
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 July 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.702142
This article is part of the Research TopicCurrent Trends in Exploiting Molecular Signaling in Bacteria-Host CrosstalkView all 30 articles
 Robin Smyth1
Robin Smyth1 Jim Sun1,2*
Jim Sun1,2*The global antimicrobial resistance crisis poses a significant threat to humankind in the coming decades. Challenges associated with the development of novel antibiotics underscore the urgent need to develop alternative treatment strategies to combat bacterial infections. Host-directed therapy is a promising new therapeutic strategy that aims to boost the host immune response to bacteria rather than target the pathogen itself, thereby circumventing the development of antibiotic resistance. However, host-directed therapy depends on the identification of druggable host targets or proteins with key functions in antibacterial defense. Protein Kinase R (PKR) is a well-characterized human kinase with established roles in cancer, metabolic disorders, neurodegeneration, and antiviral defense. However, its role in antibacterial defense has been surprisingly underappreciated. Although the canonical role of PKR is to inhibit protein translation during viral infection, this kinase senses and responds to multiple types of cellular stress by regulating cell-signaling pathways involved in inflammation, cell death, and autophagy – mechanisms that are all critical for a protective host response against bacterial pathogens. Indeed, there is accumulating evidence to demonstrate that PKR contributes significantly to the immune response to a variety of bacterial pathogens. Importantly, there are existing pharmacological modulators of PKR that are well-tolerated in animals, indicating that PKR is a feasible target for host-directed therapy. In this review, we provide an overview of immune cell functions regulated by PKR and summarize the current knowledge on the role and functions of PKR in bacterial infections. We also review the non-canonical activators of PKR and speculate on the potential mechanisms that trigger activation of PKR during bacterial infection. Finally, we provide an overview of existing pharmacological modulators of PKR that could be explored as novel treatment strategies for bacterial infections.
Interferon-induced, double-stranded RNA-activated protein kinase, also known as protein kinase R (PKR), is a ubiquitously and constitutively expressed serine-threonine kinase that is specifically found in vertebrate cells (1). PKR is encoded in humans by the EIF2AK2 gene located on chromosome 2 and is 551 amino acids in length (2, 3). This kinase senses and responds to multiple types of cellular stress by regulating cell-signaling pathways involved in inflammation, cell death, and autophagy. As such, dysregulation of PKR expression or activation has been linked to multiple human diseases, including neurodegeneration, cancer, metabolic disorders, and viral infections [reviewed in-depth previously: (4–6)]. In particular, the most well-characterized function of PKR is to sense viral double-stranded RNA (dsRNA) for its canonical role in antiviral defense (7).
The best characterized transcriptional inducers of PKR are type I interferons (IFN). Type I IFN are produced in response to pathogen-associated molecular patterns (PAMPs) and signal through the IFNα/IFNβ receptor (IFNAR) to induce transcription of numerous genes that assist in antiviral defense [reviewed in-depth previously (8)]. These genes are referred to as interferon-stimulated genes (ISGs) and include EIF2AK2. Indeed, the PKR promoter contains an interferon-stimulated response element (ISRE), thus prompting transcription of the EIF2AK2 gene in response to type I IFN signaling (9). The PKR promoter also contains a kinase conserved sequence upstream of the ISRE, which possesses binding sites for the transcription factors Sp1 and Sp3 (9, 10). Sp1 and Sp3 cooperatively activate basal PKR expression in the absence of IFN stimulation (10). The canonical activator of PKR is viral dsRNA (7); however, PKR can also be activated in response to a variety of stress signals, including serum starvation and endoplasmic reticulum (ER) stress (11). This activation of PKR in the absence of viral dsRNA is mediated by PKR protein activator (PACT). PACT is phosphorylated under cellular stress and physically interacts with PKR to trigger its activation (11). The more well-characterized roles of PKR include regulation of protein translation and apoptosis in response to viral infection, controlling cell proliferation and differentiation, and supressing tumour growth [reviewed in-depth previously: (4, 12)].
Structurally, PKR has a C-terminal kinase domain and an N-terminal dsRNA binding domain (dsRBD). The dsRBD consists of two dsRNA binding motifs (dsRBM1 and dsRBM2), both of which are required for the high-affinity interaction with viral dsRNA (13). Recognition and binding of dsRNA by the dsRBMs triggers homodimerization of PKR and its subsequent autophosphorylation (14, 15). PKR is autophosphorylated at multiple serine and threonine residues, including Thr446 and Thr451, which are consistently phosphorylated during PKR activation (14, 16). Once activated, PKR phosphorylates serine 51 (Ser51) on the alpha subunit of eukaryotic initiation factor-2 (EIF2α). PKR belongs to a family of four EIF2α kinases, all of which share the same substrate. The other three EIF2α kinases are heme-regulated inhibitor, PKR-like endoplasmic reticulum kinase (PERK), and general control non-depressible 2 (GCN2), which are activated by heme depletion (17), ER stress (18), and amino acid starvation (19), respectively.
Phosphorylation of EIF2α by PKR or any of the other three EIF2α kinases results in inhibition of protein translation. Mammalian EIF2 is critical for initiating polypeptide chain synthesis since it promotes the delivery of initiator methionyl transfer RNA (Met-tRNAi) to the 40S ribosome. EIF2α binds Met-tRNAi in a GTP-dependent manner, forming a ternary complex that interacts with the 40S subunit. Following Met-tRNAi delivery, EIF5 promotes GTP hydrolysis of EIF2-GTP, triggering the release of EIF2-GDP from the 48S initiation complex. EIF2-GDP must be regenerated to EIF2-GTP by the GTP exchange factor EIF2B, since EIF2-GDP is inactive. When Ser51 on EIF2α is phosphorylated, the affinity of EIF2 for EIF2B is increased up to 100-fold (20). Consequently, phosphorylated EIF2α competes with EIF2-GDP for binding of EIF2B. This competitive inhibition prevents the regeneration of active EIF2-GTP and as such, initiation of translation is substantially reduced (21). Functionally, this mechanism prevents the translation of both cellular and viral messenger RNA (mRNA), thereby inhibiting viral replication.
Although the canonical role of PKR is to inhibit protein translation during viral infection [reviewed in-depth previously: (12, 22–24)], PKR is in fact a versatile kinase that controls signal transduction pathways to mediate transcription and cellular processes. Given that PKR regulates critical immune cell functions in inflammation, cell death, and autophagy – processes that are critical for host immunity against bacterial infections – it is logical to expect that the role of PKR extends beyond that of antiviral defense. Surprisingly, the role of PKR in antibacterial defense is understudied and underappreciated relative to its role in antiviral defense. However, there is accumulating evidence demonstrating that PKR contributes significantly to the immune response to a variety of bacterial infections. This review provides an overview of immune cell functions regulated by PKR and includes an exhaustive summary of the current knowledge on the role and function of PKR in pathogenic bacterial infections. Specifically, we organized the sections by grouping bacteria under Gram-positive, Gram-negative, or mycobacteria, and included every report that we could find which linked PKR to pathogenic bacteria that cause human disease. We also review the non-canonical activators of PKR and speculate on the potential mechanisms that trigger PKR activation during bacterial infection. Finally, we provide an overview of existing pharmacological modulators of PKR that could be explored for treatment of bacterial infections.
PKR regulates inflammation by activating multiple downstream effectors. One mechanism utilized by PKR to regulate inflammation is by activating mitogen-activated protein kinases (MAPK) such as p38 and c-Jun N-terminal kinase (JNK) (25–27) (Figure 1). p38 and JNK trigger activating transcription factor-2 (ATF2) and c-Jun to induce the expression of proinflammatory cytokines such as interleukin (IL)-1β and tumour necrosis factor-alpha (TNFα) (28, 29) (Figure 1). JNK is activated by MAPK kinase (MKK)4 or MKK7, whereas p38 is activated by MKK3 or MKK6. Depletion of PKR by stable knockdown impaired the phosphorylation of JNK and p38 in response to dsRNA or a mutant strain of vaccinia virus (25). Another group observed that PKR expression was required for full activation of JNK and p38 in response to polyinosinic:polycytidylic acid [poly(I:C)], lipopolysaccharide (LPS), IL-1β, and TNFα (26). In that same study, deletion of PKR in mouse embryonic fibroblasts (MEFs) was observed to inhibit MKK4 and MKK3/6 phosphorylation in response to the same stimuli. Interestingly, PKR deletion did not impact p38 or JNK activation in response to stressors that impact cellular components on a global scale, such as ultraviolet radiation, osmotic shock, and heat shock. The PKR-dependent stress stimuli were limited to pro-inflammatory ligands that bind distinct receptors, i.e. PKR as a receptor for dsRNA, CD14 and toll-like receptor (TLR)-4 for LPS, and the respective cytokine receptors for IL-1β and TNFα. Thus, it is suggested that PKR mediates activation of p38 and JNK in response to “receptor-mediated” pro-inflammatory stress stimuli, but not in response to “globally acting” stressors (26). Results from a different study support the observation that PKR activates p38 by acting upstream of MKK6 (27).
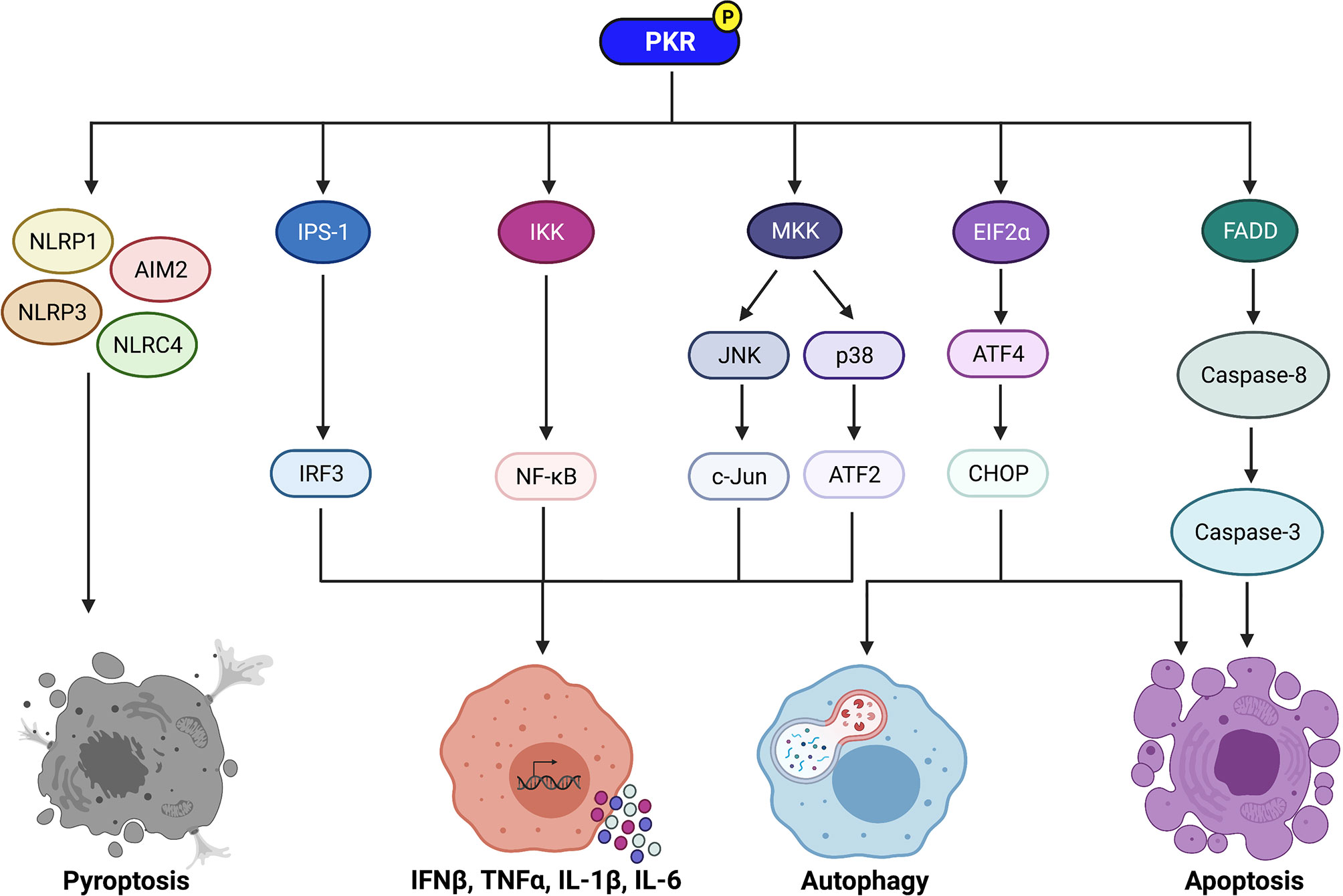
Figure 1 Signaling pathways regulated by PKR to control immune cell functions. PKR regulates downstream effectors such as IPS-1, IKK, and MKK to activate IRF3, NF-κB, and the MAP kinases JNK and p38, respectively. IRF3 induces transcription of IFNβ, whereas NF-κB induces transcription of pro-inflammatory cytokines such as TNFα, IL-1β, and IL-6. Active JNK and p38 trigger c-Jun and ATF2 activation, respectively, which also induce transcription of pro-inflammatory cytokines. PKR also plays a role in induction of pyroptosis via activation of the NLRP1, AIM2, NLRP3, and NLRC4 inflammasomes. Phosphorylation of EIF2α by PKR leads to increased translation of ATF4, which then increases expression of CHOP. ATF4 and CHOP trigger activation of autophagy by inducing transcription of essential autophagy genes. In addition, CHOP promotes apoptosis during periods of prolonged cellular stress. PKR can also induce apoptosis independently of EIF2α phosphorylation via activation of the FADD/caspase-8/caspase-3 pathway. Created with BioRender.com.
PKR can also regulate inflammation through its effects on NF-κB (Figure 1). PKR indirectly activates NF-κB by activating IκB kinase (IKK) (30). Active IKK targets IκB, a negative regulator of NF-κB, for proteasomal degradation, thereby triggering its dissociation from NF-κB. NF-κB is then free to translocate to the nucleus, where it induces transcription of genes encoding pro-inflammatory cytokines such as IL-1, IL-6, and TNFα (30, 31) (Figure 1). PKR triggers NF-κB activation in response to poly(I:C) and viral infection (30, 32, 33). While PKR physically associates with IKK (31), it remains unclear whether PKR is a structural or catalytic component in the activation of IKK (4, 12). Furthermore, although there are numerous reports of PKR activating NF-κB, there exists some contradictory evidence. Indeed, two independent studies have reported that PKR deficiency results in normal or only slightly decreased NF-κB activity in response to TNFα (34, 35). These findings suggest that PKR may play an important role in activating NF-κB in response to certain stimuli (e.g. dsRNA), but not others (e.g. TNFα treatment).
A third pathway by which PKR mediates inflammation is inducing type I IFN (Figure 1). The canonical function of type I IFN is antiviral defense as they can directly limit intracellular viral replication and induce antiviral responses from T cells, natural killer cells, and B cells [reviewed in-depth previously: (36)]. However, there is increasing evidence to show that type I IFN also play a role in regulating inflammation, since they can alter the production of both pro- and anti-inflammatory mediators. For example, IFNβ treatment has been observed to increase MCP-1 and IP-10 production via STAT1 activation, which are crucial chemoattractants that recruit immune cells to the site of infection (37). On the other hand, IFNβ treatment also increases the production of the anti-inflammatory cytokine IL-10 and inhibits the secretion of pro-inflammatory cytokines such as IL-6 and TNFα via STAT3 activation in LPS-stimulated cells (37). In addition, type I IFN have been shown to regulate inflammation by controlling inflammasome activation (38). Interestingly, IFNβ was shown to enhance AIM2-dependent IL-1β secretion in response to Francisella tularenis or Listeria monocytogenes infection (39, 40) and mediates caspase-11-dependent pyroptosis during Escherichia coli and Salmonella Typhimurium infection (41, 42). These findings provide evidence that type I IFN regulate inflammation in the context of bacterial infections.
PKR activates IFNβ promoter stimulator-1 (IPS-1) signaling, which induces interferon regulatory factor 3 (IRF3) and subsequent transcription of IFNβ (43) (Figure 1). Numerous studies have found that PKR deficiency impairs IFNβ production upon stimulation with poly(I:C) or viral infection (44). Curiously, Schultz and colleagues observed that IFNβ transcription was highly induced in PKR-deficient cells following encephalomyocarditis virus (EMCV) infection, but little or no IFNβ protein was produced (45). This suggested that PKR impacts the post-transcriptional regulation of IFNβ production. Indeed, further investigation revealed that IFNβ transcripts produced in EMCV-infected PKR-deficient cells completely lack a poly(A)tail (45). This indicates that PKR is required for the integrity of IFNβ mRNA and its translation into functional protein. As such, PKR has reported roles in increasing both transcription and translation of IFNβ. Since type I IFN modulates inflammation, the ability of PKR to induce type I IFN is another mechanism that allows the kinase to regulate the inflammatory response.
Collectively, PKR plays a central role in regulating inflammation through numerous downstream effectors, including MAPK p38 and JNK, NF-κB, and type I IFN (Figure 1). Although acute inflammation assists with microbial clearance, chronic inflammation can result in tissue damage and deleterious effects to the host. In addition, many of the signaling pathways that regulate inflammation also have key roles in the control of cell death, a major cellular response that has consequences for bacterial infections. Inflammation must therefore be tightly regulated to achieve an optimal outcome for the host during bacterial infection.
As mentioned in the previous section, inflammation and cell death share common signaling pathways and are tightly intertwined. It is thus unsurprising that PKR has reported roles in regulating cell death pathways such as apoptosis and pyroptosis. PKR was first shown to induce apoptosis in 1994, when expression of the kinase in HeLa cells triggered rapid apoptosis, an effect that was not observed in cells expressing a mutant form of PKR (46). Later studies using MEFs from PKR knockout mice or NIH3T3 cells expressing a catalytically inactive PKR mutant reinforced the finding that PKR plays a pro-apoptotic role during cellular stress (47, 48). The ability to induce apoptosis of virus-infected cells is now a well-known function of PKR (4, 49). Indeed, PKR regulates apoptosis in response to numerous viruses, including poxviruses, influenza, and EMCV (50–52). Importantly, PKR can induce apoptosis in the absence of viral infection, such as in response to LPS, TNFα, serum starvation, or ER stress (47, 48, 53, 54), suggesting a role for this kinase in non-viral contexts. PKR-dependent apoptosis in the absence of viral infection is reported to rely on PACT, which triggers PKR activation in response to numerous stressors such as serum withdrawal and ER stress (11, 55).
PKR regulates apoptosis through EIF2α-dependent mechanisms (32, 47) (Figure 1). Phosphorylation of EIF2α results in repression of global protein translation but preferential translation of activating transcription factor-4 (ATF4) mRNA (56). ATF4 increases expression of C/EBP homologous protein (CHOP), a transcription factor that promotes apoptosis during periods of prolonged cellular stress. CHOP induces apoptosis by increasing expression of the pro-apoptotic protein Bim and decreasing expression of the anti-apoptotic protein Bcl-2 (57, 58). PKR can also induce apoptosis independently of EIF2α phosphorylation. One such mechanism is through activation of the FADD/caspase-8/caspase-3 pathway (59–61) (Figure 1). In addition, NF-κB, ATF-3, and p53 are downstream effectors of PKR that are speculated to contribute to PKR-mediated apoptosis (4, 12), although the exact mechanisms remain unclear.
In addition to apoptosis, PKR is reported to regulate inflammasome activation and pyroptosis (Figure 1). Lu and colleagues observed that activation of PKR was triggered by multiple inflammasome activators, and deletion of PKR inhibited high mobility group protein B1 (HMGB1) release, IL-1β secretion, and caspase-1 activation in response to inflammasome-inducing stimuli (62). Importantly, deletion of PKR also prevented cell death of macrophages treated with inflammasome activators. In the same study, PKR was shown to physically associate with NLRP1, NLRP3, NLRC4, and AIM2 inflammasomes. A non-phosphorylatable PKR mutant failed to bind NLRP3 and was unable to activate the inflammasome, indicating that phosphorylated PKR physically interacts with inflammasomes to induce their activation (62).
However, the role of PKR in pyroptosis remains controversial, as a study by Yim et al. reported that PKR represses inflammasome activation (63). Nigericin-treated peritoneal macrophages from PKR knockout or kinase-dead PKR mice resulted in elevated levels of IL-1β and IL-18, and enhanced caspase-1 activity. Ablation of PKR expression or kinase activity also promoted the expression or assembly of inflammasome components such as NLRP3 and pro-IL-1β. Since EIF2α phosphorylation impairs protein translation, the authors speculated that PKR represses translation of inflammasome constituents by its kinase activity on EIF2α. Indeed, pre-treatment of peritoneal macrophages with the small molecule ISRIB, which counteracts the effect of EIF2α phosphorylation, increased the expression of pro-IL-1β. Yim and colleagues attribute the discrepancy between their findings and those from Lu et al. to a difference in the mouse models used (63). Notably, Lu et al. used mice from a mixed 129Sv/BALB/c background, which have attenuated inflammasome activity due to diminished caspase-11 expression (64). To complicate matters further, He et al. showed that PKR is dispensable for inflammasome activity (65). However, the in vitro differentiation of mouse macrophages from this study is considered to be the source of the discrepancy compared to findings from the Yim et al. study, where primary macrophages were used without further manipulation in vitro (63). Altogether, these conflicting reports show that the animal and cellular model is a key determinant in whether PKR mediates inflammasome activation and pyroptotic cell death.
Nevertheless, the role of PKR in cell death has major implications for bacterial infections because the mode of cell death in bacteria-infected cells influences the outcome of infection. For example, apoptotic cell death is generally considered as a pro-host response during Mycobacterium tuberculosis infection, since it enhances cross-priming of T cells and limits inflammation (66). In contrast, pyroptotic cell death is considered as an anti-host response during M. tuberculosis infection, since it results in bacterial dissemination and host tissue damage (67, 68). As such, the ability of PKR to regulate cell death pathways such as apoptosis and pyroptosis is pertinent for host immunity against bacterial pathogens.
PKR has been shown to induce autophagy, which may be a mechanism to balance its role in activating cell death and inflammation. Autophagy is a homeostatic process that generates nutrients by degrading cytoplasmic constituents, and this pathway is speculated to be protective against cell death (69). Although the canonical targets of autophagic degradation are organelles and proteins, it is now known that the autophagy pathway can selectively target pathogens for degradation in a process termed xenophagy (70, 71). Indeed, the autophagy pathway has been shown to degrade intracellular bacteria, viruses, and parasites (70–73). In the context of bacterial infections, selective autophagy allows for progressive elimination of bacteria (73), decreased bacterial burden (71), and improved control of inflammation (74). As such, autophagy is a critical pathway in antibacterial defense.
There is increasing evidence for PKR’s role in autophagy. An initial study by Tallóczy and colleagues found that EIF2α phosphorylation by the yeast EIF2α kinase GCN2 was essential for starvation-induced autophagy of yeast cells (75). Expression of PKR in GCN2-disrupted yeast rescued autophagy in these cells, indicating a role for PKR in induction of autophagy. A follow-up study by the same group showed that PKR can induce autophagy during viral infection (76). Infection of PKR knockout MEFs with a Herpes Simplex Virus-1 (HSV-1) mutant lacking the PKR-inhibiting virulence factor ICP34.5 significantly inhibited colocalization of virions with autophagosomes and resulted in increased viral titres compared to wild-type cells (76). More recently, Ogolla and colleagues reported that PKR induces autophagy in RAW264.7 macrophages during infection with the parasite Toxoplasma gondii (77). Indeed, PKR expression was required for LC3 accumulation around the parasite and lysosomal fusion with vacuole-containing T. gondii in macrophages, which are crucial events during selective autophagy. The autophagy-inducing role of PKR during T. gondii appears to be critical for controlling infection, as PKR knockout mice exhibited higher parasite loads compared to wild-type mice (77).
The mechanism of PKR-dependent induction of autophagy is likely through phosphorylation of EIF2α (Figure 1). Indeed, Tallóczy and colleagues observed that MEFs expressing a non-phosphorylatable EIF2α mutant displayed diminished xenophagic degradation of HSV-1 proteins to the same extent as PKR knockout MEFs, and the viral titres were equivalent between the two cell-lines (76). This indicates that PKR-induced autophagic degradation of HSV-1 is mediated through phosphorylation of EIF2α. Phosphorylation of EIF2α increases the expression of transcription factors ATF4 and CHOP, which then induce transcription of essential autophagy genes such as Map1lc3b, Atg12, Atg3, Atg7, and Becn1 (78). It is also possible that PKR induces autophagy through its downstream effects on MAPK p38 and JNK. Although the specific mechanisms by which p38 and JNK activate autophagy during bacterial infection have not yet been elucidated, these MAPK are reported to induce autophagy during starvation through indirect effects on Beclin-1, a crucial protein in the autophagy pathway. Specifically, p38 activates MK2 and MK3, which results in the phosphorylation of Beclin-1 (79). In contrast, JNK phosphorylates a negative regulator of Beclin-1, Bcl-2 (80), to trigger its dissociation from Beclin-1 (81). Given the critical role of autophagy against intracellular bacterial pathogens, the ability of PKR to trigger autophagy is likely a key cellular response to certain bacterial pathogens.
Staphylococcus aureus is a Gram-positive, facultative intracellular bacterium that is commonly found in the upper respiratory tract and skin flora of humans. Although S. aureus is typically a commensal bacterium, it can become an opportunistic pathogen and cause a range of illnesses with varying severity, including cellulitis, osteomyelitis, pneumonia, and meningitis. S. aureus was initially characterized as an extracellular bacterium. However, it is now understood that S. aureus is phagocytosed by neutrophils and macrophages, where it manipulates the phagosome maturation pathway to avoid lysosomal degradation (82, 83). S. aureus secretes a pore-forming toxin known as α-toxin, which assists S. aureus in escaping from macrophage phagosomes (82) and leads to activation of the autophagy pathway (84). PKR was first suspected to play a role during S. aureus infection when Kloft and colleagues observed that autophagy is activated in α-toxin-treated HaCaT cells and that phosphorylation of EIF2α was required for the accumulation of autophagosomes (85). Further examination revealed that both PKR and GCN2 are phosphorylated in response to α-toxin, whereas PERK is not. These findings suggest that PKR- and/or GCN2-mediated phosphorylation of EIF2α activates autophagy during S. aureus infection (85). However, further investigation is required to determine which of these EIF2α kinases is responsible for this effect. Interestingly, PKR might also play a proapoptotic role during S. aureus infection, although the current evidence is limited. Treatment of cardiac cells with RNA extracted from S. aureus was shown to trigger PKR activation and induce apoptosis, whereas cells treated with a PKR inhibitor were resistant to apoptosis (86) (Table 1). S. aureus RNA induced cleavage of capase-8, caspase-3, and caspase-9, an effect that was prevented following treatment with a PKR inhibitor (86). As such, PKR may activate caspase-8- and caspase-9-mediated apoptosis during S. aureus infection. Although the findings from the aforementioned studies suggest a role for PKR in the antibacterial response to S. aureus, neither of these studies examined the overall effect of PKR expression on the control of bacterial replication. Furthermore, these studies did not use live S. aureus infection, but instead examined the effect of bacterial RNA or α-toxin. As such, future investigation is required to determine whether PKR plays a role during infection with the live bacterium.

Table 1 Role of PKR in different bacterial infections.
Bacillus anthracis is a Gram-positive, extracellular bacterium that is the causative agent of anthrax. B. anthracis secretes a major virulence factor known as lethal toxin, which enters host cells and cleaves MAPK kinases to impair MAPK signaling pathways (99). In doing so, lethal toxin disrupts crucial processes such as proliferation, survival, and inflammation in host cells. Anthrax lethal toxin has been shown to trigger phosphorylation of PKR in murine peritoneal macrophages (62), which suggests that PKR would be activated by B. anthracis infection. The role of PKR during B. anthracis infection remains unclear; however, there is evidence to suggest that PKR regulates cell death during infection with this bacterium. Indeed, Hsu and colleagues observed that deletion of PKR in bone marrow-derived macrophages (BMDMs) infected with live B. anthracis had markedly reduced apoptosis levels compared to wild-type macrophages (87) (Table 1). Further examination into the mechanism revealed that PKR is required for TLR4-dependent apoptosis of B. anthracis-infected macrophages (87). Hett and colleagues provided further evidence that PKR regulates TLR4-dependent apoptosis in response to B. anthracis, since pharmacological inhibition of PKR protected LPS-sensitized macrophages from apoptosis in response to lethal toxin (88). Interestingly, Lu and colleagues observed that PKR was required for inflammasome activation in lethal toxin-treated mouse peritoneal macrophages, as indicated by impaired caspase-1 activation, IL-1β cleavage, and HMGB1 release in PKR-deficient cells (62) (Table 1). Furthermore, PKR deficiency protected macrophages from lethal toxin-induced cytotoxicity. Consistent with the findings from Lu et al., Hett and colleagues reported that PKR is required for pyroptosis in response to lethal toxin challenge (88). Indeed, PKR knockdown protected J774 macrophages from cell death following treatment with lethal toxin, and was accompanied by reduced caspase-1 activity and IL-18 secretion (Table 1). Interestingly, lethal toxin was not observed to induce PKR phosphorylation, and treatment with pharmacological inhibitors of PKR did not protect macrophages from lethal toxin-induced cell death. These findings indicate that the catalytic activity of PKR is not required for PKR-dependent pyroptosis in response to lethal toxin. As such, the authors speculate that PKR mediates activation of pyroptosis through physical interactions with inflammasomes (88). Since PKR regulates inflammasome activation in response to anthrax lethal toxin, it is possible that PKR would have the same activity in response to infection with live B. anthracis. However, findings from studies focusing on one virulence factor in vitro at the expense of studying whole organism infections with the live bacterium must be interpreted with caution. For example, Kang and colleagues reported that B. anthracis spores and lethal toxin induce IL-1β via functionally distinct pathways, demonstrating that different components of the same bacterium can mediate different signaling pathways (100). In fact, spore-induced IL-1β was observed to limit B. anthracis infection, whereas lethal toxin-induced IL-1β enabled B. anthracis to escape host defenses (100). Furthermore, although PKR expression was reported by Hsu et al. to be required for TLR4-dependent macrophage apoptosis in response to live B. anthracis infection, Moayeri and colleagues reported later that same year that lethal toxin-mediated lethality in mice was independent of TLR4 function (101). Altogether, these studies emphasize the need for in vivo studies with the live bacterium, since experiments focusing on single components/virulence factors of a bacterium in vitro can produce different results. In addition, the overall effect of PKR expression on bacterial burden during live B. anthracis infection has not been investigated.
Listeria monocytogenes is a Gram-positive, facultative intracellular bacterium that causes listeriosis, which can manifest as sepsis, meningitis, pneumonia, urinary tract infections, and gastroenteritis (102). This bacterium has been shown to induce phosphorylation of EIF2α in the murine macrophage cell-line RAW264.7, which indicates that PKR or another EIF2α kinase is activated during Listeria infection (103). Expression of a non-phosphorylatable mutant of EIF2α in MEFs resulted in increased bacterial invasion, suggesting an important role for EIF2α kinases in the antibacterial response to Listeria (103). Indeed, our group observed that L. monocytogenes infection triggers increased levels of total and phosphorylated PKR protein in the human macrophage cell-line THP-1 (98). Since L. monocytogenes is established to invade the cytosol and initiate a type I IFN response, and type I IFN signaling induces PKR transcription, Valderrama and colleagues examined the effect of L. monocytogenes infection on EIF2AK2 (PKR) mRNA levels (89). As expected, L. monocytogenes infection resulted in increased PKR transcription levels in murine bone marrow-derived dendritic cells (BMDCs) and BMDMs. Interestingly, an LLO knockout strain of L. monocytogenes, which lacks the LLO virulence factor required for phagosome escape, was able to induce transcription of PKR, although not to the same extent as wild-type L. monocytogenes (89). This indicates that cytosolic localization of L. monocytogenes is not required to induce transcription of PKR. Further investigation revealed that murine myeloid cells treated with a PKR inhibitor have reduced expression of CHOP following L. monocytogenes infection (89) (Table 1). CHOP expression is increased following EIF2α phosphorylation and induces a number of effects such as proinflammatory cytokine secretion and inflammasome activation (104–107). As such, the authors speculate that PKR-dependent activation of CHOP triggers an inflammatory response to L. monocytogenes infection. However, this inflammatory role of PKR during Listeria infection may be harmful to the host, since CHOP knockout mice had decreased splenic cell death, decreased bacterial proliferation, and better survival compared to wild-type mice (89). Nevertheless, the direct effects of PKR modulation on L. monocytogenes survival and host outcome have yet to be studied.
Salmonella enterica serovar Typhimurium is a Gram-negative, facultative intracellular bacterium that causes gastroenteritis in humans. Salmonella Typhimurium can infect both humans and animals, and infection is commonly acquired by consuming contaminated food products. S. Typhimurium infection has been observed to increase mRNA levels of EIF2AK2 in MEFs (108), and we and others have reported that S. Typhimurium increases total and phosphorylated PKR protein levels in macrophages (87, 98). These findings suggest that PKR plays a role in the antibacterial response to Salmonella. Indeed, PKR has been shown to regulate host cell death during Salmonella infection. For example, Hsu et al. reported that PKR knockout BMDMs are resistant to apoptosis induced by S. Typhimurium infection in comparison to wild-type macrophages (87) (Table 1). Macrophages expressing a non-phosphorylatable mutant of EIF2α were also less susceptible to Salmonella-induced apoptosis, although a residual apoptotic response in these macrophages suggested the existence of another PKR-dependent pro-apoptotic pathway. The authors examined the levels of Salmonella-induced apoptosis in IRF3 knockout macrophages, and these macrophages were also resistant to Salmonella-induced apoptosis (87). As such, PKR may regulate two pro-apoptotic pathways during Salmonella infection, one involving EIF2α and the other involving IRF3. Interestingly, the apoptotic response to Salmonella was considerably reduced in BMDMs from TLR4-deficient mice, indicating that PKR is required for TLR4-dependent apoptosis during Salmonella infection (87). PKR is also reported to play a role in pyroptosis activation during Salmonella infection. Lu and colleagues observed that PKR deficiency in S. Typhimurium-infected murine peritoneal macrophages significantly inhibited caspase-1 activation, IL-1β cleavage, and HMGB1 secretion, as well as Salmonella-induced cell death (62) (Table 1). PKR was shown to physically associate with inflammasomes in response to a variety of pyroptosis-inducing stimuli, and PKR expression was required for inflammasome activation. However, the specific mechanism of inflammasome activation by PKR remains unknown. Taken together, the findings from Hsu et al. and Lu et al. indicate that PKR is an important mediator of both apoptosis and pyroptosis in Salmonella-infected macrophages. However, the overall effect of PKR on bacterial survival remains to be elucidated.
Lastly, findings from Yeung and colleagues loosely suggest a role for PKR in the proper functioning of Salmonella-infected macrophages (109). The outcome of Salmonella infection is largely influenced by how the bacteria initially interact with macrophages, however the human macrophage factors required for Salmonella uptake are incompletely understood. A genome-scale CRISPR knockout library screening of THP-1 macrophages to identify loss-of-function mutations conferring resistance to Salmonella uptake identified NHLRC2, a gene involved in actin dynamics (109). NHLRC2 mutant macrophages were hyperinflammatory, unable to interact and phagocytose S. Typhimurium, and exhibited atypical morphology. Interestingly, PKR was shown to physically associate with NHLRC2, and NHLRC2 knockout macrophages had reduced PKR expression (109). Since PKR expression appears to be linked to NHLRC2 expression, and these two proteins physically interact, perhaps PKR contributes to NHLRC2-dependent uptake of Salmonella and the proper functioning of infected macrophages.
Escherichia coli is a Gram-negative bacterium that can either be harmless or pathogenic depending on the particular strain. Some strains of E. coli are part of the normal intestinal microbiota, whereas other strains can cause diarrhea, urinary tract infections, or other illnesses. There have been a few studies examining the effect of PKR deletion during E. coli infection both in vitro and in the mouse model. Lu et al. reported that PKR expression and phosphorylation is triggered in murine peritoneal macrophages following E. coli infection in vitro (62). It was observed that E. coli-induced pyroptosis was severely impaired in infected PKR knockout macrophages, as indicated by decreased cell death and impaired IL-1β production (Table 1). Furthermore, transfection with E. coli RNA in BMDCs significantly activated capase-1 and stimulated IL-1β cleavage in wild-type cells but not PKR knockout cells (62) (Table 1). Lu and colleagues also performed in vivo experiments and observed that serum IL-1β, IL-18, and HMGB1 levels were significantly reduced in E. coli-infected PKR knockout mice compared to control mice. Notably, PKR knockout mice had significantly lower titers of E. coli in the spleen and peritoneal cavity compared to control mice (62) (Table 1). Altogether, the findings from this study suggest that PKR plays a role in inflammasome activation during E. coli infection, and PKR expression appears to be conducive for E. coli persistence. However, it is difficult to interpret the results from these in vivo studies since non-virulent E. coli was used, as reflected by >109 CFU challenge doses (62). In contrast, a recent study from the same group using virulent E. coli showed that genetic deficiency or pharmacological inhibition of PKR did not affect bacterial loads in E. coli-infected mice (90) (Table 1). The discrepancy in findings between these two studies was suggested by the authors to be due to differences in the route of E. coli infection (90). In the first report, Lu et al. performed intraperitoneal infection, whereas the second study used intravenous infection (62, 90). Overall, more careful in vivo studies using virulent E. coli (typically reflected by dose challenges of 107 CFU or lower) must be performed before conclusions can be drawn.
A role for PKR in inflammasome activation was also reported by Poon and colleagues (91). This group observed that PKR knockout mice had diminished peripheral inflammatory responses following subcutaneous E. coli challenge. Indeed, PKR deficiency resulted in decreased IL-1β mRNA expression in the livers of infected mice (91) (Table 1). However, PKR deletion had no effect on the bacterial burden of E. coli-infected mice. Interestingly, while the core components of sickness (anorexia and motor impairments) were comparable between E.coli-infected wild-type and PKR knockout mice, the behavioural components of sickness – including reduced burrowing, exploratory activity deficits, and social withdrawal – were only observed in PKR knockout mice (91).
Finally, PKR might also regulate apoptotic cell death during E. coli infection. E. coli RNA was shown to activate PKR and induce apoptosis of cardiac cells, an effect that was blocked following pharmacological PKR inhibition (86) (Table 1). Altogether, the findings from the studies discussed above suggest that PKR regulates cell death pathways such as pyroptosis and apoptosis during E. coli infection. However, the effect of PKR expression on bacterial burden remains unclear, as there are discrepancies between the current findings.
Yersinia pseudotuberculosis is a Gram-negative, extracellular bacterium that causes Far East scarlet-like fever in humans. PKR is suggested to play a role in regulating inflammation and macrophage apoptosis during Y. pseudotuberculosis infection. Shrethsa and colleagues observed that EIF2α is phosphorylated in Y. pseudotuberculosis-infected RAW264.7 macrophages, and that phosphorylated EIF2α opposed bacterial invasion (103). Furthermore, phosphorylation of EIF2α was required for Yersinia-induced NF-κB activation and TNFα expression in MEFs. These findings indicated that PKR or another EIF2α kinase is involved in antibacterial defense against Y. pseudotuberculosis. Indeed, expression of the Yersinia virulence factor YopJ, which is known to inhibit EIF2α signaling in response to various stress stimuli, was also shown to inhibit PKR signalling in MEFs (103). This finding suggests that PKR is the kinase responsible for phosphorylating EIF2α during Y. pseudotuberculosis infection. PKR might also play a role in Yersinia-induced apoptosis, since deletion of PKR in infected BMDMs substantially impaired macrophage apoptosis when compared to wild-type macrophages (87) (Table 1). The apoptotic response to Y. pseudotuberculosis was considerably reduced in BMDMs from TLR4-deficient mice, suggesting that PKR is required for TLR4-dependent apoptosis during Yersinia infection (103). The effect of PKR activity on the survival of Y. pseudotuberculosis has yet to be determined.
Chlamydia trachomatis is a Gram-negative, obligate intracellular bacterium that is the causative agent of chlamydia. There is growing evidence to suggest that PKR is involved in the immune response to C. trachomatis. Shrestha and colleagues first reported that expression of a non-phosphorylatable mutant of EIF2α increased C. trachomatis invasion in MEFs, indicating that PKR or another EIF2α kinase assists in antibacterial defense against Chlamydia (103). They later reported that PKR knockout MEFs had increased C. trachomatis invasion levels compared to wild-type cells, with invasion levels similar to the levels observed in cells expressing the non-phosphorylatable EIF2α mutant (92) (Table 1). These findings indicate that PKR plays a role in the antibacterial response to C. trachomatis. Indeed, a later study revealed that C. trachomatis infection triggers phosphorylation of PKR in human monocyte-derived dendritic cells and murine BMDMs, and this activation of PKR is required for C. trachomatis-induced IFNβ production (93) (Table 1). This suggests that PKR plays a role in regulating inflammation during Chlamydia infection, but whether this PKR-dependent induction of IFNβ is ultimately conducive or detrimental to bacterial survival was not explored. However, findings from Qiu et al. indicate that the type I IFN-inducing role of PKR is conducive for C. trachomatis infection (110). IFNAR knockout mice were more resistant to Chlamydia infection compared to wild-type mice, as indicated by a smaller decrease in body weight, lower bacterial burden, and milder lung pathology in infected mice. The increased resistance to C. trachomatis infection in the knockout mice was attributed to higher numbers of bactericidal macrophages in the lung resulting from decreased macrophage apoptosis. Notably, these knockout mice had lower expression of PKR. Since PKR plays a proapoptotic role during viral infection, the authors speculate that type I IFN indirectly promote C. trachomatis infection by activating PKR, thus resulting in macrophage apoptosis and increased bacterial persistence (110). Additional experimentation is required to assess whether PKR plays a proapoptotic role during C. trachomatis infection. Altogether, PKR is activated by Chlamydia infection, where it then induces IFNβ production and limits bacterial invasion. Further investigation is necessary to determine whether PKR activity in response to Chlamydia infection is ultimately beneficial or harmful to the host.
Legionella pneumophilia is a Gram-negative, facultative intracellular bacterium that is the causative agent of Legionnaires’ disease. To examine the role of PKR during L. pneumophilia infection, Mallama and colleagues generated PKR knockdown cells using the human U937 macrophage-like cell-line (94). PKR deficiency impaired IL-6 secretion in response to L. pneumophilia infection (Table 1), a critical cytokine in antibacterial defense against this bacterium. As such, Mallama et al. concluded that PKR expression is required for an optimal cytokine response to L. pneumophilia infection (94). However, PKR deficiency did not affect the intracellular burden of L. pneumophilia in macrophages (Table 1). As such, although PKR appears to regulate inflammation during L. pneumophilia infection, the kinase may be dispensable for bacterial control.
Mycobacterium is a genus comprising over 190 species of bacteria. Mycobacteria possess an atypical outer membrane structure and organization comprised of mycolic acids, arabinogalactan, and numerous unique glycolipids (111). This unique cell wall contributes to the robustness of mycobacterial species and their natural tolerance to many antibiotics (112). As such, mycobacteria do not stain Gram-positive or Gram-negative, which contributes to their phylogenetic ambiguity. Instead, mycobacterial species are classified as acid-fast bacteria due to their ability to resist acid or ethanol-based decolorization procedures during staining. The most notorious species of mycobacteria relevant for human disease is Mycobacterium tuberculosis, a facultative intracellular bacterium that causes tuberculosis (TB) disease. Mycobacteria can be broadly classified into three major groups: the Mycobacterium tuberculosis complex, which comprises mycobacteria that cause TB disease (e.g. M. tuberculosis and Mycobacterium bovis); Mycobacterium leprae, which causes leprosy; and non-tuberculosis mycobacteria, which includes all other mycobacteria that do not cause TB or leprosy. There is growing evidence that PKR plays a role in the immune response to mycobacteria. For example, one study revealed that PKR phosphorylation is triggered in human monocytes infected with bacillus Calmette-Guérin (BCG), a live attenuated form of M. bovis used for TB vaccination (95). Pharmacological inhibition of PKR decreased mRNA and protein levels of crucial anti-BCG cytokines in infected monocytes, including TNFα, IL-6, and IL-10 (Table 1). PKR inhibition also prevented the binding of NF-κB to DNA and impaired downstream activation of the MAPK ERK1/2 in treated monocytes. As such, the authors speculate that PKR induces anti-BCG cytokine production via downstream activation of ERK1/2 and NF-κB (95). Other studies have shown that EIF2AK2 mRNA increases during infection with BCG and M. tuberculosis (113, 114). The findings that PKR expression and activation is triggered by mycobacterial infections suggest that PKR plays a role in the immune response to mycobacteria. However, the effect of PKR on mycobacterial burden was not examined in these studies.
Since PKR plays a pro-apoptotic role during viral infection, Wu and colleagues examined the effect of PKR deletion on macrophage apoptosis and bacterial burden during M. tuberculosis infection (115). While they initially reported that PKR deficiency in mice enhances macrophage apoptosis and decreases M. tuberculosis burden in the lungs (115), there was a discrepancy between the genetic backgrounds of the mutant and control mice used in the study (116). A follow-up study led by Mundhra and colleagues using mutant and control mice from the same genetic background revealed that PKR deficiency had no effect on apoptosis or M. tuberculosis burden (96, 116) (Table 1). Therefore, PKR was concluded to be dispensable during M. tuberculosis infection. In contrast, Ranjbar and colleagues demonstrated that PKR expression and activation is triggered during M. tuberculosis infection in THP-1 monocytes, and PKR deletion in M. tuberculosis-infected macrophages increased the bacterial burden (97) (Table 1). Although Ranjbar and colleagues observed an effect of PKR modulation on M. tuberculosis burden, the specific mechanism regulated by PKR to restrict M. tuberculosis growth was not investigated. As such, our group sought to examine the effect of PKR modulation on the intracellular survival of M. tuberculosis and characterize the specific mechanism(s) involved. Consistent with the findings from Ranjbar and colleagues, we showed that PKR expression and activation is triggered in M. tuberculosis-infected THP-1 and primary human macrophages, and deletion of PKR increases intracellular M. tuberculosis survival compared to control macrophages (98) (Table 1). Strikingly, we also observed that genetic overexpression of PKR decreases the intracellular survival of M. tuberculosis by nearly 80% compared to control macrophages (Table 1). Immunological profiling of infected macrophages overexpressing PKR showed increased production of IP-10 and reduced production of IL-6, two cytokines that are reported to activate and inhibit IFNγ-dependent autophagy, respectively (117, 118). Indeed, we determined that the ability of PKR overexpression to limit intracellular M. tuberculosis survival is due to the induction of selective autophagy (98) (Table 1). Although our group did not elucidate the events downstream of PKR activation that led to induction of autophagy, the mechanism is likely through the phosphorylation of EIF2α and downstream induction of ATF4. However, MAPK p38 and JNK have been shown to be important for induction of autophagy during M. tuberculosis infection, and IL-6 inhibits MAPK phosphorylation to block autophagy in M. tuberculosis-infected macrophages (118). Importantly, PKR activates p38 and JNK (25–27), and macrophages overexpressing PKR had reduced production of IL-6 (98). As such, it is possible that autophagy induction by PKR is dependent on a mechanism involving MAPK, whether by a direct effect of PKR on MAPK activation or an indirect effect from decreased IL-6 production. We did not observe an effect of PKR modulation on apoptosis or overall cell death of M. tuberculosis-infected macrophages (98), consistent with the findings from Mundhra and colleagues (96). Taken together, the results from these studies indicate that PKR plays a critical role in the antibacterial response to mycobacteria. The ability of PKR to limit the intracellular survival of M. tuberculosis in macrophages appears to be through selective autophagy induction, rather than by regulating apoptosis.
It is worth noting that the overall effect of PKR during M. tuberculosis infection may be context dependent. Although our findings and the results from Ranjbar et al. indicate that PKR limits M. tuberculosis survival in vitro (97, 98), a recent report using sst1-susceptible mice suggests that PKR contributes to macrophage necrosis in TB granulomas (119). sst1-susceptible mice develop necrotic inflammatory lung lesions similar to human TB granulomas, whereas their congenic B6 counterparts do not (120). PKR phosphorylation was increased in TNFα-treated macrophages extracted from sst1-susceptible mice compared to macrophages extracted from B6 mice (119). PKR-mediated phosphorylation of EIF2α led to hyperinduction of ATF3 and integrated stress response (ISR)-target genes in these macrophages. Importantly, pharmacological inhibition of the ISR prevented the development of necrosis in lung granulomas of M. tuberculosis-infected sst1-susceptible mice and reduced the bacterial burden (119). This finding suggests that PKR contributes to necrosis of granulomas and subsequent lung pathology during TB disease. Further investigation of the effects of PKR modulation in vivo is required to assess whether PKR activity is ultimately beneficial or harmful to the host during M. tuberculosis infection.
Non-tuberculosis mycobacteria (NTM) are mycobacteria that do not cause TB or leprosy. However, these mycobacteria can still cause illness in humans and animals, such as pulmonary disease resembling TB, lymphadenitis, and skin disease (121). NTM are mostly environmental bacteria and can be found in water and soil. Although most studies on PKR during mycobacterial infection used M. tuberculosis or BCG, there is limited evidence to indicate that PKR also plays a role during NTM infections. For instance, Madhvi et al. recently reported that EIF2AK2 mRNA increases during infection with the non-pathogenic Mycobacterium smegmatis (114). In addition, there is evidence to loosely suggest that PKR plays a role in the immune response to Mycobacterium ulcerans, the causative agent of the tropical disease Buruli ulcer. M. ulcerans secretes an exotoxin virulence factor known as mycolactone, which triggers apoptosis of host cells (122). Cells that are exposed to mycolactone can persist for several days through the induction of autophagy before succumbing to apoptosis. However, chronic exposure to mycolactone causes cell death (123). Ogbechi and colleagues observed that MEFs with a deletion of both PERK and GCN2 (Perk-/-Gcn2-/-) succumbed to apoptosis faster than wild-type cells, which was attributed to an inability of these cells to induce autophagy (123). Furthermore, mycolactone treatment increased protein levels of ATF4 and CHOP and triggered phosphorylation of PKR, PERK, GCN2, and EIF2α. Taken together, these findings suggest that mycolactone triggers phosphorylation of EIF2α kinases to activate the EIF2α-ATF4-CHOP pathway, which results in induction of autophagy during short-term exposure to the exotoxin, and induction of apoptosis during chronic exposure. Although the effect of PKR deletion on mycolactone-induced autophagy and apoptosis was not examined, residual Atf4 expression was observed in Perk-/-Gcn2-/- MEFs (123). This suggests that PKR contributes to the phosphorylation of EIF2α and downstream induction of Atf4 expression in response to mycolactone treatment. As such, it is possible that PKR plays a role in regulating autophagy and apoptosis during infection with M. ulcerans, although further experimentation using the live bacterium is required.
Although viral dsRNA is the canonical activator of PKR, recent studies have shown that PKR can also be activated by bacterial RNA. PKR was first suspected to be activated by bacteria when it was observed that purified PKR from E. coli cells is in a phosphorylated state and must be dephosphorylated to make the kinase responsive to RNA (124, 125). This suggested that endogenous bacterial products could trigger PKR activation. In 2012, Bleiblo and colleagues identified bacterial RNA as a ligand recognized by PKR (86). It was observed that total RNA extracted from E. coli and S. aureus potently activated PKR in cardiac cells in a dose-dependent manner, whereas human RNA did not. In vitro PKR binding assays showed that the bacterial RNA directly bound the purified PKR, suggesting that bacterial RNA possesses structural features that can directly activate PKR (86) (Figure 2). A later study from the same group demonstrated that removal of the base-paired secondary structures of the bacterial RNA by RNase digestion hinders the activation of PKR, indicating that the double-stranded structures of bacterial RNA are required to fully activate PKR (126). More recently, Hull and colleagues investigated the specific features of bacterial RNA that can activate PKR (127, 128). The Bacillus subtilis trp 5’-UTR was identified as an activator of PKR. The trp 5’UTR has multiple structural RNA elements representative of many bacterial mRNAs, including a terminator, 5’-stem-loop, and Shine-Dalgarno hairpin. These elements were shown to potently activate PKR. In a follow-up study by Hull and colleagues, three more functional bacterial RNAs were tested for their ability to activate PKR: the Vc2 riboswitch from Vibrio cholerae, the glmS riboswitch-ribozyme from B. anthracis, and the twister ribozyme from Clostridia bolteae (128). Most constructs derived from these RNAs were able to activate PKR, provided they were long enough to form sufficient RNA structure. These findings demonstrate that PKR can be activated by numerous RNA elements from a wide range of bacteria, including both Gram-positive and Gram-negative bacteria.
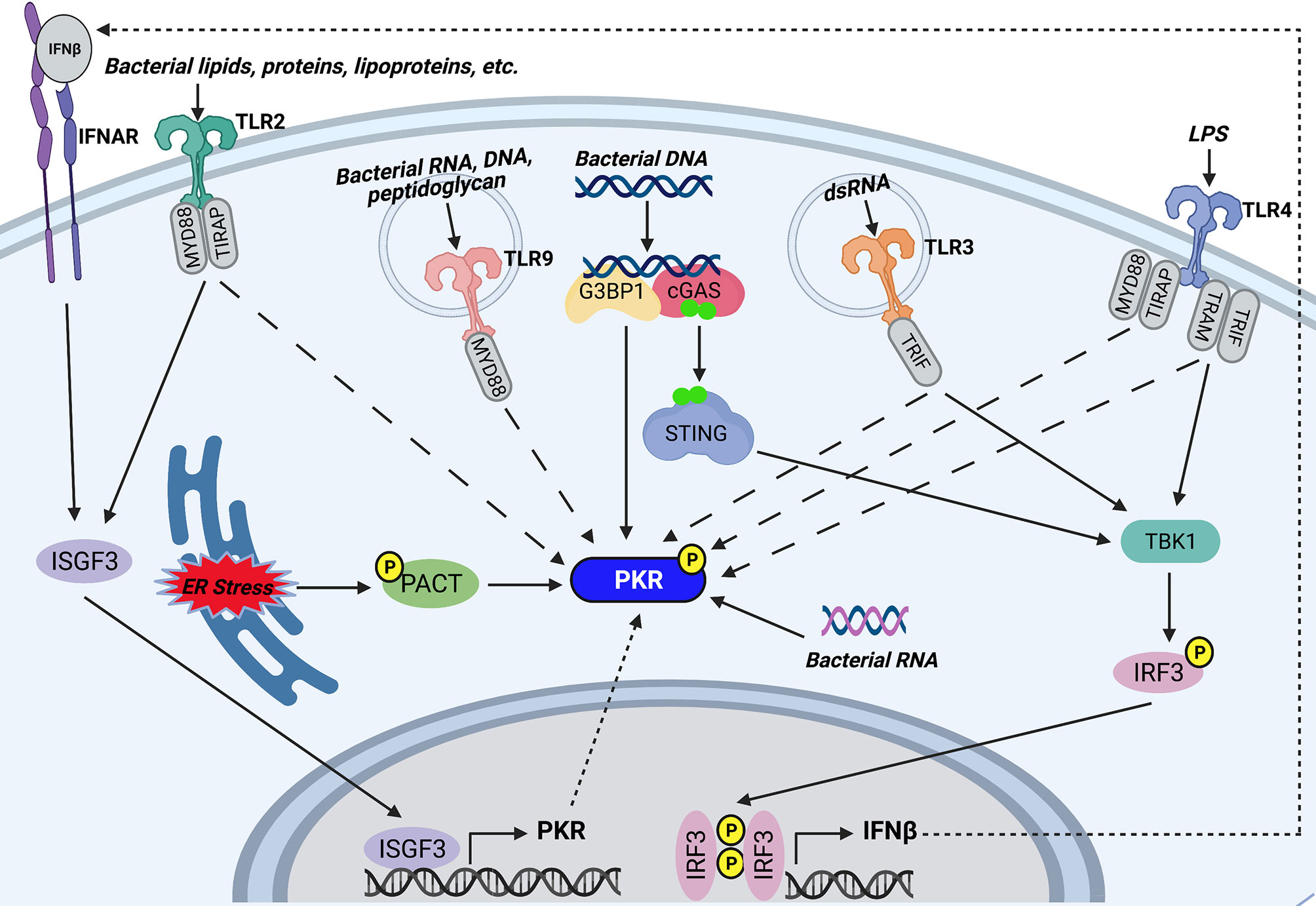
Figure 2 Potential mechanisms of PKR activation during bacterial infections. TLR2 and TLR4 on the host cell surface recognize bacterial lipids, proteins, and lipoproteins, or lipopolysaccharide (LPS), respectively. Endosomal TLR9 recognizes CpG motifs found in bacterial RNA, DNA, and peptidoglycan, whereas endosomal TLR3 is activated by bacterial dsRNA. TLR2, TLR4, TLR9, and TLR3 trigger PKR phosphorylation by unknown mechanisms. Furthermore, TLR3 and TLR4 activate TBK1 by their adaptor proteins (TRIF for TLR3 and TRAM and TRIF for TLR4) which goes on to induce phosphorylation and dimerization of IRF3. IRF3 translocates to the nucleus and induces transcription of IFNβ. IFNβ signals through IFNAR to trigger assembly and nuclear translocation of ISGF3, which regulates the PKR promoter to induce transcription of PKR. TLR2 signaling also triggers association of ISGF3 with the PKR promoter. As such, TLR3, TLR4, and TLR2 may induce PKR transcription by downstream activation of ISGF3. Cytosolic bacterial DNA is recognized by cGAS, which associates with G3BP1. G3BP1 co-localizes with PKR and may directly phosphorylate the kinase. Recognition of bacterial DNA triggers cGAS to synthesize the secondary messenger cyclic GMP-AMP. GMP-AMP triggers activation and dimerization of STING, which in turn activates the TBK1-IRF3-IFNβ-ISGF3 axis to induce PKR transcription. In addition, certain bacteria induce ER stress, which triggers phosphorylation of PACT, a cellular activator of PKR. Phosphorylation of PACT enhances its association with PKR and leads to PKR activation. Finally, cytosolic bacterial RNA can directly bind PKR and trigger its activation. Created with BioRender.com.
The finding that PKR is required for AIM2 inflammasome activation in response to DNA transfection suggested that PKR can be activated by double-stranded DNA (dsDNA) (62), but PKR does not bind dsDNA or DNA/RNA hybrid strands (129). Nevertheless, DNA may activate PKR indirectly. Indeed, one study showed that transfection of HeLa cells with exogenous DNA led to phosphorylation of both PKR and EIF2α (130). This effect was dependent on recognition of dsDNA by the cytosolic DNA sensor cGAS. It was also observed that PKR and G3BP1, an RNA/DNA and RNA/RNA helicase, co-localize with cGAS following DNA transfection and are both required for cGAS sensing of intracellular vaccinia virus DNA. Interestingly, G3BP1 was required for PKR phosphorylation. As such, the authors of this study suggest that after interacting with DNA-bound cGAS, G3BP1 activates PKR (130) (Figure 2). It is note-worthy that the cGAS/STING/TBK-1 axis stimulated by cytosolic dsDNA leads to activation of IRF3, which induces transcription of IFNβ (131). IFNβ triggers assembly and nuclear translocation of the transcription factor interferon-stimulated gene factor 3 (ISGF3), which regulates the PKR promoter at the ISRE (132). Therefore, it is also possible that the DNA-sensing pathway indirectly induces PKR transcription by triggering IFNβ production and downstream ISGF3 activation (Figure 2). Altogether, we speculate that the cytosolic DNA sensing pathway is a potential mechanism that triggers PKR expression and phosphorylation during bacterial infection.
Pathogens that are unable to perforate the phagosome, such as BCG and LLO-deficient L. monocytogenes, can trigger PKR phosphorylation and mRNA expression (89, 95). This suggests that PKR can be activated/induced in the absence of cytosolic nucleic acids. Indeed, there is accumulating evidence to support that PKR is activated by TLR signaling. PKR is reported to be activated in response to TLR4 and TLR2, which are cell surface TLRs that mainly recognize microbial membrane components such as lipids, lipoproteins, and proteins. In 2001, Horng and colleagues observed that PKR is phosphorylated following treatment with LPS, a TLR4 agonist (133). Since then, there have been numerous reports of PKR activation occurring in a TLR4-dependent manner (87, 93, 134–138). TLR4 signaling has also been shown to increase EIF2AK2 mRNA and protein levels (135). As such, we speculate that bacteria can trigger downstream PKR phosphorylation and expression by activating TLR4 signaling (Figure 2). Indeed, a link between TLR4 signaling and PKR activity has been reported during infection with bacteria such as S. Typhimurium, Y. pseudotuberculosis, B. anthracis, and C. trachomatis (87, 93). Importantly, both heat-treated and gamma-irradiated attenuated C. trachomatis can activate PKR to the same extent as live bacteria, indicating that intracellular bacterial replication or secretion of heat-labile bacterial products are not responsible for PKR activation (93). In contrast, inhibition of TLR4 prevented PKR activation (93). Altogether, these findings provide evidence that heat stable LPS is the likely bacterial product responsible for PKR activation during C. trachomatis infection. Indeed, heat-treating LPS did not impair its ability to activate PKR (93). Interestingly, PKR appears to be activated by different adaptor proteins depending on the specific TLR4 agonist (93, 133). C. trachomatis infection is unable to activate PKR in the presence of a MyD88 inhibitor but is unaffected by a TRIF inhibitor, whereas LPS is unable to activate PKR in the presence of a TRIF inhibitor but is unaffected by a MyD88 inhibitor (93). TLR2 signaling can also trigger PKR phosphorylation (Figure 2). Pam3CSK4, a TLR2 agonist, triggers PKR phosphorylation (134) in addition to increasing PKR mRNA and protein levels (135). Furthermore, PKR is activated in a TLR2-dependent manner following parasitic infection with Leishmania amazonensis (139).
PKR phosphorylation can also be triggered downstream of TLR3 and TLR9, which are endosomal TLRs that mainly recognize nucleic acids (Figure 2). Indeed, a kinase-inactive PKR mutant inhibited poly(I:C)-induced TLR3-mediated activation of NF-κB, suggesting that PKR is a downstream component of TLR3 signaling (140). A later study confirmed that PKR is activated by TLR3 signaling, since poly(I:C)-induced phosphorylation of PKR in human neuroblastoma cells was impaired in TLR3-deficient cells (141). There is limited evidence to suggest that PKR is activated in response to TLR9 signaling. PKR was shown to be phosphorylated by CpG motifs present in bacterial DNA, dsRNA, and peptidoglycans (133). CpG engages the TLR9 receptor, indicating that PKR is activated downstream of TLR9 (Figure 2). As such, TLR9 signaling could be yet another mechanism through which PKR indirectly senses bacterial DNA without a requirement for phagosome perforation. Overall, PKR appears to be a downstream component shared by at least four TLRs.
The specific mechanism of PKR activation downstream of TLR signaling remains unclear. However, Perkins and colleagues observed that TLR4 and TLR3 agonists trigger the phosphorylation of both PKR and IRF3 (138). IRF3 induces transcription of IFNβ, which triggers assembly and nuclear translocation of ISGF3. As mentioned in the previous section, ISGF3 regulates the PKR promoter (132); therefore, it is possible that TLR4 and TLR3 signaling induces PKR by triggering IFNβ production and downstream ISGF3 activation (Figure 2). Furthermore, chromatin immunoprecipitation assays revealed that L. amazonensis infection, which activates PKR through TLR2 signaling, triggered binding of ISGF3 elements to the PKR promoter, an event that did not occur in TLR2 knockout macrophages (139). Therefore, ISGF3 may be a common link between TLR2-, TLR4-, and TLR3-dependent induction of PKR (Figure 2).
There are numerous reports that PKR is activated in response to ER stress (142–144). Indeed, PKR has been shown to play a significant role in sustained ER stress-induced apoptosis (54). ER stress leads to downstream PKR activation by triggering the phosphorylation of PACT (54), a cellular activator of PKR that activates the kinase in the absence of dsRNA (55). Phosphorylation of PACT increases its association with PKR and leads to PKR activation. Certain bacteria, including Pseudomonas aeruginosa, Helicobacter pylori, and Coxiella burnetti, are reported to trigger ER stress in infected host cells (145–148). Although the phosphorylation state of PKR was not examined in these studies, PERK and EIF2α were shown to be phosphorylated in response to infection with these bacteria. It is possible that PKR is also activated in response to these bacteria and contributes to the phosphorylation of EIF2α. Indeed, one of the groups suggested that future work should assess whether PKR plays a role in the ER stress response during C. burnetti infection (146). As such, we speculate that infection with certain bacterial pathogens induces ER stress, which leads to PKR activation via PACT (Figure 2).
Due to the emergence of antibiotic-resistant bacteria such as M. tuberculosis and S. aureus, the development of alternative therapies for certain bacterial infections is urgently required. Host-directed therapy (HDT) is a promising option, since this strategy aims to boost the host immune response to a particular bacterium rather than target the bacterium itself, thus circumventing the development of antibiotic resistance. Since there is growing evidence that PKR plays a role in the host immune response to bacterial pathogens, pharmacological modulation of PKR could be a promising strategy for HDT against various bacterial infections.
There are multiple pharmacological activators of PKR in various stages of development (Table 2). Bozepinib is a small antitumor agent that both upregulates and activates PKR (149). Bozepinib has shown promise in pre-clinical studies since it induces apoptosis in breast and colon cancer cells (149, 150). Although the specific effects of bozepinib have not been assessed in vivo, it has been observed that bozepinib treatment does not cause acute toxicity in mice (150). The mechanism through which bozepinib induces and activates PKR remains unknown. Nitazoxanide (NTZ) is another drug that triggers PKR phosphorylation (151, 152). NTZ is an FDA-approved broad-spectrum antiparasitic drug. The typical use of NTZ is the treatment of cryptosporidiosis infection, however clinical trials have demonstrated efficacy and safety of NTZ in treating viral infections such as influenza and hepatitis C (188, 189). NTZ has been shown to deplete intracellular calcium stores, thereby raising levels of cytosolic calcium. This calcium mobilisation disrupts ER/Golgi glycoprotein trafficking and induces ER stress, thus triggering PKR phosphorylation (152). Several in vivo studies conducted in animal models have investigated the effects of NTZ in disease contexts such as viral infections, protozoan infections, cancer, Parkinson’s disease, neuroinflammation, and bacterial infections (Table 2) (153–166). Indeed, in vivo studies have shown that NTZ is effective in treating bacterial pathogens such as C. difficile, E. coli, M. leprae, and M. tuberculosis (153–156, 159). Interestingly, NTZ is reported to exert significant bactericidal activity directly against both replicating and non-replicating M. tuberculosis (190), and was recently evaluated for treatment of TB in a phase II clinical trial (191). In addition, Ranjbar and colleagues recently reported that NTZ treatment enhanced M. tuberculosis-induced EIF2AK2 mRNA expression in THP-1 cells, and NTZ treatment reduced M. tuberculosis burden in THP-1 and human peripheral blood mononuclear cells (97) (Table 1). As such, NTZ is a PKR activator that holds promise for being repurposed as a host-directed antibacterial drug. The synthetic compound BEPP [1H-benzimidazole1-ethanol,2,3-dihydro-2-imino-a-(phenoxymethyl)-3-(phenylmethyl)-,monohydrochloride] is another PKR activator that increases PKR and EIF2α phosphorylation in a dose-dependent manner in MEFs (167). Interestingly, BEPP was shown to induce PKR-dependent apoptosis and effectively inhibited vaccinia virus replication in MEFs (167). However, the effects of BEPP have not been studied in vivo, and the mechanism of action of BEPP on PKR activity is unknown. Lastly, 3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-5,7-dihydroxy-4H-chromen-4-one (DHBDC) is a dual activator of PKR and PERK (168). DHBDC was shown to induce the phosphorylation of EIF2α, which was blocked by siRNAs targeting PKR and PERK. The mechanism by which DHBDC activates PKR remains unknown, and the effects of this compound have not been assessed in vivo. Both BEPP and DHBDC are commercially available for research use.
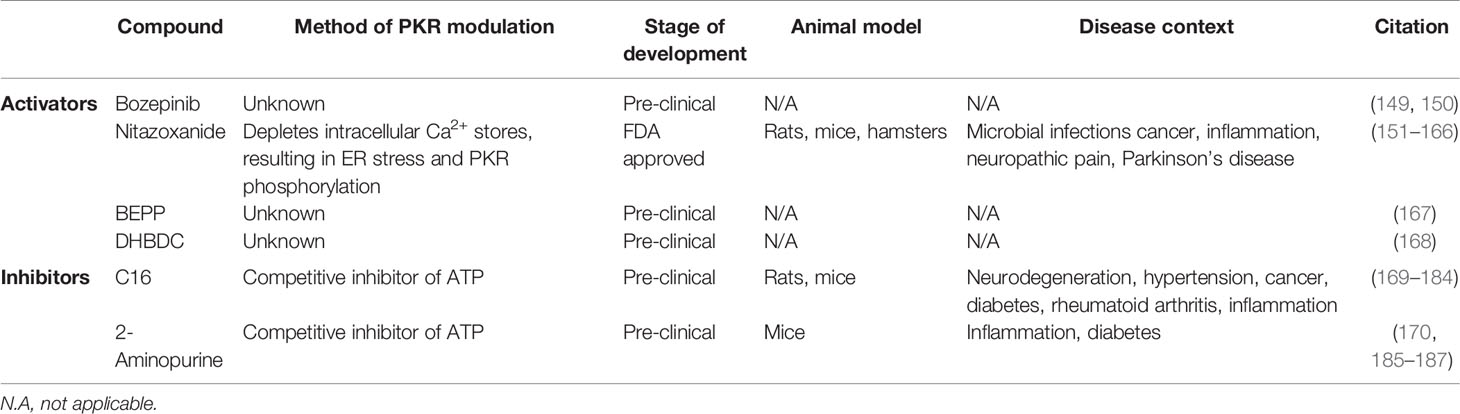
Table 2 Pharmacological modulators of PKR.
There are currently two pharmacological inhibitors of PKR being investigated in pre-clinical studies: imidazole-oxindole C16 and 2-aminopurine (2-AP) (Table 2). Both C16 and 2-AP compete for ATP at the ATP binding site of PKR, thus inhibiting PKR autophosphorylation and kinase activity (169, 185). The most widely-used PKR inhibitor is C16, also known as PKRi or Imoxin (169). C16 has been shown to inhibit PKR phosphorylation in vitro and in the mouse model (169–171). The effects of this compound have been examined in numerous in vitro studies (5). Furthermore, several in vivo studies using mice and rats have examined the effect of C16 in disease contexts such as inflammation, neurodegeneration, obesity, hypertension, cancer, and diabetes (170–184) (Table 2). Importantly, numerous groups have shown that C16 is protective of LPS-induced pathogenesis in mice, including acute lung injury, bone destruction, skeletal muscle atrophy, and acute kidney injury (177–181). Since LPS is a major cell wall component of Gram-negative bacteria, these findings suggest that C16 could protect against excessive inflammation and tissue damage during bacterial infections. However, it is also possible that the anti-inflammatory effect of C16 treatment could exacerbate disease progression in the context of live bacterial infections due to the dampening of the immune response. 2-AP is a less potent and less specific PKR inhibitor (185). In mouse models, 2-AP has been shown to prevent sepsis induced by cecal ligation puncture or endotoxin challenge (186, 187). Furthermore, 2-AP treatment reduces adipose tissue inflammation and improves insulin sensitivity in insulin-resistant obese mice (170). Since 2-AP has anti-inflammatory effects, this drug holds promise in treating bacterial infections where excessive inflammation is conducive for the pathogen. Both C16 and 2-AP are commercially available for research use.
There are numerous reports demonstrating that PKR plays an important role during bacterial infections (Table 1), which suggests that pharmacological modulation of PKR could be a promising strategy for host-directed therapy. However, the vast majority of these studies were conducted in vitro. Out of the 14 studies listed in Table 1, only 4 studies were conducted in vivo (62, 90, 91, 96). Notably, these studies only examined the bacterial burden in organs of infected mice and did not assess the overall survival of the animals. Indeed, while there exist numerous pharmacological activators and inhibitors of PKR, only NTZ has been tested in vivo in the context of bacterial infections (Table 2). As such, extensive in vivo experimentation must be conducted before promoting the use of pharmacological PKR modulation as a therapeutic intervention against bacterial pathogens. It will be critical to demonstrate that pharmacological modulation of PKR has the ability to improve host survival during bacterial infection in animal models. Furthermore, in vivo studies must be conducted to identify any limitations of using pharmacological modulators of PKR.
One potential challenge of inhibiting PKR is that interventions that dampen the inflammatory response can sometimes enhance the susceptibility of the host to lethal bacterial infection. For example, C3H/HeJ mice, which have a defective LPS response, are highly susceptible to E. coli and S. Typhimurium infection (192). Since PKR expression is demonstrated to be important for inflammasome activation and induction of pro-inflammatory cytokines in response to bacterial infections (Table 1), it is possible that pharmacological inhibition of PKR would ultimately be disadvantageous for the host. Another concern is the fact that PKR activity affects many different signaling pathways. This means that inhibiting or activating PKR-dependent pathways involved in the immune response to bacterial pathogens may also disrupt other PKR-dependent pathways that are essential for other functions. Indeed, dysregulation of PKR has been linked to numerous diseases, including neurodegeneration, cancer, and metabolic disorders (4–6). Furthermore, PKR regulates different immune functions depending on the context of the bacterial infection (Table 1), which suggests that the kinase can play either a pro- or anti-host role during bacterial infection contingent on the specific bacterium involved. As such, it is possible that clinicians would be unable to administer pharmacological PKR modulators to patients until the specific pathogen was identified, thus delaying the initiation of host-directed therapy until it is potentially too late. Altogether, these potential challenges highlight the necessity of evaluating the effects of PKR modulators in vivo.
There is growing evidence demonstrating that PKR plays key roles during infection with various bacterial pathogens (Table 1). Indeed, current literature clearly demonstrates a role for PKR in regulating selective autophagy, cell death, and inflammation during bacterial infections. In response to both Gram-positive and Gram-negative bacterial infections, PKR expression has been shown to be important for inflammasome activation, pyroptosis, and apoptosis. In contrast, PKR is not observed to regulate cell death pathways during mycobacterial infection, but is instead reported to induce selective autophagy. PKR has also been shown to regulate cytokine production in response to mycobacteria, Gram-positive, and Gram-negative bacteria. We speculate that the varying functions of PKR during bacterial infections is due to the specific bacterium involved, since bacterial pathogens have methods of manipulating host immune responses to their advantage. For instance, PKR expression is required for macrophage apoptosis during B. anthracis infection (87), but PKR modulation does not impact apoptosis during M. tuberculosis infection (98, 116). This is likely explained by the fact that a major virulence mechanism of B. anthracis is to induce rapid cell death of host cells (193, 194), whereas M. tuberculosis inhibits macrophage apoptosis to allow it to persist undetected within the phagocyte (195). Most studies thus far have focused on the role of PKR in cell death during bacterial infection. Although it is important to investigate the function of PKR during cell death, we contend that this should not be the sole focus, since PKR has recently been shown to induce selective autophagy during bacterial and parasitic infection (77, 98). As such, it will be important for future studies to investigate the function of PKR in selective autophagy of intracellular bacteria such as L. monocytogenes and S. enterica, since autophagy is reported to play a role in antibacterial defense against these pathogens (196–198).
Although PKR has critical functions in bacterial defense, whether the kinase is ultimately protective or detrimental to the host remains to be clarified. For instance, deletion of PKR led to decreased bacterial burden in organs of E. coli-infected mice (62) but resulted in increased bacterial burden in M. tuberculosis-infected macrophages (97, 98). Whether PKR plays a pro- or anti-host role during bacterial infection is likely dependent on the specific bacterium involved, and whether the bacterium establishes acute versus chronic infection. For example, S. enterica causes acute infection, and a strong inflammatory response in the early stage of infection is generally considered to assist with bacterial clearance (199, 200). In contrast, M. tuberculosis establishes chronic infection, therefore excessive and prolonged inflammation during infection with this bacterium can be harmful to the host (201). As such, we speculate that the function of PKR in inflammasome activation would be beneficial to the host during S. enterica infection, but harmful to the host during M. tuberculosis infection. Similarly, PKR is established to induce IFNβ production, which can be a pro- or anti-host function depending on the bacterial context. Indeed, IFNβ production is detrimental to the host during L. monocytogenes infection due to its role in inducing macrophage apoptosis (202), but protective for the host during L. pneumophilia infection since it promotes itaconic acid production (203).
Unfortunately, most of the existing studies of PKR in the context of bacterial infections examined either the effect of PKR on a particular host signaling pathway, or the effect of PKR expression on bacterial burden, but rarely were both examined within the same study (Table 1). Furthermore, only a limited number of studies have examined the effect of PKR modulation on bacterial burden, with only a select few that included in vivo experiments (Table 1). In vivo studies will be critical in determining whether PKR is ultimately protective or detrimental to the host during infection with a given bacterium. Importantly, in vivo experimentation of PKR is feasible given that pharmacological approaches have been vetted in other disease models (Table 2) and both genetic overexpression and deletion of PKR is well-tolerated in mice (204, 205). Both inhibition and activation of PKR by pharmacological compounds (Table 2) should be actively pursued given that modulation in either direction could be specifically harnessed for treatment of specific bacterial diseases. As such, future studies should examine the effects of pharmacological modulation of PKR on bacterial burden, morbidity, and mortality of bacteria-infected mice to assess the suitability and feasibility of targeting PKR as a novel treatment strategy. At the same time, it will also be important to determine the specific mechanism regulated by PKR during bacterial infection, since it will link the specific functional pathway to whether PKR is ultimately protective or detrimental to the host.
There also exists a knowledge gap on the specific downstream effectors mediated by PKR in response to various bacteria. For example, although our group observed that PKR induces autophagic degradation of M. tuberculosis, the downstream pathway activated by PKR in this context was not elucidated (98). PKR is reported to mediate the activation of numerous downstream effectors, including MAPK, ATF2, NF-κB, IPS-1, IRF3, ATF4, and CHOP (Figure 1). As such, there are many potential mechanisms controlled by PKR to regulate cellular processes such as autophagy, inflammation, and cell death in response to bacterial infections. Future studies should elucidate the specific downstream components regulated by PKR during the immune response to bacterial pathogens.
The upstream signaling events that trigger PKR activation during bacterial infection also need to be determined. Although the canonical activator of PKR is viral dsRNA, the ability of certain phagosome-restricted bacteria to activate PKR suggests the existence of alternative activating mechanisms (89, 95). In this review, we discussed non-canonical activators of PKR – including TLR signaling, ER stress, and bacterial nucleic acids – and speculated on potential mechanisms that trigger PKR activation during bacterial infection (Figure 2). However, although exogenous treatment of cells with TLR agonists, bacterial RNA, or chemical inducers of ER stress can activate PKR, there is a lack of evidence to show that live bacteria trigger PKR activation through these specific mechanisms. Future studies examining PKR activity during bacterial infection should strive to characterize the mechanism responsible for activating PKR in this context.
In summary, PKR undoubtedly plays key roles during bacterial infections, as multiple studies have shown that PKR regulates critical immune cell functions such as inflammation, apoptosis, pyroptosis, and autophagy. Increasing our knowledge on the role of PKR during bacterial infections is important since it could lead to the development of host-directed therapies for antibiotic-resistant bacteria. Regardless of whether PKR activity is ultimately beneficial or detrimental to the host, modulating its expression or activity holds promise as a novel treatment strategy for bacterial infections.
● PKR regulates cell death in both Gram-positive and Gram-negative bacterial infections by inducing apoptosis and activating the inflammasome to trigger pyroptosis.
● PKR regulates the production of multiple cytokines with key roles in antibacterial defense, including Type I IFNs, IL-1β, IL-6, IL-10, IL-18, and TNFα.
● PKR induces selective autophagy during M. tuberculosis infection.
● Conflicting reports exist on whether PKR is protective or detrimental to the host. This is likely due to (i) different bacterial pathogens involved, (ii) the specific infection model used for each study, and (iii) the specific mechanistic pathway at play (i.e. cell death vs. autophagy).
● The mechanisms of PKR activation during bacterial infection remain to be elucidated, but may involve bacterial nucleic acids, TLR signaling, or ER stress.
● Pharmacological modulation of PKR holds promise as an alternative treatment strategy for bacterial infections, but extensive in vivo studies must be conducted to assess the effects of PKR modulation on host survival and identify potential off-target effects.
RS and JS wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Canadian Institutes of Health Research (CIHR) PJT-162424, the National Sanitarium Association Scholar’s Program, and the Natural Sciences and Engineering Research Council of Canada (NSERC) RGPIN-2020-04032 to JS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Taniuchi S, Miyake M, Tsugawa K, Oyadomari M, Oyadomari S. Integrated Stress Response of Vertebrates Is Regulated by Four eIF2α Kinases. Sci Rep (2016) 6:1–11. doi: 10.1038/srep32886
2. Barber GN, Edelhoff S, Katze MG, Disteche CM. Chromosomal Assignment of the Interferon-Inducible Double-Stranded RNA-Dependent Protein Kinase (PRKR) to Human Chromosome 2p21-P22 and Mouse Chromosome 17 E2. Genomics (1993) 16:765–7. doi: 10.1006/geno.1993.1262
3. Meurs E, Chong K, Galabru J, Thomas NSB, Kerr IM, Williams BRG, et al. Molecular Cloning and Characterization of the Human Double-Stranded RNA-Activated Protein Kinase Induced by Interferon. Cell (1990) 62:379–90. doi: 10.1016/0092-8674(90)90374-N
4. García MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, et al. Impact of Protein Kinase PKR in Cell Biology: From Antiviral to Antiproliferative Action. Microbiol Mol Biol Rev (2006) 70:1032–60. doi: 10.1128/mmbr.00027-06
5. Gal-Ben-Ari S, Barrera I, Ehrlich M, Rosenblum K. PKR: A Kinase to Remember. Front Mol Neurosci (2019) 11:480. doi: 10.3389/fnmol.2018.00480
6. Watanabe T, Imamura T, Hiasa Y. Roles of Protein Kinase R in Cancer: Potential as a Therapeutic Target. Cancer Sci (2018) 109:919–25. doi: 10.1111/cas.13551
7. Galabru J, Hovanessians A. Autophosphorylation of the Protein Kinase Dependent on Double-Stranded RNA. J Biol Chem (1987) 262:15538–44. doi: 10.1016/S0021-9258(18)47759-9
8. Taniguchi T, Takaoka A. The Interferon-α/β System in Antiviral Responses: A Multimodal Machinery of Gene Regulation by the IRF Family of Transcription Factors. Curr Opin Immunol (2002) 14:111–6. doi: 10.1016/S0952-7915(01)00305-3
9. Kuhen KL, Samuel CE. Isolation of the Interferon-Inducible RNA-Dependent Protein Kinase Pkr Promoter and Identification of a Novel DNA Element Within the 5’-Flanking Region of Human and Mouse Pkr Genes. Virology (1997) 227:119–30. doi: 10.1006/viro.1996.8306
10. Das S, Ward SV, Tacke RS, Suske G, Samuel CE. Activation of the RNA-Dependent Protein Kinase PKR Promoter in the Absence of Interferon is Dependent Upon Sp Proteins. J Biol Chem (2006) 281:3244–53. doi: 10.1074/jbc.M510612200
11. Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a Stress-Modulated Cellular Activator of Interferon-Induced Double-Stranded RNA-Activated Protein Kinase, PKR. J Biol Chem (2000) 275:37993–8. doi: 10.1074/jbc.M004762200
12. García MA, Meurs EF, Esteban M. The dsRNA Protein Kinase PKR: Virus and Cell Control. Biochimie (2007) 89:799–811. doi: 10.1016/j.biochi.2007.03.001
13. Kuhen KL, Shen X, Carlisle ER, Richardson AL, Weier HUG, Tanaka H, et al. Structural Organization of the Human Gene (PKR) Encoding an Interferon- Inducible RNA-Dependent Protein Kinase (PKR) and Differences From its Mouse Homolog. Genomics (1996) 36:197–201. doi: 10.1006/geno.1996.0446
14. Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, et al. Mechanistic Link Between PKR Dimerization, Autophosphorylation, and eIF2α Substrate Recognition. Cell (2005) 122:901–13. doi: 10.1016/j.cell.2005.06.041
15. Lemaire PA, Lary J, Cole JL. Mechanism of PKR Activation: Dimerization and Kinase Activation in the Absence of Double-Stranded RNA. J Mol Biol (2005) 345:81–90. doi: 10.1016/j.jmb.2004.10.031
16. Zhang F, Romano PR, Nagamura-Inoue T, Tian B, Dever TE, Mathews MB, et al. Binding of Double-Stranded RNA to Protein Kinase PKR is Required for Dimerization and Promotes Critical Autophosphorylation Events in the Activation Loop. J Biol Chem (2001) 276:24946–58. doi: 10.1074/jbc.M102108200
17. Singh R, And R, London IM. Regulation of Protein Synthesis in Rabbit Reticulocyte Lysates: Purification and Initial Characterization of the Cyclic 3’:5’-AMP Independent Protein Kinase of the Heme-Regulated Translational Inhibitor (Phosphorylation of Met-tRNAf Binding Factor). Proc Natl Acad Sci USA (1976) . 73:4349–54. doi: 10.1073/pnas.73.12.4349
18. Harding HP, Zhang Y, Ron D. Protein Translation and Folding are Coupled by an Endoplasmic- Reticulum-Resident Kinase. Nature (1999) 397:271–4. doi: 10.1038/16729
19. Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG. Phosphorylation of Initiation Factor 2α by Protein Kinase GCN2 Mediates Gene-Specific Translational Control of GCN4 in Yeast. Cell (1992) 68:585–96. doi: 10.1016/0092-8674(92)90193-G
20. Sudhakar A, Ramachandran A, Ghosh S, Hasnain SE, Kaufman RJ, Ramaiah KVA. Phosphorylation of Serine 51 in Initiation Factor 2α (eIF2α) Promotes Complex Formation Between eIF2α(P) and eIF2B and Causes Inhibition in the Guanine Nucleotide Exchange Activity of eIF2B. Biochemistry (2000) 39:12929–38. doi: 10.1021/bi0008682
21. Gordiyenko Y, Llácer JL, Ramakrishnan V. Structural Basis for the Inhibition of Translation Through eIF2α Phosphorylation. Nat Commun (2019) 10:1–11. doi: 10.1038/s41467-019-10606-1
22. Liu Y, Wang M, Cheng A, Yang Q, Wu Y, Jia R, et al. The Role of Host eIF2α in Viral Infection. Virol J (2020) 17:112. doi: 10.1186/s12985-020-01362-6
23. Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2α Kinases: Their Structures and Functions. Cell Mol Life Sci (2013) 70:3493–511. doi: 10.1007/s00018-012-1252-6
24. Dalet A, Gatti E, Pierre P. Integration of PKR-Dependent Translation Inhibition With Innate Immunity is Required for a Coordinated Anti-Viral Response. FEBS Lett (2015) 589:1539–45. doi: 10.1016/j.febslet.2015.05.006
25. Zhang P, Langland JO, Jacobs BL, Samuel CE. Protein Kinase PKR-Dependent Activation of Mitogen-Activated Protein Kinases Occurs Through Mitochondrial Adapter IPS-1 and is Antagonized by Vaccinia Virus E3L. J Virol (2009) 83:5718–25. doi: 10.1128/jvi.00224-09
26. Goh KC, DeVeer MJ, Williams BRG. The Protein Kinase PKR is Required for P38 MAPK Activation and the Innate Immune Response to Bacterial Endotoxin. EMBO J (2000) 19:4292–7. doi: 10.1093/emboj/19.16.4292
27. Silva AM, Whitmore M, Xu Z, Jiang Z, Li X, Williams BRG. Protein Kinase R (PKR) Interacts With and Activates Mitogen-Activated Protein Kinase Kinase 6 (MKK6) in Response to Double-Stranded RNA Stimulation. J Biol Chem (2004) 279:37670–6. doi: 10.1074/jbc.M406554200
28. Reimold AM, Kim J, Finberg R, Glimcher LH. Decreased Immediate Inflammatory Gene Induction in Activating Transcription Factor-2 Mutant Mice. Int Immunol (2001) 13:241–8. doi: 10.1093/intimm/13.2.241
29. Kirsch K, Zeke A, Tőke O, Sok P, Sethi A, Sebő A, et al. Co-Regulation of the Transcription Controlling ATF2 Phosphoswitch by JNK and P38. Nat Commun (2020) 11:1–15. doi: 10.1038/s41467-020-19582-3
30. Gil J, Alcamí J, Esteban M. Activation of NF-κB by the dsRNA-Dependent Protein Kinase, PKR Involves the IκB Kinase Complex. Oncogene (2000) 19:1369–78. doi: 10.1038/sj.onc.1203448
31. Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. PKR Stimulates NF-κb Irrespective of its Kinase Function by Interacting With the IκB Kinase Complex. Mol Cell Biol (2000) 20:4532–42. doi: 10.1128/mcb.20.13.4532-4542.2000
32. Gil J, Alcamí J, Esteban M. Induction of Apoptosis by Double-Stranded-RNA-Dependent Protein Kinase (PKR) Involves the α Subunit of Eukaryotic Translation Initiation Factor 2 and NF-κB. Mol Cell Biol (1999) 19:4653–63. doi: 10.1128/mcb.19.7.4653
33. Yang YL, Reis LFL, Paylovic J, Aguzzi S, Schäfer R, Kumar A, et al. Deficient Signaling in Mice Devoid of Double-Stranded RNA-Dependent Protein Kinase. EMBO J (1995) 14:6095–106. doi: 10.1002/j.1460-2075.1995.tb00300.x
34. Zhang P, Samuel CE. Protein Kinase PKR Plays a Stimulus- and Virus-Dependent Role in Apoptotic Death and Virus Multiplication in Human Cells. J Virol (2007) 81:8192–200. doi: 10.1128/jvi.00426-07
35. Maran A, Maitra RK, Kumar A, Dong B, Xiao W, Li G, et al. Blockage of NF-κB Signaling by Selective Ablation of an mRNA Target by 2-5A Antisense Chimeras. Science (1994) 265:789–92. doi: 10.1126/science.7914032
36. Trinchieri G. Type I Interferon: Friend or Foe? J Exp Med (2010) 207:2053–63. doi: 10.1084/jem.20101664
37. Bolívar S, Anfossi R, Humeres C, Vivar R, Boza P, Muñoz C, et al. IFN-β Plays Both Pro- and Anti-Inflammatory Roles in the Rat Cardiac Fibroblast Through Differential STAT Protein Activation. Front Pharmacol (2018) 9:1368. doi: 10.3389/fphar.2018.01368
38. Kopitar-Jerala N. The Role of Interferons in Inflammation and Inflammasome Activation. Front Immunol (2017) 8:873. doi: 10.3389/fimmu.2017.00873
39. Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, et al. Absent in Melanoma 2 is Required for Innate Immune Recognition of Francisella Tularensis. Proc Natl Acad Sci USA (2010) 107:9771–6. doi: 10.1073/pnas.1003738107
40. Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I Interferon Signaling is Required for Activation of the Inflammasome During Francisella Infection. J Exp Med (2007) 204:987–94. doi: 10.1084/jem.20062665
41. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 Increases Susceptibility to Salmonella Infection in the Absence of Caspase-1. Nature (2012) 490:288–91. doi: 10.1038/nature11419
42. Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell (2012) 150:606–19. doi: 10.1016/j.cell.2012.07.007
43. Zhang P, Samuel CE. Induction of Protein Kinase PKR-Dependent Activation of Interferon Regulatory Factor 3 by Vaccinia Virus Occurs Through Adapter IPS-1 Signaling. J Biol Chem (2008) 283:34580–7. doi: 10.1074/jbc.M807029200
44. Munir M, Berg M. The Multiple Faces of Protein Kinase R in Antiviral Defense. Virulence (2013) 4:85–9. doi: 10.4161/viru.23134
45. Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, et al. Protein Kinase R Contributes to Immunity Against Specific Viruses by Regulating Interferon mRNA Integrity. Cell Host Microbe (2010) 7:354–61. doi: 10.1016/j.chom.2010.04.007
46. Lee SB, Esteban M. The Interferon-Induced Double-Stranded RNA-Activated Protein Kinase Induces Apoptosis. Virology (1994) 199:491–6. doi: 10.1006/viro.1994.1151
47. Srivastava SP, Kumar KU, Kaufman RJ. Phosphorylation of Eukaryotic Translation Initiation Factor 2 Mediates Apoptosis in Response to Activation of the Double-Stranded RNA-Dependent Protein Kinase. J Biol Chem (1998) 273:2416–23. doi: 10.1074/jbc.273.4.2416
48. Der SD, Yang YL, Weissmann C, Williams BRG. A Double-Stranded RNA-Activated Protein Kinase-Dependent Pathway Mediating Stress-Induced Apoptosis. Proc Natl Acad Sci USA (1997) 94:3279–83. doi: 10.1073/pnas.94.7.3279
49. Williams BRG. PKR; A Sentinel Kinase for Cellular Stress. Oncogene (1999) 18:6112–20. doi: 10.1038/sj.onc.1203127
50. Kibler KV, Shors T, Perkins KB, Zeman CC, Banaszak MP, Biesterfeldt J, et al. Double-Stranded RNA is a Trigger for Apoptosis in Vaccinia Virus-Infected Cells. J Virol (1997) 71:1992–2003. doi: 10.1128/jvi.71.3.1992-2003.1997
51. Takizawa T, Ohashi K, Nakanishi Y. Possible Involvement of Double-Stranded RNA-Activated Protein Kinase in Cell Death by Influenza Virus Infection. J Virol (1996) 70:8128–32. doi: 10.1128/jvi.70.11.8128-8132.1996
52. Yeung MC, Chang DL, Camantigue RE, Lau AS. Inhibitory Role of the Host Apoptogenic Gene PKR in the Establishment of Persistent Infection by Encephalomyocarditis Virus in U937 Cells. Proc Natl Acad Sci USA (1999) 96:11860–5. doi: 10.1073/pnas.96.21.11860
53. Yeung MC, Liu J, Lau AS. An Essential Role for the Interferon-Inducible, Double-Stranded RNA-Activated Protein Kinase PKR in the Tumor Necrosis Factor-Induced Apoptosis in U937 Cells. Proc Natl Acad Sci USA (1996) 93:12451–5. doi: 10.1073/pnas.93.22.12451
54. Lee E-S, Yoon C-H, Kim Y-S, Bae Y-S. The Double-Strand RNA-Dependent Protein Kinase PKR Plays a Significant Role in a Sustained ER Stress-Induced Apoptosis. FEBS Lett (2007) 581:4325–32. doi: 10.1016/j.febslet.2007.08.001
55. Patel RC, Sen GC. PACT, a Protein Activator of the Interferon-Induced Protein Kinase, PKR. EMBO J (1998) 17:4379–90. doi: 10.1093/emboj/17.15.4379
56. Vattem KM, Wek RC. Reinitiation Involving Upstream ORFs Regulates ATF4 mRNA Translation in Mammalian Cells. Proc Natl Acad Sci USA (2004) 101:11269–74. doi: 10.1073/pnas.0400541101
57. Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell (2007) 129:1337–49. doi: 10.1016/j.cell.2007.04.027
58. Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, et al. Identification of Novel Stress-Induced Genes Downstream of Chop. EMBO J (1998) 17:3619–30. doi: 10.1093/emboj/17.13.3619
59. Gil J, Esteban M. The Interferon-Induced Protein Kinase (PKR), Triggers Apoptosis Through FADD-Mediated Activation of Caspase 8 in a Manner Independent of Fas and TNF-α Receptors. Oncogene (2000) 19:3665–74. doi: 10.1038/sj.onc.1203710
60. Von Roretz C, Gallouzi IE. Protein Kinase RNA/FADD/caspase-8 Pathway Mediates the Proapoptotic Activity of the RNA-Binding Protein Human Antigen R (Hur). J Biol Chem (2010) 285:16806–13. doi: 10.1074/jbc.M109.087320
61. Balachandran S, Kim CN, Yeh WC, Mak TW, Bhalla K, Barber GN. Activation of the dsRNA-Dependent Protein Kinase, PKR, Induces Apoptosis Through FADD-Mediated Death Signaling. EMBO J (1998) 17:6888–902. doi: 10.1093/emboj/17.23.6888
62. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, et al. Novel Role of PKR in Inflammasome Activation and HMGB1 Release. Nature (2012) 488:670–4. doi: 10.1038/nature11290
63. Yim HCH, Wang D, Yu L, White CL, Faber PW, Williams BRG, et al. The Kinase Activity of PKR Represses Inflammasome Activity. Cell Res (2016) 26:367–79. doi: 10.1038/cr.2016.11
64. Kayagaki N, Warming S, Lamkanfi M, Vande WL, Louie S, Dong J, et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
65. He Y, Franchi L, Núñez G. The Protein Kinase PKR is Critical for LPS-Induced iNOS Production But Dispensable for Inflammasome Activation in Macrophages. Eur J Immunol (2013) 43:1147–52. doi: 10.1002/eji.201243187
66. Moraco AH, Kornfeld H. Cell Death and Autophagy in Tuberculosis. Semin Immunol (2014) 26:497–511. doi: 10.1016/j.smim.2014.10.001
67. Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, et al. Plasma Membrane Damage Causes NLRP3 Activation and Pyroptosis During Mycobacterium Tuberculosis Infection. Nat Commun (2020) 11:2270. doi: 10.1038/s41467-020-16143-6
68. Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric Oxide Controls the Immunopathology of Tuberculosis by Inhibiting NLRP3 Inflammasome-Dependent Processing of IL-1β. Nat Immunol (2013) 14:52–60. doi: 10.1038/ni.2474
69. Yonekawa T, Thorburn A. Autophagy and Cell Death. Essays Biochem (2013) 55:105–17. doi: 10.1042/BSE0550105
70. Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy Defends Cells Against Invading Group A Streptococcus. Science (2004) 306:1037–40. doi: 10.1126/science.1103966
71. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a Defense Mechanism Inhibiting BCG and Mycobacterium Tuberculosis Survival in Infected Macrophages. Cell (2004) 119:753–66. doi: 10.1016/j.cell.2004.11.038
72. Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 Induces Macrophage Anti-Toxoplasma Gondii Activity by Triggering Autophagy-Dependent Fusion of Pathogen-Containing Vacuoles and Lysosomes. J Clin Invest (2006) 116:2366–77. doi: 10.1172/JCI28796
73. Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM Induces Autophagy to Eliminate Intracellular Mycobacteria. Science (2006) 313:1438–41. doi: 10.1126/science.1129577
74. Zhang Q, Sun J, Wang Y, He W, Wang L, Zheng Y, et al. Antimycobacterial and Anti-Inflammatory Mechanisms of Baicalin Via Induced Autophagy in Macrophages Infected With Mycobacterium Tuberculosis. Front Microbiol (2017) 8:2142. doi: 10.3389/fmicb.2017.02142
75. Tallóczy Z, Jiang W, Virgin HW IV, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of Starvation- and Virus-Induced Autophagy by the elF2α Kinase Signaling Pathway. Proc Natl Acad Sci USA (2002) 99:190–5. doi: 10.1073/pnas.012485299
76. Tallóczy Z, Virgin HW IV, Levine B. PKR-Dependent Autophagic Degradation of Herpes Simplex Virus Type 1. Autophagy (2006) 2:24–9. doi: 10.4161/auto.2176
77. Ogolla PS, Portillo J-AC, White CL, Patel K, Lamb B, Sen GC, et al. The Protein Kinase Double-Stranded RNA-Dependent (PKR) Enhances Protection Against Disease Cause by a non-Viral Pathogen. PloS Pathog (2013) 9:e1003557. doi: 10.1371/journal.ppat.1003557
78. B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2α/ATF4 Pathway is Essential for Stress-Induced Autophagy Gene Expression. Nucleic Acids Res (2013) 41:7683–99. doi: 10.1093/nar/gkt563
79. Wei Y, An Z, Zou Z, Sumpter R, Su M, Zang X, et al. The Stress-Responsive Kinases MAPKAPK2/MAPKAPK3 Activate Starvation-Induced Autophagy Through Beclin 1 Phosphorylation. Elife (2015) 40:e05289. doi: 10.7554/eLife.05289
80. Pattingre S, Tassa A, Qu X, Garuti R, Xiao HL, Mizushima N, et al. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell (2005) 122:927–39. doi: 10.1016/j.cell.2005.07.002
81. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol Cell (2008) 30:678–88. doi: 10.1016/j.molcel.2008.06.001
82. Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, et al. A Potential New Pathway for Staphylococcus Aureus Dissemination: The Silent Survival of S. Aureus Phagocytosed by Human Monocyte-Derived Macrophages. PloS One (2008) 3:e1409. doi: 10.1371/journal.pone.0001409
83. Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus Aureus Inside Neutrophils Contributes to Infection. J Immunol (2000) 164:3713–22. doi: 10.4049/jimmunol.164.7.3713
84. Mestre MB, Fader CM, Sola C, Colombo MI. α-Hemolysin is Required for the Activation of the Autophagic Pathway in Staphylococcus Aureus-Infected Cells. Autophagy (2010) 6:110–25. doi: 10.4161/auto.6.1.10698
85. Kloft N, Neukirch C, Bobkiewicz W, Veerachato G, Busch T, Von Hoven G, et al. Pro-Autophagic Signal Induction by Bacterial Pore-Forming Toxins. Med Microbiol Immunol (2010) 199:299–309. doi: 10.1007/s00430-010-0163-0
86. Bleiblo F, Michael P, Brabant D, Ramana CV, Tai TC, Saleh M, et al. Bacterial RNA Induces Myocyte Cellular Dysfunction Through the Activation of PKR. J Thorac Dis (2012) 4:114–25. doi: 10.3978/j.issn.2072-1439.2012.01.07
87. Hsu LC, Park JM, Zhang K, Luo JL, Maeda S, Kaufman RJ, et al. The Protein Kinase PKR is Required for Macrophage Apoptosis After Activation of Toll-Like Receptor 4. Nature (2004) 428:341–5. doi: 10.1038/nature02405
88. Hett EC, Slater LH, Mark KG, Kawate T, Monks BG, Stutz A, et al. Chemical Genetics Reveals a Kinase-Independent Role for Protein Kinase R in Pyroptosis. Nat Chem Biol (2013) 9:398–405. doi: 10.1038/nchembio.1236
89. Valderrama C, Clark A, Urano F, Unanue ER, Carrero JA. Listeria Monocytogenes Induces an Interferon-Enhanced Activation of the Integrated Stress Response That is Detrimental for Resolution of Infection in Mice. Eur J Immunol (2017) 47:830–40. doi: 10.1002/eji.201646856
90. Yang Y, Xie L, Zhong Y, Zhong X, Meng R, Xue Q, et al. Double-Stranded RNA Dependent Kinase R Regulates Antibacterial Immunity in Sepsis. J Innate Immun (2021) 13:26–37. doi: 10.1159/000507932
91. Poon DCH, Ho YS, You R, Tse HL, Chiu K, Chang RCC. PKR Deficiency Alters E. Coli-Induced Sickness Behaviors But Does Not Exacerbate Neuroimmune Responses or Bacterial Load. J Neuroinflamm (2015) 12:212. doi: 10.1186/s12974-015-0433-2
92. Shrestha N, Boucher J, Bahnan W, Clark ES, Rosqvist R, Fields KA, et al. The Host-Encoded Heme Regulated Inhibitor (HRI) Facilitates Virulence-Associated Activities of Bacterial Pathogens. PloS One (2013) 8:68754. doi: 10.1371/journal.pone.0068754
93. Webster SJ, Ellis L, O’Brien LM, Tyrrell B, Fitzmaurice TJ, Elder MJ, et al. Ire1α Mediates PKR Activation in Response to Chlamydia Trachomatis Infection. Microbes Infect (2016) 18:472–83. doi: 10.1016/j.micinf.2016.03.010
94. Mallama CA, McCoy-Simandle K, Cianciotto NP. The Type II Secretion System of Legionella Pneumophila Dampens the MyD88 and Toll-Like Receptor 2 Signaling Pathway in Infected Human Macrophages. Infect Immun (2017) 85:e00897–16. doi: 10.1128/IAI.00897-16
95. Cheung BKW, Lee DCW, Li JCB, Lau Y-L, Lau ASY. A Role for Double-Stranded RNA-Activated Protein Kinase PKR in Mycobacterium-Induced Cytokine Expression. J Immunol (2005) 175:7218–25. doi: 10.4049/jimmunol.175.11.7218
96. Mundhra S, Bryk R, Hawryluk N, Zhang T, Jiang X, Nathan CF. Evidence for Dispensability of Protein Kinase R in Host Control of Tuberculosis. Eur J Immunol (2018) 48:612–20. doi: 10.1002/eji.201747180
97. Ranjbar S, Haridas V, Nambu A, Jasenosky LD, Sadhukhan S, Ebert TS, et al. Cytoplasmic RNA Sensor Pathways and Nitazoxanide Broadly Inhibit Intracellular Mycobacterium Tuberculosis Growth. iScience (2019) 22:299–313. doi: 10.1016/j.isci.2019.11.001
98. Smyth R, Berton S, Rajabalee N, Chan T, Sun J. Protein Kinase R Restricts the Intracellular Survival of Mycobacterium Tuberculosis by Promoting Selective Autophagy. Front Microbiol (2021) 11:613963. doi: 10.3389/fmicb.2020.613963
99. Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, et al. Proteolytic Inactivation of MAP-Kinase-Kinase by Anthrax Lethal Factor. Science (1998) 280:734–7. doi: 10.1126/science.280.5364.734
100. Kang TJ, Basu S, Zhang L, Thomas KE, Vogel SN, Baillie L, et al. Bacillus Anthracis Spores and Lethal Toxin Induce IL-1β Via Functionally Distinct Signaling Pathways. Eur J Immunol (2008) 38:1574–84. doi: 10.1002/eji.200838141
101. Moayeri M, Martinez NW, Wiggins J, Young HA, Leppla SH. Mouse Susceptibility to Anthrax Lethal Toxin is Influenced by Genetic Factors in Addition to Those Controlling Macrophage Sensitivity. Infect Immun (2004) 72:4439–47. doi: 10.1128/IAI.72.8.4439-4447.2004
102. Skogberg K, Syrjanen J, Jahkola M, Renkonen O-V, Paavonen J, Ahonen J, et al. Clinical Presentation and Outcome of Listeriosis in Patients With and Without Immunosuppressive Therapy. Clin Infect Dis (1992) 14:815–21. doi: 10.1093/clinids/14.4.815
103. Shrestha N, Bahnan W, Wiley DJ, Barber G, Fields KA, Schesser K. Eukaryotic Initiation Factor 2 (eIF2) Signaling Regulates Proinflammatory Cytokine Expression and Bacterial Invasion. J Biol Chem (2012) 287:28738–44. doi: 10.1074/jbc.M112.375915
104. Namba T, Tanaka KI, Ito Y, Ishihara T, Hoshino T, Gotoh T, et al. Positive Role of CCAAT/enhancer-Binding Protein Homologous Protein, a Transcription Factor Involved in the Endoplasmic Reticulum Stress Response in the Development of Colitis. Am J Pathol (2009) 174:1786–98. doi: 10.2353/ajpath.2009.080864
105. Suyama K, Ohmuraya M, Hirota M, Ozaki N, Ida S, Endo M, et al. C/EBP Homologous Protein is Crucial for the Acceleration of Experimental Pancreatitis. Biochem Biophys Res Commun (2008) 367:176–82. doi: 10.1016/j.bbrc.2007.12.132
106. Park SH, Choi HJ, Yang H, Do KH, Kim J, Lee DW, et al. Endoplasmic Reticulum Stress-Activated C/EBP Homologous Protein Enhances Nuclear Factor-κB Signals Via Repression of Peroxisome Proliferator-Activated Receptor. J Biol Chem (2010) 285:35330–9. doi: 10.1074/jbc.M110.136259
107. Lebeaupin C, Proics E, De Bieville CHD, Rousseau D, Bonnafous S, Patouraux S, et al. ER Stress Induces NLRP3 Inflammasome Activation and Hepatocyte Death. Cell Death Dis (2015) 6:e1879–9. doi: 10.1038/cddis.2015.248
108. Shtrichman R, Heithoff DM, Mahan MJ, Samuel CE. Tissue Selectivity of Interferon-Stimulated Gene Expression in Mice Infected With Dam+ Versus Dam- Salmonella Enterica Serovar Typhimurium Strains. Infect Immun (2002) 70:5579–88. doi: 10.1128/IAI.70.10.5579-5588.2002
109. Yeung ATY, Choi YH, Lee AHY, Hale C, Ponstingl H, Pickard D, et al. A Genome-Wide Knockout Screen in Human Macrophages Idetified Host Factors Modulating Salmonella Infection. MBio (2019) 10:e02169–19. doi: 10.1128/mBio.02169-19
110. Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, et al. Type I Ifns Enhance Susceptibility to Chlamydia Muridarum Lung Infection by Enhancing Apoptosis of Local Macrophages. J Immunol (2008) 181:2092–102. doi: 10.4049/jimmunol.181.3.2092
111. Dulberger CL, Rubin EJ, Boutte CC. The Mycobacterial Cell Envelope — a Moving Target. Nat Rev Microbiol (2020) 18:47–59. doi: 10.1038/s41579-019-0273-7
112. Barkan D, Liu Z, Sacchettini JC, Glickman MS. Mycolic Acid Cyclopropanation is Essential for Viability, Drug Resistance, and Cell Wall Integrity of Mycobacterium Tuberculosis. Chem Biol (2009) 16:499–509. doi: 10.1016/j.chembiol.2009.04.001
113. Zhou X, Yang J, Zhang Z, Zhang L, Zhu B, Lie L, et al. Different Signaling Pathways Define Different Interferon-Stimulated Gene Expression During Mycobacteria Infection in Macrophages. Int J Mol Sci (2019) 20:663. doi: 10.3390/ijms20030663
114. Madhvi A, Mishra H, Chegou NN, Tromp G, Van Heerden CJ, Pietersen RD, et al. Distinct Host-Immune Response Toward Species Related Intracellular Mycobacterial Killing: A Transcriptomic Study. Virulence (2020) 11:170–82. doi: 10.1080/21505594.2020.1726561
115. Wu K, Koo J, Jiang X, Chen R, Cohen SN, Nathan C. Improved Control of Tuberculosis and Activation of Macrophages in Mice Lacking Protein Kinase R. PloS One (2012) 7:e30512. doi: 10.1371/journal.pone.0030512
116. Wu K, Koo J, Jiang X, Chen R, Cohen SN, Nathan C. Erratum: Improved Control of Tuberculosis and Activation of Macrophages in Mice Lacking Protein Kinase R (Plos One (2012) 7:E30512 Doi: 10.1371/Journal.Pone.0205424). PloS One (2018) 13:e0205424. doi: 10.1371/journal.pone.0205424
117. Chu LY, Hsueh YC, Cheng HL, Wu KK. Cytokine-Induced Autophagy Promotes Long-Term VCAM-1 But Not ICAM-1 Expression by Degrading Late-Phase IκBα. Sci Rep (2017) 7:12472. doi: 10.1038/s41598-017-12641-8
118. Dutta RK, Kathania M, Raje M, Majumdar S. IL-6 Inhibits IFN-γ Induced Autophagy in Mycobacterium Tuberculosis H37Rv Infected Macrophages. Int J Biochem Cell Biol (2012) 44:942–54. doi: 10.1016/j.biocel.2012.02.021
119. Bhattacharya B, Xiao S, Chatterjee S, Urbanowski M, Ordonez A, Ihms EA, et al. The Integrated Stress Response Mediates Necrosis in Murine Mycobacterium Tuberculosis Granulomas. J Clin Invest (2021) 131:e130319. doi: 10.1172/JCI130319
120. Pichugin AV, Yan BS, Sloutsky A, Kobzik L, Kramnik I. Dominant Role of the Sst1 Locus in Pathogenesis of Necrotizing Lung Granulomas During Chronic Tuberculosis Infection and Reactivation in Genetically Resistant Hosts. Am J Pathol (2009) 174:2190–201. doi: 10.2353/ajpath.2009.081075
121. Lai CC, Hsueh PR. Diseases Caused by Nontuberculous Mycobacteria in Asia. Future Microbiol (2014) 9:93–106. doi: 10.2217/fmb.13.138
122. George KM, Pascopella L, Welty DM, Small PLC. A Mycobacterium Ulcerans Toxin, Mycolactone, Causes Apoptosis in Guinea Pig Ulcers and Tissue Culture Cells. Infect Immun (2000) 68:877–83. doi: 10.1128/IAI.68.2.877-883.2000
123. Ogbechi J, Hall BS, Sbarrato T, Taunton J, Willis AE, Wek RC, et al. Inhibition of Sec61-Dependent Translocation by Mycolactone Uncouples the Integrated Stress Response From ER Stress, Driving Cytotoxicity Via Translational Activation of ATF4. Cell Death Dis (2018) 9:1–15. doi: 10.1038/s41419-018-0427-y
124. Matsui T, Tanihara K, Date T. Expression of Unphosphorylated Form of Human Double-Stranded RNA-Activated Protein Kinase in Escherichia Coli. Biochem Biophys Res Commun (2001) 284:798–807. doi: 10.1006/bbrc.2001.5039
125. Zheng X, Bevilacqua PC. Activation of the Protein Kinase PKR by Short Double-Stranded RNAs With Single-Stranded Tails. RNA (2004) 10:1934–45. doi: 10.1261/rna.7150804
126. Bleiblo F, Michael P, Brabant D, Ramana CV, Tai T, Saleh M, et al. JAK Kinases are Required for the Bacterial RNA and Poly I:C Induced Tyrosine Phosphorylation of PKR. Int J Clin Exp Med (2013) 6:16–25.
127. Hull CM, Bevilacqua PC. Mechanistic Analysis of Activation of the Innate Immune Sensor PKR by Bacterial Rna. J Mol Biol (2015) 427:3501–15. doi: 10.1016/j.jmb.2015.05.018
128. Hull CM, Anmangandla A, Bevilacqua PC. Bacterial Riboswitches and Ribozymes Potently Activate the Human Innate Immune Sensor PKR. ACS Chem Biol (2016) 11:1118–27. doi: 10.1021/acschembio.6b00081
129. Bevilacqua PC, Cech TR. Minor-Groove Recognition of Double-Stranded RNA by the Double-Stranded RNA-Binding Domain From the RNA-Activated Protein Kinase PKR. Biochemistry (1996) 35:9983–94. doi: 10.1021/bi9607259
130. Hu S, Sun H, Yin L, Li J, Mei S, Xu F, et al. PKR-Dependent Cytosolic cGAS Foci are Necessary for Intracellular DNA Sensing. Sci Signal (2019) 12:eaav7934. doi: 10.1126/scisignal.aav7934
131. Tanaka Y, Chen ZJ. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci Signal (2012) 5:ra20. doi: 10.1126/scisignal.2002521
132. Ward SV, Samuel CE. Regulation of the Interferon-Inducible PKR Kinase Gene: The KCS Element is a Constitutive Promoter Element That Functions in Concert With the Interferon-Stimulated Response Element. Virology (2002) 296:136–46. doi: 10.1006/viro.2002.1356
133. Horng T, Barton GM, Medzhitov R. Tirap: An Adapter Molecule in the Toll Signaling Pathway. Nat Immunol (2001) 2:835–41. doi: 10.1038/ni0901-835
134. Cabanski M, Steinmüller M, Marsh LM, Surdziel E, Seeger W, Lohmeyer J. PKR Regulates TLR2/TLR4-Dependent Signaling in Murine Alveolar Macrophages. Am J Respir Cell Mol Biol (2008) 38:26–31. doi: 10.1165/rcmb.2007-0010OC
135. Sales PCM, Williams BRG, Silva AM. Regulation of Double-Stranded RNA Dependent Protein Kinase Expression and Attenuation of Protein Synthesis Induced by Bacterial Toll-Like Receptors Agonists in the Absence of Interferon. J Interf Cytokine Res (2012) 32:495–504. doi: 10.1089/jir.2012.0019
136. Faria MS, Calegari-Silva TC, De Carvalho Vivarini A, Mottram JC, Lopes UG, Lima APCA. Role of Protein Kinase R in the Killing of Leishmania Major by Macrophages in Response to Neutrophil Elastase and TLR4 Via TNFα: And IFNβ. FASEB J (2014) 28:3050–63. doi: 10.1096/fj.13-245126
137. Li W, Zhu S, Li J, D’Amore J, D’Angelo J, Yang H, et al. Serum Amyloid a Stimulates PKR Expression and HMGB1 Release Possibly Through TLR4/RAGE Receptors. Mol Med (2015) 21:515–25. doi: 10.2119/molmed.2015.00109
138. Perkins DJ, Qureshi N, Vogel SN. A Toll-Like Receptor-Responsive Kinase, Protein Kinase R, is Inactivated in Endotoxin Tolerance Through Differential K63/K48 Ubiquitination. MBio (2010) 1:e00239–10. doi: 10.1128/mBio.00239-10
139. Carvalho Vivarini Á, Meirelles Santos Pereira R, Teixeira KLD, Calegari-Silva TC, Bellio M, Laurenti MD, et al. Human Cutaneous Leishmaniasis: Interferon-Dependent Expression of Double-Stranded RNA-Dependent Protein Kinase (PKR). via TLR2. FASEB J (2011) 25:4162–73. doi: 10.1096/fj.11-185165
140. Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BRG, Li X. Poly(Di·Dc)-Induced Toll-Like Receptor 3 (TLR3)-Mediated Activation of Nfκb and MAP Kinase is Through an Interleukin-1 Receptor-Associated Kinase (IRAK)-Independent Pathway Employing the Signaling Components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem (2003) 278:16713–9. doi: 10.1074/jbc.M300562200
141. Chuang JH, Chuang HC, Huang CC, Wu CL, Du YY, Kung ML, et al. Differential Toll-Like Receptor 3 (TLR3) Expression and Apoptotic Response to TLR3 Agonist in Human Neuroblastoma Cells. J BioMed Sci (2011) 18:65. doi: 10.1186/1423-0127-18-65
142. Singh M, Fowlkes V, Handy I, Patel CV, Patel RC. Essential Role of PACT-Mediated PKR Activation in Tunicamycin-Induced Apoptosis. J Mol Biol (2009) 385:457–68. doi: 10.1016/j.jmb.2008.10.068
143. Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, et al. Double-Stranded RNA-Dependent Protein Kinase Links Pathogen Sensing With Stress and Metabolic Homeostasis. Cell (2010) 140:338–48. doi: 10.1016/j.cell.2010.01.001
144. Vaughn LS, Bragg DC, Sharma N, Camargos S, Cardoso F, Patel RC. Altered Activation of Protein Kinase PKR and Enhanced Apoptosis in Dystonia Cells Carrying a Mutation in PKR Activator Protein PACT. J Biol Chem (2015) 290:22543–57. doi: 10.1074/jbc.M115.669408
145. Akazawa Y, Isomoto H, Matsushima K, Kanda T, Minami H, Yamaghchi N, et al. Endoplasmic Reticulum Stress Contributes to Helicobacter Pylori VacA-Induced Apoptosis. PloS One (2013) 8:e82322. doi: 10.1371/journal.pone.0082322
146. Brann KR, Fullerton MS, Voth DE. Coxiella Burnetii Requires Host Eukaryotic Initiation Factor 2α Activity for Efficient Intracellular Replication. Infect Immun (2020) 88:e00096–20. doi: 10.1128/IAI.00096-20
147. Halder P, Datta C, Kumar R, Sharma AK, Basu J, Kundu M. The Secreted Antigen, HP0175, of Helicobacter Pylori Links the Unfolded Protein Response (UPR) to Autophagy in Gastric Epithelial Cells. Cell Microbiol (2015) 17:714–29. doi: 10.1111/cmi.12396
148. Bedi B, Lin KC, Maurice NM, Yuan Z, Bijli K, Koval M, et al. UPR Modulation of Host Immunity by Pseudomonas Aeruginosa in Cystic Fibrosis. Clin Sci (2020) 134:1911–34. doi: 10.1042/CS20200066
149. Marchal JA, Carrasco E, Ramirez A, Jiménez G, Olmedo C, Peran M, et al. Bozepinib, a Novel Small Antitumor Agent, Induces PKR-Mediated Apoptosis and Synergizes With IFNα Triggering Apoptosis, Autophagy and Senescence. Drug Des Devel Ther (2013) 7:1301–13. doi: 10.2147/DDDT.S51354
150. López-Cara LC, Conejo-García A, Marchal JA, MacChione G, Cruz-López O, Boulaiz H, et al. New (RS)-Benzoxazepin-Purines With Antitumour Activity: The Chiral Switch From (RS)-2,6-Dichloro-9-[1-(P-Nitrobenzenesulfonyl)-1,2,3,5-Tetrahydro-4,1- Benzoxazepin-3-Yl]-9h-Purine. Eur J Med Chem (2011) 46:249–58. doi: 10.1016/j.ejmech.2010.11.011
151. Elazar M, Liu M, McKenna SA, Liu P, Gehrig EA, Puglisi JD, et al. The Anti-Hepatitis C Agent Nitazoxanide Induces Phosphorylation of Eukaryotic Initiation Factor 2α Via Protein Kinase Activated by Double-Stranded RNA Activation. Gastroenterology (2009) 137:1827–35. doi: 10.1053/j.gastro.2009.07.056
152. Ashiru O, Howe JD, Butters TD. Nitazoxanide, an Antiviral Thiazolide, Depletes ATP-Sensitive Intracellular Ca2+ Stores. Virology (2014) 462–463:135–48. doi: 10.1016/j.virol.2014.05.015
153. Bolick DT, Roche JK, Hontecillas R, Bassaganya-Riera J, Nataro JP, Guerrant RL. Enteroaggregative Escherichia Coli Strain in a Novel Weaned Mouse Model: Exacerbation by Malnutrition, Biofilm as a Virulence Factor and Treatment by Nitazoxanide. J Med Microbiol (2013) 62:896–905. doi: 10.1099/jmm.0.046300-0
154. Gupta A, Meena J, Sharma D, Gupta P, Gupta UD, Kumar S, et al. Inhalable Particles for “Pincer Therapeutics” Targeting Nitazoxanide as Bactericidal and Host-Directed Agent to Macrophages in a Mouse Model of Tuberculosis. Mol Pharm (2016) 13:3247–55. doi: 10.1021/acs.molpharmaceut.6b00459
155. McVay CS, Rolfe RD. In Vitro and In Vivo Activities of Nitazoxanide Against Clostridium Difficile. Antimicrob Agents Chemother (2000) 44:2254–8. doi: 10.1128/AAC.44.9.2254-2258.2000
156. Warren CA, Van Opstal EJ, Riggins MS, Li Y, Moore JH, Kolling GL, et al. Vancomycin Treatment’s Association With Delayed Intestinal Tissue Injury, Clostridial Overgrowth, and Recurrence of Clostridium Difficile Infection in Mice. Antimicrob Agents Chemother (2013) 57:689–96. doi: 10.1128/AAC.00877-12
157. Rossignol JF. Nitazoxanide: A First-In-Class Broad-Spectrum Antiviral Agent. Antiviral Res (2014) 110:94–103. doi: 10.1016/j.antiviral.2014.07.014
158. Hemphill A, Mueller J, Esposito M. Nitazoxanide, a Broad-Spectrum Thiazolide Anti-Infective Agent for the Treatment of Gastrointestinal Infections. Expert Opin Pharmacother (2006) 7:953–64. doi: 10.1517/14656566.7.7.953
159. Bailey MA, Na H, Duthie MS, Gillis TP, Lahiri R, Parish T. Nitazoxanide is Active Against Mycobacterium Leprae. PloS One (2017) 12:e0184107. doi: 10.1371/journal.pone.0184107
160. Wang X, Shen C, Liu Z, Peng F, Chen X, Yang G, et al. Nitazoxanide, an Antiprotozoal Drug, Inhibits Late-Stage Autophagy and Promotes ING1-Induced Cell Cycle Arrest in Glioblastoma. Cell Death Dis (2018) 9:1032. doi: 10.1038/s41419-018-1058-z
161. Kiehl IGA, Riccetto E, Salustiano ACC, Ossick MV, Ferrari KL, Assalin HB, et al. Boosting Bladder Cancer Treatment by Intravesical Nitazoxanide and Bacillus Calmette-Guérin Association. World J Urol (2021) 39:1187–94. doi: 10.1007/s00345-020-03294-w
162. Pal AK, Nandave M, Kaithwas G. Chemoprophylactic Activity of Nitazoxanide in Experimental Model of Mammary Gland Carcinoma in Rats. 3 Biotech (2020) 10:338. doi: 10.1007/s13205-020-02332-z
163. Senkowski W, Zhang X, Olofsson MH, Isacson R, Höglund U, Gustafsson M, et al. Three-Dimensional Cell Culture-Based Screening Identifies the Anthelmintic Drug Nitazoxanide as a Candidate for Treatment of Colorectal Cancer. Mol Cancer Ther (2015) 14:1504–16. doi: 10.1158/1535-7163.MCT-14-0792
164. Ai N, Wood RD, Welsh WJ. Identification of Nitazoxanide as a Group I Metabotropic Glutamate Receptor Negative Modulator for the Treatment of Neuropathic Pain: An in Silico Drug Repositioning Study. Pharm Res (2015) 32:2798–807. doi: 10.1007/s11095-015-1665-7
165. Amireddy N, Puttapaka SN, Vinnakota RL, Ravuri HG, Thonda S, Kalivendi SV. The Unintended Mitochondrial Uncoupling Effects of the FDA-Approved Anti-Helminth Drug Nitazoxanide Mitigates Experimental Parkinsonism in Mice. J Biol Chem (2017) 292:15731–43. doi: 10.1074/jbc.M117.791863
166. Hong SK, Kim HJ, Song CS, Choi IS, Lee JB, Park SY. Nitazoxanide Suppresses IL-6 Production in LPS-Stimulated Mouse Macrophages and TG-Injected Mice. Int Immunopharmacol (2012) 13:23–7. doi: 10.1016/j.intimp.2012.03.002
167. Hu W, Hofstetter W, Wei X, Guo W, Zhou Y, Pataer A, et al. Double-Stranded RNA-Dependent Protein Kinase-Dependent Apoptosis Induction by a Novel Small Compound. J Pharmacol Exp Ther (2009) 328:866–72. doi: 10.1124/jpet.108.141754
168. Bai H, Chen T, Ming J, Sun H, Cao P, Fusco DN, et al. Dual Activators of Protein Kinase R (PKR) and Protein Kinase R-Like Kinase (PERK) Identify Common and Divergent Catalytic Targets. ChemBioChem (2013) 14:1255–62. doi: 10.1002/cbic.201300177
169. Jammi NV, Whitby LR, Beal PA. Small Molecule Inhibitors of the RNA-Dependent Protein Kinase. Biochem Biophys Res Commun (2003) 308:50–7. doi: 10.1016/S0006-291X(03)01318-4
170. Nakamura T, Arduini A, Baccaro B, Furuhashi M, Hotamisligil GS. Small-Molecule Inhibitors of PKR Improve Glucose Homeostasis in Obese Diabetic Mice. Diabetes (2014) 63:526–34. doi: 10.2337/db13-1019
171. Ingrand S, Barrier L, Lafay-Chebassier C, Fauconneau B, Page G, Hugon J. The Oxindole/Imidazole Derivative C16 Reduces In Vivo Brain PKR Activation. FEBS Lett (2007) 581:4473–8. doi: 10.1016/j.febslet.2007.08.022
172. Watanabe T, Ninomiya H, Saitou T, Takanezawa S, Yamamoto S, Imai Y, et al. Therapeutic Effects of the PKR Inhibitor C16 Suppressing Tumor Proliferation and Angiogenesis in Hepatocellular Carcinoma In Vitro and In Vivo. Sci Rep (2020) 10:5133. doi: 10.1038/s41598-020-61579-x
173. Kalra J, Bhat A, Jadhav KKB, Dhar A. Up-Regulation of PKR Pathway Contributes to L-NAME Induced Hypertension and Renal Damage. Heliyon (2020) 6:e05463. doi: 10.1016/j.heliyon.2020.e05463
174. Kalra J, Dasari D, Bhat A, Mangali S, Goyal SG, Jadhav KB, et al. PKR Inhibitor Imoxin Prevents Hypertension, Endothelial Dysfunction and Cardiac and Vascular Remodelling in L-NAME-Treated Rats. Life Sci (2020) 262:118436. doi: 10.1016/j.lfs.2020.118436
175. Wang WJ, Yin SJ, Rong RQ. PKR and HMGB1 Expression and Function in Rheumatoid Arthritis. Genet Mol Res (2015) 14:17864–70. doi: 10.4238/2015.December.22.11
176. Tronel C, Page G, Bodard S, Chalon S, Antier D. The Specific PKR Inhibitor C16 Prevents Apoptosis and IL-1β Production in an Acute Excitotoxic Rat Model With a Neuroinflammatory Component. Neurochem Int (2014) 64:73–83. doi: 10.1016/j.neuint.2013.10.012
177. Li R, Shang Y, Yu Y, Zhou T, Xiong W, Zou X. High-Mobility Group Box 1 Protein Participates in Acute Lung Injury by Activating Protein Kinase R and Inducing M1 Polarization. Life Sci (2020) 246:117415. doi: 10.1016/j.lfs.2020.117415
178. Li Y, Xiao J, Tan Y, Wang J, Zhang Y, Deng X, et al. Inhibition of PKR Ameliorates Lipopolysaccharide-Induced Acute Lung Injury by Suppressing NF-κB Pathway in Mice. Immunopharmacol Immunotoxicol (2017) 39:165–72. doi: 10.1080/08923973.2017.1303839
179. Teramachi J, Inagaki Y, Shinohara H, Okamura H, Yang D, Ochiai K, et al. PKR Regulates LPS-Induced Osteoclast Formation and Bone Destruction In Vitro and In Vivo. Oral Dis (2017) 23:181–8. doi: 10.1111/odi.12592
180. Zhou J, Zhang F, Lin H, Quan M, Yang Y, Lv Y, et al. The Protein Kinase R Inhibitor C16 Alleviates Sepsis-Induced Acute Kidney Injury Through Modulation of the NF-kB and NLR Family Pyrin Domain-Containing 3 (NLPR3) Pyroptosis Signal Pathways. Med Sci Monit (2020) 26:e926254. doi: 10.12659/MSM.926254
181. Valentine RJ, Jefferson MA, Kohut ML, Eo H. Imoxin Attenuates LPS-Induced Inflammation and MuRF1 Expression in Mouse Skeletal Muscle. Physiol Rep (2018) 6:e13941. doi: 10.14814/phy2.13941
182. Hwang KD, Bak MS, Kim SJ, Rhee S, Lee YS. Restoring Synaptic Plasticity and Memory in Mouse Models of Alzheimer’s Disease by PKR Inhibition. Mol Brain (2017) 10:57. doi: 10.1186/s13041-017-0338-3
183. Xiao J, Tan Y, Li Y, Luo Y. The Specific Protein Kinase R (PKR) Inhibitor C16 Protects Neonatal Hypoxia-Ischemia Brain Damages by Inhibiting Neuroinflammation in a Neonatal Rat Model. Med Sci Monit (2016) 22:5074–81. doi: 10.12659/MSM.898139
184. Segev Y, Barrera I, Ounallah-Saad H, Wibrand K, Sporild I, Livne A, et al. PKR Inhibition Rescues Memory Deficit and Atf4 Overexpression in Apoe ϵ4 Human Replacement Mice. J Neurosci (2015) 35:12986–93. doi: 10.1523/JNEUROSCI.5241-14.2015
185. Hu Y, Conway TW. 2-Aminopurine Inhibits the Double-Stranded RNA-Dependent Protein Kinase Both In Vitro and In Vivo. J Interferon Res (1993) 13:323–8. doi: 10.1089/jir.1993.13.323
186. Supinski GS, Callahan LA. Double-Stranded RNA-Dependent Protein Kinase Activation Modulates Endotoxin-Induced Diaphragm Weakness. J Appl Physiol (2011) 110:199–205. doi: 10.1152/japplphysiol.01203.2009
187. Qiu CF, Wu JF, Pei F, Wang LH, Mei MH, Ouyang B, et al. Effect of Inhibiting the Activity of Double-Stranded RNA-Dependent Protein Kinase in Sepsis Mice. Zhonghua Yi Xue Za Zhi (2020) 100:1033–7. doi: 10.3760/cma.j.cn112137-20190825-01888
188. Rossignol JF, Elfert A, El-Gohary Y, Keeffe EB. Improved Virologic Response in Chronic Hepatitis C Genotype 4 Treated With Nitazoxanide, Peginterferon, and Ribavirin. Gastroenterology (2009) 136:856–62. doi: 10.1053/j.gastro.2008.11.037
189. Haffizulla J, Hartman A, Hoppers M, Resnick H, Samudrala S, Ginocchio C, et al. Effect of Nitazoxanide in Adults and Adolescents With Acute Uncomplicated Influenza: A Double-Blind, Randomised, Placebo-Controlled, Phase 2b/3 Trial. Lancet Infect Dis (2014) 14:609–18. doi: 10.1016/S1473-3099(14)70717-0
190. Shigyo K, Ocheretina O, Merveille YM, Johnson WD, Pape JW, Nathan CF, et al. Efficacy of Nitazoxanide Against Clinical Isolates of Mycobacterium Tuberculosis. Antimicrob Agents Chemother (2013) 57:2834–7. doi: 10.1128/AAC.02542-12
191. Walsh KF, McAulay K, Lee MH, Vilbrun SC, Mathurin L, Francois DJ, et al. Early Bactericidal Activity Trial of Nitazoxanide for Pulmonary Tuberculosis. Antimicrob Agents Chemother (2020) 64:e01956–19. doi: 10.1128/AAC.01956-19
192. Hagberg L, Briles DE, Edén CS. Evidence for Separate Genetic Defects in C3H/HeJ and C3HeB/FeJ Mice, That Affect Susceptibility to Gram-Negative Infections. J Immunol (1985) 134:4118–22.
193. Popov SG, Villasmil R, Bernardi J, Grene E, Cardwell J, Wu A, et al. Lethal Toxin of Bacillus Anthracis Causes Apoptosis of Macrophages. Biochem Biophys Res Commun (2002) 293:349–55. doi: 10.1016/S0006-291X(02)00227-9
194. Alileche A, Serfass ER, Muehlbauer SM, Porcelli SA, Brojatsch J. Anthrax Lethal Toxin-Mediated Killing of Human and Murine Dendritic Cells Impairs the Adaptive Immune Response. PloS Pathog (2005) 1:e19. doi: 10.1371/journal.ppat.0010019
195. Schaaf K, Smith SR, Duverger A, Wagner F, Wolschendorf F, Westfall AO, et al. Mycobacterium Tuberculosis Exploits the PPM1A Signaling Pathway to Block Host Macrophage Apoptosis. Sci Rep (2017) 7:42101. doi: 10.1038/srep42101
196. Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy Controls Salmonella Infection in Response to Damage to the Salmonella-Containing Vacuole. J Biol Chem (2006) 281:11374–83. doi: 10.1074/jbc.M509157200
197. Py BF, Lipinski MM, Yuan J. Autophagy Limits Listeria Monocytogenes Intracellular Growth in the Early Phase of Primary Infection. Autophagy (2007) 3:117–25. doi: 10.4161/auto.3618
198. Matsuzawa T, Kim B-H, Shenoy AR, Kamitani S, Miyake M, MacMicking JD. Ifn-γ Elicits Macrophage Autophagy Via the P38 MAPK Signaling Pathway. J Immunol (2012) 189:813–8. doi: 10.4049/jimmunol.1102041
199. Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-Mediated Activation of Interleukin-1β (Il-1β) and IL-18 Contributes to Innate Immune Defenses Against Salmonella Enterica Serovar Typhimurium Infection. Infect Immun (2006) 74:4922–6. doi: 10.1128/IAI.00417-06
200. Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, et al. Role of the Caspase-1 Inflammasome in Salmonella Typhimurium Pathogenesis. J Exp Med (2006) 203:1407–12. doi: 10.1084/jem.20060206
201. Ravimohan S, Kornfeld H, Weissman D, Bisson GP. Tuberculosis and Lung Damage: From Epidemiology to Pathophysiology. Eur Respir Rev (2018) 27:170077. doi: 10.1183/16000617.0077-2017
202. Carrero JA, Calderon B, Unanue ER. Type I Interferon Sensitizes Lymphocytes to Apoptosis and Reduces Resistance to Listeria Infection. J Exp Med (2004) 200:535–40. doi: 10.1084/jem.20040769
203. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. Ifns Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection Via IRG1-Derived Itaconic Acid. PloS Pathog (2016) 12:e1005408. doi: 10.1371/journal.ppat.1005408
204. Ladiges W, Morton J, Hopkins H, Wilson R, Filley G, Ware C, et al. Expression of Human PKR Protein Kinase in Transgenic Mice. J Interf Cytokine Res (2002) 22:329–34. doi: 10.1089/107999002753675758
Keywords: Protein Kinase R, bacterial infection, macrophage signaling, antibacterial defense, EIF2AK2, cell death, autophagy, inflammation
Citation: Smyth R and Sun J (2021) Protein Kinase R in Bacterial Infections: Friend or Foe? Front. Immunol. 12:702142. doi: 10.3389/fimmu.2021.702142
Received: 29 April 2021; Accepted: 28 June 2021;
Published: 08 July 2021.
Edited by:
Monica Cartelle Gestal, Louisiana State University Health Shreveport, United StatesReviewed by:
Som G. Nanjappa, University of Illinois at Urbana-Champaign, United StatesCopyright © 2021 Smyth and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jim Sun, amltLnN1bkB1b3R0YXdhLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.