 Danny Zipris
Danny Zipris- Innate Biotechnologies LLC, Denver, CO, United States
Type 1 diabetes (T1D) is a proinflammatory pathology that leads to the specific destruction of insulin producing β-cells and hyperglycaemia. Much of the knowledge about type 1 diabetes (T1D) has focused on mechanisms of disease progression such as adaptive immune cells and the cytokines that control their function, whereas mechanisms linked with the initiation of the disease remain unknown. It has been hypothesized that in addition to genetics, environmental factors play a pivotal role in triggering β-cell autoimmunity. The BioBreeding Diabetes Resistant (BBDR) and LEW1.WR1 rats have been used to decipher the mechanisms that lead to virus-induced T1D. Both animals develop β-cell inflammation and hyperglycemia upon infection with the parvovirus Kilham Rat Virus (KRV). Our earlier in vitro and in vivo studies indicated that KRV-induced innate immune upregulation early in the disease course plays a causal role in triggering β-cell inflammation and destruction. Furthermore, we recently found for the first time that infection with KRV induces inflammation in visceral adipose tissue (VAT) detectable as early as day 1 post-infection prior to insulitis and hyperglycemia. The proinflammatory response in VAT is associated with macrophage recruitment, proinflammatory cytokine and chemokine upregulation, endoplasmic reticulum (ER) and oxidative stress responses, apoptosis, and downregulation of adipokines and molecules that mediate insulin signaling. Downregulation of inflammation suppresses VAT inflammation and T1D development. These observations are strikingly reminiscent of data from obesity and type 2 diabetes (T2D) in which VAT inflammation is believed to play a causal role in disease mechanisms. We propose that VAT inflammation and dysfunction may be linked with the mechanism of T1D progression.
Introduction
Type 1 diabetes (T1D) is a multi-step proinflammatory pathology that culminates in the specific destruction of islet β-cells and lack of insulin secretion (1–3). The Centers for Disease Control and Prevention have estimated that ~1.25 million Americans are currently living with T1D and 40,000 new cases of T1D are being diagnosed in the U.S each year and it is estimated that five million Americans will live with T1D by mid-century (4).
It is thought that both genetic and environmental factors are key players in the mechanism that triggers diabetes (5–7). The risk for T1D development is substantially increased in relatives of T1D patients, since ~6% of children of a diabetic parent, 5% of siblings and 50% of monozygotic twins develop T1D compared to only 0.4% in the general population (8, 9). More than 50 T1D genetic risk loci have been identified to be associated with disease progression (10).
There is ample evidence from humans and animals supporting the notion that the environment plays a key role in mechanisms that trigger β-cell autoimmunity (11–18), and viruses have been postulated to play a pivotal role in these mechanisms (16, 17, 19–27). Due to ethical reasons, it is almost impossible to establish a causal role for microbial infections in triggering T1D, or address virus-induced disease mechanisms in humans. Furthermore, identifying microbes involved in triggering T1D may be hindered since by the time T1D is detected, the individual might have been infected with multiple viruses and the virus triggering the disease might have been cleared (28, 29). We have therefore used the BBDR and LEW1.WR1 rat models that develop T1D following infection with Kilham Rat Virus (KRV) (30) to identify how infections lead to β-cell inflammation and destruction.
Emerging evidence suggests that inflammation plays a key role in triggering numerous inflammatory disorders (31–35). We recently hypothesized that innate immune upregulation is associated with promoting virus-induced T1D (30, 36–43). Our recent data provided for the first time evidence linking inflammation in VAT with mechanisms of T1D (44). Inflammation in VAT is detectable soon after infection prior to insulitis and hyperglycemia and is characterized by infiltration of macrophage to the site of inflammation and proinflammatory cytokine and chemokine upregulation and tissue dysfunction (44). On the basis of these observations, we hypothesize that VAT inflammation and dysfunction may be associated with T1D mechanisms.
Kilham Rat Virus
KRV is a rat-specific virus environmentally ubiquitous and a member of the Parvoviridea, a virus group of small single‐stranded DNA viruses with an average genome size of 5 Kbp encapsidated by protein in an icosahedral non‐enveloped particle (45). This virus group infects various animal species, including humans (46) and rodents (47). KRV encodes three overlapping structural proteins, VP1, VP2, and VP3, and two overlapping nonstructural proteins, NS1 and NS2 (47). There human parvovirus B19 has been linked with pro-inflammatory autoimmune disorders like acute myocarditis (48, 49), rheumatoid arthritis (50), systemic lupus erythematosus (51), and Sjögren’s syndrome, as well as other autoimmune conditions (50). Infection with B19 has been associated with the appearance of elevated levels of autoantibodies against nuclear antigens and double‐stranded DNA (50). KRV infection can occur in natural environment leading to T1D without the need for virus injection (52). Known routes by which KRV transmission may occur are direct contact, aerosol, and oral (52).
Rat Models of Virus-Induced T1D
There are two inbred rat strains that have been most used to address virus-induced T1D mechanisms, the BBDR and LEW1.WR1 rats. These animals are the only genetically un-manipulated animal models in which infection with a virus triggers anti-ß-cell autoimmunity (41). BBDR rats have normal levels and function of peripheral T cells (53, 54), and spontaneous diabetes does not develop in viral antibody–free BBDR rats (55). However, insulitis, hyperglycemia, and severe ketosis occur in animals after inducing innate immunity with Poly(I:C) plus elimination of regulatory ART2+ T cells (55), or following virus infection (52). T1D in the BBDR rat is mediated by the immune system since the transfer of lymph nodes from animals with diabetes to RT1u MHC compatible T cell deficient WAG nu/nu rats results in diabetes progression (56).
The LEW1.WR1 rat has also normal levels and function of T lymphocytes (57). The LEW1.WR1 rat has a higher degree of disease penetrance compared with that of BBDR rats as evidenced by the observation that elimination of ART2.1+ cells by itself can result in diabetes (57). As seen in the BBDR rat, KRV infection leads to hyperglycemia by specific loss of islet ß-cells, glycosuria, ketonuria, and polyuria (55, 57).
Infecting LEW1.WR1 and BBDR rats with KRV leads to specific β-cell inflammation, islet cell death and permanent T1D occurring following insulitis, 2-4 weeks following virus inoculation with disease rate of ~20 and 60%, respectively (30, 34, 35, 52). It is noteworthy that the ability of virus infection to trigger T1D or inflammation in the rat is not limited to KRV, since β-cell autoimmunity in the rat can be triggered by two other viruses, rat CMV (58). Furthermore, Poly I:C, a synthetic analogue of double stranded RNA which mimics viral infection, synergizes with low KRV titers, that by themselves do not induce T1D, on disease progression (41). Because double stranded RNA molecules can be expressed by different viruses, this may suggest that microbes other than KRV could also be associated with initiating T1D development (44). Indeed, multiple viruses have been hypothesized to be involved in triggering human T1D (5, 6, 16, 17, 19–26, 59).
A key factor linked with the mechanism leading to T1D in both animals and humans is likely to be linked with proinflammatory pathways that can potentially be upregulated by different virus groups (60). It is therefore plausible to hypothesize that while a human KRV homologue may not necessarily be involved in triggering T1D in humans, viruses that induce proinflammatory pathways similar to those induced by KRV may be linked with promoting β–cell autoimmunity in genetically susceptible individuals. Identifying mechanisms of KRV-induced T1D in rat models of virus-induced T1D could therefore provide valuable data on mechanisms mediating the human disease.
The relevance of the BBDR and LEW1.WR1 rat models to the human disease is supported by data from our laboratory and others. T1D in the rat better resembles the human disorder than the mouse model with respect to histopathology (61). Similar to the rat, there is no significant infiltration of immune cells around the islet (“peri-insulitis”) prior to disease onset and insulitis is morphologically mild and more similar to that detected in human T1D (62–64). As seen in humans, disease in the rat is not influenced by gender (65) and is MHC-dependent (61, 66).
The mechanism of T1D in the LEW1.WR1 rat is believed to be fundamentally different than that leading to T1D in the NOD mouse. In contrast to the rat, T1D development in the mouse is not dependent on microbial infections as germ-free mice retain the ability to develop disease (67). While β-cell autoimmunity in the mouse appears to be independent of the MyD88 signaling pathway (68), our studies demonstrated that the disease in the rat is mediated via the MyD88-TLR9 signaling axis (40). Finally, innate immune activation with exogenous activators of TLR2, TLR3/MDA-5, TLR4, TLR7/8, and TLR9, and exacerbates T1D in the rat (41, 69), but protects NOD mice from β–cell autoimmunity (70–73).
Innate Immunity and Inflammation
Inflammation is a physiological reaction of the innate immune system to microbial infection or tissue injury leading to the secretion of numerous inflammatory mediators, such as cytokines and chemokines, which orchestrate cellular defense mechanisms and injured tissue repair (74, 75). In contrast to adaptive immunity that identifies antigenic molecules using highly specific receptors expressed on T and B lymphocytes, inflammation is the less specific arm of the immune system (76).
Innate immune sentinel cells such as dendritic cells (DCs), macrophages, and neutrophils and recognize invading microbes via pattern recognition receptors (PRRs) activating downstream innate immune pathways aiming to eliminate infections (77, 78). A key PRR group is the Toll‐like receptors (TLRs) family each member of which recognizes a different type of conserved pathogen-associated molecular patterns (PAMPs), such as TLR2 that senses cell wall molecules of gram-positive bacteria lipoteichoic acid and TLR3 and TLR9 that sense double stranded RNA and microbial DNA, respectively reviewed in refs. (79–88). Recognition of PAMPs by PRRs induces proinflammatory responses and activation of host defense mechanisms (79–88). The interaction of TLRs with their agonists induces in addition to proinflammatory cytokine and chemokine responses, the expression of MHC Class II and costimulatory molecules on antigen presenting cells (APCs), thus enabling these cells to effectively activate antigen-specific T cells to specifically attack invading pathogenic microbes (79–88). In addition to sentinel cells, innate immunity also has a humoral arm comprised of pattern recognition molecules (PRMs), such as lectin, ficolins, pentraxins, and the complement component C1q (89, 90).
Role of Inflammation in KRV-Induced T1D
Infection with KRV induces a global innate immune upregulation detected in various lymphoid organs, such as the spleen, pancreatic lymph nodes, Peyer’s patches and thymus involving the induction of numerous proinflammatory cytokines, including IL-1ß, IFN-γ, and IL-12 3-5 days after infection, prior to insulitis and diabetes (30, 34, 35, 66). The rats develop humoral and cellular anti-KRV responses and clear the virus (91). We proposed that KRV-induced inflammation is associated with mechanisms of disease development (30, 36–43). We were the first to implicate TLR signaling in T1D progression (40, 41). We demonstrated that innate immune activation with ligands of TLRs synergizes with KRV infection on T1D development (41). Furthermore, we observed that the highly homologous H-1 parvovirus does not activate the innate immune system and fails to induce diabetes development in the BBDR rat (41). Our in vivo studies have shown that blocking IL-1 signaling with IL-1RA (39), or suppressing inflammation with a number of immunomodulatory agents, such as steroids (69), histone deacetylase inhibitor (38), antibiotics (30), or short chain fatty acids (92) prevents diabetes. Our hypothesis on the role of innate immunity in T1D is further supported by earlier data implicating TLR9 pathways in KRV-induced T1D mechanisms (40). We demonstrated that in vitro KRV-induced innate immunity is blocked by inhibitors of TLR9 and blockers of PKR and NF-κB (40). Finally, pharmacological suppression of TLR9 in vivo prevents T1D (40).
COVID-19 and T1D
Given that COVID-19 induces robust inflammation in infected individuals, it has recently been hypothesized that this virus could potentially drive T1D via mechanisms associated in part with immune upregulation (93, 94). The data on the ability of COVID-19 to induce autoimmunity are mixed and clear evidence that COVID-19 activates anti-β–cell autoimmunity is not yet available (94–103). Moreover, the observations implicating COVID-19 in T1D development are based primarily on anecdotal data (95–101). Because hyperglycemia is only the end stage of the anti-islet autoimmune process that may start many years prior to disease onset (104–106), long-term follow up epidemiological studies will be required to determine whether COVID-19 infection increases the risk for T1D development in genetically-susceptible individuals.
KRV-Induced Inflammation in Visceral Adipose Tissue (VAT)
In the course of our studies on the role of inflammation in virus-induced T1D, we observed that infection of LEW1.WR1 rats with KRV leads to inflammation in VAT detectable as early as day 1 post-infection, long before β-cell inflammation and hyperglycemia. This inflammation is characterized by an influx of CD68+ macrophages into VAT seen in the interstitial space surrounding adipocytes in KRV-infected animals but not control rats injected with PBS. In sharp contrast to VAT, subcutaneous adipose tissue (SAT) was observed to be free of cell infiltration (44). Activation of innate immunity with Poly (I:C) in the absence of virus also induces VAT inflammation. Because i.p. injection of KRV induces inflammation in proximal and distal organs, and since Poly (I:C) itself, in the absence of virus, can induce VAT inflammation, it is unlikely that the route of virus inoculation or site of infection play a critical role in triggering VAT inflammation. Unlike VAT, the exocrine tissue and islets from day 5-infected rats are insulitis-free, whereas ß-cells from day-14-infected animals are inflamed or show signs of tissue destruction (44). KRV induces the expression of virus transcripts and proinflammatory cytokines such as IL-1ß, IL-6, and IL-12p40 and chemokines in VAT in vivo and in purified adipocytes in vitro (44). Furthermore, KRV induces ER and oxidative stress response and activation of apoptotic pathways in infected VAT in vivo (44). KRV also downregulated the expression of adipokines and genes associated with mediating insulin signaling in VAT (44). Brief therapy with dexamethasone early in the disease course (days 1-5) prevents VAT inflammation and T1D. Based on these data, we hypothesized that VAT inflammation and dysfunction may be linked with early mechanisms of virus-induced disease development.
Role of Adipose Tissue in Glucose Metabolism and Immunity
There are several types of adipose tissue, i.e. white adipose tissue (WAT), brown adipose tissue (BAT) and beige adipose tissue (107). WAT is the most abundant fat accounting for 5% to 50% of human body weight (107). It plays a key role in metabolic homeostasis by storing fat for long-term survival and by functioning as an endocrine organ (107–109). WAT is a main source of many adipokines, peptides or proteins with hormone-like properties that regulate metabolic homeostasis through local paracrine effects and endocrine effects (107–109). The metabolic characteristics of WAT is determined by its location in the body, commonly classified into subcutaneous fat and visceral fat depots (107–109). Adipose tissue can release and respond to cytokines and may therefore exert immune modulatory functions on non-adipose tissues (107). The discovery of leptin and adiponectin was the first indication that adipose tissue is an endocrine organ with the ability to regulate systemic energy homeostasis and glucose metabolism as well as mediate immunity. The metabolic effects of leptin and adiponectin on target tissues were observed to be robust (110).
Why KRV induces inflammation in VAT and not SAT is unclear. It may be that this is the result of differences in the function of VAT versus SAT. SAT is less active metabolically than VAT (111). It has been shown that adipocytes of VAT undergo more lipolysis than SAT and therefore contribute larger amounts of fatty acids to the circulation (111–113). On the other hand, SAT is considered to have a better capability of storing fatty acids, implying that it could store energy in periods of excess nutrition and supply fatty acids in periods of starvation (111).
Leptin has been suggested to play a key role in T2D development (reviewed in ref. 104). The long form of the leptin receptor (ObRb) capable of intracellular signaling is expressed in ß-cells, and exogenous leptin inhibits insulin production and secretion from human islets implying a direct action of leptin on β-cell function (105, 106, 114–118). Furthermore, mice deficient of leptin have increased appetite, weight gain, insulin resistance and diabetes, conditions that can be improved with leptin therapy (104, 119–124). In addition to its role in controlling energy balance, leptin can also influence immune functions reviewed in ref. (119). Indeed, macrophages express the leptin receptor (119) and leptin can increase the proliferation of monocytes and induce the expression of inflammatory cytokines such as TNF-α and IL-6 and other surface activation molecules (125).
Adiponectin has been shown to have beneficial effects on insulin sensitivity (110, 126) and β-cell regeneration in mice with STZ-induced diabetes (127). Adiponectin has also been demonstrated to protect ß-cells from the detrimental effects of free fatty acids (128) via as yet unidentified mechanisms (118). Adiponectin is an endogenous insulin sensitizer in the skeletal muscle and liver, and administering mice with adiponectin results in lower blood glucose levels and the reversal of insulin resistance in mouse models of obesity (119). The receptor for adiponectin is expressed in macrophages, and adiponectin can suppress the production of TNF-α and IL-6 and induce the production of the anti-inflammatory mediators IL-10 and IL-1 receptor antagonist (119). Mice deficient in adiponectin have increased numbers of activated M1 macrophages in their adipose tissue with increased production of TNF-α, IL-6, and MCP-1 (119).
Crosstalk Between Innate Immunity and Glucose Metabolism
The hypothesis that there is interplay between the innate immune system and glucose metabolism emerged after it was observed that administering low doses of lipopolysaccharide (LPS) leads to hyperglycemia mediated primarily by IL-1 pathways (129). Innate immune mediators such as IL-1 may play a beneficial role in maintain a normal glucose homeostasis by inducing insulin secretion and biosynthesis and β-cell proliferation reviewed in ref. (130).
In obesity, increased fat mass can result in adipocyte hypertrophy, hypoxia, death and ER stress response reviewed in refs. (119, 130). The adipose tissue death and dysfunction lead to the induction of chronic inflammation associated with the expression of proinflammatory cytokines such as IL-1, IL-6 and TNF-α and chemokines such as MCP-1 in adipocytes. MCP-1 and other chemokines released by adipocytes and immune cells in fat tissue further promote infiltration of monocytes and other immune cells into adipose tissue (130–132). Macrophages are the most abundant innate immune cells infiltrating and accumulating into adipose tissue of obese individuals (133).
Chronic inflammation in adipose tissue is believed to play a key role in the development of insulin resistance that is a hallmark of T2D in obese individuals reviewed in ref. (133). Insulin resistance may culminate in aberrant glucose uptake and glycogen synthesis (134). Consequently, ß-cells attempt to compensate for insulin resistance by increasing insulin secretion to restore normal glucose homeostasis (134). A further decline in insulin sensitivity makes the ß-cells exhausted, leading to hyperglycemia and T2D (135).
VAT Inflammation and Dysfunction in KRV-Induced T1D Versus T2D
The underlying mechanisms and pathways critically involved in KRV-induced inflammation and T1D remain to be identified. The data from our laboratory implicating VAT inflammation and dysfunction in T1D development are highly reminiscent of observations from obesity and T2D in which VAT inflammation and dysfunction have been hypothesized to play a causal role in mechanisms that result in islet damage and diabetes progression (136–143). Although the level of the proinflammatory response detected in VAT from infected LEW1.WR1 rats is substantially greater than that typically seen in adipose tissue from T2D (44, 136, 137, 141, 142, 144–155), one cannot ignore the remarkable commonalities between inflammation observed in KRV-induced T1D versus T2D. Most notably, in both conditions, VAT is targeted by the innate immune system. Moreover, VAT inflammation in both disorders is linked with 1) macrophage infiltration into VAT, 2) expression of proinflammatory cytokines such as IL-1, IL-6 and TNF-α and as well as chemokines such as CCL2, CCL5, and CXCL-10, 3) oxidative stress response, 4) apoptosis, 5) adipocyte death, and 6) tissue dysfunction (136, 137, 141, 142, 144–155).
Role of VAT Inflammation and Dysfunction in Virus-Induced T1D
Whether and how VAT inflammation and dysfunction play a role in KRV-induced T1D mechanisms remain to be further elucidated. We propose a model that may explain how VAT inflammation and dysfunction lead to T1D (see model in Figure 1). We hypothesize that infection with KRV results in VAT infection and TLR-induced macrophage activation and infiltration into VAT. Inflammation in VAT associated with a robust proinflammatory cytokine response may lead to adipose tissue hypoxia, ER and oxidative stress responses and apoptosis and consequently aberrant adipokine expression (118, 119, 133, 156, 157). In Obesity, free fatty acids, and lipid intermediates synergistically induce adverse effects on both β-cell mass and function contributing to the progressive loss of functional β-cell mass reviewed in ref. (118). Likewise, circulating factors such as cytokines released from inflamed tissues such as adipose tissue and activated innate immune cells can adversely affect ß-cells by impairing their functions and limiting cell mass (118). In a similar manner, KRV-induced excessive lipolysis resulting from adipocyte death can result in excess of free fatty acids in the circulation, which can induce lipotoxicity. KRV-induced proinflammatory cytokines such as IL-1ß can enter the circulation and from there to the pancreas where it may exert toxic effects on islets, potentially leading to metabolic and cellular stress in ß-cells (136, 158). Furthermore, a rise in glucose levels in the microenvironment of ß-cells can activate the inflammasome in pancreatic ß-cells, further increasing the expression of IL-1ß (136, 158). Consequently, IL-1ß released from ß-cells may trigger the recruitment and activation of innate immune cells, which may then release more IL-1ß. IL-1ß in the islet microenvironment can exacerbate ß-cell dysfunction, and trigger apoptosis in ß-cells (30, 36, 40, 136, 158). Finally, islet impairment and damage may ultimately signal innate and adaptive immunity to attack and destroy ß–cells leading to permanent hyperglycemia (159–162).
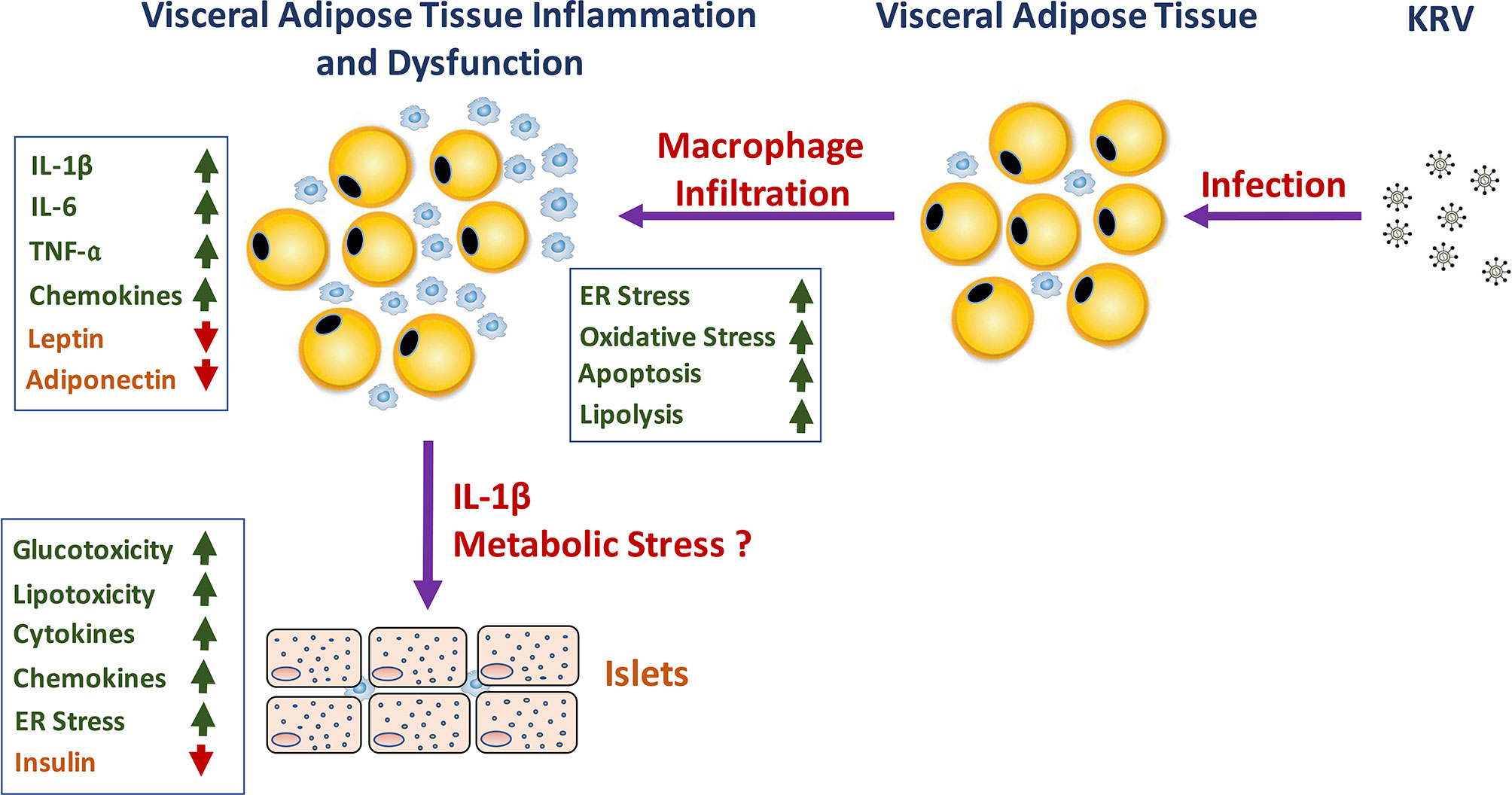
Figure 1 A schematic representation of the hypothesized role of KRV-induced VAT inflammation and dysfunction in the mechanism of KRV-induced T1D.
Conclusions and Future Perspectives
Earlier data demonstrated that the mechanism of KRV-induced T1D is associated with innate immune activation early in the disease course. We recently reported that infection with KRV results in VAT inflammation and dysfunction detected soon after infection. There are marked similarities between inflammation detected in VAT from infected LEW1.WR1 rats and inflammation detected in VAT from T2D patients. Whether as found in T2D, a cause-and-effect relationship exists between VAT inflammation and islet autoimmunity remains to be determined. As discussed in this Review, there is crosstalk between the innate immune system and glucose metabolism. We propose a paradigm by which virus-induced global innate immunity resulting in proinflammatory cytokine and chemokine upregulation and aberrant adipokine profile and lipolysis in VAT lead to metabolic stress and β–cells inflammation and destruction. Future studies will identify the interplay between the innate immune system and metabolic pathways and its role in triggering virus-induced disease. Identification of critical metabolic and immune pathways linked with β–cell autoimmunity will open new avenues for the development of targeted therapies for disease amelioration.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
DZ was employed by Innate Biotechnologies, LLC.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schranz DB, Lernmark A. Immunology in Diabetes: An Update. Diabetes Metab Rev (1998) 14(1):3–29. doi: 10.1002/(SICI)1099-0895(199803)14:1<3::AID-DMR206>3.0.CO;2-T
2. Seissler J, de Sonnaville JJ, Morgenthaler NG, Steinbrenner H, Glawe D, Khoo-Morgenthaler UY, et al. Immunological Heterogeneity in Type I Diabetes: Presence of Distinct Autoantibody Patterns in Patients With Acute Onset and Slowly Progressive Disease. Diabetologia (1998) 41(8):891–7. doi: 10.1007/s001250051004
3. Srikanta S, Ganda OP, Jackson RA, Gleason RE, Kaldany A, Garovoy MR, et al. Type I Diabetes Mellitus in Monozygotic Twins: Chronic Progressive Beta Cell Dysfunction. Ann Intern Med (1983) 99(3):320–6. doi: 10.7326/0003-4819-99-3-320
4. Prevention CfDCa. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States. Atlanta, GA: US Department of Health and Human Services (2014).
5. Jun HS, Yoon JW. The Role of Viruses in Type I Diabetes: Two Distinct Cellular and Molecular Pathogenic Mechanisms of Virus-Induced Diabetes in Animals. Diabetologia (2001) 44(3):271–85. doi: 10.1007/s001250051614
6. Jun HS, Yoon JW. A New Took at Viruses in Type 1 Diabetes. Diabetes-Metab Res (2003) 19(1):8–31. doi: 10.1002/dmrr.337
7. Soltesz G, Patterson CC, Dahlquist G. Worldwide Childhood Type 1 Diabetes Incidence–What Can We Learn From Epidemiology? Pediatr Diabetes (2007) 8(Suppl 6):6–14. doi: 10.1111/j.1399-5448.2007.00280.x
8. Redondo MJ, Rewers M, Yu L, Garg S, Pilcher CC, Elliott RB, et al. Genetic Determination of Islet Cell Autoimmunity in Monozygotic Twin, Dizygotic Twin, and Non-Twin Siblings of Patients With Type 1 Diabetes: Prospective Twin Study. BMJ (1999) 318(7185):698–702. doi: 10.1136/bmj.318.7185.698
9. Beyan H, Riese H, Hawa MI, Beretta G, Davidson HW, Hutton JC, et al. Glycotoxin and Autoantibodies Are Additive Environmentally Determined Predictors of Type 1 Diabetes. Population Study (2012) 61(5):1192–8. doi: 10.2337/db11-0971
10. Størling J, Pociot F. Type 1 Diabetes Candidate Genes Linked to Pancreatic Islet Cell Inflammation and Beta-Cell Apoptosis. Genes (2017) 8(2):72. doi: 10.3390/genes8020072
11. Filippi CM, von Herrath MG. Viral Trigger for Type 1 Diabetes: Pros and Cons. Diabetes (2008) 57(11):2863–71. doi: 10.2337/db07-1023
12. Zipris D. Epidemiology of Type 1 Diabetes and What Animal Models Teach Us About the Role of Viruses in Disease Mechanisms. Clin Immunol (2009) 131(1):11–23. doi: 10.1016/j.clim.2008.12.011
13. Hu Y, Wong FS, Wen L. Antibiotics, Gut Microbiota, Environment in Early Life and Type 1 Diabetes. Pharmacol Res (2017) 119:219–26. doi: 10.1016/j.phrs.2017.01.034
14. Stene LC, Gale EAM. The Prenatal Environment and Type 1 Diabetes. Diabetologia (2013) 56(9):1888–97. doi: 10.1007/s00125-013-2929-6
15. Gale EAM. The Rise of Childhood Type 1 Diabetes in the 20th Century. Diabetes (2002) 51(12):3353–61. doi: 10.2337/diabetes.51.12.3353
16. Kondrashova A, Hyöty H. Role of Viruses and Other Microbes in the Pathogenesis of Type 1 Diabetes. Int Rev Immunol (2014) 33(4):284–95. doi: 10.3109/08830185.2014.889130
17. Op de Beeck A, Eizirik DL. Viral Infections in Type 1 Diabetes Mellitus — Why the β Cells? Nat Rev Endocrinol (2016) 12:263. doi: 10.1038/nrendo.2016.30
18. Forlenza GP, Rewers M. The Epidemic of Type 1 Diabetes: What Is It Telling Us? Curr Opin Endocrinol Diabetes Obes (2011) 18(4):248–51. doi: 10.1097/MED.0b013e32834872ce
19. Lonnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, et al. Enterovirus Infection as a Risk Factor for Beta-Cell Autoimmunity in a Prospectively Observed Birth Cohort: The Finnish Diabetes Prediction and Prevention Study. Diabetes (2000) 49(8):1314–8. doi: 10.2337/diabetes.49.8.1314
20. Lönnrot M, Lynch KF, Elding Larsson H, Lernmark Å, Rewers MJ, Törn C, et al. Respiratory Infections Are Temporally Associated With Initiation of Type 1 Diabetes Autoimmunity: The TEDDY Study. Diabetologia (2017) 60(10):1931–40. doi: 10.1007/s00125-017-4365-5
21. Richardson SJ, Morgan NG, Foulis AK. Pancreatic Pathology in Type 1 Diabetes Mellitus. Endocr Pathol (2014) 25(1):80–92. doi: 10.1007/s12022-014-9297-8
22. Hober D, Sauter P. Pathogenesis of Type 1 Diabetes Mellitus: Interplay Between Enterovirus and Host. Nat Rev Endocrinol (2010) 6:279. doi: 10.1038/nrendo.2010.27
23. Ghazarian L, Diana J, Simoni Y, Beaudoin L, Lehuen A. Prevention or Acceleration of Type 1 Diabetes by Viruses. Cell Mol Life Sci (2012) 70(2):239–55. doi: 10.1007/s00018-012-1042-1
24. Drescher KM, von Herrath M, Tracy S. Enteroviruses, Hygiene and Type 1 Diabetes: Toward a Preventive Vaccine. Rev Med Virol (2015) 25(1):19–32. doi: 10.1002/rmv.1815
25. Imagawa A, Hanafusa T. Fulminant Type 1 Diabetes—An Important Subtype in East Asia. Diabetes Metab Res Rev (2011) 27(8):959–64. doi: 10.1002/dmrr.1236
26. Gamble DR, Taylor KW. Seasonal Incidence of Diabetes Mellitus. Br Med J (1969) 3(5671):631–3. doi: 10.1136/bmj.3.5671.631
27. Schneider DA, von Herrath MG. Potential Viral Pathogenic Mechanism in Human Type 1 Diabetes. Diabetologia (2014) 57(10):2009–18. doi: 10.1007/s00125-014-3340-7
28. Christen U, von Herrath MG. Infections and Autoimmunity–Good or Bad? J Immunol (2005) 174(12):7481–6. doi: 10.4049/jimmunol.174.12.7481
29. Jaeckel E, Manns M, von Herrath M. Viruses and Diabetes. Ann N Y Acad Sci (2002) 958:7–25. doi: 10.1111/j.1749-6632.2002.tb02943.x
30. Hara N, Alkanani AK, Ir D, Robertson CE, Wagner BD, Frank DN, et al. Prevention of Virus-Induced Type 1 Diabetes With Antibiotic Therapy. J Immunol (2012) 189(8):3805–14. doi: 10.4049/jimmunol.1201257
31. Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The Prevalence of Enteroviral Capsid Protein Vp1 Immunostaining in Pancreatic Islets in Human Type 1 Diabetes. Diabetologia (2009) 52(6):1143–51. doi: 10.1007/s00125-009-1276-0
32. Roep BO, Kleijwegt FS, Van Halteren AGS, Bonato V, Boggi U, Vendrame F, et al. Islet Inflammation and CXCL10 in Recent-Onset Type 1 Diabetes. Clin Exp Immunol (2010) 159(3):338–43. doi: 10.1111/j.1365-2249.2009.04087.x
33. Tanaka S, Nishida Y, Aida K, Maruyama T, Shimada A, Suzuki M, et al. Enterovirus Infection, CXC Chemokine Ligand 10 (CXCL10), and CXCR3 Circuit: A Mechanism of Accelerated Beta-Cell Failure in Fulminant Type 1 Diabetes. Diabetes (2009) 58(10):2285–91. doi: 10.2337/db09-0091
34. Zipris D. Toll-Like Receptors and Type 1 Diabetes. Adv Exp Med Biol (2010) 654:585–610. doi: 10.1007/978-90-481-3271-3_25
35. Zipris D. Innate Immunity in Type 1 Diabetes. Diabetes Metab Res Rev (2011) 27(8):824–9. doi: 10.1002/dmrr.1256
36. Alkanani AK, Hara N, Gianani R, Zipris D. Kilham Rat Virus-Induced Type 1 Diabetes Involves Beta Cell Infection and Intra-Islet JAK–STAT Activation Prior to Insulitis. Virology (2014) 468–470:19–27. doi: 10.1016/j.virol.2014.07.041
37. Alkanani AK, Hara N, Lien E, Ir D, Kotter CV, Robertson CE, et al. Induction of Diabetes in the RIP-B7.1 Mouse Model Is Critically Dependent on TLR3 and MyD88 Pathways and Is Associated With Alterations in the Intestinal Microbiome. Diabetes (2014) 63(2):619–31. doi: 10.2337/db13-1007
38. Hara N, Alkanani A, Dinarello C, Zipris D. Histone Deacetylase Inhibitor Suppresses Virus-Induced Proinflammatory Responses and Type 1 Diabetes. J Mol Med (2013) 92:1–10. doi: 10.1007/s00109-013-1078-1
39. Hara N, Alkanani AK, Dinarello CA, Zipris D. Modulation of Virus-Induced Innate Immunity and Type 1 Diabetes by IL-1 Blockade. Innate Immun (2014) 20(6):574–84. doi: 10.1177/1753425913502242
40. Zipris D, Lien E, Nair A, Xie JX, Greiner DL, Mordes JP, et al. TLR9-Signaling Pathways Are Involved in Kilham Rat Virus-Induced Autoimmune Diabetes in the Biobreeding Diabetes-Resistant Rat. J Immunol (2007) 178(2):693–701. doi: 10.4049/jimmunol.178.2.693
41. Zipris D, Lien E, Xie JX, Greiner DL, Mordes JP, Rossini AA. Tlr Activation Synergizes With Kilham Rat Virus Infection to Induce Diabetes in BBDR Rats. J Immunol (2005) 174(1):131–42. doi: 10.4049/jimmunol.174.1.131
42. Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD, et al. Alterations in Intestinal Microbiota Correlate With Susceptibility to Type 1 Diabetes. Diabetes (2015) 64:3510–20. doi: 10.2337/db14-1847
43. Meyers A, Shah R, Gottlieb P, Zipris D. Altered Toll-Like Receptor Signaling Pathways in Human Type 1 Diabetes. J Mol Med (2010) 88(12):1221–31. doi: 10.1007/s00109-010-0666-6
44. Needell JC, Brown MN, Zipris D. Involvement of Adipose Tissue Inflammation and Dysfunction in Virus-Induced Type 1 Diabetes. J Endocrinol (2018) 238(1):61. doi: 10.1530/JOE-18-0131
45. Brown DD, Salzman LA. Sequence Homology Between the Structural Proteins of Kilham Rat Virus. J Virol (1984) 49(3):1018–20. doi: 10.1128/jvi.49.3.1018-1020.1984
46. Cherry JD. Parvovirus Infections in Children and Adults. Adv Pediatr (1999) 46:245–69. doi: 10.1203/00006450-199908000-00019
47. Jacoby RO, Ball-Goodrich LJ, Besselsen DG, McKisic MD, Riley LK, Smith AL. Rodent Parvovirus Infections. Lab Anim Sci (1996) 46(4):370–80.
48. Bultmann BD, Klingel K, Sotlar K, Bock CT, Baba HA, Sauter M, et al. Fatal Parvovirus B19-Associated Myocarditis Clinically Mimicking Ischemic Heart Disease: An Endothelial Cell-Mediated Disease. Hum Pathol (2003) 34(1):92–5. doi: 10.1053/hupa.2003.48
49. Schowengerdt KO, Ni J, Denfield SW, Gajarski RJ, Bowles NE, Rosenthal G, et al. Association of Parvovirus B19 Genome in Children With Myocarditis and Cardiac Allograft Rejection: Diagnosis Using the Polymerase Chain Reaction. Circulation (1997) 96(10):3549–54. doi: 10.1161/01.CIR.96.10.3549
50. Tsay GJ, Zouali M. Unscrambling the Role of Human Parvovirus B19 Signaling in Systemic Autoimmunity. Biochem Pharmacol (2006) 72(11):1453–9. doi: 10.1016/j.bcp.2006.04.023
51. Nesher G, Rubinow A, Sonnenblick M. Efficacy and Adverse Effects of Different Corticosteroid Dose Regimens in Temporal Arteritis: A Retrospective Study. Clin Exp Rheumatol (1997) 15(3):303–6.
52. Guberski DL, Thomas VA, Shek WR, Like AA, Handler ES, Rossini AA, et al. Induction of Type I Diabetes by Kilham’s Rat Virus in Diabetes-Resistant BB/Wor Rats. Science (1991) 254(5034):1010–3. doi: 10.1126/science.1658938
53. Rossini AA, Handler ES, Mordes JP, Greiner DL. Human Autoimmune Diabetes Mellitus: Lessons From BB Rats and NOD Mice–Caveat Emptor. Clin Immunol Immunopathol (1995) 74(1):2–9. doi: 10.1006/clin.1995.1002
54. Greiner DL, Rossini AA, Mordes JP. Translating Data From Animal Models Into Methods for Preventing Human Autoimmune Diabetes Mellitus: Caveat Emptor and Primum non Nocere. Clin Immunol (2001) 100(2):134–43. doi: 10.1006/clim.2001.5075
55. Ellerman KE, Richards CA, Guberski DL, Shek WR, Like AA. Kilham Rat Triggers T-Cell-Dependent Autoimmune Diabetes in Multiple Strains of Rat. Diabetes (1996) 45(5):557–62. doi: 10.2337/diabetes.45.5.557
56. McKeever U, Mordes JP, Greiner DL, Appel MC, Rozing J, Handler ES, et al. Adoptive Transfer of Autoimmune Diabetes and Thyroiditis to Athymic Rats. Proc Natl Acad Sci USA (1990) 87(19):7618–22. doi: 10.1073/pnas.87.19.7618
57. Mordes JP, Guberski DL, Leif JH, Woda BA, Flanagan JF, Greiner DL, et al. Lew.1WR1 Rats Develop Autoimmune Diabetes Spontaneously and in Response to Environmental Perturbation. Diabetes (2005) 54(9):2727–33. doi: 10.2337/diabetes.54.9.2727
58. Tirabassi RS, Guberski DL, Blankenhorn EP, Leif JH, Woda BA, Liu Z, et al. Infection With Viruses From Several Families Triggers Autoimmune Diabetes in LEW.1WR1 Rats. Diabetes (2009) 59(1):110. doi: 10.2337/db09-0255
59. Ji-Won Y, Hee-Sook J. Viruses in Type 1 Diabetes: Brief Review. ILAR J (2004) 45(3):343–8. doi: 10.1093/ilar.45.3.343
60. Carty M, Bowie AG. Recent Insights Into the Role of Toll-like Receptors in Viral Infection. Clin Exp Immunol (2010) 161(3):397–406. doi: 10.1111/j.1365-2249.2010.04196.x
61. Mordes JP, Bortell R, Blankenhorn EP, Rossini AA, Greiner DL. Rat Models of Type 1 Diabetes: Genetics, Environment, and Autoimmunity. ILAR J (2004) 45(3):278–91. doi: 10.1093/ilar.45.3.278
62. Mordes JP, Zipris D, Liu Z, Blankenhorn EP. Viruses and Autoimmune Diabetes in Rats. In: Taylor K, Hyöty H, Toniolo A, Zuckerman JA, editors. Diabetes and Viruses. New York, NY: Springer New York (2013). p. 57–70. doi: 10.1007/978-1-4614-4051-2_7
63. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes (2016) 65(3):719–31. doi: 10.2337/db15-0779
64. Krogvold L, Wiberg A, Edwin B, Buanes T, Jahnsen FL, Hanssen KF, et al. Insulitis and Characterisation of Infiltrating T Cells in Surgical Pancreatic Tail Resections From Patients at Onset of Type 1 Diabetes. Diabetologia (2016) 59(3):492–501. doi: 10.1007/s00125-015-3820-4
65. Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender Bias in Autoimmunity Is Influenced by Microbiota. Immunity (2013) 39(2):400–12. doi: 10.1016/j.immuni.2013.08.013
66. Chung YH, Jun HS, Son M, Bao M, Bae HY, Kang Y, et al. Cellular and Molecular Mechanism for Kilham Rat Virus-Induced Autoimmune Diabetes in DR-BB Rats. J Immunol (2000) 165(5):2866–76. doi: 10.4049/jimmunol.165.5.2866
67. Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota Regulates Type 1 Diabetes Through Toll-like Receptors. Proc Natl Acad Sci USA (2015) 112(32):9973–7. doi: 10.1073/pnas.1508740112
68. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate Immunity and Intestinal Microbiota in the Development of Type 1 Diabetes. Nature (2008) 455(7216):1109–13. doi: 10.1038/nature07336
69. Londono P, Komura A, Hara N, Zipris D. Brief Dexamethasone Treatment During Acute Infection Prevents Virus-Induced Autoimmune Diabetes. Clin Immunol (2010) 135(3):401–11. doi: 10.1016/j.clim.2010.01.007
70. Sai P, Rivereau AS. Prevention of Diabetes in the Nonobese Diabetic Mouse by Oral Immunological Treatments. Comparative Efficiency of Human Insulin and Two Bacterial Antigens, Lipopolysacharide From Escherichia Coli and Glycoprotein Extract From Klebsiella Pneumoniae. Diabetes Metab (1996) 22(5):341–8.
71. Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-Activated B Cells Down-Regulate Th1 Immunity and Prevent Autoimmune Diabetes in Nonobese Diabetic Mice. J Immunol (2001) 167(2):1081–9. doi: 10.4049/jimmunol.167.2.1081
72. Quintana FJ, Rotem A, Carmi P, Cohen IR. Vaccination With Empty Plasmid DNA or CpG Oligonucleotide Inhibits Diabetes in Nonobese Diabetic Mice: Modulation of Spontaneous 60-Kda Heat Shock Protein Autoimmunity. J Immunol (2000) 165(11):6148–55. doi: 10.4049/jimmunol.165.11.6148
73. Aumeunier A, Grela F, Ramadan A, Pham Van L, Bardel E, Gomez Alcala A, et al. Systemic Toll-Like Receptor Stimulation Suppresses Experimental Allergic Asthma and Autoimmune Diabetes in NOD Mice. PloS One (2010) 5(7):e11484. doi: 10.1371/journal.pone.0011484
74. Pasare C, Medzhitov R. Toll-Dependent Control Mechanisms of CD4 T Cell Activation. Immunity (2004) 21(5):733–41. doi: 10.1016/j.immuni.2004.10.006
75. Barton GM, Medzhitov R. Toll-Like Receptors and Their Ligands. Curr Top Microbiol Immunol (2002) 270:81–92. doi: 10.1007/978-3-642-59430-4_5
76. Ferrero-Miliani L, Nielsen OH, Andersen PS, Girardin SE. Chronic Inflammation: Importance of NOD2 and NALP3 in Interleukin-1β Generation. Clin Exp Immunol (2007) 147(2):227–35. doi: 10.1111/j.1365-2249.2006.03261.x
77. Kawai T, Akira S. Toll-Like Receptors and Their Crosstalk With Other Innate Receptors in Infection and Immunity. Immunity (2011) 34(5):637–50. doi: 10.1016/j.immuni.2011.05.006
78. Hajishengallis G, Lambris JD. More Than Complementing Tolls: Complement–Toll-Like Receptor Synergy and Crosstalk in Innate Immunity and Inflammation. Immunol Rev (2016) 274(1):233–44. doi: 10.1111/imr.12467
79. Iwasaki A, Medzhitov R. Toll-Like Receptor Control of the Adaptive Immune Responses. Nat Immunol (2004) 5(10):987–95. doi: 10.1038/ni1112
80. Iwasaki A, Medzhitov R. Control of Adaptive Immunity by the Innate Immune System. Nat Immunol (2015) 16(4):343–53. doi: 10.1038/ni.3123
81. Janeway CA Jr, Medzhitov R. Innate Immune Recognition. Annu Rev Immunol (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
82. Medzhitov R. Toll-Like Receptors and Innate Immunity. Nat Rev Immunol (2001) 1(2):135–45. doi: 10.1038/35100529
83. Medzhitov R. Recognition of Microorganisms and Activation of the Immune Response. Nature (2007) 449(7164):819–26. doi: 10.1038/nature06246
84. Medzhitov R. Origin and Physiological Roles of Inflammation. Nature (2008) 454(7203):428–35. doi: 10.1038/nature07201
85. Medzhitov R, Janeway C Jr. Innate Immunity. N Engl J Med (2000) 343(5):338–44. doi: 10.1056/NEJM200008033430506
86. Medzhitov R, Janeway C Jr. Innate Immune Recognition: Mechanisms and Pathways. Immunol Rev (2000) 173:89–97. doi: 10.1034/j.1600-065X.2000.917309.x
87. Medzhitov R, Janeway C Jr. The Toll Receptor Family and Microbial Recognition. Trends Microbiol (2000) 8(10):452–6. doi: 10.1016/S0966-842X(00)01845-X
88. Beutler BA. Tlrs and Innate Immunity. Blood (2009) 113(7):1399–407. doi: 10.1182/blood-2008-07-019307
89. Bottazzi B, Doni A, Garlanda C, Mantovani A. An Integrated View of Humoral Innate Immunity: Pentraxins as a Paradigm. Annu Rev Immunol (2010) 28(1):157–83. doi: 10.1146/annurev-immunol-030409-101305
90. Holmskov U, Thiel S, Jensenius JC. Collectins and Ficolins: Humoral Lectins of the Innate Immune Defense. Annu Rev Immunol (2003) 21(1):547–78. doi: 10.1146/annurev.immunol.21.120601.140954
91. Zipris D, Hillebrands JL, Welsh RM, Rozing J, Xie JX, Mordes JP, et al. Infections That Induce Autoimmune Diabetes in BBDR Rats Modulate CD4+CD25+ T Cell Populations. J Immunol (2003) 170(7):3592–602. doi: 10.4049/jimmunol.170.7.3592
92. Needell JC, Ir D, Robertson CE, Kroehl ME, Frank DN, Zipris D. Maternal Treatment With Short-Chain Fatty Acids Modulates the Intestinal Microbiota and Immunity and Ameliorates Type 1 Diabetes in the Offspring. PloS One (2017) 12(9):e0183786. doi: 10.1371/journal.pone.0183786
93. Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, et al. A Human Pluripotent Stem Cell-Based Platform to Study SARS-Cov-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell (2020) 27(1):125–36.e7 doi: 10.1016/j.stem.2020.06.015.
94. Tittel SR, Rosenbauer J, Kamrath C, Ziegler J, Reschke F, Hammersen J, et al. Did the COVID-19 Lockdown Affect the Incidence of Pediatric Type 1 Diabetes in Germany? Diabetes Care (2020) 43(11):e172–3. doi: 10.2337/dc20-1633
95. Unsworth R, Wallace S, Oliver NS, Yeung S, Kshirsagar A, Naidu H, et al. New-Onset Type 1 Diabetes in Children During COVID-19: Multicenter Regional Findings in the U. K. Diabetes Care (2020) 43(11):e170–1. doi: 10.2337/dc20-1551
96. Marchand L, Pecquet M, Luyton C. Type 1 Diabetes Onset Triggered by COVID-19. Acta Diabetol (2020) 57(10):1265–6. doi: 10.1007/s00592-020-01570-0
97. Sathish T, Chandrika Anton M. Newly Diagnosed Diabetes in Patients With Mild to Moderate COVID-19. Diabetes Metab Syndrome: Clin Res Rev (2021) 15(2):569–71. doi: 10.1016/j.dsx.2021.02.034
98. Rabizadeh S, Hajmiri M, Rajab A, Emadi Kouchak H, Nakhjavani M. Severe Diabetic Ketoacidosis and Coronavirus Disease 2019 (COVID-19) Infection in a Teenage Patient With Newly Diagnosed Diabetes. J Pediatr Endocrinol Metab (2020) 33(9):1241–3. doi: 10.1515/jpem-2020-0296
99. Dehghani Firouzabadi F, Dehghani Firouzabadi M, Moosaie F, Rafiee S, Esteghamati A. Type 1 Diabetic Manifestations in a Young Man Triggered by COVID-19. Clin Case Rep (2021) 9(6):e04211. doi: 10.1002/ccr3.4211
100. Chee YJ, Ng SJH, Yeoh E. Diabetic Ketoacidosis Precipitated by Covid-19 in a Patient With Newly Diagnosed Diabetes Mellitus. Diabetes Res Clin Pract (2020) 164:108166–. doi: 10.1016/j.diabres.2020.108166
101. Heaney AI, Griffin GD, Simon EL. Newly Diagnosed Diabetes and Diabetic Ketoacidosis Precipitated by COVID-19 Infection. Am J Emergency Med (2020) 38(11):2491.e3–.e4. doi: 10.1016/j.ajem.2020.05.114
102. Boddu SK, Aurangabadkar G, Kuchay MS. New Onset Diabetes, Type 1 Diabetes and COVID-19. Diabetes Metab Syndrome: Clin Res Rev (2020) 14(6):2211–7. doi: 10.1016/j.dsx.2020.11.012
103. Marchand L, Luyton C. Response to Comment on Marchand Et al. Type 1 Diabetes Onset Triggered by COVID-19; and Follow-Up of the Case. Acta Diabetol (2021) 58(9):1285–6. doi: 10.1007/s00592-021-01762-2
104. Meek TH, Morton GJ. The Role of Leptin in Diabetes: Metabolic Effects. Diabetologia (2016) 59(5):928–32. doi: 10.1007/s00125-016-3898-3
105. Laubner K, Kieffer TJ, Lam NT, Niu X, Jakob F, Seufert J. Inhibition of Preproinsulin Gene Expression by Leptin Induction of Suppressor of Cytokine Signaling 3 in Pancreatic β-Cells. Diabetes (2005) 54(12):3410–7. doi: 10.2337/diabetes.54.12.3410
106. Seufert J, Kieffer TJ, Leech CA, Holz GG, Moritz W, Ricordi C, et al. Leptin Suppression of Insulin Secretion and Gene Expression in Human Pancreatic Islets: Implications for the Development of Adipogenic Diabetes Mellitus. J Clin Endocrinol Metab (1999) 84(2):670–6. doi: 10.1210/jc.84.2.670
107. Zhu Q, Scherer PE. Immunologic and Endocrine Functions of Adipose Tissue: Implications for Kidney Disease. Nat Rev Nephrol (2017) 14:105. doi: 10.1038/nrneph.2017.157
108. Kajimura S. Advances in the Understanding of Adipose Tissue Biology. Nat Rev Endocrinol (2017) 13:69. doi: 10.1038/nrendo.2016.211
109. Wallberg-Henriksson H, Zierath JR. Exercise Remodels Subcutaneous Fat Tissue and Improves Metabolism. Nat Rev Endocrinol (2015) 11:198. doi: 10.1038/nrendo.2015.24
110. Stern JH, Rutkowski JM, Scherer PE. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis Through Adipose Tissue Crosstalk. Cell Metab (2016) 23(5):770–84. doi: 10.1016/j.cmet.2016.04.011
111. Bjørndal B, Burri L, Staalesen V, Skorve J, Berge RK. Different Adipose Depots: Their Role in the Development of Metabolic Syndrome and Mitochondrial Response to Hypolipidemic Agents. J Obes (2011) 2011:490650. doi: 10.1155/2011/490650
112. Wajchenberg BL. Subcutaneous and Visceral Adipose Tissue: Their Relation to the Metabolic Syndrome. Endocr Rev (2000) 21(6):697–738. doi: 10.1210/edrv.21.6.0415
113. Hajer GR, van Haeften TW, Visseren FLJ. Adipose Tissue Dysfunction in Obesity, Diabetes, and Vascular Diseases. Eur Heart J (2008) 29(24):2959–71. doi: 10.1093/eurheartj/ehn387
114. Kulkarni RN, Wang ZL, Wang RM, Hurley JD, Smith DM, Ghatei MA, et al. Leptin Rapidly Suppresses Insulin Release From Insulinoma Cells, Rat and Human Islets and, In Vivo, in Mice. J Clin Invest (1997) 100(11):2729–36. doi: 10.1172/JCI119818
115. Kieffer TJ, Heller RS, Habener JF. Leptin Receptors Expressed on Pancreatic β-Cells. Biochem Biophys Res Commun (1996) 224(2):522–7. doi: 10.1006/bbrc.1996.1059
116. Fehmann HC, Berghöfer P, Brandhorst D, Brandhorst H, Hering B, Bretzel RG, et al. Leptin Inhibition of Insulin Secretion From Isolated Human Islets. Acta Diabetol (1997) 34(4):249–52. doi: 10.1007/s005920050083
117. Emilsson V, Liu Y-L, Cawthorne MA, Morton NM, Davenport M. Expression of the Functional Leptin Receptor mRNA in Pancreatic Islets and Direct Inhibitory Action of Leptin on Insulin Secretion. Diabetes (1997) 46(2):313–6. doi: 10.2337/diabetes.46.2.313
118. Tanabe K, Amo-Shiinoki K, Hatanaka M, Tanizawa Y. Interorgan Crosstalk Contributing to β-Cell Dysfunction. J Diabetes Res (2017) 2017:8. doi: 10.1155/2017/3605178
119. Taylor EB. The Complex Role of Adipokines in Obesity, Inflammation, and Autoimmunity. Clin Sci (2021) 135(6):731–52. doi: 10.1042/CS20200895
120. Dubuc PU. The Development of Obesity, Hyperinsulinemia, and Hyperglycemia in Ob/Ob Mice. Metab - Clin Exp (1976) 25(12):1567–74. doi: 10.1016/0026-0495(76)90109-8
121. Friedman JM, Halaas JL. Leptin and the Regulation of Body Weight in Mammals. Nature (1998) 395(6704):763–70. doi: 10.1038/27376
122. Wyse BM, Dulin WE. The Influence of Age and Dietary Conditions on Diabetes in the Db Mouse. Diabetologia (1970) 6(3):268–73. doi: 10.1007/BF01212237
123. Dunmore SJ, Brown JEP. The Role of Adipokines in Beta-Cell Failure of Type 2 Diabetes. J Endocrinol (2013) 216(1):T37–45. doi: 10.1530/JOE-12-0278
124. Morioka T, Asilmaz E, Hu J, Dishinger JF, Kurpad AJ, Elias CF, et al. Disruption of Leptin Receptor Expression in the Pancreas Directly Affects β Cell Growth and Function in Mice. J Clin Invest (2007) 117(10):2860–8. doi: 10.1172/JCI30910
125. Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human Leptin Stimulates Proliferation and Activation of Human Circulating Monocytes. Cell Immunol (1999) 194(1):6–11. doi: 10.1006/cimm.1999.1490
126. Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The Adipocyte-Secreted Protein Acrp30 Enhances Hepatic Insulin Action. Nat Med (2001) 7:947. doi: 10.1038/90992
127. Ye RS, Holland WL, Gordillo R, Wang M, Wang QA, Shao ML, et al. Adiponectin Is Essential for Lipid Homeostasis and Survival Under Insulin Deficiency and Promotes Beta-Cell Regeneration. Elife (2014) 3:e03851. doi: 10.7554/eLife.03851
128. Wang QA, Ye R, Wang M, Scherer PE. Adiponectin-Mediated Antilipotoxic Effects in Regenerating Pancreatic Islets. Endocrinology (2015) 156(6):2019–28. doi: 10.1210/en.2015-1066
129. Del Rey A, Besedovsky HO. Metabolic and Neuroendocrine Effects of Pro-Inflammatory Cytokines. Eur J Clin Invest (1992) 22 Suppl 1:10–5.
130. Donath MY, Dinarello CA, Mandrup-Poulsen T. Targeting Innate Immune Mediators in Type 1 and Type 2 Diabetes. Nat Rev Immunol (2019) 19:734–6. doi: 10.1038/s41577-019-0213-9
131. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J Clin Invest (2003) 112(12):1821–30. doi: 10.1172/JCI200319451
132. Ye J. Emerging Role of Adipose Tissue Hypoxia in Obesity and Insulin Resistance. Int J Obes (2009) 33(1):54–66. doi: 10.1038/ijo.2008.229
133. Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front Physiol (2020) 10:1607. doi: 10.3389/fphys.2019.01607
134. Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuñiga FA. Association Between Insulin Resistance and the Development of Cardiovascular Disease. Cardiovasc Diabetol (2018) 17(1):122. doi: 10.1186/s12933-018-0762-4
135. Shulman GI. Cellular Mechanisms of Insulin Resistance. J Clin Invest (2000) 106(2):171–6. doi: 10.1172/JCI10583
136. Donath MY, Shoelson SE. Type 2 Diabetes as an Inflammatory Disease. Nat Rev Immunol (2011) 11(2):98–107. doi: 10.1038/nri2925
137. Donath MY. Targeting Inflammation in the Treatment of Type 2 Diabetes: Time to Start. Nat Rev Drug Discovery (2014) 13(6):465–76. doi: 10.1038/nrd4275
138. Herder C, Dalmas E, Böni-Schnetzler M, Donath MY. The IL-1 Pathway in Type 2 Diabetes and Cardiovascular Complications. Trends Endocrinol Metab (2015) 26(10):551–63. doi: 10.1016/j.tem.2015.08.001
139. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor Antagonist in Type 2 Diabetes Mellitus. N Engl J Med (2007) 356(15):1517–26. doi: 10.1056/NEJMoa065213
140. Mittendorfer B. Origins of Metabolic Complications in Obesity: Adipose Tissue and Free Fatty Acid Trafficking. Curr Opin Clin Nutr Metab Care (2011) 14(6):535–41. doi: 10.1097/MCO.0b013e32834ad8b6
141. Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, et al. Mechanisms of β-Cell Death in Type 2 Diabetes. Diabetes (2005) 54(suppl 2):S108–S13. doi: 10.2337/diabetes.54.suppl_2.S108
142. Donath MY, Mandrup-Poulsen T. The Use of interleukin-1-receptor Antagonists in the Treatment of Diabetes Mellitus. Nat Clin Pract Endocrinol Metab (2008) 4(5):240–1. doi: 10.1038/ncpendmet0783
143. Olefsky JM, Glass CK. Macrophages, Inflammation, and Insulin Resistance. Annu Rev Physiol (2010) 72(1):219–46. doi: 10.1146/annurev-physiol-021909-135846
144. O’Rourke RW. Inflammation in Obesity-Related Disease. Surgery (2009) 145(3):255–9. doi: 10.1016/j.surg.2008.08.038
145. Tilg H, Moschen AR. Adipocytokines: Mediators Linking Adipose Tissue, Inflammation and Immunity. Nat Rev Immunol (2006) 6(10):772–83. doi: 10.1038/nri1937
146. de Luca C, Olefsky JM. Inflammation and Insulin Resistance. FEBS Lett (2008) 582(1):97–105. doi: 10.1016/j.febslet.2007.11.057
147. Donath MY. When Metabolism Met Immunology. Nat Immunol (2013) 14(5):421–2. doi: 10.1038/ni.2591
148. Iyer A, Fairlie DP, Prins JB, Hammock BD, Brown L. Inflammatory Lipid Mediators in Adipocyte Function and Obesity. Nat Rev Endocrinol (2010) 6(2):71–82. doi: 10.1038/nrendo.2009.264
149. Houstis N, Rosen ED, Lander ES. Reactive Oxygen Species Have a Causal Role in Multiple Forms of Insulin Resistance. Nature (2006) 440:944. doi: 10.1038/nature04634
150. Nishikawa T, Edelstein D, Du XL, Yamagishi S-I, Matsumura T, Kaneda Y, et al. Normalizing Mitochondrial Superoxide Production Blocks Three Pathways of Hyperglycaemic Damage. Nature (2000) 404:787. doi: 10.1038/35008121
151. Hotamisligil GS. Inflammation, Metaflammation and Immunometabolic Disorders. Nature (2017) 542(7640):177–85. doi: 10.1038/nature21363
152. Hotamisligil GS. Inflammation and Metabolic Disorders. Nature (2006) 444(7121):860–7. doi: 10.1038/nature05485
153. Özcan U, Cao Q, Yilmaz E, Lee A-H, Iwakoshi NN, Özdelen E, et al. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science (2004) 306(5695):457–61. doi: 10.1126/science.1103160
154. Keane KN, Cruzat VF, Carlessi R, de Bittencourt PIH, Newsholme P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and β-Cell Dysfunction. Oxid Med Cell Longev (2015) 2015:181643. doi: 10.1155/2015/181643
155. Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, et al. Insulin Resistance Causes Inflammation in Adipose Tissue. J Clin Invest (2018) 128(4):1538–50. doi: 10.1172/JCI96139
156. Donath MY. Inflammation as a Sensor of Metabolic Stress in Obesity and Type 2 Diabetes. Endocrinology (2011) 152(11):4005–6. doi: 10.1210/en.2011-1691
157. Gonzalez LL, Garrie K, Turner MD. Type 2 Diabetes – An Autoinflammatory Disease Driven by Metabolic Stress. Biochim Biophys Acta (BBA) - Mol Basis Dis (2018) 1864(11):3805–23. doi: 10.1016/j.bbadis.2018.08.034
158. Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA. Islet Inflammation in Type 2 Diabetes: From Metabolic Stress to Therapy. Diabetes Care (2008) 31(Supplement 2):S161–4. doi: 10.2337/dc08-s243
159. Oh YS. Mechanistic Insights Into Pancreatic Beta-Cell Mass Regulation by Glucose and Free Fatty Acids. Anat Cell Biol (2015) 48(1):16–24. doi: 10.5115/acb.2015.48.1.16
160. Oh YS, Bae GD, Baek DJ, Park E-Y, Jun H-S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front Endocrinol (Lausanne) (2018) 9:384. doi: 10.3389/fendo.2018.00384
161. Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, et al. Islet β-Cell Endoplasmic Reticulum Stress Precedes the Onset of Type 1 Diabetes in the Nonobese Diabetic Mouse Model. Diabetes (2012) 61(4):818–27. doi: 10.2337/db11-1293
Keywords: type 1 diabetes, Kilham rat virus, Inflammation, visceral adipose tissue (VAT), beta cells
Citation: Zipris D (2021) Visceral Adipose Tissue: A New Target Organ in Virus-Induced Type 1 Diabetes. Front. Immunol. 12:702506. doi: 10.3389/fimmu.2021.702506
Received: 29 April 2021; Accepted: 19 July 2021;
Published: 04 August 2021.
Edited by:
Li Wen, Yale University, United StatesReviewed by:
Aditi Arun Narsale, San Diego Biomedical Research Institute, United StatesAloysius Klingelhutz, The University of Iowa, United States
Copyright © 2021 Zipris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danny Zipris, emRhbm55QGlubmF0ZWJpb3RlY2hub2xvZ2llcy5jb20=