 Johannes Fessler
Johannes Fessler Stefano Angiari
Stefano Angiari
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 14 July 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.706434
This article is part of the Research Topic Cell-Cell Interactions Controlling Neuronal Functionality in Health and Disease View all 12 articles
Immunosenescence is a state of dysregulated leukocyte function characterised by arrested cell cycle, telomere shortening, expression of markers of cellular stress, and secretion of pro-inflammatory mediators. Immunosenescence principally develops during aging, but it may also be induced in other pathological settings, such as chronic viral infections and autoimmune diseases. Appearance of senescent immune cells has been shown to potentially cause chronic inflammation and tissue damage, suggesting an important role for this process in organismal homeostasis. In particular, the presence of senescent T lymphocytes has been reported in neurological diseases, with some works pointing towards a direct connection between T cell senescence, inflammation and neuronal damage. In this minireview, we provide an overview on the role of T cell senescence in neurological disorders, in particular in multiple sclerosis and Alzheimer disease. We also discuss recent literature investigating how metabolic remodelling controls the development of a senescence phenotype in T cells. Targeting metabolic pathways involved in the induction of senescent T cells may indeed represent a novel approach to limit their inflammatory activity and prevent neuroinflammation and neurodegeneration.
Aging is a natural process that has to deal with a multitude of challenges, and loss of organismal homeostasis during aging is frequently associated with increased susceptibility to infection, cancer, cardiovascular disease and autoimmunity in the elderly (1–4). A dysregulation of the immune system, ‘termed immunosenescence’, is a hallmark of the aging process, and is thought to play a central role in the higher likelihood of developing such pathologies. Senescent immune cells display a dysfunctional immune profile, including altered cytokine production, limited proliferative capacity and reduced chemotactic and phagocytic potential (5). Immunosenescence is typically associated with chronological age, and it is tightly connected to the process of ‘inflammaging’, i.e. the chronic and systemic low-grade inflammation observed in the elderly (6). However, premature immunosenescence, in particular in T lymphocytes, has also been observed in patients with chronic viral infections and in autoimmune diseases (7–10). Thus, it is essential to appreciate those influences in the context of immunosenescent manifestations.
Age represents the main risk factor for the development of several neurological disorders, in particular chronic neurodegenerative pathologies such as Alzheimer disease (AD) and Parkinson disease (PD), where inflammaging is proposed to play a relevant role in the disease course (11). Blood-borne immune cells infiltrating the central nervous system (CNS) and causing direct or indirect neuronal damage also have a central role in CNS pathologies such as multiple sclerosis (MS), traumatic brain injury (TBI), ischemic stroke, AD and PD (12–16). Among immune cells, T lymphocytes represent key players in neurological diseases, where they cause detrimental inflammation taking place in the CNS, but also participate in regulatory mechanisms aimed at protecting neurons from the inflammatory damage (12–16). Intriguingly, an increasing number of studies reported the presence of T cell subsets with a senescent-like phenotype in patients with neurological disorders (senescent T cells, sTC). Here, we summarise our knowledge on the role of sTC, in particular CD4+ and CD8+ conventional T cells, in neuroinflammation and neurodegeneration. We also provide an overview of recent works showing how intracellular metabolic reprogramming may modulate the development of a senescent phenotype in T lymphocytes.
T cell senescence is characterised by a decline in naïve T cell number and clonal diversity, which are mainly caused by age-associated thymic atrophy and reduced homeostatic proliferation of naïve-resting T cells. sTC also show loss of their proliferative capacity upon T cell receptor (TCR) reactivation, accelerated telomeric erosion, and accumulation of DNA damage. Throughout the development of a senescent phenotype, T cells downregulate co-stimulatory markers like CD28, while up-regulating natural killer cell-associated molecules, including the killer lectin receptor G1 (KLRG1) (8, 17, 18). These alterations induce a refractoriness of sTC to TCR-mediated activation, but, in parallel, they potentially augment antigen-independent effector functions. Finally, sTC display a pathogenic phenotype characterised by the secretion of several pro-inflammatory mediators, such as the cytokines tumour necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ), collectively known as senescence-associated secretory phenotype (SASP) (8, 17, 18) (Figure 1A). sTC may thus support the systemic low-grade inflammation observed in the elderly, i.e. inflammaging. However, to what extent sTC are involved in neurological diseases is still uncertain.
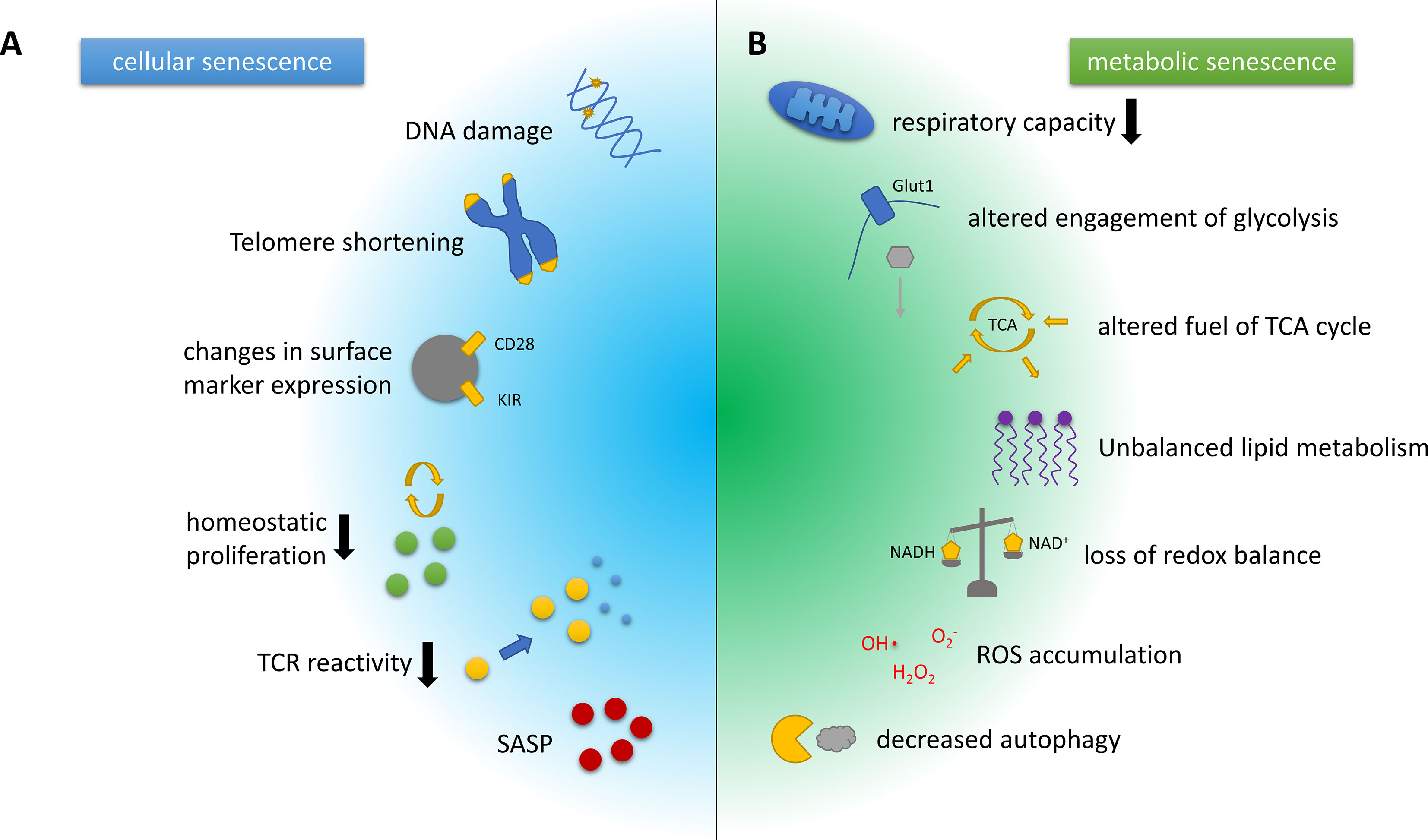
Figure 1 Hallmarks of cellular and metabolic senescence and/or aging in T cells. (A) Key characteristics of cellular senescence include DNA damage and telomere erosion that contribute to genomic instability and dysregulation of the epigenome. Phenotypically, T cell senescence is associated with a loss of surface CD28 expression and the upregulation of innate T cell markers such as the killer cell immunoglobulin-like receptors (KIRs). The involution of the thymus increases the homeostatic pressure on T cells. Senescent T cells, however, show a diminished capacity for homeostatic proliferation, and additionally feature reduced T cell receptor (TCR) reactivity. Another hallmark of T cell senescence is the production of pro-inflammatory mediators, collectively known as senescence-associated secretory phenotype (SASP). (B) Key metabolic characteristics associated with T cell senescence and aging often closely interwoven and include a decline in the respiratory capacity or efficacy and substantially altered engagement of glycolysis, as well as alterations in the pathways used to fuel the tricarboxylic acid (TCA) cycle, which are likely context- and cell subset-dependent. The same holds true for the dysbalanced lipid metabolism observed in senescent/aged T cells. In addition, ratios of coenzymes for metabolic reactions such as NAD/NADH that are crucial for balancing the cellular redox state are shifted in senescent cells. Furthermore, reactive oxygen species (ROS) accumulate and eventually cause DNA and protein damage. Damaged organelles, cell membranes and proteins are usually degraded by autophagy, another pivotal mechanism that is attenuated by the aging or the senescent process.
Development of a senescent phenotype in T cells may be relevant in MS, an autoimmune disease characterised by CNS infiltration of peripherally activated immune cells that cause neuroinflammation, neuronal death and disability (19). In particular, myelin-specific T cells are key players in the disease, and recent works highlighted the potential involvement of sTC, in particular CD4+ T cells, in MS (Table 1). Accelerated thymic involution was reported in MS patients with either relapsing-remitting (RRMS) or primary progressive (PPMS) MS forms (20), and initial works also found an increased percentage of CD4+CD28- T cells in a subset of MS patients (mainly RRMS), preferentially producing IFN-γ (21). CD4+CD28- T cells were subsequently detected in MS brain biopsies, where they displayed a cytotoxic phenotype and expressed CX3C chemokine receptor 1 (CX3CR1), which binds to chemokine (C-X3-C motif) ligand 1 (CX3CL1) (22). As CX3CL1 is upregulated in the cerebrospinal fluid (CSF) and brain of MS patients, compared to healthy controls, the authors speculated a CX3CL1-mediated recruitment of these highly inflammatory cells in the MS brain. Importantly, some CD4+CX3CL1+ brain cells where in close proximity to cleaved caspase-3 (cCASP3)+ oligodendrocytes, suggesting that they may cause direct oligodendrocyte death and demyelination (22). Another recent work performed a longitudinal analysis of several markers in T cells from blood, CSF and brain samples obtained post-mortem from patients with advanced disease (mainly progressive MS) (23). Authors found that CD8+ T cells in white matter lesions displayed a more chronically activated, effector memory-like profile, compared to their blood counterparts. They also proposed that chronic reactivation in the brain, confirmed by limited TCR diversity in the CD8+ cell population, was caused, at least in part, by specific reactivity against Epstein Barr virus-infected B cells (23). These T cells also presented a cytotoxic phenotype, and in some cases they co-localised with cCASP3+ cells in the brain, indicating putative cytotoxic activity in situ. However, the expression of the senescent marker CD57 was comparable between blood-derived T cells and brain T cells (23), which might exclude induction of cell senescence in such chronically activated, brain-infiltrating T cells.
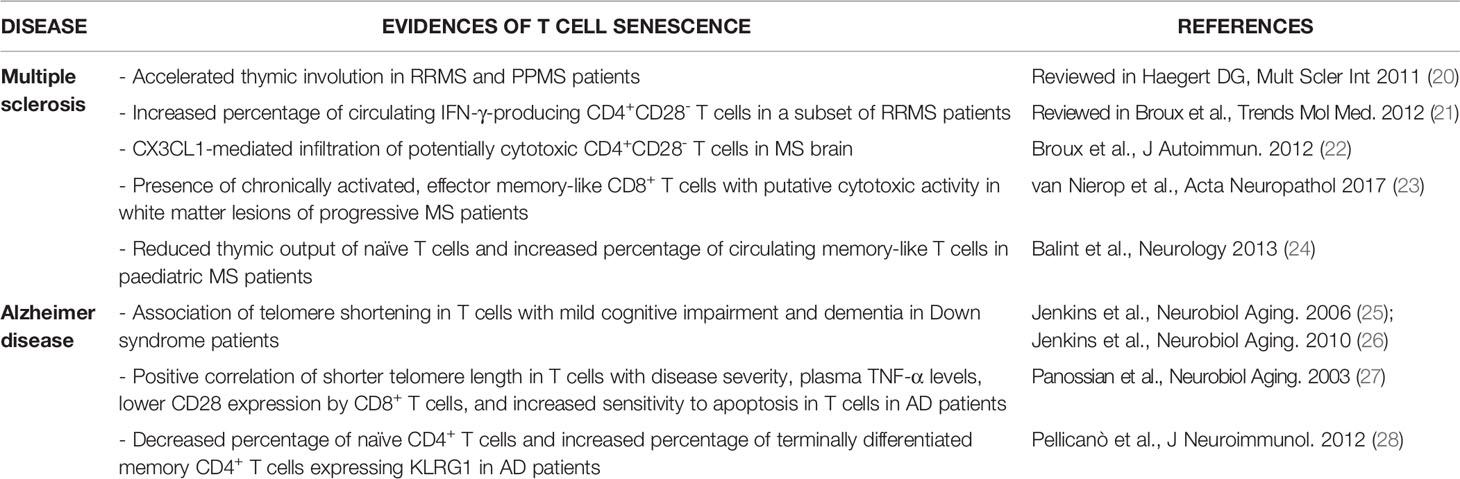
Table 1 Summary of previous works suggesting the presence of senescent T cells in MS and AD patients.
T cell senescence was also suggested to play a role in AD, a multifactorial neurodegenerative disorder characterised by progressive neuronal death and development of dementia (Table 1). AD has long been viewed as a ‘pure’ neurodegenerative disease, but recent work highlighted the importance of both local and peripheral immune system, including T cells, in disease onset and progression (29, 30). Telomere shortening in T cells, a marker of proliferative senescence, was initially associated with signs of mild cognitive impairment and dementia in Down syndrome patients, which develop premature AD-like dementia (25, 26). Shorter telomere length in T cells was also reported in AD patients, where it directly correlated with disease severity, higher plasma TNF-α levels, lower CD28 expression by CD8+ T cells, and increased sensitivity to apoptosis in T cells (27). These data suggest that dysfunctional, senescent T cells may associate with higher disease burden and systemic inflammation in AD. Supporting this view, another work showed decreased percentage of naïve CD4+ T cells and increased percentage of terminally differentiated memory CD4+ T cells expressing KLRG1 in AD patients, compared to aged-matched controls, while CD8+ T cell phenotype was unchanged (28). Lack of CD8+ sTC was lately confirmed by another study (31), suggesting that the senescent phenotype might be restricted to CD4+ T cells in AD individuals. However, T cell senescence was recently not confirmed in patients with AD or vascular dementia (32), and conflicting results have also been reported in other forms of dementia and neurodegeneration. Indeed, while a trend towards an increased number of circulating senescent-like CD8+CD45RA+CCR7- T cells was observed in dementia with Lewy bodies (33), a recent work found reduced numbers of senescent/terminally differentiated CD8+ T cells in the blood of PD patients (34). Thus, the role of sTC in neurodegeneration and dementia is still unclear.
The involvement of senescent T cells in other neurological conditions is still unknown. Several studies associated signs of leukocyte senescence like telomere shortening, oxidative stress, and reduced lymphoproliferative potential with onset or severity of other neurological disorders, such as PD (35), ischemic stroke (36) and amyotrophic lateral sclerosis (37). However, such works didn’t discriminate between different immune cell populations, and whether T cell senescence plays a role in these diseases has not been investigated. Interestingly, reversed CD4:CD8 ratio, increased percentage of effector memory and reduced numbers of naïve CD4+ T cells, all potential signs of T cell senescence, were correlated with the presence of chronic viral infections and cognitive dysfunctions in old individuals (38–40). Similarly, expansion of CD8+CD28- T cells was associated with worse cognitive performances in patients with rheumatoid arthritis (41), and higher numbers of memory CD4+ T cells and CD8+ sTC negatively correlated with cognitive impairment in systemic lupus erythematous patients (42). These works support the idea that persistent T cell activation and maturation observed during chronic viral infections and autoimmune diseases may generate detrimental sTC that cause or sustain cognitive impairment, most likely through pro-inflammatory mechanisms. Additionally, a shift towards a memory/effector-like phenotype in T cells was detected in patients after spinal cord injury (43), possibly caused by the sustained inflammation accompanying the acute damage. Similarly, in a mouse model of TBI, the concussion injury induced an acute lymphopenic response, with reduced thymic size and reduced number of circulating T cells (44). Noteworthy, some of these effects were maintained chronically (60 days after trauma), with T cells also developing into a more pro-inflammatory phenotype and CD4+ T cells displaying an effector/memory-like polarisation (44). These studies suggest that acute CNS injury may cause premature T cell senescence, which would eventually sustain the detrimental systemic inflammatory response.
It is now well established that intracellular metabolic remodelling plays a key role in the activation and engagement of effector functions in T cells (45). T cell metabolism has been extensively investigated in the last few years, including the metabolic profile of T cells from elderly individuals, but relatively little is known about the metabolic remodelling controlling T cell senescence (Figure 1B). Also, it remains unclear if such changes are a cause of T cell dysfunction or are rather a by-product of the aging process.
Several studies found mitochondrial alterations in senescent lymphocyte subsets or aged T cells (46–50). Ron-Harel and colleagues reported declined mitochondrial mass and reduced basal and maximal respiratory capacity in T cells from old mice (47). Interestingly, aged naïve T cells also showed lower glycolytic activity, as well as low levels of central carbon intermediates in glycolysis, pentose phosphate pathway, and tricarboxylic acid (TCA) cycle, suggesting and overall slower metabolism, respiration and protein synthesis (47). Upon activation, a specific deficit in the induction of enzymes of one-carbon metabolism in aged cells was shown, that potentially accounts for impaired T cell activation, such as the diminished response to vaccination observed in the elderly (47, 51). Another study showed that mitochondrial proteins involved in the electron transport chain were elevated, but at the same time mitochondrial respiration was impaired in total CD4+ T cells from older individuals. The authors also noted a significantly higher number of autophagosomes containing undegraded mitochondria, and thus suggested a defective mitochondrial turnover by autophagy, which may trigger chronic inflammation (49). Defective autophagy and mitochondrial bioenergetics in CD4+ T cells from older individuals were recently confirmed, and associated with redox imbalance (50). These metabolic alterations furthermore led to a specific proinflammatory profile of IL-17 producing (Th17) cells in older individuals, suggesting that Th17 cells are a pivotal driver of inflammaging. Noteworthy, treatment with the drug metformin enhanced autophagy and normalised mitochondrial function to attenuate age‐associated inflammation (50).
Aged, resting cells feature other major metabolic alterations, such as enhanced basal activation of the phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. Lymphocytes of patients carrying dominant-activating mutations in PI3K exhibited an aged phenotype (accumulation of senescent or terminally differentiated cells), in combination with augmented mTOR signalling and glycolysis, and cellular malfunction (52). Intriguingly, treatment with the mTOR inhibitor rapamycin reversed the immunosenescent and dysfunctional effects in these patients (52). Chronic PI3K/Akt/mTOR pathway activation may also be caused by chronic infections in humans, where T cells upregulate glucose transporter 1 (Glut1) and increase their glycolytic activity, which ultimately leads to CD4+ T cell depletion in vivo (53, 54). Given that chronic viral infections are known to induce sTC (7), this signalling axis may be relevant in infection-induced T cell senescence. A recent work also reported that T cells from old mice relied heavily on glutaminolysis, and are potentially involved in Th1 and Th17-driven alloimmune responses (55). Treatment with an inhibitor of glutaminolysis prolonged allograft survival specifically in old recipients, whereas in young animals, additional inhibition of glycolysis and oxidative phosphorylation (OXPHOS) was needed to achieve the same effect. Of note, immunosuppressive capacities of glutaminolysis inhibition were specific to CD4+ T cells, and depletion of CD8+ T cells did not alter transplant outcome (55). Memory CD4+ T cells from aged individuals were also shown to upregulate fatty acid β-oxidation (FAO)-coupled mitochondrial respiration, with this process being mediated via upregulation of sirtuin-1, a nicotinamide adenine dinucleotide (NAD)+-dependent protein deacetylase, that leads to increased carnitine palmitoyl transferase (CPT1a) transcription and maintains a more lipid-catabolic state (56). However, another study showed an age-related loss of sirtuin-1 in human CD8+CD28- T cells, that potentially contributes to metabolic reprogramming towards an enhanced glycolytic capacity (57). Interestingly, constitutive activation of glycolytic flux was reported to limit memory development in CD8+ T cells. Accordingly, blocking glycolytic metabolism promoted the generation of long-lived, functional memory cells. Enforcing glycolysis, on the other hand, drives CD8+ T-cells towards a terminally differentiated state (58). These findings suggest that high glycolytic rates may be involved in the induction of terminally differentiated/sTC.
In an elegant study, Lanna et al. observed that senescent CD4+CD27−CD28− T lymphocytes exhibit endogenously elevated p38 phosphorylation, triggered by the intracellular metabolic sensor 5’ adenosine monophosphate-activated protein kinase (AMPK) (59). In this context, AMPK responds to endogenous DNA damage and also to a fall of intracellular energy levels, thus highlighting an ‘intra-sensory’ pathway for p38 activation that senses intracellular changes such as glucose deprivation and genotoxic stress. Triggering this pathway leads to inhibition of T cell proliferation and telomerase activity, two typical features of senescent CD4+ T cells (59). The same group also found that terminally differentiated human CD8+ T cells (TEMRA) have decreased numbers of mitochondria and fail to efficiently upregulate glycolysis or OXPHOS following TCR activation, although showing high production of reactive oxygen species (48). These TEMRA cells also showed elevated levels of p38 MAPK, and inhibition of p38 MAPK signalling elevated mitochondrial biogenesis and fitness. In addition, p38 MAPK blockade also induced an increase in autophagy through enhanced interactions between p38 interacting protein (p38IP) and autophagy protein 9 (ATG9) to compensate for the heightened energy demand (48). Of note, the polyamine spermidine is capable of inducing autophagy and undergoes an age-dependent decline (60). Recently, it was shown that spermidine modulates T cell differentiation towards a regulatory phenotype and dietary supplementation with spermidine reduced pathology in a mouse model of T cell transfer-induced colitis (61). Moreover, in a study in humans, spermidine supplementation was able to recover the autophagic flux and cellular functionality in T cells from old donors (62). Interestingly, spermidine was reported to reverse cellular senescence of B cells (63), an effect that has not yet been addressed in T cells.
Despite recent evidence suggesting an involvement of sTC in neurological diseases, many questions are still open. First, most studies analysing parameters such as telomere length and distribution of specific T cell subset are mainly observational, and do not investigate in detail the functional consequences of the observed changes. Second, identification of bona fide sTC may be tricky, due to overlapping expression of some senescence markers in effector memory vs terminally differentiated vs sTC (17, 18). Third, it is still not completely clear why patients with neurological disorders would accumulate sTC, compared to aged-matched individuals. Apart from the age-related reduction in the naïve T cells compartment, the main hypothesis is that circulating T cells undergo continuous antigen-specific or cytokine-induced re-activation, due to low grade systemic inflammation and/or to the presence of persistent CNS-derived antigens. This process would eventually reduce the extent of T cell receptor diversity, and increase the amount of terminally differentiated and potentially exhausted and senescent lymphocytes. Nonetheless, how immunosenescence develops and contributes to neuroinflammation and neurodegeneration remains unclear. Strikingly, a study showed that paediatric MS patients displayed signs of premature T cell senescence, with reduced number of circulating naïve T cells recently emigrated from the thymus, and increased percentage of memory-like cells, resembling the profile of adult individuals (24). This work may support the presence of an early antigen-specific (autoimmune) response inducing T cell maturation/activation, but might also suggest that appearance of a senescent/aged phenotype in T cells could predispose to MS development. Another aspect to consider is the recently hypothesised effect of disease-modifying therapies (DMT) on immunosenescence. Some anti-inflammatory DMT used to treat neuroinflammatory conditions, for example MS, may indeed not only limit the activation capacity of the immune system, but also induce signs of premature senescence of the immune system, such as reduced T cell output from the thymus (64, 65). These effects may differ between different DMT, potentially due to their intrinsic mechanisms of action (65). Thus, these DMT may be partially responsible for the appearance of severe side effects in aged individuals, such as viral reactivation and CNS inflammation and demyelination in MS patients (64, 65), again highlighting the importance of finely balancing aging of the immune system for optimal organismal homeostasis.
An increasing amount of studies showed metabolic alterations in circulating leukocytes in diseases of the CNS, with such metabolic modulation potentially playing a role in the pathogenic activity of different immune cell populations (66). However, direct comparisons of immunosenescence and immunometabolism in neurological diseases is lacking, even though the two features may be strongly interwoven. As an example, a recent work showed that T cells with dysfunctional mitochondria due to mitochondrial transcription factor A (TFAM) deficiency act as accelerators of senescence, and furthermore incite multiple aging-related features including neurological inflammation (67). This supports the notion that alterations of metabolic pathways in immune cells may directly cause inflammation-induced neurodegeneration. Importantly, there is a multitude of potential approaches to counteract age-associated immune cell malfunctions by metabolic intervention, including supplementation of age-limited nutrients such as formate and glycine, and reinforcement of autophagy with spermidine (47, 62). Of note, a recent study reported that mTOR inhibitor therapy in elderly humans decreased the incidence of infections, improved influenza vaccination responses and up-regulated antiviral immunity (68), thus highlighting the potential clinical relevance of such metabolic approaches in age-related immune dysfunctions. Given the therapeutic potential of immunometabolic intervention in neurological diseases (66), this strategy may indeed represent a brand-new approach to limit T cell senescence and dampen T cell-induced inflammation in CNS disorders and aging.
Both authors equally contributed to literature search and manuscript writing, and approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. El Chakhtoura NG, Bonomo RA, Jump RLP. Influence of Aging and Environment on Presentation of Infection in Older Adults. Infect Dis Clin North Am (2017) 31(4):593–608. doi: 10.1016/j.idc.2017.07.017
2. Rodgers JL, Jones J, Bolleddu SI, Vanthenapalli S, Rodgers LE, Shah K, et al. Cardiovascular Risks Associated With Gender and Aging. J Cardiovasc Dev Dis (2019) 6(2):19. doi: 10.3390/jcdd6020019
3. Ray D, Yung R. Immune Senescence, Epigenetics and Autoimmunity. Clin Immunol (2018) 196:59–63. doi: 10.1016/j.clim.2018.04.002
4. Zinger A, Cho WC, Ben-Yehuda A. Cancer and Aging - the Inflammatory Connection. Aging Dis (2017) 8(5):611–27. doi: 10.14336/AD.2016.1230
5. Xu W, Wong G, Hwang YY, Larbi A. The Untwining of Immunosenescence and Aging. Semin Immunopathol (2020) 42(5):559–72. doi: 10.1007/s00281-020-00824-x
6. Xia S, Zhang X, Zheng S, Khanabdali R, Kalionis B, Wu J, et al. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J Immunol Res (2016) 2016:8426874. doi: 10.1155/2016/8426874
7. Bellon M, Nicot C. Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses (2017) 9(10):289. doi: 10.3390/v9100289
8. Weyand CM, Yang Z, Goronzy JJ. T-Cell Aging in Rheumatoid Arthritis. Curr Opin Rheumatol (2014) 26(1):93–100. doi: 10.1097/BOR.0000000000000011
9. Fessler J, Raicht A, Husic R, Ficjan A, Duftner C, Schwinger W, et al. Premature Senescence of T-Cell Subsets in Axial Spondyloarthritis. Ann Rheum Dis (2016) 75(4):748–54. doi: 10.1136/annrheumdis-2014-206119
10. Fessler J, Fasching P, Raicht A, Hammerl S, Weber J, Lackner A, et al. Lymphopenia in Primary Sjögren’s Syndrome Is Associated With Premature Aging of Naïve CD4+ T Cells. Rheumatol (Oxford) (2021) 60(2):588–97. doi: 10.1093/rheumatology/keaa105
11. Zuo L, Prather ER, Stetskiv M, Garrison DE, Meade JR, Peace TI, et al. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int J Mol Sci (2019) 20(18):4472. doi: 10.3390/ijms20184472
12. Schwartz M, Deczkowska A. Neurological Disease as a Failure of Brain-Immune Crosstalk: The Multiple Faces of Neuroinflammation. Trends Immunol (2016) 37(10):668–79. doi: 10.1016/j.it.2016.08.001
13. Needham EJ, Helmy A, Zanier ER, Jones JL, Coles AJ, Menon DK. The Immunological Response to Traumatic Brain Injury. J Neuroimmunol (2019) 332:112–25. doi: 10.1016/j.jneuroim.2019.04.005
14. Tan JSY, Chao YX, Rötzschke O, Tan EK. New Insights Into Immune-Mediated Mechanisms in Parkinson’s Disease. Int J Mol Sci (2020) 21(23):9302. doi: 10.3390/ijms21239302
15. Levard D, Buendia I, Lanquetin A, Glavan M, Vivien D, Rubio M. Filling the Gaps on Stroke Research: Focus on Inflammation and Immunity. Brain Behav Immun (2021) 91:649–67. doi: 10.1016/j.bbi.2020.09.025
16. Rossi B, Santos-Lima B, Terrabuio E, Zenaro E, Constantin G. Common Peripheral Immunity Mechanisms in Multiple Sclerosis and Alzheimer’s Disease. Front Immunol (2021) 12:639369. doi: 10.3389/fimmu.2021.639369
17. Covre LP, De Maeyer RPH, Gomes DCO, Akbar AN. The Role of Senescent T Cells in Immunopathology. Aging Cell (2020) 19(12):e13272. doi: 10.1111/acel.13272
18. Goronzy JJ, Weyand CM. Mechanisms Underlying T Cell Ageing. Nat Rev Immunol (2019) 19(9):573–83. doi: 10.1038/s41577-019-0180-1
19. Sospedra M, Martin R. Immunology of Multiple Sclerosis. Semin Neurol (2016) 36(2):115–27. doi: 10.1055/s-0036-1579739
20. Haegert DG. Multiple Sclerosis: A Disorder of Altered T-Cell Homeostasis. Mult Scler Int (2011) 2011:461304. doi: 10.1155/2011/461304
21. Broux B, Markovic-Plese S, Stinissen P, Hellings N. Pathogenic Features of CD4+CD28- T Cells in Immune Disorders. Trends Mol Med (2012) 18(8):446–53. doi: 10.1016/j.molmed.2012.06.003
22. Broux B, Pannemans K, Zhang X, Markovic-Plese S, Broekmans T, Eijnde BO, et al. CX(3)CR1 Drives Cytotoxic CD4(+)CD28(-) T Cells Into the Brain of Multiple Sclerosis Patients. J Autoimmun (2012) 38(1):10–9. doi: 10.1016/j.jaut.2011.11.006
23. van Nierop GP, van Luijn MM, Michels SS, Melief MJ, Janssen M, Langerak AW, et al. Phenotypic and Functional Characterization of T Cells in White Matter Lesions of Multiple Sclerosis Patients. Acta Neuropathol (2017) 134(3):383–401. doi: 10.1007/s00401-017-1744-4
24. Balint B, Haas J, Schwarz A, Jarius S, Fürwentsches A, Engelhardt K, et al. T-Cell Homeostasis in Pediatric Multiple Sclerosis: Old Cells in Young Patients. Neurology (2013) 81(9):784–92. doi: 10.1212/WNL.0b013e3182a2ce0e
25. Jenkins EC, Velinov MT, Ye L, Gu H, Li S, Jenkins EC Jr, et al. Telomere Shortening in T Lymphocytes of Older Individuals With Down Syndrome and Dementia. Neurobiol Aging (2006) 27(7):941–5. doi: 10.1016/j.neurobiolaging.2005.05.021
26. Jenkins EC, Ye L, Gu H, Ni SA, Velinov M, Pang D, et al. Shorter Telomeres may Indicate Dementia Status in Older Individuals With Down Syndrome. Neurobiol Aging (2010) 31(5):765–71. doi: 10.1016/j.neurobiolaging.2008.06.001
27. Panossian LA, Porter VR, Valenzuela HF, Zhu X, Reback E, Masterman D, et al. Telomere Shortening in T Cells Correlates With Alzheimer’s Disease Status. Neurobiol Aging (2003) 24(1):77–84. doi: 10.1016/s0197-4580(02)00043-x
28. Pellicanò M, Larbi A, Goldeck D, Colonna-Romano G, Buffa S, Bulati M, et al. Immune Profiling of Alzheimer Patients. J Neuroimmunol (2012) 242(1-2):52–9. doi: 10.1016/j.jneuroim.2011.11.005
29. Jevtic S, Sengar AS, Salter MW, McLaurin J. The Role of the Immune System in Alzheimer Disease: Etiology and Treatment. Ageing Res Rev (2017) 40:84–94. doi: 10.1016/j.arr.2017.08.005
30. Lindestam Arlehamn CS, Garretti F, Sulzer D, Sette A. Roles for the Adaptive Immune System in Parkinson’s and Alzheimer’s Diseases. Curr Opin Immunol (2019) 59:115–20. doi: 10.1016/j.coi.2019.07.004
31. Westman G, Lidehall AK, Magnusson P, Ingelsson M, Kilander L, Lannfelt L, et al. Decreased Proportion of Cytomegalovirus Specific CD8 T-Cells But No Signs of General Immunosenescence in Alzheimer’s Disease. PLoS One (2013) 8(10):e77921. doi: 10.1371/journal.pone.0077921
32. D’Angelo C, Goldeck D, Pawelec G, Gaspari L, Di Iorio A, Paganelli R. Exploratory Study on Immune Phenotypes in Alzheimer’s Disease and Vascular Dementia. Eur J Neurol (2020) 27(10):1887–94. doi: 10.1111/ene.14360
33. Amin J, Boche D, Clough Z, Teeling J, Williams A, Gao Y, et al. Peripheral Immunophenotype in Dementia With Lewy Bodies and Alzheimer’s Disease: An Observational Clinical Study. J Neurol Neurosurg Psychiatry (2020) 91(11):1219–26. doi: 10.1136/jnnp-2020-323603
34. Williams-Gray CH, Wijeyekoon RS, Scott KM, Hayat S, Barker RA, Jones JL. Abnormalities of Age-Related T Cell Senescence in Parkinson’s Disease. J Neuroinflamm (2018) 15(1):166. doi: 10.1186/s12974-018-1206-5
35. Vida C, Kobayashi H, Garrido A, Martínez de Toda I, Carro E, Molina JA, et al. Lymphoproliferation Impairment and Oxidative Stress in Blood Cells From Early Parkinson’s Disease Patients. Int J Mol Sci (2019) 20(3):771. doi: 10.3390/ijms20030771
36. Zhang W, Chen Y, Wang Y, Liu P, Zhang M, Zhang C, et al. Short Telomere Length in Blood Leucocytes Contributes to the Presence of Atherothrombotic Stroke and Haemorrhagic Stroke and Risk of Post-Stroke Death. Clin Sci (Lond) (2013) 125(1):27–36. doi: 10.1042/CS20120691
37. De Felice B, Annunziata A, Fiorentino G, Manfellotto F, D’Alessandro R, Marino R, et al. Telomerase Expression in Amyotrophic Lateral Sclerosis (ALS) Patients. J Hum Genet (2014) 59(10):555–61. doi: 10.1038/jhg.2014.72
38. Luz Correa B, Ornaghi AP, Cerutti Muller G, Engroff P, Pestana Lopes R, Gomes da Silva Filho I, et al. The Inverted CD4:CD8 Ratio Is Associated With Cytomegalovirus, Poor Cognitive and Functional States in Older Adults. Neuroimmunomodulation (2014) 21(4):206–12. doi: 10.1159/000356827
39. Vescovini R, Biasini C, Telera AR, Basaglia M, Stella A, Magalini F, et al. Intense Antiextracellular Adaptive Immune Response to Human Cytomegalovirus in Very Old Subjects With Impaired Health and Cognitive and Functional Status. J Immunol (2010) 184(6):3242–9. doi: 10.4049/jimmunol.0902890
40. Moro-García MA, Alonso-Arias R, López-Vázquez A, Suárez-García FM, Solano-Jaurrieta JJ, Baltar J, et al. Relationship Between Functional Ability in Older People, Immune System Status, and Intensity of Response to CMV. Age (Dordr) (2012) 34(2):479–95. doi: 10.1007/s11357-011-9240-6
41. Petersen LE, Grassi-Oliveira R, Siara T, dos Santos SG, Ilha M, de Nardi T, et al. Premature Immunosenescence Is Associated With Memory Dysfunction in Rheumatoid Arthritis. Neuroimmunomodulation (2015) 22(3):130–7. doi: 10.1159/000358437
42. Kalim H, Pratama MZ, Mahardini E, Winoto ES, Krisna PA, Handono K. Accelerated Immune Aging was Correlated With Lupus-Associated Brain Fog in Reproductive-Age Systemic Lupus Erythematosus Patients. Int J Rheum Dis (2020) 23(5):620–6. doi: 10.1111/1756-185X.13816
43. Pavlicek D, Krebs J, Capossela S, Bertolo A, Engelhardt B, Pannek J, et al. Immunosenescence in Persons With Spinal Cord Injury in Relation to Urinary Tract Infections -a Cross-Sectional Study. Immun Ageing (2017) 14:22. doi: 10.1186/s12979-017-0103-6
44. Ritzel RM, Doran SJ, Barrett JP, Henry RJ, Ma EL, Faden AI, et al. Chronic Alterations in Systemic Immune Function After Traumatic Brain Injury. J Neurotrauma (2018) 35(13):1419–36. doi: 10.1089/neu.2017.5399
45. Geltink RIK, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
46. Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, et al. Epigenomics of Human CD8 T Cell Differentiation and Aging. Sci Immunol (2017) 2(8):eaag0192. doi: 10.1126/sciimmunol.aag0192
47. Ron-Harel N, Notarangelo G, Ghergurovich JM, Paulo JA, Sage PT, Santos D, et al. Defective Respiration and One-Carbon Metabolism Contribute to Impaired Naïve T Cell Activation in Aged Mice. Proc Natl Acad Sci U S A (2018) 115(52):13347–52. doi: 10.1073/pnas.1804149115
48. Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ, et al. P38 Signaling Inhibits Mtorc1-Independent Autophagy in Senescent Human CD8+T Cells. J Clin Invest (2014) 124(9):4004–16. doi: 10.1172/JCI75051
49. Bektas A, Schurman SH, Gonzalez-Freire M, Dunn CA, Singh AK, Macian F, et al. Age-Associated Changes in Human CD4+ T Cells Point to Mitochondrial Dysfunction Consequent to Impaired Autophagy. Aging (Albany NY) (2019) 11(21):9234–63. doi: 10.18632/aging.102438
50. Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab (2020) 32(1):44–55.e6. doi: 10.1016/j.cmet.2020.04.015
51. Gustafson CE, Kim C, Weyand CM, Goronzy JJ. Influence of Immune Aging on Vaccine Responses. J Allergy Clin Immunol (2020) 145(5):1309–21. doi: 10.1016/j.jaci.2020.03.017
52. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-Activating Germline Mutations in the Gene Encoding the PI(3)K Catalytic Subunit P110δ Result in T Cell Senescence and Human Immunodeficiency. Nat Immunol (2014) 15(1):88–97. doi: 10.1038/ni.2771
53. Palmer CS, Ostrowski M, Gouillou M, Tsai L, Yu D, Zhou J, et al. Increased Glucose Metabolic Activity Is Associated With CD4+ T-Cell Activation and Depletion During Chronic HIV Infection. AIDS (2014) 28(3):297–309. doi: 10.1097/QAD.0000000000000128
54. Palmer CS, Duette GA, Wagner MCE, Henstridge DC, Saleh S, Pereira C, et al. Metabolically Active CD4+ T Cells Expressing Glut1 and OX40 Preferentially Harbor HIV During in vitro infection. FEBS Lett (2017) 591(20):3319–32. doi: 10.1002/1873-3468.12843
55. Nian Y, Iske J, Maenosono R, Minami K, Heinbokel T, Quante M, et al. Targeting Age-Specific Changes in CD4+ T Cell Metabolism Ameliorates Alloimmune Responses and Prolongs Graft Survival. Aging Cell (2021) 20(2):e13299. doi: 10.1111/acel.13299
56. Yanes RE, Zhang H, Shen Y, Weyand CM, Goronzy JJ. Metabolic Reprogramming in Memory CD4 T Cell Responses of Old Adults. Clin Immunol (2019) 207:58–67. doi: 10.1016/j.clim.2019.07.003
57. Jeng MY, Hull PA, Fei M, Kwon HS, Tsou CL, Kasler H, et al. Metabolic Reprogramming of Human CD8+ Memory T Cells Through Loss of SIRT1. J Exp Med (2018) 215(1):51–62. doi: 10.1084/jem.20161066
58. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting Glycolytic Metabolism Enhances CD8+ T Cell Memory and Antitumor Function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/JCI69589
59. Lanna A, Henson SM, Escors D, Akbar AN. The Kinase P38 Activated by the Metabolic Regulator AMPK and Scaffold TAB1 Drives the Senescence of Human T Cells. Nat Immunol (2014) 15(10):965–72. doi: 10.1038/ni.2981
60. Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of Autophagy by Spermidine Promotes Longevity. Nat Cell Biol (2009) 11(11):1305–14. doi: 10.1038/ncb1975
61. Carriche GM, Almeida L, Stüve P, Velasquez L, Dhillon-LaBrooy A, Roy U, et al. Regulating T-Cell Differentiation Through the Polyamine Spermidine. J Allergy Clin Immunol (2021) 147(1):335–48.e11. doi: 10.1016/j.jaci.2020.04.037
62. Alsaleh G, Panse I, Swadling L, Zhang H, Richter FC, Meyer A, et al. Autophagy in T Cells From Aged Donors Is Maintained by Spermidine and Correlates With Function and Vaccine Responses. Elife (2020) 9:e57950. doi: 10.7554/eLife.57950
63. Zhang H, Alsaleh G, Feltham J, Sun Y, Napolitano G, Riffelmacher T, et al. Polyamines Control Eif5a Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol Cell (2019) 76(1):110–25.e9. doi: 10.1016/j.molcel.2019.08.005
64. Schweitzer F, Laurent S, Fink GR, Barnett MH, Reddel S, Hartung HP, et al. Age and the Risks of High-Efficacy Disease Modifying Drugs in Multiple Sclerosis. Curr Opin Neurol (2019) 32(3):305–12. doi: 10.1097/WCO.0000000000000701
65. Paghera S, Sottini A, Previcini V, Capra R, Imberti L. Age-Related Lymphocyte Output During Disease-Modifying Therapies for Multiple Sclerosis. Drugs Aging (2020) 37(10):739–46. doi: 10.1007/s40266-020-00789-4
66. Runtsch MC, Ferrara G, Angiari S. Metabolic Determinants of Leukocyte Pathogenicity in Neurological Diseases. J Neurochem (2020) 1–23. doi: 10.1111/jnc.15169
67. Desdín-Micó G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabandé-Rodríguez E, et al. T Cells With Dysfunctional Mitochondria Induce Multimorbidity and Premature Senescence. Science (2020) 368(6497):1371–76. doi: 10.1126/science.aax0860
Keywords: immunosenescence, T cell, neuroinflammation, neurodegeneration, immunometabolism
Citation: Fessler J and Angiari S (2021) The Role of T Cell Senescence in Neurological Diseases and Its Regulation by Cellular Metabolism. Front. Immunol. 12:706434. doi: 10.3389/fimmu.2021.706434
Received: 07 May 2021; Accepted: 28 June 2021;
Published: 14 July 2021.
Edited by:
Jorge Correale, Fundación Para la Lucha Contra las Enfermedades Neurológicas de la Infancia (FLENI), ArgentinaReviewed by:
Maria Celica Ysrraelit, Fundación Para la Lucha Contra las Enfermedades Neurológicas de la Infancia (FLENI), ArgentinaCopyright © 2021 Fessler and Angiari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johannes Fessler, am9oYW5uZXMuZmVzc2xlckBtZWR1bmlncmF6LmF0; Stefano Angiari, c3RlZmFuby5hbmdpYXJpQG1lZHVuaWdyYXouYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.