 Vladimir Mulens-Arias
Vladimir Mulens-Arias José Manuel Rojas
José Manuel Rojas Domingo F. Barber
Domingo F. Barber- 1Department of Immunology and Oncology, and NanoBiomedicine Initiative, Centro Nacional de Biotecnología (CNB)-CSIC, Madrid, Spain
- 2Centro de Investigación en Sanidad Animal, Centro Nacional Instituto de Investigación y Tecnología Agraria y Alimentaria (CISA-INIA)-CSIC, Valdeolmos, Madrid, Spain
The synthesis and functionalization of iron oxide nanoparticles (IONPs) is versatile, which has enhanced the interest in studying them as theranostic agents over recent years. As IONPs begin to be used for different biomedical applications, it is important to know how they affect the immune system and its different cell types, especially their interaction with the macrophages that are involved in their clearance. How immune cells respond to therapeutic interventions can condition the systemic and local tissue response, and hence, the final therapeutic outcome. Thus, it is fundamental to understand the effects that IONPs have on the immune response, especially in cancer immunotherapy. The biological effects of IONPs may be the result of intrinsic features of their iron oxide core, inducing reactive oxygen species (ROS) and modulating intracellular redox and iron metabolism. Alternatively, their effects are driven by the nanoparticle coating, for example, through cell membrane receptor engagement. Indeed, exploiting these properties of IONPs could lead to the development of innovative therapies. In this review, after a presentation of the elements that make up the tumor immunological microenvironment, we will review and discuss what is currently known about the immunomodulatory mechanisms triggered by IONPs, mainly focusing on macrophage polarization and reprogramming. Consequently, we will discuss the implications of these findings in the context of plausible therapeutic scenarios for cancer immunotherapy.
Introduction
The highly innovative field of nanotheranostics has been expanding now for more than two decades, with easy-to-scale nanomaterials emerging as potential candidates to treat a variety of pathologies, such as cancer (1–4), autoimmune diseases (5, 6) or neurodegenerative disorders (7, 8). The therapeutic interest in nanomaterials, and particularly in nanoparticles, is in part kindled by the chemical and physical versatility of these materials. Nanoparticles can be functionalized with targeting moieties (9) or drugs (10), and their surface can be built for specific biomolecule release using molecular domains responsive to stimuli like pH (11, 12) or reactive oxygen species (ROS (13, 14). In addition, they also possess physical properties associated with their core that can be exploited, such as magnetism (15) and plasmon coupling (16).
Iron oxide nanoparticles (IONPs) are of particular therapeutic interest due to their magnetic properties and their flexibility for surface functionalization. IONPs have been used as contrast agents and as heat-inducers through the application of an external magnetic field (17, 18). Their versatility in terms of surface functionalization means they can target diverse molecules and they can be used to ensure the correct localized delivery of different cargos, such as drugs, RNAs, cytokines or antibodies (15). Importantly, IONPs also exhibit intrinsic biological activity in cellular systems, including the immune system, which can be exploited to broaden their therapeutic potential. This review will first outline the main characteristics of the tumor microenvironment (TME), emphasizing the influence of tumor-associated macrophages (TAMs), and subsequently, we will address the impact that IONPs have on macrophage reprogramming and the implications of this for cancer immunotherapy.
Immunological Tumor Microenvironment
Cancer is a complex and heterogeneous disease that involves the dysregulation of various cell processes, such as metabolism (19), proliferation (20), intracellular pH dynamics (21), redox signaling (22), and migration/invasion (23, 24). The complexity of this disease is also reflected by the different ecosystems that constitute a permissive TME (25, 26). A close inspection of the TME reveals a network of cellular and non-cellular components that provide the signals that control tumor cell survival, proliferation, angiogenesis, immune evasion and metastasis. We can divide the TME landscape into three ecosystems: 1) the cellular compartment; 2) the soluble factors; and 3) the extracellular matrix (ECM: Figure 1 and Table 1). The tumor niche is a very dynamic 3D structure in which stromal cells play a crucial role in regulating different stages of tumor development and in which there is also an intricate interplay among these cells. The TME cell ecosystem also includes a plethora of non-immune stromal cell types, such as cancer-associated fibroblasts (CAFs (54), mesenchymal stem cells (MSCs), pericytes, adipocytes, endothelial and vascular cells. Notably, these cells exhibit a high degree of plasticity and they may originate through trans-differentiation. For instance, breast cancer CAFs may stem from resident fibroblasts, from breast epithelial cells via the epithelial-to-mesenchymal transition (EMT) or from pericytes in the perivascular niche (55, 56). CAFs may also be derived from bone marrow-derived mesenchymal stem cells (BM-MSCs), as PDGFR-α−, CD45−, CD34− BM-MSCs are recruited into primary breast tumors to differentiate into α-SMA+, PDGFR-α−, CD45−, CD34− CAFs (57). This fact highlights the complex transcriptional reprogramming that many stromal cells go through, suggesting that the cellular ecosystem in the TME is in constant transcriptional flux (58, 59). Indeed, this dynamic transcriptional program is likely to constantly redefine the immunological landscape of the TME.
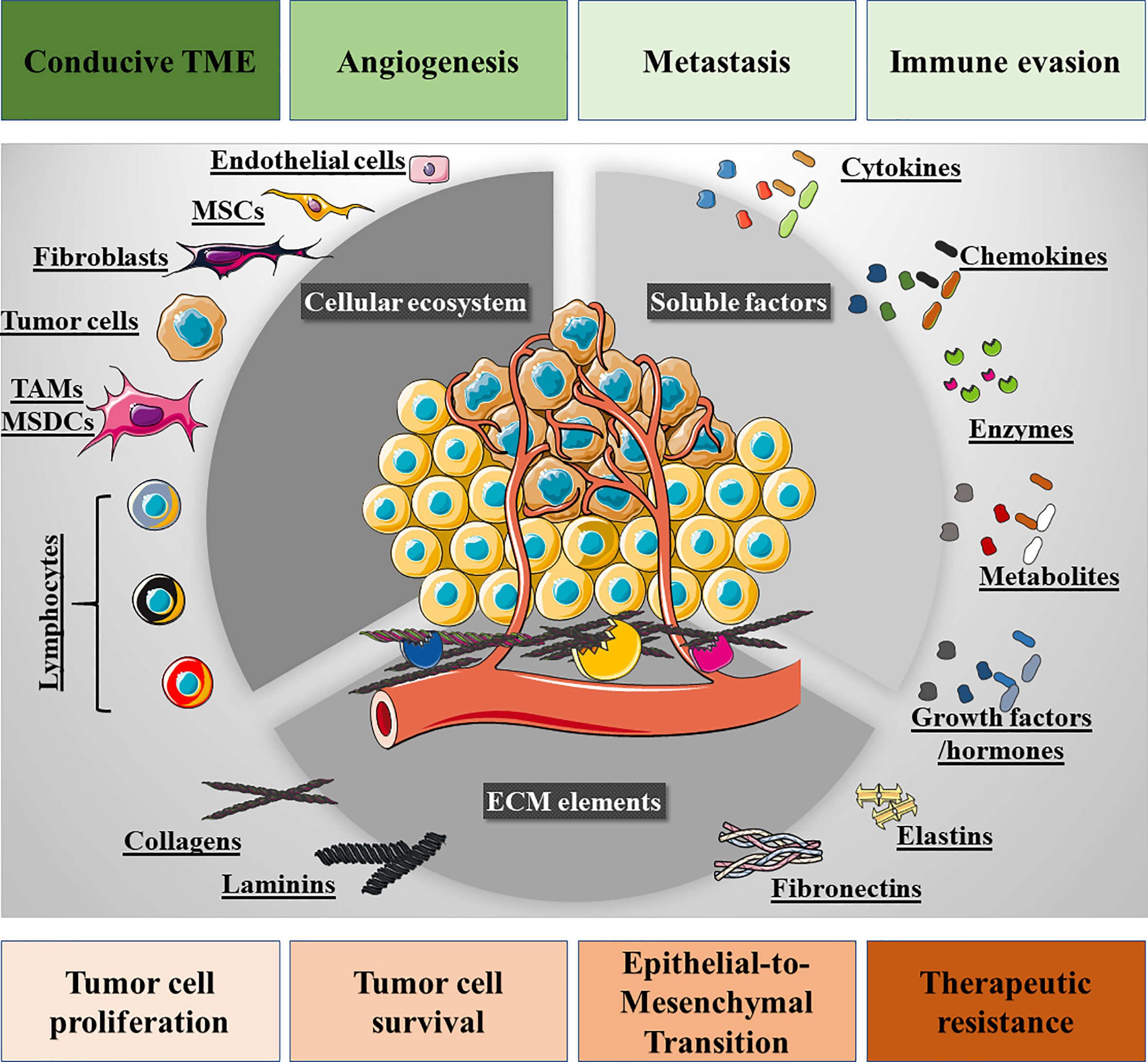
Figure 1 Overview of the tumor microenvironment (TME). Three ecosystems contribute to the TME: firstly, the cellular ecosystem that is composed of immune cells (lymphoid and myeloid), fibroblasts, mesenchymal stem cells (MSCs), pericytes, endothelial cells, and tumor cells. Secondly, the cell-to-cell membrane interactions and soluble secreted factors that participate in the intricate interplay among these cells, e.g., cytokines, chemokines, growth factors, hormones, proteolytic enzymes, and metabolites. Thirdly, the extracellular matrix (ECM) bed on which the cellular ecosystem resides, also providing biological signals to the tumor and stromal cells through ECM-derived peptides and the structural domains of its proteins. The interplay of these signaling networks and ecosystems promotes tumor cell proliferation, survival, epithelial-to-mesenchymal transition, drug resistance and loco-regional modulation, such that the TME is conducive to tumor cell invasion and metastatic spreading, angiogenesis and immune cell evasion.
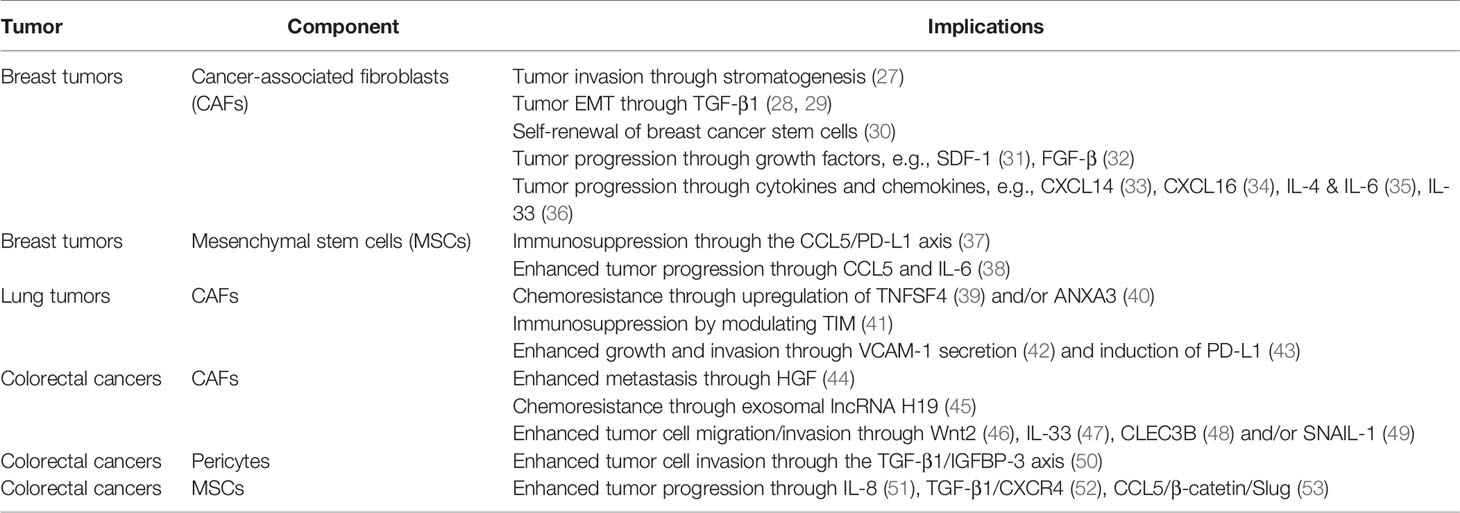
Table 1 Examples of TME ecosystems and their implications in the progression of three significant cancers: breast, lung and colorectal.
The TME is also comprised of tumor-infiltrating immune cells, both innate immune cells (monocytes, macrophages, and NK cells) and adaptive immune cells (T and B cells), that define the tumor immune microenvironment (TIME). Dynamic communication takes place within this ecosystem that are mediated by cell-to-cell contacts and cell-derived soluble factors. The intermediates derived from stromal and tumor cells, such as cytokines, chemokines, and ROS, promote immune evasion by inducing CD8+ T cell anergy/exhaustion, T regulatory (Tregs) cells, suppressor dendritic cells (DCs), and M2 macrophage differentiation (60). As a result, tumors escape immune surveillance and they adopt a metastatic phenotype through modulation of the EMT, enhanced angiogenesis and ECM degradation.
The non-cellular TME network is comprised of ECM components [e.g., collagens (61), fibronectin (62), elastin (63), and laminin (64)], and soluble cellular derivatives [e.g., cytokines, chemokines (65), hormones (66), metabolites (67, 68) and growth factors (69)]. This non-cellular network is responsible for cell-to-cell crosstalk, ultimately shaping the pro-malignant environment.
However, the immunological landscape within the TME has emerged as a crucial variable for cancer progression and treatment, and understanding the TIME has become a critical step in designing efficient immunotherapies for cancer. Indeed, the TIME defines the prognosis of cancer patients (70, 71) and their therapeutic response to immunotherapies like checkpoint inhibitors (72, 73), T-cell transfer (74), or therapeutic vaccines (75). Driven by tumor cell plasticity, the TIME is a dynamic system where diverse innate and adaptive immune cells co-exist, continually changing over time in response to the reprogramming of tumor cell transcription (Figure 2). To better comprehend the TIME’s influence on cancer prognosis, the TIME can be divided into the T cell-inflammatory microenvironment and non-T cell-inflammatory microenvironment. The first of these is characterized by the infiltration of T cell subsets and macrophages, whereas the second is mainly composed of TAMs. Of all immune cells, TAMs play a pivotal role in defining the tumor immunological landscape and thus, they have been the target of various therapeutic approaches.
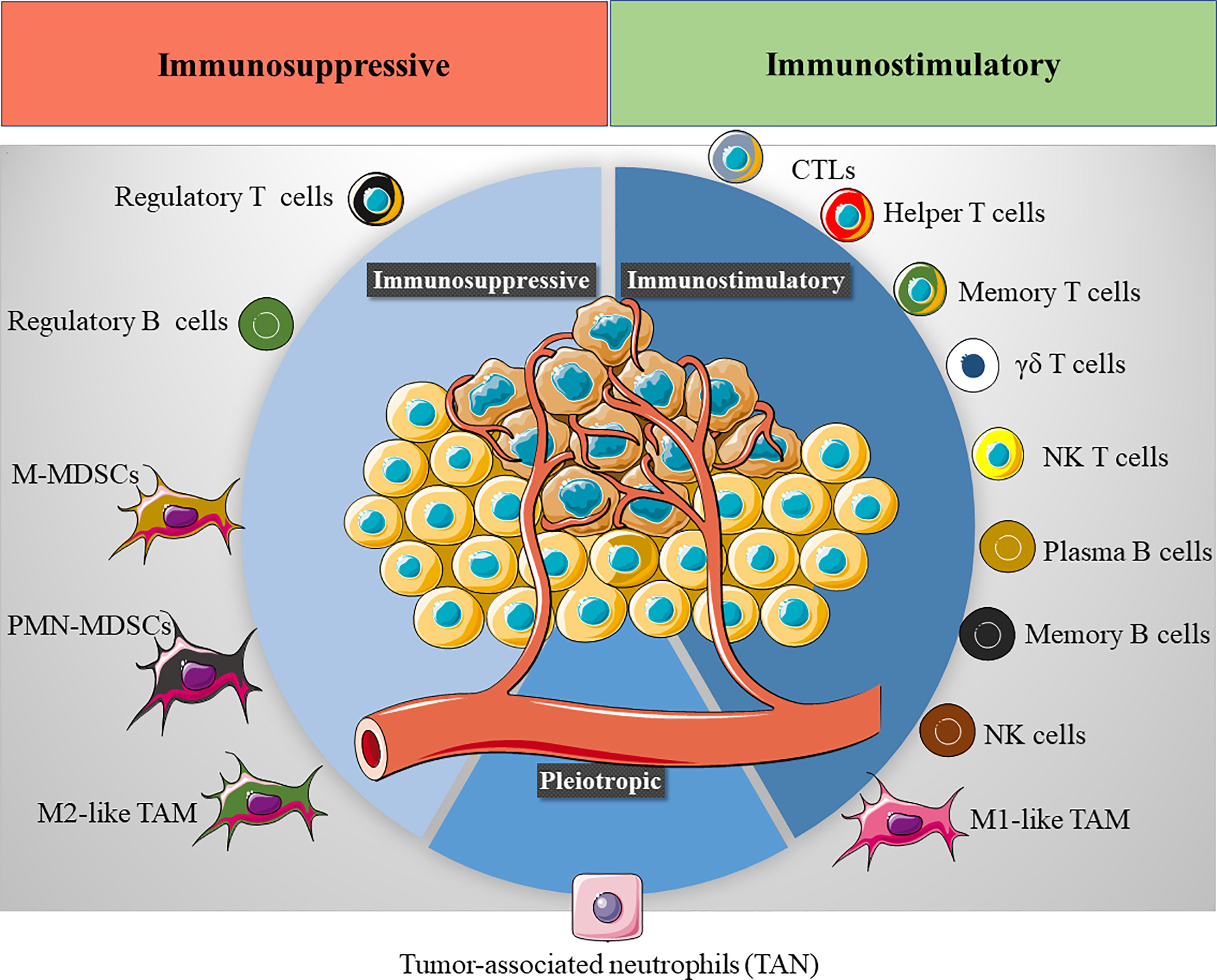
Figure 2 The tumor immune microenvironment (TIME). Several immune cells are found in the TIME, exhibiting either an immunostimulatory (CTLs, cytotoxic T cells, helper T cells, memory T cells, γδ T cells, NK T cells, plasma B cells, memory B cells, NK cells and M1-like TAMs) or immunosuppressive phenotype (Tregs cells, regulatory B cells, M-MDSCs, monocytic monocyte-derived suppressor cells, PMN-MDSCs, polymorphonuclear monocyte-derived suppressor cells and M2-like TAMs). The final immunological response in the TME will depend on the balance between these immunomodulatory populations.
Immunosuppressive Tumor-Associated Macrophages
TAMs are tumor-enriched immunosuppressor cells that exert a pivotal influence on tumor progression and metastasis. Since their first description 30 years ago (76), TAMs have been characterized as potent pro-tumorigenic agents that act primarily by modulating the natural (and induced) anti-tumor response, ECM remodeling, and inducing angiogenesis, not only leading to tumor cell survival and proliferation but also, to their dissemination (Figure 3). It is currently accepted that the TAM phenotype resembles the alternatively activated macrophage M2 phenotype (Arginase 1+, CD163+, CD206+, CD209+, FIZZ1+, and Ym1/2+), which can be subdivided into four subtypes: M2a, M2b, M2c, and M2d (77). These subtypes are generated by the stimuli triggering macrophage differentiation and some specific phenotypic markers (Table 2). However, it is generally accepted that TAMs retain a high degree of plasticity, permitting several different subtypes to co-exist simultaneously and their trans-differentiation into each different subtype depending on the TME signals available.
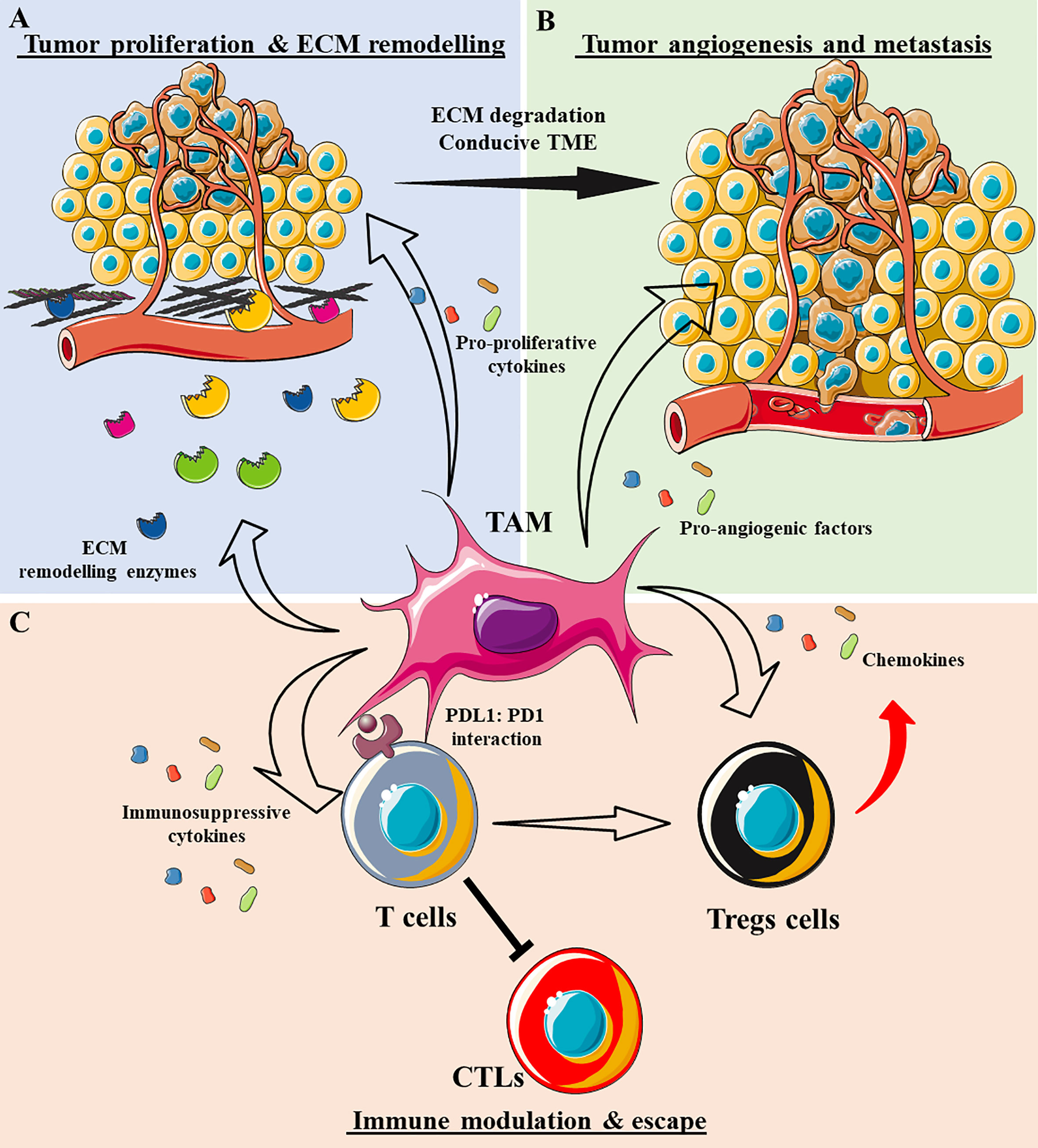
Figure 3 The role of tumor-associated macrophages (TAMs) in shaping the tumor microenvironment (TME). (A) TAMs secrete a plethora of enzymes that degrade ECM components, such as metalloproteinases (MMPs), cathepsins, disintegrin and metalloproteinase (ADAM)-family proteases, and tissue inhibitors of metalloproteinases (TIMPs). As a result, the ECM becomes destructured and conducive to tumor cell invasion. TAMs also secrete cytokines that support tumor cell proliferation, e.g., TGF-β1, IL-10, IL-6, IL-1β, and EGF. (B) TAMs secrete various pro-angiogenic factors that induce vessel formation, e.g., VEGF-A, bFGF, IL-6, and TNFα. Together with ECM degradation, tumor angiogenesis permits the systemic dissemination of tumor cells. (C) TAMs adopt an immunosuppressive phenotype by secreting many anti-inflammatory cytokines/chemokines, e.g., IL-10, TGF-β1, CCL17, CCL18, and CCL22, inhibiting cytotoxic T cells (CTLs) and attracting or differentiating T cells into regulatory T cells. TAMs can also exhaust CTLs by direct engagement of anti-inflammatory cognates receptors like PD1-PD-L1.

Table 2 | M2 macrophage subtypes and their involvement in tumor development.
In general, blood monocytes infiltrate the TME, and along with the tumor-resident macrophages, they represent a source of TAMs. In this context, tumor cells shape the macrophage’s immunosuppressive phenotype by secreting anti-inflammatory interleukins and other metabolites. The TAMs then inhibit tumor-infiltrating T cells directly through receptor-ligand cognates [e.g., PD-1:PD-L1 (84)] or by releasing anti-inflammatory cytokines (IL-10, TGF-β1, and IL-6). Concomitantly, the TAMs can produce different proteolytic enzymes such as metalloproteinases (MMPs), cathepsins, and disintegrin and metalloproteinase-like proteases (ADAMs), thereby producing a profound ECM remodeling. Consequently, the ECM becomes conducive to invasion, and it facilitates tumor cell dissemination into the surrounding tissue and peripheral circulation. TAMs can further enhance tumor invasiveness by inducing angiogenesis, mediated by various cytokines and growth factors like VEGF-A (85) and IL-8 (86). Since TAMs are involved in tumor progression, the induction of a specific phenotype that switches these cells towards a pro-immunogenic profile has been proposed as an attractive therapeutic tool to enhance local anti-tumor immune responses.
The modulation of TAM activity is a plausible and promising therapeutic approach to combat tumors when combined with cancer immunotherapies. Indeed, multiple drugs that modulate the pro-tumor activity of TAMs have been tested, including bisphosphonates (87) and zoledronic acid (88) in particular, or chemotherapeutic drugs like docetaxel and cyclophosphamide (89). While zoledronic acid can revert the M2 TAM phenotype in breast tumors into an M1-like phenotype or induce TAM apoptosis, the chemotherapeutic drugs can promote an M1-phenotype that secretes pro-inflammatory cytokines like IL-12, thus driving an anti-tumor effect. In this context, nanoparticles that modulate TAM activity, particularly IONPs, provide new and innovative tools to prolong anti-tumor responses in situ.
Intrinsic Modulation of the TIME by Iron Oxide Nanoparticles (IONPs)
IONPs have been studied extensively as an effective magnetic nanocarrier for various cargos, such as drugs (15), cytokines (90, 91), siRNAs (92), and adjuvants (93). There are several motives for the increasing interest in IONPs as nanocarriers. First, the IONP core responds to an external electromagnetic field that permits their use in applications like magnetic targeting, magnetic resonance imaging (MRI) or the induction of local hyperthermia. Second, mammalian cells have efficient iron metabolism that can prevent the cells from suffering iron-related toxicity. Third, the IONP surface provides a chemical interface that can be easily modified with a number of polymers and moieties, which when combined with the high surface-to-volume ratio, facilitate the delivery of wide range of cargoes.
However, IONPs also produce interesting intrinsic biological effects that provide added therapeutic benefits to IONP-based nanomedicines. We demonstrated that polyethyleneimine (PEI)-coated IONPs can inhibit the migration and invasion of tumor cells (94), and impair angiogenesis (95). More importantly, the intrinsic biological effects of IONPs arise from their surface coating and the surrounding protein corona, as well as the free intracellular iron derived from IONP degradation. While IONP surface microdomains are primarily involved in the nanoparticle’s interaction with cell membrane receptors, soluble factors, and intracellular components, the released intracellular iron actively changes the intracellular redox status through the Fenton reaction (96), modulating several iron-regulated genes. Since macrophages contribute to the TIME, their interaction with IONPs can define the theranostic outcome and provide an invaluable tool to reprogram the phenotype of TAMs. The most recent findings on how IONPs affect macrophage activation are summarized in Table 3.

Table 3 | Example of the effects of iron oxide nanoparticles on macrophage polarization.
To understand how IONPs affect macrophage polarization, we have to consider the internalization process as at least three different steps, during which IONPs can engage with different signaling cascades: 1) IONP interaction with the cell membrane; 2) endocytosis and endolysosomal trafficking; and 3) IONP degradation. In each step the IONPs are exposed to diverse biological milieu and ultimately, this determines the indirect or direct engagement that drives macrophage transcriptional reprogramming and shifts in phenotype. This effect on transcriptional reprogramming of macrophages has been assessed by several groups whereby key transcription factors such as STAT family (107) and c-Fos/c-Jun complex (107) are upregulated upon IONP treatment. Noteworthy, IONPs appear to induce a variety of transcription factors related to MAPK pathways and the innate response, including the TLR-AP-1 signaling pathway (108). This complex reprogramming was revealed by Liu Y et al., who observed that the DMSA-coated IONPs engaged the activation of the signaling pathways mentioned above (107). Therefore, the IONPs can trigger a multifactorial transcription reprogramming of macrophages where several signaling pathways are involved.
It is important to note that among the transcriptional reprogramming that IONPs can induce in macrophages, some are related to cell death processes such as apoptosis, ferroptosis, and autophagy. The balance between all signaling pathways activated by a particular IONPs will determine the macrophage fate. In this review, we focus on the transcriptional reprogramming of macrophage response in terms of the immune response and suggest other comprehensive and recent studies on the toxicity of IONPs that can be more thorough in this sense (109, 110).
The coating of IONPs influences their interaction with cell membrane-associated proteins like receptors, thereby triggering signaling cascades that can activate macrophages. As such, IONPs with a positively charged coating consistently polarize macrophages towards a M1-like phenotype. Indeed, when macrophages are treated with PEI-coated IONPs, a straightforward program of M1 activation occurs, enhancing co-stimulatory receptors like CD40, CD80, and CD86, along with the secretion of the pro-inflammatory cytokine, IL-12 (101). When analyzing the transcriptional reprogramming induced by PEI-coated IONPs, several pro-inflammatory genes were seen to be upregulated (i.e., Il1b, Tnfa, Ccl2 and Il6). However, the most exciting finding was the involvement of the toll-like receptor 4 (TLR-4) in PEI-coated IONP-induced macrophage activation (101). The PEI polymer appears to engage TLR-4 activation, stimulating the mitogen-activating protein kinase (MAPK). Two commercially available IONPs (carboxydextran-coated Resovist and carboxylmethyl-dextran coated feraheme) have also been demonstrated to induce macrophage activation through TLR-4 engagement, indicating that different IONP coatings can activate macrophages in this way, although activation by these IONPs induces autophagy (102). Other effects of IONPs were at least partly associated with different TLRs, including the cell membrane TLR2, TLR4, and TLR6, and the intracellular TLR8. Indeed, IONP size influences TLR activation as a relatively small IONP (10 nm) can enhance TLR2, TLR6, TLR4, and TLR8-induced cytokine secretion in peripheral blood, whereas a larger IONP (30 nm) only affects TLR2 and TLR6-dependent cytokine secretion (108). Although a direct interaction between the IONPs and the cell surface TLRs has yet to be demonstrated, the dependence of cytokine enhancement on the formation of a complex between TLR4/MD2 and the CD14 co-receptor suggests that a physical interaction between the TLR4 complex and IONPs could be responsible for the increase in TLR4 activity. However, elsewhere IONPs were shown to interfere with TLR4 agonist activation, suggesting that this mechanism could depend on the type of IONP (111).
In addition, it has been shown that IONPs with opposite surface charges promote similar macrophage repolarization. Two opposite charged IONPs induced an M1-like phenotype in RAW 264.7 macrophages, although negatively charged IONPs appeared to be more potent in promoting this effect (98) and neutral IONPs have a negligible impact. The crucial role that such M1-differentiated macrophages can play within the TIME was also addressed and there was significant tumor growth retardation when IONP+ or IONP- treated macrophages were co-inoculated with HT1080 human fibrosarcoma cells, reflecting the anti-tumor effect of these repolarized M1-like macrophages (98).
IONP morphology also plays a critical role in determining the degree of macrophage activation. Using IONPs with four distinct morphologies (octopod, plate, cube, and spherical), yet with a comparable aspect ratios and surface charge, the IONPs with an octopod or plate morphology were seen to significantly activate the inflammasome, as measured by IL-1β secretion (112). More importantly, this dependence on morphology appeared to be related to the nanoparticle’s capacity to induce ROS production. IONP size also affected the extent of inflammasome activation in macrophages, with spherical IONPs of ~30 nm inducing significantly more IL-1β release than larger spherical IONPs of ~80 and 120 nm (113). ROS production appears to be a common molecular mechanism for the effect of IONPs on macrophage activation, although this result also seems to depend on lysosomal destabilization and may reflect another common phenomenon. The involvement of ROS in IONP-induced macrophage activation is related to the central role these metabolites play in macrophage cell biology as short-lived second messengers. ROS mediate the oxidation of thiol groups in several proteins, altering their structure and hence, their function. The MAPK pathway is ROS-sensitive and it regulates several biological processes like cell proliferation, apoptosis, and the innate immune response. In this regard, ROS have been implicated in the induction and maintenance of an M1-like status of macrophages through the activation of NFκB and p38 MAPK signaling. In the former situation, ROS trigger the phosphorylation of the NFκB inhibitor, IκB, thereby activating NFκB (114). In the latter, ROS induce the phosphorylation of the apoptosis signaling-regulating kinase 1 (ASK1) and the downstream activation of the p38 MAPK (115). However, ROS can either activate or inhibit NFκB in a context-dependent manner, highlighting the need to characterize the effect of IONP-triggered ROS production on NFκB activation in a cell-type and context-dependent manner (116). In addition to MAPK, the phosphoinositide-3 kinase (PI3K) is also regulated by ROS, sensitizing macrophages to hormone, cytokine, and growth factor signaling (117).
IONP phagocytosis can lead to autophagy, as is the case for the two FDA-approved IONPs, resovist and ferumoxytol that induce the appearance of an early autophagic vacuole and eventually, IONPs-containing double-membrane autophagic vacuoles, small internal vesicles, and cellular and membrane debris (102). These events were accompanied by the accumulation of LC3 puncta and overexpression of the p62/SQSTM1-positive sequestosome (118–120). In accordance with the involvement of TLRs in this effect, the TLR4-p38-Nrf2 pathway appears to mediate IONP-induced autophagy as opposed to the classic autophagy machinery dependent on ATG5/12. Indeed, pre-treatment with the TLR4 signaling inhibitor, CLI-095, prevented IONP-loaded macrophages from inducing the aforementioned structural changes (102).
Importantly, each macrophage phenotype expresses different factors involved in iron metabolism, reflected in their distinct iron sensitivity (121). For instance, M2-polarized THP1 macrophages internalize significantly more IONPs than M1-polarized and M0 macrophages, leading to a higher T1 signal in M2 macrophages and a higher T2* signal in M0 macrophages (122). In turn, internalized IONPs could also exert effects on polarization and iron metabolism. Indeed, our group demonstrated that DMSA-, APS-, and aminodextran-coated IONPs shifted iron metabolism towards an iron-sequestering status in M2-like macrophages (103). In the light of the above, we can propose a general overview of the events induced by IONPs that precipitates macrophage activation (Figure 4).
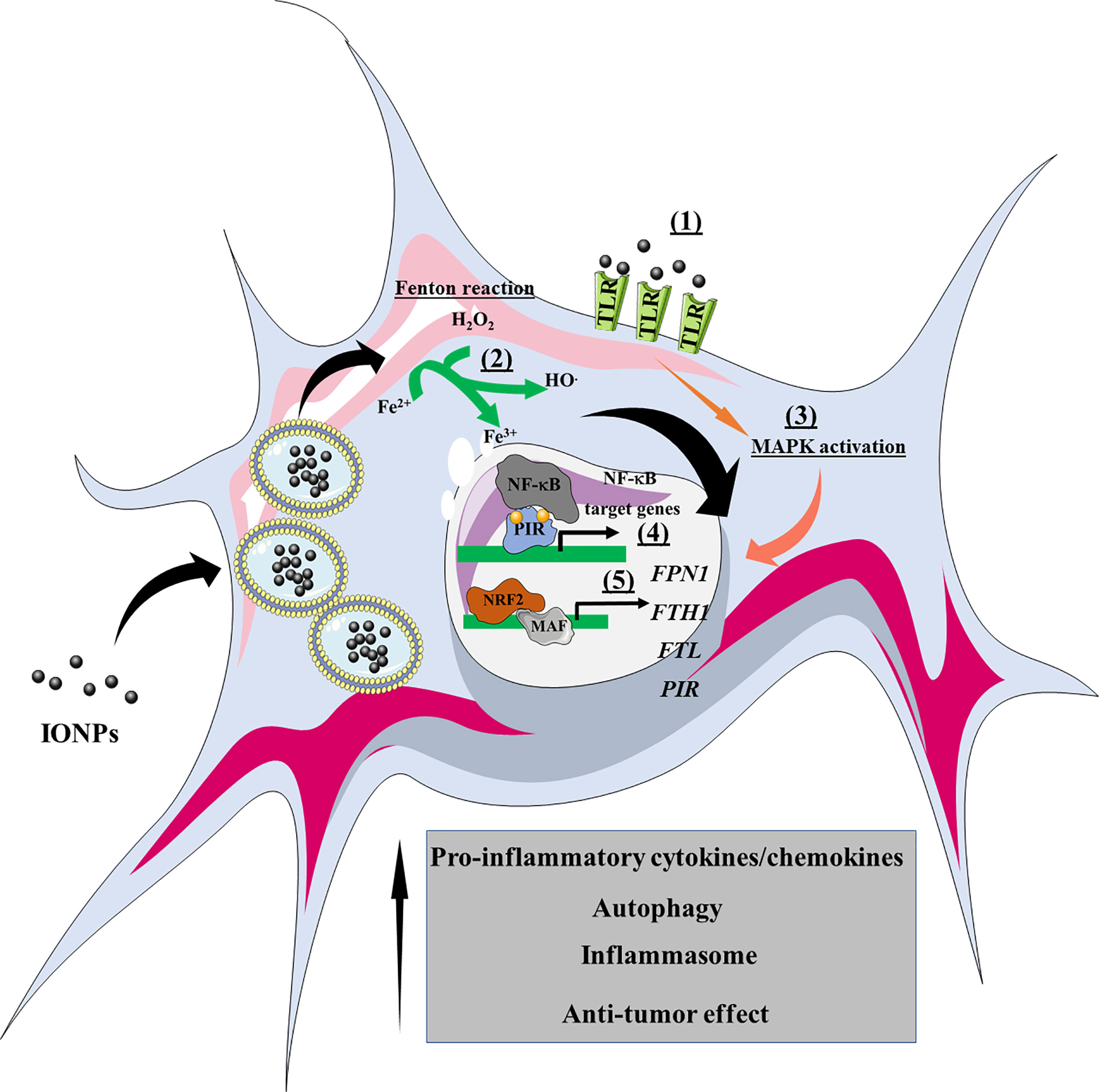
Figure 4 Overview of the effects of IONPs on macrophage polarization. The IONPs can interact with cell surface receptors such as TLRs (1), leading to activation of the MAPK signaling pathway. Once internalized by macrophages, the IONPs are enclosed within endolysosomes where they are biodegraded. Consequently, atomic iron is released into the cytoplasm, where it engages the Fenton reaction and produces ROS (2). As a result, transcriptional reprogramming is triggered, such as that involving NF-κB (e.g., cytokines, chemokines) and NRF2 target genes (e.g., iron metabolism). NRF2, nuclear factor (erythroid-derived 2)-like 2; PIR, Pirin; FPN1, ferroportin-1; FTH1, ferritin heavy chain; FTL, ferritin light chain; MAPK, mitogen-activated protein kinases; MAF, musculoaponeurotic fibrosarcoma.
IONPs have also been used to track microglia and assayed as a potential nanocarrier in brain tumors. Microglia are highly phagocytic cells found entirely in the central nervous system (CNS) where they protect the nervous tissue from debris and damaged CNS structures and from viruses, microorganisms, and tumors (123–126). Therefore, like macrophages, microglia can phagocytose IONPs and react to them. In this sense, Wu HY et al. found that the carboxydextran-coated IONP (Resovist™) counteract the LPS-induced microglia activation by directly decrease IL-1β secretion (127), suggesting IONPs can protect CNS from an exacerbated inflammation. However, other reports pinpoint the involvement of IONPs in recruiting and activating microglia in CNS structures such as the olfactory bulb, hippocampus, and striatum. Indeed, Wang Y et al. found that Fe2O3 IONPs administered intranasally promote the recruitment of microglia into the above CNS structures and induced microglia activation and proliferation, with ROS and nitric oxide (NO) production, as a possible defense mechanism against foreign particulates (128). Thus, IONPs appear to change CNS immunological microenvironment toward an inflammatory or anti-inflammatory phenotype, highlighting the need to comprehend these effects in the context of brain tumors.
Therapeutic Iron Oxide Nanoparticle-Enabled Modulation of TIME
We have discussed the activation of macrophages by IONPs and the molecular mechanisms mediating these effects. Considering the intrinsic biological activity of IONPs on macrophages, their application in therapeutic and prophylactic vaccination schemes has emerged as an attractive therapeutic approach to treat cancer. This approach relies on the possibility of combining the carrier capacity of IONPs with their by-stander activation of macrophages within the TIME. A general overview of IONP-based vaccine designs highlights the use of IONPs as an antigen carrier (primarily associated with the tumor cells)with the possible addition of adjuvant and/or a targeting moiety.
The use of IONPs as an antigen carrier in a vaccination schedule takes advantage of the intrinsic capacity of the IONPs to drive macrophages or DCs towards a pro-inflammatory phenotype. Consequently, antigen internalization, intracellular processing, and restricted major histocompatibility complex (MHC) presentation to T cells within an inflammatory microenvironment will elicit a robust immune response against the antigen-expressing tumor cells. A simple vaccine formulation has been tested by loading ovalbumin (OVA) onto IONPs, demonstrating that this formulation could activate bone marrow-derived dendritic cells (BMDCs) and RAW 264.7 macrophages. However, the most exciting finding was that prophylactic or therapeutic injection of three doses of this preparation delayed OVA-expressing B16 tumor cell growth.
Interestingly, OVA-coated IONPs effectively prevented lung metastasis from OVA-expressing cells (129). Likewise, the sole conjugation of OVA alone with IONPs was sufficient to elicit potent DC and macrophage activation, and to reduce the OVA-expressing CT26 tumor burden in vivo (130). This anti-tumor effect appeared to be mediated by the induction of pro-inflammatory cytokines like IL-6, TNF-α, and IFN-γ.
Other studies have addressed the potential of the IONPs as carriers of tumor-associated antigens in vaccine designs. For example, the administration of self-assembled MUC1 lipo(glycol)peptide-coated IONPs elicited a strong antibody response, prompting an antibody profile able to recognize the MUC1-expressing tumor cell line, MCF7 (131). In this scenario, the anti-tumor effect seems more likely to be related to the enhanced activation of plasma B cells due to the high number of lipo(glycol)peptide copies presented on the IONP surface. However, we cannot rule out a direct effect on macrophages or DCs.
It is desirable that macrophage-based anti-tumor therapy induces naive macrophages to adopt a M1 phenotype and that it switches the resident M2 program into a M1 phenotype, ensuring a pro-inflammatory and anti-tumor TIME. It was seen that hyaluronic acid-modified IONPs or bare IONPs trigger the production of ROS and pro-inflammatory cytokines (132). Consequently, IONP-treated macrophages exerted an anti-tumor effect on the murine 4T1 breast-tumor cell line in a cell contact-independent manner, inducing active caspase 3 and inhibiting cell proliferation. Notably, hyaluronic acid-modified IONPs induced M1 macrophages resistant to M2-inducing factors and M2-to-M1 macrophage reversion (132). IONP intracellular degradation also increases the labile iron pool, providing another element that can modulate the TIME. It was shown that red blood cells (RBCs) were responsible for the presence of iron-loaded macrophages nesting in the invasive margins of non-small lung cell tumors, which were in turn correlated with a smaller tumor size (133). Indeed, hemolytic RBCs triggered TAM polarization toward a M1-like phenotype, as evident by the expression of M1 marker transcripts (Il6, Nos2, and Tnfa) and their increased anti-tumor activity (133). More importantly, IONPs injected intravenously in Lewis lung carcinoma (LLC)-bearing mice accumulated within F4/80 macrophages and reduced tumor growth, indicating that these IONPs have a similar effect reverting M2 macrophages to a M1 phenotype (133).
Advantages have also been reported when a combination of antigen-coated IONPs and adjuvant-coated IONPs is used therapeutically. While IONPs were initially used as antigen carriers, adjuvant and nanoparticle association enhanced the adjuvant effect on the respective signaling pathway. Indeed, co-delivery of polyIC-R837@mPEG-PL-OA-IONPs (as TLR3-7 agonists) and OVA@mPEG-PL-OA-IONPs (as antigen) delayed tumor growth in OVA-expressing B16-bearing animals and led to tumor-free survival in some individuals, probably through an enhanced agonist effect on TLR signaling. The increase in the ferroptosis process induced by IONP-derived iron further promoted an antitumoral TME, indicating that the IONPs provide not only transport but also an intrinsic potential to change the TME toward an anti-tumor phenotype (134). Table 4 summarizes the most recent approaches using IONPs in anti-tumor vaccination regimens.

Table 4 Use of IONPs in vaccine formulation.
Conclusions
IONPs have been studied intensively in recent decades to exploit their magnetic and surface chemical features. However, only recently has attention been drawn to their intrinsic immunomodulatory properties, especially their effects on macrophages. These effects are particularly important in the context of cancer immunotherapy as IONPs can provide an efficient vehicle for antigen delivery and elicit a potent immune response, reprogramming TAMs toward an immunogenic phenotype. Two main molecular mechanisms can explain the intrinsic immunomodulatory effect of IONPs: 1) the production of ROS and consequently, the modulation of redox-sensitive signaling pathways; and 2) the direct engagement and activation of immune response-related receptors, such as TLRs, inducing transcriptional reprogramming in macrophages. The use of IONPs can provide a reliable platform to reprogram the typical M2-TAM phenotype toward a pro-immunogenic phenotype, synergizing with currently used immunotherapies like checkpoint inhibitors to mount a potent anti-tumor immune response both locally and systemically.
Author Contributions
VM-A, JR, and DB conceived and designed the review. VM-A and JR wrote sections of the manuscript, and prepared the figures and tables. DB coordinated, critically revised, modified and completed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
VM-A is a post-doctoral scholar working under a Juan de la Cierva-Incorporación Contract (IJCI-2017-31447) from the Spanish Ministry of Science and Innovation. The European Commission-funded VetBioNet INFRAIA-731014 project supports JR. This work was supported in part by grants from the Spanish Ministry of Science and Innovation (SAF-2017-82223-R and PID-2020-112685RB-100 to DB). DFB group is part of the Network “Nanotechnology in Translational Hyperthermia” (HIPERNANO, RED2018-102626-T) supported by the Spanish Ministry of Science and Innovation.
Conflict of Interest
The authors declare that their research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the laboratories led by Domingo F Barber (CNB-CSIC, Madrid) and María del Puerto Morales (ICMM-CSIC, Madrid) for helpful comments and discussion. The authors are also grateful to Mark Sefton (BiomedRed SL) for English language editing of the manuscript.
References
1. Habibi N, Quevedo DF, Gregory JV, Lahann J. Emerging Methods in Therapeutics Using Multifunctional Nanoparticles. WIREs Nanomed Nanobiotechnol (2020) e1625. doi: 10.1002/wnan.1625
2. Khan AU, Khan M, Cho MH, Khan MM. Selected Nanotechnologies and Nanostructures for Drug Delivery, Nanomedicine and Cure. Bioprocess Biosyst Eng (2020) 43:1339–57. doi: 10.1007/s00449-020-02330-8
3. Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in Cancer Therapy: Challenges, Opportunities, and Clinical Applications. J Controlled Release (2015) 200:138–57. doi: 10.1016/j.jconrel.2014.12.030
4. Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and Strategies in Anti-Cancer Nanomedicine Development: An Industry Perspective. Adv Drug Delivery Rev (2017) 108:25–38. doi: 10.1016/j.addr.2016.04.025
5. Gharagozloo M, Majewski S, Foldvari M. Therapeutic Applications of Nanomedicine in Autoimmune Diseases: From Immunosuppression to Tolerance Induction. Nanomed: Nanotechnol Biol Med (2015) 11:1003–18. doi: 10.1016/j.nano.2014.12.003
6. Qamar N, Arif A, Bhatti A, John P. Nanomedicine: An Emerging Era of Theranostics and Therapeutics for Rheumatoid Arthritis. Rheumatology (2019) 58:1715–21. doi: 10.1093/rheumatology/kez286
7. Poovaiah N, Davoudi Z, Peng H, Schlichtmann B, Mallapragada S, Narasimhan B, et al. Treatment of Neurodegenerative Disorders Through the Blood–Brain Barrier Using Nanocarriers. Nanoscale (2018) 10:16962–83. doi: 10.1039/C8NR04073G
8. Ramanathan S, Archunan G, Sivakumar M, Tamil Selvan S, Fred AL, Kumar S, et al. Theranostic Applications of Nanoparticles in Neurodegenerative Disorders. Int J Nanomed (2018) 13:5561–76. doi: 10.2147/IJN.S149022
9. Steichen SD, Caldorera-Moore M, Peppas NA. A Review of Current Nanoparticle and Targeting Moieties for the Delivery of Cancer Therapeutics. Eur J Pharm Sci (2013) 48:416–27. doi: 10.1016/j.ejps.2012.12.006
10. Amreddy N, Babu A, Muralidharan R, Panneerselvam J, Srivastava A, Ahmed R, et al. “Chapter Five - Recent Advances in Nanoparticle-Based Cancer Drug and Gene Delivery”. In: Tew KD, Fisher PB, editors. Advances in Cancer Research. Academic Press. (2018) 137 p. 115–70. doi: 10.1016/bs.acr.2017.11.003
11. Deirram N, Zhang C, Kermaniyan SS, Johnston APR, Such GK. Ph-Responsive Polymer Nanoparticles for Drug Delivery. Macromol Rapid Commun (2019) 40:1800917. doi: 10.1002/marc.201800917
12. Karimi M, Eslami M, Sahandi-Zangabad P, Mirab F, Farajisafiloo N, Shafaei Z, et al. pH-Sensitive Stimulus-Responsive Nanocarriers for Targeted Delivery of Therapeutic Agents. WIREs Nanomed Nanobiotechnol (2016) 8:696–716. doi: 10.1002/wnan.1389
13. Xu X, Saw PE, Tao W, Li Y, Ji X, Bhasin S, et al. Ros-Responsive Polyprodrug Nanoparticles for Triggered Drug Delivery and Effective Cancer Therapy. Adv Mater (2017) 29:1700141. doi: 10.1002/adma.201700141
14. Lv X, Zhu Y, Ghandehari H, Yu A, Wang Y. An ROS-responsive and Self-Accelerating Drug Release Nanoplatform for Overcoming Multidrug Resistance. Chem Commun (2019) 55:3383–6. doi: 10.1039/C9CC00358D
15. Vangijzegem T, Stanicki D, Laurent S. Magnetic Iron Oxide Nanoparticles for Drug Delivery: Applications and Characteristics. Expert Opin Drug Delivery (2019) 16:69–78. doi: 10.1080/17425247.2019.1554647
16. Xia H, Gao Y, Yin L, Cheng X, Wang A, Zhao M, et al. Light-Triggered Covalent Coupling of Gold Nanoparticles for Photothermal Cancer Therapy. ChemBioChem (2019) 20:667–71. doi: 10.1002/cbic.201800648
17. Berry SL, Walker K, Hoskins C, Telling ND, Price HP. Nanoparticle-Mediated Magnetic Hyperthermia is an Effective Method for Killing the Human-Infective Protozoan Parasite Leishmania Mexicana In Vitro. Sci Rep (2019) 9:1059. doi: 10.1038/s41598-018-37670-9
18. Faraj AA, Luciani N, Kolosnjaj-Tabi J, Mattar E, Clement O, Wilhelm C, et al. Real-Time High-Resolution Magnetic Resonance Tracking of Macrophage Subpopulations in a Murine Inflammation Model: A Pilot Study With a Commercially Available Cryogenic Probe. Contrast Media Mol Imaging (2013) 8:193–203. doi: 10.1002/cmmi.1516
19. Nagarajan SR, Butler LM, Hoy AJ. The Diversity and Breadth of Cancer Cell Fatty Acid Metabolism. Cancer Metab (2021) 9:2. doi: 10.1186/s40170-020-00237-2
20. Chen Q, Gao J, Zhao Y, Hou R. Long non-Coding RNA Lbx2-AS1 Enhances Glioma Proliferation Through Downregulating Microrna-491-5p. Cancer Cell Int (2020) 20:411. doi: 10.1186/s12935-020-01433-2
21. Czowski BJ, Romero-Moreno R, Trull KJ, White KA. Cancer and Ph Dynamics: Transcriptional Regulation, Proteostasis, and the Need for New Molecular Tools. Cancers (Basel) (2020) 12:2760. doi: 10.3390/cancers12102760
22. Kaweme NM, Zhou S, Changwe GJ, Zhou F. The Significant Role of Redox System in Myeloid Leukemia: From Pathogenesis to Therapeutic Applications. Biomark Res (2020) 8:63–3. doi: 10.1186/s40364-020-00242-z
23. Ruan Z, Deng H, Liang M, Xu Z, Lai M, Ren H, et al. Overexpression of Long non-Coding RNA00355 Enhances Proliferation, Chemotaxis, and Metastasis in Colon Cancer Via Promoting GTF2B-mediated Itga2. Transl Oncol (2021) 14:100947–7. doi: 10.1016/j.tranon.2020.100947
24. Hudson LG, Gillette JM, Kang H, Rivera MR, Wandinger-Ness A. Ovarian Tumor Microenvironment Signaling: Convergence on the Rac1 Gtpase. Cancers (Basel) (2018) 10:358. doi: 10.3390/cancers10100358
25. Bhat AA, Yousuf P, Wani NA, Rizwan A, Chauhan SS, Siddiqi MA, et al. Tumor Microenvironment: An Evil Nexus Promoting Aggressive Head and Neck Squamous Cell Carcinoma and Avenue for Targeted Therapy. Signal Transduct Targeted Ther (2021) 6:12. doi: 10.1038/s41392-020-00419-w
26. Song SG, Kim S, Koh J, Yim J, Han B, Kim YA, et al. Comparative Analysis of the Tumor Immune-Microenvironment of Primary and Brain Metastases of non-Small-Cell Lung Cancer Reveals Organ-Specific and EGFR Mutation-Dependent Unique Immune Landscape. Cancer Immunol Immunother (2021). doi: 10.1007/s00262-020-02840-0
27. Giatromanolaki A, Sivridis E, Koukourakis MI. The Pathology of Tumor Stromatogenesis. null (2007) 6:639–45. doi: 10.4161/cbt.6.5.4198
28. Lebrun J-J. The Dual Role of Tgfβ in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol Biol (2012) 2012:381428–8. doi: 10.5402/2012/381428
29. Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. Tgf-β: Duality of Function Between Tumor Prevention and Carcinogenesis. J Natl Cancer Inst (2014) 106:djt369–9. doi: 10.1093/jnci/djt369
30. Wang B, Xi C, Liu M, Sun H, Liu S, Song L, et al. Breast Fibroblasts in Both Cancer and Normal Tissues Induce Phenotypic Transformation of Breast Cancer Stem Cells: A Preliminary Study. PeerJ (2018) 6:e4805–5. doi: 10.7717/peerj.4805
31. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis Through Elevated Sdf-1/CXCL12 Secretion. Cell (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
32. Suh J, Kim D-H, Lee Y-H, Jang J-H, Surh Y-J. Fibroblast Growth factor-2, Derived From Cancer-Associated Fibroblasts, Stimulates Growth and Progression of Human Breast Cancer Cells Via FGFR1 Signaling. Mol Carcinogene (2020) 59:1028–40. doi: 10.1002/mc.23233
33. Sjöberg E, Meyrath M, Milde L, Herrera M, Lövrot J, Hägerstrand D, et al. A Novel Ackr2-Dependent Role of Fibroblast-Derived CXCL14 in Epithelial-to-Mesenchymal Transition and Metastasis of Breast Cancer. Clin Cancer Res (2019) 25:3702. doi: 10.1158/1078-0432.CCR-18-1294
34. Allaoui R, Bergenfelz C, Mohlin S, Hagerling C, Salari K, Werb Z, et al. Cancer-Associated Fibroblast-Secreted CXCL16 Attracts Monocytes to Promote Stroma Activation in Triple-Negative Breast Cancers. Nat Commun (2016) 7:13050–0. doi: 10.1038/ncomms13050
35. Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer Associated Fibroblasts Promote Tumor Growth and Metastasis by Modulating the Tumor Immune Microenvironment in a 4T1 Murine Breast Cancer Model. PloS One (2009) 4:e7965–5. doi: 10.1371/journal.pone.0007965
36. Shani O, Vorobyov T, Monteran L, Lavie D, Cohen N, Raz Y, et al. Fibroblast-Derived IL33 Facilitates Breast Cancer Metastasis by Modifying the Immune Microenvironment and Driving Type 2 Immunity. Cancer Res (2020) 80:5317. doi: 10.1158/0008-5472.CAN-20-2116
37. Aboulkheyr Es H, Bigdeli B, Zhand S, Aref AR, Thiery JP, Warkiani ME. Mesenchymal Stem Cells Induce PD-L1 Expression Through the Secretion of CCL5 in Breast Cancer Cells. J Cell Physiol (2020) 3918–28. doi: 10.1002/jcp.30135
38. Gallo M, Frezzetti D, Roma C, Chicchinelli N, Barbieri A, Arra C, et al. RANTES and IL-6 Cooperate in Inducing a More Aggressive Phenotype in Breast Cancer Cells. Oncotarget (2018) 9:17543–53. doi: 10.18632/oncotarget.24784
39. Li Y, Chen Y, Miao L, Wang Y, Yu M, Yan X, et al. Stress-Induced Upregulation of TNFSF4 in Cancer-Associated Fibroblast Facilitates Chemoresistance of Lung Adenocarcinoma Through Inhibiting Apoptosis of Tumor Cells. Cancer Lett (2021) 497:212–20. doi: 10.1016/j.canlet.2020.10.032
40. Wang L, Li X, Ren Y, Geng H, Zhang Q, Cao L, et al. Cancer-Associated Fibroblasts Contribute to Cisplatin Resistance by Modulating ANXA3 in Lung Cancer Cells. Cancer Sci (2019) 110:1609–20. doi: 10.1111/cas.13998
41. Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, et al. Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing Ros-Generating Monocytic Mdscs in Lung Squamous Cell Carcinoma. Cancer Immunol Res (2020) 8:436. doi: 10.1158/2326-6066.CIR-19-0507
42. Zhou Z, Zhou Q, Wu X, Xu S, Hu X, Tao X, et al. VCAM-1 Secreted From Cancer-Associated Fibroblasts Enhances the Growth and Invasion of Lung Cancer Cells Through AKT and MAPK Signaling. Cancer Lett (2020) 473:62–73. doi: 10.1016/j.canlet.2019.12.039
43. Inoue C, Miki Y, Saito R, Hata S, Abe J, Sato I, et al. Pd-L1 Induction by Cancer-Associated Fibroblast-Derived Factors in Lung Adenocarcinoma Cells. Cancers (Basel) (2019) 11:1257. doi: 10.3390/cancers11091257
44. Zhang R, Qi F, Shao S, Li G, Feng Y. Human Colorectal Cancer-Derived Carcinoma Associated Fibroblasts Promote CD44-mediated Adhesion of Colorectal Cancer Cells to Endothelial Cells by Secretion of HGF. Cancer Cell Int (2019) 19:192–2. doi: 10.1186/s12935-019-0914-y
45. Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, et al. Carcinoma-Associated Fibroblasts Promote the Stemness and Chemoresistance of Colorectal Cancer by Transferring Exosomal lncRNA H19. Theranostics (2018) 8:3932–48. doi: 10.7150/thno.25541
46. Aizawa T, Karasawa H, Funayama R, Shirota M, Suzuki T, Maeda S, et al. Cancer-Associated Fibroblasts Secrete Wnt2 to Promote Cancer Progression in Colorectal Cancer. Cancer Med (2019) 8:6370–82. doi: 10.1002/cam4.2523
47. Landskron G, de la Fuente López M, Dubois-Camacho K, Díaz-Jiménez D, Orellana-Serradell O, Romero D, et al. Interleukin 33/ST2 Axis Components Are Associated to Desmoplasia, a Metastasis-Related Factor in Colorectal Cancer. Front Immunol (2019) 10:1394. doi: 10.3389/fimmu.2019.01394
48. Zhu H-F, Zhang X-H, Gu C-S, Zhong Y, Long T, Ma Y-D, et al. Cancer-Associated Fibroblasts Promote Colorectal Cancer Progression by Secreting CLEC3B. Cancer Biol Ther (2019) 20:967–78. doi: 10.1080/15384047.2019.1591122
49. Stanisavljevic J, Loubat-Casanovas J, Herrera M, Luque T, Peña R, Lluch A, et al. Snail1-Expressing Fibroblasts in the Tumor Microenvironment Display Mechanical Properties That Support Metastasis. Cancer Res (2015) 75:284. doi: 10.1158/0008-5472.CAN-14-1903
50. Navarro R, Tapia-Galisteo A, Martín-García L, Tarín C, Corbacho C, Gómez-López G, et al. Tgf-β-Induced IGFBP-3 is a Key Paracrine Factor From Activated Pericytes That Promotes Colorectal Cancer Cell Migration and Invasion. Mol Oncol (2020) 14:2609–28. doi: 10.1002/1878-0261.12779
51. Ma X, Liu J, Yang X, Fang K, Zheng P, Liang X, et al. Mesenchymal Stem Cells Maintain the Stemness of Colon Cancer Stem Cells Via interleukin-8/mitogen-activated Protein Kinase Signaling Pathway. Exp Biol Med (Maywood) (2020) 245:562–75. doi: 10.1177/1535370220910690
52. Tan H-X, Xiao Z-G, Huang T, Fang Z-X, Liu Y, Huang Z-C. Cxcr4/Tgf-β1 Mediated Self-Differentiation of Human Mesenchymal Stem Cells to Carcinoma-Associated Fibroblasts and Promoted Colorectal Carcinoma Development. Cancer Biol Ther (2020) 21:248–57. doi: 10.1080/15384047.2019.1685156
53. Chen K, Liu Q, Tsang LL, Ye Q, Chan HC, Sun Y, et al. Human MSCs Promotes Colorectal Cancer Epithelial-Mesenchymal Transition and Progression Via CCL5/β-Catenin/Slug Pathway. Cell Death Dis (2017) 8:e2819–9. doi: 10.1038/cddis.2017.138
54. Giorello MB, Borzone FR, Labovsky V, Piccioni FV, Chasseing NA. Cancer-Associated Fibroblasts in the Breast Tumor Microenvironment. J Mammary Gland Biol Neoplasia (2021). doi: 10.1007/s10911-020-09475-y
55. Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, et al. Spatially and Functionally Distinct Subclasses of Breast Cancer-Associated Fibroblasts Revealed by Single Cell RNA Sequencing. Nat Commun (2018) 9:5150. doi: 10.1038/s41467-018-07582-3
56. Elwakeel E, Brüggemann M, Fink AF, Schulz MH, Schmid T, Savai R, et al. Phenotypic Plasticity of Fibroblasts During Mammary Carcinoma Development. Int J Mol Sci (2019) 20:4438. doi: 10.3390/ijms20184438
57. Raz Y, Cohen N, Shani O, Bell RE, Novitskiy SV, Abramovitz L, et al. Bone Marrow-Derived Fibroblasts are a Functionally Distinct Stromal Cell Population in Breast Cancer. J Exp Med (2018) 215:3075–93. doi: 10.1084/jem.20180818
58. Hass R, von der Ohe J, Ungefroren H. Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness. Cancers (Basel) (2020) 12:3716. doi: 10.3390/cancers12123716
59. Najafi M, Mortezaee K, Majidpoor J. Stromal Reprogramming: A Target for Tumor Therapy. Life Sci (2019) 239:117049. doi: 10.1016/j.lfs.2019.117049
60. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front Oncol (2020) 10:188. doi: 10.3389/fonc.2020.00188
61. Chen D, Liu Z, Liu W, Fu M, Jiang W, Xu S, et al. Predicting Postoperative Peritoneal Metastasis in Gastric Cancer With Serosal Invasion Using a Collagen Nomogram. Nat Commun (2021) 12:179. doi: 10.1038/s41467-020-20429-0
62. Zhong C, Tao B, Tang F, Yang X, Peng T, You J, et al. Remodeling Cancer Stemness by Collagen/Fibronectin Via the AKT and CDC42 Signaling Pathway Crosstalk in Glioma. Theranostics (2021) 11:1991–2005. doi: 10.7150/thno.50613
63. Li J, Xu X, Jiang Y, Hansbro NG, Hansbro PM, Xu J, et al. Elastin is a Key Factor of Tumor Development in Colorectal Cancer. BMC Cancer (2020) 20:217–7. doi: 10.1186/s12885-020-6686-x
64. Huang C, Chen J. Laminin−332 Mediates Proliferation, Apoptosis, Invasion, Migration and Epithelial−to−Mesenchymal Transition in Pancreatic Ductal Adenocarcinoma. Mol Med Rep (2021) 23:11. doi: 10.3892/mmr.2020.11649
65. Hussain S, Peng B, Cherian M, Song JW, Ahirwar DK, Ganju RK. The Roles of Stroma-Derived Chemokine in Different Stages of Cancer Metastases. Front Immunol (2020) 11:598532. doi: 10.3389/fimmu.2020.598532
66. Contreras-Zárate MJ, Cittelly DM. Sex Steroid Hormone Function in the Brain Niche: Implications for Brain Metastatic Colonization and Progression. Cancer Rep (2020) n/a:e1241. doi: 10.1002/cnr2.1241
67. Li X, Berg NK, Mills T, Zhang K, Eltzschig HK, Yuan X. Adenosine at the Interphase of Hypoxia and Inflammation in Lung Injury. Front Immunol (2021) 11:604944. doi: 10.3389/fimmu.2020.604944
68. Haferkamp S, Drexler K, Federlin M, Schlitt HJ, Berneburg M, Adamski J, et al. Extracellular Citrate Fuels Cancer Cell Metabolism and Growth. Front Cell Dev Biol (2020) 8:602476. doi: 10.3389/fcell.2020.602476
69. Souza JL, Martins-Cardoso K, Guimarães IS, de Melo AC, Lopes AH, Monteiro RQ, et al. Interplay Between EGFR and the Platelet-Activating Factor/Paf Receptor Signaling Axis Mediates Aggressive Behavior of Cervical Cancer. Front Oncol (2020) 10:557280. doi: 10.3389/fonc.2020.557280
70. Miyake M, Hori S, Owari T, Oda Y, Tatsumi Y, Nakai Y, et al. Clinical Impact of Tumor-Infiltrating Lymphocytes and PD-L1-Positive Cells as Prognostic and Predictive Biomarkers in Urological Malignancies and Retroperitoneal Sarcoma. Cancers (Basel) (2020) 12:3153. doi: 10.3390/cancers12113153
71. Fusco N, Vaira V, Righi I, Sajjadi E, Venetis K, Lopez G, et al. Characterization of the Immune Microenvironment in Malignant Pleural Mesothelioma Reveals Prognostic Subgroups of Patients. Lung Cancer (2020) 150:53–61. doi: 10.1016/j.lungcan.2020.09.026
72. Vilariño N, Bruna J, Bosch-Barrera J, Valiente M, Nadal E. Immunotherapy in NSCLC Patients With Brain Metastases. Understanding Brain Tumor Microenvironment and Dissecting Outcomes From Immune Checkpoint Blockade in the Clinic. Cancer Treat Rev (2020) 89:1–10. doi: 10.1016/j.ctrv.2020.102067
73. Baba Y, Nomoto D, Okadome K, Ishimoto T, Iwatsuki M, Miyamoto Y, et al. Tumor Immune Microenvironment and Immune Checkpoint Inhibitors in Esophageal Squamous Cell Carcinoma. Cancer Sci (2020) 111:3132–41. doi: 10.1111/cas.14541
74. Effern M, Glodde N, Braun M, Liebing J, Boll HN, Yong M, et al. Adoptive T Cell Therapy Targeting Different Gene Products Reveals Diverse and Context-Dependent Immune Evasion in Melanoma. Immunity (2020) 53:564–580.e9. doi: 10.1016/j.immuni.2020.07.007
75. Osborne N, Sundseth R, Burks J, Cao H, Liu X, Kroemer AH, et al. Gastrin Vaccine Improves Response to Immune Checkpoint Antibody in Murine Pancreatic Cancer by Altering the Tumor Microenvironment. Cancer Immunol Immunother (2019) 68:1635–48. doi: 10.1007/s00262-019-02398-6
76. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 Potently Enhances Murine Macrophage Mannose Receptor Activity: A Marker of Alternative Immunologic Macrophage Activation. J Exp Med (1992) 176:287–92. doi: 10.1084/jem.176.1.287
77. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The Adenosine-Dependent Angiogenic Switch of Macrophages to an M2-like Phenotype is Independent of Interleukin-4 Receptor Alpha (IL-4Rα) Signaling. Inflammation (2013) 36:921–31. doi: 10.1007/s10753-013-9621-3
78. Little AC, Pathanjeli P, Wu Z, Bao L, Goo LE, Yates JA, et al. Il-4/Il-13 Stimulated Macrophages Enhance Breast Cancer Invasion Via Rho-Gtpase Regulation of Synergistic Vegf/Ccl-18 Signaling. Front Oncol (2019) 9:456. doi: 10.3389/fonc.2019.00456
79. Grugan KD, McCabe FL, Kinder M, Greenplate AR, Harman BC, Ekert JE, et al. Tumor-Associated Macrophages Promote Invasion While Retaining Fc-Dependent Anti-Tumor Function. J Immunol (2012) 189:5457. doi: 10.4049/jimmunol.1201889
80. Liu Y, Ji X, Kang N, Zhou J, Liang X, Li J, et al. Tumor Necrosis Factor α Inhibition Overcomes Immunosuppressive M2b Macrophage-Induced Bevacizumab Resistance in Triple-Negative Breast Cancer. Cell Death Dis (2020) 11:993–3. doi: 10.1038/s41419-020-03161-x
81. Steitz AM, Steffes A, Finkernagel F, Unger A, Sommerfeld L, Jansen JM, et al. Tumor-Associated Macrophages Promote Ovarian Cancer Cell Migration by Secreting Transforming Growth Factor Beta Induced (TGFBI) and Tenascin C. Cell Death Dis (2020) 11:249–9. doi: 10.1038/s41419-020-2438-8
82. Sousa S, Brion R, Lintunen M, Kronqvist P, Sandholm J, Mönkkönen J, et al. Human Breast Cancer Cells Educate Macrophages Toward the M2 Activation Status. Breast Cancer Res (2015) 17:101–1. doi: 10.1186/s13058-015-0621-0
83. Cao W, Peters JH, Nieman D, Sharma M, Watson T, Yu J. Macrophage Subtype Predicts Lymph Node Metastasis in Oesophageal Adenocarcinoma and Promotes Cancer Cell Invasion In Vitro. Br J Cancer (2015) 113:738–46. doi: 10.1038/bjc.2015.292
84. Shima T, Shimoda M, Shigenobu T, Ohtsuka T, Nishimura T, Emoto K, et al. Infiltration of Tumor-Associated Macrophages is Involved in Tumor Programmed Death-Ligand 1 Expression in Early Lung Adenocarcinoma. Cancer Sci (2020) 111:727–38. doi: 10.1111/cas.14272
85. Verheul HMW, Pinedo HM. The Role of Vascular Endothelial Growth Factor (VEGF) in Tumor Angiogenesis and Early Clinical Development of VEGFReceptor Kinase Inhibitors. Clin Breast Cancer (2000) 1:S80–4. doi: 10.3816/CBC.2000.s.015
86. Tan PHS, Chia SS, Toh SL, Goh JCH, Nathan SS. The Dominant Role of IL-8 as an Angiogenic Driver in a Three-Dimensional Physiological Tumor Construct for Drug Testing. Tissue Eng Part A (2014) 20:1758–66. doi: 10.1089/ten.TEA.2013.0245
87. Rogers TL, Holen I. Tumour Macrophages as Potential Targets of Bisphosphonates. J Transl Med (2011) 9:177–7. doi: 10.1186/1479-5876-9-177
88. Veltman JD, Lambers MEH, van Nimwegen M, Hendriks RW, Hoogsteden HC, Hegmans JPJJ, et al. Zoledronic Acid Impairs Myeloid Differentiation to Tumour-Associated Macrophages in Mesothelioma. Br J Cancer (2010) 103:629–41. doi: 10.1038/sj.bjc.6605814
89. Khan KA, Ponce de Léon JL, Benguigui M, Xu P, Chow A, Cruz-Muñoz W, et al. Immunostimulatory and Anti-Tumor Metronomic Cyclophosphamide Regimens Assessed in Primary Orthotopic and Metastatic Murine Breast Cancer. NPJ Breast Cancer (2020) 6:29. doi: 10.1038/s41523-020-0171-1
90. Mejías R, Pérez-Yagüe S, Gutiérrez L, Cabrera LI, Spada R, Acedo P, et al. Dimercaptosuccinic Acid-Coated Magnetite Nanoparticles for Magnetically Guided In Vivo Delivery of Interferon Gamma for Cancer Immunotherapy. Biomaterials (2011) 32:2938–52. doi: 10.1016/j.biomaterials.2011.01.008
91. Jin L, Wang Q, Chen J, Wang Z, Xin H, Zhang D. Efficient Delivery of Therapeutic siRNA by Fe(3)O(4) Magnetic Nanoparticles Into Oral Cancer Cells. Pharmaceutics (2019) 11:615. doi: 10.3390/pharmaceutics11110615
92. Cristofolini T, Dalmina M, Sierra JA, Silva AH, Pasa AA, Pittella F, et al. Multifunctional Hybrid Nanoparticles as Magnetic Delivery Systems for siRNA Targeting the HER2 Gene in Breast Cancer Cells. Mater Sci Eng: C (2020) 109:110555. doi: 10.1016/j.msec.2019.110555
93. Bocanegra Gondan AI, Ruiz-de-Angulo A, Zabaleta A, Gómez Blanco N, Cobaleda-Siles BM, García-Granda MJ, et al. Effective Cancer Immunotherapy in Mice by polyIC-imiquimod Complexes and Engineered Magnetic Nanoparticles. Biomaterials (2018) 170:95–115. doi: 10.1016/j.biomaterials.2018.04.003
94. Mulens-Arias V, Rojas JM, Pérez-Yagüe S, Morales M del P, Barber DF. Polyethylenimine-Coated SPION Exhibits Potential Intrinsic Anti-Metastatic Properties Inhibiting Migration and Invasion of Pancreatic Tumor Cells. J Controlled Release (2015) 216:78–92. doi: 10.1016/j.jconrel.2015.08.009
95. Mulens-Arias V, Rojas JM, Sanz-Ortega L, Portilla Y, Pérez-Yagüe S, Barber DF. Polyethylenimine-Coated Superparamagnetic Iron Oxide Nanoparticles Impair In Vitro and In Vivo Angiogenesis. Nanomed: Nanotechnol Biol Med (2019) 21:102063. doi: 10.1016/j.nano.2019.102063
96. Akatsuka S, Yamashita Y, Ohara H, Liu Y-T, Izumiya M, Abe K, et al. Fenton Reaction Induced Cancer in Wild Type Rats Recapitulates Genomic Alterations Observed in Human Cancer. PloS One (2012) 7:e43403–3. doi: 10.1371/journal.pone.0043403
97. Zhou Y, Que K-T, Tang H-M, Zhang P, Fu Q-M, Liu Z-J. Anti-CD206 Antibody-Conjugated Fe(3)O(4)-based PLGA Nanoparticles Selectively Promote Tumor-Associated Macrophages to Polarize to the Pro-Inflammatory Subtype. Oncol Lett (2020) 20:298–8. doi: 10.3892/ol.2020.12161
98. Zhang W, Cao S, Liang S, Tan CH, Luo B, Xu X, et al. Differently Charged Super-Paramagnetic Iron Oxide Nanoparticles Preferentially Induced M1-Like Phenotype of Macrophages. Front Bioeng Biotechnol (2020) 8:537. doi: 10.3389/fbioe.2020.00537
99. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron Oxide Nanoparticles Inhibit Tumour Growth by Inducing Pro-Inflammatory Macrophage Polarization in Tumour Tissues. Nat Nanotechnol (2016) 11:986–94. doi: 10.1038/nnano.2016.168
100. Cheng J, Zhang Q, Fan S, Zhang A, Liu B, Hong Y, et al. The Vacuolization of Macrophages Induced by Large Amounts of Inorganic Nanoparticle Uptake to Enhance the Immune Response. Nanoscale (2019) 11:22849–59. doi: 10.1039/C9NR08261A
101. Mulens-Arias V, Rojas JM, Pérez-Yagüe S, Morales MP, Barber DF. Polyethylenimine-Coated SPIONs Trigger Macrophage Activation Through TLR-4 Signaling and ROS Production and Modulate Podosome Dynamics. Biomaterials (2015) 52:494–506. doi: 10.1016/j.biomaterials.2015.02.068
102. Jin R, Liu L, Zhu W, Li D, Yang L, Duan J, et al. Iron Oxide Nanoparticles Promote Macrophage Autophagy and Inflammatory Response Through Activation of Toll-Like Receptor-4 Signaling. Biomaterials (2019) 203:23–30. doi: 10.1016/j.biomaterials.2019.02.026
103. Rojas JM, Sanz-Ortega L, Mulens-Arias V, Gutiérrez L, Pérez-Yagüe S, Barber DF. Superparamagnetic Iron Oxide Nanoparticle Uptake Alters M2 Macrophage Phenotype, Iron Metabolism, Migration and Invasion. Nanomed: Nanotechnol Biol Med (2016) 12:1127–38. doi: 10.1016/j.nano.2015.11.020
104. Laskar A, Eilertsen J, Li W, Yuan X-M. SPION Primes THP1 Derived M2 Macrophages Towards M1-like Macrophages. Biochem Biophys Res Commun (2013) 441:737–42. doi: 10.1016/j.bbrc.2013.10.115
105. Chen Y, Zeng Z, Ying H, Wu C, Chen S. Superparamagnetic Iron Oxide Nanoparticles Attenuate Lipopolysaccharide-Induced Inflammatory Responses Through Modulation of Toll-Like Receptor 4 Expression. J Appl Toxicol (2020) 40:1067–75. doi: 10.1002/jat.3967
106. Dalzon B, Torres A, Reymond S, Gallet B, Saint-Antonin F, Collin-Faure V, et al. Influences of Nanoparticles Characteristics on the Cellular Responses: The Example of Iron Oxide and Macrophages. Nanomater (Basel) (2020) 10:E266. doi: 10.3390/nano10020266
107. Liu Y, Chen Z, Gu N, Wang J. Effects of DMSA-coated Fe3O4 Magnetic Nanoparticles on Global Gene Expression of Mouse Macrophage RAW264.7 Cells. Toxicol Lett (2011) 205:130–9. doi: 10.1016/j.toxlet.2011.05.1031
108. Wolf-Grosse S, Mollnes TE, Ali S, Stenvik J, Nilsen AM. Iron Oxide Nanoparticles Enhance Toll-like Receptor-Induced Cytokines in a Particle Size- and Actin-Dependent Manner in Human Blood. Nanomedicine (2018) 13:1773–85. doi: 10.2217/nnm-2017-0362
109. Frtús A, Smolková B, Uzhytchak M, Lunova M, Jirsa M, Kubinová Š, et al. Analyzing the Mechanisms of Iron Oxide Nanoparticles Interactions With Cells: A Road From Failure to Success in Clinical Applications. J Controlled Release (2020) 328:59–77. doi: 10.1016/j.jconrel.2020.08.036
110. Malhotra N, Lee J-S, Liman RAD, Ruallo JMS, Villaflores OB, Ger T-R, et al. Potential Toxicity of Iron Oxide Magnetic Nanoparticles: A Review. Molecules (2020) 25:3159. doi: 10.3390/molecules25143159
111. Grosse S, Stenvik J, Nilsen AM. Iron Oxide Nanoparticles Modulate Lipopolysaccharide-Induced Inflammatory Responses in Primary Human Monocytes. Int J Nanomed (2016) 11:4625–42. doi: 10.2147/IJN.S113425
112. Liu L, Sha R, Yang L, Zhao X, Zhu Y, Gao J, et al. Impact of Morphology on Iron Oxide Nanoparticles-Induced Inflammasome Activation in Macrophages. ACS Appl Mater Interfaces (2018) 10:41197–206. doi: 10.1021/acsami.8b17474
113. Chen S, Chen S, Zeng Y, Lin L, Wu C, Ke Y, et al. Size-Dependent Superparamagnetic Iron Oxide Nanoparticles Dictate Interleukin-1β Release From Mouse Bone Marrow-Derived Macrophages. J Appl Toxicol (2018) 38:978–86. doi: 10.1002/jat.3606
114. Narayanan A, Amaya M, Voss K, Chung M, Benedict A, Sampey G, et al. Reactive Oxygen Species Activate Nfκb (p65) and p53 and Induce Apoptosis in RVFV Infected Liver Cells. Virology (2014) 449:270–86. doi: 10.1016/j.virol.2013.11.023
115. Wang Y, Zeigler MM, Lam GK, Hunter MG, Eubank TD, Khramtsov VV, et al. The Role of the NADPH Oxidase Complex, P38 MAPK, and Akt in Regulating Human Monocyte/Macrophage Survival. Am J Respir Cell Mol Biol (2007) 36:68–77. doi: 10.1165/rcmb.2006-0165OC
116. Lingappan K. Nf-κb in Oxidative Stress. Curr Opin Toxicol (2018) 7:81–6. doi: 10.1016/j.cotox.2017.11.002
117. Schieber M, Chandel NS. Ros Function in Redox Signaling and Oxidative Stress. Curr Biol (2014) 24:R453–62. doi: 10.1016/j.cub.2014.03.034
118. Liu X, Gal J, Zhu H. Sequestosome 1/p62: A Multi-Domain Protein With Multi-Faceted Functions. Front Biol (2012) 7:189–201. doi: 10.1007/s11515-012-1217-z
119. Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/P62 Is a Polyubiquitin Chain Binding Protein Involved in Ubiquitin Proteasome Degradation. Mol Cell Biol (2004) 24:8055. doi: 10.1128/MCB.24.18.8055-8068.2004
120. Yang H, Ni H-M, Guo F, Ding Y, Shi Y-H, Lahiri P, et al. Sequestosome 1/P62 Protein is Associated With Autophagic Removal of Excess Hepatic Endoplasmic Reticulum in Mice. J Biol Chem (2016) 291:18663–74. doi: 10.1074/jbc.M116.739821
121. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential Regulation of Iron Homeostasis During Human Macrophage Polarized Activation. Eur J Immunol (2010) 40:824–35. doi: 10.1002/eji.200939889
122. Zini C, Venneri MA, Miglietta S, Caruso D, Porta N, Isidori AM, et al. USPIO-Labeling in M1 and M2-polarized Macrophages: An In Vitro Study Using a Clinical Magnetic Resonance Scanner. J Cell Physiol (2018) 233:5823–8. doi: 10.1002/jcp.26360
123. Bolte AC, Lukens JR. Neuroimmune Cleanup Crews in Brain Injury. Trends Immunol (2021) 42:480–94. doi: 10.1016/j.it.2021.04.003
124. Cayre M, Falque M, Mercier O, Magalon K, Durbec P. Myelin Repair: From Animal Models to Humans. Front Cell Neurosci (2021) 15:604865. doi: 10.3389/fncel.2021.604865
125. Stoessel MB, Majewska AK. Little Cells of the Little Brain: Microglia in Cerebellar Development and Function. Trends Neurosci (2021) doi: 10.1016/j.tins.2021.04.001
126. Lanza M, Casili G, Campolo M, Paterniti I, Colarossi C, Mare M, et al. Immunomodulatory Effect of Microglia-Released Cytokines in Gliomas. Brain Sci (2021) 11:466. doi: 10.3390/brainsci11040466
127. Wu H-Y, Chung M-C, Wang C-C, Huang C-H, Liang H-J, Jan T-R. Iron Oxide Nanoparticles Suppress the Production of IL-1beta Via the Secretory Lysosomal Pathway in Murine Microglial Cells. Particle Fibre Toxicol (2013) 10:46. doi: 10.1186/1743-8977-10-46
128. Wang Y, Wang B, Zhu M-T, Li M, Wang H-J, Wang M, et al. Microglial Activation, Recruitment and Phagocytosis as Linked Phenomena in Ferric Oxide Nanoparticle Exposure. Toxicol Lett (2011) 205:26–37. doi: 10.1016/j.toxlet.2011.05.001
129. Luo L, Iqbal MZ, Liu C, Xing J, Akakuru OU, Fang Q, et al. Engineered Nano-Immunopotentiators Efficiently Promote Cancer Immunotherapy for Inhibiting and Preventing Lung Metastasis of Melanoma. Biomaterials (2019) 223:119464. doi: 10.1016/j.biomaterials.2019.119464
130. Zhao Y, Zhao X, Cheng Y, Guo X, Yuan W. Iron Oxide Nanoparticles-Based Vaccine Delivery for Cancer Treatment. Mol Pharmaceut (2018) 15:1791–9. doi: 10.1021/acs.molpharmaceut.7b01103
131. Sungsuwan S, Yin Z, Huang X. Lipopeptide-Coated Iron Oxide Nanoparticles as Potential Glycoconjugate-Based Synthetic Anticancer Vaccines. ACS Appl Mater Interfaces (2015) 17535–44. doi: 10.1021/acsami.5b05497
132. Li C-X, Zhang Y, Dong X, Zhang L, Liu M-D, Li B, et al. Artificially Reprogrammed Macrophages as Tumor-Tropic Immunosuppression-Resistant Biologics to Realize Therapeutics Production and Immune Activation. Adv Mater (2019) 31:1807211. doi: 10.1002/adma.201807211
133. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron Induces Anti-Tumor Activity in Tumor-Associated Macrophages. Front Immunol (2017) 8:1479. doi: 10.3389/fimmu.2017.01479
134. Ruiz-de-Angulo A, Bilbao-Asensio M, Cronin J, Evans SJ, Clift MJD, Llop J, et al. Chemically Programmed Vaccines: Iron Catalysis in Nanoparticles Enhances Combination Immunotherapy and Immunotherapy-Promoted Tumor Ferroptosis. iScience (2020) 23:1–18. doi: 10.1016/j.isci.2020.101499
135. Shen L, Krauthäuser S, Fischer K, Hobernik D, Abassi Y, Dzionek A, et al. Vaccination With Trifunctional Nanoparticles That Address CD8+ Dendritic Cells Inhibits Growth of Established Melanoma. Nanomedicine (2016) 11:2647–62. doi: 10.2217/nnm-2016-0174
136. Shevtsov MA, Nikolaev BP, Yakovleva LY, Parr MA, Marchenko YY, Eliseev I, et al. 70-kDa Heat Shock Protein Coated Magnetic Nanocarriers as a Nanovaccine for Induction of Anti-Tumor Immune Response in Experimental Glioma. J Controlled Release (2015) 220:329–40. doi: 10.1016/j.jconrel.2015.10.051
137. Traini G, Ruiz-de-Angulo A, Blanco-Canosa JB, Zamacola Bascarán K, Molinaro A, Silipo A, et al. Cancer Immunotherapy of TLR4 Agonist–Antigen Constructs Enhanced With Pathogen-Mimicking Magnetite Nanoparticles and Checkpoint Blockade of PD-L1. Small (2019) 15:1803993. doi: 10.1002/smll.201803993
Keywords: iron oxide nanoparticles, nanoparticle–macrophage interaction, macrophage polarization, tumor associated macrophages, therapeutic applications
Citation: Mulens-Arias V, Rojas JM and Barber DF (2021) The Use of Iron Oxide Nanoparticles to Reprogram Macrophage Responses and the Immunological Tumor Microenvironment. Front. Immunol. 12:693709. doi: 10.3389/fimmu.2021.693709
Received: 11 April 2021; Accepted: 24 May 2021;
Published: 09 June 2021.
Edited by:
David Pozo, University of Seville, SpainReviewed by:
Diana Caballero-Hernández, Universidad Autónoma de Nuevo León, MexicoCeren Ciraci, Istanbul Technical University, Turkey
Copyright © 2021 Mulens-Arias, Rojas and Barber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domingo F. Barber, ZGZiYXJiZXJAY25iLmNzaWMuZXM=