 Liyun Xu
Liyun Xu Wen Liu
Wen Liu Fuxiang Bai
Fuxiang Bai Yong Xu
Yong Xu Xiaohong Liang
Xiaohong Liang Chunhong Ma
Chunhong Ma Lifen Gao
Lifen Gao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 December 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.708978
This article is part of the Research Topic The Effects of Enriched Environment on NK cells and Macrophages and Their Underlying Mechanisms View all 7 articles
Fatty liver disease, characterized by excessive inflammation and lipid deposition, is becoming one of the most prevalent liver metabolic diseases worldwide owing to the increasing global incidence of obesity. However, the underlying mechanisms of fatty liver disease are poorly understood. Accumulating evidence suggests that hepatic macrophages, specifically Kupffer cells (KCs), act as key players in the progression of fatty liver disease. Thus, it is essential to examine the current evidence of the roles of hepatic macrophages (both KCs and monocyte-derived macrophages). In this review, we primarily address the heterogeneities and multiple patterns of hepatic macrophages participating in the pathogenesis of fatty liver disease, including Toll-like receptors (TLRs), NLRP3 inflammasome, lipotoxicity, glucotoxicity, metabolic reprogramming, interaction with surrounding cells in the liver, and iron poisoning. A better understanding of the diverse roles of hepatic macrophages in the development of fatty liver disease may provide a more specific and promising macrophage-targeting therapeutic strategy for inflammatory liver diseases.
Fatty liver disease was proposed by Schaffner et al. in 1986 (1), characterized by the over-deposition of cytoplasmic triglycerides (TGs), as lipid droplets, in the liver. Initially, fatty liver disease is classified into two categories: alcoholic fatty liver disease and non-alcoholic fatty liver disease (NAFLD). With the improvement of living standards and changes in dietary habits in recent years, risk factors, including but not limited to overweight, type 2 diabetes, and a sedentary lifestyle, have facilitated the occurrence of NAFLD as one of the most prevalent chronic liver diseases in the world, affecting 20–30% of the general population (2). Recently, NAFLD has been renamed as a consensus and appropriate nomenclature for metabolically associated fatty liver disease (MAFLD) (3), characterized by cytoplasmic TG vacuole deposition exceeding 5% of hepatocytes in the absence of other recognized sources of fatty liver (eg., drugs and viral infection) (4). MAFLD represents a broad term encompassing a spectrum of pathological conditions. The first stage is steatosis, as the most common type, and may progress from steatohepatitis to advanced liver fibrosis, cirrhosis, and ultimately hepatocellular carcinoma. Without external intervention, fatty liver disease is becoming the leading determinant of liver transplantation, liver-related morbidity, and mortality (5). Recently, the “multiple hits” hypothesis, which includes hits coming from the liver, adipose tissue adipocytokines, and gut microbiota, provides a more accurate explanation of the pathogenesis of fatty liver disease (6). In this process, hepatic macrophage-derived inflammatory mediators play key roles in the pathogenesis of fatty liver disease. However, the biochemical events involved in fatty liver disease have not been well explored due to an incomplete understanding of the complicated pathogenesis. Thus, it is important to elucidate and explore the precise pathogenesis of fatty liver disease, which may provide macrophage-targeting immune therapeutic strategies for the intervention of fatty liver disease.
The liver is not only a central organ of energy metabolism, but it also acts as an immune organ, which is rich in multiple immune cells; thus, macrophages are abundantly found in the liver. Innate immunocytes and related effector factors play an indispensable role in the development of fatty liver disease. Immunoregulation in the liver mainly occurs at the hepatic sinusoid capillary junction of the hepatic portal vein and hepatic artery branches (7), where Kupffer cells (KCs) are in close contact with hormones, bacterial endotoxins, metabolites, and immune complexes carried from the hepatic circulation. Macrophages form a highly active, dynamic, and complex immune network system that plays various vital roles in fatty liver disease progression. Three major distinct origins of macrophage subpopulations exist in the liver: yolk sac-derived tissue-resident macrophages–KCs, monocyte-derived macrophages (MDMs)/myeloid-derived mononuclear macrophages, and liver capsular macrophages (LCMs). Although the sources of hepatic macrophages show obvious heterogeneity in the liver, it is difficult to distinguish these populations based on the existing techniques. Moreover, a consensus has not been reached on classification markers, and MDMs sometimes are known to switch to KCs under pathological conditions; therefore, in this review, we have not distinguished them strictly, and most hepatic macrophages are referred to as KCs.
Over the past few years, several reviews (Baffy G. J Hepatol. 2009; Lanthier N. World J Hepatol. 2015; Lefere S et al. JHEP Reports 2019; Chen J et al. Int J Biol Sci. 2020) have summarized the key roles of macrophages in fatty liver disease in different ways, including the heterogeneity, recruitment of macrophages, and crosstalk between macrophages and metabolic stimuli, etc. (8–11). Since hepatic macrophages contribute to both inflammation and tissue homeostasis, this review provides a comprehensive update of hepatic macrophages in fatty liver disease, mainly focusing on the origins, heterogeneities, and pathways of hepatic macrophages in the pathogenesis of fatty liver disease. We expect that this review will broaden the understanding of the association between hepatic macrophages and fatty liver disease, which would shed new light on the potential application of macrophages in the intervention of fatty liver disease.
Hepatic macrophages, consisting of resident KCs, MDMs, and LCMs, display a remarkable heterogeneity (Table 1). The origins of liver macrophages differ greatly, and MDMs can switch to KCs. KC was named by the German anatomist Karl Wilhelm von Kupffer. In 1997, Naito et al. found that KCs originated almost exclusively from yolk sac-derived erythromyeloid progenitors in the liver of mice during the first 9.5–12.5 days after the start of embryogenesis (12, 13). These findings were confirmed by using the cell tracer technique (14). As a subpopulation of liver-resident macrophages, KCs possess self-renewal properties depending on M-CSF signaling and exert strong phagocytosis and efferocytosis (15). The recruited monocytes rapidly differentiate into pro-inflammatory M1-like macrophages in high-fat diet (HFD)-fed mice (16).
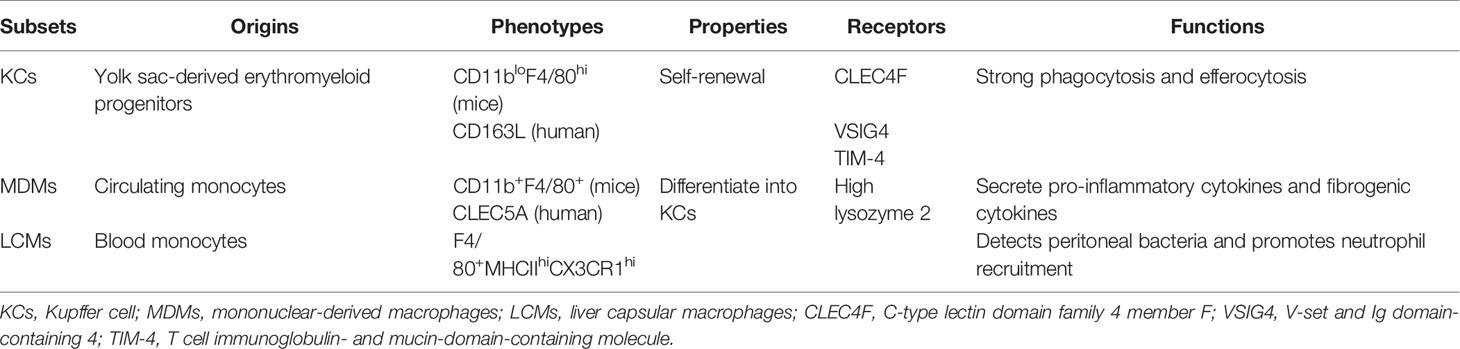
Table 1 Characteristics of the liver macrophage subsets.
Infiltrating MDMs are derived from circulating monocytes. Two major populations of circulating monocytes exist in mice: lymphocyte antigen 6C+ (Ly6C+) high (Ly6Chi)-expressing monocytes are present in the bone marrow, and Ly6C low (Ly6Clo)-expressing monocytes are derived from the spleen (17, 18). Recent evidence shows that CD11b+F4/80+ macrophages originate from infiltrating monocytes, while CD11bloF4/80hi macrophages are derived from resident KCs and mature monocytes (19, 20).
The monocytes in mice are marked with Ly6Chi and CC-chemokine receptor 2hi (CCR2hi) (21). Several studies have suggested that infiltrated Ly6Chi monocytes in early murine steatohepatitis are mainly identified by chemokine receptors, pattern recognition receptors (PRRs), and cytokine secretion (22). Ly6Clo monocytes are characterized by their scavenger receptors (23). Recently, single-cell RNA sequencing has suggested that KCs are characterized by increased C-type lectin domain family 4 member F, V-set and Ig domain-containing 4, and T-cell immunoglobulin- and mucin-domain-containing molecule, whereas MDMs are mainly identified by high lysozyme 2 in the liver of murine steatohepatitis (24).
Another liver-resident macrophage subset is the LCM; distinct from KCs ontogenetically and phenotypically, LCMs are replenished from blood monocytes and are identified as F4/80+MHCIIhiCX3CR1hi, which detects peritoneal bacteria and promotes neutrophil recruitment to the capsule (25). However, there is no consensus on the specific marker of hepatic macrophages; thus, further investigation is required to clarify the sources of macrophages.
Although KCs and MDMs show controversial markers, it should be noted that KCs and MDMs in the liver are not immutable. Under severe hepatic damage, MDMs can differentiate into KCs when KCs are depleted (26). In the livers of Western diet-induced MAFLD mice, the recruited monocytes could also be differentiated into a distinct population of KCs termed hepatic lipid-associated macrophages, characterized by osteopontin expression and a similar capacity of lipid metabolism to that in adipose tissue (27). At the early stage of liver injury, CC-chemokine ligand 2 (CCL2), which is secreted by KCs, triggers circulating Ly6Chi monocytes with CCR2 recruitment into the liver, and the recruited MDMs further secrete pro-inflammatory cytokines and fibrogenic cytokines, accelerating the progression of fibrosis (28). In the later phase of liver injury, the improved phagocytic activity of macrophages facilitates Ly6Chi macrophage differentiation into the Ly6Clo macrophage subset and induces extracellular matrix degradation by matrix metalloproteinases (MMPs) (29). However, another study demonstrated contrasting results that Ly6Clo macrophages are derived from the spleen and are not switched from Ly6Chi (23). In addition, peritoneal macrophage infiltration is also manifested in liver injury (30). However, the mechanisms of peritoneal macrophage recruitment remain poorly understood.
Monocytes in humans can be identified by CD14 and CD162 (31). In humans, the surface markers of MDMs and KCs are CLEC5A and CD163L, respectively (32). In patients with fatty liver disease, a marked increase in the number of hepatic macrophages occurs gradually with the aggravation of steatosis and inflammation (15), which is mainly attributed to the extensive infiltration of CD11b+Ly6C+ monocytes into the liver.
Macrophages are the most plastic cells in the hematopoietic system and show great functional diversity. Liver macrophages can switch their phenotype towards pro-inflammatory (classically activated macrophages, designated M1-like macrophages) or anti-inflammatory (alternatively activated macrophages, designated M2-like macrophages) in response to various signals, such as cytokines, fatty acids, endotoxins, metabolites, and danger-/pathogen-associated molecular patterns (DAMPs/PAMPs) (Table 2). Thus, liver macrophages may display a variety of or even completely opposite roles in different diseases and even in different stages of the same disease.

Table 2 Comparison of M1- and M2-like macrophages.
In vitro, hepatic macrophages are skewed towards M1, similar to that of macrophages exposed to lipopolysaccharide (LPS), IL-12, IFN-γ, TNF-α, or GM-CSF. Activated M1-like macrophages produce a set of pro-inflammatory mediators (e.g., IL-1β, IL-12, TNF-α, CCL2, and CCL5) and increased reactive oxygen species (ROS) and nitric oxide (NO) intermediates, displaying an IL-12hiIL-23hiIL-10lo phenotype, and exert pro-inflammatory, anti-tumor, and anti-bacterial effects. In contrast, M2-like macrophages, which are primed by IL-4, IL-13, IL-33, or IL-14, release IL-10, IL-4, IL-13, and TGF-β cytokines, displaying an IL-12loIL-23loIL-10hi phenotype, triggering an anti-inflammatory response and tissue repair.
In the microenvironment of fatty liver disease, cytokines and various kinds of fatty acids regulate macrophage differentiation. The saturated fatty acid palmitic acid (PA) induces pro-inflammatory M1-like macrophage polarization through hypoxia-inducible factor 1α, identified by increased TNF-α and IL-6 production, whereas the unsaturated fatty acid oleic acid (OA) promotes anti-inflammatory M2-like macrophage differentiation, characterized by the increased expression of arginase-1, type 2 mannose receptor, and IL-10 (33). Furthermore, probiotic (eg., Lactobacillus paracasei) administration also increases the number of M2-like macrophages in the liver of murine steatohepatitis and alleviates steatosis (34). In addition, macrophage polarization differs between mouse strains. In C57BL/6 mice with fatty liver disease, steatosis promotes the secretion of IL-1β, which is beneficial for M1-like macrophage polarization, whereas in BALB/c mice, steatosis mainly induces M2-like macrophage responses (35).
In vivo, hepatic macrophages are stimulated by endotoxins, cytokines, lipids, and other metabolites; thus, phenotypes may change dynamically with the progress and development of fatty liver disease (36). In methionine- and choline-deficient (MCD) diet-induced murine steatohepatitis, a phenotypic switch is observed from M1- to M2-like macrophages, accompanied by a shift in cytokine levels (37). Some studies have also shown that hepatic macrophages seem to express biomarkers of both M1- and M2-like macrophages simultaneously in the process of liver injury (38). The evidence mentioned above suggests that macrophage polarization is a highly plastic physiological process in the progression of fatty liver disease.
Several studies have revealed that M1-like macrophages promote hepatocyte steatosis and insulin resistance (IR), whereas M2-like macrophages show the opposite effect (39). Compared with BALB/c mice, C57BL/6 mice fed an MCD diet display more severe lipid deposition and inflammation in the liver, while M2-like macrophage polarization induced by pharmaceuticals partially inhibits lipid deposition and apoptosis in hepatocytes (40). Moreover, Arg-2-/- mice develop steatosis spontaneously and exhibit the characteristics of steatohepatitis without HFD induction (41). In murine fatty liver disease, M1-like macrophages promote TG synthesis by increasing the activity of diacylglycerol (DAG) transferase (19), promoting liver inflammation through vascular cell adhesion molecule-1, intercellular adhesion molecule-1 (ICAM-1), and TNF-α (42) and by inhibiting fatty acid oxidation by peroxisome proliferator-activated receptor α (PPARα) (43). Notably, IL-10 secreted by M2-like macrophages leads to the apoptosis of M1-like macrophages and senescence of hepatocytes (40). A study of HFD-induced fatty liver disease in mice showed that macrophages with cytokine deficiency (IL-4, IL-10, and IFN-γ) are prone to polarization to the M2-like phenotype, which aggravates liver inflammation and fibrosis (44). However, it has also been reported that, in patients with steatohepatitis, differentiated M2-like macrophages increase the risk of liver fibrosis but do not promote liver tissue repair (23).
These data together highlight that the regulatory roles of M1- and M2-like macrophages are not uniform in fatty liver disease.
Normally, KCs contribute to maintaining tissue homeostasis by expressing low levels of major histocompatibility complex II molecules and co-stimulatory molecules (45), high levels of programmed cell death ligand 1, and inhibitory cytokines IL-10 and TGF-β. KCs promote regulatory T cells to facilitate immune tolerance in the liver (46, 47). In contrast, KCs also recognize extrinsic antigens to induce immune responses through PRRs and complements (48). KC depletion by treatment with clodronate-encapsulated liposomes or gadolinium chloride rapidly alleviated steatosis and inflammation in fatty liver disease, probably due to the decreased expression of inflammatory cytokines and fibrosis-related genes, and diminished insulin resistance in hepatocytes (43). However, Clementi AH et al. found that, in a diet-induced obese mice model, KC ablation increased hepatic steatosis, STAT3 signaling, and additional hepatic TG accumulation (49).
Hepatic macrophages play various roles in the different stages of fatty liver disease. In the early stage of hepatic injury, Ly6Chi inflammatory monocytes and neutrophils are recruited to the liver by KCs and differentiate into CD11b+F4/80+ M1-like macrophages. During acute inflammation, KCs can degrade the extracellular matrix and repair tissue injuries. During the repair period, macrophages selectively differentiate into the M2-like phenotype, which promotes fibrosis progression by secreting IL-13 and TGF-β. In summary, these findings highlight that KCs play a complex role and show functional plasticity in the progression of fatty liver disease.
Hepatic macrophages are interacted with other cells and reprogrammed under pathologic conditions. In fatty liver disease, distinctly heterogeneous populations of macrophages can recognize extracellular stimuli through PRRs, including membrane-bound Toll-like receptors (TLRs) and cytoplasmic nucleotide-binding oligomerization domain-like receptors (NLRs), resulting in the secretion of a large amount of inflammatory cytokines, chemokines, and other reactive molecules such as ROS and NO (50). In addition, macrophages could also participate in the progression of fatty liver disease through lipotoxicity, glucotoxicity, and iron poisoning (Figure 1).
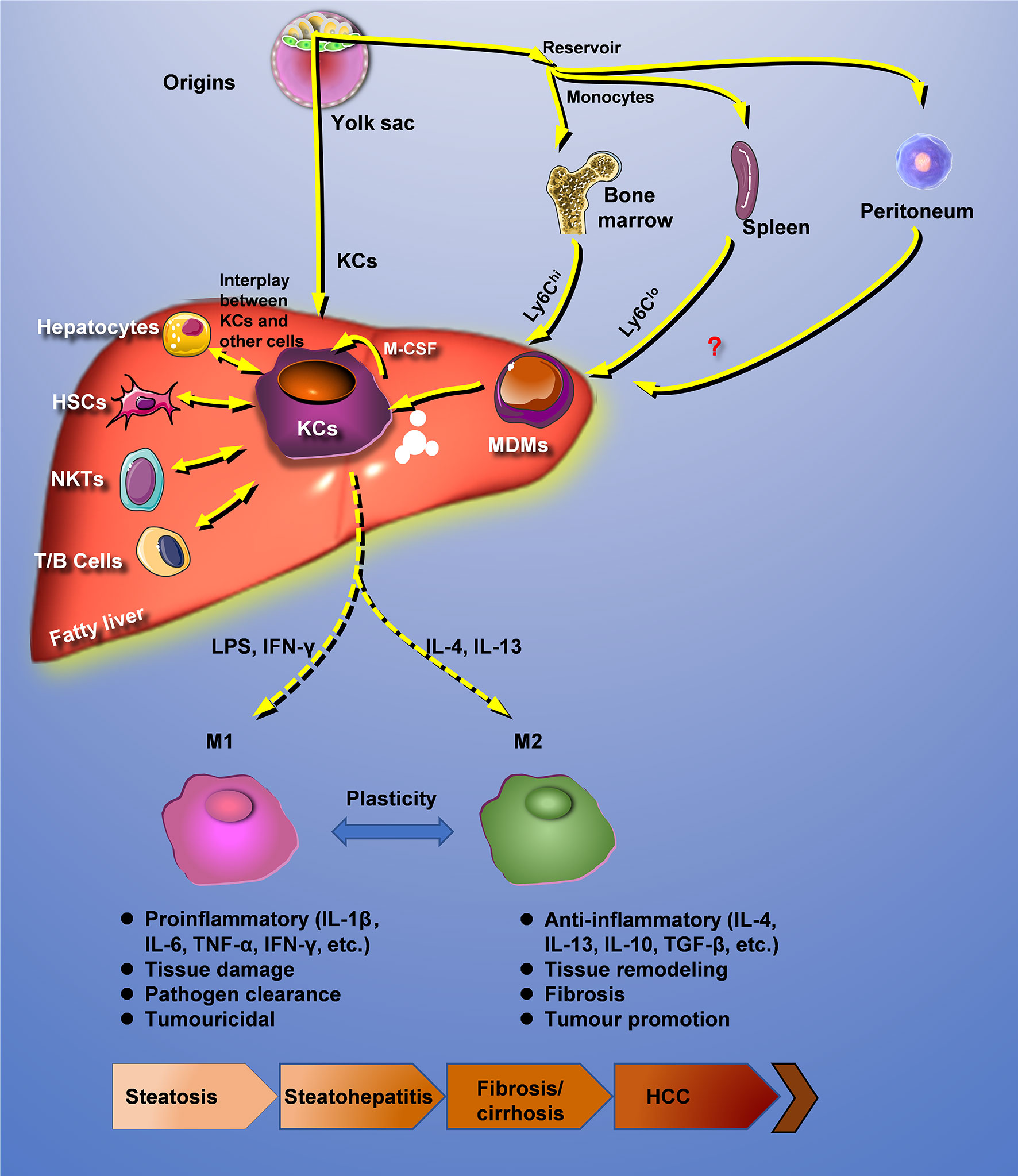
Figure 1 Crosstalk with other cells and reprogramming of hepatic macrophages. Under pathologic conditions, hepatic macrophages are interacted with other cells and reprogrammed. HCC, hepatocellular carcinoma; HSCs, hepatic stellate cells; KCs, Kupffer cells; MDMs, mononuclear-derived macrophages.
KCs are located in the center of hepatic sinusoids; therefore, it is possible that KCs are in intimate contact with the surrounding immune and parenchymal cells in the liver (51). KCs affect lipid metabolism in hepatic parenchyma cells through cell–cell crosstalk. In patients with fatty liver at an early stage, an increase in portal macrophages in liver biopsy sections was the earliest change detected (52). In an HFD-induced murine fatty liver disease, IL-1β released from KCs promotes hepatic steatosis by inhibiting PPARα activity in hepatocytes (47). The released TNF-α activates Caspase-8 in hepatocytes and triggers hepatocyte apoptosis by binding to TNF receptor 1 (53). The increased circulating IL-6 contributes to the development of obesity-associated IR in hepatocytes (54). Additionally, KCs can engulf apoptotic hepatocytes via efferocytic clearance triggered by phosphatidylserine (PS) signals. Recently, our group reported that the PS receptors T cell immunoglobulin mucin domain-containing molecule 3 (Tim-3) and Tim-4 are elevated in liver macrophages in murine steatohepatitis, and their knockout leads to an increased production of ROS, IL-1β, and IL-18 in macrophages, with aggravation of steatosis and inflammation. However, the underlying mechanisms of macrophage-mediated efferocytosis in fatty liver disease require further exploration (55, 56).
KCs can also regulate hepatic stellate cell (HSC) activation by cytokines and chemokines. A co-culture system of macrophages and hepatocytes suggests that macrophages activate the NF-κB signaling pathway in HSCs via the increased secretion of IL-1β, TNF-α, and IL-6 and the upregulated expression of the tissue inhibitor of metalloproteinase 1 in HSCs, which promotes the progression of hepatic fibrosis (57). Activated KCs display a strong acceleration of CC chemokine-induced HSC migration and recruitment through CCL2 and CCL5 production (58). KCs also activate HSCs through TGF-β and platelet-derived growth factor (PDGF), which increases the expression of collagen and accelerates fibrosis progression by trans-differentiating HSCs into myofibroblast phenotype (59–61).
In murine steatohepatitis models, KC secretion of the chemokines CCL2, CXCL10, and TNF-related apoptosis-inducing ligand (TRAIL) is upregulated to recruit monocytes and to trigger monocyte differentiation into KCs. This process is characterized by the over-expression of MMP-12, MMP-13, and insulin growth factor 1 to ameliorate liver injury and fibrosis by degrading the matrix (62, 63). The ability of KCs from HFD-induced mice to recruit CD4+ T lymphocytes and B cells is increased (64). Hepatic macrophages promote neutrophil adhesion to liver sinusoidal endothelial cells (LSECs) via TLR4 (65) and increase neutrophil recruitment via chemokines, such as CXCL1, CXCL2, and CXCL8 (66). KCs can also affect the number and activation of natural killer T (NKT) cells in various ways. KC-derived chemokine CXCL16 recruits CXCR6-expressing NKT cells to accentuate inflammation and fibrosis in the liver (46). The interaction between Tim-3+ KCs and Gal-9 promotes NKT cell proliferation by IL-15 secretion in an HFD-induced murine fatty liver disease (67). In addition, IL-12 released by KCs is associated with the reduction of hepatic NKT cells in the liver of choline-deficient diet-induced mice (68). In addition, PA exposure-activated macrophages present exogenous antigens to NKT cells through CD1d, resulting in excessive activation and apoptosis of NKT cells in HFD-fed mice (69).
It is evidenced that KCs can regulate the function of other cells in the liver. However, they are also influenced by hepatocytes and surrounding cells in the microenvironment of fatty liver disease. Hepatic macrophages internalize extracellular vesicles (EVs). Lipotoxic hepatocyte-derived EVs are enriched with CXCL10, which induces the hepatic recruitment of monocytes, depending on JNK and mixed lineage kinase 3 pathway, in addition to ceramide-containing EVs (70, 71). Moreover, lipotoxic hepatocytes can release active integrin β1-containing EVs to mediate monocyte adhesion to LSECs and inflammation in murine steatohepatitis (72). The injured hepatocytes can also release HMGB1-containing EVs, which mediate mitochondrial damage through the TLR4-JNK pathway and induce inflammation by activating KCs (73). In addition, CCL2 released from lipotoxic hepatocytes recruits monocytes to the liver by binding to CCR2 on monocytes in the process of liver injury (69, 70). The leaked cholesterol crystals from apoptotic hepatocytes can be engulfed by KCs and activate the NLRP3 inflammasome in KCs, causing the production of pro-inflammatory cytokines (74). KC-mediated clearance of apoptotic bodies formed by hepatocytes promotes the production of death receptors, including Fas ligand and TNF-α, which further induces hepatocyte apoptosis, depending on a positive feedback loop (75). Damaged hepatocyte-derived mtDNA could be sensed by the stimulator of IFN genes (STING) in KCs to increase TNF-α and IL-6 production in MCD and HFD-induced murine steatohepatitis models (76). ATP released from damaged hepatocytes promotes NLRP3 inflammasome activation and IL-1β and IL-18 release by the P2X7 receptor on KCs (77). LSECs facilitate the hepatic recruitment of monocytes through the increased production of CCL2, and they could also display anti-inflammatory properties to prevent KC activation in the progression of fatty liver disease (78). In low-density lipoprotein receptor-deficient mice, increased myeloperoxidase secreted by neutrophils causes toxicity to macrophages and aggravates inflammation and insulin resistance (79). Consistent with this, myeloperoxidase deficiency reduces liver inflammation and improves IR in murine fatty liver disease (23). In addition, ROS and growth factors released from neutrophils enhance the M1-like macrophage function in promoting fibrosis by activating HSCs (80). Moreover, single-cell RNA sequencing results showed that activated HSCs regulate the functions of macrophages via HSC-derived stellakines, such as CCL11, CCL2, and CXCL2, in the livers of murine steatohepatitis (81).
Taken together, these findings indicate that macrophages participate in fatty liver disease by regulating the liver parenchymal cells, HSCs, and recruitment of monocytes and NKT cells. Conversely, infiltrating neutrophils and damaged hepatocytes also activate macrophages by secreting factors, which further aggravates the progression of fatty liver disease.
In fatty liver disease, macrophages require metabolic reprogramming to meet the demands for energy and biosynthesis during the process of activation, while changes in metabolic patterns could switch the phenotype of macrophages. A glucose metabolic shift occurs during macrophage polarization. When exposed to LPS and IFN-γ, macrophages are polarized into the M1 phenotype, accompanied by enhanced glycolysis, increased lactic acid production, and activation of the pentose phosphate pathway (PPP). When stimulated by IL-4, IL-13, IL-10, or glucocorticoids, macrophages differentiate into the M2 phenotype and secrete the anti-inflammatory factor IL-10, which results in augmented oxidative phosphorylation (82, 83). A lipid metabolic shift also occurs during macrophage polarization. In vitro studies have shown that IL-4 treatment increases the fatty acid intake and fatty acid oxidation of macrophages; however, IFN-γ and LPS stimulation decreases fatty acid uptake and fatty acid oxidation (84).
In fatty liver disease, the regulation of lipid metabolism determines the macrophage phenotype. Saturated fatty acids promote macrophage differentiation toward the M1 phenotype by activating the NF-κB pathway and increase lipid synthesis by activating sterol regulatory element binding protein-1c (SREBP-1c) in fatty liver disease (85, 86). Fatty acids can regulate lipid metabolism by activating the nuclear transcription factor PPARs. Myeloid-specific PPARδ knockout mice display increased IR and the occurrence of hepatitis by inhibiting macrophage transition to the M2 phenotype (87). Recent evidence shows that hepatic retinoic acid receptor-related orphan receptor-α (ROR-α) promotes macrophage differentiation to the M2 phenotype through kruppel-like factor 4. Moreover, ROR-α-specific knockout macrophages aggravate lipid deposition in HFD-fed mice (88). Increasing evidence indicate that metabolic reprogramming can ameliorate steatosis by switching macrophages to the M2 phenotype (Figure 1).
TLRs mainly recognize bacterial products derived from components of intestinal bacteria, such as LPS and peptidoglycan. The microenvironment of fatty liver disease upregulates TLR4 expression, increases intestinal permeability, and leads to a significant increase in serum LPS. These events lead to the activation of the MyD88-NF-κB signaling pathway and promote inflammatory cytokine secretion in both humans and mice with fatty liver disease (89–91). Moreover, KCs isolated from HFD-fed mice are more sensitive to LPS-induced activation (92).
High-mobility group protein B1 (HMGB1) can trigger TLR4 signaling pathway by promoting p38 phosphorylation, NF-κB translocation, TNF-α release, and polarization of M1-like macrophages (93, 94). The abundance of free fatty acids (FFAs) in the microenvironment of fatty liver disease activates inflammatory signaling pathways in KCs in a TLR2- and TLR4-dependent manner (95–97). Moreover, FFAs can also activate KCs by binding to TLR4 indirectly via fetuin-A (98, 99). The knockout of MyD88 attenuates steatosis and hepatitis induced by a choline-deficient amino acid-defined (CDAA) diet in mice (100). HFD-induced fatty liver disease in mice with macrophage-specific IKK-β deficiency displays over-activation of the NF-κB pathway, insulin resistance, and hepatitis (101). IKK2 inhibition of the NF-κB pathway alleviates steatosis and inflammatory responses in murine steatohepatitis (102).
Unlike the surface receptors TLR2 and TLR4, TLR9 is confined primarily to the endosomes of macrophages. In HFD-fed mice, an increased level of mtDNA released from damaged hepatocytes is responsible for the activation of macrophage populations via TLR9 activation (103). The unmethylated CpG motif-containing bacterial DNA could also bind to TLR9 in KCs and promote IL-1β secretion. In CDAA diet-induced murine steatohepatitis, TLR9 knockout relieves hepatic steatosis, inflammation, and fibrosis by suppressing IL-1β secreted by KCs rather than hepatocytes and hepatic stellate cells (100). TLR9 deficiency also suppresses lipid deposition in HFD-fed mice (103).
Taken together, these results suggest that lipids activate macrophages through TLR pathways, thus promoting the development of fatty liver disease (Figure 2). Therefore, intervention of the TLR pathway might be an ideal strategy for the treatment of fatty liver disease in the future.
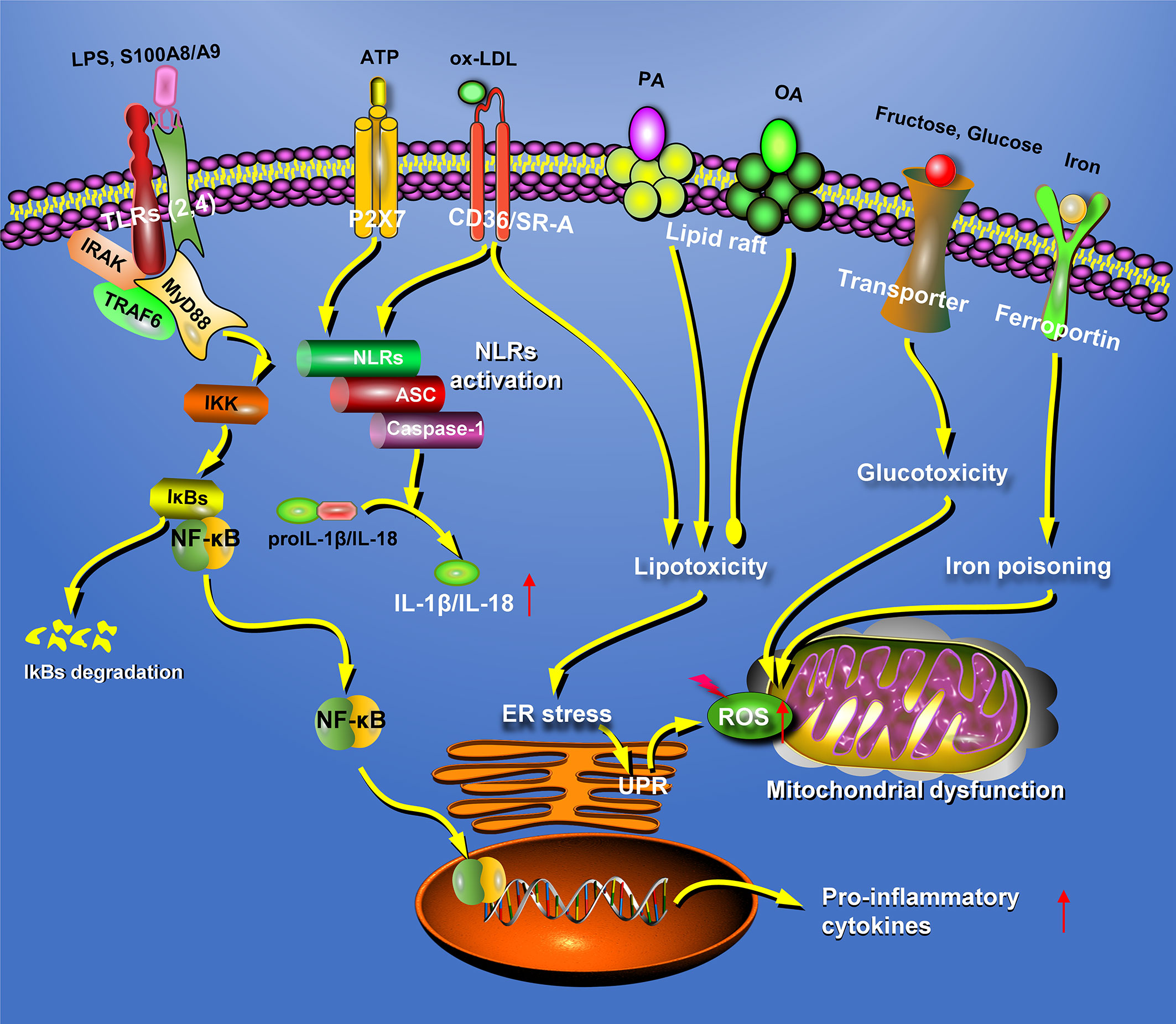
Figure 2 Hepatic macrophages participate in the pathogenesis of fatty liver disease in many different patterns. In fatty liver disease, the macrophages can recognize extracellular stimuli through pattern recognition receptors, including TLRs and NLRs, resulting in the secretion of inflammatory factors. In addition, macrophages could also participate in the progression of NAFLD through lipotoxicity, glucotoxicity, and iron poisoning. ATP, adenosine triphosphate; ER, endoplasmic reticulum; NLRs, nucleotide-binding oligomerization domain-like receptors; OA, oleic acid; PA, palmitic acid; P2X7, P2X purinoceptor 7; ROS, reactive oxygen species; S100A8, S100 calcium-binding proteins A8; SR-A, scavenger receptor-A; TLRs, Toll-like receptors; UPR, unfolded protein response.
NLRs are components of inflammasomes in the cytoplasm, and the responses to danger signals lead to inflammasome activation and IL-1β and IL-18 secretion. The NLRP3 inflammasome is currently the most extensively studied and best-characterized inflammasome in macrophages. Moreover, KCs are considered to be the main cell type responsible for NLRP3 inflammasome activation in the liver. It has been reported that Caspase-1 activity and mature IL-1β levels are significantly increased in steatohepatitis models (104), and NLRP3 inflammasome activation aggravates hepatic steatosis, liver inflammation, and fibrogenesis, supporting the contribution of NLRP3 inflammasome to pathogenesis of fatty liver disease (105). Accordingly, the inhibition of NLRP3 inflammasome by related gene knockout or specific inhibitors has been proposed as an effective therapeutic option for fatty liver disease (106, 107).
Classical NLRP3 inflammasome activation requires active Caspase-1. In this process, adenosine triphosphate (ATP) and P2X purinoceptor 7 (P2X7) receptors on KCs mediate the assembly of the NLRP3 inflammasome. Once activated, pro-Caspase-1 permits auto-cleavage and forms an active Caspase-1 p10/p20 tetramer, which cleaves pro-IL-1β and pro-IL-18 to generate mature IL-1β and IL-18. It has been reported that non-classical NLRP3 inflammasome activation also exists in KCs, requiring active Caspase-11. In this process, LPS from the cytoplasm binds directly to pro-Caspase-11 (108), or LPS binds to TLR4 in the endosome, which promotes pro-Caspase-11 synthesis through the TRAF3-IRF3-IFN pathway (109), resulting in the production of active Caspase-11 and Gsdmd. Active Gsdmd induces KC pyroptosis and promotes the secretion of IL-1β and IL-18 (108, 110–112). Caspase-1 and -11 depletion in macrophages attenuate liver inflammation by reducing the formation of cholesterol crystals and increasing cholesterol effusion (113). Notably, recent studies have shown that inflammasomes are released from virus-infected macrophages, which could provide a novel approach for preventing chronic inflammation (114).
In addition to TLRs and NLRs, hepatic macrophages are also regulated by other receptors. Recently, it was reported that bile acid (a regulator of glycolipid metabolism) inhibits inflammasome activation by binding to transmembrane G protein-coupled receptor 5 on KCs and upregulates the production of anti-inflammatory cytokines (115). In a high-fat- and high-cholesterol-diet-induced murine fatty liver disease, dietary advanced glycation end-product (AGE) activates the MyD88-NF-κB signaling pathway in KCs by upregulating the receptor for AGE (RAGE) (116). S100 calcium-binding protein A8 (S100A8) and S100A9 promotes inflammatory responses in macrophages in both RAGE- and TLR4-dependent manners (117, 118). Docosahexaenoic acid activates PPARγ and AMPKα by binding to GPCR120 on macrophages and decreases the expression of pro-inflammatory genes by suppressing the NF-κB pathway (119–121). Taken together, these data suggest that activation of the NLRP3 inflammasome can aggravate hepatic steatosis (Figure 2).
Hepatic macrophages participate in the development of fatty liver disease by secreting multiple pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α. In mice with fatty liver disease, KCs are a major source of IL-1β. Enhanced IL-1β from activated M1-like macrophages could promote hepatic inflammation by upregulating ICAM-1 in LSECs to attract more neutrophils into the liver (122). IL-1β promotes steatosis by inhibiting fatty acid oxidation via PPARα and promotes TG synthesis, accumulation and lipid droplet formation in hepatocytes (43, 123). IL-1β can also promote hepatocyte apoptosis and aggravate liver fibrosis by activating NF-κB in HSCs (100). Increased IL-6 levels result in an enhanced risk of insulin resistance in patients with fatty liver disease (124). KCs secrete TGF-β and PDGF, which are potent mitogenic factors of HSCs and are vital for hepatic fibrosis progression (125). High levels of TNF-α contribute to inflammasome activation through the NF-κB pathway and aggravate inflammatory injury, insulin resistance, and steatosis (126, 127). Under FFA stimulation or an HFD-induced microenvironment of fatty liver disease, activated KCs release more TNF-α and IFN-γ (128). TNF-α increases hepatic cholesterol synthesis and suppresses its elimination, which results in a dramatic increase in LDL cholesterol and a decrease in HDL cholesterol (129, 130), while a TNF-α blocking antibody alleviates hepatic steatosis in ob/ob mice that have a leptin deficiency (131). In patients with fatty liver disease, TNF-α and IL-8 released from myeloid-derived immune cells, including KCs, DCs, and neutrophils, are positively correlated with the severity of fatty liver disease (132). However, clinical evidences show that treatment with a TNF-α-specific blocking antibody-CDP571 could not alleviate the symptoms of metabolic diseases (133), which may be related to the multiple sources of TNF-α from KCs (57), DCs (134), neutrophils (135), and broad effects in fatty liver disease. The above-mentioned data indicate that a large number of inflammatory cytokines secreted by activated KCs participate in the progression of fatty liver disease.
Activated KCs also participate in fatty liver disease by secreting a variety of chemokines to recruit mononuclear cells to the liver. Levels of CCL2 and CCL19 are increased in the serum of patients with fatty liver disease (136). In murine steatohepatitis, CCL2 binding to CCR2 on Ly6C+ monocytes (137) or CCL1 binding to CCR8 (10) results in more Ly6C+ monocyte recruitment into the liver, which further promotes the progression of hepatitis and liver fibrosis. Therefore, the infiltration of Ly6C+ monocytes has been identified as a key factor in the progression of steatohepatitis and hepatic fibrosis in mice (138, 139). CCR2-/- or CCR2 inhibitor treatment in mice alleviates steatosis, inflammatory cell infiltration, and fibrosis (98, 99). It was also confirmed that the proportion of macrophages in the liver decreased by approximately 80% following the reinfusion of CCR2-/- monocytes (140). CCL2/CCR2 has been identified as chemokines that promote monocyte infiltration into the injured liver; therefore, CCL2/CCR2 is likely to be applied in clinics in the treatment of fatty liver disease in the future. In addition to CCL2, TRAIL, which is secreted by KCs, is also involved in the recruitment of monocytes (141). Monocytes are also recruited by CXCR3, CXCL10, or ceramide through sphingosine 1-phosphate in murine steatohepatitis (142–144).
These results indicate that activated KCs secrete many pro-inflammatory factors that aggravate the development of fatty liver disease (Figure 2).
Under physiological conditions, lipids are responsible for maintaining intracellular metabolism, cell communication, inflammation regulation, and the membrane structural integrity in fatty liver disease. When the rate of fatty acid uptake and synthesis exceeds the rate of fatty acid removal, fatty acids induce cellular stress and lipid toxicity. The term “lipid toxicity” was proposed by Unger for the first time in 1994 when he described cell damage in the muscle of patients with type 2 diabetes and metabolic syndrome induced by toxic lipid molecules (145).
Increased TG levels and the upregulated expression of fatty acid synthesis-related genes, including carbohydrate response element binding protein 1 (CHREBP1), PPARγ, fatty acid synthase, fatty acid binding protein 2, fatty acid transporter 5, and DAG acyltransferase, lead to an excessive accumulation of lipids in KCs (64). Although TG accounts for the largest proportion of these over-synthesized lipids, TG is almost a non-lipotoxic molecule (146, 147). Although lipidomic analysis revealed that the total accumulation of non-toxic TG in the liver of HFD mice was significantly higher than that observed in ND mice, there was no significant difference in the deposition of TG in KCs. In murine steatohepatitis, TG synthesis inhibition ameliorates hepatic steatosis but aggravates liver injury and fibrosis (146).
A relatively small proportion of lipids, including saturated fatty acids, free cholesterol, DAG, ceramide, lysophosphatidylcholine, and bile acid, can be lipotoxic to KCs and hepatocytes (148–152). A sustained toxic lipid accumulation in KCs disrupts the structure of lipid rafts in the plasma and mitochondrial membrane and results in oxidative and endoplasmic reticulum (ER) stress. Under toxic lipid exposure, macrophages are polarized towards the M1 phenotype, with high levels of inflammatory cytokines (such as TNF-α, IL-6, and IL-1β) and secretion of chemokines (such as CCL2, CCL5, and CXCL10) (145). The most abundant fatty acids found in food and fatty liver are saturated fatty acids (PAs) and monounsaturated fatty acids (OA). The excessive accumulation of PA and FFAs in KCs activates the inflammatory signaling pathway (95), induces ER stress and mitochondrial injury, and increases ROS levels (153). However, OA and polyunsaturated fatty acids, such as omega-3 and omega-6, attenuate inflammation and lipotoxicity (154, 155). Saturated fatty acid-induced lipotoxicity in KCs exacerbates the development of fatty liver disease. Short-chain fatty acids enhance fatty acid oxidation and inhibit steatosis progression through PPARγ (156).
Normally, FFAs can be oxidized in the mitochondria, peroxisomes, and microsomes to produce ROS. In fatty liver disease, steatosis increases the efflux of FFAs into the liver, leading to elevated fatty acid β-oxidation and ROS production in the mitochondria. However, excessive ROS leads to mitochondrial dysfunction by reacting with polyunsaturated fatty acids in the mitochondrial membrane, which results in mitochondrial membrane injury, superoxide dismutase inactivation, mitochondrial DNA mutations, and fragmentation (157, 158). In addition, accumulation of misfolded proteins in the ER causes dysfunction and ER stress, which triggers the activation of the unfolded protein response (UPR). In patients with fatty liver disease, increased ER stress activates UPR through transducers inositol-requiring enzyme 1, protein kinase R-like kinase, and activating transcription factor 6, which promotes the expression of p53, release of cytochrome C from the mitochondria, and the activation of NF-κB, JNK, and CEBP signaling pathways in KCs, resulting in IR and apoptosis (159, 160).
In HFD-fed mice, the accumulation of toxic lipids in KCs, such as free cholesterol, cholesterol ester, DAG, and ceramide, is much higher than that in ND mice. Accumulating evidence indicate that lipotoxicity caused by excessive cholesterol accumulation in KCs leads to foam-like cell formation and accelerated fatty liver disease development from simple steatosis to steatohepatitis. Cholesterol uptake by KCs occurs in two ways: LDL receptor (LDLR)-mediated endocytosis and modified LDL uptake by scavenger receptors (SRs). The native LDLs binding to LDLR on KCs promote the lysosomal degradation of LDL into free cholesterol. Increased levels of free cholesterol reduce the intake of cholesterol by inhibiting LDLR and producing lipid-loaded foam-like KCs containing cholesterol crystals. Modified LDLs, such as ox-LDL, are ingested by SR-A and CD36 on KCs, resulting in excessive cholesterol accumulation in lysosomes, NLRP3 inflammasome activation, and foam-like KCs in NASH (161). Moreover, intracellular cholesterol does not regulate SR-A expression, which further increases the number of foam-like KCs and accelerates the development of fatty liver disease. Foam-like KCs secrete chemokines to recruit monocytes and neutrophils and TNF-α and TGF-β to activate hepatic stellate cells, which transform them into myofibroblasts, resulting in hepatic fibrosis (162, 163).
The storage of cholesterol in the mitochondria is increased in human steatohepatitis as evidenced by upregulated mitochondrial cholesterol transporters and steroidogenic acute regulatory protein, but a small amount of cholesterol is found in the cell membrane and ER (164). In rats fed a choline-deficient high-cholesterol diet, mitochondrial function is impaired by the accumulation of free cholesterol and increased sensitivity to TNF-α, leading to Fas-mediated liver injury (165). Cholesterol-lowering agents, such as 2-hydroxypropyl-β-cyclodextrin, could further promote cholesterol efflux from lysosomes to alleviate liver inflammation in murine steatohepatitis (166). In Ldlr−/− mice, SRA-/-/Cd36-/- bone marrow transplantation partially alleviated high-fat- and high-cholesterol-diet-induced inflammation and fibrosis (167). Steatohepatitis induced by an HFD diet combined with ox-LDL also illustrates the important role of ox-LDL in the progression of fatty liver disease. Taken together, the long-term accumulation of toxic lipids in macrophages accelerates the progression of fatty liver disease (Figure 2).
Fatty liver disease progression is associated not only with lipids but also with sugar and glucose transporters (GLUTs), which affect the activity or phenotype switching of macrophages. Glucose and fructose are the two most dominant monosaccharides. However, only fructose is metabolized in the liver (168) and displays a stronger lipogenesis effect (10%) than glucose (2%) (169). Compared with healthy individuals, patients with fatty liver disease have a higher fructose intake (170). A high-sugar diet could further fuel HFD-induced fatty liver disease progression. Evidence reveals that fructose can promote the progression of fatty liver disease by regulating lipase activity, increasing intestinal permeability and motility via TLR4 on KCs (171) and enhancing the interaction with thioredoxin-interacting protein in macrophages. Enhanced nuclear transcription factor SREBP-1c and CHREBP1 promote the de novo synthesis of lipids in the liver (172). Thioredoxin is shuttled into the mitochondria to mediate NLRP3 inflammasome activation and IL-1β, IL-18, and ROS production (173).
In fatty liver disease, increased glucose transporter GLUT1 promotes M1-like macrophage polarization by upregulating the PPP (174). In addition, hypoxia increases glucose uptake by GLUT3 in macrophages, which increases the de novo synthesis and deposition of lipids and promotes the progression of fatty liver disease (175). In summary, high glucose levels polarize macrophages to the pro-inflammatory M1 phenotype and promote the progression of fatty liver disease (Figure 2).
Hepatic iron overload contributes to hepatic inflammation by increasing hepatic cytokine expression in a HFD plus high-iron-induced rat model (176), while hepatic iron depletion by deferoxamine treatment in ob/ob mice improved hepatic steatosis by upregulating lipid metabolism-related genes as well as reducing free radical formation and pro-inflammatory cytokines (177). Although iron overload leads to macrophage polarization toward the M1 phenotype and a significant decrease in prominent regulators of M2 activation, such as PGC-1β, PPARγ, and KLF4, and reduced phosphorylation of STAT6 (178), iron-overloaded hepatic macrophages activate a novel signaling pathway partially consisting of MEK1-TAK1-PI3K-IκB kinase (179).
In a study of 849 patients with steatohepatitis, more than 34% of patients displayed high amounts of iron deposition in liver biopsies (180), and approximately 34% of patients with fatty liver disease showed a dysfunction in the metabolism of iron overload (181). In addition, serum transferrin levels are increased in patients with fatty liver disease (182). Abnormal phagocytosis of erythrocytes by hepatic KCs possibly leads to the accumulation of hemoglobin iron in the liver and triggers oxidative stress (183).
Collectively, these findings suggest that iron poisoning of hepatic macrophages is involved in fatty liver disease (Figure 2), but the mechanisms of iron accumulation in KCs remain unclear and require further investigation.
Several studies in both human and animal models have shown that hepatic macrophages play a central role in the development and progression of fatty liver disease. In the microenvironment of fatty liver disease, any signal of DAMPs, PAMPs, lipotoxicity, or glucotoxicity could trigger KC activation or polarization through the TLR or NLR signaling pathways, resulting in the increased secretion of inflammatory cytokines and chemokines and imbalanced metabolic reprogramming. The released cytokines facilitate the communication between KCs and other cells, including parenchymal cells, HSCs, NK cells, and NKT cells, and the activation of these cells in the liver, while secreted chemokines foster more monocyte infiltration into the liver, where they can differentiate into KCs in a positive loop manner. In addition, metabolic reprogramming leads to disorders in glycolysis, lipid synthesis, and iron metabolism. All these abnormalities collectively contribute to steatosis, inflammation, and fibrogenesis in the development of fatty liver disease. The current strategies for targeting macrophages to treat fatty liver disease mainly include the inhibition of macrophage activation (e.g., via inhibiting the inflammasome assembly), regulation of macrophage polarization (e.g., via promoting polarization into the M2 phenotype through nanoparticles), inhibition of monocyte recruitment and infiltration (e.g., via suppressing the expression of chemokines like CCL2, CCL10, or CCL3), and amelioration of toxic lipid accumulation (e.g., via promoting lipolysis, FFA efflux, and transformation to nontoxic TG) (184–186). Various medications currently targeting macrophage for fatty liver disease are under clinical evaluation in humans. These medications include cenicriviroc, selonsertib, emricasan, GR-MD-02, IMM-124E, JKB-121, SGM-1019, tropifexor, GS-0976, GS-9674, and lanifibranor (9, 10). Further in-depth investigation of hepatic macrophages will help develop novel strategies for the treatment of fatty liver disease and related chronic liver diseases in the future.
LX and WL wrote and revised the manuscript. FB, YX, XL, and CM collected the related papers and helped to draft and revise the manuscript. LG participated in the design of the manuscript and was the major contributor. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (nos. 81971480, 81670520, and 81902921), the joint fund project of the Natural Science Foundation of Shandong Province (ZR2019LZL013), the Taishan Scholarship (no. tspd20181201), and the Shandong Provincial Key Innovation project (no. 2018YFJH0503). The authors would like to express their thanks for the support from the Collaborative Innovation Center of Technology and Equipment for Biological Diagnosis and Therapy in the Universities of Shandong.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Williams C, Stengel J, Asike M, Torres D, Shaw J, Contreras M, et al. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology (2011) 140:124–31. doi: 10.1053/j.gastro.2010.09.038
3. Eslam M, Sanyal A, George J, Panel I. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology (2020) 158:1999–2014. e1. doi: 10.1053/j.gastro.2019.11.312
4. Cohen JC, Horton JD, Hobbs HH. Human Fatty Liver Disease: Old Questions and New Insights. Science (2011) 332:519–1523. doi: 10.1126/science.1204265
5. Moore JB. Non-Alcoholic Fatty Liver Disease: The Hepatic Consequence of Obesity and the Metabolic Syndrome. Proc Nutr Soc (2010) 69:211–20. doi: 10.1017/S0029665110000030
6. Buzzetti E, Pinzani M, Tsochatzis EA. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism (2016) 65:1038–48. doi: 10.1016/j.metabol.2015.12.012
7. Smedsrod B, Bleser PJ, Braet F, Lovisetti P, Vanderkerken K, Wisse E, et al. Cell Biology of Liver Endothelial and Kupffer Cells. Gut (1994) 35:1509–16. doi: 10.1136/gut.35.11.1509
8. Baffy G. Kupffer Cells in Non-Alcoholic Fatty Liver Disease: The Emerging View. J Hepatol (2009) 51:212–23. doi: 10.1016/j.jhep.2009.03.008
9. Lanthier N. Targeting Kuffer Cells in Non-Alcoholic Fatty Liver Disease/Non-Alcohlic Steatohepatitis: Why and How? World J Hepatol (2015) 7:2184–8. doi: 10.4254/wjh.v7.i19.2184
10. Tacke F, Lefere S. Macrophages in Obesity and Non-Alcoholic Fatty Liver Disease:Crosstalk With Metabolism. JHEP Rep (2019) 1:30–43. doi: 10.1016/j.jhepr.2019.02.004
11. Chen JJ, Deng XY, Liu YJ, Tan QH, Huang GD, Che QS, et al. Kupffer Cells in Non-Alcoholic Fatty Liver Disease: Friend or Foe? Int J Biol Sci (2020) 16:2367–78. doi: 10.7150/ijbs.47143
12. Naito M, Hasegawa G, Takahashi K. Development, Differentiation, and Maturation of Kupffer Cells. Microsc Res Tech (1997) 39:350–64. doi: 10.1002/(SICI)1097-0029(19971115)39:4<350::AID-JEMT5>3.0.CO;2-L
13. Perdiguero EG, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-Resident Macrophages Originate From Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature (2015) 518:547–51. doi: 10.1038/nature13989
14. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages Under Homeostasis. Immunity (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
15. Park JW, Jeong G, Kim SJ, Kim MK, Park SM. Predictors Reflecting the Pathological Severity of Non-Alcoholic Fatty Liver Disease: Comprehensive Study of Clinical and Immunohistochemical Findings in Younger Asian Patients. J Gastroenterol Hepatol (2007) 22:491–7. doi: 10.1111/j.1440-1746.2006.04758.x
16. Deng Z, Liu Y, Liu C, Xiang XY, Wang JH, Cheng ZQ, et al. Immature Myeloid Cells Induced by a High-Fat Diet Contribute to Liver Inflammation. Hepatol (Baltimore Md) (2009) 50:1412–20. doi: 10.1002/hep.23148
17. Serbina NV, Pamer EG. Monocyte Emigration From Bone Marrow During Bacterial Infection Requires Signals Mediated by Chemokine Receptor CCR2. Nat Immunol (2006) 7:311–7. doi: 10.1038/ni1309
18. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science (2009) 325:612–6. doi: 10.1126/science.1175202
19. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic Recruitment of the Inflammatory Gr1+ Monocyte Subset Upon Liver Injury Promotes Hepatic Fibrosis. Hepatology (2009) 50:261–74. doi: 10.1002/hep.22950
20. Bartneck M, Ritz T, Keul HA, Wambach M, Bornemann J, Gbureck U, et al. Peptide-Functionalized Gold Nanorods Increase Liver Injury in Hepatitis. ACS Nano (2012) 6:8767–77. doi: 10.1021/nn302502u
21. Tacke F, Randolph GJ. Migratory Fate and Differentiation of Blood Monocyte Subsets. Immunobiology (2016) 211:609–18. doi: 10.1016/j.imbio.2006.05.025
22. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-C Motif) Receptor 2-Positive Monocytes Aggravate the Early Phase of Acetaminophen-Induced Acute Liver Injury. Hepatology (2016) 64:1667–82. doi: 10.1002/hep.28682
23. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased Hepatic Myeloperoxidase Activity in Obese Subjects With Nonalcoholic Steatohepatitis. Am J Pathol (2009) 175:1473–82. doi: 10.2353/ajpath.2009.080999
24. Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T. Et al. Myeloid Cells in Liver and Bone Marrow Acquire a Functionally Distinct Inflammatory Phenotype During Obesity-Related Steatohepatitis. Gut (2020) 69:551–63. doi: 10.1136/gutjnl-2019-318382
25. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity (2017) 47(2):374–88.e6. doi: 10.1016/j.immuni.2017.07.018
26. Scott CL, Zheng F, Baetselier PD, Martens L, Saeys Y, Prijck SD, et al. Bone Marrow-Derived Monocytes Give Rise to Self-Renewing and Fully Differentiated Kupffer Cells. Nat Commun (2016) 7:10321. doi: 10.1038/ncomms10321
27. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct From Kupffer Cells in the Fatty Liver. Immunity (2020) 53:1–17. doi: 10.1016/j.immuni.2020.08.004
28. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic Inhibition of Inflammatory Monocyte Recruitment Reduces Steatohepatitis and Liver Fibrosis. Hepatol (Baltimore Md) (2018) 67:1270–83. doi: 10.1002/hep.29544
29. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective Depletion of Macrophages Reveals Distinct, Opposing Roles During Liver Injury and Repair. J Clin Invest (2005) 115:56–65. doi: 10.1172/JCI22675
30. Wang J, Kubes P. A Reservoir of Mature Cavity Macrophages That Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell (2016) 165:668–78. doi: 10.1016/j.cell.2016.03.009
31. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Reinhard H, et al. Comparison of Gene Expression Profiles Between Human and Mouse Monocyte Subsets. Blood (2010) 115:e10–19. doi: 10.1182/blood-2009-07-235028
32. Gonzalez-Dominguez E, Samaniego R, Flores-Sevilla JL, Campos-Campos SF, Gomez-Campos G, Salas A, et al. CD163L1 and CLEC5A Discriminate Subsets of Human Resident and Inflammatory Macrophages In Vivo. J Leukoc Biol (2015) 98:453–66. doi: 10.1189/jlb.3HI1114-531R
33. Luo WJ, Xu QY, Wang Q, Wu HM, Hua J. Effect of Modulation of PPAR-Gamma Activity on Kupffer Cells M1/M2 Polarization in the Development of Non-Alcoholic Fatty Liver Disease. Sci Rep (2017) 7:44612. doi: 10.1038/srep44612
34. Sohn W, Jun DW, Lee KN, Lee HL, Lee OY, Choi HS, et al. Lactobacillus Paracasei Induces M2-Dominant Kupffer Cell Polarization in a Mouse Model of Nonalcoholic Steatohepatitis. Dig Dis Sci (2015) 60:3340–50. doi: 10.1007/s10620-015-3770-1
35. Wu HM, Ni XX, Xu QY, Wang Q, Li XY, Hua J. Regulation of Lipid-Induced Macrophage Polarization Through Modulating Peroxisome Proliferator-Activated Receptor-Gamma Activity Affects Hepatic Lipid Metabolism via a Toll-Like Receptor 4/NF-κb Signaling Pathway. J Gastroenterol Hepatol (2020) 35:1998–2008. doi: 10.1111/jgh.15025
36. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-shaul H, Merad M, et al. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell (2014) 159:1312–26. doi: 10.1016/j.cell.2014.11.018
37. Jindal A, Bruzzi S, Sutti S, Locatelli I, Bozzola C, Paternostro C, et al. Fat-Laden Macrophages Modulate Lobular Inflammation in Nonalcoholic Steatohepatitis (NASH). Exp Mol Pathol (2015) 99:155–62. doi: 10.1016/j.yexmp.2015.06.015
38. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C Expression Identifies the Recruited Macrophage Phenotype, Which Orchestrates the Regression of Murine Liver Fibrosis. Proc Natl Acad Sci USA (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
39. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
40. Wan JH, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, et al. M2 Kupffer Cells Promote M1 Kupffer Cell Apoptosis: A Protective Mechanism Against Alcoholic and Nonalcoholic Fatty Liver Disease. Hepatology (2014) 59:130–42. doi: 10.1002/hep.26607
41. Navarro LA, Wree A, Povero D, Berk MP, Eguchi A, Ghosh S, et al. Arginase 2 Deficiency Results in Spontaneous Steatohepatitis: A Novel Link Between Innate Immune Activation and Hepatic De Novo Lipogenesis. J Hepatol (2015) 62:412–20. doi: 10.1016/j.jhep.2014.09.015
42. Tomita K, Tamiya G, Ando S, Chiyo T, Mizutani A, Toda K, et al. Tumour Necrosis Factor Alpha Signalling Through Activation of Kupffer Cells Plays an Essential Role in Liver Fibrosis of Non-Alcoholic Steatohepatitis in Mice. Gut (2006) 55:415–24. doi: 10.1136/gut.2005.071118
43. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, Rooijen NV, et al. Kupffer Cells Promote Hepatic Steatosis via Interleukin-1beta-Dependent Suppression of Peroxisome Proliferator-Activated Receptor Alpha Activity. Hepatol (Baltimore Md) (2010) 51:511–22. doi: 10.1002/hep.23337
44. Hart KM, Fabre T, Sciurba JC, Gieseck3rd RL, Borthwick LA, Vannella KM, et al. Type 2 Immunity Is Protective in Metabolic Disease But Exacerbates NAFLD Collaboratively With TGF-Beta. Sci Transl Med (2017) 9:eaal3694. doi: 10.1126/scitranslmed.aal3694
45. You Q, Cheng LL, Kedl RM, Ju C. Mechanism of T Cell Tolerance Induction by Murine Hepatic Kupffer Cells. Hepatology (2008) 48:978–90. doi: 10.1002/hep.22395
46. Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic Regulatory T Cells and Kupffer Cells Are Crucial Mediators of Systemic T Cell Tolerance to Antigens Targeting Murine Liver. Hepatology (2009) 50:612–21. doi: 10.1002/hep.23043
47. Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell-Specific Expression of Transforming Growth Factor-Beta in Rat Liver. Evidence for Autocrine Regulation of Hepatocyte Proliferation. J Clin Invest (1995) 96:447–55. doi: 10.1172/JCI118055
48. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver Inflammation Abrogates Immunological Tolerance Induced by Kupffer Cells. Hepatology (2015) 62:279–91. doi: 10.1002/hep.27793
49. Clementi AH, Gaudy AM, Rooijen NV, Pierce RH, Mooney RA. Loss of Kupffer Cells in Diet-Induced Obesity Is Associated With Increased Hepatic Steatosis, STAT3 Signaling, and Further Decreases in Insulin Signaling. Biochim Biophys Acta (2009) 1792:1062–72. doi: 10.1016/j.bbadis.2009.08.007
50. Wehr A, Baeck C, Heymann F, Niemietz NL, Martin C, Zimmermann HW, et al. Chemokine Receptor CXCR6-Dependent Hepatic NK T Cell Accumulation Promotes Inflammation and Liver Fibrosis. J Immunol (2013) 190:5226–36. doi: 10.4049/jimmunol.1202909
51. Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott C, Martens L, et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity (2019) 51:638–54.e639. doi: 10.1016/j.immuni.2019.08.017
52. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The Portal Inflammatory Infiltrate and Ductular Reaction in Human Nonalcoholic Fatty Liver Disease. Hepatology (2014) 59:1393–405. doi: 10.1002/hep.26937
53. Hirsova CP, Gores GJ. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cell Mol Gastroenterol Hepatol (2015) 1:17–27. doi: 10.1016/j.jcmgh.2014.11.005
54. Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 Induces Cellular Insulin Resistance in Hepatocytes. Diabetes (2002) 51:3391–9. doi: 10.2337/diabetes.51.12.3391
55. Du XH, Wu ZC, Xu Y, Liu Y, Liu W, Wang TX, et al. Increased Tim-3 Expression Alleviates Liver Injury by Regulating Macrophage Activation in MCD-Induced NASH Mice. Cell Mol Immunol (2019) 16:878–86. doi: 10.1038/s41423-018-0032-0
56. Liu W, Bai FX, Wang HX, Liang Y, Du XH, Liu C, et al. Tim-4 Inhibits NLRP3 Inflammasome via the LKB1/AMPKalpha Pathway in Macrophages. J Immunol (2019) 203:990–1000. doi: 10.4049/jimmunol.1900117
57. Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic Stellate Cells in Liver Development, Regeneration, and Cancer. J Clin Invest (2013) 123:1902–10. doi: 10.1172/JCI66369
58. Seki E, Minicis SD, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, et al. CCR1 and CCR5 Promote Hepatic Fibrosis in Mice. J Clin Invest (2009) 119:1858–70. doi: 10.1172/jci37444
59. Pradere JP, Kluwe J, Minicis SD, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic Macrophages But Not Dendritic Cells Contribute to Liver Fibrosis by Promoting the Survival of Activated Hepatic Stellate Cells in Mice. Hepatology (2013) 58:1461–73. doi: 10.1002/hep.26429
60. Minicis SD, Seki E, Uchinami H, Kluwe J, Zhang YH, Brenner DA, et al. Gene Expression Profiles During Hepatic Stellate Cell Activation in Culture and In Vivo. Gastroenterology (2007) 132:1937–46. doi: 10.1053/j.gastro.2007.02.033
61. Kiagiadaki F, Kampa M, Voumvouraki A, Castanas E, Kouroumalis E, Notas G. Activin-A Causes Hepatic Stellate Cell Activation via the Induction of TNFalpha and TGFbeta in Kupffer Cells. Biochim Biophys Acta Mol Basis Dis (2018) 1864:891–9. doi: 10.1016/j.bbadis.2017.12.031
62. Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, et al. Scar-Associated Macrophages Are a Major Source of Hepatic Matrix Metalloproteinase-13 and Facilitate the Resolution of Murine Hepatic Fibrosis. J Immunol (2007) 178:5288–95. doi: 10.4049/jimmunol.178.8.5288
63. Pellicoro A, Aucott RL, Ramachandran P, Robson AJ, Fallowfield JA, Snowdon JA, et al. Elastin Accumulation Is Regulated at the Level of Degradation by Macrophage Metalloelastase (MMP-12) During Experimental Liver Fibrosis. Hepatology (2012) 55:1965–75. doi: 10.1002/hep.25567
64. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, et al. Toxic Lipids Stored by Kupffer Cells Correlates With Their Pro-Inflammatory Phenotype at an Early Stage of Steatohepatitis. J Hepatol (2012) 57:141–9. doi: 10.1016/j.jhep.2012.02.028
65. Nakamoto N, Kanai T. Role of Toll-Like Receptors in Immune Activation and Tolerance in the Liver. Front Immunol (2014) 5:2211. doi: 10.3389/fimmu.2014.002211
66. Marra F, Tacke F. Roles for Chemokines in Liver Disease. Gastroenterology (2014) 147:577–94.e571. doi: 10.1053/j.gastro.2014.06.043
67. Tang ZH, Liang SW, Potter J, Jiang X, Mao HQ, Li ZP. Tim-3/Galectin-9 Regulate the Homeostasis of Hepatic NKT Cells in a Murine Model of Nonalcoholic Fatty Liver Disease. J Immunol (2013) 190:1788–96. doi: 10.4049/jimmunol.1202814
68. Kremer M, Thomas E, Milton RJ, Perry AW, Rooijen NV, Wheeker MD, et al. Kupffer Cell and Interleukin-12-Dependent Loss of Natural Killer T Cells in Hepatosteatosis. Hepatology (2010) 51:130–41. doi: 10.1002/hep.23292
69. Tang TF, Sui YH, Lian M, Li ZP, Hua J. Pro-Inflammatory Activated Kupffer Cells by Lipids Induce Hepatic NKT Cells Deficiency Through Activation-Induced Cell Death. PloS One (2013) 8:e81949. doi: 10.1371/journal.pone.0081949
70. Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes Release Ceramide-Enriched Pro-Inflammatory Extracellular Vesicles in an IRE1alpha-Dependent Manner. J Lipid Res (2016) 57:233–45. doi: 10.1194/jlr.M063412
71. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed Lineage Kinase 3 Mediates Release of C-X-C Motif Ligand 10-Bearing Chemotactic Extracellular Vesicles From Lipotoxic Hepatocytes. Hepatology (2016) 63:731–44. doi: 10.1002/hep.28252
72. Guo QQ, Furuta K, Lucien F, Sanchez LH, Hirsova P, Krishnan A, et al. Integrin Beta1-Enriched Extracellular Vesicles Mediate Monocyte Adhesion and Promote Liver Inflammation in Murine NASH. J Hepatol (2019) 71:1193–205. doi: 10.1016/j.jhep.2019.07.019
73. Gan LT, Rooyen DM, Koina ME, McCuskey RS, Teoh N, Farrell GC. Hepatocyte Free Cholesterol Lipotoxicity Results From JNK1-Mediated Mitochondrial Injury and Is HMGB1 and TLR4-Dependent. J Hepatol (2014) 61:1376–84. doi: 10.1016/j.jhep.2014.07.024
74. Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol Crystallization Within Hepatocyte Lipid Droplets and Its Role in Murine NASH. J Lipid Res (2017) 58:1067–79. doi: 10.1194/jlr.M072454
75. Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer Cell Engulfment of Apoptotic Bodies Stimulates Death Ligand and Cytokine Expression. Hepatology (2003) 38:1188–98. doi: 10.1053/jhep.2003.50472
76. Yu YS, Liu Y, An WS, Song JW, Zhang YF, Zhao XX, et al. STING-Mediated Inflammation in Kupffer Cells Contributes to Progression of Nonalcoholic Steatohepatitis. J Clin Invest (2019) 129:546–55. doi: 10.1172/JCI121842
77. Ishimaru M, Yusuke N, Tsukimoto M, Harada H, Takenouchi T, Kitani H, et al. Purinergic Signaling via P2Y Receptors Up-Mediates IL-6 Production by Liver Macrophages/Kupffer Cells. J Toxicol Sci (2014) 39:413–23. doi: 10.2131/jts.39.413
78. Hammoutene A, Rautou PE. Role of Liver Sinusoidal Endothelial Cells in Non-Alcoholic Fatty Liver Disease. J Hepatol (2019) 70:1278–91. doi: 10.1016/j.jhep.2019.02.012
79. Rensen SS, Bieghs V, Xanthoulea S, Arifianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-Derived Myeloperoxidase Aggravates Non-Alcoholic Steatohepatitis in Low-Density Lipoprotein Receptor-Deficient Mice. PloS One (2012) 7:e52411. doi: 10.1371/journal.pone.0052411
80. Kubes P, Mehal WZ. Sterile Inflammation in the Liver. Gastroenterology (2012) 143:1158–72. doi: 10.1053/j.gastro.2012.09.008
81. Xiong XL, Kuang H, Ansari S, Liu TY, Gong JK, Wang S, et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell (2019) 75:644–60.e645. doi: 10.1016/j.molcel.2019.07.028
82. Biswas SK, Mantovani A. Macrophage Plasticity and Interaction With Lymphocyte Subsets: Cancer as a Paradigm. Nat Immunol (2010) 11:889–96. doi: 10.1038/ni.1937
83. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Frezza C, et al. Succinate Is an Inflammatory Signal That Induces IL-1beta Through HIF-1alpha. Nature (2013) 496:238–42. doi: 10.1038/nature11986
84. Odegaard JI, Chawla A. Alternative Macrophage Activation and Metabolism. Annu Rev Pathol (2011) 6:275–97. doi: 10.1146/ananurev-pathol-011110-130138
85. Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Yong SG, et al. Linking Lipid Metabolism to the Innate Immune Response in Macrophages Through Sterol Regulatory Element Binding Protein-1a. Cell Metab (2011) 13:540–9. doi: 10.1016/j.cmet.2011.04.001
86. Hubler MJ, Kennedy AJ. Role of Lipids in the Metabolism and Activation of Immune Cells. J Nutr Biochem (2016) 34:1–7.02. doi: 10.1016/j.jnutbio.2015.11.002
87. Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-Derived Th2 Cytokines and Myeloid PPARdelta Regulate Macrophage Polarization and Insulin Sensitivity. Cell Metab (2008) 7:485–95. doi: 10.1016/j.cmet.2008.04.002
88. Han YH, Kim HJ, Na H, Nam MW, Kim JY, Kim JS, et al. RORalpha Induces KLF4-Mediated M2 Polarization in the Liver Macrophages That Protect Against Nonalcoholic Steatohepatitis. Cell Rep (2017) 20:124–35. doi: 10.1016/j.celrep.2017.06.017
89. Kono H, Karmarkar D, Iwakura Y, Rock KL. Identification of the Cellular Sensor That Stimulates the Inflammatory Response to Sterile Cell Death. J Immunol (2010) 184:4470–8. doi: 10.4049/jimmunol.0902485
90. Vespasiani-Gentilucci U, Carotti S, Perrone G, Mazzarelli C, Galati G, Onetti-Muda A, et al. Hepatic Toll-Like Receptor 4 Expression Is Associated With Portal Inflammation and Fibrosis in Patients With NAFLD. Liver Int Off J Int Assoc Study Liver (2015) 35:569–81. doi: 10.1111/liv.12531
91. Rivera CA, Adegboyega P, Rooijen NV, Tagalicud A, Allman M, Wallace M. Toll-Like Receptor-4 Signaling and Kupffer Cells Play Pivotal Roles in the Pathogenesis of Non-Alcoholic Steatohepatitis. J Hepatol (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
92. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer Cells in the Liver. Compr Physiol (2013) 3:785–97. doi: 10.1002/cphy.c120026
93. Qin YH, Dai SM, Tang GS, Zhang J, Ren D, Wang ZW, et al. HMGB1 Enhances the Proinflammatory Activity of Lipopolysaccharide by Promoting the Phosphorylation of MAPK P38 Through Receptor for Advanced Glycation End Products. J Immunol (2009) 183:6244–50. doi: 10.4049/jimmunol.0900390
94. Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, et al. Nuclear Factor High-Mobility Group Box1 Mediating the Activation of Toll-Like Receptor 4 Signaling in Hepatocytes in the Early Stage of Nonalcoholic Fatty Liver Disease in Mice. Hepatology (2011) 54:1620–30. doi: 10.1002/hep.24552
95. Ramadori P, Kroy D, Streetz KL. Immunoregulation by Lipids During the Development of Non-Alcoholic Steatohepatitis. Hepatobiliary Surg Nutr (2015) 4:11–23. doi: 10.3978/j.issn.2304-3881.2015.01.02
96. Miura K, Yang L, Rooijen NV, Brenner DA, Ohnishi H, Seki E. Toll-Like Receptor 2 and Palmitic Acid Cooperatively Contribute to the Development of Nonalcoholic Steatohepatitis Through Inflammasome Activation in Mice. Hepatology (2013) 57:577–89. doi: 10.1002/hep.26081
97. Lee JY, Ye JP, Gao ZG, Youn HS, Lee WH, Zhao L, et al. Reciprocal Modulation of Toll-Like Receptor-4 Signaling Pathways Involving MyD88 and Phosphatidylinositol 3-Kinase/AKT by Saturated and Polyunsaturated Fatty Acids. J Biol Chem (2003) 278:37041–51. doi: 10.1074/jbc.M305213200
98. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A Acts as an Endogenous Ligand of TLR4 to Promote Lipid-Induced Insulin Resistance. Nat Med (2012) 18:1279–85. doi: 10.1038/nm.2851
99. Erridge C, Samani NJ. Saturated Fatty Acids Do Not Directly Stimulate Toll-Like Receptor Signaling. Arterioscler Thromb Vasc Biol (2009) 29:1944–9. doi: 10.1161/ATVBAHA.109.194050
100. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-Like Receptor 9 Promotes Steatohepatitis by Induction of Interleukin-1beta in Mice. Gastroenterology (2010) 139:323–4e327. doi: 10.1053/j.gastro.2010.03.052
101. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of Liver Kupffer Cells Prevents the Development of Diet-Induced Hepatic Steatosis and Insulin Resistance. Diabetes (2010) 59:347–57. doi: 10.2337/db09-0016
102. Beraza N, Malato Y, Borght SV, Liedtke C, Wasmuth HE, Vos RD, et al. Pharmacological IKK2 Inhibition Blocks Liver Steatosis and Initiation of Non-Alcoholic Steatohepatitis. Gut (2008) 57:655–63. doi: 10.1136/gut.2007.134288
103. Garcia-Martinez I, Santoro N, Chen YL, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte Mitochondrial DNA Drives Nonalcoholic Steatohepatitis by Activation of TLR9. J Clin Invest (2016) 126:859–64. doi: 10.1172/JCI83885
104. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty Acid and Endotoxin Activate Inflammasomes in Mouse Hepatocytes That Release Danger Signals to Stimulate Immune Cells. Hepatology (2011) 54:133–44. doi: 10.1002/hep.24341
105. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
106. Mridha AR, Wree A, Robertson AA, Yeh M, Johnson CD, Rooyen DM, et al. NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J Hepatol (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
107. Yang G, Lee HE, Lee JY. A Pharmacological Inhibitor of NLRP3 Inflammasome Prevents Non-Alcoholic Fatty Liver Disease in a Mouse Model Induced by High Fat Diet. Sci Rep (2016) 6:24399. doi: 10.1038/srep24399
108. Kayagaki N, Wong MT, Stowe IB, Ranjani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science (2013) 341:1246–9. doi: 10.1126/science.1240248
109. Shi JJ, Zhao Y, Wang YP, Gao WQ, Ding JJ, Li P, et al. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
110. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science (2013) 339:975–8. doi: 10.1126/science.1230751
111. Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
112. Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell (2012) 150:606–19. doi: 10.1016/j.cell.2012.07.007
113. Hendrikx T, Bieghs V, Walenbergh SM, Gorp PJ, Verheyen F, Jeurissen ML, et al. Macrophage Specific Caspase-1/11 Deficiency Protects Against Cholesterol Crystallization and Hepatic Inflammation in Hyperlipidemic Mice. PloS One (2013) 8:e78792. doi: 10.1371/journal.pone.0078792
114. Tzeng TC, Schattgen S, Monks B, Wang DH, Cerny A, Latz E, et al. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks During In Vivo Infection. Cell Rep (2016) 16:571–82. doi: 10.1016/j.celrep.2016.06.011
115. Guo CS, Xie SJ, Chi ZX, Zhang JH, Liu YY, Zhang L, et al. Bile Acids Control Inflammation and Metabolic Disorder Through Inhibition of NLRP3 Inflammasome. Immunity (2016) 45:802–16. doi: 10.1016/j.immuni.2016.09.008
116. Leung C, Herath CB, Jia ZY, Andrikopoulos S, Brown BE, Davies MJ, et al. Dietary Advanced Glycation End-Products Aggravate Non-Alcoholic Fatty Liver Disease. World J Gastroenterol (2016) 22:8026–40. doi: 10.3748/wjg.v22.i35.8026
117. Fassl SK, Austermann J, Papantonopoulou O, Riemenschneider M, Xue J, Berthloot D, et al. Transcriptome Assessment Reveals a Dominant Role for TLR4 in the Activation of Human Monocytes by the Alarmin MRP8. J Immunol (2015) 194:575–83. doi: 10.4049/jimmunol.1401085
118. Xia C, Braunstein Z, Toomey AC, Zhong JX, Rao XQ. S100 Proteins As an Important Regulator of Macrophage Inflammation. Front Immunol (2017) 8:1908. doi: 10.3389/fimmu.2017.01908
119. Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, Balint BL, et al. Structural Basis for the Activation of PPARgamma by Oxidized Fatty Acids. Nat Struct Mol Biol (2008) 15:924–31. doi: 10.1038/nsmb.1474
120. Xue BZ, Yang ZG, Wang XF, Shi H. Omega-3 Polyunsaturated Fatty Acids Antagonize Macrophage Inflammation via Activation of AMPK/SIRT1 Pathway. PloS One (2012) 7:e45990. doi: 10.1371/journal.pone.0045990
121. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan WQ, et al. GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-Inflammatory and Insulin-Sensitizing Effects. Cell (2010) 142:687–98. doi: 10.1016/j.cell.2010.07.041
122. Ito S, Yukawa T, Uetake S, Yamauchi M. Serum Intercellular Adhesion Molecule-1 in Patients With Nonalcoholic Steatohepatitis: Comparision With Alcoholic Hepatitis. Alcohol Clin Exp Res (2007) 31(1Suppl):S83–7. doi: 10.1111/j.1530-0277.2006.00292.x
123. Negrin KA, Flach RJ, DiStefano MT, Matevossian A, Friedline RH, Jung DY, et al. IL-1 Signaling in Obesity-Induced Hepatic Lipogenesis and Steatosis. PloS One (2014) 9:e107265. doi: 10.1371/journal.pone.0107265
124. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-Reactive Protein, Interleukin 6, and Risk of Developing Type 2 Diabetes Mellitus. JAMA (2001) 286:327–34. doi: 10.1001/jama.286.3.327
125. Dongiovanni P, Romeo S, Valenti L. Hepatocellular Carcinoma in Nonalcoholic Fatty Liver: Role of Environmental and Genetic Factors. World J Gastroenterol (2014) 20:12945–55. doi: 10.3748/wjg.v20.i36.12945
126. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer Cells Trigger Nonalcoholic Steatohepatitis Development in Diet-Induced Mouse Model Through Tumor Necrosis Factor-Alpha Production. J Biol Chem (2012) 287:40161–72. doi: 10.1074/jbc.M112.417014
127. Stojsavljevic S, Palcic MG, Jukic LV, Duvnjak LS, Duvnjak M. Adipokines and Proinflammatory Cytokines, the Key Mediators in the Pathogenesis of Nonalcoholic Fatty Liver Disease. World J Gastroenterol (2014) 20:18070–91. doi: 10.3748/wjg.v20.i48.18070
128. Bergheim I, Weber S, Vos M, Kramer S, Volynets V, Kaserounni S, et al. Antibiotics Protect Against Fructose-Induced Hepatic Lipid Accumulation in Mice: Role of Endotoxin. J Hepatol (2008) 48:983–92. doi: 10.1016/j.jhep.2008.01.035
129. Tacer KF, Kuzman D, Seliskar M, Pompon D, Rozman D. TNF-Alpha Interferes With Lipid Homeostasis and Activates Acute and Proatherogenic Processes. Physiol Genomics (2007) 31:216–27. doi: 10.1152/physiolgenomics.00264.2006
130. Tacer KF, Pompon D, Rozman D. Adaptation of Cholesterol Synthesis to Fasting and TNF-Alpha: Profiling Cholesterol Intermediates in the Liver, Brain, and Testis. J Steroid Biochem Mol Biol (2010) 121:619–25. doi: 10.1016/j.jsbmb.2010.02.026
131. Li ZP, Yang SQ, Lin HZ, Huang JW, Watkins PA, Moser AB, et al. Probiotics and Antibodies to TNF Inhibit Inflammatory Activity and Improve Nonalcoholic Fatty Liver Disease. Hepatology (2003) 37:343–50. doi: 10.1053/jhep.2003.50048
132. Plessis JD, Korf H, Pelt JV, Windmolders P, Elst IV, Verrijken A, et al. Pro-Inflammatory Cytokines But Not Endotoxin-Related Parameters Associate With Disease Severity in Patients With NAFLD. PloS One (2016) 11:e0166048. doi: 10.1371/journal.pone.0166048
133. Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Effects of an Engineered Human Anti-TNF-Alpha Antibody (CDP571) on Insulin Sensitivity and Glycemic Control in Patients With NIDDM. Diabetes (1996) 45:881–5. doi: 10.2337/diab.45.7.881
134. Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, et al. Dendritic Cells Limit Fibroinflammatory Injury in Nonalcoholic Steatohepatitis in Mice. Hepatology (2013) 58:589–602. doi: 10.1002/hep.26267
135. Talukdar S, Oh DY, Bandyopadhyay G, Li DM, Xu JF, McNelis J, et al. Neutrophils Mediate Insulin Resistance in Mice Fed a High-Fat Diet Through Secreted Elastase. Nat Med (2012) 18:1407–12. doi: 10.1038/nm.2885
136. Haukeland JW, Damas JK, Konopski Z, Loberg EM, Haaland T, Goverud I, et al. Systemic Inflammation in Nonalcoholic Fatty Liver Disease Is Characterized by Elevated Levels of CCL2. J Hepatol (2006) 44:1167–74. doi: 10.1016/j.jhep.2006.02.011
137. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological Inhibition of the Chemokine CCL2 (MCP-1) Diminishes Liver Macrophage Infiltration and Steatohepatitis in Chronic Hepatic Injury. Gut (2012) 61:416–26. doi: 10.1136/gutjnl-2011-300304
138. Reid DT, Reyes JL, McDonald BA, Vo T, Reimer RA, Eksteen B. Kupffer Cells Undergo Fundamental Changes During the Development of Experimental NASH and Are Critical in Initiating Liver Damage and Inflammation. PloS One (2016) 11:e0159524. doi: 10.1371/journal.pone.0159524
139. Miura K, Yang L, Rooijen NV, Ohnishi H, Seki E. Hepatic Recruitment of Macrophages Promotes Nonalcoholic Steatohepatitis Through CCR2. Am J Physiol Gastrointest Liver Physiol (2012) 302:G1310–1321. doi: 10.1152/ajpgi.00365.2011
140. Oh DY, Morinaga H, Talukdar S, Bae EJ, Olefsky JM. Increased Macrophage Migration Into Adipose Tissue in Obese Mice. Diabetes (2012) 61. doi: 10.2337/db11-0860
141. Idrissova L, Malhi H, Werneburg NW, LeBrasseur NK, Bronk SF, Fingas C, et al. TRAIL Receptor Deletion in Mice Suppresses the Inflammation of Nutrient Excess. J Hepatol (2015) 62:1156–63. doi: 10.1016/j.jhep.2014.11.033
142. Krenkel O, Tacke F. Liver Macrophages in Tissue Homeostasis and Disease. Nat Rev Immunol (2017) 17:306–21. doi: 10.1038/nri.2017.11
143. Zhang X, Han JQ, Man K, Li XX, Du JH, Chu ES, et al. CXC Chemokine Receptor 3 Promotes Steatohepatitis in Mice Through Mediating Inflammatory Cytokines, Macrophages and Autophagy. J Hepatol (2016) 64:160–70. doi: 10.1016/j.jhep.2015.09.005
144. Mauer AS, Hirsova P, Maiers JL, Shah VH, Malhi H. Inhibition of Sphingosine 1-Phosphate Signaling Ameliorates Murine Nonalcoholic Steatohepatitis. Am J Physiol Gastrointest Liver Physiol (2017) 312:G300–13. doi: 10.1152/ajpgi.00222.2016
145. Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH. Beta-Cell Lipotoxicity in the Pathogenesis of Non-Insulin-Dependent Diabetes Mellitus of Obese Rats: Impairment in Adipocyte-Beta-Cell Relationships. Proc Natl Acad Sci USA (1994) 91:10878–82. doi: 10.1073/pnas.91.23.10878
146. Yamaguchi K, Yang L, McCall S, Huang JW, Yu XX, Pandey S, et al. Inhibiting Triglyceride Synthesis Improves Hepatic Steatosis But Exacerbates Liver Damage and Fibrosis in Obese Mice With Nonalcoholic Steatohepatitis. Hepatology (2007) 45:1366–74. doi: 10.1002/hep.21655
147. Wouters K, Bilsen MV, Gorp PJ, Bieghs V, Lutjohann D, Kerksiek A, et al. Intrahepatic Cholesterol Influences Progression, Inhibition and Reversal of Non-Alcoholic Steatohepatitis in Hyperlipidemic Mice. FEBS Lett (2010) 584:1001–5. doi: 10.1016/j.febslet.2010.01.046
148. Kim EJ, Kim BH, Seo HS, Lee YJ, Kim HH, Son HH, et al. Cholesterol-Induced Non-Alcoholic Fatty Liver Disease and Atherosclerosis Aggravated by Systemic Inflammation. PloS One (2014) 9:e97841. doi: 10.1371/journal.pone.0097841
149. Hendrikx T, Walenbergh SM, Hofker MH, Shiri-Sverdlov R. Lysosomal Cholesterol Accumulation: Driver on the Road to Inflammation During Atherosclerosis and Non-Alcoholic Steatohepatitis. Obes Rev (2014) 15:424–33. doi: 10.1111/obr.12159
150. Kumashiro N, Erion DM, Zhang DY, Kahn M, Beddow SA, Chu X, et al. Cellular Mechanism of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Proc Natl Acad Sci USA (2011) 108:16381–5. doi: 10.1073/pnas.1113359108
151. Chaurasia B, Summers SA. Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol Metab (2015) 26:538–50. doi: 10.1016/j.tem.2015.07.006
152. Neuschwander-Tetri BA. Hepatic Lipotoxicity and the Pathogenesis of Nonalcoholic Steatohepatitis: The Central Role of Nontriglyceride Fatty Acid Metabolites. Hepatology (2010) 52:774–88. doi: 10.1002/hep.23719
153. Marra F, Svegliati-Baroni G. Lipotoxicity and the Gut-Liver Axis in NASH Pathogenesis. J Hepatol (2018) 68:280–95. doi: 10.1016/j.jhep.2017.11.014
154. Hegazy M, Elsayed NM, Ali HM, Hassan HG, Rashed L. Diabetes Mellitus, Nonalcoholic Fatty Liver Disease, and Conjugated Linoleic Acid (Omega 6): What Is the Link? J Diabetes Res (2019) 2019:5267025. doi: 10.1155/2019/5267025
155. Scorletti E, Byrne CD. Omega-3 Fatty Acids, Hepatic Lipid Metabolism, and Nonalcoholic Fatty Liver Disease. Annu Rev Nutr (2013) 33:231–48. doi: 10.1146/annurev-nutr-071812-161230
156. Besten GD, Bleeker A, Gerding A, Eunen KV, Havinga R, Dijk TH, et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes (2015) 64:2398–408. doi: 10.2337/db14-1213
157. Kang JH. Modification and Inactivation of Cu, Zn-Superoxide Dismutase by the Lipid Peroxidation Product, Acrolein. BMB Rep (2013) 46:555–60. doi: 10.5483/bmbrep.2013.46.11.138
158. Pigeolet E, Corbisier P, Houbion A, Lambert D, Michiels C, Raes M, et al. Glutathione Peroxidase, Superoxide Dismutase, and Catalase Inactivation by Peroxides and Oxygen Derived Free Radicals. Mech Ageing Dev (1990) 51:283–97. doi: 10.1016/0047-6374(90)90078-t
159. Passos E, Ascensao A, Martins MJ, Magalhaes J. Endoplasmic Reticulum Stress Response in Non-Alcoholic Steatohepatitis: The Possible Role of Physical Exercise. Metabolism (2015) 64:780–92. doi: 10.1016/j.metabol.2015.02.003
160. Ashraf NU, Sheikh TA. Endoplasmic Reticulum Stress and Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Free Radic Res (2015) 49:1405–18. doi: 10.3109/10715762.2015.1078461
161. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger Receptors Class A-I/II and CD36 Are the Principal Receptors Responsible for the Uptake of Modified Low Density Lipoprotein Leading to Lipid Loading in Macrophages. J Biol Chem (2002) 277:49982–8. doi: 10.1074/jbc.M209649200
162. Ioannou GN, Rooyen DM, Savard C, Haigh WG, Yeh MM, Teoh NC, et al. Cholesterol-Lowering Drugs Cause Dissolution of Cholesterol Crystals and Disperse Kupffer Cell Crown-Like Structures During Resolution of NASH. J Lipid Res (2015) 56:277–85. doi: 10.1194/jlr.M053785
163. Wallace MC, Friedman SL, Mann DA. Emerging and Disease-Specific Mechanisms of Hepatic Stellate Cell Activation. Semin Liver Dis (2015) 35:107–18. doi: 10.1055/s-0035-1550060
164. Caballero F, Fernandez A, Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C. Enhanced Free Cholesterol, SREBP-2 and StAR Expression in Human NASH. J Hepatol (2009) 50:789–96. doi: 10.1016/j.jhep.2008.12.016
165. Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial Free Cholesterol Loading Sensitizes to TNF- and Fas-Mediated Steatohepatitis. Cell Metab (2006) 4:185–98. doi: 10.1016/j.cmet.2006.07.006
166. Liu B, Ramirez CM, Miller AM, Repa JJ, Turley SD, Dietschy JM, et al. Cyclodextrin Overcomes the Transport Defect in Nearly Every Organ of NPC1 Mice Leading to Excretion of Sequestered Cholesterol as Bile Acid. J Lipid Res (2010) 51:933–44. doi: 10.1194/jlr.M000257
167. Bieghs V, Wouters K, Gorp PJ, Gijbels MJ, Winther MP, Binder CJ, et al. Role of Scavenger Receptor A and CD36 in Diet-Induced Nonalcoholic Steatohepatitis in Hyperlipidemic Mice. Gastroenterology (2010) 138:2477–2486,2486 e2471-2473. doi: 10.1053/j.gastro.2010.02.051
168. Lustig RH. Fructose: It’s "Alcohol Without the Buzz". Adv Nutr (2013) 4:226–35. doi: 10.3945/an.112.002998
169. Faeh D, Minehira K, Schwarz JM, Periasamy R, Park S, Tappy L. Effect of Fructose Overfeeding and Fish Oil Administration on Hepatic De Novo Lipogenesis and Insulin Sensitivity in Healthy Men. Diabetes (2005) 54:1907–13. doi: 10.2337/diabetes.54.7.1907
170. Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, et al. Fructose Consumption as a Risk Factor for Non-Alcoholic Fatty Liver Disease. J Hepatol (2008) 48:993–9. doi: 10.1016/j.jhep.2008.02.011
171. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage Activation and Polarization. Front Biosci (2008) 13:453–61. doi: 10.2741/2692
172. Postic C, Girard J. Contribution of De Novo Fatty Acid Synthesis to Hepatic Steatosis and Insulin Resistance: Lessons From Genetically Engineered Mice. J Clin Invest (2008) 118:829–38. doi: 10.1172/JCI34275
173. Choe JY, Kim SK. Quercetin and Ascorbic Acid Suppress Fructose-Induced NLRP3 Inflammasome Activation by Blocking Intracellular Shuttling of TXNIP in Human Macrophage Cell Lines. Inflammation (2017) 40:980–94. doi: 10.1007/s10753-017-0542-4
174. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic Reprogramming of Macrophages: Glucose Transporter 1 (GLUT1)-Mediated Glucose Metabolism Drives a Proinflammatory Phenotype. J Biol Chem (2014) 289:7884–96. doi: 10.1074/jbc.M113.522037
175. Li L, Liu B, Haversen L, Lu E, Magnusson LU, Stahlman M, et al. The Importance of GLUT3 for De Novo Lipogenesis in Hypoxia-Induced Lipid Loading of Human Macrophages. PloS One (2012) 7:e42360. doi: 10.1371/journal.pone.0042360
176. Atarashi M, Izawa T, Miyagi R, Ohji A, Hashimoto A, Kuwamura M, et al. Dietary Iron Supplementation Alters Hepatic Inflammation in a Rat Model of Nonalcoholic Steatohepatitis. Nutrients (2018) 10:175. doi: 10.3390/nu10020175
177. Xue H, Chen D, Zhong YK, Zhou ZD, Fang SX, Li MY, et al. Deferoxamine Ameliorates Hepatosteatosis via Several Mechanisms in Ob/Ob Mice. Ann New York Acad Sci (2016) 1375:52–65. doi: 10.1111/nyas.13174
178. Handa P, Thomas S, Morgan-Stevenson V, Maliken BD, Gochanour E, Boukhar S, et al. Iron Alters Macrophage Polarization Status and Leads to Steatohepatitis and Fibrogenesis. J Leukocyte Biol (2019) 105:1015–26. doi: 10.1002/JLB.3A0318-108R
179. Chen L, Xiong S, She H, Lin SW, Wang JH, Tsukamoto H. Iron Causes Interactions of TAK1, P21ras, and Phosphatidylinositol 3-Kinase in Caveolae to Activate IkappaB Kinase in Hepatic Macrophages. J Biol Chem (2007) 282:5582–8. doi: 10.1074/jbc.M609273200
180. Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, et al. Relationship Between the Pattern of Hepatic Iron Deposition and Histological Severity in Nonalcoholic Fatty Liver Disease. Hepatology (2011) 53:448–57. doi: 10.1002/hep.24038
181. Marmur J, Beshara S, Eggertsen G, Onelov L, Albiin N, Danielsson O, et al. Hepcidin Levels Correlate to Liver Iron Content, But Not Steatohepatitis, in Non-Alcoholic Fatty Liver Disease. BMC Gastroenterol (2018) 18:78. doi: 10.1186/s12876-018-0804-0
182. Kowdley KV, Belt P, Wilson LA, Yeh MM, Neuschwander-Tetri BA, Chalasani N, et al. Serum Ferritin Is an Independent Predictor of Histologic Severity and Advanced Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Hepatology (2012) 55:77–85. doi: 10.1002/hep.24706
183. Lotowska JM, Sobaniec-Lotowska ME, Lebensztejn DM. The Role of Kupffer Cells in the Morphogenesis of Nonalcoholic Steatohepatitis - Ultrastructural Findings. The First Report in Pediatric Patients. Scand J Gastroenterol (2013) 48:352–7. doi: 10.3109/00365521.2012.746390
184. Szabo G, Petrasek J. Inflammasome Activation and Function in Liver Disease. Nat Rev Gastroenterol Hepatol (2015) 12:387–400. doi: 10.1038/nrgastro.201594
185. Ergen C, Heymann F, Rawashdeh WA, Gremse F, Bartneck M, Panzer U, et al. Targeting Distinct Myeloid Cell Populations In Vivo Using Polymers, Liposomes and Microbubbles. Biomaterials (2017) 114:106–20. doi: 10.1016/j.biomaterials.2016.11.009
Keywords: macrophages, NAFLD, TLRs, NLRP3 inflammasome, lipid toxicity
Citation: Xu L, Liu W, Bai F, Xu Y, Liang X, Ma C and Gao L (2021) Hepatic Macrophage as a Key Player in Fatty Liver Disease. Front. Immunol. 12:708978. doi: 10.3389/fimmu.2021.708978
Received: 13 May 2021; Accepted: 16 November 2021;
Published: 09 December 2021.
Edited by:
Yoshiro Kobayashi, Toho University, JapanReviewed by:
Marco Di Gioia, Boston Children’s Hospital and Harvard Medical School, United StatesCopyright © 2021 Xu, Liu, Bai, Xu, Liang, Ma and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lifen Gao, Z2xmZmxnQHNkdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.