
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 03 September 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.703574
This article is part of the Research Topic The Versatile Role of Nicotinamide Adenine Dinucleotide in Immunity View all 10 articles
 Natalie Baum1†
Natalie Baum1† Marie Eggers1†
Marie Eggers1† Julia Koenigsdorf1,2
Julia Koenigsdorf1,2 Stephan Menzel1,3
Stephan Menzel1,3 Julia Hambach1,2Tobias Staehler1
Julia Hambach1,2Tobias Staehler1 Ralf Fliegert4Frederike Kulow4Gerhard Adam2
Ralf Fliegert4Frederike Kulow4Gerhard Adam2 Friedrich Haag1
Friedrich Haag1 Peter Bannas2*‡
Peter Bannas2*‡ Friedrich Koch-Nolte1*‡
Friedrich Koch-Nolte1*‡CD38 is the major NAD+-hydrolyzing ecto-enzyme in most mammals. As a type II transmembrane protein, CD38 is also a promising target for the immunotherapy of multiple myeloma (MM). Nanobodies are single immunoglobulin variable domains from heavy chain antibodies that naturally occur in camelids. Using phage display technology, we isolated 13 mouse CD38-specific nanobodies from immunized llamas and produced these as recombinant chimeric mouse IgG2a heavy chain antibodies (hcAbs). Sequence analysis assigned these hcAbs to five distinct families that bind to three non-overlapping epitopes of CD38. Members of families 4 and 5 inhibit the GDPR-cyclase activity of CD38. Members of families 2, 4 and 5 effectively induce complement-dependent cytotoxicity against CD38-expressing tumor cell lines, while all families effectively induce antibody dependent cellular cytotoxicity. Our hcAbs present unique tools to assess cytotoxicity mechanisms of CD38-specific hcAbs in vivo against tumor cells and potential off-target effects on normal cells expressing CD38 in syngeneic mouse tumor models, i.e. in a fully immunocompetent background.
NAD+ is released as an endogenous danger signal from cells during inflammation (1, 2). CD38, a 43 kDa type II transmembrane protein consisting of a short intracellular N-terminal domain, a transmembrane helix and a long C-terminal extracellular catalytic domain, is the major NAD+-hydrolyzing ecto-enzyme of mammals (3–6). NAD+-hydrolysis by CD38 limits the availability of NAD+ for extracellular-ADP-ribosyltransferases (7, 8), and generates the Ca2+-mobilizing metabolites ADP-ribose and cyclic ADP-ribose (9) that can be further hydrolyzed to immunosuppressive adenosine by other ecto-enzymes (10). CD38 is highly expressed in hematological malignancies including multiple myeloma (11, 12). It has been proposed that the enzymatic activity of CD38 contributes to a microenvironment favourable for tumor survival in the bone marrow niche (13, 14).
CD38 represents a promising target for monoclonal antibody (mAb)-based immunotherapy of multiple myeloma (MM) (11, 15, 16). Several CD38-specific mAbs, including daratumumab and isatuximab, have shown encouraging results in the clinic (17–20). The anti-tumor effects of these mAbs presumably reflect their ability to induce immune effector functions, such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) (21). However, these antibodies may also induce the depletion of CD38-expressing NK cells and may have other off-target effects on normal cells expressing CD38 (22, 23). Moreover, the use of mAbs has disadvantages that include limited tissue penetration due to their large size of approximately 150 kD (24, 25).
Nanobodies are recombinant, single antigen-binding immunoglobulin variable domains (designated VHH) derived from naturally occurring camelid heavy chain antibodies (hcAbs) (26, 27). Nanobodies have several advantages over conventional antibodies, including a 10-fold smaller size (15 kDa vs. 150 kDa) (28, 29). To endow immune-effector functions, nanobodies can be fused to the hinge, CH2, and CH3 domains of a conventional mouse or human IgG antibody to generate nanobody-based chimeric hcAbs (30). These chimeric hcAbs lack the CH1 domain and the light chain, resulting in approximately half the molecular size of a conventional antibody (75 kDa vs. 150 kDa) (30).
Both, nanobodies and hcAbs are emerging as promising theranostic molecules (31–34). For example, we have recently shown that human CD38-specific hcAbs can be used to effectively target human MM cells in xenograft mouse models of systemic human lymphoma (35). Lack of reactivity with mouse CD38, however, makes it difficult to understand and assess potential off-target effects of such therapeutic antibodies on immune cells that endogenously express CD38. Substituting three amino acid residues in the CH2 domain of mouse IgG2a or human IgG1 (L234A, L235A, P329G) eliminates complement dependent cytotoxicity (CDC) as well as CD16-mediated antibody dependent cellular toxicity (ADCC) (36). These so-called LALA-PG mutants retain the thermostability and pharmacokinetics of the parental IgG (36).
We aimed to develop mouse CD38-specific nanobodies and hcAbs, to assess their binding epitopes, and to evaluate their capacity to induce cytotoxicity against tumor cells expressing CD38 in vitro as a basis for future in vivo studies of syngeneic MM models in immunocompetent mice.
BALB/c and C57BL/6 mice were obtained from The Jackson Laboratory or Charles River. Cd38-/- mice (3) were back-crossed onto the BALB/c and C57BL/6 backgrounds for 8 –12 generations. The mouse EL4 (C57BL76N lymphoma, ATCC TIB-39) and MOPC 315 (BALB/C myeloma, ATCC TIB-23) cell lines were obtained from the American Type Culture Collection. EL4 and MOPC 315 cells were cultured in RPMI-1640 medium (Gibco, Life Technologies, Paisley, UK) supplemented with 2 mM sodium pyruvate (Gibco), 2 mM L-glutamine (Gibco) and 10% (v/v) fetal calf serum (Gibco). Human HEK cells (ATCC CRL-1573) were transiently co-transfected with expression vectors for nuclear GFP and mouse CD38 (gene ID: 12494) or human CD38 (gene ID: 952) (37). The human NK-92 cell line (ACC 488) was obtained from the DSMZ German Collection of Microorganisms and Cell Cultures. NK-92 cells were stably transduced with mouse CD16 by retroviral transduction using the pSF91 retroviral vector (35). The sequence for CD16, i.e. the ectodomain of FcγRIII fused to the transmembrane and cytosolic domains of FcϵRI, was kindly provided by B. Clémenceau (Nantes, France). NK-92 cells were cultured in alpha MEM culture medium (Gibco) supplemented with 10% FCS (Gibco), 10% horse serum (Gibco), 100 IU/mL IL2 (Proleukin, Novartis) and 2 mM L-glutamine (Gibco). Primary spleen cells were obtained from wild type and Cd38-/- mice by passing spleen cell suspensions through a 70 µm cell strainer.
Two llamas were immunized subcutaneously by ballistic cDNA immunization with an expression vector encoding the full-length open reading frame of mouse CD38. The VHH repertoire was PCR-amplified from peripheral blood lymphocytes and cloned into the pHEN2 phagemid vector as described previously (37). Selection of specific phages was performed by sequential panning of the phage library on primary splenocytes obtained from Cd38-/- and WT mice. Following extensive washing, bound phages were eluted by trypsinization. Plasmid DNA was isolated from single colonies and subjected to sequence analyses using pHEN2-specific forward and reverse primers (37).
The coding region of selected nanobodies was subcloned using NcoI/PciI and NotI into the pCSE2.5 vector (38) (kindly provided by Thomas Schirrmann, Braunschweig, Germany) upstream of either a chimeric His6x-Myc epitope tag, the coding region for the hinge and Fc domains of mouse IgG2a, or the corresponding coding region for the LALA-PG mutant (36) of mouse IgG2a (gene ID: 404711). Recombinant myc-his tagged nanobodies and chimeric nanobody-mouse IgG2c heavy chain antibodies were produced in transiently transfected HEK-6E cells (39) (kindly provided by Ives Durocher, Ottowa, Canada) cultivated in serum-free medium. Six days post transfection, supernatants were harvested and cleared by centrifugation at 4000 rpm for 10 min. Nanobodies in cell supernatants were quantified by SDS-PAGE and Coomassie staining relative to marker proteins of known quantity as described previously (37). Yields typically ranged from 0.5–3 μg Nb or hcAb per 10 µl of HEK-6E cell supernatant. Myc-His tagged nanobodies were purified by immobilized metal affinity chromatography using Ni-NTA agarose (Sigma, St Louis, MO), hcAbs by affinity chromatography on protein A immobilized on sepharose beads (GE Healthcare) (37).
The extracellular domain of mouse CD38 (aa 45–304) with intact glycosylation sites was produced as a secretory protein with a chimeric His6x-Myc epitope tag in the pCSE2.5 vector. The tagged protein was purified using immobilized metal affinity chromatography (IMAC). Affinity of hcAbs to recombinant mouse CD38 was determined by BLI-technology using a fortéBIO BLItz instrument. Assays were performed at 20°C with running buffer (PBS, 0.01% (m/v) BSA, 0.002% (v/v) Tween-20). Protein A sensors were hydrated in running buffer and loaded until saturation with hcAbs at 10 µg/ml. After washing, purified mouse CD38 (1.8 µM) was allowed to associate for 120 seconds on immobilized hcAbs, followed by dissociation for 120 seconds. Respective binding curves were referenced against antibody-loaded sensors receiving only buffer for association and dissociation steps. Curve fitting and affinity calculations were performed using Graph Pad Prism (version 7) using non-linear regression and the build-in “association then dissociation” method.
Purified hcAbs were conjugated via amino groups to Alexa Fluor647-fluorochrome according to the manufacturer’s instructions (Molecular Probes, Thermo Fisher Scientific). For epitope mapping analyses, EL4 cells were pre-incubated with a saturating concentration (100 nM) of unconjugated hcAbs for 30 min at 4°C, followed by addition of Alexa Fluor647-conjugated hcAbs (10 nM) and further incubation for 20 min at 4°C. Cells were washed and analyzed by flow cytometry on a BD-FACS Canto. Data was analyzed using the FlowJo software (Treestar). The percentage of cross-blockade was calculated from mean fluorescence intensities (MFI) as follows: (MFI in the absence of competing Abs – MFI in the presence of competing Abs): (MFI in the presence of competing Abs) x 100. Spleen cells were pre-incubated with Fc-block (BioXcell, clone 2.4G2) to minimize unspecific binding to Fc-receptors. Cells were then incubated with Alexa Fluor647-conjugated hcAbs, FITC-conjugated anti-B220 (BD biosciences, clone RA3-6B2), and Alexa Fluor 750 as a viability dye (ThermoFischer). Gating was performed on Alexa Fluor 750-low cells (live cells).
EL4 cells (3 x 105 cells/well) were incubated at 37°C in the dark for 20 min with hcAbs (10 or 100 µg/ml) or araF-NAD (10 µM) before fluorescence measurements. After recording for 20 cycles, NGD+ (80 µM, Sigma, St Louis, MO) was added, followed by further incubation in the dark at 37°C. Production of cGDPR was monitored continuously for 50 min at 410 nm (emission wavelength) with the excitation wavelength set to 300 nm, using a Tecan Infinite M 200 microplate fluorimeter (37). Readings (EX300/EM410) from wells without cells were subtracted from all sample readings and values were plotted as Relative Fluorescence Units (RFU) vs. time. The rate of cGDPR production was calculated as the slope of the curves (RFU/s) during the linear phase of the reaction, i.e. t = 500-1200 s.
To analyze the complement-dependent cytotoxicity (CDC) mediated by hcAbs, EL4 or MOPC 315 cells were incubated for 10 min at 4°C with hcAbs before addition of guinea pig serum (25% v/v) as a source of complement. Cells were incubated for 120 minutes at 37°C, washed and resuspended in PBS/0.2% BSA/propidium iodide before analysis by flow cytometry (35).
To analyze antibody dependent cellular cytotoxicity (ADCC) mediated by NK-92 cells, EL4 or MOPC 315 cells were incubated for 10 min at 4°C with hcAbs before addition of NK92 cells. In order to distinguish NK92 effector cells from target cells, NK92 cells were prelabelled with eFluor 450 (ThermoFisher) for 20 min at 4°C and washed three times before addition of hcAb-treated target cells at an effector to target ratio [E:T] of 3:1. Cells were co-incubated for 3 hours at 37°C, washed, and resuspended in PBS/0.2% BSA/propidium iodide before flow cytometry (35). In order to quantify cytotoxicity against target cells, gating was performed on eFluor 450-negative cells. Dead target cells were quantified using uptake of propidium iodide (PI) and decrease in forward scatter (FCS) as indicators of cell lysis. As negative controls, hcAbs carrying the ADCC and CDC abrogating LALA-PG mutations (36) were used.
Data were analyzed using GraphPad Prism version 7.00 (GraphPad Software). For enzyme inhibition assays, statistical significance was calculated using one-way ANOVA followed by a Bonferroni post hoc test for multiple comparisons. with P< 0.05 (*), P < 0.01 (**), P < 0.001 (***), P < 0.0001 (****). Data for CDC and ADCC assays in bar diagrams represent the mean ± SD from three independent experiments.
We cloned the VHH-repertoire from blood lymphocytes of two llamas immunized (40) with a mouse CD38-encoding cDNA expression vector into M13 phage display libraries. Selection of CD38-specific phages was achieved by first panning the libraries on cells lacking CD38, i.e. splenocytes from CD38-deficient mice and YAC-1 lymphoma cells to remove unspecific binders. Libraries were then panned on cells expressing high levels of CD38, i.e. splenocytes from WT mice and EL4 thymoma cells. Sequencing of selected clones revealed 13 distinct clones (JK3, JK5, JK13, JK16, NB3, NB7, NB11, NB22, NB24, NB28, NB32, NB38, NB40 and NB42) that can be subgrouped into five distinct nanobody families based on sequence similarities in the framework and complementarity determining regions, with CDR3 lengths ranging from three to 13 amino acid residues (Table 1).
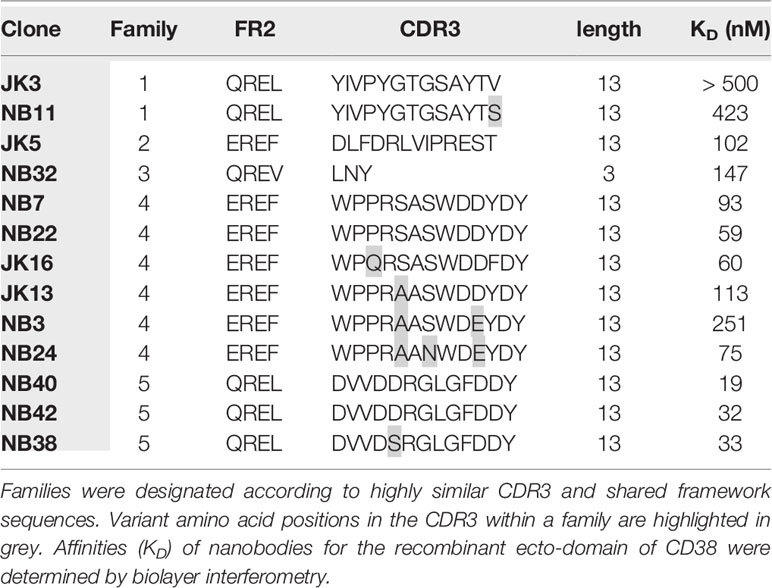
Table 1 Characteristics of mouse CD38-specific nanobodies.
In order to generate nanobody-based mouse heavy chain antibodies (hcAbs), the VHH-coding region was fused to the hinge, CH2 and CH3 domains of mouse IgG2a. These hcAbs were produced as secreted recombinant proteins in transiently transfected HEK cells grown in serum-free medium. Affinities of the hcAbs were determined by biolayer interferometry using the recombinant ecto-domain of mouse CD38 (Table 1). The results reveal moderate affinities in the two and three digit nanomolar range; family 1 nanobodies had the lowest affinities (> 400 nM), family 5 nanobodies the highest affinities (19-33 nM). In order to verify the specificity of the selected antibodies, we performed flow cytometry analyses of HEK cells co-transfected with GFP and either mouse CD38 or human CD38 (Figure 1A). The results show specific binding of the selected hcAbs to mouse CD38 but not to human CD38. Flow cytometry analyses of splenocytes obtained from wild type and from CD38-deficient mice confirmed the specific binding of all hcAbs to native mouse CD38 (Figure 1B).
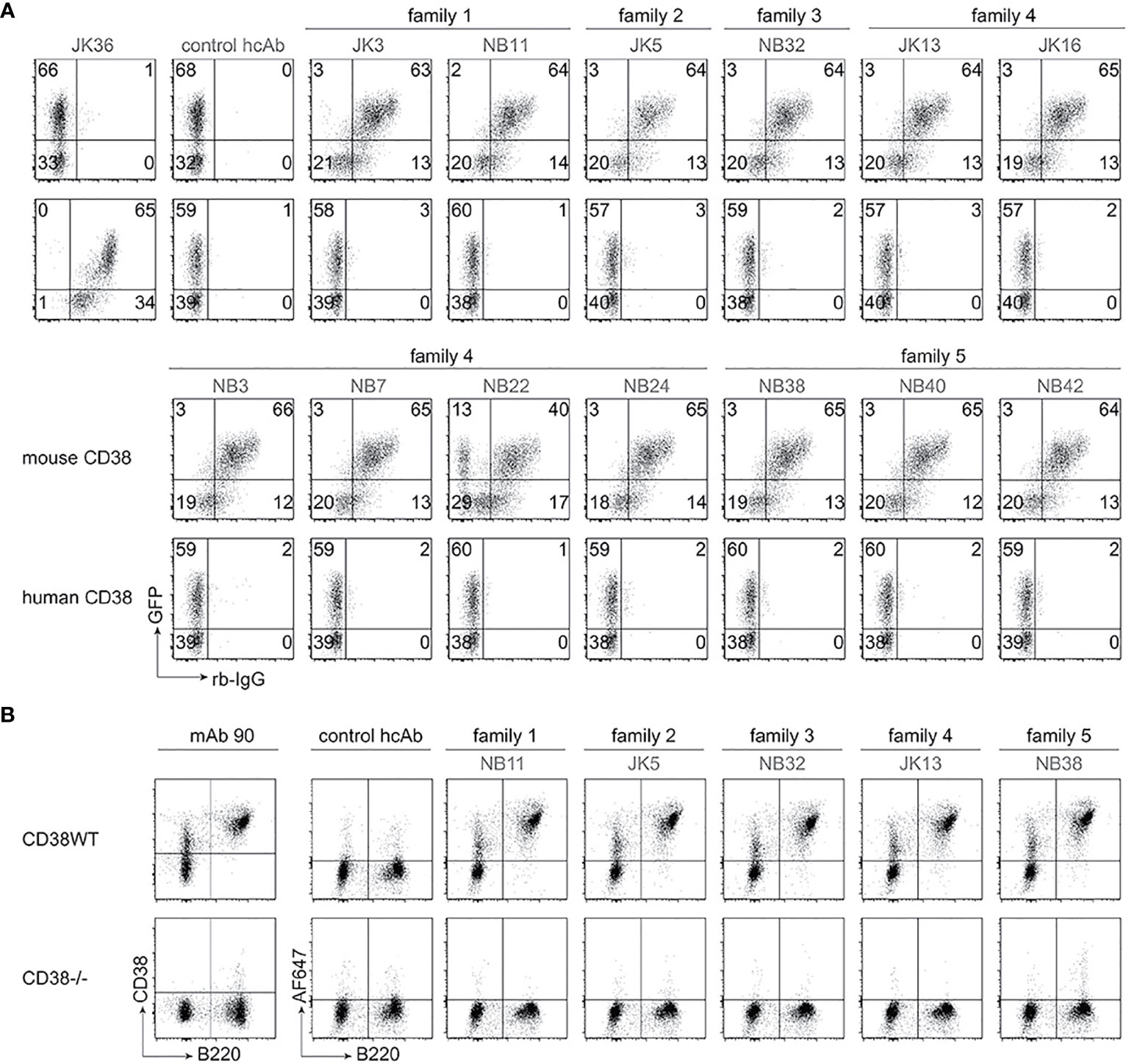
Figure 1 Selected heavy chain antibodies specifically recognize mouse CD38. (A) HEK cells were transiently co-transfected with expression constructs for GFP and either mouse CD38 (top rows) or human CD38 (bottom rows). Twenty-four hours after transfection, cells were incubated sequentially with selected nanobody-rabbit IgG hcAbs and APC-conjugated anti-rabbit IgG. Control stainings were performed with an isotype control hcAb and with human CD38-specific JK36-hcAb. Numbers indicate the percentage of cells in the respective quadrants. Data are representative of two independent experiments. (B) Splenocytes from wildtype (WT) and Cd38-/- mice were incubated with Alexa Fluor 647-conjugated hcAbs, a FITC-conjugated B220-specific mAb, and Alexa Fluor 750 as vitality dye. Control stainings were performed with an isotype control hcAb and a conventional mouse CD38-specific mAb (clone 90). Gating was performed on live (Alexa Fluor 750-low) cells. Data are representative of two independent experiments.
Next, we aimed to assess whether the identified anti-mouse CD38-specific hcAbs recognize overlapping or distinct epitopes on mouse CD38. To this end, we performed cross blockade flow cytometry analyses. We monitored the binding of Alexa Fluor647-conjugated hcAbs (JK3-hcAb, JK5-hcAb, JK13-hcAb, JK16-hcAb) to EL4 thymoma cells in the presence of excess unlabelled hcAbs (Table 2). The results show that the selected hcAbs fall into three distinct binding groups. Group 1 hcAbs block binding of hcAbs derived from nanobodies JK3 and NB11 (both family 1). These nanobodies recognize an overlapping epitope designated epitope 1. Group 2 hcAbs block binding of hcAbs derived from nanobodies JK5 (family 2) and NB32 (family 3). These nanobodies recognize an overlapping epitope designated epitope 2. Group 3 hcAbs block binding of hcAbs derived from nanobodies JK13, JK16, NB3, NB7, NB22, NB24 (family 4) and NB38, NB40, and NB42 (family 5). These nanobodies recognize an overlapping epitope designated epitope 3. Remarkably NB32-hcAb (epitope 2), which has the shortest CDR3, seemed to enhance binding of hcAbs that bind to the epitope 1 (JK3-hcAb) and epitope 3 (JK13-hcAb, JK16-hcAb). Note that JK13-hcAb and JK16-hcAb (both epitope 3) blocked binding of JK3-hcAb (epitope 1), suggesting that these hcAbs either sterically interfere with binding of JK3-hcAb or alter the conformation of mouse CD38 so as to inhibit binding of JK3-hcAb.
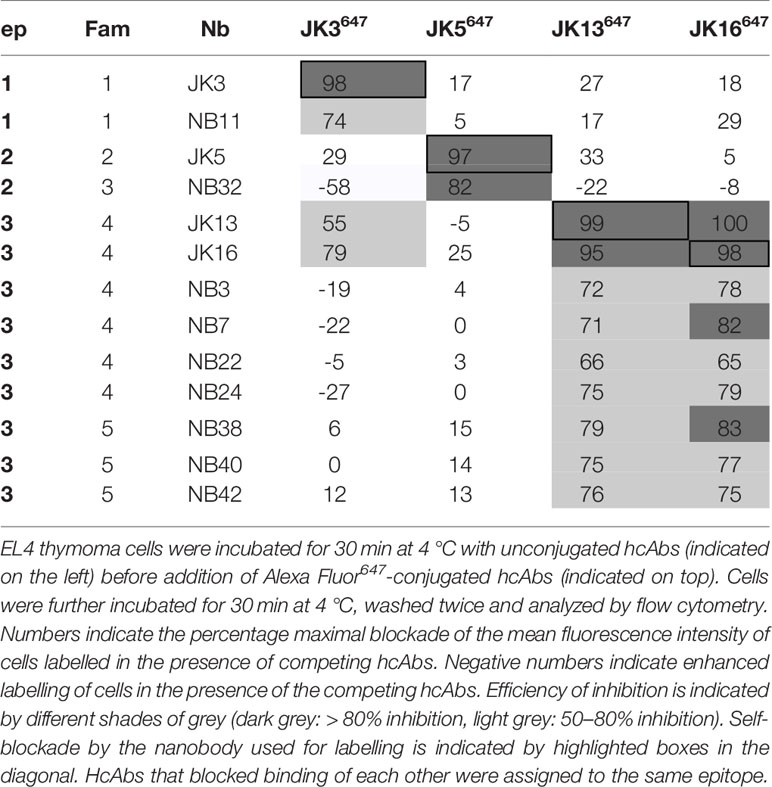
Table 2 Epitope mapping of nanobody-based mouse CD38-specific hcAbs.
To analyze the potential functional effects of the selected hcAbs on the enzyme activity of mouse CD38 on cells, we employed a fluorometric GDPR cyclase assay (37, 41–43). This assay uses NGD+ (which carries a guanine nucleobase instead of adenine) instead of NAD+ as substrate. We incubated CD38-positive EL4 cells with either CD38-specific hcAbs for 15 min before addition of NGD+ and monitored the increased fluorescence of the product cGDPR by fluorimetry (44) (Figure 2). The results showed a continuous increase of cGDPR during incubation of EL4 cells with NGD+ in the absence of antibodies. As a control, we used the NAD+-analogue nicotinamide 2-deoxy-2-fluoroarabinoside adenine dinucleotide (araF-NAD+), a highly specific inhibitor of mouse CD38 (7, 45). Addition of araF-NAD+ effectively abrogated the increase of cGDPR, indicating that the increased fluorescence is largely due to CD38 on the surface of EL4 cells. Addition of hcAb from family 1 (JK3-hcAb), family 2 (JK5-hcAb), and family 3 (NB32-hcAb) had little if any effect on the enzyme activity of EL4 cells (Figures 2A–C). Addition of family 4 hcAbs (hcAbs NB3, NB7, NB22, NB24) showed significant, but varying levels of enzyme inhibition (Figure 2D). The most potent inhibitory effect of the GDPR cyclase activity was observed for all tested hcAbs from family 5 (hcAbs NB38, NB40, NB42) (Figure 2E). Figure 2F allows for direct visual comparison of the inhibitory effects of all tested hcAbs and demonstrates the dose dependency of the hcAbs used.
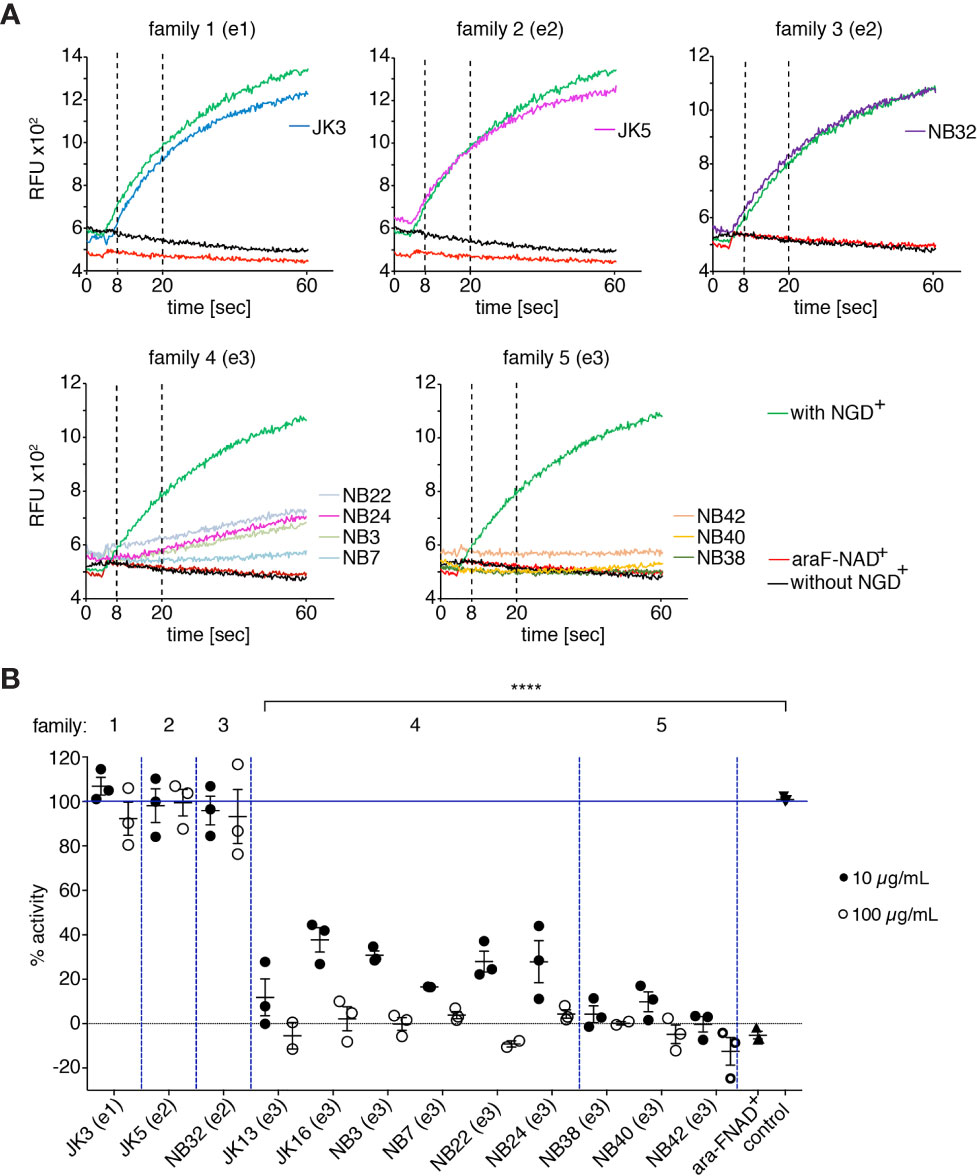
Figure 2 hcAbs of families 4 and 5 inhibit the GDPR-cyclase activity of mouse CD38. EL4 thymoma cells were incubated with the indicated hcAbs or 10 μM araF-NAD+ for 15 min at 37°C before fluorescence measurements with a microplate reader. After the first 20 cycles, 50 μM NGD+ was added and kinetic fluorescence reading (ex/em: 300/410 nm) was continued for 60 min. (A) Representative fluorimetry plots for cells incubated with the indicated hcAbs. Controls included cells incubated with (green) or without (black) NGD+, and cells incubated with NGD+ and araF-NAD+ (red). Vertical dotted lines at 8 and 20 min depict the time points used for calculation of slopes depicted in (B). (B) Each dot indicates the slope of the curve during the linear phase (t = 8-20 min), relative to the slope of the curve obtained from control cells incubated with NGD+ alone (n = 3). Statistical significance was calculated using one-way ANOVA followed by a Bonferroni post hoc test for multiple comparisons. ****p < 0.0001. Data are representative of three independent experiments.
To analyze the capacity of mouse CD38-specific hcAbs to induce ADCC, EL4 thymoma cells or MOPC 315 myeloma cells were used as target cells and NK-92 cells stably transfected with mouse Fc-receptor III (CD16) were used as effector cells. Flow cytometry analyses confirmed high expression of CD38 by EL4 and MOPC 315 cells (Figures 3 A, B, panel 1), while human NK-92 cells were negative for mouse CD38 (not shown). To monitor ADCC, EL4 and MOPC 315 cells were incubated with eFluor 450-labelled NK92 cells in the absence or presence of Nb-based mouse IgG2a hcAbs. To evaluate cell death, we monitored uptake of propidium iodide and decrease in forward scatter by flow cytometry (Figures 3 A, B, panels 2–4). The results show that all analyzed hcAbs mediated effective killing of EL4 and MOPC 315 target cells. In contrast, only background levels of cell death were observed in the presence of mouse IgG2a hcAbs carrying the three LALA-PG mutations that abrogate binding to Fc-receptors (36).
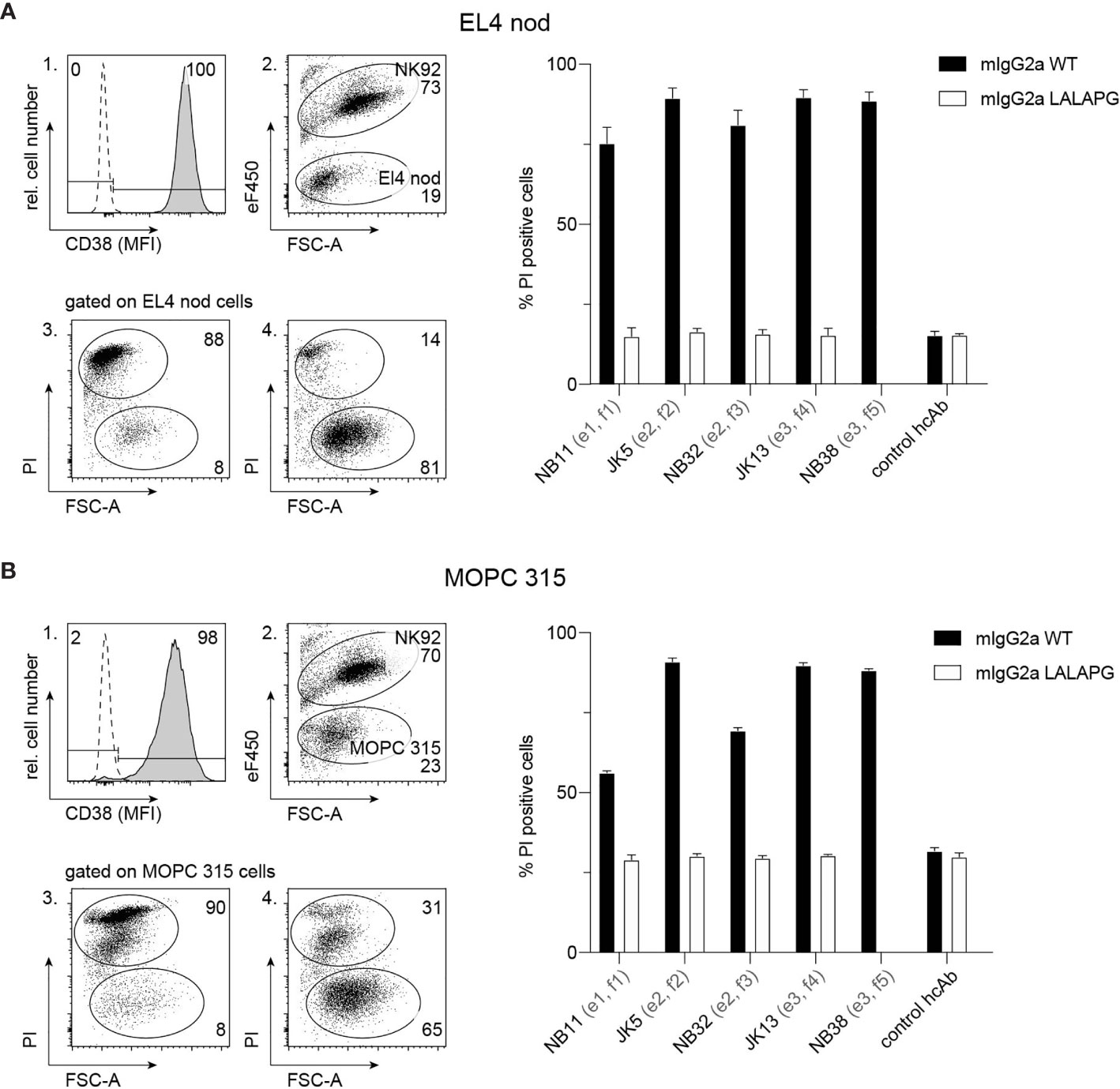
Figure 3 hcAbs of all five families effectively induce antibody-dependent cellular cytotoxicity against CD38-expressing thymoma and myeloma cell lines. Cell surface expression of CD38 by EL4 thymoma (A) and MOPC 315 myeloma (B) cells was assessed by flow cytometry using a fluorochrome-conjugated mouse CD38-specific mAb and an isotype control (open histogram) (panel 1). To assess the capacity of hcAbs to induce ADCC, EL4 cells (A) or MOPC 315 cells (B) were co-cultured with eFluor 450-labelled NK-92 cells for 3h at 37°C at an effector to target ratio of 3:1 in the presence of CD38-specific mouse IgG2a hcAbs from all five nanobody families. As controls, we used the same mouse IgG2a hcAbs carrying the LALA-PG mutation that abrogate FcR-binding and a non-binding control hcAb. Cells were then incubated with propidium iodide (PI) and analyzed by flow cytometry. Representative dot plots illustrate the clear separation of eFluor 450-postive effector cells and eFluor 450-negative target cells (panel 2). To assess cytotoxicity against target cells, gating was performed on eFluor 450-negative cells (panels 3, 4). Representative dot plots in panels 3 and 4 illustrate the clear distinction of dead target cells (PI-positive, low forward scatter/FSC-A) from live target cells (PI-negative, FSChigh). Panel 3 shows the results of cells incubated with JK5 hcAb, panel 4 shows the results of cells incubated with the isotype control hcAb. Numbers in panels 2-4 indicate the percentage of cells in the indicated gated populations. Epitopes and nanobody families are indicated in parentheses behind the nanobody names. Data in bar diagrams represent mean ± SD from three independent experiments.
To analyze the potential of hcAbs to induce CDC, we incubated EL4 and MOPC 315 cells with saturating amounts of mouse IgG2a hcAbs and guinea pig serum as a source of complement. As indicators of cell lysis, we monitored permeabilization of cells to the DNA-staining dye propidium iodide and decrease in forward scatter using flow cytometry (Figure 4). The results show that hcAbs JK5, JK13, and NB38 from families 2, 4 and 5 mediate effective CDC of EL4 and MOPC 315 target cells. In contrast, only background levels of cell death were observed when incubations were performed in the presence of the LALA-PG mutants of the corresponding hcAbs that abrogate binding of complement factor C1q (36).
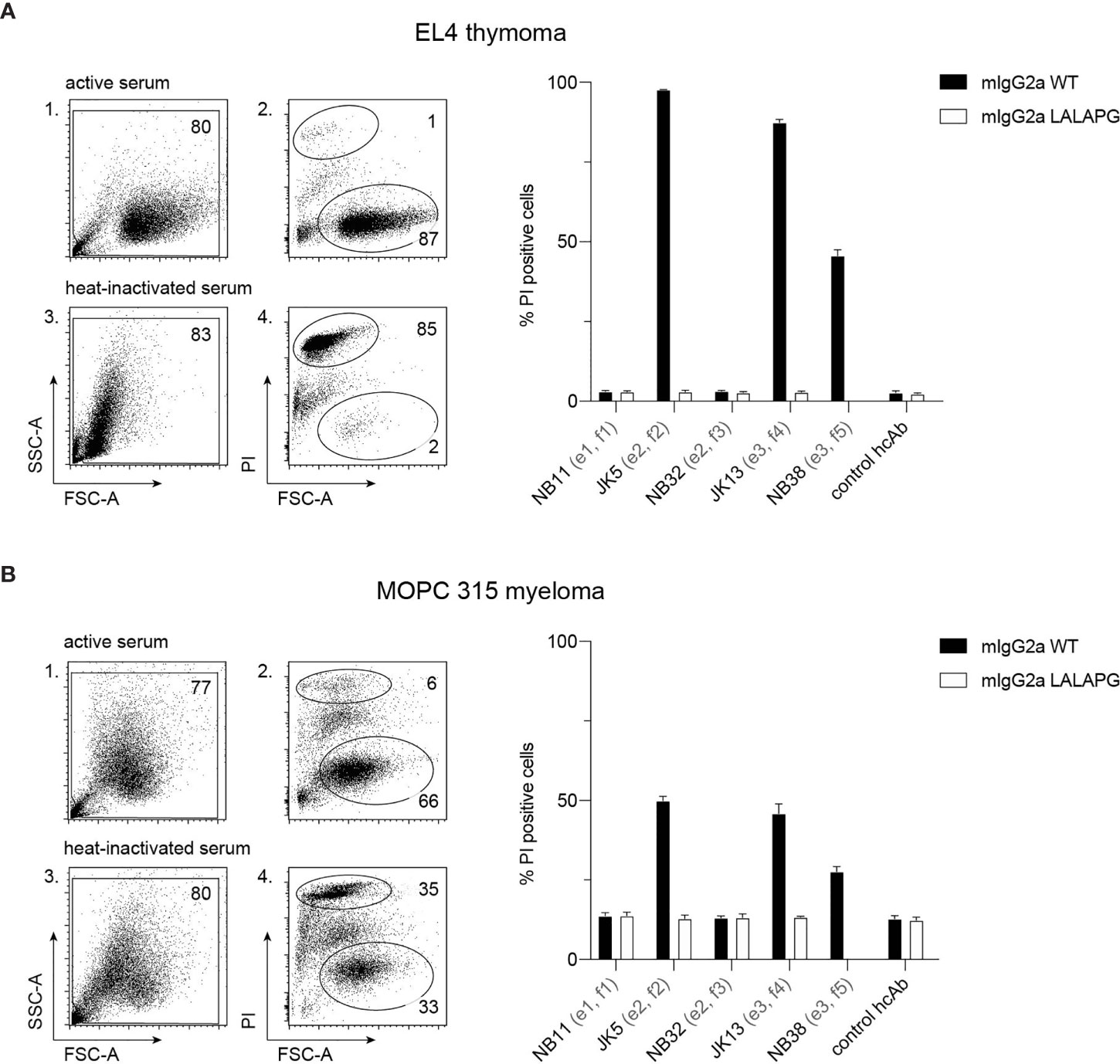
Figure 4 hcAbs of families 2, 4, and 5 mediate complement-dependent cytotoxicity of cells against CD38-expressing thymoma and myeloma cell lines. EL4 thymoma (A) and MOPC 315 myeloma (B) cells were incubated for 120 min at 37°C with the indicated CD38-specific mouse IgG2a hcAbs in the presence of 25% guinea pig serum as a source of complement. The same serum was pretreated for 10 min at 70°C to inactivate complement components and used as a control (heat-inactivated serum). As additional controls, we used LALA-PG mutant hcAbs (with abrogated C1q-binding) and a non-binding control hcAb. Cells were stained with propidium iodide and analyzed by flow cytometry to quantify the percentage of dead (PI-positive, FSClow) cells. Gating was performed to exclude cellular debris with very low FSC and low SSC. Numbers in panels 1-4 indicate the percentage of cells in the indicated gated populations. Data in the bar diagrams represent mean ± SD from three independent experiments.
From immunized llamas, we selected 13 mouse CD38-specific hcAbs that derive from five nanobody families, each of which carries a common framework region and a highly similar CDR3. These hcAbs bind specifically to three distinct epitopes of murine CD38.
All epitope 3-directed hcAbs (i.e., families 4 and 5) inhibited the GDPR-cyclase activity in a dose dependent fashion. In contrast, hcAbs of families 1, 2 and 3 had little if any effect on the GDPR-cyclase activity. This NGD+-based assay is commonly used to assess the effect of antibodies on CD38 enzyme activity (37, 43, 46). A limitation of this assay is that it only allows an estimate of the allosteric inhibitory effect of antibodies on the GDPR-cyclase, but not necessarily on the ADPR-cyclase or NAD-glycohydrolase activities of CD38. It is possible that nanobodies might affect cyclase and NADase activities differentially. Indeed, in a recent study we found that both, daratumumab and human CD38-specific hcAb 1067, inhibited the GDPR-cyclase and ADPR-cyclase activities of human CD38 (47), while neither daratumumab nor hcAb 1067 had any detectable effect on the NADase activity of CD38. It has been proposed that CD38 contributes to shaping an immunosuppressive tumor microenvironment (TME) by fuelling the conversion of NAD+ to immunosuppressive adenosine (14, 48–50). Since inhibition of the NADase activity of CD38 is more relevant in this context than inhibition of its cyclase activities, there remains a need for better CD38-inhibitory antibodies.
The CD38-specific hcAbs from all five families, irrespective of their binding epitopes induced potent ADCC of murine cell lines. This in line with the results of our previous studies showing that human CD38-specific nanobody-based hcAbs potently induced ADCC of several human lymphoma and myeloma cell lines, including LP-1 myeloma, CA-46 and Daudi Burkitt lymphoma (35, 51).
Members of hcAbs families 2, 4 and 5, but not of families 1 and 3 also mediated CDC. These findings differ from those of our previous studies with hcAbs directed against human CD38, in which we found that the 22 nanobody-based hcAbs directed against 3 different epitopes of human CD38 showed little if any capacity to induce CDC against different human CD38 expressing lymphoma cell lines (35, 51). Similar findings were reported for a panel of 42 human mAbs, of which only a single Ab, daratumumab, was able to induce CDC (35, 52).
The ability of daratumumab to induce CDC was strongly potentiated by point mutations of residue E345 or E430 that facilitate oligomerization of CD38-bound antibodies into ordered hexamers on the cell surface (53, 54). Similarly introduction of the E345R mutation also markedly enhanced the CDC potency of human CD38-specific hcAbs (51).
New structural insights regarding the CDC-inducing potency have recently been obtained for antibodies directed against the B-cell membrane protein CD20 (54). These Abs have been subdivided into two groups, which either recruit complement effectively (type I) or not (type II). A cryo-electron microscopy analysis of the Fab fragments of such antibodies in complex with full length dimeric CD20 found that the CD20 dimer bound only one Fab arm of the type II mAb obinutuzumab, but two Fab fragments of the type I mAbs rituximab and ofatumumab (55). These findings indicate that type I antibodies act as molecular seeds that allow formation of oligomeric complexes, while type II antibodies preclude recruitment of additional complexes. Since CD38 can also form dimers and oligomers (6, 56), it is tempting to speculate that mouse CD38-specific hcAbs of families 2, 4, and 5 can similarly act as molecular seeds that facilitate formation of oligomeric complexes of CD38 on the cell surface and thereby enhance CDC.
In vivo studies are ultimately needed to assess the potential therapeutic efficacy of the hcAbs reported here in mouse myeloma models. Of note, we have previously shown that nanobody-based hcAbs can achieve therapeutic efficacy in vivo in xenograft mouse models using our previously generated hcAbs directed against human CD38 (35). These human CD38-specific hcAbs reduced the growth of a systemic lymphoma and prolonged the survival of tumor bearing SCID mice. The specificity for murine CD38 makes our hcAbs unique tools to simultaneously assess the cytotoxicity mechanisms of CD38-specific hcAbs in vivo against tumor cells and their potential off-target effects on normal cells expressing CD38 in syngeneic mouse tumor models, i.e. in a fully immunocompetent background.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The animal study was reviewed and approved by the animal welfare commission (Amt für Verbraucherschutz, Lebensmittelsicherheit und Veterinärwesen Hamburg, Nr. A8a/694).
PB and FK-N conceived the project. All authors established experimental procedures. NB, PB, and FK-N wrote the manuscript. All authors contributed to the article and approved the submitted version.
Supported by grants SFB1328-A05 and Z02 from the Deutsche Forschungsgemeinschaft to RF, FH, and FK-N and by a grant from the Hamburger Krebsgesellschaft to ME.
FH and FKN receive a share of antibody sales via MediGate GmbH, a wholly owned subsidiary of the University Medical Center Hamburg-Eppendorf. PB and FKN are coinventors on a patent application on CD38-specific nanobodies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Bianca Morgenrot and Sarah Hewald, UKE central animal facility, and Fabienne Seyfried, Dorte Wendt, Lynn Diekow and Anna Josephine Gebhardt, Institute of Immunology, for excellent technical assistance.
ADCC, antibody dependent cellular cytotoxicity; CDC, complement-dependent cytotoxicity; CDR, complementarity determining region; Fc, crystallizing fragment; hcAb, heavy chain antibody, Ig immunoglobulin; kDa, kilodalton; MM, multiple myeloma; NAD+, nicotinamide adenine dinucleotide; mAb, monoclonal antibody; Nb, nanobody; VHH, variable domain of a camelid heavy chain antibody.
1. Adriouch S, Haag F, Boyer O, Seman M, Koch-Nolte F. Extracellular NAD(+): A Danger Signal Hindering Regulatory T Cells. Microbes Infect (2012) 14(14):1284–92. doi: 10.1016/j.micinf.2012.05.011
2. Haag F, Adriouch S, Brass A, Jung C, Moller S, Scheuplein F, et al. Extracellular NAD and ATP: Partners in Immune Cell Modulation. Purinergic Signal (2007) 3(1-2):71–81. doi: 10.1007/s11302-006-9038-7
3. Cockayne DA, Muchamuel T, Grimaldi JC, Muller-Steffner H, Randall TD, Lund FE, et al. Mice Deficient for the Ecto-Nicotinamide Adenine Dinucleotide Glycohydrolase CD38 Exhibit Altered Humoral Immune Responses. Blood (1998) 92(4):1324–33. doi: 10.1182/blood.V92.4.1324.416k26_1324_1333
4. Lund FE, Cockayne DA, Randall TD, Solvason N, Schuber F, Howard MC, et al. CD38: A New Paradigm in Lymphocyte Activation and Signal Transduction. Immunol Rev (1998) 161:79–93. doi: 10.1111/j.1600-065X.1998.tb01573.x
5. Young GS, Choleris E, Lund FE, Kirkland JB. Decreased cADPR and Increased NAD+ in the Cd38-/- Mouse. Biochem Biophys Res Commun (2006) 346(1):188–92. doi: 10.1016/j.bbrc.2006.05.100
6. Hara-Yokoyama M, Kukimoto-Niino M, Terasawa K, Harumiya S, Podyma-Inoue KA, Hino N, et al. Tetrameric Interaction of the Ectoenzyme CD38 on the Cell Surface Enables Its Catalytic and Raft-Association Activities. Structure (2012) 20(9):1585–95. doi: 10.1016/j.str.2012.06.017
7. Krebs C, Adriouch S, Braasch F, Koestner W, Leiter EH, Seman M, et al. CD38 Controls ADP-Ribosyltransferase-2-Catalyzed ADP-Ribosylation of T Cell Surface Proteins. J Immunol (2005) 174(6):3298–305. doi: 10.4049/jimmunol.174.6.3298
8. Chen J, Reifsnyder YG, Schott PC, Lee WH, Osborne CH, Scheuplein M, et al. Targeted Disruption of CD38 Accelerates Autoimmune Diabetes in NOD/Lt Mice by Enhancing Autoimmunity in an ADP-Ribosyltransferase 2-Dependent Fashion. J Immunol (2006) 176(8):4590–9. doi: 10.4049/jimmunol.176.8.4590
9. Schuber F, Lund FE. Structure and Enzymology of ADP-Ribosyl Cyclases: Conserved Enzymes That Produce Multiple Calcium Mobilizing Metabolites. Curr Mol Med (2004) 4(3):249–61. doi: 10.2174/1566524043360708
10. Lund FE. Signaling Properties of CD38 in the Mouse Immune System: Enzyme-Dependent and -Independent Roles in Immunity. Mol Med (2006) 12(11-12):328–33. doi: 10.2119/2006-00099.Lund
11. Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V, Malavasi F. CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma. Front Immunol (2018) 9:2722. doi: 10.3389/fimmu.2018.02722
12. Calabretta E, Carlo-Stella C. The Many Facets of CD38 in Lymphoma: From Tumor-Microenvironment Cell Interactions to Acquired Resistance to Immunotherapy. Cells (2020) 9(4):802. doi: 10.3390/cells9040802
13. Chillemi A, Quarona V, Antonioli L, Ferrari D, Horenstein AL, Malavasi F. Roles and Modalities of Ectonucleotidases in Remodeling the Multiple Myeloma Niche. Front Immunol (2017) 8:305. doi: 10.3389/fimmu.2017.00305
14. Konen JM, Fradette JJ, Gibbons DL. The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors. Cells (2019) 9(1):52. doi: 10.3390/cells9010052
15. van de Donk N, Richardson PG, Malavasi F. CD38 Antibodies in Multiple Myeloma: Back to the Future. Blood (2018) 131(1):13–29. doi: 10.1182/blood-2017-06-740944
16. Bonello F, D'Agostino M, Moscvin M, Cerrato C, Boccadoro M, Gay F. CD38 as an Immunotherapeutic Target in Multiple Myeloma. Expert Opin Biol Ther (2018) 18(12):1209–21. doi: 10.1080/14712598.2018.1544240
17. Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 With Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med (2015) 373(13):1207–19. doi: 10.1056/NEJMoa1506348
18. Frerichs KA, Nagy NA, Lindenbergh PL, Bosman P, Marin Soto J, Broekmans M, et al. CD38-Targeting Antibodies in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Expert Rev Clin Immunol (2018) 14(3):197–206. doi: 10.1080/1744666X.2018.1443809
19. Moreno L, Perez C, Zabaleta A, Manrique I, Alignani D, Ajona D, et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin Cancer Res (2019) 25(10):3176–87. doi: 10.1158/1078-0432.CCR-18-1597
20. Chong LL, Soon YY, Soekojo CY, Ooi M, Chng WJ, de Mel S. Daratumumab-Based Induction Therapy for Multiple Myeloma: A Systematic Review and Meta-Analysis. Crit Rev Oncol Hematol (2021) 159:103211. doi: 10.1016/j.critrevonc.2020.103211
21. Phipps C, Chen Y, Gopalakrishnan S, Tan D. Daratumumab and its Potential in the Treatment of Multiple Myeloma: Overview of the Preclinical and Clinical Development. Ther Adv Hematol (2015) 6(3):120–7. doi: 10.1177/2040620715572295
22. Wang Y, Zhang Y, Hughes T, Zhang J, Caligiuri MA, Benson DM, et al. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin Cancer Res (2018) 24(16):4006–17. doi: 10.1158/1078-0432.CCR-17-3117
23. Adams HC 3rd, Stevenaert F, Krejcik J, Van der Borght K, Smets T, Bald J, et al. High-Parameter Mass Cytometry Evaluation of Relapsed/Refractory Multiple Myeloma Patients Treated With Daratumumab Demonstrates Immune Modulation as a Novel Mechanism of Action. Cytometry A (2019) 95(3):279–89. doi: 10.1002/cyto.a.23693
24. Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, de Haard HJ, et al. Improved Tumor Targeting of Anti-Epidermal Growth Factor Receptor Nanobodies Through Albumin Binding: Taking Advantage of Modular Nanobody Technology. Mol Cancer Ther (2008) 7(8):2288–97. doi: 10.1158/1535-7163.MCT-07-2384
25. Bannas P, Lenz A, Kunick V, Well L, Fumey W, Rissiek B, et al. Molecular Imaging of Tumors With Nanobodies and Antibodies: Timing and Dosage are Crucial Factors for Improved In Vivo Detection. Contrast Media Mol Imaging (2015) 10(5):367–78. doi: 10.1002/cmmi.1637
26. Muyldermans S. Nanobodies: Natural Single-Domain Antibodies. Annu Rev Biochem (2013) 82:775–97. doi: 10.1146/annurev-biochem-063011-092449
27. Muyldermans S. Applications of Nanobodies. Annu Rev Anim Biosci (2021) 9:401–21. doi: 10.1146/annurev-animal-021419-083831
28. Wesolowski J, Alzogaray V, Reyelt J, Unger M, Juarez K, Urrutia M, et al. Single Domain Antibodies: Promising Experimental and Therapeutic Tools in Infection and Immunity. Med Microbiol Immunol (2009) 198(3):157–74. doi: 10.1007/s00430-009-0116-7
29. Ingram JR, Schmidt FI, Ploegh HL. Exploiting Nanobodies’ Singular Traits. Annu Rev Immunol (2018) 36:695–715. doi: 10.1146/annurev-immunol-042617-053327
30. Bannas P, Hambach J, Koch-Nolte F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front Immunol (2017) 8:1603. doi: 10.3389/fimmu.2017.01603
31. Liu Q, Jiang L, Li K, Li H, Lv G, Lin J, et al. Immuno-PET Imaging of (68)Ga-Labelled Nanobody Nb109 for Dynamic Monitoring the PD-L1 Expression in Cancers. Cancer Immunol Immunother (2021) 70:1721–33. doi: 10.1007/s00262-020-02818-y
32. Hu Y, Liu C, Muyldermans S. Nanobody-Based Delivery Systems for Diagnosis and Targeted Tumor Therapy. Front Immunol (2017) 8:1442. doi: 10.3389/fimmu.2017.01442
33. Bannas P, Well L, Lenz A, Rissiek B, Haag F, Schmid J, et al. In Vivo Near-Infrared Fluorescence Targeting of T Cells: Comparison of Nanobodies and Conventional Monoclonal Antibodies. Contrast Media Mol Imaging (2014) 9(2):135–42. doi: 10.1002/cmmi.1548
34. Verhaar ER, Woodham AW, Ploegh HL. Nanobodies in Cancer. Semin Immunol (2020) p:101425. doi: 10.1016/j.smim.2020.101425
35. Schriewer L, Schutze K, Petry K, Hambach J, Fumey W, Koenigsdorf J, et al. Nanobody-Based CD38-Specific Heavy Chain Antibodies Induce Killing of Multiple Myeloma and Other Hematological Malignancies. Theranostics (2020) 10(6):2645–58. doi: 10.7150/thno.38533
36. Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang YL, et al. Effector-Attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J Biol Chem (2017) 292(9):3900–8. doi: 10.1074/jbc.M116.767749
37. Fumey W, Koenigsdorf J, Kunick V, Menzel S, Schutze K, Unger M, et al. Nanobodies Effectively Modulate the Enzymatic Activity of CD38 and Allow Specific Imaging of CD38(+) Tumors in Mouse Models In Vivo. Sci Rep (2017) 7(1):14289. doi: 10.1038/s41598-017-14112-6
38. Jager V, Bussow K, Wagner A, Weber S, Hust M, Frenzel A, et al. High Level Transient Production of Recombinant Antibodies and Antibody Fusion Proteins in HEK293 Cells. BMC Biotechnol (2013) 13:52. doi: 10.1186/1472-6750-13-52
39. Zhang J, MacKenzie R, Durocher Y. Production of Chimeric Heavy-Chain Antibodies. Methods Mol Biol (2009) 525:323–36, xv. doi: 10.1007/978-1-59745-554-1_17
40. Koch-Nolte F, Glowacki G, Bannas P, Braasch F, Dubberke G, Ortolan E, et al. Use of Genetic Immunization to Raise Antibodies Recognizing Toxin-Related Cell Surface ADP-Ribosyltransferases in Native Conformation. Cell Immunol (2005) 236(1):66–71. doi: 10.1016/j.cellimm.2005.08.033
41. Horenstein AL, Bracci C, Morandi F, Malavasi F. CD38 in Adenosinergic Pathways and Metabolic Re-Programming in Human Multiple Myeloma Cells: In-Tandem Insights From Basic Science to Therapy. Front Immunol (2019) 10:760. doi: 10.3389/fimmu.2019.00760
42. Martin TG, Corzo K, Chiron M, Velde HV, Abbadessa G, Campana F, et al. Therapeutic Opportunities With Pharmacological Inhibition of CD38 With Isatuximab. Cells (2019) 8(12). doi: 10.3390/cells8121522
43. van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, et al. Monoclonal Antibodies Targeting CD38 in Hematological Malignancies and Beyond. Immunol Rev (2016) 270(1):95–112. doi: 10.1111/imr.12389
44. Graeff RM, Walseth TF, Hill HK, Lee HC. Fluorescent Analogs of Cyclic ADP-Ribose: Synthesis, Spectral Characterization, and Use. Biochemistry (1996) 35(2):379–86. doi: 10.1021/bi952083f
45. Muller-Steffner HM, Malver O, Hosie L, Oppenheimer NJ, Schuber F. Slow-Binding Inhibition of NAD+ Glycohydrolase by Arabino Analogues of Beta-NAD. J Biol Chem (1992) 267(14):9606–11. doi: 10.1016/S0021-9258(19)50133-8
46. Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher VS, Vallee F, et al. SAR650984, A Novel Humanized CD38-Targeting Antibody, Demonstrates Potent Antitumor Activity in Models of Multiple Myeloma and Other CD38+ Hematologic Malignancies. Clin Cancer Res (2014) 20(17):4574–83. doi: 10.1158/1078-0432.CCR-14-0695
47. Baum N, Fliegert R, Bauche A, Hambach J, Menzel S, Haag F, et al. Koch-Nolte, Daratumumab and Nanobody-Based Heavy Chain Antibodies Inhibit the ADPR Cyclase But Not the NAD. Cancers (Basel) (2020) 13(1):76. doi: 10.3390/cancers13010076
48. Vaisitti T, Arruga F, Guerra G, Deaglio S. Ectonucleotidases in Blood Malignancies: A Tale of Surface Markers and Therapeutic Targets. Front Immunol (2019) 10:2301. doi: 10.3389/fimmu.2019.02301
49. Morandi F, Morandi B, Horenstein AL, Chillemi A, Quarona V, Zaccarello G, et al. A Non-Canonical Adenosinergic Pathway Led by CD38 in Human Melanoma Cells Induces Suppression of T Cell Proliferation. Oncotarget (2015) 6(28):25602–18. doi: 10.18632/oncotarget.4693
50. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 Ectoenzymatic Pathway Independent of CD39 Drives a Novel Adenosinergic Loop in Human T Lymphocytes. Oncoimmunology (2013) 2(9):e26246. doi: 10.4161/onci.26246
51. Schutze K, Petry K, Hambach J, Schuster N, Fumey W, Schriewer L, et al. CD38-Specific Biparatopic Heavy Chain Antibodies Display Potent Complement-Dependent Cytotoxicity Against Multiple Myeloma Cells. Front Immunol (2018) 9:2553. doi: 10.3389/fimmu.2018.02553
52. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J Immunol (2011) 186(3):1840–8. doi: 10.4049/jimmunol.1003032
53. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science (2014) 343(6176):1260–3. doi: 10.1126/science.1248943
54. de Jong RN, Beurskens FJ, Verploegen S, Strumane K, van Kampen MD, Voorhorst M, et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PloS Biol (2016) 14(1):e1002344. doi: 10.1371/journal.pbio.1002344
55. Kumar A, Planchais C, Fronzes R, Mouquet H, Reyes N. Binding Mechanisms of Therapeutic Antibodies to Human CD20. Science (2020) 369(6505):793–9. doi: 10.1126/science.abb8008
Keywords: CD38, NAD+, antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, multiple myeloma, nanobody, heavy chain antibody, antibody engineering
Citation: Baum N, Eggers M, Koenigsdorf J, Menzel S, Hambach J, Staehler T, Fliegert R, Kulow F, Adam G, Haag F, Bannas P and Koch-Nolte F (2021) Mouse CD38-Specific Heavy Chain Antibodies Inhibit CD38 GDPR-Cyclase Activity and Mediate Cytotoxicity Against Tumor Cells. Front. Immunol. 12:703574. doi: 10.3389/fimmu.2021.703574
Received: 30 April 2021; Accepted: 04 August 2021;
Published: 03 September 2021.
Edited by:
Santina Bruzzone, University of Genoa, ItalyReviewed by:
Michele Cea, University of Genoa, ItalyCopyright © 2021 Baum, Eggers, Koenigsdorf, Menzel, Hambach, Staehler, Fliegert, Kulow, Adam, Haag, Bannas and Koch-Nolte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friedrich Koch-Nolte, bm9sdGVAdWtlLmRl; Peter Bannas, cC5iYW5uYXNAdWtlLmRl
†These authors share first authorship
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.