 Mercedes Beatriz Fuertes
Mercedes Beatriz Fuertes Carolina Inés Domaica
Carolina Inés Domaica Norberto Walter Zwirner
Norberto Walter Zwirner
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 July 2021
Sec. NK and Innate Lymphoid Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.713158
This article is part of the Research Topic The Interaction of NKG2D and its Ligands in Health and Diseases View all 6 articles
Immune checkpoint inhibitors (ICI) revolutionized the field of immuno-oncology and opened new avenues towards the development of novel assets to achieve durable immune control of cancer. Yet, the presence of tumor immune evasion mechanisms represents a challenge for the development of efficient treatment options. Therefore, combination therapies are taking the center of the stage in immuno-oncology. Such combination therapies should boost anti-tumor immune responses and/or target tumor immune escape mechanisms, especially those created by major players in the tumor microenvironment (TME) such as tumor-associated macrophages (TAM). Natural killer (NK) cells were recently positioned at the forefront of many immunotherapy strategies, and several new approaches are being designed to fully exploit NK cell antitumor potential. One of the most relevant NK cell-activating receptors is NKG2D, a receptor that recognizes 8 different NKG2D ligands (NKG2DL), including MICA and MICB. MICA and MICB are poorly expressed on normal cells but become upregulated on the surface of damaged, transformed or infected cells as a result of post-transcriptional or post-translational mechanisms and intracellular pathways. Their engagement of NKG2D triggers NK cell effector functions. Also, MICA/B are polymorphic and such polymorphism affects functional responses through regulation of their cell-surface expression, intracellular trafficking, shedding of soluble immunosuppressive isoforms, or the affinity of NKG2D interaction. Although immunotherapeutic approaches that target the NKG2D-NKG2DL axis are under investigation, several tumor immune escape mechanisms account for reduced cell surface expression of NKG2DL and contribute to tumor immune escape. Also, NKG2DL polymorphism determines functional NKG2D-dependent responses, thus representing an additional challenge for leveraging NKG2DL in immuno-oncology. In this review, we discuss strategies to boost MICA/B expression and/or inhibit their shedding and propose that combination strategies that target MICA/B with antibodies and strategies aimed at promoting their upregulation on tumor cells or at reprograming TAM into pro-inflammatory macrophages and remodeling of the TME, emerge as frontrunners in immuno-oncology because they may unleash the antitumor effector functions of NK cells and cytotoxic CD8 T cells (CTL). Pursuing several of these pipelines might lead to innovative modalities of immunotherapy for the treatment of a wide range of cancer patients.
The therapeutic alternatives to treat tumors received a formidable boost when immune checkpoint inhibitors (ICI) were developed (1). These assets revolutionized the field of immuno-oncology (I-O), leading to successful treatment of liquid and solid tumors. ICI are monoclonal antibodies (mAb) whose mechanism of action involve the blockade or interference with cell surface receptors that mediate inhibitory signals in cells of the immune system, mainly T lymphocytes but also natural killer (NK) cells (2, 3). However, ICI also have a dark side that comprises a low frequency of responding patients, their adverse effects (4) and, more recently described and still ill-characterized, the occurrence of hyper-progression of the tumor (5–7). An additional promising option is to combine ICI with other anti-tumor compounds. Combination of anti-CTLA4 and anti-PD-1 or anti-PD-L1 mAb has shown promising results in several tumors (8). Moreover, novel strategies, molecular targets and cell-based therapies keep emerging as alternatives to improve the efficacy of the treatment options for cancer patients, positioning the field of I-O at the top in the investment and competition for academic institutions and pharmaceutical/biotech companies.
NK cells and CTL constitute the most relevant effector cells that mediate tumor cell elimination through their cytolytic activity and a proinflammatory function. Cytolytic activity is exerted though the secretory pathway and the expression of death receptors (9). Inflammatory activity is exerted through the secretion of cytokines, mainly IFN-γ and TNF, and chemokines such as CCL5, XCL1 and XCL2 (10, 11). Evidence obtained some years ago (12), and recent studies that established that NK cell frequency, infiltration and function are associated with improved patient survival (13–16) demonstrated that NK cells play a crucial role in tumor immunity. Remarkably, although NK cells were considered rapid but short-lived responders to virus-infected and tumor cells, this idea has been challenged by the identification of NK cells that undergo clonal expansion and acquire features of long- lived memory cells, a hallmark of adaptive T and B lymphocytes. The molecular and cellular mechanisms that drive adaptive NK-cell expansion and activity are being elucidated (17). These concepts strengthened even more the interest in exploiting NK cells´ immunotherapeutic potential to combat cancer in oncologic patients (18, 19).
Furthermore, NK cells became progressively positioned at the forefront of current immunotherapy strategies (18, 19). Many new compounds, including but not limited to mAb are being developed to fully exploit their antitumor potential (20). Also, it is currently possible to produce large amounts of NK cells suitable for adoptive transfer to patients. A recapitulation of NK cell-based therapies in I-O indicates that most of these approaches fall within one of the following categories: a) in vitro expansion and activation; b) adoptive transfer of allogeneic NK cells; c) generation of chimeric antigen receptor modified NK cells (CAR-NK) and d) administration of mAb or other bioactive compounds that regulate NK cell activity against tumors (21). Some success in the treatment of liquid tumors has been achieved using these NK cell-based strategies (22–27). While in vitro expansion and activation of autologous NK cells, and adoptive transfer of allogeneic NK cells have yielded variable degrees of success with liquid tumors, high hopes have been put on the generation and use of CAR-NK. This is because CAR-NK cells have several advantages over CAR-T cells such as a shorter half-life (and a subsequent better opportunity to control eventual side effects), a lack of induction of cytokine release syndrome (CRS, often severe and/or fatal in patients that received CAR-T cells), and the possibility of preparing off-the-shelf CAR-NK cells for the treatment of multiple patients (28–30). However, the landscape is quite different for solid tumors mostly because NK cells must face the formidable task of overcoming the immunosuppressive TME to avoid becoming exhausted and dysfunctional (31, 32). Also, even if NK cell can overcome this hostile environment, their weak capacity to infiltrate solid tumors is another of the reasons that explain the low success of NK cell-based therapies to treat solid tumors (28, 29). Thus, adoptive transfer of NK cells might require the combination with additional strategies to bolster an effective anti-tumor NK cell function. Combination with ICI emerge as attractive possibilities but, in view of our current knowledge about dysfunctional NK cells, other molecules such as TIM-3, TIGIT and LAG-3 are taking the center of the stage in I-O, as their blockade, knock down or knock out results in a better tumor eradication in different models (33).
The possibility of promoting NK cell effector functions through immunotherapeutic manipulation is further supported by data that indicate that NK cells respond to ICI. Single-cell RNA sequencing (scRNAseq) data indicate that tumor NK cell infiltration is associated with better patient outcomes in several different cancer types (13, 15) and that NK cell infiltration contributes to a robust ICI response (10, 14). Also, scRNAseq and CYTOF revealed that ICI induced a significant remodeling of lymphoid and myeloid cells in the TME, and this effect was dependent on IFN-γ (34). Accordingly, there is a considerable interest in harnessing antitumor NK cell effector functions through the development of novel cancer immunotherapies (21, 35). Many companies currently have NK cell pipelines in their portfolios mainly intended to foster NK cell effector functions in cancer patients using novel ICI or immunomodulatory agents (35–37). However, these strategies face the challenge of having to overcome the decline in NK cell activity due to tumor immune escape mechanisms. In addition, in ccRCC, an RNAseq analysis demonstrated that expression of NK cell-associated receptors and molecules, and some other ligands recognized by these receptors affect overall survival (38). These findings sustain the necessity of a deeper exploration of the TME as a major contributor to NK cell (dys)function and the characterization of tumor-specific factors and mechanisms that regulate NK cell activity. Additionally, a big question is whether it is feasible to reinvigorate dysfunctional tumor-infiltrating NK cells (TINK) or to eliminate/deplete them and create a niche for the recruitment of newly activated, fully functional NK cells through the administration of immunotherapeutic agents to the patient.
NK cells detect tumor cells through a collection of germline-encoded activating receptors whose actions are counterbalanced by another group of inhibitory receptors (28, 39, 40). Thus, NK cells develop effector functions when signals triggered by activating receptors override signals triggered by inhibitory receptors. NK cell activity is also regulated by cytokines, especially those produced by myeloid cells such as IL-12, IL-23, IL-27, IL-15, IL-18 and TGF-β (41–44) and by agonists of several Toll-like receptors (TLR) such as TLR3, TLR7 and TLR9 that are expressed by NK cells (45–47). Therefore, the integration of activating and inhibitory signals present in their environment dramatically determines NK cells’ capacity to mobilize effector functions.
Besides CD16 (FcγIIIa receptor) that recognizes Fc fractions of several IgG subclasses and is responsible for the antibody-dependent cell-mediated cytotoxicity (ADCC) (48), the Natural Cytotoxicity Receptors (NCR) NKp30 (CD337, the product of the ncr3 gene), NKp44 (CD336, the product of the ncr2 gene), NKp46 (CD335, the product of the ncr1 gene) and NKp80 (the product of the klrf1 gene), together with DNAM-1 (CD226) and NKG2D (CD314, the product of the klrk1 gene) emerged as a major activating receptors (17, 37, 49–52). In vivo blockade of NKG2D or NKG2D knock out mice leads to an increased susceptibility to spontaneous tumor development and tumor progression (53, 54). Therefore, efforts are underway to capitalize on NKG2D ligands (NKG2DL) as molecular targets in I-O. In humans, 8 different NKG2DL have been described (51, 55). The first known NKG2DL were the proteins encoded by the MHC class I-chain related genes A and B (MICA and MICB), also called PERB11.1 and PERB11.2 respectively (56, 57). Both genes map within the MHC, are highly polymorphic (58, 59) and the alleles are expressed in a codominant manner (60). The MICA and MICB proteins encoded by most alleles consist of three extracellular domains, one transmembrane domain and a cytoplasmic tail. An exception is the MICA*008 allele (the most frequent in different populations) that harbors an insertion in exon 5 that introduces a shift in the reading frame, encoding a truncated protein with a partial transmembrane domain and no cytoplasmic tail. Nonetheless, MICA*008 is stably expressed on the cell surface of different cells. MICA and MICB are also highly glycosylated. Due to their polymorphic nature, MICA and MICB constitute targets of the immune response against allogeneic transplants and patients with kidney, hearth and lung transplant rejection exhibit anti-MICA/B Ab in serum (61–66).
Although the significance of the polymorphism of MICA and MICB remains ill-defined, associations between alleles and autoimmune diseases and cancer has been widely reported (67–75). Also, a differential regulation of cell surface expression of MICA isoforms has been observed upon infection with cytomegalovirus (76), suggesting that resistance to infectious agents could be a driving force for the selection of several MICA alleles in the population. Dimorphism at position 129, which maps to the α2 domain of MICA, affects NKG2D recognition. Alleles with Met at position 129 trigger stronger NKG2D signaling and subsequent NKG2D-dependent effector functions than alleles with Val, but, at the same time, MICA-129Met isoforms promote a more intense downregulation of NKG2D than MICA-129Val isoforms (77). MICA-129Met homozygosity confers susceptibility to inflammatory bowel disease (78), while MICA-129Val homozygosity leads to faster progression of multiple myeloma (MM) (79) and also plays a role in susceptibility for breast cancer development (80). Therefore, although the majority of the polymorphic positions in MICA are not exposed to its contact area with NKG2D (58, 81), such polymorphism may affect other aspects of MICA and determine the chances of success of strategies aimed at leveraging MICA as molecular target in I-O.
The existence of MICA-null haplotypes in individuals without particular susceptibility to infectious or autoimmune diseases or cancer indicates some redundancy in the biological function of MICA (82–85). Likely, this is because there are additional ligands for NKG2D. Besides MICB, which shares a homology of above 80% with MICA, currently we know six additional NKG2DL but with a homology with MICA and MICB that is below 25%. These additional NKG2DL are members of the UL-16 binding protein (ULBP) family, also known as Retinoic Acid Early Transcripts (RAET) 1. Therefore, they were named ULBP-1 (or RAET1I), ULBP-2 (or RAET1H), ULBP-3 (or RAET1N), ULBP-4 (or RAET1E), ULBP-5 (or RAET1G) and ULBP-6 (or RAET1L) (51, 55, 86–89).
It has been considered for some time that MICA and MICB are not expressed or are weakly expressed in normal cells with very few exceptions such as the luminal side of the intestinal epithelium (56, 90–93). However, it was observed later that MICA and MICB transcripts could be detected in most normal tissues with the exception of the central nervous system (94). However, as NKG2DL experience post-translational modifications that regulate their expression as cell surface molecules (95–97), their cell surface expression on normal cells remains controversial. We demonstrated that premalignant quiescent melanocytic nevi, benign lesions of the skin and normal skin do not express MICA, in opposition to a primary recently diagnosed melanoma (98). Also, expression of MICA/B in many tumors and normal epithelia was recently reported, but with a predominant intracellular localization and low cell surface expression (99). The reasons for these discrepancies are not quite well understood but different technical approaches to assess cell surface MICA expression may produce discrepant results. As MICA colocalizes with some intracellular markers, these results indicate that there is an intracellular pool of MICA. Moreover, cell surface expression should be better analyzed by flow cytometry instead of tissue microscopy to generate compelling information. Accordingly, we observed that several melanoma cell lines and metastatic melanomas display an intracellular pool of MICA but only some of them exhibit cell surface MICA (100).
MICA is expressed in a wide variety of tumors (91, 101–110). RNA sequencing (seq) data indicate that MICA is the NKG2DL that exhibits the highest expression in, from example, lung, colorectal, stomach, liver and breast cancers (111). Also, MICA exhibits a very low tumor mutational burden, suggesting that its expression is not subject to DNA editing to confer some kind of adaptive advantage to tumors. Although over-expression of NKG2DL may represent a valid strategy to limit tumor progression (112–114), tumors display escape strategies that subvert the biological function of NKG2D (115, 116). The underlying mechanisms involves the proteolytic shedding of MICA and other NKG2DL induced by tumor-secreted metalloproteases (MMP) (115–117) or secretion in exosomes (118). Released soluble MICA (sMICA) and soluble MICB (sMICB) can thereafter bind to NKG2D and induce its down-modulation and degradation, subverting NKG2D-dependent effector functions of NK cells and facilitating tumor immune escape (110, 115, 116). This dual role of MICA/B is schematically represented in Figure 1. However, recent data indicates that the suppressive activity of sMICA on NK cells is not due to the down-regulation of NKG2D but to a blockade of NKG2D by sMICA (119). In addition, other mechanisms account for low cell surface expression of MICA and impaired recognition by NKG2D, as we have demonstrated previously (100).
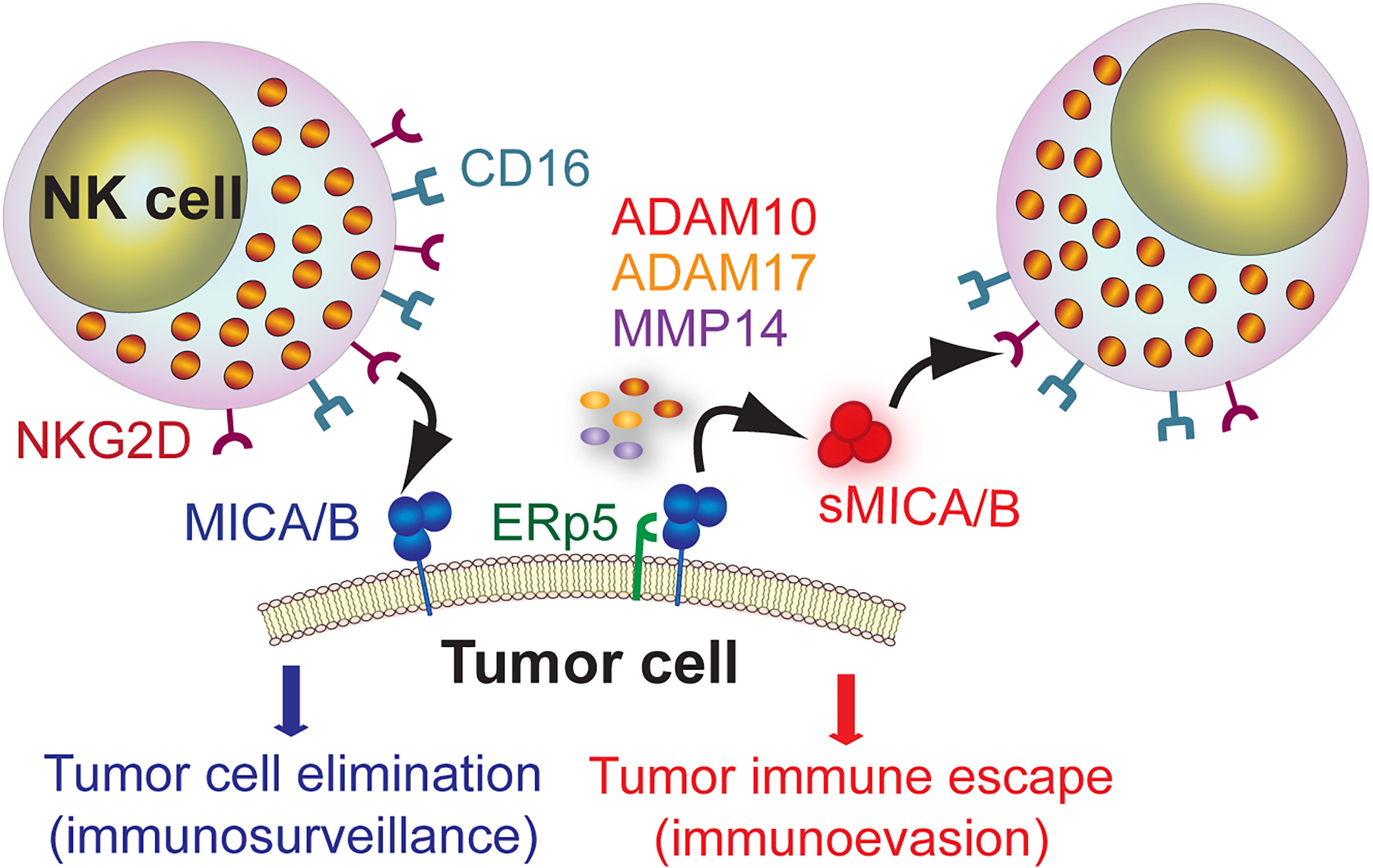
Figure 1 Dual role of MICA/B as target molecule for immunosurveillance by NK cells and as mediator of tumor immune escape. MICA/B expressed on the cell surface of tumor cells can be recognized by NK cells through NKG2D and promote a cytotoxic response that leads to tumor cell elimination (immunosurveillance). However, MICA/B can associate with the ERp5 chaperone and, through a proteolytic cleavage mediated by ADAM10, ADAM17 and MMP14, generate sMICA/B that promote NKG2D down-regulation and impairment of NK cell-effector functions, thus facilitating tumor immune escape (immunoevasion).
The different UBLP or RAET1 also have been shown to be over-expressed on tumors (mainly on leukemic blasts) and mobilize NKG2D-dependent NK cell effector functions (110, 120–126). These additional NKG2DL, unlike MICA and MICB, consist of two extracellular domains. ULBP-4 and ULBP-5 also carry a transmembrane domain and a cytoplasmic tail. However, ULBP-1, ULBP-2, ULBP-3 and ULBP-6 are anchored to glycosylphosphatidylinositol (GPI) of the cell membrane (127) and ULBP-5 can also generate a GPI-anchored form (128). Also, ULBP can generate soluble forms with immunosuppressive activity due to a shedding process mediated by MMP, by phosphatidylinositol phospholipase C or in exosomes (129–132).
Moreover, NKG2D plays an important role during immunosurveillance in patients with acute myelogenous leukemia (AML) (133). AML mortality is mostly due to recurrence caused by chemotherapy-resistant leukemia stem cells (LSC), which, in contrast to bulk AML cells, are highly leukemogenic in immunodeficient mice (134). NKG2DL are broadly expressed on patient-derived bulk AML cells but not on LSC. However, when LSC differentiate into mature AML cells, they upregulate NKG2DL expression, and these NKG2DL-expressing AML cells, in opposition to LSC, become susceptible to NK cells in vitro. Therefore, LSC may preclude NK cell effector functions through suppression of NKG2DL expression and contribute to the AML burden through a continuous differentiation into NKG2DL+ AML blasts. Hence, therapy-induced upregulation of NKG2DL in LSC from AML may promote the reinstatement of susceptibility to NK cells.
The reason for the multiplicity of NKG2DL remains speculative. Although all of them seem to be over-expressed by different tumor cells, more knowledge has been obtained for MICA and MICB. These two NKG2DL also stand out because potential therapeutic strategies targeting MICA and MICB but not therapeutic strategies targeting UBLP emerged during the last years. Therefore, we focused this review mainly on MICA and MICB as targets in I-O.
Given the formidable challenge of overcoming the immunosuppressive TME to leverage NK cell-based immunotherapies for solid tumors (28, 29), it becomes necessary to elucidate the regulatory circuits that shut down NK cell effector functions in this particular niche. NK cells can function as drivers of tumor inflammation, which leads to tumor immune cell infiltration and promotes the conversion of “cold” tumors into “hot” tumors, subsequently conferring better responsiveness to ICI (10). NK cells´ critical functions to induce an effective tumor immunity depend on a successful crosstalk with conventional dendritic cells (cDC1) and the production of the chemokines CCL5 and XCL1, and that tumor-derived prostaglandin E2 (PGE2) interferes with this reciprocal stimulatory loop (11). In addition, early accumulation of IFN-γ-producing TINK promote the remodeling of the TME and unleash CTL-mediated tumor eradication, a circuit that is interfered by tumor-derived prostaglandin E2 (PGE2) acting via EP2 and EP4 receptors on NK cells (135). Moreover, only tumors responsive to ICI exhibited an inflammatory gene expression signature after ICI treatment accompanied by increased infiltration by NK cells that displayed an activated phenotype and produced IFN-γ, and that were critical for this response (136).
Moreover, additional mechanisms and mediators that affect NK cell recruitment, activation and display of optimal effector functions against solid tumors in the TME are known (137–141). TGF-β is a major negative regulator of NK cell effector function and tumor elimination ability (142–148). In LNT-229 glioma cells, it has been observed that an autocrine circuit that involves TGF-β and MMP production promotes shedding of MICA and ULBP-2 and negatively affects the expression of these NKG2DL on the tumor cell surface (149). TGF-β also negatively affects expression of NKp30 and NKG2D on human NK cells stimulated with IL-2 and NK cell-mediated cytotoxicity (150). TGF-β also impacts on NK cell metabolism, inducing a reduced glycolysis and oxidative phosphorylation that dampens NK cell effector functions (151). These effects can be mediated by different forms (soluble or membrane-bound) of TGF-β produced by regulatory T cells (152, 153), anti-inflammatory macrophages (44, 154) or by myeloid-derived suppressor cells (MDSC) (155). Consequently, it has been suggested that novel therapies that interfere with TGF-β may trigger NKG2D-dependent NK cell-mediated tumor elimination (143). Among them, bintrafusp alfa (M7824, a bifunctional fusion protein targeting TGF-beta and PD-L1) and galunisertib have shown promising results. Bintrafusp alfa has been demonstrated to revert TGF-β-mediated suppressive effects on NK cells and is currently explored in different clinical trials (156). Galunisertib, a SMAD2 inhibitor, has been shown to facilitate NK cell activation and effector function against neuroblastomas, and enhance ADCC by dinutuximab (157). Also, it has been observed recently that glioblastomas are infiltrated by NK cells that display an altered phenotype and impaired effector functions. Moreover, treatment of mice engrafted with glioblastoma stem cells (GSC) with allogeneic NK cells and galunisertib or Ly2019761 prevented the development of such dysfunctional NK cells and allowed a better control of tumor growth, indicating that TGF-β was responsible for this effect (158). These data highlight that the TME strongly affects NK cell effector functions and that they can be reinvigorated by therapeutic intervention. MDSC also produce indoleamine 2,3-dioxygenase (IDO), and enzyme that catabolizes tryptophan and participates in a metabolic pathway that generates kynurenine, and this pathway impairs NK cell effector functions (159, 160). Another soluble mediator that blunts NK cell effector function is PGE2, which is abundant in the TME of several tumors (11, 161–168).
Another factor that contributes to NK cell dysfunction is chronic stimulation. Mice with ubiquitous enforced expression of MICA that triggered chronic stimulation through NKG2D exhibited NK cells with reduced expression of NKG2D, and these NK cells exhibited an impaired NKG2D-dependent NK cell-mediated cytotoxicity and accelerated growth of a MICA-expressing melanoma (169, 170). In humans, it has been observed that chronic HCMV infection leads to an increased frequency of CD57+NKG2C+ peripheral blood NK cells (PBNK) that display rapid and robust effector function upon restimulation. However, their chronic stimulation induced high expression of the co-inhibitory receptors LAG-3 and PD-1, and these chronically stimulated NK cells were dysfunctional due to epigenetic reprograming and alterations in DNA methylation (171). Therefore, chronic stimulation of NK cells through several activating receptors, including NKG2D, leads to a functional impairment that can impact negatively on NK cell-mediated immunosurveillance.
TAM constitute major components of the TME and players of tumor immune escape. Strategies aimed at targeting TAM to promote their elimination or reprogramming may lead to better clinical outcome especially in patients with solid tumors (172). Interference with TAM-mediated immunosuppression targeting the scavenger receptor MARCO with a blocking mAb lead to decreased tumor vascularization, metabolic reprogramming of TAM, and an efficient activation of NK cell cytotoxic effector functions, and this effect also synergized with ICI (173). Therefore, targeting TAM can improve tumor immunity through restoration of NK cell activation and effector functions, further supporting the idea that strategies that convert “cold” tumors into “hot” tumors through manipulation of TAM and NK cells might constitute forefront strategies in I-O.
The possibility to phenotypically characterize dysfunctional TINK generated a renewed interest in targeting co-inhibitory receptors to reinvigorate NK cells in cancer patients using current or novel ICI (174). Targeting the PD-1/PD-L1 axis, CD96 (TACTILE), NKG2A, TIGIT, TIM-3 and LAG-3 have emerged as forefront alternatives because they are usually over-expressed in dysfunctional NK cells (33, 52, 175–179). Although the revolution in I-O achieved with ICI was originally attributed to an enhanced T cell-mediated antitumor response, increasing evidence demonstrates that NK cells also express PD-1 and PD-L1, and that they constitute targets of ICI that results in a reinvigoration of anti-tumor NK cell effector functions. ICI induced a CTL- and NK cell-mediated tumor growth control accompanied by a weakened suppressive immune cell infiltrate in the TME in a murine model of glioblastoma (180). Moreover, increased frequencies of PD-1+ NK cells with heightened expression of PD-1 were observed in PBNK and TINK from patients with several gastrointestinal tumors, and higher expression correlated with impaired survival in some cases. These NK cells exhibited impaired IFN-γ production and degranulation, after in vitro exposure to ICI led to a functional reinvigoration, and in vivo treatment with ICI of nude mice xenografted with a human esophageal squamous cell carcinoma caused NK cell-dependent delayed tumor growth (181). TINK from transplantable, spontaneous, and genetically induced tumors also contained a high frequency of PD-1+ NK cells and suppressed IFN-γ production and cytotoxicity in vitro but ICI treatment resulted in a restoration of NK cell response that was essential for the therapeutic effect ICI (182). ICI also exert effects beyond their blocking activity as an anti-PD-L1 mAb such as avelumab, but no atezolizumab, can trigger ADCC by PBNK from healthy donors against triple negative breast cancer (TNBC) tumor cell lines in vitro (183). Also, ADCC triggered by anti-PD-L1 mAb against multiple carcinoma cell lines could be enhanced by histone deacetylase inhibitors (HDACi) because they augmented the expression of PD-L1, NKG2DL and other NK cell activating ligands and death receptors on target cells (184). Chemotherapeutic agents can increase the susceptibility of nasopharyngeal carcinoma cell lines to NK cell-mediated cytotoxicity, and such effect was enhanced by an anti-PD-1 blocking mAb because the chemotherapeutic agents also stimulated up-regulation of PD-1 on NK cells and PD-L1 on the target cell lines (185). In head and neck cancer patients, PBNK contain a higher frequency of PD-1+ cells, and ICI treatment with an anti-PD-1 mAb promotes heightened cetuximab-mediated NK cell activation (186). In non−small cell lung cancer patients, TINK exhibit functional defects accompanied by a higher frequency of PD-1+ cells, while in vitro treatment with ICI reverted such NK cell dysfunction (187).
Targeting other co-inhibitory receptors on NK cells is also under investigation. TIGIT is a co-inhibitory receptor that emerged as ICI candidate because it is over-expressed on exhausted TINK and tumor-infiltrating T cells and is responsible for such functional exhaustion. Blockade of TIGIT induced a NK cell reinvigoration, restored an efficient tumor immunity, and also enhanced the efficacy of therapy with ICI against PD-L1 (188). CD96 is another co-inhibitory receptor expressed on NK cells that, through binding to CD155 expressed on tumor cells, limits NK cell effector functions (189). Patients with hepatocellular carcinoma that present reduced disease-free survival have dysfunctional (exhausted) TINK with a higher frequency of CD96+ cells and increased expression of CD96, but blockade of CD96 restores NK cell-mediated effector functions (190). NKG2A is another inhibitory receptor that associates with CD94 and negatively regulates NK cell functions. High expression of NKG2A and of its ligand HLA-E can be detected in tumor tissue of hepatocellular carcinoma patients, and NKG2A-expressing TINK exhibit features of exhausted cells and are associated with a poor prognosis (191). NKG2A and HLA-E are overexpressed in several other human cancers including head and neck, colorectal, ovarian, endometrial and cervical cancers. Blocking NKG2A with Monalizumab (a humanized anti-NKG2A mAb) enhances tumor immunity in combination with ICI against PD-L1 by promoting NK cell and CTL cell effector functions in mice and humans and enhances ADCC in combination with Cetuximab against a head and neck carcinoma cell line, indicating that its mechanism of action directly impacts on NK cell effector functions (192–194). Also, patients with chronic lymphocytic leukemia (CLL) exhibit an upregulation of LAG-3 on leukemic blasts, NK cells and T lymphocytes, accompanied by high amounts of soluble LAG-3 (sLAG-3) in plasma, that correlated with impaired outcome. However, in vitro exposure of peripheral blood mononuclear cells to relatlimab, an anti-LAG-3 blocking mAb under evaluation in several clinical trials, depleted leukemic cells and restored NK cell- and T cell-effector functions (179). Conversely, the use of a blocking mAb against the inhibitory receptors KIR2DL1, KIR2DL2 and KIR2DL3 named Lirilumab (195, 196) did not show the expected clinical efficacy (197, 198), indicating that target co-inhibitory receptors need to be carefully selected.
Altogether, the described evidences demonstrate that NK cells are dysfunctional in cancer patients and that their reinvigoration appears as feasible, and these findings are motorizing the development of novel ICI aimed at leveraging NK cell mediated effector functions (177). However, targeting co-inhibitory receptors has a dark side as treatment with ICI can trigger many cytokine-mediated immune-related adverse events (irAE) that can be severe and require interruption of ICI treatment and/or complementary treatment with other compounds (199–203). Moreover, the description of patients with accelerated tumor growth after treatment with ICI (hyperprogression) due to ill-defined mechanisms is an additional concern (5–7). These drawbacks are further complicated by the lack of validated predictive biomarkers that would permit the selection of patients that optimally respond to ICI with minimal or no irAE and no hyperprogression (204, 205). Therefore, the development of novel ICI against additional target molecules requires a deep risk mitigation to avoid irAE and hyperprogression.
ADCC is one of the major NK cell-mediated effector functions and therapeutic efficacy of several mAb currently used to treat cancer patients depend on their ability to induce ADCC (48, 183, 206–215). Therefore, selection of appropriate molecular targets expressed by tumor cells is a crucial step towards the development of successful immunotherapies to treat cancer patients. As MICA is the NKG2DL more widely overexpressed in tumors, it is reasonable to consider MICA as a frontrunner candidate as target molecule for I-O strategies. A seminal finding about MICA as target in I-O came from the observation that some melanoma patients that received ipilimumab (an anti-CTLA4 mAb) and autologous tumor cells engineered to produce GM-CSF, spontaneously developed anti-MICA Ab that promoted clearing of sMICA from plasma and opsonization of tumor cells for dendritic cell cross-presentation. These effects were associated with a restoration of the expression of NKG2D on NK cells and CTL, a recovery of NKG2D-dependent NK cell effector functions and a better outcome (216, 217). Later, it was observed that MM patients, in contrast to patients with monoclonal gammopathy of undetermined significance (MGUS), also exhibit high titers of anti-MICA Ab that antagonize with the suppressive effects of sMICA and stimulate dendritic cell cross-presentation of malignant plasma cells (108). The serendipitous appearance of anti-MICA Ab with a therapeutic effect prompted us to develop a strategy to actively induce such Ab. To this end, we generated a chimeric protein consisting of the ectodomain of MICA fused to a bacterial immunogenic protein that exhibits adjuvant properties (Brucella lumazine synthase, BLS), used this chimeric protein (named BLS-MICA) for the induction of anti-MICA Ab in tumor-bearing hosts, and investigated their therapeutic activity and mechanism of action. BLS-MICA elicited high titers of anti-MICA Ab in mice that in vitro recognized MICA naturally expressed on the cell surface of human tumor cells and on MICA-transduced mouse tumor cells. Prophylactic active immunization with BLS-MICA significantly delayed the growth of MICA-expressing tumors in part due to the ability to promote scavenging of sMICA from mouse sera. Passive immunization experiments demonstrated that such effect was mediated by anti-MICA Ab that mediated in vitro and in vivo ADCC, and that tilted the balance of tumor-infiltrating cells towards an anti-tumoral/pro-inflammatory phenotype characterized by an increased presence of TAM with an M1-skewed phenotype and antigen-experienced CTL (111). Therefore, immunization with BLS-MICA induced therapeutic anti-MICA Ab that constitute a “two-in-one” strategy as they promote tumor elimination by ADCC and interfere with a tumor immune escape through scavenging of sMICA.
Targeting the NKG2D pathway has also been approached using different chimeric proteins where the ectodomain of MICA was fused to single chain Fv (scFv) against other molecules that promoted NK cell-mediated anti-tumor effects (218–220). Although these approaches have shown some preliminary interesting effects, compared to anti-MICA Ab-based strategies, they only promote the bridging between NK cells and tumor cells and do not target, for example, suppressive sMICA, which limits their competitive landscape.
In addition, several anti-MIC mAb also are showing promising preclinical results. A mAb that targets sMICA (B10G5) demonstrated therapeutic effects, alone or combined with an anti-CTLA-4 mAb in mice challenged with MIC transgenic TRAMP (transgenic adenocarcinoma of the mouse prostate) cells. The mechanism of action of this mAb involves ADCC that results in a revitalization of CTL- and Th1-mediated anti-tumor immunity, and remodeling of the TME (221, 222). Other anti-MICA/B mAb that target the α3 domain and inhibit proteolytic shedding were generated. One of these mAb (7C6) inhibited MICA and MICB shedding by human cancer cells, delayed the growth of mouse melanoma and colon carcinoma engineered to express MICA, reduced human melanoma metastases in a humanized mouse model and its mechanism of action involves the stimulation of NKG2D- and CD16-dependent NK cell-mediated effector functions (223). Also, 7C6 acts synergistically with the HDACi panobinostat through stabilizing tumor cell surface MICA/B expression (224) and exhibits synergy with human cytokine-induced NK cells (CIK) in vitro (225). A different humanized anti-MICA/B mAb stimulates NK cell-mediated cytotoxicity in vitro against primary hepatocellular carcinoma cells (226). In addition, three novel mAb (5E10, 7G10 and 6E1) that recognize the α3 domain of MICA interfere with the immunosuppressive activity of sMICA on human NK cells and stimulate their activation in an Fc-dependent manner due to the formation of immune complexes with sMICA (119). Additionally, the therapeutic efficacy of mAb-mediated neutralization of sMICA (222) or MICA shedding (223) has been shown to negatively affect tumor growth in mouse models.
Accordingly, Ab that target MICA/B should trigger ADCC against MICA/B-expressing tumor cells to stimulate tumor cell elimination and, simultaneously, promote the formation of immune complexes with sMICA/B to facilitate their clearance by macrophages and interfere with the tumor immune escape mechanism mediated by sMICA/B. Such Ab will likely display therapeutic activity in patients with tumors that express MICA/B on their cell surface, tumors that shed significant amounts of sMICA or both. This therapeutic opportunity for anti-MICA/B Ab is schematically represented in Figure 2. Currently, we cannot anticipate if Ab-based therapies against MICA/B may trigger antigen down-modulation on tumor cells and/or the selection of resistant tumor cells that lost expression of NKG2DL because of a selective killing of tumor cells that express the target molecule. We also cannot anticipate if Ab against NKG2DL promote off target and side/unwanted effects in humans. Also, currently there is not a good estimate about the relative relevance of tumor cell surface-expressed MICA/B vs sMICA/B during tumor immunity. The relative expression of MICA/B on the tumor cell surface and the shedding of sMICA/B will probably vary from tumor to tumor and be associated with the ability of each tumor to produce sMICA/B (either by the action of MMP or released in exosomes) and to sustain cell surface MICA/B expression in an immunocompetent host (to resist NK cell effector functions). Therefore, targeting both forms of MICA/B (soluble or cell surface-expressed) with Ab-based strategies emerges as a “two-in-one” strategy that can boost ADCC (immunosurveillance) and that can interfere with the immunosuppressive effect of sMICA/B (tumor immune escape).
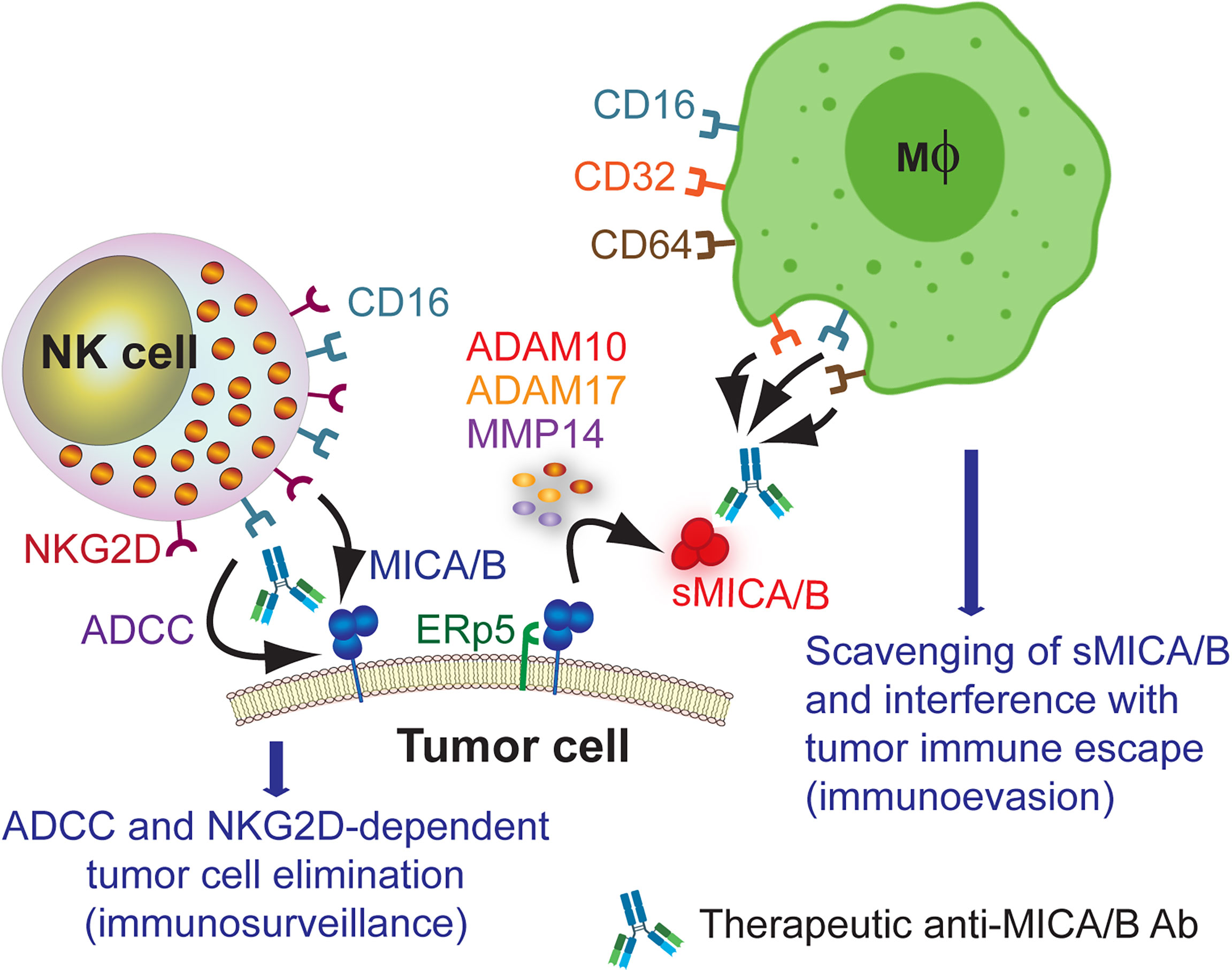
Figure 2 Therapeutic opportunity for anti-MICA/B Ab. Administration of anti-MICA/B Ab may trigger CD16-dependent ADCC by NK cells when these Ab recognize cell surface-expressed MICA/B, contributing to tumor cell elimination. Ab that do not interfere with the binding of NKG2D to MICA/B also would trigger NKG2D-dependent NK cell-mediated cytotoxicity, further contributing to tumor cell elimination. Moreover, recognition of sMICA/B by these therapeutic anti-MICA/B Ab would lead to the formation of immune complexes that would be removed by macrophages upon recognition through CD16, CD32 and CD64. This scavenging of sMICA/B will consequently interfere with tumor immune escape (immunoevasion).
Targeting NKG2DL has also been addressed using chimeric antigen receptor (CAR) T cells (CAR-T) or NK cells (CAR-NK). The similarities and differences in using CAR-T or CAR-NK cells have been reviewed recently and one aspect that stands out is that CAR-NK cells appear superior to CAR-T cells to treat hematological malignancies (30). Notably, a big success represents the use of CAR-NK cells directed against CD19, as they induce tumor regression without the development of major toxic effects in the majority of patients with relapsed or refractory CD19-positive leukemias or lymphomas (24). However, treatment of solid tumors with CAR-T or CAR-NK cells has not yielded the expected results, mainly because they must face the immunosuppressive TME (29). Some CAR-T cells engineered to express NKG2D with increased effector functions against cell lines in vitro and against xenografted human or syngeneic mouse tumor cell lines in vivo have been developed (227–238). Also, CAR-NK cells that express the ectodomain of human NKG2D fused with DAP12 exhibit augmented NK cell-mediated cytotoxicity against several solid tumor cell lines in vitro and mediate a therapeutic effect in vivo in NSG mice challenged with human tumor cell lines. Moreover, in three human patients with colorectal cancer, these CAR-NK cells mediated tumor regression and patient improvement, highlighting their potential therapeutic utility in solid tumors (239). Although clinical trials for CAR-T and CAR-NK are underway. we don´t know their outcome yet (details can be found at clinicaltrials.gov searching for “CAR-T cells” or “CAR-NK” cells and “NKG2D”). Nevertheless, problems and disadvantages of the CAR-T or the CAR-NK cell approaches are the necessity of promoting the correct migration of the injected cells to the tumors, the long-term persistence of these cells with preserved anti-tumor effector functions, the risk of inducing off target effects, the elevated cost of these alternatives, and the fact that they do not target sMICA/B and consequently, their biological activity may also become vanished in patients with high sMICA/B. In addition, CAR-T cells have the risk of inducing CRS. Therefore, approaches that target the NKG2D-NKG2DL axis, especially MICA/B, with Ab appear superior or with better chance to move to the clinic.
The current trend towards the use of combination therapies with ICI (240) certainly creates a new landscape to capitalize the therapeutic utility of anti-MICA/B Ab through combination with other agents. Leveraging MICA/B in I-O represents a promising alternative, but these strategies must face the formidable task of having to overcome the suppressive TME including the negative regulatory circuits imposed by TAM. Among recently emerged combination therapies to treat cancer patients, small molecules are taking the center of the stage (241). Several therapies targeting selected molecules and/or mechanisms can be envisaged to be rationally combined with Ab against MICA/B and, consequently, leverage the therapeutic effect of these anti-MICA/B Ab. These strategies would convert “cold” tumors into “hot” tumors through manipulation of NK cell effector functions, the TME (242) and/or TAM (243). In addition, administration of IL-15 leads to huge NK cell expansion in human patients, a fact that might exploit the immunotherapeutic potential of NK cells further (244). Promising candidates that could be combined with anti-MICA/B Ab are described in the following sub-sections.
NKG2DL expression is controlled by the DNA damage response pathway (DDR) (245), but epigenetic remodelers such as HDACi also triggered upregulated expression of MICA/B (246, 247). Some HDACi such as Trichostatin A (TSA), suberanilohydroxamic acid (SAHA or vorinostat), PXD101 (belinostat), LBH589 (panobinostat) and LAQ824 (dacinostat) are broad spectrum HDACi; others such as valproic acid (VPA), sodium butyrate (NaB), trapoxin and apicidin exhibit specificity for certain groups of HDAC; and others such as MS-275 (entinostat) and FR901228 (romidepsin, depsipeptide or FK228) exhibit high specificity for certain HDAC (248, 249). HDACi exert antiproliferative effects through the induction of cell-cycle arrest, apoptosis, and autophagy, but it has been observed that TSA (247, 250–253), SAHA (247, 254–257), belinostat (247), VPA (246, 251, 252, 254, 258–270), NaB (251, 252, 255, 257, 258, 261, 271), romidepsin (247) and entinostat (255, 257, 269) trigger up-regulation of MICA/B in different cell lines derived from liquid and solid tumors. In most cases, it was demonstrated that HDACi induce upregulation of MICA/B accompanied by a higher NKG2D-dependent, NK cell-mediated cytotoxicity against HDACi-treated tumor cells (246, 247, 250, 251, 253, 255, 257–259, 261–266, 271). In other cases, NKG2D-dependent, CD8 T cell-mediated (260) or γδ T cell-mediated cytotoxicity (270) against HDACi-treated tumor cells was observed. Cooperative effect between HDACi and other compounds such as hydroxyurea (263), gemcitabine (268), All-Trans-Retinoic Acid (ATRA) (260) to promote increased expression of MICA/B was also observed. Moreover, impaired NK cell recognition of vemurafenib-treated BRAFV600E mutant melanoma cells is due to a drug-induced down-regulation of MICA and CD155, while treatment with NaB promotes a recovered surface expression of MICA and NK cell degranulation (272). However, most studies were performed in vitro, and only tumor cells were exposed to HDACi. Studies in xenografted immunodeficient mice injected with human tumor cell lines demonstrated that treatment with TSA or VPA and HDACi-mediated upregulation of MICA/B resulted in a delayed tumor growth when they were adoptively treated with cytokine-induced killer cells (251) or with NK-92 cells (266). Moreover, chronic exposure to vorinostat and HDACi-mediated upregulation of MICA/B resulted in a reduced tumorigenic capacity of human colon adenocarcinoma cells xenografted in nude mice (256). Besides, several deleterious effects of HDACi on NK cells were described, such as downregulation of activating receptors and a negative impact on NK cell effector functions (252, 254, 267, 269). Therefore, a delicate selection of HDACi with selective specificity for certain HDAC may lead to an up-regulation of MICA/B without compromising NK cell´s ability to detect tumor cells and mobilize effector functions. Accordingly, VPA down-regulated NKG2D expression and NK cell degranulation but entinostat, which is highly selective for class I HDAC, induced an up-regulated expression of NKG2D and increased NK cell degranulation (269). Moreover, apicidin has been shown to promote the upregulation of ADAM10, one of the MMP involved in MICA/B shedding (273). Therefore, HDACi may induce heightened expression of MICA/B but some of them also increase the amount of sMICA/B. Overall, integrating data about the effect of HDACi on MICA/B expression, on NKG2D expression and on NK cell effector functions, and taking into consideration the few HDACi approved for the treatment of human patients, vorinostsat, belinostat and entinostat emerge as frontrunners to be used to induced heightened expression of MICA/B and sensitize tumor cells to anti-MICA/B Ab.
Upregulation of MICA/B expression can also be achieved with Bortezomib, a proteasome inhibitor that is used to treat patients with MM. MM results from the progression to malignancy of MGUS and MICA expression on malignant plasma cells is higher in MGUS than in MM. However, MM, but not MGUS patients exhibit high sMICA, which is associated with lower expression of NKG2D and NK cell dysfunction in MM patients. Bortezomib promoted upregulated expression of MICA in some MM cells and enhanced the therapeutic effect of anti-MICA Ab that these patients generated spontaneously. Therefore, Bortezomib can cooperate with anti-MICA Ab to exert a therapeutic effect (108). Increased expression of MICA upon in vitro or ex vivo exposure of MM cells to Bortezomib, accompanied by a subsequent stimulation of NK cell effector functions was also observed by other authors (274, 275). The effect of Bortezomib on MICA expression was also detected on melanoma cell lines (100) and B cell acute lymphocytic leukemias (265), while this drug also promoted the upregulation of MICB on human lung cancer, hepatoma and melanoma cell lines, resulting in an improved NKG2D-dependent NK cell-mediated cytotoxicity (276, 277).
Other drugs used to treat patients with cancer such as sorafenib and sunitinib (278) and 5-fluorouracil (279) affect MICA/B expression. Also, doxorubicin and melphalan, two drugs used to treat patients with MM, induced the expression of MICA/B on cell lines and patient-derived plasmablasts through activation of the DDR pathway, and this effect stimulated heightened NK cell degranulation (274). Another drug that induced MICA/B expression is temozolomide (TMZ), a drug used in some patients with glioblastomas. TMZ upregulated the expression of MICA/B in vitro and in vivo in murine and human glioblastoma models, and this effect facilitated a NGK2D-dependent elimination of the glioblastoma cells (280).
In summary, several drugs currently used to treat patients with different types of cancer might stimulate upregulation of MICA/B which may lead to a subsequent increased sensitivity of tumor cells to the therapeutic effects mediated by anti-MICA/B Ab (ADCC through CD16) and to NK cells through NKG2D. The search for assets that promote upregulated expression of MICA is particularly relevant because tumors evolve under immunological pressure of the host, and such pressure may promote the emergence of tumors with low cell surface expression of MICA/B. Also, MICA/MICB exhibit a very low mutational burden (https://portal.gdc.cancer.gov/), indicating that drug-induced enforced MICA/B expression on tumor cells and targeting with Ab would hardly face the problem of the emergence of escape variants. These effects of DDR inducers, HDACi and proteasome inhibitors on MICA/B expression and their impact on the efficacy of anti-MICA/B Ab are schematically depicted in Figure 3 and several candidate drugs that are approved, in phase II or late-stage clinical are listed in Table 1.

Figure 3 Leveraging anti-MICA/B Ab therapeutic efficacy through combination therapies with DDR inducers, HDACi and proteasome inhibitors. The use of DDR inducers, HDACi and proteasome inhibitors may lead to an increased MICA/B synthesis, reduced degradation, and its consequent accumulation on the cell surface. This effect may result in an improved CD16-dependent ADCC of anti-MICA/B Ab, and a recovery of NKG2D-dependent NK cell-mediated cytotoxicity against tumor cells, facilitating the reinstatement of an efficient tumor cell elimination.
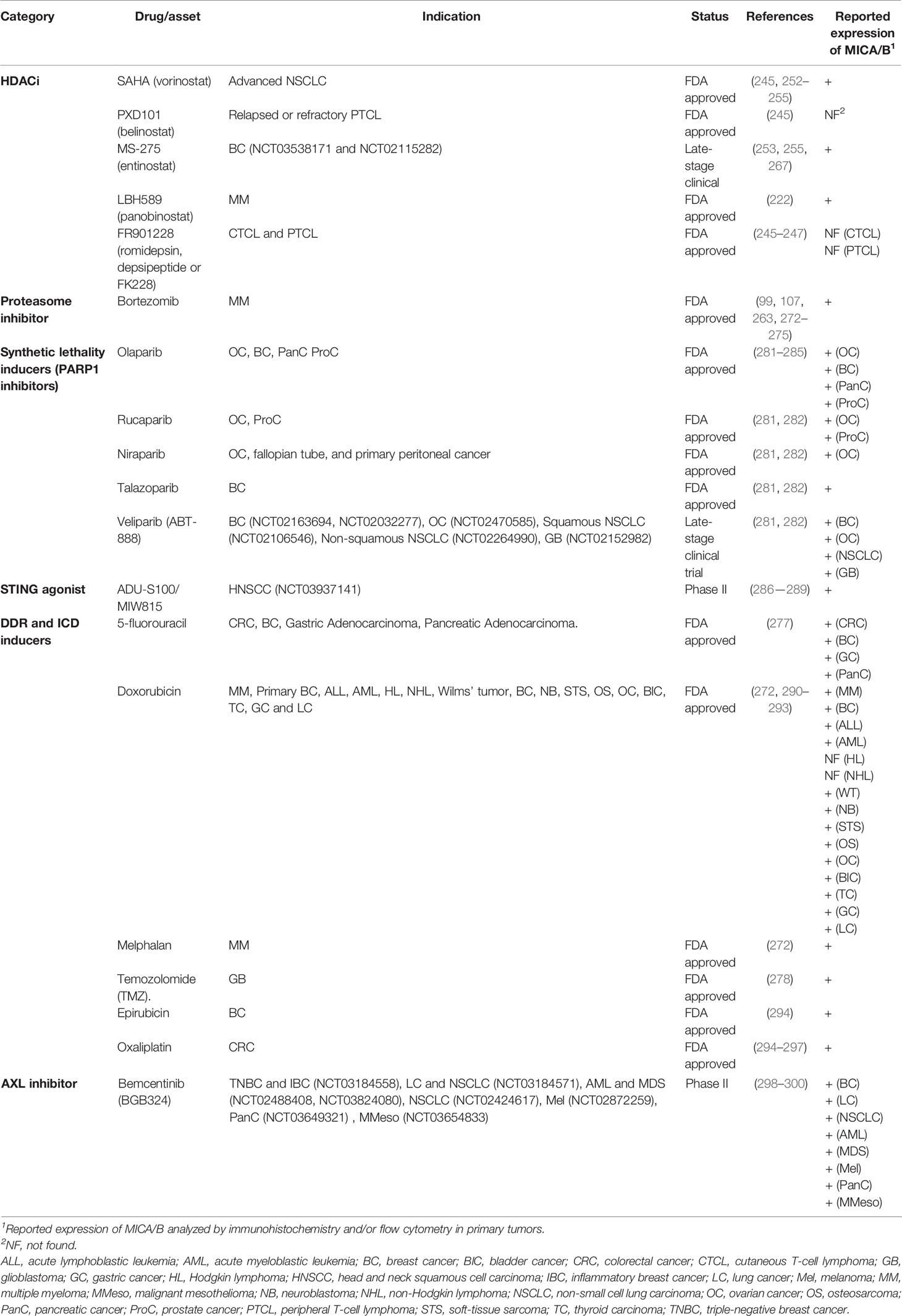
Table 1 Approved, phase II and late-stage clinical drugs that might be combined with anti-MICA/B Ab.
Shedding of MICA/B depends on proteolytic cleavage mediated by several MMP such as ADAM10, ADAM17 and MMP14 (117, 301, 302) or secretion in exosomes (118, 301). Therefore, blocking the shedding of MICA/B would interfere with the tumor immune escape driven by these soluble forms of MICA/B, resulting also in a restoration of cell surface expression of MICA/B and improved NKG2D-dependent NK cell effector functions (251, 279, 303). Increased cell surface expression of MICA/B would also likely contribute to a better responsiveness to anti-MICA/B Ab and enhanced ADCC. Therefore, MMP inhibitors (MMPI) constitute attractive candidates, which is further supported by the finding that mouse prostate tumors engineered to express a shedding-resistant noncleavable MICB did not grow when implanted into SCID mice but treatment of the animals with an NKG2D blocking mAb led to the development of tumors (304). Several MMPI such as MMPI-I (116, 305), MMPI-II (117), MMPI III (117, 271), MMPI-IV (306), batimastat or BB94 (117, 307), GW280264X (117, 301), GI1254023X (117), ilomastat or GM6001 (117, 301, 308), URB597 (309), periostat or doxycycline (310) and some ADAM-10 selective inhibitors (311, 312) can inhibit MICA/B shedding in vitro, resulting in a heightened cell surface expression of the NKG2DL and an increase in NK cell-mediated cytotoxicity. However, among these compounds, only periostat is currently in clinical trials for different types of tumors (313). An alternative approach to inhibit MICA/B shedding is the use of the 7C6 mAb (223) and the 6E1 mAb (119), both of which recognize the α3 domain of MICA/B, inhibit their proteolytic shedding, and lead to increased cell surface expression of MICA/B. Moreover, the B10G5 mAb inhibited the shedding of MICB but its effect on cell surface expression of MICB remains unknown (281, 282). Overall, pharmacologic or mAb-mediated inhibition of MICA/B shedding constitutes another opportunity to combine with anti-MICA/B Ab to induce improved ADCC and tumor cell elimination, as depicted schematically in Figure 4 and mentioned in Table 1.
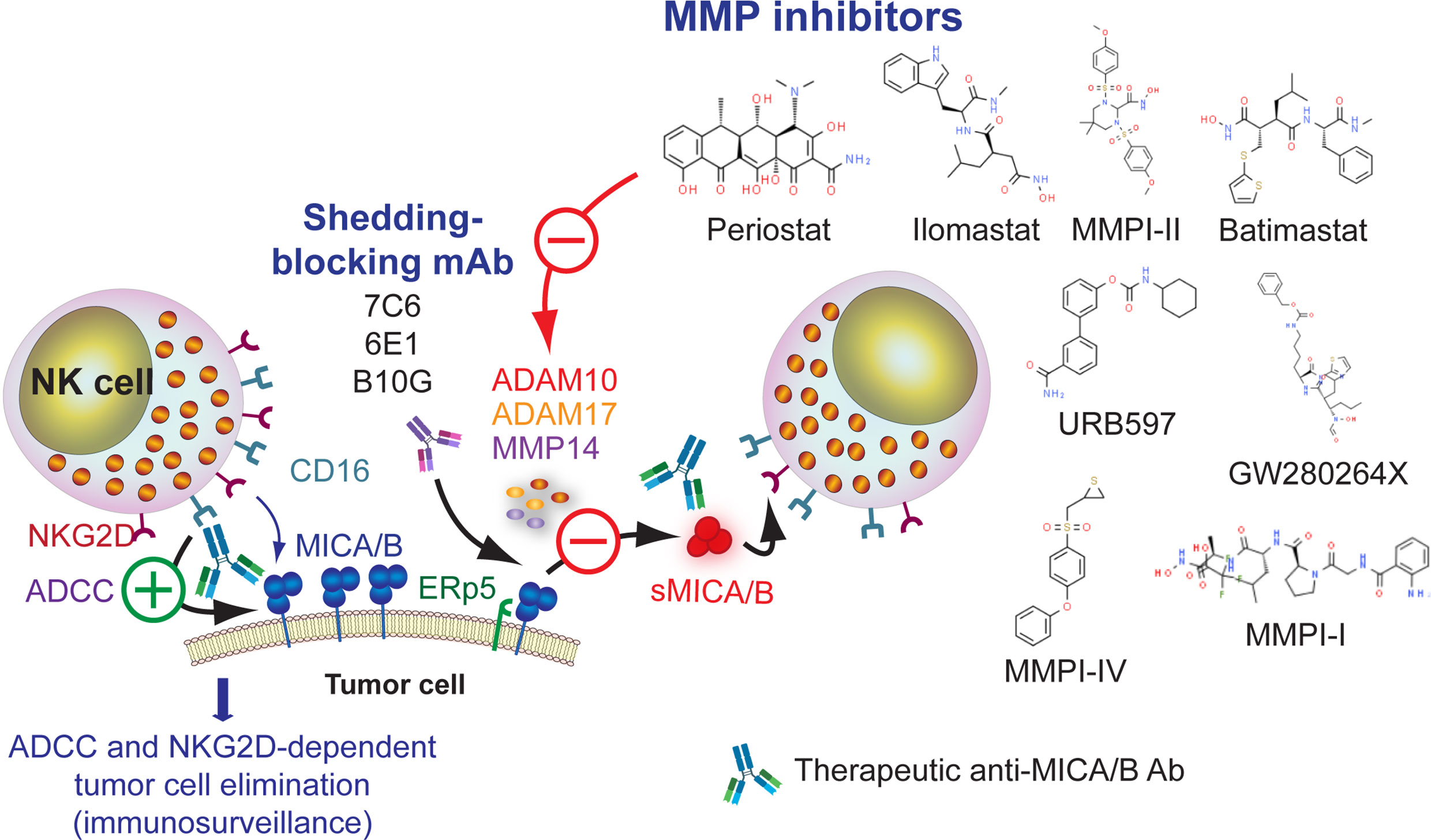
Figure 4 Leveraging anti-MICA/B Ab therapeutic efficacy through combination therapies with pharmacologic inhibition of MMP or with mAb that block MICA/B shedding. Inhibition of MMP with small molecules (detailed in the figure) may prevent MICA/B shedding and lead to a subsequent accumulation of MICA/B on the cell surface. A similar effect can be achieved using mAb that interfere with MICA/B shedding. In both cases, inhibition of MICA/B shedding may lead to an improved CD16-dependent ADCC of anti-MICA/B Ab, and a recovery of NKG2D-dependent NK cell-mediated cytotoxicity against tumor cells.
Synthetic lethality inducers such as olaparib, rucaparib, niraparib, talazoparib, veliparib and others exert a therapeutic effect due to their ability to inhibit PARP1, a key enzyme in DNA repair and the preservation of genome integrity (283, 314). However, PARP1 inhibitors also can foster tumor immunity because they activate the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon (IFN) genes (STING) pathway in tumor cells (315, 316). Thus, there are many efforts underway to explore the combination of PARP1 inhibitors with ICI in cancer patients (284, 317–320). PARP1 inhibition has also been demonstrated to affect the NKG2D/NKG2DL axis because PARP1 is involved in the repression of NKG2DL (mainly MICA and MICB) in LSC in patients with AML. Pre-treatment of AML cells with the PARP1 inhibitor AG-14361 resulted in a reduced leukemogenic activity in NSG mice, and administration of AG-14361 to NSG mice xenografted with human AML followed by administration of human NK cells, inhibited leukemogenesis in an NKG2D-dependen manner (133). Therefore, inhibition of PARP1 unleashes NKG2DL expression on AML cells that in turn become more susceptible to NKG2D-dependent, NK cell-mediated effector functions and efficient tumor cell elimination. PARP1 inhibition with olaparib also increases dendritic cell (DC) activation and CTL infiltration in mouse BRCA1-deficient ovarian tumors (315) and in triple negative breast cancer cells (285) through activation of cGAS–STING in tumor cells. Combination of PARP inhibition and CSF-1R blockade enhanced anti-tumor immunity and prolonged survival of BRCA-deficient tumors in vivo, indicating that PARP inhibition affects TAM suppressive activity in the TME (321). In addition, combination of cetuximab (anti-EGFR) or avelumab (anti-PD-L1) with olaparib demonstrated that PARP1 inhibition fosters NK cell-mediated ADCC (322). Overall, these results provide a solid rationale for the development of combination therapies between PARP1 inhibitors and Ab that target MICA/B, as depicted schematically in Figure 5 and several candidate drugs that are approved, in phase II or late-stage clinical are listed in Table 1.
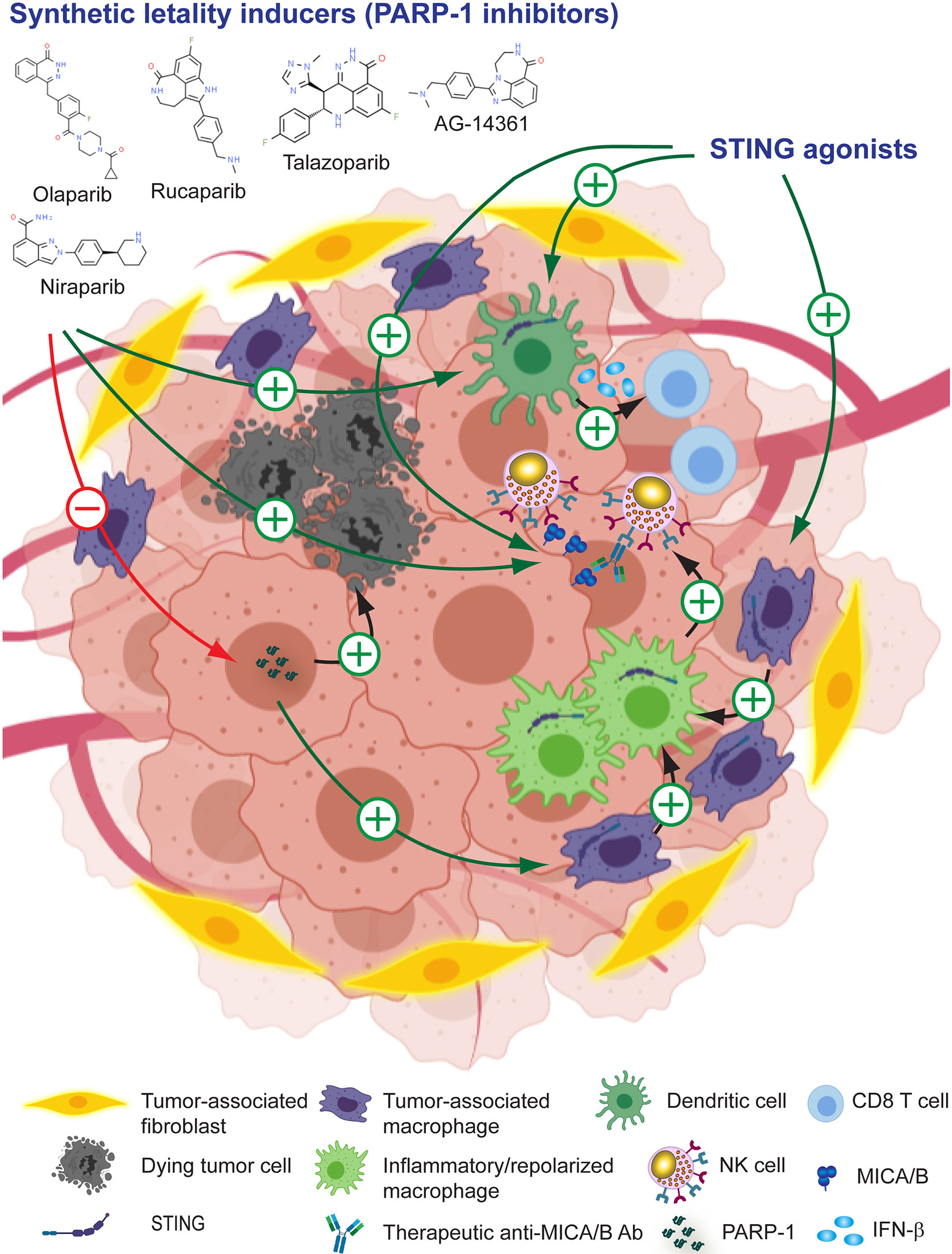
Figure 5 Leveraging anti-MICA/B Ab therapeutic efficacy through combination therapies with pharmacologic PARP1 inhibitors or STING agonists. The use of PARP1 inhibitors can promote tumor cell death and unleash the activation of STING. Immunogenic cell death and STING activation induce the remodeling of the TME, resulting in a heightened production of IFN-β by DC and CTL-mediated tumor eradication, and a reprogramming of TAM into pro-inflammatory macrophages. These pro-inflammatory macrophages, instead of inhibiting NK cells, might now promote efficient NK cell effector functions. In addition, PARP1 inhibition and STING activation might promote increased expression of MICA/B, resulting in an improved CD16-dependent ADCC of anti-MICA/B Ab, and a recovery of NKG2D-dependent NK cell-mediated cytotoxicity against tumor cells. Together, these effects may contribute to foster an efficient tumor cell elimination.
Downstream effects of PARP1 inhibition involves the activation of the cGAS-STING pathway (285, 315), and STING enhances NK cell activation and tumor immunity through the production of type I IFN (323–326). Thus, agonists of STING may reconfigure the TME into a proinflammatory milieu and promote the conversion of “cold” tumors into “hot” tumors in part due to an effect on TAM repolarization (327–330). STING activation also cooperates with ICI to foster antitumor immunity (331–333) via TAM reprogramming (333). Also, STING agonists can stimulate NK cell-mediated clearance of CD8 T cell-resistant tumors (325) and mobilize tumor-infiltrating myeloid cells to produce IFN-β which in turn activates NK cells (286). In addition, the HDACi entinostat triggers up-regulated expression of MICA/B on tumor cells (255, 257, 269) and activation of STING (334). Therefore, its therapeutic effect could be due to a remodeling of the TME due to a STING-mediated proinflammatory response with an associated repolarization of TAM, and the induction of the expression of MICA/B that would promote improved NKG2D-dependent NK cell effector functions and ADCC mediated by anti-MICA/B Ab. Several available STING agonists such as cyclic dinucleotides (CDN) and their derivates (2´,3´cGAMP, 3´,3´cGAMP, cAIM-derived CDN, cGAMP-derived and others) are currently being tested as monotherapy or in combination with other assets (335). Additional promising STING agonists with antitumor activity have been developed, one of which can be administered orally (287, 336). Also, as the activation of cGAS/STING pathway induces the up-regulation of mouse NKG2DL (289), STING emerges as another attractive molecular target in I-O to leverage NKG2D-dependent NK cell-mediated anti-tumor effects, and to be combined with Ab against MICA/B to manipulate the TME and catalyze tumor immunity, as depicted schematically in Figure 5 and mentioned in Table 1.
Drugs that induce ICD such as anthracyclines (doxorubicin, epirubicin, oxaliplatin and others) can trigger an effective antitumor immune response that suppresses tumor growth in mice because they make tumor cells immunogenic. This process involves the mobilization of calreticulin (CRT) to the tumor cell surface, the secretion and extracellular accumulation of ATP and several alarmins such as HMGB1 and annexin A1, and the production of type I IFN which, acting together, contribute to the inflammatory response that remodels the TME, suppress TAM and negatively affect tumor growth (290–293, 295, 337–339). ICD can also be induced by other immunomodulatory compounds such as agonists of RIG-I (340, 341) or STING (288, 342) because they promote the release or expression of several DAMP by tumor cells. ICD treatment of tumors not only turns them intrinsically more immunogenic. Oxaliplatin also promotes the production of T-cell-recruiting chemokines by TAM, resulting in superior CAR-T cell infiltration, remodeling of the TME, and an improved response to ICI (296). Accordingly, many clinical trials currently explore ICD-triggering compounds, alone or combined with ICI, to foster tumor immunity through the stimulation of the immunogenicity of dying tumor cells (294). Moreover, oxaliplatin itself stimulates the upregulation of MICA/B on cancer cells and susceptibility to NK cell-mediated cytotoxicity (297). Therefore, ICD inducers might also be combined with anti-MICA/B Ab to enhance ADCC against MICA/B expressed on tumor cells mediated by NK cells and to prevent their exhaustion through an effect that involves the remodeling of the TME with an associated repolarization of TAM. Moreover, NK cells can themselves promote ICD of tumor cells (343), ICD and NK cell-mediated tumor elimination might be therapeutically amplified to promote a self-perpetuation of the antitumor immune response. For example, CRT exposure on AML blasts is associated with better NK cell-mediated cytotoxicity, and this effect depends on CD11c+CD14high cells that become better “helpers” for NK cell activation upon exposure to CRT (337). These concepts indicate that NK cells, through the induction of ICD of tumor cells, can spark-ignite a whole TME remodeling that involves TAM reprograming. Therefore, strategies that potentiate NK ICD and NK cell effector functions such as the use of ICD inducers combined with anti-MICA/B Ab appear as promising alternatives to reinstate tumor immunity, as depicted schematically in Figure 6 and several candidate drugs that are approved, in phase II or late-stage clinical are listed in Table 1.
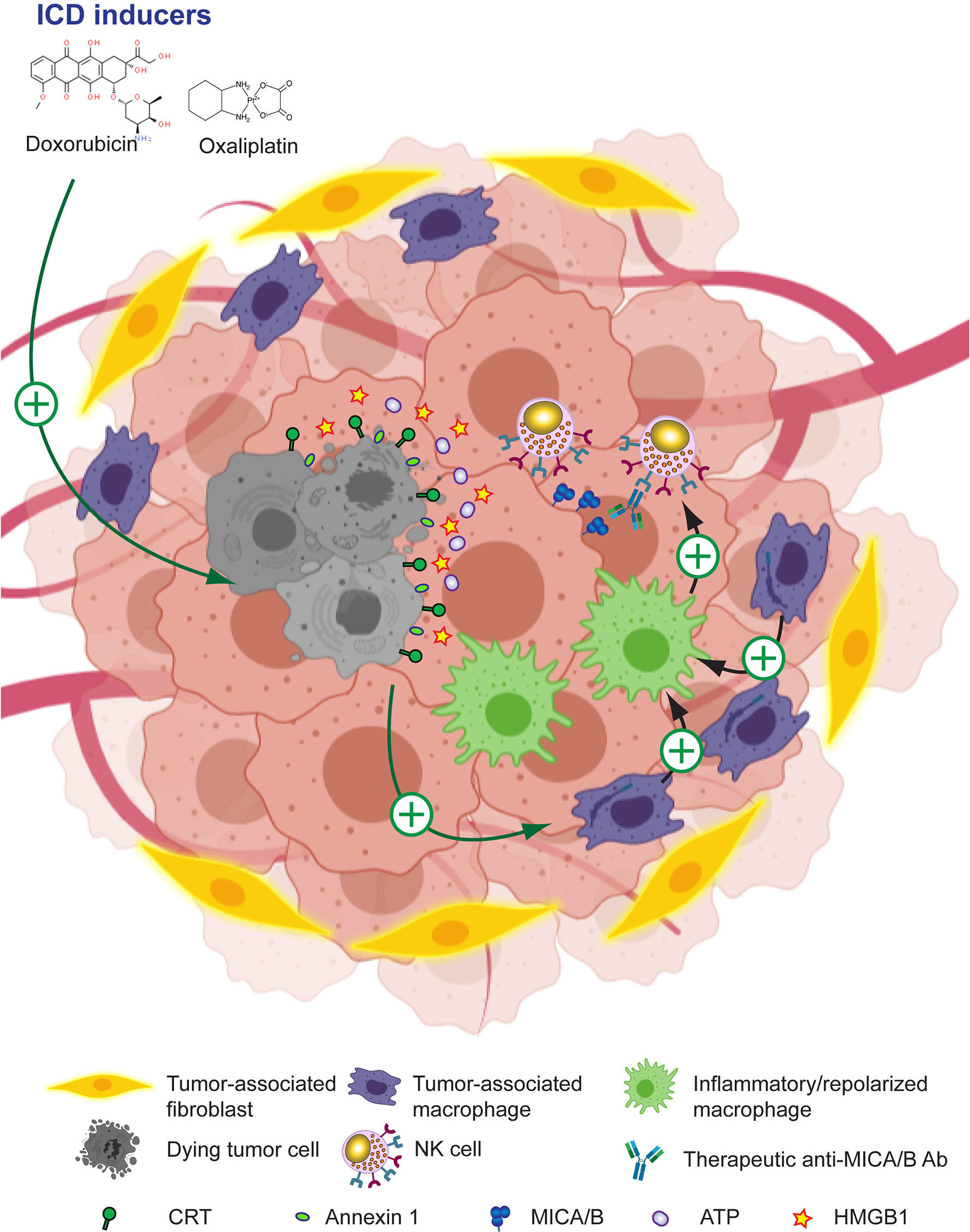
Figure 6 Leveraging anti-MICA/B Ab therapeutic efficacy through combination therapies with ICD inducers. The use of ICD inducers can promote immunogenic tumor cell death with the subsequent expression of CRT and annexin 1 on the tumor cell surface, and the secretion of ATP and alarmins such as HMGB1. Together, these effects result in the remodeling of the TME and a reprogramming of TAM into pro-inflammatory macrophages. These pro-inflammatory macrophages, instead of inhibiting NK cells, might now promote efficient NK cell effector functions such as improved CD16-dependent ADCC of anti-MICA/B Ab, and a recovery of NKG2D-dependent NK cell-mediated cytotoxicity against tumor cells. Together, these effects may contribute to foster an efficient tumor cell elimination.
TAM reprogramming into proinflammatory macrophages restores NK cell effector functions in vitro and in vivo (301–305). To capitalize on TAM reprogramming, several alternatives are being explored. Besides STING agonists and ICD inducers, as discussed earlier, there are additional emerging alternatives. Tyro, Axl and MerTK receptor tyrosine kinases (TAMRTK) constitute important players for the homeostasis of the immune response because they participate in the resolution of inflammation, contribute to the clearance of apoptotic cell debris, the restoration of vascular integrity and regulate the magnitude of the immune response (344). Therefore, interference with TAMRTK function can lead to chronic inflammatory and autoimmunity. Cancer cells coopt regulatory circuits evolutionarily generated to maintain tissue homeostasis in order to resist growth under immunological pressure and promote tumor immune escape. Accordingly, altered expression and signaling of TAMRTK is involved in tumor progression (298, 300) and inhibition of TYRO3 conferred responsiveness to ICI because TYRO3 promotes the development and accumulation of suppressive TAM (345). Also, blockade of MerTK on TAM triggered P2X7R-dependent activation of STING by tumor-derived cGAMP, stimulated a type I IFN response that reshaped the TME, promoted T cell activation and synergized with ICI, contributing to an efficient antitumor immunity (346). Accordingly, Tyro3 or MerTK inhibition interferes with suppressive circuits that are driven by the TME and TAM. Therefore, efforts are being made to develop TAMRTK inhibitors (298–300, 347). Also, it has been demonstrated that TAM exhibit upregulated expression of folate receptor beta (FRβ) within the TME, and that targeting FRβ+ TAM with folate coupled to a TLR7 agonist reduced their immunosuppressive activity, stimulated CD8+ T-cell infiltration and repolarized macrophages into proinflammatory cells, which negatively impacted on tumor growth and metastasis, and improved overall survival (348). Overall, these results indicate that targeting TAM with TAMRTK inhibitors or other compounds emerge as promising alternatives to reinstate tumor immunity either used as monotherapy or combined with ICI (347). In addition, combination of Tyro3 or MerTK inhibition with anti-MICA/B Ab might synergistically reprogram immunosuppressive TAM, interfere with NK cell exhaustion, and potentiate tumor immunity. Overall, these results provide a solid rationale for the development of combination therapies between pharmacologic reprogramming of TAM and Ab that target MICA/B, as depicted schematically in Figure 7 and mentioned in Table 1.
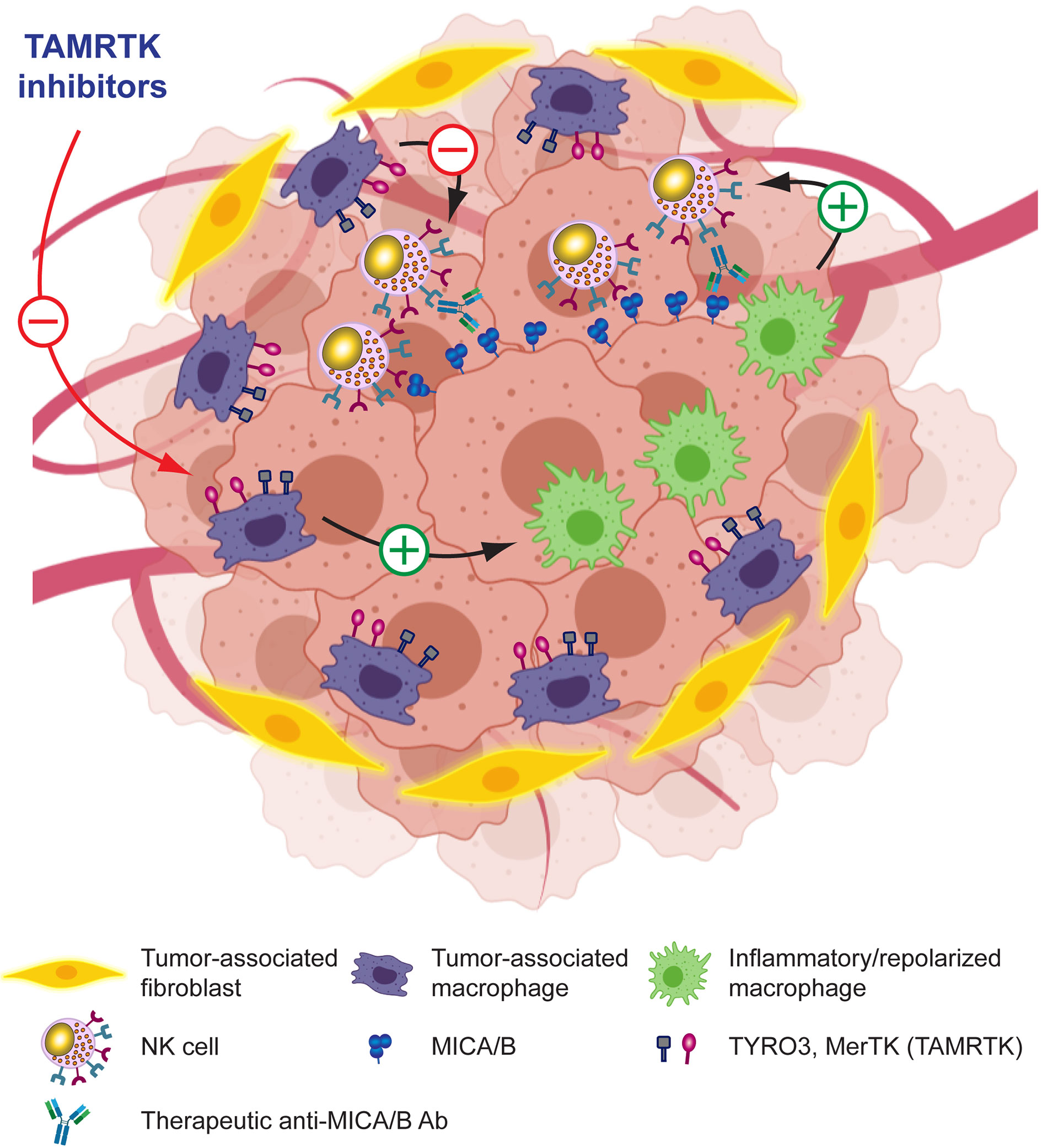
Figure 7 Leveraging anti-MICA/B Ab therapeutic efficacy through combination therapies with molecules that target TAM. Pharmacologic blockade of receptors involved in TAM-mediated immunosuppression in the TME may promote reprogramming of TAM into pro-inflammatory macrophages and a subsequent remodeling of the TME. These pro-inflammatory macrophages, instead of inhibiting NK cells, might now promote efficient NK cell effector functions such as improved CD16-dependent ADCC of anti-MICA/B Ab, and a recovery of NKG2D-dependent NK cell-mediated cytotoxicity against tumor cells. Together, these effects may contribute to foster an efficient tumor cell elimination.
NK cells are currently an important component of several pipelines in different pharma/biotech companies aimed at exploiting their therapeutic potential in I-O. Some initial success in the treatment of liquid tumors has been achieved, but solid tumors represent another level of difficulty imposed, among other factors, by the necessity to overcome the suppressive TME in which TAM play a major role. Also, the selection of novel target molecules and therapeutic modalities in I-O is another critical aspect. In this review, we revised data that position MICA and MICB as attractive targets in I-O to leverage NKG2D-dependent NK cell-mediated anti-tumor effects and catalyze tumor immunity. Ab-based strategies emerge as superior to cell-based alternatives because they appear safer, cheaper and of wider application than cell-based therapies such as CAR-T cells or CAR-NK cells. However, as MICA and MICB are polymorphic, therapeutic Ab should target monomorphic/conserved regions. Cell surface MICA/B can be targeted with Ab, either monoclonal or polyclonal, to promote tumor cell elimination through ADCC. Moreover, their soluble counterparts involved in tumor immune escape can be targeted with Ab to promote clearance of immune complexes by macrophages and reinstate tumor immunity. Thus, Ab-based strategies constitute “two-in-one” therapeutic options that might be further fostered through combination with other assets, in a field where combination strategies are taking the center of the stage. Current evidence indicates that such anti-MICA/B Ab can be either monoclonal or polyclonal (for example, induced by immunization with immunogens such as BLS-MICA). Along this review, we presented frontrunner alternatives to combine with these anti-MICA/B Ab such as the use of drugs that can promote upregulation of MICA/B on tumor cells, blockers of sMICA/B shedding, synthetic lethality inducers that target PARP1, agonists of STING, drugs that induce ICD and molecules that target TAM. Directly or indirectly, all these strategies leverage MICA/B in I-O. Overall, most of these strategies may contribute, in a direct or in an indirect manner, to tumor elimination through restoration of NK cell activation and effector functions, and to subsequently convert “cold” tumors into “hot” tumors. Also, some of the discussed strategies such as the use of drugs that trigger upregulation of MICA/B, that promote a remodeling of the TME and/or that affect TAM may also foster CAR-T/CAR-NK cell-based therapies aimed at targeting MICA/B. A central question that remains unanswered is whether it is better to reinvigorate dysfunctional TINK or to eliminate/deplete them and create a niche for the recruitment of newly activated, fully functional NK cells through the administration of immunotherapeutic agents to the patient. In any case, pursuing the addressed pipelines might lead to innovative modalities of immunotherapy for the treatment of a wide range of cancer patients. Moreover, although we focused the state of the art and perspectives on the control of primary tumors and considering that NK cells also play an important role in the detection and eradication of tumor cells within the circulation and limiting metastasis (349–351), the approaches proposed may also impact on the ability of NK cells to suppress metastasis.
NZ conceived and designed the review and wrote the manuscript. MF and CD provided critical inputs and ideas and corrected the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded with grants from the National Agency for Promotion of Science and Technology from Argentina (ANPCYT), the National Research Council of Argentina (CONICET) and the Trust in Science Program from GlaxoSmithKline (GSK), all to NZ. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. We also thank to Fundación Williams and Fundación René Barón for providing financial assistance (donations) to our laboratory.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Some figures or part of them were created with Biorender.com.
1. Hoos A. Development of Immuno-Oncology Drugs - From CTLA4 to PD1 to the Next Generations. Nat Rev Drug Discov (2016) 15:235–47. doi: 10.1038/nrd.2015.35
2. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060
3. Chamoto K, Hatae R, Honjo T. Current Issues and Perspectives in PD-1 Blockade Cancer Immunotherapy. Int J Clin Oncol (2020) 25:790–800. doi: 10.1007/s10147-019-01588-7
4. Kalbasi A, Ribas A. Tumour-Intrinsic Resistance to Immune Checkpoint Blockade. Nat Rev Immunol (2020) 20:25–39. doi: 10.1038/s41577-019-0218-4
5. Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria J-C, et al. Hyperprogressive Disease: Recognizing a Novel Pattern to Improve Patient Management. Nat Rev Clin Oncol (2018) 15:748–62. doi: 10.1038/s41571-018-0111-2
6. Camelliti S, Le Noci V, Bianchi F, Moscheni C, Arnaboldi F, Gagliano N, et al. Mechanisms of Hyperprogressive Disease After Immune Checkpoint Inhibitor Therapy: What We (Don’t) Know. J Exp Clin Cancer Res (2020) 39:236. doi: 10.1186/s13046-020-01721-9
7. Denis M, Duruisseaux M, Brevet M, Dumontet C. How Can Immune Checkpoint Inhibitors Cause Hyperprogression in Solid Tumors? Front Immunol (2020) 11:492. doi: 10.3389/fimmu.2020.00492
8. Sharma P, Allison JP. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies With Curative Potential. Cell (2015) 161:205–14. doi: 10.1016/j.cell.2015.03.030
9. Cantoni C, Wurzer H, Thomas C, Vitale M. Escape of Tumor Cells From the NK Cell Cytotoxic Activity. J Leukoc Biol (2020) 108:1339–60. doi: 10.1002/JLB.2MR0820-652R
10. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A Natural Killer-Dendritic Cell Axis Defines Checkpoint Therapy-Responsive Tumor Microenvironments. Nat Med (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
11. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. Nk Cells Stimulate Recruitment of cDC1 Into the Tumor Microenvironment Promoting Cancer Immune Control. Cell (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
12. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural Cytotoxic Activity of Peripheral-Blood Lymphocytes and Cancer Incidence: An 11-Year Follow-Up Study of a General Population. Lancet (2000) 356:1795–9. doi: 10.1016/S0140-6736(00)03231-1
13. Cursons J, Souza-Fonseca-Guimaraes F, Foroutan M, Anderson A, Hollande F, Hediyeh-Zadeh S, et al. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol Res (2019) 7:1162–74. doi: 10.1158/2326-6066.CIR-18-0500
14. Lee H, Quek C, Silva I, Tasker A, Batten M, Rizos H, et al. Integrated Molecular and Immunophenotypic Analysis of NK Cells in Anti-PD-1 Treated Metastatic Melanoma Patients. Oncoimmunology (2019) 8:e1537581. doi: 10.1080/2162402X.2018.1537581
15. Li B, Jiang Y, Li G, Fisher GA, Li R. Natural Killer Cell and Stroma Abundance Are Independently Prognostic and Predict Gastric Cancer Chemotherapy Benefit. JCI Insight (2020) 5:e136570. doi: 10.1172/jci.insight.136570
16. Gil M, Kim KE. Interleukin-18 Is a Prognostic Biomarker Correlated With CD8+ T Cell and Natural Killer Cell Infiltration in Skin Cutaneous Melanoma. J Clin Med (2019) 8:1993. doi: 10.3390/jcm8111993
17. Mujal AM, Delconte RB, Sun JC. Natural Killer Cells: From Innate to Adaptive Features. Annu Rev Immunol (2021) 39:417–47. doi: 10.1146/annurev-immunol-101819-074948
18. Hodgins JJ, Khan ST, Park MM, Auer RC, Ardolino M. Killers 2.0: NK Cell Therapies at the Forefront of Cancer Control. J Clin Invest (2019) 129:3499–510. doi: 10.1172/JCI129338
19. Myers JA, Miller JS. Exploring the NK Cell Platform for Cancer Immunotherapy. Nat Rev Clin Oncol (2021) 18:85–100. doi: 10.1038/s41571-020-0426-7
20. Demaria O, Gauthier L, Debroas G, Vivier E. Natural Killer Cell Engagers in Cancer Immunotherapy: Next Generation of Immuno-Oncology Treatments. Eur J Immunol (2021). doi: 10.1002/eji.202048953
21. Guillerey C, Huntington ND, Smyth MJ. Targeting Natural Killer Cells in Cancer Immunotherapy. Nat Immunol (2016) 17:1025–36. doi: 10.1038/ni.3518
22. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science (2002) 295:2097–100. doi: 10.1126/science.1068440
23. Benson DM, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a Novel Anti-Inhibitory KIR Antibody, and Lenalidomide Combine to Enhance the Natural Killer Cell Versus Multiple Myeloma Effect. Blood (2011) 118:6387–91. doi: 10.1182/blood-2011-06-360255
24. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
25. Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful Transfer of Alloreactive Haploidentical KIR Ligand-Mismatched Natural Killer Cells After Infusion in Elderly High Risk Acute Myeloid Leukemia Patients. Blood (2011) 118:3273–9. doi: 10.1182/blood-2011-01-329508
26. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful Adoptive Transfer and In Vivo Expansion of Human Haploidentical NK Cells in Patients With Cancer. Blood (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
27. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. J Clin Oncol (2010) 28:955–9. doi: 10.1200/JCO.2009.24.4590
28. Shimasaki N, Jain A, Campana D. NK Cells for Cancer Immunotherapy. Nat Rev Drug Discov (2020) 19:200–18. doi: 10.1038/s41573-019-0052-1
29. Navin I, Lam MT, Parihar R. Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment. Cancers (2020) 12. doi: 10.3390/cancers12123871
30. Daher M, Rezvani K. Outlook for New Car-Based Therapies With a Focus on CAR NK Cells: What Lies Beyond Car-Engineered T Cells in the Race Against Cancer. Cancer Discov (2020) 11:45–58. doi: 10.1158/2159-8290.CD-20-0556
31. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive Transfer of Autologous Natural Killer Cells Leads to High Levels of Circulating Natural Killer Cells But Does Not Mediate Tumor Regression. Clin Cancer Res (2011) 17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347
32. Merino AM, Kim H, Miller JS, Cichocki F. Unraveling Exhaustion in Adaptive and Conventional NK Cells. J Leukoc Biol (2020) 108:1361–8. doi: 10.1002/JLB.4MR0620-091R
33. Schnell A, Bod L, Madi A, Kuchroo VK. The Yin and Yang of Co-Inhibitory Receptors: Toward Anti-Tumor Immunity Without Autoimmunity. Cell Res (2020) 30:285–99. doi: 10.1038/s41422-020-0277-x
34. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling During Successful Immune-Checkpoint Cancer Therapy. Cell (2018) 175:1443. doi: 10.1016/j.cell.2018.11.003
35. Souza-Fonseca-Guimaraes F, Cursons J, Huntington ND. The Emergence of Natural Killer Cells as a Major Target in Cancer Immunotherapy. Trends Immunol (2019) 40:142–58. doi: 10.1016/j.it.2018.12.003
36. Huntington ND, Vosshenrich CA, Di Santo JP. Developmental Pathways That Generate Natural-Killer-Cell Diversity in Mice and Humans. Nat Rev Immunol (2007) 7:703–14. doi: 10.1038/nri2154
37. Morvan MG, Lanier LL. NK Cells and Cancer: You can Teach Innate Cells New Tricks. Nat Rev Cancer (2016) 16:7–19. doi: 10.1038/nrc.2015.5
38. Huntington ND, Cursons J, Rautela J. The Cancer-Natural Killer Cell Immunity Cycle. Nat Rev Cancer (2020) 20:437–54. doi: 10.1038/s41568-020-0272-z
39. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of Natural Killer Cells. Nat Immunol (2008) 9:503–10. doi: 10.1038/ni1582
40. Kruse PH, Matta J, Ugolini S, Vivier E. Natural Cytotoxicity Receptors and Their Ligands. Immunol Cell Biol (2014) 92:221–9. doi: 10.1038/icb.2013.98
41. Ziblat A, Domaica CI, Spallanzani RG, Iraolagoitia XL, Rossi LE, Avila DE, et al. IL-27 Stimulates Human NK-cell Effector Functions and Primes NK Cells for IL-18 Responsiveness. Eur J Immunol (2015) 45:192–202. doi: 10.1002/eji.201444699
42. Ziblat A, Nunez SY, Raffo Iraolagoitia XL, Spallanzani RG, Torres NI, Sierra JM, et al. Interleukin (IL)-23 Stimulates IFN-Gamma Secretion by CD56(bright) Natural Killer Cells and Enhances IL-18-Driven Dendritic Cells Activation. Front Immunol (2017) 8:1959. doi: 10.3389/fimmu.2017.01959
43. Zwirner NW, Ziblat A. Regulation of NK Cell Activation and Effector Functions by the IL-12 Family of Cytokines: The Case of IL-27. Front Immunol (2017) 8:25. doi: 10.3389/fimmu.2017.00025
44. Nuñez SY, Ziblat A, Secchiari F, Torres NI, Sierra JM, Raffo Iraolagoitia XL, et al. Human M2 Macrophages Limit NK Cell Effector Functions Through Secretion of TGF-β and Engagement of CD85j. J Immunol (2018) 200:1008–15. doi: 10.4049/jimmunol.1700737
45. Sivori S, Falco M, Della Chiesa M, Carlomagno S, Vitale M, Moretta L, et al. Cpg and Double-Stranded RNA Trigger Human NK Cells by Toll-Like Receptors: Induction of Cytokine Release and Cytotoxicity Against Tumors and Dendritic Cells. Proc Natl Acad Sci USA (2004) 101:10116–21. doi: 10.1073/pnas.0403744101
46. Girart MV, Fuertes MB, Domaica CI, Rossi LE, Zwirner NW. Engagement of TLR3, TLR7, and NKG2D Regulate IFN-Gamma Secretion But Not NKG2D-Mediated Cytotoxicity by Human NK Cells Stimulated With Suboptimal Doses of IL-12. J Immunol (2007) 179:3472–9. doi: 10.4049/jimmunol.179.6.3472
47. Hart OM, Athie-Morales V, O’Connor GM, Gardiner CM. TLR7/8-Mediated Activation of Human NK Cells Results in Accessory Cell-Dependent IFN-Gamma Production. J Immunol (2005) 175:1636–42. doi: 10.4049/jimmunol.175.3.1636
48. Gauthier M, Laroye C, Bensoussan D, Boura C, Decot V. Natural Killer Cells and Monoclonal Antibodies: Two Partners for Successful Antibody Dependent Cytotoxicity Against Tumor Cells. Crit Rev Oncol Hematol (2021) 160:103261. doi: 10.1016/j.critrevonc.2021.103261
49. Wensveen FM, Jelencic V, Polic B. Nkg2d: A Master Regulator of Immune Cell Responsiveness. Front Immunol (2018) 9:441. doi: 10.3389/fimmu.2018.00441
50. Bléry M, Vivier E. NKG2D–MICA Interaction: A Paradigm Shift in Innate Recognition. J Immunol (2018) 200:2229. doi: 10.4049/jimmunol.1800176
51. Lanier LL. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol Res (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
52. Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L. The Immune Checkpoint PD-1 in Natural Killer Cells: Expression, Function and Targeting in Tumour Immunotherapy. Cancers (2020) 12:3285. doi: 10.3390/cancers12113285
53. Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D Function Protects the Host From Tumor Initiation. J Exp Med (2005) 202:583–8. doi: 10.1084/jem.20050994
54. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-Deficient Mice Are Defective in Tumor Surveillance in Models of Spontaneous Malignancy. Immunity (2008) 28:571–80. doi: 10.1016/j.immuni.2008.02.016
55. Zhang J, Basher F, Wu JD. Nkg2d Ligands in Tumor Immunity: Two Sides of a Coin. Front Immunol (2015) 6:97. doi: 10.3389/fimmu.2015.00097
56. Bahram S, Bresnahan M, Geraghty DE, Spies T. A Second Lineage of Mammalian Major Histocompatibility Complex Class I Genes. Proc Natl Acad Sci USA (1994) 91:6259–63. doi: 10.1073/pnas.91.14.6259
57. Leelayuwat C, Townend DC, Degli-Esposti MA, Abraham LJ, Dawkins RL. A New Polymorphic and Multicopy MHC Gene Family Related to Nonmammalian Class I. Immunogenetics (1994) 40:339–51. doi: 10.1007/BF01246675
58. Stephens HA. MICA And MICB Genes: Can the Enigma of Their Polymorphism Be Resolved? Trends Immunol (2001) 22:378–85. doi: 10.1016/s1471-4906(01)01960-3
59. Baranwal AK, Mehra NK. Major Histocompatibility Complex Class I Chain-Related A (MICA) Molecules: Relevance in Solid Organ Transplantation. Front Immunol (2017) 8:182. doi: 10.3389/fimmu.2017.00182
60. Molinero LL, Marcos CY, Mirbaha F, Fainboim L, Stastny P, Zwirner NW. Codominant Expression of the Polymorphic MICA Alloantigens Encoded by Genes in the HLA Region. Eur J Immunogenet (2002) 29:315–9. doi: 10.1046/j.1365-2370.2002.00274.x
61. Zwirner NW, Marcos CY, Mirbaha F, Zou Y, Stastny P. Identification of MICA as a New Polymorphic Alloantigen Recognized by Antibodies in Sera of Organ Transplant Recipients. Hum Immunol (2000) 61:917–24. doi: 10.1016/S0198-8859(00)00162-2
62. Sumitran-Holgersson S, Wilczek HE, Holgersson J, Söderström K. Identification of the Nonclassical HLA Molecules, MICA, as Targets for Humoral Immunity Associated With Irreversible Rejection of Kidney Allografts. Transplantation (2002) 74:268–77. doi: 10.1097/00007890-200207270-00019
63. Suárez-Alvarez B, López-Vázquez A, Gonzalez MZ, Fdez-Morera JL, Díaz-Molina B, Blanco-Gelaz MA, et al. The Relationship of Anti-MICA Antibodies and MICA Expression With Heart Allograft Rejection. Am J Transplant (2007) 7:1842–8. doi: 10.1111/j.1600-6143.2007.01838.x
64. Zou Y, Stastny P, Süsal C, Döhler B, Opelz G. Antibodies Against MICA Antigens and Kidney-Transplant Rejection. N Engl J Med (2007) 357:1293–300. doi: 10.1056/NEJMoa067160
65. Zhang Q, Cecka JM, Gjertson DW, Ge P, Rose ML, Patel JK, et al. HLA and MICA: Targets of Antibody-Mediated Rejection in Heart Transplantation. Transplantation (2011) 91:1153–8. doi: 10.1097/TP.0b013e3182157d60
66. Ming Y, Hu J, Luo Q, Ding X, Luo W, Zhuang Q, et al. Acute Antibody-Mediated Rejection in Presence of MICA-DSA and Successful Renal Re-Transplant With Negative-MICA Virtual Crossmatch. PloS One (2015) 10:e0127861. doi: 10.1371/journal.pone.0127861
67. Gambelunghe G, Falorni A, Ghaderi M, Laureti S, Tortoioli C, Santeusanio F, et al. Microsatellite Polymorphism of the MHC Class I Chain-Related (MIC-A and MIC-B) Genes Marks the Risk for Autoimmune Addison’s Disease. J Clin Endocrinol Metab (1999) 84:3701–7. doi: 10.1210/jcem.84.10.6069
68. Tay GK, Hui J, Gaudieri S, Schmitt-Egenolf M, Martinez OP, Leelayuwat C, et al. PERB11 (MIC): A Polymorphic MHC Gene Is Expressed in Skin and Single Nucleotide Polymorphisms Are Associated With Psoriasis. Clin Exp Immunol (2000) 119:553–8. doi: 10.1046/j.1365-2249.2000.01140.x
69. González S, Martínez-Borra J, López-Vázquez A, García-Fernández S, Torre-Alonso JC, López-Larrea C. MICA Rather Than MICB, TNFA, or HLA-DRB1 Is Associated With Susceptibility to Psoriatic Arthritis. J Rheumatol (2002) 29:973–8.
70. Gambelunghe G, Gerli R, Bocci EB, Del Sindaco P, Ghaderi M, Sanjeevi CB, et al. Contribution of MHC Class I Chain-Related A (MICA) Gene Polymorphism to Genetic Susceptibility for Systemic Lupus Erythematosus. Rheumatology (2005) 44:287–92. doi: 10.1093/rheumatology/keh459
71. Jumnainsong A, Romphruk AV, Jearanaikoon P, Klumkrathok K, Romphruk A, Luanrattanakorn S, et al. Association of Polymorphic Extracellular Domains of MICA With Cervical Cancer in Northeastern Thai Population. Tissue Antigens (2007) 69:326–33. doi: 10.1111/j.1399-0039.2006.00754.x
72. Gambelunghe G, Brozzetti A, Ghaderi M, Candeloro P, Tortoioli C, Falorni A. MICA Gene Polymorphism in the Pathogenesis of Type 1 Diabetes. Ann NY Acad Sci (2007) 1110:92–8. doi: 10.1196/annals.1423.011
73. Zhou X, Wang J, Zou H, Ward MM, Weisman MH, Espitia MG, et al. MICA, a Gene Contributing Strong Susceptibility to Ankylosing Spondylitis. Ann Rheum Dis (2014) 73:1552–7. doi: 10.1136/annrheumdis-2013-203352
74. Martinez-Chamorro A, Moreno A, Gómez-García M, Cabello MJ, Martin J, Lopez-Nevot MÁ. MICA*A4 Protects Against Ulcerative Colitis, Whereas MICA*A5.1 Is Associated With Abscess Formation and Age of Onset. Clin Exp Immunol (2016) 184:323–31. doi: 10.1111/cei.12786
75. Toledo-Stuardo K, Ribeiro CH, Canals A, Garate V, Rodríguez A, Tello S, et al. Major Histocompatibility Complex Class I-Related Chain A (MICA) Allelic Variants Associate With Susceptibility and Prognosis of Gastric Cancer. Front Immunol (2021) 12:645528. doi: 10.3389/fimmu.2021.645528
76. Zou Y, Bresnahan W, Taylor RT, Stastny P. Effect of Human Cytomegalovirus on Expression of MHC Class I-Related Chains a. J Immunol (2005) 174:3098–104. doi: 10.4049/jimmunol.174.5.3098
77. Isernhagen A, Malzahn D, Bickeböller H, Dressel R. Impact of the MICA-129Met/Val Dimorphism on NKG2D-Mediated Biological Functions and Disease Risks. Front Immunol (2016) 7:588. doi: 10.3389/fimmu.2016.00588
78. López-Hernández R, Valdés M, Lucas D, Campillo JA, Martínez-Garcia P, Salama H, et al. Association Analysis of MICA Gene Polymorphism and MICA-129 Dimorphism With Inflammatory Bowel Disease Susceptibility in a Spanish Population. Hum Immunol (2010) 71:512–4. doi: 10.1016/j.humimm.2010.02.003
79. Zingoni A, Vulpis E, Cecere F, Amendola MG, Fuerst D, Saribekyan T, et al. Mica-129 Dimorphism and Soluble MICA Are Associated With the Progression of Multiple Myeloma. Front Immunol (2018) 9:926. doi: 10.3389/fimmu.2018.00926
80. Ouni N, Ben Chaaben A, Kablouti G, Lajnef M, Ayari F, Abaza H, et al. Mica-129met/Val Polymorphism Is Associated With Early-Onset Breast Cancer Risk. Immunol Invest (2017) 46:603–14. doi: 10.1080/08820139.2017.1336175
81. Li P, Willie ST, Bauer S, Morris DL, Spies T, Strong RK. Crystal Structure of the MHC Class I Homolog MIC-A, a Gammadelta T Cell Ligand. Immunity (1999) 10:577–84. doi: 10.1016/S1074-7613(00)80057-6
82. Ota M, Bahram S, Katsuyama Y, Saito S, Nose Y, Sada M, et al. On the MICA deleted-MICB Null, HLA-B*4801 Haplotype. Tissue Antigens (2000) 56:268–71. doi: 10.1034/j.1399-0039.2000.560309.x
83. Komatsu-Wakui M, Tokunaga K, Ishikawa Y, Kashiwase K, Moriyama S, Tsuchiya N, et al. MIC-A Polymorphism in Japanese and a MIC-A-MIC-B Null Haplotype. Immunogenetics (1999) 49:620–8. doi: 10.1007/s002510050658
84. Komatsu-Wakui M, Tokunaga K, Ishikawa Y, Leelayuwat C, Kashiwase K, Tanaka H, et al. Wide Distribution of the MICA-MICB Null Haplotype in East Asians. Tissue Antigens (2001) 57:1–8. doi: 10.1034/j.1399-0039.2001.057001001.x
85. Aida K, Russomando G, Kikuchi M, Candia N, Franco L, Almiron M, et al. High Frequency of MIC Null Haplotype (HLA-B48-MICA-Del-MICB*0107 N) in the Angaite Amerindian Community in Paraguay. Immunogenetics (2002) 54:439–41. doi: 10.1007/s00251-002-0485-1
86. Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. Ulbps, Novel MHC Class I-Related Molecules, Bind to CMV Glycoprotein UL16 and Stimulate NK Cytotoxicity Through the NKG2D Receptor. Immunity (2001) 14:123–33. doi: 10.1016/S1074-7613(01)00095-4
87. Jan Chalupny N, Sutherland CL, Lawrence WA, Rein-Weston A, Cosman D. ULBP4 Is a Novel Ligand for Human NKG2D. Biochem Biophys Res Commun (2003) 305:129–35. doi: 10.1016/S0006-291X(03)00714-9
88. Eagle RA, Traherne JA, Hair JR, Jafferji I, Trowsdale J. ULBP6/RAET1L Is an Additional Human NKG2D Ligand. Eur J Immunol (2009) 39:3207–16. doi: 10.1002/eji.200939502
89. Bacon L, Eagle RA, Meyer M, Easom N, Young NT, Trowsdale J. Two Human ULBP/RAET1 Molecules With Transmembrane Regions Are Ligands for NKG2D. J Immunol (2004) 173:1078–84. doi: 10.4049/jimmunol.173.2.1078
90. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell Stress-Regulated Human Major Histocompatibility Complex Class I Gene Expressed in Gastrointestinal Epithelium. Proc Natl Acad Sci USA (1996) 93:12445–50. doi: 10.1073/pnas.93.22.12445
91. Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad Tumor-Associated Expression and Recognition by Tumor-Derived Gamma Delta T Cells of MICA and MICB. Proc Natl Acad Sci USA (1999) 96:6879–84. doi: 10.1073/pnas.96.12.6879
92. Zwirner NW, Fernandez-Vina MA, Stastny P. MICA, a New Polymorphic HLA-Related Antigen, Is Expressed Mainly by Keratinocytes, Endothelial Cells, and Monocytes. Immunogenetics (1998) 47:139–48. doi: 10.1007/s002510050339
93. Zwirner NW, Dole K, Stastny P. Differential Surface Expression of MICA by Endothelial Cells, Fibroblasts, Keratinocytes, and Monocytes. Hum Immunol (1999) 60:323–30. doi: 10.1016/S0198-8859(98)00128-1
94. Schrambach S, Ardizzone M, Leymarie V, Sibilia J, Bahram S. In Vivo Expression Pattern of MICA and MICB and Its Relevance to Auto-Immunity and Cancer. PloS One (2007) 2:e518. doi: 10.1371/journal.pone.0000518
95. Ashiru O, López-Cobo S, Fernández-Messina L, Pontes-Quero S, Pandolfi R, Reyburn HT, et al. A GPI Anchor Explains the Unique Biological Features of the Common NKG2D-Ligand Allele MICA*008. Biochem J (2013) 454:295–302. doi: 10.1042/BJ20130194
96. Mellergaard M, Skovbakke SL, Schneider CL, Lauridsen F, Andresen L, Jensen H, et al. N-Glycosylation of Asparagine 8 Regulates Surface Expression of Major Histocompatibility Complex Class I Chain-Related Protein A (MICA) Alleles Dependent on Threonine 24. J Biol Chem (2014) 289:20078–91. doi: 10.1074/jbc.M114.573238
97. Fang L, Gong J, Wang Y, Liu R, Li Z, Wang Z, et al. MICA/B Expression Is Inhibited by Unfolded Protein Response and Associated With Poor Prognosis in Human Hepatocellular Carcinoma. J Exp Clin Cancer Res (2014) 33:76. doi: 10.1186/s13046-014-0076-7
98. Fuertes MB, Rossi LE, Peralta CM, Cabrera HN, Allevato MA, Zwirner NW. Premalignant Quiescent Melanocytic Nevi Do Not Express the MHC Class I Chain-Related Protein A. Medicina (2011) 71:357–60.
99. Ghadially H, Brown L, Lloyd C, Lewis L, Lewis A, Dillon J, et al. MHC Class I Chain-Related Protein A and B (MICA and MICB) Are Predominantly Expressed Intracellularly in Tumour and Normal Tissue. Br J Cancer (2017) 116:1208–17. doi: 10.1038/bjc.2017.79
100. Fuertes MB, Girart MV, Molinero LL, Domaica CI, Rossi LE, Barrio MM, et al. Intracellular Retention of the NKG2D Ligand MHC Class I Chain-Related Gene A in Human Melanomas Confers Immune Privilege and Prevents NK Cell-Mediated Cytotoxicity. J Immunol (2008) 180:4606–14. doi: 10.4049/jimmunol.180.7.4606
101. Schmiedel D, Mandelboim O. Nkg2d Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front Immunol (2018) 9:2040. doi: 10.3389/fimmu.2018.02040
102. Zhang X, Yan L, Jiao W, Ren J, Xing N, Zhang Y, et al. The Clinical and Biological Significance of MICA in Clear Cell Renal Cell Carcinoma Patients. Tumour Biol (2016) 37:2153–9. doi: 10.1007/s13277-015-4041-7
103. Ribeiro CH, Kramm K, Galvez-Jiron F, Pola V, Bustamante M, Contreras HR, et al. Clinical Significance of Tumor Expression of Major Histocompatibility Complex Class I-Related Chains A and B (MICA/B) in Gastric Cancer Patients. Oncol Rep (2016) 35:1309–17. doi: 10.3892/or.2015.4510
104. Okita R, Yukawa T, Nojima Y, Maeda A, Saisho S, Shimizu K, et al. MHC Class I Chain-Related Molecule A and B Expression Is Upregulated by Cisplatin and Associated With Good Prognosis in Patients With Non-Small Cell Lung Cancer. Cancer Immunol Immunother (2016) 65:499–509. doi: 10.1007/s00262-016-1814-9
105. Wang B, Wang Q, Wang Z, Jiang J, Yu SC, Ping YF, et al. Metastatic Consequences of Immune Escape From NK Cell Cytotoxicity by Human Breast Cancer Stem Cells. Cancer Res (2014) 74:5746–57. doi: 10.1158/0008-5472.CAN-13-2563
106. Dambrauskas Z, Svensson H, Joshi M, Hyltander A, Naredi P, Iresjo BM. Expression of Major Histocompatibility Complex Class I-Related Chain A/B (MICA/B) in Pancreatic Carcinoma. Int J Oncol (2014) 44:99–104. doi: 10.3892/ijo.2013.2156
107. Sconocchia G, Spagnoli GC, Del Principe D, Ferrone S, Anselmi M, Wongsena W, et al. Defective Infiltration of Natural Killer Cells in MICA/B-Positive Renal Cell Carcinoma Involves Beta(2)-Integrin-Mediated Interaction. Neoplasia (2009) 11:662–71. doi: 10.1593/neo.09296
108. Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, et al. MHC Class I Chain-Related Protein A Antibodies and Shedding Are Associated With the Progression of Multiple Myeloma. Proc Natl Acad Sci USA (2008) 105:1285–90. doi: 10.1073/pnas.0711293105
109. Watson NF, Spendlove I, Madjd Z, McGilvray R, Green AR, Ellis IO, et al. Expression of the Stress-Related MHC Class I Chain-Related Protein MICA Is an Indicator of Good Prognosis in Colorectal Cancer Patients. Int J Cancer (2005) 118:1445–52. doi: 10.1002/ijc.21510
110. Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG, et al. Functional Expression and Release of Ligands for the Activating Immunoreceptor NKG2D in Leukemia. Blood (2003) 102:1389–96. doi: 10.1182/blood-2003-01-0019
111. Torres N, Regge MV, Secchiari F, Friedrich AD, Spallanzani RG, Raffo Iraolagoitia XL, et al. Restoration of Antitumor Immunity Through Anti-MICA Antibodies Elicited With a Chimeric Protein. J Immunother Cancer (2020) 8:e000233. doi: 10.1136/jitc-2019-000233
112. Cerwenka A, Baron JL, Lanier LL. Ectopic Expression of Retinoic Acid Early Inducible-1 Gene (RAE-1) Permits Natural Killer Cell-Mediated Rejection of a MHC Class I-Bearing Tumor In Vivo. Proc Natl Acad Sci USA (2001) 98:11521–6. doi: 10.1073/pnas.201238598
113. Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 Ligands of the NKG2D Receptor Stimulate Tumour Immunity. Nature (2001) 413:165–71. doi: 10.1038/35093109
114. Friese MA, Platten M, Lutz SZ, Naumann U, Aulwurm S, Bischof F, et al. MICA/NKG2D-Mediated Immunogene Therapy of Experimental Gliomas. Cancer Res (2003) 63:8996–9006.
115. Groh V, Wu J, Yee C, Spies T. Tumour-Derived Soluble MIC Ligands Impair Expression of NKG2D and T-Cell Activation. Nature (2002) 419:734–8. doi: 10.1038/nature01112
116. Salih HR, Rammensee H-G, Steinle A. Down-Regulation of MICA on Human Tumors by Proteolytic Shedding. J Immunol (2002) 169:4098–102. doi: 10.4049/jimmunol.169.8.4098
117. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-Associated MICA Is Shed by ADAM Proteases. Cancer Res (2008) 68:6368–76. doi: 10.1158/0008-5472.CAN-07-6768
118. Ashiru O, Boutet P, Fernández-Messina L, Agüera-González S, Skepper JN, Valés-Gómez M, et al. Natural Killer Cell Cytotoxicity Is Suppressed by Exposure to the Human NKG2D Ligand MICA*008 That Is Shed by Tumor Cells in Exosomes. Cancer Res (2010) 70:481–9. doi: 10.1158/0008-5472.CAN-09-1688
119. Du C, Bevers J, Cook R, Lombana TN, Rajasekaran K, Matsumoto M, et al. MICA Immune Complex Formed With Alpha 3 Domain-Specific Antibody Activates Human NK Cells in a Fc-Dependent Manner. J Immunother Cancer (2019) 7:207. doi: 10.1186/s40425-019-0687-9
120. Pende D, Rivera P, Marcenaro S, Chang CC, Biassoni R, Conte R, et al. Major Histocompatibility Complex Class I-Related Chain A and UL16-Binding Protein Expression on Tumor Cell Lines of Different Histotypes: Analysis of Tumor Susceptibility to NKG2D-Dependent Natural Killer Cell Cytotoxicity. Cancer Res (2002) 62:6178–86.
121. Sutherland CL, Rabinovich B, Chalupny NJ, Brawand P, Miller R, Cosman D. ULBPs, Human Ligands of the NKG2D Receptor, Stimulate Tumor Immunity With Enhancement by IL-15. Blood (2006) 108:1313–9. doi: 10.1182/blood-2005-11-011320
122. McGilvray RW, Eagle RA, Watson NFS, Al-Attar A, Ball G, Jafferji I, et al. NKG2D Ligand Expression in Human Colorectal Cancer Reveals Associations With Prognosis and Evidence for Immunoediting. Clin Cancer Res (2009) 15:6993–7002. doi: 10.1158/1078-0432.CCR-09-0991
123. Dulphy N, Berrou J, Campillo JA, Bagot M, Bensussan A, Toubert A. NKG2D Ligands Expression and NKG2D-Mediated NK Activity in Sezary Patients. J Invest Dermatol (2009) 129:359–64. doi: 10.1038/jid.2008.256
124. McGilvray RW, Eagle RA, Rolland P, Jafferji I, Trowsdale J, Durrant LG. ULBP2 and RAET1E NKG2D Ligands Are Independent Predictors of Poor Prognosis in Ovarian Cancer Patients. Int J Cancer (2010) 127:1412–20. doi: 10.1002/ijc.25156
125. Nanbakhsh A, Pochon C, Mallavialle A, Amsellem S, Bourhis JH, Chouaib S. c-Myc Regulates Expression of NKG2D Ligands ULBP1/2/3 in AML and Modulates Their Susceptibility to NK-Mediated Lysis. Blood (2014) 123:3585–95. doi: 10.1182/blood-2013-11-536219
126. Tsukagoshi M, Wada S, Yokobori T, Altan B, Ishii N, Watanabe A, et al. Overexpression of Natural Killer Group 2 Member D Ligands Predicts Favorable Prognosis in Cholangiocarcinoma. Cancer Sci (2016) 107:116–22. doi: 10.1111/cas.12853
127. Fernández-Messina L, Reyburn H, Vales-Gomez M. Human NKG2D-Ligands: Cell Biology Strategies to Ensure Immune Recognition. Front Immunol (2012) 3:299. doi: 10.3389/fimmu.2012.00299
128. Ohashi M, Eagle RA, Trowsdale J. Post-Translational Modification of the NKG2D Ligand RAET1G Leads to Cell Surface Expression of a Glycosylphosphatidylinositol-Linked Isoform. J Biol Chem (2010) 285:16408–15. doi: 10.1074/jbc.M109.077636
129. Waldhauer I, Steinle A. Proteolytic Release of Soluble UL16-Binding Protein 2 From Tumor Cells. Cancer Res (2006) 66:2520–6. doi: 10.1158/0008-5472.CAN-05-2520
130. Song H, Kim J, Cosman D, Choi I. Soluble ULBP Suppresses Natural Killer Cell Activity Via Down-Regulating NKG2D Expression. Cell Immunol (2006) 239:22–30. doi: 10.1016/j.cellimm.2006.03.002
131. Cao W, Xi X, Hao Z, Li W, Kong Y, Cui L, et al. RAET1E2, a Soluble Isoform of the UL16-Binding Protein RAET1E Produced by Tumor Cells, Inhibits NKG2D-Mediated NK Cytotoxicity. J Biol Chem (2007) 282:18922–8. doi: 10.1074/jbc.M702504200
132. Fernandez-Messina L, Ashiru O, Boutet P, Aguera-Gonzalez S, Skepper JN, Reyburn HT, et al. Differential Mechanisms of Shedding of the Glycosylphosphatidylinositol (GPI)-Anchored NKG2D Ligands. J Biol Chem (2010) 285:8543–51. doi: 10.1074/jbc.M109.045906
133. Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D Ligands Defines Leukaemia Stem Cells and Mediates Their Immune Evasion. Nature (2019) 572:254–9. doi: 10.1038/s41586-019-1410-1
134. Bonnet D, Dick JE. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat Med (1997) 3:730–7. doi: 10.1038/nm0797-730
135. Bonavita E, Bromley CP, Jonsson G, Pelly VS, Sahoo S, Walwyn-Brown K, et al. Antagonistic Inflammatory Phenotypes Dictate Tumor Fate and Response to Immune Checkpoint Blockade. Immunity (2020) 53:1215–29. doi: 10.1016/j.immuni.2020.10.020
136. Zemek RM, De Jong E, Chin WL, Schuster IS, Fear VS, Casey TH, et al. Sensitization to Immune Checkpoint Blockade Through Activation of a STAT1/NK Axis in the Tumor Microenvironment. Sci Transl Med (2019) 11:eaav7816. doi: 10.1126/scitranslmed.aav7816
137. Komohara Y, Hasita H, Ohnishi K, Fujiwara Y, Suzu S, Eto M, et al. Macrophage Infiltration and Its Prognostic Relevance in Clear Cell Renal Cell Carcinoma. Cancer Sci (2011) 102:1424–31. doi: 10.1111/j.1349-7006.2011.01945.x
138. Prinz PU, Mendler AN, Brech D, Masouris I, Oberneder R, Noessner E. NK-Cell Dysfunction in Human Renal Carcinoma Reveals Diacylglycerol Kinase as Key Regulator and Target for Therapeutic Intervention. Int J Cancer (2014) 135:1832–41. doi: 10.1002/ijc.28837
139. Russick J, Torset C, Hemery E, Cremer I. NK Cells in the Tumor Microenvironment: Prognostic and Theranostic Impact. Recent Advances and Trends. Semin Immunol (2020) 48:101407. doi: 10.1016/j.smim.2020.101407
140. Russick J, Joubert P-E, Gillard-Bocquet M, Torset C, Meylan M, Petitprez F, et al. Natural Killer Cells in the Human Lung Tumor Microenvironment Display Immune Inhibitory Functions. J Immunother Cancer (2020) 8:e001054. doi: 10.1136/jitc-2020-001054
141. Riggan L, Shah S, O’Sullivan TE. Arrested Development: Suppression of NK Cell Function in the Tumor Microenvironment. Clin Transl Immunol (2021) 10:e1238. doi: 10.1002/cti2.1238
142. Regis S, Dondero A, Caliendo F, Bottino C, Castriconi R. NK Cell Function Regulation by TGF-β-Induced Epigenetic Mechanisms. Front Immunol (2020) 11:311. doi: 10.3389/fimmu.2020.00311
143. Lazarova M, Steinle A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front Immunol (2019) 10:2689. doi: 10.3389/fimmu.2019.02689
144. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta Utilizes SMAD3 to Inhibit CD16-Mediated IFN-Gamma Production and Antibody-Dependent Cellular Cytotoxicity in Human NK Cells. J Immunol (2008) 181:3784–92. doi: 10.4049/jimmunol.181.6.3784
145. Donatelli SS, Zhou J-M, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. Tgf-β-Inducible microRNA-183 Silences Tumor-Associated Natural Killer Cells. Proc Natl Acad Sci USA (2014) 111:4203–8. doi: 10.1073/pnas.1319269111
146. Viel S, Marçais A, Guimaraes FS-F, Loftus R, Rabilloud J, Grau M, et al. TGF-β Inhibits the Activation and Functions of NK Cells by Repressing the mTOR Pathway. Sci Signal (2016) 9:ra19. doi: 10.1126/scisignal.aad1884
147. Dasgupta S, Bhattacharya-Chatterjee M, O’Malley BW, Chatterjee SK. Inhibition of NK Cell Activity Through TGF-Beta 1 by Down-Regulation of NKG2D in a Murine Model of Head and Neck Cancer. J Immunol (2005) 175:5541–50. doi: 10.4049/jimmunol.175.8.5541
148. Peng L-S, Zhang J-Y, Teng Y-S, Zhao Y-L, Wang T-T, Mao F-Y, et al. Tumor-Associated Monocytes/Macrophages Impair NK-Cell Function Via TGFβ1 in Human Gastric Cancer. Cancer Immunol Res (2017) 5:248–56. doi: 10.1158/2326-6066.CIR-16-0152
149. Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, et al. TGF-Beta and Metalloproteinases Differentially Suppress NKG2D Ligand Surface Expression on Malignant Glioma Cells. Brain: J Neurol (2006) 129:2416–25. doi: 10.1093/brain/awl205
150. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming Growth Factor Beta 1 Inhibits Expression of NKp30 and NKG2D Receptors: Consequences for the NK-Mediated Killing of Dendritic Cells. Proc Natl Acad Sci USA (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
151. Slattery K, Woods E, Zaiatz-Bittencourt V, Marks S, Chew S, Conroy M, et al. Tgfβ Drives NK Cell Metabolic Dysfunction in Human Metastatic Breast Cancer. J Immunother Cancer (2021) 9:e002044. doi: 10.1136/jitc-2020-002044
152. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ Regulatory T Cells Inhibit Natural Killer Cell Functions in a Transforming Growth Factor-Beta-Dependent Manner. J Exp Med (2005) 202:1075–85. doi: 10.1084/jem.20051511
153. Smyth MJ, Teng MWL, Swann J, Kyparissoudis K, Godfrey DI, Hayakawa Y. CD4+CD25+ T Regulatory Cells Suppress NK Cell-Mediated Immunotherapy of Cancer. J Immunol (2006) 176:1582–7. doi: 10.4049/jimmunol.176.3.1582
154. Brownlie D, Doughty-Shenton D, Yh Soong D, Nixon C, O Carragher N, M Carlin L, et al. Metastasis-Associated Macrophages Constrain Antitumor Capability of Natural Killer Cells in the Metastatic Site at Least Partially by Membrane Bound Transforming Growth Factor β. J Immunother Cancer (2021) 9:e001740. doi: 10.1136/jitc-2020-001740
155. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells Through Membrane-Bound TGF-beta1. J Immunol (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
156. Lind H, Gameiro SR, Jochems C, Donahue RN, Strauss J, Gulley JL, et al. Dual Targeting of TGF-β and PD-L1 Via a Bifunctional Anti-PD-L1/TGF-βRII Agent: Status of Preclinical and Clinical Advances. J Immunother Cancer (2020) 8:e000433. doi: 10.1136/jitc-2019-000433
157. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFβr1 Blockade With Galunisertib (Ly2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (ch14.18) With Natural Killer Cells. Clin Cancer Res (2017) 23:804–13. doi: 10.1158/1078-0432.CCR-16-1743
158. Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the αv Integrin-TGF-β Axis Improves Natural Killer Cell Function Against Glioblastoma Stem Cells. J Clin Invest (2021), in press, 142116. doi: 10.1172/JCI142116
159. Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The Tryptophan Catabolite L-Kynurenine Inhibits the Surface Expression of NKp46- and NKG2D-Activating Receptors and Regulates NK-cell Function. Blood (2006) 108:4118–25. doi: 10.1182/blood-2006-03-006700
160. Spallanzani RG, Dalotto-Moreno T, Raffo Iraolagoitia XL, Ziblat A, Domaica CI, Avila DE, et al. Expansion of CD11b(+)Ly6G (+)Ly6C (Int) Cells Driven by Medroxyprogesterone Acetate in Mice Bearing Breast Tumors Restrains NK Cell Effector Functions. Cancer Immunol Immunother (2013) 62:1781–95. doi: 10.1007/s00262-013-1483-x
161. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E(2) (PGE2) Suppresses Natural Killer Cell Function Primarily Through the PGE(2) Receptor EP4. Cancer Immunol Immunother (2011) 60:1577–86. doi: 10.1007/s00262-011-1064-9
162. Holt DM, Ma X, Kundu N, Collin PD, Fulton AM. Modulation of Host Natural Killer Cell Functions in Breast Cancer Via Prostaglandin E2 Receptors EP2 and EP4. J Immunother (2012) 35:179–88. doi: 10.1097/CJI.0b013e318247a5e9
163. Van Elssen CH, Vanderlocht J, Oth T, Senden-Gijsbers BL, Germeraad WT, Bos GM. Inflammation-Restraining Effects of Prostaglandin E2 on Natural Killer-Dendritic Cell (NK-DC) Interaction Are Imprinted During DC Maturation. Blood (2011) 118:2473–82. doi: 10.1182/blood-2010-09-307835
164. Ma X, Holt D, Kundu N, Reader J, Goloubeva O, Take Y, et al. A Prostaglandin E (PGE) Receptor EP4 Antagonist Protects Natural Killer Cells From PGE2-Mediated Immunosuppression and Inhibits Breast Cancer Metastasis. Oncoimmunology (2013) 2:e22647. doi: 10.4161/onci.22647
165. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of Tumor-Derived Prostaglandin-E2 Blocks the Induction of Myeloid-Derived Suppressor Cells and Recovers Natural Killer Cell Activity. Clin Cancer Res (2014) 20:4096–106. doi: 10.1158/1078-0432.CCR-14-0635
166. Park A, Lee Y, Kim MS, Kang YJ, Park Y-J, Jung H, et al. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front Immunol (2018) 9:1859. doi: 10.3389/fimmu.2018.01859
167. Wong JL, Obermajer N, Odunsi K, Edwards RP, Kalinski P. Synergistic COX2 Induction by IFNγ and TNFα Self-Limits Type-1 Immunity in the Human Tumor Microenvironment. Cancer Immunol Res (2016) 4:303–11. doi: 10.1158/2326-6066.CIR-15-0157
168. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 Blockade Suppresses Gliomagenesis by Inhibiting Myeloid-Derived Suppressor Cells. Cancer Res (2011) 71:2664–74. doi: 10.1158/0008-5472.CAN-10-3055
169. Wiemann K, Mittrucker H-W, Feger U, Welte SA, Yokoyama WM, Spies T, et al. Systemic NKG2D Down-Regulation Impairs NK and CD8 T Cell Responses In Vivo. J Immunol (2005) 175:720–9. doi: 10.4049/jimmunol.175.2.720
170. Koch C, Kim Y, Zöller T, Born C, Steinle A. Chronic NKG2D Engagement In Vivo Differentially Impacts NK Cell Responsiveness by Activating NK Receptors. Front Immunol (2017) 8:1466. doi: 10.3389/fimmu.2017.01466
171. Merino A, Zhang B, Dougherty P, Luo X, Wang J, Blazar BR, et al. Chronic Stimulation Drives Human NK Cell Dysfunction and Epigenetic Reprograming. J Clin Invest (2019) 129:3770–85. doi: 10.1172/JCI125916
172. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
173. Eisinger S, Sarhan D, Boura VF, Ibarlucea-Benitez I, Tyystjärvi S, Oliynyk G, et al. Targeting a Scavenger Receptor on Tumor-Associated Macrophages Activates Tumor Cell Killing by Natural Killer Cells. Proc Natl Acad Sci USA (2020) 117:32005–16. doi: 10.1073/pnas.2015343117
174. Muntasell A, Ochoa MC, Cordeiro L, Berraondo P, López-Díaz de Cerio A, Cabo M, et al. Targeting NK-Cell Checkpoints for Cancer Immunotherapy. Curr Opin Immunol (2017) 45:73–81. doi: 10.1016/j.coi.2017.01.003
175. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: New Checkpoint Receptor Targets for Cancer Immunotherapy. Immunol Rev (2017) 276:112–20. doi: 10.1111/imr.12518
176. Mayes PA, Hance KW, Hoos A. The Promise and Challenges of Immune Agonist Antibody Development in Cancer. Nat Rev Drug Discov (2018) 17:509–27. doi: 10.1038/nrd.2018.75
177. Bi J, Tian Z. Nk Cell Dysfunction and Checkpoint Immunotherapy. Front Immunol (2019) 10:1999. doi: 10.3389/fimmu.2019.01999
178. Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers (2019) 11:877. doi: 10.3390/cancers11060877
179. Sordo-Bahamonde C, Lorenzo-Herrero S, González-Rodríguez AP, Payer ÁR, González-García E, López-Soto A, et al. LAG-3 Blockade With Relatlimab (Bms-986016) Restores Anti-Leukemic Responses in Chronic Lymphocytic Leukemia. Cancers (2021) 13:2112. doi: 10.3390/cancers13092112
180. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res (2016) 4:124–35. doi: 10.1158/2326-6066.CIR-15-0151
181. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased Expression of Programmed Cell Death Protein 1 on NK Cells Inhibits NK-Cell-Mediated Anti-Tumor Function and Indicates Poor Prognosis in Digestive Cancers. Oncogene (2017) 36:6143–53. doi: 10.1038/onc.2017.209
182. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault M-C, Trevino TN, et al. Contribution of NK Cells to Immunotherapy Mediated by PD-1/PD-L1 Blockade. J Clin Invest (2018) 128:4654–68. doi: 10.1172/JCI99317
183. Juliá EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an IgG1 Anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front Immunol (2018) 9:2140. doi: 10.3389/fimmu.2018.02140
184. Hicks KC, Fantini M, Donahue RN, Schwab A, Knudson KM, Tritsch SR, et al. Epigenetic Priming of Both Tumor and NK Cells Augments Antibody-Dependent Cellular Cytotoxicity Elicited by the Anti-PD-L1 Antibody Avelumab Against Multiple Carcinoma Cell Types. Oncoimmunology (2018) 7:e1466018. doi: 10.1080/2162402X.2018.1466018
185. Makowska A, Meier S, Shen L, Busson P, Baloche V, Kontny U. Anti-PD-1 Antibody Increases NK Cell Cytotoxicity Towards Nasopharyngeal Carcinoma Cells in the Context of Chemotherapy-Induced Upregulation of PD-1 and PD-L1. Cancer Immunol Immunother (2021) 70:323–36. doi: 10.1007/s00262-020-02681-x
186. Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. Pd-L1 Mediates Dysfunction in Activated PD-1+ NK Cells in Head and Neck Cancer Patients. Cancer Immunol Res (2018) 6:1548–60. doi: 10.1158/2326-6066.CIR-18-0062
187. Trefny MP, Kaiser M, Stanczak MA, Herzig P, Savic S, Wiese M, et al. PD-1+ Natural Killer Cells in Human Non-Small Cell Lung Cancer Can Be Activated by PD-1/PD-L1 Blockade. Cancer Immunol Immunother (2020) 69:1505–17. doi: 10.1007/s00262-020-02558-z
188. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the Checkpoint Receptor TIGIT Prevents NK Cell Exhaustion and Elicits Potent Anti-Tumor Immunity. Nat Immunol (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
189. Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The Receptors CD96 and CD226 Oppose Each Other in the Regulation of Natural Killer Cell Functions. Nat Immunol (2014) 15:431–8. doi: 10.1038/ni.2850
190. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology (2019) 70:168–83. doi: 10.1002/hep.30347
191. Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A Expression Contributes to NK Cell Exhaustion and Predicts a Poor Prognosis of Patients With Liver Cancer. Oncoimmunology (2017) 6:e1264562. doi: 10.1080/2162402X.2016.1264562
192. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A Mab Is a Checkpoint Inhibitor That Promotes Anti-Tumor Immunity by Unleashing Both T and NK Cells. Cell (2018) 175:1731–43.e13. doi: 10.1016/j.cell.2018.10.014
193. van Hall T, André P, Horowitz A, Ruan DF, Borst L, Zerbib R, et al. Monalizumab: Inhibiting the Novel Immune Checkpoint NKG2A. J Immunother Cancer (2019) 7:263. doi: 10.1186/s40425-019-0761-3
194. Borst L, van der Burg SH, van Hall T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin Cancer Res (2020) 26:5549–56. doi: 10.1158/1078-0432.CCR-19-2095
195. Romagné F, André P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical Characterization of 1-7F9, a Novel Human anti-KIR Receptor Therapeutic Antibody That Augments Natural Killer-Mediated Killing of Tumor Cells. Blood (2009) 114:2667–77. doi: 10.1182/blood-2009-02-206532
196. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR Antibody Enhancement of Anti-Lymphoma Activity of Natural Killer Cells as Monotherapy and in Combination With anti-CD20 Antibodies. Blood (2014) 123:678–86. doi: 10.1182/blood-2013-08-519199
197. Korde N, Carlsten M, Lee M-J, Minter A, Tan E, Kwok M, et al. A Phase II Trial of pan-KIR2D Blockade With IPH2101 in Smoldering Multiple Myeloma. Haematologica (2014) 99:e81–3. doi: 10.3324/haematol.2013.103085
198. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint Inhibition of KIR2D With the Monoclonal Antibody IPH2101 Induces Contraction and Hyporesponsiveness of NK Cells in Patients With Myeloma. Clin Cancer Res (2016) 22:5211–22. doi: 10.1158/1078-0432.CCR-16-1108
199. Kang JH, Bluestone JA, Young A. Predicting and Preventing Immune Checkpoint Inhibitor Toxicity: Targeting Cytokines. Trends Immunol (2021) 42:293–311. doi: 10.1016/j.it.2021.02.006
200. Dougan M, Pietropaolo M. Time to Dissect the Autoimmune Etiology of Cancer Antibody Immunotherapy. J Clin Invest (2020) 130:51–61. doi: 10.1172/JCI131194
201. Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chávez A, Keegan N, Khamashta MA, et al. Immune-Related Adverse Events of Checkpoint Inhibitors. Nat Rev Dis Primers (2020) 6:38. doi: 10.1038/s41572-020-0160-6
202. Perdigoto AL, Kluger H, Herold KC. Adverse Events Induced by Immune Checkpoint Inhibitors. Curr Opin Immunol (2021) 69:29–38. doi: 10.1016/j.coi.2021.02.002
203. Bagchi S, Yuan R, Engleman EG. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
204. Jia X-H, Geng L-Y, Jiang P-P, Xu H, Nan K-J, Yao Y, et al. The Biomarkers Related to Immune Related Adverse Events Caused by Immune Checkpoint Inhibitors. J Exp Clin Cancer Res (2020) 39:284. doi: 10.1186/s13046-020-01749-x
205. Xu Y, Fu Y, Zhu B, Wang J, Zhang B. Predictive Biomarkers of Immune Checkpoint Inhibitors-Related Toxicities. Front Immunol (2020) 11:2023. doi: 10.3389/fimmu.2020.02023
206. Muntasell A, Cabo M, Servitja S, Tusquets I, Martínez-García M, Rovira A, et al. Interplay Between Natural Killer Cells and Anti-HER2 Antibodies: Perspectives for Breast Cancer Immunotherapy. Front Immunol (2017) 8:1544. doi: 10.3389/fimmu.2017.01544
207. Ochoa MC, Minute L, Rodriguez I, Garasa S, Perez-Ruiz E, Inoges S, et al. Antibody-Dependent Cell Cytotoxicity: Immunotherapy Strategies Enhancing Effector NK Cells. Immunol Cell Biol (2017) 95:347–55. doi: 10.1038/icb.2017.6
208. Zhang M, Wen B, Anton OM, Yao Z, Dubois S, Ju W, et al. IL-15 Enhanced Antibody-Dependent Cellular Cytotoxicity Mediated by NK Cells and Macrophages. Proc Natl Acad Sci USA (2018) 115:E10915–24. doi: 10.1073/pnas.1811615115
209. McMichael EL, Benner B, Atwal LS, Courtney NB, Mo X, Davis ME, et al. A Phase I/Ii Trial of Cetuximab in Combination With Interleukin-12 Administered to Patients With Unresectable Primary or Recurrent Head and Neck Squamous Cell Carcinoma. Clin Cancer Res (2019) 25:4955–65. doi: 10.1158/1078-0432.CCR-18-2108
210. Schmied BJ, Lutz MS, Riegg F, Zekri L, Heitmann JS, Bühring H-J, et al. Induction of NK Cell Reactivity Against B-Cell Acute Lymphoblastic Leukemia by an Fc-Optimized FLT3 Antibody. Cancers (2019) 11:1966. doi: 10.3390/cancers11121966
211. Dixon KJ, Wu J, Walcheck B. Engineering Anti-Tumor Monoclonal Antibodies and Fc Receptors to Enhance ADCC by Human Nk Cells. Cancers (2021) 13:312. doi: 10.3390/cancers13020312
212. Yu T, Chaganty B, Lin L, Xing L, Ramakrishnan B, Wen K, et al. VIS832, a Novel CD138-Targeting Monoclonal Antibody, Potently Induces Killing of Human Multiple Myeloma and Further Synergizes With IMiDs or Bortezomib In Vitro and In Vivo. Blood Cancer J (2020) 10:110. doi: 10.1038/s41408-020-00378-z
213. Zhu C, Song Z, Wang A, Srinivasan S, Yang G, Greco R, et al. Isatuximab Acts Through Fc-Dependent, Independent, and Direct Pathways to Kill Multiple Myeloma Cells. Front Immunol (2020) 11:1771. doi: 10.3389/fimmu.2020.01771
214. Mani R, Rajgolikar G, Nunes J, Zapolnik K, Wasmuth R, Mo X, et al. Fc-Engineered Anti-CD33 Monoclonal Antibody Potentiates Cytotoxicity of Membrane-Bound Interleukin-21 Expanded Natural Killer Cells in Acute Myeloid Leukemia. Cytotherapy (2020) 22:369–76. doi: 10.1016/j.jcyt.2020.02.001
215. Casneuf T, Xu XS, Adams HC, Axel AE, Chiu C, Khan I, et al. Effects of Daratumumab on Natural Killer Cells and Impact on Clinical Outcomes in Relapsed or Refractory Multiple Myeloma. Blood Adv (2017) 1:2105–14. doi: 10.1182/bloodadvances.2017006866
216. Jinushi M, Hodi FS, Dranoff G. Therapy-Induced Antibodies to MHC Class I Chain-Related Protein A Antagonize Immune Suppression and Stimulate Antitumor Cytotoxicity. Proc Natl Acad Sci USA (2006) 103:9190–5. doi: 10.1073/pnas.0603503103
217. Dranoff G. Targets of Protective Tumor Immunity. Ann N Y Acad Sci (2009) 1174:74–80. doi: 10.1111/j.1749-6632.2009.04938.x
218. Acheampong DO, Tang M, Wang Y, Zhao X, Xie W, Chen Z, et al. A Novel Fusion Antibody Exhibits Antiangiogenic Activity and Stimulates NK Cell-Mediated Immune Surveillance Through Fused Nkg2d Ligand. J Immunother (2017) 40:94–103. doi: 10.1097/CJI.0000000000000157
219. Wang T, Sun F, Wang Y, Jiang J, Pan M, Yuan M, et al. NKG2D Immunoligand rG7S-MICA Enhances Nk Cell-Mediated Immunosurveillance in Colorectal Carcinoma. J Immunother (2018) 41:109–17. doi: 10.1097/CJI.0000000000000215
220. Zou Y, Luo W, Guo J, Luo Q, Deng M, Lu Z, et al. NK Cell-Mediated Anti-Leukemia Cytotoxicity Is Enhanced Using a NKG2D Ligand MICA and Anti-CD20 scFv Chimeric Protein. Eur J Immunol (2018) 48:1750–63. doi: 10.1002/eji.201847550
221. Lu S, Zhang J, Liu D, Li G, Staveley-O’Carroll KF, Li Z, et al. Nonblocking Monoclonal Antibody Targeting Soluble MIC Revamps Endogenous Innate and Adaptive Antitumor Responses and Eliminates Primary and Metastatic Tumors. Clin Cancer Res (2015) 21:4819–30. doi: 10.1158/1078-0432.CCR-15-0845
222. Zhang J, Liu D, Li G, Staveley-O’Carroll KF, Graff JN, Li Z, et al. Antibody-Mediated Neutralization of Soluble MIC Significantly Enhances CTLA4 Blockade Therapy. Sci Adv (2017) 3:e1602133. doi: 10.1126/sciadv.1602133
223. Ferrari de Andrade L, Tay RE, Pan D, Luoma AM, Ito Y, Badrinath S, et al. Antibody-Mediated Inhibition of MICA and MICB Shedding Promotes NK Cell-Driven Tumor Immunity. Science (2018) 359:1537–42. doi: 10.1126/science.aao0505
224. Ferrari de Andrade L, Kumar S, Luoma AM, Ito Y, Alves da Silva PH, Pan D, et al. Inhibition of MICA and MICB Shedding Elicits NK-Cell-Mediated Immunity Against Tumors Resistant to Cytotoxic T Cells. Cancer Immunol Res (2020) 8:769–80. doi: 10.1158/2326-6066.CIR-19-0483
225. Wu X, Zhang Y, Li Y, Schmidt-Wolf IGH. Increase of Antitumoral Effects of Cytokine-Induced Killer Cells by Antibody-Mediated Inhibition of MICA Shedding. Cancers (2020) 12:1818. doi: 10.3390/cancers12071818
226. Mantovani S, Varchetta S, Mele D, Donadon M, Torzilli G, Soldani C, et al. An Anti-MICA/B Antibody and IL-15 Rescue Altered NKG2D-Dependent NK Cell Responses in Hepatocellular Carcinoma. Cancers (2020) 12:3583. doi: 10.3390/cancers12123583
227. Tao K, He M, Tao F, Xu G, Ye M, Zheng Y, et al. Development of NKG2D-based Chimeric Antigen Receptor-T Cells for Gastric Cancer Treatment. Cancer Chemother Pharmacol (2018) 82:815–27. doi: 10.1007/s00280-018-3670-0
228. Han Y, Xie W, Song D-G, Powell DJ. Control of Triple-Negative Breast Cancer Using Ex Vivo Self-Enriched, Costimulated NKG2D CAR T Cells. J Hematol Oncol (2018) 11:92. doi: 10.1186/s13045-018-0635-z
229. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. Nkg2d-Based Car T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res (2018) 78:1031–43. doi: 10.1158/0008-5472.CAN-17-1788
230. Spear P, Barber A, Rynda-Apple A, Sentman CL. Nkg2d CAR T-Cell Therapy Inhibits the Growth of NKG2D Ligand Heterogeneous Tumors. Immunol Cell Biol (2013) 91:435–40. doi: 10.1038/icb.2013.17
231. He C, Zhou Y, Li Z, Farooq MA, Ajmal I, Zhang H, et al. Co-Expression of IL-7 Improves NKG2D-Based Car T Cell Therapy on Prostate Cancer by Enhancing the Expansion and Inhibiting the Apoptosis and Exhaustion. Cancers (2020) 12:1969. doi: 10.3390/cancers12071969
232. Sun B, Yang D, Dai H, Liu X, Jia R, Cui X, et al. Eradication of Hepatocellular Carcinoma by NKG2D-Based CAR-T Cells. Cancer Immunol Res (2019) 7:1813–23. doi: 10.1158/2326-6066.CIR-19-0026
233. Yang D, Sun B, Dai H, Li W, Shi L, Zhang P, et al. T Cells Expressing NKG2D Chimeric Antigen Receptors Efficiently Eliminate Glioblastoma and Cancer Stem Cells. J Immunother Cancer (2019) 7:171. doi: 10.1186/s40425-019-0642-9
234. Deng X, Gao F, Li N, Li Q, Zhou Y, Yang T, et al. Antitumor Activity of NKG2D CAR-T Cells Against Human Colorectal Cancer Cells In Vitro and In Vivo. Am J Cancer Res (2019) 9:945–58.
235. Driouk L, Gicobi JK, Kamihara Y, Rutherford K, Dranoff G, Ritz J, et al. Chimeric Antigen Receptor T Cells Targeting NKG2D-Ligands Show Robust Efficacy Against Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. Front Immunol (2020) 11:580328. doi: 10.3389/fimmu.2020.580328
236. Cook WJ, Choi Y, Gacerez A, Bailey-Kellogg C, Sentman CL. A Chimeric Antigen Receptor That Binds to a Conserved Site on MICA. Immunohorizons (2020) 4:597–607. doi: 10.4049/immunohorizons.2000041
237. Ng Y-Y, Tay JCK, Li Z, Wang J, Zhu J, Wang S. T Cells Expressing NKG2D CAR With a DAP12 Signaling Domain Stimulate Lower Cytokine Production While Effective in Tumor Eradication. Mol Ther (2021) 29:75–85. doi: 10.1016/j.ymthe.2020.08.016
238. Zhang Y, Li X, Zhang J, Mao L. Novel Cellular Immunotherapy Using NKG2D CAR-T for the Treatment of Cervical Cancer. BioMed Pharmacother (2020) 131:110562. doi: 10.1016/j.biopha.2020.110562
239. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther (2019) 27:1114–25. doi: 10.1016/j.ymthe.2019.03.011
240. Upadhaya S, Neftelino ST, Hodge JP, Oliva C, Campbell JR, Yu JX. Combinations Take Centre Stage in PD1/PDL1 Inhibitor Clinical Trials. Nat Rev Drug Discov (2020) 20:168–9. doi: 10.1038/d41573-020-00204-y
241. Adams JL, Smothers J, Srinivasan R, Hoos A. Big Opportunities for Small Molecules in Immuno-Oncology. Nat Rev Drug Discov (2015) 14:603–22. doi: 10.1038/nrd4596
242. Ngiow SF, Young A. Re-Education of the Tumor Microenvironment With Targeted Therapies and Immunotherapies. Front Immunol (2020) 11:1633. doi: 10.3389/fimmu.2020.01633
243. Liu KX, Joshi S. “Re-Educating” Tumor Associated Macrophages as a Novel Immunotherapy Strategy for Neuroblastoma. Front Immunol (2020) 11:1947. doi: 10.3389/fimmu.2020.01947
244. Dubois SP, Miljkovic MD, Fleisher TA, Pittaluga S, Hsu-Albert J, Bryant BR, et al. Short-Course IL-15 Given as a Continuous Infusion Led to a Massive Expansion of Effective NK Cells: Implications for Combination Therapy With Antitumor Antibodies. J Immunother Cancer (2021) 9:e002193. doi: 10.1136/jitc-2020-002193
245. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA Damage Pathway Regulates Innate Immune System Ligands of the NKG2D Receptor. Nature (2005) 436:1186–90. doi: 10.1038/nature03884
246. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural Killer Cell-Mediated Lysis of Hepatoma Cells Via Specific Induction of NKG2D Ligands by the Histone Deacetylase Inhibitor Sodium Valproate. Cancer Res (2005) 65:6321–9. doi: 10.1158/0008-5472.CAN-04-4252
247. Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer Cells Become Susceptible to Natural Killer Cell Killing After Exposure to Histone Deacetylase Inhibitors Due to Glycogen Synthase Kinase-3-Dependent Expression of MHC Class I-Related Chain A and B. Cancer Res (2005) 65:11136–45. doi: 10.1158/0008-5472.CAN-05-0599
248. Bolden JE, Peart MJ, Johnstone RW. Anticancer Activities of Histone Deacetylase Inhibitors. Nat Rev Drug Discov (2006) 5:769–84. doi: 10.1038/nrd2133
249. Minucci S, Pelicci PG. Histone Deacetylase Inhibitors and the Promise of Epigenetic (and More) Treatments for Cancer. Nat Rev Cancer (2006) 6:38–51. doi: 10.1038/nrc1779
250. Kato N, Tanaka J, Sugita J, Toubai T, Miura Y, Ibata M, et al. Regulation of the Expression of MHC Class I-Related Chain A, B (MICA, MICB) Via Chromatin Remodeling and Its Impact on the Susceptibility of Leukemic Cells to the Cytotoxicity of NKG2D-Expressing Cells. Leukemia (2007) 21:2103–8. doi: 10.1038/sj.leu.2404862
251. Huang B, Sikorski R, Sampath P, Thorne SH. Modulation of NKG2D-ligand Cell Surface Expression Enhances Immune Cell Therapy of Cancer. J Immunother (2011) 34:289–96. doi: 10.1097/CJI.0b013e31820e1b0d
252. Rossi LE, Avila DE, Spallanzani RG, Ziblat A, Fuertes MB, Lapyckyj L, et al. Histone Deacetylase Inhibitors Impair NK Cell Viability and Effector Functions Through Inhibition of Activation and Receptor Expression. J Leukoc Biol (2012) 91:321–31. doi: 10.1189/jlb.0711339
253. Höring E, Podlech O, Silkenstedt B, Rota IA, Adamopoulou E, Naumann U. The Histone Deacetylase Inhibitor Trichostatin a Promotes Apoptosis and Antitumor Immunity in Glioblastoma Cells. Anticancer Res (2013) 33:1351–60.
254. Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J. Histone Deacetylase Inhibitors Suppress Natural Killer Cell Cytolytic Activity. FEBS Lett (2007) 581:1317–22. doi: 10.1016/j.febslet.2007.02.045
255. Schmudde M, Braun A, Pende D, Sonnemann J, Klier U, Beck JF, et al. Histone Deacetylase Inhibitors Sensitize Tumour Cells for Cytotoxic Effects of Natural Killer Cells. Cancer Lett (2008) 272:110–21. doi: 10.1016/j.canlet.2008.06.027
256. Bressan A, Bigioni M, Bellarosa D, Nardelli F, Irrissuto C, Maggi CA, et al. Induction of a Less Aggressive Phenotype in Human Colon Carcinoma HCT116 Cells by Chronic Exposure to HDAC Inhibitor SAHA. Oncol Rep (2010) 24:1249–55. doi: 10.3892/or_00000979
257. Berghuis D, Schilham MW, Vos HI, Santos SJ, Kloess S, Buddingh’ EP, et al. Histone Deacetylase Inhibitors Enhance Expression of NKG2D Ligands in Ewing Sarcoma and Sensitize for Natural Killer Cell-Mediated Cytolysis. Clin Sarcoma Res (2012) 2:8. doi: 10.1186/2045-3329-2-8
258. Valés-Gómez M, Chisholm SE, Cassady-Cain RL, Roda-Navarro P, Reyburn HT. Selective Induction of Expression of a Ligand for the NKG2D Receptor by Proteasome Inhibitors. Cancer Res (2008) 68:1546–54. doi: 10.1158/0008-5472.CAN-07-2973
259. Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, et al. NKG2D Ligand Expression in AML Increases in Response to HDAC Inhibitor Valproic Acid and Contributes to Allorecognition by NK-Cell Lines With Single KIR-HLA Class I Specificities. Blood (2008) 111:1428–36. doi: 10.1182/blood-2007-07-101311
260. Poggi A, Catellani S, Garuti A, Pierri I, Gobbi M, Zocchi MR. Effective In Vivo Induction of NKG2D Ligands in Acute Myeloid Leukaemias by All-Trans-Retinoic Acid or Sodium Valproate. Leukemia (2009) 23:641–8. doi: 10.1038/leu.2008.354
261. Zhang C, Wang Y, Zhou Z, Zhang J, Tian Z. Sodium Butyrate Upregulates Expression of NKG2D Ligand MICA/B in HeLa and HepG2 Cell Lines and Increases Their Susceptibility to NK Lysis. Cancer Immunol Immunother (2009) 58:1275–85. doi: 10.1007/s00262-008-0645-8
262. Yamanegi K, Yamane J, Kobayashi K, Kato-Kogoe N, Ohyama H, Nakasho K, et al. Sodium Valproate, a Histone Deacetylase Inhibitor, Augments the Expression of Cell-Surface NKG2D Ligands, MICA/B, Without Increasing Their Soluble Forms to Enhance Susceptibility of Human Osteosarcoma Cells to NK Cell-Mediated Cytotoxicity. Oncol Rep (2010) 24:1621–7. doi: 10.3892/or_00001026
263. Lu X, Ohata K, Kondo Y, Espinoza JL, Qi Z, Nakao S. Hydroxyurea Upregulates NKG2D Ligand Expression in Myeloid Leukemia Cells Synergistically With Valproic Acid and Potentially Enhances Susceptibility of Leukemic Cells to Natural Killer Cell-Mediated Cytolysis. Cancer Sci (2010) 101:609–15. doi: 10.1111/j.1349-7006.2009.01439.x
264. Wu X, Tao Y, Hou J, Meng X, Shi J. Valproic Acid Upregulates NKG2D Ligand Expression Through an ERK-Dependent Mechanism and Potentially Enhances NK Cell-Mediated Lysis of Myeloma. Neoplasia (2012) 14:1178–89. doi: 10.1593/neo.121236
265. Jardine L, Hambleton S, Bigley V, Pagan S, Wang X-N, Collin M. Sensitizing Primary Acute Lymphoblastic Leukemia to Natural Killer Cell Recognition by Induction of NKG2D Ligands. Leuk Lymphoma (2013) 54:167–73. doi: 10.3109/10428194.2012.708026
266. Shi P, Yin T, Zhou F, Cui P, Gou S, Wang C. Valproic Acid Sensitizes Pancreatic Cancer Cells to Natural Killer Cell-Mediated Lysis by Upregulating MICA and MICB Via the PI3K/Akt Signaling Pathway. BMC Cancer (2014) 14:370. doi: 10.1186/1471-2407-14-370
267. Shi X, Li M, Cui M, Niu C, Xu J, Zhou L, et al. Epigenetic Suppression of the Antitumor Cytotoxicity of NK Cells by Histone Deacetylase Inhibitor Valproic Acid. Am J Cancer Res (2016) 6:600–14.
268. Miyashita T, Miki K, Kamigaki T, Makino I, Tajima H, Nakanuma S, et al. Low-Dose Valproic Acid With Low-Dose Gemcitabine Augments MHC Class I-Related Chain A/B Expression Without Inducing the Release of Soluble MHC Class I-Related Chain A/B. Oncol Lett (2017) 14:5918–26. doi: 10.3892/ol.2017.6943
269. Ni L, Wang L, Yao C, Ni Z, Liu F, Gong C, et al. The Histone Deacetylase Inhibitor Valproic Acid Inhibits NKG2D Expression in Natural Killer Cells Through Suppression of STAT3 and HDAC3. Sci Rep (2017) 7:45266. doi: 10.1038/srep45266
270. Bhat J, Dubin S, Dananberg A, Quabius ES, Fritsch J, Dowds CM, et al. Histone Deacetylase Inhibitor Modulates Nkg2d Receptor Expression and Memory Phenotype of Human Gamma/Delta T Cells Upon Interaction With Tumor Cells. Front Immunol (2019) 10:569. doi: 10.3389/fimmu.2019.00569
271. Nwangwu CA, Weiher H, Schmidt-Wolf IGH. Increase of CIK Cell Efficacy by Upregulating Cell Surface MICA and Inhibition of NKG2D Ligand Shedding in Multiple Myeloma. Hematol Oncol (2017) 35:719–25. doi: 10.1002/hon.2326
272. López-Cobo S, Pieper N, Campos-Silva C, García-Cuesta EM, Reyburn HT, Paschen A, et al. Impaired NK Cell Recognition of Vemurafenib-Treated Melanoma Cells Is Overcome by Simultaneous Application of Histone Deacetylase Inhibitors. Oncoimmunology (2018) 7:e1392426. doi: 10.1080/2162402X.2017.1392426
273. Hu X-T, Zhu B-L, Zhao L-G, Wang J-W, Liu L, Lai Y-J, et al. Histone Deacetylase Inhibitor Apicidin Increases Expression of the α-Secretase ADAM10 Through Transcription Factor USF1-Mediated Mechanisms. FASEB J (2017) 31:1482–93. doi: 10.1096/fj.201600961RR
274. Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. Atm-ATR-Dependent Up-Regulation of DNAM-1 and NKG2D Ligands on Multiple Myeloma Cells by Therapeutic Agents Results in Enhanced NK-Cell Susceptibility and Is Associated With a Senescent Phenotype. Blood (2009) 113:3503–11. doi: 10.1182/blood-2008-08-173914
275. Niu C, Jin H, Li M, Zhu S, Zhou L, Jin F, et al. Low-Dose Bortezomib Increases the Expression of NKG2D and DNAM-1 Ligands and Enhances Induced NK and γδ T Cell-Mediated Lysis in Multiple Myeloma. Oncotarget (2017) 8:5954–64. doi: 10.18632/oncotarget.13979
276. Armeanu S, Krusch M, Baltz KM, Weiss TS, Smirnow I, Steinle A, et al. Direct and Natural Killer Cell-Mediated Antitumor Effects of Low-Dose Bortezomib in Hepatocellular Carcinoma. Clin Cancer Res (2008) 14:3520–8. doi: 10.1158/1078-0432.CCR-07-4744
277. Luo D, Dong X-W, Yan B, Liu M, Xue T-H, Liu H, et al. MG132 Selectively Upregulates MICB Through the DNA Damage Response Pathway in A549 Cells. Mol Med Rep (2019) 19:213–20. doi: 10.3892/mmr.2018.9676
278. Huang Y, Wang Y, Li Y, Guo K, He Y. Role of Sorafenib and Sunitinib in the Induction of Expressions of NKG2D Ligands in Nasopharyngeal Carcinoma With High Expression of ABCG2. J Cancer Res Clin Oncol (2011) 137:829–37. doi: 10.1007/s00432-010-0944-2
279. Ke M, Wang H, Zhou Y, Li J, Liu Y, Zhang M, et al. SEP Enhanced the Antitumor Activity of 5-Fluorouracil by Up-Regulating NKG2D/MICA and Reversed Immune Suppression Via Inhibiting ROS and Caspase-3 in Mice. Oncotarget (2016) 7:49509–26. doi: 10.18632/oncotarget.10375
280. Weiss T, Schneider H, Silginer M, Steinle A, Pruschy M, Polić B, et al. NKG2D-Dependent Antitumor Effects of Chemotherapy and Radiotherapy Against Glioblastoma. Clin Cancer Res (2018) 24:882–95. doi: 10.1158/1078-0432.CCR-17-1766
281. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. Perturbation of NK Cell Peripheral Homeostasis Accelerates Prostate Carcinoma Metastasis. J Clin Invest (2013) 123:4410–22. doi: 10.1172/JCI69369
282. Wu J. Antibody Targeting Soluble NKG2D Ligand sMIC Refuels and Invigorates the Endogenous Immune System to Fight Cancer. Oncoimmunology (2016) 5:e1095434. doi: 10.1080/2162402X.2015.1095434
283. Curtin NJ, Szabo C. Poly(ADP-Ribose) Polymerase Inhibition: Past, Present and Future. Nat Rev Drug Discov (2020) 19:711–36. doi: 10.1038/s41573-020-0076-6
284. Appleton KM, Elrod AK, Lassahn KA, Shuford S, Holmes LM, DesRochers TM. Pd-1/Pd-L1 Checkpoint Inhibitors in Combination With Olaparib Display Antitumor Activity in Ovarian Cancer Patient-Derived Three-Dimensional Spheroid Cultures. Cancer Immunol Immunother (2021) 70:843–56. doi: 10.1007/s00262-021-02849-z
285. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. Parp Inhibitor Efficacy Depends on CD8+ T-Cell Recruitment Via Intratumoral Sting Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov (2019) 9:722–37. doi: 10.1158/2159-8290.CD-18-1218
286. Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived Cgamp Triggers a STING-Mediated Interferon Response in Non-Tumor Cells to Activate the NK Cell Response. Immunity (2018) 49:754–63. doi: 10.1016/j.immuni.2018.09.016
287. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang S-Y, et al. Design of Amidobenzimidazole STING Receptor Agonists With Systemic Activity. Nature (2018) 564:439–43. doi: 10.1038/s41586-018-0705-y
288. Wang-Bishop L, Wehbe M, Shae D, James J, Hacker BC, Garland K, et al. Potent STING Activation Stimulates Immunogenic Cell Death to Enhance Antitumor Immunity in Neuroblastoma. J Immunother Cancer (2020) 8:e000282. doi: 10.1136/jitc-2019-000282
289. Lam AR, Le Bert N, Ho SS, Shen YJ, Tang ML, Xiong GM, et al. RAE1 Ligands for the NKG2D Receptor Are Regulated by STING-Dependent DNA Sensor Pathways in Lymphoma. Cancer Res (2014) 74:2193–203. doi: 10.1158/0008-5472.CAN-13-1703
290. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J Exp Med (2005) 202:1691–701. doi: 10.1084/jem.20050915
291. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat Med (2007) 13:54–61. doi: 10.1038/nm1523
292. Obeid M, Panaretakis T, Tesniere A, Joza N, Tufi R, Apetoh L, et al. Leveraging the Immune System During Chemotherapy: Moving Calreticulin to the Cell Surface Converts Apoptotic Death From “Silent” to Immunogenic. Cancer Res (2007) 67:7941–4. doi: 10.1158/0008-5472.CAN-07-1622
293. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat Rev Immunol (2017) 17:97–111. doi: 10.1038/nri.2016.107
294. Vanmeerbeek I, Sprooten J, De Ruysscher D, Tejpar S, Vandenberghe P, Fucikova J, et al. Trial Watch: Chemotherapy-Induced Immunogenic Cell Death in Immuno-Oncology. Oncoimmunology (2020) 9:1703449. doi: 10.1080/2162402X.2019.1703449
295. Fucikova J, Kline JP, Galluzzi L, Spisek R. Calreticulin Arms NK Cells Against Leukemia. Oncoimmunology (2020) 9:1671763. doi: 10.1080/2162402X.2019.1671763
296. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy When Combined With Checkpoint Blockade. Cancer Cell (2021) 39:193–208.e10. doi: 10.1016/j.ccell.2020.11.005
297. Siew Y-Y, Neo S-Y, Yew H-C, Lim S-W, Ng Y-C, Lew S-M, et al. Oxaliplatin Regulates Expression of Stress Ligands in Ovarian Cancer Cells and Modulates Their Susceptibility to Natural Killer Cell-Mediated Cytotoxicity. Int Immunol (2015) 27:621–32. doi: 10.1093/intimm/dxv041
298. Akalu YT, Rothlin CV, Ghosh S. TAM Receptor Tyrosine Kinases as Emerging Targets of Innate Immune Checkpoint Blockade for Cancer Therapy. Immunol Rev (2017) 276:165–77. doi: 10.1111/imr.12522
299. Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM Receptors): Implications for Macrophages in the Tumor Microenvironment. Mol Cancer (2019) 18:94. doi: 10.1186/s12943-019-1022-2
300. Aehnlich P, Powell RM, Peeters MJW, Rahbech A, Thor Straten P. Tam Receptor Inhibition-Implications for Cancer and the Immune System. Cancers (Basel) (2021) 13:1195. doi: 10.3390/cancers13061195
301. Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Furst D, et al. Shedding of Endogenous MHC Class I-Related Chain Molecules A and B From Different Human Tumor Entities: Heterogeneous Involvement of the “A Disintegrin and Metalloproteases” 10 and 17. Int J Cancer (2013) 133:1557–66. doi: 10.1002/ijc.28174
302. Liu G, Atteridge CL, Wang X, Lundgren AD, Wu JD. The Membrane Type Matrix Metalloproteinase MMP14 Mediates Constitutive Shedding of MHC Class I Chain-Related Molecule A Independent of A Disintegrin and Metalloproteinases. J Immunol (2010) 184:3346–50. doi: 10.4049/jimmunol.0903789
303. Zingoni A, Vulpis E, Nardone I, Soriani A, Fionda C, Cippitelli M, et al. Targeting NKG2D and NKp30 Ligands Shedding to Improve Nk Cell-Based Immunotherapy. Crit Rev Immunol (2016) 36:445–60. doi: 10.1615/CritRevImmunol.2017020166
304. Wu JD, Atteridge CL, Wang X, Seya T, Plymate SR. Obstructing Shedding of the Immunostimulatory MHC Class I Chain-Related Gene B Prevents Tumor Formation. Clin Cancer Res (2009) 15:632–40. doi: 10.1158/1078-0432.CCR-08-1305
305. Sun D, Wang X, Zhang H, Deng L, Zhang Y. MMP9 Mediates MICA Shedding in Human Osteosarcomas. Cell Biol Int (2011) 35:569–74. doi: 10.1042/CBI20100431
306. Le Maux Chansac B, Missé D, Richon C, Vergnon I, Kubin M, Soria J-C, et al. Potentiation of NK Cell-Mediated Cytotoxicity in Human Lung Adenocarcinoma: Role of NKG2D-Dependent Pathway. Int Immunol (2008) 20:801–10. doi: 10.1093/intimm/dxn038
307. Boutet P, Agüera-González S, Atkinson S, Pennington CJ, Edwards DR, Murphy G, et al. Cutting Edge: The Metalloproteinase ADAM17/TNF-Alpha-Converting Enzyme Regulates Proteolytic Shedding of the MHC Class I-Related Chain B Protein. J Immunol (2009) 182:49–53. doi: 10.4049/jimmunol.182.1.49
308. Shiraishi K, Mimura K, Kua LF, Koh V, Siang LK, Nakajima S, et al. Inhibition of MMP Activity Can Restore NKG2D Ligand Expression in Gastric Cancer, Leading to Improved NK Cell Susceptibility. J Gastroenterol (2016) 51:1101–11. doi: 10.1007/s00535-016-1197-x
309. Sekiba K, Otsuka M, Seimiya T, Tanaka E, Funato K, Miyakawa Y, et al. The Fatty-Acid Amide Hydrolase Inhibitor URB597 Inhibits MICA/B Shedding. Sci Rep (2020) 10:15556. doi: 10.1038/s41598-020-72688-y
310. Tang H, Sampath P, Yan X, Thorne SH. Potential for Enhanced Therapeutic Activity of Biological Cancer Therapies With Doxycycline Combination. Gene Ther (2013) 20:770–8. doi: 10.1038/gt.2012.96
311. Camodeca C, Nuti E, Tepshi L, Boero S, Tuccinardi T, Stura EA, et al. Discovery of a New Selective Inhibitor of A Disintegrin And Metalloprotease 10 (Adam-10) Able to Reduce the Shedding of NKG2D Ligands in Hodgkin’s Lymphoma Cell Models. Eur J Med Chem (2016) 111:193–201. doi: 10.1016/j.ejmech.2016.01.053
312. Zocchi MR, Camodeca C, Nuti E, Rossello A, Venè R, Tosetti F, et al. ADAM10 New Selective Inhibitors Reduce NKG2D Ligand Release Sensitizing Hodgkin Lymphoma Cells to NKG2D-Mediated Killing. Oncoimmunology (2016) 5:e1123367. doi: 10.1080/2162402X.2015.1123367
313. Raeeszadeh-Sarmazdeh M, Do LD, Hritz BG. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells (2020) 9:1313. doi: 10.3390/cells9051313
314. Ashworth A, Lord CJ. Synthetic Lethal Therapies for Cancer: What’s Next After PARP Inhibitors? Nat Rev Clin Oncol (2018) 15:564–76. doi: 10.1038/s41571-018-0055-6
315. Ding L, Kim H-J, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. Parp Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep (2018) 25:2972–80. doi: 10.1016/j.celrep.2018.11.054
316. Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. Parpi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of Brcaness. Cancer Res (2019) 79:311–9. doi: 10.1158/0008-5472.CAN-18-1003
317. Stewart RA, Pilié PG, Yap TA. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res (2018) 78:6717–25. doi: 10.1158/0008-5472.CAN-18-2652
318. Lee EK, Konstantinopoulos PA. Combined PARP and Immune Checkpoint Inhibition in Ovarian Cancer. Trends Cancer (2019) 5:524–8. doi: 10.1016/j.trecan.2019.06.004
319. Goncalves A, Mezni E, Bertucci F. Combining Poly(ADP-Ribose) Polymerase Inhibitors and Immune Checkpoint Inhibitors in Breast Cancer: Rationale and Preliminary Clinical Results. Curr Opin Oncol (2020) 32:585–93. doi: 10.1097/CCO.0000000000000680
320. Patel PS, Algouneh A, Hakem R. Exploiting Synthetic Lethality to Target BRCA1/2-Deficient Tumors: Where We Stand. Oncogene (2021) 40:3001–14. doi: 10.1038/s41388-021-01744-2
321. Mehta AK, Cheney EM, Hartl CA, Pantelidou C, Oliwa M, Castrillon JA, et al. Targeting Immunosuppressive Macrophages Overcomes PARP Inhibitor Resistance in BRCA1-Associated Triple-Negative Breast Cancer. Nat Cancer (2021) 2:66–82. doi: 10.1038/s43018-020-00148-7
322. Fenerty KE, Padget M, Wolfson B, Gameiro SR, Su Z, Lee JH, et al. Immunotherapy Utilizing the Combination of Natural Killer- and Antibody Dependent Cellular Cytotoxicity (ADCC)-Mediating Agents With Poly (ADP-Ribose) Polymerase (PARP) Inhibition. J Immunother Cancer (2018) 6:133. doi: 10.1186/s40425-018-0445-4
323. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host Type I IFN Signals Are Required for Antitumor CD8+ T Cell Responses Through CD8{alpha}+ Dendritic Cells. J Exp Med (2011) 208:2005–16. doi: 10.1084/jem.20101159
324. Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I Interferon Response and Innate Immune Sensing of Cancer. Trends Immunol (2013) 34:67–73. doi: 10.1016/j.it.2012.10.004
325. Nicolai CJ, Wolf N, Chang I-C, Kirn G, Marcus A, Ndubaku CO, et al. NK Cells Mediate Clearance of CD8+ T Cell-Resistant Tumors in Response to STING Agonists. Sci Immunol (2020) 5:eaaz2738. doi: 10.1126/sciimmunol.aaz2738
326. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
327. Downey CM, Aghaei M, Schwendener RA, Jirik FR. DMXAA Causes Tumor Site-Specific Vascular Disruption in Murine Non-Small Cell Lung Cancer, and Like the Endogenous Non-Canonical Cyclic Dinucleotide STING Agonist, 2’3’-cGAMP, Induces M2 Macrophage Repolarization. PloS One (2014) 9:e99988. doi: 10.1371/journal.pone.0099988
328. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep (2015) 11:1018–30. doi: 10.1016/j.celrep.2015.04.031
329. Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, et al. Intratumoral Administration of cGAMP Transiently Accumulates Potent Macrophages for Anti-Tumor Immunity at a Mouse Tumor Site. Cancer Immunol Immunother (2017) 66:705–16. doi: 10.1007/s00262-017-1975-1
330. Zhang C-X, Ye S-B, Ni J-J, Cai T-T, Liu Y-N, Huang D-J, et al. STING Signaling Remodels the Tumor Microenvironment by Antagonizing Myeloid-Derived Suppressor Cell Expansion. Cell Death Differ (2019) 26:2314–28. doi: 10.1038/s41418-019-0302-0
331. Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING Activation With T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol Res (2017) 5:676–84. doi: 10.1158/2326-6066.CIR-17-0049
332. Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING Agonist Formulated Cancer Vaccines Can Cure Established Tumors Resistant to PD-1 Blockade. Sci Transl Med (2015) 7:283ra52. doi: 10.1126/scitranslmed.aaa4306
333. Dahal LN, Dou L, Hussain K, Liu R, Earley A, Cox KL, et al. Sting Activation Reverses Lymphoma-Mediated Resistance to Antibody Immunotherapy. Cancer Res (2017) 77:3619–31. doi: 10.1158/0008-5472.CAN-16-2784
334. Idso JM, Lao S, Schloemer NJ, Knipstein J, Burns R, Thakar MS, et al. Entinostat Augments NK Cell Functions Via Epigenetic Upregulation of IFIT1-STING-STAT4 Pathway. Oncotarget (2020) 11:1799–815. doi: 10.18632/oncotarget.27546
335. Naour JL, Zitvogel L, Galluzzi L, Vacchelli E, Kroemer G. Trial Watch: STING Agonists in Cancer Therapy. OncoImmunology (2020) 9:1777624. doi: 10.1080/2162402X.2020.1777624
336. Pan B-S, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. An Orally Available Non-Nucleotide STING Agonist With Antitumor Activity. Science (2020) 369:eaba6098. doi: 10.1126/science.aba6098
337. Truxova I, Kasikova L, Salek C, Hensler M, Lysak D, Holicek P, et al. Calreticulin Exposure on Malignant Blasts Correlates With Improved Natural Killer Cell-Mediated Cytotoxicity in Acute Myeloid Leukemia Patients. Haematologica (2020) 105:1868–78. doi: 10.3324/haematol.2019.223933
338. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis (2020) 11:1013. doi: 10.1038/s41419-020-03221-2
339. Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G. Immunogenic Tumor Cell Death for Optimal Anticancer Therapy: The Calreticulin Exposure Pathway. Clin Cancer Res (2010) 16:3100–4. doi: 10.1158/1078-0432.CCR-09-2891
340. Elion DL, Jacobson ME, Hicks DJ, Rahman B, Sanchez V, Gonzales-Ericsson PI, et al. Therapeutically Active Rig-I Agonist Induces Immunogenic Tumor Cell Killing in Breast Cancers. Cancer Res (2018) 78:6183–95. doi: 10.1158/0008-5472.CAN-18-0730
341. Castiello L, Zevini A, Vulpis E, Muscolini M, Ferrari M, Palermo E, et al. An Optimized Retinoic Acid-Inducible Gene I Agonist M8 Induces Immunogenic Cell Death Markers in Human Cancer Cells and Dendritic Cell Activation. Cancer Immunol Immunother (2019) 68:1479–92. doi: 10.1007/s00262-019-02380-2
342. Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, et al. Magnitude of Therapeutic STING Activation Determines Cd8+ T Cell-Mediated Anti-tumor Immunity. Cell Rep (2018) 25:3074–3085.e5. doi: 10.1016/j.celrep.2018.11.047
343. Minute L, Teijeira A, Sanchez-Paulete AR, Ochoa MC, Alvarez M, Otano I, et al. Cellular Cytotoxicity Is a Form of Immunogenic Cell Death. J Immunother Cancer (2020) 8:e000325. doi: 10.1136/jitc-2019-000325
344. Rothlin CV, Carrera-Silva EA, Bosurgi L, Ghosh S. TAM Receptor Signaling in Immune Homeostasis. Annu Rev Immunol (2015) 33:355–91. doi: 10.1146/annurev-immunol-032414-112103
345. Jiang Z, Lim S-O, Yan M, Hsu JL, Yao J, Wei Y, et al. TYRO3 Induces Anti–PD-1/PD-L1 Therapy Resistance by Limiting Innate Immunity and Tumoral Ferroptosis. J Clin Invest (2021) 131:139434. doi: 10.1172/JCI139434
346. Zhou Y, Fei M, Zhang G, Liang W-C, Lin W, Wu Y, et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2x7r-Dependent STING Activation by Tumor-Derived Cgamp. Immunity (2020) 52:357–73.e9. doi: 10.1016/j.immuni.2020.01.014
347. Yokoyama Y, Lew ED, Seelige R, Tindall EA, Walsh C, Fagan PC, et al. Immuno-Oncological Efficacy of RXDX-106, a Novel Tam (Tyro3, AXL, Mer) Family Small-Molecule Kinase Inhibitor. Cancer Res (2019) 79:1996–2008. doi: 10.1158/0008-5472.CAN-18-2022
348. Cresswell GM, Wang B, Kischuk EM, Broman MM, Alfar RA, Vickman RE, et al. Folate Receptor Beta Designates Immunosuppressive Tumor-Associated Myeloid Cells That Can Be Reprogrammed With Folate-Targeted Drugs. Cancer Res (2021) 81:671–84. doi: 10.1158/0008-5472.CAN-20-1414
349. Sathe P, Delconte RB, Souza-Fonseca-Guimaraes F, Seillet C, Chopin M, Vandenberg CJ, et al. Innate Immunodeficiency Following Genetic Ablation of Mcl1 in Natural Killer Cells. Nat Commun (2014) 5:4539. doi: 10.1038/ncomms5539
350. López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
Keywords: NK cells, NKG2D, MICA, tumor immunity, immuno-oncology
Citation: Fuertes MB, Domaica CI and Zwirner NW (2021) Leveraging NKG2D Ligands in Immuno-Oncology. Front. Immunol. 12:713158. doi: 10.3389/fimmu.2021.713158
Received: 21 May 2021; Accepted: 02 July 2021;
Published: 29 July 2021.
Edited by:
Chanvit Leelayuwat, Khon Kaen University, ThailandReviewed by:
Jörg Wischhusen, University Hospital Würzburg, GermanyCopyright © 2021 Fuertes, Domaica and Zwirner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Norberto Walter Zwirner, bnp3aXJuZXJAaWJ5bWUuY29uY2V0Lmdvdi5hcg==, orcid.org/0000-0001-7098-359X
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.