 Miranda D. Chávez
Miranda D. Chávez Hubert M. Tse
Hubert M. Tse
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 July 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.703972
This article is part of the Research Topic The Role of Reactive Oxygen Species in Protective Immunity View all 13 articles
Mitochondrial dysfunction resulting in oxidative stress could be associated with tissue and cell damage common in many T cell-mediated autoimmune diseases. Autoreactive CD4 T cell effector subsets (Th1,Th17) driving these diseases require increased glycolytic metabolism to upregulate key transcription factors (TF) like T-bet and RORγt that drive differentiation and proinflammatory responses. However, research in immunometabolism has demonstrated that mitochondrial-derived reactive oxygen species (ROS) act as signaling molecules contributing to T cell fate and function. Eliminating autoreactive T cells by targeting glycolysis or ROS production is a potential strategy to inhibit autoreactive T cell activation without compromising systemic immune function. Additionally, increasing self-tolerance by promoting functional immunosuppressive CD4 T regulatory (Treg) cells is another alternative therapeutic for autoimmune disease. Tregs require increased ROS and oxidative phosphorylation (OxPhos) for Foxp3 TF expression, differentiation, and anti-inflammatory IL-10 cytokine synthesis. Decreasing glycolytic activity or increasing glutathione and superoxide dismutase antioxidant activity can also be beneficial in inhibiting cytotoxic CD8 T cell effector responses. Current treatment options for T cell-mediated autoimmune diseases such as Type 1 diabetes (T1D), multiple sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE) include global immunosuppression, antibodies to deplete immune cells, and anti-cytokine therapy. While effective in diminishing autoreactive T cells, they can also compromise other immune responses resulting in increased susceptibility to other diseases and complications. The impact of mitochondrial-derived ROS and immunometabolism reprogramming in autoreactive T cell differentiation could be a potential target for T cell-mediated autoimmune diseases. Exploiting these pathways may delay autoimmune responses in T1D.
Failure to maintain self-tolerance leads to autoreactive T cells that recognize systemic or organ-specific self-antigens and subsequently, the development of autoimmunity (1). Mechanisms that result in decreased regulatory T cell (Treg) numbers and/or function could be contributing to self-tolerance failure. Tregs are essential in maintaining self-tolerance by secreting immunosuppressive/anti-inflammatory cytokines including IL-10 and TGF-β, and expression of the inhibitory receptor, CTLA-4 (2, 3) (Figure 1). Re-establishing self-tolerance by increasing Treg cell numbers and/or increasing Treg immune suppression function may inhibit autoreactive T cell effector responses and delay the progression of T cell-mediated autoimmune diseases (4). CD4 T cell subsets associated with T cell-mediated autoimmune diseases include T helper (Th) Th17 and Th1 cells.
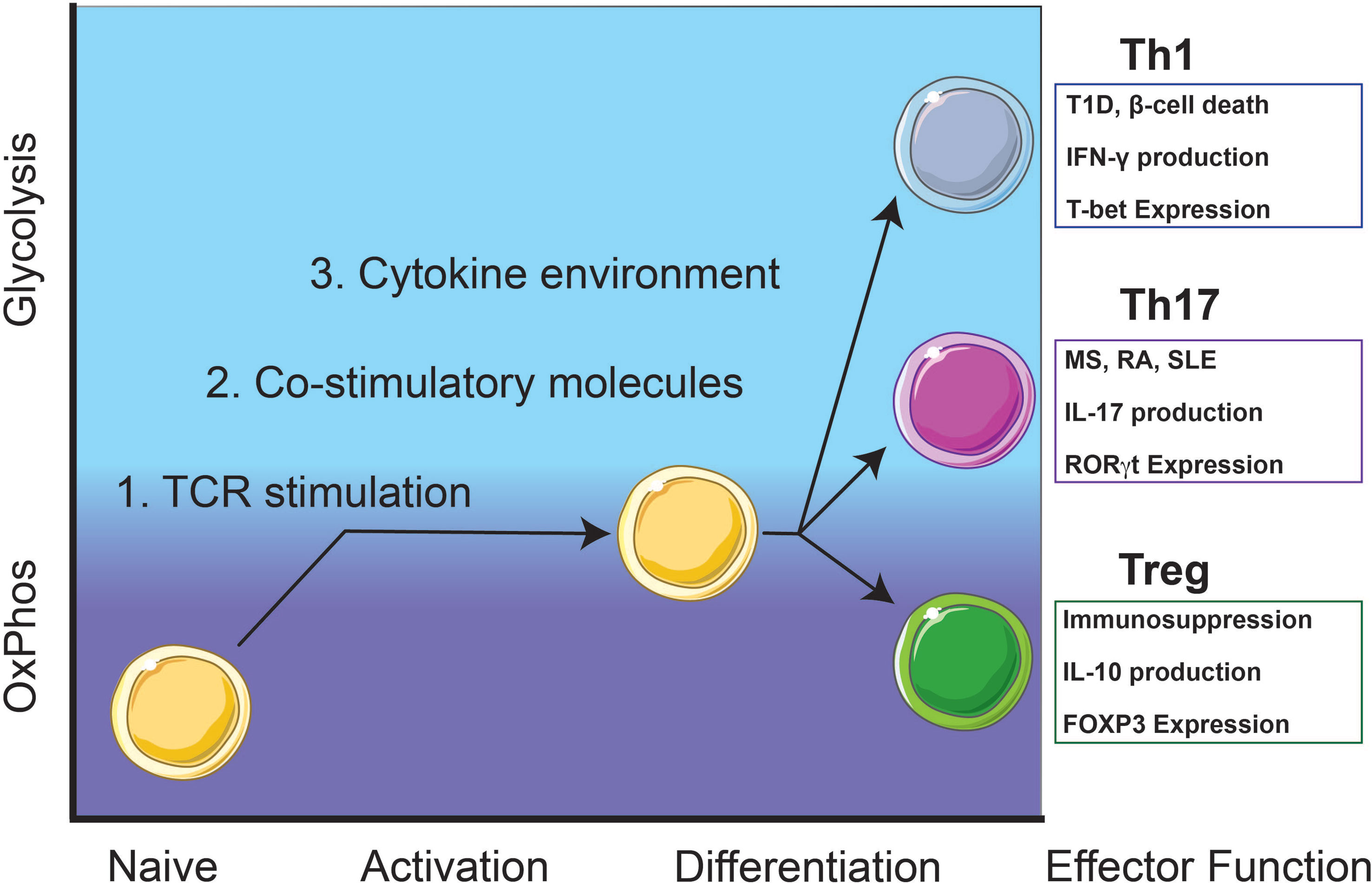
Figure 1 Autoreactive Th1 and Th17 T cell responses rely on glycolysis while immunosuppressive Treg cells utilize oxidative phosphorylation (OxPhos). Th1 cells contribute to β-cell death and destruction in type 1 diabetes (T1D) and Th17 cells contribute to pathogenesis of other T-cell mediated diseases including multiple sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE). Upon activation, naïve T cells will metabolically shift from OxPhos-dependence to a balance between glycolysis and OxPhos. Throughout early activation and differentiation, this balance is maintained until a commitment toward an effector function is achieved. Once fully differentiated, autoreactive Th1 and Th17 cells utilize glycolysis for homeostasis and maintenance while immunosuppressive Treg cells rely on OxPhos.
Generally, Th17 cells mediate immunity to extracellular pathogens and are characterized by the expression of the transcription factor (TF) RORγt and the production of cytokines such as interleukin (IL)-17A/F (5) (Figure 1). Th17 cells are involved in autoimmune-mediated diseases such as multiple sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE) (6, 7) (Figure 1). Th1 T cell responses are responsible for cell-mediated immunity and are characterized by the expression of the TF T-bet and the production of IFN-γ (8) (Figure 1). In Type 1 diabetes (T1D), CD4 T cell Th1 cytokine responses and CD8 cytotoxic T cell responses contribute to inflammation and destruction of insulin-producing β-cells (9) (Figure 1).
Current strategies to treat T cell-mediated autoimmune diseases include the use of low dose IL-2, mTOR inhibition (rapamycin), T cell depletion antibodies (teplizumab, anti-thymocyte globulin), or cytokine neutralizing antibodies/soluble receptor proteins (Infliximab, Etanercept) (10–12). These treatments are successful in temporarily diminishing disease pathogenesis; however, these reagents do not provide a permanent cure for autoimmune diseases. Therefore, further research is required to fully understand the mechanisms involved in autoreactive T cell differentiation in autoimmune disease development. One mechanism that is involved in T cell fate and function includes cellular metabolism and specifically, the interplay between signaling pathways involved in T cell differentiation and metabolic reprogramming to determine T cell effector responses (13, 14).
Immunometabolism has garnered extensive attention in recent years due to the reliance of specific metabolic pathways necessary for efficient T cell activation and differentiation of T cell subsets. Immunometabolic pathways of interest includes glycolysis, oxidative phosphorylation (OxPhos), and the contribution of mitochondrial-derived reactive oxygen species (mtROS) to mediate autoreactive T cell activation and differentiation by functioning as signaling molecules (7, 15, 16). Therefore, reprogramming T cell metabolism by targeting mtROS may counteract autoreactive T cell function and proinflammatory responses (17). This review will discuss the impact of mtROS and its potential target for immunometabolism reprogramming therapy in the context of T cell-mediated autoimmune diseases.
Initially, metabolically quiescent naïve CD4 T cells rely on OxPhos to meet all their metabolic needs (18) (Figure 1). During activation, T cells undergo a shift in metabolism away from OxPhos by increasing glycolysis. T cell activation requires three main signals from antigen-presenting cells (APC); antigen presentation on major histocompatibility complex (MHC), co-stimulation, and cytokines/reactive oxygen species (ROS) (16, 19). CD4 T cell activation begins with an interaction between the T cell receptor (TCR) and peptide loaded on MHC class-II which initiates metabolic reprograming on naïve CD4 T cells (20). Co-stimulatory receptors present on both APCs and T cells will provide the second signal necessary for T cell activation. Finally, cytokines and ROS present in the local environment will provide a proinflammatory third signal to begin T cell differentiation toward specific effector responses (16, 21). Each signal lays the foundation for potential T cell fates and drives the shift in metabolism.
TCR stimulation increases hydrogen peroxide signaling to support the upregulation of interleukin (IL)-2 (22, 23). The TCR is comprised of α and β chains and CD3 accessory chains that cumulatively bridge extracellular antigen presentation by APCs to facilitate into intracellular signaling (19, 24). After TCR stimulation, there is an influx of CD3-dependant store-operated Ca2+ entry (SOCE) that induces the accumulation of mitochondria near the TCR (23) (Figures 2A, B). Ca2+ influx into localized mitochondria influences IL-2 secretion by impacting the production of hydrogen peroxide (22) (Figures 2C, D). Due to its ability to stimulate differentiation and proliferation of T cells, production of IL-2 is a key cytokine for T cell activation (24). Deficiency in SOCE channels, such as Ca2+ release-activated Ca2+, results in a lack of IL-2 production that is potentially due to decreased hydrogen peroxide synthesis (25).
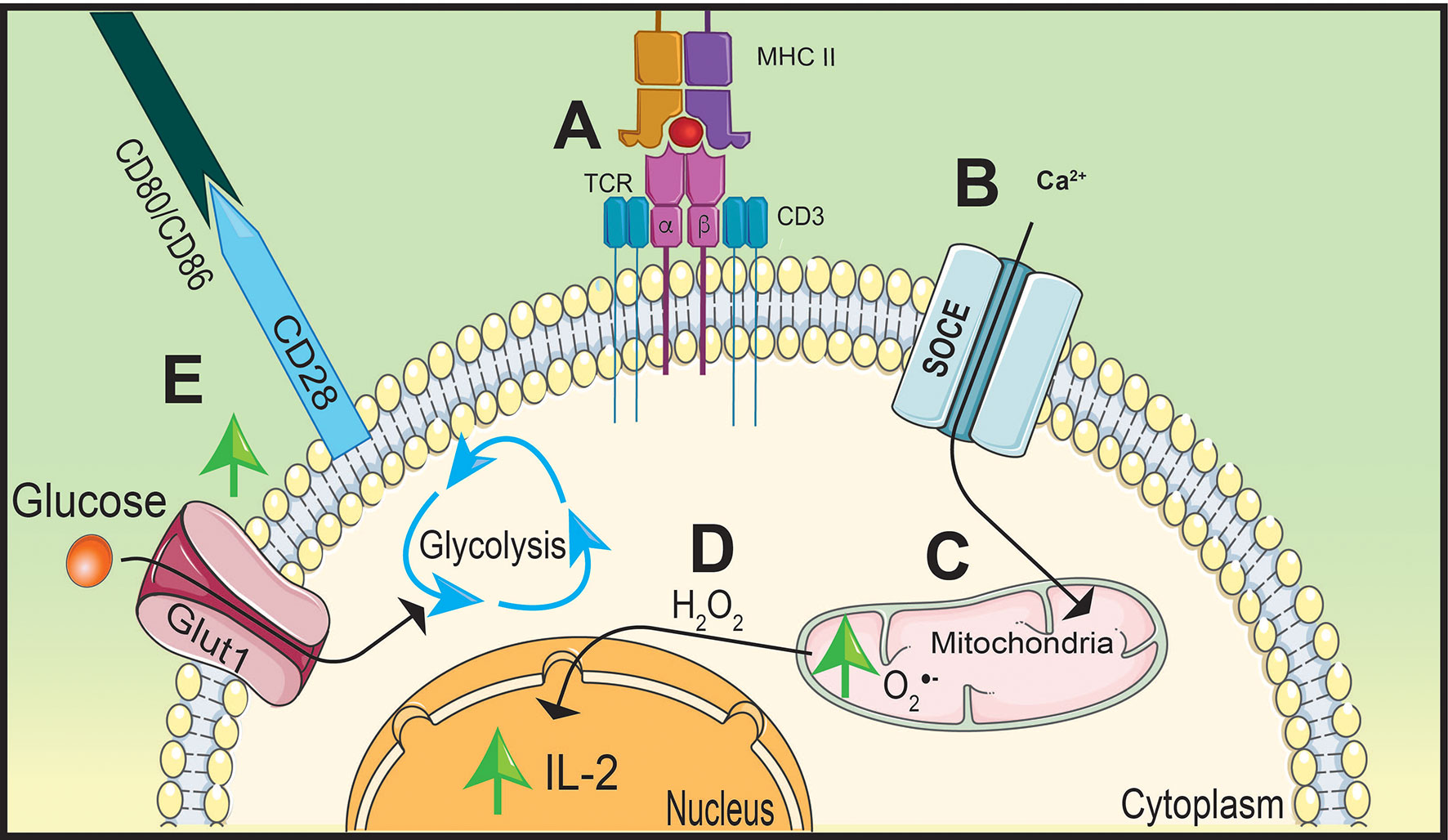
Figure 2 Stimulation of TCR and CD28 causes a metabolic shift in naïve CD4 T cells. T cell receptor (TCR) stimulation by peptide presented on major histocompatibility complex-II (MHC-II) (A) will increase calcium (Ca2+) entry into the cytoplasm through store operated calcium entry (SOCE) channels (B) increasing mitochondrial-derived superoxide generation (C). Oxidative phosphorylation (OxPhos) will promote interleukin (IL)-2 expression by hydrogen peroxide (H2O2) signaling (D). Simultaneously, CD28 will upregulate Glut1 expression on the cell membrane increasing glucose uptake shifting metabolism away from OxPhos to glycolysis to support rapid proliferation (E).
While Ca2+ influx is important to promote IL-2 production, the essential signaling molecule is hydrogen peroxide. This concept is supported by Ca2+-independent nuclear factor kappa-B (NF-κB) signaling, shown to be activated by hydrogen peroxide, significantly inhibiting IL-2 promoter regions as a result of blocking Ca2+ and/or ROS synthesis (26, 27). Similar to NF-κB signaling, hydrogen peroxide also mediates nuclear factor of activated T cells (NFAT) nuclear translocation and interaction with AP-1 to induce IL-2 production (28). These reports highlight the importance of Ca2+ influx and mtROS production to facilitate IL-2 production and T cell activation. However, optimal T cell activation also requires co-stimulatory molecule interactions in order to induce multiple signaling pathways and shift metabolism away from OxPhos to initiate T cell effector differentiation (29).
Co-stimulatory molecule, CD28, will increase glycolytic activity within the T cell to promote rapid proliferation (30, 31). Engagement of the TCR with the MHC-II/peptide complex forms an immunological synapse between the APC and the CD4 T cell by recruiting co-stimulatory molecules organized on lipid rafts on the cell membrane (32). This includes CD28 on the T cell which will interact with CD80 and CD86 on the APC (19). In order to maximize glycolytic potential, activated T cells will upregulate glucose transporter 1 (Glut1) to the cell membrane following co-stimulation and engage in aerobic glycolysis to ensure glucose will be utilized despite the presence or availability of oxygen (33, 34) (Figure 2E). CD28 will also upregulate signaling pathways that support T cell differentiation and glycolysis (31). One key pathway that is essential for T cell activation and proliferation is mitogen-activated protein kinase (MAPK) (30).
Within this pathway, extracellular signal-regulated kinase (ERK) signaling can regulate cell proliferation and differentiation by promoting glycolysis, facilitating the switch from OxPhos (35). While TCR stimulation will initiate ERK signaling in T cells, CD28 influences ERK activation by recruiting the appropriate kinases, such as lymphocyte-specific protein tyrosine kinase to enhance the signaling cascade (31). ERK phosphorylation in naïve T cells negatively regulates TCR-induced Ca2+ influx reducing OxPhos and promoting glycolysis (36). High glucose and glycolytic activity within the cell will support ERK/MAPK signaling, increase cell proliferation, differentiation, and prevent apoptosis (30, 37). The kinetics of T cell activation would be greatly diminished without CD28-dependent effects on ERK signaling and may result in weakened T cell responses (31).
In order to ensure a strong T cell response, CD28 stimulation also increases expression of pro-survival proteins including BCL-XL to prevent cell death in rapidly expanding T cells (38). Under non-apoptotic conditions, including T cell activation and expansion, overexpression of BCL-XL will result in mitochondrial depolarization and halting OxPhos (39). BCL-XL is able to influence OxPhos by reducing intracellular Ca2+ channel type 1 inositol 1,4,5-triphosphate receptor, which decreases the magnitude and duration of Ca2+ release following TCR signal transduction (39). BCL-XL will also redirect cytosolic Ca2+ away from the mitochondria through ER sequestration (39). Once co-stimulation has induced a metabolic state that is balanced between OxPhos and glycolysis; cytokines and ROS present in the immune environment will prime activated T cells for differentiation into specific effector subsets and responses.
The efficacy of both TCR and co-stimulatory signaling is greatly influenced by cytokines and ROS present during T cell activation (16, 21). Cytokine signaling can function in a paracrine and autocrine manner to influence APC and T cell activation (40). Downstream signaling pathways such as mTOR, NFAT, and NF-κB involved in T cell activation and differentiation are not only altered by the concentration of cytokines, but also by their kinetic expression as well (13, 21). Prior to activation, IL-7 is an essential cytokine for naïve T cell survival and homeostasis due to its inactivation of pro-apoptotic proteins (41, 42). Upon stimulation, T cells will secrete IL-2 to promote expansion and prime T cells for differentiation (41). An autoreactive Th1 effector response will result from a cytokine environment comprised of IFN-γ, IL-1β, TNF-α, IL-6, and IL-12 (43). IL-17, IL-23, and IL-21 cytokines result in an autoreactive Th17 effector response; while TGF-β and IL-10 will result in the promotion of self-tolerance (43).
Traditionally it was thought that cytokines were solely responsible for driving T cell differentiation toward specific effector responses. However, superoxide may also be contributing to differentiation by activating redox-dependent signaling pathways associated with certain T cell effector functions (44). Past research has demonstrated that even under conditions which utilized cytokines to skew toward a proinflammatory response, the reduction of ROS results in decreased NF-κB-regulated proinflammatory cytokine production (45). Additionally, the decrease in ROS, specifically superoxide, in both APC and T cells not only decreased proinflammatory IFN-γ synthesis, but also T-bet expression as well (16). These data accentuate the indispensable role of ROS generation in effector T cell responses.
Due to the role of ROS in cellular homeostasis, irregular ROS generation contributes to disease pathology making it a prime target for metabolic reprograming therapies (46). ROS including superoxide are mainly generated by the mitochondria, but may also be produced by NADPH oxidase enzymes (47, 48). Antioxidants present within the cell and extracellular environment regulate ROS to prevent damage to the cells caused by oxidative stress (49). Oxidative stress and mitochondrial dysfunction are common pathologies normally found in T cell-mediated autoimmune diseases (49, 50), indicating that increasing our understanding of oxidative stress and ROS regulation may have potential for the development of novel therapies to mitigate autoimmune dysfunction.
ROS are generated throughout the cell as a result of various redox reactions (47, 48). The mitochondria are one of the main cellular sources of ROS synthesis due to electrons leaking from the election transport chain (ETC) and interacting with oxygen to generate superoxide (47) (Figure 3A). Pyruvate generated from glycolysis will migrate into the mitochondrial matrix, be converted to acetyl-CoA, and feed into the citric acid cycle (CAC) (51). Succinate dehydrogenase, also known as complex II, is the only membrane-bound component of the CAC and a complex within the ETC (52). Electron movement creates a proton gradient between the mitochondrial matrix and the inner membrane space driving the phosphorylation of adenosine diphosphate to adenosine triphosphate (ATP) by ATP synthase (53). Superoxide generation is not limited to the mitochondria, it can also be generated by NADPH oxidase enzymes on the plasma membrane (48) (Figure 3B).
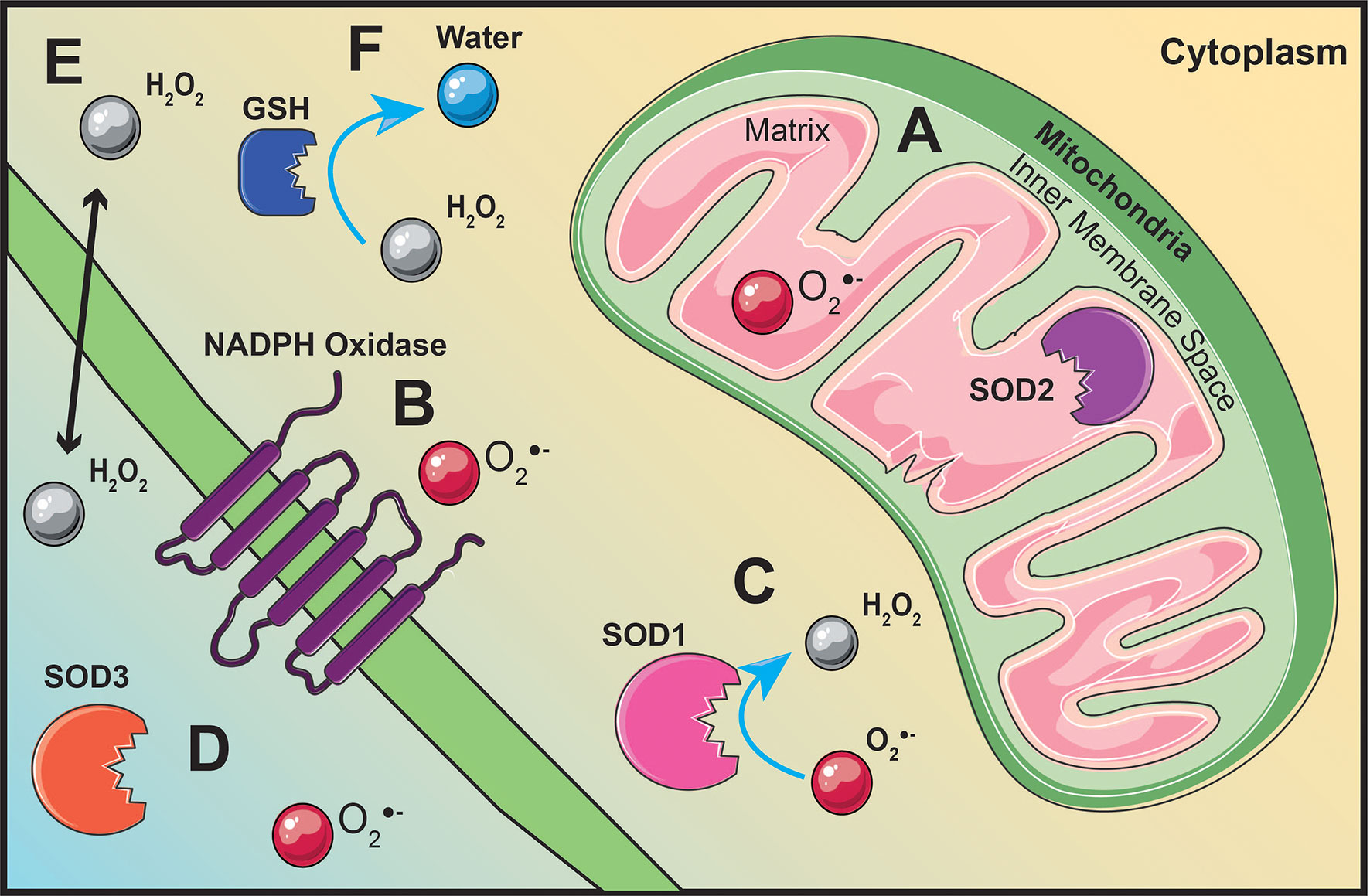
Figure 3 ROS production originates from various locations throughout the cell. Superoxide is generated within the mitochondrial matrix (A) or by membrane bound NADPH oxidases (B). Superoxide dismutase (SOD) located within the mitochondrial matrix (A), within the cytoplasm (C), or extracellularly (D) will convert superoxide into hydrogen peroxide (H2O2) (C). Hydrogen peroxide is able to act as signaling molecule within the cell or extracellular due to its ability to diffuse across the cell membrane (E). Glutathione (GSH), located throughout the cytoplasm, will regulate hydrogen peroxide levels by converting hydrogen peroxide into water (F).
Excess levels of superoxide can be toxic to the cell; therefore, superoxide dismutase (SOD) enzymes act to dismutate superoxide into hydrogen peroxide and molecular oxygen (46). There are three different isoforms of SOD located within and outside the cell. Copper- and zinc-containing SOD (Cu/Zn SOD, SOD1) is mainly localized in the cytoplasm (54) (Figure 3C). Manganese-containing SOD (MnSOD, SOD2) resides within the mitochondrial matrix (54) (Figure 3A). Similar to SOD2, extracellular SOD (SOD3) also contains manganese, except it is localized outside the cell (54) (Figure 3D). Regardless of its location, enzymatic regulation of superoxide by SOD will generate hydrogen peroxide as a result of its reaction (48) (Figure 3C). Unlike superoxide, hydrogen peroxide is stable, diffusible, and can regulate redox-dependent signaling pathways (55) (Figure 3E). Hydrogen peroxide can modulate pathway activation by inhibiting phosphatases, activating tyrosine kinases, and transcription factor activation (55). Catalase, glutathione peroxidase, and thioredoxin peroxidase are the main enzymes involved in maintaining hydrogen peroxide levels for optimal cell function and signaling (56).
A common factor in several mitochondrial-related diseases such as diabetes, obesity, and neurodegenerative diseases is oxidative stress (50). Oxidative stress is defined by an imbalance of ROS and antioxidant activity that could lead to cell and tissue damage (49). Excess levels of mtROS, specifically superoxide, can lead to cell damage and mitochondrial-dependent apoptosis (57). Under oxidative stress, a pore is created in the inner mitochondrial membrane, halting OxPhos, leaking cytochrome C into the cytoplasm, and inducing apoptosis as a result of DNA fragmentation and ultimately ending in cell death (57, 58). However, not all ROS production is detrimental to cells, as low levels of hydrogen peroxide can activate pro-survival proteins such as Bcl-2 (57).
Mitochondrial dysfunction and the generation of oxidative stress could contribute to disease pathology in SLE, T1D, and RA (4, 50, 59–61). Increased antioxidant activity in patients with SLE improved mitochondrial metabolism, decreased mtROS, and reduced inflammation (59). In T1D, ROS synthesis can influence autoreactive T cell activation and directly mediate β-cell damage partly due to weak antioxidant defenses in β-cells (60). An inadequate antioxidant defense may contribute to oxidative stress within the β-cell, influencing its dysfunction and immunogenicity in T1D (62). Oxidative stress due to mitochondrial dysfunction could also enhance inflammatory responses in RA (61). Analysis of genes and protein-protein interactions in human patients with RA revealed that numerous mitochondrial-related pathways influence RA pathogenesis (61).
Mitochondrial DNA (mtDNA) deletions may manifest in a variety of syndromes, diseases, or phenotypes varying in severity (63, 64). mtDNA encodes for 13 proteins that are essential for OxPhos (64). In humans, it was revealed that the single nucleotide polymorphism (SNP) nt13708A in mtDNA significantly increased susceptibility to MS and SLE (65, 66). This SNP is in the MT-ND5 gene, which encodes a subunit of complex I in the ETC (65, 66). While both studies identified nt13708A, the functional consequences of this SNP remain unknown. However, not all mtDNA SNPs result in increased disease susceptibility. In T1D, the C5173A SNP in the mt-ND2 gene provided protection against disease development (67). Further characterization of mt-Nd2a, an allelic variant of mt-Nd2, exhibited a reduction in ROS generated from complex I (67). These findings demonstrate that SNPs in mtDNA can modulate disease by altering OxPhos. Further studies on the effects of mtDNA deletions on OxPhos or superoxide generation could provide insight into the development of autoimmunity as a result of mitochondrial dysfunction.
Understanding the redox-dependent effects on immunometabolism and the generation of proinflammatory T cell responses needs to be further defined as a potential therapeutic strategy. The field of immunometabolism will define the molecular and cellular contributors that connect metabolism and immune cell function (14). Decades of research have shown nutrient availability and its effects on immune responses. Yet, only the last decade has provided insight on how metabolism coupled with ROS is a determinant of adaptive immune cell phenotype and effector responses.
Inflammatory responses prime Th1/Th17 differentiation by promoting a metabolic environment toward glycolysis. An inflammatory response often creates an environment depleted of nutrients and oxygen (68). Coupled with proinflammatory cytokines that will induce the activation of TF necessary for Th1/Th17 differentiation, the lack of oxygen results in increased glycolysis and skewed T cell differentiation (69–71). In addition to an enhanced glycolytic environment, ROS synthesis will stabilize RORγt TF expression to promote Th17 differentiation (72, 73). Increasing our understanding of the effects of glycolysis and ROS synthesis in Th1 and Th17 CD4 T cell differentiation could give rise to novel targets for delaying T cell-mediated autoimmune diseases.
Th1 induction begins with APCs secreting IL-12, a key cytokine in Th1 differentiation, to upregulate proinflammatory cytokine IFN-γ secretion and TF T-bet expression (74). Deficiency in IL-12 will reduce autoreactive Th1 responses, including IFN-γ production, as a result of disrupting a feedback loop in activated T cells (75). The induction of this feedback loop is key to Th1 differentiation since IFN-γ is a proinflammatory cytokine that will upregulate other cytokines necessary for Th1 differentiation including IL-12 (70, 71).
Th1 differentiation is primarily driven by an upregulation of glycolysis to support expression of T-bet and IFN-γ (69–71). Differentiating T cells engaged in aerobic glycolysis preferentially translate Ifng mRNA versus those using OxPhos (76). Pyruvate, generated by glycolysis, is utilized by lactate dehydrogenase A, which also supports aerobic glycolysis in activated T cells in order to promote proinflammatory IFN-γ production (69). This suggests that glycolytic upregulation will promote proinflammatory cytokines that shift naïve CD4 T cell activation toward Th1 differentiation (69–71). Therefore, it is not surprising that cytokine-producing Th1 cells are diminished when the inability to transport glucose for glycolysis is facilitated by a Glut-1 deficiency (77). In T1D, there is an abundance of glucose, which could be contributing to an environment that preferentially drives Th1 differentiation.
IFN-γ production by autoreactive Th1 cells is involved in the destruction of insulin-secreting β-cells in T1D (78) (Figure 1). During insulitis, APCs infiltrate islets and migrate to peripheral lymphoid tissues where both naïve autoreactive CD8 and CD4 T cells are activated (9). Following activation, T cells migrate back to the islets located within the pancreas (79). Once in the pancreas, activated Th1 cells produce IL-2 and IFN-γ to mature cytotoxic CD8 T cell responses (80). The presence of other cytokines, such as IL-1β, TNF-α, and IFN-γ, promote Th1 differentiation in addition to inhibiting insulin secretion and inducing β-cell apoptosis (81, 82). Although both CD8 and CD4 T cells contribute to β-cell destruction, a deficiency of CD4 T cells protects against spontaneous T1D development in NOD mice (83). Therefore, reducing glycolysis and/or ROS production in autoreactive Th1 CD4 T cells represents a potential therapeutic strategy for delaying T1D.
Insulin is a key hormone that is secreted from β-cells in response to high glucose, impacting metabolism by signaling cells to uptake glucose and undergo glycolysis (84). Insulin signaling requires insulin-induced hydrogen peroxide generation to maintain autophosphorylation of insulin-stimulated insulin receptor (73). Inhibition of superoxide generation by complex II of the ETC prevents hydrogen peroxide generation after insulin stimulation, disrupting insulin signaling and glucose uptake (85). Decreasing glycolytic signaling in T cells by regulating hydrogen peroxide to decrease glucose uptake may be another immunotherapy used to orchestrate a metabolic blockage. The use of the glycolytic inhibitor 2-deoxy glucose (2-DG), has shown promise in diminishing autoreactive CD4 T cell responses without harming other aspects of the immune system (34). This would be beneficial compared to other potential treatment options, such as rapamycin, which acts as a universal immunosuppressant targeting APCs and T cells (86). Rapamycin is a mTOR inhibitor that would prevent all cells from being able to monitor for DNA damage, synthesize proteins, and coordinate an appropriate response to intracellular stress if administered systemically (87). This could alter immune function, lipid homeostasis, and muscle mass homeostasis resulting in increased susceptibility to other diseases (87). Therefore, there is a need for the development of targeted metabolic therapies specific for autoreactive T cells without impairing the ability of the immune system to mount defenses against microbial pathogens.
Th17 cells, like Th1 cells, also rely on glycolysis for their differentiation. Currently, there are studies showing the promise of this type of metabolic targeting therapy in Th17-mediated diseases including MS (88). Suppression of glycolysis via deletion of Glut1 has been shown to protect mice from Th1 and Th17 cell-driven diseases such as colitis and inflammatory bowel disease by decreasing the expansion and survival of T cells (77). A possible target for diminishing glycolysis in Th17 cytokine responses would be decreasing hypoxia-inducible factor (HIF)-1α expression. HIF-1α is a TF that will contribute to metabolic programming of activated T cells by mediating a switch from OxPhos to aerobic glycolysis (89, 90). HIF-1α will specifically stimulate Th17 differentiation by binding to the RORγt promoter to increase IL-17A production (89). Prostaglandin E2 (PGE2) will boost glycolytic activity by stabilizing HIF-1α (91), while simultaneously skewing T cells away from a Th1 phenotype by inhibiting IFN-γ production (92). Prevention of hypoxic environments that will foster glycolytic activity via PGE2 inhibition is a current treatment option for Th17-mediated autoimmune diseases including RA (92).
In MS, pathogenesis is driven by pathogenic Th17 cells interacting with the central nervous system (CNS) (93) (Figure 1). Naïve T cells are primed outside the CNS by APCs, cross the blood-brain barrier, and produce proinflammatory responses that result in the destruction of myelin and axons (93). Experimental autoimmune encephalomyelitis (EAE) is a mouse model used to study MS and studies have shown that increased ROS during pathogenic Th17 differentiation is another possible metabolic target for MS (94). Inflammatory Th17 responses are promoted by hydrogen peroxide stabilizing insulin receptor signaling to activate HIF-1α (72, 73). T cells deficient in insulin receptor signaling will have a diminished inflammatory response when activated, demonstrating the importance of insulin receptor stabilization by hydrogen peroxide for pathogenic Th17 differentiation (72, 95). Additionally, hydrogen peroxide contributes to Th17 differentiation by activating HIF target genes and stabilizing HIF-1α proteins (96). By inhibiting mitochondrial OxPhos during Th17 activation, mTOR signaling and the expression of basic leucin zipper transcriptional factor ATF-like (BATF) will be decreased (97). Without BATF to increase chromatin accessibility of Th17 transcription factors, mice displayed a resistance to EAE without affecting Th1/Th2 effector responses (98). This observation highlights the promise of metabolic therapies targeting ROS production in autoreactive Th17 cells, but immunometabolic reprogramming can also affect Treg differentiation as another potential avenue to delay EAE disease onset (97).
It has been debated whether the loss of Treg function and/or the number of Treg cells is a major contributor in T cell-mediated autoimmune diseases such as T1D, MS, and RA (99). Tregs have a broad set of functions including ensuring tolerance to autoantigens, limiting excess immune responses, and providing homeostasis of various tissues (100). Their development is similar to conventional T cells, but they are unique in their underlying self-reactivity during thymic selection, allowing them to suppress other CD4 T cell effector populations (2, 101). Strategies to increase the number of Tregs and enhance immunosuppressive Treg function are ideal approaches to restore peripheral tolerance in autoimmune diseases.
Therefore, studies to determine how immunometabolism contributes to Treg homeostasis, differentiation, and function are worthy research endeavors. Loss of the anti-apoptotic factor, Bcl-2, did not alter the quality or quantity of Tregs isolated from mice (102). However, increased expression of Bcl-2 by IL-7 enables natural Tregs to survive and proliferate properly while they circulate between secondary lymphoid organs (100). Bcl-2 is known to increase mtROS superoxide production in complexes I and III within the ETC (103). Tregs deficient in complex III-derived superoxide are able to maintain stable Foxp3 expression, cell proliferation, and survival, yet their suppressive capacity is lost (104, 105). This paralleled loss of function emphasizes the importance of proper mitochondrial metabolism throughout T cell activation and homeostasis.
There is a close developmental relationship between Treg and Th17 cells. TGF-β alone will induce Treg differentiation, however if IL-6 is present, Th17 cells will be preferentially induced (5). IL-6 will promote glucose uptake and metabolism (106, 107). This is due to IL-6 terminating Foxp3-mediated inhibition of Th17 TF RORγt (107). Foxp3 is a lineage-specific TF for Treg cells (3) (Figure 1). In the absence of other proinflammatory cytokines, Foxp3 inhibits Th17 differentiation by blocking RORγt activity in activated T cells (108). IL-2 negatively regulates IL-17 production while driving Treg proliferation, differentiation, and function (109, 110). In SLE, due to the reduction of IL-2, there is a decrease in the Treg population (111). In the absence of HIF-1α, a Th17 promoting factor, Th17 cytokine responses will be dampened and Foxp3 Treg differentiation will be elicited instead (97). This could be due to the absence of the Foxp3 ubiquitin-mediated degradation pathway that is regulated by HIF-1α (112). HIF-1α increases RORγt expression while simultaneously tagging Foxp3 protein for ubiquitination (89).
Myelocytomatosis oncogene (Myc) is a TF that drives metabolic reprogramming toward glycolysis (34). Expression of Foxp3 suppresses glycolysis by binding to the Myc promotor and suppressing Myc gene expression (34). Diminishing glycolysis via Glut1deficiency does not affect Treg differentiation or cell numbers (77) since Treg cells utilize fatty acid oxidation (FAO) for Treg differentiation (113). By suppressing glycolysis, Foxp3 reactivates OxPhos, which is essential for Treg metabolism, stability, and function (Figure 1).
Similar to Tregs, Th2 cells upregulate fatty acid lipid metabolism and inhibition of these metabolic pathways will reduce a Th2-mediated response (114). The key role of OxPhos in Th2 differentiation is highlighted by a study that showed that reduced mtROS produced by complex-I inhibited anti-CD3-induced IL-2 and IL-4 expression and stunted Th2 differentiation (27). The production of IL-4 aids in the differentiation of naïve T cells toward the Th2 phenotype by inducing expression of the key TF Gata3 (8, 115). Expression of Gata3 is sufficient to induce the Th2 phenotype because it creates a positive feedback loop by inducing IL-4, which maintains Th2 cell identity (114). Increased activity of the kinase mTOR is present in all effector Th lineages in order to coordinate cell proliferation and metabolism (87). Unique to Th2 cells is the increase in fatty acid uptake by peroxisome proliferator activated receptor gamma (PPAR-γ) that also regulates fatty acid metabolism (114). Both IL-4 and mTOR promote PPAR-γ in Th2 cells and its absence will not only affect OxPhos, but Th2 function as well (114). Unfortunately, Th2 differentiation is diminished in individuals taking rapamycin due to its inhibitory effects on mTOR activity (87). Loss of Th2 function via decreased mTOR could be associated with decreased PPAR-γ binding to key genes such as Gata3, Il5, and Stat5 (116).
Th2 cells protect from parasitic helminths infections and stimulate repair of damaged tissue repair (117). While there are no Th2-mediated autoimmune diseases, modulating the balance between Th1 and Th2 cells can affect autoimmune responses in animal models of RA (118). Treatment of arthritic mice with IL-4 suppressed disease, but did not reverse disease progression (118). Similar success was also observed in mouse models of T1D that skewed T cell responses toward a Th2 phenotype in an effort to prevent the activation of autoreactive Th1 cytokine responses (119). The efficacy of Th2 skewing has not been investigated in mouse models of MS or SLE. However, the loss of the nuclear factor erythroid 2-related factor 2 (NRF2) TF involved in inducing an antioxidant response, diminished Th2 cytokine responses and concomitantly, increased Th1 differentiation and SLE (120). While Th2 skewing may delay autoimmunity, Th2 cells also contribute to asthma and allergy. Allergen immunotherapy involves continuous exposure to allergens at increasing concentrations and is the only well-established treatment for asthma that targets Th2 cells (121). While the mechanism is still undefined, allergen immunotherapy has been shown to skew T cell differentiation away from a Th2 phenotype toward a Treg or Th1 phenotype (122).
Similar to CD4 T cells, CD8 T cell activation requires antigen presentation, co-stimulatory molecule interactions, and the synthesis of cytokines and ROS (123, 124). However, unlike CD4 T cells, MHC-I is required for CD8 TCR stimulation, and deficiency in co-stimulatory molecules such as CD40L and CD28 does not result in anergy or decreased proliferation (125). Additionally, despite an increase in glycolysis to support clonal expansion, a deficiency in Glut1 expression does not affect CD8 T cell proliferation after activation (77, 126). Rather, their role in viral clearance is maximized by proinflammatory cytokines such as IFN-γ and IL-12, because it shifts activation toward short-lived effector cell (SLEC) populations (123).
The two main subsets of CD8 T cells are SLEC and memory precursor effector cells (MPEC) (123). Following TCR stimulation, mitochondrial fusion occurs in CD8 T cells to support increased OxPhos that promote cell proliferation (127, 128). Halting OxPhos via the deletion of AMP-activated protein kinase (AMPK) promotes the activation of the mTOR pathway and subsequently, defective CD8 T cell memory differentiation (129, 130). Inhibition of mTOR through AMPK activity improves the quality of CD8 memory T cells by accelerating effector to memory transition without affecting CD8 T cell effector function (129, 131). The effect of AMPK on CD8 T cell differentiation resulted in an interest in AMPK as a possible target for CD8 T cell memory generation. AMPK, in addition to inhibition of growth regulator mTOR, also regulates mitochondrial fission/fusion and promotes mitochondrial biogenesis (132). Due to memory T cells having an increased mitochondrial mass, AMPK regulation of mitochondrial biogenesis could contribute to increasing mitochondrial mass promoting memory T cells rather than cytotoxic CD8 T cells (133).
In T1D, APCs such as macrophages and dendritic cells are the first immune cells that infiltrate islets and contribute to pancreatic β-cell destruction (134). Macrophages activate Th1 CD4 T cells to produce IL-2 and IFN-γ to facilitate the differentiation of CD8 T cells to become cytotoxic (80). IL-1β secretion by APCs can function as a proinflammatory third signal for T cell maturation and enhance antigen-specific CD8 T cell effector function by increasing cytotoxic activity and IFN-γ production (135). If MHC-I is not expressed on β-cells, mice were protected from diabetes development (134). Therefore, strategies that can inhibit CD8 T cell effector responses would be beneficial as a T1D therapeutic. Recent investigations have tested the efficacy of 2-DG to inhibit glycolysis and autoreactive CD8 T cell cytolytic responses in T1D (136). Inhibition of glycolysis decreased islet infiltration of pathogenic CD8 T cells (136), but the effects of 2-DG on cytotoxic CD8 T cell effector responses were not determined.
In MS, CD8 T cells are a viable target for therapy (137). Pathogenic CD8 T cells have been located in MS plaques, cerebrospinal fluid, and demyelinated axons (137). Mutations in MHC-I, along with gradual upregulation of MHC-I on various cell types throughout disease progression are associated with increased risk for MS and may also contribute to the increased ratio of CD8 to CD4 T cells in MS brain lesions (138). The accumulation of the glycolytic byproduct, lactate, in the cerebrospinal fluid of MS patients compared to controls has recently been considered an indicator of disease progression (139). Promoting a metabolic shift to decrease glycolytic activity may provide protection from disease progression. Lactic acid is able to promote OxPhos by inducing glycolysis/OxPhos interconversion to suppress cytotoxic CD8 T cell proliferation and cytokine production (140, 141). These biomarkers and different metabolic indicators for disease may all be therapeutic options worth experimentally perusing in order to decrease pathogenic CD8 T cell activity.
Antioxidants are effective in decreasing oxidative stress and loss of antioxidant activity may also be involved in autoimmune diseases. Glutathione (GSH) is a tripeptide antioxidant consisting of glutamate, cysteine, and glycine, located within the cytoplasm and utilized by the cell to detoxify ROS such as hydrogen peroxide (142, 143) (Figure 3F). The reductive capability of the sulfhydryl group contained within the cysteine makes GSH a pivotal component of protecting the cell from oxidative damage and stress resulting from excess ROS (143). Decreased activation of the NRF2 pathway, which regulates antioxidant genes including GSH, is observed across many T cell-mediated diseases (144). This is most likely due to GSH supporting activation-induced metabolic reprograming in T cells. By decreasing ROS, GSH reinforces glycolytic metabolic reprogramming that is required by inflammatory Th1 and Th17 cytokine responses (145). Studies have also shown that a deficiency in GSH results in impaired IFN-γ and IL-17 cytokine production (146).
Hyperglycemia is associated with increased oxidative stress, free radical production, suppression of antioxidant function, and is also a common complication in T1D and type 2 diabetes (T2D) (49, 147, 148). Furthermore, increased cytosolic hydrogen peroxide generation in pancreatic islets as a result of decreased GSH and SOD in individuals with T2D leads to impaired insulin secretion (149, 150). Children diagnosed with T1D show significantly decreased GSH antioxidant capacity compared to age-matched healthy children (147, 151). Therefore, it is encouraging that NRF2 activators have shown a great deal of promise as a therapeutic in pre-clinical trials for diabetic complications associated with increased oxidative stress (152).
GSH deficiency is also prevalent in MS (153). Compared to adjacent white matter, there is a significant reduction in GSH in MS plagues (153). GSH delivery to the CNS is difficult due to the inability to cross the blood-brain barrier, short half-life, and the requirement of high doses to achieve a therapeutic effect (154). While oral GSH did not impact disease progression, there are clinical trials to investigate NRF2 activation as a potential therapeutic in MS to increase downstream expression of antioxidants such as GSH (154). A phase 3 clinical trial for MS demonstrated that oral treatment with delayed-release dimethyl fumarate (DMF) was effective in activating NRF2, but further studies are needed to determine if GSH levels were increased and if MS could be delayed following DMF treatment (155).
In mouse models of RA, overexpression of SOD3 suppressed proinflammatory cytokine production and disease development (156). Unfortunately, human translation is limited due to a short half-life and instability of SOD (157). Interestingly, saliva taken from individuals with SLE displayed decreased presence of SOD compared to healthy controls (158). Therefore, it has been proposed that decreased SOD activity could alternatively be utilized as a biomarker for SLE (158). Other investigators have employed SOD mimetics in mouse models of T1D to increase stability and longevity of antioxidant activity in an effort to delay pancreatic β-cell destruction (159). The use of SOD mimetics can delay the adoptive transfer of T1D with a diabetogenic CD4 T cell clone that was partly due to a decrease in T cell proliferation and IFN-γ production (160). Similar results were also observed when CD8 T cells were treated with a SOD mimetic resulting in decreased proliferation, cytokine production and cytolytic effector responses (124). The ability of SOD mimetics to delay T1D progression and inhibit autoreactive T cell responses suggests these novel antioxidants may also be applicable for the treatment of SLE and RA in future studies.
Increasing our understanding of the role of immunometabolism in T cell-mediated autoimmune diseases may provide novel insights into the redox-dependent mechanisms involved in immune dysregulation. Specifically, determining the homeostatic role of glycolysis and OxPhos in naïve and effector T cells may bridge the knowledge gap of metabolic reprogramming in T cell activation and differentiation. T1D, MS, and RA are autoimmune diseases that would benefit from metabolic reprogramming of autoreactive T cells to alleviate disease pathogenesis. Metabolically targeting glycolysis to decrease Th1 cytokine responses in T1D may prevent T cell-mediated β-cell destruction. Similarly, inhibiting glycolysis by targeting HIF-1α stabilizers such as hydrogen peroxide and PGE2 can also dampen Th17 cytokine responses in MS to delay neuronal damage. Decreasing glycolysis is not only effective in limiting autoreactive CD4 T cell responses, but can also diminish cytotoxic CD8 T cell effector responses in T1D and MS. Increasing our knowledge of the contribution of mtROS in T1D, MS, RA, and SLE and the effect of antioxidants to diminish mtROS may provide a novel immunotherapeutic approach to limit T cell-mediated autoimmunity in the near future.
MC and HT conceived, wrote, and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by an NIH/NIDDK R01 DK099550, JDRF award SRA-2016-270-S-B, JDRF award 2-SRA-2019-692-S-B, NIH/NIDDK R56 DK126456, and NIH/NIDDK R01 DK127497.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Jessie Bara, Katie Heath, Cailin Kellum, KaLia Burnette, Jared Taylor, Joseph Feduska, Samuel Blum, and Shekinah Philips for critical reading of the manuscript. Artwork in figures were transformed and adapted from Servier Medical Art.
1. Lin X, Lu L. B Cell-Mediated Autoimmune Diseases. Adv Exp Med Biol (2020) 1254:145–60. doi: 10.1007/978-981-15-3532-1_11
2. Corthay A. How do Regulatory T Cells Work? Scand J Immunol (2009) 70(4):326–36. doi: 10.1111/j.1365-3083.2009.02308.x
3. Rudensky AY. Regulatory T Cells and Foxp3. Immunol Rev (2011) 241(1):260–8. doi: 10.1111/j.1600-065X.2011.01018.x
4. Kamradt T, Mitchison NA. Tolerance and Autoimmunity. N Engl J Med (2001) 344(9):655–64. doi: 10.1056/NEJM200103013440907
5. Crome SQ, Wang AY, Levings MK. Translational Mini-Review Series on Th17 Cells: Function and Regulation of Human T Helper 17 Cells in Health and Disease. Clin Exp Immunol (2010) 159(2):109–19. doi: 10.1111/j.1365-2249.2009.04037.x
6. Shah K, Lee WW, Lee SH, Kim SH, Kang SW, Craft J, et al. Dysregulated Balance of Th17 and Th1 Cells in Systemic Lupus Erythematosus. Arthritis Res Ther (2010) 12(2):R53. doi: 10.1186/ar2964
7. Kaufmann U, Kahlfuss S, Yang J, Ivanova E, Koralov SB, Feske S. Calcium Signaling Controls Pathogenic Th17 Cell-Mediated Inflammation by Regulating Mitochondrial Function. Cell Metab (2019) 29(5):1104–18.e6. doi: 10.1016/j.cmet.2019.01.019
8. Romagnani S. Th1/Th2 Cells. Inflammation Bowel Dis (1999) 5(4):285–94. doi: 10.1097/00054725-199911000-00009
9. Pugliese A. Autoreactive T Cells in Type 1 Diabetes. J Clin Invest (2017) 127(8):2881–91. doi: 10.1172/JCI94549
10. Nepom GT, Ehlers M, Mandrup-Poulsen T. Anti-Cytokine Therapies in T1D: Concepts and Strategies. Clin Immunol (2013) 149(3):279–85. doi: 10.1016/j.clim.2013.02.003
11. Vojdani A, Lambert J, Kellermann G. The Role of Th17 in Neuroimmune Disorders: A Target for CAM Therapy. Part III Evid Based Complement Alternat Med (2011) 2011:548086. doi: 10.1093/ecam/nep062
12. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and Clinical Effects of Low-Dose Interleukin-2 Across 11 Autoimmune Diseases in a Single, Open Clinical Trial. Ann Rheum Dis (2019) 78(2):209–17. doi: 10.1136/annrheumdis-2018-214229
13. Pinkoski MJ, Waterhouse NJ, Green DR. Mitochondria, Apoptosis and Autoimmunity. Curr Dir Autoimmun (2006) 9:55–73. doi: 10.1159/000090772
14. Hotamisligil GS. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity (2017) 47(3):406–20. doi: 10.1016/j.immuni.2017.08.009
15. Abimannan T, Peroumal D, Parida JR, Barik PK, Padhan P, Devadas S. Oxidative Stress Modulates the Cytokine Response of Differentiated Th17 and Th1 Cells. Free Radic Biol Med (2016) 99:352–63. doi: 10.1016/j.freeradbiomed.2016.08.026
16. Padgett LE, Tse HM. NADPH Oxidase-Derived Superoxide Provides a Third Signal for CD4 T Cell Effector Responses. J Immunol (2016) 197(5):1733–42. doi: 10.4049/jimmunol.1502581
17. Pålsson-McDermott EM, O’Neill LAJ. Targeting Immunometabolism as an Anti-Inflammatory Strategy. Cell Res (2020) 30(4):300–14. doi: 10.1038/s41422-020-0291-z
18. Rashida Gnanaprakasam JN, Wu R, Wang R. Metabolic Reprogramming in Modulating T Cell Reactive Oxygen Species Generation and Antioxidant Capacity. Front Immunol (2018) 9:1075. doi: 10.3389/fimmu.2018.01075
19. Chaplin DD. Overview of the Immune Response. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S3–23. doi: 10.1016/j.jaci.2009.12.980
20. Couture A, Garnier A, Docagne F, Boyer O, Vivien D, Le-Mauff B, et al. HLA-Class II Artificial Antigen Presenting Cells in CD4(+) T Cell-Based Immunotherapy. Front Immunol (2019) 10:1081. doi: 10.3389/fimmu.2019.01081
21. Barberis M, Helikar T, Verbruggen P. Simulation of Stimulation: Cytokine Dosage and Cell Cycle Crosstalk Driving Timing-Dependent T Cell Differentiation. Front Physiol (2018) 9:879. doi: 10.3389/fphys.2018.00879
22. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria Are Required for Antigen-Specific T Cell Activation Through Reactive Oxygen Species Signaling. Immunity (2013) 38(2):225–36. doi: 10.1016/j.immuni.2012.10.020
23. Schwindling C, Quintana A, Krause E, Hoth M. Mitochondria Positioning Controls Local Calcium Influx in T Cells. J Immunol (2010) 184(1):184–90. doi: 10.4049/jimmunol.0902872
24. Ross SH, Cantrell DA. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu Rev Immunol (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
25. Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, et al. Store-Operated Ca(2+) Entry Controls Clonal Expansion of T Cells Through Metabolic Reprogramming. Immunity (2017) 47(4):664–79.e6. doi: 10.1016/j.immuni.2017.09.003
26. Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-kappaB in the Nervous System. Biochim Biophys Acta (2005) 1745(3):287–99. doi: 10.1016/j.bbamcr.2005.05.009
27. Kamiński MM, Sauer SW, Klemke C-D, Süss D, Okun JG, Krammer PH, et al. Mitochondrial Reactive Oxygen Species Control T Cell Activation by Regulating IL-2 and IL-4 Expression: Mechanism of Ciprofloxacin-Mediated Immunosuppression. J Immunol (2010) 184(9):4827–41. doi: 10.4049/jimmunol.0901662
28. Smith-Garvin JE, Koretzky GA, Jordan MS. T Cell Activation. Annu Rev Immunol (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
29. Courtney AH, Lo WL, Weiss A. TCR Signaling: Mechanisms of Initiation and Propagation. Trends Biochem Sci (2018) 43(2):108–23. doi: 10.1016/j.tibs.2017.11.008
30. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp Ther Med (2020) 19(3):1997–2007. doi: 10.3892/etm.2020.8454
31. Rohrs JA, Siegler EL, Wang P, Finley SD. ERK Activation in CAR T Cells Is Amplified by CD28-Mediated Increase in CD3ζ Phosphorylation. iScience (2020) 23(4):101023. doi: 10.1016/j.isci.2020.101023
32. Boomer JS, Green JM. An Enigmatic Tail of CD28 Signaling. Cold Spring Harb Perspect Biol (2010) 2(8):a002436. doi: 10.1101/cshperspect.a002436
33. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J Immunol (2008) 180(7):4476–86. doi: 10.4049/jimmunol.180.7.4476
34. Martins CP, Piganelli JD. Targeting T Cell Metabolism to Combat Autoimmunity: Implications for the Future of Type 1 Diabetes Therapeutics. Immunometabolism (2020) 2(2):e200010. doi: 10.20900/immunometab20200010
35. Wei X, Zhang Y, Li C, Ai K, Li K, Li H, et al. The Evolutionarily Conserved MAPK/Erk Signaling Promotes Ancestral T-Cell Immunity in Fish via C-Myc-Mediated Glycolysis. J Biol Chem (2020) 295(10):3000–16. doi: 10.1074/jbc.RA119.012231
36. Adachi K, Davis MM. T-Cell Receptor Ligation Induces Distinct Signaling Pathways in Naive vs. Antigen-Experienced T Cells. Proc Natl Acad Sci USA (2011) 108(4):1549–54. doi: 10.1073/pnas.1017340108
37. Han J, Zhang L, Guo H, Wysham WZ, Roque DR, Willson AK, et al. Glucose Promotes Cell Proliferation, Glucose Uptake and Invasion in Endometrial Cancer Cells via AMPK/mTOR/S6 and MAPK Signaling. Gynecol Oncol (2015) 138(3):668–75. doi: 10.1016/j.ygyno.2015.06.036
38. Wu LX, La Rose J, Chen L, Neale C, Mak T, Okkenhaug K, et al. CD28 Regulates the Translation of Bcl-xL via the Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Pathway. J Immunol (2005) 174(1):180–94. doi: 10.4049/jimmunol.174.1.180
39. Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB. Bcl-X(L) Affects Ca(2+) Homeostasis by Altering Expression of Inositol 1,4,5-Trisphosphate Receptors. Proc Natl Acad Sci USA (2002) 99(15):9830–5. doi: 10.1073/pnas.152571899
40. Leonard WJ, Lin JX. Cytokine Receptor Signaling Pathways. J Allergy Clin Immunol (2000) 105(5):877–88. doi: 10.1067/mai.2000.106899
41. Surh CD, Sprent J. Homeostasis of Naive and Memory T Cells. Immunity (2008) 29(6):848–62. doi: 10.1016/j.immuni.2008.11.002
42. Li WQ, Jiang Q, Khaled AR, Keller JR, Durum SK. Interleukin-7 Inactivates the Pro-Apoptotic Protein Bad Promoting T Cell Survival. J Biol Chem (2004) 279(28):29160–6. doi: 10.1074/jbc.M401656200
43. Moudgil KD, Choubey D. Cytokines in Autoimmunity: Role in Induction, Regulation, and Treatment. J Interferon Cytokine Res (2011) 31(10):695–703. doi: 10.1089/jir.2011.0065
44. Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, et al. Superoxide Production by Macrophages and T Cells Is Critical for the Induction of Autoreactivity and Type 1 Diabetes. Diabetes (2011) 60(8):2144–51. doi: 10.2337/db10-1222
45. Tse HM, Milton MJ, Piganelli JD. Mechanistic Analysis of the Immunomodulatory Effects of a Catalytic Antioxidant on Antigen-Presenting Cells: Implication for Their Use in Targeting Oxidation-Reduction Reactions in Innate Immunity. Free Radic Biol Med (2004) 36(2):233–47. doi: 10.1016/j.freeradbiomed.2003.10.029
46. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res (2018) 122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401
47. Turrens JF. Mitochondrial Formation of Reactive Oxygen Species. J Physiol (2003) 552(Pt 2):335–44. doi: 10.1113/jphysiol.2003.049478
48. Hayyan M, Hashim MA, AlNashef IM. Superoxide Ion: Generation and Chemical Implications. Chem Rev (2016) 116(5):3029–85. doi: 10.1021/acs.chemrev.5b00407
49. Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. Oxidative Stress, Aging, and Diseases. Clin Interv Aging (2018) 13:757–72. doi: 10.2147/CIA.S158513
50. Sheu SS, Nauduri D, Anders MW. Targeting Antioxidants to Mitochondria: A New Therapeutic Direction. Biochim Biophys Acta (2006) 1762(2):256–65. doi: 10.1016/j.bbadis.2005.10.007
51. Shi L, Tu BP. Acetyl-CoA and the Regulation of Metabolism: Mechanisms and Consequences. Curr Opin Cell Biol (2015) 33:125–31. doi: 10.1016/j.ceb.2015.02.003
52. Cecchini G. Function and Structure of Complex II of the Respiratory Chain. Annu Rev Biochem (2003) 72(1):77–109. doi: 10.1146/annurev.biochem.72.121801.161700
53. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int J Mol Med (2019) 44(1):3–15. doi: 10.3892/ijmm.2019.4188
54. Holley AK, Bakthavatchalu V, Velez-Roman JM, St Clair DK. Manganese Superoxide Dismutase: Guardian of the Powerhouse. Int J Mol Sci (2011) 12(10):7114–62. doi: 10.3390/ijms12107114
55. Gough DR, Cotter TG. Hydrogen Peroxide: A Jekyll and Hyde Signalling Molecule. Cell Death Dis (2011) 2(10):e213. doi: 10.1038/cddis.2011.96
56. Veal EA, Day AM, Morgan BA. Hydrogen Peroxide Sensing and Signaling. Mol Cell (2007) 26(1):1–14. doi: 10.1016/j.molcel.2007.03.016
57. Redza-Dutordoir M, Averill-Bates DA. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim Biophys Acta (2016) 1863(12):2977–92. doi: 10.1016/j.bbamcr.2016.09.012
58. Halestrap AP. What Is the Mitochondrial Permeability Transition Pore? J Mol Cell Cardiol (2009) 46(6):821–31. doi: 10.1016/j.yjmcc.2009.02.021
59. Blanco LP, Pedersen HL, Wang X, Lightfoot YL, Seto N, Carmona-Rivera C, et al. Improved Mitochondrial Metabolism and Reduced Inflammation Following Attenuation of Murine Lupus With Coenzyme Q10 Analog Idebenone. Arthritis Rheumatol (2020) 72(3):454–64. doi: 10.1002/art.41128
60. Chen J, Stimpson SE, Fernandez-Bueno GA, Mathews CE. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid Redox Signal (2018) 29(14):1361–72. doi: 10.1089/ars.2017.7346
61. Panga V, Kallor AA, Nair A, Harshan S, Raghunathan S. Mitochondrial Dysfunction in Rheumatoid Arthritis: A Comprehensive Analysis by Integrating Gene Expression, Protein-Protein Interactions and Gene Ontology Data. PloS One (2019) 14(11):e0224632. doi: 10.1371/journal.pone.0224632
62. Morgan D, Oliveira-Emilio HR, Keane D, Hirata AE, Santos da Rocha M, Bordin S, et al. Glucose, Palmitate and Pro-Inflammatory Cytokines Modulate Production and Activity of a Phagocyte-Like NADPH Oxidase in Rat Pancreatic Islets and a Clonal Beta Cell Line. Diabetologia (2007) 50(2):359–69. doi: 10.1007/s00125-006-0462-6
63. Goldstein A, Falk MJ. Mitochondrial DNA Deletion Syndromes. In: GeneReviews(®). Seattle, WA: University of Washington, Seattle (1993). Copyright © 1993-2021, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.
64. Pitceathly RD, Rahman S, Hanna MG. Single Deletions in Mitochondrial DNA–molecular Mechanisms and Disease Phenotypes in Clinical Practice. Neuromuscul Disord (2012) 22(7):577–86. doi: 10.1016/j.nmd.2012.03.009
65. Yu X, Koczan D, Sulonen AM, Akkad DA, Kroner A, Comabella M, et al. mtDNA Nt13708a Variant Increases the Risk of Multiple Sclerosis. PloS One (2008) 3(2):e1530. doi: 10.1371/journal.pone.0001530
66. Jönsen A, Yu X, Truedsson L, Nived O, Sturfelt G, Ibrahim S, et al. Mitochondrial DNA Polymorphisms Are Associated With Susceptibility and Phenotype of Systemic Lupus Erythematosus. Lupus (2009) 18(4):309–12. doi: 10.1177/0961203308097477
67. Gusdon AM, Votyakova TV, Mathews CE. Mt-Nd2a Suppresses Reactive Oxygen Species Production by Mitochondrial Complexes I and III. J Biol Chem (2008) 283(16):10690–7. doi: 10.1074/jbc.M708801200
68. Dumitru C, Kabat AM, Maloy KJ. Metabolic Adaptations of CD4(+) T Cells in Inflammatory Disease. Front Immunol (2018) 9:540. doi: 10.3389/fimmu.2018.00540
69. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic Glycolysis Promotes T Helper 1 Cell Differentiation Through an Epigenetic Mechanism. Science (2016) 354(6311):481–4. doi: 10.1126/science.aaf6284
70. Smeltz RB, Chen J, Ehrhardt R, Shevach EM. Role of IFN-Gamma in Th1 Differentiation: IFN-Gamma Regulates IL-18R Alpha Expression by Preventing the Negative Effects of IL-4 and by Inducing/Maintaining IL-12 Receptor Beta 2 Expression. J Immunol (2002) 168(12):6165–72. doi: 10.4049/jimmunol.168.12.6165
71. Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T Cell Responses: Naive to Memory and Everything in Between. Adv Physiol Educ (2013) 37(4):273–83. doi: 10.1152/advan.00066.2013
72. Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Bürgel T. Normoxic Induction of the Hypoxia-Inducible Factor 1alpha by Insulin and Interleukin-1beta Involves the Phosphatidylinositol 3-Kinase Pathway. FEBS Lett (2002) 512(1-3):157–62. doi: 10.1016/S0014-5793(02)02247-0
73. Storozhevykh TP, Senilova YE, Persiyantseva NA, Pinelis VG, Pomytkin IA. Mitochondrial Respiratory Chain Is Involved in Insulin-Stimulated Hydrogen Peroxide Production and Plays an Integral Role in Insulin Receptor Autophosphorylation in Neurons. BMC Neurosci (2007) 8:84. doi: 10.1186/1471-2202-8-84
74. Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 Signaling Is Required for Th1 Development. J Immunol (2004) 172(1):61–9. doi: 10.4049/jimmunol.172.1.61
75. Eriksson U, Kurrer MO, Sebald W, Brombacher F, Kopf M. Dual Role of the IL-12/IFN-Gamma Axis in the Development of Autoimmune Myocarditis: Induction by IL-12 and Protection by IFN-Gamma. J Immunol (2001) 167(9):5464–9. doi: 10.4049/jimmunol.167.9.5464
76. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
77. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The Glucose Transporter Glut1 Is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab (2014) 20(1):61–72. doi: 10.1016/j.cmet.2014.05.004
78. Walker LS, von Herrath M. CD4 T Cell Differentiation in Type 1 Diabetes. Clin Exp Immunol (2016) 183(1):16–29. doi: 10.1111/cei.12672
79. Marrero I, Vong A, Dai Y, Davies JD. T Cell Populations in the Pancreatic Lymph Node Naturally and Consistently Expand and Contract in NOD Mice as Disease Progresses. Mol Immunol (2012) 52(1):9–18. doi: 10.1016/j.molimm.2012.04.004
80. Yoon JW, Jun HS. Autoimmune Destruction of Pancreatic Beta Cells. Am J Ther (2005) 12(6):580–91. doi: 10.1097/01.mjt.0000178767.67857.63
81. Chang I, Cho N, Kim S, Kim JY, Kim E, Woo JE, et al. Role of Calcium in Pancreatic Islet Cell Death by IFN-Gamma/TNF-Alpha. J Immunol (2004) 172(11):7008–14. doi: 10.4049/jimmunol.172.11.7008
82. Thomas HE, Darwiche R, Corbett JA, Kay TW. Interleukin-1 Plus Gamma-Interferon-Induced Pancreatic Beta-Cell Dysfunction Is Mediated by Beta-Cell Nitric Oxide Production. Diabetes (2002) 51(2):311–6. doi: 10.2337/diabetes.51.2.311
83. Burrack AL, Martinov T, Fife BT. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front Endocrinol (Lausanne) (2017) 8:343. doi: 10.3389/fendo.2017.00343
84. De Meyts P. The Insulin Receptor and Its Signal Transduction Network. In: Endotext. South Dartmouth (MA: MDText.com (2000). Inc.Copyright © 2000-2021, MDText.com, Inc.
85. Pomytkin IA. H2O2 Signalling Pathway: A Possible Bridge Between Insulin Receptor and Mitochondria. Curr Neuropharmacol (2012) 10(4):311–20. doi: 10.2174/157015912804499492
86. Baroja-Mazo A, Revilla-Nuin B, Ramírez P, Pons JA. Immunosuppressive Potency of Mechanistic Target of Rapamycin Inhibitors in Solid-Organ Transplantation. World J Transplant (2016) 6(1):183–92. doi: 10.5500/wjt.v6.i1.183
87. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
88. Heidker RM, Emerson MR, LeVine SM. Metabolic Pathways as Possible Therapeutic Targets for Progressive Multiple Sclerosis. Neural Regener Res (2017) 12(8):1262–7. doi: 10.4103/1673-5374.213542
89. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) Balance by Hypoxia-Inducible Factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
90. Cho SH, Raybuck AL, Blagih J, Kemboi E, Haase VH, Jones RG, et al. Hypoxia-Inducible Factors in CD4(+) T Cells Promote Metabolism, Switch Cytokine Secretion, and T Cell Help in Humoral Immunity. Proc Natl Acad Sci USA (2019) 116(18):8975–84. doi: 10.1073/pnas.1811702116
91. Liu XH, Kirschenbaum A, Lu M, Yao S, Dosoretz A, Holland JF, et al. Prostaglandin E2 Induces Hypoxia-Inducible Factor-1alpha Stabilization and Nuclear Localization in a Human Prostate Cancer Cell Line. J Biol Chem (2002) 277(51):50081–6. doi: 10.1074/jbc.M201095200
92. Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, et al. Prostaglandin E2 Regulates Th17 Cell Differentiation and Function Through Cyclic AMP and EP2/EP4 Receptor Signaling. J Exp Med (2009) 206(3):535–48. doi: 10.1084/jem.20082293
93. Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis (MS). Br J Pharmacol (2011) 164(4):1079–106. doi: 10.1111/j.1476-5381.2011.01302.x
94. Fu G, Xu Q, Qiu Y, Jin X, Xu T, Dong S, et al. Suppression of Th17 Cell Differentiation by Misshapen/NIK-Related Kinase MINK1. J Exp Med (2017) 214(5):1453–69. doi: 10.1084/jem.20161120
95. Tsai S, Clemente-Casares X, Zhou AC, Lei H, Ahn JJ, Chan YT, et al. Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity During Inflammation and Infection. Cell Metab (2018) 28(6):922–34.e4. doi: 10.1016/j.cmet.2018.08.003
96. Pouysségur J, Mechta-Grigoriou F. Redox Regulation of the Hypoxia-Inducible Factor. Biol Chem (2006) 387(10-11):1337–46. doi: 10.1515/BC.2006.167
97. Shin B, Benavides GA, Geng J, Koralov SB, Hu H, Darley-Usmar VM, et al. Mitochondrial Oxidative Phosphorylation Regulates the Fate Decision Between Pathogenic Th17 and Regulatory T Cells. Cell Rep (2020) 30(6):1898–909.e4. doi: 10.1016/j.celrep.2020.01.022
98. Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, et al. The AP-1 Transcription Factor Batf Controls T(H)17 Differentiation. Nature (2009) 460(7253):405–9. doi: 10.1038/nature08114
99. Long SA, Buckner JH. CD4+FOXP3+ T Regulatory Cells in Human Autoimmunity: More Than a Numbers Game. J Immunol (2011) 187(5):2061–6. doi: 10.4049/jimmunol.1003224
100. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
101. Wyss L, Stadinski BD, King CG, Schallenberg S, McCarthy NI, Lee JY, et al. Affinity for Self Antigen Selects Treg Cells With Distinct Functional Properties. Nat Immunol (2016) 17(9):1093–101. doi: 10.1038/ni.3522
102. Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, et al. Antiapoptotic Mcl-1 Is Critical for the Survival and Niche-Filling Capacity of Foxp3⁺ Regulatory T Cells. Nat Immunol (2013) 14(9):959–65. doi: 10.1038/ni.2649
103. Kang J, Chong SJ, Ooi VZ, Vali S, Kumar A, Kapoor S, et al. Overexpression of Bcl-2 Induces STAT-3 Activation via an Increase in Mitochondrial Superoxide. Oncotarget (2015) 6(33):34191–205. doi: 10.18632/oncotarget.5763
104. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martínez-Reyes I, et al. Mitochondrial Complex III Is Essential for Suppressive Function of Regulatory T Cells. Nature (2019) 565(7740):495–9. doi: 10.1038/s41586-018-0846-z
105. Bleier L, Dröse S. Superoxide Generation by Complex III: From Mechanistic Rationales to Functional Consequences. Biochim Biophys Acta (2013) 1827(11-12):1320–31. doi: 10.1016/j.bbabio.2012.12.002
106. Glund S, Deshmukh A, Long YC, Moller T, Koistinen HA, Caidahl K, et al. Interleukin-6 Directly Increases Glucose Metabolism in Resting Human Skeletal Muscle. Diabetes (2007) 56(6):1630–7. doi: 10.2337/db06-1733
107. Bhaumik S, Basu R. Cellular and Molecular Dynamics of Th17 Differentiation and Its Developmental Plasticity in the Intestinal Immune Response. Front Immunol (2017) 8:254. doi: 10.3389/fimmu.2017.00254
108. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-Beta-Induced Foxp3 Inhibits T(H)17 Cell Differentiation by Antagonizing RORgammat Function. Nature (2008) 453(7192):236–40. doi: 10.1038/nature06878
109. Ghelani A, Bates D, Conner K, Wu MZ, Lu J, Hu YL, et al. Defining the Threshold IL-2 Signal Required for Induction of Selective Treg Cell Responses Using Engineered IL-2 Muteins. Front Immunol (2020) 11:1106. doi: 10.3389/fimmu.2020.01106
110. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 Signaling via STAT5 Constrains T Helper 17 Cell Generation. Immunity (2007) 26(3):371–81. doi: 10.1016/j.immuni.2007.02.009
111. Lieberman LA, Tsokos GC. The IL-2 Defect in Systemic Lupus Erythematosus Disease has an Expansive Effect on Host Immunity. J BioMed Biotechnol (2010) 2010:740619. doi: 10.1155/2010/740619
112. Barbi J, Pardoll DM, Pan F. Ubiquitin-Dependent Regulation of Foxp3 and Treg Function. Immunol Rev (2015) 266(1):27–45. doi: 10.1111/imr.12312
113. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
114. Stark JM, Tibbitt CA, Coquet JM. The Metabolic Requirements of Th2 Cell Differentiation. Front Immunol (2019) 10:2318. doi: 10.3389/fimmu.2019.02318
115. Zhang Y, Zhang Y, Gu W, Sun B. TH1/TH2 Cell Differentiation and Molecular Signals. Adv Exp Med Biol (2014) 841:15–44. doi: 10.1007/978-94-017-9487-9_2
116. Henriksson J, Chen X, Gomes T, Ullah U, Meyer KB, Miragaia R, et al. Genome-Wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk Between Activation and Differentiation. Cell (2019) 176(4):882–96.e18. doi: 10.1016/j.cell.2018.11.044
117. Walker JA, McKenzie ANJ. T(H)2 Cell Development and Function. Nat Rev Immunol (2018) 18(2):121–33. doi: 10.1038/nri.2017.118
118. Joosten LA, Lubberts E, Helsen MM, Saxne T, Coenen-de Roo CJ, Heinegård D, et al. Protection Against Cartilage and Bone Destruction by Systemic Interleukin-4 Treatment in Established Murine Type II Collagen-Induced Arthritis. Arthritis Res (1999) 1(1):81–91. doi: 10.1186/ar14
119. Lin MS, Tse HM, Delmastro MM, Bertera S, Wong CT, Lakomy R, et al. A Multivalent Vaccine for Type 1 Diabetes Skews T Cell Subsets to Th2 Phenotype in NOD Mice. Immunol Res (2011) 50(2-3):213–20. doi: 10.1007/s12026-011-8215-0
120. Rockwell CE, Zhang M, Fields PE, Klaassen CD. Th2 Skewing by Activation of Nrf2 in CD4(+) T Cells. J Immunol (2012) 188(4):1630–7. doi: 10.4049/jimmunol.1101712
121. Caramori G, Groneberg D, Ito K, Casolari P, Adcock IM, Papi A. New Drugs Targeting Th2 Lymphocytes in Asthma. J Occup Med Toxicol (2008) 3 Suppl 1(Suppl 1):S6. doi: 10.1186/1745-6673-3-S1-S6
122. Akdis CA, Akdis M. Mechanisms of Allergen-Specific Immunotherapy and Immune Tolerance to Allergens. World Allergy Organ J (2015) 8(1):17. doi: 10.1186/s40413-015-0063-2
123. Zhang N, Bevan MJ. CD8(+) T Cells: Foot Soldiers of the Immune System. Immunity (2011) 35(2):161–8. doi: 10.1016/j.immuni.2011.07.010
124. Sklavos MM, Tse HM, Piganelli JD. Redox Modulation Inhibits CD8 T Cell Effector Function. Free Radic Biol Med (2008) 45(10):1477–86. doi: 10.1016/j.freeradbiomed.2008.08.023
125. Seder RA, Ahmed R. Similarities and Differences in CD4+ and CD8+ Effector and Memory T Cell Generation. Nat Immunol (2003) 4(9):835–42. doi: 10.1038/ni969
126. Buck MD, O’Sullivan D, Pearce EL. T Cell Metabolism Drives Immunity. J Exp Med (2015) 212(9):1345–60. doi: 10.1084/jem.20151159
127. Yao CH, Wang R, Wang Y, Kung CP, Weber JD, Patti GJ. Mitochondrial Fusion Supports Increased Oxidative Phosphorylation During Cell Proliferation. Elife (2019) 8:e41351. doi: 10.7554/eLife.41351
128. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial Dynamics Controls T Cell Fate Through Metabolic Programming. Cell (2016) 166(1):63–76. doi: 10.1016/j.cell.2016.05.035
129. Zhang L, Romero P. Metabolic Control of CD8(+) T Cell Fate Decisions and Antitumor Immunity. Trends Mol Med (2018) 24(1):30–48. doi: 10.1016/j.molmed.2017.11.005
130. Phan AT, Doedens AL, Palazon A, Tyrakis PA, Cheung KP, Johnson RS, et al. Constitutive Glycolytic Metabolism Supports CD8(+) T Cell Effector Memory Differentiation During Viral Infection. Immunity (2016) 45(5):1024–37. doi: 10.1016/j.immuni.2016.10.017
131. Araki K, Youngblood B, Ahmed R. The Role of mTOR in Memory CD8 T-Cell Differentiation. Immunol Rev (2010) 235(1):234–43. doi: 10.1111/j.0105-2896.2010.00898.x
132. Herzig S, Shaw RJ. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat Rev Mol Cell Biol (2018) 19(2):121–35. doi: 10.1038/nrm.2017.95
133. van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 Memory T Cells Have a Bioenergetic Advantage That Underlies Their Rapid Recall Ability. Proc Natl Acad Sci USA (2013) 110(35):14336–41. doi: 10.1073/pnas.1221740110
134. Tsai S, Shameli A, Santamaria P. CD8+ T Cells in Type 1 Diabetes. Adv Immunol (2008) 100:79–124. doi: 10.1016/S0065-2776(08)00804-3
135. Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 Enhances Expansion, Effector Function, Tissue Localization, and Memory Response of Antigen-Specific CD8 T Cells. J Exp Med (2013) 210(3):491–502. doi: 10.1084/jem.20122006
136. Garyu JW, Uduman M, Stewart A, Rui J, Deng S, Shenson J, et al. Characterization of Diabetogenic CD8+ T Cells: Immune Therapy With Metabolic Blockade. J Biol Chem (2016) 291(21):11230–40. doi: 10.1074/jbc.M115.713362
137. Huseby ES, Huseby PG, Shah S, Smith R, Stadinski BD. Pathogenic CD8 T Cells in Multiple Sclerosis and Its Experimental Models. Front Immunol (2012) 3:64. doi: 10.3389/fimmu.2012.00064
138. Salou M, Nicol B, Garcia A, Laplaud DA. Involvement of CD8(+) T Cells in Multiple Sclerosis. Front Immunol (2015) 6:604. doi: 10.3389/fimmu.2015.00604
139. Albanese M, Zagaglia S, Landi D, Boffa L, Nicoletti CG, Marciani MG, et al. Cerebrospinal Fluid Lactate Is Associated With Multiple Sclerosis Disease Progression. J Neuroinflamm (2016) 13:36. doi: 10.1186/s12974-016-0502-1
140. Duan K, Liu ZJ, Hu SQ, Huo HY, Xu ZR, Ruan JF, et al. Lactic Acid Induces Lactate Transport and Glycolysis/OXPHOS Interconversion in Glioblastoma. Biochem Biophys Res Commun (2018) 503(2):888–94. doi: 10.1016/j.bbrc.2018.06.092
141. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
142. Buettner GR. Superoxide Dismutase in Redox Biology: The Roles of Superoxide and Hydrogen Peroxide. Anticancer Agents Med Chem (2011) 11(4):341–6. doi: 10.2174/187152011795677544
143. Forman HJ, Zhang H, Rinna A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol Aspects Med (2009) 30(1-2):1–12. doi: 10.1016/j.mam.2008.08.006
144. Angajala A, Lim S, Phillips JB, Kim JH, Yates C, You Z, et al. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front Immunol (2018) 9:1605. doi: 10.3389/fimmu.2018.01605
145. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity (2017) 46(6):1089–90. doi: 10.1016/j.immuni.2017.06.009
146. Yarosz EL, Chang CH. The Role of Reactive Oxygen Species in Regulating T Cell-Mediated Immunity and Disease. Immune Netw (2018) 18(1):e14. doi: 10.4110/in.2018.18.e14
147. Castro-Correia C, Maia ML, Norberto S, Costa-Santos C, Barroso MF, Carvalho A, et al. Can Antioxidative Status Be Involved in Type 1 Diabetes? J Clin Med Res (2017) 9(12):998–1001. doi: 10.14740/jocmr3120w
148. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular Death, Reactive Oxygen Species (ROS) and Diabetic Complications. Cell Death Dis (2018) 9(2):119. doi: 10.1038/s41419-017-0135-z
149. Vilas-Boas EA, Nalbach L, Ampofo E, Lucena CF, Naudet L, Ortis F, et al. Transient NADPH Oxidase 2-Dependent H(2)O(2) Production Drives Early Palmitate-Induced Lipotoxicity in Pancreatic Islets. Free Radic Biol Med (2021) 162:1–13. doi: 10.1016/j.freeradbiomed.2020.11.023
150. Dworzański J, Strycharz-Dudziak M, Kliszczewska E, Kiełczykowska M, Dworzańska A, Drop B, et al. Glutathione Peroxidase (GPx) and Superoxide Dismutase (SOD) Activity in Patients With Diabetes Mellitus Type 2 Infected With Epstein-Barr Virus. PloS One (2020) 15(3):e0230374. doi: 10.1371/journal.pone.0230374
151. Likidlilid A, Patchanans N, Poldee S, Peerapatdit T. Glutathione and Glutathione Peroxidase in Type 1 Diabetic Patients. J Med Assoc Thai (2007) 90(9):1759–67. doi: 10.1016/S1567-5688(08)70953-X
152. de Haan JB. Nrf2 Activators as Attractive Therapeutics for Diabetic Nephropathy. Diabetes (2011) 60(11):2683–4. doi: 10.2337/db11-1072
153. van Meeteren ME, Teunissen CE, Dijkstra CD, van Tol EA. Antioxidants and Polyunsaturated Fatty Acids in Multiple Sclerosis. Eur J Clin Nutr (2005) 59(12):1347–61. doi: 10.1038/sj.ejcn.1602255
154. Carvalho AN, Lim JL, Nijland PG, Witte ME, Van Horssen J. Glutathione in Multiple Sclerosis: More Than Just an Antioxidant? Mult Scler (2014) 20(11):1425–31. doi: 10.1177/1352458514533400
155. Gopal S, Mikulskis A, Gold R, Fox RJ, Dawson KT, Amaravadi L. Evidence of Activation of the Nrf2 Pathway in Multiple Sclerosis Patients Treated With Delayed-Release Dimethyl Fumarate in the Phase 3 DEFINE and CONFIRM Studies. Mult Scler (2017) 23(14):1875–83. doi: 10.1177/1352458517690617
156. Yu DH, Yi JK, Yuh HS, Park S, Kim HJ, Bae KB, et al. Over-Expression of Extracellular Superoxide Dismutase in Mouse Synovial Tissue Attenuates the Inflammatory Arthritis. Exp Mol Med (2012) 44(9):529–35. doi: 10.3858/emm.2012.44.9.060
157. Karlsson K, Sandström J, Edlund A, Edlund T, Marklund SL. Pharmacokinetics of Extracellular-Superoxide Dismutase in the Vascular System. Free Radic Biol Med (1993) 14(2):185–90. doi: 10.1016/0891-5849(93)90009-j
158. Zaieni SH, Derakhshan Z, Sariri R. Alternations of Salivary Antioxidant Enzymes in Systemic Lupus Erythematosus. Lupus (2015) 24(13):1400–5. doi: 10.1177/0961203315593170
159. Coudriet GM, Delmastro-Greenwood MM, Previte DM, Marré ML, O’Connor EC, Novak EA, et al. Treatment With a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes. Antioxidants (Basel) (2017) 6(4):85. doi: 10.3390/antiox6040085
Keywords: autoimmunity, T cell, immunometabolism, mitochondria, reactive oxygen species
Citation: Chávez MD and Tse HM (2021) Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell-Mediated Autoimmune Diseases. Front. Immunol. 12:703972. doi: 10.3389/fimmu.2021.703972
Received: 01 May 2021; Accepted: 18 June 2021;
Published: 01 July 2021.
Edited by:
Michael Walch, Université de Fribourg, SwitzerlandReviewed by:
Marc S. Horwitz, University of British Columbia, CanadaCopyright © 2021 Chávez and Tse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hubert M. Tse, aHRzZUB1YWIuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.