-1β deficiency: in one study Il1b KO was sufficient to phenocopy Il1r1 KO (155); in another study the double cytokine KO only reduced pulmonary Mtb CFU control (196); in a third study only double cytokine KO, not single, shortened survival like Il1r1 KO (197). Lastly, heterozygous deficiency of IL-1R antagonist, overexpressed in mice carrying the Sst1 (super susceptibility to tuberculosis 1) locus, almost completely rescued these mice from their type-I IFN driven early mortality and excessive pulmonary CFU burden during Mtb Erdman strain infection, again highlighting the protective effect of IL-1R signalling (198). Thus, MyD88-dependent cytokine-receptor systems can be critical for Mtb control in mice.
In mice lacking TNF receptor, or treated with anti-TNF antibodies, mice succumbed to uncontrolled Mtb Erdman strain i.v. infection in about a month while WT controls all survived past 125 days (199). This result has been replicated in Tnf KO mice in numerous studies over the years (118, 160, 197). TNF receptor deficient mice died approximately as rapidly as Tnf KO, even if the receptor KO was only on myeloid cells; lymphoid cell receptor KO did not differ from WT (200). Thus, the TNF pathway is critical for Mtb control in mice to prevent rapid death. The importance of TNF with Mtb infection in humans was demonstrated when anti-TNF treatment was associated with the emergence of TB in patients receiving this treatment for other reasons (201).
Similarly, IFN-γ signalling has been known to be critical for control of Mtb in animal models for decades (202, 203). IFN-γ from CD4+ T cells in particular is necessary for survival, and animals lacking IFN-γ from just CD4+ T cells succumb after two months post aerosol infection; however IFN-γ’s role was mostly extrapulmonary with a limited role in the lungs (204). IL-12p40, upon which IFN-γ is partly dependent, has also been knocked-out in mice and resulted in uncontrolled replication of Mtb (Erdman strain, administered i.v.) and mortality within 1.5 months compared to WT mice which lived “to old age” (205). Human mutations in IFN-γ or IL-12 pathway genes causing impaired IFN-γ-mediated immunity result in Mendelian Susceptibility to Mycobacterial Disease, which manifests as childhood BCG dissemination or non-tuberculous mycobacterial infection, and occasionally Mtb infection later in those who live (206).
Cytokine and PRR signalling on their own do not have direct bactericidal effects – they are thought to modulate innate defense mechanisms and instruct adaptive immunity. The endgame of bacteriologic control are the host’s killing mechanisms, which in macrophages include low phagosomal pH, digestive enzymes like lysozymes, and reactive oxygen species. As an example, the well-studied nitric oxide is produced by NOS2 to attack Mtb. Mice lacking NOS2 all died within 50 days of i.v. infection with 105 CFU Erdman strain while WT median survival was about 150 days (207). In a separate study, aerosol infection with 100 CFU H37Rv strain similarly resulted in death before day 50 associated with increased Mtb burden (208). Thus, effectors like nitric oxide are irreplaceable for control of Mtb and host survival.
Genetic epidemiology studies have provided only a handful of PRR and non-PRR genes as global risk factors for TB. This lack of success is in striking contrast to leprosy, the second most common mycobacterial disease in humans (209). Strain diversity of Mtb compared to M. leprae might have played a role; however, the most likely cause for the lack of consistent results is phenotypic heterogeneity among TB cases. Most studies define TB as a single entity combining cases regardless of their clinical and biological characteristics. While this approach has worked for leprosy (210, 211), in other instances combining all leprosy cases has proven troublesome due to the presence of well-defined endophenotypes (212, 213). Common endophenotypes in leprosy are excessive host inflammatory responses, so-called lepra reactions, that sub-divide the overall group of patients. Endophenotypes can result in misclassification of genetic effects (213, 214). Indeed, the genetic associations can be in opposite direction between endophenotype and disease per se (212, 215).
Genetic modulators with opposing effects on unrecognized endophenotypes and clinically defined TB might be difficult to detect even in studies with very large sample sizes. This raises the question if similar, perhaps more complex endophenotypes, underlie the disappointing results from TB genetic studies. Specifically, considering the impact of PPR genes on intermediary immune phenotypes in the mouse, it is conceivable that PPR polymorphism may yet have a role to play in the genetics of TB pathogenesis. Heterogeneity among cases appears to be predominant in large scale genetic studies in TB and the existence of TB endotypes has been proposed (216). Recent advances in molecular and analytical techniques have allowed the identification of at least two TB endotypes through unbiased clustering of transcriptional changes in distinct molecular pathways (217). One endophenotype presented immune exhaustion resulting in poor prognosis compared to the second endophenotype.
What remains unclear is to what extent TB endophenotypes represent the continued progression of TB pathogenesis or if they are distinct forms of the same disease. More studies will be necessary to settle this question. Such future studies need to focus on defining endophenotypes with the full weight of omics approaches, keeping in mind that these better-resolution phenotypes may represent kinetic entities. Such a ‘systems-medicine’ definition of TB, in excess of clinical and microbiological data, is expected to improve power for efficient mapping of endophenotypes. Molecular (RNA, proteins and metabolites) and immune (cellular) phenotyping using blood can provide information for dissociating TB cases into endophenotypes. This is a two-step approach, where first identification of interindividual molecular/cellular similarities is done prior to the genetic study. How to deal with the genetic study in the second step would depend on the groups, but could be either a continuous phenotype or stratified by endophenotype. Using an omics signature would overcome the limitations where patients are clinically similar but the genetic cause of TB is not the same. Clinical heterogeneity with Mtb infection that is ambiguous (e.g. placement on a spectrum from TST positive to active TB) can be better-defined or bypassed with non-biased omics data. However, independently of the nature of TB endotypes, it is now clear that heterogeneity may impact on genetic studies of TB and perhaps shed new light on the role of PPR polymorphisms.
PRRs appear to be important for immunologic responses but have a more subtle role in control of Mtb and the course of TB. We hypothesize that this is partly due to the redundancy of many PRRs sensing different Mtb MAMPs. Amongst this redundancy, however, there may be unique immunological adjustments performed by specific PRRs. In contrast, genes that produce products mediating distinct effects, like IFN-γ, IL-12, nitric oxide and TNF are clearly essential to the host’s survival. Although we can consider PRRs ‘less important’ than effectors, this prompts an interesting question: Is this a situation of reduced selective pressure, which explains human PRR diversity? It is imaginable that the immunological outcome performed by an orchestra of PRRs can be quite varied as individual PRR activities are tuned differently by genetics. By contrast, altering the potency of an effector like IFN-γ would directly correlate with Mtb control, and therefore selection would be purifying.
Human genetic association studies of TB have yielded but a few promising leads. Animal and cellular human data clearly demonstrate that PRRs affect immunity during Mtb infection, despite small and/or delayed survival and bacteriologic phenotypes in PRR KO mice. Thus, PRR mutation in humans might manifest in endophenotypes of Mtb infection – states of altered immunity wherein the progression of TB may possess subtly different parameters. Defining such endophenotypes of Mtb infection through molecular and immunological profiling of patients may provide a roadmap on which to trace the effects of PRR variation on the course of TB.
Literature searches were performed on the Medline database with Pubmed and results were collected and curated using Endnote X9 (Clarivate Analytics, USA). The text, table and figure were created with Microsoft Word, Excel and PowerPoint, respectively.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
J-YD is supported by a Canadian Institutes of Health Research (CIHR) Canada Graduate Scholarship – Master’s Program, Fonds de Recherche du Québec – Santé (FRQ-S) Doctoral Training Award, RI-MUHC studentships and scholarships from the McGill Department of Microbiology and Immunology. MB: CIHR foundation grant (FDN-148362). ES CIHR foundation grant (FDN-143332) grant by NIH (1R01AI124349).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BCG, Bacille Calmette-Guérin, attenuated M. bovis vaccine against Mtb; CFUs, Colony-forming units, a measure of the amount of bacteria; CLR, C-type lectin receptor; Collectins, Soluble CLR; KO, Knockout, removal of a gene from a genome; LAM, Lipoarabinomannan, mannosylated = ManLAM, uncapped = araLAM; LPS, Lipopolysaccharide, a.k.a. endotoxin, MAMP from Gram negative bacteria; MAMP, Microbe-associated molecular pattern, recognized by host PRR(s); MDP, Muramyl dipeptide; MHC, Major histocompatibility complex; Mtb, Mycobacterium tuberculosis, causative agent of TB; NLR, NOD-like receptor; NOD, Nucleotide-binding oligomerization domain-containing; PIMs, phosphatidylinositol mannosides; PRR, Pattern recognition receptor, recognize MAMP(s); TB, Tuberculosis, the disease caused by Mtb; TDM, Trehalose dimycolate; TLR, Toll-like receptor; TST, Tuberculin skin test, indicates adaptive T-cell response to mycobacterial antigens.
1. Houben RM, Dodd PJ. The Global Burden of Latent Tuberculosis Infection: A Re-Estimation Using Mathematical Modelling. PloS Med (2016) 13(10):e1002152. doi: 10.1371/journal.pmed.1002152
2. Behr MA, Edelstein PH, Ramakrishnan L. Revisiting the Timetable of Tuberculosis. BMJ (2018) 362:k2738. doi: 10.1136/bmj.k2738
3. Bos KI, Harkins KM, Herbig A, Coscolla M, Weber N, Comas I, et al. Pre-Columbian Mycobacterial Genomes Reveal Seals as a Source of New World Human Tuberculosis. Nature (2014) 514(7523):494–7. doi: 10.1038/nature13591
4. Menardo F, Duchêne S, Brites D, Gagneux S. The Molecular Clock of Mycobacterium Tuberculosis. PloS Pathog (2019) 15(9):e1008067. doi: 10.1371/journal.ppat.1008067
5. Gagneux S. Host-Pathogen Coevolution in Human Tuberculosis. Philos Trans R Soc Lond B Biol Sci (2012) 367(1590):850–9. doi: 10.1098/rstb.2011.0316
6. Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, et al. Tuberculosis and Impaired IL-23-Dependent IFN-γ Immunity in Humans Homozygous for a Common TYK2 Missense Variant. Sci Immunol (2018) 3(30):eaau8714. doi: 10.1126/sciimmunol.aau8714
7. Kerner G, Laval G, Patin E, Boisson-Dupuis S, Abel L, Casanova JL, et al. Human Ancient DNA Analyses Reveal the High Burden of Tuberculosis in Europeans Over the Last 2,000 Years. Am J Hum Genet (2021) 108(3):517–24. doi: 10.1016/j.ajhg.2021.02.009
8. Kerner G, Ramirez-Alejo N, Seeleuthner Y, Yang R, Ogishi M, Cobat A, et al. Homozygosity for TYK2 P1104A Underlies Tuberculosis in About 1% of Patients in a Cohort of European Ancestry. Proc Natl Acad Sci USA (2019) 116(21):10430–4. doi: 10.1073/pnas.1903561116
9. Möller M, Kinnear CJ, Orlova M, Kroon EE, van Helden PD, Schurr E, et al. Genetic Resistance to Mycobacterium Tuberculosis Infection and Disease. Front Immunol (2018) 9:2219(2219). doi: 10.3389/fimmu.2018.02219
10. Luo Y, Suliman S, Asgari S, Amariuta T, Baglaenko Y, Martínez-Bonet M, et al. Early Progression to Active Tuberculosis is a Highly Heritable Trait Driven by 3q23 in Peruvians. Nat Commun (2019) 10(1):3765. doi: 10.1038/s41467-019-11664-1
11. Napier G, Campino S, Merid Y, Abebe M, Woldeamanuel Y, Aseffa A, et al. Robust Barcoding and Identification of Mycobacterium Tuberculosis Lineages for Epidemiological and Clinical Studies. Genome Med (2020) 12(1):114. doi: 10.1186/s13073-020-00817-3
12. Ngabonziza JCS, Loiseau C, Marceau M, Jouet A, Menardo F, Tzfadia O, et al. A Sister Lineage of the Mycobacterium Tuberculosis Complex Discovered in the African Great Lakes Region. Nat Commun (2020) 11(1):2917. doi: 10.1038/s41467-020-16626-6
13. Coscolla M, Gagneux S, Menardo F, Loiseau C, Ruiz-Rodriguez P, Borrell S, et al. Phylogenomics of Mycobacterium Africanum Reveals a New Lineage and a Complex Evolutionary History. Microb Genom (2021) 7(2):000477. doi: 10.1099/mgen.0.000477
14. Bottai D, Frigui W, Sayes F, Di Luca M, Spadoni D, Pawlik A, et al. TbD1 Deletion as a Driver of the Evolutionary Success of Modern Epidemic Mycobacterium Tuberculosis Lineages. Nat Commun (2020) 11(1):684. doi: 10.1038/s41467-020-14508-5
15. Ernst JD. Antigenic Variation and Immune Escape in the MTBC. Adv Exp Med Biol (2017) 1019:171–90. doi: 10.1007/978-3-319-64371-7_9
16. Kroon EE, Kinnear CJ, Orlova M, Fischinger S, Shin S, Boolay S, et al. An Observational Study Identifying Highly Tuberculosis-Exposed, HIV-1-Positive But Persistently TB, Tuberculin and IGRA Negative Persons With M. tuberculosis specific antibodies in Cape Town, South Africa. EBioMedicine (2020) 61:103053. doi: 10.1016/j.ebiom.2020.103053
17. Joosten SA, Ottenhoff THM, Lewinsohn DM, Hoft DF, Moody DB, Seshadri C. Harnessing Donor Unrestricted T-Cells for New Vaccines Against Tuberculosis. Vaccine (2019) 37(23):3022–30. doi: 10.1016/j.vaccine.2019.04.050
18. Ruibal P, Voogd L, Joosten SA, Ottenhoff THM. The Role of Donor-Unrestricted T-Cells, Innate Lymphoid Cells, and NK Cells in Anti-Mycobacterial Immunity. Immunol Rev (2021) 301(1):30–47. doi: 10.1111/imr.12948
19. Janeway CA. Approaching the Asymptote? Evolution and Revolution in Immunology. Cold Spring Harb Symp Quant Biol (1989) 54:1–13. doi: 10.1101/SQB.1989.054.01.003
20. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The Dorsoventral Regulatory Gene Cassette Spätzle/Toll/cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell (1996) 86(6):973–83. doi: 10.1016/s0092-8674(00)80172-5
21. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A Human Homologue of the Drosophila Toll Protein Signals Activation of Adaptive Immunity. Nature (1997) 388(6640):394–7. doi: 10.1038/41131
22. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science (1998) 282(5396):2085–8. doi: 10.1126/science.282.5396.2085
23. Qureshi ST, Larivière L, Leveque G, Clermont S, Moore KJ, Gros P, et al. Endotoxin-Tolerant Mice Have Mutations in Toll-Like Receptor 4 (Tlr4). J Exp Med (1999) 189(4):615–25. doi: 10.1084/jem.189.4.615
24. Means TK, Lien E, Yoshimura A, Wang S, Golenbock DT, Fenton MJ. The CD14 Ligands Lipoarabinomannan and Lipopolysaccharide Differ in Their Requirement for Toll-Like Receptors. J Immunol (1999) 163(12):6748–55.
25. Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ. Human Toll-Like Receptors Mediate Cellular Activation by Mycobacterium Tuberculosis. J Immunol (1999) 163(7):3920–7.
26. Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, et al. Host Defense Mechanisms Triggered by Microbial Lipoproteins Through Toll-Like Receptors. Science (1999) 285(5428):732–6. doi: 10.1126/science.285.5428.732
27. Jones BW, Means TK, Heldwein KA, Keen MA, Hill PJ, Belisle JT, et al. Different Toll-Like Receptor Agonists Induce Distinct Macrophage Responses. J Leukoc Biol (2001) 69(6):1036–44.
28. Gilleron M, Quesniaux VF, Puzo G. Acylation State of the Phosphatidylinositol Hexamannosides From Mycobacterium Bovis Bacillus Calmette Guerin and Mycobacterium Tuberculosis H37Rv and its Implication in Toll-Like Receptor Response. J Biol Chem (2003) 278(32):29880–9. doi: 10.1074/jbc.M303446200
29. Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH. Mycobacterium Tuberculosis LprG (Rv1411c): A Novel TLR-2 Ligand That Inhibits Human Macrophage Class II MHC Antigen Processing. J Immunol (2004) 173(4):2660–8. doi: 10.4049/jimmunol.173.4.2660
30. Pecora ND, Gehring AJ, Canaday DH, Boom WH, Harding CV. Mycobacterium Tuberculosis LprA is a Lipoprotein Agonist of TLR2 That Regulates Innate Immunity and APC Function. J Immunol (2006) 177(1):422–9. doi: 10.4049/jimmunol.177.1.422
31. Drage MG, Pecora ND, Hise AG, Febbraio M, Silverstein RL, Golenbock DT, et al. TLR2 and its Co-Receptors Determine Responses of Macrophages and Dendritic Cells to Lipoproteins of Mycobacterium Tuberculosis. Cell Immunol (2009) 258(1):29–37. doi: 10.1016/j.cellimm.2009.03.008
32. Prados-Rosales R, Baena A, Martinez LR, Luque-Garcia J, Kalscheuer R, Veeraraghavan U, et al. Mycobacteria Release Active Membrane Vesicles That Modulate Immune Responses in a TLR2-Dependent Manner in Mice. J Clin Invest (2011) 121(4):1471–83. doi: 10.1172/jci44261
33. Blanc L, Gilleron M, Prandi J, Song OR, Jang MS, Gicquel B, et al. Mycobacterium Tuberculosis Inhibits Human Innate Immune Responses via the Production of TLR2 Antagonist Glycolipids. Proc Natl Acad Sci USA (2017) 114(42):11205–10. doi: 10.1073/pnas.1707840114
34. Cehovin A, Coates AR, Hu Y, Riffo-Vasquez Y, Tormay P, Botanch C, et al. Comparison of the Moonlighting Actions of the Two Highly Homologous Chaperonin 60 Proteins of Mycobacterium Tuberculosis. Infect Immun (2010) 78(7):3196–206. doi: 10.1128/iai.01379-09
35. Bai W, Liu H, Ji Q, Zhou Y, Liang L, Zheng R, et al. TLR3 Regulates Mycobacterial RNA-Induced IL-10 Production Through the PI3K/AKT Signaling Pathway. Cell Signal (2014) 26(5):942–50. doi: 10.1016/j.cellsig.2014.01.015
36. Keegan C, Krutzik S, Schenk M, Scumpia PO, Lu J, Pang YLJ, et al. Mycobacterium Tuberculosis Transfer RNA Induces IL-12p70 via Synergistic Activation of Pattern Recognition Receptors Within a Cell Network. J Immunol (2018) 200(9):3244–58. doi: 10.4049/jimmunol.1701733
37. Pawar K, Shigematsu M, Sharbati S, Kirino Y. Infection-Induced 5’-Half Molecules of Trnahisgug Activate Toll-Like Receptor 7. PloS Biol (2020) 18(12):e3000982. doi: 10.1371/journal.pbio.3000982
38. Matsumoto S, Matsumoto M, Umemori K, Ozeki Y, Furugen M, Tatsuo T, et al. DNA Augments Antigenicity of Mycobacterial DNA-Binding Protein 1 and Confers Protection Against Mycobacterium Tuberculosis Infection in Mice. J Immunol (2005) 175(1):441–9. doi: 10.4049/jimmunol.175.1.441
39. Ruiz A, Guzmán-Beltrán S, Carreto-Binaghi LE, Gonzalez Y, Juárez E. DNA From Virulent M. Tuberculosis Induces TNF-α Production and Autophagy in M1 Polarized Macrophages. Microb Pathog (2019) 132:166–77. doi: 10.1016/j.micpath.2019.04.041
40. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
41. Brown GD, Willment JA, Whitehead L. C-Type Lectins in Immunity and Homeostasis. Nat Rev Immunol (2018) 18(6):374–89. doi: 10.1038/s41577-018-0004-8
42. Schlesinger LS. Macrophage Phagocytosis of Virulent But Not Attenuated Strains of Mycobacterium Tuberculosis is Mediated by Mannose Receptors in Addition to Complement Receptors. J Immunol (1993) 150(7):2920–30.
43. Schlesinger LS, Hull SR, Kaufman TM. Binding of the Terminal Mannosyl Units of Lipoarabinomannan From a Virulent Strain of Mycobacterium Tuberculosis to Human Macrophages. J Immunol (1994) 152(8):4070–9.
44. Rajaram MV, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS. Mycobacterium Tuberculosis Activates Human Macrophage Peroxisome Proliferator-Activated Receptor Gamma Linking Mannose Receptor Recognition to Regulation of Immune Responses. J Immunol (2010) 185(2):929–42. doi: 10.4049/jimmunol.1000866
45. Guirado E, Rajaram MV, Chawla A, Daigle J, La Perle KM, Arnett E, et al. Deletion of Pparγ in Lung Macrophages Provides an Immunoprotective Response Against M. tuberculosis infection in mice. Tuberculosis (Edinb) (2018) 111:170–7. doi: 10.1016/j.tube.2018.06.012
46. Tailleux L, Schwartz O, Herrmann JL, Pivert E, Jackson M, Amara A, et al. DC-SIGN is the Major Mycobacterium Tuberculosis Receptor on Human Dendritic Cells. J Exp Med (2003) 197(1):121–7. doi: 10.1084/jem.20021468
47. Maeda N, Nigou J, Herrmann JL, Jackson M, Amara A, Lagrange PH, et al. The Cell Surface Receptor DC-SIGN Discriminates Between Mycobacterium Species Through Selective Recognition of the Mannose Caps on Lipoarabinomannan. J Biol Chem (2003) 278(8):5513–6. doi: 10.1074/jbc.C200586200
48. Driessen NN, Ummels R, Maaskant JJ, Gurcha SS, Besra GS, Ainge GD, et al. Role of Phosphatidylinositol Mannosides in the Interaction Between Mycobacteria and DC-SIGN. Infect Immun (2009) 77(10):4538–47. doi: 10.1128/iai.01256-08
49. Geurtsen J, Chedammi S, Mesters J, Cot M, Driessen NN, Sambou T, et al. Identification of Mycobacterial Alpha-Glucan as a Novel Ligand for DC-SIGN: Involvement of Mycobacterial Capsular Polysaccharides in Host Immune Modulation. J Immunol (2009) 183(8):5221–31. doi: 10.4049/jimmunol.0900768
50. Koppel EA, Ludwig IS, Hernandez MS, Lowary TL, Gadikota RR, Tuzikov AB, et al. Identification of the Mycobacterial Carbohydrate Structure That Binds the C-Type Lectins DC-SIGN, L-SIGN and SIGNR1. Immunobiology (2004) 209(1-2):117–27. doi: 10.1016/j.imbio.2004.03.003
51. Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TB. C-Type Lectin DC-SIGN Modulates Toll-Like Receptor Signaling via Raf-1 Kinase-Dependent Acetylation of Transcription Factor NF-Kappab. Immunity (2007) 26(5):605–16. doi: 10.1016/j.immuni.2007.03.012
52. Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Geijtenbeek TB. Carbohydrate-Specific Signaling Through the DC-SIGN Signalosome Tailors Immunity to Mycobacterium Tuberculosis, HIV-1 and Helicobacter Pylori. Nat Immunol (2009) 10(10):1081–8. doi: 10.1038/ni.1778
53. Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, et al. Direct Recognition of the Mycobacterial Glycolipid, Trehalose Dimycolate, by C-Type Lectin Mincle. J Exp Med (2009) 206(13):2879–88. doi: 10.1084/jem.20091750
54. Ostrop J, Jozefowski K, Zimmermann S, Hofmann K, Strasser E, Lepenies B, et al. Contribution of MINCLE-SYK Signaling to Activation of Primary Human APCs by Mycobacterial Cord Factor and the Novel Adjuvant TDB. J Immunol (2015) 195(5):2417–28. doi: 10.4049/jimmunol.1500102
55. Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, et al. The Adaptor Molecule CARD9 is Essential for Tuberculosis Control. J Exp Med (2010) 207(4):777–92. doi: 10.1084/jem.20090067
56. Miyake Y, Toyonaga K, Mori D, Kakuta S, Hoshino Y, Oyamada A, et al. C-Type Lectin MCL is an Fcrγ-Coupled Receptor That Mediates the Adjuvanticity of Mycobacterial Cord Factor. Immunity (2013) 38(5):1050–62. doi: 10.1016/j.immuni.2013.03.010
57. Miyake Y, Oh-hora M, Yamasaki S. C-Type Lectin Receptor MCL Facilitates Mincle Expression and Signaling Through Complex Formation. J Immunol (2015) 194(11):5366–74. doi: 10.4049/jimmunol.1402429
58. Kerscher B, Wilson GJ, Reid DM, Mori D, Taylor JA, Besra GS, et al. Mycobacterial Receptor, Clec4d (CLECSF8, MCL), is Coregulated With Mincle and Upregulated on Mouse Myeloid Cells Following Microbial Challenge. Eur J Immunol (2016) 46(2):381–9. doi: 10.1002/eji.201545858
59. Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL, Brown GD, et al. Dectin-1 Interaction With Mycobacterium Tuberculosis Leads to Enhanced IL-12p40 Production by Splenic Dendritic Cells. J Immunol (2007) 179(6):3463–71. doi: 10.4049/jimmunol.179.6.3463
60. Das R, Koo MS, Kim BH, Jacob ST, Subbian S, Yao J, et al. Macrophage Migration Inhibitory Factor (MIF) is a Critical Mediator of the Innate Immune Response to Mycobacterium Tuberculosis. Proc Natl Acad Sci U.S.A. (2013) 110(32):E2997–3006. doi: 10.1073/pnas.1301128110
61. Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, et al. Dectin-2 is a Direct Receptor for Mannose-Capped Lipoarabinomannan of Mycobacteria. Immunity (2014) 41(3):402–13. doi: 10.1016/j.immuni.2014.08.005
62. Decout A, Silva-Gomes S, Drocourt D, Blattes E, Rivière M, Prandi J, et al. Deciphering the Molecular Basis of Mycobacteria and Lipoglycan Recognition by the C-Type Lectin Dectin-2. Sci Rep (2018) 8(1):16840. doi: 10.1038/s41598-018-35393-5
63. Toyonaga K, Torigoe S, Motomura Y, Kamichi T, Hayashi JM, Morita YS, et al. C-Type Lectin Receptor DCAR Recognizes Mycobacterial Phosphatidyl-Inositol Mannosides to Promote a Th1 Response During Infection. Immunity (2016) 45(6):1245–57. doi: 10.1016/j.immuni.2016.10.012
64. Downing JF, Pasula R, Wright JR, Twigg HL 3rd, Martin WJ 2nd. Surfactant Protein a Promotes Attachment of Mycobacterium Tuberculosis to Alveolar Macrophages During Infection With Human Immunodeficiency Virus. Proc Natl Acad Sci U.S.A. (1995) 92(11):4848–52. doi: 10.1073/pnas.92.11.4848
65. Gaynor CD, McCormack FX, Voelker DR, McGowan SE, Schlesinger LS. Pulmonary Surfactant Protein A Mediates Enhanced Phagocytosis of Mycobacterium Tuberculosis by a Direct Interaction With Human Macrophages. J Immunol (1995) 155(11):5343–51.
66. Pasula R, Wright JR, Kachel DL, Martin WJ 2nd. Surfactant Protein A Suppresses Reactive Nitrogen Intermediates by Alveolar Macrophages in Response to Mycobacterium Tuberculosis. J Clin Invest (1999) 103(4):483–90. doi: 10.1172/jci2991
67. Sidobre S, Nigou J, Puzo G, Rivière M. Lipoglycans are Putative Ligands for the Human Pulmonary Surfactant Protein A Attachment to Mycobacteria. Crit role Lipids Lectin Carbohydrate Recognit J Biol Chem (2000) 275(4):2415–22. doi: 10.1074/jbc.275.4.2415
68. Ragas A, Roussel L, Puzo G, Rivière M. The Mycobacterium Tuberculosis Cell-Surface Glycoprotein Apa as a Potential Adhesin to Colonize Target Cells via the Innate Immune System Pulmonary C-Type Lectin Surfactant Protein a. J Biol Chem (2007) 282(8):5133–42. doi: 10.1074/jbc.M610183200
69. Ferguson JS, Voelker DR, McCormack FX, Schlesinger LS. Surfactant Protein D Binds to Mycobacterium Tuberculosis Bacilli and Lipoarabinomannan via Carbohydrate-Lectin Interactions Resulting in Reduced Phagocytosis of the Bacteria by Macrophages. J Immunol (1999) 163(1):312–21.
70. Ferguson JS, Voelker DR, Ufnar JA, Dawson AJ, Schlesinger LS. Surfactant Protein D Inhibition of Human Macrophage Uptake of Mycobacterium Tuberculosis is Independent of Bacterial Agglutination. J Immunol (2002) 168(3):1309–14. doi: 10.4049/jimmunol.168.3.1309
71. Ferguson JS, Martin JL, Azad AK, McCarthy TR, Kang PB, Voelker DR, et al. Surfactant Protein D Increases Fusion of Mycobacterium Tuberculosis-Containing Phagosomes With Lysosomes in Human Macrophages. Infect Immun (2006) 74(12):7005–9. doi: 10.1128/iai.01402-06
72. Garred P, Harboe M, Oettinger T, Koch C, Svejgaard A. Dual Role of Mannan-Binding Protein in Infections: Another Case of Heterosis? Eur J Immunogenet (1994) 21(2):125–31. doi: 10.1111/j.1744-313x.1994.tb00183.x
73. Luo F, Sun X, Wang Y, Wang Q, Wu Y, Pan Q, et al. Ficolin-2 Defends Against Virulent Mycobacteria Tuberculosis Infection In Vivo, and its Insufficiency is Associated With Infection in Humans. PloS One (2013) 8(9):e73859. doi: 10.1371/journal.pone.0073859
74. Bartlomiejczyk MA, Swierzko AS, Brzostek A, Dziadek J, Cedzynski M. Interaction of Lectin Pathway of Complement-Activating Pattern Recognition Molecules With Mycobacteria. Clin Exp Immunol (2014) 178(2):310–9. doi: 10.1111/cei.12416
75. Polotsky VY, Belisle JT, Mikusova K, Ezekowitz RA, Joiner KA. Interaction of Human Mannose-Binding Protein With Mycobacterium Avium. J Infect Dis (1997) 175(5):1159–68. doi: 10.1086/520354
76. Świerzko AS, Bartłomiejczyk MA, Brzostek A, Łukasiewicz J, Michalski M, Dziadek J, et al. Mycobacterial Antigen 85 Complex (Ag85) as a Target for Ficolins and Mannose-Binding Lectin. Int J Med Microbiol (2016) 306(4):212–21. doi: 10.1016/j.ijmm.2016.04.004
77. Zhong Y, Kinio A, Saleh M. Functions of NOD-Like Receptors in Human Diseases. Front Immunol (2013) 4:333. doi: 10.3389/fimmu.2013.00333
78. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a General Sensor of Peptidoglycan Through Muramyl Dipeptide (MDP) Detection. J Biol Chem (2003) 278(11):8869–72. doi: 10.1074/jbc.C200651200
79. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host Recognition of Bacterial Muramyl Dipeptide Mediated Through NOD2. Implications Crohn’s Disease J Biol Chem (2003) 278(8):5509–12. doi: 10.1074/jbc.C200673200
80. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An Essential Role for NOD1 in Host Recognition of Bacterial Peptidoglycan Containing Diaminopimelic Acid. Nat Immunol (2003) 4(7):702–7. doi: 10.1038/ni945
81. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jéhanno M, Viala J, et al. Nod1 Detects a Unique Muropeptide From Gram-Negative Bacterial Peptidoglycan. Science (2003) 300(5625):1584–7. doi: 10.1126/science.1084677
82. Lee JY, Hwang EH, Kim DJ, Oh SM, Lee KB, Shin SJ, et al. The Role of Nucleotide-Binding Oligomerization Domain 1 During Cytokine Production by Macrophages in Response to Mycobacterium Tuberculosis Infection. Immunobiology (2016) 221(1):70–5. doi: 10.1016/j.imbio.2015.07.020
83. Mahapatra S, Crick DC, McNeil MR, Brennan PJ. Unique Structural Features of the Peptidoglycan of Mycobacterium Leprae. J Bacteriol (2008) 190(2):655–61. doi: 10.1128/JB.00982-07
84. Essers L, Schoop HJ. Evidence for the Incorporation of Molecular Oxygen, a Pathway in Biosynthesis of N-Glycolylmuramic Acid in Mycobacterium Phlei. Biochim Biophys Acta (1978) 544:180–4. doi: 10.1016/0304-4165(78)90221-0
85. Raymond JB, Mahapatra S, Crick DC, Pavelka MS Jr. Identification of the namH Gene, Encoding the Hydroxylase Responsible for the N-Glycolylation of the Mycobacterial Peptidoglycan. J Biol Chem (2005) 280(1):326–33. doi: 10.1074/jbc.M411006200
86. Coulombe F, Divangahi M, Veyrier F, de Leseleuc L, Gleason JL, Yang Y, et al. Increased NOD2-Mediated Recognition of N-Glycolyl Muramyl Dipeptide. J Exp Med (2009) 206(8):1709–16. doi: 10.1084/jem.20081779
87. Hansen JM, Golchin SA, Veyrier FJ, Domenech P, Boneca IG, Azad AK, et al. N-Glycolylated Peptidoglycan Contributes to the Immunogenicity But Not Pathogenicity of Mycobacterium Tuberculosis. J Infect Dis (2014) 209(7):1045–54. doi: 10.1093/infdis/jit622
88. Dubé J-Y, McIntosh F, Zarruk JG, David S, Nigou J, Behr MA. Synthetic Mycobacterial Molecular Patterns Partially Complete Freund’s Adjuvant. Sci Rep (2020) 10(1):5874. doi: 10.1038/s41598-020-62543-5
89. Gandotra S, Jang S, Murray PJ, Salgame P, Ehrt S. Nucleotide-Binding Oligomerization Domain Protein 2-Deficient Mice Control Infection With Mycobacterium Tuberculosis. Infect Immun (2007) 75(11):5127–34. doi: 10.1128/IAI.00458-07
90. Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. NOD2-Deficient Mice Have Impaired Resistance to Mycobacterium Tuberculosis Infection Through Defective Innate and Adaptive Immunity. J Immunol (2008) 181(10):7157–65. doi: 10.4049/jimmunol.181.10.7157
91. Landes MB, Rajaram MV, Nguyen H, Schlesinger LS. Role for NOD2 in Mycobacterium Tuberculosis-Induced iNOS Expression and NO Production in Human Macrophages. J Leukoc Biol (2015) 97(6):1111–9. doi: 10.1189/jlb.3A1114-557R
92. Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, de Jong DJ, Langenberg DM, et al. NOD2 and Toll-Like Receptors are Nonredundant Recognition Systems of Mycobacterium Tuberculosis. PloS Pathog (2005) 1(3):279–85. doi: 10.1371/journal.ppat.0010034
93. Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, et al. Plasma Membrane Damage Causes NLRP3 Activation and Pyroptosis During Mycobacterium Tuberculosis Infection. Nat Commun (2020) 11(1):2270. doi: 10.1038/s41467-020-16143-6
94. Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, et al. Critical Role of AIM2 in Mycobacterium Tuberculosis Infection. Int Immunol (2012) 24(10):637–44. doi: 10.1093/intimm/dxs062
95. Watson RO, Manzanillo PS, Cox JS. Extracellular M. Tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell (2012) 150(4):803–15. doi: 10.1016/j.cell.2012.06.040
96. van der Vaart M, Korbee CJ, Lamers GE, Tengeler AC, Hosseini R, Haks MC, et al. The DNA Damage-Regulated Autophagy Modulator DRAM1 Links Mycobacterial Recognition via TLR-MYD88 to Autophagic Defense [Corrected]. Cell Host Microbe (2014) 15(6):753–67. doi: 10.1016/j.chom.2014.05.005
97. Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium Tuberculosis. Cell Host Microbe (2015) 17(6):820–8. doi: 10.1016/j.chom.2015.05.005
98. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium Tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses Through ESX-1. Cell Host Microbe (2015) 17(6):799–810. doi: 10.1016/j.chom.2015.05.003
99. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The Cytosolic Sensor cGAS Detects Mycobacterium Tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe (2015) 17(6):811–9. doi: 10.1016/j.chom.2015.05.004
100. Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, et al. A Bacterial Cyclic Dinucleotide Activates the Cytosolic Surveillance Pathway and Mediates Innate Resistance to Tuberculosis. Nat Med (2015) 21(4):401–6. doi: 10.1038/nm.3813
101. Yan S, Shen H, Lian Q, Jin W, Zhang R, Lin X, et al. Deficiency of the AIM2-ASC Signal Uncovers the STING-Driven Overreactive Response of Type I IFN and Reciprocal Depression of Protective IFN-γ Immunity in Mycobacterial Infection. J Immunol (2018) 200(3):1016–26. doi: 10.4049/jimmunol.1701177
102. Patten DA, Shetty S. More Than Just a Removal Service: Scavenger Receptors in Leukocyte Trafficking. Front Immunol (2018) 9:2904. doi: 10.3389/fimmu.2018.02904
103. Pombinho R, Sousa S, Cabanes D. Scavenger Receptors: Promiscuous Players During Microbial Pathogenesis. Crit Rev Microbiol (2018) 44(6):685–700. doi: 10.1080/1040841X.2018.1493716
104. Zimmerli S, Edwards S, Ernst JD. Selective Receptor Blockade During Phagocytosis Does Not Alter the Survival and Growth of Mycobacterium Tuberculosis in Human Macrophages. Am J Respir Cell Mol Biol (1996) 15(6):760–70. doi: 10.1165/ajrcmb.15.6.8969271
105. Bowdish DM, Sakamoto K, Kim MJ, Kroos M, Mukhopadhyay S, Leifer CA, et al. MARCO, TLR2, and CD14 are Required for Macrophage Cytokine Responses to Mycobacterial Trehalose Dimycolate and Mycobacterium Tuberculosis. PloS Pathog (2009) 5(6):e1000474. doi: 10.1371/journal.ppat.1000474
106. Benard EL, Roobol SJ, Spaink HP, Meijer AH. Phagocytosis of Mycobacteria by Zebrafish Macrophages is Dependent on the Scavenger Receptor Marco, a Key Control Factor of Pro-Inflammatory Signalling. Dev Comp Immunol (2014) 47(2):223–33. doi: 10.1016/j.dci.2014.07.022
107. Khan A, Mann L, Papanna R, Lyu MA, Singh CR, Olson S, et al. Mesenchymal Stem Cells Internalize Mycobacterium Tuberculosis Through Scavenger Receptors and Restrict Bacterial Growth Through Autophagy. Sci Rep (2017) 7(1):15010. doi: 10.1038/s41598-017-15290-z
108. Shi S, Blumenthal A, Hickey CM, Gandotra S, Levy D, Ehrt S. Expression of Many Immunologically Important Genes in Mycobacterium Tuberculosis-Infected Macrophages is Independent of Both TLR2 and TLR4 But Dependent on IFN-Alphabeta Receptor and STAT1. J Immunol (2005) 175(5):3318–28. doi: 10.4049/jimmunol.175.5.3318
109. Ozeki Y, Tsutsui H, Kawada N, Suzuki H, Kataoka M, Kodama T, et al. Macrophage Scavenger Receptor Down-Regulates Mycobacterial Cord Factor-Induced Proinflammatory Cytokine Production by Alveolar and Hepatic Macrophages. Microb Pathog (2006) 40(4):171–6. doi: 10.1016/j.micpath.2005.12.006
110. Schäfer G, Guler R, Murray G, Brombacher F, Brown GD. The Role of Scavenger Receptor B1 in Infection With Mycobacterium Tuberculosis in a Murine Model. PloS One (2009) 4(12):e8448. doi: 10.1371/journal.pone.0008448
111. Khan HS, Nair VR, Ruhl CR, Alvarez-Arguedas S, Galvan Rendiz JL, Franco LH, et al. Identification of Scavenger Receptor B1 as the Airway Microfold Cell Receptor for Mycobacterium Tuberculosis. Elife (2020) 9:e52551. doi: 10.7554/eLife.52551
112. Philips JA, Rubin EJ, Perrimon N. Drosophila RNAi Screen Reveals CD36 Family Member Required for Mycobacterial Infection. Science (2005) 309(5738):1251–3. doi: 10.1126/science.1116006
113. Dodd CE, Pyle CJ, Glowinski R, Rajaram MV, Schlesinger LS. CD36-Mediated Uptake of Surfactant Lipids by Human Macrophages Promotes Intracellular Growth of Mycobacterium Tuberculosis. J Immunol (2016) 197(12):4727–35. doi: 10.4049/jimmunol.1600856
114. Hawkes M, Li X, Crockett M, Diassiti A, Finney C, Min-Oo G, et al. CD36 Deficiency Attenuates Experimental Mycobacterial Infection. BMC Infect Dis (2010) 10:299. doi: 10.1186/1471-2334-10-299
115. Sattler N, Bosmani C, Barisch C, Guého A, Gopaldass N, Dias M, et al. Functions of the Dictyostelium LIMP-2 and CD36 Homologues in Bacteria Uptake, Phagolysosome Biogenesis and Host Cell Defence. J Cell Sci (2018) 131(17):jcs218040. doi: 10.1242/jcs.218040
116. Reiling N, Hölscher C, Fehrenbach A, Kröger S, Kirschning CJ, Goyert S, et al. Cutting Edge: Toll-Like Receptor (TLR)2- and TLR4-Mediated Pathogen Recognition in Resistance to Airborne Infection With Mycobacterium Tuberculosis. J Immunol (2002) 169(7):3480–4. doi: 10.4049/jimmunol.169.7.3480
117. Sugawara I, Yamada H, Li C, Mizuno S, Takeuchi O, Akira S. Mycobacterial Infection in TLR2 and TLR6 Knockout Mice. Microbiol Immunol (2003) 47(5):327–36. doi: 10.1111/j.1348-0421.2003.tb03404.x
118. Drennan MB, Nicolle D, Quesniaux VJ, Jacobs M, Allie N, Mpagi J, et al. Toll-Like Receptor 2-Deficient Mice Succumb to Mycobacterium Tuberculosis Infection. Am J Pathol (2004) 164(1):49–57. doi: 10.1016/s0002-9440(10)63095-7
119. Báfica A, Scanga CA, Schito ML, Hieny S, Sher A. Cutting Edge: In Vivo Induction of Integrated HIV-1 Expression by Mycobacteria is Critically Dependent on Toll-Like Receptor 2. J Immunol (2003) 171(3):1123–7. doi: 10.4049/jimmunol.171.3.1123
120. Tjärnlund A, Guirado E, Julián E, Cardona PJ, Fernández C. Determinant Role for Toll-Like Receptor Signalling in Acute Mycobacterial Infection in the Respiratory Tract. Microbes Infect (2006) 8(7):1790–800. doi: 10.1016/j.micinf.2006.02.017
121. Hölscher C, Reiling N, Schaible UE, Hölscher A, Bathmann C, Korbel D, et al. Containment of Aerogenic Mycobacterium Tuberculosis Infection in Mice Does Not Require MyD88 Adaptor Function for TLR2, -4 and -9. Eur J Immunol (2008) 38(3):680–94. doi: 10.1002/eji.200736458
122. Carlos D, Frantz FG, Souza-Júnior DA, Jamur MC, Oliver C, Ramos SG, et al. TLR2-Dependent Mast Cell Activation Contributes to the Control of Mycobacterium Tuberculosis Infection. Microbes Infect (2009) 11(8-9):770–8. doi: 10.1016/j.micinf.2009.04.025
123. Teixeira-Coelho M, Cruz A, Carmona J, Sousa C, Ramos-Pereira D, Saraiva AL, et al. TLR2 Deficiency by Compromising P19 (IL-23) Expression Limits Th 17 Cell Responses to Mycobacterium Tuberculosis. Int Immunol (2011) 23(2):89–96. doi: 10.1093/intimm/dxq459
124. Choi HH, Kim KK, Kim KD, Kim HJ, Jo EK, Song CH. Effects of Mycobacterial Infection on Proliferation of Hematopoietic Precursor Cells. Microbes Infect (2011) 13(14-15):1252–60. doi: 10.1016/j.micinf.2011.08.001
125. McBride A, Bhatt K, Salgame P. Development of a Secondary Immune Response to Mycobacterium Tuberculosis is Independent of Toll-Like Receptor 2. Infect Immun (2011) 79(3):1118–23. doi: 10.1128/iai.01076-10
126. McBride A, Konowich J, Salgame P. Host Defense and Recruitment of Foxp3⁺ T Regulatory Cells to the Lungs in Chronic Mycobacterium Tuberculosis Infection Requires Toll-Like Receptor 2. PloS Pathog (2013) 9(6):e1003397. doi: 10.1371/journal.ppat.1003397
127. Konowich J, Gopalakrishnan A, Dietzold J, Verma S, Bhatt K, Rafi W, et al. Divergent Functions of TLR2 on Hematopoietic and Nonhematopoietic Cells During Chronic Mycobacterium Tuberculosis Infection. J Immunol (2017) 198(2):741–8. doi: 10.4049/jimmunol.1601651
128. Gopalakrishnan A, Dietzold J, Verma S, Bhagavathula M, Salgame P. Toll-Like Receptor 2 Prevents Neutrophil-Driven Immunopathology During Infection With Mycobacterium Tuberculosis by Curtailing CXCL5 Production. Infect Immun (2019) 87(3):e00760-18. doi: 10.1128/iai.00760-18
129. Blumenthal A, Kobayashi T, Pierini LM, Banaei N, Ernst JD, Miyake K, et al. RP105 Facilitates Macrophage Activation by Mycobacterium Tuberculosis Lipoproteins. Cell Host Microbe (2009) 5(1):35–46. doi: 10.1016/j.chom.2008.12.002
130. Abel B, Thieblemont N, Quesniaux VJ, Brown N, Mpagi J, Miyake K, et al. Toll-Like Receptor 4 Expression is Required to Control Chronic Mycobacterium Tuberculosis Infection in Mice. J Immunol (2002) 169(6):3155–62. doi: 10.4049/jimmunol.169.6.3155
131. Kamath AB, Alt J, Debbabi H, Behar SM. Toll-Like Receptor 4-Defective C3H/HeJ Mice are Not More Susceptible Than Other C3H Substrains to Infection With Mycobacterium Tuberculosis. Infect Immun (2003) 71(7):4112–8. doi: 10.1128/iai.71.7.4112-4118.2003
132. Shim TS, Turner OC, Orme IM. Toll-Like Receptor 4 Plays No Role in Susceptibility of Mice to Mycobacterium Tuberculosis Infection. Tuberculosis (Edinb) (2003) 83(6):367–71. doi: 10.1016/s1472-9792(03)00071-4
133. Branger J, Leemans JC, Florquin S, Weijer S, Speelman P, van der Poll T. Toll-Like Receptor 4 Plays a Protective Role in Pulmonary Tuberculosis in Mice. Int Immunol (2004) 16(3):509–16. doi: 10.1093/intimm/dxh052
134. Park J, Kim H, Kwon KW, Choi HH, Kang SM, Hong JJ, et al. Toll-Like Receptor 4 Signaling-Mediated Responses are Critically Engaged in Optimal Host Protection Against Highly Virulent Mycobacterium Tuberculosis K Infection. Virulence (2020) 11(1):430–45. doi: 10.1080/21505594.2020.1766401
135. Wieland CW, van der Windt GJ, Wiersinga WJ, Florquin S, van der Poll T. CD14 Contributes to Pulmonary Inflammation and Mortality During Murine Tuberculosis. Immunology (2008) 125(2):272–9. doi: 10.1111/j.1365-2567.2008.02840.x
136. Branger J, Leemans JC, Florquin S, Speelman P, Golenbock DT, van der Poll T. Lipopolysaccharide Binding Protein-Deficient Mice Have a Normal Defense Against Pulmonary Mycobacterial Infection. Clin Immunol (2005) 116(2):174–81. doi: 10.1016/j.clim.2005.03.014
137. Gopalakrishnan A, Dietzold J, Salgame P. Vaccine-Mediated Immunity to Experimental Mycobacterium Tuberculosis is Not Impaired in the Absence of Toll-Like Receptor 9. Cell Immunol (2016) 302:11–8. doi: 10.1016/j.cellimm.2015.12.009
138. Wieland CW, Koppel EA, den Dunnen J, Florquin S, McKenzie AN, van Kooyk Y, et al. Mice Lacking SIGNR1 Have Stronger T Helper 1 Responses to Mycobacterium Tuberculosis. Microbes Infect (2007) 9(2):134–41. doi: 10.1016/j.micinf.2006.10.018
139. Tanne A, Ma B, Boudou F, Tailleux L, Botella H, Badell E, et al. A Murine DC-SIGN Homologue Contributes to Early Host Defense Against Mycobacterium Tuberculosis. J Exp Med (2009) 206(10):2205–20. doi: 10.1084/jem.20090188
140. Court N, Vasseur V, Vacher R, Frémond C, Shebzukhov Y, Yeremeev VV, et al. Partial Redundancy of the Pattern Recognition Receptors, Scavenger Receptors, and C-Type Lectins for the Long-Term Control of Mycobacterium Tuberculosis Infection. J Immunol (2010) 184(12):7057–70. doi: 10.4049/jimmunol.1000164
141. Heitmann L, Schoenen H, Ehlers S, Lang R, Holscher C. Mincle is Not Essential for Controlling Mycobacterium Tuberculosis Infection. Immunobiology (2013) 218(4):506–16. doi: 10.1016/j.imbio.2012.06.005
142. Lee WB, Kang JS, Yan JJ, Lee MS, Jeon BY, Cho SN, et al. Neutrophils Promote Mycobacterial Trehalose Dimycolate-Induced Lung Inflammation via the Mincle Pathway. PloS Pathog (2012) 8(4):e1002614. doi: 10.1371/journal.ppat.1002614
143. Wilson GJ, Marakalala MJ, Hoving JC, van Laarhoven A, Drummond RA, Kerscher B, et al. The C-Type Lectin Receptor CLECSF8/CLEC4D is a Key Component of Anti-Mycobacterial Immunity. Cell Host Microbe (2015) 17(2):252–9. doi: 10.1016/j.chom.2015.01.004
144. Marakalala MJ, Guler R, Matika L, Murray G, Jacobs M, Brombacher F, et al. The Syk/CARD9-Coupled Receptor Dectin-1 is Not Required for Host Resistance to Mycobacterium Tuberculosis in Mice. Microbes Infect (2011) 13(2):198–201. doi: 10.1016/j.micinf.2010.10.013
145. Lemos MP, McKinney J, Rhee KY. Dispensability of Surfactant Proteins A and D in Immune Control of Mycobacterium Tuberculosis Infection Following Aerosol Challenge of Mice. Infect Immun (2011) 79(3):1077–85. doi: 10.1128/iai.00286-10
146. McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, et al. Granuloma Formation and Host Defense in Chronic Mycobacterium Tuberculosis Infection Requires PYCARD/ASC But Not NLRP3 or Caspase-1. PloS One (2010) 5(8):e12320. doi: 10.1371/journal.pone.0012320
147. Walter K, Hölscher C, Tschopp J, Ehlers S. NALP3 is Not Necessary for Early Protection Against Experimental Tuberculosis. Immunobiology (2010) 215(9-10):804–11. doi: 10.1016/j.imbio.2010.05.015
148. Dorhoi A, Nouailles G, Jörg S, Hagens K, Heinemann E, Pradl L, et al. Activation of the NLRP3 Inflammasome by Mycobacterium Tuberculosis is Uncoupled From Susceptibility to Active Tuberculosis. Eur J Immunol (2012) 42(2):374–84. doi: 10.1002/eji.201141548
149. Allen IC, McElvania-TeKippe E, Wilson JE, Lich JD, Arthur JC, Sullivan JT, et al. Characterization of NLRP12 During the In Vivo Host Immune Response to Klebsiella Pneumoniae and Mycobacterium Tuberculosis. PloS One (2013) 8(4):e60842. doi: 10.1371/journal.pone.0060842
150. Hu S, Du X, Huang Y, Fu Y, Yang Y, Zhan X, et al. NLRC3 Negatively Regulates CD4+ T Cells and Impacts Protective Immunity During Mycobacterium Tuberculosis Infection. PloS Pathog (2018) 14(8):e1007266. doi: 10.1371/journal.ppat.1007266
151. Marinho FV, Benmerzoug S, Rose S, Campos PC, Marques JT, Báfica A, et al. The cGAS/STING Pathway Is Important for Dendritic Cell Activation But Is Not Essential to Induce Protective Immunity Against Mycobacterium Tuberculosis Infection. J Innate Immun (2018) 10(3):239–52. doi: 10.1159/000488952
152. Sever-Chroneos Z, Tvinnereim A, Hunter RL, Chroneos ZC. Prolonged Survival of Scavenger Receptor Class A-Deficient Mice From Pulmonary Mycobacterium Tuberculosis Infection. Tuberculosis (Edinb) (2011) 91 Suppl 1(Suppl 1):S69–74. doi: 10.1016/j.tube.2011.10.014
153. Hu C, Mayadas-Norton T, Tanaka K, Chan J, Salgame P. Mycobacterium Tuberculosis Infection in Complement Receptor 3-Deficient Mice. J Immunol (2000) 165(5):2596–602. doi: 10.4049/jimmunol.165.5.2596
154. Melo MD, Catchpole IR, Haggar G, Stokes RW. Utilization of CD11b Knockout Mice to Characterize the Role of Complement Receptor 3 (CR3, CD11b/CD18) in the Growth of Mycobacterium Tuberculosis in Macrophages. Cell Immunol (2000) 205(1):13–23. doi: 10.1006/cimm.2000.1710
155. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, et al. Cutting Edge: Caspase-1 Independent IL-1β Production Is Critical for Host Resistance to Mycobacterium Tuberculosis and Does Not Require TLR Signaling In Vivo. J Immunol (2010) 184(7):3326–30. doi: 10.4049/jimmunol.0904189
156. Flynn JL, Gideon HP, Mattila JT, Lin PL. Immunology Studies in non-Human Primate Models of Tuberculosis. Immunol Rev (2015) 264(1):60–73. doi: 10.1111/imr.12258
157. Plumlee CR, Duffy FJ, Gern BH, Delahaye JL, Cohen SB, Stoltzfus CR, et al. Ultra-Low Dose Aerosol Infection of Mice With Mycobacterium Tuberculosis More Closely Models Human Tuberculosis. Cell Host Microbe (2021) 29(1):68–82.e5. doi: 10.1016/j.chom.2020.10.003
158. Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 Regulates Th1 Responses and Cooperates With TLR2 in Mediating Optimal Resistance to Mycobacterium Tuberculosis. J Exp Med (2005) 202(12):1715–24. doi: 10.1084/jem.20051782
159. Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, et al. IL-1 Receptor-Mediated Signal is an Essential Component of MyD88-Dependent Innate Response to Mycobacterium Tuberculosis Infection. J Immunol (2007) 179(2):1178–89. doi: 10.4049/jimmunol.179.2.1178
160. Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium Tuberculosis Infection Despite Adaptive Immune Response in the Absence of Myd88. J Clin Invest (2004) 114(12):1790–9. doi: 10.1172/jci21027
161. Sugawara I, Yamada H, Mizuno S, Takeda K, Akira S. Mycobacterial Infection in MyD88-Deficient Mice. Microbiol Immunol (2003) 47(11):841–7. doi: 10.1111/j.1348-0421.2003.tb03450.x
162. Quintana-Murci L. Human Immunology Through the Lens of Evolutionary Genetics. Cell (2019) 177(1):184–99. doi: 10.1016/j.cell.2019.02.033
163. Zeberg H, Paabo S. The Major Genetic Risk Factor for Severe COVID-19 is Inherited From Neanderthals. Nature (2020) 587(7835):610–2. doi: 10.1038/s41586-020-2818-3
164. Barreiro LB, Quintana-Murci L. From Evolutionary Genetics to Human Immunology: How Selection Shapes Host Defence Genes. Nat Rev Genet (2010) 11(1):17–30. doi: 10.1038/nrg2698
165. Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, et al. Evolutionary Dynamics of Human Toll-Like Receptors and Their Different Contributions to Host Defense. PloS Genet (2009) 5(7):e1000562. doi: 10.1371/journal.pgen.1000562
166. Ferwerda B, McCall MB, Alonso S, Giamarellos-Bourboulis EJ, Mouktaroudi M, Izagirre N, et al. TLR4 Polymorphisms, Infectious Diseases, and Evolutionary Pressure During Migration of Modern Humans. Proc Natl Acad Sci USA (2007) 104(42):16645–50. doi: 10.1073/pnas.0704828104
167. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B Cell Responses to RNA-Related Antigens Due to TLR7 Gene Duplication. Science (2006) 312(5780):1669–72. doi: 10.1126/science.1124978
168. Krieg AM, Vollmer J. Toll-Like Receptors 7, 8, and 9: Linking Innate Immunity to Autoimmunity. Immunol Rev (2007) 220:251–69. doi: 10.1111/j.1600-065X.2007.00572.x
169. Celhar T, Magalhaes R, Fairhurst AM. TLR7 and TLR9 in SLE: When Sensing Self Goes Wrong. Immunol Res (2012) 53(1-3):58–77. doi: 10.1007/s12026-012-8270-1
170. Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, et al. Genome-Wide Patterns of Selection in 230 Ancient Eurasians. Nature (2015) 528(7583):499–503. doi: 10.1038/nature16152
171. Wong SH, Gochhait S, Malhotra D, Pettersson FH, Teo YY, Khor CC, et al. Leprosy and the Adaptation of Human Toll-Like Receptor 1. PloS Pathog (2010) 6:e1000979. doi: 10.1371/journal.ppat.1000979
172. Ma X, Liu Y, Gowen BB, Graviss EA, Clark AG, Musser JM. Full-Exon Resequencing Reveals Toll-Like Receptor Variants Contribute to Human Susceptibility to Tuberculosis Disease. PloS One (2007) 2(12):e1318. doi: 10.1371/journal.pone.0001318
173. Uciechowski P, Imhoff H, Lange C, Meyer CG, Browne EN, Kirsten DK, et al. Susceptibility to Tuberculosis is Associated With TLR1 Polymorphisms Resulting in a Lack of TLR1 Cell Surface Expression. J Leukoc Biol (2011) 90(2):377–88. doi: 10.1189/jlb.0409233
174. Hawn TR, Misch EA, Dunstan SJ, Thwaites GE, Lan NT, Quy HT, et al. A Common Human TLR1 Polymorphism Regulates the Innate Immune Response to Lipopeptides. Eur J Immunol (2007) 37(8):2280–9. doi: 10.1002/eji.200737034
175. Marques Cde S, Brito-de-Souza VN, Guerreiro LT, Martins JH, Amaral EP, Cardoso CC, et al. Toll-Like Receptor 1 N248S Single-Nucleotide Polymorphism is Associated With Leprosy Risk and Regulates Immune Activation During Mycobacterial Infection. J Infect Dis (2013) 208(1):120–9. doi: 10.1093/infdis/jit133
176. Mortaz E, Adcock IM, Tabarsi P, Masjedi MR, Mansouri D, Velayati AA, et al. Interaction of Pattern Recognition Receptors With Mycobacterium Tuberculosis. J Clin Immunol (2015) 35(1):1–10. doi: 10.1007/s10875-014-0103-7
177. Jin X, Yin S, Zhang Y, Chen X. Association Between TLR2 Arg677Trp Polymorphism and Tuberculosis Susceptibility: A Meta-Analysis. Microb Pathog (2020) 144:104173. doi: 10.1016/j.micpath.2020.104173
178. Cubillos-Angulo JM, Arriaga MB, Silva EC, Müller BLA, Ramalho DMP, Fukutani KF, et al. Polymorphisms in TLR4 and TNFA and Risk of Mycobacterium Tuberculosis Infection and Development of Active Disease in Contacts of Tuberculosis Cases in Brazil: A Prospective Cohort Study. Clin Infect Dis (2019) 69(6):1027–35. doi: 10.1093/cid/ciy1001
179. Graustein AD, Horne DJ, Arentz M, Bang ND, Chau TT, Thwaites GE, et al. TLR9 Gene Region Polymorphisms and Susceptibility to Tuberculosis in Vietnam. Tuberculosis (Edinb) (2015) 95(2):190–6. doi: 10.1016/j.tube.2014.12.009
180. Velez DR, Wejse C, Stryjewski ME, Abbate E, Hulme WF, Myers JL, et al. Variants in Toll-Like Receptors 2 and 9 Influence Susceptibility to Pulmonary Tuberculosis in Caucasians, African-Americans, and West Africans. Hum Genet (2010) 127(1):65–73. doi: 10.1007/s00439-009-0741-7
181. Vasseur E, Boniotto M, Patin E, Laval G, Quach H, Manry J, et al. The Evolutionary Landscape of Cytosolic Microbial Sensors in Humans. Am J Hum Genet (2012) 91(1):27–37. doi: 10.1016/j.ajhg.2012.05.008
182. Wang C, Chen ZL, Pan ZF, Wei LL, Xu DD, Jiang TT, et al. NOD2 Polymorphisms and Pulmonary Tuberculosis Susceptibility: A Systematic Review and Meta-Analysis. Int J Biol Sci (2013) 10(1):103–8. doi: 10.7150/ijbs.7585
183. Austin CM, Ma X, Graviss EA. Common Nonsynonymous Polymorphisms in the NOD2 Gene are Associated With Resistance or Susceptibility to Tuberculosis Disease in African Americans. J Infect Dis (2008) 197(12):1713–6. doi: 10.1086/588384
184. Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu NY, et al. Functional Variants in the LRRK2 Gene Confer Shared Effects on Risk for Crohn’s Disease and Parkinson’s Disease. Sci Transl Med (2018) 10(423):eaai7795. doi: 10.1126/scitranslmed.aai7795
185. Ravimohan S, Nfanyana K, Tamuhla N, Tiemessen CT, Weissman D, Bisson GP. Common Variation in NLRP3 Is Associated With Early Death and Elevated Inflammasome Biomarkers Among Advanced HIV/TB Co-Infected Patients in Botswana. Open Forum Infect Dis (2018) 5(5):ofy075. doi: 10.1093/ofid/ofy075
186. Barreiro LB, Patin E, Neyrolles O, Cann HM, Gicquel B, Quintana-Murci L. The Heritage of Pathogen Pressures and Ancient Demography in the Human Innate-Immunity CD209/CD209L Region. Am J Hum Genet (2005) 77(5):869–86. doi: 10.1086/497613
187. Barreiro LB, Neyrolles O, Babb CL, Tailleux L, Quach H, McElreavey K, et al. Promoter Variation in the DC-SIGN-Encoding Gene CD209 is Associated With Tuberculosis. PloS Med (2006) 3(2):e20. doi: 10.1371/journal.pmed.0030020
188. Vannberg FO, Chapman SJ, Khor CC, Tosh K, Floyd S, Jackson-Sillah D, et al. CD209 Genetic Polymorphism and Tuberculosis Disease. PloS One (2008) 3(1):e1388. doi: 10.1371/journal.pone.0001388
189. Ben-Ali M, Barreiro LB, Chabbou A, Haltiti R, Braham E, Neyrolles O, et al. Promoter and Neck Region Length Variation of DC-SIGN is Not Associated With Susceptibility to Tuberculosis in Tunisian Patients. Hum Immunol (2007) 68(11):908–12. doi: 10.1016/j.humimm.2007.09.003
190. Kobayashi K, Yuliwulandari R, Yanai H, Lien LT, Hang NT, Hijikata M, et al. Association of CD209 Polymorphisms With Tuberculosis in an Indonesian Population. Hum Immunol (2011) 72(9):741–5. doi: 10.1016/j.humimm.2011.04.004
191. da Silva RC, Segat L, da Cruz HL, Schindler HC, Montenegro LM, Crovella S, et al. Association of CD209 and CD209L Polymorphisms With Tuberculosis Infection in a Northeastern Brazilian Population. Mol Biol Rep (2014) 41(8):5449–57. doi: 10.1007/s11033-014-3416-y
192. Xu DD, Wang C, Jiang F, Wei LL, Shi LY, Yu XM, et al. Association of the FCN2 Gene Single Nucleotide Polymorphisms With Susceptibility to Pulmonary Tuberculosis. PloS One (2015) 10(9):e0138356. doi: 10.1371/journal.pone.0138356
193. Li Y, You EQ, Lin WH, Liu XN, Shen DP, Zhang XL, et al. Association of Ficolin-1 and Ficolin-3 Gene Variation and Pulmonary Tuberculosis Susceptibility in a Chinese Population. J Clin Lab Anal (2021) 35(4):e23732. doi: 10.1002/jcla.23732
194. Juffermans NP, Florquin S, Camoglio L, Verbon A, Kolk AH, Speelman P, et al. Interleukin-1 Signaling is Essential for Host Defense During Murine Pulmonary Tuberculosis. J Infect Dis (2000) 182(3):902–8. doi: 10.1086/315771
195. Sugawara I, Yamada H, Hua S, Mizuno S. Role of Interleukin (IL)-1 Type 1 Receptor in Mycobacterial Infection. Microbiol Immunol (2001) 45(11):743–50. doi: 10.1111/j.1348-0421.2001.tb01310.x
196. Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I. Protective Role of Interleukin-1 in Mycobacterial Infection in IL-1 α/β Double-Knockout Mice. Lab Invest (2000) 80(5):759–67. doi: 10.1038/labinvest.3780079
197. Bourigault ML, Segueni N, Rose S, Court N, Vacher R, Vasseur V, et al. Relative Contribution of IL-1α, IL-1β and TNF to the Host Response to Mycobacterium Tuberculosis and Attenuated M. bovis BCG Immun Inflammation Dis (2013) 1(1):47–62. doi: 10.1002/iid3.9
198. Ji DX, Yamashiro LH, Chen KJ, Mukaida N, Kramnik I, Darwin KH, et al. Type I Interferon-Driven Susceptibility to Mycobacterium Tuberculosis is Mediated by IL-1ra. Nat Microbiol (2019) 4(12):2128–35. doi: 10.1038/s41564-019-0578-3
199. Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor Necrosis Factor-Alpha is Required in the Protective Immune Response Against Mycobacterium Tuberculosis in Mice. Immunity (1995) 2(6):561–72. doi: 10.1016/1074-7613(95)90001-2
200. Segueni N, Benmerzoug S, Rose S, Gauthier A, Bourigault ML, Reverchon F, et al. Innate Myeloid Cell TNFR1 Mediates First Line Defence Against Primary Mycobacterium Tuberculosis Infection. Sci Rep (2016) 6:22454. doi: 10.1038/srep22454
201. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis Associated With Infliximab, a Tumor Necrosis Factor Alpha-Neutralizing Agent. N Engl J Med (2001) 345(15):1098–104. doi: 10.1056/NEJMoa011110
202. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated Tuberculosis in Interferon Gamma Gene-Disrupted Mice. J Exp Med (1993) 178(6):2243–7. doi: 10.1084/jem.178.6.2243
203. Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An Essential Role for Interferon Gamma in Resistance to Mycobacterium Tuberculosis Infection. J Exp Med (1993) 178(6):2249–54. doi: 10.1084/jem.178.6.2249
204. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T Cell-Derived IFN-Gamma Plays a Minimal Role in Control of Pulmonary Mycobacterium Tuberculosis Infection and Must Be Actively Repressed by PD-1 to Prevent Lethal Disease. PloS Pathog (2016) 12(5):e1005667. doi: 10.1371/journal.ppat.1005667
205. Cooper AM, Magram J, Ferrante J, Orme IM. Interleukin 12 (IL-12) is Crucial to the Development of Protective Immunity in Mice Intravenously Infected With Mycobacterium Tuberculosis. J Exp Med (1997) 186(1):39–45. doi: 10.1084/jem.186.1.39
206. Döffinger R, Dupuis S, Picard C, Fieschi C, Feinberg J, Barcenas-Morales G, et al. Inherited Disorders of IL-12- and IFNgamma-Mediated Immunity: A Molecular Genetics Update. Mol Immunol (2002) 38(12-13):903–9. doi: 10.1016/s0161-5890(02)00017-2
207. MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of Nitric Oxide Synthase as a Protective Locus Against Tuberculosis. Proc Natl Acad Sci (1997) 94(10):5243–8. doi: 10.1073/pnas.94.10.5243
208. Jung YJ, LaCourse R, Ryan L, North RJ. Virulent But Not Avirulent Mycobacterium Tuberculosis can Evade the Growth Inhibitory Action of a T Helper 1-Dependent, Nitric Oxide Synthase 2-Independent Defense in Mice. J Exp Med (2002) 196(7):991–8. doi: 10.1084/jem.20021186
209. Fava VM, Dallmann-Sauer M, Schurr E. Genetics of Leprosy: Today and Beyond. Hum Genet (2020) 139(6-7):835–46. doi: 10.1007/s00439-019-02087-5
210. Zhang F, Liu H, Chen S, Low H, Sun L, Cui Y, et al. Identification of Two New Loci at IL23R and RAB32 That Influence Susceptibility to Leprosy. Nat Genet (2011) 43(12):1247–51. doi: 10.1038/ng.973
211. Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, et al. Genomewide Association Study of Leprosy. N Engl J Med (2009) 361(27):2609–18. doi: 10.1056/NEJMoa0903753
212. Fava VM, Cobat A, Gzara C, Alcais A, Abel L, Schurr E. Reply to Zhang et al.: The Differential Role of LRRK2 Variants in Nested Leprosy Phenotypes. Proc Natl Acad Sci USA (2020) 117(19):10124–5. doi: 10.1073/pnas.2002654117
213. Fava VM, Cobat A, Van Thuc N, Latini AC, Stefani MM, Belone AF, et al. Association of TNFSF8 Regulatory Variants With Excessive Inflammatory Responses But Not Leprosy Per Se. J Infect Dis (2015) 211(6):968–77. doi: 10.1093/infdis/jiu566
214. Fava VM, Sales-Marques C, Alcais A, Moraes MO, Schurr E. Age-Dependent Association of TNFSF15/TNFSF8 Variants and Leprosy Type 1 Reaction. Front Immunol (2017) 8:155. doi: 10.3389/fimmu.2017.00155
215. Fava VM, Xu YZ, Lettre G, Van Thuc N, Orlova M, Thai VH, et al. Pleiotropic Effects for Parkin and LRRK2 in Leprosy Type-1 Reactions and Parkinson’s Disease. Proc Natl Acad Sci USA (2019) 116(31):15616–24. doi: 10.1073/pnas.1901805116
216. Lange C, Aarnoutse R, Chesov D, van Crevel R, Gillespie SH, Grobbel HP, et al. Perspective for Precision Medicine for Tuberculosis. Front Immunol (2020) 11:566608. doi: 10.3389/fimmu.2020.566608
Keywords: mycobacterium tuberculosis, tuberculosis, pattern recognition receptor (PRR), genetic association studies (GAS), C-type lectin receptors (CLRs), NOD-like receptors (NLRs), toll-like receptors (TLR), microbe associated molecular pattern (MAMP)
Citation: Dubé J-Y, Fava VM, Schurr E and Behr MA (2021) Underwhelming or Misunderstood? Genetic Variability of Pattern Recognition Receptors in Immune Responses and Resistance to Mycobacterium tuberculosis. Front. Immunol. 12:714808. doi: 10.3389/fimmu.2021.714808
Received: 25 May 2021; Accepted: 17 June 2021;
Published: 30 June 2021.
Edited by:
Chetan Seshadri, University of Washington, United StatesReviewed by:
Sarah Dunstan, The University of Melbourne, AustraliaCopyright © 2021 Dubé, Fava, Schurr and Behr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Yves Dubé, amVhbi15dmVzLmR1YmVAbWFpbC5tY2dpbGwuY2E=; Marcel A. Behr, bWFyY2VsLmJlaHJAbWNnaWxsLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.