 Yingman Liu
Yingman Liu Wenxuan Huang2
Wenxuan Huang2- 1Department of Periodontics, School and Hospital of Stomatology, China Medical University, Liaoning Provincial Key Laboratory of Oral Diseases, Shenyang, China
- 2School of Stomatology, Shenyang Medical College, Shenyang, China
- 3Department of Oral Biology, School and Hospital of Stomatology, China Medical University, Liaoning Provincial Key Laboratory of Oral Diseases, Shenyang, China
- 4Science Experiment Center, China Medical University, Shenyang, China
Periodontal disease, a common inflammatory disease, is considered a hazardous factor that contributes to the development of diseases of the digestive system as well as other systems. The bridge between periodontitis and systemic diseases is believed to be periodontal pathogens. The intestine, as part of the lower gastrointestinal tract, has a close connection with the oral cavity. Within the intestine, the intestinal barrier acts as a multifunctional system including microbial, mucous, physical and immune barrier. The intestinal barrier forms the body’s first line of defense against external pathogens; its breakdown can lead to pathological changes in the gut and other organs or systems. Reports in the literature have described how oral periodontal pathogens and pathobiont-reactive immune cells can transmigrate to the intestinal mucosa, causing the destruction of intestinal barrier homeostasis. Such findings might lead to novel ideas for investigating the relationship between periodontal disease and other systemic diseases. This review summarizes studies on the effects of periodontal pathogens on the intestinal barrier, which might contribute to understanding the link between periodontitis and gastrointestinal diseases.
Introduction
As the most important digestive organ and largest immune organ in the human body, the intestine not only absorbs nutrients and water but also maintains the harmonious coexistence of symbionts. To keep such a balance between symbionts, the human intestine has evolved unique regional immune characteristics that have resulted in an intestinal defense barrier known as the “gut barrier” (1). The intestinal barrier is equipped with an intricate multilayer system that is made up of resident gut microbiota, a mucous layer, a monolayer of intestinal epithelial cells, and a complex network of immune cells (2, 3). These components form microbial, chemical, physical, and immune barriers that work both simultaneously and sequentially. Multilayer gut barriers are organized into two main parts: an upper physical part and a lower functional part. The upper physical part, the superficial layer of the intestinal barrier, frustrates bacterial adhesion and regulates the diffusion of materials from the lumen to the underlying tissue. The lower functional part, the deep layer of the intestinal barrier, distinguishes commensal bacteria from pathogens; it is responsible for immune suppression to counter commensals and the immune response to pathogenic microbes and components (4, 5). It has been shown that perturbations to the integrity of the intestinal barrier can aggravate a variety of diseases: for example, arthritis, hepatic disorders, diabetes and inflammatory bowel disease (IBD) (6, 7).
Periodontal disease, a chronic inflammatory disease involving the destruction of gingival tissue and the resorption of alveolar bone, is manipulated by the intricate interaction between the host’s immune system and microbial infection. In the course of the initiation and progression of periodontal disease, a dramatic shift occurs in the subgingival ecology as it changes from a symbiotic to dysfunctional microbial community composed mostly of anaerobic genera. The keystone periodontal pathogens, including Porphyromonas gingivalis, Fusobacterium nucleatum, and Aggregatibacter actinomycetemcomitans, to name a few, are predominant bacterial species of the anaerobic microbiota. The keystone periodontal pathogens are enriched in virulence factors and have adapted to thrive in an inflammatory environment (8, 9). It is recognized that the presence of pathogenic bacteria in periodontal disease correlates with the incidence and progression of several systemic diseases such as cardiovascular disease, diabetes, Alzheimer’s disease, and arthritis (10, 11). Reports in the literature (12–14) have proposed that periodontal pathogens might induce multiple changes in the microbiota community, barrier function, and immune system of the gut, which lead to an increased risk of many systemic diseases associated with low-grade inflammation. Therefore, to explore the multifaceted role of periodontal pathogens in the intestinal barrier might help us understand the mechanism by which periodontitis exerts an impact on systemic diseases.
The Possible Routes by Which Periodontal Pathogens Influence the Intestine
The Gastrointestinal Route
The gastrointestinal route is a widely approved route that the oral bacteria travel into intestine. Similar to the gut, the oral cavity is also a major microbial reservoir. The mouth separates internal and external environments and is where bacteria enter the gastrointestinal tract, along with saliva, food, and water (15). The physiological secretion of saliva occurs in the oral cavity at the rate of 0.75~1.5L per day from mainly three pairs of salivary glands (16). Saliva is sterile when excreted into the mouth; however, saliva becomes contaminated with a conglomerate of bacteria after they are shed from the oral surface (17, 18). Under physiological conditions, the salivary flora rarely arrived to the gut because of the protection from the gastric acid and alkaline bile. However, the salivary flora of patients with periodontitis is remarkably distinct from that of orally healthy controls. For example, the salivary abundance of P. gingivalis was significantly higher in patients with periodontal disease (19, 20). According to a quantitative analysis, it was estimated that patients with severe periodontitis swallowed approximately 1012~1013 P. gingivalis bacteria per day (21–23). Because of it is acid resistant, P. gingivalis might pass through the stomach and reach the intestinal tract, destroying intestinal homeostasis (24). This hypothesis was further confirmed by the discovery of P. gingivalis in the intestinal tract. Arimatsu and his colleagues ferreted out P. gingivalis in the jejunum and ileum of P. gingivalis-treated mice one hour after oral administration; its proportion in the microbial flora was diminished by 3 hours. At 16 hours, P. gingivalis was discovered in the colon (25). Another study also described how P. gingivalis was detected in both fecal and cecal samples (26).
Of course the majority of oral bacteria are eliminated by the gastric acid and alkaline bile. However, the impairment of the physiological protection can allow microbiota translocation and communication. For example, the gut colonization by oral bacteria is significantly augmented in patients who have achlorhydria-related gastric dysfunction. Besides, the transmission probability of oral bacteria, including periodontal pathogens, from oral cavity to intestine, is increased in patients with systemic diseases such as colorectal cancer and IBD (13, 27).
Blood Route
The hematogenous route is one of the important routes for oral bacteria dissemination to other organs. The extremely well vascularization and gingival ulceration in periodontal pockets in the connective tissues of the periodontium allow the periodontal pathogens to readily enter the bloodstream (9, 28). Plaque accumulation and gingival inflammation significantly increase the prevalence of bacteraemia. The theory also supported by the studies that periodontal pathogens such as P. gingivalis and F. nucleatum were detected in the blood of experimental periodontitis models and traveled to other organs such as liver, spleen and vascular samples (29, 30). A study proved that the hematogenous route appeared much more resultful than the digestive tract for fusobacteria trafficking to colorectal cancer (CRC) (31). It was biologically more plausible that oral fusobacteria spread to CRC through hematogenous route, because Fusobacteria was absent from biofilms in the colonic mucous layer of adenomas of familial adenomatous polyposis patients and some sporadic CRC patients (32).
Immune Cell Migration Route
Given that oral and colonic mucosae are physically correlative, it is undoubtable that oral bacteria transmigrate to the lower gastrointestinal tract and presumably trigger pathogenic immune responses (33). John Bienenstock presented a concept of “common mucosal immune system” and speculated that the mucosal immune system might be a system-wide “organ” in which the immune cells distributed throughout the body could interplay among different mucosal tissues (34). In addition, it has been shown that immune cells derived from oral draining lymph nodes are able to migrate to other lymphoid tissues, including but not limited to the gut (35). Consequently, it was likely that periodontal inflammation given rise to the advent of periodontal pathobiont-reactive T cells in the lymph nodes draining the oral cavity. The pathobiont-reactive T cells were then incited by periodontal pathogens to migrate from the oral mucosa to the gut where they activated intestinal immune cells and caused intestinal inflammation (36).
Both bacterial ectopic colonization and lymph node drainage of immune cells have been recognized as the ways by which periodontal pathogens disrupt intestinal barrier homeostasis (Figure 1). However, we still don’t know whether periodontal bacteria travel to the gut as free bacteria or by means of erythrocytes or lymphocytes. It is also unclear how the periodontal pathogens travelling to the gastrointestinal tract can survive the gastric acid and alkaline bile. The mechanism that regulates the immune cell migration between oral and colonic mucosae is also to be illuminated.
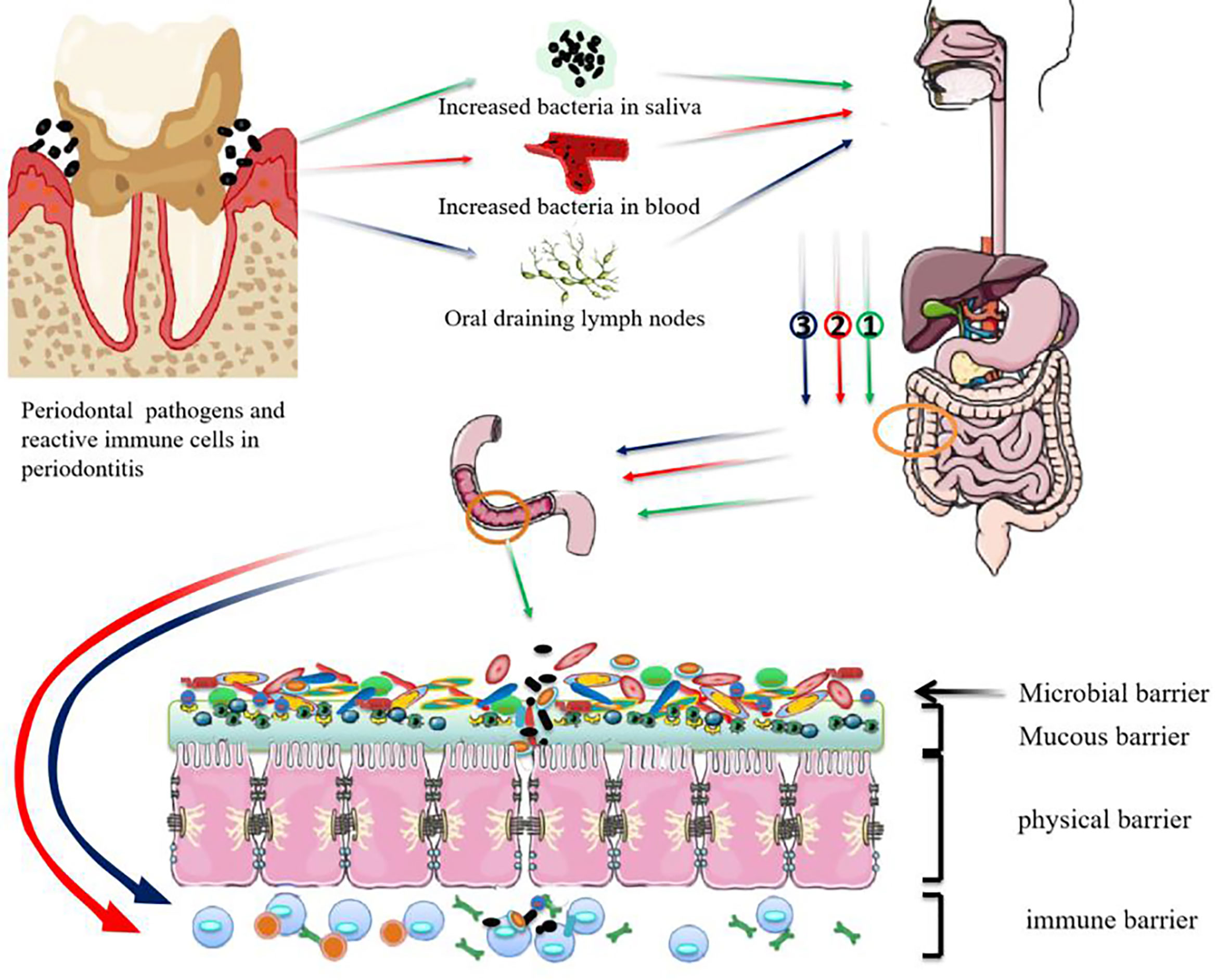
Figure 1 Possible mechanisms for periodontal pathogens damaging the intestinal barrier. ① Swallowed periodontal pathogens may transmigrate to the lower gastrointestinal tract and disrupt intestinal homeostasis. ② periodontal pathogens may disseminate to intestinal tissues through the bloodstream and take part in the progression of intestinal diseases. ③ Pathobiont-reactive immune cells activated by periodontal pathogens may transmigrate from the oral draining lymph nodes to the gut where they activate intestinal immune cells.
Effects of Periodontal Pathogens on Intestinal Microbial Barrier
Composition and Function of Intestinal Microbiota
Microbiota denotes the entire population of microorganisms that colonizes a specific region, including not just bacteria but also other microbes such as fungi, protozoans, viruses, and archaea (37, 38). The majority of microorganisms residing in humans are found in the gut, forming the largest microbial ecosystem of the body. Culture-based studies have led to the proposal for a core microbiota since nearly all healthy grown-ups have been found to consistently share the majority of gut bacterial species (39). The four core bacterial phyla are present in the adult human gut: Bacteroidetes and Firmicutes are the most predominant, followed distantly by Actinobacteria and Proteobacteria (40). Despite the uniformity of these main components of the intestinal microbiota, their relative proportions, as well as the other species present, vary strikingly across individuals. Over a person’s lifespan, subtle temporal and spatial variations occur in the distribution of intestinal microorganisms (41). It is generally believed that the development of the microbiota begins from birth (42, 43) and only reaches a relatively distinct, stable microbiota community by about two or three years of age (44). Spatially, the distribution of the gut microbiota is stratified along not only a longitudinal but also transverse axis. In longitudinal axis, from the small intestine to the colon, facultative anaerobes are thought to survive in the small intestine while a dense and diverse community of anaerobes has been shown to dominate the colon. In the transverse axis, from the mucous layer to lumen, the composition of the microbiota is significantly dissimilar in the mucous layer and feces/lumen. For example, Bacteroidetes appears to be more abundant in fecal/luminal samples than in the mucus. Compared with the gut lumen, Firmicutes, specifically Clostridium cluster XIVa, are mainly enriched in the mucous layer.
As an involved ecological community, the microbiota in our gut assists us to stay healthy by exerting essential multivarious functions. Presently, the intestinal microbiota exhibits three major functions (45, 46): (i) metabolic activities with respect to salvaging energy and absorbing nutrients; (ii) trophic effects on intestinal epithelial cells, and the immune system and function; and (iii) shield of the colonized hosts against the attack by alien microorganisms. The co-evolution of the host and microbes contributes to many host physiological processes. However, impaired regulation in the microbiota may generate diseases such as intestinal inflammation and cancer. It has been clearly shown in animal experiments that the microbiota functions in the pathogenesis of IBD and intestinal neoplasia (47, 48). In support of this, in the absence of the commensal microbiota under gnotobiotic conditions, experimental animals did not develop colitis though they did in conventional environments (49). Additionally, the carcinogenic agent, azoxymethane, induced colon tumors in colitis-susceptible interleukin (IL)-10 knockout mice colonized with certain commensal bacteria but failed to do so in gnotobiotic IL-10 knockout mice (50). The duration and severity of chronic colitis are the significant risk factors for colitis-associated colon cancer (CAC). The IL-10 knockout mice develop spontaneously colitis on account of the intolerance to intestinal bacteria and microbiota-induced activation of effector T cells. Thus the gnotobiotic IL-10 knockout mice failed to develop the colitis and subsequent CAC. Besides, the intestinal bacteria such as Bacteroides fragilis, E. coli, and so on, promoted the proliferation of tumor cells and the development of colon cancer via activating diverse signaling routes (51, 52).
Effect of Periodontal Pathogens on Intestinal Microbiota
As periodontal pathogens translocate to the intestine, the concomitant variation in gut microbiota was affirmed. Firmicutes was reduced and Bacteroides increased at the phylum level after P. gingivalis was given to C57BL/6N mice. Because P. gingivalis was not detected in the blood and the proportion of P. gingivalis in the gut was very low, systemic inflammation might have occurred because of variation in the gut microbiota induced by P. gingivalis. Sasaki et al. found that the composition of the intestinal microbiota was also altered by intravenous injection of inactivated P. gingivalis in mice, although sonicated P. gingivalis did not directly reach the gut (53). Changes included that the phyla Tenericutes and Proteobacteria tended to decrease. Unsurprisingly, findings in a clinical case-control study were consistent with those in animal experiments. Lourenco et al. compared the intestinal flora of individuals with healthy periodontium, gingivitis, or periodontitis. It was found that the variation in intestinal flora was significant, with decreased microbial diversity and a high Firmicutes/Bacteroides ratio associated with disease severity. In addition, periodontally healthy and diseased groups could be distinguished based on the composition of the microbiome in stool samples. The identification of the gut microbiota, especially the Mogibacteriaceae and Ruminococcaceae families and the genus Prevotella, might be able to aid in the classification of diseased patients (54).
The alteration in intestinal flora caused by periodontal disease is not entirely consistent. In terms of the two major phyla in human intestinal flora, a tendency for decreased Bacteroides and increased Firmicutes in a P. gingivalis-administered mouse group in comparison to a control mouse group was shown (12). However, a contrary finding has been described in the literature: that is, a high Bacteroides/Firmicutes ratio was reported (26, 55). Ohtsu administered P. gingivalis to streptozotocin-induced diabetic mice and found no noticeable change in the abundance of intestinal Bacteroides (56). Contradictory tendencies for Firmicutes and Bacteroides have also been reported in several systemic inflammatory conditions, such as rheumatoid arthritis (RA) (57, 58) and obesity (59, 60) as well as others. Hence, confounding factors, including diet, obesity, and sampling sites, have been involved in alterations of intestinal flora caused by periodontal pathogens.
The gut microbiota can be disturbed by the massive long-term oral ingestion of periodontal pathogens, which is deemed a potential mechanism that links periodontitis to inflammatory systemic diseases. In a study by Komazaki et al., C57BL/6J mice orally administered Aggregatibacter actinomycetemcomitans for 12 weeks presented with impaired glucose tolerance, insulin resistance, and a perturbation of the gut flora (61). Specifically, in A. actinomycetemcomitans–administered mice, the genus Turicibacter was underrepresented. This genus reportedly produced butyric acid, which improved insulin sensitivity. Coincidentally, in C57BL/6J mice fed a high-fat diet, recipients of oral P. gingivalis showed inhibited glucose uptake in skeletal muscles, with higher tumor necrosis factor (TNF)-α expression and lower insulin signaling. At the same time, Turicibacter was underrepresented in the gut of mice treated with oral P. gingivalis. These findings suggest that the administration of periodontal bacteria may negatively affect insulin resistance by decreasing the abundance of Turicibacter in the gut, thereby decreasing insulin sensitivity.
In addition to metabolic disorders, it is also considered that the “oral-gut” axis was connected to neuroinflammation. According to epidemiological studies, periodontitis was recognized as a risk factor for cognitive impairment. Scholars speculated that chronic periodontitis evoked a neuroinflammatory response, as well as subsequent neurodegeneration and cognitive decline because of a disorder in a “microbiota–gut–brain” axis (62). Moreover, in mice with chronic periodontitis, alteration of the microbiota at the phylum and genus levels was significantly found in saliva and feces. Meanwhile, intestinal and blood–brain barriers were destroyed, and endotoxin and pro-inflammatory cytokines at the mRNA level were upregulated systemically and in the brain. One study detected a systemic inflammatory response after changes in the intestinal flora in P. gingivalis-administered mice, which supported the idea that the perturbation of intestinal microbiota by periodontal pathogens might be a mechanism connecting periodontal disease and systemic diseases (26). These findings suggested indirectly that periodontal pathogens might travel to the intestinal tract and alter the composition of the intestinal flora (Table 1).
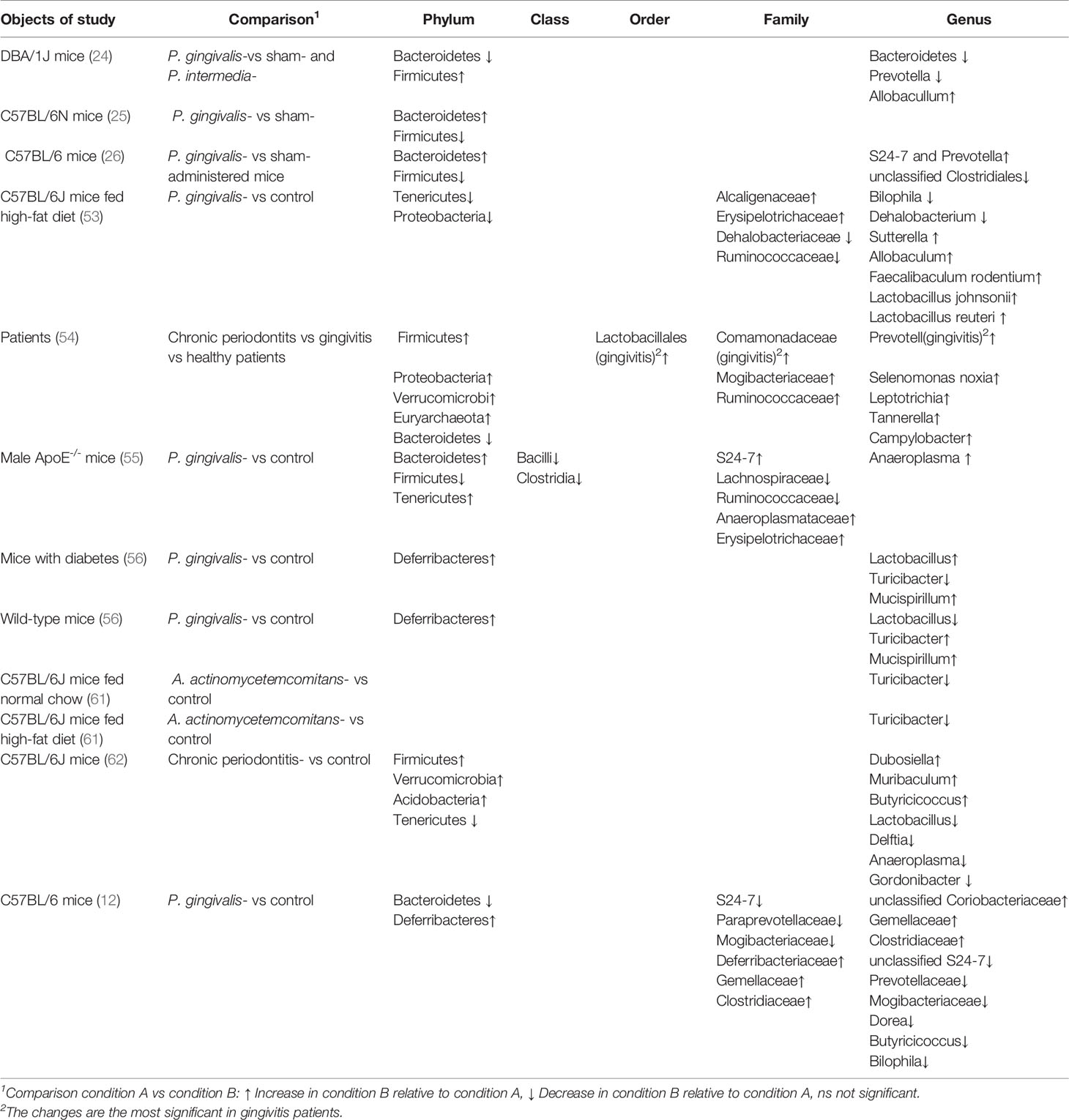
Table 1 Main studies about the effects of periodontal pathogens on the intestinal microbiota.
Effect of Periodontal Pathogens on Intestinal Mucous Barrier
The mucous barrier, an often ignored part of the intestinal barrier system, is the first physical defense that separates bacteria in the lumen from the epithelium. The main building blocks of the mucous barrier are highly glycosylated mucin proteins that form a gel-like sieve structure overlying the intestinal epithelium. The mucin 2 (MUC2) secreted by goblet cells is the most abundant mucin protein in small and large intestine (63). The small intestine only has one mucous layer, whereas the colon has two layers: a loose outer layer and a dense inner layer. The loose outer layer allows the colonization of indigenous or transient microorganisms. The dense inner, devoid of bacteria, is firmly adherent to the epithelium (64–66). The mucous components of the gut barrier are fortified by glycoproteins, antimicrobial peptides (AMPs) and proteins including lysozyme. These AMPs including alpha-defensins, lysozyme and Reg3 proteins are secreted by Paneth cells and enterocytes to protect the epithelium from a variety of insults (67, 68). Thus, the mucous layer is also a chemical barrier (69). Secretory IgA (SIgA) molecules represent another important constituent of the intestinal mucous surface. IgA is produced by local plasma cells in the mucosal lamina propria and then transported into gut secretions by the polymeric immunoglobulin receptor (pIgR). In secretions, the extracellular domain of pIgR released by proteolytic cleavage covalently bounds to IgA to form the SIgA complex. MUC2 and pIgR act coordinately to localize SIgA in the outer mucous layer (70). In the outer mucous layer, SIgA binds bacteria-derived and food-derived antigens to retain toxins or bacteria within the lumen (67).
If mucin production or processing is defective, the mucous layer will fail to restrict bacterial access to the epithelia. MUC2 is important in maintaining the integrity of intestinal barrier, as MUC2 knock-out mice spontaneously developed inflammation and colon cancer (71–73). Gingipains are a group of complex arginine- or lysine-specific cysteine proteinases (also widely known as RgpA, RgpB, and Kgp) that have been acknowledged as one of the essential virulence factors of P. gingivalis. They are potent “trypsin-like” proteinases, capable of cleaving or degrading a series of host proteins including cytokines, extracellular matrix proteins, plasma proteins and host cell surface proteins (74). A study demonstrated that RgpB secreted by P. gingivalis was able to cleave MUC2 at a specific site where it would disrupt the MUC2 polymeric network (75). Animal study findings have revealed that AMPs are crucial in the prevention and clearance of intestinal pathogens (76–78). However, P. gingivalis is able to circumvent and even manipulate AMPs, which has been demonstrated in periodontal disease and atherosclerosis (79). These detrimental effects of P. gingivalis on mucous layer may contribute to the further disruption of intestinal homeostasis.
Effect of Periodontal Pathogens on the Intestinal Physical Barrier
Composition and Function of the Intestinal Physical Barrier
Underneath the mucous layer is the intestinal physical barrier. It is mainly made up of intestinal epithelial cells and tight junctions (TJs) by which epithelial cells are linked together. The integrity and regenerative capacity of the intestinal epithelium are known to be the structural basis of the intestinal physical barrier. The specialized epithelium is composed of a monolayer of columnar cells that forms the largest barrier that connects the internal tract with a changing external environment (80). TJs play several vital roles in the intestinal barrier. By controlling intestinal epithelial permeability, TJs are major players in the functions of the intestinal physical barrier. They are located at the most apical–lateral position between adjacent epithelial cells and consist of multiple proteins, including occludin, claudins, zona occludens (ZOs), and adhesion molecules. Occludin is essential for the maintenance of the homeostasis of the intestinal epithelial barrier (81). The decreased expression of occludin means an increase in gut permeability (82). At present, at least 27 different claudin proteins have been detected, forming a skeleton structure of TJs. Mounting evidence has highlighted how different claudin proteins play different roles in the intestinal barrier. For example, claudins -1, -3, -4, -7, and -8 reduce the transport of solutes and water, enhancing the barrier effect, while claudins -2, -10, -12, and -15 do the opposite (83). The ZO family, including ZO-1, ZO-2, and ZO-3, is the most studied. ZO proteins act as scaffolds by connecting with other transmembrane proteins (84).
An intestinal physical barrier that regulates the flow of nutrients, ions and water between the lumen and underlying tissues restricts contact between the host and intraluminal exogenous antigens and microbes (85). The destruction of an intestinal physical barrier and an increase in intestinal epithelial permeability are common pathophysiological mechanisms in diverse diseases (84, 86, 87), such as Parkinson’s disease (88), liver cirrhosis (89), and IBD (90), among others. These diseases present with common alterations in the content and distribution of proteins in the physical barrier. If the content or distribution of the proteins is maladjusted, three effects might ensue: (i) a damaged paracellular pathway; (ii) enhanced paracellular transport of solutes and water; and (iii) increased permeability to macromolecules (91). A disruption of the physical barrier structure can be both a cause and an effect of disease (92).
Effect of Periodontal Pathogens on the Intestinal Physical Barrier
The intestinal physical barrier can be regulated by a variety of factors. Research on the effect of periodontal pathogens on the intestinal physical barrier has mainly focused on the destruction of TJ proteins induced by P. gingivalis. The lipopolysaccharide (LPS) from P. gingivalis, known as endotoxin, elicits a broad spectrum of biological responses. In the gut, LPS has been identified as a major irritant that causes intestinal barrier dysfunction. Studies have shown that an instant increase in the permeability of the small intestine for 3 hours in mice could be induced by a high-dose intravenous LPS injection (93); this was most pronounced in the ileum (94). Nakajima et al. found that tight junction protein-1 (ZO-1) and occludin were downregulated at the mRNA level after a single oral administration of P. gingivalis, with elevated endotoxin found in the serum (26).
What’s more, Tsuzuno and co-workers concluded that the expression of ZO-1 in the intestine of mice treated with P. gingivalis was dramatically decreased at the protein rather than mRNA level (95). The possible involvement of gingipains in the degradation of intestinal ZO-1 protein was suggested by the use of gingipain-deficient P. gingivalis. Based on the ability of P. gingivalis to invade epithelial cells, a possible mechanism whereby the bacterium might penetrate mucus and intestinal epithelial cells, then degrade ZO-1 using gingipains was proposed (96).
In addition, TJ components are regulated by proinflammatory cytokines driven by the oral administration of P. gingivalis (97, 98). Oral P. gingivalis induced serum IL-17A, with increased IL-1β and TNF-α found in the colon, and decreased the TJ protein, ZO-1, in R1441G mice (99). P. gingivalis–induced IL-17A was less likely to originate from intestinal peripheral lymphoid tissues since IL-17A was not found in the colon. Oral P. gingivalis was found to impair intestinal epithelium permeability and induced an augmentation of peripheral IL-17A. Downregulated TJs and upregulated proinflammatory cytokines were observed in the ileum of mice orally administered P. gingivalis in a research study by Arimatsu (25). P. gingivalis was not found in the blood, so the increased endotoxins might have been derived from intestinal flora because of the destroyed barrier in the intestinal epithelium. Flak and colleagues (100) verified the hypothesis that P. gingivalis could aggravate joint inflammation by the dysregulation of gut barrier function. They found that in mice with inflammatory arthritis, the inoculation of P. gingivalis raised the endotoxin concentration and the whole bacterial load of the inner mucous layer and lamina propria of the colon, which were presumably related to a downregulation of the TJ molecule, ZO-1.
Meilian et al. declared that the oral administration of P. gingivalis was insufficient to represent the effects of periodontitis because of the failure to initiate periodontal lesions. Thus, they evaluated the gut mechanical barrier in mice with experimental periodontitis induced by P. gingivalis–adhered tooth ligatures. In accordance with the published literature, expression levels of TJ proteins changed in the ileum and the epithelial barrier became damaged in mice with experimental periodontitis (101).
From the above, it can be concluded that virulent factors from P. gingivalis and the intestinal inflammation the bacterium causes might induce enhanced intestinal permeability in the gut epithelium (Table 2).
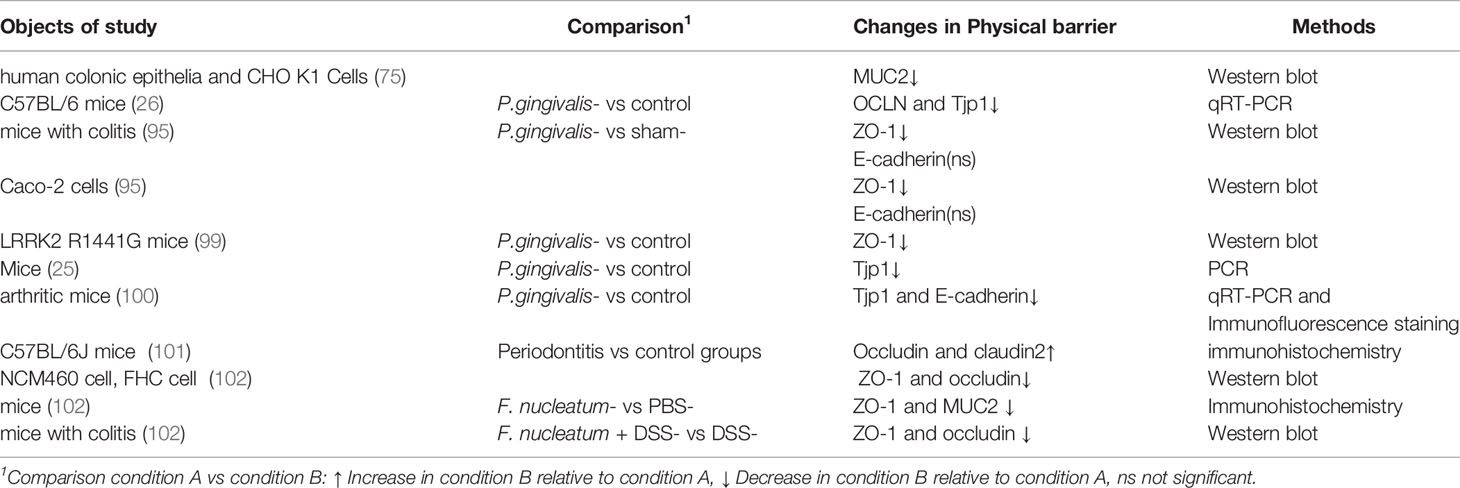
Table 2 Main studies about the effects of periodontal pathogens on the intestinal physical barrier.
Effect of Periodontal Pathogens on the Intestinal Immune Barrier
Composition and Function of the Intestinal Immune Barrier
The surface of the human gastrointestinal tract is estimated to be about 200–400 m2, forming the largest surface for interplay between humans and the external environment. The intestinal tract, as one of the largest lymphoid organs, possesses up to 70% of the body’s total immunocytes (103). The immune barrier in intestine is formed by innate and adaptive immunity. In the epithelium and lamina propria, the initiation of the innate immune response is triggered by recognition of pathogen-associated molecular patterns (PAMPs) by pathogen-recognition receptors (PRRs) (e.g., toll-like receptors (TLRs), nucleotide-binding oligomerization domain proteins, retinoic-acid-inducible gene-I, C-type lectins) (104). The PRRs, localized either within endosomes or in the cell surface, can be expressed by intestinal epithelial cells, stromal cells, dendritic cells, macrophages and B and T cells (105). The cells of the intestinal adaptive immune system are distributed regularly throughout the mucosa in the form of organized lymphoid tissue structures of gut-associated lymphoid tissue (GALT), or lamina propria, with intraepithelial cell populations (106). Such intestinal immune tissue can principally be grouped into inductive and effector sites. Priming adaptive immune cell responses in the intestine are located principally in the organized lymphoid structures of GALT and draining lymph nodes. Quantities of effector immune cells pervade the lamina propria and overlying epithelium (107). Particulate antigens are taken and transported by inductive immune cells from the lumen into an underlying dendritic cell (DC)-rich domain, where particulate antigens can be presented to effector immune cells (108).
The functions of the intestinal immune barrier are two-fold: (i) the rapid recognition of antigens, such as foreign microorganisms and toxins, and the induction of an immune response; and (ii) an immune tolerance to nutrients or harmless antigens such as probiotics. The intestinal immune system must orchestrate a complex balance between immune activation versus tolerance to maintain intestinal homeostasis. A breach of this balance can result in the occurrence of intestinal (109) and parenteral diseases (110).
Effect of Periodontal Pathogens on the Intestinal Immune Barrier
Effect of F. nucleatum on Intestinal Immune Barrier
F. nucleatum, identified as a normal commensal bacterium that generally exists on tooth surfaces of healthy individuals, bridges different bacterial species to form tooth plaque (111). F. nucleatum, as an opportunistic pathogen, can ectopically colonize extra-oral sites under unhealthy conditions of the body and is associated with a wide range of human diseases. Virulence-related factors from F. nucleatum stimulate the progression of intestinal cancer and negatively influence therapeutic efficacy through interactions with the local immune system (112). In support of this, a high concentration of F. nucleatum was found in colorectal carcinoma tissues (113). Moreover, the intestinal F. nucleatum of patients with colorectal cancer possibly originates from the oral cavity (114). F. nucleatum engages with intestinal tumorigenesis by a number of mechanisms. F. nucleatum binds to tumor cells and protects them from the cytotoxicity of natural killer cells via an interaction between the Fusobacterial Fap2 protein and the T-cell immunoglobulin and ITIM domain (TIGIT) receptor expressed on all natural killer cells in humans (115). In mice fed F. nucleatum and having tumors, myeloid-derived immune cells that can impede T-cell proliferation and evoke T-cell apoptosis were expanded by the bacterium (116, 117). Herein, F. nucleatum results in the immune escape of tumor cells by modulating the tumor immune microenvironment. F. nucleatum also can target tumor cell itself and activate TLR4/MYD88/NF-κB, E-cadherin/β-catenin and other pathways to promote the proliferation of colorectal cancer cells (118, 119). In the microenvironment of cancers, F. nucleatum infection increases M2 macrophage polarization through a TLR4-dependent mechanism to enhance tumor growth (120). In contrary, a study showed that Autoinducer-2 of F. nucleatum promoted microphage migration and M1 polarization (121). Macrophages polarize to distinct functional phenotypes: an M1- or M2-phenotype according to the microenvironment. M1-type polarization is pro-inflammatory, releasing inducible nitric oxide synthase, TNF-α, and IL-8. M2-type polarization is anti-inflammatory, and tumor-promoting, releasing IL-4, IL-10 and transforming growth factor (TGF)-β (121, 122). The contradictory results may be due to a variety of virulent factors of F. nucleatum contributing to the development of cancer by different mechanisms (121). Furthermore, several studies (102, 123, 124) demonstrated that F. nucleatum also facilitated the intestinal inflammatory disease via acting on the gut immunity.
Effect of P. gingivalis on Intestinal Immune Barrier
In addition to F. nucleatum, an increasing number of studies have shown that P. gingivalis initiates the inflammatory response in both the periodontium and other tissues (9). A histopathological analysis indicated that intestinal inflammation was identified by an increased infiltration of macrophages, neutrophils, lymphocytes, and plasma cells in the lamina propria of the gastrointestinal tract in mice with experimental periodontal disease (125). In a study on the relationship between periodontitis and systemic inflammation, Palioto et al. developed experimental models of periodontitis in C57BL/6 mice by oral gavage, ligature, or a combination of oral gavage and ligature. It was demonstrated that the expression levels of inflammatory and epigenetic markers were elevated, which included interleukin-18 receptor 1 (IL-18R1, inflammatory marker), DNA methyltransferases 3b (DNMT3b, de novo methylation marker), and B and T lymphocyte attenuator (BTLA) in the gut and serum of mice with experimental periodontitis (126). Periodontitis is recognized as an environmental trigger of RA in which P. gingivalis may be involved in its pathogenesis (127). Most RA cases are associated with the activation of bacterial citrullination and anti-cyclic citrullinated peptide antibodies. Therefore, P. gingivalis, as the only known periodontal pathogen synthesizing peptidylarginine deiminase (PPAD) that is capable of inducing the citrullination of fibrinogen and vimentin, has attracted much attention (128). However, during inflammatory arthritis, PPAD deletion in P. gingivalis mainly exacerbated P. gingivalis–induced joint inflammation. Therefore, it is questioned whether PPAD synthesized by P. gingivalis influences the progression of RA (129–131). A hypothesis was proposed that immune responses in the gut that were elicited with P. gingivalis inoculation exacerbated RA (132). T helper 17 (Th17) cells have been proven to be involved in the progression of RA by regulating the adaptive immune response, particularly at mucosal surfaces in the intestine (133). Periodontal pathogens skew the type II collagen–characteristic T-cell immune response in lymph tissues drawing arthritic joints toward the Th17 phenotype (134). And in a study (24) where P. gingivalis was orally administered to mice with RA, the proportion of Th17 cells was significantly elevated in mesenteric lymphocytes and Peyer’s patches. Therefore, the association of periodontal disease with RA might be due to effects on intestinal immune responses exerted by ingested P. gingivalis.
Clinical studies revealed that Porphyromonas was enriched in the stool samples and colorectal mucosa of colorectal cancer patients (135, 136). P. gingivalis, as the member of Porphyromonas, might be associated with the colorectal cancer. In vitro, P. gingivalis changed the tumor immune environment by selectively expanding myeloid-derived immune cells and induced CRC development by activating hematopoietic nucleotide-binding oligomerization domain receptor and pyrin domain containing 3 (NLRP3) (137).
Collectively, periodontal pathogens can induce or inhibit the intestinal immune response via a variety of pathways. A breakdown in the integrity of the gut immune barrier exerts a threat to normal physiological functions in humans (Table 3).
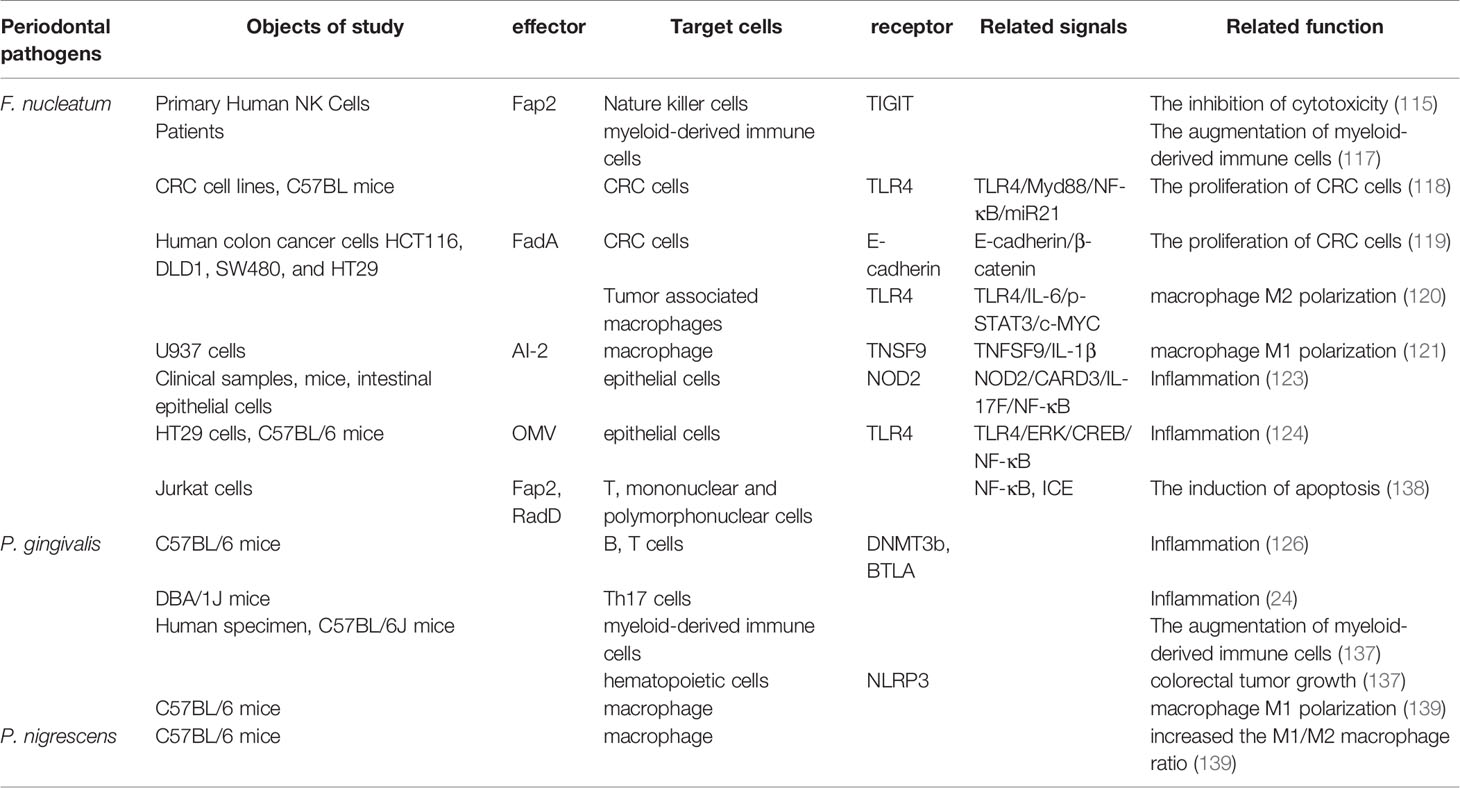
Table 3 Main studies about the effects of periodontal pathogens on the intestinal immune barrier.
Conclusions
In the gut, microbial, immune, and physical barriers interact with each other. For example, the intestinal flora is crucial to the development of host immunity; in turn, the immune system regulates the intestinal flora by immune tolerance and immune rejection (140, 141). The ectopic colonization of periodontal pathogens can cause an intestinal micro-ecological imbalance and inflammation. Meanwhile, virulence factors from periodontal pathogens can destroy the physical intestinal epithelial barrier, allowing bacteria and metabolites of the intestinal lumen to leak into the bloodstream. In addition, periodontal pathogens directly or indirectly regulate the immune system and destroy homeostasis (125, 139).
Recent studies have indicated that the periodontal pathogens, pathogenic to the intestinal barrier, are associated with the progression of a variety of intestinal diseases. However, information on the mechanisms by which periodontal pathogens disrupt the intestinal epithelial barrier is still far from sufficient. Because many diseases pertain to a compromised function of intestinal barrier (81, 142, 143), future studies that dissect the specific mechanisms of periodontal pathogens affecting the intestinal barrier will yield new insights into the significance of periodontal treatment for the prevention and cure of gastrointestinal and systemic diseases.
Author Contributions
YL and YK made substantial contributions to the conception and design of the study. YL, WH, JW, JM, MZ, XL, JL, and YK participated in drafting the article. YL and YK gave final approval of the version to be submitted and revised versions.
Funding
Natural Science Foundation of Liaoning Province (20180551232) and Science and Technology Project of Shenyang (F16-102-4-00).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wells JM, Brummer RJ, Derrien M, MacDonald TT, Troost F, Cani PD, et al. Homeostasis of the Gut Barrier and Potential Biomarkers. Am J Physiol Gastrointest Liver Physiol (2017) 312:G171–93. doi: 10.1152/ajpgi.00048.2015
2. König J, Wells J, Cani PD, García-Ródenas CL, MacDonald T, Mercenier A, et al. Human Intestinal Barrier Function in Health and Disease. Clin Transl Gastroenterol (2016) 7:e196. doi: 10.1038/ctg.2016.54
3. Cardoso-Silva D, Delbue D, Itzlinger A, Moerkens R, Withoff S, Branchi F, et al. Intestinal Barrier Function in Gluten-Related Disorders. Nutrients (2019) 11:2325. doi: 10.3390/nu11102325
4. Chen P. Enteric Dysbiosis, Gut Barrier and Liver Disease. Hepatobiliary Surg Nutr (2018) 7:38–40. doi: 10.21037/hbsn.2017.12.06
5. Scaldaferri F, Pizzoferrato M, Gerardi V, Lopetuso L, Gasbarrini A. The Gut Barrier: New Acquisitions and Therapeutic Approaches. J Clin Gastroenterol (2012) 46 Suppl:S12–7. doi: 10.1097/MCG.0b013e31826ae849
6. Chopyk DM, Grakoui A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology (2020) 159:849–63. doi: 10.1053/j.gastro.2020.04.077
7. Meddings J. The Significance of the Gut Barrier in Disease. Gut (2008) 57:438–40. doi: 10.1136/gut.2007.143172
8. Bartold PM, Van Dyke TE. An Appraisal of the Role of Specific Bacteria in the Initial Pathogenesis of Periodontitis. J Clin Periodontol (2019) 46:6–11. doi: 10.1111/jcpe.13046
9. Hajishengallis G. Periodontitis: From Microbial Immune Subversion to Systemic Inflammation. Nat Rev Immunol (2015) 15:30–44. doi: 10.1038/nri3785
10. Bui FQ, Almeida-da-Silva CLC, Huynh B, Trinh A, Liu J, Woodward J, et al. Association Between Periodontal Pathogens and Systemic Disease. Biomed J (2019) 42:27–35. doi: 10.1016/j.bj.2018.12.001
11. Cullinan MP, Seymour GJ. Periodontal Disease and Systemic Illness: Will the Evidence Ever be Enough? Periodontology (2013) 2000 62:271–86. doi: 10.1111/prd.12007
12. Kato T, Yamazaki K, Nakajima M, Date Y, Kikuchi J, Hase K, et al. Oral Administration of Porphyromonas Gingivalis Alters the Gut Microbiome and Serum Metabolome. mSphere (2018) 3:e00460–18. doi: 10.1128/mSphere.00460-18
13. Kitamoto S, Nagao-Kitamoto H, Hein R, Schmidt TM, Kamada N. The Bacterial Connection Between the Oral Cavity and the Gut Diseases. J Dental Res (2020) 99:1021–9. doi: 10.1177/0022034520924633
14. Acharya C, Sahingur SE, Bajaj JS. Microbiota, Cirrhosis, and the Emerging Oral-Gut-Liver Axis. JCI Insight (2017) 2:e94416. doi: 10.1172/jci.insight.94416
15. Lira-Junior R, Boström EA. Oral-Gut Connection: One Step Closer to an Integrated View of the Gastrointestinal Tract? Mucosal Immunol (2018) 11:316–8. doi: 10.1038/mi.2017.116
16. Martina E, Campanati A, Diotallevi F, Offidani A. Saliva and Oral Diseases. J Clin Med (2020) 9:466. doi: 10.3390/jcm9020466
17. Schrøder SA, Bardow A, Eickhardt-Dalbøge S, Johansen HK, Homøe P. Is Parotid Saliva Sterile on Entry to the Oral Cavity? Acta Otolaryngol (2017) 137:762–4. doi: 10.1080/00016489.2016.1272002
18. Belstrøm D. The Salivary Microbiota in Health and Disease. J Oral Microbiol (2020) 12:1723975. doi: 10.1080/20002297.2020.1723975
19. Damgaard C, Danielsen AK, Enevold C, Massarenti L, Nielsen CH, Holmstrup P, et al. Porphyromonas Gingivalis in Saliva Associates With Chronic and Aggressive Periodontitis. J Oral Microbiol (2019) 11:1653123. doi: 10.1080/20002297.2019.1653123
20. Chen C, Hemme C, Beleno J, Shi ZJ, Ning D, Qin Y, et al. Oral Microbiota of Periodontal Health and Disease and Their Changes After Nonsurgical Periodontal Therapy. ISME J (2018) 12:1210–24. doi: 10.1038/s41396-017-0037-1
21. Boutaga K, Savelkoul PH, Winkel EG, van Winkelhoff AJ. Comparison of Subgingival Bacterial Sampling With Oral Lavage for Detection and Quantification of Periodontal Pathogens by Real-Time Polymerase Chain Reaction. J Periodontol (2007) 78:79–86. doi: 10.1902/jop.2007.060078
22. Saygun I, Nizam N, Keskiner I, Bal V, Kubar A, Acikel C, et al. Salivary Infectious Agents and Periodontal Disease Status. J Periodontal Res (2011) 46:235–9. doi: 10.1111/j.1600-0765.2010.01335.x
23. He J, Huang W, Pan Z, Cui H, Qi G, Zhou X, et al. Quantitative Analysis of Microbiota in Saliva, Supragingival, and Subgingival Plaque of Chinese Adults With Chronic Periodontitis. Clin Oral Investig (2012) 16:1579–88. doi: 10.1007/s00784-011-0654-4
24. Sato K, Takahashi N, Kato T, Matsuda Y, Yokoji M, Yamada M, et al. Aggravation of Collagen-Induced Arthritis by Orally Administered Porphyromonas Gingivalis Through Modulation of the Gut Microbiota and Gut Immune System. Sci Rep (2017) 7:6955. doi: 10.1038/s41598-017-07196-7
25. Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima M, Ryder MI, et al. Oral Pathobiont Induces Systemic Inflammation and Metabolic Changes Associated With Alteration of Gut Microbiota. Sci Rep (2014) 4:4828. doi: 10.1038/srep04828
26. Nakajima M, Arimatsu K, Kato T, Matsuda Y, Minagawa T, Takahashi N, et al. Oral Administration of P. Gingivalis Induces Dysbiosis of Gut Microbiota and Impaired Barrier Function Leading to Dissemination of Enterobacteria to the Liver. PloS One (2015) 10:e0134234. doi: 10.1371/journal.pone.0134234
27. Sohn J, Sun Y, Genco RJ, Kirkwood KL. The Periodontal Microenvironment: A Potential Reservoir for Intestinal Pathobionts in Crohn’s Disease. Curr Oral Health Rep (2020) 7:37–44. doi: 10.1007/s40496-020-00251-9
28. Deshpande RG, Khan MB, Genco CA. Invasion of Aortic and Heart Endothelial Cells by Porphyromonas Gingivalis. Infect Immun (1998) 66:5337–43. doi: 10.1128/IAI.66.11.5337-5343.1998
29. Velsko IM, Chukkapalli SS, Rivera-Kweh MF, Chen H, Zheng D, Bhattacharyya I, et al. Fusobacterium Nucleatum Alters Atherosclerosis Risk Factors and Enhances Inflammatory Markers With an Atheroprotective Immune Response in ApoE(null) Mice. PloS One (2015) 10:e0129795. doi: 10.1371/journal.pone.0129795
30. Figuero E, Lindahl C, Marín MJ, Renvert S, Herrera D, Ohlsson O, et al. Quantification of Periodontal Pathogens in Vascular, Blood, and Subgingival Samples From Patients With Peripheral Arterial Disease or Abdominal Aortic Aneurysms. J Periodontol (2014) 85:1182–93. doi: 10.1902/jop.2014.130604
31. Abed J, Maalouf N, Manson AL, Earl AM, Parhi L, Emgård JEM, et al. Colon Cancer-Associated Fusobacterium Nucleatum May Originate From the Oral Cavity and Reach Colon Tumors Via the Circulatory System. Front Cell Infect Microbiol (2020) 10:400. doi: 10.3389/fcimb.2020.00400
32. Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients With Familial Adenomatous Polyposis Harbor Colonic Biofilms Containing Tumorigenic Bacteria. Science (2018) 359:592–7. doi: 10.1126/science.aah3648
33. Li B, Ge Y, Cheng L, Zeng B, Yu J, Peng X, et al. Oral Bacteria Colonize and Compete With Gut Microbiota in Gnotobiotic Mice. Int J Oral Sci (2019) 11:10. doi: 10.1038/s41368-018-0043-9
34. Wang J, Tian Z. How Lung Infection Leads to Gut Injury. Oncotarget (2015) 6:42394–5. doi: 10.18632/oncotarget.6470
35. Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D. Endoscopic Photoconversion Reveals Unexpectedly Broad Leukocyte Trafficking to and From the Gut. Proc Natl Acad Sci USA (2014) 111:6696–701. doi: 10.1073/pnas.1405634111
36. Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG 3rd, Hayashi A, Imai J, et al. The Intermucosal Connection Between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell (2020) 182:447–62.e14. doi: 10.1016/j.cell.2020.05.048
37. Dias AM, Cordeiro G, Estevinho MM, Veiga R, Figueira L, Reina-Couto M, et al. Gut Bacterial Microbiome Composition and Statin Intake-A Systematic Review. Pharmacol Res Perspect (2020) 8:e00601. doi: 10.1002/prp2.601
38. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature (2010) 464:59–65. doi: 10.1038/nature08821
39. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature (2012) 489:220–30. doi: 10.1038/nature11550
40. Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, et al. Towards the Human Intestinal Microbiota Phylogenetic Core. Environ Microbiol (2009) 11:2574–84. doi: 10.1111/j.1462-2920.2009.01982.x
41. Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar Reddy D. Role of the Normal Gut Microbiota. World J Gastroenterol (2015) 21:8787–803. doi: 10.3748/wjg.v21.i29.8787
42. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the Human Infant Intestinal Microbiota. PloS Biol (2007) 5:e177. doi: 10.1371/journal.pbio.0050177
43. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of Microbial Consortia in the Developing Infant Gut Microbiome. Proc Natl Acad Sci USA (2011) 108 Suppl 1:4578–85. doi: 10.1073/pnas.1000081107
44. Voreades N, Kozil A, Weir TL. Diet and the Development of the Human Intestinal Microbiome. Front Microbiol (2014) 5:494. doi: 10.3389/fmicb.2014.00494
45. Guarner F, Malagelada JR. Gut Flora in Health and Disease. Lancet (Lond Engl) (2003) 361:512–9. doi: 10.1016/S0140-6736(03)12489-0
46. Robles Alonso V, Guarner F. Linking the Gut Microbiota to Human Health. Br J Nutr (2013) 109 Suppl 2:S21–6. doi: 10.1017/S0007114512005235
47. Liu Z, Cao AT, Cong Y. Microbiota Regulation of Inflammatory Bowel Disease and Colorectal Cancer. Semin Cancer Biol (2013) 23:543–52. doi: 10.1016/j.semcancer.2013.09.002
48. Pickard JM, Zeng MY, Caruso R, Núñez G. Gut Microbiota: Role in Pathogen Colonization, Immune Responses, and Inflammatory Disease. Immunol Rev (2017) 279:70–89. doi: 10.1111/imr.12567
49. Sartor RB. Microbial Influences in Inflammatory Bowel Diseases. Gastroenterology (2008) 134:577–94. doi: 10.1053/j.gastro.2007.11.059
50. Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the Intestinal Microbiota Alters Colitis-Associated Colorectal Cancer Susceptibility. PloS One (2009) 4:e6026. doi: 10.1371/journal.pone.0006026
51. Torres-Maravilla E, Boucard AS, Mohseni AH, Taghinezhad SS, Cortes-Perez NG, Bermúdez-Humarán LG. Role of Gut Microbiota and Probiotics in Colorectal Cancer: Onset and Progression Microorganisms (2021) 9(5):1021. doi: 10.3390/microorganisms9051021
52. Tomkovich S, Yang Y, Winglee K, Gauthier J, Mühlbauer M, Sun X, et al. Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer Res (2017) 77:2620–32. doi: 10.1158/0008-5472.CAN-16-3472
53. Sasaki N, Katagiri S, Komazaki R, Watanabe K, Maekawa S, Shiba T, et al. Endotoxemia by Porphyromonas Gingivalis Injection Aggravates Non-Alcoholic Fatty Liver Disease, Disrupts Glucose/Lipid Metabolism, and Alters Gut Microbiota in Mice. Front Microbiol (2018) 9:2470. doi: 10.3389/fmicb.2018.02470
54. Lourenvarsigmao TGB, Spencer SJ, Alm EJ, Colombo APV. Defining the Gut Microbiota in Individuals With Periodontal Diseases: An Exploratory Study. J Oral Microbiol (2018) 10:1487741. doi: 10.1080/20002297.2018.1487741
55. Kramer CD, Simas AM, He X, Ingalls RR, Weinberg EO, Genco CA. Distinct Roles for Dietary Lipids and Porphyromonas Gingivalis Infection on Atherosclerosis Progression and the Gut Microbiota. Anaerobe (2017) 45:19–30. doi: 10.1016/j.anaerobe.2017.04.011
56. Ohtsu A, Takeuchi Y, Katagiri S, Suda W, Maekawa S, Shiba T, et al. Influence of Porphyromonas Gingivalis in Gut Microbiota of Streptozotocin-Induced Diabetic Mice. Oral Dis (2019) 25:868–80. doi: 10.1111/odi.13044
57. Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An Expansion of Rare Lineage Intestinal Microbes Characterizes Rheumatoid Arthritis. Genome Med (2016) 8:43. doi: 10.1186/s13073-016-0299-7
58. Jeong Y, Kim JW, You HJ, Park SJ, Lee J, Ju JH, et al. Gut Microbial Composition and Function Are Altered in Patients With Early Rheumatoid Arthritis. J Clin Med (2019) 8:693. doi: 10.3390/jcm8050693
59. Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity Alters Gut Microbial Ecology. Proc Natl Acad Sci USA (2005) 102:11070–5. doi: 10.1073/pnas.0504978102
60. Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, et al. Human Colonic Microbiota Associated With Diet, Obesity and Weight Loss. Int J Obes (Lond) (2008) 32:1720–4. doi: 10.1038/ijo.2008.155
61. Komazaki R, Katagiri S, Takahashi H, Maekawa S, Shiba T, Takeuchi Y, et al. Periodontal Pathogenic Bacteria, Aggregatibacter Actinomycetemcomitans Affect Non-Alcoholic Fatty Liver Disease by Altering Gut Microbiota and Glucose Metabolism. Sci Rep (2017) 7:13950. doi: 10.1038/s41598-017-14260-9
62. Xue L, Zou X, Yang XQ, Peng F, Yu DK, Du JR. Chronic Periodontitis Induces Microbiota-Gut-Brain Axis Disorders and Cognitive Impairment in Mice. Exp Neurol (2020) 326:113176. doi: 10.1016/j.expneurol.2020.113176
63. Vancamelbeke M, Vermeire S. The Intestinal Barrier: A Fundamental Role in Health and Disease. Expert Rev Gastroenterol Hepatol (2017) 11:821–34. doi: 10.1080/17474124.2017.1343143
64. Capaldo CT, Powell DN, Kalman D. Layered Defense: How Mucus and Tight Junctions Seal the Intestinal Barrier. J Mol Med (Berl) (2017) 95:927–34. doi: 10.1007/s00109-017-1557-x
65. Pelaseyed T, Bergström JH, Gustafsson JK, Ermund A, Birchenough GM, Schütte A, et al. The Mucus and Mucins of the Goblet Cells and Enterocytes Provide the First Defense Line of the Gastrointestinal Tract and Interact With the Immune System. Immunol Rev (2014) 260:8–20. doi: 10.1111/imr.12182
66. Johansson ME, Sjövall H, Hansson GC. The Gastrointestinal Mucus System in Health and Disease. Nat Rev Gastroenterol Hepatol (2013) 10:352–61. doi: 10.1038/nrgastro.2013.35
67. Zhang K, Hornef MW, Dupont A. The Intestinal Epithelium as Guardian of Gut Barrier Integrity. Cell Microbiol (2015) 17:1561–9. doi: 10.1111/cmi.12501
68. Takiishi T, Fenero CIM, Câmara NOS. Intestinal Barrier and Gut Microbiota: Shaping Our Immune Responses Throughout Life. Tissue barriers (2017) 5:e1373208. doi: 10.1080/21688370.2017.1373208
69. Chelakkot C, Ghim J, Ryu SH. Mechanisms Regulating Intestinal Barrier Integrity and Its Pathological Implications. Exp Mol Med (2018) 50:1–9. doi: 10.1038/s12276-018-0126-x
70. Rogier EW, Frantz AL, Bruno ME, Kaetzel CS. Secretory IgA Is Concentrated in the Outer Layer of Colonic Mucus Along With Gut Bacteria. Pathogens (2014) 3:390–403. doi: 10.3390/pathogens3020390
71. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The Inner of the Two Muc2 Mucin-Dependent Mucus Layers in Colon Is Devoid of Bacteria. Proc Natl Acad Sci USA (2008) 105:15064–9. doi: 10.1073/pnas.0803124105
72. Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, et al. Colorectal Cancer in Mice Genetically Deficient in the Mucin Muc2. Science (2002) 295:1726–9. doi: 10.1126/science.1069094
73. Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-Deficient Mice Spontaneously Develop Colitis, Indicating That MUC2 Is Critical for Colonic Protection. Gastroenterology (2006) 131:117–29. doi: 10.1053/j.gastro.2006.04.020
74. Zhang R, Yang J, Wu J, Sun WB, Liu Y. Effect of Deletion of the rgpA Gene on Selected Virulence of Porphyromonas Gingivalis. J Dent Sci (2016) 11:279–86. doi: 10.1016/j.jds.2016.03.004
75. van der Post S, Subramani DB, Bäckström M, Johansson MEV, Vester-Christensen MB, Mandel U, et al. Site-Specific O-Glycosylation on the MUC2 Mucin Protein Inhibits Cleavage by the Porphyromonas Gingivalis Secreted Cysteine Protease (RgpB). J Biol Chem (2013) 288:14636–46. doi: 10.1074/jbc.M113.459479
76. Natividad JM, Verdu EF. Modulation of Intestinal Barrier by Intestinal Microbiota: Pathological and Therapeutic Implications. Pharmacol Res (2013) 69:42–51. doi: 10.1016/j.phrs.2012.10.007
77. Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjöberg J, Amir E, et al. Enteric Defensins Are Essential Regulators of Intestinal Microbial Ecology. Nat Immunol (2010) 11:76–83. doi: 10.1038/ni.1825
78. Brandl K, Plitas G, Schnabl B, DeMatteo RP, Pamer EG. MyD88-Mediated Signals Induce the Bactericidal Lectin RegIII Gamma and Protect Mice Against Intestinal Listeria Monocytogenes Infection. J Exp Med (2007) 204:1891–900. doi: 10.1084/jem.20070563
79. Hussain M, Stover CM, Dupont A. P. Gingivalis in Periodontal Disease and Atherosclerosis - Scenes of Action for Antimicrobial Peptides and Complement. Front Immunol (2015) 6:45. doi: 10.3389/fimmu.2015.00045
80. Peterson LW, Artis D. Intestinal Epithelial Cells: Regulators of Barrier Function and Immune Homeostasis. Nat Rev Immunol (2014) 14:141–53. doi: 10.1038/nri3608
81. Odenwald MA, Turner JR. The Intestinal Epithelial Barrier: A Therapeutic Target? Nat Rev Gastroenterol Hepatol (2017) 14:9–21. doi: 10.1038/nrgastro.2016.169
82. Cummins PM. Occludin: One Protein, Many Forms. Mol Cell Biol (2012) 32:242–50. doi: 10.1128/MCB.06029-11
83. Barmeyer C, Schulzke JD, Fromm M. Claudin-Related Intestinal Diseases. Semin Cell Dev Biol (2015) 42:30–8. doi: 10.1016/j.semcdb.2015.05.006
84. Lechuga S, Ivanov AI. Disruption of the Epithelial Barrier During Intestinal Inflammation: Quest for New Molecules and Mechanisms. Biochim Biophys Acta Mol Cell Res (2017) 1864:1183–94. doi: 10.1016/j.bbamcr.2017.03.007
85. Luissint AC, Parkos CA, Nusrat A. Inflammation and the Intestinal Barrier: Leukocyte-Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology (2016) 151:616–32. doi: 10.1053/j.gastro.2016.07.008
86. Nagpal R, Yadav H. Bacterial Translocation From the Gut to the Distant Organs: An Overview. Ann Nutr Metab (2017) 71 (Suppl 1):11–6. doi: 10.1159/000479918
87. Vandenbroucke RE, Dejonckheere E, Van Hauwermeiren F, Lodens S, De Rycke R, Van Wonterghem E, et al. Matrix Metalloproteinase 13 Modulates Intestinal Epithelial Barrier Integrity in Inflammatory Diseases by Activating TNF. EMBO Mol Med (2013) 5:1000–16. doi: 10.1002/emmm.201202100
88. van ISCD, Derkinderen P. The Intestinal Barrier in Parkinson’s Disease: Current State of Knowledge. J Parkinsons Dis (2019) 9:S323–s329. doi: 10.3233/JPD-191707
89. Feng D, Zou J, Su D, Mai H, Zhang S, Li P, et al. Curcumin Prevents High-Fat Diet-Induced Hepatic Steatosis in ApoE(-/-) Mice by Improving Intestinal Barrier Function and Reducing Endotoxin and Liver TLR4/NF-κb Inflammation. Nutr Metab (2019) 16:79. doi: 10.1186/s12986-019-0410-3
90. Ramos GP, Papadakis KA. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clinic Proc (2019) 94:155–65. doi: 10.1016/j.mayocp.2018.09.013
91. Krug SM, Schulzke JD, Fromm M. Tight Junction, Selective Permeability, and Related Diseases. Semin Cell Dev Biol (2014) 36:166–76. doi: 10.1016/j.semcdb.2014.09.002
92. Marchiando AM, Graham WV, Turner JR. Epithelial Barriers in Homeostasis and Disease. Annu Rev Pathol (2010) 5:119–44. doi: 10.1146/annurev.pathol.4.110807.092135
93. Guo S, Nighot M, Al-Sadi R, Alhmoud T, Nighot P, Ma TY. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and Myd88. J Immunol (Baltimore Md: 1950) (2015) 195:4999–5010. doi: 10.4049/jimmunol.1402598
94. Yue C, Ma B, Zhao Y, Li Q, Li J. Lipopolysaccharide-Induced Bacterial Translocation Is Intestine Site-Specific and Associates With Intestinal Mucosal Inflammation. Inflammation (2012) 35:1880–8. doi: 10.1007/s10753-012-9510-1
95. Tsuzuno T, Takahashi N, Yamada-Hara M, Yokoji-Takeuchi M, Sulijaya B, Aoki-Nonaka Y, et al. Ingestion of Porphyromonas Gingivalis Exacerbates Colitis Via Intestinal Epithelial Barrier Disruption in Mice. J Periodontal Res (2021) 56:275–88. doi: 10.1111/jre.12816
96. Amano A. Disruption of Epithelial Barrier and Impairment of Cellular Function by Porphyromonas Gingivalis. Front Biosci (2007) 12:3965–74. doi: 10.2741/2363
97. Edelblum KL, Turner JR. The Tight Junction in Inflammatory Disease: Communication Breakdown. Curr Opin Pharmacol (2009) 9:715–20. doi: 10.1016/j.coph.2009.06.022
98. López-Posadas R, Neurath MF, Atreya I. Molecular Pathways Driving Disease-Specific Alterations of Intestinal Epithelial Cells. Cell Mol Life Sci (2017) 74:803–26. doi: 10.1007/s00018-016-2363-2
99. Feng YK, Wu QL, Peng YW, Liang FY, You HJ, Feng YW, et al. Gingivalis Impairs Gut Permeability and Mediates Immune Responses Associated With Neurodegeneration in LRRK2 R1441G Mice. J Neuroinflamm (2020) 17:347. doi: 10.1186/s12974-020-02027-5
100. Flak MB, Colas RA, Muñoz-Atienza E, Curtis MA, Dalli J, Pitzalis C. Inflammatory Arthritis Disrupts Gut Resolution Mechanisms, Promoting Barrier Breakdown by Porphyromonas Gingivalis. JCI Insight (2019) 4:e125191. doi: 10.1172/jci.insight.125191
101. Meilian X, Ting Y, Ying Z, Xin H, Baoyi X, Dongying X, et al. Periodontal Inflammation Affects the Mechanical and Immune Barrier Functions of Mice Gut. Hua Xi Kou Qiang Yi Xue Za Zhi (2016) 34:414–8. doi: 10.7518/hxkq.2016.04.019
102. Cao P, Chen Y, Chen Y, Su W, Zhan N, Dong W. Fusobacterium Nucleatum Activates Endoplasmic Reticulum Stress to Promote Crohn’s Disease Development Via the Upregulation of CARD3 Expression. Front Pharmacol (2020) 11:106. doi: 10.3389/fphar.2020.00106
103. Jung C, Hugot JP, Barreau F. Peyer’s Patches: The Immune Sensors of the Intestine. Int J Inflamm (2010) 2010:823710. doi: 10.4061/2010/823710
104. Wang L, Zhu L, Qin S. Gut Microbiota Modulation on Intestinal Mucosal Adaptive Immunity. J Immunol Res (2019) 2019:4735040. doi: 10.1155/2019/4735040
105. Broom LJ, Kogut MH. Gut Immunity: Its Development and Reasons and Opportunities for Modulation in Monogastric Production Animals. Anim Health Res Rev (2018) 19:46–52. doi: 10.1017/S1466252318000026
106. Makala LH, Nishikawa Y, Suzuki N, Nagasawa H. Immunology. Antigen-Presenting Cells in the Gut. J Biomed Sci (2004) 11:130–41. doi: 10.1007/BF02256556
107. Brandtzaeg P, Pabst R. Let’s Go Mucosal: Communication on Slippery Ground. Trends Immunol (2004) 25:570–7. doi: 10.1016/j.it.2004.09.005
108. Mowat AM, Agace WW. Regional Specialization Within the Intestinal Immune System. Nat Rev Immunol (2014) 14:667–85. doi: 10.1038/nri3738
109. Neurath MF. Targeting Immune Cell Circuits and Trafficking in Inflammatory Bowel Disease. Nat Immunol (2019) 20:970–9. doi: 10.1038/s41590-019-0415-0
110. Winer DA, Winer S, Dranse HJ, Lam TK. Immunologic Impact of the Intestine in Metabolic Disease. J Clin Invest (2017) 127:33–42. doi: 10.1172/JCI88879
111. Huh JW, Roh TY. Opportunistic Detection of Fusobacterium Nucleatum as a Marker for the Early Gut Microbial Dysbiosis. BMC Microbiol (2020) 20:208. doi: 10.1186/s12866-020-01887-4
112. Sun CH, Li BB, Wang B, Zhao J, Zhang XY, Li TT, et al. The Role of Fusobacterium Nucleatum in Colorectal Cancer: From Carcinogenesis to Clinical Management. Chronic Dis Trans Med (2019) 5:178–87. doi: 10.1016/j.cdtm.2019.09.001
113. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium Nucleatum Infection Is Prevalent in Human Colorectal Carcinoma. Genome Res (2012) 22:299–306. doi: 10.1101/gr.126516.111
114. Komiya Y, Shimomura Y, Higurashi T, Sugi Y, Arimoto J, Umezawa S, et al. Patients With Colorectal Cancer Have Identical Strains of Fusobacterium Nucleatum in Their Colorectal Cancer and Oral Cavity. Gut (2019) 68:1335–7. doi: 10.1136/gutjnl-2018-316661
115. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 Protein of Fusobacterium Nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors From Immune Cell Attack. Immunity (2015) 42:344–55. doi: 10.1016/j.immuni.2015.01.010
116. Nosho K, Sukawa Y, Adachi Y, Ito M, Mitsuhashi K, Kurihara H, et al. Association of Fusobacterium Nucleatum With Immunity and Molecular Alterations in Colorectal Cancer. World J Gastroenterol (2016) 22:557–66. doi: 10.3748/wjg.v22.i2.557
117. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium Nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe (2013) 14:207–15. doi: 10.1016/j.chom.2013.07.007
118. Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium Nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-κB, and Up-Regulating Expression of MicroRNA-21. Gastroenterology (2017) 152:851–66.e24. doi: 10.1053/j.gastro.2016.11.018
119. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium Nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling Via Its FadA Adhesin. Cell Host Microbe (2013) 14:195–206. doi: 10.1016/j.chom.2013.07.012
120. Chen T, Li Q, Wu J, Wu Y, Peng W, Li H, et al. Fusobacterium Nucleatum Promotes M2 Polarization of Macrophages in the Microenvironment of Colorectal Tumours Via a TLR4-Dependent Mechanism. Cancer Immunol Immunother (2018) 67:1635–46. doi: 10.1007/s00262-018-2233-x
121. Wu J, Li K, Peng W, Li H, Li Q, Wang X, et al. Autoinducer-2 of Fusobacterium Nucleatum Promotes Macrophage M1 Polarization Via TNFSF9/IL-1β Signaling. Int Immunopharmacol (2019) 74:105724. doi: 10.1016/j.intimp.2019.105724
122. Zhong J, Wang H, Chen W, Sun Z, Chen J, Xu Y, et al. Ubiquitylation of MFHAS1 by the Ubiquitin Ligase Praja2 Promotes M1 Macrophage Polarization by Activating JNK and P38 Pathways. Cell Death Dis (2017) 8:e2763. doi: 10.1038/cddis.2017.102
123. Chen Y, Chen Y, Cao P, Su W, Zhan N, Dong W. Fusobacterium Nucleatum Facilitates Ulcerative Colitis Through Activating IL-17F Signaling to NF-κB Via the Upregulation of CARD3 Expression. J Pathol (2020) 250:170–82. doi: 10.1002/path.5358
124. Engevik MA, Danhof HA, Ruan W, Engevik AC, Chang-Graham AL, Engevik KA, et al. Fusobacterium Nucleatum Secretes Outer Membrane Vesicles and Promotes Intestinal Inflammation. mBio (2021) 12:e02706–20. doi: 10.1128/mBio.02706-20
125. Gatej SM, Bright R, Weyrich LS, Marino V, Christophersen CT, Gibson RJ, et al. Probiotic Lactobacillus Rhamnosus GG Protects Against P. Gingivalis And F. Nucleatum Gut Dysbiosis. J Int Acad Periodontol (2020) 22:18–27.
126. Palioto DB, Finoti LS, Kinane DF, Benakanakere M. Epigenetic and Inflammatory Events in Experimental Periodontitis Following Systemic Microbial Challenge. J Clin Periodontol (2019) 46:819–29. doi: 10.1111/jcpe.13151
127. Arvikar SL, Collier DS, Fisher MC, Unizony S, Cohen GL, McHugh G, et al. Clinical Correlations With Porphyromonas Gingivalis Antibody Responses in Patients With Early Rheumatoid Arthritis. Arthritis Res Ther (2013) 15:R109. doi: 10.1186/ar4289
128. Jenning M, Marklein B, Ytterberg J, Zubarev RA, Joshua V, van Schaardenburg D, et al. Bacterial Citrullinated Epitopes Generated by Porphyromonas Gingivalis Infection-A Missing Link for ACPA Production. Ann Rheum Dis (2020) 79:1194–202. doi: 10.1136/annrheumdis-2019-216919
129. Konig MF, Paracha AS, Moni M, Bingham CO 3rd, Andrade F. Defining the Role of Porphyromonas Gingivalis Peptidylarginine Deiminase (PPAD) in Rheumatoid Arthritis Through the Study of PPAD Biology. Ann Rheum Dis (2015) 74:2054–61. doi: 10.1136/annrheumdis-2014-205385
130. Munenaga S, Ouhara K, Hamamoto Y, Kajiya M, Takeda K, Yamasaki S, et al. The Involvement of C5a in the Progression of Experimental Arthritis With Porphyromonas Gingivalis Infection in SKG Mice. Arthritis Res Ther (2018) 20:247. doi: 10.1186/s13075-018-1744-3
131. Muñoz-Atienza E, Flak MB, Sirr J, Paramonov NA, Aduse-Opoku J, Pitzalis C, et al. Gingivalis Autocitrullinome Is Not a Target for ACPA in Early Rheumatoid Arthritis. J Dental Res (2020) 99:456–62. doi: 10.1177/0022034519898144
132. Hamamoto Y, Ouhara K, Munenaga S, Shoji M, Ozawa T, Hisatsune J, et al. Effect of Porphyromonas Gingivalis Infection on Gut Dysbiosis and Resultant Arthritis Exacerbation in Mouse Model. Arthritis Res Ther (2020) 22:249. doi: 10.1186/s13075-020-02348-z
133. Yang P, Qian FY, Zhang MF, Xu AL, Wang X, Jiang BP, et al. Th17 Cell Pathogenicity and Plasticity in Rheumatoid Arthritis. J Leukoc Biol (2019) 106:1233–40. doi: 10.1002/JLB.4RU0619-197R
134. de Aquino SG, Abdollahi-Roodsaz S, Koenders MI, van de Loo FA, Pruijn GJ, Marijnissen RJ, et al. Periodontal Pathogens Directly Promote Autoimmune Experimental Arthritis by Inducing a TLR2- and IL-1-Driven Th17 Response. J Immunol (Baltimore Md: 1950) (2014) 192:4103–11. doi: 10.4049/jimmunol.1301970
135. Zackular JP, Rogers MA, Ruffin M, Schloss PD. The Human Gut Microbiome as a Screening Tool for Colorectal Cancer. Cancer Prev Res (Phila) (2014) 7:1112–21. doi: 10.1158/1940-6207.CAPR-14-0129
136. Chen W, Liu F, Ling Z, Tong X, Xiang C. Human Intestinal Lumen and Mucosa-Associated Microbiota in Patients With Colorectal Cancer. PloS One (2012) 7:e39743. doi: 10.1371/journal.pone.0039743
137. Wang X, Jia Y, Wen L, Mu W, Wu X, Liu T, et al. Porphyromonas Gingivalis Promotes Colorectal Carcinoma by Activating the Hematopoietic NLRP3 Inflammasome. Cancer Res (2021) 81:2745–59. doi: 10.1158/0008-5472.CAN-20-3827
138. Kaplan CW, Ma X, Paranjpe A, Jewett A, Lux R, Kinder-Haake S, et al. Fusobacterium Nucleatum Outer Membrane Proteins Fap2 and RadD Induce Cell Death in Human Lymphocytes. Infect Immun (2010) 78:4773–8. doi: 10.1128/IAI.00567-10
139. Kobayashi R, Ogawa Y, Hashizume-Takizawa T, Kurita-Ochiai T. Oral Bacteria Affect the Gut Microbiome and Intestinal Immunity. Pathog Dis (2020) 78:ftaa024. doi: 10.1093/femspd/ftaa024
140. Brown EM, Kenny DJ, Xavier RJ. Gut Microbiota Regulation of T Cells During Inflammation and Autoimmunity. Annu Rev Immunol (2019) 37:599–624. doi: 10.1146/annurev-immunol-042718-041841
141. Zhou B, Yuan Y, Zhang S, Guo C, Li X, Li G, et al. Intestinal Flora and Disease Mutually Shape the Regional Immune System in the Intestinal Tract. Front Immunol (2020) 11:575. doi: 10.3389/fimmu.2020.00575
142. Albillos A, de Gottardi A, Rescigno M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J Hepatol (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
Keywords: periodontal pathogens, intestinal barrier, gut microbiota, tight junction, immune barrier
Citation: Liu Y, Huang W, Wang J, Ma J, Zhang M, Lu X, Liu J and Kou Y (2021) Multifaceted Impacts of Periodontal Pathogens in Disorders of the Intestinal Barrier. Front. Immunol. 12:693479. doi: 10.3389/fimmu.2021.693479
Received: 11 April 2021; Accepted: 12 July 2021;
Published: 27 July 2021.
Edited by:
Laurel L. Lenz, University of Colorado, United StatesReviewed by:
Massimo Costalonga, University of Minnesota Twin Cities, United StatesKeith Kirkwood, University at Buffalo, United States
Copyright © 2021 Liu, Huang, Wang, Ma, Zhang, Lu, Liu and Kou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yurong Kou, eXJrb3VAY211LmVkdS5jbg==