 Yael Dinur-Schejter
Yael Dinur-Schejter Irina Zaidman1,2
Irina Zaidman1,2 Polina Stepensky
Polina Stepensky- 1Faculty of Medicine, Hebrew University of Jerusalem, Jerusalem, Israel
- 2The Bone Marrow Transplantation and Cancer Immunotherapy Department, Hadassah Ein Kerem Medical Center, Jerusalem, Israel
- 3Allergy and Clinical Immunology Unit, Hadassah Ein-Kerem Medical Center, Jerusalem, Israel
- 4Monique and Jacques Roboh Department of Genetic Research, Hadassah Ein Kerem Medical Center, Jerusalem, Israel
Adaptor molecules lack enzymatic and transcriptional activities. Instead, they exert their function by linking multiple proteins into intricate complexes, allowing for transmitting and fine-tuning of signals. Many adaptor molecules play a crucial role in T-cell signaling, following engagement of the T-cell receptor (TCR). In this review, we focus on Linker of Activation of T cells (LAT) and SH2 domain-containing leukocyte protein of 76 KDa (SLP-76). Monogenic defects in these adaptor proteins, with known roles in T-cell signaling, have been described as the cause of human inborn errors of immunity (IEI). We describe the current knowledge based on defects in cell lines, murine models and human patients. Germline mutations in Adhesion and degranulation adaptor protein (ADAP), have not resulted in a T-cell defect.
Introduction
Engagement of the T cell receptor (TCR) triggers a signaling cascade responsible for T-cell activation, maturation and differentiation. Fine tuning of this complex multi-protein cascade enables discriminating different signals based on strength and duration. In the thymus, this process allows for positive selection. In the periphery, weak, self-peptide-MHC survival signals are differentiated from strong foreign-peptide-MHC activating signals (1). Moreover, signaling strength is crucial for determining T-cell fate (2).
Following engagement of the TCR, immunoreceptor tyrosine-based activation motifs (ITAMs) on CD3 cytoplasmic tails and ζ chains are phosphorylated by LCK, leading to the phosphorylation of ZAP70, which in turn phosphorylates LAT, SLP-76 and CD6 (1, 3–5) (Figure 1). Phosphorylation of the 4 distal tyrosine residues of LAT leads to the assembly of the LAT signalosome, including PLCγ1, Itk, SLP-76, Gads and Grb-2 (5–7). This signalosome mediates downstream events which are crucial for T-cell activation, including Calcium mobilization, Erk and NFAT activation, CD69 expression, and cytoskeletal organization (6).
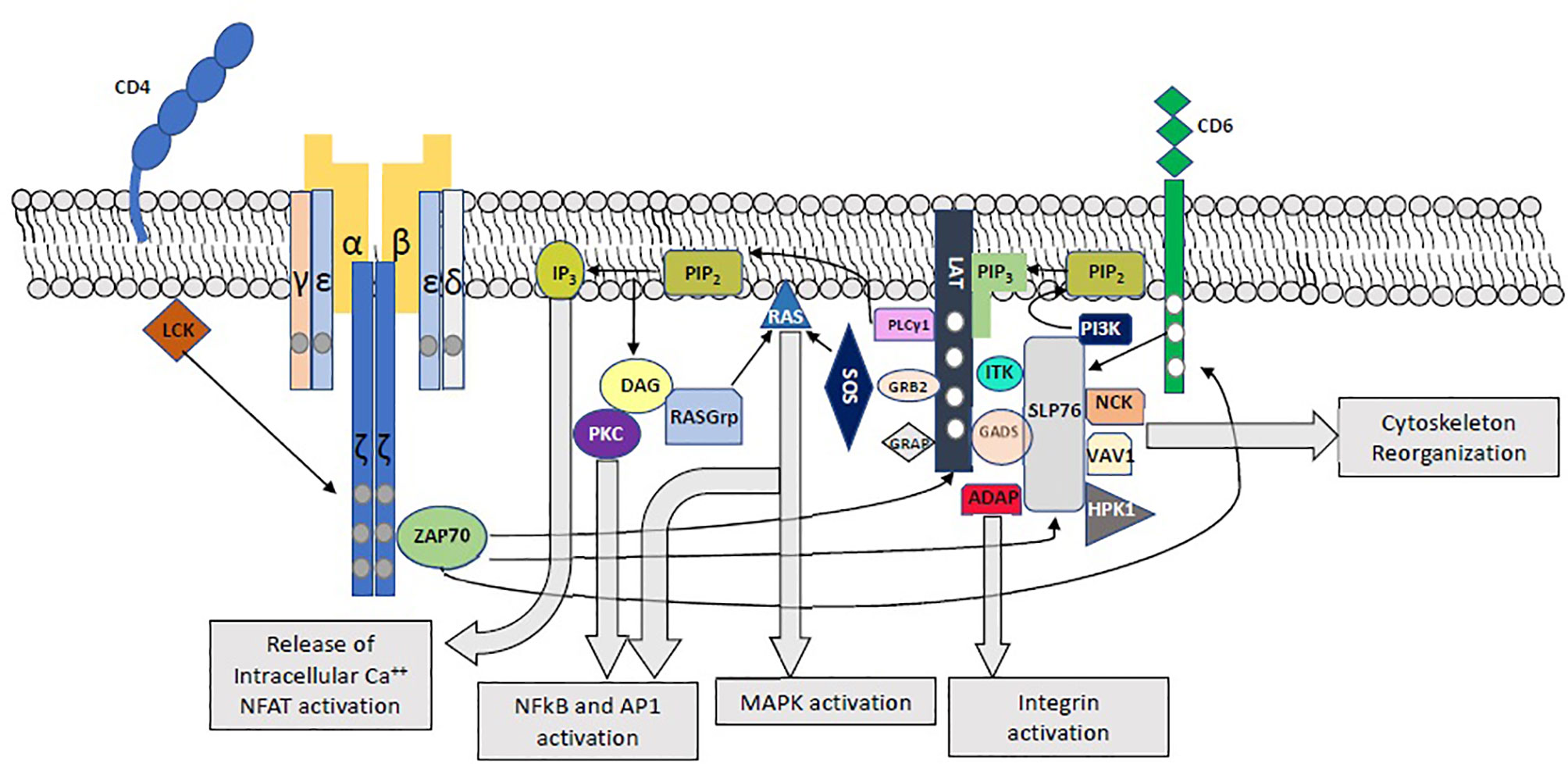
Figure 1 Proximal T-Cell Activation Signaling. The main proximal signaling events following engagement of the TCR are depicted, including the assembly of the LAT and SLP-76 signalosomes and their downstream effects.
Adaptor molecules lack both enzymatic and transcriptional activities. Through multiple interaction domains they function as modular scaffolds for the formation of multiprotein signaling complexes and have a vital role in transmitting and fine-tuning of T-cell activation. It is not surprising, therefore, to find human germline defects in these molecules that result in immune dysregulation.
Newly recognized inborn errors of immunity (IEI) due to monogenic mutations in several key adaptors in T-cell signaling have shed light on their function in humans. In this review, we summarize the current knowledge on adaptors in T-cell signaling for which human monogenic defects have been described, and discuss lessons learnt from comparison of cell lines, murine models and humans.
Linker For Activation Of T Cells
LAT is expressed in T-, mast, NK- and immature B-cells, as well as megakaryocytes and platelets (6). This protein, with a short extracellular domain, a single transmembrane domain, and a long intracellular region, serves as a crucial nucleating factor for multiprotein signaling complexes. It plays a central role in T-cell activation downstream of the TCR, by recruiting kinases, effectors, and other adaptors into highly regulated signal transduction pathways. Upon TCR engagement, LAT is phosphorylated primarily by ZAP70, but also by Itk and Lck (6) in 4 conserved tyrosine residues (Y132, Y171 Y191 and Y226). Phosphorylated LAT binds to PLCγ1, SLP-76 (via Gads) and Grb-2, leading to the assembly of the LAT signalosomes (1, 5–7)(Figure 1). Phosphorylated tyrosine residues show predilection towards specific binding proteins. In this way, the Y132 residue binds PLCγ1 with greater affinity, while Grb2, Gads and Grap associate with the distal LAT phosphotyrosines Y171, Y191 and Y226 (6, 8–11). Nevertheless, each binding site specificity is not insulated, and the different SH2-containing LAT binding proteins show cooperative interactions: while Gads-SLP-76 binding stabilizes the LAT-PLCγ1 association and PLCγ1 activation through recruitment of Itk (12, 13), PLCγ1 stabilizes the binding of Grb2 to LAT. The cooperative interaction of LAT, Sos1 and Grb2, as well as LAT-Gads-SLP-76-ADAP allow for the assembly of macromolecular LAT signaling complexes, which are essential for T-cell signaling (6, 11, 14–16).
Once activated, PLCγ1 hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to produce inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). DAG stimulates RasGrp, which in turn activates ERK1/2, and protein kinase C (PKC), leading to activation of the NFκB pathway. IP3 promotes the release of intracellular Ca2+ stores, leading to extracellular Ca2+ influx and NFAT activation. Grb2, through its constitutive association with Sos1 and Cbl, is involved in Ras and MAP kinase activation (6, 17). Moreover, by binding multiple Grb2 molecules to LAT, the Grb2-Sos1-LAT interaction mediates oligomerization of LAT. Importantly, LAT oligomerization was shown to be of greater significance for T-cell signaling under limiting stimulating conditions (14, 15).
SLP-76, another crucial adaptor, is recruited to the LAT signalosome via its constitutive interactions with Gads. In addition to its contribution to LAT- PLCγ1 interaction and PLCγ1 activation, this adaptor is involved in integrin and cytoskeletal function as well as downstream T-cell activation through multiple interactions with Nck, Vav1, Rac1, ADAP, Shb and p85 (5, 6, 17–19). Importantly, SLP-76 is also activated in a LAT-independent manner by phosphorylated CD6 (1, 20), and following integrin activation (21).
LAT Murine Models
A complete LAT knockout murine model exhibited early arrest of thymocyte development in the double negative 3 (DN3) stage (6, 22). A LAT4YF knock-in model, deficient in the 4 distal tyrosines showed a similar phenotype, indicating the importance of these 4 residues to pre-TCR signaling and T-cell development (23). In contrast, a point mutation in the Y136 residue (LATY136F, corresponding to human Y132 residue), impaired LAT binding to PLCγ1, Ca2+ flux and NFAT signaling, while the effect on ERK phosphorylation was variable (17, 24–27). Affected T-cells show severe, yet incomplete arrest in their development. Affected mice developed a polyclonal, Th2 lymphoproliferative disorder, with secondary massive expansion of eosinophils and B cells, multiorgan inflammation, autoimmune nephritis and fibrosis (24, 28–32). Aberrant T-cells exhibited an effector phenotype, with reduced proliferation in response to TCR stimulation, reduced FasL-mediated apoptosis, and a TCRloCD5hi phenotype, indicative of an abnormal survival and proliferation of autoreactive T-cells (24, 25, 30). In a knock-in murine model of the Y175, Y195 and Y235 residues (LATY7/8/9F corresponding to the human Y171, Y191 and Y226 residues), a complete arrest of αβ-T cell alongside partial arrest of γδ T-cell development caused a similar albeit slightly delayed-onset lymphoproliferative Th2 disease (33). When the LATY136F knock-in was crossed with TCRβ-/- mice, a similar phenotype to the LATY7/8/9F knock-in ensued, which demonstrates the importance of the LAT- PLCγ1 to both αβ and γδ T-cell maturation (29). Since recruitment of Grb2 and Gads is important for activation of PLCγ1, the difference between the LATY136F and LATY7/8/9F phenotypes can be attributed to differences in signal strength and the lower level of signaling required for γδ T-cell maturation (17).
While LATY136F Tregs were non-functional, using adoptive transfer experiments with floxed genes, autoimmune phenotype in LAT mutated mice was proven to be intrinsic to effector CD4+ cells (27, 32) and independent of thymic development and of the distal 3 LAT tyrosine residues (1, 25, 27). Presumably, both the peripheral ablation of LAT in normally developed T-cells and the occurrence of peripheral LATY136F T-cells enable a weak tonic TCR-dependent signal, leading to continued positive LAT-independent signaling events, including PI3K and SLP-76 activation, which is unopposed by a normal negative-feedback loop. This results in the emergence and expansion of an abnormal, autoreactive polyclonal Th2 subset (17, 25). Such negative feedback signals include Gab2-dependent SHP-2 activation and competitive inhibition of SLP-76 (34), Grap inhibition of ERK activation (35), SHIP-1-Dok1/2 mediated inhibition of Akt and Zap70 (36, 37), THEMIS-SHP1-Grb2 mediated regulation of proximal-TCR signaling, and LAT negative feedback of ZAP70 and CD3ζ phosphorylation (38), PTPN7 (38) and HPK-1 (39).
LAT Inborn Errors of Immunity
Recently, human inborn errors of immunity (IEI) caused by monogenic defects in LAT have been described, with varying phenotypes: Bacchelli et al. (40) described 5 patients from a single consanguineous pedigree with severe combined immunodeficiency (SCID). All patients presented in early infancy with recurrent infections and failure to thrive (FTT), extremely low (<300 cells/mm3) T cell counts and absent T-cell proliferative response to phytohemagglutinin (PHA) and normal B and NK-cell counts. In one patient, increased γδ T-cell count was suspected. While all patients underwent hematopoietic stem cell transplantation (HSCT) using various donor types and conditioning regimens, 3/5 (60%) died of transplant related complications. A homozygous LAT c.44_45insT p.Leu16AlafsX28 mutation, associated with a complete loss of protein expression was found. Mutant LAT reconstitution in Jurkat cell lines was unable to restore post-stimulation CD69 expression, Ca2+ flux and downstream phosphorylation of SLP-76 and Vav1. TCR-induced apoptosis was severely reduced in LAT-deficient T-cell lines and was not restored following reconstitution with the LAT-mutant.
Keller et al. (41) described 3 siblings of a consanguineous family with a homozygous LAT c.268_269del mutation, which resulted in a premature stop-codon eliminating all major intracellular phosphorylation sites. While patients also presented in early infancy with recurrent infection including CMV viremia and recurrent pneumoniae resulting in bronchiectasis, their phenotype was notable for severe autoimmune cytopenias, anti-ADAMTS13+ microangiopathic hemolytic anemia, lymphoproliferation and an expansion of Th2-like effector T-cells, as well as elevated γδ T-cell counts, reminiscent of the partially deficient murine models (24, 33). Patients developed progressive hypogammaglobulinemia and CD4 and B cell lymphopenia with reductions in naïve CD4 and CD8 cells and reduced CD3 expression. TCR-dependent proliferation and activation was abrogated. While 2 patients died at 9 and 2 years of age of disseminated CMV infection and thrombotic thrombocytopenic purpura (TTP), respectively, one patient underwent a successful HSCT and is currently well.
Considering the elimination of all 4 major known phosphorylation sites, and the complete block in T-cell development in the equivalent murine model (23), it was somewhat surprising that Ca2+ mobilization and IκBα degradation, both downstream of PLCγ1 activation were normal in patients’ T cells, despite being affected in Jurkat cell lines (8, 41). While ERK phosphorylation was absent in patients’ CD4CD45Ro cells, ITK phosphorylation was normal both in patient’s cells and Jurkat-cell lines, in contrast to a previous report (42). The authors attribute the difference to the presence of a yet unknown LAT-replacing adaptor which is absent in Jurkat-cell lines. Indeed, the importance of LAT-independent TCR-signaling has been recognized previously (7, 20, 25, 38, 43–46). CD6 is known to recruit SLP-76, Gads, Grb2, Vav1 and SHIP1 independently of LAT (20, 47, 48), and is therefore a potential candidate for LAT-independent Itk phosphorylation and Calcium flux. While induced CD6 expression in Jurkat cell lines did not rescue Ca2+ mobilization in LAT-mutated cell-line, it is possible that this is because of lack of expression of the CD6 ligand (48). Possibly, CD6 or another LAT-substitute is responsible for Ca2+ mobilization downstream of PLCγ1, but is unable to replace ERK activation, which requires both PLCγ1 activation and an intact LAT-Grb2-Sos1 complex (6, 49). Another possible explanation lies in the partial rescue of LAT-dependent signaling via LCK binding to LAT upstream of the mutation site (50). Possibly, the impact of such binding is stronger in primary T-cells compared to Jurkat cell lines. Loss of RasGrp-induced ERK activation in turn, may have resulted in aberrant tonic T-cell signaling and basal TCRα levels in naïve CD4 cell (51), affecting LAT’s regulatory role in maintaining T-cell homeostasis. This is supported by the observation of low CD3 expression in LAT-mutated mice, Jurkat cell lines and human cells (41, 51, 52), as well as by the similar phenotype observed in RasGrp deficient patients (53). In this way, we postulate, that while LAT-independent signals are sufficient to allow for partial αβ and γδ T-cell development in the thymus, abnormally weak signal in the periphery results in aberrant tonic T-cell signaling and the development of a population of activated, dysregulated CD4+ cell.
The current concept for LAT function, is that LAT has both positive and negative roles in T-cell signaling: in the thymus, LAT is responsible for pre-TCR signaling and positive selection, and so complete loss of protein expression results in arrest of T-cell development. In the periphery, however, LAT has a dual role: on one hand it acts as a positive regulator of T-cell activation, including early T-cell activation, immune synapse development and cytoskeletal changes. On the other hand, LAT-dependent inhibition augments T-cell signaling and is involved in maintaining T-cell homeostasis (6, 51). Moreover, it is known that LAT functions as a central hub for the creation of multiple microclusters which then assemble into condensates. Recently, using affinity purification with mass spectrometry, LAT microclusters were proven to be heterogeneous. Many abortive or partially functional LAT signalosomes accompany the fully functional, high order signalosome. As such, it was shown, that 30 seconds after TCR engagement, LAT-SHIP1 containing signalosomes are much more abundant than LAT-SLP-76 signalosomes (47). Therefore, it is possible, that the different LAT signalosome isoforms have distinct functions, and that the net result of TCR-engagement depends on the combined output of these higher order LAT-signalosome-containing condensates. In this way, a partial deletion in LAT could potentially alter the composition of LAT-microclusters and attenuate the TCR signal.
SH2 Domain-Containing Leukocyte Protein Of 76 KDa
SLP-76 is expressed in T cells, platelets, neutrophils, mast cells, macrophages and NK cells (54). It exerts its function through four distinct domains: an amino-terminal sterile α motif (SAM) domain is responsible for ACK1 binding and oligomerization (55, 56), followed by three tyrosine phosphorylation motifs, responsible for binding of multiple effectors, including Vav1, Nck, Itk and p85, thus promoting signal transduction and cytoskeletal organization (54). A central proline-rich domain includes the binding site for Gads, Grb2 and PLCγ1, and is responsible for the recruitment of PLCγ1 to the LAT signalosome and its activation by Itk (57). A C-terminal SH2 domain is involved in integrin function and the formation of LAT microclusters via ADAP (11, 16, 21, 58–60), as well as a negative-feedback loop through binding of HPK1 (11), and CD6 interaction (61).
An SLP-76 deficient Jurkat-cell line (denoted J14) demonstrated the importance of SLP-76 in PLCγ1 activation, intracellular Ca2+ flux, activation of the Ras, NFAT and AP1 pathways and early T-cell activation events, such as CD69 expression (62). Mutations in the three N-terminal tyrosine residues of SLP-76 (denoted SLP-76Y3F), as well as mutations of the Gads-binding site, all showed reduced Ca2+-dependent NFAT activation, ERK1/2 and PLCγ1 phosphorylation. A mutation in the SLP-76 SH2 domain, however, affected PLCγ1 phosphorylation to the same extent, while NFAT activation was variably affected and ERK phosphorylation was comparable to wild type (60, 63).
While there is a 60% perinatal mortality rate among SLP-76 deficient mice, the remaining suffer from defects in T-cell development, as well as mast cell, neutrophil, platelet and vascular defects (54, 64–67). SLP-76 deficient T-cells show arrested thymic development at the DN3 stage, in a similar manner to LAT-deficient mice. An SLP-76-N-Terminal domain depleted murine model showed a similar phenotype to the SLP-76-/- model (68). On the other hand, knock-in mutations in the N-terminal and proline-rich domains, including the SLP-76Y3F, SLP-76Y112/128F, SLP-76Y145F knock-ins, and selective deletions of the Gads-binding site all resulted in varying degrees of aberrant, yet not obliterated thymic differentiation, impaired calcium flux, actin polymerization and PLCγ1 and ERK phosphorylation (58, 68, 69). Deletion of the SH2 domain results in a milder impairment in thymocyte development, near normal Ca2+ flux and ERK activation, but defective T-cell proliferation and activation, reminiscent of the ADAP-deficient murine model (19, 68, 69).
SLP-76 Inborn Errors of Immunity
Recently, a single Palestinian patient was described with a novel homozygous mutation c.957+1G>A; p.K309FSx17 in SLP-76, affecting a donor splice site and resulting in skipping of exon 14 and deletion of the C-terminal domain (70). Clinically, the patient presented in early infancy with a combination of autoimmune and lymphoproliferative manifestations, CMV viremia, skin disease, Aspergillus fumigatus brain abscesses and local BCGitis. His immune phenotype included a skewed CD4:CD8 ratio, clonal expansion of central memory CD4+ cells and terminally differentiated CD8+ cells, and a skewed T-cell repertoire, alongside a severe neutrophil defect, an NK functional defect and arrest of B-cell development. A defect in platelet aggregation led to a petechial rash. The patient underwent a haplo-identical HSCT at the age of 10 months. However, he died in the immediate post-HSCT period of transplant related complications.
While there was no protein expression in patient’s peripheral blood mononuclear cells (PBMCs), there was somewhat lower expression of the SLP-76 in J14 reconstituted with the mutant protein. The differential expression between patient cells and reconstituted Jurkat cell lines could be attributed to fixed promoter-driven cDNA expression in Jurkat cell lines, as well as lower expression of SLP-76 in patient’s clonally expanded, terminally differentiated cells. The authors concluded that the mutation results in a hypomorphic, unstable yet partially functional protein, leading to reduced TCR-dependent ERK, S6 and PLCγ1 phosphorylation, abnormal Ca2+ flux and poor upregulation of CD69, CD25 and CD98. This hypothesis is supported by the hypomorphic phenotype associated with SH2-domain defects (54, 63). Partial T-cell developmental arrest and immune dysregulation were also noted in a murine model of ~90% reduced SLP-76 levels (71), pointing to the possible association between reduced SLP-76 signaling and immune dysregulation. Interestingly, in contrast to the murine phenotype, B-cell arrest was evident in the patient. SLP-76 is known to be involved in pre-B cell signaling (72). Other defects in pre-BCR signaling also show a discrepancy between the mouse and human phenotype, perhaps pointing to the more stringent requirements for human pre-BCR signaling as compared to mice (73).
Adhesion And Degranulation Adaptor Protein
ADAP, also termed FYB and SLP130, is expressed in T-cells and myeloid cells. This protein contains a proline-rich region and an SH3-like domain, both of which bind SKAP55. A phosphotyrosine motifs rich domain is responsible for binding FYN and SLP-76, and an Ena/VASP-homology1 (EVH1)-binding domain binds Ena/VASP family proteins (74). Upon T-cell activation, ADAP is phosphorylated at 3 tyrosine residues by FYN, followed by binding to SLP-76 through its SH2 domain (6, 59, 74). Through its association with SLP-76, ADAP contributes to the cross-linking of LAT molecules into microclusters and amplification of proximal signaling events (11, 16). This protein has also been implicated in regulation of the assembly of the CBM (CARMA1/Bcl10/MALT1) complex, leading to NFkB activation (19), as well as integrin activation (19, 21, 74, 75). In CD8+ cells, ADAP was associated with increased PD1 expression and reduced anti-tumor immunity (76), pointing to a possible regulatory role (77). ADAP-/- mice show moderate thrombocytopenia and mildly decreased thymocyte numbers (78). Despite normal proximal TCR-signaling events, ADAP-/- T cells have abnormal activation and proliferation and impaired LFA-1 clustering in response to TCR stimulation. It is somewhat surprising then, that ADAP deficiency in humans, which is associated with congenital autosomal-recessive small-platelet thrombocytopenia (CARST) (79–82), has no known immune defect. Activation-dependent, raft recruited ADAP-like phosphoprotein (ARAP) shares sequence homology with ADAP and activates integrin in a TCR- and SLP-76-dependent manner (83). Therefore, it is a possible candidate for rescuing ADAP function in T-cells.
Conclusion
The function of adaptor molecules in T-cell signaling has been thoroughly investigated. However, recent discoveries of human inborn errors of immunity have raised further questions regarding the differential function and compensatory mechanisms in T-cell signaling complexes between murine models, Jurkat cells and patients. Further research is needed to answer these questions.
Author Contributions
YD-S wrote the paper. PS initiated the idea for review and reviewed the paper. IZ reviewed the paper. HM-S reviewed the paper. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Malissen B, Grégoire C, Malissen M, Roncagalli R. Integrative Biology of T Cell Activation. Nat Immunol (2014) 15:790–7. Nature Publishing Group;. doi: 10.1038/ni.2959
2. VanPanhuys N, Klauschen F, Germain RN. T-Cell-Receptor-Dependent Signal Intensity Dominantly Controls CD4+ T Cell Polarization In Vivo. Immunity (2014) 41(1):63–74. doi: 10.1016/j.immuni.2014.06.003
3. Smith-Garvin JE, Koretzky GA, Jordan MS. T Cell Activation. Annu Rev Immunol (2009) 27(1):591–619. doi: 10.1146/annurev.immunol.021908.132706
4. Malissen B, Bongrand P. Early T Cell Activation: Integrating Biochemical, Structural, and Biophysical Cues. Annu Rev Immunol (2015) 33(1):539–61. doi: 10.1146/annurev-immunol-032414-112158
5. Leo A, Wienands J, Baier G, Horejsi V, Schraven B. Adapters in Lymphocyte Signaling. J Clin Invest (2002) 109(3):301–9. doi: 10.1172/JCI0214942
6. Balagopalan L, Coussens NP, Sherman E, Samelson LE, Sommers CL. The LAT Story: A Tale of Cooperativity, Coordination, and Choreography. Cold Spring Harbor Perspect Biol (2010) 2:1–21. doi: 10.1101/cshperspect.a005512
7. Roncagalli R, Mingueneau M, Grégoire C, Malissen M, Malissen B. LAT Signaling Pathology: An “Autoimmune” Condition Without T Cell Self-Reactivity. Trends Immunol (2010) 31:253–9. doi: 10.1016/j.it.2010.05.001
8. Zhang W, Trible RP, Zhu M, Liu SK, McGlade CJ, Samelson LE. Association of Grb2, Gads, and Phospholipase C-γ1 With Phosphorylated LAT Tyrosine Residues: Effect of LAT Tyrosine Mutations on T Cell Antigen Receptor-Mediated Signaling. J Biol Chem (2000) 275(30):23355–61. doi: 10.1074/jbc.M000404200
9. Zhu M, Janssen E, Zhang W. Minimal Requirement of Tyrosine Residues of Linker for Activation of T Cells in TCR Signaling and Thymocyte Development. J Immunol (2003) 170(1):325–33. doi: 10.4049/jimmunol.170.1.325
10. Lin J, Weiss A. Identification of the Minimal Tyrosine Residues Required for Linker for Activation of T Cell Function. J Biol Chem (2001) 276(31):29588–95. doi: 10.1074/jbc.M102221200
11. Yablonski D. Bridging the Gap: Modulatory Roles of the Grb2-Family Adaptor, Gads, in Cellular and Allergic Immune Responses. Front Immunol NLM (Medline) (2019) 10:1704. doi: 10.3389/fimmu.2019.01704
12. Braiman A, Barda-Saad M, Sommers CL, Samelson LE. Recruitment and Activation of Plcγ1 in T Cells: A New Insight Into Old Domains. EMBO J (2006) 25(4):774–84. doi: 10.1038/sj.emboj.7600978
13. Devkota S, Joseph RE, Min L, Bruce Fulton D, Andreotti AH. Scaffold Protein SLP-76 Primes Plcγ1 for Activation by ITK-Mediated Phosphorylation. J Mol Biol (2015) 427(17):2734–47. doi: 10.1016/j.jmb.2015.04.012
14. Houtman JCD, Yamaguchi H, Barda-Saad M, Braiman A, Bowden B, Appella E, et al. Oligomerization of Signaling Complexes by the Multipoint Binding of GRB2 to Both LAT and SOS1. Nat Struct Mol Biol (2006) 13(9):798–805. doi: 10.1038/nsmb1133
15. Nag A, Monine MI, Faeder JR, Goldstein B. Aggregation of Membrane Proteins by Cytosolic Cross-Linkers: Theory and Simulation of the LAT-Grb2-SOS1 System. Biophys J (2009) 96(7):2604–23. doi: 10.1016/j.bpj.2009.01.019
16. Lewis JB, Scangarello FA, Murphy JM, Eidell KP, Sodipo MO, Ophir MJ, et al. ADAP Is an Upstream Regulator That Precedes SLP-76 at Sites of TCR Engagement and Stabilizes Signaling Microclusters. J Cell Sci (2018) 131(21):1–17. doi: 10.1242/jcs.215517
17. Malissen B, Aguado E, Malissen M. Role of the LAT Adaptor in T-Cell Development and Th2 Differentiation. Adv Immunol (2005) 87:1–25. doi: 10.1016/S0065-2776(05)87001-4
18. Wipa P, Paensuwan P, Ngoenkam J, Woessner NM, Minguet S, Schamel WW, et al. Actin Polymerization Regulates Recruitment of Nck to CD3ϵ Upon T-Cell Receptor Triggering. Immunology (2020) 159(3):298–308. doi: 10.1111/imm.13146
19. Burns JC, Corbo E, Degen J, Gohil M, Anterasian C, Schraven B, et al. The SLP-76 Src Homology 2 Domain Is Required for T Cell Development and Activation. J Immunol (2011) 187(9):4459–66. doi: 10.4049/jimmunol.0903379
20. Roncagalli R, Hauri S, Fiore F, Liang Y, Chen Z, Sansoni A, et al. Quantitative Proteomics Analysis of Signalosome Dynamics in Primary T Cells Identifies the Surface Receptor CD6 as a Lat Adaptor-Independent TCR Signaling Hub. Nat Immunol (2014) 15(4):384–92. doi: 10.1038/ni.2843
21. Baker RG, Hsu CJ, Lee D, Jordan MS, Maltzman JS, Hammer DA, et al. The Adapter Protein SLP-76 Mediates “Outside-In” Integrin Signaling and Function in T Cells. Mol Cell Biol (2009) 29(20):5578–89. doi: 10.1128/MCB.00283-09
22. Zhang W, Sommers CL, Burshtyn DN, Stebbins CC, DeJarnette JB, Trible RP, et al. Essential Role of LAT in T Cell Development. Immunity (1999) 10(3):323–32. doi: 10.1016/S1074-7613(00)80032-1
23. Sommers CL, Menon RK, Grinberg A, Zhang W, Samelson LE, Love PE. Knock-In Mutation of the Distal Four Tyrosines of Linker for Activation of T Cells Blocks Murine T Cell Development. J Exp Med (2001) 194(2):135–42. doi: 10.1084/jem.194.2.135
24. Sommers CL, Park CS, Lee J, Feng C, Fuller CL, Grinberg A, et al. A LAT Mutation That Inhibits T Cell Development Yet Induces Lymphoproliferation. Science (2002) 296(5575):2040–3. doi: 10.1126/science.1069066
25. Mingueneau M, Roncagalli R, Grégoire C, Kissenpfennig A, Miazek A, Archambaud C, et al. Loss of the LAT Adaptor Converts Antigen-Responsive T Cells Into Pathogenic Effectors That Function Independently of the T Cell Receptor. Immunity (2009) 31(2):197–208. doi: 10.1016/j.immuni.2009.05.013
26. Sommers CL, Lee J, Steiner KL, Gurson JM, DePersis CL, El-Khoury D, et al. Mutation of the Phospholipase C-γ1-Binding Site of LAT Affects Both Positive and Negative Thymocyte Selection. J Exp Med (2005) 201(7):1125–34. doi: 10.1084/jem.20041869
27. Chuck MI, Zhu M, Shen S, Zhang W. The Role of the LAT–PLC-γ1 Interaction in T Regulatory Cell Function. J Immunol (2010) 184(5):2476–86. doi: 10.4049/jimmunol.0902876
28. Yamada K, Zuka M, Ito K, Mizuguchi K, Kakuchi Y, Onoe T, et al. LatY136F Knock-in Mouse Model for Human IgG4-Related Disease. PloS One (20180) 13(6):1–14. doi: 10.1371/journal.pone.0198417
29. Sullivan SA, Zhu M, Bao S, Lewis CA, Ou-Yang C, Zhang W. The Role of LAT–Plcγ1 Interaction in γδ T Cell Development and Homeostasis. J Immunol (2014) 192(6):2865–74. doi: 10.4049/jimmunol.1302493
30. Aguado E, Richelme S, Nuñez-Cruz S, Miazek A, Mura AM, Richelme M, et al. Induction of T Helper Type 2 Immunity by a Point Mutation in the LAT Adaptor. Science (2002) 296(5575):2036–40. doi: 10.1126/science.1069057
31. Genton C, Wang Y, Izui S, Malissen B, Delsol G, Fournié GJ, et al. The Th2 Lymphoproliferation Developing in Lat Y136F Mutant Mice Triggers Polyclonal B Cell Activation and Systemic Autoimmunity. J Immunol (2006) 177(4):2285–93. doi: 10.4049/jimmunol.177.4.2285
32. Wang Y, Kissenpfennig A, Mingueneau M, Richelme S, Perrin P, Chevrier S, et al. Th2 Lymphoproliferative Disorder of Lat Y136F Mutant Mice Unfolds Independently of TCR-MHC Engagement and Is Insensitive to the Action of Foxp3 + Regulatory T Cells. J Immunol (2008) 180(3):1565–75. doi: 10.4049/jimmunol.180.3.1565
33. Nuñez-Cruz S, Aguado E, Richelme S, Chetaille B, Mura AM, Richelme M, et al. LAT Regulates γδ T Cell Homeostasis and Differentiation. Nat Immunol (2003) 4(10):999–1008. doi: 10.1038/ni977
34. Yamasaki S, Nishida K, Sakuma M, Berry D, McGlade CJ, Hirano T, et al. Gads/Grb2-Mediated Association With LAT Is Critical for the Inhibitory Function of Gab2 in T Cells. Mol Cell Biol (2003) 23(7):2515–29. doi: 10.1128/MCB.23.7.2515-2529.2003
35. Shen R, Ouyang Y-B, Qu C-K, Alonso A, Sperzel L, Mustelin T, et al. Grap Negatively Regulates T-Cell Receptor-Elicited Lymphocyte Proliferation and Interleukin-2 Induction. Mol Cell Biol (2002) 22(10):3230–6. doi: 10.1128/MCB.22.10.3230-3236.2002
36. Dong S, Corre B, Foulon E, Dufour E, Veillette A, Acuto O, et al. T Cell Receptor for Antigen Induces Linker for Activation of T Cell-Dependent Activation of a Negative Signaling Complex Involving Dok-2, SHIP-1, and Grb-2. J Exp Med (2006) 203(11):2509–18. doi: 10.1084/jem.20060650
37. Yasuda T, Bundo K, Hino A, Honda K, Inoue A, Shirakata M, et al. Dok-1 and Dok-2 Are Negative Regulators of T Cell Receptor Signaling. Int Immunol (2007) 19(4):487–95. doi: 10.1093/intimm/dxm015
38. Salek M, McGowan S, Trudgian DC, Dushek O, de Wet B, Efstathiou G, et al. Quantitative Phosphoproteome Analysis Unveils LAT as a Modulator of CD3ζ and ZAP-70 Tyrosine Phosphorylation. PloS One (2013) 8(10):1–9. doi: 10.1371/journal.pone.0077423
39. Liou J, Kiefer F, Dang A, Hashimoto A, Cobb MH, Kurosaki T, et al. HPK1 is Activated by Lymphocyte Antigen Receptors and Negatively Regulates AP-1. Immunity (2000) 12(4):399–408. doi: 10.1016/S1074-7613(00)80192-2
40. Bacchelli C, Moretti FA, Carmo M, Adams S, Stanescu HC, Pearce K, et al. Mutations in Linker for Activation of T Cells (LAT) Lead to a Novel Form of Severe Combined Immunodeficiency. J Allergy Clin Immunol (2017) 139(2):634–642.e5. doi: 10.1016/j.jaci.2016.05.036
41. Keller B, Zaidman I, Yousefi OS, Hershkovitz D, Stein J, Unger S, et al. Early Onset Combined Immunodeficiency and Autoimmunity in Patients With Loss-of-Function Mutation in LAT. J Exp Med (2016) 213(7):1185–99. doi: 10.1084/jem.20151110
42. Shan X, Wange RL. Itk/Emt/Tsk Activation in Response to CD3 Cross-Linking in Jurkat T Cells Requires ZAP-70 and Lat and Is Independent of Membrane Recruitment. J Biol Chem (1999) 274(41):29323–30. doi: 10.1074/jbc.274.41.29323
43. Shan X, Balakir R, Criado G, Wood JS, Seminario M-C, Madrenas J, et al. ZAP-70-Independent Ca2+ Mobilization and Erk Activation in Jurkat T Cells in Response to T-Cell Antigen Receptor Ligation. Mol Cell Biol (2001) 21(21):7137–49. doi: 10.1128/MCB.21.21.7137-7149.2001
44. Ku GM, Yablonski D, Manser E, Lim L, Weiss A. A PAK1-PIX-PKL Complex is Activated by the T-Cell Receptor Independent of Nck, Slp-76 and LAT. EMBO J (2001) 20(3):457–65. doi: 10.1093/emboj/20.3.457
45. Chiang YJ, Sommers CL, Jordan MS, Gu H, Samelson LE, Koretzky GA, et al. Inactivation of C-Cbl Reverses Neonatal Lethality and T Cell Developmental Arrest of SLP-76-Deficient Mice. J Exp Med (2004) 200(1):25–34. doi: 10.1084/jem.20040262
46. Graham LJ, Verí MC, DeBell KE, Noviello C, Rawat R, Jen S, et al. 70z/3 Cbl Induces Plcγ1 Activation in T Lymphocytes via an Alternate Lat- and Slp-76-Independent Signaling Mechanism. Oncogene (2003) 22(16):2493–503. doi: 10.1038/sj.onc.1206318
47. Mori D, Grégoire C, Voisinne G, Celis-Gutierrez J, Aussel R, Girard L, et al. The T Cell CD6 Receptor Operates a Multitask Signalosome With Opposite Functions in T Cell Activation. J Exp Med (2021) 218(2):1–19. doi: 10.1084/jem.20201011
48. Breuning J, Brown MH. T Cell Costimulation by CD6 Is Dependent on Bivalent Binding of a GADS/SLP-76 Complex. Mol Cell Biol (2017) 37(11):1–12. doi: 10.1128/MCB.00071-17
49. Poltorak M, Meinert I, Stone JC, Schraven B, Simeoni L. Sos1 Regulates Sustained TCR-Mediated Erk Activation. Eur J Immunol (2014) 44(5):1535–40. doi: 10.1002/eji.201344046
50. Lo WL, Shah NH, Ahsan N, Horkova V, Stepanek O, Salomon AR, et al. Lck Promotes Zap70-Dependent LAT Phosphorylation by Bridging Zap70 to LAT. Nat Immunol (2018) 19(7):733–41. doi: 10.1038/s41590-018-0131-1
51. Markegard E, Trager E, Yang C wen O, Zhang W, Weiss A, Roose JP. Basal LAT-Diacylglycerol-Rasgrp1 Signals in T Cells Maintain Tcrα Gene Expression. PloS One (2011) 6(9):1–9. doi: 10.1371/journal.pone.0025540
52. Archambaud C, Sansoni A, Mingueneau M, Devilard E, Delsol G, Malissen B, et al. STAT6 Deletion Converts the Th2 Inflammatory Pathology Afflicting Lat Y136F Mice Into a Lymphoproliferative Disorder Involving Th1 and CD8 Effector T Cells. J Immunol (2009) 182(5):2680–9. doi: 10.4049/jimmunol.0803257
53. Somekh I, Marquardt B, Liu Y, Rohlfs M, Hollizeck S, Karakukcu M, et al. Novel Mutations in RASGRP1 Are Associated With Immunodeficiency, Immune Dysregulation, and EBV-Induced Lymphoma. J Clin Immunol (2018) 38(6):699–710. doi: 10.1007/s10875-018-0533-8
54. Koretzky GA, Abtahian F, Silverman MA. SLP76 and SLP65: Complex Regulation of Signalling in Lymphocytes and Beyond [Internet]. Vol. 6, Nature Reviews Immunology. Nat Rev Immunol (2006) 6:67–78. doi: 10.1038/nri1750
55. Liu H, Thaker YR, Stagg L, Schneider H, Ladbury JE, Rudd CE. SLP-76 Sterile α Motif (SAM) and Individual H5 α Helix Mediate Oligomer Formation for Microclusters and T-Cell Activation. J Biol Chem (2013) 288(41):29539–49. doi: 10.1074/jbc.M112.424846
56. Thaker YR, Recino A, Raab M, Jabeen A, Wallberg M, Fernandez N, et al. Activated Cdc42-Associated Kinase 1 (ACK1) Binds the Sterile - Motif (SAM) Domain of the Adaptor SLP-76 and Phosphorylates Proximal Tyrosines. J Biol Chem (2017) 292(15):6281–90. doi: 10.1074/jbc.M116.759555
57. Samelson LE. Signal Transduction Mediated by the T Cell Antigen Receptor: The Role of Adapter Proteins. Annu Rev Immunol (2003) 20:371–94. doi: 10.1146/annurev.immunol.20.092601.111357
58. Jordan MS, Koretzky GA. Coordination of Receptor Signaling in Multiple Hematopoietic Cell Lineages by the Adaptor Protein SLP-76. Cold Spring Harb Perspect Biol (2010) 2(4):a002501. doi: 10.1101/cshperspect.a002501
59. Coussens NP, Hayashi R, Brown PH, Balagopalan L, Balbo A, Akpan I, et al. Multipoint Binding of the SLP-76 SH2 Domain to ADAP Is Critical for Oligomerization of SLP-76 Signaling Complexes in Stimulated T Cells. Mol Cell Biol (2013) 33(21):4140–51. doi: 10.1128/MCB.00410-13
60. Bunnell SC, Singer AL, Hong DI, Jacque BH, Jordan MS, Seminario M-C, et al. Persistence of Cooperatively Stabilized Signaling Clusters Drives T-Cell Activation. Mol Cell Biol (2006) 26(19):7155–66. doi: 10.1128/MCB.00507-06
61. Hassan NJ, Simmonds SJ, Clarkson NG, Hanrahan S, Puklavec MJ, Bomb M, et al. CD6 Regulates T-Cell Responses Through Activation-Dependent Recruitment of the Positive Regulator SLP-76. Mol Cell Biol (2006) 26(17):6727–38. doi: 10.1128/MCB.00688-06
62. Yablonski D, Kuhne MR, Kadlecek T, Weiss A. Uncoupling of Nonreceptor Tyrosine Kinases From PLC-γ1 in an SLP-76- Deficient T Cell. Science (1998) 281(5375):413–6. doi: 10.1126/science.281.5375.413
63. Yablonski D, Kadlecek T, Weiss A. Identification of a Phospholipase C-γ1 (PLC-γ1) SH3 Domain-Binding Site in SLP-76 Required for T-Cell Receptor-Mediated Activation of PLC-γ1 and NFAT. Mol Cell Biol (2001) 21(13):4208–18. doi: 10.1128/MCB.21.13.4208-4218.2001
64. Clements JL, Yang B, Ross-Barta SE, Eliason SL, Hrstka RF, Williamson RA, et al. Requirement for the Leukocyte-Specific Adapter Protein SLP-76 for Normal T Cell Development. Science (1998) 281(5375):416–9. doi: 10.1126/science.281.5375.416
65. Pivniouk V, Tsitsikov E, Swinton P, Rathbun G, Alt FW, Geha RS. Impaired Viability and Profound Block in Thymocyte Development in Mice Lacking the Adaptor Protein SLP-76. Cell (1998) 94(2):229–38. doi: 10.1016/S0092-8674(00)81422-1
66. Clements JL, Lee JR, Gross B, Yang B, Olson JD, Sandra A, et al. Fetal Hemorrhage and Platelet Dysfunction in SLP-76-Deficient Mice. J Clin Invest (1999) 103(1):19–25. doi: 10.1172/JCI5317
67. Newbrough SA, Mocsai A, Clemens RA, Wu JN, Silverman MA, Singer AL, et al. SLP-76 Regulates Fcγ Receptor and Integrin Signaling in Neutrophils. Immunity (2003) 19(5):761–9. doi: 10.1016/S1074-7613(03)00305-4
68. Kumar L, Pivniouk V, de la Fuente MA, Laouini D, Geha RS. Differential Role of SLP-76 Domains in T Cell Development and Function. Proc Natl Acad Sci United States America (2002) 99(2):884–9. doi: 10.1073/pnas.022619199
69. Myung PS, Derimanov GS, Jordan MS, Punt JA, Liu QH, Judd BA, et al. Differential Requirement for SLP-76 Domains in T Cell Development and Function. Immunity (2001) 15(6):1011–26. doi: 10.1016/S1074-7613(01)00253-9
70. Lev A, Lee YN, Sun G, Hallumi E, Simon AJ, Zrihen KS, et al. Inherited SLP76 Deficiency in Humans Causes Severe Combined Immunodeficiency, Neutrophil and Platelet Defects. J Exp Med (2021) 218(3):1–14. doi: 10.1084/jem.20201062
71. Siggs OM, Miosge LA, Daley SR, Asquith K, Foster PS, Liston A, et al. Quantitative Reduction of the TCR Adapter Protein SLP-76 Unbalances Immunity and Immune Regulation. J Immunol (2015) 194(6):2587–95. doi: 10.4049/jimmunol.1400326
72. Su YW, Jumaa H. LAT Links the Pre-BCR to Calcium Signaling. Immunity (2003) 19(2):295–305. doi: 10.1016/S1074-7613(03)00202-4
73. Conley ME, Rohrer J, Rapalus L, Boylin EC, Minegishi Y. Defects in Early B-Cell Development: Comparing the Consequences of Abnormalities in Pre-BCR Signaling in the Human and the Mouse. Immunol Rev (2000) 178:75–90. Blackwell Munksgaard. doi: 10.1034/j.1600-065X.2000.17809.x
74. Griffiths EK, Penninger JM. Communication Between the TCR and Integrins: Role of the Molecular Adapter ADAP/Fyb/Slap. Curr Opin Immunol (2002) 14:317–22. Elsevier Current Trends. doi: 10.1016/S0952-7915(02)00334-5
75. Horn J, Wang X, Reichardt P, Stradal TE, Warnecke N, Simeoni L, et al. Src Homology 2-Domain Containing Leukocyte-Specific Phosphoprotein of 76 kDa Is Mandatory for TCR-Mediated Inside-Out Signaling, But Dispensable for CXCR4-Mediated LFA-1 Activation, Adhesion, and Migration of T Cells. J Immunol (2009) 183(9):5756–67. doi: 10.4049/jimmunol.0900649
76. Li C, Li W, Xiao J, Jiao S, Teng F, Xue S, et al. ADAP and SKAP 55 Deficiency Suppresses PD -1 Expression in CD 8 + Cytotoxic T Lymphocytes for Enhanced Anti-Tumor Immunotherapy. EMBO Mol Med (2015) 7(6):754–69. doi: 10.15252/emmm.201404578
77. Fiege JK, Burbach BJ, Shimizu Y. Negative Regulation of Memory Phenotype CD8 T Cell Conversion by Adhesion and Degranulation–Promoting Adapter Protein. J Immunol (2015) 195(7):3119–28. doi: 10.4049/jimmunol.1402670
78. Peterson EJ, Woods ML, Dmowski SA, Derimanov G, Jordan MS, Wu JN, et al. Coupling of the TCR to Integrin Activation by SLAP-130/Fyb. Science (2001) 293(5538):2263–5. doi: 10.1126/science.1063486
79. Spindler M, van Eeuwijk JMM, Schurr Y, Nurden P, Nieswandt B, Stegner D, et al. ADAP Deficiency Impairs Megakaryocyte Polarization With Ectopic Proplatelet Release and Causes Microthrombocytopenia. Blood (2018) 132(6):635–46. doi: 10.1182/blood-2018-01-829259
80. Levin C, Koren A, Pretorius E, Rosenberg N, Shenkman B, Hauschner H, et al. Deleterious Mutation in the FYB Gene Is Associated With Congenital Autosomal Recessive Small-Platelet Thrombocytopenia. J Thromb Haemostasis (2015) 13(7):1285–92. doi: 10.1111/jth.12966
81. Levin C, Zalman L, Tamary H, Krasnov T, Khayat M, Shalev S, et al. Small-Platelet Thrombocytopenia in a Family With Autosomal Recessive Inheritance Pattern. Pediatr Blood Cancer (2013) 60(10):E128–30. doi: 10.1002/pbc.24581
82. Hamamy H, Makrythanasis P, Al-Allawi N, Muhsin AA, Antonarakis SE. Recessive Thrombocytopenia Likely Due to a Homozygous Pathogenic Variant in the FYB Gene: Case Report. BMC Med Genet (2014) 15(1):1–5. doi: 10.1186/s12881-014-0135-0
Keywords: adaptor molecules, ADAP, LAT, SLP-76, T-cell signaling, primary immune deficiency
Citation: Dinur-Schejter Y, Zaidman I, Mor-Shaked H and Stepensky P (2021) The Clinical Aspect of Adaptor Molecules in T Cell Signaling: Lessons Learnt From Inborn Errors of Immunity. Front. Immunol. 12:701704. doi: 10.3389/fimmu.2021.701704
Received: 28 April 2021; Accepted: 26 July 2021;
Published: 12 August 2021.
Edited by:
Anne Spurkland, University of Oslo, NorwayReviewed by:
Martha Jordan, University of Pennsylvania, United StatesStephen Charles Bunnell, Tufts University School of Medicine, United States
Copyright © 2021 Dinur-Schejter, Zaidman, Mor-Shaked and Stepensky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yael Dinur-Schejter, eWFlbHNjaEBoYWRhc3NhaC5vcmcuaWw=