 Akihiro Shimba1,2*
Akihiro Shimba1,2* Koichi Ikuta
Koichi Ikuta- 1Laboratory of Immune Regulation, Department of Virus Research, Institute for Frontier Life and Medical Sciences, Kyoto University, Kyoto, Japan
- 2Department of Human Health Sciences, Graduate School of Medicine, Kyoto University, Kyoto, Japan
- 3Graduate School of Biostudies, Kyoto University, Kyoto, Japan
Glucocorticoids (GCs) are a class of steroid hormones secreted from the adrenal cortex. Their production is controlled by circadian rhythm and stress, the latter of which includes physical restraint, hunger, and inflammation. Importantly, GCs have various effects on immunity, metabolism, and cognition, including pleiotropic effects on the immune system. In general, GCs have strong anti-inflammatory and immunosuppressive effects. Indeed, they suppress inflammatory cytokine expression and cell-mediated immunity, leading to increased risks of some infections. However, recent studies have shown that endogenous GCs induced by the diurnal cycle and dietary restriction enhance immune responses against some infections by promoting the survival, redistribution, and response of T and B cells via cytokine and chemokine receptors. Furthermore, although GCs are reported to reduce expression of Th2 cytokines, GCs enhance type 2 immunity and IL-17-associated immunity in some stress conditions. Taken together, GCs have both immunoenhancing and immunosuppressive effects on the immune system.
Introduction
Glucocorticoids (GCs) are a class of steroid hormones with multiple functions. GCs not only regulate functions of the brain, liver, muscle, and bone, they also exert immunoregulatory effects (1). In general, they have strong anti-inflammatory and immunosuppressive effects and are commonly used to treat allergies, autoimmunity conditions, and inflammation by suppressing the expression of inflammatory cytokines, increasing immunosuppressive proteins, and inducing the apoptosis of lymphocytes (1–3). Especially, GCs strongly inhibit cell-mediated immune responses against cancer and infection (4–6). However, recent studies have reported that GCs, when driven by the diurnal cycle or dietary restriction (DR), enhance immune responses by inducing lymphocyte homing to the lymphoid organs (7, 8). Furthermore, stress-induced GCs have the potential to aggravate inflammation by promoting the differentiation and function of Th17 cells (9, 10). Thus, GCs play important roles both in immune responses against infection and cancer and in triggering inflammation. In this review, we will discuss the immunoenhancing and immunosuppressive functions of GCs, which depend on the immune microenvironment.
Production and Action of GCs
In steady state, GC production is controlled by circadian rhythm through multiple steps (1). First, the suprachiasmatic nucleus (SCN) stimulate the paraventricular nucleus (PVN) of the hypothalamus, which secretes corticotropin-releasing hormone (CRH). The circadian rhythm of the SCN is regulated by transcriptional-translational feedback loop (TTFL) of the molecular circadian clock comprising positive, negative, and accessory loops (11). In addition, light input from retina controls the TTFL in the SCN to synchronize the intrinsic circadian rhythm with environmental light/dark cycle. Next, CRH goes on to stimulate the anterior pituitary to produce adrenocorticotropic hormone (ACTH) into the blood. ACTH induces the expression of the enzyme 11β-hydroxylase, which catalyzes the synthesis of corticosterone and cortisol in the adrenal cortex. Lastly, secreted GCs suppress CRH production from PVN by negative feedback (12). Due to the induction and suppression of GC production, serum GC levels exhibit diurnal oscillation, with a peak at early morning and a nadir at night in diurnal animals like humans, but the opposite in nocturnal animals like rodents. Moreover, psychological, physical, and nutritional stresses induce high levels of GCs. Adrenergic neurons in the locus coeruleus (LC) of the brain stem sense the stress and produce noradrenaline to stimulate CRH-releasing neurons (13, 14). In addition to neuronal signals, inflammatory cytokines such as IL-1, IL-2, IL6, IL-12, TNF-α, and IFN-γ stimulate the hypothalamus-PVN axis and induce GC production (15).
After the production of GCs from the adrenal cortex, bioavailability of GCs is regulated via corticosteroid binding globulin (CBG) and corticosteroid 11β-dehydrogenase (11β-HSD) in peripheral organs (16). Because GCs are hydrophobic molecules, GCs require a transporter and CBG acts as a buffer and carrier. Neutrophil elastase induces cleavage of CBG to lead the delivery of GCs to cells. As the expression level of CBG also follow the circadian rhythm (17), this might contribute to the circadian oscillation of bioavailability of GCs. After delivery of GCs, active cortisol and inactive cortisone are interconverted by two isozymes of 11β-HSD, 11β-HSD1 and 11β-HSD2, in each organ (16). The 11β-HSD1 mainly metabolizes cortisone to cortisol, while the 11β-HSD2 converts cortisol to cortisone. Several papers reported that inflammation induced 11β-HSD1 expression in tissues via TNF-α in rheumatoid arthritis (RA), colitis, and chronic kidney disease, suggesting that inflammation augments effects of GCs by induction of 11β-HSD1 (18–21). Taken together, not only the hypothalamus-PVN axis but also CBG and 11β-HSD control the effects of GC via circadian rhythm and inflammation.
GCs exert their effects through complicated mechanisms (22). In general, GCs bind to glucocorticoid receptor (GR) in the cytoplasm (23), which induces the dimerization of GR and its translocation into the nucleus. There, GR acts as a transcription factor that promotes or suppresses the transcription of target genes by binding to specific DNA sequences known as glucocorticoid-response elements (GREs). In some cases, GR represses transcription by binding to negative GREs (nGREs). Binding of GR monomer to nGRE recruits transcriptional co-repressors, which suppresses transcriptional activation by NF-κB nearby, without direct interaction to NF-κB (22). In other case, GR monomer directly interacts with DNA-bound NF-κB and AP-1 and tethers transcriptional corepressors, without DNA binding of GR (24, 25). Transcriptional induction by GR, however, might be more important than transcriptional repression via nGREs and tethering. First, GCs induce the transcription of IκBα, A20, DUSP1, and GILZ, negative regulators of NF-κB and AP-1, which inhibits macrophage activation by LPS (26). Second, GC-induced transcription might snatch transcriptional coactivators and chromatin remodeling factors from the enhancers in target genes of inflammatory cytokines and reduce their transcription (2).
Regulation of Immune-Related Gene Expression by GR
GR represses the production of inflammatory cytokines and proteins such as IL-6, C3, and TSLP by binding to nGREs and recruiting the corepressors NCOR2 and HDAC2 (27). In addition, GR induces the expression of immunosuppressive molecules such as IκBα, A20 (TNFAIP3), DUSP1, and GILZ. IκBα binds to NF-κB and blocks the activation of NF-κB (28). The A20/TAX1BP1 deubiquitinase complex inhibits the ubiquitination and degradation of RIP1 and enhances the degradation of E2 enzyme Ubc13, which suppresses NF-κB activation (28). The phosphatase DUSP1 suppresses the MAPK pathway including p38, JNK, and ERK (29, 30). DUSP1 dephosphorylates ERK and inhibits the activation of p38 and JNK, which reduces the expression of inflammatory cytokines and chemokines such as IL-1β, IL-6, GM-CSF, CCL2, CXCL1, and CXCL2. GILZ suppresses NF-κB by preventing nuclear transport of NF-κB p65 subunit and inhibits MAPK signaling by directly binding to Ras and Raf-1 (31–33). Thus, these molecules induced by GCs attenuate inflammation by inhibiting NF-κB and MAPK cascades.
Furthermore, NF-κB and GR might cooperatively control gene expression. Vollmer et al. reported that induction of DUSP-1 by GR agonist was enhanced by LPS stimulation in macrophages (34). In addition, Kadiyala et al. reported that GR and NF-κB cooperatively bound to the enhancer of the A20 locus and induced A20 expression (35). However, Rao et al. reported that increase of GR-binding sites in HeLa cells after stimulation with TNF-α and a GR agonist (~1,000 sites) was much smaller than all GR-binding sites after GR agonist stimulation (~8,700 sites) and NF-κB binding sites after TNF-α stimulation (~12,000 sites), suggesting that GR-NF-κB interaction might be partial in GR repression mechanisms (36).
On the other hand, Oh and colleagues suggested that inflammation is not critical for immunosuppressive functions of GR. They analyzed the transcription and chromatinscape of macrophages with dexamethasone (DEX) treatment before and after LPS-stimulation (26). They found that DEX treatment after LPS stimulation showed similar gene expression profile to DEX treatment before LPS stimulation. DEX treatment before and after LPS stimulation showed the upregulation of NF-κB and AP-1 inhibitors such as IκBα, A20, DUSP1, GILZ, which were critical for extinguishing inflammation. Thus, these results suggest that the induction of immunoregulatory factors by GR is important for immune regulation, independently of nGREs and tethering.
To induce transcription, GR binding to palindromic GRE might not be necessary. Schiller et al. performed ChIP-seq analysis with a human osteosarcoma cell line expressing GR with a mutation in dimerization domain and found that DNA-binding of the mutant GR was mostly overlapped with that of wild-type GR (37). It suggests that GR monomer and widespread GRE half-sites are enough for effects of GR. Sasse et al. performed global run-on sequencing (GRO-seq) in human airway epithelial cells to detect nascent RNA and found that GR repressed transcription within 10 minutes (38). By integrating the results of ChIP-seq and GRO-seq, they found that rapid repression of TNF-induced genes by GR did not require local GR-binding to canonical GREs in TNF-induced gene loci. Moreover, GR rapidly changed the accessibility of TNF-induced gene enhancers. Based on these observations, Gerber et al. proposed the squelching model that GR binding to GREs might snatch transcriptional coactivators and chromatin remodeling factors from the enhancers of TNF-induced genes and reduce their transcription of the TNF-induced genes (2). Taken together, transcriptional regulation mediated by GR is highly diversified.
Immunosuppressive Effects of GCs
Endogenous GCs suppress inflammatory responses via innate immune cells and stromal cells in mouse disease models. As for the innate immune cells, GR-deficient macrophages express higher levels of inflammatory molecules, such as IL-6, TNF-α, and COX-2, through the overactivation of p38 MAPK after stimulation with LPS, leading to higher mortality (39). In contrast to the suppression of inflammatory macrophages, GCs enhance the differentiation of tissue-repair macrophages (40, 41). Galuppo et al. reported that LysM-Cre GR-deficient mice exhibited higher mortality and impaired tissue repair in a myocardial infarction model (42). In addition, Ly6Clow monocyte-derived macrophages in the infarcted myocardium of GR-deficient mice were reduced in number and expressed lower levels of genes related with neovascularization, collagen degradation, and scar formation. At the same time, the expression of the inflammatory chemokine CCL5 was upregulated. Thus, endogenous GCs suppress inflammatory macrophages but enhance suppressive macrophages.
Endogenous GCs suppress the maturation and function of dendritic cells (DCs). Li et al. reported that GR-deficient DCs secrete large amounts of inflammatory cytokines, such as IL-1β, IL-12, and TNF-α, which increased IFN-γ production in NK cells and caused higher mortality of GR-deficient mice (43). Elftman et al. showed that the expression of B7 and MHC class II, the maturation markers of DCs, was attenuated by GC treatment in vitro and in vivo, suggesting that GCs suppress DC maturation (44). Thus, GC-treated DCs failed to activate CD8 T cells in herpes simplex virus infection. On the other hand, DEX promotes the differentiation of IL-10-producing tolerogenic DCs (45). Hodrea et al. reported that DEX enhances phagocytosis of human DCs by inducing the expression of the apoptophagocytic genes, ADORA3 (adenosine receptor guiding macrophages to apoptotic cells), CD14, and MERTK (phagocytic receptor for apoptotic cells) (46). Therefore, endogenous and exogenous GCs suppress immunoenhancing function but enhance immunosuppressive function of DCs.
Like innate immune cells, GCs suppress cytokine production in stromal cells and alleviate colitis and asthma. Aranda et al. showed that intestinal epithelial cell-specific villin-Cre GR-deficient mice exacerbated DSS-induced colitis by increase of the neutrophil-recruiting chemokines, CXCL1, CXCL5, and CCL5 (47). Klassen et al. reported that OVA-induced allergic asthma in the lung was alleviated by DEX but that GR deficiency in the airway epithelium (SPC-Cre GR-deficient mice), but not in T cells nor DCs, canceled the suppressive effect of DEX (48). Gibbs et al. reported that GR represses the expression of the neutrophil-recruiting chemokine CXCL5 via nGRE in the CXCL5 promoter in an LPS-induced lung inflammation model (49). Indeed, adrenalectomized mice exhibited higher levels of CXCL5 at night, when the GC concentration is high, with a loss of circadian changes in the neutrophil accumulation in the lung. By contrast, Ince et al. reported that airway club cell-specific GR-deficient mice showed normal oscillations of LPS-induced neutrophil homing to the lung despite the loss of the circadian rhythm of CXCL5 and IL-6 (50). In addition, macrophage-specific LysM-Cre GR-deficient mice showed no diurnal change of CXCL5 and TNF-α but normal change in neutrophil count in the lung. These findings demonstrate that GCs at physiological concentrations suppress inflammation by affecting both immune cells and stromal cells. However, further studies are needed to understand how GCs control neutrophil homing to the lung. In contrast to suppression by GCs, it was also reported that DEX promoted the expression of TLR2 and soluble leukocyte protease inhibitor (SLPI), an antimicrobial molecule, in airway epithelial cells, suggesting that exogenous GCs contribute to immune response against bacterial infection (51–53). Interestingly, DEX-induced DUSP1 promoted IL-1β-driven TLR2 induction by inhibiting p38- and JNK-mediated negative feedback of TLR2 expression (52, 54, 55). Furthermore, DEX-induced DUSP1 maintained IL-1β-induced IRF1 upregulation and IRF-dependent CXCL10 expression. Thus, endogenous and exogenous GCs might support the first defense of lung epithelium but suppress excessive inflammation after infection.
GCs strongly impair the cell-mediated immunity mediated by IFN-γ-producing type-1 helper T (Th1) cells, CD8 T cells, and NK cells. Blotta et al. reported that DEX suppressed IL-12 production in human monocytes and impair IFN-γ production in NK cells (56). Moreover, Quatrini et al. reported that endogenous GCs directly suppressed the immune response of NK cells (57). GR-deficient NK cells from NK cell-specific Ncr1-Cre GR-deficient mice produced higher amounts of IFN-γ after stimulation with IL-12 and IL-18 in vitro. In addition, these mice exhibited increased IFN-γ production and higher lethality after the administration of LPS without any change in IL-6 or TNF-α. Furthermore, GR-deficient NK cells induced excessive inflammation and the lethality of mice with mouse cytomegalovirus (MCMV) infection, because GR deficiency decreased PD-1 expression and elevated IFN-γ production (4).
Like NK cells, endogenous GCs suppress the differentiation and IFN-γ expression of T cells. They also reduce the expression of IL-12R in T cells (58, 59). In addition, GR interacts with T-bet to inhibit its DNA-binding activity (59). Kugler et al. reported that, in a Toxoplasma infection model, T cell-specific Lck-Cre GR-deficient mice exhibited Th1 cells that had normal differentiation but also produced abnormally high levels of IFN-γ and TNF-α, leading to higher mortality (5). Thus, GCs suppress IFN-γ production by Th1 cells and thereby prevent excessive inflammation. GCs also cause dysfunctional CD8 T cells. Acharya et al. found that monocyte-macrophage lineage cells in tumors expressed the enzyme for steroidogenesis, Cyp11a1. and produced local GCs (60). In addition, exhausted CD8 T cells in tumors showed higher expressions of GR. In a MC38-OVA tumor model, GR-deficient CD8 T cells expressed higher IL-2, TNF-α, and IFN-γ; blocked tumor-growth and were less exhausted. These data suggest that GCs produced by monocyte-macrophage lineage cells promote the exhaustion of CD8 T cells and impair the immune surveillance against tumors. Taken together, endogenous GCs suppress excessive inflammation and cell-mediated immunity via innate and cytotoxic immune cells.
The Circadian Rhythm of T Cell Immunity Is Controlled by Endogenous Oscillating GCs
Although GCs strongly repress inflammatory cytokine production, past studies have reported that GCs induced the expression of receptors for IL-2, IL-6, IFN-γ, GM-CSF, and TNF-α, implicating positive effects by GCs on immunity (61). In addition, Franchimont et al. reported that human blood T cells stimulated with DEX increased the transcription of genes related with cell proliferation (Mt1l, Mt1e, and Mt1b), metabolism (Sat1, Vdr), oxidation damage (Ido1), and cell surface receptors (Il1r2, Il7r) (62). IL-7 is a member of the common γ-chain cytokine family and binds to the complex of the IL-7Rα-chain and common γ-chain. IL-7 supports the development, survival, and proliferation of T cells, B cells, and innate lymphoid cells (ILCs) (63–65). In addition, IL-7R signaling protects T cells from the apoptosis induced by GCs (62). Indeed, DEX administration increased IL-7Rα expression in human T-acute lymphoblastic leukemia cells and augmented Bcl-2 expression by IL-7, which protected leukemia cells from apoptosis (66). Like IL-7Rα, GCs also induce CXCR4 expression in immune and stromal cells. GCs elevated the CXCR4 expression in T and B lymphocytes, granulocytes, and monocytes in mouse and human (67–71). Interestingly, Leigh et al. reported that GCs enhanced CXCR4 expression in human bronchial cells, whereas Carolina et al. showed that GCs reduced CXCR4 expression in lung endothelial progenitors, implying a complicated regulation of CXCR4 expression by GCs (72, 73). Thus, GC may enhance T cell immunity by inducing IL-7Rα and CXCR4.
In general, how GCs induce IL-7Rα expression on T cells is well studied. There exists a non-coding conserved sequence 1 (CNS-1) 3.6 kb upstream of the IL-7Rα promoter, the deletion of which prevented the IL-7Rα induction by GCs in mice (74, 75). The CNS-1 region contains two GRE motifs conserved between human and mouse. To investigate the biological significance of the IL-7R induction by GCs, mice harboring point mutations in the two GREs of CNS-1 (GREm mice) as well as T cell-specific CD4-Cre GR-deficient mice have been generated (7). The IL-7R expression on T cells was elevated at night and reduced at daytime in control mice, consistent with the diurnal fluctuation of GCs. Moreover, T cells in the control mice accumulated in the spleen, lymph nodes, and Peyer’s patches at nighttime, but circulated more in peripheral blood at daytime. However, the diurnal fluctuation of IL-7R expression and T cell numbers in the blood and lymphoid organs was abolished in CD4-Cre GR-deficient mice and GREm mice. This oscillation seems to be regulated partly by CXCR4, because CXCR4 expression was also induced by GCs and IL-7R. Therefore, the GC-IL-7R axis controls the diurnal oscillation of T cell distribution between the blood and lymphoid organs by regulating CXCR4 expression (Figure 1).
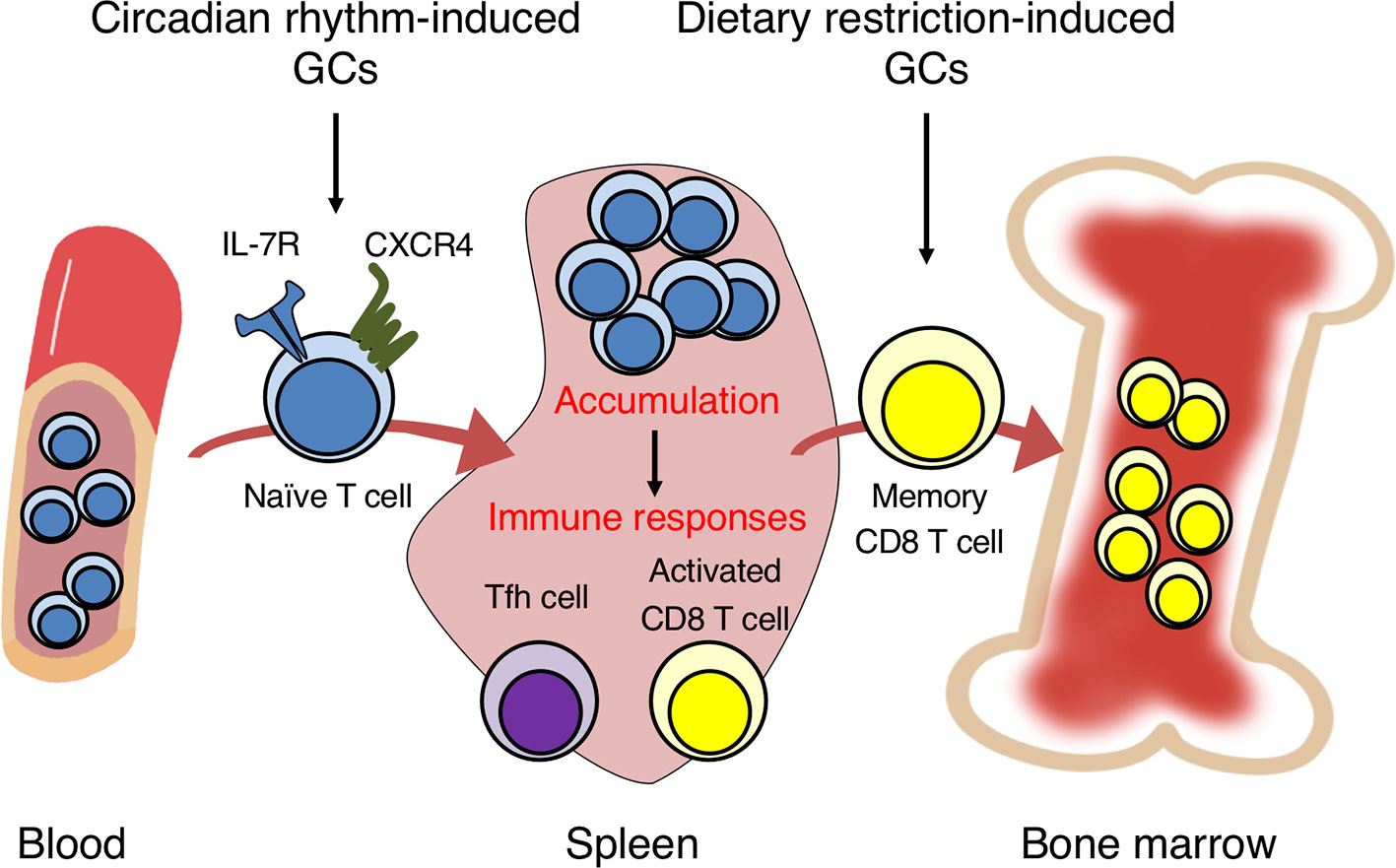
Figure 1 Glucocorticoids in circadian rhythm and dietary restriction induce the migration of naïve and memory T cells into the spleen and bone marrow. GCs induced by circadian rhythm promote the homing of naïve T cells into secondary lymphoid organs from peripheral blood by inducing the expression of IL-7R and CXCR4. The T cell accumulation induces strong immune responses by activated CD8 T and follicular helper T (Tfh) cells against bacterial infection and soluble antigens. GCs induced by dietary restriction trigger the egress of memory CD8 T cells from secondary lymphoid organs and homing into the bone marrow. The accumulation in the bone marrow enhances the survival and response of memory CD8 T cells.
The accumulation of T cells in the lymphoid organs by GCs and IL-7R may enhance immune responses. The infection of control mice with Listeria monocytogenes at nighttime induced antigen-specific effector CD8 T cells more efficiently than at daytime (7). By contrast, the increase of effector CD8 T cells with nighttime infection was not observed in CD4-Cre GR-deficient or GREm mice, suggesting that the diurnal surge of GCs at night enhances the CD8 T cell response against bacterial infection. Similarly, immunization with soluble antigens at night enhanced the generation of follicular helper T (Tfh) cells, germinal center B cells, and class-switched B cells, an effect lost in the mutant mice. Furthermore, previous studies reported that GR affects the differentiation and function of helper T cell subsets (76). GCs strongly suppress the function of Th1 cells but promote the function of Th2 cells. Ramirez et al. found that primed CD4 T cells pretreated with DEX in vitro produced a large amount of IL-4 and IL-13 (77). Consistently, GR-deficient CD4 T cells produced lower levels of IL-4 and IL-13 in Th2-skewed culture (7). Thus, endogenous GCs promote the differentiation and function of Th2 cells. Taken together, oscillating GCs induced by the circadian rhythm have immunoenhancing effects on immunity.
Like T cells, GCs induced by the circadian rhythm enhance B cell responses. Cain et al. reported that the diurnal induction of CXCR4 in B cells was impaired in B cell-specific mb1-Cre GR-deficient mice, which attenuated B cell homing into the bone marrow (69). Additionally, B cell numbers in the blood of GR-deficient mice lost their normal diurnal change, indicating that GCs control the diurnal change of B cell recirculation between the bone marrow and blood via CXCR4 induction. IgG production after immunization with the T-independent antigen NP-Ficoll was impaired in GR-deficient mice, suggesting that endogenous GCs per se might promote the activation of B cells. Furthermore, it is reported that adrenergic signaling and the clock gene Bmal1 control the diurnal change of B cell recirculation between the blood and lymph nodes and that B cell retention in the lymph nodes enhances B cell responses (78, 79). Therefore, CXCR4 induction by GCs might trigger B cell homing to the lymph nodes and bone marrow and elevate the responses of B cells. Taken together, GCs induced by the circadian rhythm enhance B cell responses through the accumulation of lymphocytes in lymphoid organs and the induction of Th2 and Tfh cell differentiation (Table 1).
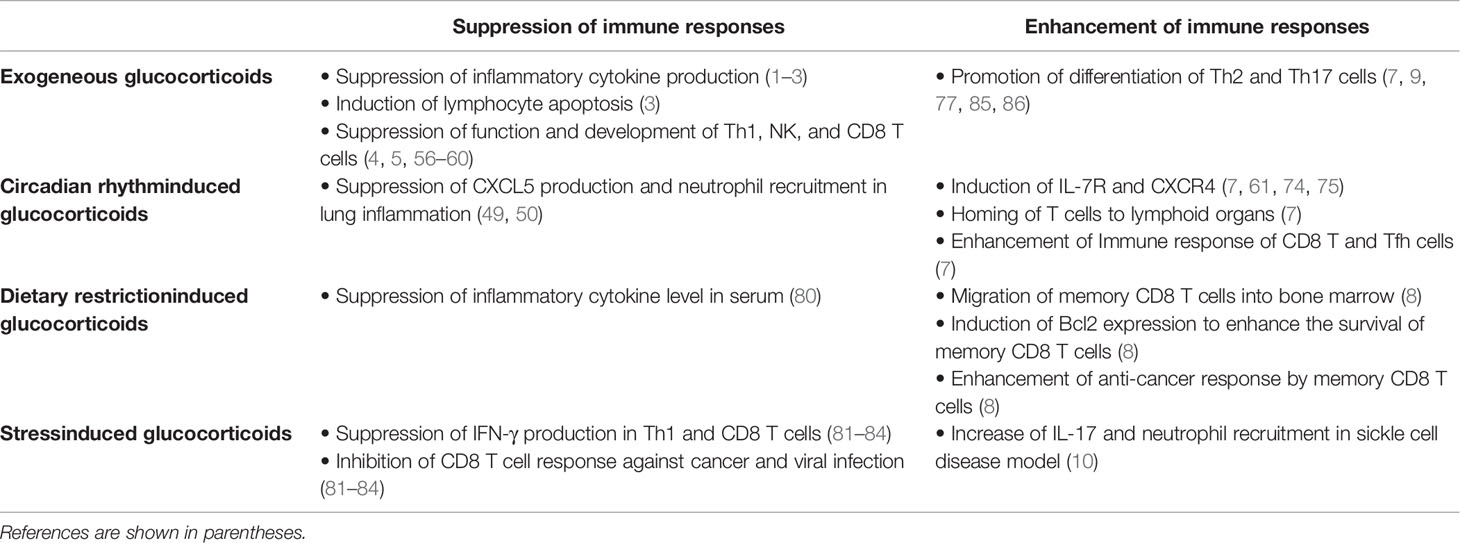
Table 1 A summary of the pleiotropic effects of glucocorticoids in different conditions.
GCs Under Dietary Restriction (DR) Promote Memory CD8 T Cell Response
DR contributes to longevity and reduces inflammation and cancer (80, 87). Because immune cells consume large amount of energy in inflammation, M1 macrophages and effector T cells activate glycolytic and lipogenic metabolic pathways for rapid ATP synthesis (88). Thus, the undernutrition might impair the maintenance and response of leukocytes. DR reduced the mass of thymus and spleen and induced reversible lymphopenia of T, B, and NK cells (89, 90). DR also suppressed the PI3K/Akt/mTOR signaling via activation of AMPK and sirtuin, which impaired the function of effector T cells and M1 macrophages (91). In addition, Yang et al. reported that serum TNF-α levels decreased with elevated cortisol under calorie restriction (CR), suggesting that CR suppresses inflammation by inducing GC production (80). On the other hand, DR enhances the cytotoxicity of CD8 T cells. Di Biase et al. reported that a fasting-mimicking diet increased common lymphoid progenitors in the bone marrow and tumor-infiltrating CD8 T cells in a breast cancer model (92). Therefore, DR promotes lymphopoiesis and cell-mediated immunity. In line with that study, it is reported that diet affects cell-mediated immune responses by inducing GCs. Collins et al. reported that GCs induced by DR enhanced the maintenance and function of memory CD8 T cells (8). DR triggered the migration of memory CD8 T cells from secondary lymphoid organs into the bone marrow, but this effect was abolished in adrenalectomized mice and T cell-specific Lck-Cre GR-deficient mice. In addition, the accumulation of memory CD8 T cells in the bone marrow by DR was impaired in CXCR4- and S1PR1-deficient mice, suggesting that GCs might induce the egress of memory CD8 T cells from secondary lymphoid organs via S1PR1 and drive homing to the bone marrow via CXCR4 (Figure 1). Interestingly, DR reduced mTOR signaling in memory CD8 T cells and changed gene expressions associated with heat-shock protein chaperone binding and the regulation of protein folding. It also induced amino-acid deprivation and the cellular response to rapamycin, suggesting that DR changes the metabolic state of memory CD8 T cells to quiescent. In addition, DR and DEX increased Bcl-2 expression in memory CD8 T cells. Finally, memory CD8 T cells efficiently responded to bacterial infection and tumors during DR. Together, these reports demonstrate that GCs induced by DR enhance the long-term maintenance of memory CD8 T cells, which promotes cytotoxic responses against infection and tumors.
As described above, GCs produced by monocyte-macrophage lineage cells in the tumor microenvironment suppress the function of effector CD8 T cells, whereas DR-induced GCs enhance the maintenance and response of memory CD8 T cells (8, 60). Thus, GCs seem to exert different effects on immune cells, probably by factors supplied from the microenvironment. Circadian rhythm- and DR-induced GCs trigger the homing of IL-7R-positive CD8 T cells into lymphoid organs, which supply pro-survival factors and enhance the maintenance and response of T cells. By contrast, GCs produced in the tumor microenvironment accelerate the dysfunction of IL-7R-negative effector CD8 T cells. Therefore, the supply of IL-7 from the microenvironment may determine whether the GC effects are positive or negative.
Stress Suppresses Cell-Mediated Immunity But Enhances Inflammatory Diseases
Because stress-induced GCs suppress cell-mediated immunity, stress might exacerbate viral infection and tumor growth. Steelman et al. reported that restraint stress reduced IFN-γ-expressing CD4 T and CD8 T cells and their T-bet expression following Theiler’s murine encephalomyelitis virus (TMEV) infection (81). In addition, treatment with a GC antagonist alleviated the clinical manifestations induced by the restraint stress. Elftman et al. found that restraint stress impaired the expression of granzyme B and IFN-γ in CD8 T cells following herpes simplex virus (HSV) infection (82). However, this effect was alleviated if the infection was in T cell-specific Lck-Cre GR-deficient mice, suggesting that stress-induced GCs suppress the production of granzyme B and IFN-γ by CD8 T cells via GR. These findings indicate that stress exacerbates the viral infection by suppressing cell-mediated immunity via GCs. Like viral infection, stress caused by chronic sleep restriction, forced swimming, and abdominal surgery promoted the progression of cancers (83, 84). Hong et al. reported the relationship between perinatal stress and cell-mediated immunity (6). The perinatal exposure of fetal mice to DEX as a stress model diminished CD8 T cell response against tumors in adulthood. Thus, exposure to stress-induced GCs during pregnancy appear to cause dysfunction of anti-tumor response by CD8 T cells in offspring after birth. Overall, stress-induced GCs might impair cell-mediated immunity over the lifetime.
Although stress suppresses cell-mediated immunity, it also triggers chronic inflammation and autoimmunity. Qiu et al. reported that restraint stress enhanced tissue destruction in a DNBS-induced colitis model (93). The colitis was ameliorated in CD4-deficient mice, suggesting that helper T cell function is critical for the stress-induced tissue destruction. Arima et al. reported that wet bedding and restraint stress triggered upper gastrointestinal bleeding in mice transferred with myelin oligodendrocyte glycoprotein (MOG) peptide-primed pathogenic T cells (94). Because Th17 cells play a critical role in experimental autoimmune encephalomyelitis (EAE), the stress might enhance Th17 cell function. de Castro Kroner et al. reported that GCs elevated RAR-related orphan receptor C (RORC) expression by inhibiting IL-2 secretion in human T cells (85). In addition, GCs induced the expression of defensins and CCL20. In mouse T cells, GCs promoted the IL-23-dependent differentiation of Th17 cells in vitro but not if IL-6 and TGF-β were also present (9). In addition, Marchetti et al. reported that transgenic (Tg) mice expressing GR antisense RNA exhibited less severe neurological inflammation (86). Thus, GCs have the potential to enhance the function of Th17 cells. Furthermore, Xu et al. showed that restraint stress increased serum IL-17 and aged neutrophils, which exacerbated vaso-occlusive episodes via microbiota in a sickle cell disease model (10). Treatment with metyrapone, an inhibitor of GC production, alleviated the leukocyte recruitment and inflammation under stress. Overall, stress-induced GCs aggravate inflammation by promoting the differentiation and function of Th17 cells via microbiota.
Although GCs exert some anti-inflammatory effects in neutrophils, such as reduction in COX-2 and iNOS expression and superoxide release (95–97), GCs also promote the development and function of neutrophils. First, GCs enhanced the development and proliferation of neutrophils in bone marrow and induced neutrophilia in peripheral blood (98, 99). Second, GCs promote survival of neutrophils. GCs inhibited spontaneous apoptosis of human neutrophils in vitro (100, 101). Chang et al. reported that DEX treatment reduced the expression of Fas and caspase-8 in neutrophils (102). Furthermore, Bouterse et al. found that the expression of a pro-apoptotic factor Bak was reduced in DEX-treated neutrophils (103). Third, GCs elevated the expression of the IL-1R in neutrophils and enhanced IL-1β-triggered inflammation (104). Thus, GCs might augment tissue inflammation by enhancing the functions of neutrophils.
Stress-induced GCs control inflammation also via Treg cells. Harpaz et al. found that chronic variable stress reduced the number of Treg cells and increased the susceptibility to EAE (105). In addition, the administration of mifepristone, an antagonist of GCs, blocked the stress-induced exacerbation of EAE. On the other hand, it is reported that GCs also promote Treg cell function. GC stimulation upregulates the expression of Foxp3, IL-10, TGF-β, and CTLA4 (106–108). Rocamora-Reverte et al. reported that GR-deficient Treg cells failed to suppress tissue damage in a colitis model in Treg-specific Foxp3-Cre GR-deficient mice (109). Furthermore, Engler et al. reported that the onset of EAE was delayed in pregnant mice, because progesterone binding to GR increased the number of Treg cells (110). Treatment with a GC antagonist and T cell-specific Lck-Cre GR-deletion blocked the activation of Treg cells. However, it remains unknown whether stress enhances inflammation by blocking Treg cells even though GCs enhance Treg function. One possibility is that an unknown factor might block the GC-enhanced Treg function to promote inflammation under stress. Thus, because stress-induced GCs may affect both Th17 and Treg cells, it remains unclear whether GCs enhance inflammation via Th17 cells or suppress it via Treg cells. Taken together, stress-induced GCs seem to have pleiotropic effects either to attenuate cell-mediated immunity or to aggravate inflammation, depending on the cell types and the disease models (Table 1).
GC Resistance in Inflammatory Diseases
GCs are well used for therapy of allergic and autoimmune diseases. GCs mitigate the symptoms in psoriasis and multiple sclerosis, whereas some patients with ulcerative colitis (UC) and asthma are refractory to treatment with GCs (111, 112). However, the mechanism of this GC resistance is unclear. One explanation is that GCs might promote survival of immune cells and enhance immune responses. As explained above, GCs have the potential to augment immune responses of Th2 cells, Th17 cells, and neutrophils, which exacerbates tissue damage. Second, some immune cells escape from the suppressive effects of GCs by reducing the amounts of GCs and GR in the cytoplasm. Ramesh et al. reported that human Th17 cells expressed multiple drug resistance 1 (MDR1), a membrane efflux pump with broad substrate specificity, which reduces the sensitivity to GCs (113). Paugh et al. found that caspase-1 activated by NLRP3 inflammasome cleaved GR (114). Third, some inflammatory cytokines such as IFN-γ, IL-17, IL-4, and TNF-α canceled the suppressive effects of GCs (115–119). These cytokines diminish GC-induced apoptosis of immune cells and blunt the repression of inflammatory cytokine production by GCs. Thus, both the enhancing effects of GCs and the cancelation of GC effects might contribute to the GC resistance in the treatment of inflammatory diseases. Manipulation of these mechanisms will facilitate to overcome the GC resistance and contribute to cure allergic and autoimmune diseases.
Conclusions
This review summarizes the immunoenhancing and immunosuppressive effects of GCs (Table 1). Exogeneous and stress-induced GCs suppress the IFN-γ production and exhaustion of CD8 T cells, whereas GCs under circadian rhythm or DR enhance the maintenance and activation of naïve and memory CD8 T cells. In addition, GCs strongly suppress the function of Th1 cells and enhance the differentiation of Th2 and Th17 cells. Thus, the effects of GCs on immunity can be positive or negative depending on the tissue and cell type. The immunoenhancing effects of GCs possibly depend on the microenvironment because GCs trigger T cell homing to lymphoid organs, which supply pro-survival cytokines. Further studies are required to understand how GCs control the interaction between immune cells and the microenvironment. Moreover, the effects of GCs might depend on different GR-target genes in different cell types. To address this question, it is necessary to investigate GR-induced changes in gene transcription, DNA binding, and chromatin accessibility in immune and stromal cells. Revealing the pleiotropic effects of GCs will help understand how GCs trigger immune dysfunction and chronic inflammation and maximize the therapeutic effects of GCs in refractory allergies and autoimmune diseases.
Author Contributions
AS wrote the first draft of the manuscript. AE and KI modified, revised, and approved the submitted version. All authors contributed to the article andapproved the submitted version.
Funding
This work was supported by JSPS KAKENHI Grant Numbers 20K21525 (KI) and 20K16280 (AS). AS was supported by the Shimizu Foundation for Immunology and Neuroscience grant for 2016, by a grant from the Takeda Science Foundation, and by a grant from the Ichiro Kanehara Foundation for the Promotion of Medical Sciences and Medical care for 2018. AE was supported by the PhD Scholarship (Kibou Project) from Japanese Society for Immunology. KI was supported by a grant from the Naito Foundation and by a grant from the Uehara Memorial Foundation. It was also supported by the Joint Usage Research Center program of the Institute for Frontier Life and Medical Sciences, Kyoto University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cain DW, Cidlowski JA. Immune Regulation by Glucocorticoids. Nat Rev Immunol (2017) 17:233–47. doi: 10.1038/nri.2017.1
2. Gerber AN, Newton R, Sasse SK. Repression of Transcription by the Glucocorticoid Receptor: A Parsimonious Model for the Genomics Era. J Biol Chem (2021) 296:100687. doi: 10.1016/j.jbc.2021.100687
3. Herold MJ, McPherson KG, Reichardt HM. Glucocorticoids in T Cell Apoptosis and Function. Cell Mol Life Sci (2006) 63:60–72. doi: 10.1007/s00018-005-5390-y
4. Quatrini L, Wieduwild E, Escaliere B, Filtjens J, Chasson L, Laprie C, et al. Endogenous Glucocorticoids Control Host Resistance to Viral Infection Through the Tissue-Specific Regulation of PD-1 Expression on NK Cells. Nat Immunol (2018) 19:954–62. doi: 10.1038/s41590-018-0185-0
5. Kugler DG, Mittelstadt PR, Ashwell JD, Sher A, Jankovic D. CD4+ T Cells Are Trigger and Target of the Glucocorticoid Response That Prevents Lethal Immunopathology in Toxoplasma Infection. J Exp Med (2013) 210:1919–27. doi: 10.1084/jem.20122300
6. Hong JY, Lim J, Carvalho F, Cho JY, Vaidyanathan B, Yu S, et al. Medzhitov. Long-Term Programming of CD8 T Cell Immunity by Perinatal Exposure to Glucocorticoids. Cell (2020) 180:847–61. doi: 10.1016/j.cell.2020.02.018
7. Shimba A, Cui GW, Tani-ichi S, Ogawa M, Abe S, Okazaki F, et al. Glucocorticoids Drive Diurnal Oscillations in T Cell Distribution and Responses by Inducing Interleukin-7 Receptor and CXCR4. Immunity (2018) 48:286–98. doi: 10.1016/j.immuni.2018.01.004
8. Collins N, Han SJ, Enamorado M, Link VM, Huang B, Moseman EA, et al. The Bone Marrow Protects and Optimizes Immunological Memory During Dietary Restriction. Cell (2019) 178:1088–101. doi: 10.1016/j.cell.2019.07.049
9. Zhao J, Lloyd CM, Noble A. Th17 Responses in Chronic Allergic Airway Inflammation Abrogate Regulatory T-Cell-Mediated Tolerance and Contribute to Airway Remodeling. Mucosal Immunol (2013) 6:335–46. doi: 10.1038/mi.2012.76
10. Xu CL, Lee SK, Zhang DC, Frenette PS. The Gut Microbiome Regulates Psychological-Stress-Induced Inflammation. Immunity (2020) 53:417–28. doi: 10.1016/j.immuni.2020.06.025
11. Fisk AS, Tam SKE, Brown LA, Vyazovskiy VV, Bannerman DM, Peirson SN. Light and Cognition: Roles for Circadian Rhythms, Sleep, and Arousal. Front Neurol (2018) 9:56. doi: 10.3389/fneur.2018.00056
12. Gjerstad JK, Lightman SL, Spiga F. Role of Glucocorticoid Negative Feedback in the Regulation of HPA Axis Pulsatility. Stress (2018) 21:403–16. doi: 10.1080/10253890.2018.1470238
13. Dunn AJ, Swiergiel AH. The Role of Corticotropin-Releasing Factor and Noradrenaline in Stress-Related Responses, and the Inter-Relationships Between the Two Systems. Eur J Pharmacol (2008) 583:186–93. doi: 10.1016/j.ejphar.2007.11.069
14. McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, et al. CRH Engagement of the Locus Coeruleus Noradrenergic System Mediates Stress-Induced Anxiety. Neuron (2015) 87:605–20. doi: 10.1016/j.neuron.2015.07.002
15. Besedovsky L, Born LJ, Lange T. Blockade of Mineralocorticoid Receptors Enhances Naive T-Helper Cell Counts During Early Sleep in Humans. Brain Behav Immun (2012) 26:1116–21. doi: 10.1016/j.bbi.2012.07.016
16. Perogamvros I, Ray DW, Trainer PJ. Regulation of Cortisol Bioavailability-Effects on Hormone Measurement and Action. Nat Rev Endocrinol (2012) 8:717–27. doi: 10.1038/nrendo.2012.134
17. Melin J, Hartung N, Parra-Guillen ZP, Whitaker MJ, Ross RJ, Kloft C. The Circadian Rhythm of Corticosteroid-Binding Globulin has Little Impact on Cortisol Exposure After Hydrocortisone Dosing. Clin Endocrinol (2019) 91:33–40. doi: 10.1111/cen.13969
18. Nanus DE, Filer AD, Hughes B, Fisher BA, Taylor PC, Stewart PM, et al. TNFα Regulates Cortisol Metabolism In Vivo in Patients With Inflammatory Arthritis. Ann Rheum Dis (2015) 74:464–9. doi: 10.1136/annrheumdis-2013-203926
19. Hardy R, Rabbitt EH, Filer A, Emery P, Hewison M, Stewart PM, et al. Local and Systemic Glucocorticoid Metabolism in Inflammatory Arthritis. Ann Rheum Dis (2008) 67:1204–10. doi: 10.1136/ard.2008.090662
20. Stegk JP, Ebert B, Martin HJ, Maser E. Expression Profiles of Human 11β-Hydroxysteroid Dehydrogenases Type 1 and Type 2 in Inflammatory Bowel Diseases. Mol Cell Endocrinol (2009) 25:104–8. doi: 10.1016/j.mce.2008.10.030
21. Sagmeister MS, Taylor AE, Fenton A, Wall NA, Chanouzas D, Nightingale PG, et al. Glucocorticoid Activation by 11β-Hydroxysteroid Dehydrogenase Enzymes in Relation to Inflammation and Glycaemic Control in Chronic Kidney Disease: A Cross-Sectional Study. Clin Endocrinol (2019) 90:241–9. doi: 10.1111/cen.13889
22. Hardy RS, Raza K, Cooper MS. Therapeutic Glucocorticoids: Mechanisms of Actions in Rheumatic Diseases. Nat Rev Rheumatol (2020) 16:133–44. doi: 10.1038/s41584-020-0371-y
23. Vandevyver S, Dejager L, Libert C. Comprehensive Overview of the Structure and Regulation of the Glucocorticoid Receptor. Endocr Rev (2014) 35:671–93. doi: 10.1210/er.2014-1010
24. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, et al. How Glucocorticoid Receptors Modulate the Activity of Other Transcription Factors: A Scope Beyond Tethering. Mol Cell Endocrinol (2013) 380:41–54. doi: 10.1016/j.mce.2012.12.014
25. Li MD. Yang XY. A Retrospective on Nuclear Receptor Regulation of Inflammation: Lessons From GR and PPARs. PPPAR Res (2011) 2011:742785. doi: 10.1155/2011/742785
26. Oh KS, Patel H, Gottschalk RA, Lee WS, Baek S, Fraser IDC, et al. Sung. Anti-Inflammatory Chromatinscape Suggests Alternative Mechanisms of Glucocorticoid Receptor Action. Immunity (2017) 47:298–309. doi: 10.1016/j.immuni.2017.07.012
27. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread Negative Response Elements Mediate Direct Repression by Agonist-Liganded Glucocorticoid Receptor. Cell (2011) 145:224–41. doi: 10.1016/j.cell.2011.03.027
28. Shembade N, Harhaj EW. Regulation of NF-κB Signaling by the A20 Deubiquitinase. Cell Mol Immunol (2012) 9:123–30. doi: 10.1038/cmi.2011.59
29. Abraham SM, Clark AR. Dual-Specificity Phosphatase 1: A Critical Regulator of Innate Immune Responses. Biochem Soc Trans (2006) 34:1018–23. doi: 10.1042/bst0341018
30. Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato ACB. Glucocorticoids Inhibit MAP Kinase via Increased Expression and Decreased Degradation of MKP-1. EMBO J (2001) 20:7108–16. doi: 10.1093/emboj/20.24.7108
31. Cannarile L, Delfino DV, Adorisio S, Riccardi C, Ayroldi E. Implicating the Role of GILZ in Glucocorticoid Modulation of T-Cell Activation. Front Immunol (2019) 10:1823. doi: 10.3389/fimmu.2019.01823
32. Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, et al. Modulation of T-Cell Activation by the Glucocorticoid-Induced Leucine Zipper Factor via Inhibition of Nuclear Factor κB. Blood (2001) 98:743–53. doi: 10.1182/blood.V98.3.743
33. Ronchetti S, Migliorati G, Riccardi C. GILZ as a Mediator of the Anti-Inflammatory Effects of Glucocorticoids. Front Endocrinol (2015) 6:170. doi: 10.3389/fendo.2015.00170
34. Vollmer TR, Stockhausen A, Zhang JZ. Anti-Inflammatory Effects of Mapracorat, a Novel Selective Glucocorticoid Receptor Agonist, Is Partially Mediated by MAP Kinase Phosphatase-1 (MKP-1). J Biol Chem (2012) 287:35212–21. doi: 10.1074/jbc.M112.400671
35. Kadiyala V, Sasse SK, Altonsy MO, Berman R, Chu HW, Phang TL, et al. Cistrome-Based Cooperation Between Airway Epithelial Glucocorticoid Receptor and NF-κB Orchestrates Anti-Inflammatory Effects. J Biol Chem (2016) 291:12673–87. doi: 10.1074/jbc.M116.721217
36. Rao NAS, McCalman MT, Moulos P, Francoijs KJ, Chatziioannou A, Kolisis FN, et al. Coactivation of GR and NFκB Alters the Repertoire of Their Binding Sites and Target Genes. Genome Res (2011) 21:1404–16. doi: 10.1101/gr.118042.110
37. Schiller BJ, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. Glucocorticoid Receptor Binds Half Sites as a Monomer and Regulates Specific Target Genes. Genome Biol (2014) 15:418. doi: 10.1186/s13059-014-0418-y
38. Sasse SK, Gruca M, Allen MA, Kadiyala V, Song TY, Gally F, et al. Nascent Transcript Analysis of Glucocorticoid Crosstalk With TNF Defines Primary and Cooperative Inflammatory Repression. Genome Res (2019) 29:1753–65. doi: 10.1101/gr.248187.119
39. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage Glucocorticoid Receptors Regulate Toll-Like Receptor 4-Mediated Inflammatory Responses by Selective Inhibition of P38 MAP Kinase. Blood (2007) 109:4313–9. doi: 10.1182/blood-2006-10-048215
40. Desgeorges T, Caratti G, Mounier R, Tuckermann J, Chazaud B. Glucocorticoids Shape Macrophage Phenotype for Tissue Repair. Front Immunol (2019) 10:1591. doi: 10.3389/fimmu.2019.01591
41. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: Their Role, Activation and Polarization in Pulmonary Diseases. Immunobiology (2018) 223:383–96. doi: 10.1016/j.imbio.2017.11.001
42. Galuppo P, Vettorazzi S, Hovelmann J, Scholz CJ, Tuckermann JP, Bauersachs J, et al. The Glucocorticoid Receptor in Monocyte-Derived Macrophages Is Critical for Cardiac Infarct Repair and Remodeling. FASEB J (2017) 31:5122–32. doi: 10.1096/fj.201700317R
43. Li CYC, Munitic I, Mittelstadt PR, Castro E, Ashwell JD. Suppression of Dendritic Cell-Derived IL-12 by Endogenous Glucocorticoids Is Protective in LPS-Induced Sepsis. PloS Biol (2015) 13:e1002269. doi: 10.1371/journal.pbio.1002269
44. Elftman MD, Norbury CC, Bonneau RH, Truckenmiller ME. Corticosterone Impairs Dendritic Cell Maturation and Function. Immunology (2007) 122:279–90. doi: 10.1111/j.1365-2567.2007.02637.x
45. Chamorro S, Garcia-Vallejo JJ, Unger WWJ, Fernandes RJ, Bruijns SCM, Laban S, et al. TLR Triggering on Tolerogenic Dendritic Cells Results in TLR2 Up-Regulation and a Reduced Proinflammatory Immune Program. J Immunol (2009) 183:2984–94. doi: 10.4049/jimmunol.0801155
46. Hodrea J, Majai G, Doro Z, Zahuczky G, Pap A, Rajnavolgyi E, et al. The Glucocorticoid Dexamethasone Programs Human Dendritic Cells for Enhanced Phagocytosis of Apoptotic Neutrophils and Inflammatory Response. J Leukoc Biol (2012) 91:127–36. doi: 10.1189/jlb.0511243
47. Muzzi C, Watanabe N, Twomey E, Meers GK, Reichardt HM, Bohnenberger H, et al. The Glucocorticoid Receptor in Intestinal Epithelial Cells Alleviates Colitis and Associated Colorectal Cancer in Mice. Cell Mol Gastroenterol Hepatol (2021) 11:1505–18. doi: 10.1016/j.jcmgh.2020.12.006
48. Klassen C, Karabinskaya A, Dejager L, Vettorazzi S, Van Moorleghem J, Luhder F, et al. Airway Epithelial Cells Are Crucial Targets of Glucocorticoids in a Mouse Model of Allergic Asthma. J Immunol (2017) 199:48–61. doi: 10.4049/jimmunol.1601691
49. Gibbs J, Ince L, Matthews L, Mei JJ, Bell T, Yang N, et al. An Epithelial Circadian Clock Controls Pulmonary Inflammation and Glucocorticoid Action. Nat Med (2014) 20:919–26. doi: 10.1038/nm.3599
50. Ince LM, Zhang ZG, Beesley S, Vonslow RM, Saer B, Matthews LC, et al. Circadian Variation in Pulmonary Inflammatory Responses Is Independent of Rhythmic Glucocorticoid Signaling in Airway Epithelial Cells. FASEB J (2019) 33:126–39. doi: 10.1096/fj.201800026RR
51. Silverstein R, Johnson DC. Endogenous Versus Exogenous Glucocorticoid Responses to Experimental Bacterial Sepsis. J Leukoc Biol (2003) 73:417–27. doi: 10.1189/jlb.0702379
52. Imasato A, Desbois-Mouthon C, Han JH, Kai H, Cato ACB, Akira S, et al. Inhibition of P38 MAPK by Glucocorticoids via Induction of MAPK Phosphatase-1 Enhances Nontypeable Haemophilus Influenzae-Induced Expression of Toll-Like Receptor 2. J Biol Chem (2002) 277:47444–50. doi: 10.1074/jbc.M208140200
53. Abbinantenissen JM, Simpson LG, Leikauf GD. Corticosteroids Increase Secretory Leucocyte Protease Inhibitor Transcript Levels in Airway Epitherial Cells. Am J Physiol (1995) 268:601–6. doi: 10.1152/ajplung.1995.268.4.L601
54. Sakai A, Han JH, Cato ACB, Akira S, Li JD. Glucocorticoids Synergize With IL-1β to Induce TLR2 Expression via MAP Kinase Phosphatase-1-Dependent Dual Inhibition of MAPK JNK and P38 in Epithelial Cells. BMC Mol Biol (2004) 5:2. doi: 10.1186/14712199-5-2
55. Shah S, King EM, Mostafa MM, Altonsy MO, Newton R. DUSP1 Maintains IRF1 and Leads to Increased Expression of IRF1-Dependent Genes: A Mechanism Promoting Glucocorticoid Insensitivity. J Biol Chem (2016) 291:21802–16. doi: 10.1074/jbc.M116.728964
56. Blotta MH, DeKruyff RH, Umetsu DT. Corticosteroids Inhibit IL-12 Production in Human Monocytes and Enhance Their Capacity to Induce IL-4 Synthesis in CD4+ Lymphocytes. J Immunol (1997) 158:5589–95.
57. Quatrini L, Wieduwild E, Guia S, Bernat C, Glaichenhaus N, Vivier E, et al. Host Resistance to Endotoxic Shock Requires the Neuroendocrine Regulation of Group 1 Innate Lymphoid Cells. J Exp Med (2017) 214:3531–41. doi: 10.1084/jem.20171048
58. Franchimont D, Galon J, Gadina M, Visconti R, Zhou YJ, Aringer M, et al. Inhibition of Th1 Immune Response by Glucocorticoids, Dexamethasone Selectively Inhibits IL-12-Induced Stat4 Phosphorylation in T Lymphocytes. J Immunol (2000) 164:1768–74. doi: 10.4049/jimmunol.164.4.1768
59. Liberman AC, Refojo D, Druker J, Toscano M, Rein T, Holsboer F, et al. The Activated Glucocorticoid Receptor Inhibits the Transcription Factor T-Bet by Direct Protein-Protein Interaction. FASEB J (2007) 21:1177–88. doi: 10.1096/fj.06-7452com
60. Acharya N, Madi A, Zhang HY, Klapholz M, Escobar G, Dulberg S, et al. Endogenous Glucocorticoid Signaling Regulates CD8+ T Cell Differentiation and Development of Dysfunction in the Tumor Microenvironment. Immunity (2020) 53:658–71. doi: 10.1016/j.immuni.2020.08.005
61. Wiegers GJ, Reul J. Induction of Cytokine Receptors by Glucocorticoids: Functional and Pathological Significance. Trends Pharmacol Sci (1998) 19:317–21. doi: 10.1016/s0165-6147(98)01229-2
62. Franchimont D, Galon J, Vacchio MS, Fan S, Visconti R, Frucht DM. Positive Effects of Glucocorticoids on T Cell Function by Up-Regulation of IL-7 Receptor α. J Immunol (2002) 168:2212–8. doi: 10.4049/jimmunol.168.5.2212
63. Mazzucchelli R, Durum SK. Interleukin-7 Receptor Expression: Intelligent Design. Nat Rev Immunol (2007) 7:144–54. doi: 10.1038/nri2023
64. Tani-ichi S, Shimba A, Wagatsuma K, Miyachi H, Kitano S, Imai K, et al. Interleukin-7 Receptor Controls Development and Maturation of Late Stages of Thymocyte Subpopulations. Proc Natl Acad Sci USA (2013) 110:612–7. doi: 10.1073/pnas.1219242110
65. Diefenbach A, Colonna M, Koyasu S. Development, Differentiation, and Diversity of Innate Lymphoid Cells. Immunity (2014) 41:354–65. doi: 10.1016/j.immuni.2014.09.005
66. Meyer LK, Huang BJ, Delgado-Martin C, Roy RP, Hechmer A, Wandler AM, et al. Glucocorticoids Paradoxically Facilitate Steroid Resistance in T Cell Acute Lymphoblastic Leukemias and Thymocytes. J Clin Invest (2020) 130:863–76. doi: 10.1172/jci130189
67. Dimitrov S, Benedict C, Heutling D, Westermann J, Born J, Lange T. Cortisol and Epinephrine Control Opposing Circadian Rhythms in T Cell Subsets. Blood (2009) 113:5134–43. doi: 10.1182/blood-2008-11-190769
68. Besedovsky L, Born J, Lange T. Endogenous Glucocorticoid Receptor Signaling Drives Rhythmic Changes in Human T-Cell Subset Numbers and the Expression of the Chemokine Receptor CXCR4. FASEB J (2014) 28:67–75. doi: 10.1096/fj.13-237958
69. Cain DW, Bortner CD, Diaz-Jimenez D, Petrillo MG, Gruver-Yates A, Cidlowski JA. Murine Glucocorticoid Receptors Orchestrate B Cell Migration Selectively Between Bone Marrow and Blood. J Immunol (2020) 205:619–29. doi: 10.4049/jimmunol.1901135
70. Nagase H, Miyamasu M, Yamaguchi M, Kawasaki H, Ohta K, Yamamoto K, et al. Glucocorticoids Preferentially Upregulate Functional CXCR4 Expression in Eosinophils. J Allergy Clin Immunol (2000) 106:1132–9. doi: 10.1067/mai.2000.110923
71. Heideveld E, Hampton-O’Neil LA, Cross SJ, van Alphen FPJ, van den Biggerlaar M, Toye AM, et al. Glucocorticoids Induce Differentiation of Monocytes Towards Macrophages That Share Functional and Phenotypical Aspects With Erythroblastic Island Macrophages. Haematological (2018) 103:395–405. doi: 10.3324/haematol.2017.179341
72. Leigh R, Mostafa MM, King EM, CRider CF, Shah S, Dumonceaux C, et al. An Inhaled Dose of Budesonide Induces Genes Involved in Transcription and Signaling in the Human Airways: Enhancement of Anti- and Proinflammatory Effector Genes. Pharmacol Res Perspectl (2016) 4:e00243. doi: 10.1002/prp2.243
73. Carolina E, Kato T, Khanh VC, Moriguchi K, Yamashita T, Takeuchi K, et al. Glucocorticoid Impaired the Wound Healing Ability of Endothelial Progenitor Cells by Reducing the Expression of CXCR4 in the PGE2 Pathway. Front Med (2018) 5:276. doi: 10.3389/fmed.2018.00276
74. Lee HC, Shibata H, Ogawa S, Maki K, Ikuta K. Transcriptional Regulation of the Mouse IL-7 Receptor α Promoter by Glucocorticoid Receptor. J Immunol (2005) 174:7800–6. doi: 10.4049/jimmunol.174.12.7800
75. Abe A, Tani-ichi S, Shitara S, Cui G, Yamada H, Miyachi H, et al. An Enhancer of the IL-7 Receptor α-Chain Locus Controls IL-7 Receptor Expression and Maintenance of Peripheral T Cells. J Immunol (2015) 195:3129–38. doi: 10.4049/jimmunol.1302447
76. Elenkov IJ. Glucocorticoids and the Th1/Th2 Balance. Ann NY Acad Sci (2004) 1024:138–46. doi: 10.1196/annals.1321.010
77. Ramirez F, Fowell DJ, Puklavec M, Simmonds S, Mason D. Glucocorticoids Promote a Th2 Cytokine Response by CD4+ T Cells In Vitro. J Immunol (1996) 156:2406–12.
78. Suzuki K, Hayano Y, Nakai A, Furuta F, Noda M. Adrenergic Control of the Adaptive Immune Response by Diurnal Lymphocyte Recirculation Through Lymph Nodes. J Exp Med (2016) 213:2567–74. doi: 10.1084/jem.20160723
79. Druzd D, Matveeva O, Ince L, Harrison U, He WY, Schmal C, et al. Lymphocyte Circadian Clocks Control Lymph Node Trafficking and Adaptive Immune Responses. Immunity (2017) 46:120–32. doi: 10.1016/j.immuni.2016.12.011
80. Yang L, Licastro D, Cava E, Veronese N, Spelta F, Rizza W, et al. Long-Term Calorie Restriction Enhances Cellular Quality-Control Processes in Human Skeletal Muscle. Cell Rep (2016) 14:422–8. doi: 10.1016/j.celrep.2015.12.042
81. Steelman AJ, Dean DD, Young CR, Smith R, Prentice TW, Meagher MW, et al. Restraint Stress Modulates Virus Specific Adaptive Immunity During Acute Theiler’s Virus Infection. Brain Behav Immun (2009) 23:830–43. doi: 10.1016/j.bbi.2009.03.010
82. Elftman MD, Hunzeker JT, Mellinger JC, Bonneau RH, Norbury CC, Truckenmiller ME. Stress-Induced Glucocorticoids at the Eliest Stages of Herpes Simplex Virus-1 Infection Suppress Subsequent Antiviral Immunity, Implicating Impaired Dendritic Cell Function. J Immunol (2010) 184:1867–75. doi: 10.4049/jimmunol.0902469
83. De Lorenzo BHP, Brito R, Leal TP, Garcia NP, dos Santos RMM, Alvares-Saraiva AM, et al. Chronic Sleep Restriction Impairs the Antitumor Immune Response in Mice. Neuroimmunomodulation (2018) 25:59–67. doi: 10.1159/000490352
84. Ben-Eliyahu S, Page GG, Yirmiya R, Shakhar G. Evidence That Stress and Surgical Interventions Promote Tumor Development by Suppressing Natural Killer Cell Activity. Int J Cancer (1999) 80:880–8. doi: 10.1002/(sici)1097-0215(19990315)80:6<880::aid-ijc14>3.0.co;2-y
85. de Castro Kroner J, Knoke K, Kofler DM, Steiger J, Fabri M. Glucocorticoids Promote Intrinsic Human TH17 Differentiation. J Allergy Clin Immunol (2018) 142:1669–73. doi: 10.1016/j.jaci.2018.07.019
86. Marchetti B, Morale MC, Testa N, Tirolo C, Caniglia S, Amor S, et al. Stress, the Immune System and Vulnerability to Degenerative Disorders of the Central Nervous System in Transgenic Mice Expressing Glucocorticoid Receptor Antisense RNA. Brain Res Rev (2001) 37:259–72. doi: 10.1016/s0165-0173(01)00130-8
87. Pistollato F, Forbes-Hernandez TY, Iglesias RC, Ruiz R, Elexpuru-Zabaleta M, Dominguez I, et al. Effects of Caloric Restriction on Immunosurveillance, Microbiota and Cancer Cell Phenotype: Possible Implications for Cancer Treatment. Semin Cancer Biol (2020) 30:S1104–579X(20)30255-8. doi: 10.1016/j.semcancer.2020.11.017
88. Childs CE, Calder PC, Miles EA. Diet and Immune Function. Nutrients (2019) 11:1933. doi: 10.3390/nu11081933
89. Xu DL, Hu XK, Tian YF. Effect of Temperature and Food Restriction on Immune Function in Striped Hamsters (Cricetulus Barabensis). J Exp Biol (2017) 220:2187–95. doi: 10.1242/jeb.153601
90. Contreras NA, Fontana L, Tosti V, Nikolich-Zugich J. Calorie Restriction Induces Reversible Lymphopenia and Lymphoid Organ Atrophy Due to Cell Redistribution. Geroscience (2018) 40:279–91. doi: 10.1007/s11357-018-0022-2
91. Okawa T, Nagai M, Hase K. Dietary Intervention Impacts Immune Cell Functions and Dynamics by Inducing Metabolic Rewiring. Front Immunol (2021) 11:623989. doi: 10.3389/fimmu.2020.623989
92. Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell (2016) 30:136–46. doi: 10.1016/j.ccell.2016.06.005
93. Qiu BS, Vallance BA, Blennerhassett PA, Collins SM. The Role of CD4+ Lymphocytes in the Susceptibility of Mice to Stress-Induced Reactivation of Experimental Colitis. Nat Med (1999) 5:1178–82. doi: 10.1038/13503
94. Arima Y, Ohki T, Nishikawa N, Higuchi K, Ota M, Tanaka Y, et al. Brain Micro-Inflammation at Specific Vessels Dysregulates Organ-Homeostasis via the Activation of a New Neural Circuit. Elife (2017) 6:e25517. doi: 10.7554/eLife.25517
95. Ignacchiti MDC, Sesti-Costa R, Marchi LF, Chedraoui-Silva S, Mantovani B. Effect of Academic Psychological Stress in Post-Graduate Students: The Modulatory Role of Cortisol on Superoxide Release by Neutrophils. Stress (2011) 14:290–300. doi: 10.3109/10253890.2010.545459
96. Satoh S, Oishi K, Iwagaki A, Senba M, Akaike T, Akiyama M, et al. Dexamethasone Impairs Pulmonary Defence Against Pseudomonas Aeruginosa Through Suppressing iNOS Gene Expression and Peroxynitrite Production in Mice. Clin Exp Immunol (2001) 126:266–73. doi: 10.1046/j.1365-2249.2001.01656.x
97. Maloney CG, Kutchera WA, Albertine KH, McIntyre TM, Prescott SM, Zimmerman GA. Inflammatory Agonists Induce Cyclooxygenase Type 2 Expression by Human Neutrophils. J Immunol (1998) 160:1402–10.
98. Trottier MD, Newsted MM, King LE, Fraker PJ. Natural Glucocorticoids Induce Expansion of All Developmental Stages of Murine Bone Marrow Granulocytes Without Inhibiting Function. Proc Natl Acad Sci USA (2008) 105:2028–33. doi: 10.1073/pnas.0712003105
99. Mishler JM, Emerson PM. Development of Neutrophilia by Serially Increasing Doses of Dexamethasone. Br J Haematol (1977) 36:249–57. doi: 10.1111/j.1365-2141.1977.tb00646.x
100. Saffar AS, Dragon S, Ezzati P, Shan L, Gounni AS. Phosphatidylinositol 3-Kinase and P38 Mitogen-Activated Protein Kinase Regulate Induction of Mcl-1 and Survival in Glucocorticoid-Treated Human Neutrophils. J Allergy Clin Immunol (2008) 121:492–8. doi: 10.1016/j.jaci.2007.10.003
101. Cox G. Glucocorticoid Treatment Inhibits Apoptosis in Human Neutrophils. Separation of Survival and Activation Outcomes. J Immunol (1995) 154:14719–4725.
102. Chang LC, Madsen SA, Toelboell T, Weber PSD, Burton JL. Effects of Glucocorticoids on Fas Gene Expression in Bovine Blood Neutrophils. J Endocrinol (2004) 183:569–83. doi: 10.1677/joe.1.05822
103. Madsen-Bouterse SA, Rosa GJM, Burton JL. Glucocorticoid Modulation of Bcl-2 Family Members A1 and Bak During Delayed Spontaneous Apoptosis of Bovine Blood Neutrophils. Endocrinology (2006) 147:3826–34. doi: 10.1210/en.2006-0142
104. Shieh JH, Peterson RHF, Moore MAS. Cytokines and Dexamethasone Modulation of IL-1 Receptors on Human Neutrophils In Vitro. J Immunol (1993) 150:3515–24.
105. Harpaz I, Abutbul S, Nemirovsky A, Gal R, Cohen H, Monsonego A. Chronic Exposure to Stress Predisposes to Higher Autoimmune Susceptibility in C57BL/6 Mice: Glucocorticoids as a Double-Edged Sword. Eur J Immunol (2013) 43:758–69. doi: 10.1002/eji.201242613
106. Karagiannidis C, Akdis M, Holopainen P, Woolley NJ, Hense G, Ruckert B, et al. Glucocorticoids Upregulate FOXP3 Expression and Regulatory T Cells in Asthma. J Allergy Clin Immunol (2004) 114:1425–33. doi: 10.1016/j.jaci.2004.07.014
107. Ugor E, Prenek L, Pap R, Berta G, Ernszt D, Najbauer J, et al. Glucocorticoid Hormone Treatment Enhances the Cytokine Production of Regulatory T Cells by Upregulation of Foxp3 Expression. Immunobiology (2018) 223:422–31. doi: 10.1016/j.imbio.2017.10.010
108. Chen X, Murakami T, Oppenheim JJ, Howard DMZ. Differential Response of Murine CD4+CD25+ and CD4+CD25– T Cells to Dexamethasone-Induced Cell Death. Eur J Immunol (2004) 34:859–69. doi: 10.1002/eji.200324506
109. Rocamora-Reverte L, Tuzlak S, von Raffay L, Tisch M, Fiegl H, Drach M, et al. Glucocorticoid Receptor-Deficient Foxp3+ Regulatory T Cells Fail to Control Experimental Inflammatory Bowel Disease. Front Immunol (2019) 10:47. doi: 10.3389/fimmu.2019.0047
110. Engler JB, Kursawe N, Solano ME, Patas K, Wehrmann S, Heckmann N, et al. Glucocorticoid Receptor in T Cells Mediates Protection From Autoimmunity in Pregnancy. Proc Natl Acad Sci USA (2017) 114:181–90. doi: 10.1073/pnas.1617115114
111. Banuelos J, Cao Y, Shin SC, Lu NZ. Immunopathology Alters Th17 Cell Glucocorticoid Sensitivity. Allergy (2017) 72:331–41. doi: 10.1111/all.13051
112. Leung DYM, Bloom JW. Update on Glucocorticoid Action and Resistance. J Allergy Clin Inmmunol (2003) 111:3–23. doi: 10.1067/mai.2003.97
113. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, et al. Pro-Inflammatory Human Th17 Cells Selectively Express P-Glycoprotein and Are Refractory to Glucocorticoids. J Exp Med (2014) 211:89–104. doi: 10.1084/jem.20130301
114. Paugh SW, Bonten EJ, Savic D, Ramsey LB, Thierfelder WE, Gurung P, et al. NALP3 Inflammasome Upregulation and CASP1 Cleavage of the Glucocorticoid Receptor Cause Glucocorticoid Resistance in Leukemia Cells. Nat Genet (2015) 47:607–14. doi: 10.1038/ng.3283
115. Britt RD, Thompson MA, Sasse S, Pabelick CM, Gerber AN, Prakash YS. Th1 Cytokines TNF-α and IFN-γ Promote Corticosteroid Resistance in Developing Human Airway Smooth Muscle. Am J Physiol Lung Cell Mol Physiol (2019) 316:L71–81. doi: 10.1152/ajplung.00547.2017
116. Chambers ES, Nanzer AM, Pfeffer PE, Richards DF, Timms PM, Martineau AR, et al. Distinct Endotypes of Steroid-Resistant Asthma Characterized by IL-17Ahigh and IFN-γHigh Immunophenotypes: Potential Benefits of Calcitriol. J Allergy Clim Immunol (2015) 136:628–37. doi: 10.1016/j.jaci.2015.01.026
117. Nguyen TH, Maltby S, Tay HL, Eyers F, Foster PS, Yang M. Identification of IFN-γ and IL-27 as Critical Regulators of Respiratory Syncytial Virus-Induced Exacerbation of Allergic Airways Disease in a Mouse Model. J Immunol (2018) 200:237–47. doi: 10.4049/jimmunol.1601950
118. Pazdrak K, Straub C, Maroto R, Stafford S, White WI, Calhoun WJ, et al. Cytokine-Induced Glucocorticoid Resistance From Eosinophil Activation: Protein Phosphatase 5 Modulation of Glucocorticoid Receptor Phosphorylation and Signaling. J Immunol (2016) 197:3782–91. doi: 10.4049/jimmunol.1601029
Keywords: glucocorticoids, circadian rhythm, stress, cell-mediated immunity, IL-7 receptor
Citation: Shimba A, Ejima A and Ikuta K (2021) Pleiotropic Effects of Glucocorticoids on the Immune System in Circadian Rhythm and Stress. Front. Immunol. 12:706951. doi: 10.3389/fimmu.2021.706951
Received: 08 May 2021; Accepted: 22 September 2021;
Published: 08 October 2021.
Edited by:
Detlef Neumann, Hannover Medical School, GermanyReviewed by:
Rowan Hardy, University of Birmingham, United KingdomRobert Newton, University of Calgary, Canada
Copyright © 2021 Shimba, Ejima and Ikuta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akihiro Shimba, c2hpbWJhLmFraWhpcm8uN3dAa3lvdG8tdS5hYy5qcA==; Koichi Ikuta, aWt1dGEua29pY2hpLjZjQGt5b3RvLXUuYWMuanA=