 Sohei Makita1
Sohei Makita1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 01 July 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.711633
This article is part of the Research Topic Post-Transcriptional Regulation of Immune Responses View all 11 articles
Post-transcriptional regulation is involved in the regulation of many inflammatory genes. Zinc finger protein 36 (ZFP36) family proteins are RNA-binding proteins involved in messenger RNA (mRNA) metabolism pathways. The ZFP36 family is composed of ZFP36 (also known as tristetraprolin, TTP), ZFP36L1, ZFP36L2, and ZFP36L3 (only in rodents). The ZFP36 family proteins contain two tandemly repeated CCCH-type zinc-finger motifs, bind to adenine uridine-rich elements in the 3’-untranslated regions (3’ UTR) of specific mRNA, and lead to target mRNA decay. Although the ZFP36 family members are structurally similar, they are known to play distinct functions and regulate different target mRNAs, probably due to their cell-type-specific expression patterns. For instance, ZFP36 has been well-known to function as an anti-inflammatory modulator in murine models of systemic inflammatory diseases by down-regulating the production of various pro-inflammatory cytokines, including TNF-α. Meanwhile, ZFP36L1 is required for the maintenance of the marginal-zone B cell compartment. Recently, we found that ZFP36L2 reduces the expression of Ikzf2 (encoding HELIOS) and suppresses regulatory T cell function. This review summarizes the current understanding of the post-transcriptional regulation of immunological responses and inflammatory diseases by RNA-binding ZFP36 family proteins.
For many years, the importance of post-transcriptional regulation of mRNAs has not been fully recognized in the immune system. However, with the advance in functional analyses of RNA-binding proteins (RBPs), the importance of post-transcriptional regulation in immune system regulation has come to the fore. RBPs are critical effectors of gene expression of many genes and form regulatory networks to maintain cell homeostasis. RBPs recognize target RNA with the RNA-recognition domain (1). RBPs also have binding domains with other proteins, and these interactions enable them to fulfill their regulatory functions (2).
Recent analyses have shown that RBPs are remarkably involved in regulating various cell type-specific functions (3). Among RBPs, ZFP36 family proteins including ZFP36, known as tristetraprolin (TTP), are characterized by the presence of one or more CCCH-type zinc finger domain(s) that contain three cysteines (C) and one histidine (H) residues. ZFP36 family proteins bind to adenylate-uridylate-rich elements (AREs) in the 3’-untranslated region (3’ UTR) of a target mRNA, leading to the decay of the mRNA (4). Although the ZFP36 family members are structurally similar, they play different roles and regulate different target mRNAs, probably due to their cell type-specific expression patterns (5). For instance, ZFP36 plays a significant role in regulating immune responses and inflammatory diseases by inhibiting the production of various inflammatory cytokines such as TNF-α in macrophages (6).
ZFP36L1 is known to be required for the maintenance of the marginal zone B cell compartment by limiting the expression of the transcription factors such as Kruppel-like factor 2 (KLF2) and interferon regulatory factor 8 (IRF8) (7). We have recently reported that ZFP36L2 down-regulates the expression of Ikzf2 (encoding HELIOS) and suppresses the function of induced regulatory T cells (iTregs) (8). In this review, we discuss our current understanding of post-transcriptional regulation in immune responses by RNA-binding ZFP36 family proteins. We also discuss the control of those protein expressions as potential therapeutic strategies for human inflammatory diseases.
RBPs recognize cis-elements or specific structures in the 5’ UTR, 3’ UTR, or intron of mRNA at each step of RNA metabolism (9). Adenylate-uridylate-rich elements (AU-rich elements; AREs) characterized by AUUUA nucleotide repeats are present in the 3’ UTRs of many cytokines, chemokines, and proto-oncogenes (3), and ARE-binding RBPs, including ZFP36, human antigen R (HuR)/ELAVL1, AU-rich RNA binding factor 1 (AUF1), T-cell interleukin-1 (TIA-1)/TIA-associated protein (TIAR), and KH-type splicing regulatory protein (KSRP), regulate the degradation and translation of target mRNA (3). In contrast, several other RBPs such as Roquin, regulatory RNase (Regnase), and AT-rich interactive domain-containing protein 5a (Arid5a) recognize the stem-loop structure of the 3’ UTR (3). Thus, RBPs can interact with specific RNA sequences and structures and interact with them to regulate target mRNAs positively or negatively (3).
ZFP36 family is composed of three proteins (ZFP36 (TTP), ZFP36L1, and ZFP36L2) in humans and most other mammals, while the fourth subtype, ZFP36L3, is expressed in the yolk sac and placenta of rodents (10). The ZFP36 family members are known to have three essential domains: An N-terminal nuclear export sequence (NES), a central tandem Cys-Cys-Cys-His (CCCH) zinc finger domain, and a C-terminal CNOT1 binding domain (9). Although ZFP36 family members are structurally similar to each other, each ZFP36 family member is thought to have different functions, as it has been shown in both immune and non-immune cells that each ZFP36 family member is expressed in different cell-type and is differently controlled upon stimulation (8, 11).
Among ZFP36 family members, the molecular mechanisms of the post-transcriptional regulation are most intensively investigated for ZFP36 (9). The C-terminal motif of ZFP36 binds directly to the central domain of CNOT1, which is the core subunit of the CCR4-NOT complex, and the ZFP36-CCR4-NOT complex plays a crucial role in ZFP36-mediated deadenylation of target mRNAs (12). The deadenylation is thought to be important for rapid mRNA degradation and to be induced in small nests of the cytoplasm (called processing bodies) containing many enzymes (13). Under stress conditions, ZFP36-bound mRNAs are recruited to stress granules, and the translation repressor, TIA-1, prevents translation in stress granules (14). In addition, ZFP36 has been shown to facilitate the degradation of selected mRNAs by transporting them from stress granules to processing bodies (15, 16). Taken together, although the precise mechanism of mRNA turnover by ZFP36 is still unclear, various factors such as the CCR4-NOT complex seem to be essential for the regulation of ZFP36-mediated decay of mRNAs (Figure 1).
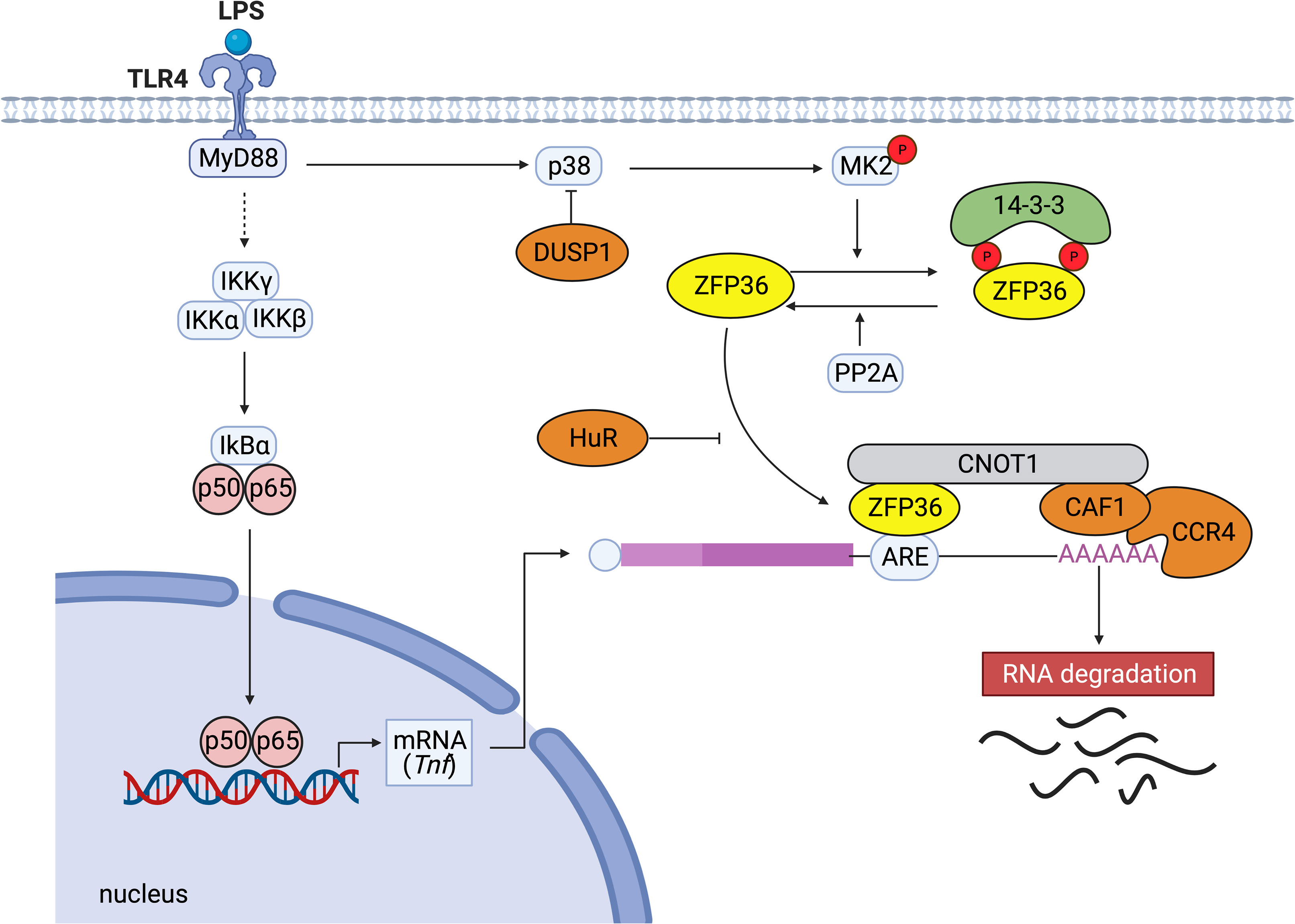
Figure 1 Typical post-transcriptional regulation by ZFP36. When LPS activates TLR4, the downstream NF-κB kinase (IKK) complexes (IKKγ, IKKα, IKKβ) are activated. Subsequently, IκBα is phosphorylated and degraded. The released NF-κB migrates to the nucleus and activates the expression of genes such as TNF. ZFP36 binds to the ARE in the 3’ UTR of Tnf mRNA and promotes the decay of the target mRNA by recruiting the CCR4-CAF1-CNOT1 complex. ZFP36 binds directly to the central domain of CNOT1, the core subunit of the CCR4-NOT complex. Conversely, the binding of stabilizing proteins such as human antigen R (HuR) that compete with destabilizing factors inhibits ARE-mediated RNA degradation. p38 MAPK activates MK2, which phosphorylates two serine residues of ZFP36 (S60 and S186 in humans, S52 and S178 in mice). Dual-specificity phosphatase DUSP1 dephosphorylates p38 MAPK. Phosphorylation of ZFP36 promotes its binding to 14-3-3 proteins, resulting in stabilization of target mRNAs. Serine/threonine PP2A dephosphorylates ZFP36 and releases 14-3-3 proteins from ZFP36.
With respect to the molecular mechanisms to regulate ZFP36 expression, it has been shown that ZFP36 autoregulates its expression via interaction with AREs in 3’ UTR of its mRNA (17). Experimental deletion of ARE from Zfp36 mRNA has been shown to free ZFP36 from autoinhibition or repression by other ARE-binding proteins and increase the abundance of ZFP36 (18). Regarding the second mechanism to regulate ZFP36 function, phosphorylation is reported to be involved in the stabilization and inactivation of ZFP36. ZFP36 is phosphorylated by multiple kinases such as ERK, p38 MAPK, JNK, and AKT (19). MAPK-activated protein kinase 2 (MK2) is activated by p38 MAPK and phosphorylates ZFP36 at two serine residues (S60 and S186 in humans, and S52 and S178 in mice) (20, 21). Phosphorylated ZFP36 is more stable than unphosphorylated ZFP36, and the phosphorylated ZFP36 accumulates until p38 MAPK activity is reduced (22, 23). Moreover, the phosphorylation of ZFP36 promotes its binding to 14-3-3 proteins, and the resultant ZFP36-14-3-3 complex does not recruit the CNOT deadenylase complex (21, 22). Therefore, phosphorylated ZFP36 seems to lose its ability to degrade mRNA.
Dual-specificity phosphatase 1 (DUSP1) is known to dephosphorylate and inactivate MAPK superfamily members such as JNKs, p38a, and p38b MAPKs, and then DUSP1 and ZFP36 cooperate to regulate inflammation (23). The loss of DUSP1 leads to ZFP36 phosphorylation and accumulation of inactive ZFP36, and the production of TNF-α and IL-10 is enhanced in Dusp1-deficient bone marrow-derived macrophages (23). Moreover, protein phosphatase 2A (PP2A) activation has been shown to induce dephosphorylation and activation of ZFP36 (21).
In terms of the other mechanisms preventing ZFP36 function, HuR competes with ZFP36 for the AREs in the 3’ UTR of Il6 mRNA and stabilizes it (24). In addition, ZFP36 is polyubiquitinated by TNF receptor-associated factor 2 (TRAF2), and the polyubiquitination appears to be specifically necessary for its function for JNK activation (25). These studies suggest that multiple mechanisms in immune responses regulate the expression and function of ZFP36, and various kinases affect the activation and stability of ZFP36 in response to different environmental cues (Figure 1).
It is well known that mRNAs encoding cytokines such as TNF-α have short half-lives and decay via AREs (26). ZFP36 down-regulates TNF-α production by directly binding to the ARE in the 3’ UTR of Tnf mRNA and promoting Tnf mRNA decay by recruiting the CCR4-NOT deadenylase complex (6). Meanwhile, ZFP36 expression is induced by TNF-α-mediated signaling. Thus, ZFP36 acts as one component of a negative feedback loop that regulates TNF-α production by destabilizing Tnf mRNA (27). In accordance with this finding, ZFP36-deficient mice develop a complex syndrome of inflammatory arthritis, dermatitis, cachexia, autoimmunity, and bone marrow hyperplasia, which resemble the phenotypes due to excessive TNF-α production in vivo just like the phenomena observed in TNF-transgenic mice (28, 29).
Not only TNF-α but also IL-6 is well-known to be a multifunctional pro-inflammatory cytokine that plays a critical role in various diseases, and its expression is tightly regulated at both the transcriptional and post-transcriptional levels (4). There are five AREs in the 3’ UTR of murine Il6 mRNA, and ZFP36 is shown to bind to ARE2, ARE3, and ARE4 in the 3’ UTR region to promote Il6 mRNA degradation (30).
Surprisingly, the mRNA of IL-10, which is one of the representative anti-inflammatory cytokines, was also identified as a target of ZFP36. Consistent with studies regarding TNF-α and IL-6, Il10 mRNA degradation was induced by the binding of ZFP36 to ARE in its 3’ UTR (31). Furthermore, IL-10 induces the ZFP36 expression in macrophages by activating STAT3 (32). Thus, IL-10-mediated ZFP36 induction seems a part of the negative feedback loop to regulate IL-10 production to terminate anti-inflammatory signals. Interestingly, Schaljo et al. have reported that IL-10 reduces TNF-α expression in LPS-activated bone marrow-derived murine macrophages in part through the induction of ZFP36 (33). Together, it is suggested that ZFP36-mediated post-transcriptional regulatory mechanisms control both the initiation and resolution of inflammatory responses in multiple mechanisms.
With respect to the roles of ZFP36 in T cell-mediated immune responses, Moore et al. have recently shown that using a lymphocytic choriomeningitis virus (LCMV) infection model, virus-specific expansion and recession of T cells is accelerated, and LCMV clearance is enhanced by the absence of ZFP36 (34), suggesting that ZFP36 restrains T cells and slows down the immune responses.
Taken together, ZFP36 regulates immune responses in various immune cells through many mechanisms.
Similar to ZFP36, ZFP36L1 interacts with AREs in the 3’ UTR of mRNAs to attenuate the expression of the corresponding genes (35). Regarding the role of ZFP36L1 in post-transcriptional regulation, it has recently been demonstrated that ZFP36L1 expressed in B cells has an essential function in maintaining a population of marginal zone B cells by limiting the expression of KLF2 and IRF8 (7). Although the precise roles of ZFP36L1 in germinal center responses and immune memory remain unclear, it has been reported that ZFP36L1 expressed in B cells promotes the migration of antibody-secreting cells from secondary lymphoid organs to survival niches in the bone marrow by restricting the expression of G protein-coupled receptor kinase 2 (GRK2) and integrin chains α4 and β1, facilitating the long-term establishment of antibody-secreting cells (36).
In developing B cells, because the expression of recombination activating gene 2 (RAG2) protein is restricted to the G0-G1 phase of the cell cycle (37–39), quiescence is essential for promoting variable-diversity-joining (VDJ) recombination. Recently, Galloway et al. have shown that in developing B cells, both ZFP36L1 and ZFP36L2 are important for maintaining quiescence before expressing pre-B cell receptor (pre-BCR) and for the re-establishment of quiescence after expansion by the pre-BCR (40). Importantly, double-deficiency of ZFP36L1 and ZFP36L2 in T-cell lineage in mice causes the arrest of thymopoiesis at the double-negative stage and develops T cell acute lymphoblastic leukemia (T-ALL) due to aberrant activation of Notch signaling (41). In contrast, the single deletion of ZFP36L1 or ZFP36L2 in T-cell lineage does not result in T-ALL (41). These findings suggest that ZFP36L1 and ZFP36L2 play both redundant and non-redundant roles in lymphocyte differentiation.
How ZFP36L2 alters the function of T cells is not fully understood yet. We have recently shown that ZFP36L2 is highly expressed in naive CD4+ T cells, and ZFP36L2 expression in CD4+ T cells is rapidly reduced by the stimulation via the T cell receptor (8). In addition, we found that ZFP36L2 expression levels in iTregs are significantly lower than those in naive CD4+ T cells (8). Moreover, we found that ZFP36L2 directly binds to AREs in 3’ UTR of Ikzf2 mRNA, resulting in its degradation of Ikzf2 mRNA and down-regulation of iTreg function (Figure 2) (8). These results indicate that ZFP36L2 also promotes post-transcriptional regulation of immune responses and regulates immune cell function.
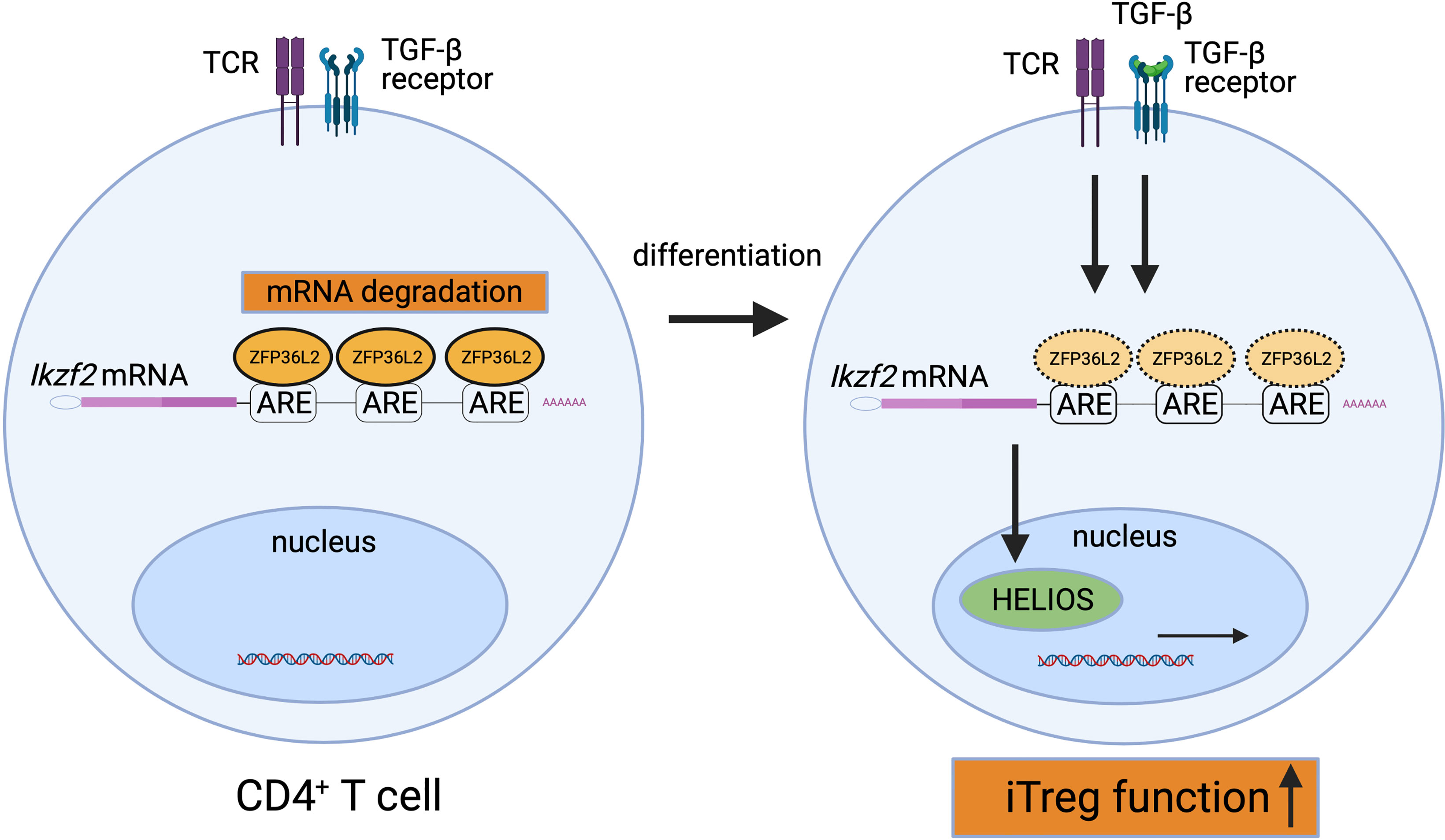
Figure 2 Suppression of HELIOS expression by ZFP36L2. ZFP36L2 is highly expressed in naïve CD4+ T cells and can degrade Ikzf2 mRNAs by binding ARE in the 3’ UTR. Upon TCR stimulation, ZFP36L2 expression is rapidly reduced. Consequently, the transcribed Ikzf2 mRNA is stabilized, HELIOS is firmly produced, CD4+ T cells differentiate and mature into iTregs possessing sufficient suppressive capacity.
Genome-wide association studies (GWAS) have highlighted the association of ZFP36 family members with pathogenic mechanisms in various autoimmune diseases. Twenty-eight single-nucleotide polymorphisms (SNPs) in the ZFP36 gene were found in patients with autoimmune disorders such as rheumatoid arthritis (RA), psoriasis, multiple sclerosis (MS), and juvenile idiopathic arthritis (JIA) (42).
Interestingly, one SNP called ZFP36*8 variant has been shown to be significantly associated with RA in African Americans (42). Suzuki et al. have reported that compared with AA/AG genotypes, GG genotype in ZFP36 promoter region SNP, in which promoter activity is lower than that with AA/AG genotypes, is associated with age at onset, duration, disease progression, and infliximab usage in Japanese RA patients (43).
It is not yet clear how ZFP36 is involved in the pathogenesis of human diseases. It has been reported that ZFP36 is highly expressed in synovial tissues of RA patients and inflamed mucosal tissues of inflammatory bowel disease (IBD) (44–46). In the rheumatoid synovium, ZFP36 is detected in macrophages, vascular endothelial cells, and fibroblasts (45). Interestingly, ZFP36/TNF gene expression ratio in synovial tissues correlates inversely with CRP (44). These findings suggest that inappropriate TTP production in response to increased TNF-α may be one factor that contributes to the pathogenesis of RA.
GWAS have also revealed that the ZFP36L1 region is significantly associated with RA, JIA, Crohn’s disease, celiac disease, and type 1 diabetes (47, 48). In addition, ZFP36L2 is identified as a susceptibility gene of MS, and its expression is decreased in MS patients compared to healthy subjects (49). Similarly, gene expression levels of ZFP36L2 in peripheral blood mononuclear cells are significantly lower in SLE patients than those in healthy controls (50). Therefore, the variants of ZFP36, ZFP36L1, and ZFP36L2 or dysregulation of those expressions may be involved in developing various inflammatory diseases in humans.
Regarding the association with allergic diseases, a comprehensive transcriptome analysis has shown that ZFP36 expression in peripheral blood leucocytes is lower in persistent asthma children than in healthy children (51), suggesting that the reduction of ZFP36 gene expression may be associated with asthma in children. Moreover, Leigh et al. have reported that budesonide inhalation induces various gene expressions including ZFP36 in bronchial tissues and whole blood cells in healthy subjects (52), indicating that inhaled corticosteroids may provide anti-inflammatory effects by inducing ZFP36 expression in both immune cells and non-immune cells. On the other hand, the expression of ZFP36L1 in bronchoalveolar lavage cells is higher in patients with steroid-resistant asthma than that in patients with steroid-sensitive asthma (53). Hansel et al. have reported that ZFP36L2 expression in peripheral blood CD4+ T cells is significantly higher in severe asthma patients than in mild asthma patients (54). Although further studies are required, these findings suggest that ZFP36 family proteins in immune cells and bronchial structural cells may contribute to the development of allergic airway inflammation and the sensitivity to inhaled corticosteroids.
The forced expression of ZFP36 family proteins in peripheral tissues or immune cells could be novel therapeutic approaches for inflammatory diseases in humans (55). It has been reported that adenoviral overexpression of ZFP36 results in protection against bone loss and reduced inflammatory cell infiltration in experimental periodontitis in rats (56). Consistent with these findings, ZFP36-delta ARE mice, in which the stability of ZFP36 mRNA is enhanced by the deletion of a 136-base instability motif in the 3’ UTR of ZFP36 mRNA, show the increased levels of ZFP36 expression in tissues (57) and are protected from anti-type II collagen antibody-induced arthritis, imiquimod-induced dermatitis, and experimental autoimmune encephalomyelitis (57). These findings suggest that sustained ZFP36 expression or activation is helpful in developing therapeutic strategies against inflammatory diseases.
As discussed in this review, the p38 MAPK pathway inactivates ZFP36 via the phosphorylation of two serine residues in mice and humans (20), while ZFP36 is dephosphorylated and activated by PP2A (58). Importantly, Ross et al. have reported that in vivo administration of PP2A agonists such as COG1410 (an apolipoprotein E peptide mimetic) or AAL(s) (a lipid derivative of the immunosuppressant FTY720 (Fingolimod)) activates ZFP36 through the dephosphorylation and ameliorates experimental murine arthritis models (45). Although the precise roles of ZFP36L2 in T cell function and inflammatory diseases remain to be elucidated, we have reported that ZFP36L2 reduces HELIOS expression in iTregs and suppresses iTreg function (Figure 2) (8). Thus, the reduction of ZFP36L2 expression in iTregs could be an attractive strategy for developing adoptive antigen-specific iTreg therapy.
RBPs, including ZFP36 family proteins, are essential for post-transcriptional regulation in RNA metabolism. Recent studies have uncovered that gene polymorphism of ZFP36 family members is associated with various autoimmune diseases and that the dysregulation of stabilization or inactivation by phosphorylation of ZFP36 family proteins could be involved in the pathogenesis of inflammatory diseases. However, it remains to be elucidated if there is functional redundancy and interaction among these family molecules for post-transcriptional regulation of immune responses. Therefore, a better understanding of the post-transcriptional processes mediated by each of the ZFP36 family members will be necessary to develop a novel therapeutic strategy for chronic inflammatory diseases.
SM, HT, and HN planned and wrote the MS. All authors contributed to the article and approved the submitted version.
This work was supported in part by Grants-in-Aids for Scientific research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), LGS (Leading Graduate School at Chiba University) Program, Global Prominent Research (Chiba University), Takeda Science Foundation, and GSK Japan Research Grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, et al. Insights Into RNA Biology From an Atlas of Mammalian mRNA-Binding Proteins. Cell (2012) 149(6):1393–406. doi: 10.1016/j.cell.2012.04.031
2. Lunde BM, Moore C, Varani G. RNA-Binding Proteins: Modular Design for Efficient Function. Nat Rev Mol Cell Biol (2007) 8(6):479–90. doi: 10.1038/nrm2178
3. Akira S, Maeda K. Control of RNA Stability in Immunity. Annu Rev Immunol (2021) 39:481–509. doi: 10.1146/annurev-immunol-101819-075147
4. Fu M, Blackshear PJ. RNA-Binding Proteins in Immune Regulation: A Focus on CCCH Zinc Finger Proteins. Nat Rev Immunol (2017) 17(2):130–43. doi: 10.1038/nri.2016.129
5. Turner M, Diaz-Munoz MD. RNA-Binding Proteins Control Gene Expression and Cell Fate in the Immune System. Nat Immunol (2018) 19(2):120–9. doi: 10.1038/s41590-017-0028-4
6. Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Evidence That Tristetraprolin Binds to AU-Rich Elements and Promotes the Deadenylation and Destabilization of Tumor Necrosis Factor Alpha mRNA. Mol Cell Biol (1999) 19(6):4311–23. doi: 10.1128/MCB.19.6.4311
7. Newman R, Ahlfors H, Saveliev A, Galloway A, Hodson DJ, Williams R, et al. Maintenance of the Marginal-Zone B Cell Compartment Specifically Requires the RNA-Binding Protein ZFP36L1. Nat Immunol (2017) 18(6):683–93. doi: 10.1038/ni.3724
8. Makita S, Takatori H, Iwata A, Tanaka S, Furuta S, Ikeda K, et al. RNA-Binding Protein ZFP36L2 Downregulates Helios Expression and Suppresses the Function of Regulatory T Cells. Front Immunol (2020) 11:1291. doi: 10.3389/fimmu.2020.01291
9. Kafasla P, Skliris A, Kontoyiannis DL. Post-Transcriptional Coordination of Immunological Responses by RNA-Binding Proteins. Nat Immunol (2014) 15(6):492–502. doi: 10.1038/ni.2884
10. Stumpo DJ, Trempus CS, Tucker CJ, Huang W, Li L, Kluckman K, et al. Deficiency of the Placenta- and Yolk Sac-Specific Tristetraprolin Family Member ZFP36L3 Identifies Likely mRNA Targets and an Unexpected Link to Placental Iron Metabolism. Development (2016) 143(8):1424–33. doi: 10.1242/dev.130369
11. Hacker C, Valchanova R, Adams S, Munz B. ZFP36L1 is Regulated by Growth Factors and Cytokines in Keratinocytes and Influences Their VEGF Production. Growth Factors (2010) 28(3):178–90. doi: 10.3109/08977190903578660
12. Fabian MR, Frank F, Rouya C, Siddiqui N, Lai WS, Karetnikov A, et al. Structural Basis for the Recruitment of the Human CCR4-NOT Deadenylase Complex by Tristetraprolin. Nat Struct Mol Biol (2013) 20(6):735–9. doi: 10.1038/nsmb.2572
13. Anderson P, Kedersha N. RNA Granules: Post-Transcriptional and Epigenetic Modulators of Gene Expression. Nat Rev Mol Cell Biol (2009) 10(6):430–6. doi: 10.1038/nrm2694
14. Protter DSW, Parker R. Principles and Properties of Stress Granules. Trends Cell Biol (2016) 26(9):668–79. doi: 10.1016/j.tcb.2016.05.004
15. Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis Suppressor Genes TIA-1 and TTP Dampen the Expression of Tumor Necrosis Factor Alpha, Cyclooxygenase 2, and Inflammatory Arthritis. Proc Natl Acad Sci USA (2004) 101(7):2011–6. doi: 10.1073/pnas.0400148101
16. Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, et al. Stress Granules and Processing Bodies are Dynamically Linked Sites of mRNP Remodeling. J Cell Biol (2005) 169(6):871–84. doi: 10.1083/jcb.200502088
17. Brooks SA, Connolly JE, Rigby WF. The Role of mRNA Turnover in the Regulation of Tristetraprolin Expression: Evidence for an Extracellular Signal-Regulated Kinase-Specific, AU-Rich Element-Dependent, Autoregulatory Pathway. J Immunol (2004) 172(12):7263–71. doi: 10.4049/jimmunol.172.12.7263
18. Tang T, Scambler TE, Smallie T, Cunliffe HE, Ross EA, Rosner DR, et al. Macrophage Responses to Lipopolysaccharide are Modulated by a Feedback Loop Involving Prostaglandin E2, Dual Specificity Phosphatase 1 and Tristetraprolin. Sci Rep (2017) 7(1):4350. doi: 10.1038/s41598-017-04100-1
19. Clark AR, Dean JL. The Control of Inflammation via the Phosphorylation and Dephosphorylation of Tristetraprolin: A Tale of Two Phosphatases. Biochem Soc Trans (2016) 44(5):1321–37. doi: 10.1042/BST20160166
20. Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, et al. MAPKAP Kinase 2 Phosphorylates Tristetraprolin on In Vivo Sites Including Ser178, a Site Required for 14-3-3 Binding. J Biol Chem (2004) 279(11):10176–84. doi: 10.1074/jbc.M310486200
21. Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, et al. MK2-Induced Tristetraprolin:14-3-3 Complexes Prevent Stress Granule Association and ARE-mRNA Decay. EMBO J (2004) 23(6):1313–24. doi: 10.1038/sj.emboj.7600163
22. Tiedje C, Diaz-Munoz MD, Trulley P, Ahlfors H, Laass K, Blackshear PJ, et al. The RNA-Binding Protein TTP is a Global Post-Transcriptional Regulator of Feedback Control in Inflammation. Nucleic Acids Res (2016) 44(15):7418–40. doi: 10.1093/nar/gkw474
23. Smallie T, Ross EA, Ammit AJ, Cunliffe HE, Tang T, Rosner DR, et al. Dual-Specificity Phosphatase 1 and Tristetraprolin Cooperate To Regulate Macrophage Responses to Lipopolysaccharide. J Immunol (2015) 195(1):277–88. doi: 10.4049/jimmunol.1402830
24. Shi JX, Su X, Xu J, Zhang WY, Shi Y. HuR Post-Transcriptionally Regulates TNF-Alpha-Induced IL-6 Expression in Human Pulmonary Microvascular Endothelial Cells Mainly via Tristetraprolin. Respir Physiol Neurobiol (2012) 181(2):154–61. doi: 10.1016/j.resp.2012.02.011
25. Resch U, Cuapio A, Sturtzel C, Hofer E, de Martin R, Holper-Schichl YM. Polyubiquitinated Tristetraprolin Protects From TNF-Induced, Caspase-Mediated Apoptosis. J Biol Chem (2014) 289(36):25088–100. doi: 10.1074/jbc.M114.563312
26. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/Off Regulation of TNF Biosynthesis in Mice Lacking TNF AU-Rich Elements: Implications for Joint and Gut-Associated Immunopathologies. Immunity (1999) 10(3):387–98. doi: 10.1016/S1074-7613(00)80038-2
27. Carballo E, Lai WS, Blackshear PJ. Feedback Inhibition of Macrophage Tumor Necrosis Factor-Alpha Production by Tristetraprolin. Science (1998) 281(5379):1001–5. doi: 10.1126/science.281.5379.1001
28. Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, et al. A Pathogenetic Role for TNF Alpha in the Syndrome of Cachexia, Arthritis, and Autoimmunity Resulting From Tristetraprolin (TTP) Deficiency. Immunity (1996) 4(5):445–54. doi: 10.1016/S1074-7613(00)80411-2
29. Carballo E, Blackshear PJ. Roles of Tumor Necrosis Factor-Alpha Receptor Subtypes in the Pathogenesis of the Tristetraprolin-Deficiency Syndrome. Blood (2001) 98(8):2389–95. doi: 10.1182/blood.V98.8.2389
30. Zhao W, Liu M, D’Silva NJ, Kirkwood KL. Tristetraprolin Regulates Interleukin-6 Expression Through P38 MAPK-Dependent Affinity Changes With mRNA 3’ Untranslated Region. J Interferon Cytokine Res (2011) 31(8):629–37. doi: 10.1089/jir.2010.0154
31. Stoecklin G, Tenenbaum SA, Mayo T, Chittur SV, George AD, Baroni TE, et al. Genome-Wide Analysis Identifies Interleukin-10 mRNA as Target of Tristetraprolin. J Biol Chem (2008) 283(17):11689–99. doi: 10.1074/jbc.M709657200
32. Gaba A, Grivennikov SI, Do MV, Stumpo DJ, Blackshear PJ, Karin M. Cutting Edge: IL-10-Mediated Tristetraprolin Induction is Part of a Feedback Loop That Controls Macrophage STAT3 Activation and Cytokine Production. J Immunol (2012) 189(5):2089–93. doi: 10.4049/jimmunol.1201126
33. Schaljo B, Kratochvill F, Gratz N, Sadzak I, Sauer I, Hammer M, et al. Tristetraprolin is Required for Full Anti-Inflammatory Response of Murine Macrophages to IL-10. J Immunol (2009) 183(2):1197–206. doi: 10.4049/jimmunol.0803883
34. Moore MJ, Blachere NE, Fak JJ, Park CY, Sawicka K, Parveen S, et al. ZFP36 RNA-Binding Proteins Restrain T Cell Activation and Anti-Viral Immunity. Elife (2018) 7:e33057. doi: 10.7554/eLife.33057
35. Brooks SA, Blackshear PJ. Tristetraprolin (TTP): Interactions With mRNA and Proteins, and Current Thoughts on Mechanisms of Action. Biochim Biophys Acta (2013) 1829(6-7):666–79. doi: 10.1016/j.bbagrm.2013.02.003
36. Saveliev A, Bell SE, Turner M. Efficient Homing of Antibody-Secreting Cells to the Bone Marrow Requires RNA-Binding Protein ZFP36L1. J Exp Med (2021) 218(3):e20200504. doi: 10.1084/jem.20200504
37. Zhang L, Reynolds TL, Shan X, Desiderio S. Coupling of V(D)J Recombination to the Cell Cycle Suppresses Genomic Instability and Lymphoid Tumorigenesis. Immunity (2011) 34(2):163–74. doi: 10.1016/j.immuni.2011.02.003
38. Johnson K, Chaumeil J, Micsinai M, Wang JM, Ramsey LB, Baracho GV, et al. IL-7 Functionally Segregates the Pro-B Cell Stage by Regulating Transcription of Recombination Mediators Across Cell Cycle. J Immunol (2012) 188(12):6084–92. doi: 10.4049/jimmunol.1200368
39. Bendall SC, Davis KL, Amir e-A, Tadmor MD, Simonds EF, Chen TJ, et al. Single-Cell Trajectory Detection Uncovers Progression and Regulatory Coordination in Human B Cell Development. Cell (2014) 157(3):714–25. doi: 10.1016/j.cell.2014.04.005
40. Galloway A, Saveliev A, Lukasiak S, Hodson DJ, Bolland D, Balmanno K, et al. RNA-Binding Proteins ZFP36L1 and ZFP36L2 Promote Cell Quiescence. Science (2016) 352(6284):453–9. doi: 10.1126/science.aad5978
41. Hodson DJ, Janas ML, Galloway A, Bell SE, Andrews S, Li CM, et al. Deletion of the RNA-Binding Proteins ZFP36L1 and ZFP36L2 Leads to Perturbed Thymic Development and T Lymphoblastic Leukemia. Nat Immunol (2010) 11(8):717–24. doi: 10.1038/ni.1901
42. Carrick DM, Chulada P, Donn R, Fabris M, McNicholl J, Whitworth W, et al. Genetic Variations in ZFP36 and Their Possible Relationship to Autoimmune Diseases. J Autoimmun (2006) 26(3):182–96. doi: 10.1016/j.jaut.2006.01.004
43. Suzuki T, Tsutsumi A, Suzuki H, Suzuki E, Sugihara M, Muraki Y, et al. Tristetraprolin (TTP) Gene Polymorphisms in Patients With Rheumatoid Arthritis and Healthy Individuals. Mod Rheumatol (2008) 18(5):472–9. doi: 10.3109/s10165-008-0085-5
44. Tsutsumi A, Suzuki E, Adachi Y, Murata H, Goto D, Kojo S, et al. Expression of Tristetraprolin (G0S24) mRNA, a Regulator of Tumor Necrosis Factor-Alpha Production, in Synovial Tissues of Patients With Rheumatoid Arthritis. J Rheumatol (2004) 31(6):1044–9.
45. Ross EA, Naylor AJ, O’Neil JD, Crowley T, Ridley ML, Crowe J, et al. Treatment of Inflammatory Arthritis via Targeting of Tristetraprolin, a Master Regulator of Pro-Inflammatory Gene Expression. Ann Rheum Dis (2017) 76(3):612–9. doi: 10.1136/annrheumdis-2016-209424
46. Di Silvestre A, Lucafò M, Pugnetti L, Bramuzzo M, Stocco G, Barbi E, et al. Role of Tristetraprolin Phosphorylation in Paediatric Patients With Inflammatory Bowel Disease. World J Gastroenterol (2019) 25(39):5918–25. doi: 10.3748/wjg.v25.i39.5918
47. Hinks A, Cobb J, Marion MC, Prahalad S, Sudman M, Bowes J, et al. Dense Genotyping of Immune-Related Disease Regions Identifies 14 New Susceptibility Loci for Juvenile Idiopathic Arthritis. Nat Genet (2013) 45(6):664–9. doi: 10.1038/ng.2614
48. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-Wide Meta-Analysis Increases to 71 the Number of Confirmed Crohn’s Disease Susceptibility Loci. Nat Genet (2010) 42(12):1118–25. doi: 10.1038/ng.717
49. Parnell GP, Gatt PN, Krupa M, Nickles D, McKay FC, Schibeci SD, et al. The Autoimmune Disease-Associated Transcription Factors EOMES and TBX21 are Dysregulated in Multiple Sclerosis and Define a Molecular Subtype of Disease. Clin Immunol (2014) 151(1):16–24. doi: 10.1016/j.clim.2014.01.003
50. Mandel M, Gurevich M, Pauzner R, Kaminski N, Achiron A. Autoimmunity Gene Expression Portrait: Specific Signature That Intersects or Differentiates Between Multiple Sclerosis and Systemic Lupus Erythematosus. Clin Exp Immunol (2004) 138(1):164–70. doi: 10.1111/j.1365-2249.2004.02587.x
51. Persson H, Kwon AT, Ramilowski JA, Silberberg G, Soderhall C, Orsmark-Pietras C, et al. Transcriptome Analysis of Controlled and Therapy-Resistant Childhood Asthma Reveals Distinct Gene Expression Profiles. J Allergy Clin Immunol (2015) 136(3):638–48. doi: 10.1016/j.jaci.2015.02.026
52. Leigh R, Mostafa MM, King EM, Rider CF, Shah S, Dumonceaux C, et al. An Inhaled Dose of Budesonide Induces Genes Involved in Transcription and Signaling in the Human Airways: Enhancement of Anti- and Proinflammatory Effector Genes. Pharmacol Res Perspect (2016) 4(4):e00243. doi: 10.1002/prp2.243
53. Alrashoudi RH, Crane IJ, Wilson HM, Al-Alwan M, Alajez NM. Gene Expression Data Analysis Identifies Multiple Deregulated Pathways in Patients With Asthma. Biosci Rep (2018) 38(6):BSR20180548. doi: 10.1042/BSR20180548
54. Hansel NN, Hilmer SC, Georas SN, Cope LM, Guo J, Irizarry RA, et al. Oligonucleotide-Microarray Analysis of Peripheral-Blood Lymphocytes in Severe Asthma. J Lab Clin Med (2005) 145(5):263–74. doi: 10.1016/j.lab.2005.02.010
55. Patial S, Blackshear PJ. Tristetraprolin as a Therapeutic Target in Inflammatory Disease. Trends Pharmacol Sci (2016) 37(10):811–21. doi: 10.1016/j.tips.2016.07.002
56. Patil CS, Liu M, Zhao W, Coatney DD, Li F, VanTubergen EA, et al. Targeting mRNA Stability Arrests Inflammatory Bone Loss. Mol Ther (2008) 16(10):1657–64. doi: 10.1038/mt.2008.163
57. Patial S, Curtis AD 2nd, Lai WS, Stumpo DJ, Hill GD, Flake GP, et al. Enhanced Stability of Tristetraprolin mRNA Protects Mice Against Immune-Mediated Inflammatory Pathologies. Proc Natl Acad Sci U S A (2016) 113(7):1865–70. doi: 10.1073/pnas.1519906113
58. Sun L, Stoecklin G, Van Way S, Hinkovska-Galcheva V, Guo RF, Anderson P, et al. Tristetraprolin (TTP)-14-3-3 Complex Formation Protects TTP From Dephosphorylation by Protein Phosphatase 2a and Stabilizes Tumor Necrosis Factor-Alpha mRNA. J Biol Chem (2007) 282(6):3766–77. doi: 10.1074/jbc.M607347200
Keywords: tristetraprolin, zinc finger protein 36, zinc finger protein 36-like 1, zinc finger protein 36-like 2, RNA-binding protein, untranslated region, AU-rich element
Citation: Makita S, Takatori H and Nakajima H (2021) Post-Transcriptional Regulation of Immune Responses and Inflammatory Diseases by RNA-Binding ZFP36 Family Proteins. Front. Immunol. 12:711633. doi: 10.3389/fimmu.2021.711633
Received: 18 May 2021; Accepted: 17 June 2021;
Published: 01 July 2021.
Edited by:
Osamu Takeuchi, Kyoto University, JapanReviewed by:
Taishin Akiyama, RIKEN Yokohama, JapanCopyright © 2021 Makita, Takatori and Nakajima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroaki Takatori, takatorih@faculty.chiba-u.jp; Hiroshi Nakajima, nakajimh@faculty.chiba-u.jp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.