
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 30 August 2021
Sec. Vaccines and Molecular Therapeutics
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.713276
This article is part of the Research TopicInduction of Immune Tolerance: Addressing Unmet Medical Need in Immune Mediated Diseases and Immune Responses to BiologicsView all 16 articles
 Mengjun Zhang1,2†
Mengjun Zhang1,2† Yuanqiang Wang3†Xiangqian Li2Gang Meng4Xiaoling Chen2Lina Wang5Zhihua Lin3
Yuanqiang Wang3†Xiangqian Li2Gang Meng4Xiaoling Chen2Lina Wang5Zhihua Lin3 Li Wang2*
Li Wang2*Autoreactive CD8+ T cells play an indispensable key role in the destruction of pancreatic islet β-cells and the initiation of type 1 diabetes (T1D). Insulin is an essential β-cell autoantigen in T1D. An HLA-A*0201-restricted epitope of insulin A chain (mInsA2-10) is an immunodominant ligand for autoreactive CD8+ T cells in NOD.β2mnull.HHD mice. Altered peptide ligands (APLs) carrying amino acid substitutions at T cell receptor (TCR) contact positions within an epitope are potential to modulate autoimmune responses via triggering altered TCR signaling. Here, we used a molecular simulation strategy to guide the generation of APL candidates by substitution of L-amino acids with D-amino acids at potential TCR contact residues (positions 4 and 6) of mInsA2-10, named mInsA2-10DQ4 and mInsA2-10DC6, respectively. We found that administration of mInsA2-10DQ4, but not DC6, significantly suppressed the development of T1D in NOD.β2mnull.HHD mice. Mechanistically, treatment with mInsA2-10DQ4 not only notably eliminated mInsA2-10 autoreactive CD8+ T cell responses but also prevented the infiltration of CD4+ T and CD8+ T cells, as well as the inflammatory responses in the pancreas of NOD.β2mnull.HHD mice. This study provides a new strategy for the development of APL vaccines for T1D prevention.
Type 1 diabetes (T1D) is a spontaneous organ-specific autoimmune disease characterized by T cell-mediated elimination of insulin-producing pancreatic islet β-cells. Given the close association between major histocompatibility complex (MHC) class II molecules and type 1 diabetes has been early found (1), β-cell autoreactive CD4+ T cells were the most intensively studied in both humans and NOD mice (2). However, the importance of β-cell autoantigen-specific CD8+ T cells in the pathogenesis of type 1 diabetes has been heightened by multiple studies in NOD mice (3–5). Either CD8α or β2-microglobulin-deficient NOD mice do not develop diabetes (6). Similarly, diabetes does not occur in NOD mice depleted of CD8+ T cells by antibody treatment (7). Human leukocyte antigen-A*0201 (HLA-A*0201), one of the most commonly expressed MHC class I allele in Caucasians and Asians (50%), has been also indicated to contribute to the susceptibility to T1D (8). NOD.β2mnull.HHD mice, carrying human HLA-A*0201 but no murine MHC class I molecules, show significantly accelerated T1D onset (8). Thus, induction of β-cell autoreactive CD8+ T-cell tolerance has been considered as a promising approach for the prevention of T1D (9).
Insulin is a primary β-cell autoantigen that initiates spontaneous T1D in both NOD mice and human (10), so induction of insulin-autoreactive T-cell tolerance can lead to prevention of T1D (11). Multiple CD8+ T cell epitopes of insulin such as A chain 2-10 (mInsA2-10) and B chain 5-14 (mInsB5-14), which are identified to be HLA-A*0201-restricted and immunodominant in NOD.β2mnull.HHD mice with high potential T1D relevance, represent important candidates of CD8+ T cell targets in human T1D patients.
Altered peptide ligands (APLs) carrying amino acid substitutions at T cell receptor (TCR) or MHC contact positions can trigger altered TCR signaling events and are suggested as useful tools to modulate autoimmune responses. For examples, administration of APLs of a known immunodominant CD8+ T cell epitope with partial agonist activity (12) or nanoparticles coated with APL-MHCs complexes (13) has been shown to effectively induce autoreactive CD8+ T cell tolerance and prevent T1D in NOD mice. We recently found that repeated treatment of APL of mInsB5–14 with histidine to phenylalanine substitution at the potential TCR contact site (p6) prevents T1D via selectively expanding a tiny population of CD8+CD25+Foxp3+ regulatory T cells in humanized NOD mice (14).
The introduction of D-amino acids into the sequence of peptides is widely used to improve the stability and prevent peptides from proteolytic degradation (15). On the other hand, scanning with D-amino acid has been previously exploited for molecular analysis of MHC binding or/and TCR interacting residues within a T cell epitope, since the corresponding single D-amino acid substitution minimizes the influence of charges of each original residue as well as the size and molecular weight of the native peptide (16–18). Although these limited earlier studies have shown that T cell priming capacity of a T cell epitope can be altered by introducing D-amino acid at selected residues, whether APLs derived from D-amino acid substitution in TCR contact residues of native peptide are suitable for prevention of autoimmune diseases has been rarely considered. Here, we designed two APL candidates by in silico-assisted substitution of L-amino acids with D-amino acids at positions 4 and 6 of mInsA2-10, respectively, which are in close contact with the TCR and potentially important for the recognition and response of specific T cells. We found that these two APLs, mInsA2-10DQ4 and DC6, significantly inhibited the native mInsA2-10 peptide-induced proliferation of splenocytes from NOD.β2mnull.HHD mice in vitro. However, in vivo administration of mInsA2-10DQ4 but not DC6 notably reduced the insulitis and effectively delayed the development of T1D in NOD.β2mnull.HHD mice. Mechanistically, systemic treatment with mInsA2-10DQ4 blinded autoreactive CD8+ T cell responses toward to mInsA2-10 and reduced the infiltration of both autoreactive CD4+ T and CD8+ T cells into the pancreas in NOD.β2mnull.HHD mice.
NOD.β2mnull.HHD mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All mice were bred and maintained in specific pathogen-free facilities and handled according to “Principles of Laboratory Animal Care and Use in Research” (Ministry of Health, Beijing, China). All experimental protocols were approved by the Animal Ethics Committee of the Army Medical University (Third Military Medical University).
Blood glucose was monitored using a glucometer (OneTouch Ultral; LifeScan, Milpitas, CA, USA) at weekly intervals, beginning at 10 weeks of age. Diabetes was defined as two consecutive blood glucose values above 11.1 mM.
We constructed the HLA-A*0201/mInsA2-10(IVDQCCTSI) complex based on the crystal structure (PDB ID: 3MRE) by mutating of epitope, which was used as template to build other complexes by replacing the L-amino acid of mInsA2-10 with the corresponding D-amino acid at the assigned position (listed in Table 1). Subsequently, the epitopes complexed to HLA-A*0201 were put into a cubic box with 0.15 M NaCl solution and kept the ensemble neutral, then 120 ns (100 ns for HLA-A*0201/epitope) molecular dynamics simulation was performed with ff14SB force filed by AMBER 16 (19, 20). The root means square deviation (RMSD) of backbone (Cα, C, N, O of main chain) between sampling and initial conformation of HLA-A*0201 and epitopes was used to monitor the states of MD simulation. The ensemble reached the stable state while the RMSD was fluctuating at any number for a long time (more than 20 ns), then the MD simulation was ended. The binding free energy between epitopes (included mInsA2-10) and HLA-A*0201 was calculated with molecular mechanics/Poisson Boltzmann surface area (MM/PBSA) method for the stable conformations extracted from last 20 ns trajectory, which were used to evaluate their potential affinity (21). Sequentially, we averaged the number of hydrogen bonds between epitopes and HLA-A*0201 and the solvent accessible surface area (SASA) of epitope with the LCPO model for stable conformations (22).

Table 1 The distance (P1–P9), average hydrogen bonds, and binding energy of epitopes.
Synthetic peptides mInsA2-10 (IVDQCCTSI), OVA257-264 (SIINFEKL), HIV pol476-484 (ILKEPVHGV), mInsA2-10DC6 (IVDQC(D-Cys)TSI), and mInsA2-10DQ4 (IVD(D-Gln)CCTSI) were synthesized with purity >95% at Chinese Peptide Company (Hangzhou, China). Cohorts of 4-week-old female NOD.β2mnull.HHD mice were intraperitoneally injected with 100 μg (1 μg/μl) in PBS, and this procedure was repeated every week until the sixth injection.
Pancreatic tissues from 12-week-old non-diabetic female NOD.β2mnull.HHD mice immunized with different peptides (10 mice per group) were fixed in 10% neutral-buffered formalin, and the paraffin-embedded samples were stained with hematoxylin and eosin (H&E). A minimum of 10 islets from each mouse were microscopically observed by two different observers, and insulitis scoring was performed according to the following criteria: 0, no infiltration; 1, peri-insulitis; 2, insulitis with <50% islet area infiltrated; 3, insulitis with >50% islet area infiltrated.
T2 cells (1×106 cells/ml) were incubated with each peptide (50 µg/ml) in serum-free RPMI 1640 medium supplemented with 3 µg/ml β-2-microglobulin (Sigma-Aldrich, St. Louis, MO, USA) for 16 h at 37°C. Then cells were washed and stained with anti-HLA-A2 mAb BB7.2 (purified in-house from the hybridoma obtained from ATCC), followed by incubation with FITC-conjugated goat anti-mouse IgG (Beyotime, Jiangsu, China), and analyzed using an FACSAria™ instrument (BD Bioscience, Franklin Lakes, NJ, USA).
ELISPOT plates were precoated with anti-mouse mAb (MabTech, Stockholm, Sweden) overnight at 4°C, and blocked with RPMI 1640 plus 10% FBS (HyClone Corp., Logan, UT, USA). CD8+ T cells were purified from splenocytes of 12-week-old non-diabetic female NOD.β2mnull.HHD mice treated with or without different peptides using the EasySep mouse CD8+ T cell isolation kit (StemCell Technologies, Vancouver, Canada). Purified CD8+ T cells (purity>90%, 2×105 cells/well) were incubated with each peptide (50 µg/ml)-pulsed T2 cells (1×104 cells/well) for 24 h at 37°C. After incubation, cells were removed and plates were processed according to the IFN-γ ELISPOT kit (MabTech) manufacturer’s instructions. Spots were counted using a spot reader system (Saizhi, Beijing, China).
Splenocytes (1×106 cells/ml) freshly isolated from 12-week-old non-diabetic female NOD.β2mnull.HHD mice were co-cultured with 10 μg/ml indicated peptides and 10 U/ml recombinant murine interleukin 2 (rmIL-2; Peprotech, Rocky Hill, NJ, USA), followed by twice weekly rmIL-2. After incubation at 37°C for 72 h, [3H] thymidine (1 μCi/well) was added for an additional 16 h of culture, and uptake of [3H] thymidine was determined using a liquid scintillation counter (Beckman Coulter, Brea, CA, USA).
Pancreatic biopsy samples (n=6) from indicated peptide-treated NOD.β2mnull.HHD mice at 12 weeks of age were lysed in Trizol reagent (Invitrogen, Carlsbad, CA, USA), and total RNA was extracted according to the manufacturer’s instruction. About 500 ng of total RNA was used for reverse transcription using a PrimeScript® RT reagent Kit (TaKaRa, Shiga, Japan) in a volume of 10 μl, and products were detected using a SYBR® Premix Ex Tap™ Kit (TaKaRa). Data were collected and quantitatively analyzed on an M×3000 P Real-Time PCR System (Stratagene, Austin, TX, USA). Values were normalized using β-actin as an endogenous internal standard. The sequences used were as follows: mouse IL-6, Sense 5’-TAGTCCTTCCTACCCCAATTTCC-3’, Anti-sense 5’-TTGGTCCTTAGCCACTCCTTC-3’; mouse IL-1β, Sense 5’-GCAACTGTTCCTGAACTCAACT-3’, Anti-sense 5’-ATCTTTTGGGGTCCGTCAACT-3’; mouse TNF-α, Sense 5’-CACGCTCTTCTGTCTACTGAAC-3’, Anti-sense 5’-ATCTGAGTGTGAGGGTCTGG-3’; mouse IFN-γ, Sense 5’-TCAAGTGGCATAGATGTGGAAG-3’, Anti-sense 5’-CGCTTATGTTGTTGCTGATGG-3’; mouse IL-17, Sense 5’- ATCTGTGTCTCTGATGCTGTTG-3’, Anti-sense 5’- AACGGTTGAGGTAGTCTGAGG-3’; mouse β-actin, Sense 5’-GAGACCTTCAACACCCCAGC-3’, Anti-sense 5’-ATGTCACGCACGATTTCCC-3’.
Mice were euthanized and systemically perfused by injection of physiological saline into the left heart ventricle. After removal of all visible pancreatic lymph nodes, the pancreases were cut into tiny pieces and then digested in HBSS containing 1 mg/ml collagenase IV and 1.25 μg/ml DNase (Sigma), by shaking (200 rpm) at 37°C for 15 min. Single-cell suspensions were collected after diluting the enzyme with ice-cold HBSS containing 2% FCS. Aggregates were further digested with 0.5 mg/ml collagenase IV and 1 μg/ml DNase for 10 min and 0.25 mg/ml collagenase IV and 1 μg/ml DNase for 6 min. Single-cell suspensions were washed three times, and pancreas-infiltration cells were isolated by percoll density-gradient centrifugation according to the manufacturer’s instructions, and then resuspended in medium.
Single-cell suspensions were made from spleens and pancreas-infiltrating immune cells of 12-week-old non-diabetic female NOD.β2mnull.HHD mice immunized with different peptides and resuspended in RPMI 1640 medium. Fluorochrome-conjugated antibodies specific for surface markers used in this study were anti-CD3-FITC (145-2C11), anti-CD4-PE (GK1.5), and anti-CD8-PerCP-Cy5.5 (53-6.7) (eBiosciences, San Diego, CA, USA). Events were collected on the BD Acurri C6 flow cytometer and analyzed with FlowJo software.
Paired t-test was used to compare autoreactive CD8+ T cell responses to control peptide or mInsA2-10 of an individual within a certain treatment group. A non-parametric ANOVA (Kruskal-Wallis test) followed by a Dunn’s test was performed to analyze differences among the groups, and a log-rank test was used to assess the cumulative incidence of diabetes. Other statistical analyses were conducted by the two-tailed Student’s t-test. P<0.05 were considered statistically significant.
We firstly verified the immunodominance of HLA-A*0201-restricted native mInsA2-10 peptide in 12-week-old non-diabetic female NOD.β2mnull.HHD mice. As expected, IFN-γ ELISPOT analysis revealed that potent CD8+ T-cell responses against mInsA2-10 were indeed present in 12-week-old non-diabetic female NOD.β2mnull.HHD mice (Figures 1A, B). So we questioned whether systemic administration of the native mInsA2-10 peptide could protect from T1D in NOD.β2mnull.HHD mice. However, mInsA2-10 peptide showed no protective activity (Figure 1C), and this encouraged us to explore the APLs of mInsA2-10 peptide with antidiabetic activity.
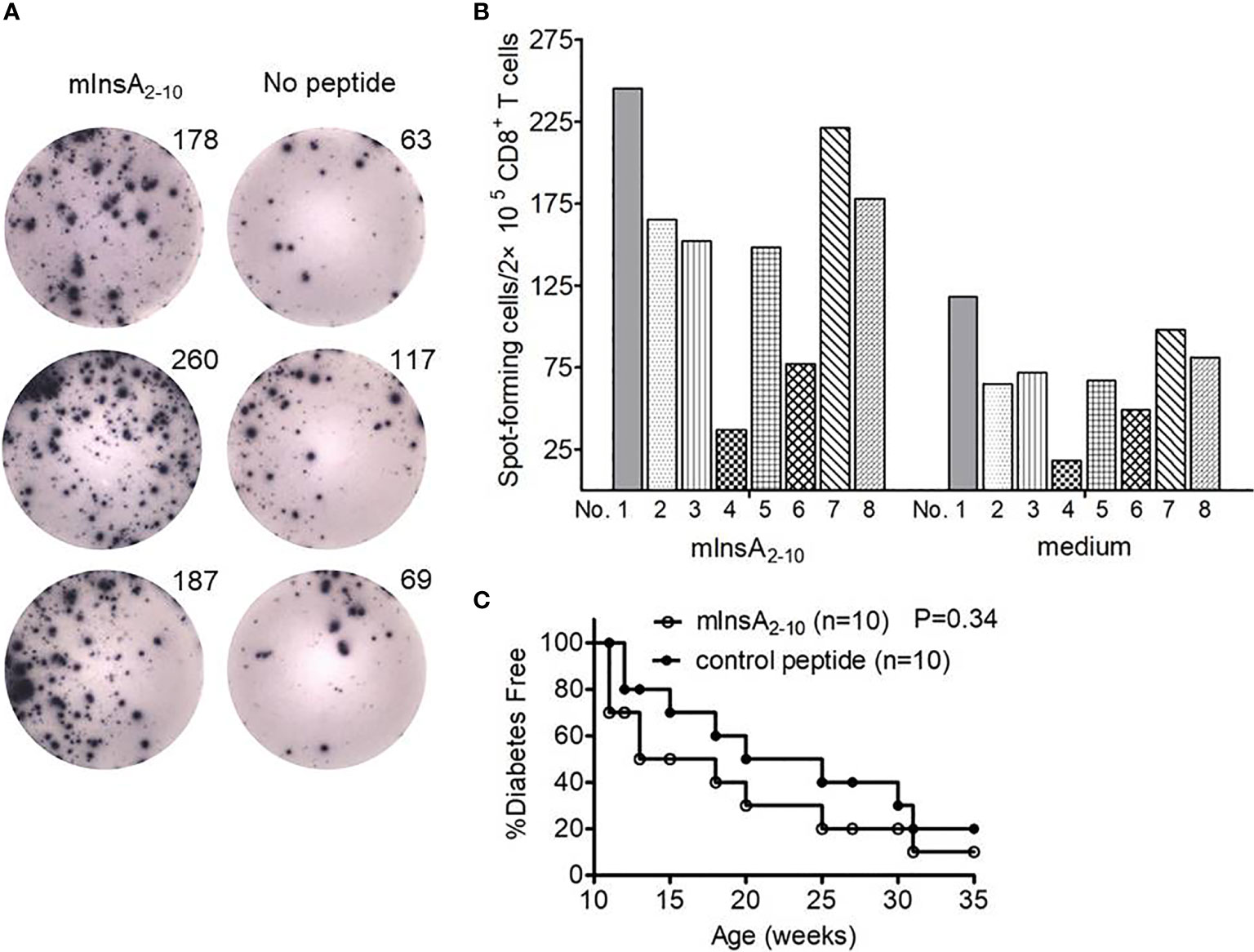
Figure 1 The mInsA2-10 epitope treatment could not prevent the development of T1D in NOD.β2mnull.HHD mice. (A) Representative IFN-γ ELISPOT assay demonstrated splenic CD8+ T cell responses to T2 cell-loaded mInsA2-10 (10 μg/ml), or no peptide in 12-week-old non-diabetic female NOD.β2mnull.HHD mice. (B) The average number of peptide-specific IFN-γ-positive spots per 2×105 splenic CD8+ T cells in triplicate cultures was calculated for each indicated peptides (10 μg/ml) or no peptide from eight separate experiments. SDs are not shown to avoid excessive visual clutter. (C) Percentage of female NOD.β2mnull.HHD mice developing diabetes after 100 μg mInsA2-10 i.p. weekly injections from 4 to 9 weeks of age (white circles, n = 10) vs. mice that were treated with OVA257-264 (black circles, n = 10).
To define the potential TCR contact residues in mInsA2-10, we constructed the HLA-A*0201/mInsA2-10(IVDQCCTSI) complex based on the crystal structure (PDB ID: 3MRE) by in silico replacing of epitope. Figure 2A showed amino acid residues at positions 4 and 6 were bulged out of the binding groove and potentially important for the interaction with TCR. Therefore, we generated two APL candidates via in silico-assisted replacing L-amino acid of mInsA2-10 at positions 4 and 6 by the corresponding D-isomer, respectively. Then, 120 ns MD simulation for the complex of HLA-A*0201/each peptide was performed, and the average structures (200 conformations) for the last 20 ns MD simulation were used to analyze their binding mode. The distance (Cα) between amino acids at positions 1 and 9, RMSD, binding energy, and the number of hydrogen bonds between each peptide and HLA-A*0201 (Table 1), as well as the solvent accessible surface area (Table 2) and the conformation comparison between APL candidates and mInsA2-10, were analyzed. The RMSD plot showed that both HLA-A*0201 and mInsA2-10 remained stable after 80 ns MD simulation, with the fluctuation around 1.8 Å. While HLA-A*0201 molecule complexed with APL DQ4 and DC6 reached stable after 90 ns (Figures 2B, C) shows mInsA2-10 may stably bind to HLA-A*0201 through strong hydrophilic interaction. Notably, the predicted binding free energy of mInsA2-10DQ4 to HLA-A*0201 was very similar to that of mInsA2-10 to HLA-A*0201. Whereas, the predicted binding free energy between DC6 and HLA-A*0201 was increased (Table 1), indicating that the binding strength between DC6 and HLA-A* 0201 might be weakened. Thus, these results suggested that the substitution of D amino acid at position 4 rather than position 6 might have minor effect on the binding ability of peptide to HLA-A*0201 molecule.
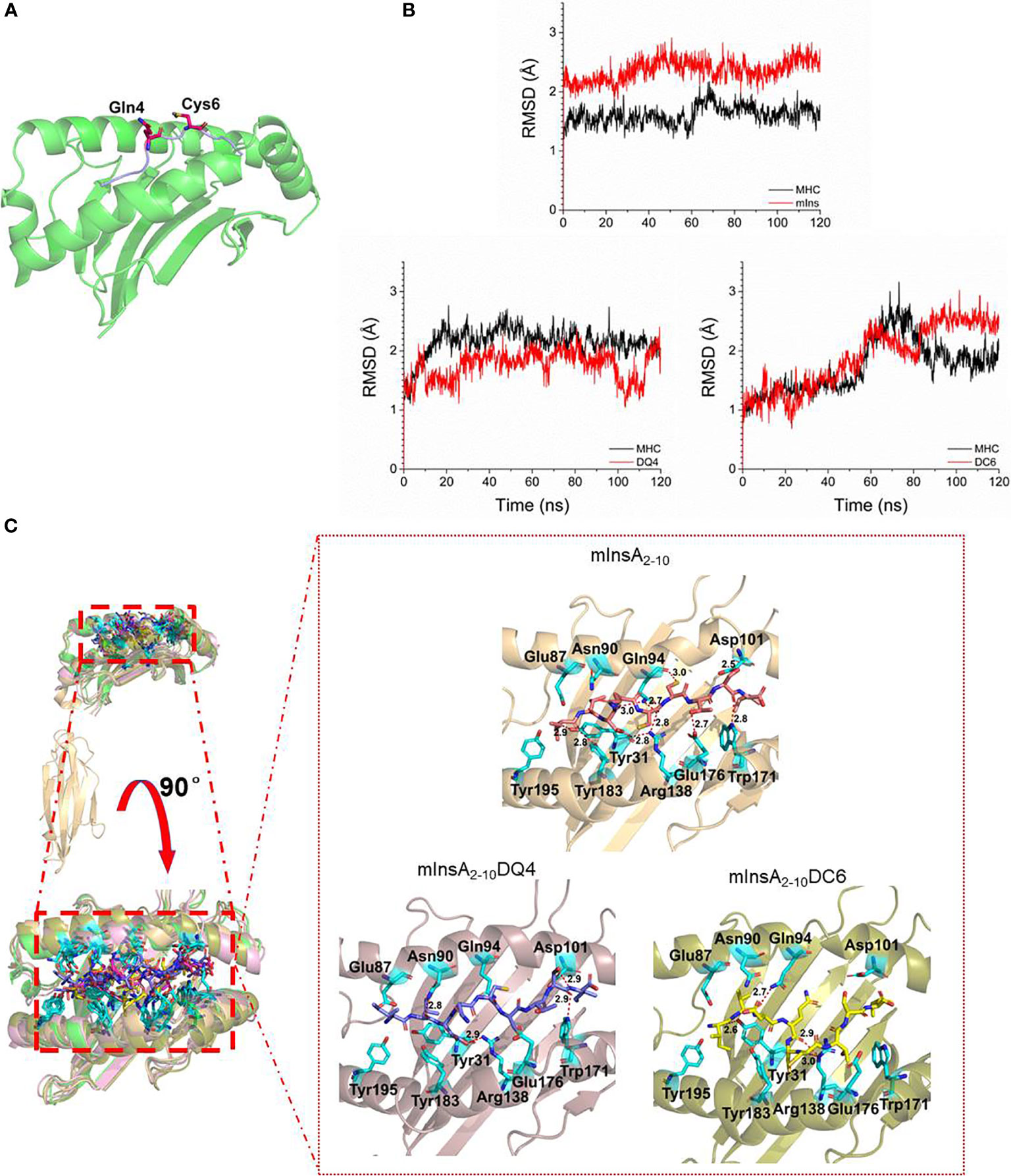
Figure 2 Design of a potential antagonist peptide for mInsA2-10. (A) The modeled structure of the HLA-A*0201/mInsA2-10 complex showed that residues Gln 4 and Cys 6 bulged out of the binding groove, which were the potential TCR contact residues. (B) The RMSD of simulation of complex HLA-A*0201/epitope, the stable conformation for these three complexes were achieved through 120 ns MD simulation, and the RMSD fluctuated near 2Å. (C) The binding mode between HLA-A*0201 and epitopes, these epitopes bound to HLA-A*0201 through stronger hydrogen bonds, but mInsA2-10 had the most hydrogen bonds.
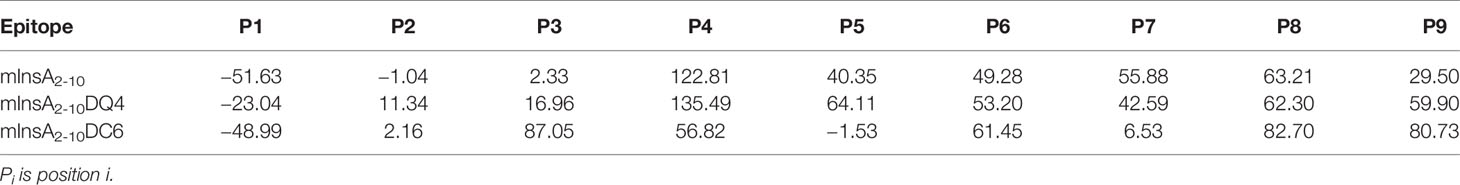
Table 2 The solvent accessible surface area (SASA) of residues of epitopes.
The binding affinity of the native peptide mInsA2-10 and the two APLs for HLA-A*0201 molecule was evaluated in vitro using a T2-cell-peptide binding test. Both mInsA2-10 and mInsA2-10 DQ4 showed a strong HLA-A*0201-binding affinity similar to the positive control HIVpol476-484. Whereas mInsA2-10DC6 showed a relatively weaker binding affinity to HLA-A*0201 (Figures 3A, B). These results confirmed that the substitution of D-amino acid at position 4 rather than position 6 did not change the binding ability of APL to HLA-A*0201. To assess whether mInsA2-10DQ4 and DC6 displayed any stimulating or inhibiting activity toward mInsA2-10-reactive T cell population, the proliferation of splenocytes from 12-week-old non-diabetic female NOD.β2mnull.HHD mice was tested upon the stimulation with mInsA2-10 alone or plus each APL. When compared to the stimulation with mInsA2-10 alone, the proliferation of splenocytes decreased significantly upon stimulation with mInsA2-10 plus either mInsA2-10 DQ4 or DC6 (Figure 3C). Thus, these preliminary data indicated that both mInsA2-10DQ4 and DC6 displayed inhibitory effects on mInsA2-10-stimulated lymphocyte proliferation in vitro.
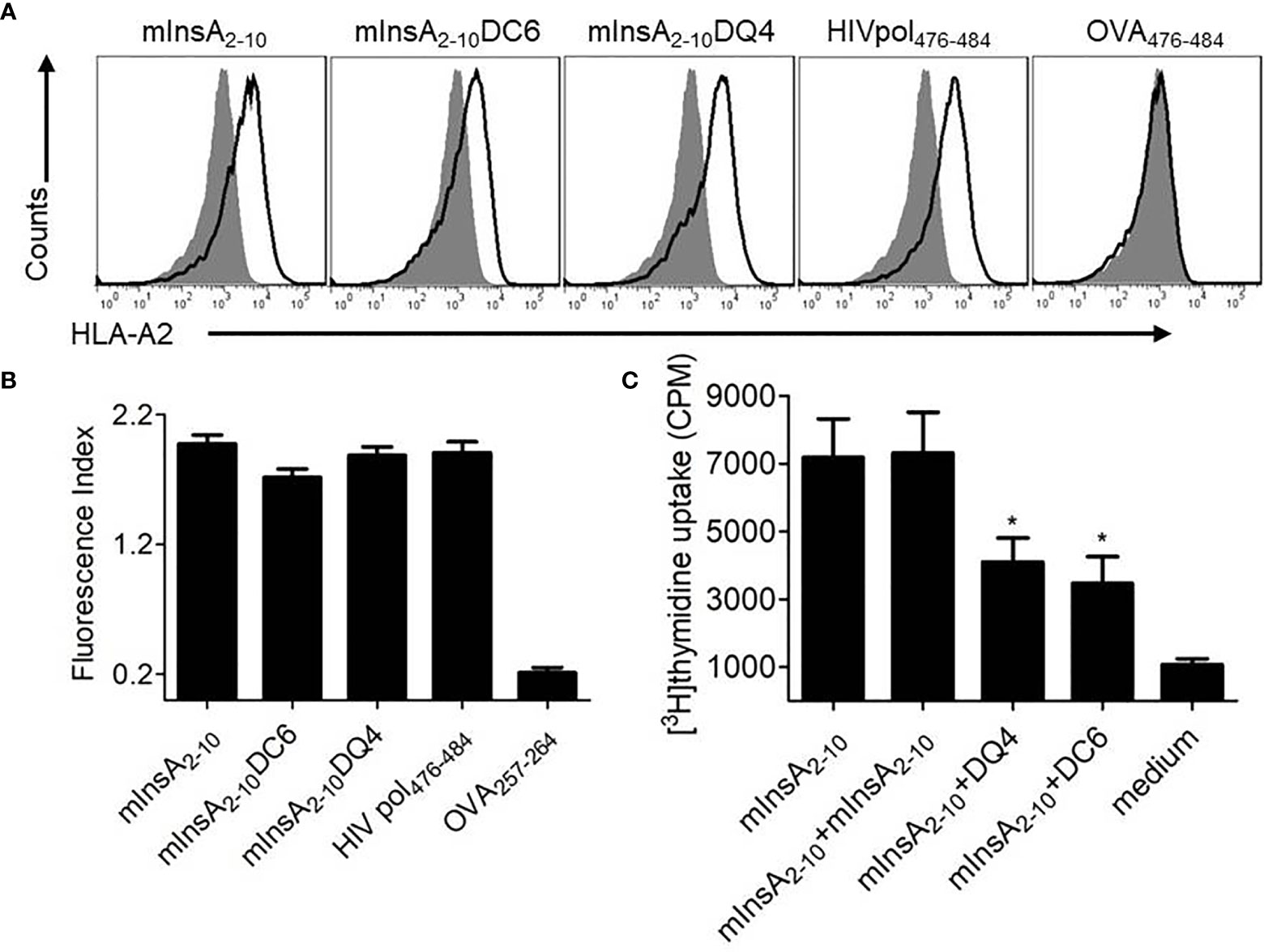
Figure 3 Selection of a promising antagonist peptide for mInsA2-10. (A) T2 cells were incubated with or without the indicated peptides (50 μg/ml), and the stabilization of surface HLA-A2 molecules was detected by flow cytometry. The HLA-A*0201-binding peptide HIV Pol476-484 was used as a positive control. The H-2Kb-binding peptide OVA257-264 was used as a negative control. Filled histograms, no peptide; open histograms, plus peptide. (B) The binding affinity was presented as the fluorescent index (FI) that was calculated as follows: FI = (mean fluorescence intensity with the given peptide − mean fluorescence intensity without peptide)/(mean fluorescence intensity without peptide). Bars represent the mean ± SEM of three independent experiments. (C) Splenocytes were treated with 10 μg/ml mInsA2-10 plus 10 μg/ml indicated peptides for 72 h in 96-well plates (2×105 per well) at 37°C. The cell proliferation was measured by [3H] thymidine incorporation. Bars represent the mean ± SEM of seven independent experiments. *P < 0.05.
To investigate whether the two APLs had antidiabetic activity in vivo, female NOD.β2mnull.HHD mice were injected intraperitoneally with soluble peptide mInsA2-10, mInsA2-10DC6, mInsA2-10DQ4, or control peptide (OVA257-264) in PBS, respectively, starting at 4 weeks of age. Consistently, native mInsA2-10 peptide treatment had no protective effect on T1D. Interestingly, administration of mInsA2-10DQ4, but not DC6, significantly suppressed the development of T1D compared with the control group (p=0.0086) (Figure 4A). Histopathological analysis of mInsA2-10DQ4-treated female NOD.β2mnull.HHD mice at 12 weeks showed less insulitis, compared to age-matched non-diabetic NOD.β2mnull.HHD mice of mInsA2-10, mInsA2-10DC6, or control peptide treatment (Figures 4B, C). These results indicated that the treatment of mInsA2-10DQ4 reduces insulitis and prevents the development of T1D in NOD.β2mnull.HHD mice.
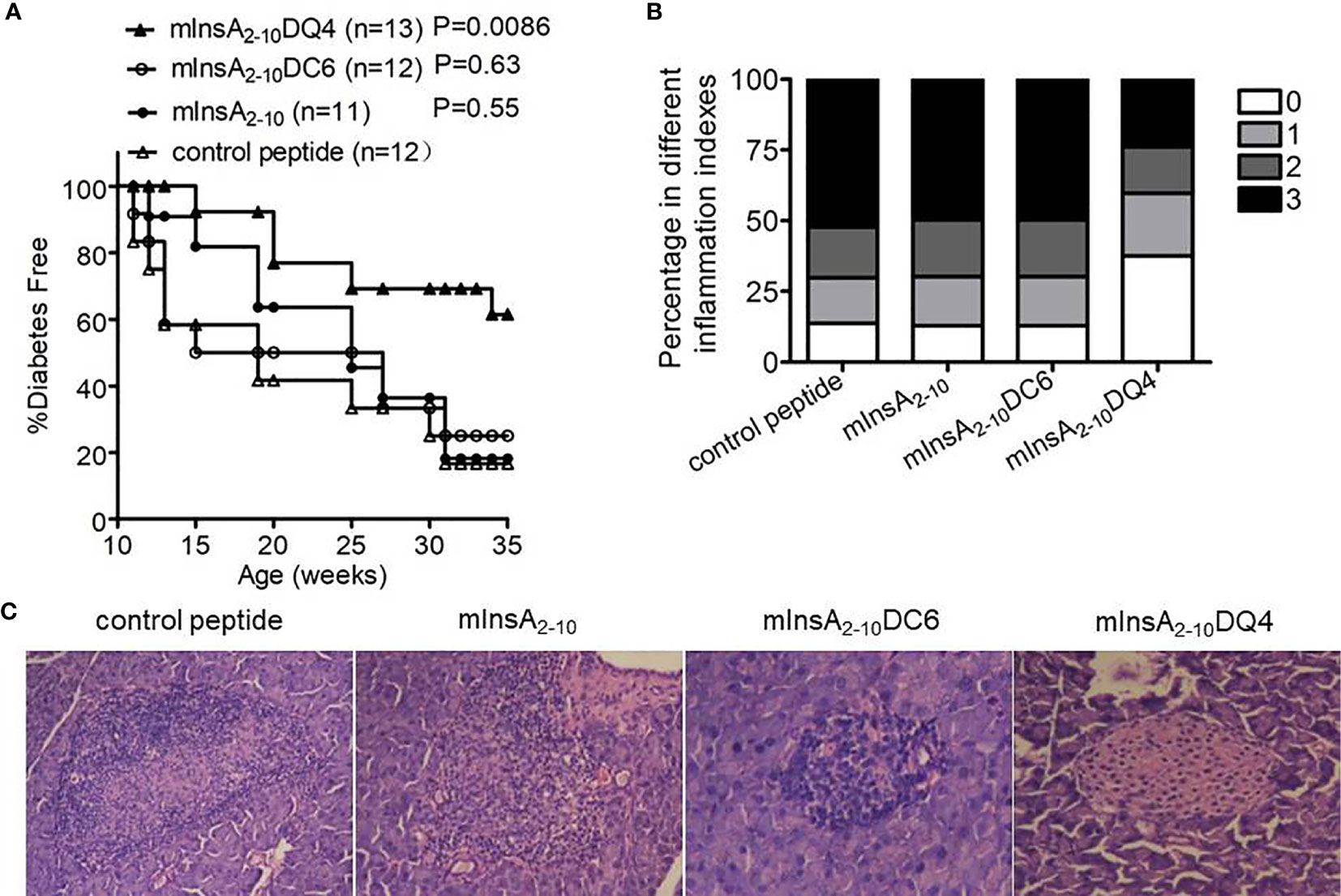
Figure 4 The treatment with mInsA2-10DQ4 prevented diabetes development in NOD.β2mnull.HHD mice. NOD.β2mnull.HHD female mice were injected intraperitoneally (once weekly) at 4–9 weeks of age with 100 μg of peptide mInsA2-10, mInsA2-10DQ4, mInsA2-10DC6, or OVA257-264. (A) Mice were monitored for diabetes development. (B) Histopathological evaluation of pancreatic sections from indicated peptide-treated NOD.β2mnull.HHD mice at 12 weeks of age. (C) Representative hematoxylin and eosin–stained paraffin-embedded pancreas sections (200 × magnification) are shown.
To further determine whether mInsA2-10DQ4 treatment could induce mInsA2-10-specific CD8+ T cell tolerance in vivo, we analyzed mInsA2-10-pulsed T2 cells stimulated IFN-γ spots forming by purified splenic CD8+ T cells from NOD.β2mnull.HHD mice treated with mInsA2-10DQ4, mInsA2-10, or control peptide. As shown in Figures 5A ,B, when compared with control peptide-loaded T2 cells, the stimulation of T2 cells pulsed with mInsA2-10 significantly increased IFN-γ spot formation of splenic CD8+ T cells in both control peptide and mInsA2-10-treated NOD.β2mnull.HHD mice. In contrast, T2 cells pulsed with mInsA2-10 failed to increase IFN-γ spot formation of splenic CD8+ T cells in mInsA2-10DQ4-treated NOD.β2mnull.HHD mice. These results together indicated that treatment with mInsA2-10DQ4 blinded peripheral mInsA2-10-autoreactive CD8+ T cell responses in NOD.β2mnull.HHD mice.
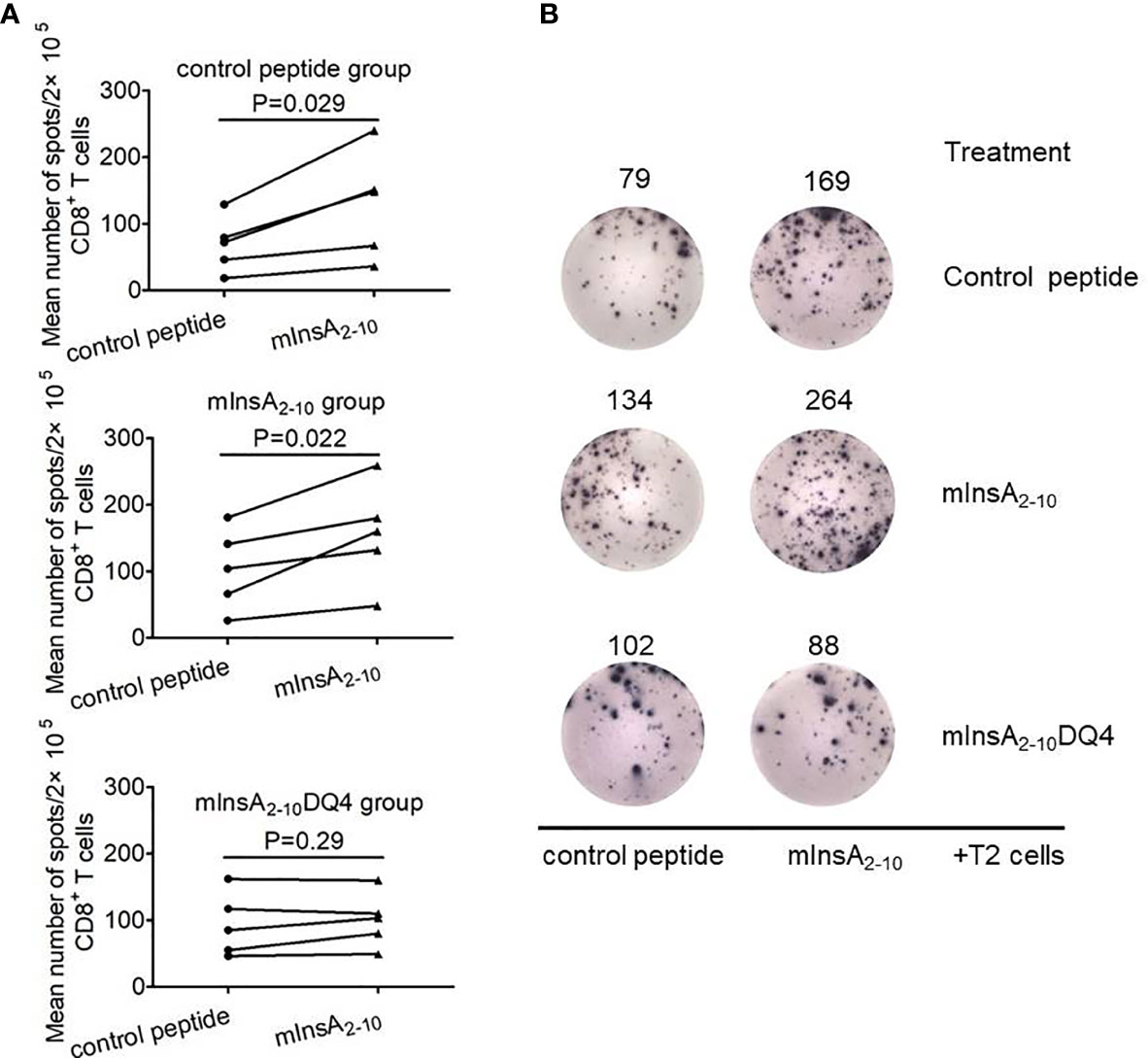
Figure 5 mInsA2-10DQ4 treatment resulted in loss of mInsA2-10 autoreactive CD8+ T cell responses. (A) Statistical analysis of the average number of control peptide or mInsA2-10-specific IFN-γ-positive spots per 2×105 splenic CD8+ T cells in triplicate cultures with a treatment group (n = 5) are shown. Significance was determined by a paired t-test. (B) Representative IFN-γ ELISPOT assay demonstrated splenic CD8+ T cell responses to T2 cell loaded with mInsA2-10 in each peptide-treated group of non-diabetic NOD.β2mnull.HHD mice at 12 weeks of age.
Since insulin has been considered as an only essential autoantigen to initiate spontaneous T1D and the elimination of insulin-reactive T cells can block epitopes expansion and subsequent destruction of β cells by other autoreactive T cells in NOD mice, we questioned whether the treatment of mInsA2-10DQ4 prevented the infiltration of CD4+ and CD8+ T cells and inflammatory responses in the pancreas of NOD.β2mnull.HHD mice. Strikingly, both the frequency and absolute number of pancreas-infiltrating CD4+ T and CD8+ T cells were markedly reduced in mInsA2-10DQ4-treated NOD.β2mnull.HHD mice (CD4+ T cells, 18.5 ± 1.7%, 12,757.4 ± 2,556.6; CD8+ T cells, 1.6 ± 0.3%, 1,172.2 ± 206.1) compared with those detected in mInsA2-10-treated (CD4+ T cells, 29.3 ± 4.0%, 24,215.2 ± 2,994.9; CD8+ T cells, 3.3 ± 0.4%, 2,005.8 ± 326.6) or control peptide-treated (CD4+ T cells, 29.2 ± 5.7%, 23,309.2 ± 2,171.7; CD8+ T cells, 3.2 ± 0.5%, 2,015.2 ± 314.6) NOD.β2mnull.HHD mice (Figures 6A, B). As expected, the mRNA levels of the proinflammatory cytokines IL-6, IL-1β, TNF-α, IFN-γ, and IL-17 significantly decreased in the pancreas of mInsA2-10DQ4-treated NOD.β2mnull.HHD mice, when compared with NOD.β2mnull.HHD mice that received mInsA2-10 or control peptide injection (Figure 6C). Thus, these data suggested that the treatment of mInsA2-10DQ4 reduces the aggregation of autoreactive CD4+ T and CD8+ T cells and inflammatory responses in the pancreas of NOD.β2mnull.HHD mice.
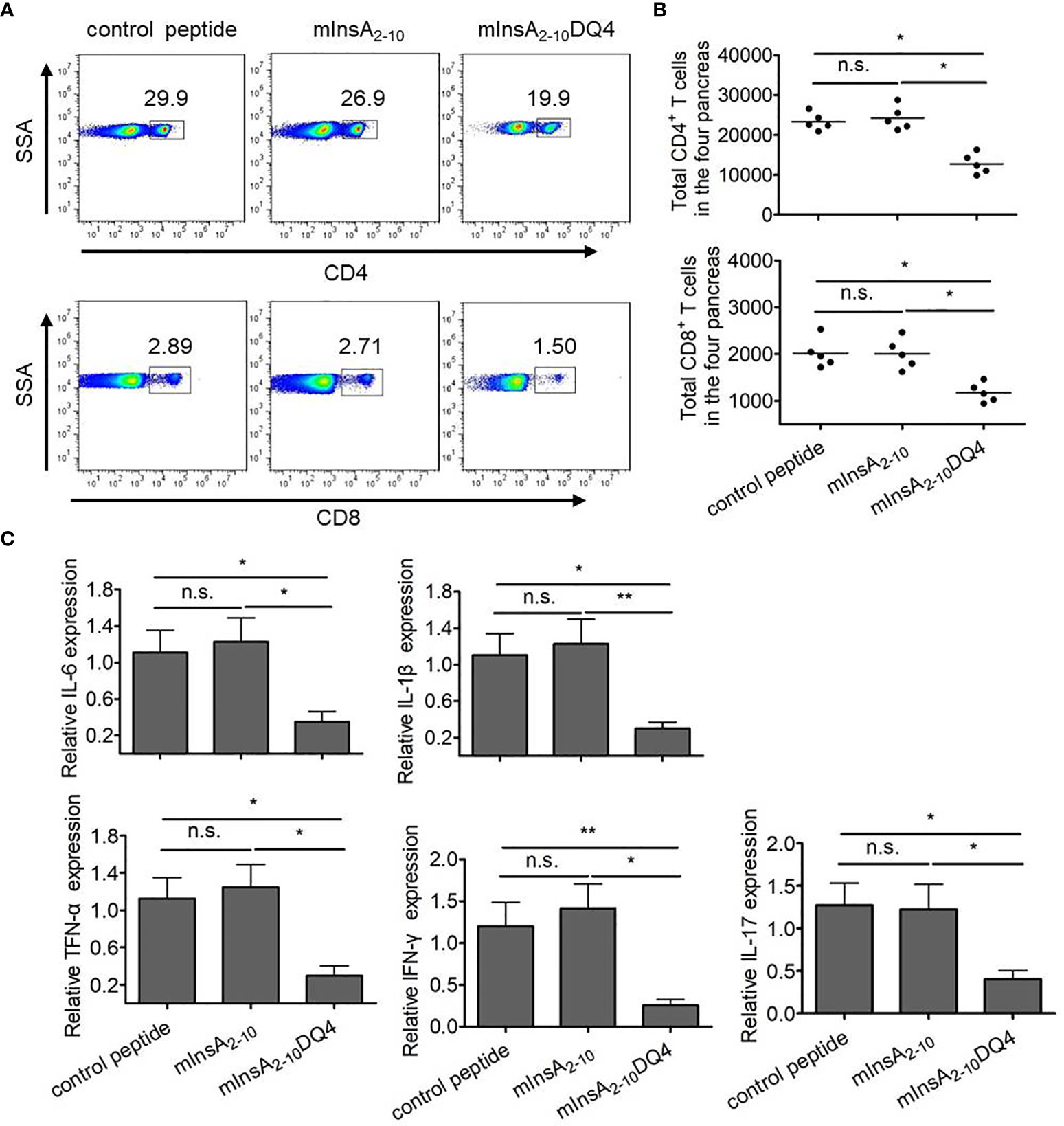
Figure 6 Treatment with mInsA2-10DQ4 notably reduced frequencies and absolute numbers of pancreas-infiltrating CD4+ T and CD8+ T cells in NOD.β2mnull.HHD mice. (A) Representative fluorescence-activated cell sorting (FACS) histogram plots for pancreas-infiltrating CD4+ T and CD8+ T cells from four mice. (B) The results for absolute numbers of these pancreas-infiltrating CD4+ T and CD8+ T cells are expressed as the mean ± SD. Each symbol in (B) represents a sample of pooled pancreas-infiltrating CD4+ T and CD8+ T cells from four mice. (C) Total RNA was extracted from the pancreas in each peptide-treated group of non-diabetic mice (n = 6). The mRNA expression levels of these cytokines were quantified by real-time RT-PCR. The data are presented as fold-change compared to the mRNA levels expressed in pancreas from control peptide-treated mice. *P < 0.05 and **P < 0.01. n.s. indicates no significance.
T1D in both NOD mice and humans is an organ-specific autoimmune disease resulting from selective damage of pancreatic β cells by autoreactive T lymphocytes. Autoreactive CD8+ T cells, which play an indispensable key role in initiation and progression of T1D, are recommended as ideal targets for the prevention of T1D. In this study, we generated an APL, mInsA2-10DQ4, derived from D-amino acid substitution at a potential TCR contact site of an HLA-A*0201-restricted immunodominant insulin epitope. Treatment with mInsA2-10DQ4 significantly suppressed the development of T1D in NOD.β2mnull.HHD mice via inducing mInsA2-10 autoreactive CD8+ T cell tolerance and preventing the subsequent infiltration of CD4+ T and CD8+ T cells as well as inflammatory responses in the pancreas in NOD.β2mnull.HHD mice.
It has been reported that the modification of peptides with D-amino acids instead of natural L-amino acids can not only preserve the feasible recognition properties but also significantly improve the stability and half-life of peptides (23, 24). Most of the available literatures reported that the replacement of all L-amino acid residue in T cell epitope by their D-enantiomers results in normal or reversed (retro) amide linkage (25, 26). A recent study reported that a retro-inverso-D-amino acid-based insulin B-chain (9–23) peptide blocked the presentation of native InsB9-23 by HLA-DQ8 to autoreactive T-cells, suggesting that D-amino acid peptides may be an innovative treatment for T1D (27). Whereas, relatively few studies have been reported on a single amino acid substitution by the respective D-enantiomer in T cell epitope. For example, the individual substitutions by the corresponding D-amino acid at most positions within an I-Ed-restricted 13-mer snake toxin epitope greatly diminished its T-cell stimulating activity (17). A modified H-2Dd-restricted epitope of HIV-1 IIIB envelope glycoprotein P18-I10 (RGPGRAFVTI) with a single amino acid substitution by the respective D-enantiomer at positions 324F, 325V, 326T, or 327I markedly reduced the cytotoxic activity of P18-I10-specific murine CD8+ T lymphocytes (18).
Subtle changes at the TCR contact residues can dramatically alter the downstream signaling events, leading to T-cell anergy, apoptosis, or high activation (28). Since amino acid residues at positions 4 and 6 of mInsA2-10 were predicted to be in close contact with the TCR, we generated two APL candidates via in silico-assisted single D-amino acid substitution at the two positions, respectively. Our in silico analysis predicted that single D-amino acid substitution at position 4 rather than position 6 might have minor influence on the binding affinity of epitope to HLA-A*0201. These simulation results were supported by the peptide binding assays, which showed that DQ4 had a similar HLA-A*0201-binding affinity with that of native peptide, whereas DC6 displayed a relatively weaker binding ability to HLA-A*0201. Although both two APLs showed antagonistic effects against natural peptide-stimulated T cell proliferation in vitro, only mInsA2-10DQ4, but not DC6, exhibited in vivo antidiabetic effects in NOD.β2mnull.HHD mice. A plausible explanation is that APL DC6 has a reduced binding ability to HLA-A*0201 relative to its parent peptide, and it is difficult for DC6 to competitively bind to HLA-A*0201 in vivo and therefore cannot be an effective antagonist. Whereas, mInsA2-10DQ4 not only has a similar HLA-A*0201-binding affinity with mInsA2-10 but also has a theoretically stronger stability in vivo than mInsA2-10. Therefore, DQ4 could effectively antagonizes the natural peptide in vivo. The metabolic dynamics and stability of DQ4 need further study.
We did not further study the mechanisms by which DQ4 induced mInsA2-10-autoreactive T-cell tolerance in humanized NOD mice. APLs with amino acid substitutions at TCR contact positions are suggested as useful tools to modulate T cell responses induced by the native peptide. For example, APLs of insulin B15-23 (LYLVCGERG) G6H and R8L, which were generated by one natural amino acid substitution at position 6 and position 8 (TCR contact sites), respectively, showed the antagonist activity of the highly pathogenic insulin B15-23-reactive CD8+ T cell clone G9C8 in cytotoxicity and IFN-γ production assays (29). Another study indicated that a superagonist APL with an amino acid substitution at position 6 TCR contact site proved more effective than the native peptide in blocking autoimmune diabetes by a decreased accumulation of pathogenic CD8+ T cells in the pancreas (30). We hypothesized that DQ4 with an increased stability and modified TCR contact site could provide an altered antigen stimulation signal to the native mInsA2-10-autoreactive T cells, leading to induce their anergy or apoptosis, or change their functional state in vivo. However, the specific molecular and cellular mechanisms need to be further studied.
At least two studies have reported the presence of specific T cell responses to human proinsulin (PPI) 90-99 or InsA1-10 (GIVEQCCTSI) in T1D patients (31, 32). Thus, human InsA2-10 is likely to be an immunogenic target for diabetic patients. The sequence of mInsA2-10 (IVDQCCTSI) is highly consistent with that of hInsA2-10 (IVEQCCTSI) with only one amino acid difference at position 3, suggesting the possibility of cross-reactivity between the two peptides in T1D patients, but further studies are needed. We also did not detect whether mInsA2-10 DQ4 can antagonize T cell responses towards hInsA2-10 in PBMC of T1D patients. Therefore, further studies are needed to determine whether DQ4 has potential clinical application value.
As we know, prediction of T1D onset is possible, and prevention is now a goal in T1D. T1D vaccines based on islet autoantigens are considered to be one of such strategies to prevent or delay the occurrence of T1D by modulating autoimmune responses towards pancreatic islet antigens and prevent further destruction of pancreatic β-cells in T1D-susceptible individuals or preclinical individuals (33). Interestingly, our findings confirmed again that insulin is the key autoantigen for T1D initiation, since the administration of DQ4 not only eliminated peripheral mInsA2-10-autoreactive T cells response but also reduced the subsequent infiltration of CD4+ T and CD8+ T cells as well as inflammatory responses in the pancreas in NOD.β2mnull.HHD mice. Consistent with this, administration of chemically fixed splenic antigen-presenting cells coupled with intact insulin or the dominant insulin epitopes, but not epitopes of other autoantigens, protected 4–6-week-old NOD mice from development of T1D, which also indicated insulin is the key initiating autoantigen (34). Therefore, induction of insulin-autoreactive T cell immune tolerance in prediabetic mice may reduce the autoimmune insulitis and damage of islets, and prevent the subsequent epitope spreading and the activation and recruitment of other autoreactive T cells. In this sense, T1D vaccine targeting insulin, such as APLs of insulin epitopes, has potential value in the prevention of T1D when applied to the high-risk individuals whose islet autoimmunity has not yet occurred.
In conclusion, this present study describes that an APL designed by in silico-assisted single D-amino acid substitution at the potential TCR contact site displayed a protective effect against T1D in humanized NOD mice. This study provides a new strategy for the development of APL vaccines for T1D prevention.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the Animal Ethics Committee of the Army Medical University (Third Military Medical University).
MZ and YW performed main experiments and data analysis and drafted the manuscript. XL, GM, XC, and LNW performed experiments and/or analyzed data. ZL revised the manuscript critically for important intellectual content. LW carried out the project design, guided the research process, supervised and managed it, completed the manuscript, and provided research funds. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Nos. 31771002, 81871301, and 82071825) and the National Key Project for Research & Development of China (No. 2016YFA0502204).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kanagawa O, Martin SM, Vaupel BA, Carrasco-Marin E, Unanue ER. Autoreactivity of T Cells From Nonobese Diabetic Mice: An I-Ag7-Dependent Reaction. Proc Natl Acad Sci USA (1998) 95(4):1721–4. doi: 10.1073/pnas.95.4.1721
2. Haskins K, Cooke A. CD4 T Cells and Their Antigens in the Pathogenesis of Autoimmune Diabetes. Curr Opin Immunol (2011) 23(6):739–45. doi: 10.1016/j.coi.2011.08.004
3. Katz J, Benoist C, Mathis D. Major Histocompatibility Complex Class I Molecules Are Required for the Development of Insulitis in Non-Obese Diabetic Mice. Eur J Immunol (1993) 23(12):3358–60. doi: 10.1002/eji.1830231244
4. Delovitch TL, Singh B. The Nonobese Diabetic Mouse as a Model of Autoimmune Diabetes: Immune Dysregulation Gets the NOD. Immunity (1997) 7(6):727–38. doi: 10.1016/S1074-7613(00)80392-1
5. Liblau RS, Wong FS, Mars LT, Santamaria P. Autoreactive CD8 T Cells in Organ-Specific Autoimmunity: Emerging Targets for Therapeutic Intervention. Immunity (2002) 17(1):1–6. doi: 10.1016/S1074-7613(02)00338-2
6. Sumida T, Furukawa M, Sakamoto A, Namekawa T, Maeda T, Zijlstra M, et al. Prevention of Insulitis and Diabetes in Beta 2-Microglobulin-Deficient non-Obese Diabetic Mice. Int Immunol (1994) 6(9):1445–9. doi: 10.1093/intimm/6.9.1445
7. Marks DI, Iannoni B, Coote MA, Mandel MA, Fox RM. Prevention of Cyclophosphamide-Induced and Spontaneous Diabetes in NOD Mice by Syngeneic Splenocytes Treated With Cytotoxic Drugs. Autoimmunity (1991) 11(2):73–9. doi: 10.3109/08916939109035137
8. Takaki T, Marron MP, Mathews CE, Guttmann ST, Bottino R, Trucco M, et al. HLA-A*0201-Restricted T Cells From Humanized NOD Mice Recognize Autoantigens of Potential Clinical Relevance to Type 1 Diabetes. J Immunol (2006) 176(5):3257–65. doi: 10.4049/jimmunol.176.5.3257
9. Niens M, Grier AE, Marron M, Kay TW, Greiner DL, Serreze DV. Prevention of "Humanized" Diabetogenic CD8 T-Cell Responses in HLA-Transgenic NOD Mice by a Multipeptide Coupled-Cell Approach. Diabetes (2011) 60(4):1229–36. doi: 10.2337/db10-1523
10. Wilson DB. Immunology: Insulin Auto-Antigenicity in Type 1 Diabetes. Nature (2005) 438(7067):E5; discussion E5–6. doi: 10.1038/nature04423
11. Homann D, Eisenbarth GS. An Immunologic Homunculus for Type 1 Diabetes. J Clin Invest (2006) 116(5):1212–5. doi: 10.1172/JCI28506
12. Han B, Serra P, Amrani A, Yamanouchi J, Maree AF, Edelstein-Keshet L, et al. Prevention of Diabetes by Manipulation of Anti-IGRP Autoimmunity: High Efficiency of a Low-Affinity Peptide. Nat Med (2005) 11(6):645–52. doi: 10.1038/nm1250
13. Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, et al. Reversal of Autoimmunity by Boosting Memory-Like Autoregulatory T Cells. Immunity (2010) 32(4):568–80. doi: 10.1016/j.immuni.2010.03.015
14. Zhang M, Wang S, Guo B, Meng G, Shu C, Mai W, et al. An Altered CD8(+) T Cell Epitope of Insulin Prevents Type 1 Diabetes in Humanized NOD Mice. Cell Mol Immunol (2019) 16(6):590–601. doi: 10.1038/s41423-018-0058-3
15. Hamamoto K, Kida Y, Zhang Y, Shimizu T, Kuwano K. Antimicrobial Activity and Stability to Proteolysis of Small Linear Cationic Peptides With D-Amino Acid Substitutions. Microbiol Immunol (2002) 46(11):741–9. doi: 10.1111/j.1348-0421.2002.tb02759.x
16. Hernandez JF, Cretin F, Lombard-Platet S, Salvi JP, Walchshofer N, Gerlier D, et al. Critical Residue Combinations Dictate Peptide Presentation by MHC Class II Molecules. Peptides (1994) 15(4):583–90. doi: 10.1016/0196-9781(94)90080-9
17. Maillere B, Mourier G, Cotton J, Herve M, Leroy S, Menez A. Probing Immunogenicity of a T Cell Epitope by L-Alanine and D-Amino Acid Scanning. Mol Immunol (1995) 32(14-15):1073–80. doi: 10.1016/0161-5890(95)00073-9
18. Nakagawa Y, Kikuchi H, Takahashi H. Molecular Analysis of TCR and Peptide/MHC Interaction Using P18-I10-Derived Peptides With a Single D-Amino Acid Substitution. Biophys J (2007) 92(7):2570–82. doi: 10.1529/biophysj.106.095208
19. Salomon-Ferrer R, Gotz AW, Poole D, Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations With AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput (2013) 9(9):3878–88. doi: 10.1021/ct400314y
20. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. Ff14sb: Improving the Accuracy of Protein Side Chain and Backbone Parameters From Ff99sb. J Chem Theory Comput (2015) 11(8):3696–713. doi: 10.1021/acs.jctc.5b00255
21. Wang J, Hou T. Develop and Test a Solvent Accessible Surface Area-Based Model in Conformational Entropy Calculations. J Chem Inf Model (2012) 52(5):1199–212. doi: 10.1021/ci300064d
22. Bashford D, Case DA. Generalized Born Models of Macromolecular Solvation Effects. Annu Rev Phys Chem (2000) 51:129–52. doi: 10.1146/annurev.physchem.51.1.129
23. Tugyi R, Uray K, Ivan D, Fellinger E, Perkins A, Hudecz F. Partial D-Amino Acid Substitution: Improved Enzymatic Stability and Preserved Ab Recognition of a MUC2 Epitope Peptide. Proc Natl Acad Sci USA (2005) 102(2):413–8. doi: 10.1073/pnas.0407677102
24. Chen S, Gfeller D, Buth SA, Michielin O, Leiman PG, Heinis C. Improving Binding Affinity and Stability of Peptide Ligands by Substituting Glycines With D-Amino Acids. Chembiochem (2013) 14(11):1316–22. doi: 10.1002/cbic.201300228
25. Bartnes K, Hannestad K, Guichard G, Briand JP. A Retro-Inverso Analog Mimics the Cognate Peptide Epitope of a CD4+ T Cell Clone. Eur J Immunol (1997) 27(6):1387–91. doi: 10.1002/eji.1830270614
26. Van Regenmortel MH, Muller S. D-Peptides as Immunogens and Diagnostic Reagents. Curr Opin Biotechnol (1998) 9(4):377–82. doi: 10.1016/s0958-1669(98)80011-6
27. Lombardi A, Concepcion E, Hou H, Arib H, Mezei M, Osman R, et al. Retro-Inverso D-Peptides as a Novel Targeted Immunotherapy for Type 1 Diabetes. J Autoimmun (2020) 115:102543. doi: 10.1016/j.jaut.2020.102543
28. Shang X, Wang L, Niu W, Meng G, Fu X, Ni B, et al. Rational Optimization of Tumor Epitopes Using in Silico Analysis-Assisted Substitution of TCR Contact Residues. Eur J Immunol (2009) 39(8):2248–58. doi: 10.1002/eji.200939338
29. Petrich de Marquesini LG, Moustakas AK, Thomas IJ, Wen L, Papadopoulos GK, Wong FS. Functional Inhibition Related to Structure of a Highly Potent Insulin-Specific CD8 T Cell Clone Using Altered Peptide Ligands. Eur J Immunol (2008) 38(1):240–9. doi: 10.1002/eji.200737762
30. Hartemann-Heurtier A, Mars LT, Bercovici N, Desbois S, Cambouris C, Piaggio E, et al. An Altered Self-Peptide With Superagonist Activity Blocks a CD8-Mediated Mouse Model of Type 1 Diabetes. J Immunol (2004) 172(2):915–22. doi: 10.4049/jimmunol.172.2.915
31. Rathmann S, Rajasalu T, Rosinger S, Schlosser M, Eiermann T, Boehm BO, et al. Preproinsulin-Specific CD8+ T Cells Secrete IFNgamma in Human Type 1 Diabetes. Ann NY Acad Sci (2004) 1037:22–5. doi: 10.1196/annals.1337.004
32. Baker C, Marquesini LGPD, Bishop AJ, Hedges AJ, Dayan CM, Wong FS. Human CD8 Responses to a Complete Epitope Set From Preproinsulin: Implications for Approaches to Epitope Discovery. J Clin Immunol (2008) 28(4):350–60. doi: 10.1007/s10875-008-9177-4
33. Desai S, Buchade S, Chitlange S, Sharma H, Bhombe D, Shewale S, et al. Vaccines For Type 1 Diabetes: Prevention or Reversal? Curr Diabetes Rev (2021) 17(1):30–6. doi: 10.2174/1573399816666200330145501
Keywords: type 1 diabetes, altered peptide ligand, D-amino acid substitution, mInsA2-10, NOD.β2mnull.HHD mice
Citation: Zhang M, Wang Y, Li X, Meng G, Chen X, Wang L, Lin Z and Wang L (2021) A Single L/D-Substitution at Q4 of the mInsA2-10 Epitope Prevents Type 1 Diabetes in Humanized NOD Mice. Front. Immunol. 12:713276. doi: 10.3389/fimmu.2021.713276
Received: 22 May 2021; Accepted: 09 August 2021;
Published: 30 August 2021.
Edited by:
Sophie Tourdot, Pfizer, United StatesCopyright © 2021 Zhang, Wang, Li, Meng, Chen, Wang, Lin and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Wang, MTMwNjg1ODA5N0BxcS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.