 Takahide Ara
Takahide Ara Daigo Hashimoto
Daigo Hashimoto
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 August 2021
Sec. Alloimmunity and Transplantation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.713631
This article is part of the Research Topic Mouse Models of Hematopoietic Stem Cell Transplantation View all 11 articles
Prophylaxis for and treatment of graft-versus-host disease (GVHD) are essential for successful allogeneic hematopoietic stem cell transplantation (allo-SCT) and mainly consist of immunosuppressants such as calcineurin inhibitors. However, profound immunosuppression can lead to tumor relapse and infectious complications, which emphasizes the necessity of developing novel management strategies for GVHD. Emerging evidence has revealed that tissue-specific mechanisms maintaining tissue homeostasis and promoting tissue tolerance to combat GVHD are damaged after allo-SCT, resulting in exacerbation and treatment refractoriness of GVHD. In the gastrointestinal tract, epithelial regeneration derived from intestinal stem cells (ISCs), a microenvironment that maintains healthy gut microbiota, and physical and chemical mucosal barrier functions against pathogens are damaged by conditioning regimens and/or GVHD. The administration of growth factors for cells that maintain intestinal homeostasis, such as interleukin-22 (IL-22) for ISCs, R-spondin 1 (R-Spo1) for ISCs and Paneth cells, and interleukin-25 (IL-25) for goblet cells, mitigates murine GVHD. In this review, we summarize recent advances in the understanding of GVHD-induced tissue damage and emerging strategies for the management of GVHD.
Mature epithelial cells in the gut, skin, and liver have long been recognized as the primary target of acute graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplantation (allo-SCT). Mature epithelial cells in the gut are composed of functionally distinct populations, including enterocytes, Paneth cells, goblet cells, tuft cells, and enteroendocrine cells. Each of these epithelial populations contributes to the maintenance of tissue homeostasis (Table 1). Thus, injury of these epithelial cells results in alteration of the tissue microenvironment and disruption of tissue homeostasis, potentially amplifying GVHD-induced tissue damage. Furthermore, emerging evidence indicates that adult tissue stem cells are primarily targeted by GVHD, which decreases tissue resilience in GVHD target organs (5, 7, 19). Here, we review recent advances in the understanding the cellular and molecular mechanisms of GVHD-induced tissue damage and disruption of the tissue microenvironment. This review mainly focuses on gastrointestinal GVHD, while recent findings on the injury of tissue stem cells in the other organs are also summarized.
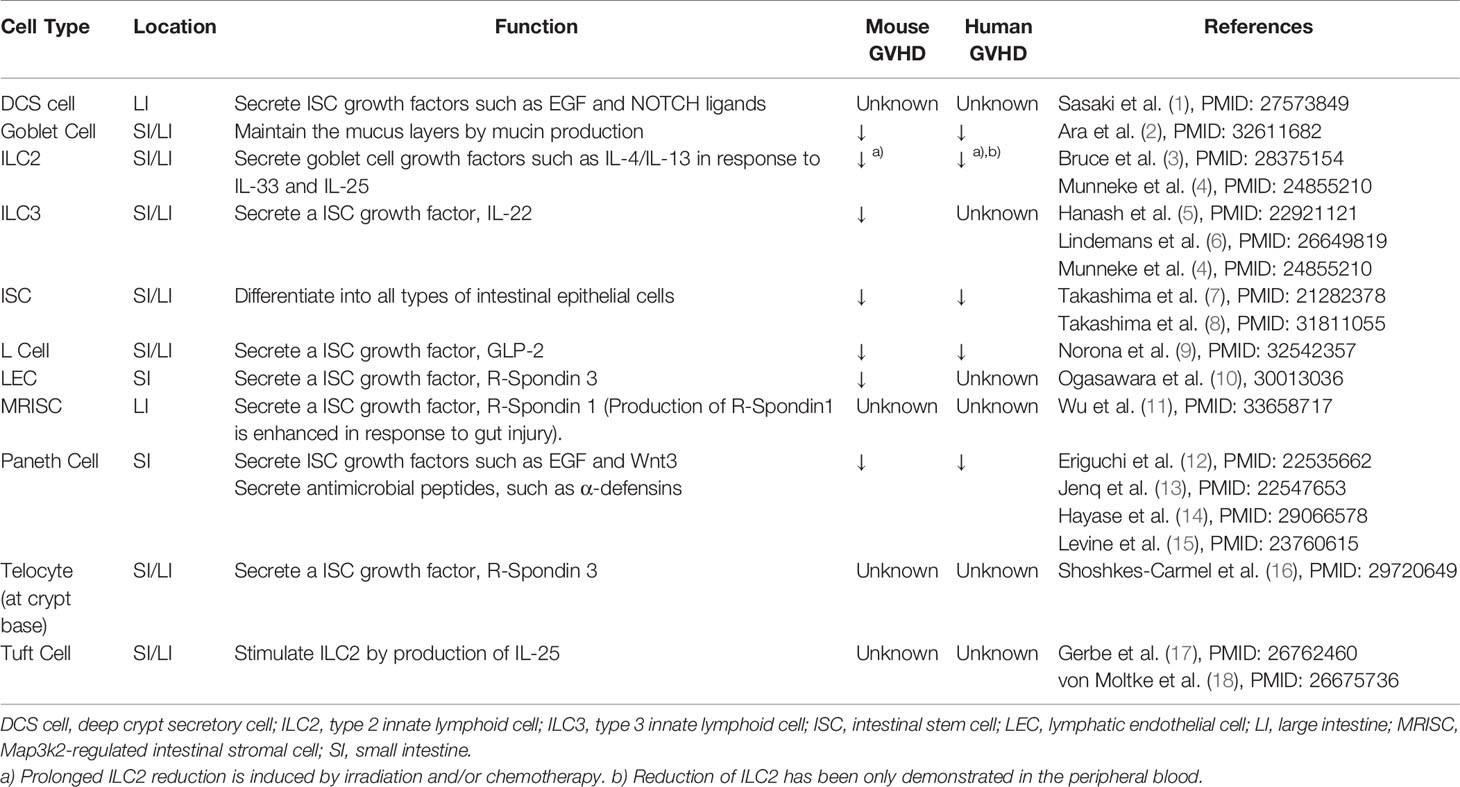
Table 1 Intestinal cells that maintain intestinal homeostasis.
It has been recognized that bacterial and fungal pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide and α-mannan, play a critical role in initiating GVHD (20–23). PAMPs enhance production of proinflammatory cytokines, host alloantigen presentation, and infiltration of innate cells into the gastrointestinal tracts early after conditioning (21, 24, 25). Recent advances revealed the critical role of sterile damage-associated molecular patterns (DAMPs) in pathophysiology of GVHD. Tissue damage induced by conditioning chemotherapy and/or irradiation promotes release of DAMPs from damaged cells and initiates the inflammatory cascade which culminates in expansion of donor alloreactive T cells and development of acute GVHD. DAMPs are comprised of various molecules that are sequestrated in the cells in the steady state, while released into the extracellular space by cellular damages. Extracellular ATP activates host antigen presenting cells and inflammatory monocytes via the purinergic P2X7 and P2Y2 receptors, respectively, that exaggerates mouse GVHD (26, 27). It has been shown that lack of nucleotide-binding oligomerization domain–like (NOD) receptor protein 3 (NLRP3), a known target of ATP/P2X7 receptor signaling, in the recipient mice ameliorated GVHD, suggesting that ATP exaggerates GVHD via activation of NLRP3 inflammasome (28). ATP-induced NLRP3 activation in myeloid-derived suppressor cells reduces anti-GVHD effects of these cells after adoptive transfer (29). Another NLRP3 activator, uric acid is released into the extracellular space after conditioning and exaggerates GVHD (28). Interleukin-33 (IL-33) is released from epithelial cells after injury and promotes effector T-cell differentiation of donor T cells, that results in the exaggeration of GVHD (30, 31). Heparan sulfate and high-mobility group box 1 protein bind to toll like receptor 4 and induce GVHD after allo-SCT (32, 33).
DAMPs and PAMPs primarily activate myeloid inflammatory cells such as neutrophils and monocytes, and antigen presenting cells such as dendritic cells and macrophages. Conditioning-induced tissue damage promotes accumulation of host neutrophils and production of reactive oxygen species in the gastrointestinal tract, that in turn amplifies the tissue injury (25). Interestingly, neutrophils accumulated in the gastrointestinal tract early after conditioning migrate to mesenteric lymph nodes and promote activation of host antigen presenting cells and donor T cells (34). It has been shown that donor neutrophils also exaggerate GVHD (35). In patients’ samples, higher density of neutrophil infiltration in the gut was associated with worse outcomes of GVHD, further emphasizing critical role of neutrophils in pathophysiology of acute GVHD (36). Monocytes and inflammatory macrophages also contribute to development of GVHD by producing proinflammatory cytokines in response to DAMPs and PAMPs and promoting activation of donor T cells (23, 27, 37). Importantly, IL-12 produced from monocytes and macrophages after irradiation enhances antigen presentation by host non-hematopoietic cells and exaggerates GVHD (24). On the other hand, host tissue resident macrophage, the ontogenetically independent population from monocytes and inflammatory macrophages, plays a protective role against GVHD by suppressing donor T cell expansion (38–40).
Histological features of intestinal GVHD include epithelial apoptosis, crypt degeneration, and mucosal sloughing, as well as inflammatory cell infiltration (41). Early preclinical studies pointed out that proliferation of crypt cells was enhanced in less severe GVHD, while more severe GVHD abrogated crypt cell proliferation in association with villus atrophy and loss of the crypt; these findings indicated that severe GVHD targets putative tissue stem cells residing in the intestinal crypt (42). More recently, leucine-rich-repeat-containing G protein-coupled receptor 5 (LGR5) was found to be a unique marker for cycling intestinal stem cells (ISCs) residing at the crypt base of the small intestine and colon (43) (Figure 1). In the steady state, approximately 10 cells are produced every hour in each crypt and migrate to the villus tip in 2-3 days, and a lineage-tracing study using the LGR5-Cre reporter system revealed that LGR5+ ISCs give rise to all gut epithelial lineages (43, 44). Depletion of LGR5+ ISCs in the mice, in which diphtheria toxin receptor (DTR) was specifically expressed in LGR5+ cells, significantly delayed epithelial regeneration after irradiation-induced intestinal damage, suggesting that LGR5+ ISCs are important also for the regenerative process after gut injury (45). Adoptive transfer of eGFP-specific TCR-transgenic T cells (Jedi T cells) depleted LGR5-eGFP+ ISCs and profoundly impaired the regenerative response after irradiation, suggesting that ISCs are susceptible to T cell-mediated injury (46–48). Furthermore, the crypt base region is the primary site infiltrated by donor T cells after allo-SCT; donor T cells migrate to the crypt base region in a MAdCAM-1-dependent manner as early as day 4 after murine allo-SCT, suggesting that ISCs are the primary target of gut GVHD (49).
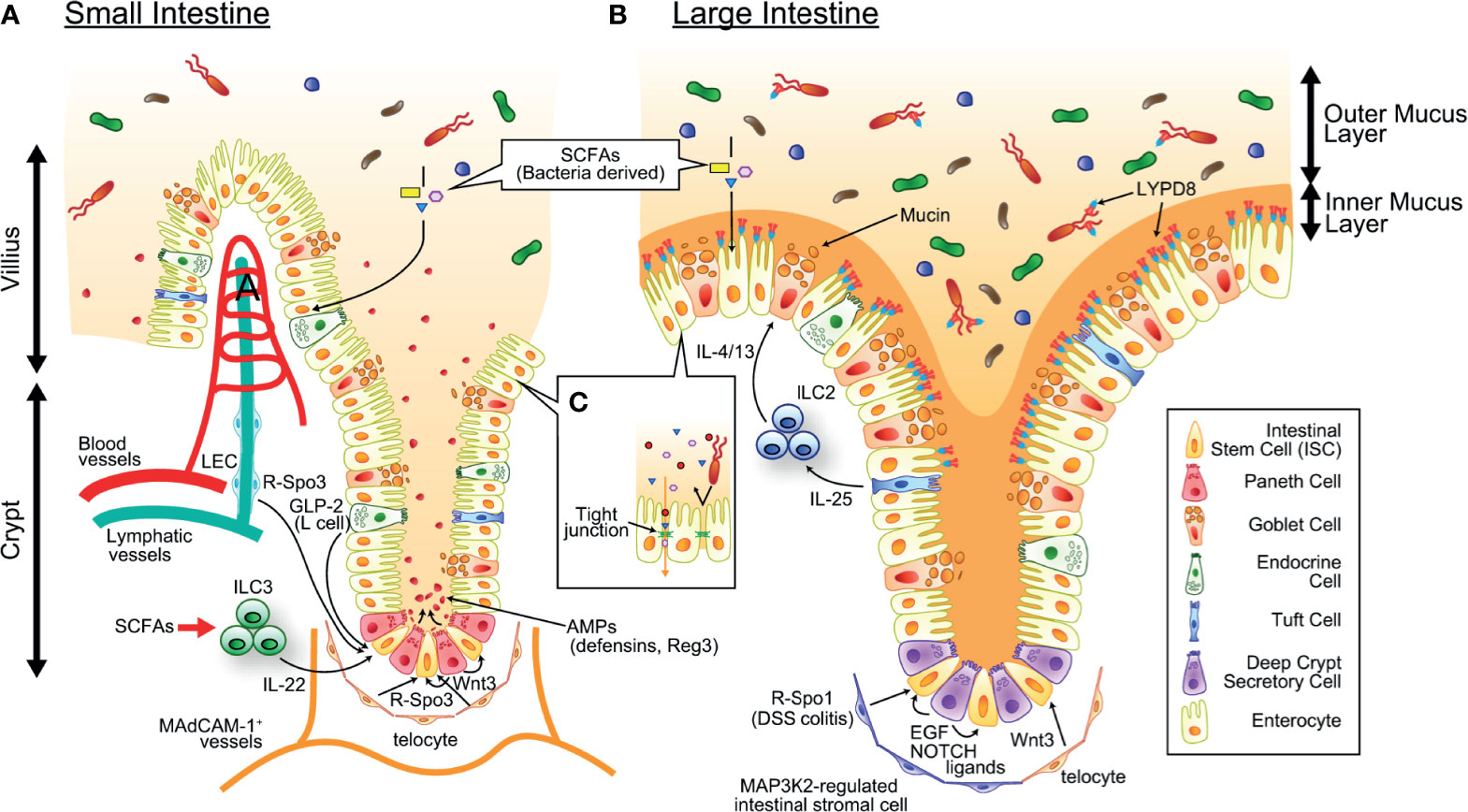
Figure 1 The mechanism maintaining intestinal homeostasis. ISCs residing at the crypt base give rise to all cell lineages in the epithelium and are supported by growth factors produced by definitive and putative niche components. SCFAs produced by commensal bacteria serves as energy source of intestinal epithelial cells. (A) In the small intestine, Paneth cells and telocytes produce Wnt3, telocytes and LECs produce R-Spo3, L cells produce GLP2, and ILC3s (green round cells in the figure) produce IL-22. Paneth cells also produce a large amount of AMPs such as α-defensins and REG3, and maintain healthy intestinal microbiota. (B) In the colon, deep crypt secretory cells produce EGF and NOTCH ligands, telocytes produce WNT3, and MAP3K2-regulated intestinal stromal cells produce R-Spo1. There are tremendous numbers of bacteria in the colonic lumen, which is segregated from epithelial cells by the inner mucus layer containing mucins produced by goblet cells and antimicrobial molecules such as REG3 and LYPD8 produced by enterocytes. IL-25 produced from Tuft cells stimulates ILC2s (blue round cells in figure) to secrete goblet cell growth factors such as IL-4 and IL-13. SCFAs produced by commensal bacteria serves as energy source of intestinal epithelial cells. (C) The intestinal epithelial tight junctions exhibit both size and charge selectivity and regulate the selective paracellular permeability, inhibiting penetration of bacteria and bacterial components while permitting the passage of water, ions, and small molecules. AMP, antimicrobial peptide; EGF, epithelial growth factor; GLP-2, Glucagon-like peptide 2; LEC, lymphatic endothelial cell; ILC2/3, type 2/3 innate lymphoid cell; IL-4/13/22/25, interleukin-4/13/22/25; ISC, intestinal stem cell; LYPD8, Ly6/PLAUR domain-containing protein 8; R-Spo1/3, R-spondin 1/3; SCFA, short-chain fatty acid.
A landmark study by Takashima et al. demonstrated that ISCs marked by another ISC-specific marker, olfactomedin-4 (Olfm4), are targeted by intestinal GVHD (7) (Figure 2). The reduction of LGR5+ ISCs in intestinal GVHD was then confirmed using a LGR5 reporter system (5). Due to the rapid turnover of gut epithelial cells, depletion of cycling ISCs in the crypt in intestinal GVHD soon leads to villus atrophy and causes refractory colitis (44). In the small intestine, quiescent Bmi1+ stem cells exist at four cell diameters above the base of the crypt and are called +4 stem cells (50). These cells are activated only after severe gut injury or depletion of LGR5+ ISCs and differentiate into all types of epithelial cells, including LGR5+ ISCs (51, 52). However, the fate and role of this second stem cell population in GVHD remain to be clarified. The mechanisms by which GVHD causes injury of LGR5+ ISCs have been studied intensively using a gut organoid culture system. Single LGR5+ ISCs isolated from the intestine give rise to crypt–villus organoids containing all differentiated cell types of the intestinal epithelium without the support of niche cells (53). Coculture of intestinal organoids with activated T cells induced caspase-3/caspase-7 cleavage and apoptosis of LGR5+ ISCs in the organoid, while IFN-γ blockade prevented T cell-mediated injury of the organoids, indicating that activated T cells damage LGR5+ ISCs in an IFN-γ-dependent manner (8, 54). In mouse models of allo-SCT, significantly more LGR5+ ISCs persisted after transplantation with IFN-γ-deficient donor T cells than after transplantation with wild-type (WT) donor T cells (54). Furthermore, administration of IFN-γ significantly reduced LGR5+ ISCs in the mice conditioned with total body irradiation, while it induced a proliferative response in the crypt in nonirradiated mice (54). Thus, IFN-γ seems to be more harmful for ISCs in the presence of genotoxic stress, such as irradiation, in vivo, while a high concentration of IFN-γ alone could induce apoptosis of ISCs in vitro. Alternatively, radiosensitive niche components could protect ISCs from IFN-γ in vivo to some extent.
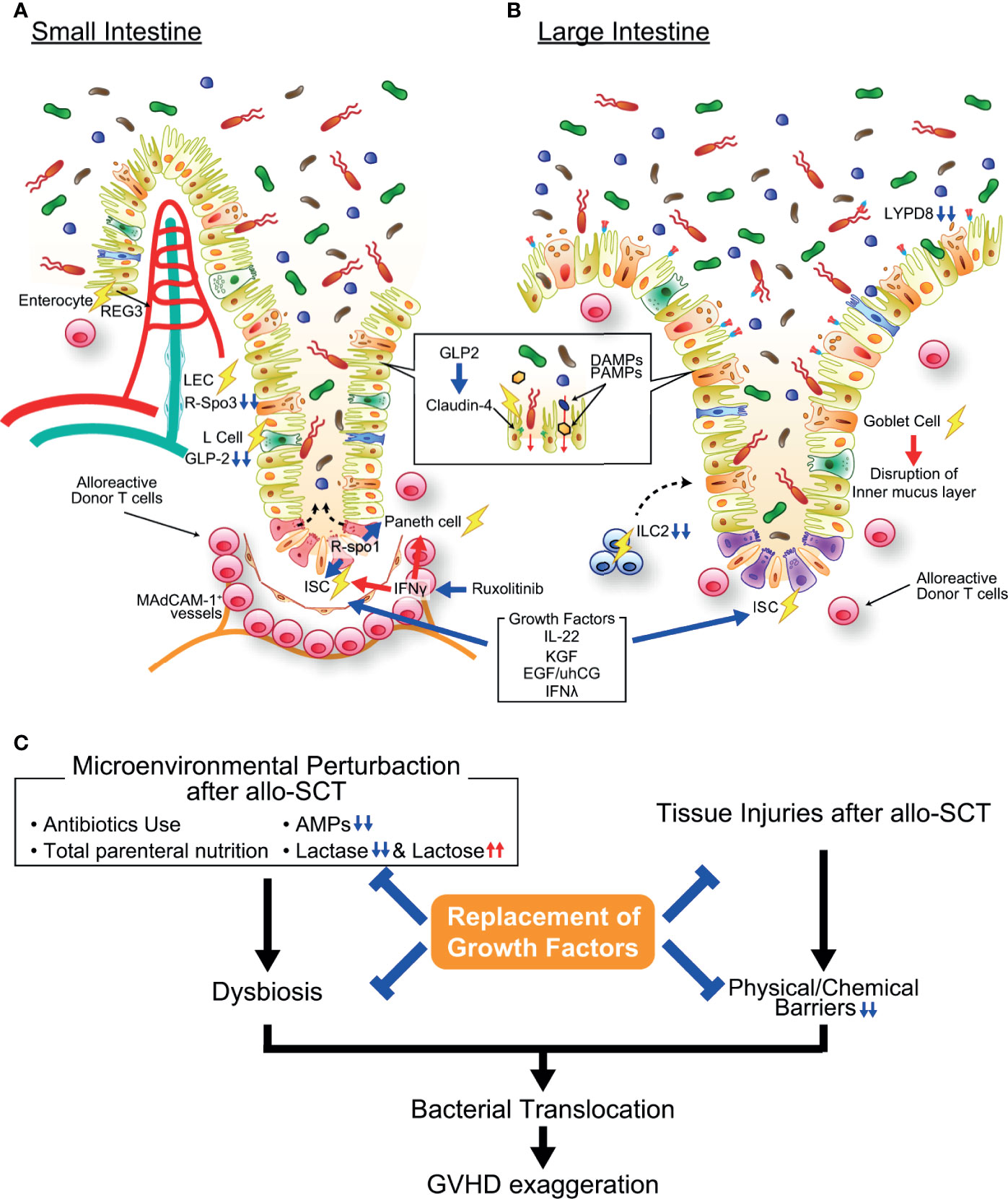
Figure 2 Pathophysiology of gastrointestinal graft-versus-host disease (GVHD). (A) In the small intestine, activated alloreactive donor T cells (pink round cells in figure) migrate to the crypt base region early after allogeneic transplantation in a MAdCAM-1-dependent manner and damage ISCs, resulting in impairment of mature intestinal epithelial cell regeneration. Paneth cell injury causes the reduction of AMP production and loss of function as an ISC niche. IFN-γ plays an important role in both ISC and Paneth cell injury in GVHD, and ruxolitinib protects ISCs and Paneth cells against GVHD. Moreover, growth factors of ISCs such as R-Spondin 3, IL-22, and GLP-2 are reduced in the intestine due to GVHD-induced reduction of LECs, ILC3s, and L cells. The expression of tight junction molecules such as claudin-4 are also reduced in GVHD, resulting in disruption of intestinal epithelial barrier function. (B) In the large intestine, goblet cell injury in GVHD results in disruption of the mucus layers bleaching both chemical and mechanical barrier functions of the intestinal mucosa. ILC2s, producer of goblet cell growth factors, are profoundly depleted by conditioning radiotherapy or chemotherapy, likely inhibiting regeneration of goblet cells. (C) Microenvironmental perturbation after allo-SCT induced by administration of antibiotics and/or total parenteral nutrition, reduction of AMP production, and lactose malabsorption leads to intestinal dysbiosis, frequently accompanying Enterococcus domination. Dysbiosis and disruption of barrier function of the intestinal mucosa enhance bacterial translocation, further exaggerating GVHD. Replacement of growth factors for ISCs, Paneth cells, and goblet cells ameliorate GVHD. Allo-SCT, allogeneic hematopoietic stem cell transplantation; DAMP, damage-associated molecular pattern; EGF, epidermal growth factor; IFN-γ, interferon-γ; KGF, keratinocyte growth factor; LYPD8, Ly6/PLAUR domain-containing protein 8; PAMP, pathogen-associated molecular pattern; REG, regenerating islet-derived protein; R-Spo1, R-spondin1; uhCG, urinary-derived human chorionic gonadotropin.
The ISC niche, which provides survival and growth factors for ISCs, is also targeted by GVHD (Figures 1 and 2). Interleukin-22 (IL-22) produced by type 3 innate lymphoid cells (ILC3s) is a well-described growth factor of LGR5+ ISCs (5). Total body irradiation (TBI) enhances IL-22 production from radioresistant ILC3s in an interleukin-23 (IL-23)-dependent manner, which is believed to promote regeneration of epithelial cells from radiation-induced damage. IL-22-producing ILC3s persisted after syngeneic bone marrow transplantation, while they were depleted after mouse allogeneic transplant, indicating that GVHD targets ILC3s. A reduction in IL-22 producing ILC3s in GVHD is associated with prolonged depletion of ISCs and exacerbation of gut GVHD.
Crypt bases have enriched transcription of Wnt target genes, and Paneth cells produce high levels of Wnt3, suggesting that Paneth cells are an ISC niche component (55, 56). Although the survival and proliferation of LGR5+ ISCs were not affected in Paneth cell-deficient mice in the steady state, Paneth cells may protect ISCs against gut injury (57). Because Paneth cells are also susceptible to IFN-γ-induced apoptosis in GVHD, regeneration of the gut epithelium from ISCs could be further disturbed in intestinal GVHD (12, 13). On the other hand, Paneth cell-derived Wnt3 is redundant with that produced from subepithelial telocytes (16, 58–60). It remains to be clarified whether telocytes are targeted by GVHD. In the colon, which is devoid of Paneth cells, deep crypt secretory (DCS) cells residing at the crypt base act as the niche for LGR5+ ISCs by producing NOTCH ligands and epidermal growth factor (EGF) (1). While DCS cells do not produce Wnt ligands, stromal tissues surrounding colonic crypts produce Wnt ligands and support colonic ISCs (58). It also remains to be clarified whether DCS cell are targeted by GVHD. R-spondins are the ligands of LGR4, LGR5, and LGR6 and enhance Wnt/β-catenin signaling by preventing ubiquitination and degradation of the Wnt receptor Frizzled (61, 62). The R-spondin family is composed of four molecules. R-Spo1-R-Spo4 share a similar structure, and each of these four molecules can bind to LGR4, LGR5 and LGR6 (63). We found that R-Spo3 is the major molecule produced in the small intestine (10). Although it has been reported that mesenchymal cells, including telocytes, produce R-Spo3, we found that CD90+CD31+podoplanin+ lymphatic endothelial cells are the main producers of R-Spo3 in the intestine (10, 16). Importantly, both the R-Spo3 production and absolute numbers of lymphatic endothelial cells are significantly reduced in GVHD (10). On the other hand, it remains to be clarified whether R-Spo3-producing telocytes are targeted by GVHD. The importance of R-Spo3 was also demonstrated in an antibody-mediated inhibition study, in which administration of anti-R-Spo3 antibodies alone reduced LGR5+ ISCs in naïve mice and suppressed the regenerative response after irradiation (64). Although this study showed that anti-R-Spo2 antibodies and anti-R-Spo3 antibodies work synergistically in the depletion of LGR5+ ISCs, the cellular source of R-Spo2 in the intestine remains to be clarified. Interestingly, recent study showed that Map3k2-regulated intestinal stromal cells (MRISCs) residing around the crypt base enhance production of R-Spo1 in response to dextran sodium sulfate (DSS)-induced colitis and protect colonic ISCs (11). Map3k2-deficient mice are more susceptible to DSS-induced colitis compared with wild type controls, further emphasizing a protective role of MRISCs against inflammation of the colon (11). These findings suggest that there are distinct ISC niche systems in the small intestine and the colon, and further studies are required to assess the fate of these ISC niches in GVHD.
Strategies that protect ISCs or induce their regeneration could be therapeutic options for GVHD that avoid strengthening immune suppression, which could lead to infection or leukemia relapse. As mentioned above, the reduction in IL-22 produced by ILC3s in GVHD leads to depletion of ISCs. IL-22 induces the proliferation and differentiation of ISCs and inhibits the apoptosis of ISCs after genotoxic stress (65). Replacement of IL-22 by administration of F-652, a recombinant fusion protein consisting of an rhIL-22 dimer and Fc fusion protein, after mouse allogeneic bone marrow transplantation enhanced the recovery of ISCs, increased epithelial regeneration, and ameliorated GVHD (6). However, the potential benefit of IL-22 could be limited because IL-22 secreted from donor T cells has been shown to aggravate GVHD by reducing Tregs and enhancing inflammatory responses (66–68). It has been suggested that IL-22 induces Th1 cell infiltration in the gastrointestinal tract via a host type I interferon dependent manner (69). Thus, the safety and efficacy of IL-22 replacement therapy must be evaluated in clinical studies; F-652 is now being tested for the treatment of lower gastrointestinal acute GVHD (NCT02406651). Because ILC3s produce IL-22 in response to bacterial metabolites such as short-chain fatty acids (SCFAs), probiotics that produce SCFAs could be used for GVHD prophylaxis (70).
Administration of R-spondins is also promising for GVHD prophylaxis, as this strategy protects ISCs against mouse GVHD. Recombinant human R-Spo1 (rhR-Spo1) was found to stimulate the proliferation of epithelial cells in the intestinal crypt (71). Subsequently, it was shown that rhR-Spo1 expands ISCs in naïve mice and mice undergoing allo-SCT. Importantly, rhR-Spo1 administered in the peritransplant period protects ISCs against GVHD and ameliorated GVHD after allo-SCT in TBI-conditioned mice (7). In contrast, rhR-Spo1 does not impact the severity of GVHD after allo-SCT without conditioning, potentially indicating synergistic effects of TBI and T-cell-derived IFN-γ on ILC injury (7, 54). Administration of a Robo ligand, Slit2 works synergistically with R-Spo1 in preventing ISC loss after chemoradiotherapy, suggesting that this combination could be useful for GVHD prophylaxis (72).
Type III interferon plays a protective role against gastrointestinal GVHD. Type III interferon family was discovered in 2003 and consists of four molecules, IFN-λ1 (IL-29), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and IFN-λ4 (73). Among them, IFN-λ2 and IFN-λ3 are expressed in both humans and mice, while IFN-λ1 gene is a pseudogene in mice, and IFN-λ4 gene is absent in mice. IFN-λ receptor consists of two chains, including a unique subunit, IFN-λ receptor 1 (IFNLR1) and common IL-10 receptor-β (IL-10RB) chain, which is shared with cytokines of the IL-10 family. IFNLR1 is preferentially expressed in gastrointestinal epithelium, suggesting that IFNλ is a key effector cytokine in mucosal immunity (74, 75). Recently, Henden and colleagues showed that IFNλ treatment improves the proliferative and regenerative capacity of LGR5+ ISCs independently of IL-22 and ameliorates murine GVHD (76). Since, pegylated recombinant IL-29 is being developed as an adjunctive therapy for Hepatitis C, this agent may be rapidly testable for clinical GVHD (77).
Ruxolitinib, a JAK1/2 inhibitor, has been shown to ameliorate mouse and human GVHD and has been approved by the Food and Drug Administration (FDA) in the United States for the treatment of steroid-refractory acute GVHD (78, 79). Ruxolitinib profoundly suppresses T cell activation, proliferation, and differentiation toward T helper 1 (Th1), Th17 and cytotoxic T cells (79). Given the critical role of IFN-γ in ISC injury, it has been tested if ruxolitinib could protect ISCs against GVHD by inhibiting JAK1/2-STAT1 pathway, an indispensable pathway in IFN-γ receptor signaling. Organoid culture systems have demonstrated that allogeneic T cells induce apoptosis of organoids and ISCs in an IFN-γ-dependent manner (8). Ruxolitinib protected ISCs and Paneth cells in organoids from IFN-γ and allogeneic T cells (8, 54). Furthermore, ruxolitinib prevented IFN-γ-induced ISC injury after syngeneic SCT, indicating that ruxolitinib protects ISCs independent of suppression of allogeneic T cell activation (54). These ISC-targeting strategies for GVHD prophylaxis and treatment are promising and could promote regeneration of all types of intestinal epithelial cells after GVHD-mediated injury (Figure 2).
Tissue stem cells in other target organs, such as the skin and liver, could be involved in GVHD pathophysiology. The fate of skin stem cells in acute cutaneous GVHD has been studied. Multiple tissue stem and/or progenitor populations of epithelial cells have been identified in the skin. The bulge of hair follicles has long been recognized to foster tissue stem cells because long-lived label-retaining cells exist in the hair bulge (80). More recently, it became possible to identify hair follicle stem cells (HFSCs) in the lower part of the bulge as CD34+, cytokeratin 15 (CK15)+, and LGR5+ cells using flow cytometric or immunofluorescent studies (81–83). These HFSCs alone can regenerate all structures of hair follicles and hair shafts and contribute to regeneration of the epidermis after skin injury (19, 82, 84). In addition to HFSCs, LGR6+ stem cells residing directly above the bulge and leucine-rich repeats and immunoglobulin-like domains 1 (Lrig1)+ stem cells in the isthmus maintain the upper pilosebaceous units (85, 86). Other than stem cells in the hair follicles, there are CK15+ epidermal progenitor and/or stem cells in the rete-like prominences (RLPs) of mouse tongues, a surrogate of human epidermal rete ridges of the skin.
Early studies demonstrated that donor T cells primarily migrate to stem cell-rich parts of the skin, such as mouse RLPs, human rete ridges, and the bulge of hair follicles, suggesting that skin stem cells could be targeted by GVHD (87–90). Among multiple stem cell populations, CK15+ stem and/or progenitor cells in mouse RLPs have been shown to undergo cytokine-induced apoptosis in cutaneous GVHD (90–92). Recently, we found that LGR5+ HFSCs were significantly reduced in mouse cutaneous GVHD, in association with reduced numbers of hair follicles, alopecia, and delayed wound healing (19). This finding was rather surprising because a previous study showed that injection of eGFP-specific Jedi T cells did not deplete LGR5-eGFP+ HFSCs, suggesting that these LGR5+ HFSCs are immune privileged (47). This discrepancy suggests that HFSCs are not inherently immune privileged and that the environment and/or HFSC niche protect HFSCs against immune-mediated injury. An extensive inflammatory environment or disruption of the HFSC niche could be responsible for HFSC damage in cutaneous GVHD. One of the HFSC niche components, subcutaneous fat, which acts as regulator of hair cycling and energy reservoir for HFSCs, becomes atrophic in cutaneous GVHD, which can lead to a reduction of LGR5+ HFSCs (19, 93–95). Although the mechanism by which GVHD depletes LGR5+ HFSCs and CK15+ RLP stem cells remains to be clarified, it is worth of note that topical administration of ruxolitinib protects these stem cells from mouse GVHD (19). On the other hand, topical corticosteroids demonstrate direct toxicity that leads to depletion of HFSCs after syngeneic and allogeneic SCT, even though topical steroids dramatically reduce donor T cell infiltration to the skin in cutaneous GVHD. Protection of LGR5+ HFSCs with topical ruxolitinib was associated with suppression of alopecia and enhancement of wound healing after allo-SCT, while topical corticosteroid was not (19). Based on its protective effects on both ISCs and skin stem cells, ruxolitinib could be an ideal therapeutic agent for GVHD (78, 79). The fate of other stem cell populations in the skin, such as LGR6+ stem cells and Lrig1+ stem cells in the hair follicles, remains to be clarified (85, 86).
The liver, another major target organ in acute GVHD, is a highly regenerative organ, and there are two main epithelial populations: hepatocytes and biliary epithelial cells (BECs). Lineage-tracing studies have shown that there are stem and/or progenitor populations of hepatocytes that maintain the hepatocyte pool in steady states, for example, studies in Axin2-Cre/ER reporter mice (96). However, some of these lineage-tracing strains have aberrant proliferation of labeled hepatocytes, possibly due to deletion of exons of the target molecule, which potentially leads to overestimation of the contribution of labeled cells to tissue regeneration after liver injury (97). More recently, it has been shown that mid-lobular hepatocytes cycle and maintain whole hepatocytes in the liver, except glutamine synthetase (GS)-expressing hepatocytes facing the central vein, which are maintained independently from other hepatocytes (97, 98). In certain contexts of liver injury, LGR5+ and Sox9+ hepatocytes endowed with the potential to differentiate both into hepatocytes and BECs emerge, and BECs proliferate and contribute to the reconstitution of hepatocytes after severe liver injury (99–103). Because jaundice and biliary dysfunction are the cardinal features of liver GVHD, it should be more important to study BEC stem cells rather than stem cells of hepatocytes. Huch et al. found that single LGR5+ cells isolated from the hepatic duct give rise to liver organoids that can be differentiated into both hepatocytes and BECs (99). The fate of BEC stem cells needs to be clarified in future studies.
Intestinal dysbiosis is frequently observed after allo-SCT and is associated with exacerbation of GVHD and transplantation-related death (13, 104, 105). Multiple factors, such as antibiotics and total parenteral nutrition, can lead to dysbiosis after allo-SCT (Figure 2 and Table 1). In addition, GVHD-induced tissue injury can generate a microenvironment related to dysbiosis. α-Defensins, major antimicrobial peptides (AMPs) produced from Paneth cells, exert potent bactericidal effects on pathogenic bacteria that occupy a minor proportion of the healthy microbiota but are minimally effective on nonpathogenic commensals that dominate the healthy gut microbiota (106, 107). Paneth cells are highly sensitive to GVHD, and α-defensin production is profoundly decreased in GVHD (12, 13, 108). This reduction is mediated by IFN-γ signaling, and ruxolitinib can protect Paneth cells against GVHD (54). R-Spo1, a growth factor of ISCs, is also a potent inducer of Paneth cell differentiation from ISCs, and we found that administration of rhR-Spo1 induced expansion of Paneth cells in naïve mice, leading to marked elevation of fecal levels of α-defensins such as cryptdin-1 (Crp-1) and cryptdin-4 (Crp-4) (14). In mouse GVHD, peritransplant administration of R-Spo1 protects not only ISCs but also Paneth cells, resulting in preserved α-defensin production and prevention of intestinal dysbiosis after allo-SCT (14). Short-term oral administration of Crp-4 to allogeneic recipient mice temporally mitigated intestinal dysbiosis and inflammation in the gut after allo-SCT, while dysbiosis developed after cessation of Crp-4 treatment, indicating that long-term administration of Crp-4, until Paneth cell regeneration, is required for the prevention of dysbiosis after allo-SCT (14). Paneth cell numbers in duodenal biopsies from transplanted patients are negatively related to gut GVHD severity, further emphasizing the protective role of Paneth cells against GVHD (15).
REG3, another major AMP in the intestine, is produced by intestinal epithelial cells, including Paneth cells and enterocytes, and diffuses into the inner mucus layer, segregating luminal bacteria from the gut epithelium (109, 110). In mouse models of allo-SCT, the expression levels of REG3γ, the mouse homolog of human REG3α, in the small intestine were significantly reduced in GVHD, and REG3γ leaked from the gut to the blood, leading to elevation of plasma levels of REG3γ (111, 112). In clinical allo-SCT, the plasma levels of REG3α and ST2 are now widely appreciated as diagnostic and prognostic biomarkers of acute GVHD (113–115). In mouse models of steroid-refractory GVHD, it has been shown that IL-22 produced by donor Th/Tc22 cells stimulates REG3γ production in the intestine, and excess REG3γ leads to dysbiosis and exacerbation of GVHD (67). Thus, REG3γ could be a therapeutic target for treating steroid-refractory GVHD.
Enterococcus domination, defined as a status in which 30% or more of all the bacteria in the fecal microbiota are enterococci, develops frequently after allo-SCT and is associated with blood stream infection, development and exacerbation of GVHD, and GVHD-related death after allo-SCT (105, 116–119). Because the presence of the VanA gene in fecal samples from allo-SCT recipients has been associated with Enterococcus domination, antibiotics likely contribute to the development of Enterococcus domination (119). However, Enterococcus domination is also observed after murine allo-SCT in which no antibiotics are used, indicating that antibiotics are not the only reason for Enterococcus domination and that GVHD may induce a microenvironment suitable for the expansion of enterococci. The growth of enterococci is strictly dependent on lactose, and the expression of lactase, a critical enzyme for the absorption of lactose from the diet, in the intestine is reduced in GVHD (119, 120). The reduction in lactase in GVHD leads to ineffective absorption and an increase in lactose availability in the gut lumen, leading to enterococcal expansion. Importantly, the lactose intolerance allele is associated with the persistence of Enterococcus domination after the cessation of antibiotics. These data suggest that a lactose-free diet or lactase administration could be used for prophylactic treatment of Enterococcus domination, which could improve the outcomes of allo-SCT. In addition to antibiotic administration and lactase reduction, reduction of α-defensins, which exert potent bactericidal effects on Enterococcus, could contribute to enterococcal expansion after allo-SCT (107).
The intestinal mucosa has the complex task of acting as a semipermeable barrier that allows the absorption of nutrients and water while limiting the transport of potentially harmful microbes and microbial components. Sheets of gut epithelial cells are bound to each other via tight junctions, acting as a physical barrier against luminal components (Figure 1). Conditioning and allogeneic T cell responses damage epithelial cells (Table 1), leading to the loss of the physical barrier function of the mucosal epithelium against bacteria and bacterial components , which fosters an environment prone to GVHD development (121). Thus, epithelial growth factors have been proposed as therapeutic options for acute GVHD (Figure 2). Keratinocyte growth factor (KGF) promotes the proliferation and differentiation of epithelial cells. It was suggested that ISCs are also supported by KGF; however, whether KGF protects ISCs against GVHD has not been explored using specific ISC markers, such as LGR5 (122, 123). Although it has been reported that KGF ameliorates murine gut GVHD (124, 125), human recombinant KGF did not demonstrate significant beneficial effects on the incidence and severity of GVHD in randomized clinical trials (126–128). Although the reason for this discrepancy between preclinical and clinical studies is not fully understood, it has been suggested that KGF could exert more potent anti-GVHD effects in recipients conditioned with TBI alone than in those conditioned with TBI in combination with cytotoxic agents; the latter strategy is used in the clinical setting (126).
Glucagon-like peptide 2 (GLP-2) is another growth factor of gut epithelial cells, and administration of a GLP-2 analog protects ISCs against irradiation-induced injury (129). GLP-2-producing enteroendocrine L cells are targeted by GVHD, and reduction of L cells in the patients’ colon is associated with worse outcome after allo-SCT. Because GLP-2 is inactivated by DPP-4, a DPP-4-resistant GLP-2 analog, teduglutide, was tested for a GVHD prophylaxis. Peritransplant administration of teduglutide protected ISCs and Paneth cells against GVHD, and prolonged survival after mouse allo-SCT (9). Furthermore, GLP-2 and GLP-2 analogues enhance the expression of tight junction molecules such as claudin-4, possibly enhancing intestinal barrier function in GVHD (9, 130). A clinical trial in which teduglutide is tested for treatment of short bowel syndrome (NCT04733066) is ongoing, and future clinical studies are required to test if teduglutide could protect patients against GVHD. Interestingly, a small-scale phase II study demonstrated that peritransplant administration of high-dose sitagliptin, a DPP-4 inhibitor, prevented the onset of acute GVHD (131). Although it is most likely that DPP-4 inhibition prevents GVHD by suppressing donor T cell activation (132), DPP-4 inhibition may mitigate damage to the intestinal epithelium by inhibiting GLP-2 degradation (133). The impact of DPP-4 inhibitors on GVHD-induced damage to gut epithelial cells needs to be clarified in future studies.
In rodent models of radiation colitis, administration of EGF enhanced gut epithelial regeneration (134, 135). In a phase I clinical trial, it has been shown that administration of a urinary-derived human chorionic gonadotropin (uhCG) agent containing abundant EGF was safe and possibly effective for the treatment of high-risk or steroid-refractory acute GVHD (136). This agent may improve GVHD via EGF-induced protection of gut epithelial cells, while the Treg expansion observed after administration of this agent could contribute to GVHD suppression, too. This inexpensive and commercially available uhCG agent will be studied in phase II and III trials.
In mouse GVHD, the TNF-α/MLCK210 axis increases tight junction permeability to larger molecules (137). IFN-γ also regulates tight junction permeability (138). Thus, GVHD actively increases permeability through tight junctions, which promotes the absorption of bacterial components, further recruiting donor T cells and propagating GVHD (137). Prevention of increase of tight junction permeability could be another prophylactic strategy against GVHD.
Loss of commensals in intestinal microbiota after allo-SCT leads to the reduction of bacterial metabolites which contribute to maintenance of tissue homeostasis. Among these metabolites, butyrate is mainly produced by commensal anaerobes such as Clostridia and Blautia, and mitigates harmful immune reactions by promoting differentiation of regulatory T cells (139). Microbiota-derived butyrate is also taken up by intestinal epithelial cells through G-protein coupled receptor, GPR43 and serves as a major energy source of intestinal epithelial cells. Butyrate acts as a histone deacetylase (HDAC) inhibitor and promotes tricarboxylic acid cycling, improving integrity of barrier function of intestinal mucosa (140, 141). Thus, dysbiosis with the reduction of butyrogenic bacteria reduces butyrate in the intestinal epithelial cells and impairs the resilience of the gut epithelium after allo-SCT (142). Probiotics containing butyrogenic bacteria or prebiotics containing butyrogenic fibers and starch are promising therapeutic options against mouse and human GVHD (140, 143). The urinary levels of 3-indoxyl sulfate (3-IS) are positively correlated with the abundances of Lachnospiraceae and Ruminococcaceae in the gut microbiota, and higher levels of urinary 3-IS predicts better survival after allo-SCT. Although the direct role of 3-IS in GVHD remains to be clarified, 3-IS could act as a ligand for aryl hydrocarbon receptor, the critical receptor for maintenance of intestinal epithelial barrier function and production of AMPs (144–146).
The intestinal mucus layer constitutes a critical barrier that segregates millions of microbes and environmental antigens in the gut lumen from the host immune system (Figure 1). The mucus layer serves as the first line of innate defense, and gel-forming mucins secreted by goblet cells form the basic scaffold of the mucus layer. Mice lacking the Muc2 gene, encoding the major gel-forming mucin in the intestine, are devoid of mucus layers and prone to developing severe colitis, suggesting that direct contact between luminal bacteria and the intestinal mucosa triggers inflammation (147, 148). The large intestine has a system with two mucus layers; the inner mucus layer is enriched with antimicrobial molecules (AMMs), such as Ly6/Plaur domain-containing 8 (LYPD8), and devoid of bacteria, suggesting that the mucus layer also acts as a chemical barrier against luminal bacteria (149, 150).
As noted above, GVHD-induced Paneth cell injury and lactose malabsorption together with other factors, such as antibiotic administration and induction of total parenteral nutrition, lead to intestinal dysbiosis after allo-SCT. In such a situation, the mucosal barrier must act as the final line of defense against pathogenic bacteria expanding in the gut lumen. However, GVHD leads to a reduction in intestinal goblet cells, which results in disruption of the mucus layer due to its rapid turnover (Figure 2 and Table 1); the mucus layer is renewed every 1 to 2 hours by newly produced mucus from goblet cells (151–153). Recently, we studied the role of goblet cells and the inner mucus layer in the pathophysiology of acute GVHD using mouse models of acute GVHD (2). First, we confirmed that goblet cells were profoundly and persistently reduced in the colon after allo-SCT, which led to disruption of the colonic two-layered mucus system in allogeneic recipients in association with enhanced bacterial translocation, elevated plasma levels of proinflammatory cytokines, and exacerbation of GVHD. Although the mechanism by which GVHD targets goblet cells remains to be clarified, it is possible that goblet cells are reduced due to GVHD-induced depletion of ISCs considering the rapid turnover of goblet cells (3 to 7 days) (154). In the steady state and after parasite infections, ILC2s produce growth factors of goblet cells, such as interleukin 13 (IL-13), in response to interleukin-25 (IL-25) secreted from Tuft cells (17, 18). We found that pretransplant administration of IL-25 expanded goblet cells that persisted after GVHD, preventing bacterial translocation, elevation of proinflammatory cytokines, and exacerbation of GVHD (2). Conditioning TBI and chemotherapy lead to prolonged depletion of ILC2s in mice and humans (3, 4), which could further reduce goblet cells or impair regeneration of these cells. Bruce et al. showed that donor ILC2 infusion promotes IL-13 production by ILC2s and enhances the survival of donor myeloid suppressor cells, suppresses donor T cell production of proinflammatory cytokines, and reduces GVHD (3). Although this study demonstrated that transfer of donor ILC2s improves intestinal epithelial integrity, the impact on goblet cells was not addressed. Deficiency of NOD-like receptor family pyrin domain-containing 6 (NLRP6), the critical molecule for goblet cell secretion of mucus, in nonhematopoietic cells of recipients mitigates goblet cell injury after allo-SCT and ameliorates intestinal GVHD, suggesting that NLRP6 is another target molecule for protection of goblet cells after allo-SCT (155).
LYPD8 is produced by enterocytes in the colon and enriched in the inner mucus layer (149). LYPD8 binds to flagellated bacteria such as Escherichia coli and prevents bacterial translocation by inhibiting bacterial motility. Based on these findings, we studied the protective role of LYPD8 in murine GVHD using LYPD8-deficient mice as recipients (2). First, we found that disruption of the inner mucus layer in allogeneic recipients led to disappearance of the LYPD8-rich layer in the mucus layer. Next, we found that bacterial translocation was dramatically enhanced in LYPD8-deficient recipients compared to WT recipients after allo-SCT, in association with exacerbation of GVHD. Furthermore, goblet cell expansion using IL-25 did not ameliorate GVHD in LYPD8-deficient recipients, suggesting that the mucus layer containing LYPD8 is critical for goblet cell-mediated GVHD suppression (2).
The discovery of specific markers for tissue stem cells has enabled us to study the fate of tissue stem cells in mouse GVHD, and we found that ISCs and HFSCs are targeted by GVHD. Furthermore, niche components that support tissue stem cells are also damaged after allo-SCT, likely inhibiting the recovery of tissue stem cells after GVHD-mediated injury. Emerging evidence also indicates that human and mouse GVHD targets specific epithelial populations, such as Paneth cells, L cells, and goblet cells, resulting in disruption of tissue homeostasis (Figure 2 and Table 1). Strategies to promote recovery of tissue stem cells and maintenance of the tissue microenvironment are promising adjuncts to standard immunosuppressive GVHD prophylaxis and treatment, which may enable the separation of GVHD and graft-versus-leukemia effects.
There remain many unanswered questions in this field. Although the existence of LGR5+ ISCs is also demonstrated in the human intestine, the fate of LGR5+ tissue stem cells in the intestine and skin after human allo-SCT remains to be clarified (156). The role and fate of BEC stem cells need to be studied both in human and mouse liver GVHD. Furthermore, the role of tissue stem cells in pathophysiology of chronic GVHD has not been well studied, and studies about intestinal dysbiosis in chronic GVHD has only just begun (157). Although it has been shown that protection of intestinal stem cells, Paneth cells, or goblet cells represents a promising anti-GVHD treatment, these strategies have been tested only in mouse models of GVHD in a prophylactic manner. It should be tested if these strategies are also useful for treatment of established GVHD.
All authors contributed to the article and approved the submitted version.
This study was supported by JSPS KAKENHI (21K16259 to TA, 21K08409 to DH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sasaki N, Sachs N, Wiebrands K, Ellenbroek SI, Fumagalli A, Lyubimova A, et al. Reg4+ Deep Crypt Secretory Cells Function as Epithelial Niche for Lgr5+ Stem Cells in Colon. Proc Natl Acad Sci USA (2016) 113(37):E5399–407. doi: 10.1073/pnas.1607327113
2. Ara T, Hashimoto D, Hayase E, Noizat C, Kikuchi R, Hasegawa Y, et al. Intestinal Goblet Cells Protect Against GVHD After Allogeneic Stem Cell Transplantation via Lypd8. Sci Transl Med (2020) 12(550):eaaw0720. doi: 10.1126/scitranslmed.aaw0720
3. Bruce DW, Stefanski HE, Vincent BG, Dant TA, Reisdorf S, Bommiasamy H, et al. Type 2 Innate Lymphoid Cells Treat and Prevent Acute Gastrointestinal Graft-Versus-Host Disease. J Clin Invest (2017) 127(5):1813–25. doi: 10.1172/JCI91816
4. Munneke JM, Bjorklund AT, Mjosberg JM, Garming-Legert K, Bernink JH, Blom B, et al. Activated Innate Lymphoid Cells Are Associated With a Reduced Susceptibility to Graft-Versus-Host Disease. Blood (2014) 124(5):812–21. doi: 10.1182/blood-2013-11-536888
5. Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, et al. Interleukin-22 Protects Intestinal Stem Cells From Immune-Mediated Tissue Damage and Regulates Sensitivity to Graft Versus Host Disease. Immunity (2012) 37(2):339–50. doi: 10.1016/j.immuni.2012.05.028
6. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 Promotes Intestinal-Stem-Cell-Mediated Epithelial Regeneration. Nature (2015) 528(7583):560–4. doi: 10.1038/nature16460
7. Takashima S, Kadowaki M, Aoyama K, Koyama M, Oshima T, Tomizuka K, et al. The Wnt Agonist R-Spondin1 Regulates Systemic Graft-Versus-Host Disease by Protecting Intestinal Stem Cells. J Exp Med (2011) 208(2):285–94. doi: 10.1084/jem.20101559
8. Takashima S, Martin ML, Jansen SA, Fu Y, Bos J, Chandra D, et al. T Cell-Derived Interferon-Gamma Programs Stem Cell Death in Immune-Mediated Intestinal Damage. Sci Immunol (2019) 4(42):eaay8556. doi: 10.1126/sciimmunol.aay8556
9. Norona J, Apostolova P, Schmidt D, Ihlemann R, Reischmann N, Taylor G, et al. Glucagon-Like Peptide 2 for Intestinal Stem Cell and Paneth Cell Repair During Graft-Versus-Host Disease in Mice and Humans. Blood (2020) 136(12):1442–55. doi: 10.1182/blood.2020005957
10. Ogasawara R, Hashimoto D, Kimura S, Hayase E, Ara T, Takahashi S, et al. Intestinal Lymphatic Endothelial Cells Produce R-Spondin3. Sci Rep (2018) 8(1):10719. doi: 10.1038/s41598-018-29100-7
11. Wu N, Sun H, Zhao X, Zhang Y, Tan J, Qi Y, et al. MAP3K2-Regulated Intestinal Stromal Cells Define a Distinct Stem Cell Niche. Nature (2021) 592(7855):606–10. doi: 10.1038/s41586-021-03283-y
12. Eriguchi Y, Takashima S, Oka H, Shimoji S, Nakamura K, Uryu H, et al. Graft-Versus-Host Disease Disrupts Intestinal Microbial Ecology by Inhibiting Paneth Cell Production of Alpha-Defensins. Blood (2012) 120(1):223–31. doi: 10.1182/blood-2011-12-401166
13. Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, et al. Regulation of Intestinal Inflammation by Microbiota Following Allogeneic Bone Marrow Transplantation. J Exp Med (2012) 209(5):903–11. doi: 10.1084/jem.20112408
14. Hayase E, Hashimoto D, Nakamura K, Noizat C, Ogasawara R, Takahashi S, et al. R-Spondin1 Expands Paneth Cells and Prevents Dysbiosis Induced by Graft-Versus-Host Disease. J Exp Med (2017) 214(12):3507–18. doi: 10.1084/jem.20170418
15. Levine JE, Huber E, Hammer ST, Harris AC, Greenson JK, Braun TM, et al. Low Paneth Cell Numbers at Onset of Gastrointestinal GVHD Identify Patients at High Risk for Non-Relapse Mortality. Blood (2013) 122(8):1505–9. doi: 10.1182/blood-2013-02-485813
16. Shoshkes-Carmel M, Wang YJ, Wangensteen KJ, Toth B, Kondo A, Massasa EE, et al. Subepithelial Telocytes Are an Important Source of Wnts That Supports Intestinal Crypts. Nature (2018) 557(7704):242–6. doi: 10.1038/s41586-018-0084-4
17. Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, et al. Intestinal Epithelial Tuft Cells Initiate Type 2 Mucosal Immunity to Helminth Parasites. Nature (2016) 529(7585):226–30. doi: 10.1038/nature16527
18. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-Cell-Derived IL-25 Regulates an Intestinal ILC2-Epithelial Response Circuit. Nature (2016) 529(7585):221–5. doi: 10.1038/nature16161
19. Takahashi S, Hashimoto D, Hayase E, Ogasawara R, Ohigashi H, Ara T, et al. Ruxolitinib Protects Skin Stem Cells and Maintains Skin Homeostasis in Murine Graft-Versus-Host Disease. Blood (2018) 131(18):2074–85. doi: 10.1182/blood-2017-06-792614
20. van Bekkum DW, Roodenburg J, Heidt PJ, van der Waaij D. Mitigation of Secondary Disease of Allogeneic Mouse Radiation Chimeras by Modification of the Intestinal Microflora. J Natl Cancer Inst (1974) 52(2):401–4. doi: 10.1093/jnci/52.2.401
21. Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J Jr, et al. Tumor Necrosis Factor- Alpha Production to Lipopolysaccharide Stimulation by Donor Cells Predicts the Severity of Experimental Acute Graft-Versus-Host Disease. J Clin Invest (1998) 102(10):1882–91. doi: 10.1172/JCI4285
22. Cooke KR, Gerbitz A, Crawford JM, Teshima T, Hill GR, Tesolin A, et al. LPS Antagonism Reduces Graft-Versus-Host Disease and Preserves Graft-Versus-Leukemia Activity After Experimental Bone Marrow Transplantation. J Clin Invest (2001) 107(12):1581–9. doi: 10.1172/JCI12156
23. Uryu H, Hashimoto D, Kato K, Hayase E, Matsuoka S, Ogasawara R, et al. Alpha-Mannan Induces Th17-Mediated Pulmonary Graft-Versus-Host Disease in Mice. Blood (2015) 125(19):3014–23. doi: 10.1182/blood-2014-12-615781
24. Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hulsdunker J, Varelias A, et al. MHC Class II Antigen Presentation by the Intestinal Epithelium Initiates Graft-Versus-Host Disease and Is Influenced by the Microbiota. Immunity (2019) 51(5):885–98.e7. doi: 10.1016/j.immuni.2019.08.011
25. Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, Mocsai A, et al. Neutrophil Granulocytes Recruited Upon Translocation of Intestinal Bacteria Enhance Graft-Versus-Host Disease via Tissue Damage. Nat Med (2014) 20(6):648–54. doi: 10.1038/nm.3517
26. Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, et al. Graft-Versus-Host Disease Is Enhanced by Extracellular ATP Activating P2X7R. Nat Med (2010) 16(12):1434–8. doi: 10.1038/nm.2242
27. Klambt V, Wohlfeil SA, Schwab L, Hulsdunker J, Ayata K, Apostolova P, et al. A Novel Function for P2Y2 in Myeloid Recipient-Derived Cells During Graft-Versus-Host Disease. J Immunol (2015) 195(12):5795–804. doi: 10.4049/jimmunol.1501357
28. Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, Guarda G, et al. The Nlrp3 Inflammasome Regulates Acute Graft-Versus-Host Disease. J Exp Med (2013) 210(10):1899–910. doi: 10.1084/jem.20130084
29. Koehn BH, Saha A, McDonald-Hyman C, Loschi M, Thangavelu G, Ma L, et al. Danger-Associated Extracellular ATP Counters MDSC Therapeutic Efficacy in Acute GVHD. Blood (2019) 134(19):1670–82. doi: 10.1182/blood.2019001950
30. Reichenbach DK, Schwarze V, Matta BM, Tkachev V, Lieberknecht E, Liu Q, et al. The IL-33/ST2 Axis Augments Effector T-Cell Responses During Acute GVHD. Blood (2015) 125(20):3183–92. doi: 10.1182/blood-2014-10-606830
31. Zhang J, Ramadan AM, Griesenauer B, Li W, Turner MJ, Liu C, et al. ST2 Blockade Reduces Sst2-Producing T Cells While Maintaining Protective Mst2-Expressing T Cells During Graft-Versus-Host Disease. Sci Transl Med (2015) 7(308):308ra160. doi: 10.1126/scitranslmed.aab0166
32. Brennan TV, Lin L, Huang X, Cardona DM, Li Z, Dredge K, et al. Heparan Sulfate, an Endogenous TLR4 Agonist, Promotes Acute GVHD After Allogeneic Stem Cell Transplantation. Blood (2012) 120(14):2899–908. doi: 10.1182/blood-2011-07-368720
33. Im KI, Kim N, Lim JY, Nam YS, Lee ES, Kim EJ, et al. The Free Radical Scavenger NecroX-7 Attenuates Acute Graft-Versus-Host Disease via Reciprocal Regulation of Th1/Regulatory T Cells and Inhibition of HMGB1 Release. J Immunol (2015) 194(11):5223–32. doi: 10.4049/jimmunol.1402609
34. Hulsdunker J, Ottmuller KJ, Neeff HP, Koyama M, Gao Z, Thomas OS, et al. Neutrophils Provide Cellular Communication Between Ileum and Mesenteric Lymph Nodes at Graft-Versus-Host Disease Onset. Blood (2018) 131(16):1858–69. doi: 10.1182/blood-2017-10-812891
35. Giroux M, Delisle JS, Gauthier SD, Heinonen KM, Hinsinger J, Houde B, et al. SMAD3 Prevents Graft-Versus-Host Disease by Restraining Th1 Differentiation and Granulocyte-Mediated Tissue Damage. Blood (2011) 117(5):1734–44. doi: 10.1182/blood-2010-05-287649
36. Socie G, Mary JY, Lemann M, Daneshpouy M, Guardiola P, Meignin V, et al. Prognostic Value of Apoptotic Cells and Infiltrating Neutrophils in Graft-Versus-Host Disease of the Gastrointestinal Tract in Humans: TNF and Fas Expression. Blood (2004) 103(1):50–7. doi: 10.1182/blood-2003-03-0909
37. Reinhardt K, Foell D, Vogl T, Mezger M, Wittkowski H, Fend F, et al. Monocyte-Induced Development of Th17 Cells and the Release of S100 Proteins Are Involved in the Pathogenesis of Graft-Versus-Host Disease. J Immunol (2014) 193(7):3355–65. doi: 10.4049/jimmunol.1400983
38. MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An Antibody Against the Colony-Stimulating Factor 1 Receptor Depletes the Resident Subset of Monocytes and Tissue- and Tumor-Associated Macrophages But Does Not Inhibit Inflammation. Blood (2010) 116(19):3955–63. doi: 10.1182/blood-2010-02-266296
39. Hashimoto D, Chow A, Greter M, Saenger Y, Kwan WH, Leboeuf M, et al. Pretransplant CSF-1 Therapy Expands Recipient Macrophages and Ameliorates GVHD After Allogeneic Hematopoietic Cell Transplantation. J Exp Med (2011) 208(5):1069–82. doi: 10.1084/jem.20101709
40. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-Resident Macrophages Self-Maintain Locally Throughout Adult Life With Minimal Contribution From Circulating Monocytes. Immunity (2013) 38(4):792–804. doi: 10.1016/j.immuni.2013.04.004
41. Washington K, Bentley RC, Green A, Olson J, Treem WR, Krigman HR. Gastric Graft-Versus-Host Disease: A Blinded Histologic Study. Am J Surg Pathol (1997) 21(9):1037–46. doi: 10.1097/00000478-199709000-00008
42. Mowat AM, Felstein MV, Borland A, Parrott DM. Experimental Studies of Immunologically Mediated Enteropathy. Development of Cell Mediated Immunity and Intestinal Pathology During a Graft-Versus-Host Reaction in Irradiated Mice. Gut (1988) 29(7):949–56. doi: 10.1136/gut.29.7.949
43. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of Stem Cells in Small Intestine and Colon by Marker Gene Lgr5. Nature (2007) 449(7165):1003–7. doi: 10.1038/nature06196
44. Mowat A, Socie G. Intestianl Graft-vs.-Host Disease. In: Mowat A, Socie G. editors. Graft vs. Host Disease. United States: CRC Press (2004). pp. 279–327.
45. Metcalfe C, Kljavin NM, Ybarra R, de Sauvage FJ. Lgr5+ Stem Cells Are Indispensable for Radiation-Induced Intestinal Regeneration. Cell Stem Cell (2014) 14(2):149–59. doi: 10.1016/j.stem.2013.11.008
46. Agudo J, Ruzo A, Park ES, Sweeney R, Kana V, Wu M, et al. GFP-Specific CD8 T Cells Enable Targeted Cell Depletion and Visualization of T-Cell Interactions. Nat Biotechnol (2015) 33(12):1287–92. doi: 10.1038/nbt.3386
47. Agudo J, Park ES, Rose SA, Alibo E, Sweeney R, Dhainaut M, et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity (2018) 48(2):271–85.e5. doi: 10.1016/j.immuni.2018.02.001
48. Sherman SE, Agudo J. Immune-Mediated Specific Depletion of Intestinal Stem Cells. Methods Mol Biol (2020) 2171:25–39. doi: 10.1007/978-1-0716-0747-3_2
49. Fu YY, Egorova A, Sobieski C, Kuttiyara J, Calafiore M, Takashima S, et al. T Cell Recruitment to the Intestinal Stem Cell Compartment Drives Immune-Mediated Intestinal Damage After Allogeneic Transplantation. Immunity (2019) 51(1):90–103.e3. doi: 10.1016/j.immuni.2019.06.003
50. Li L, Clevers H. Coexistence of Quiescent and Active Adult Stem Cells in Mammals. Science (2010) 327(5965):542–5. doi: 10.1126/science.1180794
51. Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, et al. A Reserve Stem Cell Population in Small Intestine Renders Lgr5-Positive Cells Dispensable. Nature (2011) 478(7368):255–9. doi: 10.1038/nature10408
52. Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, et al. The Intestinal Stem Cell Markers Bmi1 and Lgr5 Identify Two Functionally Distinct Populations. Proc Natl Acad Sci USA (2012) 109(2):466–71. doi: 10.1073/pnas.1118857109
53. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures In Vitro Without a Mesenchymal Niche. Nature (2009) 459(7244):262–5. doi: 10.1038/nature07935
54. Eriguchi Y, Nakamura K, Yokoi Y, Sugimoto R, Takahashi S, Hashimoto D, et al. Essential Role of IFN-Gamma in T Cell-Associated Intestinal Inflammation. JCI Insight (2018) 3(18):e121886. doi: 10.1172/jci.insight.121886
55. Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, et al. Paneth Cells Constitute the Niche for Lgr5 Stem Cells in Intestinal Crypts. Nature (2011) 469(7330):415–8. doi: 10.1038/nature09637
56. Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DV, et al. Visualization of a Short-Range Wnt Gradient in the Intestinal Stem-Cell Niche. Nature (2016) 530(7590):340–3. doi: 10.1038/nature16937
57. Kim TH, Escudero S, Shivdasani RA. Intact Function of Lgr5 Receptor-Expressing Intestinal Stem Cells in the Absence of Paneth Cells. Proc Natl Acad Sci USA (2012) 109(10):3932–7. doi: 10.1073/pnas.1113890109
58. Farin HF, Van Es JH, Clevers H. Redundant Sources of Wnt Regulate Intestinal Stem Cells and Promote Formation of Paneth Cells. Gastroenterology (2012) 143(6):1518–29.e7. doi: 10.1053/j.gastro.2012.08.031
59. Kabiri Z, Greicius G, Madan B, Biechele S, Zhong Z, Zaribafzadeh H, et al. Stroma Provides an Intestinal Stem Cell Niche in the Absence of Epithelial Wnts. Development (2014) 141(11):2206–15. doi: 10.1242/dev.104976
60. Degirmenci B, Valenta T, Dimitrieva S, Hausmann G, Basler K. GLI1-Expressing Mesenchymal Cells Form the Essential Wnt-Secreting Niche for Colon Stem Cells. Nature (2018) 558(7710):449–53. doi: 10.1038/s41586-018-0190-3
61. Carmon KS, Gong X, Lin Q, Thomas A, Liu Q. R-Spondins Function as Ligands of the Orphan Receptors LGR4 and LGR5 to Regulate Wnt/beta-Catenin Signaling. Proc Natl Acad Sci USA (2011) 108(28):11452–7. doi: 10.1073/pnas.1106083108
62. Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, et al. ZNRF3 Promotes Wnt Receptor Turnover in an R-Spondin-Sensitive Manner. Nature (2012) 485(7397):195–200. doi: 10.1038/nature11019
63. de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, et al. Lgr5 Homologues Associate With Wnt Receptors and Mediate R-Spondin Signalling. Nature (2011) 476(7360):293–7. doi: 10.1038/nature10337
64. Storm EE, Durinck S, de Sousa e Melo F, Tremayne J, Kljavin N, Tan C, et al. Targeting PTPRK-RSPO3 Colon Tumours Promotes Differentiation and Loss of Stem-Cell Function. Nature (2016) 529(7584):97–100. doi: 10.1038/nature16466
65. Gronke K, Hernandez PP, Zimmermann J, Klose CSN, Kofoed-Branzk M, Guendel F, et al. Interleukin-22 Protects Intestinal Stem Cells Against Genotoxic Stress. Nature (2019) 566(7743):249–53. doi: 10.1038/s41586-019-0899-7
66. Couturier M, Lamarthee B, Arbez J, Renauld JC, Bossard C, Malard F, et al. IL-22 Deficiency in Donor T Cells Attenuates Murine Acute Graft-Versus-Host Disease Mortality While Sparing the Graft-Versus-Leukemia Effect. Leukemia (2013) 27(7):1527–37. doi: 10.1038/leu.2013.39
67. Song Q, Wang X, Wu X, Kang TH, Qin H, Zhao D, et al. IL-22-Dependent Dysbiosis and Mononuclear Phagocyte Depletion Contribute to Steroid-Resistant Gut Graft-Versus-Host Disease in Mice. Nat Commun (2021) 12(1):805. doi: 10.1038/s41467-021-21133-3
68. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: Immunobiology and Pathology. Annu Rev Immunol (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
69. Lamarthee B, Malard F, Gamonet C, Bossard C, Couturier M, Renauld JC, et al. Donor Interleukin-22 and Host Type I Interferon Signaling Pathway Participate in Intestinal Graft-Versus-Host Disease via STAT1 Activation and CXCL10. Mucosal Immunol (2016) 9(2):309–21. doi: 10.1038/mi.2015.61
70. Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity (2019) 51(5):871–84.e6. doi: 10.1016/j.immuni.2019.09.014
71. Kim KA, Kakitani M, Zhao J, Oshima T, Tang T, Binnerts M, et al. Mitogenic Influence of Human R-Spondin1 on the Intestinal Epithelium. Science (2005) 309(5738):1256–9. doi: 10.1126/science.1112521
72. Zhou WJ, Geng ZH, Spence JR, Geng JG. Induction of Intestinal Stem Cells by R-Spondin 1 and Slit2 Augments Chemoradioprotection. Nature (2013) 501(7465):107–11. doi: 10.1038/nature12416
73. Broggi A, Granucci F, Zanoni I. Type III Interferons: Balancing Tissue Tolerance and Resistance to Pathogen Invasion. J Exp Med (2020) 217(1):e20190295. doi: 10.1084/jem.20190295
74. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-Lambda (IFN-Lambda) Is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PLoS Pathog (2008) 4(3):e1000017. doi: 10.1371/journal.ppat.1000017
75. Ye L, Schnepf D, Staeheli P. Interferon-Lambda Orchestrates Innate and Adaptive Mucosal Immune Responses. Nat Rev Immunol (2019) 19(10):614–25. doi: 10.1038/s41577-019-0182-z
76. Henden AS, Koyama M, Robb RJ, Forero A, Kuns RD, Chang K, et al. IFNlambda Therapy Prevents Severe Gastrointestinal Graft-Versus-Host Disease. Blood (2021). doi: 10.1182/blood.2020006375 [Epub ahead of print]
77. Nelson M, Rubio R, Lazzarin A, Romanova S, Luetkemeyer A, Conway B, et al. Safety and Efficacy of Pegylated Interferon Lambda, Ribavirin, and Daclatasvir in HCV and HIV-Coinfected Patients. J Interferon Cytokine Res (2017) 37(3):103–11. doi: 10.1089/jir.2016.0082
78. Zeiser R, von Bubnoff N, Butler J, Mohty M, Niederwieser D, Or R, et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-Versus-Host Disease. N Engl J Med (2020) 382(19):1800–10. doi: 10.1056/NEJMoa1917635
79. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of Therapeutic JAK 1/2 Blockade in Graft-Versus-Host Disease. Blood (2014) 123(24):3832–42. doi: 10.1182/blood-2013-12-543736
80. Cotsarelis G, Sun TT, Lavker RM. Label-Retaining Cells Reside in the Bulge Area of Pilosebaceous Unit: Implications for Follicular Stem Cells, Hair Cycle, and Skin Carcinogenesis. Cell (1990) 61(7):1329–37. doi: 10.1016/0092-8674(90)90696-c
81. Liu Y, Lyle S, Yang Z, Cotsarelis G. Keratin 15 Promoter Targets Putative Epithelial Stem Cells in the Hair Follicle Bulge. J Invest Dermatol (2003) 121(5):963–8. doi: 10.1046/j.1523-1747.2003.12600.x
82. Jaks V, Barker N, Kasper M, van Es JH, Snippert HJ, Clevers H, et al. Lgr5 Marks Cycling, Yet Long-Lived, Hair Follicle Stem Cells. Nat Genet (2008) 40(11):1291–9. doi: 10.1038/ng.239
83. Schepeler T, Page ME, Jensen KB. Heterogeneity and Plasticity of Epidermal Stem Cells. Development (2014) 141(13):2559–67. doi: 10.1242/dev.104588
84. Ito M, Liu Y, Yang Z, Nguyen J, Liang F, Morris RJ, et al. Stem Cells in the Hair Follicle Bulge Contribute to Wound Repair But Not to Homeostasis of the Epidermis. Nat Med (2005) 11(12):1351–4. doi: 10.1038/nm1328
85. Snippert HJ, Haegebarth A, Kasper M, Jaks V, van Es JH, Barker N, et al. Lgr6 Marks Stem Cells in the Hair Follicle That Generate All Cell Lineages of the Skin. Science (2010) 327(5971):1385–9. doi: 10.1126/science.1184733
86. Page ME, Lombard P, Ng F, Gottgens B, Jensen KB. The Epidermis Comprises Autonomous Compartments Maintained by Distinct Stem Cell Populations. Cell Stem Cell (2013) 13(4):471–82. doi: 10.1016/j.stem.2013.07.010
87. Sale GE, Beauchamp M. Parafollicular Hair Bulge in Human GVHD: A Stem Cell-Rich Primary Target. Bone Marrow Transplant (1993) 11(3):223–5.
88. Sale GE, Beauchamp MD, Akiyama M. Parafollicular Bulges, But Not Hair Bulb Keratinocytes, Are Attacked in Graft-Versus-Host Disease of Human Skin. Bone Marrow Transplant (1994) 14(3):411–3.
89. Murphy GF, Lavker RM, Whitaker D, Korngold R. Cytotoxic Folliculitis in GvHD. Evidence of Follicular Stem Cell Injury and Recovery. J Cutan Pathol (1991) 18(5):309–14. doi: 10.1111/j.1600-0560.1991.tb01541.x
90. Sale GE, Shulman HM, Gallucci BB, Thomas ED. Young Rete Ridge Keratinocytes Are Preferred Targets in Cutaneous Graft-Versus-Host Disease. Am J Pathol (1985) 118(2):278–87.
91. Whitaker-Menezes D, Jones SC, Friedman TM, Korngold R, Murphy GF. An Epithelial Target Site in Experimental Graft-Versus-Host Disease and Cytokine-Mediated Cytotoxicity Is Defined by Cytokeratin 15 Expression. Biol Blood Marrow Transplant (2003) 9(9):559–70. doi: 10.1016/S1083-8791(03)00288-X
92. Zhan Q, Signoretti S, Whitaker-Menezes D, Friedman TM, Korngold R, Murphy GF. Cytokeratin15-Positive Basal Epithelial Cells Targeted in Graft-Versus-Host Disease Express a Constitutive Antiapoptotic Phenotype. J Invest Dermatol (2007) 127(1):106–15. doi: 10.1038/sj.jid.5700583
93. Plikus MV, Mayer JA, de la Cruz D, Baker RE, Maini PK, Maxson R, et al. Cyclic Dermal BMP Signalling Regulates Stem Cell Activation During Hair Regeneration. Nature (2008) 451(7176):340–4. doi: 10.1038/nature06457
94. Festa E, Fretz J, Berry R, Schmidt B, Rodeheffer M, Horowitz M, et al. Adipocyte Lineage Cells Contribute to the Skin Stem Cell Niche to Drive Hair Cycling. Cell (2011) 146(5):761–71. doi: 10.1016/j.cell.2011.07.019
95. Hsu YC, Li L, Fuchs E. Emerging Interactions Between Skin Stem Cells and Their Niches. Nat Med (2014) 20(8):847–56. doi: 10.1038/nm.3643
96. Wang B, Zhao L, Fish M, Logan CY, Nusse R. Self-Renewing Diploid Axin2(+) Cells Fuel Homeostatic Renewal of the Liver. Nature (2015) 524(7564):180–5. doi: 10.1038/nature14863
97. Wei Y, Wang YG, Jia Y, Li L, Yoon J, Zhang S, et al. Liver Homeostasis Is Maintained by Midlobular Zone 2 Hepatocytes. Science (2021) 371(6532):eabb1625. doi: 10.1126/science.abb1625
98. He L, Pu W, Liu X, Zhang Z, Han M, Li Y, et al. Proliferation Tracing Reveals Regional Hepatocyte Generation in Liver Homeostasis and Repair. Science (2021) 371(6532):eabc4346. doi: 10.1126/science.abc4346
99. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, et al. In Vitro Expansion of Single Lgr5+ Liver Stem Cells Induced by Wnt-Driven Regeneration. Nature (2013) 494(7436):247–50. doi: 10.1038/nature11826
100. Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, Umemura A, et al. Hybrid Periportal Hepatocytes Regenerate the Injured Liver Without Giving Rise to Cancer. Cell (2015) 162(4):766–79. doi: 10.1016/j.cell.2015.07.026
101. Han X, Wang Y, Pu W, Huang X, Qiu L, Li Y, et al. Lineage Tracing Reveals the Bipotency of SOX9(+) Hepatocytes During Liver Regeneration. Stem Cell Rep (2019) 12(3):624–38. doi: 10.1016/j.stemcr.2019.01.010
102. Ang CH, Hsu SH, Guo F, Tan CT, Yu VC, Visvader JE, et al. Lgr5(+) Pericentral Hepatocytes Are Self-Maintained in Normal Liver Regeneration and Susceptible to Hepatocarcinogenesis. Proc Natl Acad Sci USA (2019) 116(39):19530–40. doi: 10.1073/pnas.1908099116
103. Planas-Paz L, Orsini V, Boulter L, Calabrese D, Pikiolek M, Nigsch F, et al. The RSPO-LGR4/5-ZNRF3/RNF43 Module Controls Liver Zonation and Size. Nat Cell Biol (2016) 18(5):467–79. doi: 10.1038/ncb3337
104. Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, et al. The Effects of Intestinal Tract Bacterial Diversity on Mortality Following Allogeneic Hematopoietic Stem Cell Transplantation. Blood (2014) 124(7):1174–82. doi: 10.1182/blood-2014-02-554725
105. Peled JU, Gomes ALC, Devlin SM, Littmann ER, Taur Y, Sung AD, et al. Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N Engl J Med (2020) 382(9):822–34. doi: 10.1056/NEJMoa1900623
106. Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of Microbicidal Alpha-Defensins by Intestinal Paneth Cells in Response to Bacteria. Nat Immunol (2000) 1(2):113–8. doi: 10.1038/77783
107. Masuda K, Sakai N, Nakamura K, Yoshioka S, Ayabe T. Bactericidal Activity of Mouse Alpha-Defensin Cryptdin-4 Predominantly Affects Noncommensal Bacteria. J Innate Immun (2011) 3(3):315–26. doi: 10.1159/000322037
108. Eriguchi Y, Nakamura K, Hashimoto D, Shimoda S, Shimono N, Akashi K, et al. Decreased Secretion of Paneth Cell Alpha-Defensin in Graft-Versus-Host Disease. Transpl Infect Dis (2015) 17(5):702–6. doi: 10.1111/tid.12423
109. Bevins CL, Salzman NH. Paneth Cells, Antimicrobial Peptides and Maintenance of Intestinal Homeostasis. Nat Rev Microbiol (2011) 9(5):356–68. doi: 10.1038/nrmicro2546
110. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The Antibacterial Lectin RegIIIgamma Promotes the Spatial Segregation of Microbiota and Host in the Intestine. Science (2011) 334(6053):255–8. doi: 10.1126/science.1209791
111. Shin JH, Seeley RJ. Reg3 Proteins as Gut Hormones? Endocrinology (2019) 160(6):1506–14. doi: 10.1210/en.2019-00073
112. Zhao D, Kim YH, Jeong S, Greenson JK, Chaudhry MS, Hoepting M, et al. Survival Signal REG3alpha Prevents Crypt Apoptosis to Control Acute Gastrointestinal Graft-Versus-Host Disease. J Clin Invest (2018) 128(11):4970–9. doi: 10.1172/JCI99261
113. Ferrara JL, Harris AC, Greenson JK, Braun TM, Holler E, Teshima T, et al. Regenerating Islet-Derived 3-Alpha Is a Biomarker of Gastrointestinal Graft-Versus-Host Disease. Blood (2011) 118(25):6702–8. doi: 10.1182/blood-2011-08-375006
114. Levine JE, Braun TM, Harris AC, Holler E, Taylor A, Miller H, et al. A Prognostic Score for Acute Graft-Versus-Host Disease Based on Biomarkers: A Multicentre Study. Lancet Haematol (2015) 2(1):e21–9. doi: 10.1016/S2352-3026(14)00035-0
115. Hartwell MJ, Ozbek U, Holler E, Renteria AS, Major-Monfried H, Reddy P, et al. An Early-Biomarker Algorithm Predicts Lethal Graft-Versus-Host Disease and Survival. JCI Insight (2017) 2(3):e89798. doi: 10.1172/jci.insight.89798
116. Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, Gobourne A, et al. Intestinal Domination and the Risk of Bacteremia in Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Clin Infect Diseases (2012) 55(7):905–14. doi: 10.1093/cid/cis580
117. Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, et al. Vancomycin-Resistant Enterococcus Domination of Intestinal Microbiota Is Enabled by Antibiotic Treatment in Mice and Precedes Bloodstream Invasion in Humans. J Clin Invest (2010) 120(12):4332–41. doi: 10.1172/JCI43918
118. Holler E, Butzhammer P, Schmid K, Hundsrucker C, Koestler J, Peter K, et al. Metagenomic Analysis of the Stool Microbiome in Patients Receiving Allogeneic Stem Cell Transplantation: Loss of Diversity Is Associated With Use of Systemic Antibiotics and More Pronounced in Gastrointestinal Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2014) 20(5):640–5. doi: 10.1016/j.bbmt.2014.01.030
119. Stein-Thoeringer CK, Nichols KB, Lazrak A, Docampo MD, Slingerland AE, Slingerland JB, et al. Lactose Drives Enterococcus Expansion to Promote Graft-Versus-Host Disease. Science (2019) 366(6469):1143–9. doi: 10.1126/science.aax3760
120. Lund EK, Bruce MG, Smith MW, Ferguson A. Selective Effects of Graft-Versus-Host Reaction on Disaccharidase Expression by Mouse Jejunal Enterocytes. Clin Sci (Lond) (1986) 71(2):189–98. doi: 10.1042/cs0710189
121. Nalle SC, Kwak HA, Edelblum KL, Joseph NE, Singh G, Khramtsova GF, et al. Recipient NK Cell Inactivation and Intestinal Barrier Loss Are Required for MHC-Matched Graft-Versus-Host Disease. Sci Transl Med (2014) 6(243):243ra87. doi: 10.1126/scitranslmed.3008941
122. Housley RM, Morris CF, Boyle W, Ring B, Biltz R, Tarpley JE, et al. Keratinocyte Growth Factor Induces Proliferation of Hepatocytes and Epithelial Cells Throughout the Rat Gastrointestinal Tract. J Clin Invest (1994) 94(5):1764–77. doi: 10.1172/JCI117524
123. Khan WB, Shui C, Ning S, Knox SJ. Enhancement of Murine Intestinal Stem Cell Survival After Irradiation by Keratinocyte Growth Factor. Radiat Res (1997) 148(3):248–53. doi: 10.2307/3579609
124. Panoskaltsis-Mortari A, Lacey DL, Vallera DA, Blazar BR. Keratinocyte Growth Factor Administered Before Conditioning Ameliorates Graft-Versus-Host Disease After Allogeneic Bone Marrow Transplantation in Mice. Blood (1998) 92(10):3960–7. doi: 10.1182/blood.V92.10.3960
125. Krijanovski OI, Hill GR, Cooke KR, Teshima T, Crawford JM, Brinson YS, et al. Keratinocyte Growth Factor Separates Graft-Versus-Leukemia Effects From Graft-Versus-Host Disease. Blood (1999) 94(2):825–31. doi: 10.1182/blood.V94.2.825
126. Blazar BR, Weisdorf DJ, Defor T, Goldman A, Braun T, Silver S, et al. Phase 1/2 Randomized, Placebo-Control Trial of Palifermin to Prevent Graft-Versus-Host Disease (GVHD) After Allogeneic Hematopoietic Stem Cell Transplantation (HSCT). Blood (2006) 108(9):3216–22. doi: 10.1182/blood-2006-04-017780
127. Jagasia MH, Abonour R, Long GD, Bolwell BJ, Laport GG, Shore TB, et al. Palifermin for the Reduction of Acute GVHD: A Randomized, Double-Blind, Placebo-Controlled Trial. Bone Marrow Transplant (2012) 47(10):1350–5. doi: 10.1038/bmt.2011.261
128. Mozaffari HR, Payandeh M, Ramezani M, Sadeghi M, Mahmoudiahmadabadi M, Sharifi R. Efficacy of Palifermin on Oral Mucositis and Acute GVHD After Hematopoietic Stem Cell Transplantation (HSCT) in Hematology Malignancy Patients: A Meta-Analysis of Trials. Contemp Oncol (Pozn) (2017) 21(4):299–305. doi: 10.5114/wo.2017.72400
129. Booth C, Booth D, Williamson S, Demchyshyn LL, Potten CS. Teduglutide ([Gly2]GLP-2) Protects Small Intestinal Stem Cells From Radiation Damage. Cell Prolif (2004) 37(6):385–400. doi: 10.1111/j.1365-2184.2004.00320.x
130. Yu C, Jia G, Deng Q, Zhao H, Chen X, Liu G, et al. The Effects of Glucagon-Like Peptide-2 on the Tight Junction and Barrier Function in IPEC-J2 Cells Through Phosphatidylinositol 3-Kinase-Protein Kinase B-Mammalian Target of Rapamycin Signaling Pathway. Asian Australas J Anim Sci (2016) 29(5):731–8. doi: 10.5713/ajas.15.0415
131. Farag SS, Abu Zaid M, Schwartz JE, Thakrar TC, Blakley AJ, Abonour R, et al. Dipeptidyl Peptidase 4 Inhibition for Prophylaxis of Acute Graft-Versus-Host Disease. N Engl J Med (2021) 384(1):11–9. doi: 10.1056/NEJMoa2027372
132. Hatano R, Ohnuma K, Yamamoto J, Dang NH, Yamada T, Morimoto C. Prevention of Acute Graft-Versus-Host Disease by Humanized Anti-CD26 Monoclonal Antibody. Br J Haematol (2013) 162(2):263–77. doi: 10.1111/bjh.12378
133. Holst JJ. Glucagon and Glucagon-Like Peptides 1 and 2. Results Probl Cell Differ (2010) 50:121–35. doi: 10.1007/400_2009_35
134. McKenna KJ, Ligato S, Kauffman GL Jr., Abt AB, Stryker JA, Conter RL. Epidermal Growth Factor Enhances Intestinal Mitotic Activity and DNA Content After Acute Abdominal Radiation. Surgery (1994) 115(5):626–32.
135. Procaccino F, Reinshagen M, Hoffmann P, Zeeh JM, Lakshmanan J, McRoberts JA, et al. Protective Effect of Epidermal Growth Factor in an Experimental Model of Colitis in Rats. Gastroenterology (1994) 107(1):12–7. doi: 10.1016/0016-5085(94)90055-8
136. Holtan SG, Hoeschen AL, Cao Q, Arora M, Bachanova V, Brunstein CG, et al. Facilitating Resolution of Life-Threatening Acute GVHD With Human Chorionic Gonadotropin and Epidermal Growth Factor. Blood Adv (2020) 4(7):1284–95. doi: 10.1182/bloodadvances.2019001259
137. Nalle SC, Zuo L, Ong M, Singh G, Worthylake AM, Choi W, et al. Graft-Versus-Host Disease Propagation Depends on Increased Intestinal Epithelial Tight Junction Permeability. J Clin Invest (2019) 129(2):902–14. doi: 10.1172/JCI98554
138. Madara JL, Stafford J. Interferon-Gamma Directly Affects Barrier Function of Cultured Intestinal Epithelial Monolayers. J Clin Invest (1989) 83(2):724–7. doi: 10.1172/JCI113938
139. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
140. Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, et al. Gut Microbiome-Derived Metabolites Modulate Intestinal Epithelial Cell Damage and Mitigate Graft-Versus-Host Disease. Nat Immunol (2016) 17(5):505–13. doi: 10.1038/ni.3400
141. Fujiwara H, Docampo MD, Riwes M, Peltier D, Toubai T, Henig I, et al. Microbial Metabolite Sensor GPR43 Controls Severity of Experimental GVHD. Nat Commun (2018) 9(1):3674. doi: 10.1038/s41467-018-06048-w
142. Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, Ahr KF, et al. Intestinal Blautia Is Associated With Reduced Death From Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2015) 21(8):1373–83. doi: 10.1016/j.bbmt.2015.04.016
143. Yoshifuji K, Inamoto K, Kiridoshi Y, Takeshita K, Sasajima S, Shiraishi Y, et al. Prebiotics Protect Against Acute Graft-Versus-Host Disease and Preserve the Gut Microbiota in Stem Cell Transplantation. Blood Adv (2020) 4(19):4607–17. doi: 10.1182/bloodadvances.2020002604
144. Weber D, Oefner PJ, Hiergeist A, Koestler J, Gessner A, Weber M, et al. Low Urinary Indoxyl Sulfate Levels Early After Transplantation Reflect a Disrupted Microbiome and Are Associated With Poor Outcome. Blood (2015) 126(14):1723–8. doi: 10.1182/blood-2015-04-638858
145. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous Stimuli Maintain Intraepithelial Lymphocytes via Aryl Hydrocarbon Receptor Activation. Cell (2011) 147(3):629–40. doi: 10.1016/j.cell.2011.09.025
146. Riwes M, Reddy P. Microbial Metabolites and Graft Versus Host Disease. Am J Transplant (2018) 18(1):23–9. doi: 10.1111/ajt.14443
147. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The Inner of the Two Muc2 Mucin-Dependent Mucus Layers in Colon Is Devoid of Bacteria. Proc Natl Acad Sci USA (2008) 105(39):15064–9. doi: 10.1073/pnas.0803124105
148. Johansson ME, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H, et al. Bacteria Penetrate the Normally Impenetrable Inner Colon Mucus Layer in Both Murine Colitis Models and Patients With Ulcerative Colitis. Gut (2014) 63(2):281–91. doi: 10.1136/gutjnl-2012-303207
149. Okumura R, Kurakawa T, Nakano T, Kayama H, Kinoshita M, Motooka D, et al. Lypd8 Promotes the Segregation of Flagellated Microbiota and Colonic Epithelia. Nature (2016) 532(7597):117–21. doi: 10.1038/nature17406
150. Antoni L, Nuding S, Weller D, Gersemann M, Ott G, Wehkamp J, et al. Human Colonic Mucus Is a Reservoir for Antimicrobial Peptides. J Crohns Colitis (2013) 7(12):e652–64. doi: 10.1016/j.crohns.2013.05.006
151. Eigenbrodt ML, Eigenbrodt EH, Thiele DL. Histologic Similarity of Murine Colonic Graft-Versus-Host Disease (GVHD) to Human Colonic GVHD and Inflammatory Bowel Disease. Am J Pathol (1990) 137(5):1065–76.
152. Levy DA, Wefald A. Gut Mucosal Mast Cells and Goblet Cells During Acute Graft-Versus-Host Disease in Rats. Ann Inst Pasteur Immunol (1986) 137D(2):281–8.
153. Johansson ME. Fast Renewal of the Distal Colonic Mucus Layers by the Surface Goblet Cells as Measured by In Vivo Labeling of Mucin Glycoproteins. PLoS One (2012) 7(7):e41009. doi: 10.1371/journal.pone.0041009
154. Barker N. Adult Intestinal Stem Cells: Critical Drivers of Epithelial Homeostasis and Regeneration. Nat Rev Mol Cell Biol (2014) 15(1):19–33. doi: 10.1038/nrm3721
155. Toubai T, Fujiwara H, Rossi C, Riwes M, Tamaki H, Zajac C, et al. Host NLRP6 Exacerbates Graft-Versus-Host Disease Independent of Gut Microbial Composition. Nat Microbiol (2019) 4(5):800–12. doi: 10.1038/s41564-019-0373-1
156. Becker L, Huang Q, Mashimo H. Immunostaining of Lgr5, an Intestinal Stem Cell Marker, in Normal and Premalignant Human Gastrointestinal Tissue. ScientificWorldJournal (2008) 8:1168–76. doi: 10.1100/tsw.2008.148
Keywords: allogeneic hematopoietic stem cell transplantation, GVHD, graft-versus-host disease, intestinal stem cells, tissue stem cells, microbiota, Paneth cell, goblet cell
Citation: Ara T and Hashimoto D (2021) Novel Insights Into the Mechanism of GVHD-Induced Tissue Damage. Front. Immunol. 12:713631. doi: 10.3389/fimmu.2021.713631
Received: 23 May 2021; Accepted: 10 August 2021;
Published: 27 August 2021.
Edited by:
Christian Chabannon, Aix-Marseille Université, FranceReviewed by:
Philippe Saas, U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique (INSERM), FranceCopyright © 2021 Ara and Hashimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daigo Hashimoto, RDVoYXNoQHBvcC5tZWQuaG9rdWRhaS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.