
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 July 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.694928
This article is part of the Research Topic Negative Regulators of Innate Immunity and their Role in Host Responses to Injury and Infection View all 13 articles
 Hongbin Wang1,2,3*
Hongbin Wang1,2,3* Mengyao Liu1
Mengyao Liu1Complement C4, a key molecule in the complement system that is one of chief constituents of innate immunity for immediate recognition and elimination of invading microbes, plays an essential role for the functions of both classical (CP) and lectin (LP) complement pathways. Complement C4 is the most polymorphic protein in complement system. A plethora of research data demonstrated that individuals with C4 deficiency are prone to microbial infections and autoimmune disorders. In this review, we will discuss the diversity of complement C4 proteins and its genetic structures. In addition, the current development of the regulation of complement C4 activation and its activation derivatives will be reviewed. Moreover, the review will provide the updates on the molecule interactions of complement C4 under the circumstances of bacterial and viral infections, as well as autoimmune diseases. Lastly, more evidence will be presented to support the paradigm that links microbial infections and autoimmune disorders under the condition of the deficiency of complement C4. We provide such an updated overview that would shed light on current research of complement C4. The newly identified targets of molecular interaction will not only lead to novel hypotheses on the study of complement C4 but also assist to propose new strategies for targeting microbial infections, as well as autoimmune disorders.
Complement system plays a pivotal role in human innate immunity defending microbial infections, eliminating foreign pathogens, and maintaining tissue homeostasis. The activation of the complement system induces the increased production of cytokines, chemokines, and other innate defense molecules. In addition, complement activation fragments (e.g., anaphylatoxin C3a and C5a) significantly increased the recognition of antigens by follicular dendritic cells and B cells and induced the humoral adaptive immune response and production of antibodies and reactive T cells. Moreover, complement system functions as an effector on the clearance of soluble immune complexes and cell debris, which otherwise could induce an immune response against auto antigens and potentially trigger autoimmunity (1–3). Deficiency or dysfunction of the complement system could cause infections in adult patients (4) and also predispose individuals to autoimmune diseases, such as rheumatoid arthritis (RA), systemic sclerosis, and systemic lupus erythematosus (SLE) (5).
Complement component C4 (Mw = ~200 kDa), an essential component in complement system, plays an indispensable role in the activation of classical and lectin complement cascades. It is a disulfide-bonded three-chain glycoprotein, consisting of an α-chain (95 kDa), a β-chain (75 kDa), and a γ-chain (30 kDa) (Figure 1) (6, 7). In the process of the activation of classical complement pathway, C1q from C1 complex [C1q-(C1r)2-(C1s)2] recognizes antigen–antibody immune complexes or certain membrane-bound structures, e.g. C-reactive protein (CRP) or lipopolysaccharides (LPS), resulting in the transition from C1s zymogen to an active C1s repositions, which would be able to interact with sulfotyrosine residues on C4 (8, 9). Similar to the activation of classical complement pathway, the lectin complement pathway is activated by complex carbohydrate structures and mediated via recognition molecules as mannan binding lectin (MBL), ficolins, and collectin 10/11, leading to the activation of mannan-associated serine protease-2 (MASP-2), which relies heavily on its active sites, two complement control protein (CCP) domains, and the serine protease (SP) domain for the efficient binding and cleavage of C4 (10–12). As shown in Figure 1, the activated C1s and MASP-2 from classical and lectin pathways respectively cleaves the amino terminal part of the α-chain at a single site of complement C4 to generate C4a fragment peptide (9 kDa) and C4b (195 kDa) (13). C4b binds to target surface via its reactive thioester, which can be inactivated to an intermediate form iC4b by proteolytic cleavage by the serine protease factor I together with co-factor CD46 (14, 15). iC4b is further cleaved to thioester linked C4d (45 kDa) and soluble C4c (146 kDa), which can be used as a biomarker for complement activation from classical and lectin pathways. Both classical and lectin pathways lead to further activation of C2 to generate C3 convertase C4b2a, which will activate C3 to generate C3a and C3b. C3 convertase binds to C3b to form C5 convertase that will cleave C5 to generate C5a and C5b. C5b binds to C6, C7, C8, and C9 to form membrane attack complexes (MAC) C5b-9 that are formed on the surface of pathogen cell membranes. Comparing crystal and solution structures of C4b with its paralog C3b, their conformations are shown surprisingly conserved (16). Further study revealed that the C3 convertases (C4b2a vs. C3bBb) from the classical/lectin and alternative pathways are also strikingly similar, which is in agreement with their identical functions in the cleavage of the downstream complement proteins C3 and C5 (17).
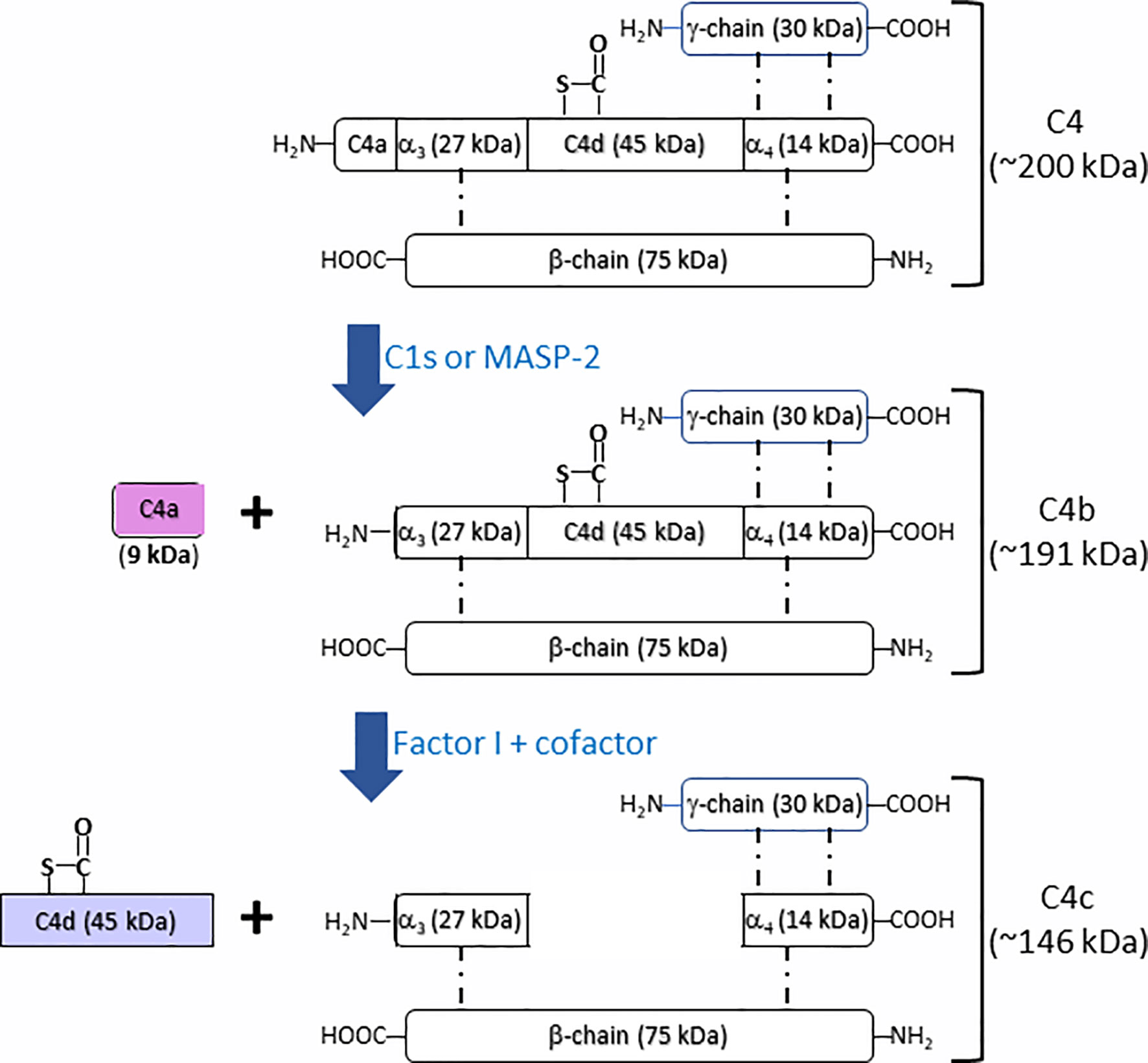
Figure 1 Schematic illustration of fragmentation of complement C4 activation. Complement C4 (~200 kDa) is activated by the serine protease C1s or MASP2 from classical and lectin complement pathway, respectively, to form the activation fragments C4a (~9 kDa) and C4b (~191 kDa). C4b (~191 kDa) is then inactivated and cleaved by the factor I together with cofactors to generate intermediate product iC4b and then further to generate the thioester linked C4d (~45 kDa) and C4c (~146 kDa).
The complete or partial deficiency of complement C4 results in the increased risk of infection and autoimmune diseases. A plethora of studies demonstrated that complement C4 plays an essential role in defensing microbial infection. It is also well established that the complete or partial deficiency of complement C4 is associated with the increased susceptibility to infections (18–23). In addition, the deficiency of complement C4 could lead to various autoimmune diseases (24–33). The reduced concentrations of C4 protein and the reduced serum complement activity occur with the active disease in SLE (25, 34), as well as in infections (35).
In this review, we will look into the updated studies on the role of complement C4 in infectious diseases and autoimmune disorders. In this way, we aim to elucidate and update the functions of complement C4 in infectious diseases and autoimmune disorders, trying to highlight the important role of complement C4 as a potential intervention target for the management of those disorders.
The human complement C4 gene (C4A and C4B genes) locus is located in the highly polymorphic major histocompatibility complex (MHC) class III gene region on chromosome 6, which could be a short form (C4S, 14.6 kb) or a long form (C4L, 21 kb), depending on the absence or the presence of the 6.36 kb endogenous retroviral sequence HERV-K(C4) in intron 9 of human C4 genes. Three quarters of C4 genes harbor the 6.36-kb endogenous retrovirus HERV-K (C4) (36). Each human C4 gene has 41 exons, which codes for a 5.4 kb transcript. C4 gene lies within a unit of four consecutive genes known as an RCCX module, which stands for the serine/threonine nuclear protein kinase RP, Complement component C4, steroid 21-hydroxylase CYP21, and extracellular matrix protein tenascin TNX (RP-C4-CYP21-TNX) (RCCX) (37–44). An elevated level of genomic copy number variations (CNV) was shown in MHC region III, supposedly to present immunologic diversity (45). The duplication of these four genes occurs as a module in the class III region of a haplotype for the MHC. The gene copy number (GCN) of C4A genes varies from 0 to 5 and GCN of C4B genes varies from 0 to 4. The highest total C4 gene dosage reported is 7 (46). It took a long time for scientists to make it clear on the genetic diversity of human complement C4. The initial model was proposed as a single locus of codominant alleles for C4A and C4B, and later two-locus or C4A-C4B models dominated the complement field for about two decades. Extensive molecular and genetic studies have now provided a clear definition of genetic structures that are responsible for C4 isotypes (C4A and C4B) protein expression. Complement C4 protein exists as two isotypes, C4A and C4B, which are encoded by the C4 genes (C4A or C4B gene), and share >99% sequence identities. Five nucleotide variations located in exon 26 confer to four isotype-specific amino acid substitutions at positions 1101–1106 (PCPVLD for C4A and LSPVIH for C4B) and the major structural and functional differences between the C4A and C4B isotypes (47). C4A is named after its acidity and migrates faster in agarose gel electrophoresis as compared to C4B that is basic and migrates slower (47–51). In addition, C4A and C4B are highly polymorphic with more than 40 different alleles, gene duplications, and “null alleles” (52–55). C4A is generally associated with the Rodgers (Rg) blood group antigens and is more reactive with immune complex or the targets containing free amino groups, whereas C4B is generally associated with the Chido (Ch) blood group antigens and is more affinity to hydroxyl groups (56). It was revealed that C4A has a longer half-life in plasma as compared to C4B, suggesting a role of C4A in the clearance of the immune complex and a role of C4B for membrane attack complex formation and the defense against bacterial pathogens (57). The individuals with long C4 genes (C4L) have lower serum levels of complement C4 as compared with short C4 genes (C4S) (36). C4 gene copy number variations (CNV) are correlated to the serum levels of complement C4 protein and low C4 GCNs predisposes individuals with various disease susceptibility (58). Low copy numbers or the deficiency of C4 genes was reported to be one of the strongest risk factors associated with several immune disorders, such as SLE, chronic central serous chorioretinopathy, Behçet’s disease, and Vogt-koyanagi-Harada disease (25, 58–63). It was also reported that the deficiency of either C4A or C4B has been associated with the increased susceptibility of infections (18, 64, 65). Interestingly, it was reported by Bay et al. that low C4 gene copy numbers (< 4 total copies of C4 genes) are associated with superior graft survival in patients transplanted with a deceased donor kidney (66). A comprehensive review on the variations of C4 genetic structures and proteins was presented by Blanchong CA et al. (36).
Other causes than genetic variations also can affect the expression or the function of complement C4. Early studies by Goldman et al. in cultured guinea pig peritoneal cells demonstrated that complement component can be regulated by short-term treatment in vivo or in vitro with monospecific antibody to individual complement components can have long-term effects on the production of those components. Antibody treatment induced specific suppression of C4 in peritoneal cell monolayers. Further studies revealed that long-term C4 suppression is actively maintained by a soluble suppressor factor (FsC4) (67–70). Most of those experiments were carried out in vitro cellular models from guinea pig. It is still unclear whether this observation can be replicated in human.
Dysregulation of classic and lectin complement pathways that complement C4 participates in can lead to complement-mediated autoimmune diseases. C4 nephritic factor (C4Nef), first described by Halbwachs et al. in 1980, is an autoantibody to C3 convertase (C4b2a). C4Nef can prolong the half-life of C3 convertase by stabilizing C4b2a and protects C4b2a against decay dissociation by C4 binding protein (C4BP). Multiple clinical studies demonstrated that C4NeF was associated with post-infectious acute glomerulonephritis, systemic lupus erythematosus, chronic proliferative glomerulonephritis and hypocomplementemic membranoproliferative glomerulonephritis (MPGN), and meningococcal disease (71–76).
A recent study by Battin et al. using in vitro binding screening demonstrated that Neuropilin-1 (NRP1) acts as a receptor for complement split products, such as C4d, C3d, and iC3b. NRP1 is a highly conserved type 1 transmembrane protein that is involved in the tumorigenesis, the development of cardiovascular, and nervous systems through the interaction with vascular endothelial growth factor (VEGF) and semaphoring 3A (Sema3A). NRP1 is also expressed in murine immune cells and serves as a marker for mouse Treg cells. Interestingly, NRP1 was demonstrated to bind C4d in a concentration-dependent and saturable manner. These data demonstrated NRP1 functions as a receptor for C4d that is covalently bound to target surfaces during complement activation, suggesting that NRP1 might be involved in regulation of the process of infections and autoimmune disorders by targeting the split product from classical or lectin complement pathway (77).
Complement C4 is involved in the activation of both classical and lectin complement pathways. The classical pathway of complement system is crucial for anti-microbial defense through anti-pathogen antibody, which recruits C1 complex and initiates a cleavage cascade involving C4, C2, C3, and C5 and accomplishing microbial clearance. In addition, lectin complement pathway is also involved in the anti-microbial defense. Recent study revealed that loss of classical pathway results in rapidly progressing septicemia and impaired macrophage activation, suggesting that the classical pathway is the dominant pathway for activation of the complement system during complement innate immunity to S. pneumoniae. In response to microbial pathogens, lectin pathway is activated as an innate immune response through direct binding to bacterial surface sugar components. In contrast, the classical pathway was an effector of adaptive immune response through activation of antibody–antigen complexes on bacterial surfaces and plays a vital role partially targeted by the binding of natural IgM to bacteria (21).
An early research work by Schifferli et al. demonstrated that C4A isoform of complement C4 was more efficient than C4B isoform in the processing of immune complexes in humans. In contrast, hemolysis by C4B isoform was more efficient than by C4A isoform, suggesting that both C4 isoforms are complementary (78). A recent study by Liesmaa et al. demonstrated that homozygous C4A deficiency in patients was associated with the increased prevalence of lymphomas, celiac disease, and autoimmune disease SLE. In the same study, homozygous C4B deficiency in patients was documented to be linked with the drug intolerance and various post-infectious symptoms. Homozygous C4B deficiency alone is not considered as a significant factor in causing invasive infection (79). From the multiple studies from different laboratories, it seems still debatable in terms of the role of homozygous C4A or C4B deficiency in infection-proneness of an individual (64, 79–82).
Complement interfering protein (CIP) expressed on the surface of group B. Streptococcus (GBS) enables cell adhesion and penetration and impedes innate and adaptive immune responses. It was found that CIP was able to interact with the human C4b ligand and to interfere with the classical- and lectin-complement pathways (83). Clinical Staphylococcus aureus (S. aureus) strains can recruit complement regulator C4-binding protein (C4BP) to S. aureus surface to inhibit C4 complement effectors through binding significant amounts of the C4BP from serum. The complex (S. aureus-bound C4BP) functions as a cofactor for factor I-mediated C4b cleavage to iC4b and C4d, which was used as a strategy by S. aureus for immune evasion (84).
A recent study revealed that gram-negative Bordetella pertussis could evade the attack from the human complement system by releasing virulent protein Vag8 of B. pertussis. Endogenously secreted and recombinant Vag8 can inhibit complement deposition on the bacterial surface at the level of C4b. The binding of C1 inhibitor (C1-inh) to C1s, C1r, and MASP-2 was disrupted by the association of Vag8 with human C1-inh, which will free active proteases to cleave C2 and C4 away from the bacterial surface, revealing a mechanism of the unique complement evasion strategy of B. pertussis (85).
An alkaline protease Alp1p secreted from A. fumigatus mycelia can facilitate early immune evasion by deactivating the complement defense in the human host, either by directly cleaving the complement components C3, C4, and C5 or by cleaving them to a form that is further fragmented by other proteases (86, 87).
Some small organisms other than microbials are also involved in the activation or the inhibition of complement systems. For examples, complement activation-inhibition substance from maggot excretions, which splits complement proteins C3 and C4 in a cation-independent manner, could provide a novel treatment for several diseases that result from the activation of complement system (88). ES-62, a protein with an N-linked glycan linked with phosphorylcholine (PCh) produced from parasitic nematodes, was bound to C-reactive protein (CRP) in normal human serum. C1q can capture ES-62-CRP to form a larger complex ES-62-CRP-C1q in serum. Following CRP interaction, ES-62 was able to deplete early complement component C4 and inhibit classical pathway activation (89). The immune evasion strategies used by those microorganisms aforementioned were summarized in Table 1.

Table 1 The evasion mechanisms of microorganisms from the attack of innate immunity of complement system by targeting complement C4.
It is now becoming apparent that microbial organisms generate various mechanisms to defend the attacks from innate immunity of complement system. Elucidation of those mechanisms will potentially provide strategies to treat microbial infectious diseases, as well as to explore the anti-complement therapeutic interventions.
There are extensive studies on the role of complement system in viral infections (93–95). A study by Bugdaci et al. was carried out in 143 patients on the relationship of serum complement C4 levels and chronic hepatitis B (CHB) infection with high transaminase level. Serum C4 levels in patients with CHB with high transaminase level were found significantly lower. In addition, Child score in patients with cirrhosis inversely correlated with C4 levels, suggesting that the levels of complement C4 in plasma significantly correlate with liver biopsy findings and may be a useful indicator of disease activity and/or damage in patients with CHB with high transaminase levels (96).
A study by the same research group of Bugdaci et al. on 100 patients with chronic hepatitis C found that complement C4 levels showed significant correlation with alanine aminotransferase but could not find any relationship between serum complement C4 level and fibrosis (97). It remains unanswered why C4 activity was significantly lower in patients with chronic hepatitis C virus (HCV) infections. One speculation could be due to excessive activation of C4 protein by the activation of classical and lectin complement pathways during HCV infections. Several studies evaluated the expression of C4 in terms of anti-HCV therapeutic response and disease progression in chronic hepatitis C (CHC) patients. The studies revealed that mRNA and protein levels of complement C4 were significantly increased after anti-HCV treatment. A positive alteration in C4 level represents as an independent predictor for treatment response and reflects viral clearance after anti-HCV therapy in CHC patients (98–100). Further studies revealed that hepatitis C virus proteins [HCV core; non-structural (NS) 5A] render transcriptional suppression of the expression of complement C4. Liver biopsy specimens from chronic HCV patients displayed significantly lower levels of complement C4 mRNA as compared to the liver tissue samples from patients with other types of liver disease. HCV core protein was found to decrease the expression of upstream stimulating factor 1 (USF-1), a transcription factor essential for basal C4 expression. In addition, HCV NS5A protein can inhibit the expression of interferon regulatory factor 1 (IRF-1), which is important for IFNγ-induced complement C4 expression. These results highlight the roles of HCV proteins in establishing a chronic infection through the regulation of innate immunity by affecting the expression of complement C4 (90).
Another study by Mawatari et al. demonstrated that HCV NS3/4A protease could cleave the γ-chain of complement C4 in a concentration-dependent manner, suggesting that complement C4 is a novel cellular substrate of HCV N3/4A protease, which reveals new insight into the mechanisms underlying persistent HCV infection (91).
Natural killer (NK) cells have been revealed to contribute to regulating complement synthesis. Studies using co-culture of NK cells (NK3.3) with human hepatoma cells (Huh7.5) expressing HCV core or NS5A protein revealed a significantly increased synthesis of complement C4 and C3 via increased specific transcription factors. The regulatory activity is mediated through a direct interaction between the hepatocyte protein major histocompatibility complex class I-related chains A and B (MICA/B) and NKG2D on NK cells. However, when NK cells were co-cultured with Huh7.5 cells infected with cell culture-grown HCV, complement C4 and C3 synthesis was impaired. MICA/B expression in HCV-infected hepatocytes was found to be repressed during co-culture because the HCV-associated expressions of NS2 and NS5B proteins can disable a crucial receptor ligand in infected hepatoma cells, resulting in the disability of infected cells to respond to stimuli from NK cells to up-regulate the expression of complement C3 and C4 (92). This piece of data revealed that HCV synthesizes the proteins that can down-regulate complement C4 expression to evade the attack from complement systems.
Flavivirus infection, such as West Nile virus (WNV) and Dengue virus (DENV), was restricted through an antibody-independent fashion. N-linked glycans on the structural proteins of flaviviruses was recognized by mannose-binding lectin (MBL), resulting in neutralization and efficient clearance via a C3- and C4-dependent mechanism that applied both the canonical and bypass complement lectin activation pathways, which recognizes terminal mannose-containing carbohydrates on the viruses (101). A recent study by Avirutnan et al. demonstrated that flavivirus non-structural (NS)1 protein from dengue virus (DENV), West Nile virus (WNV), and yellow fever virus (YFV) binds to C4 to enhance cleavage of C4 and reduce C4b deposition and C3 convertase (C4bC2a) activity that confers to immune evasion function for the viruses (93).
Interestingly, Puumala (PUUV) hantavirus triggers complement system activation via the alternative pathway, which is complement C4-independent, causing the increase of sC5b-9 and the decrease of C3. In the acute stage of PUUV infection, the level of complement activation correlates with disease severity, indicating that complement activation may contribute to the pathogenesis of acute PUUV infection (102).
A recent study by Bottermann et al. revealed a novel antiviral mechanism that is C4-dependent and late-acting complement components-independent. C4 inhibits human adenovirus infection by the deposition of cleaved C4b on capsid, which inhibits it disassembly, preventing endosomal escape and cytosolic access (103). The mechanisms that viruses applied to downregulate the expression or the function of complement C4 are summarized in Table 1.
In the midst of pandemic with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)/COVID-19. The infection involves in multiple organs and cause striking elevations in pro-inflammatory cytokines and high risk of thrombosis. Numerous postmortem studies have revealed deposits of complement fragments on interalveolar endothelial cells, high incidence of venous thromboembolism (VTE), and diffused microvascular thrombi with endothelial swelling with a thrombotic microangiopathy (TMA). Preclinical studies with SARS-CoV-1 and MERS-CoV, which have significant homology to SARS-CoV-2, confirm that complement activation is not only linked to virus related organ damage but also is possible causative (104). In mouse models with infection with MERS-CoV or SARS-CoV-1, increased tissue deposition of C5b-9, C3b, and C4d and correlation with severity of injury were observed. Given the fact that deficiency of complement C3, C4, and factor B can protect mice from virus caused by lung injury, classical, lectin, and alternative complement pathways might be involved in mediating SARS-CoV-1 or SARS-CoV–triggered lung injury (105, 106). In the few published post-mortem studies of COVID-19 patients, the increased deposits of C3b, MBL, MASP-2, C4b, and C5b-9 were observed (107, 108). It shows excessive activation of lectin pathway, which is in line with the fact that the spike protein in SARS-CoV-2 is heavily glycosylated with L-fucose and mannose, which provides recognition sites for MLB binding and causes activation of lectin pathway (109). There is no doubt that complement C4 will be hyper-activated in SARS-CoV-2 infection. Is there any relationship between the complement C4 activation and COVID-19 infection caused endothelial swelling and diffused microvascular thrombi that resemble TMA? We would speculate that the hyperactivation of lectin pathway might cause endothelial disruption that might be one of the mechanisms to induce microvascular thrombi in COVID-19 patients. It remains to be evaluated how the lectin pathway and complement C4 activations cause endothelial swelling and diffused microvascular thrombi.
A recent study by Romano et al. revealed that anti–interleukin-6 receptor monoclonal antibody (Tocilizumab) could dramatically decrease serum level of complement C4 in rheumatoid arthritis patients. Neither circulating immunocomplexes nor any patients ever displaying clinical features of immunocomplex diseases was found. The study concluded that C4 consumption is because of the direct action of the drug rather than immunocomplex-induced complement activation (110).
Histone H3 and H4 can be released from the damaged or lysed cells. One recent study by Qaddoori et al. revealed that histone H3 and H4 strongly bind to C4b region of complement C4, result in significant inhibition of classical and mannose-binding lectin pathways. Histone H3 and H4 did not affect the cleavage of C4 to C4a and C4b, indicating a possible natural feedback mechanism to prevent the excessive injury of host cells by the inhibition of complement activation by histones (111).
A recent study by Vogt et al., using highly specific antibody against a cleavage neoepitope in C4d, identified pigment epithelium-derived factor (PEDF) from synovial fluid of rheumatoid arthritis patients as an activator of classical complement pathway, which belongs to the serine proteinase inhibitor family. C1q protein can bind PEDF, in particular, head regions of C1q, which is known to interact with other activators of the classical pathway. The results suggested PEDF activated classical complement and might mediate inflammatory processes in joint (112). The interactions of virus, bacteria, and some pathological conditions causing the consumption or inhibition of complement C4 through classic or lectin complement pathway are illustrated in Figure 2.
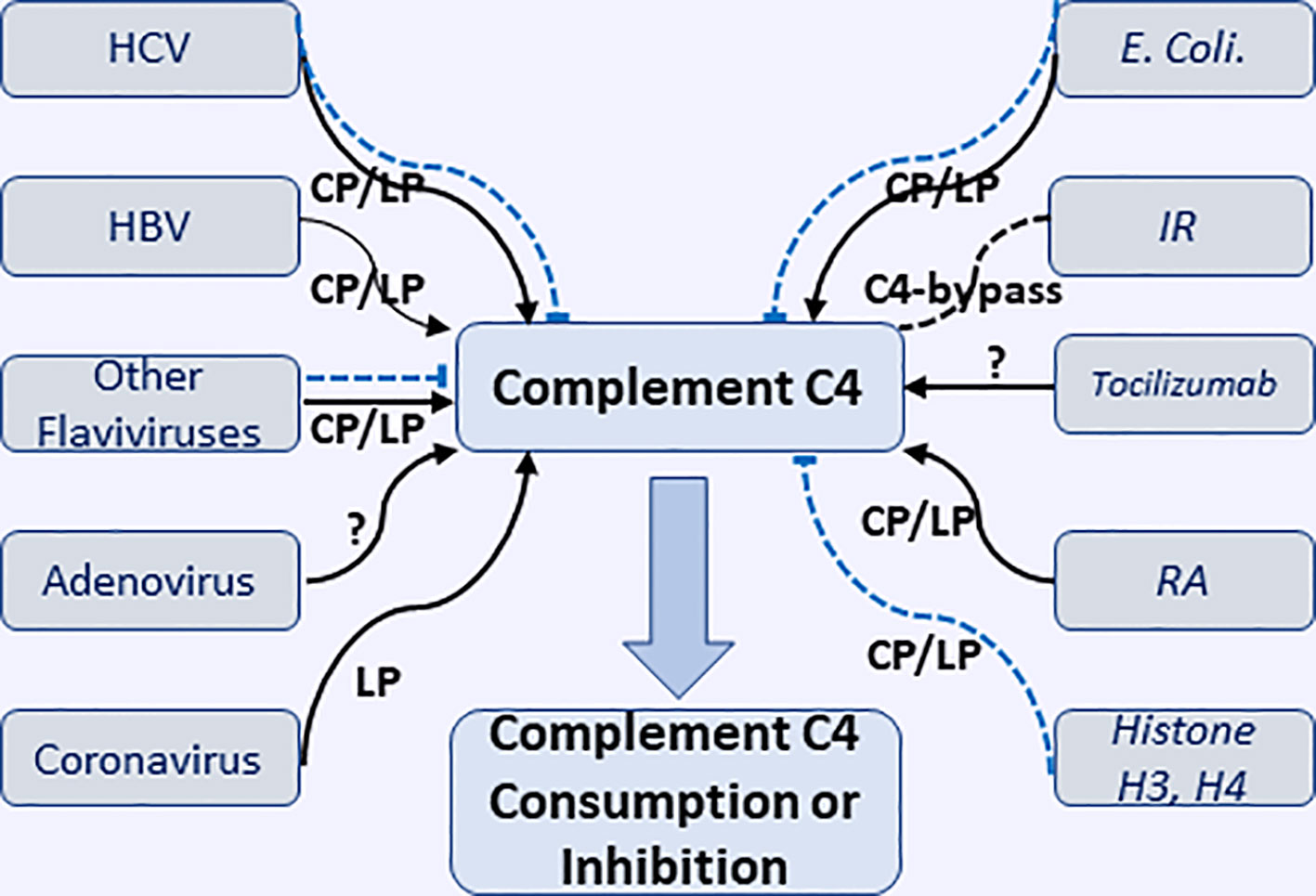
Figure 2 Interactions of various microorganisms (including viruses and E. coli.) and pathological conditions with complement C4 to cause consumption or inhibition of complement C4 through classical, lectin, and undefined pathways. HCV, hepatitis V virus; HBV, hepatitis B virus; IR, ischemia reperfusion; RA, rheumatoid arthritis. Black solid arrow represents the activation of complement C4; Blue dash line represents the inhibition of complement C4.
Using computational approach (protein-protein docking and molecular dynamics simulation), a recent study tried to understand Trypsin (Tryp)-mediated C4 activation by comparing with the co-crystalized structure of C4-MASP2. Comparative analysis of C4 alone, C4-Tryp, and C4-MASP2 discovered the impact of Tryp on C4 was like that of MASP2. These studies define the role of sessile loop in the interaction with serine domain, which could be beneficial to understand the interactions of complement C4 with other complement components (113).
It seems that sometimes the activation of three complement pathways is not clear-cut. Recent studies in mice established that the complement activation via the alternative pathway requires the presence of C4 and MBL proteins and the complement activation by Cryptococcus spp. can take place via multiple complement pathways (114, 115).
Although complement C4 does not directly participate in the activation of alternative complement pathway (AP), several early studies from the 1980s to 1990s of last century demonstrated that C4b generated from classical pathway activation could trigger the alternative pathway without involvement of complement C2 (116–118). It was reported that MBL can activate complement C3 and AP without the involvement of MASP-2, C2, or C4 (119, 120). A recent study by Tateishi and Matsushita demonstrated that upon the attachment to serogroup C-specific oligosaccharide from Salmonella, in contrast to that MBL activates the alternative pathway via a C2-bypass pathway without the involvement of MASP-2, C2 or C4, mannan-bound MBL can activate the alternative pathway via a C2-bypass pathway that requires both MASP-2 and C4, suggesting that there may be two distinct MBL-mediated C2-bypass activation of alternative complement pathway, depending on the ligands to which MBL binds (121). It seems that there are some issues related to those in vitro assays. First, those MBL preparations could possibly have the trace contamination with MASP-2. Another question is how pure the serum preparations with the depleted MBL, C2, or C4 can be. The mechanism of MLB mediated C2-bypass AP activation remains to be determined and further studies are needed to elucidate the molecular base of MBL-mediated C2-bypass pathways as indicated in the paper.
Several studies suggested that MLB complement pathway could activate C3 or C5 through C4-bypass mechanisms (119, 122–125). The study in a mouse model by Schwaeble et al. demonstrated that in the absence of complement C4, in vitro lectin pathway-mediated activation of C3 requires MASP-2, C2, and MASP-1/3. In a model of transient myocardial postischemic reperfusion injury (IRI), comparing to their wild-type littermates, infarct volumes of MASP-2-deficient mice were smaller. However, mice deficient in complement C4 were not protected, the observation implies the presence of a previously undocumented C4-bypass and lectin pathway-dependent mechanism. As monoclonal Antibody-based inhibitors of MASP-2 and MASP-2 deficiency can also protect mice from gastrointestinal IRI, suggesting the benefit of anti-MASP-2 antibody therapy in reperfusion injury and other lectin pathway-mediated disorders (126). In this study, it was unclear how the correlation between complement C3 activation by C4-bypass lectin pathway and the disease state of infarct volume. IRI may not be due to the complement C3 activation, but could be attributed by MASP-2- or MASP-1-induced direct activation of coagulation systems, leading to the formation of a fibrin clot (127, 128). Lectin pathway complement activation plays a critical role in contributing to ischemia reperfusion (IR) injury. One recent study in a mouse model corroborates the effect of MASP-2, an essential enzyme for lectin pathway, which mediates tissue injury and renal ischemia reperfusion injury independent of complement C4 (129). C2- and C4-bypass lectin pathways activation are depicted in Figure 3.
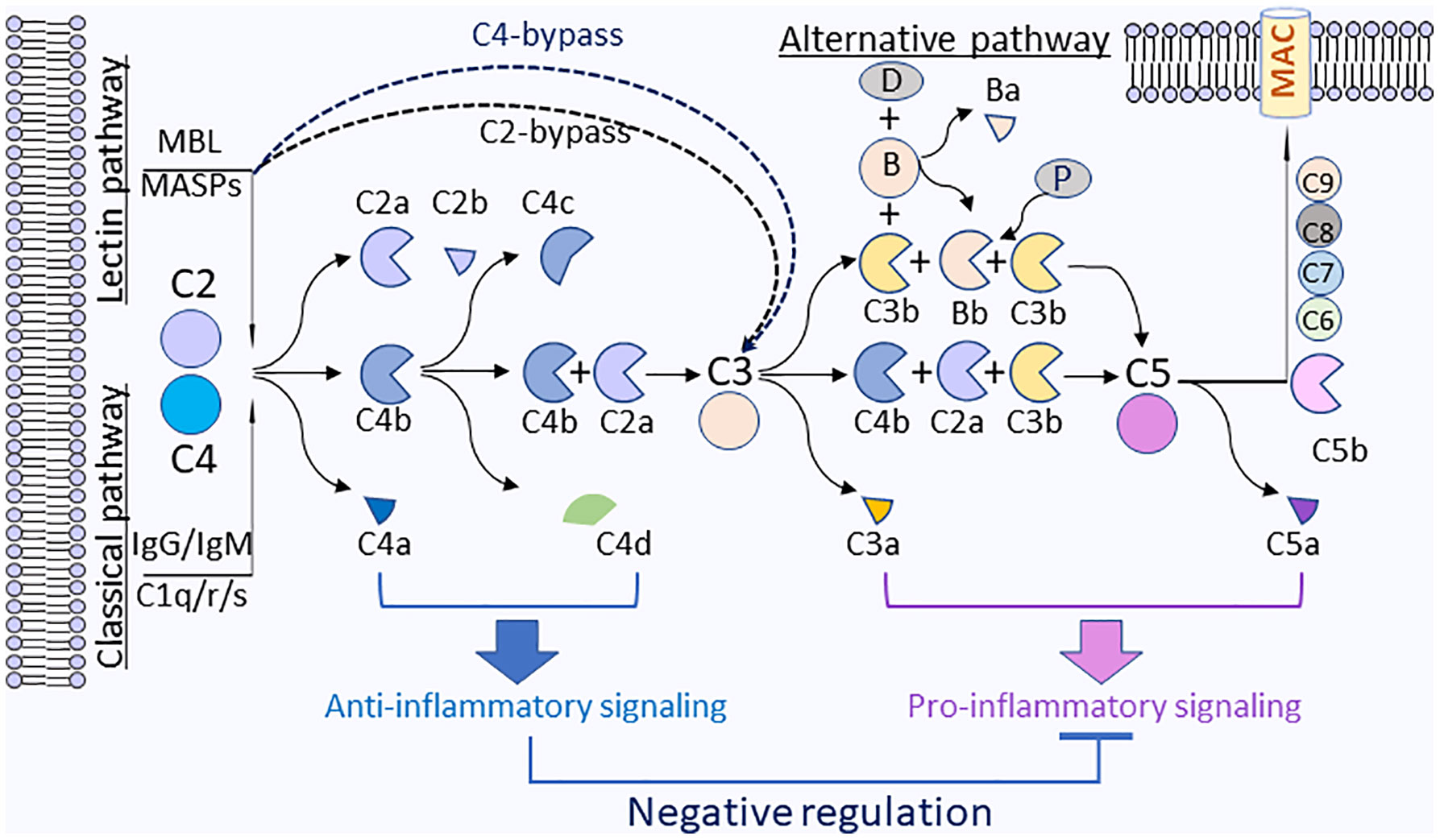
Figure 3 Anti-inflammatory functions by complement C4 activation fragments C4a and C4d. On the surface of pathogens, the activation of complement C4 is triggered through classical (antibody) or lectin (sugar) pathways that will activate C1s or MAPSs, which will rapidly cleave C4 to generate C4a and C4b. C4b will be further fragmented by factor I and cofactors to generate C4d and C4c. C4b will associate with C2a to form a complement C3 cleavage enzyme (C3 convertase), C4b2a, which will cleave C3 to generate C3a and C3b. C3b will be associated with C4b2a to converge to C5 convertase (C4bC2aC3b), which will cleave C5 to generate C5a and C5b. C5b will associate with C6, C7, C8, and C9 to form membrane attack complexes (MAC) C5b-9 on the surface of pathogens. For the alternative pathway, C3b is spontaneous C3 turnover or generated by classical or lectin pathways. C3b bound to factor B (B). The complex is converted by factor D (D) to C3-cleaving enzyme C3bBb that is stabilized by properdin (P) and further form C3bBbC3b, which can cleave C5 to generate C5a and C5b. Plasma membrane (blue) represents the surface of pathogen cells.
Although classical and the alternative pathways can still be activated, MASP-2 deficient mice fail to opsonize Streptococcus pneumoniae in the none-immune host and therefore are highly susceptible to pneumococcal infection. Mouse ficolin A, human L-ficolin, and collectin 11 in both species, but not mannan-binding lectin (MBL), are the pattern recognition molecules that drive lectin pathway activation on the surface of S. pneumoniae. pneumococcal opsonization in the absence of complement C4. This study corroborates the crucial function of MASP-2 in the lectin pathway and underlines the prominence of MBL-independent lectin pathway activation in the host defense against pneumococci (130).
Recent study in mice demonstrated that MASP-2 deposits complement C4 onto mitochondria, revealing the potential role of the complement lectin pathway in mitochondrial immune handling. These processes are speculated to be involved either in the induction of problematic inflammatory reactions or in homeostatic clearance of mitochondria (131).
As discussed above, the complement lectin pathway has a protective function against invading pathogens and plays an essential role in ischemia/reperfusion (I/R)-injury as well. The serpin C1-inhibitor and aprotinin, a Kunitz-type inhibitor can inhibit MASP-2. Recombinant tissue factor pathway inhibitor (rTFPI) was identified as a novel selective inhibitor of MASP-2, without disturbing the activity of MASP-1 or the classical pathway proteases C1s and C1r. Ex vivo assay revealed that Kunitz-2 domain in TFPI was necessary for the inhibition of MASP-2 activity. TFPI could be a therapeutic approach to constraint the tissue injury in the conditions of cerebral stroke, myocardial infarction, or solid organ transplantation (132).
C4a, one of the activation fragments of complement C4, identified in 1979, was regarded as the third anaphylatoxin although it is still under debate (133, 134). C4a has been described to possess a strong chemotaxis inhibitory effect on monocytes at the concentration as low as 10-16 M (135). C4a was also reported to inhibit C3a-induced O2·− generation in guinea pig macrophages, to produce immediate erythema/edema when injected into human skin, and to induce contraction of guinea pig ileum (133, 136). It was suggested that a function for C4a is closely related to C3a due to its ability to desensitize the action of C3a-induced contraction of guinea pig ileum (133). It was later revealed that human C4a acted as an agonist for the guinea pig but not the human C3aR receptor (137). Studies using recombinant human C4a have also demonstrated that C4a can impair C5a-induced neointima formation, reduce C3a- or C5a-mediated chemoattractant and secretagogue activities in mast cells, and prevent hyperoxic lung injury via a macrophage-dependent signaling pathway (138–140). It remains to be established how C4a can modulate the functions of monocytes/macrophages to generate anti-inflammatory effects.
C4d, another cleavage product by complement C4 activation, has long been considered as a biomarker for disease activity in autoimmune disorders or antibody-mediated allograft rejection. A recent study identified Ig-like transcript (ILT) 4 and ITL5v2 as cellular receptors for C4d and interaction of C4d with ILT4 conferred a dose-dependent inhibition of TNFα and IL-6 secretion and attenuation of intracellular [Ca2+] flux in monocytes activated via Fc-cross-linking of up to 50% as compared to control (141). ILT4 has been involved in the control of autoreactivity (142, 143), induction of transplantation tolerance (144), and maintenance of feto-maternal tolerance during pregnancy (145). Mice lack of PIR-B, the ortholog of ILT4, suffer from autoimmune glomerulonephritis (146) and exacerbated graft versus host disease (147). It appears that the interactions of complement C4 activation cleavage fragments (such as C4a and C4d) with their respective receptors plays inhibitory roles to impede inflammatory reactions induced by cytokines, chemokines, and other anaphylatoxins (C3a, C5a). One interesting paradigm will be that upon complement activation (i.e., microbial or viral infection, immunocomplex, or apoptotic debris), complement C4 activation fragments may act as regulators to maintain homeostasis and to contain downstream anaphylatoxins’ proinflammatory effects, which may trigger hyper-inflammatory reactions. The activation of complement C4 and potential immune-regulation mechanisms of split products from complement C4 are illustrated in Figure 3.
It remains incompletely understood why total deficiencies of some complement components are associated with some autoimmune diseases (148). Complement C4 plays a vital role in the activation of classical and lectin pathways and the formation of C3 convertase, which leads to the generation of the membrane attack complex (MAC) against microbes. It was reported that complement C4 is protective for autoimmune lupus disease independent of C3 in mice (30). C4(-/-) mice have significantly more IgM anti-double-strand DNA antibodies than C4(+/+) control mice (32). Increased frequency of C4 deficiency phenotypes was reported in IgA nephropathy and Henoch-Schönlein purpura (HSP) (26), insulin-dependent diabetes mellitus (IDDM) (149), systemic lupus erythematosus (SLE) (150, 151), repeated infections (152), juvenile idiopathic arthritis patients (27). glomerulonephritis (153). The lack of complement C4 can trigger inapt clearance of apoptotic debris and stimulate chronic activation of myeloid cells. The deficiency in complement component C4 also results in a breakdown in the elimination of autoreactive B-cell clones at the transitional stage, depicted by a relative increase in their response to a series of stimuli, entering into follicles, and a higher tendency to form self-reactive germinal centers (GCs), allowing the maturation and activation of self-reactive B-cell clones (154). Using two well-defined murine models to examine complement deficiency in peripheral tolerance, the study revealed that complement C4 protein and the receptors CD21/CD35 are involved in negative selection of self-reactive B lymphocytes, suggesting an immune deficiency of complement C4 predisposes mice to SLE (29). A low serum C4 level in patients with autoimmune disease may be due to ongoing disease activity associated with the consumption caused by complement activation and or it may be due to genetic factors (155). One of the questions still remains: whether and how does infections link to autoimmune disease upon the deficiency of complement C4? It is speculated that C4 deficiency would negatively affect the efficiency and progression of complement activation, decrease phagocytes functions and the clearance of apoptotic and necrotic cells (156).
A recent study by Yammani et al. demonstrated that complement C4 deficiency is a predisposing factor for streptococcus pneumonia-induced anti-dsDNA IgA autoantibody production. In a C4KO mice model, serotype 19F and virulent serotype 3 pneumococci induce systemic anti-dsDNA IgA production; interestingly which is more pronounced in female C4KO mice. Further study revealed that pneumococci pneumococcal polysaccharide (PPS) vaccination alone induced increases in anti-dsDNA IgA levels, which can be completely blocked by TLR2/4 antagonist, OxPAPC. Pam3CSK4, a TLR2 agonist, equally stimulated anti-dsDNA IgA production in C4KO mice, suggesting complement C4 plays a role in subduing autoantibody production stimulated by cross-reactive antigens and TLR2 agonists associated with S. pneumonia (33).
A complete analysis of the potential Epstein-Barr virus (EBV) peptide cross-reactome has been performed to search for peptides common to SLE-EBV and human SLE autoantigens. The study found EBV proteome can act as an immunological potential. Using publicly available databases, fifty-one SLE-related proteins were analyzed for hexapeptide sharing with EBV proteome and found 34 of hexapeptides are shared between human SLE autoantigens and EBV proteins. Interestingly, the study also revealed that peptide sharing mostly occurred with complement component C4 and Interleukin-10 (IL-10). This study demonstrated that the EBV vs. SLE autoantigens peptide overlap and powerfully supports cross-reactivity as a major mechanism in EBV-associated SLE etiopathogenesis (157). Among patients with systemic lupus erythematosus (SLE), a prevalence of HPV infection has been reported. One interesting hypothesis is that immune responses caused by HPV infection may interact with proteins that associate with SLE (158).
In lymphoid tissues and peripheral blood of C4KO mice, it was discovered with the decreased frequencies of CD4+CD25+ Tregs and reduced expression of Foxp3 and TGF-β, which are crucial for the efficient development and function of Tregs cells. Thus, the study suggested that the association of the deficiency of complement C4 in the classical complement pathway with the development of autoimmune disorder might be via the role of Tregs deficiency (159). It remains to be elucidated how the fragments generated from complement C4 activation, such as C4a and C4d, are regulating Tregs cells functions.
Complement system is essential for the maintenance of homeostasis by elimination of immune complexes, supporting self-tolerance and anti-inflammation, and promoting tissue repair (160, 161). While the complement activation of the downstream of complement C3 resulting in inflammatory molecules, such as C3a, C5a, and the membrane attack complex (MAC), plays a less important role, the early components of the classical pathway, such as C1q, C4, and C2, are more critical in maintaining homeostasis and lack of some of early components of classical pathway will predispose an individual to autoimmune disorders. Many studies have linked the complement C4 deficiency/partial deficiency with autoimmune disorders. In addition, C4 deficiency is clearly linked to the susceptibility of infections. Those observations persuade us to speculate that infection-caused inflammation needs the containment that requires the immune modulation from the contribution of complement C4, otherwise it will be aggravated under the deficiency of C4. Complement C4 is reported to be chiefly expressed in hepatocytes, but the upregulation of mRNA expression of complement C4 was observed by LPS, IFNγ, and interleukin-6 in other types of cells, indicating that infection-induced cytokines could trigger the upregulation of complement C4 expression as a feedback regulatory response. Mounting evidence supports the observation that infections may initiate and/or exacerbate autoimmune reactions (162–165), which is in line with the studies in mouse models that have established the role of complement C4 as suppressing auto-antibody production (31–33, 166). Nevertheless, the mechanisms of complement C4 involved in homeostasis still have been poorly addressed. Recent studies demonstrated that complement C4 activation fragments, like C4a and C4d, can modulate cytokines generation from macrophages probably through their respective receptors. One of possible mechanisms that complement C4 mediated homeostatic process might be via its activation fragments, which can modulate immune reactions to restrain infection-induced hyper-inflammatory reactions induced by cytokines and anaphylatoxin C3a and C5a (Figure 3).
Future studies are necessary to focus on the immune regulatory functions of C4 activation fragments, which will be explored as therapeutic targets for the treatment of infections, as well as the autoimmune disorders.
HW contributed to the conception and idea of the work. All authors contributed to the article and approved the submitted version.
Research work was supported by the seed grant (to HW) from College of Pharmacy and mini-grant (to HW) from College of Medicine, California Northstate University, Elk Grove, CA 95757, United States.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kallionpaa H, Elo LL, Laajala E, Mykkanen J, Ricano-Ponce I, Vaarma M, et al. Innate Immune Activity Is Detected Prior to Seroconversion in Children With HLA-Conferred Type 1 Diabetes Susceptibility. Diabetes (2014) 63(7):2402–14. doi: 10.2337/db13-1775
2. Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of Complement as a Molecular Adjuvant: Bridging Innate and Acquired Immunity. Science (1996) 271(5247):348–50. doi: 10.1126/science.271.5247.348
3. Nauta AJ, Daha MR, van Kooten C, Roos A. Recognition and Clearance of Apoptotic Cells: a Role for Complement and Pentraxins. Trends Immunol (2003) 24(3):148–54. doi: 10.1016/S1471-4906(03)00030-9
4. Audemard-Verger A, Descloux E, Ponard D, Deroux A, Fantin B, Fieschi C, et al. Infections Revealing Complement Deficiency in Adults: a French Nationwide Study Enrolling 41 Patients. Med (Baltimore) (2016) 95(19):e3548. doi: 10.1097/MD.0000000000003548
5. Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, et al. Complement and Autoimmunity. Immunol Res (2013) 56(2-3):477–91. doi: 10.1007/s12026-013-8422-y
6. Law SK, Dodds AW. The Internal Thioester and the Covalent Binding Properties of the Complement Proteins C3 and C4. Protein Sci (1997) 6(2):263–74. doi: 10.1002/pro.5560060201
7. Sim RB, Sim E. Autolytic Fragmentation of Complement Components C3 and C4 and Its Relationship to Covalent Binding Activity. Ann N Y Acad Sci (1983) 421:259–76. doi: 10.1111/j.1749-6632.1983.tb18114.x
8. Perry AJ, Wijeyewickrema LC, Wilmann PG, Gunzburg MJ, D’Andrea L, Irving JA, et al. A Molecular Switch Governs the Interaction Between the Human Complement Protease C1s and Its Substrate, Complement C4. J Biol Chem (2013) 288(22):15821–9. doi: 10.1074/jbc.M113.464545
9. Duncan RC, Mohlin F, Taleski D, Coetzer TH, Huntington JA, Payne RJ, et al. Identification of a Catalytic Exosite for Complement Component C4 on the Serine Protease Domain of C1s. J Immunol (2012) 189(5):2365–73. doi: 10.4049/jimmunol.1201085
10. Duncan RC, Bergstrom F, Coetzer TH, Blom AM, Wijeyewickrema LC, Pike RN. Multiple Domains of MASP-2, an Initiating Complement Protease, Are Required for Interaction With Its Substrate C4. Mol Immunol (2012) 49(4):593–600. doi: 10.1016/j.molimm.2011.10.006
11. Drentin N, Conroy P, Gunzburg MJ, Pike RN, Wijeyewickrema LC. Investigation of the Mechanism of Interaction Between Mannose-Binding Lectin-Associated Serine Protease-2 and Complement C4. Mol Immunol (2015) 67(2 Pt B):287–93. doi: 10.1016/j.molimm.2015.06.011
12. Kidmose RT, Laursen NS, Dobo J, Kjaer TR, Sirotkina S, Yatime L, et al. Structural Basis for Activation of the Complement System by Component C4 Cleavage. Proc Natl Acad Sci USA (2012) 109(38):15425–30. doi: 10.1073/pnas.1208031109
13. Pilely K, Skjoedt MO, Nielsen C, Andersen TE, Louise Aabom A, Vitved L, et al. A Specific Assay for Quantification of Human C4c by Use of an Anti-C4c Monoclonal Antibody. J Immunol Methods (2014) 405:87–96. doi: 10.1016/j.jim.2014.01.011
14. Wieser M, Francisci T, Lackner D, Buerckstuemmer T, Wasner K, Eilenberg W, et al. CD46 Knock-Out Using CRISPR/Cas9 Editing of hTERT Immortalized Human Cells Modulates Complement Activation. PloS One (2019) 14(4):e0214514. doi: 10.1371/journal.pone.0214514
15. Barilla-LaBarca ML, Liszewski MK, Lambris JD, Hourcade D, Atkinson JP. Role of Membrane Cofactor Protein (CD46) in Regulation of C4b and C3b Deposited on Cells. J Immunol (2002) 168(12):6298–304. doi: 10.4049/jimmunol.168.12.6298
16. Mortensen S, Kidmose RT, Petersen SV, Szilagyi A, Prohaszka Z, Andersen GR. Structural Basis for the Function of Complement Component C4 Within the Classical and Lectin Pathways of Complement. J Immunol (2015) 194(11):5488–96. doi: 10.4049/jimmunol.1500087
17. Mortensen S, Jensen JK, Andersen GR. Solution Structures of Complement C2 and Its C4 Complexes Propose Pathway-Specific Mechanisms for Control and Activation of the Complement Proconvertases. J Biol Chem (2016) 291(32):16494–507. doi: 10.1074/jbc.M116.722017
18. Bishof NA, Welch TR, Beischel LS. C4B Deficiency: a Risk Factor for Bacteremia With Encapsulated Organisms. J Infect Dis (1990) 162(1):248–50. doi: 10.1093/infdis/162.1.248
19. Jaatinen T, Lahti M, Ruuskanen O, Kinos R, Truedsson L, Lahesmaa R, et al. Total C4B Deficiency Due to Gene Deletion and Gene Conversion in a Patient With Severe Infections. Clin Diagn Lab Immunol (2003) 10(2):195–201. doi: 10.1128/CDLI.10.2.195-201.2003
20. Kang YS, Do Y, Lee HK, Park SH, Cheong C, Lynch RM, et al. A Dominant Complement Fixation Pathway for Pneumococcal Polysaccharides Initiated by SIGN-R1 Interacting With C1q. Cell (2006) 125(1):47–58. doi: 10.1016/j.cell.2006.01.046
21. Brown JS, Hussell T, Gilliland SM, Holden DW, Paton JC, Ehrenstein MR, et al. The Classical Pathway Is the Dominant Complement Pathway Required for Innate Immunity to Streptococcus Pneumoniae Infection in Mice. Proc Natl Acad Sci USA (2002) 99(26):16969–74. doi: 10.1073/pnas.012669199
22. Mold C, Rodic-Polic B, Du Clos TW. Protection From Streptococcus Pneumoniae Infection by C-Reactive Protein and Natural Antibody Requires Complement But Not Fc Gamma Receptors. J Immunol (2002) 168(12):6375–81. doi: 10.4049/jimmunol.168.12.6375
23. Rowe PC, McLean RH, Wood RA, Leggiadro RJ, Winkelstein JA. Association of Homozygous C4B Deficiency With Bacterial Meningitis. J Infect Dis (1989) 160(3):448–51. doi: 10.1093/infdis/160.3.448
24. Hauptmann G, Tappeiner G, Schifferli JA. Inherited Deficiency of the Fourth Component of Human Complement. Immunodefic Rev (1988) 1(1):3–22.
25. Yang Y, Chung EK, Zhou B, Lhotta K, Hebert LA, Birmingham DJ, et al. The Intricate Role of Complement Component C4 in Human Systemic Lupus Erythematosus. Curr Dir Autoimmun (2004) 7:98–132. doi: 10.1159/000075689
26. McLean RH, Wyatt RJ, Julian BA. Complement Phenotypes in Glomerulonephritis: Increased Frequency of Homozygous Null C4 Phenotypes in IgA Nephropathy and Henoch-Schonlein Purpura. Kidney Int (1984) 26(6):855–60. doi: 10.1038/ki.1984.228
27. Gilliam BE, Wolff AE, Moore TL. Partial C4 Deficiency in Juvenile Idiopathic Arthritis Patients. J Clin Rheumatol (2007) 13(5):256–60. doi: 10.1097/RHU.0b013e318156b9e3
28. Wopenka U, Thysell H, Sjoholm AG, Truedsson L. C4 Phenotypes in IgA Nephropathy: Disease Progression Associated With C4A Deficiency But Not With C4 Isotype Concentrations. Clin Nephrol (1996) 45(3):141–5.
29. Prodeus AP, Goerg S, Shen LM, Pozdnyakova OO, Chu L, Alicot EM, et al. A Critical Role for Complement in Maintenance of Self-Tolerance. Immunity (1998) 9(5):721–31. doi: 10.1016/S1074-7613(00)80669-X
30. Einav S, Pozdnyakova OO, Ma M, Carroll MC. Complement C4 Is Protective for Lupus Disease Independent of C3. J Immunol (2002) 168(3):1036–41. doi: 10.4049/jimmunol.168.3.1036
31. Chen Z, Koralov SB, Kelsoe G. Complement C4 Inhibits Systemic Autoimmunity Through a Mechanism Independent of Complement Receptors CR1 and CR2. J Exp Med (2000) 192(9):1339–52. doi: 10.1084/jem.192.9.1339
32. Paul E, Pozdnyakova OO, Mitchell E, Carroll MC. Anti-DNA Autoreactivity in C4-Deficient Mice. Eur J Immunol (2002) 32(9):2672–9. doi: 10.1002/1521-4141(200209)32:9<2672::AID-IMMU2672>3.0.CO;2-X
33. Yammani RD, Leyva MA, Jennings RN, Haas KM. C4 Deficiency Is a Predisposing Factor for Streptococcus Pneumoniae-Induced Autoantibody Production. J Immunol (2014) 193(11):5434–43. doi: 10.4049/jimmunol.1401462
34. Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, et al. Deficiencies of Human Complement Component C4A and C4B and Heterozygosity in Length Variants of RP-C4-CYP21-TNX (RCCX) Modules in Caucasians. The Load of RCCX Genetic Diversity on Major Histocompatibility Complex-Associated Disease. J Exp Med (2000) 191(12):2183–96. doi: 10.1084/jem.191.12.2183
35. Hovingh ES, van Gent M, Hamstra HJ, Demkes M, Mooi FR, Pinelli E. Emerging Bordetella Pertussis Strains Induce Enhanced Signaling of Human Pattern Recognition Receptors TLR2, NOD2 and Secretion of IL-10 by Dendritic Cells. PloS One (2017) 12(1):e0170027. doi: 10.1371/journal.pone.0170027
36. Blanchong CA, Chung EK, Rupert KL, Yang Y, Yang Z, Zhou B, et al. Genetic, Structural and Functional Diversities of Human Complement Components C4A and C4B and Their Mouse Homologues, Slp and C4. Int Immunopharmacol (2001) 1(3):365–92. doi: 10.1016/S1567-5769(01)00019-4
37. Carroll MC, Palsdottir A, Belt KT, Porter RR. Deletion of Complement C4 and Steroid 21-Hydroxylase Genes in the HLA Class III Region. EMBO J (1985) 4(10):2547–52. doi: 10.1002/j.1460-2075.1985.tb03969.x
38. Carroll MC, Campbell RD, Bentley DR, Porter RR. A Molecular Map of the Human Major Histocompatibility Complex Class III Region Linking Complement Genes C4, C2 and Factor B. Nature (1984) 307(5948):237–41. doi: 10.1038/307237a0
39. Yu CY. Molecular Genetics of the Human MHC Complement Gene Cluster. Exp Clin Immunogenet (1998) 15(4):213–30. doi: 10.1159/000019075
40. Yung Yu C, Yang Z, Blanchong CA, Miller W. The Human and Mouse MHC Class III Region: a Parade of 21 Genes at the Centromeric Segment. Immunol Today (2000) 21(7):320–8. doi: 10.1016/S0167-5699(00)01664-9
41. White PC, Grossberger D, Onufer BJ, Chaplin DD, New MI, Dupont B, et al. Two Genes Encoding Steroid 21-Hydroxylase Are Located Near the Genes Encoding the Fourth Component of Complement in Man. Proc Natl Acad Sci USA (1985) 82(4):1089–93. doi: 10.1073/pnas.82.4.1089
42. Bristow J, Tee MK, Gitelman SE, Mellon SH, Miller WL. Tenascin-X: a Novel Extracellular Matrix Protein Encoded by the Human XB Gene Overlapping P450c21B. J Cell Biol (1993) 122(1):265–78. doi: 10.1083/jcb.122.1.265
43. Shen L, Wu LC, Sanlioglu S, Chen R, Mendoza AR, Dangel AW, et al. Structure and Genetics of the Partially Duplicated Gene RP Located Immediately Upstream of the Complement C4A and the C4B Genes in the HLA Class III Region. Molecular Cloning, Exon-Intron Structure, Composite Retroposon, and Breakpoint of Gene Duplication. J Biol Chem (1994) 269(11):8466–76. doi: 10.1016/S0021-9258(17)37217-4
44. Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular Variations of the Human Major Histocompatibility Complex Class III Genes for Serine/Threonine Kinase RP, Complement Component C4, Steroid 21-Hydroxylase CYP21, and Tenascin TNX (the RCCX Module). A Mechanism for Gene Deletions and Disease Associations. J Biol Chem (1999) 274(17):12147–56. doi: 10.1074/jbc.274.17.12147
45. Olsson LM, Holmdahl R. Copy Number Variation in Autoimmunity–Importance Hidden in Complexity? Eur J Immunol (2012) 42(8):1969–76. doi: 10.1002/eji.201242601
46. Wu YL, Savelli SL, Yang Y, Zhou B, Rovin BH, Birmingham DJ, et al. Sensitive and Specific Real-Time Polymerase Chain Reaction Assays to Accurately Determine Copy Number Variations (CNVs) of Human Complement C4A, C4B, C4-Long, C4-Short, and RCCX Modules: Elucidation of C4 CNVs in 50 Consanguineous Subjects With Defined HLA Genotypes. J Immunol (2007) 179(5):3012–25. doi: 10.4049/jimmunol.179.5.3012
47. Yu CY, Belt KT, Giles CM, Campbell RD, Porter RR. Structural Basis of the Polymorphism of Human Complement Components C4A and C4B: Gene Size, Reactivity and Antigenicity. EMBO J (1986) 5(11):2873–81. doi: 10.1002/j.1460-2075.1986.tb04582.x
48. Belt KT, Yu CY, Carroll MC, Porter RR. Polymorphism of Human Complement Component C4. Immunogenetics (1985) 21(2):173–80. doi: 10.1007/BF00364869
49. Dodds AW, Law SK, Porter RR. The Purification and Properties of Some Less Common Allotypes of the Fourth Component of Human Complement. Immunogenetics (1986) 24(5):279–85. doi: 10.1007/BF00395532
50. Sim E, Cross SJ. Phenotyping of Human Complement Component C4, a Class-III HLA Antigen. Biochem J (1986) 239(3):763–7. doi: 10.1042/bj2390763
51. Awdeh ZL, Alper CA. Inherited Structural Polymorphism of the Fourth Component of Human Complement. Proc Natl Acad Sci USA (1980) 77(6):3576–80. doi: 10.1073/pnas.77.6.3576
52. Dangel AW, Mendoza AR, Baker BJ, Daniel CM, Carroll MC, Wu LC, et al. The Dichotomous Size Variation of Human Complement C4 Genes Is Mediated by a Novel Family of Endogenous Retroviruses, Which Also Establishes Species-Specific Genomic Patterns Among Old World Primates. Immunogenetics (1994) 40(6):425–36. doi: 10.1007/BF00177825
53. Yu CY. The Complete Exon-Intron Structure of a Human Complement Component C4A Gene. DNA Sequences, Polymorphism, and Linkage to the 21-Hydroxylase Gene. J Immunol (1991) 146(3):1057–66.
54. Ulgiati D, Abraham LJ. Comparative Analysis of the Disease-Associated Complement C4 Gene From the HLA-A1, B8, DR3 Haplotype. Exp Clin Immunogenet (1996) 13(1):43–54. doi: 10.1007/s002510050059
55. Schneider PM, Carroll MC, Alper CA, Rittner C, Whitehead AS, Yunis EJ, et al. Polymorphism of the Human Complement C4 and Steroid 21-Hydroxylase Genes. Restriction Fragment Length Polymorphisms Revealing Structural Deletions, Homoduplications, and Size Variants. J Clin Invest (1986) 78(3):650–7. doi: 10.1172/JCI112623
56. Yu CY, Campbell RD. Definitive RFLPs to Distinguish Between the Human Complement C4A/C4B Isotypes and the Major Rodgers/Chido Determinants: Application to the Study of C4 Null Alleles. Immunogenetics (1987) 25(6):383–90. doi: 10.1007/BF00396104
57. Dodds AW, Ren XD, Willis AC, Law SK. The Reaction Mechanism of the Internal Thioester in the Human Complement Component C4. Nature (1996) 379(6561):177–9. doi: 10.1038/379177a0
58. Juptner M, Flachsbart F, Caliebe A, Lieb W, Schreiber S, Zeuner R, et al. Low Copy Numbers of Complement C4 and Homozygous Deficiency of C4A may Predispose to Severe Disease and Earlier Disease Onset in Patients With Systemic Lupus Erythematosus. Lupus (2018) 27(4):600–9. doi: 10.1177/0961203317735187
59. Hou S, Qi J, Liao D, Fang J, Chen L, Kijlstra A, et al. High C4 Gene Copy Numbers Protects Against Vogt-Koyanagi-Harada Syndrome in Chinese Han. Br J Ophthalmol (2014) 98(12):1733–7. doi: 10.1136/bjophthalmol-2014-305596
60. Hou S, Qi J, Liao D, Zhang Q, Fang J, Zhou Y, et al. Copy Number Variations of Complement Component C4 Are Associated With Behcet’s Disease But Not With Ankylosing Spondylitis Associated With Acute Anterior Uveitis. Arthritis Rheum (2013) 65(11):2963–70. doi: 10.1002/art.38116
61. Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene Copy-Number Variation and Associated Polymorphisms of Complement Component C4 in Human Systemic Lupus Erythematosus (SLE): Low Copy Number Is a Risk Factor for and High Copy Number Is a Protective Factor Against SLE Susceptibility in European Americans. Am J Hum Genet (2007) 80(6):1037–54. doi: 10.1086/518257
62. Boteva L, Morris DL, Cortes-Hernandez J, Martin J, Vyse TJ, Fernando MM. Genetically Determined Partial Complement C4 Deficiency States Are Not Independent Risk Factors for SLE in UK and Spanish Populations. Am J Hum Genet (2012) 90(3):445–56. doi: 10.1016/j.ajhg.2012.01.012
63. Breukink MB, Schellevis RL, Boon CJ, Fauser S, Hoyng CB, den Hollander AI, et al. Genomic Copy Number Variations of the Complement Component C4B Gene Are Associated With Chronic Central Serous Chorioretinopathy. Invest Ophthalmol Vis Sci (2015) 56(9):5608–13. doi: 10.1167/iovs.15-17343
64. Kainulainen L, Peltola V, Seppanen M, Viander M, He Q, Lokki ML, et al. C4A Deficiency in Children and Adolescents With Recurrent Respiratory Infections. Hum Immunol (2012) 73(5):498–501. doi: 10.1016/j.humimm.2012.02.015
65. Jaatinen T, Ruuskanen O, Truedsson L, Lokki ML. Homozygous Deletion of the CYP21A-TNXA-RP2-C4B Gene Region Conferring C4B Deficiency Associated With Recurrent Respiratory Infections. Hum Immunol (1999) 60(8):707–14. doi: 10.1016/S0198-8859(99)00047-6
66. Bay JT, Schejbel L, Madsen HO, Sorensen SS, Hansen JM, Garred P. Low C4 Gene Copy Numbers Are Associated With Superior Graft Survival in Patients Transplanted With a Deceased Donor Kidney. Kidney Int (2013) 84(3):562–9. doi: 10.1038/ki.2013.195
67. Goldman JN, O’Rourke KS, Goldman MB. Antibody-Induced Suppression and Postsuppression Stimulation of Complement In Vitro. III. Long-Term C4 Suppression Is Actively Maintained by a Soluble Suppressor Factor (Fsc4). Cell Immunol (1985) 96(1):26–37. doi: 10.1016/0008-8749(85)90337-5
68. Goldman MB, O’Rourke KS, Becker DS, Goldman JN. Antibody-Induced Suppression and Postsuppression Stimulation of Complement In Vitro. II. Intracellular and Extracellular Changes in C4 During Long-Term C4 Suppression in Guinea Pig Splenic Fragments. J Immunol (1985) 134(5):3298–306.
69. Goldman JN, O’Rourke KS, McMannis JD, Goldman MB. Effects of Anti-C4 Antibody on Complement Production by Splenic and Peritoneal Macrophages. Complement (1988) 5(1):13–26. doi: 10.1159/000463027
70. Goldman MB, Becker DS, O’Rourke KS, Goldman JN. Enhancement by Cyclic AMP of Antibody-Induced Suppression of the Fourth Component of Complement. J Immunol (1985) 135(4):2701–6.
71. Daha MR, van Es LA. Relative Resistance of the F-42-Stabilized Classical Pathway C3 Convertase to Inactivation by C4-Binding Protein. J Immunol (1980) 125(5):2051–4.
72. Garam N, Prohaszka Z, Szilagyi A, Aigner C, Schmidt A, Gaggl M, et al. C4 Nephritic Factor in Patients With Immune-Complex-Mediated Membranoproliferative Glomerulonephritis and C3-Glomerulopathy. Orphanet J Rare Dis (2019) 14(1):247. doi: 10.1186/s13023-019-1237-8
73. Zhang Y, Meyer NC, Fervenza FC, Lau W, Keenan A, Cara-Fuentes G, et al. C4 Nephritic Factors in C3 Glomerulopathy: a Case Series. Am J Kidney Dis (2017) 70(6):834–43. doi: 10.1053/j.ajkd.2017.07.004
74. Halbwachs L, Leveille M, Lesavre P, Wattel S, Leibowitch J. Nephritic Factor of the Classical Pathway of Complement: Immunoglobulin G Autoantibody Directed Against the Classical Pathway C3 Convetase Enzyme. J Clin Invest (1980) 65(6):1249–56. doi: 10.1172/JCI109787
75. Tanuma Y, Ohi H, Watanabe S, Seki M, Hatano M. C3 Nephritic Factor and C4 Nephritic Factor in the Serum of Two Patients With Hypocomplementaemic Membranoproliferative Glomerulonephritis. Clin Exp Immunol (1989) 76(1):82–5.
76. Miller EC, Chase NM, Densen P, Hintermeyer MK, Casper JT, Atkinson JP. Autoantibody Stabilization of the Classical Pathway C3 Convertase Leading to C3 Deficiency and Neisserial Sepsis: C4 Nephritic Factor Revisited. Clin Immunol (2012) 145(3):241–50. doi: 10.1016/j.clim.2012.09.007
77. Battin C, De Sousa Linhares A, Paster W, Isenman DE, Wahrmann M, Leitner J, et al. Neuropilin-1 Acts as a Receptor for Complement Split Products. Front Immunol (2019) 10:2209. doi: 10.3389/fimmu.2019.02209
78. Schifferli JA, Hauptmann G, Paccaud JP. Complement-Mediated Adherence of Immune Complexes to Human Erythrocytes. Difference in the Requirements for C4A and C4B. FEBS Lett (1987) 213(2):415–8. doi: 10.1016/0014-5793(87)81533-8
79. Liesmaa I, Paakkanen R, Jarvinen A, Valtonen V, Lokki ML. Clinical Features of Patients With Homozygous Complement C4A or C4B Deficiency. PloS One (2018) 13(6):e0199305. doi: 10.1371/journal.pone.0199305
80. Skattum L, van Deuren M, van der Poll T, Truedsson L. Complement Deficiency States and Associated Infections. Mol Immunol (2011) 48(14):1643–55. doi: 10.1016/j.molimm.2011.05.001
81. Samano ES, Ribeiro Lde M, Gorescu RG, Rocha KC, Grumach AS. Involvement of C4 Allotypes in the Pathogenesis of Human Diseases. Rev Hosp Clin Fac Med Sao Paulo (2004) 59(3):138–44. doi: 10.1590/S0041-87812004000300009
82. Senbagavalli P, Kumar N, Kaur G, Mehra NK, Geetha ST, Ramanathan VD. Major Histocompatibility Complex Class III (C2, C4, Factor B) and C3 Gene Variants in Patients With Pulmonary Tuberculosis. Hum Immunol (2011) 72(2):173–8. doi: 10.1016/j.humimm.2010.11.002
83. Giussani S, Pietrocola G, Donnarumma D, Norais N, Speziale P, Fabbrini M, et al. The Streptococcus Agalactiae Complement Interfering Protein Combines Multiple Complement-Inhibitory Mechanisms by Interacting With Both C4 and C3 Ligands. FASEB J (2019) 33(3):4448–57. doi: 10.1096/fj.201801991R
84. Hair PS, Wagner SM, Friederich PT, Drake RR, Nyalwidhe JO, Cunnion KM. Complement Regulator C4BP Binds to Staphylococcus Aureus and Decreases Opsonization. Mol Immunol (2012) 50(4):253–61. doi: 10.1016/j.molimm.2012.01.010
85. Hovingh ES, van den Broek B, Kuipers B, Pinelli E, Rooijakkers SHM, Jongerius I. Acquisition of C1 Inhibitor by Bordetella Pertussis Virulence Associated Gene 8 Results in C2 and C4 Consumption Away From the Bacterial Surface. PloS Pathog (2017) 13(7):e1006531. doi: 10.1371/journal.ppat.1006531
86. Shende R, Wong SSW, Rapole S, Beau R, Ibrahim-Granet O, Monod M, et al. Aspergillus Fumigatus Conidial Metalloprotease Mep1p Cleaves Host Complement Proteins. J Biol Chem (2018) 293(40):15538–55. doi: 10.1074/jbc.RA117.001476
87. Behnsen J, Lessing F, Schindler S, Wartenberg D, Jacobsen ID, Thoen M, et al. Secreted Aspergillus Fumigatus Protease Alp1 Degrades Human Complement Proteins C3, C4, and C5. Infect Immun (2010) 78(8):3585–94. doi: 10.1128/IAI.01353-09
88. Cazander G, Schreurs MW, Renwarin L, Dorresteijn C, Hamann D, Jukema GN. Maggot Excretions Affect the Human Complement System. Wound Repair Regener (2012) 20(6):879–86. doi: 10.1111/j.1524-475X.2012.00850.x
89. Ahmed UK, Maller NC, Iqbal AJ, Al-Riyami L, Harnett W, Raynes JG. The Carbohydrate-Linked Phosphorylcholine of the Parasitic Nematode Product ES-62 Modulates Complement Activation. J Biol Chem (2016) 291(22):11939–53. doi: 10.1074/jbc.M115.702746
91. Mawatari S, Uto H, Ido A, Nakashima K, Suzuki T, Kanmura S, et al. Hepatitis C Virus NS3/4A Protease Inhibits Complement Activation by Cleaving Complement Component 4. PloS One (2013) 8(12):e82094. doi: 10.1371/journal.pone.0082094
90. Banerjee A, Mazumdar B, Meyer K, Di Bisceglie AM, Ray RB, Ray R. Transcriptional Repression of C4 Complement by Hepatitis C Virus Proteins. J Virol (2011) 85(9):4157–66. doi: 10.1128/JVI.02449-10
92. Kim DW, Lee SA, Kim H, Won YS, Kim BJ. Naturally Occurring Mutations in the Nonstructural Region 5B of Hepatitis C Virus (HCV) From Treatment-Naive Korean Patients Chronically Infected With HCV Genotype 1b. PloS One (2014) 9(1):e87773. doi: 10.1371/journal.pone.0087773
93. Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, et al. Antagonism of the Complement Component C4 by Flavivirus Nonstructural Protein NS1. J Exp Med (2010) 207(4):793–806. doi: 10.1084/jem.20092545
94. Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. Complement Evasion Strategies of Viruses: an Overview. Front Microbiol (2017) 8:1117. doi: 10.3389/fmicb.2017.01117
95. Mellors J, Tipton T, Longet S, Carroll M. Viral Evasion of the Complement System and Its Importance for Vaccines and Therapeutics. Front Immunol (2020) 11:1450. doi: 10.3389/fimmu.2020.01450
96. Bugdaci MS, Alkim C, Karaca C, Kesici B, Bayraktar B, Sokmen M. Could Complement C4 be an Alternative to Biopsy for Chronic Hepatitis B Histopathologic Findings? J Clin Gastroenterol (2011) 45(5):449–55. doi: 10.1097/MCG.0b013e31820f7ee5
97. Bugdaci MS, Karaca C, Alkim C, Kesici B, Bayraktar B, Sokmen M. Serum Complement C4 in Chronic Hepatitis C: Correlation With Histopathologic Findings and Disease Activity. Turk J Gastroenterol (2012) 23(1):33–7. doi: 10.4318/tjg.2012.0310
98. Chowdhury SJ, Karra VK, Bharali R, Kar P. Role of Complement Component C4 in Treatment Response and Disease Progression in Chronic Hepatitis C Patients. J Viral Hepat (2015) 22(8):671–4. doi: 10.1111/jvh.12383
99. El-Fatah Fahmy Hanno A, Mohiedeen KM, Deghedy A, Sayed R. Serum Complements C3 and C4 in Chronic HCV Infection and Their Correlation With Response to Pegylated Interferon and Ribavirin Treatment. Arab J Gastroenterol (2014) 15(2):58–62. doi: 10.1016/j.ajg.2014.04.005
100. Chang ML, Tsou YK, Hu TH, Lin CH, Lin WR, Sung CM, et al. Distinct Patterns of the Lipid Alterations Between Genotype 1 and 2 Chronic Hepatitis C Patients After Viral Clearance. PloS One (2014) 9(8):e104783. doi: 10.1371/journal.pone.0104783
101. Fuchs A, Lin TY, Beasley DW, Stover CM, Schwaeble WJ, Pierson TC, et al. Direct Complement Restriction of Flavivirus Infection Requires Glycan Recognition by Mannose-Binding Lectin. Cell Host Microbe (2010) 8(2):186–95. doi: 10.1016/j.chom.2010.07.007
102. Sane J, Laine O, Makela S, Paakkala A, Jarva H, Mustonen J, et al. Complement Activation in Puumala Hantavirus Infection Correlates With Disease Severity. Ann Med (2012) 44(5):468–75. doi: 10.3109/07853890.2011.573500
103. Bottermann M, Foss S, Caddy SL, Clift D, van Tienen LM, Vaysburd M, et al. Complement C4 Prevents Viral Infection Through Capsid Inactivation. Cell Host Microbe (2019) 25(4):617–29 e7. doi: 10.1016/j.chom.2019.02.016
104. Conway EM, Pryzdial ELG. Is the COVID-19 Thrombotic Catastrophe Complement-Connected? J Thromb Haemost (2020) 18(11):2812–22. doi: 10.1111/jth.15050
105. Jiang Y, Zhao G, Song N, Li P, Chen Y, Guo Y, et al. Blockade of the C5a-C5aR Axis Alleviates Lung Damage in Hdpp4-Transgenic Mice Infected With MERS-CoV. Emerg Microbes Infect (2018) 7(1):77. doi: 10.1038/s41426-018-0063-8
106. Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR, et al. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio (2018) 9(5):1–15. doi: 10.1128/mBio.01753-18
107. Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and Cardiac Pathology in African American Patients With COVID-19: an Autopsy Series From New Orleans. Lancet Respir Med (2020) 8(7):681–6. doi: 10.1016/S2213-2600(20)30243-5
108. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: a Report of Five Cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
109. Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science (2020) 369(6501):330–3. doi: 10.1126/science.abb9983
110. Romano C, Del Mastro A, Sellitto A, Solaro E, Esposito S, Cuomo G. Tocilizumab Reduces Complement C3 and C4 Serum Levels in Rheumatoid Arthritis Patients. Clin Rheumatol (2018) 37(6):1695–700. doi: 10.1007/s10067-018-3992-7
111. Qaddoori Y, Abrams ST, Mould P, Alhamdi Y, Christmas SE, Wang G, et al. Extracellular Histones Inhibit Complement Activation Through Interacting With Complement Component 4. J Immunol (2018) 200(12):4125–33. doi: 10.4049/jimmunol.1700779
112. Vogt LM, Talens S, Kwasniewicz E, Scavenius C, Struglics A, Enghild JJ, et al. Activation of Complement by Pigment Epithelium-Derived Factor in Rheumatoid Arthritis. J Immunol (2017) 199(3):1113–21. doi: 10.4049/jimmunol.1700018
113. Sinha VK, Sharma OP, Kumar MS. Insight Into the Intermolecular Recognition Mechanism Involved in Complement Component 4 Activation Through Serine Protease-Trypsin. J Biomol Struct Dyn (2018) 36(3):575–89. doi: 10.1080/07391102.2017.1288658
114. Mershon-Shier KL, Vasuthasawat A, Takahashi K, Morrison SL, Beenhouwer DO. In Vitro C3 Deposition on Cryptococcus Capsule Occurs via Multiple Complement Activation Pathways. Mol Immunol (2011) 48(15-16):2009–18. doi: 10.1016/j.molimm.2011.06.215
115. Takahashi M, Ishida Y, Iwaki D, Kanno K, Suzuki T, Endo Y, et al. Essential Role of Mannose-Binding Lectin-Associated Serine Protease-1 in Activation of the Complement Factor D. J Exp Med (2010) 207(1):29–37. doi: 10.1084/jem.20090633
116. Matsushita M, Okada H. Alternative Complement Pathway Activation by C4b Deposited During Classical Pathway Activation. J Immunol (1986) 136(8):2994–8.
117. Farries TC, Steuer KL, Atkinson JP. The Mechanism of Activation of the Alternative Pathway of Complement by Cell-Bound C4b. Mol Immunol (1990) 27(11):1155–61. doi: 10.1016/0161-5890(90)90104-8
118. Meri S, Pangburn MK. A Mechanism of Activation of the Alternative Complement Pathway by the Classical Pathway: Protection of C3b From Inactivation by Covalent Attachment to C4b. Eur J Immunol (1990) 20(12):2555–61. doi: 10.1002/eji.1830201205
119. Selander B, Martensson U, Weintraub A, Holmstrom E, Matsushita M, Thiel S, et al. Mannan-Binding Lectin Activates C3 and the Alternative Complement Pathway Without Involvement of C2. J Clin Invest (2006) 116(5):1425–34. doi: 10.1172/JCI25982
120. Dumestre-Perard C, Lamy B, Aldebert D, Lemaire-Vieille C, Grillot R, Brion JP, et al. Aspergillus Conidia Activate the Complement by the Mannan-Binding Lectin C2 Bypass Mechanism. J Immunol (2008) 181(10):7100–5. doi: 10.4049/jimmunol.181.10.7100
121. Tateishi K, Matsushita M. Activation of the Alternative Complement Pathway by Mannose-Binding Lectin via a C2-Bypass Pathway. Microbiol Immunol (2011) 55(11):817–21. doi: 10.1111/j.1348-0421.2011.00378.x
122. May JE, Frank MM. Hemolysis of Sheep Erythrocytes in Guinea Pig Serum Deficient in the Fourth Component of Complement. I. Antibody and Serum Requirements. J Immunol (1973) 111(6):1671–7.
123. Yang Y, Lhotta K, Chung EK, Eder P, Neumair F, Yu CY. Complete Complement Components C4A and C4B Deficiencies in Human Kidney Diseases and Systemic Lupus Erythematosus. J Immunol (2004) 173(4):2803–14. doi: 10.4049/jimmunol.173.4.2803
124. Schweinle JE, Ezekowitz RA, Tenner AJ, Kuhlman M, Joiner KA. Human Mannose-Binding Protein Activates the Alternative Complement Pathway and Enhances Serum Bactericidal Activity on a Mannose-Rich Isolate of Salmonella. J Clin Invest (1989) 84(6):1821–9. doi: 10.1172/JCI114367
125. Atkinson JP, Frank MM. Bypassing Complement: Evolutionary Lessons and Future Implications. J Clin Invest (2006) 116(5):1215–8. doi: 10.1172/JCI28622
126. Schwaeble WJ, Lynch NJ, Clark JE, Marber M, Samani NJ, Ali YM, et al. Targeting of Mannan-Binding Lectin-Associated Serine Protease-2 Confers Protection From Myocardial and Gastrointestinal Ischemia/Reperfusion Injury. Proc Natl Acad Sci USA (2011) 108(18):7523–8. doi: 10.1073/pnas.1101748108
127. Gulla KC, Gupta K, Krarup A, Gal P, Schwaeble WJ, Sim RB, et al. Activation of Mannan-Binding Lectin-Associated Serine Proteases Leads to Generation of a Fibrin Clot. Immunology (2010) 129(4):482–95. doi: 10.1111/j.1365-2567.2009.03200.x
128. Megyeri M, Mako V, Beinrohr L, Doleschall Z, Prohaszka Z, Cervenak L, et al. Complement Protease MASP-1 Activates Human Endothelial Cells: PAR4 Activation Is a Link Between Complement and Endothelial Function. J Immunol (2009) 183(5):3409–16. doi: 10.4049/jimmunol.0900879
129. Asgari E, Farrar CA, Lynch N, Ali YM, Roscher S, Stover C, et al. Mannan-Binding Lectin-Associated Serine Protease 2 Is Critical for the Development of Renal Ischemia Reperfusion Injury and Mediates Tissue Injury in the Absence of Complement C4. FASEB J (2014) 28(9):3996–4003. doi: 10.1096/fj.13-246306
130. Ali YM, Lynch NJ, Haleem KS, Fujita T, Endo Y, Hansen S, et al. The Lectin Pathway of Complement Activation Is a Critical Component of the Innate Immune Response to Pneumococcal Infection. PloS Pathog (2012) 8(7):e1002793. doi: 10.1371/journal.ppat.1002793
131. Brinkmann CR, Jensen L, Dagnaes-Hansen F, Holm IE, Endo Y, Fujita T, et al. Mitochondria and the Lectin Pathway of Complement. J Biol Chem (2013) 288(12):8016–27. doi: 10.1074/jbc.M112.430249
132. Keizer MP, Pouw RB, Kamp AM, Patiwael S, Marsman G, Hart MH, et al. TFPI Inhibits Lectin Pathway of Complement Activation by Direct Interaction With MASP-2. Eur J Immunol (2015) 45(2):544–50. doi: 10.1002/eji.201445070
133. Gorski JP, Hugli TE, Muller-Eberhard HJ. C4a: the Third Anaphylatoxin of the Human Complement System. Proc Natl Acad Sci USA (1979) 76(10):5299–302. doi: 10.1073/pnas.76.10.5299
134. Barnum SR. C4a: an Anaphylatoxin in Name Only. J Innate Immun (2015) 7(4):333–9. doi: 10.1159/000371423
135. Tsuruta T, Yamamoto T, Matsubara S, Nagasawa S, Tanase S, Tanaka J, et al. Novel Function of C4a Anaphylatoxin. Release From Monocytes of Protein Which Inhibits Monocyte Chemotaxis. Am J Pathol (1993) 142(6):1848–57.
136. Murakami Y, Yamamoto T, Imamichi T, Nagasawa S. Cellular Responses of Guinea-Pig Macrophages to C4a; Inhibition of C3a-Induced O2- Generation by C4a. Immunol Lett (1993) 36(3):301–4. doi: 10.1016/0165-2478(93)90104-A
137. Lienenklaus S, Ames RS, Tornetta MA, Sarau HM, Foley JJ, Crass T, et al. Human Anaphylatoxin C4a Is a Potent Agonist of the Guinea Pig But Not the Human C3a Receptor. J Immunol (1998) 161(5):2089–93.
138. Jiang X, Ma Y, Yu J, Li H, Xie F. Protective Effect of C4a Against Hyperoxic Lung Injury via a Macrophage-Dependent But Not a Neutrophil/Lymphocyte-Dependent Signaling Pathway. Mol Med Rep (2016) 13(2):1250–6. doi: 10.3892/mmr.2015.4651
139. Xie P, Nishiura H, Semba U, Chen J, Zhao R, Kuniyasu A, et al. Inhibitory Effects of C4a on Chemoattractant and Secretagogue Functions of the Other Anaphylatoxins via Gi Protein-Adenylyl Cyclase Inhibition Pathway in Mast Cells. Int Immunopharmacol (2012) 12(1):158–68. doi: 10.1016/j.intimp.2011.11.006
140. Zhao Y, Xu H, Yu W, Xie BD. Complement Anaphylatoxin C4a Inhibits C5a-Induced Neointima Formation Following Arterial Injury. Mol Med Rep (2014) 10(1):45–52. doi: 10.3892/mmr.2014.2176
141. Hofer J, Forster F, Isenman DE, Wahrmann M, Leitner J, Holzl MA, et al. Ig-Like Transcript 4 as a Cellular Receptor for Soluble Complement Fragment C4d. FASEB J (2016) 30(4):1492–503. doi: 10.1096/fj.15-275594
142. Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, et al. Differentiation of Type 1 T Regulatory Cells (Tr1) by Tolerogenic DC-10 Requires the IL-10-Dependent ILT4/HLA-G Pathway. Blood (2010) 116(6):935–44. doi: 10.1182/blood-2009-07-234872
143. Anderson KJ, Allen RL. Regulation of T-Cell Immunity by Leucocyte Immunoglobulin-Like Receptors: Innate Immune Receptors for Self on Antigen-Presenting Cells. Immunology (2009) 127(1):8–17. doi: 10.1111/j.1365-2567.2009.03097.x
144. Ristich V, Zhang W, Liang S, Horuzsko A. Mechanisms of Prolongation of Allograft Survival by HLA-G/ILT4-Modified Dendritic Cells. Hum Immunol (2007) 68(4):264–71. doi: 10.1016/j.humimm.2006.11.008
145. Hunt JS, Petroff MG, McIntire RH, Ober C. HLA-G and Immune Tolerance in Pregnancy. FASEB J (2005) 19(7):681–93. doi: 10.1096/fj.04-2078rev
146. Kubo T, Uchida Y, Watanabe Y, Abe M, Nakamura A, Ono M, et al. Augmented TLR9-Induced Btk Activation in PIR-B-Deficient B-1 Cells Provokes Excessive Autoantibody Production and Autoimmunity. J Exp Med (2009) 206(9):1971–82. doi: 10.1084/jem.20082392
147. Nakamura A, Kobayashi E, Takai T. Exacerbated Graft-Versus-Host Disease in Pirb-/- Mice. Nat Immunol (2004) 5(6):623–9. doi: 10.1038/ni1074
148. Ballow M, McLean RH, Yunis EJ, Awdeh ZL, O’Neill GJ, Einarson M, et al. C4 Polymorphism and HLA Linkage: Studies in a Family With Hereditary C4 Deficiency. Clin Immunol Immunopathol (1981) 20(3):354–60. doi: 10.1016/0090-1229(81)90146-X
149. Raum D, Awdeh Z, Alper CA. BF Types and the Mode of Inheritance of Insulin-Dependent Diabetes Mellitus (IDDM). Immunogenetics (1981) 12(1-2):59–74. doi: 10.1007/BF01561651
150. Fielder AH, Walport MJ, Batchelor JR, Rynes RI, Black CM, Dodi IA, et al. Family Study of the Major Histocompatibility Complex in Patients With Systemic Lupus Erythematosus: Importance of Null Alleles of C4A and C4B in Determining Disease Susceptibility. Br Med J (Clin Res Ed) (1983) 286(6363):425–8. doi: 10.1136/bmj.286.6363.425
151. Kjellman M, Laurell AB, Low B, Sjoholm AG. Homozygous Deficiency of C4 in a Child With a Lupus Erythematosus Syndrome. Clin Genet (1982) 22(6):331–9. doi: 10.1111/j.1399-0004.1982.tb01849.x
152. Mascart-Lemone F, Hauptmann G, Goetz J, Duchateau J, Delespesse G, Vray B, et al. Genetic Deficiency of C4 Presenting With Recurrent Infections and a SLE-Like Disease. Genetic and Immunologic Studies. Am J Med (1983) 75(2):295–304. doi: 10.1016/0002-9343(83)91208-1
153. Schaller JG, Gilliland BG, Ochs HD, Leddy JP, Agodoa LC, Rosenfeld SI. Severe Systemic Lupus Erythematosus With Nephritis in a Boy With Deficiency of the Fourth Component of Complement. Arthritis Rheum (1977) 20(8):1519–25. doi: 10.1002/art.1780200812
154. Chatterjee P, Agyemang AF, Alimzhanov MB, Degn S, Tsiftsoglou SA, Alicot E, et al. Complement C4 Maintains Peripheral B-Cell Tolerance in a Myeloid Cell Dependent Manner. Eur J Immunol (2013) 43(9):2441–50. doi: 10.1002/eji.201343412
155. Castley AS, Martinez OP. Molecular Analysis of Complement Component C4 Gene Copy Number. Methods Mol Biol (2012) 882:159–71. doi: 10.1007/978-1-61779-842-9_9
156. Rupert KL, Moulds JM, Yang Y, Arnett FC, Warren RW, Reveille JD, et al. The Molecular Basis of Complete Complement C4A and C4B Deficiencies in a Systemic Lupus Erythematosus Patient With Homozygous C4A and C4B Mutant Genes. J Immunol (2002) 169(3):1570–8. doi: 10.4049/jimmunol.169.3.1570
157. Kanduc D. Proteome-Wide Epstein-BarrVirus Analysis of Peptide Sharing With Human Systemic Lupus Erythematosus Autoantigens. Isr Med Assoc J (2019) 21(7):444–8.
158. Segal Y, Dahan S, Calabro M, Kanduc D, Shoenfeld Y. HPV and Systemic Lupus Erythematosus: a Mosaic of Potential Crossreactions. Immunol Res (2017) 65(2):564–71. doi: 10.1007/s12026-016-8890-y
159. Leaungwutiwong P, Ittiprasert W, Saikhun K, Tong-Ngam P, Akapirat S, Chattanadee S, et al. Impairment of CD4+CD25+ Regulatory T Cells in C4-Deficient Mice. Asian Pac J Allergy Immunol (2011) 29(3):220–8.
160. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a Key System for Immune Surveillance and Homeostasis. Nat Immunol (2010) 11(9):785–97. doi: 10.1038/ni.1923
161. Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, et al. Drusen Complement Components C3a and C5a Promote Choroidal Neovascularization. Proc Natl Acad Sci USA (2006) 103(7):2328–33. doi: 10.1073/pnas.0408835103
162. Ercolini AM, Miller SD. The Role of Infections in Autoimmune Disease. Clin Exp Immunol (2009) 155(1):1–15. doi: 10.1111/j.1365-2249.2008.03834.x
163. Getts DR, Chastain EM, Terry RL, Miller SD. Virus Infection, Antiviral Immunity, and Autoimmunity. Immunol Rev (2013) 255(1):197–209. doi: 10.1111/imr.12091
164. Barzilai O, Ram M, Shoenfeld Y. Viral Infection can Induce the Production of Autoantibodies. Curr Opin Rheumatol (2007) 19(6):636–43. doi: 10.1097/BOR.0b013e3282f0ad25
165. Sfriso P, Ghirardello A, Botsios C, Tonon M, Zen M, Bassi N, et al. Infections and Autoimmunity: the Multifaceted Relationship. J Leukoc Biol (2010) 87(3):385–95. doi: 10.1189/jlb.0709517
Keywords: complement, C4, C4a, C4d, infections, autoimmune diseases
Citation: Wang H and Liu M (2021) Complement C4, Infections, and Autoimmune Diseases. Front. Immunol. 12:694928. doi: 10.3389/fimmu.2021.694928
Received: 14 April 2021; Accepted: 21 June 2021;
Published: 14 July 2021.
Edited by:
Maciej Lech, LMU Munich University Hospital, GermanyReviewed by:
Chack-Yung Yu, The Ohio State University, United StatesCopyright © 2021 Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongbin Wang, aG9uZ2Jpbi53YW5nQGNuc3UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.