
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 29 October 2021
Sec. Nutritional Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.701341
 Rachel L. Markley1,2,3†
Rachel L. Markley1,2,3† Katherine H. Restori2†
Katherine H. Restori2† Bhuvana Katkere2
Bhuvana Katkere2 Sarah E. Sumner1,2
Sarah E. Sumner1,2 McKayla J. Nicol1,2Anastasia Tyryshkina4,5Shaneice K. Nettleford1,2David R. Williamson2
McKayla J. Nicol1,2Anastasia Tyryshkina4,5Shaneice K. Nettleford1,2David R. Williamson2 David E. Place2,6
David E. Place2,6 Kalyan K. Dewan2,7
Kalyan K. Dewan2,7 Ashley E. Shay2,8Bradley A. Carlson9
Ashley E. Shay2,8Bradley A. Carlson9 Santhosh Girirajan5
Santhosh Girirajan5 K. Sandeep Prabhu2,10
K. Sandeep Prabhu2,10 Girish S. Kirimanjeswara2,10*
Girish S. Kirimanjeswara2,10*The essential micronutrient Selenium (Se) is co-translationally incorporated as selenocysteine into proteins. Selenoproteins contain one or more selenocysteines and are vital for optimum immunity. Interestingly, many pathogenic bacteria utilize Se for various biological processes suggesting that Se may play a role in bacterial pathogenesis. A previous study had speculated that Francisella tularensis, a facultative intracellular bacterium and the causative agent of tularemia, sequesters Se by upregulating Se-metabolism genes in type II alveolar epithelial cells. Therefore, we investigated the contribution of host vs. pathogen-associated selenoproteins in bacterial disease using F. tularensis as a model organism. We found that F. tularensis was devoid of any Se utilization traits, neither incorporated elemental Se, nor exhibited Se-dependent growth. However, 100% of Se-deficient mice (0.01 ppm Se), which express low levels of selenoproteins, succumbed to F. tularensis-live vaccine strain pulmonary challenge, whereas 50% of mice on Se-supplemented (0.4 ppm Se) and 25% of mice on Se-adequate (0.1 ppm Se) diet succumbed to infection. Median survival time for Se-deficient mice was 8 days post-infection while Se-supplemented and -adequate mice was 11.5 and >14 days post-infection, respectively. Se-deficient macrophages permitted significantly higher intracellular bacterial replication than Se-supplemented macrophages ex vivo, corroborating in vivo observations. Since Francisella replicates in alveolar macrophages during the acute phase of pneumonic infection, we hypothesized that macrophage-specific host selenoproteins may restrict replication and systemic spread of bacteria. F. tularensis infection led to an increased expression of several macrophage selenoproteins, suggesting their key role in limiting bacterial replication. Upon challenge with F. tularensis, mice lacking selenoproteins in macrophages (TrspM) displayed lower survival and increased bacterial burden in the lung and systemic tissues in comparison to WT littermate controls. Furthermore, macrophages from TrspM mice were unable to restrict bacterial replication ex vivo in comparison to macrophages from littermate controls. We herein describe a novel function of host macrophage-specific selenoproteins in restriction of intracellular bacterial replication. These data suggest that host selenoproteins may be considered as novel targets for modulating immune response to control a bacterial infection.
In both prokaryotes and eukaryotes the trace element selenium (Se) is co-translationally incorporated as selenocysteine (Sec) into selenoproteins (1, 2). Selenoproteins contain one or more Sec residues and have been shown to promote fitness of several bacterial pathogens (3). Bacterial selenoprotein enzymes including formate-dehydrogenase, xanthine dehydrogenase, hydrogenase-3 (FHL complex) (4), and glycine reductase, specific to Clostridia, promote the pathogen’s growth and fitness (5). Formate-dehydrogenase (Fdh), the most common selenoprotein expressed by bacteria (6), catalyzes the reversible two-electron oxidation of formate (7). Importantly, fdh is necessary for anaerobic metabolism of many bacteria (8) such as Campylobacter jejuni, a gut-dwelling bacterium and main causative agent of food borne illness worldwide (9). Mutant strains of C. jejuni deficient in formic acid metabolism exhibited reduced fitness and caused less severe enteric infection than the parent strain (10). Therefore, ablating the function of fdh results in loss of fitness, suggesting that selenoproteins are necessary for pathogen infectivity.
While selenoproteins promote normal growth, immunity, and reproductive health in humans (11), the ability of Se- supplementation and therefore, selenoproteins of the host to limit infectious disease severity has also been demonstrated in nutritional intervention studies (12–20). Se deficiency is associated with chronic infections caused by HIV/AIDS, hepatitis C virus, and Mycobacterium tuberculosis (12–15); nutritional interventions that contain Se have benefitted patients (16–20). However, many of these studies did not test the change in expression of selenoproteins following intervention. In addition, many of the nutritional Se supplementation studies were performed in Se-limiting geographical areas and likely had limited effect on the expression of selenoproteins. Sepsis is caused by a dysregulated immune response to infection, which usually originates from gram-negative bacteria resulting in life-threatening organ dysfunction, long term morbidity, and heightened risk of mortality (21). Plasma Se levels were lower in 92% of critically ill surgical patients than the standard value upon admission to the Intensive Care Unit (ICU). All patients exhibited decreased Se levels for the duration of the ICU stay, but strikingly, lower Se plasma concentrations were observed in patients with infection, tissue damage, organ dysfunction/failure and increased ICU mortality (22). Accordingly, several studies have noted a beneficial effect of Se supplementation as an adjunct therapy in patients with sepsis (23, 24). Se supplementation, and thus enhancement of selenoprotein function, may serve as a beneficial intervention to decrease infectious disease severity and improve patient outcome.
Previously, selenoproteins have been shown to be advantageous to the host by providing protection from inflammation and mediating resolution of infection as well as regulating overt immune responses (25–27). Selenoproteins are known to play a major role in maintaining cellular redox homeostasis and regulate a variety of biological processes such as intracellular calcium signaling (28, 29). The selenoprotein thioredoxin reductase 1 (TR1) is a pyridine nucleotide-disulfide oxioreductase that reduces disulfides to free thiols (30) and negatively regulates the HIV-1 encoded transcriptional activator, Tat, resulting in decreased HIV-1 replication in human macrophages (31). The action of TR1 in limiting viral replication, is one selenoprotein-dependent mechanism that may, in part, explain the therapeutic effects of Se supplementation in HIV/AIDS patients. Furthermore, macrophage-specific selenoproteins were found to be essential for clearance of the gastrointestinal nematode parasite, Nippostrongylus brasiliensis (27). An alternatively activated, or M2, macrophage response was deemed necessary for protection against helminthic infections, and Se supplementation of macrophages induced a phenotypic change from classically activated M1 to M2 (32). Macrophage selenoproteins were also necessary to control damaging proinflammatory responses in a mouse model of acute colitis, as Se-deficient and adequate mice exhibited increased colitis severity, inflammation and poor survival when compared to Se-supplemented mice (25). Additionally, in a model of gut inflammation induced by Citrobacter rodentium, a bacterium that induces murine enterohemorrhagic Escherichia coli (EHEC) infection, Se-deficient mice had increased mortality that was associated with poor integrity of colonic epithelial barrier cells in comparison to Se-adequate and supplemented mice (26). Although selenoproteins regulate many physiological processes under steady-state and inflammatory conditions, it is currently unknown if host selenoproteins are vital for antibacterial defense via limiting their intracellular replication.
In the current report, we utilize F. tularensis as a model organism to investigate the contribution of host vs. bacterial selenoproteins to the pathogenesis of pulmonary tularemia and disease outcome. F. tularensis is a facultative, intracellular, gram-negative bacterium that has broad host range (33, 34). Among the four subspecies of F. tularensis, F. tularensis spp. tularensis is most virulent to humans, followed by F. tularensis spp. holartica, mediasiatica and F. novicida is considered more of an environmental organism that can cause disease in rodents (34). Due to the lack of an approved vaccine, pathogen virulence, severity of pulmonary infection, ease of aerosolization, and its historical use as a bioweapon, F. tularensis ssp. tularensis is categorized as a Tier I select agent by the Centers for Disease Control (35, 36). Tularemia in humans is manifested in several forms owing to the route of infection that can cause cutaneous ulcers, glandular, ocular, typhoidal or pneumonic disease (37). The pneumonic form is an acute disease, resulting from the inhalation of as few as 10 colony-forming units (CFU) of F. tularensis and may result in a mortality rate of up to 60% if untreated (33, 38). Upon inhalation, F. tularensis preferentially infects phagocytes such as alveolar macrophages (39), and after several rounds of replication, spreads to tissue macrophages and dendritic cells in the lung, liver, spleen and lymph nodes (40–42). Additional cell types such as epithelial cells, hepatocytes, B cells, and red blood cells may support F. tularensis replication during the late stages of disease, but macrophages are the primary host cells during acute infection (39–42).
Bacterial incorporation of Sec into proteins involves the products of the selA, selB, selC and selD genes (43, 44). A unique tRNA specific for the UGA codon tRNA(sec) that is encoded by selC is initially charged with serine; the seryl moiety is then converted to a selenocysteinyl moiety (45). SelA encodes selenocysteine synthase that catalyzes the conversion of serine to selenocysteine, which requires selenophosphate, as a donor provided by selenophosphate synthetase that is encoded by selD (46). Selenoprotein mRNA features a stem-loop secondary structure known as a SEleno Cysteine Insertion Sequence (SECIS) element and is located immediately downstream of a UGA codon in bacteria. However, in eukaryotes the SECIS element is located in the 3’untranslated region of the mRNA (28). For the UGA codon to be translated as Sec, a specialized translational elongation factor that is encoded by selB must interact with the selenocysteine-tRNA(sec), the SECIS element, and GTP at the ribosome (47). Thus, bacteria that translate proteins containing Sec generally require selA, selB, selC, and selD genes and their respective products. Alternatively, bacteria may utilize Se as a part of a unique 2-selenouridine (SeU) base in the wobble position of select tRNAs (48). Lastly, 2-selenouridine synthase (YbbB) can be synthesized independently of selA, selB, and selC, but is thought to require selD (48).
It was previously proposed that F. tularensis exploited host resources for the acquisition of Se for its fitness, as F. tularensis holartica Live Vaccine Strain (LVS) infection of A549 bronchial airway epithelial cells resulted in upregulation of host Se metabolism genes (49). However, in our current report, in silico sequence analysis determined that F. tularensis does not incorporate Se as a selenoprotein, a modified base, or as a cofactor. Additionally, F. tularensis does not incorporate elemental Se nor require Se for optimal in vitro growth, virulence gene expression, or in vivo infection. Se-adequate or supplemented mice exhibit greater survival from pulmonary tularemia, while Se-deficient mice succumb to F. tularensis LVS challenge. Bone-marrow derived macrophages (BMDMs) from dietary Se-adequate or Se-supplemented mice restricted bacterial replication in contrast to macrophages from Se-deficient mice. We then investigated whether the selenoproteins in macrophages were crucial in limiting the severity of pulmonary tularemia in vivo, as alveolar macrophages are the primary cells for F. tularensis replication. Transgenic mice that lack selenoproteins in macrophages (TrspM) increasingly succumbed to pulmonary tularemia with a correspondingly greater bacterial burden in systemic tissues at later stages of infection. BMDMs from TrspM mice are unable to control F. tularensis LVS replication, a phenomenon that failed to be rescued by ex vivo Se supplementation, further demonstrating the importance of functional host macrophage selenoproteins in limiting bacterial replication. Herein, we provide a first report of a novel function of host macrophage selenoproteins in restriction of intracellular bacterial replication.
Four week-old C57BL/6 mice were maintained on purified diet (Tekland diets, Envigo, Madison, WI, USA) differing only in Na2SeO3 levels (deficient diet (TD.92163, <0.01 ppm Na2SeO3), adequate diet [TD.96363, 0.1 ppm (0.1 mg/kg) Na2SeO3], or supplemented diet [TD.07326, 0.4 ppm (0.4 mg/kg) Na2SeO3)] for greater than 12 weeks as previously described (27, 32).
TrspM mice lacking macrophage-specific selenoproteins were a kind gift from Dolph L. Hatfield at the Center for Cancer Research, National Institutes of Health, Bethesda, MD. TrspM mice are Trspfl/fl that are either heterozygous or homozygous for cre-recombinase under the lysozyme M promoter as previously described (50). TrspM and littermate control, WT mice, 6-8 weeks old, were maintained on standard chow diet that contains approximately 0.2 ppm of Se, which is also considered to be an ‘adequate’ level of Se diet. All animal experiments were conducted in accordance with Institutional Animal Use and Care Committee guidelines at the Pennsylvania State University.
Bacterial stocks were generated by expansion of 1 colony-forming unit (CFU) of F. tularensis holartica LVS in Chamberlain’s defined media (CDM) as previously described (51). Cultures were serially passaged at least 4 times in CDM under deficient [0nM sodium selenite (Na2SeO3 (Sigma- Aldrich, USA)], adequate (50nM Na2SeO3) or supplemented (200nM Na2SeO3) conditions. Bacterial growth assays were performed in 96 well flat bottom plates (Costar®, Corning®, Sigma- Aldrich, USA) with a starting OD600nM of F. tularensis LVS at 0.002 in CDM in the presence of 0nM, 50nM or 200nM Na2SeO3. OD600nM was recorded every 30 min for a period of 25 hr using a Spectromax spectrophotometer (Molecular Devices, CA, USA). Bacteria were enumerated by serial dilution in PBS (Hyclone™, GE Health Care, USA) and plated every 8 hr on chocolate agar plates prepared from Mueller-Hinton agar (Becton, Dickinson and Company, NJ, USA) supplemented with 1% (w/v) bovine hemoglobin (Reel™, ThermoFisher Scientific, USA) and 0.5% (v/v) IsoVitaleX™ (Becton, Dickinson and Company, NJ, USA). Plates were incubated at 37°C in an atmosphere of 5% CO2 for 72 hr before enumeration.
Mice were anesthetized with isofluorane and intranasally inoculated with 750, 1500 or 1750 CFU F. tularensis LVS in 50μl of PBS. Intranasal infections of mice with F. tularensis Schu S4 were performed under BSL3 conditions with Institutional Biosafety Committee approval at the Pennsylvania State University. CDC approved appropriate PPEs, procedures, and biosafety protocols were followed at the CDC certified ABSL-3 facility at PSU. Body weight was measured twice daily and mice were observed for changes in body condition. Mice were euthanized by CO2 asphyxiation at day 15 p.i. or if greater than 20% of initial body weight had been lost.
For bacterial enumeration, lung and spleen were homogenized in PBS with 1.0 mm Zircon/silicon beads (RPI Research products, IL, USA) in a Bead Blaster™24 (D2400, Benchmark Scientific, NJ, USA) using 6, 1 min cycles at a power level of 7 with a 30 sec break in between cycles. Whole livers were placed in whirl bags and crushed in 1mL of PBS. Lung, liver, spleen homogenates and blood were diluted and plated on chocolate agar (Becton, Dickinson and Company, NJ, USA). Plates were incubated at 37°C in an atmosphere of 5% CO2 for 72 hr before enumeration.
In silico analysis of genes required for the synthesis and incorporation of selenocysteine (Sec) or 2-selenouridine (SeU) Data was retrieved from the REFSEQ genomic and protein databases in FASTA file format. A blastp 2.5.0+ function using the default settings was performed on known selenoprotein sequences using the reference sequences from E. coli (NC_000913.3) and D. vulargis (NC_002937.3) against F. tularensis (NC_006570.2, NC_007880.1) sequences, and this was followed by a reciprocal function (52, 53). F. tularensis genomes were interrogated for tRNA, using the tRNADB-CE6 and GtRNAdb7 databases (54, 55). E. coli K12 was used a reference using the Conserved Domain Architecture Retrieval Tool (CDART) (56). Ontology was assessed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (57).
Virulence gene expression was measured in bacteria grown to log phase in CDM with or without Se. RNA was extracted with TRIzol™ reagent (Invitrogen™, ThermoFisher Scientific, USA) and purified with an RNA isolation kit (Ambion™, ThermoFisher Scientific, USA). cDNA was prepared with an RT kit (BioRad, Hercules, CA, USA) and qPCR was performed with SYBR™ Green reaction mix (ThermoFisher Scientific, USA) using a CFX96 Touch™ Real-Time PCR Detection System (BioRad, Hercules, CA, USA). Fold change values were calculated by comparing the CT values of fopA(FTL _1328), iglC(FTL_1159 and tul4(FTL _0421) to the internal control polA(FTL _1666). Primers (Integrated DNA Technologies, USA) are listed in Supplementary Table 1.
F. tularensis LVS and E. coli (ssp. K-12) were grown in CDM or Brain Heart Infusion (BHI) (Becton, Dickinson and Company, NJ, USA) broth at 37°C overnight with agitation at 175 rpm in a final concentration of 0, 50 nM or 200 nM Na2SeO3. Cultures were washed thrice in PBS, centrifuging at 3000 × g for 20 min at 4°C. Pellets were then resuspended in 2 mL of MS grade water (Sigma-Aldrich, USA) and pulsed using a digital sonicator (Branson, Digital Sonifier®, Emerson, USA) for 3 cycles of 30 sec at an amplitude of 20. Bacterial lysates or control media were then filtered through a 0.45 μM filter and subjected to AAS to measure elemental Se by the Water Quality Laboratory, at the Penn State Institute of Energy and the Environment.
BMDMs were prepared as described previously (58). In brief, the femur and tibia were crushed through a 70 μm strainer in complete DMEM (Hyclone™, SH3008102, GE Health Care, USA), 5% fetal bovine serum (Hyclone™, Lot# AWG18462, GE Health Care, USA tested to contain a very low level of Se- 25 μg/dl), 2mM L-Glutamine (Gemini Bio-Products, CA, USA), 1.5 mM HEPES (Corning, USA), 1 mM sodium pyruvate (Hyclone™, GE Health Care, USA) and 1X nonessential amino acids (Hyclone™, GE Health Care, USA). Cells were centrifuged at 400 × g for 10 min at 25°C, the supernatant was decanted and pellet resuspended in complete DMEM with 20% (v/v) L929 conditioned media that was generated using low Se FBS as described above [containing macrophage-colony stimulating factor (M-CSF)]. Bone-marrow progenitor cells were plated at a density of 5 × 105 cells/100cm2 petri dish (VWR, USA) in deficient (0 nM), adequate (50 nM Na2SeO3) or supplemented (200 nM Na2SeO3) culture conditions. On days 3 and 5 of culture, media was aspirated and replenished with complete DMEM media containing 20% L929 with or without Na2SeO3. On day 7 of culture, BMDMs were harvested and utilized to assess Se status of diet mice by measurement of GPX1 expression or were allocated for gentamicin protection assays.
BMDMs were seeded at a density of 3-5 × 105 cells/well and infected at an MOI of 1:100 CFU in a 24 well tissue culture treated plate (CellStar®, VWR, USA). Cells were centrifuged at 300 x g at RT for 10 min and then incubated at 37°C, 5% CO2 for 20 min. Media was aspirated, fresh DMEM complete media containing gentamicin (100 μg/mL) (Gibco™, ThermoFisher Scientific, USA) was added and cells were incubated at 37°C, 5% CO2 for 1 hr to remove extracellular bacteria. BMDMs were washed thrice with PBS and lysed with a solution of 0.1% deoxycholate in PBS solution for 2-5 min at RT for CFU enumeration at 2hr post-inoculation. Remaining cells were resuspended in DMEM complete media and lysed 18 hr later. Cell lysates from both time points were serially diluted in PBS and plated for bacterial enumeration.
Three biological replicates of infected and uninfected BMDMs from gentamicin protection assays at 10hr p.i. were collected and processed for RNA isolation using a combination method incorporating both TRIzol reagent and a Purelink™ RNA Mini Kit (Invitrogen, Carlsbad, CA). Transcriptome library preparation and subsequent sequencing was done through BGI Genomics (Cambridge, MA) using their unique DNBseq platform. 100bp paired end reads were obtained at an average coverage of 35.2 million aligned reads/sample. We used FastQC for quality control assessment and STAR v. 2.7.3a (59) to align the raw sequencing reads to GRCm38.p6 using gene coordinates from the GENCODE database Release M25 (60). Duplicate reads were tagged using Picard v. 2.9.0 (61). Mapping quality was checked with transcript integrity scores using RSeQC v. 3.0.1 (62), and genes were quantified for expression using RNA-SeQC v. 2.3.5 (63). We then checked for clustering of samples with multidimensional scaling analysis and corrected for batch effects using Combat-Seq (64). Principle component analysis was performed, and batch 3 samples were removed due to poor clustering with other replicates. Genes were filtered for sufficient coverage using the edgeR function filterByExpr to filter out genes that had less than 10 reads in more than half of the samples (65). We conducted differential expression analysis on infected vs. uninfected macrophages using edgeR v. 3.30.0 (65) RNA-seq data can be accessed at BioSample accession SAMN19317215
Log-Rank Mantle-Cox tests analyzed survival data from in vivo F. tularensis LVS challenge in Se diet and TrspM mice. One-way ANOVA with Tukey’s post hoc test analyzed data from gentamycin protection assays and Bonferroni’s multiple comparison test was used to analyze relative GPX1 expression in BMDMs. Two-way ANOVA with Sidak’s post hoc test analyzed data from in vivo bacterial burden enumeration and gentamycin protection assays from BMDMs of TrspM mice. All statistical analyses were performed using Prism (version 5) (GraphPad Software, San Diego, CA, USA).
It was previously reported that F. tularensis LVS upregulates expression of genes associated with Se-metabolism in alveolar epithelial cells. These findings led to speculation that F. tularensis may sequester Se for some critical biological processes (49). We therefore investigated whether F. tularensis requires Se for any specific metabolic processes. To determine if F. tularensis can incorporate Sec into proteins, we probed F. tularensis genomes using bioinformatics tools and databases. First, a blastp2.5.0+ tool (66) was used to identify putative selenoproteins in the F. tularensis proteome. The use of protein sequences overcame an inherent codon usage bias between reference organisms and F. tularensis. The protein blastp function was enhanced by the curated domain (CD) search tool, but resulted in hits with extremely low scores and high E-values (67). Even though the search identified a potential tRNA (selC) and formate dehydrogenase (fdh) in F. tularensis, the identity was exceedingly low suggesting that the bacterium may not be able to synthesize selenocysteine.
Next, we investigated whether F. tularensis encodes tRNA(sec) using tRNADB-CE6 and GtRNAdb7 databases (54, 55). This confirmed the presence of tRNA(sec) in known Sec incorporating bacteria (E. coli and D. vulgaris), but failed to identify the tRNA(sec) in any published F. tularensis genomes, including LVS and Schu S4 (Table 1). The lack of a tRNA(sec) with the appropriate anticodon strongly suggests that F. tularensis is unable to incorporate Sec into proteins, and therefore do not express any functional selenoproteins.

Table 1 Genes required for synthesis and incorporation of selenocysteine in reference bacteria and genomes of bacteria were probed for expression of Sec biosynthesis genes and alternatively, selenouridine (SeU) synthesis, which are two of the three known mechanisms of Se utilization in prokaryotes.
To determine whether other components of the Sec incorporation machinery are present in F. tularensis, we utilized CDART and KEGG databases (56, 57). Using the amino acid sequences of E. coli selA and selB gene products as templates, CDART identified architectures comprised of two and five conserved domains, respectively. Filtering by the NCBI taxonomy tree revealed that these domain architectures were found in the genomes of other known Sec incorporating bacteria, e.g., D. vulgaris, but were absent in all F. tularensis genomes. Furthermore, individual domains specific to selenometabolism, e.g., Sec synthase N-terminal domain in selA and selB-winged helix domain in selB, were completely absent in F. tularensis, suggesting that functional copies of these genes are not present. KEGG resources also demonstrated that in all available F. tularensis genomes, selB was not among the translation elongation factors. Additionally, selA, selD, and SeU were absent in the KEGG selenocompounds metabolism pathway in F. tularensis, while they were all present in E. coli and D. vulgaris (excluding YbbB). In silico analyses of genes necessary for Sec incorporation (selA-D) or an alternative mechanism of Se utilization (YbbB) revealed that F. tularensis ssp. holartica LVS and F. tularensis ssp. tularensis Schu S4 lacked these genes, while E. coli and D. vulgaris possess the genes vital for Se utilization (Table 1).
To validate results from bioinformatics analyses, F. tularensis LVS was cultured in the absence or presence of Se to examine growth kinetics, cellular entry, intracellular replication, and in vivo infectivity. F. tularensis LVS cultures containing Se did not display altered growth kinetics in comparison to cultures deficient in Se as measured by optical density (Supplementary Figure 2A) and CFU over a period of time (Supplementary Figure 2B). Furthermore, expression of key virulence genes of F. tularensis LVS were not perturbed by addition of Se to liquid culture (Supplementary Table 2). Since macrophages are a primary site of F. tularensis replication during disease pathogenesis (39), we next asked if Se addition ex vivo could influence F. tularensis LVS entry or intracellular replication. BMDMs from Se-deficient mice were inoculated with F. tularensis LVS in the absence or presence of Se. Bacterial entry at 2 hr post-inoculation (Supplementary Figure 3A) and burden at 24 hr post-inoculation (Supplementary Figure 3B) were found to be identical between Se-deficient and supplemented conditions, indicating in vitro Se supplementation does not affect bacterial entry or replication. Additionally, Se-deficient mice were intranasally inoculated with F. tularensis LVS grown in Se-deficient or supplemented conditions to measure the ability of Se to regulate in vivo bacterial colonization and replication. Pulmonary bacterial burden was comparable at 6, 12, and 24 hr post-inoculation between Se-deficient and supplemented mice (Supplementary Figure 4). These data indicate that Se does not alter the physiology or infectivity of the pathogen, F. tularensis.
The third known mechanism of Se utilization in prokaryotes, Se incorporation as a cofactor in molybdenum hydroxylases (68), could not be examined by bioinformatics analyses. Francisella primarily utilizes copper and zinc as cofactors, but not Se as predicted by a protein database of trace element utilization (69). Nonetheless, we further confirmed that F. tularensis does not accumulate Se by this third mechanism of utilization by measuring incorporation of elemental Se. F. tularensis LVS and E. coli were grown to stationary phase under deficient (0 nM) or supplemented (200 nM Na2SeO3) conditions in defined Chamberlain’s defined media (CDM) or undefined brain-heart infusion (BHI) media, and bacterial lysates were subjected to AAS (atomic absorption spectroscopy) analysis. Indeed, Se was not incorporated into F. tularensis LVS as lysates yielded, at most, only the concentration of Se originally added to culture (Figure 1). However, E. coli had an appreciable concentration of elemental Se when cultured in Se-supplemented CDM (Figure 1A) as well as detectable levels in BHI (Figure 1B) and increased levels in Se-supplemented BHI. Taken together, in silico, gene level, growth kinetics, and AAS analyses determined that Francisella does not incorporate Se as a selenoprotein, as a modified base, or as a cofactor.
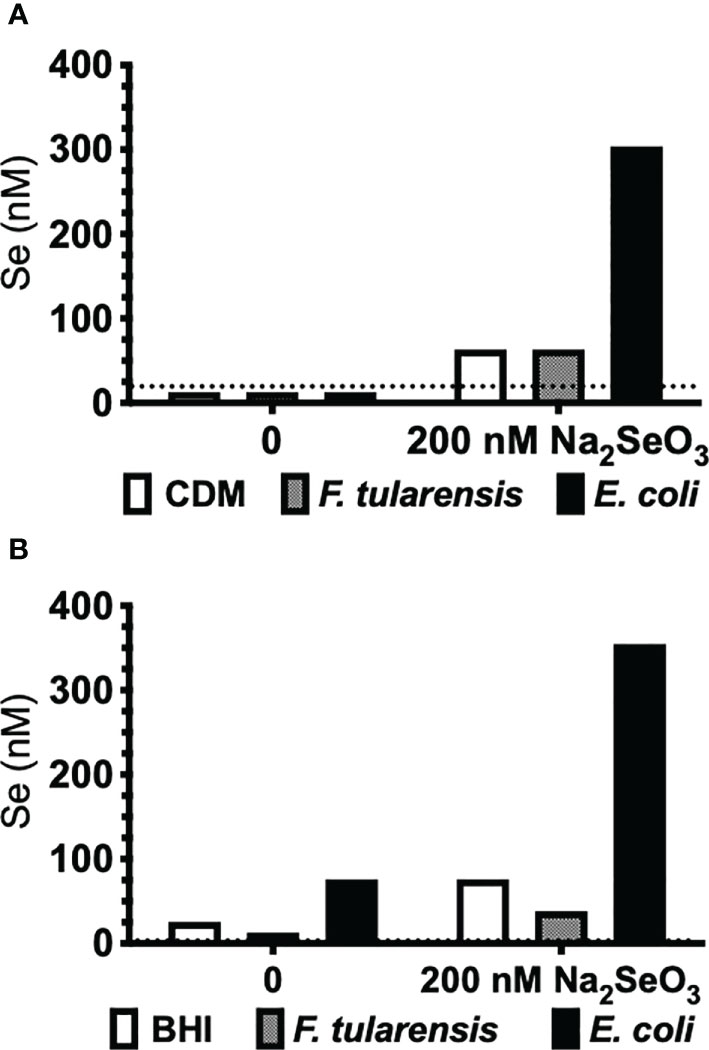
Figure 1 F. tularensis LVS does not incorporate Se. F. tularensis LVS or E. coli K12 was grown to saturation in (A) CDM or (B) undefined BHI broth in the presence (200 nM) or absence (0 nM) of Na2SeO3. Elemental Se concentration in bacterial lysates was measured by AAS. Data are representative of three independent experiments.
Next, we examined if Se influences disease pathogenesis. Alteration of dietary Se is a well-established model to change Se status and thus selenoprotein expression and function in mice (25–27, 32, 70). Mice maintained on Se-deficient, -adequate, or -supplemented purified diets for at least 12 weeks were intranasally infected with F. tularensis LVS, and body weight and survival were monitored for 14 days post-infection (p.i.). All deficient animals succumbed to infection with 1500 CFU by day 11 p.i. (Figure 2A). In contrast, adequate and supplemented mice exhibited increased survival rates (75% and 50%, respectively) and median survival for Se-deficient mice was day 8 p.i. while Se-supplemented mice was day 11.5 p.i. (Figure 2A). There was no statistical difference in the rate of survival between Se-supplemented and Se-adequate mice. Consistent with survival, Se-adequate and supplemented mice displayed decreased weight loss in comparison to surviving Se-deficient mice (Figure 2B), suggesting that Se status of the host influences F. tularensis LVS infection severity.
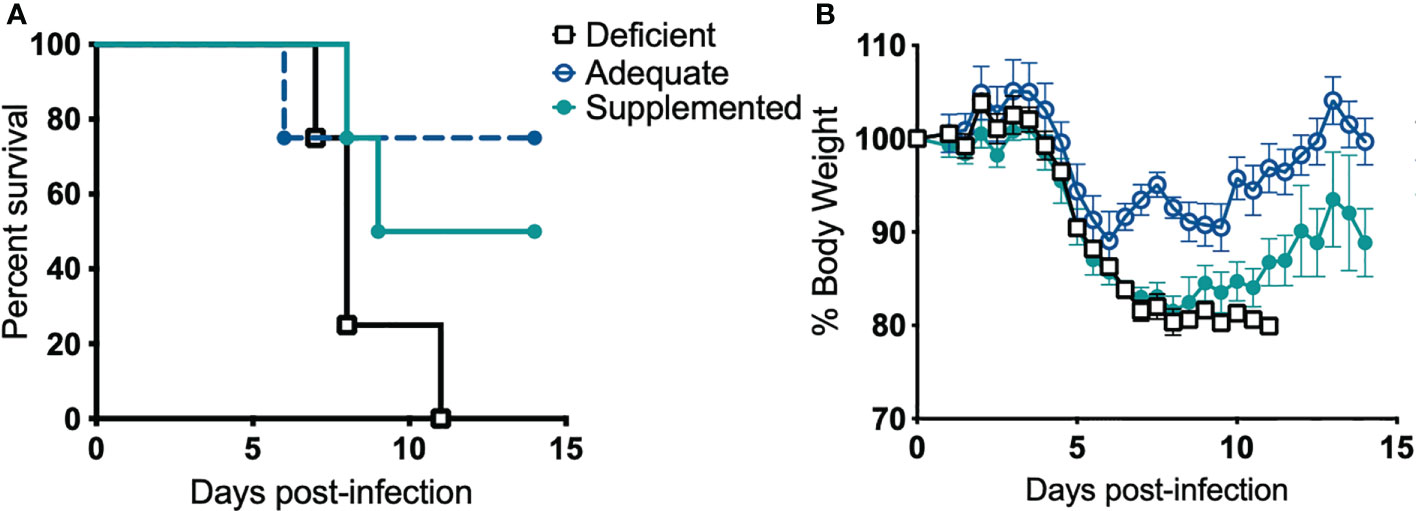
Figure 2 Dietary Se is required for survival from F. tularensis LVS challenge. Se-deficient, -adequate or -supplemented diet mice (n= 4/group) were intranasally inoculated with 1500 CFU of F. tularensis LVS and (A) survival and (B) body weight was monitored daily for 14 days. Statistical significance of survival was assessed using the Log-rank (Mantel-Cox) test. Daily weights represent the mean % body weight of the surviving animals; error bars denote +SEM. Data are representative of three independent in vivo experiments with similar outcomes.
We next determined if the observed lethality of Se-deficient mice from pulmonary tularemia was due to an inability to control bacterial replication and systemic dissemination. Since macrophages are the initial site of F. tularensis infection and replication (39), and Se is known to regulate macrophage function via selenoproteins (32, 71, 72), we hypothesized that this compartment of the immune system was compromised under Se-deficient conditions, leading to an inability to control bacterial replication. Se status of mice was first confirmed by measuring expression of the selenoprotein GPX1 in BMDMs, which was significantly greater as dietary Na2SeO3 concentration increased (Deficient vs. Adequate, ***p<0.001; Adequate vs. Supplemented ***p<0.001) (Supplementary Figure 5). To measure intracellular growth of F. tularensis LVS, a gentamicin protection assay was performed with BMDMs cultured from Se diet mice and maintained under respective deficient, adequate, or supplemented conditions ex vivo. Se-deficient macrophages had the greatest bacterial burden 24 hr p.i. compared to Se-adequate (*p<0.05) and supplemented macrophages (***p<0.001, Figure 3A). Interestingly, when the growth of bacteria was measured by normalizing CFU at 24 hr to 2 hr p.i., Se supplementation limited bacterial growth in a dose-dependent manner as less growth was observed in adequate (**p<0.01) and supplemented (***p<0.001) groups in comparison to deficient (Figure 3B). Therefore, the presence of Se restricts F. tularensis LVS intracellular growth in macrophages, which may provide an advantage to the Se-adequate and supplemented hosts during pulmonary challenge.
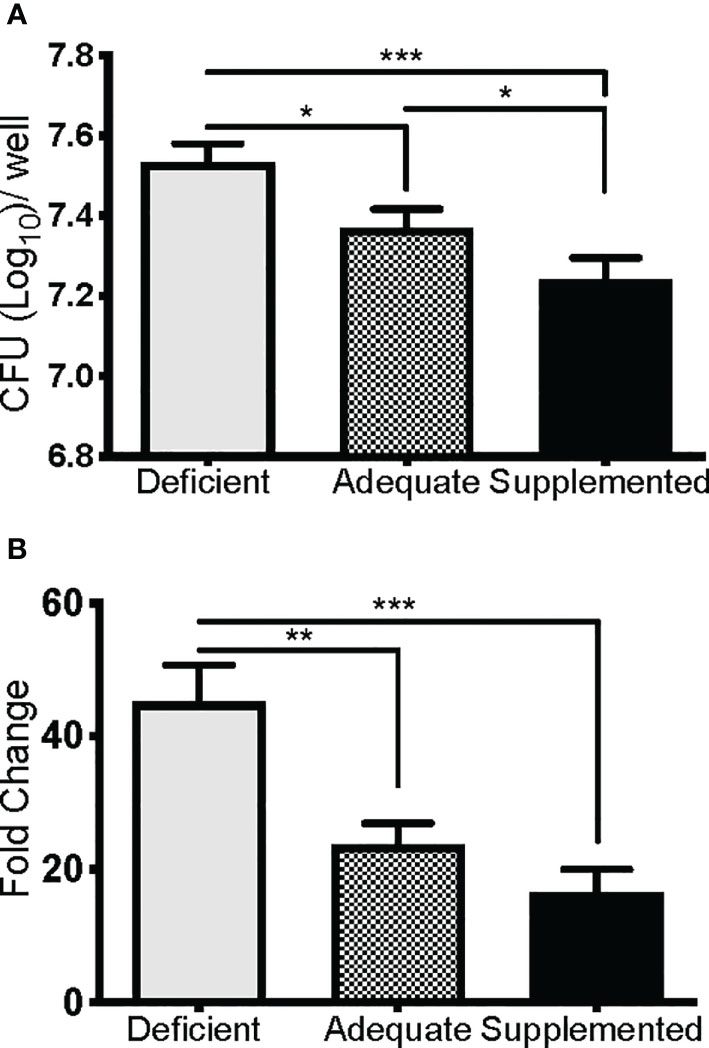
Figure 3 Se restricts F. tularensis LVS replication in macrophages. BMDMs from Se-deficient, -adequate, or -supplemented mice were maintained ex vivo under Se deficient (0 nM), adequate (50 nM) or supplemented (200 nM) conditions (n=3/one biological replicate, n=3 biological replicates/diet group), infected with F. t LVS at an MOI of 1:100. (A) Intracellular growth of bacteria was enumerated at 24 hr post-infection. (B) Bacterial growth over 24 hr represented as fold change in bacteria enumerated at 2 hr post infection. Data are depicted as the mean of three biological replicates and error bars denote +SD. Statistical significance was assessed by One-way ANOVA with Tukey’s Multiple Comparison Test (*p< 0.05) and data are representative of three independent experiments. **p<0.01, ***p<0.001.
We then determined if BMDMs supplemented with alternative selenocompounds would have similar GPX1 expression patterns, and if these compounds could also limit intracellular bacterial growth. BMDMs were supplemented with 200 nM of methylseleninic acid (MSA), selenomethionine (SeMet), Na2SeO3, or maintained under deficient conditions for 24 hr. BMDMs supplemented with MSA and Na2SeO3 had reduced intracellular growth compared to BMDMs maintained in deficient media or supplemented with SeMet (Supplementary Figure 6A). Additionally, GPX1 protein expression levels in MSA and Na2SeO3 treated BMDMs were higher than levels of deficient or SeMet treated BMDMs (Supplementary Figure 6B); thus, supporting the hypothesis that bacterial replication restriction was influenced by selenoprotein expression levels. We speculate that the supplementation of SeMet in BMDMs did not result in an increase of selenoproteins as macrophages do not express methioninase-γ-lyase (72), an enzyme required for the utilization of SeMet in the methylselenol pool (73). In summary, Se status of the host and ex vivo supplementation clearly limited bacterial growth and replication in macrophages, while suggesting an important role for host selenoproteins in the process.
Selenoproteins in macrophages are necessary for beneficial immune responses (26, 27). As described above, Se-deficient animals succumbed to F. tularensis LVS intranasal challenge (Figure 2A), Se-deficient BMDMs were more permissible to bacterial replication (Figure 3B), and intracellular growth of bacteria was inversely related to the level of macrophage selenoproteins (Supplementary Figure 6A). We therefore sought to determine the contribution of macrophage-specific selenoproteins during pulmonary tularemia in mice that lack selenoproteins in macrophages (TrspM), as previously described (44). The selenocysteinyl tRNA (Trspfl/fl) gene was disrupted using Cre/loxP system when co-expressed with Cre recombinase under the lysozyme M promoter. Mature macrophages express lysozyme M, thus resulting in a disruption of the Trsp gene product and an inability to synthesize selenoproteins. We confirmed the deletion of selenoproteins in macrophages of TrspM mice by measuring GPX1 expression in BMDMs derived from WT and TrspM mice. Indeed, GPX1 expression is absent in TrspM macrophages (Supplementary Figure 7). TrspM and WT littermate controls were intranasally infected with 750 CFU of F. tularensis LVS. TrspM mice succumbed to infection more quickly than WT mice (Figure 4A) with concordant weight loss (Figure 4B), suggesting that macrophage-specific selenoproteins are protective against pulmonary tularemia. The LD100 for TrspM mice was 1500 CFU (Supplementary Figures 8A, B) and 1750 CFU for WT mice (Supplementary Figures 8C, D), indicating that the presence of functional selenoproteins cannot protect mice from pulmonary tularemia at higher inoculating doses.
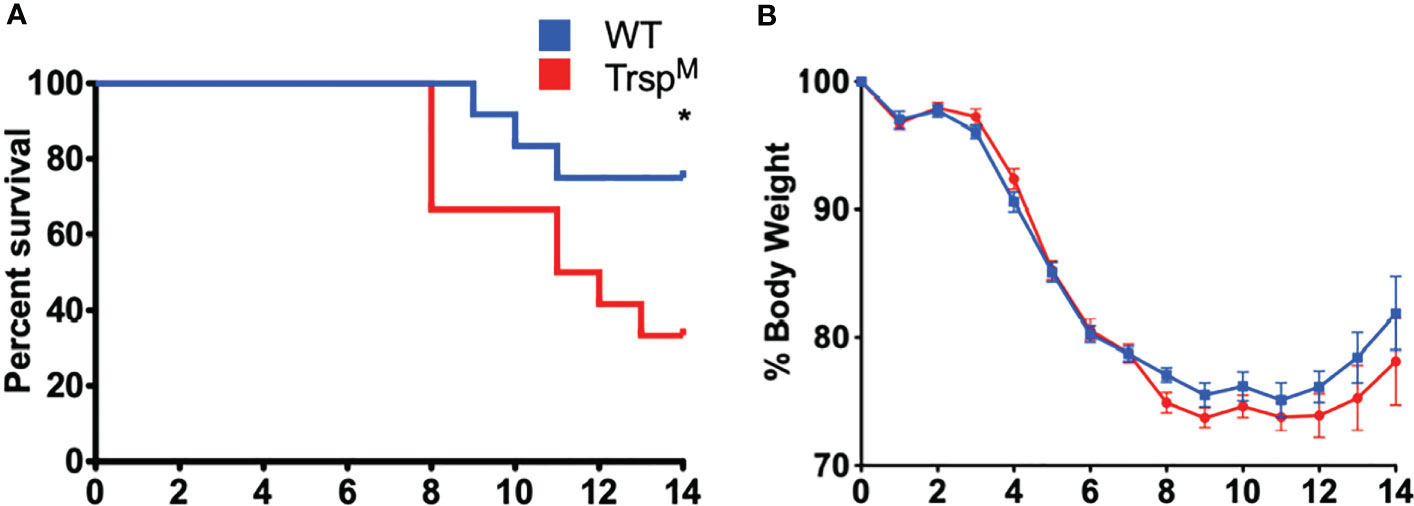
Figure 4 Macrophage selenoproteins are required for survival from F. tularensis LVS infection. Six- to eight-week-old WT and TrspM mice (n= 8-16/group) were inoculated with 750 CFU of F. tularensis LVS and (A) survival and (B) body weight were monitored for 14 days. Mice that lost greater than 20% of body weight were euthanized. Statistical significance of survival was assessed by the Log-rank (Mantel-Cox) test (*p<0.05). Weights were represented as the mean % body weight of the surviving animals. Error bars denote +/-SEM and data are representative of three independent experiments.
To further elucidate the protective contribution of macrophage-specific selenoproteins, TrspM and WT littermate controls were challenged with F. tularensis LVS, and bacterial burden was assessed in the lung, liver, blood, and spleen throughout infection. Although no differences were observed at day 3 p.i., bacterial burden was increased in TrspM mice in the liver and blood at day 5 p.i. (Figure 5B **p<0.01, 5C *p<0.05) and in the lung and the spleen at day 7 p.i. (Figure 5A *p<0.05, 5D ****p<0.0001) in comparison to WT controls. Presence of macrophage-specific selenoproteins in littermate controls also limited bacterial replication in the liver and blood on day 7 (Figure 5B ****p<0.0001, 5C **p<0.01) in comparison to TrspM mice. These data confirm that macrophage-specific selenoproteins limit bacterial replication at both the site of infection and in systemic tissues at later stages of the infection, and thus, promote survival from pulmonary tularemia challenge (Figure 4A).
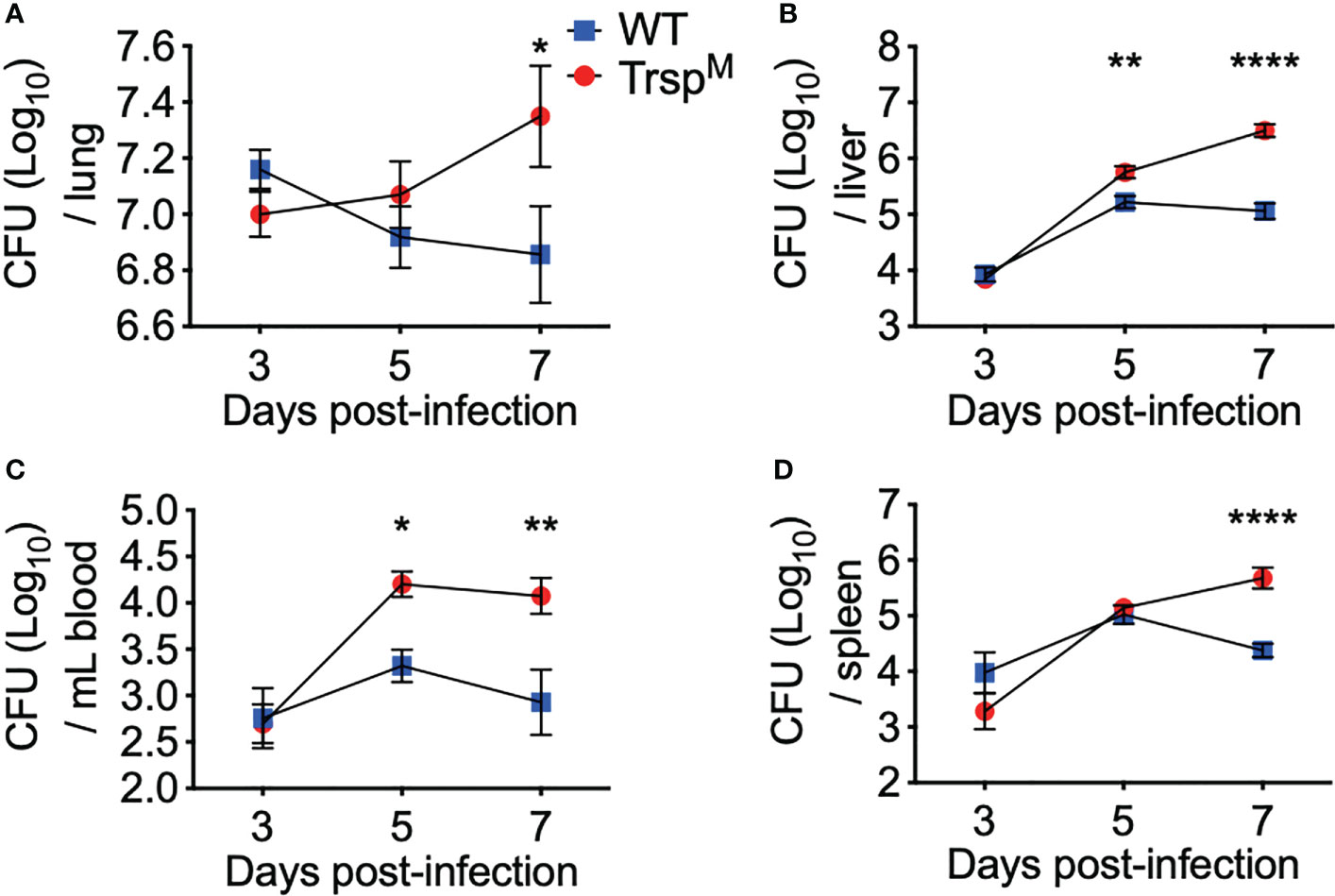
Figure 5 Macrophage selenoproteins are required for control of F. tularensis LVS infection in mice. WT (blue square) and TrspM (red circle) mice were intranasally inoculated with 750 CFU of F. tularensis LVS and at day 3, 5 or 7 p.i. mice were euthanized and bacterial burden was enumerated from the (A) lungs, (B) liver, (C) blood and (D) spleen. Statistical significance was assessed by two-way ANOVA with Sidak’s post-hoc test (*p<0.05). Data are representative of the mean and error bars +/-SEM. (ABD) Day 3 data were combined from three separate in vivo experiments (n=9/genotype), and four separate in vivo experiments at day 5 and 7 (n=14/genotype). (C) Day 3 data were combined from two separate in vivo experiments (n=6/ genotype) and day 5 and 7 data were combined from three separate in vivo experiments (n=11/ genotype). **p<0.01, ****p<0.0001.
The role of selenoproteins in direct antibacterial defense is poorly understood. Since macrophage selenoproteins limit the severity of F. tularensis infection (Figures 4, 5), it was necessary to establish if bacterial replication restriction observed in the Se dietary model (Figure 3B) was mediated in a selenoprotein-specific manner. Indeed, BMDMs from TrspM mice were more permissive to F. tularensis replication in comparison to WT littermate controls as measured by CFU (* p<0.05, Figures 6A) and fold change replication at 24 hr over 2 hr (**p<0.01, Figure 6B). Moreover, the failure to limit replication that was observed in TrspM macrophages could not be rescued by the addition of Na2SeO3 (**p<0.01 TrspM vs. WT, Figure 6B), indicating that replication restriction is strictly by a selenoprotein-mediated mechanism. Experiments with F. tularensis Schu S4 yielded similar results (data not shown) indicating that macrophage specific selenoproteins play a role in limiting the replication of virulent F. tularensis.
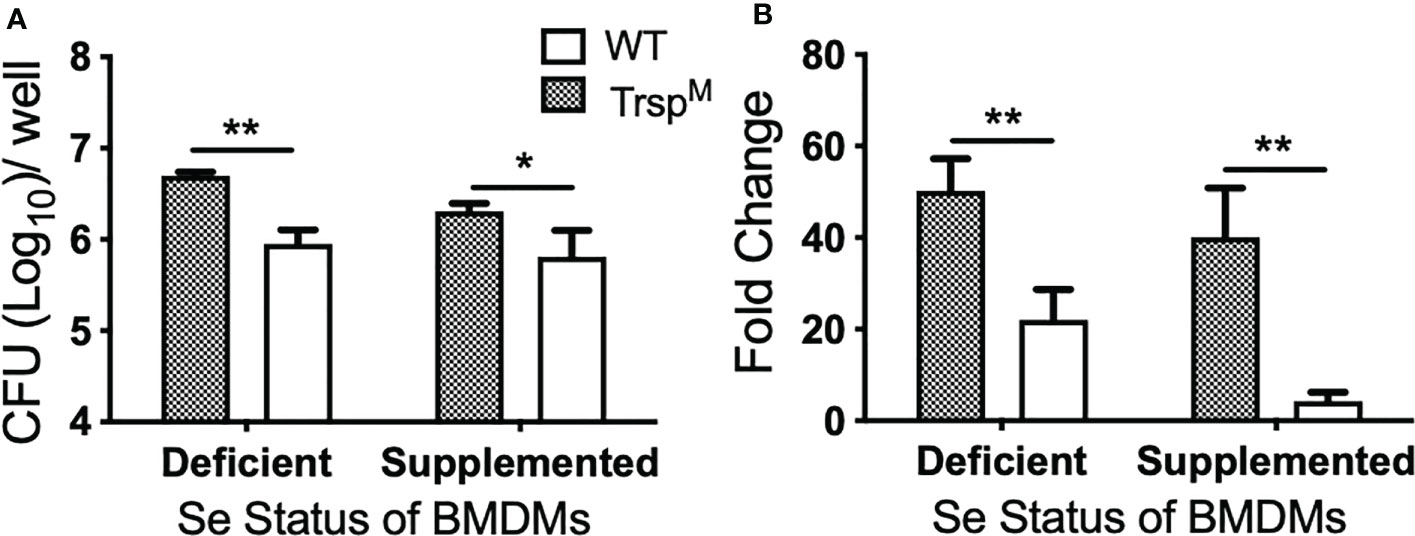
Figure 6 Macrophage selenoproteins restrict intracellular replication of F. tularensis LVS. BMDMs from WT and TrspM mice (n=3/group) were infected with F. tularensis LVS at an MOI of 1:50 and intracellular bacteria were enumerated at 24 hr p.i. (A) The number of bacteria recovered at 24 hr. Line represents the mean and error bars denote +SD. (B) Bacterial growth over 24 hr represented as fold change. Statistical significance was assessed by one-way ANOVA followed by Bonferroni’s multiple comparison test (*p<0.05). Data are representative of three experiments. **p<0.01.
Twenty-five and 24 unique selenoproteins have been identified in the human and murine genomes, respectively (28). Many of the selenoproteins have been classified according to their function into separate groups associated with redox signaling, protein folding, antioxidative capacity, and others. There are also several selenoproteins whose functions are as yet unknown (29, 74). To more clearly elucidate which selenoproteins might be important for the restriction of bacterial replication in macrophages, we performed transcriptomic analyses. Gentamicin protection assays were performed with BMDMs isolated from WT mice and infected with F. tularensis LVS. At 10hr p.i., both infected and uninfected cells were harvested and processed for RNA isolation and subsequent sequencing. RNA-seq results revealed 15,393 genes with sufficient coverage, and 1,866 genes that were differentially expressed between the infected and uninfected groups with a false discovery rate (FDR) of less than 0.05. Of the significant differentially expressed genes, six were confirmedto be selenoproteins (Figure 7). mRNA expression levels of Selenoprotein W (SelenoW), Glutathione peroxidase1 (Gpx1), SelenoM, Gpx4, Methionine sulfoxide reductase B (Msrb1), and SelenoH were all elevated in infected macrophages when compared to uninfected controls. Although not significant, an additional 14 selenoproteins were found to be differentially expressed between the groups (Figure 7, Supplementary Table 3). SelenoW, which has been suggested to have antioxidant functions as well as a potential for mediating cellular immunity (75, 76), was one of the differentially expressed selenoprotein at the RNA level. This observation was confirmed via qPCR analysis of the same infected and uninfected BMDM groups (Supplementary Figure 9). However, protein expression patterns was not significantly different between uninfected and infected cells as determined by western blot analysis in two representative selenoproteins, Gpx1 and Gpx4 (Supplementary Figure 10). Similarly, no significant difference between protein levels of SelenoW, SeleneoM, and MsrB1was observed (data not shown).
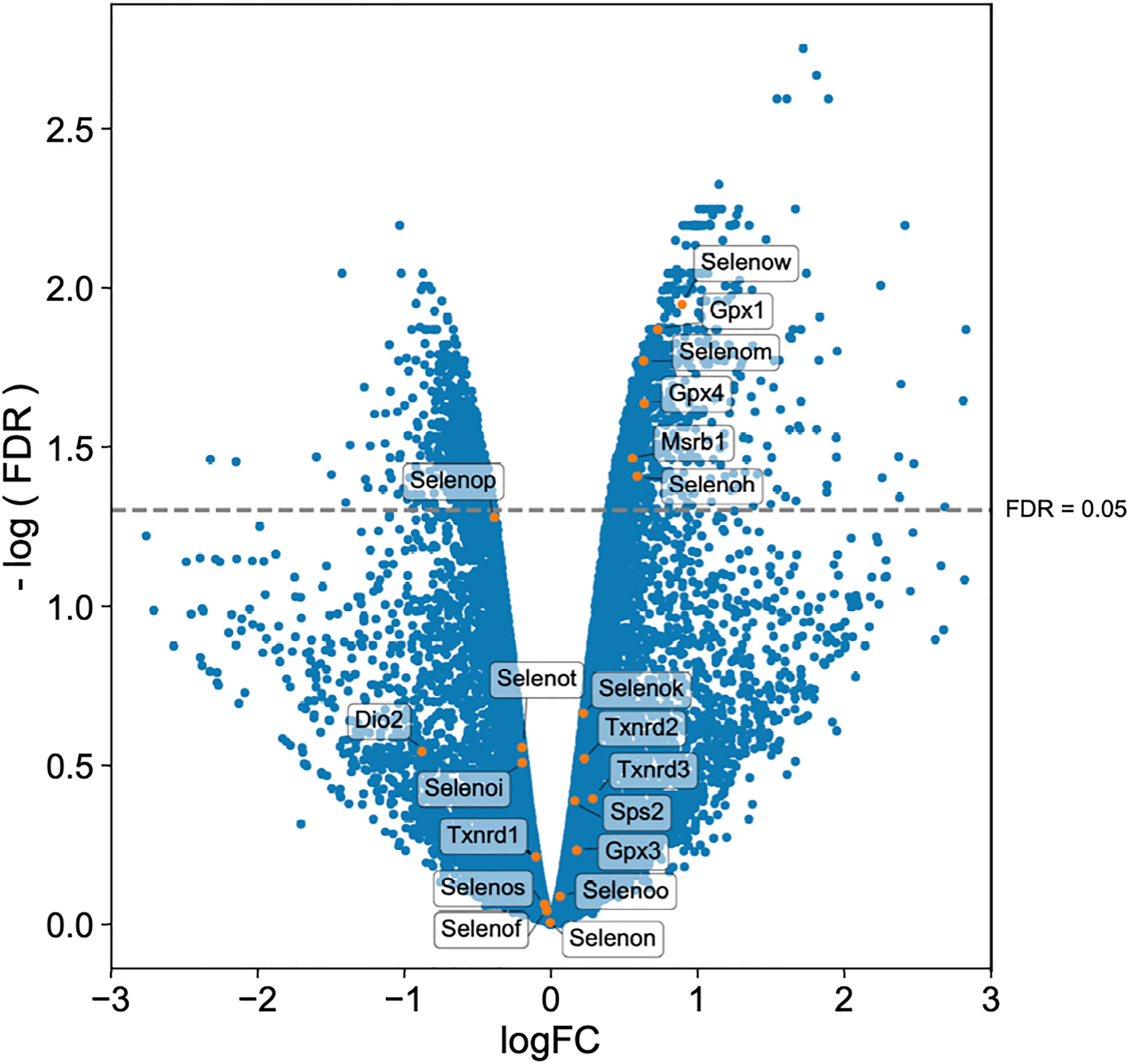
Figure 7 Selenoproteins are differentially regulated between infected and uninfected macrophages. BMDMs from WT mice (n=3/group) were infected with F. tularensis LVS at an MOI of 1:10. Cells were harvested at 10hr p.i., and processed for RNA isolation and subsequent sequencing. Volcano plot of differentially expressed genes shows that 20 of 24 murine selenoproteins are differentially expressed when comparing infected BMDMs to uninfected controls, with six selenoproteins showing significance. Differential expression analysis was conducted using edgeR v. 3.30.0 (FDR <0.05).
Several studies have utilized dietary animal models fed with various levels of Se to investigate the role of Se in infectious diseases. In a model of central nervous system listeriosis induced by the intracellular bacterium, Listeria monocytogenes, Se-deficiency led to greater central nervous system lesion development (77). In a separate murine model of listeriosis, Se-deficient mice had impaired innate immune cell responses such as decreased splenic NK cell cytotoxicity and serum proinflammatory cytokine (IL-12) production and consequently exhibited increased bacterial burden in the liver, spleen and brain in comparison to Se-adequate mice (78). Additionally, Se deficiency during Staphylococcus aureus-induced mastitis exacerbated the proinflammatory cytokine response due to suppressed PPARγ activity, and increased NF-κB activation, that resulted in increased nitric oxide levels and larger inflammatory lesions (79). In a model of bacterial induced gut inflammation, Se-deficient mice challenged with C. rodentium had decreased group 3 innate lymphoid cells and T helper 17 cells in comparison to mice with adequate or supplemented Se-status. This altered immune response at the site of infection was associated with decreased epithelial integrity and increased mortality of Se-deficient mice (26). We observed increased severity of disease in Se-deficient mice in response to pneumonic challenge of F. tularensis (Figure 2). Our findings are in agreement with previous studies suggesting that adequate dietary Se-status of the host is necessary for proper immune function and resolution of bacterial infection. In addition, for the first time our studies demonstrate Se- and selenoprotein-dependent restriction of bacterial replication in macrophages (Figure 3).
Transcriptome analysis of a bronchial airway epithelial cell line during F. tularensis LVS infection revealed upregulation of Se metabolism genes highlighting the importance of Se utilization traits. It was proposed that F. tularensis utilizes this strategy to exploit host resources for pathogen fitness (49). Surprisingly, bioinformatics (Table 1) and AAS analyses (Figure 1) revealed the inability of F. tularensis LVS to express Se utilization traits or incorporate Se into its proteome. We therefore hypothesized that the upregulation of genes associated with selenoamino acid incorporation, may be a protective mechanism employed by the host to limit bacterial replication. We then determined that Se status of the host impacts the severity of pulmonary tularemia, as Se-deficient mice succumbed to F. tularensis challenge, while animals of adequate and supplemented Se status exhibited greater survival and decreased weight loss (Figure 2). Macrophages are the initial cellular target for F. tularensis infection, replication, and expansion during the pathogenesis of tularemia (39). We demonstrated that BMDMs of adequate or supplemented Se status limited bacterial replication in comparison to Se-deficient BMDMs (Figure 3). As Se status of mice increased, so did selenoprotein expression in BMDMs (Supplementary Figure 5), providing correlative evidence that host selenoproteins may function to limit intracellular replication. F. tularensis challenge of TrspM mice revealed that macrophage-specific selenoproteins are necessary for survival from pulmonary tularemia as TrspM mice succumbed to the disease, whilst WT littermate controls survived (Figure 4). Greater bacterial burden in systemic tissues such as the spleen and liver later during infection in TrspM mice (Figure 5) suggested that absence of macrophage selenoproteins leaves the host unable to control F. tularensis replication. Indeed, BMDMs from TrspM mice permitted greater bacterial replication in comparison to WT littermate controls (Figure 6), thus demonstrating the necessity of macrophage-specific selenoproteins in limiting bacterial replication. Although bactericidal activities in macrophages have been demonstrated to be enhanced by Se-supplementation (80, 81), no studies have thus far demonstrated a specific role for macrophage-specific selenoproteins in bacterial growth.
Interestingly, RNA expression of six selenoproteins Gpx1, Gpx4, Msrb1, SelenoH, SelenoM, and SelenoW, were upregulated during F. tularensis infection of WT BMDMs (Figure 7). Gpx1 and Gpx4 have well-characterized antioxidant functions, reside in the cytoplasm and mitochondrial membrane and are ubiquitously expressed throughout the body (82). MsrB1 catalyzes reversible stereoselective methionine oxidation of the R enantiomer of oxidized methionine residues in proteins (83). As MsrB1 function decreases during Se-deficiency, innate immunity is compromised in macrophages owing to the disruption of actin polymerization-dependent processes, e.g., filopodia formation, micropinocytosis (84), and cytokine release (85). SelenoM resides in the ER membrane and although its function is unknown, it may be involved in neurodegeneration as it is most highly expressed brain tissue (86). Both SelenoH and SelenoW possess antioxidant functions, belong to the thioredoxin-like family of selenoproteins (76). SelenoW is expressed during the development of the nervous system, skeletal muscles and heart and may protect developing myoblasts from oxidative stress (87), but also mediates cell immunity (88). Contribution of these selenoproteins to antibacterial defense in macrophages is not known. Interestingly, mRNA expression of the selenoprotein genes gPx1, msrB1, selenoW and selenoH are most affected by Se availability and translation is reduced during Se-deficiency (89). This group of selenoproteins are therefore referred to as ‘stress selenoproteins’ (29) and F. tularensis infection may be inducing Se-deficiency in host cells as Se is utilized via selenoproteins to limit bacterial replication. Alternatively, F. tularensis infection leads to macrophage stress, which in turn may lead to upregulation of these selenoproteins. However, we did not observe a statistically significant increase in the expression of these selenoproteins at the protein level (Supplementary Figure 10). This may be due to the limited availability of Se in the medium due to perhaps increased utilization of Se by infected stressed cells, delayed and hierarchal expression kinetics of selenoproteins based on the availability of Se, and delayed translation of selenoproteins. Therefore, further in-depth studies are needed to establish the expression pattern and specific role of these selenoproteins during F. tularensis infection.
Previous studies have shown Se through selenoproteins alter the phenotype of macrophages from proinflammatory M1 to anti-inflammatory M2 (32), via cyclooxygenase-dependent cyclopentenone prostaglandin J2 (90) or ‘eicosanoid class switching’ (74). Se supplementation promotes selenoprotein expression in macrophages that skews the arachidonic acid pathway from pro-inflammatory mediators prostaglandin E2 and thromboxane A2 to produce prostaglandin D2 and the downstream ant-inflammatory metabolites cyclopentenone prostaglandins (74). The beneficial effects of Se and selenoproteins of macrophages mediating an M2 immune response have been observed in mouse models of Se dietary deficiency and in TrspM mice during N. brasiliensis infection as this parasite requires a strong type 2 immune response for resolution of infection (27). Additional investigations utilizing TrspM mice to determine the contribution of macrophage specific selenoproteins to both bacterial and chemically-induced colitis revealed that the selenoproteins of macrophages are essential to clear C. rodentium and resolve inflammation. TrspM mice exhibited increased mortality as a result of C. rodentium infection (26), while TrspM were unable to resolve colitis-associated inflammation regardless if they were maintained on either Se-supplemented and deficient diets (25). While the role of M2 macrophages in tularemia pathogenesis is debated, anti-inflammatory M2 macrophages have not been previously shown to be protective against F. tularensis-induced pneumonic tularemia. In fact, classically activated M1 macrophages are thought to control F. tularensis (91, 92). Therefore, we believe the inability of TrspM and Se-deficient macrophages to control F. tularensis could be attributed to mechanisms other than their failure to differentiate into M2 macrophages.
Various selenoproteins have been demonstrated to have antioxidant functions and are critical in maintaining the redox status of the host cells. LPS stimulation of Se-deficient macrophages with correspondingly low selenoprotein expression elevated total cellular oxidative tone, nitric oxide synthase, and increased nitric oxide production, while Se-supplemented macrophages dampened reactive oxygen species (ROS) (71). Several studies have shown that IFN-γ activated macrophages restrict F. tularensis replication via a nitric oxide-dependent mechanism (93–95). F. tularensis also alters antioxidant defenses and proinflammatory cytokine production to promote intracellular survival (96). Therefore, it is tempting to speculate that selenoprotein-deficient macrophages have higher ROS levels leading to reduced bacterial growth. However, nitric oxide production was similar between WT or TrspM macrophages infected with F. tularensis (data not shown). In fact, it is known that F. tularensis neutralizes ROS/reactive nitrogen species by inhibition of NADPH oxidase within the resting phagocytes, thus promoting intracellular bacterial survival (97). In addition, the oxidative stress response system of F. tularensis, comprised of superoxide dismutases (98), and an H2O2-decomposing enzyme catalase (99), are necessary for intracellular growth and virulence. Therefore, we speculate that host selenoproteins must be restricting bacterial replication during pulmonary tularemia by a distinct mechanism.
Upon intracellular infection, F. tularensis becomes phenotypically auxotrophic and requires amino acids from the host for its survival and replication (100). In order to sequester nutrients from host cells, one of the strategies that the pathogen employs is the alteration of host autophagy (101). Dietary Se-deficiency induces autophagy as demonstrated through an upregulation of autophagy associated gene 5 and Beclin 1 mRNA and protein expression, and morphological changes in autophagy vacuoles, autolysosomes, and lysosomal degradation in the immune organs of chickens in comparison to Se diet controls (102). Since Se-adequate or supplemented macrophages are skewed toward an M2 phenotype (32) and have lower mTOR activation (103, 104), a kinase necessary for induction of canonical autophagy, selenoproteins in macrophages may be inhibiting autophagy induced by F. tularensis. However, autophagy is a complex process utilized by both the host and pathogen to facilitate survival, and the mechanism of macrophage selenoprotein regulation of autophagy during F. tularensis infection remains to be fully elucidated and is currently being investigated.
Herein, we identified a novel role for host macrophage selenoproteins to limit intracellular replication of a bacteria. Our studies could have direct implications for treating infectious diseases such as tuberculosis. In addition, these findings can have implications in managing diseases caused by infectious agents, such as sepsis. Sepsis is a pathophysiologic process involving activation and dysregulation of pro-inflammatory/anti-inflammatory responses intertwined with other physiological processes. Sepsis patients suffer organ damage from a dysregulated immune response to pathogen(s) that results in increased oxidative stress (105). Due to altered hepatic Se metabolism, sepsis patients exhibit decreased plasma Se concentrations as synthesis of the protein that transports Se, selenoprotein P (SelenoP), decreases in the liver and therefore less Se is transported throughout the body (106, 107). Accordingly, a beneficial role for Se supplementation was suggested for positive outcome by relieving oxidative stress and inflammation (108). In fact, several small-scale clinical studies have demonstrated a positive prognosis when Se was given as an adjunctive therapy (109, 110). Our findings provide additional evidence that Se supplementation may help during infectious disease particularly in infections by intracellular bacteria via inhibition of bacterial replication to control infection.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA; PRJNA732368.
The animal study was reviewed and approved by The Pennsylvania State University Institutional Animal and Care Use Committee.
RM and KR planned and performed experiments, analyzed the data, supervised experiments performed by others, and wrote the manuscript. BK, SS, MN, SN, DW, DP, KD, AT, and AS performed some of the experiments and analyzed the data. SG and KP analyzed data and helped write the manuscript. BC provided critical reagents and helped write the manuscript. GK planned and supervised the study, procured funding, analyzed the data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
RLM and DRW were supported by T32AI074551, SES by T32GM108563, MJN by TL1TR002016, and AT by T32LM012415 from the National Institutes of Health. KSP and GSK were supported by USDA-NIFA grant #4771 (Accession number 0000005). In addition, SG was supported by GM121907, KSP was supported by DK077152 and GSK was supported by NIH grants AI123521 and AI77917 from the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Resources and funding from Penn State University, The Huck Institutes of Life Sciences, and the College of Agricultural Sciences are acknowledged.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.701341/full#supplementary-material
1. Stadtman TC. Selenocysteine. Annu Rev Biochem (1996) 65:83–100. doi: 10.1146/annurev.bi.65.070196.000503
2. Böck A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, et al. Selenocysteine: The 21st Amino Acid. Mol Microbiol (1991) 5:515–20. doi: 10.1111/j.1365-2958.1991.tb00722.x
3. Zhang Y, Romero H, Salinas G, Gladyshev VN. Dynamic Evolution of Selenocysteine Utilization in Bacteria: A Balance Between Selenoprotein Loss and Evolution of Selenocysteine From Redox Active Cysteine Residues. Genome Biol (2006) 7:R94. doi: 10.1186/gb-2006-7-10-r94
4. Pinske C, Sawers RG. Anaerobic Formate and Hydrogen Metabolism. EcoSal Plus (2016) 7:1–32. doi: 10.1128/ecosalplus.ESP-0011-2016
5. Axley MJ, Stadtman TC. Selenium Metabolism and Seleniuim-Dependent Enzymes in Microorganisms. Annu Rev Nutr (1989) 9:127–37. doi: 10.1146/annurev.nu.09.070189.001015
6. Peng T, Lin J, Xu YZ, Zhang Y. Comparative Genomics Reveals New Evolutionary and Ecological Patterns of Selenium Utilization in Bacteria. ISME J (2016) 10:2048–59. doi: 10.1038/ismej.2015.246
7. Ferry JG. Formate Dehydrogenase. FEMS Microbiol Lett (1990) 7:377–82. doi: 10.1111/j.1574-6968.1990.tb04940.x
8. Leonhartsberger S, Korsa I, Böck A. The Molecular Biology of Formate Metabolism in Enterobacteria. J Mol Microbiol Biotechnol (2002) 4:269–76.
9. Havelaar AH, Cawthorne A, Angulo F, Bellinger D, Corrigan T, Cravioto A, et al. WHO Initiative to Estimate the Global Burden of Foodborne Diseases. Lancet (2013) 381:S59. doi: 10.1016/S0140-6736(13)61313-6
10. Bereswill S, Fischer A, Plickert R, Haag LM, Otto B, Kühl AA, et al. Novel Murine Infection Models Provide Deep Insights Into the “Ménage À Trois” of Campylobacter Jejuni, Microbiota and Host Innate Immunity. PloS One (2011) 6:e20953. doi: 10.1371/annotation/5247af81-4595-44b7-9c3f-2e45ad85abfa
11. Rayman MP. The Importance of Selenium to Human Health. Lancet (2000) 356:233–41. doi: 10.1016/S0140-6736(00)02490-9
12. Kassu A, Yabutani T, Mahmud ZH, Mohammad A, Nguyen N, Huong BTM, et al. Alterations in Serum Levels of Trace Elements in Tuberculosis and HIV Infections. Eur J Clin Nutr (2006) 60:580–6. doi: 10.1038/sj.ejcn.1602352
13. Sheehan HB, Benetucci J, Muzzio E, Redini L, Naveira J, Segura M, et al. High Rates of Serum Selenium Deficiency Among HIV-And HCV-Infected and Uninfected Drug Users in Buenos Aires, Argentina. Public Health Nutr (2012) 15:538–45. doi: 10.1017/S1368980011002357
14. de Moraes ML, Ramalho DM de P, Delogo KN, Miranda PFC, Mesquita EDD, Oliveira HM de MG de, et al. Association Between Serum Selenium Level and Conversion of Bacteriological Tests During Antituberculosis Treatment. J Bras Pneumol (2014) 40:269–78. doi: 10.1590/S1806-37132014000300010
15. Muzembo BA, Ngatu NR, Januka K, Huang HL, Nattadech C, Suzuki T, et al. Selenium Supplementation in HIV-Infected Individuals: A Systematic Review of Randomized Controlled Trials. Clin Nutr ESPEN (2019) 34:1–7. doi: 10.1016/j.clnesp.2019.09.005
16. Groenbaek K, Friis H, Hansen M, Ring-Larsen H, Krarup HB. The Effect of Antioxidant Supplementation on Hepatitis C Viral Load, Transaminases and Oxidative Status: A Randomized Trial Among Chronic Hepatitis C Virus-Infected Patients. Eur J Gastroenterol Hepatol (2006) 18:985–9. doi: 10.1097/01.meg.0000231746.76136.4a
17. Seyedrezazadeh E, Ostadrahimi A, Mahboob S, Assadi Y, Ghaemmagami J, Pourmogaddam M. Effect of Vitamin E and Selenium Supplementation on Oxidative Stress Status in Pulmonary Tuberculosis Patients. Respirology (2008) 13:294–8. doi: 10.1111/j.1440-1843.2007.01200.x
18. Rudolph M, Kroll F, Beery M, Marinda E, Sobiecki JF, Douglas G, et al. A Pilot Study Assessing the Impact of a Fortified Supplementary Food on the Health and Well-Being of Crèche Children and Adult TB Patients in South Africa. PloS One (2013) 8:e55544. doi: 10.1371/journal.pone.0055544
19. Kawai K, Meydani SN, Urassa W, Wu D, Mugusi FM, Saathoff E, et al. Micronutrient Supplementation and T Cell-Mediated Immune Responses in Patients With Tuberculosis in Tanzania. Epidemiol Infect (2014) 142:1505–9. doi: 10.1017/S0950268813002495
20. Campa A, Baum M, Bussmann H, Martinez S, Farahani M, van Widenfelt E, et al. The Effect of Micronutrient Supplementation on Active TB Incidence Early in HIV Infection in Botswana. Nutr Diet Suppl (2017) 9:37–45. doi: 10.2147/NDS.S123545
21. Singer M, Deutschman CS, Seymour C, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA - J Am Med Assoc (2016) 315:801–10. doi: 10.1001/jama.2016.0287
22. Sakr Y, Reinhart K, Bloos F, Marx G, Russwurm S, Bauer M, et al. Time Course and Relationship Between Plasma Selenium Concentrations, Systemic Inflammatory Response, Sepsis, and Multiorgan Failure. Br J Anaesth (2007) 98:775–84. doi: 10.1093/bja/aem091
23. Kočan L, Vašková J, Vaško L, Šimonová J, Šimon R, Firment J. Selenium Adjuvant Therapy in Septic Patients Selected According to Carrico Index. Clin Biochem (2014) 47:44–50. doi: 10.1016/j.clinbiochem.2014.07.004
24. Chelkeba L, Ahmadi A, Abdollahi M, Najafi A, Ghadimi MH, Mosaed R, et al. The Effect of Parenteral Selenium on Outcomes of Mechanically Ventilated Patients Following Sepsis: A Prospective Randomized Clinical Trial. Ann Intensive Care (2015) 5:29. doi: 10.1186/s13613-015-0071-y
25. Kaushal N, Kudva AK, Patterson AD, Chiaro C, Kennett MJ, Desai D, et al. Crucial Role of Macrophage Selenoproteins in Experimental Colitis. J Immunol (2014) 193:3683–92. doi: 10.4049/jimmunol.1400347
26. Nettleford SK, Zhao L, Qian F, Herold M, Arner B, Desai D, et al. The Essential Role of Selenoproteins in the Resolution of Citrobacter Rodentium-Induced Intestinal Inflammation. Front Nutr (2020) 7. doi: 10.3389/fnut.2020.00096
27. Nelson SM, Shay AE, James JL, Carlson BA, Urban JF, Prabhu KS. Selenoprotein Expression in Macrophages Is Critical for Optimal Clearance of Parasitic Helminth Nippostrongylus Brasiliensis. J Biol Chem (2016) 291:2787–98. doi: 10.1074/jbc.M115.684738
28. Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, et al. Characterization of Mammalian Selenoproteomes. Sci (80- ) (2003) 300:1439–43. doi: 10.1126/science.1083516
29. Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: Molecular Pathways and Physiological Roles. Physiol Rev (2014) 94:739–77. doi: 10.1152/physrev.00039.2013
30. Arnér ESJ, Holmgren A. Physiological Functions of Thioredoxin and Thioredoxin Reductase. Eur J Biochem (2000) 267:6102–9. doi: doi: 10.1046/j.1432-1327.2000.01701.x
31. Kalantari P, Narayan V, Natarajan SK, Muralidhar K, Gandhi UH, Vunta H, et al. Thioredoxin Reductase-1 Negatively Regulates HIV-1 Transactivating Protein Tat-Dependent Transcription in Human Macrophages. J Biol Chem (2008) 283:33183–90. doi: 10.1074/jbc.M807403200
32. Nelson SM, Lei X, Prabhu KS. Selenium Levels Affect the IL-4–Induced Expression of Alternative Activation Markers in Murine Macrophages. J Nutr (2011) 141:1754–61. doi: 10.3945/jn.111.141176
33. Ellis J, Oyston PCF, Green M, Titball RW. Tularemia. Clin Microbiol Rev (2002) 15:631–46. doi: 10.1128/CMR.15.4.631-646.2002
34. Keim P, Johansson A, Wagner DM. Molecular Epidemiology, Evolution, and Ecology of Francisella. Ann New York Acad Sci (2007) 1105:30–66. doi: 10.1196/annals.1409.011
35. Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, et al. Tularemia as a Biological Weapon: Medical and Public Health Management. J Am Med Assoc (2001) 285:2763–73. doi: 10.1001/jama.285.21.2763
36. Oyston PCF, Sjostedt A, Titball RW. Tularaemia: Bioterrorism Defence Renews Interest in Francisella Tularensis. Nat Rev Microbiol (2004) 2:967–78. doi: 10.1038/nrmicro1045
37. Hayes E, Marshall S, Dennis D. Tularemia - United States, 1990-2000. J Am Med Assoc (2002) 51:182–4.
38. Stuart BM, Pullen RL. TULAREMIC PNEUMONIA. Am J Med Sci (1945) 210:223–36. doi: 10.1097/00000441-194508000-00013
39. Celli J, Zahrt TC. Mechanisms of Francisella Tularensis Intracellular Pathogenesis. Cold Spring Harb Perspect Med (2013) 3:a010314. doi: 10.1101/cshperspect.a010314
40. Hall JD, Craven RR, Fuller JR, Pickles RJ, Kawula TH. Francisella Tularensis Replicates Within Alveolar Type II Epithelial Cells In Vitro and In Vivo Following Inhalation. Infect Immun (2007) 75:1034–9. doi: 10.1128/IAI.01254-06
41. Law HT, Lin AEJ, Kim Y, Quach B, Nano FE, Guttman JA. Francisella Tularensis Uses Cholesterol and Clathrin-Based Endocytic Mechanisms to Invade Hepatocytes. Sci Rep (2011) 1:192. doi: 10.1038/srep00192
42. Fortier AH, Slayter MV, Ziemba R, Meltzer MS, Nacy CA. Live Vaccine Strain of Francisella Tularensis: Infection and Immunity in Mice. Infect Immun (1991) 59:2922–8. doi: 10.1128/iai.59.9.2922-2928.1991
43. Stock T, Rother M. Selenoproteins in Archaea and Gram-Positive Bacteria. Biochim Biophys Acta - Gen Subj (2009) 1790:1520–32. doi: 10.1016/j.bbagen.2009.03.022
44. Yoshizawa S, Böck A. The Many Levels of Control on Bacterial Selenoprotein Synthesis. Biochim Biophys Acta - Gen Subj (2009) 1790:1404–14. doi: 10.1016/j.bbagen.2009.03.010
45. Leinfelder W, Zehelein E, Mandrandberthelot M, Bock A. Gene for a Novel tRNA Species That Accepts L-Serine and Cotranslationally Inserts Selenocysteine. Nature (1988) 331:723–5. doi: 10.1038/331723a0
46. Forchhammer K, Leinfelder W, Boesmiller K, Veprek B, Bock A. Selenocysteine Synthase From Escherichia Coli: Nucleotide Sequence of the Gene (Sela) and Purification of the Protein. J Biol Chem (1991) 266:6318–23. doi: 10.1016/S0021-9258(18)38120-1
47. Forchhammer K, Leinfelder W, Böck A. Identification of a Novel Translation Factor Necessary for the Incorporation of Selenocysteine Into Protein. Nature (1989) 342:453–6. doi: 10.1038/342453a0
48. Romero H, Zhang Y, Gladyshev VN, Salinas G. Evolution of Selenium Utilization Traits. Genome Biol (2005) 6:R66. doi: 10.1186/gb-2005-6-8-r66
49. Bradburne CE, Verhoeven AB, Manyam GC, Chaudhry SA, Chang EL, Thach DC, et al. Temporal Transcriptional Response During Infection of Type II Alveolar Epithelial Cells With Francisella Tularensis Live Vaccine Strain (Lvs) Supports a General Host Suppression and Bacterial Uptake by Macropinocytosis. J Biol Chem (2013) 288:10780–91. doi: 10.1074/jbc.M112.362178
50. Carlson BA, Yoo MH, Tsuji PA, Gladyshev VN, Hatfield DL. Mouse Models Targeting Selenocysteine tRNA Expression for Elucidating the Role of Selenoproteins in Health and Development. Molecules (2009) 14:3509–27. doi: 10.3390/molecules14093509
51. Chamberlain RE. Evaluationm of Live Tularemia Vaccine Prepared in A Chemically Defined Medium. Appl Microbiol (1965) 13:232–5. doi: 10.1128/am.13.2.232-235.1965
52. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res (1997) 25:3389–402. doi: 10.1093/nar/25.17.3389
53. Schäffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, et al. Improving the Accuracy of PSI-BLAST Protein Database Searches With Composition-Based Statistics and Other Refinements. Nucleic Acids Res (2001) 29:2994–3005. doi: 10.1093/nar/29.14.2994
54. Abe T, Ikemura T, Sugahara J, Kanai A, Ohara Y, Uehara H, et al. TRNADB-CE 2011: TRNA Gene Database Curated Manually by Experts. Nucleic Acids Res (2011) 39:D210–3. doi: 10.1093/nar/gkq1007
55. Chan PP, Lowe TM. GtRNAdb: A Database of Transfer RNA Genes Detected in Genomic Sequence. Nucleic Acids Res (2009) 37:D93–7. doi: 10.1093/nar/gkn787
56. Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: Protein Homology by Domain Architecture. Genome Res (2002) 12:1619–26. doi: 10.1101/gr.278202
57. Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, Information, Knowledge and Principle: Back to Metabolism in KEGG. Nucleic Acids Res (2014) 42:D199–205. doi: 10.1093/nar/gkt1076
58. Goralski TDP, Dewan KK, Alumasa JN, Avanzato V, Place DE, Markley RL, et al. Inhibitors of Ribosome Rescue Arrest Growth of Francisella Tularensis at All Stages of Intracellular Replication. Antimicrob Agents Chemother (2016) 60:3276–82. doi: 10.1128/AAC.03089-15
59. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
60. Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res (2019) 47:D766–73. doi: 10.1093/nar/gky955
62. Wang L, Wang S, Li W. RSeQC: Quality Control of RNA-Seq Experiments. Bioinformatics (2012) 28:2184–5. doi: 10.1093/bioinformatics/bts356
63. Deluca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, et al. RNA-SeQC: RNA-Seq Metrics for Quality Control and Process Optimization. Bioinformatics (2012) 28:1530–2. doi: 10.1093/bioinformatics/bts196
64. Zhang Y, Parmigiani G, Johnson WE. ComBat-Seq: Batch Effect Adjustment for RNA-Seq Count Data. bioRxiv (2020) 2:Iqaa078. doi: 10.1101/2020.01.13.904730
65. Robinson MD, McCarthy DJ, Smyth GK. Edger: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics (2009) 26:139–40. doi: 10.1093/bioinformatics/btp616
66. Mahram A, Herbordt MC. Fast and Accurate NCBI BLASTP. ICS ‘10: Proceedings of the 24th ACM International Conference on Supercomputing. (2010). p. 73–82. doi: 10.1145/1810085.1810099
67. Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, Deweese-Scott C, Fong JH, et al. CDD: Specific Functional Annotation With the Conserved Domain Database. Nucleic Acids Res (2009) 37:D205–10. doi: 10.1093/nar/gkn845
68. Haft DH, Self WT. Orphan SelD Proteins and Selenium-Dependent Molybdenum Hydroxylases. Biol Direct (2008) 3:1–6. doi: 10.1186/1745-6150-3-4
69. Zhang Y, Gladyshev VN. dbTEU: A Protein Database of Trace Element Utilization. Bioinformatics (2010) 26:700–2. doi: 10.1093/bioinformatics/btp705
70. Novoselov SV, Calvisi DF, Labunskyy VM, Factor VM, Carlson BA, Fomenko DE, et al. Selenoprotein Deficiency and High Levels of Selenium Compounds can Effectively Inhibit Hepatocarcinogenesis in Transgenic Mice. Oncogene (2005) 24(54):8003–11. doi: 10.1038/sj.onc.1208940
71. Prabhu KS, Zamamiri-Davis F, Stewart JB, Thompson JT, Sordillo LM, Reddy CC. Selenium Deficiency Increases the Expression of Inducible Nitric Oxide Synthase in RAW 264.7 Macrophages: Role of Nuclear Factor-κb in Up-Regulation. Biochem J (2002) 366:203–209. doi: 10.1042/bj20020256
72. Narayan V, Ravindra KC, Liao C, Kaushal N, Carlson BA, Prabhu KS. Epigenetic Regulation of Inflammatory Gene Expression in Macrophages by Selenium. J Nutr Biochem (2015) 26:138–45. doi: 10.1016/j.jnutbio.2014.09.009
73. Suzuki KT, Kurasaki K, Suzuki N. Selenocysteine β-Lyase and Methylselenol Demethylase in the Metabolism of Se-Methylated Selenocompounds Into Selenide. Biochim Biophys Acta - Gen Subj (2007) 1770:1053–1061. doi: 10.1016/j.bbagen.2007.03.007
74. Qian F, Misra S, Prabhu KS. Selenium and Selenoproteins in Prostanoid Metabolism and Immunity. Crit Rev Biochem Mol Biol (2019) 54:484–516. doi: 10.1080/10409238.2020.1717430
75. Vendeland SC, Beilstein MA, Yeh JY, Ream W, Whanger PD. Rat Skeletal Muscle Selenoprotein W: cDNA Clone and mRNA Modulation by Dietary Selenium. Proc Natl Acad Sci USA (1995) 92:8749–205. doi: 10.1073/pnas.92.19.8749
76. Dikiy A, Novoselov SV, Fomenko DE, Sengupta A, Carlson BA, Cerny RL, et al. SelT, SelW, SelH, and Rdx 12: Genomics and Molecular Insights Into the Functions of Selenoproteins of a Novel Thioredoxin-Like Family. Biochemistry (2007) 46:6871–82. doi: 10.1021/bi602462q
77. Altimira J, Prats N, López S, Domingo M, Briones V, Domínguez L, et al. Effect of Selenium Deficiency on the Development of Central Nervous System Lesions in Murine Listeriosis. J Comp Pathol (2000) 123:104–9. doi: 10.1053/jcpa.2000.0399
78. Wang C, Wang H, Luo J, Hu Y, Wei L, Duan M, et al. Selenium Deficiency Impairs Host Innate Immune Response and Induces Susceptibility to Listeria Monocytogenes Infection. BMC Immunol (2009) 10:1–12. doi: 10.1186/1471-2172-10-55
79. Gao X, Zhang Z, Li Y, Shen P, Hu X, Cao Y, et al. Selenium Deficiency Facilitates Inflammation Following S. Aureus Infection by Regulating TLR2-Related Pathways in the Mouse Mammary Gland. Biol Trace Elem Res (2016) 172:449457. doi: 10.1007/s12011-015-0614-y
80. Aribi M, Meziane W, Habi S, Boulatika Y, Marchandin H, Aymeric JL. Macrophage Bactericidal Activities Against Staphylococcus Aureus are Enhanced In Vivo by Selenium Supplementation in a Dose-Dependent Manner. PloS One (2015) 10:e0135515. doi: 10.1371/journal.pone.0135515
81. Liu Z, Huang J, Nie Y, Qazi IH, Cao Y, Wang L, et al. Selenium Treatment Enhanced Clearance of Salmonella in Chicken Macrophages (HD11). Antioxidants (2019) 81:532. doi: 10.3390/antiox8110532
82. Diamond AM. The Subcellular Location of Selenoproteins and the Impact on Their Function. Nutrients (2015) 7:3938–48. doi: 10.3390/nu7053938
83. Kryukov GV, Kumar RA, Koc A, Sun Z, Gladyshev VN. Selenoprotein R is a Zinc-Containing Stereo-Specific Methionine Sulfoxide Reductase. Proc Natl Acad Sci USA (2002) 99:4245–50. doi: 10.1073/pnas.072603099
84. Lee BC, Péterfi Z, Hoffmann FKW, Moore RE, Kaya A, Avanesov A, et al. MsrB1 and MICALs Regulate Actin Assembly and Macrophage Function via Reversible Stereoselective Methionine Oxidation. Mol Cell (2013) 51:397–404. doi: 10.1016/j.molcel.2013.06.019
85. Lee BC, Lee SG, Choo MK, Kim JH, Lee HM, Kim S, et al. Selenoprotein MsrB1 Promotes Anti-Inflammatory Cytokine Gene Expression in Macrophages and Controls Immune Response In Vivo /631/45/612 /631/250 /38 /82 /82/80 Article. Sci Rep (2017) 7:5119. doi: 10.1038/s41598-017-05230-2
86. Korotkov KV, Novoselov SV, Hatfield DL, Gladyshev VN. Mammalian Selenoprotein in Which Selenocysteine (Sec) Incorporation Is Supported by a New Form of Sec Insertion Sequence Element. Mol Cell Biol (2002) 22:1402–11. doi: 10.1128/MCB.22.5.1402-1411.2002
87. Loflin J, Lopez N, Whanger PD, Kioussi C. Selenoprotein W During Development and Oxidative Stress. J Inorg Biochem (2006) 100:1679–84. doi: 10.1016/j.jinorgbio.2006.05.018
88. Whanger PD. Selenoprotein Expression and Function-Selenoprotein W. Biochim Biophys Acta - Gen Subj (2009) 1790:1448–52. doi: 10.1016/j.bbagen.2009.05.010
89. Howard MT, Carlson BA, Anderson CB, Hatfield DL. Translational Redefinition of UGA Codons is Regulated by Selenium Availability. J Biol Chem (2013) 288:19401–13. doi: 10.1074/jbc.M113.481051
90. Vunta H, Davis F, Palempalli UD, Bhat D, Arner RJ, Thompson JT, et al. The Anti-Inflammatory Effects of Selenium are Mediated Through 15-Deoxy-Δ12,14-Prostaglandin J2 in Macrophages. J Biol Chem (2007) 282:17964–73. doi: 10.1074/jbc.M703075200
91. Shirey KA, Cole LE, Keegan AD, Vogel SN. Francisella Tularensis Live Vaccine Strain Induces Macrophage Alternative Activation as a Survival Mechanism. J Immunol (2008) 181:4159–67. doi: 10.4049/jimmunol.181.6.4159
92. Samaniego S, Marcu KB. Ikkβ in Myeloid Cells Controls the Host Response to Lethal and Sublethal Francisella Tularensis LVS Infection. PloS One (2013) 8:e54124. doi: 10.1371/journal.pone.0054124
93. Fortier AH, Polsinelli T, Green SJ, Nacy CA. Activation of Macrophages for Destruction of Francisella Tularensis: Identification of Cytokines, Effector Cells, and Effector Molecules. Infect Immun (1992) 60:817–825. doi: 10.1128/iai.60.3.817-825.1992
94. Parsa KVL, Butchar JP, Rajaram MVS, Cremer TJ, Gunn JS, Schlesinger LS, et al. Francisella Gains a Survival Advantage Within Mononuclear Phagocytes by Suppressing the Host Ifnγ Response. Mol Immunol (2008) 45:3428–37. doi: 10.1016/j.molimm.2008.04.006
95. Edwards JA, Rockx-Brouwer D, Nair V, Celli J. Restricted Cytosolic Growth of Francisella Tularensis Subsp. Tularensis by IFN-γ Activation of Macrophages. Microbiology (2010) 156:327–39. doi: 10.1099/mic.0.031716-0
96. Rabadi SM, Sanchez BC, Varanat M, Ma Z, Catlett SV, Melendez JA, et al. Antioxidant Defenses of Francisella Tularensis Modulate Macrophage Function and Production of Proinflammatory Cytokines. J Biol Chem (2016) 291:5009–21. doi: 10.1074/jbc.M115.681478
97. McCaffrey RL, Schwartz JT, Lindemann SR, Moreland JG, Buchan BW, Jones BD, et al. Multiple Mechanisms of NADPH Oxidase Inhibition by Type A and Type B Francisella Tularensis. J Leukoc Biol (2010) 88:791–805. doi: 10.1189/jlb.1209811
98. Melillo AA, Mahawar M, Sellati TJ, Malik M, Metzger DW, Melendez JA, et al. Identification of Francisella Tularensis Live Vaccine Strain CuZn Superoxide Dismutase as Critical for Resistance to Extracellularly Generated Reactive Oxygen Species. J Bacteriol (2009) 191:6447–56. doi: 10.1128/JB.00534-09
99. Honn M, Lindgren H, Bharath GK, Sjöstedt A. Lack of OxyR and KatG Results in Extreme Susceptibility of Francisella Tularensis LVS to Oxidative Stress and Marked Attenuation In Vivo. Front Cell Infect Microbiol (2017) 191. doi: 10.3389/fcimb.2017.00014
100. Ziveri J, Barel M, Charbit A. Importance of Metabolic Adaptations in Francisella Pathogenesis. Front Cell Infect Microbiol (2017) 7. doi: 10.3389/fcimb.2017.00096
101. Best A, Abu Kwaik Y. Nutrition and Bipartite Metabolism of Intracellular Pathogens. Trends Microbiol (2019) 27:550–61. doi: 10.1016/j.tim.2018.12.012
102. Khoso PA, Pan T, Wan N, Yang Z, Liu C, Li S. Selenium Deficiency Induces Autophagy in Immune Organs of Chickens. Biol Trace Elem Res (2017) 177:159–68. doi: 10.1007/s12011-016-0860-7
103. Covarrubias AJ, Aksoylar HI, Horng T. Control of Macrophage Metabolism and Activation by mTOR and Akt Signaling. Semin Immunol (2015) 27:289–296. doi: 10.1016/j.smim.2015.08.001
104. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
105. Armstrong BA, Betzold RD, May AK. Sepsis and Septic Shock Strategies. Surg Clinics North America (2017) 97:1339–79. doi: 10.1016/j.suc.2017.07.003
106. Forceville X, Vitoux D, Gauzit R, Combes A, Lahilaire P, Chappuis P. Selenium, Systemic Immune Response Syndrome, Sepsis, and Outcome in Critically Ill Patients. Crit Care Med (1998) 26:1536–44. doi: 10.1097/00003246-199809000-00021
107. Asci A, Surmeli-Onay O, Erkekoglu P, Yigit S, Yurdakok M, Kocer-Gumusel B. Oxidant and Antioxidant Status in Neonatal Proven and Clinical Sepsis According to Selenium Status. Pediatr Int (2015) 57:1131–7. doi: 10.1111/ped.12698
108. Prauchner CA. Oxidative Stress in Sepsis: Pathophysiological Implications Justifying Antioxidant Co-Therapy. Burns (2017) 43:471–85. doi: 10.1016/j.burns.2016.09.023
109. Angstwurm MWA, Schottdorf J, Schopohl J, Gaertner R. Selenium Replacement in Patients With Severe Systemic Inflammatory Response Syndrome Improves Clinical Outcome. Crit Care Med (1999) 27:1807–13. doi: 10.1097/00003246-199909000-00017
110. Angstwurm MWA, Engelmann L, Zimmermann T, Lehmann C, Spes CH, Abel P, et al. Selenium in Intensive Care (SIC): Results of a Prospective Randomized, Placebo-Controlled, Multiple-Center Study in Patients With Severe Systemic Inflammatory Response Syndrome, Sepsis, and Septic Shock. Crit Care Med (2007) 35:118–26. doi: 10.1097/01.CCM.0000251124.83436.0E
Keywords: selenium, redox, intracellular bacteria, tularemia, innate immunity
Citation: Markley RL, Restori KH, Katkere B, Sumner SE, Nicol MJ, Tyryshkina A, Nettleford SK, Williamson DR, Place DE, Dewan KK, Shay AE, Carlson BA, Girirajan S, Prabhu KS and Kirimanjeswara GS (2021) Macrophage Selenoproteins Restrict Intracellular Replication of Francisella tularensis and Are Essential for Host Immunity. Front. Immunol. 12:701341. doi: 10.3389/fimmu.2021.701341
Received: 27 April 2021; Accepted: 24 September 2021;
Published: 29 October 2021.
Edited by:
Philip Calder, University of Southampton, United KingdomReviewed by:
Paul Copeland, Rutgers Biomedical and Health Sciences, United StatesCopyright © 2021 Markley, Restori, Katkere, Sumner, Nicol, Tyryshkina, Nettleford, Williamson, Place, Dewan, Shay, Carlson, Girirajan, Prabhu and Kirimanjeswara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Girish S. Kirimanjeswara, Z3NrMTI1QHBzdS5lZHU=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.