
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 28 July 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.692004
This article is part of the Research Topic Systems Biology Approach to the Immunology of Asthma and Allergy View all 8 articles
 Juan Rodriguez-Coira1,2,3†
Juan Rodriguez-Coira1,2,3† Alma Villaseñor1,2†
Alma Villaseñor1,2† Elena Izquierdo1
Elena Izquierdo1 Mengting Huang3
Mengting Huang3 Tomás Clive Barker-Tejeda1,2
Tomás Clive Barker-Tejeda1,2 Urszula Radzikowska3
Urszula Radzikowska3 Milena Sokolowska3*
Milena Sokolowska3* Domingo Barber1*
Domingo Barber1*There is increasing evidence that the metabolic status of T cells and macrophages is associated with severe phenotypes of chronic inflammation, including allergic inflammation. Metabolic changes in immune cells have a crucial role in their inflammatory or regulatory responses. This notion is reinforced by metabolic diseases influencing global energy metabolism, such as diabetes or obesity, which are known risk factors of severity in inflammatory conditions, due to the metabolic-associated inflammation present in these patients. Since several metabolic pathways are closely tied to T cell and macrophage differentiation, a better understanding of metabolic alterations in immune disorders could help to restore and modulate immune cell functions. This link between energy metabolism and inflammation can be studied employing animal, human or cellular models. Analytical approaches rank from classic immunological studies to integrated analysis of metabolomics, transcriptomics, and proteomics. This review summarizes the main metabolic pathways of the cells involved in the allergic reaction with a focus on T cells and macrophages and describes different models and platforms of analysis used to study the immune system and its relationship with metabolism.
The correct onset and regulation of immune responses in cancer and infectious diseases are affected by the activation of different intracellular metabolic pathways. When we talk about the sequence of metabolic events that takes place in different immunological cellular players, we refer in a general way to immunometabolism (1). There is an increasing interest in figuring out the changes in metabolic routes associated with the proliferation and control of immunological responses. Due to the SARS-CoV-2 pandemic, metabolic co-morbidities such as diabetes and obesity have dramatically been associated with an increased risk for severe disease, highlighting the relevance of energy metabolism for mounting correct immunological responses to infections (2–4). Another area where immunometabolism research has exploded in recent years is cancer. Metabolic reprogramming of the immune system by cancer cells (5) and new intervention strategies aiming to reactivate anergic cells or to block tumor-induced regulatory signals are subjects of intensive research (6–8).
Cells adapt their metabolic status by means of sensors detecting extracellular or intracellular signals. Determination of the mechanisms involved will provide new potential therapeutic targets to control inflammation. In fact, already today, different therapeutic approaches such as rapamycin, calcineurin inhibitors, or purine and pyrimidine synthesis inhibitors target T cell proliferation to alleviate autoimmune or inflammatory diseases (9).
Little is known about the intracellular metabolic changes in cells that lead to exacerbated allergic responses to innocuous antigens. The variety in exposome, from mere allergic sensitization with occasional exposure (as in the case of Hymenoptera venom allergy) to allergic perennial phenotypes in highly poly-sensitized patients makes allergic diseases an interesting and complex model to analyze the role of an altered energy metabolism in disease progression. Recent evidence suggests that metabolic reprogramming steering T cell proliferation plays an important role in the evolution from mild to severe allergic phenotypes (10, 11). In fact, systemic signatures pointing to Warburg metabolism have been described in severe allergy respiratory patients (12). Therefore, elucidating the mechanisms underlying the metabolic rewiring of immune cells might be a key to understand the evolution to severe allergic phenotypes and to develop new biomarker strategies that could lead to a personalized intervention, a clear unmet need in allergic patient management (13, 14). For this purpose, it is imperative to understand the values and limitations of the methodological approaches used to study immunometabolism as it ranges from more classical immunological and biochemical approaches like flow cytometry to the single-cell level of different -omics studies.
In the present review, we provide a holistic view of immunometabolism with a focus on allergic diseases. We provide a general view of metabolic routes at both systemic and cellular levels, with a focus on the main immunological players, and summarizing the main methodological approaches used so far.
The classical allergic reaction is an immune response characterized by the leading role of allergen-specific type 2 T helper cells (Th2) and type 2 innate lymphoid cells (ILCs), which produce their characteristic cytokines – mainly interleukin (IL)-4, -5, and -13 (15, 16). These cytokines lead to an inflammatory environment with the involvement of other cell types, mainly epithelial cells of the skin and mucosa, dendritic cells (DCs), mast cells (MCs), basophils, and eosinophils (17, 18).
The most prevalent and well-known allergic responses are type-2 reactions, which are immunoglobulin E (IgE)-dependent (i.e., allergic asthma, anaphylaxis, allergic rhinitis, atopic dermatitis, and most food allergies) (17). However, other allergic phenotypes with dominance on non-type 2 inflammation (or so-called type-2 low) and no significant IgE response are also quite frequent, such as non-IgE-mediated food allergies or type-2 low asthma (19). Their mechanisms are much less studied (20).
There are three main phases in the course of allergic inflammation: early-phase reactions, late-phase reactions, and chronic allergic inflammation (Figure 1) (21, 22).
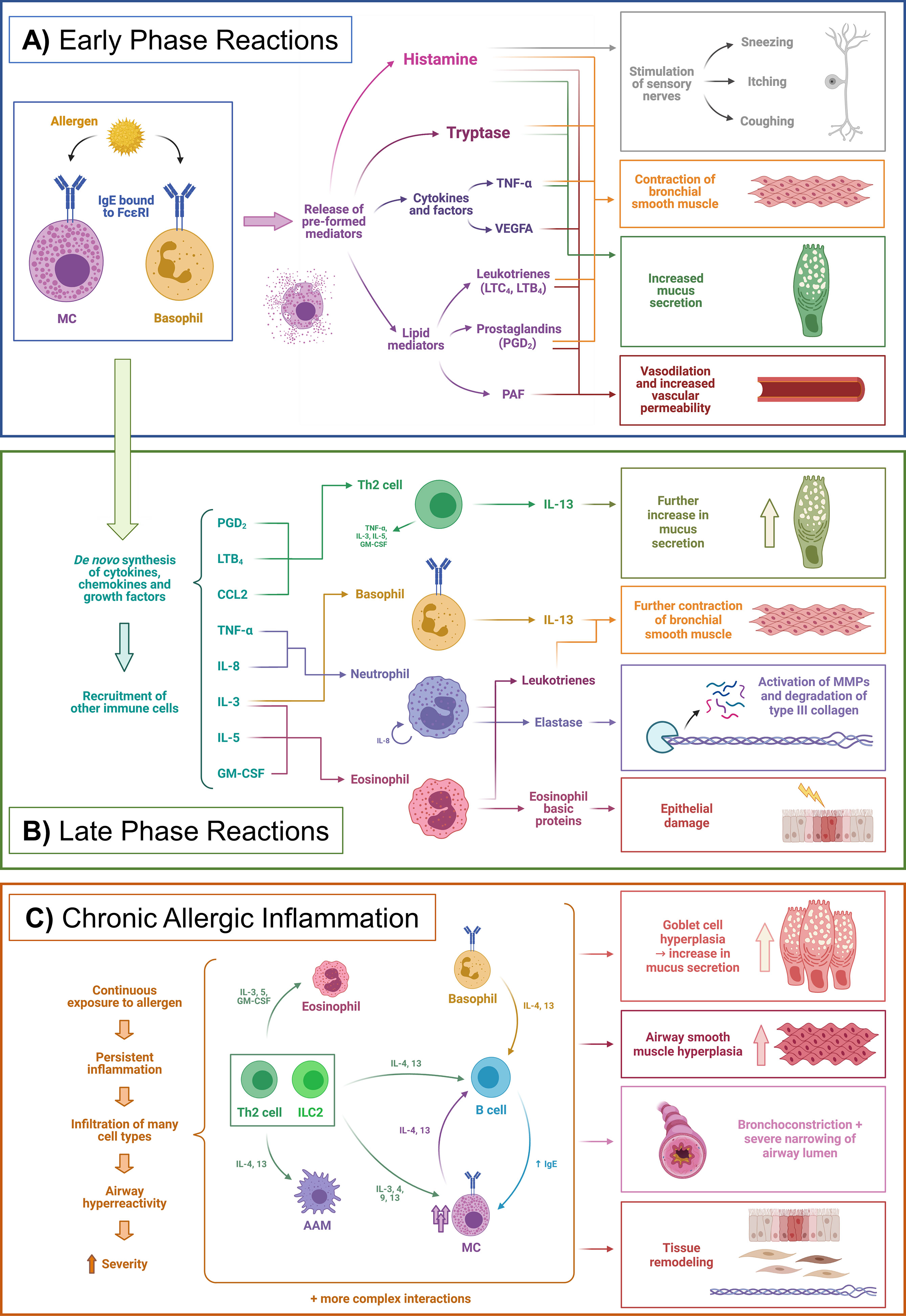
Figure 1 Phases of allergic inflammation. Example of a simplified allergic asthma model. (A) Early phase reactions, (B) Late phase reactions, (C) Chronic allergic inflammation. AAM, Alternatively activated macrophages; CCL2, Chemokine (C-C motif) ligand 2; FcεRI, High-affinity IgE receptor (Fc epsilon receptor I); GM-CSF, Granulocyte-macrophage colony-stimulating factor; IgE, Immunoglobulin E; IL, Interleukin; ILC2, Type 2 innate lymphoid cell; LTB4, Leukotriene B4; LTC4, Leukotriene C4; MC, Mast cell; MMPs, Matrix metalloproteinases; PAF, Platelet-activating factor; PGD2, Prostaglandin D2; Th2, Type 2 T helper cell; TNF-α, Tumor necrosis factor alpha. Adapted from Galli et al. Nature 2008;454 (7203),445-454. Created with BioRender.com.
Early phase reactions are characterized by the liberation of mediators from MCs – and basophils to a lesser extent (23). For this, sensitization must have previously occurred, in which the allergen has been presented to T cells by antigen-presenting cells (APCs) mainly DCs and macrophages. Briefly, APCs ingest allergenic proteins, process them to peptides, and present them via the major histocompatibility complex class II (MHC-II) to naïve T cells in the lymph nodes. Then, this presentation triggers a type 2 polarization which ultimately results in the production of allergen-specific IgE by plasma cells, which are derived from the immunoglobulin class-switch recombination and differentiation of B cells (21, 24). The role of epithelial cells as essential components of the innate immune response and specifically in allergy has sparked increasing interest and has been reviewed recently (17). This IgE is bound to the high-affinity Fc epsilon receptor I (FcεRI) on the surface of MCs and basophils. When re-exposed to the sensitized allergen, binding to the IgE triggers the release of the aforementioned mediators, which have been pre-formed and stored in cytoplasmic granules. These are mainly histamine, serine proteases such as tryptases, other enzymes, cytokines [e.g. tumor necrosis factor-alpha (TNF-α) and vascular endothelial growth factor A (VEGFA)], and many potent lipid mediators, such as prostaglandin D2 (PGD2), leukotriene B4 (LTB4) and cysteinyl leukotrienes (CysLTs) (Figure 1A) (21, 25, 26). If the mediators are released locally and in a self-limiting way, the symptoms are usually not life-threatening, whereas a rapid and massive release into the circulation may cause a severe systemic reaction called anaphylaxis (21, 27).
Apart from these pre-formed mediators, MCs also produce cytokines, chemokines, lipid mediators, and growth factors de novo when triggered by an allergen. This process is slower than the fast degranulation, and the consequences are evident in the late-phase reaction, occurring hours after the allergen challenge. This is a consequence of the recruitment of other immune cells by products such as TNF-α, IL-8, chemokine (C-C motif) ligand 2 (CCL2), CysLTs, and other chemokines. Cells recruited include Th2 cells, neutrophils, monocytes, eosinophils, and basophils (Figure 1B) (28). However, late-phase reactions do not occur in all patients and may not be clearly delimited from early-phase reactions (21).
If the exposure to the allergen persists over time, or the inflammation is not resolved adequately, the immune response evolves into chronic allergic inflammation, which is characterized by infiltration of many different types of type 2 and non-type 2 immune cells from both innate and adaptive systems. This infiltration can ultimately lead to structural changes – i.e. tissue remodeling (29) – and altered functions of the affected organs. In the case of asthma, a well-studied example of this process, the remodeling includes thickening of the airway walls, hyperplasia of goblet cells – with the subsequent increase in mucus production – epithelial injury and increased numbers of MCs, to name a few examples, leading ultimately to a state of airway hyperreactivity and a more severe phenotype (Figure 1C) (19, 21, 22, 29). Additionally, persistent IgE levels – at least in lifelong food allergies– seem to be sustained by allergen-specific long-lived memory B cells that upon reactivation with an allergen undergo class-switch recombination and replenish the IgE+ plasma cell compartment, instead of long-lived IgE+ plasma cells as was previously thought (24).
Metabolism can be defined as the sum of all chemical reactions used for sustaining life. Although every type of cell presents some specific reactions, a great amount of cell metabolism is conserved between cell types. As such, this part of the review will focus on the general metabolism used for energy production and its general regulation (Figure 2).
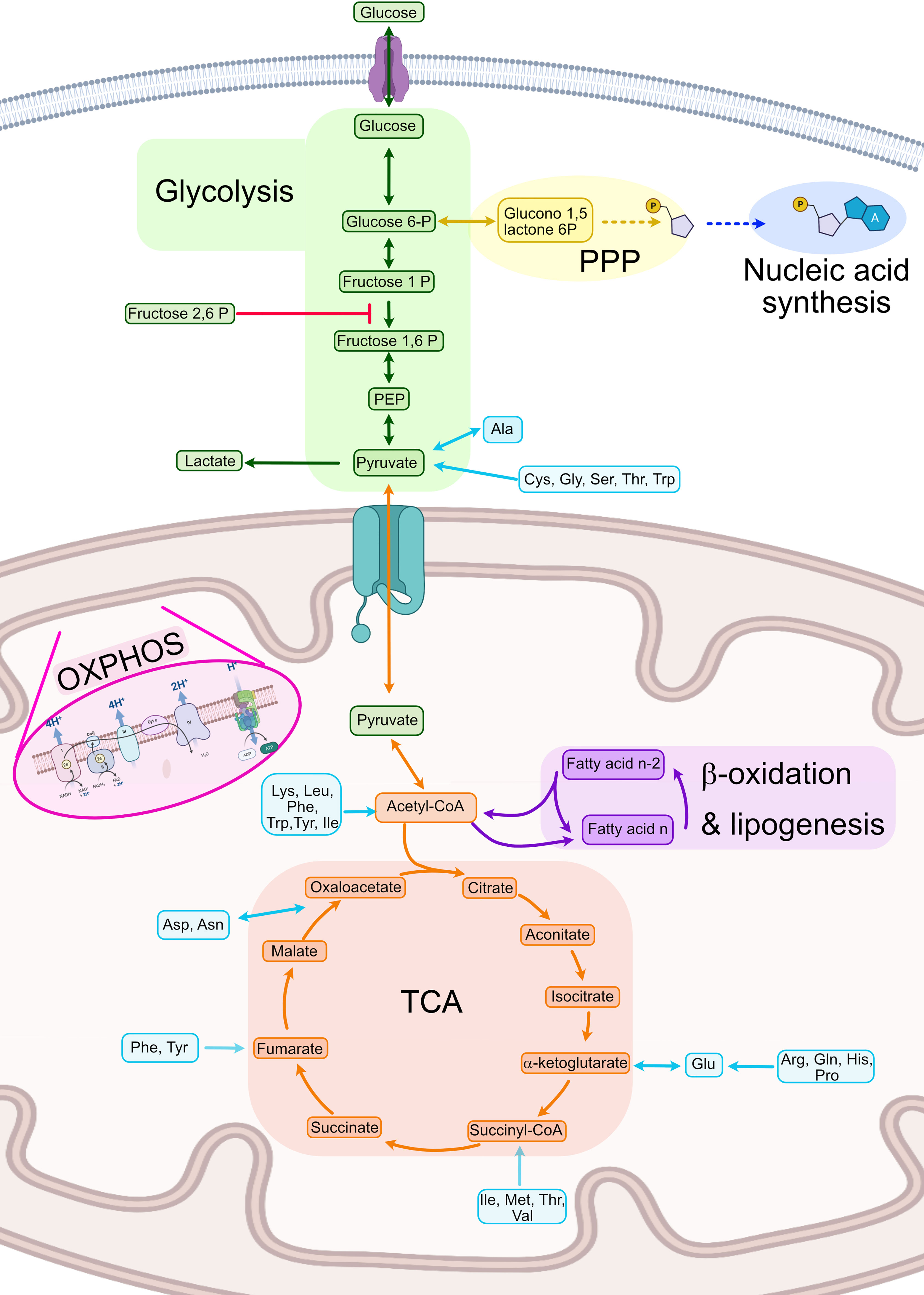
Figure 2 Summary of human cellular metabolism. In green steps from the glycolysis pathway, in yellow the PPP, in dark blue nucleic acid biosynthesis, in orange the TCA, in purple fatty acid oxidation and lipogenesis, in light blue amino acid metabolism. Ala, Alanine; Asn, Asparagine; Asp, Aspartate; Cys, Cysteine; Gln, Glutamine; Glu, Glutamate; Gly, Glycine; His, Histidine; Ile, Isoleucine; Leu, Leucine; Lys, Lysine; Met, Methionine; OXPHOS, Oxidative phosphorylation; PEP, Phosphoenol pyruvate; Phe, Phenylalanine; PPP, Pentose phosphate pathway; Pro, Proline; Ser, Serine; TCA, Tricarboxylic acid cycle Thr, Threonine; Trp, Tryptophan; Tyr, Tyrosine; Val, Valine. Created with BioRender.com.
One of the most important metabolic pathways for energy generation is glycolysis, which is a heavily controlled process focused on the complete breakdown of glucose molecules to obtain nicotinamide adenine dinucleotide (NADH) and energy. Hexokinases (HK) 1-3 are the first enzymes needed for initiating the pathway by phosphorylating glucose molecules and thus inhibiting glucose excretion from the cell (30, 31). Secondly, the reaction that converts fructose-P to fructose 1,6-bisphosphate (1,6-FBP), catalyzed by the phosphofructose kinase (PFK), is the main point of regulation of the glycolysis as it is an irreversible step (32–34). This enzyme is only activated in presence of fructose 2,6 bisphosphate (2,6-FBP), which is synthetized by PFK2 under high levels of intracellular adenosine monophosphate (AMP). Finally, the last step of the glycolysis pathway catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate and ATP by pyruvate kinase (PK) which is also regulated by 2,6-FBP (35). Glycolysis is usually used in anaerobic conditions or when fast energy is required. However, in the case of activated immune cells, it is one of the most important pathways for energy production even in the presence of oxygen, a phenomenon called aerobic glycolysis or Warburg effect (36, 37). When nucleic acid synthesis is needed, G6P can be used instead to create the pentoses needed for nucleic acid synthesis using the anabolic pathway called pentose phosphate pathway (PPP) (Figure 2).
Once pyruvate is generated, it needs to be further processed into either lactate (anaerobiosis) or acetyl coenzyme A (acetyl-CoA) to regenerate the reductive molecules generated in glycolysis (NADH). Acetyl-CoA can enter the tricarboxylic acid cycle (TCA) also known as the Krebs cycle which takes place in the mitochondria (38). This cycle is an amphibolic pathway, meaning it can participate in both anabolic and catabolic processes depending on the needs of the cell (39). TCA is a central pathway in which different metabolic processes such as glycolysis, transamination, deamination, lipogenesis, and β- oxidation converge (39). In the TCA, acetyl-CoA is introduced into the TCA by the enzyme citrate lyase, which produces citrate. This molecule will be completely metabolized in a sequence of reactions until oxaloacetate (Figure 2). This cycle is mainly regulated by the concentrations of product and the availability of substrates in each step. Conversion of acetyl-CoA into citrate is usually the limiting step. TCA takes place in the mitochondria of human cells and produces ATP and NADPH. The latter is used to fuel oxidative phosphorylation (OXPHOS) also known as mitochondrial respiration. OXPHOS is based on a series of redox reactions in the inner mitochondrial membrane by the electron transport chain complex to completely reduce O2 to water (40). In the first step, H+ ions from NADH and nicotinamide adenine dinucleotide phosphate (NADPH) are pushed into the intermembrane space generating a chemical gradient and different electric potential across the inner membrane. At the same, the electrons of those H atoms get transported along a protein transport chain in the inner mitochondrial membrane. At the end of the chain, the ATPases use the H+ electrochemical gradient to generate ATP when the H+ return to the mitochondrial matrix. These protons will be then linked to O2 and the transported electrons to synthesize H2O (41) (Figure 2).
Acetyl-CoA can also be generated in β-oxidation also known as fatty acid (FA) oxidation (FAO), in which a series of sequential reactions convert FAs into acetyl-CoA (42). First, the FAs are activated by adding the CoA molecule by an acyl-CoA dehydrogenase. Secondly, the FAs are transported into either the mitochondria or the peroxisome by carnitines (43, 44). Thirdly, they will be sequentially dehydrogenated, hydrated, and reduced, and finally the molecule of acetyl-CoA and a FA with 2 fewer carbons (n-2 FA) are generated. In lipogenesis, the opposite procedure is carried out, in which from acetyl-CoA longer acyl chains are built up by carbon pairs until the FA reaches the desired length (Figure 2).
Finally, amino acids in immune cells have other functions apart from the synthesis of proteins, such as energy consumption in T cells. Amino acids can be incorporated into the TCA cycle at different points, as described in Figure 2 (45). Specifically, ketogenic amino acids [leucine (Leu) and lysine (Lys)] enter throughout acetyl-CoA and glucogenic amino acids enter through TCA intermediates or pyruvate (46).
As the interface separating host and environment, mucosal barriers are key players actively involved in building tolerance to external particles or the development of allergic inflammation. In recent years, it has been shown that metabolic regulation of mucosal barriers is important in allergic responses. Reactive oxygen species (ROS), including hydrogen peroxide (H2O2), hydroxyl radical (OH), superoxide anion radical (O2–), and nitric oxide (NO), play an important role in antimicrobial immunity in the mucosal layer. Nevertheless, unchecked production can lead to mucosal barrier damage and dysbiosis, suggesting a new key role in inflammatory lung diseases (47–51). This can be exemplified in chronic rhinosinusitis (CRS), in which a complete metabolic rewiring of epithelial cells has been found. Specifically, ATP metabolic pathways were downregulated while carbohydrate and lipid metabolism pathways were upregulated leading to ROS accumulation. Such metabolic rewiring suggests a long-term inability to keep up with an increased metabolic demand during chronic inflammation (52). When challenged by allergens, barrier cells including bronchial epithelial cells and intestinal epithelial cells can overproduce ROS by up-regulating NADPH oxidase and activating nuclear factor kappa-light-chain-enhancer (NF-κB) signal pathway, leading to elevated IL-1, IL-5, IL-6, IL-33, thymic stromal lymphopoietin (TSLP) (53–56). Furthermore, increased intracellular levels of ROS can also lead to mitochondrial damage, generation of toxic metabolites, and oxidative DNA damage leading to cell apoptosis or autophagy weakening the epithelial barrier and promoting allergen introduction throughout the membrane (57). The allergens itself are also able to induce ROS production. For example, pollen NADPH oxidases increase ROS production leading to recruitment of neutrophils independent of the immune response (54). Not only allergens, but Th2-related cytokines such as IL-13 have been shown to promote ROS production and have been linked to increased inflammation in asthma models (58). Furthermore, superoxide dismutase (SOD), an enzyme with antioxidant protective activity, was found to be impaired in asthma patients compared to control subjects, which triggered apoptosis and shedding of airway epithelial cells and accelerated inflammatory responses (59).
Apart from oxidative homeostasis, glucose and FA metabolism are also pivotal for the maintenance and regeneration of airway epithelium. The proliferation and differentiation of airway epithelial progenitor cells need a moderate level of glucose. The blockade of glucose uptake or glycolysis disrupts airway club cells proliferation while promoting ciliated and goblet cell differentiation, failing to restore the homeostasis of the epithelial layer and promoting asthma chronification (60). Moreover, influenced by peroxisome proliferator-activated receptor gamma (PPAR-γ1, PPAR- γ2) and activator protein 1 (AP-1) transcription factors, glycolysis and OXPHOS were found to be upregulated in the epithelial cells during epithelial-mesenchymal transition (EMT) suggesting a specific metabolic rewiring during tissue remodeling (61). Lipid metabolism plays a key role in inflammation as mediators such as leukotrienes and imbalance in lipid metabolism have been associated with lung inflammation. More specifically, expression of stearoyl-CoA desaturase (SCD), an important enzyme in the synthesis of monounsaturated FAs (MUFAs), has been found to be significantly lower in epithelial cells from asthmatic patients (62). Other enzymes from FA metabolism such as acetyl carboxylase beta (ACC-β) or FA synthase (FAS) also presented distinctive expression patterns in asthmatics, indicating that the imbalance on FA metabolism in asthma patients results in airway hyper-sensitivity and reduced antiviral defense (62).
FA-binding protein 5 (FABP5) is mostly expressed in epidermal cells and plays an important protective role against excessive oxidative damage to lipids in lung infection (63). It has been shown that FABP5 can also help bronchial epithelial cells defend against cigarette smoke exposure and bacterial infection through its interaction with PPARγ (64, 65). Moreover, FABP5 positively regulates the expression of interleukin 1 receptor-like 1 (ST2) – an IL-33 receptor – in alveolar epithelial cells which suppresses excessive activation of ILC2 during allergic inflammation as well as maintaining mucosal barrier homeostasis and its anti-inflammatory function by assisting in retinoic acid (RA) generation (66–70). FABP5 and ST2 were found to be downregulated in the lung tissue of high−fat diet−fed compared to normal diet−fed mice, which might explain why obese people are more susceptible to allergic lung inflammation (70). As such, the positive effects of FABP5 should be investigated further – specifically, which FA are preferably transported.
Some lipid metabolites have a significant influence on allergy responses in mucosal barriers. 12(S)-hydroxyheptadeca-5Z,8E,10E-trienoic acid (12-HHT), one of the metabolites formed during unsaturated FA oxygenation (71), was found to be an endogenous agonist of leukotriene B4 receptor type 2 (BLT2) (72). By increasing claudin 4 (CLDN4) expression through the Gαi protein-p38 mitogen-activated protein kinase pathway, the 12-HHT–BLT2 axis is able to maintain epithelial barrier functions and prevent inflammation (73). 15-oxo-eicosatetraenoic acid (15-Oxo-ETE), synthesized by epithelial and MCs metabolism, may increase the dysregulation of arachidonic acid metabolism through the 15-lipooxygenase pathway and the severity of enhanced sinonasal disease observed in aspirin-exacerbated respiratory disease (AERD) (74).
Not only does the metabolic regulation of epithelial cells influence the host response to allergy, but also microbial and xenobiotic metabolites can influence epithelium in allergic responses. The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that regulates metabolization of xenobiotic toxicants by activating genes including phase I xenobiotic-metabolizing enzymes and phase II enzymes, such as NADPH quinone oxidoreductase 1 (75). Bronchial epithelial cells of AhR knockout mice are induced to EMT and become more susceptible to hyperoxic lung damage because of reduced antioxidant enzymes and increased inflammation (76, 77). Furthermore, in a human cohort study, a decreased expression of AhR in human bronchial epithelial cells also led to the worsening of allergic asthma symptoms (76).
In the case of microbial metabolites, short-chain FAs (SCFAs) – mainly butyrate, propionate, and acetate – can be very crucial resources for mucosal cells to provide energy (78) as well as to stimulate epithelial production of RA (79). P-cresol sulfate (PCS), a microbially-generated product of Tyr metabolism in the colonic epithelium (80), was recently found to distally protect the host against allergic airway inflammation such as asthma (81). While CCL-20 plays an important role in DC recruitment in allergy and asthma (82), PCS could bind in the interdomain pocket of epidermal growth factor receptor (EGFR) (83) and block signal transduction of toll-like receptor 4 (TLR-4) to inhibit CCL-20 production by airway epithelial cells and reduce allergic airway responses (81). Lactic acid-producing bacteria can alleviate shrimp tropomyosin-induced allergic mucosal disorders via regulation of arginine and proline metabolism and subsequent mammalian target of rapamycin (mTOR) pathway activation (84). All these studies above indicate how immunometabolism in mucosal barriers shapes the inflammatory or allergic responses.
APCs are critical for the initiation of adaptive immune responses and maintenance of peripheral tolerance. They uptake and process antigens which they later present to T cells. DCs are professional APCs contained in the peripheral blood but also DCs are found in tissues such as skin, nose, lungs, stomach, and intestine. DCs remain poorly immunogenic until allergens or pattern recognition receptor (PRR) ligands trigger their activation, a process that is linked to cell metabolism alterations (85). Multiple evidence has shown distinct metabolic requirements of each DC subset and its specific response. While resting or immature DCs obtain ATP by oxidative phosphorylation in mitochondria, TLR-activation rapidly enhances glycolysis, the accumulation of succinate, and the generation of citrate, which is used for the synthesis of FAs and prostaglandins, and to expand membranes in the endoplasmic reticulum and Golgi (86). The TLR-mediated “glycolytic burst” supports DC immune activity by increasing the synthesis and secretion of inflammatory cytokines. Conversely, tolerogenic properties of DCs rely on FAO, as it was observed by using different molecules that favor OXPHOS such as resveratrol, vitamin-D3, and dexamethasone (87, 88) and through the study of tolerogenic human DC metabolism (89). Metabolic control in DCs and monocytes has also been linked to the epigenetic changes and subsequent phenomenon of “trained immunity” – a memory, induced in cells of innate immunity such as DCs and monocytes/macrophages (90). Importantly, this phenomenon has been suggested to play an important role in the induction of tolerance to allergens during allergen-specific immunotherapy (AIT) as tolerogenic plasmacytoid DCs, CD141+ myeloid DCs, and intermediate monocytes were increased after 1 year of therapy whereas pro-inflammatory CD1c+ myeloid DCs and non-classical monocytes were reduced (91).
Food tolerance is orchestrated by intestinal DCs located in lymphoid tissue and lamina propria of the small intestine and colon (92), where dietary and gut microbiota-derived FAs have a significant influence as it has been reviewed before in detail (93). Polyunsaturated FAs (PUFAs), especially n-3 PUFAs, inhibit the pro-inflammatory phenotype of DCs. In contrast, saturated fatty acids (SFAs) increase DC maturation, activation, and T-cell stimulation properties. Moreover, gut bacterial metabolites, such as SCFAs and biogenic amines, enhance DC regulatory activity, leading to the induction of T reg cells and IL-10-secreting T cells (94). Although clinical data show contradictory results, early introduction of fish to diet (n-3 PUFAs) reduces sensitization to foods and development of allergic disease in children (95) and prevents allergic sensitization to cow’s milk protein in mice due to a reduction of classical DC type 1 (cDC1) population and an increase in Treg numbers (96). Remarkably, the type of FA ingested not only influences the protective or pro-allergenic role of DCs in food allergy. A higher risk of uncontrolled asthma has been documented in populations with increased consumption of SFAs- and n-6 PUFA-rich food, in contrast to a lower incidence of asthma in populations with higher proportions of dietary n-3 PUFAs. Similar observations have been made on allergic rhinitis and atopic dermatitis (AD) development in children, while in adults FAs influences are under debate (93, 97).
Another link between metabolism and the regulatory role of DCs in allergy has been confirmed by using rapamycin (mTOR inhibitor) and murine models of mTOR gene depletion. mTOR activation influences DC immune response by stimulating protein synthesis, glycolysis, mitochondrial functions, and lipid synthesis (98). Inhibition of mTORC1 during TLR triggering promotes IL-12 production and inhibits expression of IL-10 and interferon (IFN) type 1 by DCs. A recent study showed the role of the mTOR signaling pathway in the pathogenesis of allergic asthma proving that upon mTOR inactivation, CD11b+ DCs of the lung can skew allergic inflammation from eosinophilic Th2 to neutrophilic Th17 polarity. The mechanism for this change is dependent on DC IL-23 production and an increase in the use of FAO (99).
Additionally, sphingolipids are involved in DC maturation, activation and migration. It has been shown that blocking sphingosine 1-phosphate (S1P) receptors reduces the migration and the antigen presentation capacity of DCs, which decreased allergic asthma in mice (100). In addition, treatment with a non-phosphorylated S1P analog - 2-Amino-2-[2-(4-octyl-phenyl)-ethyl]-propane-1,3-diol hydrochloride (FTY720) - reduces in vitro and in vivo allergic respiratory features (101–103). A dysregulation of skin-S1P homeostasis has been discussed in the pathogenesis of AD. Indeed, topical application of S1P diminished DC antigen capture and abrogated DC migration which improved AD treatment (103, 104). Similarly, the topical administration of S1P in a murine contact hypersensitivity model reduced the number of DCs within the lymph node and decreased IL-6 and IFN-γ secretion (105). A summary of the aforementioned DC metabolism modulation can be found in Figure 3A.
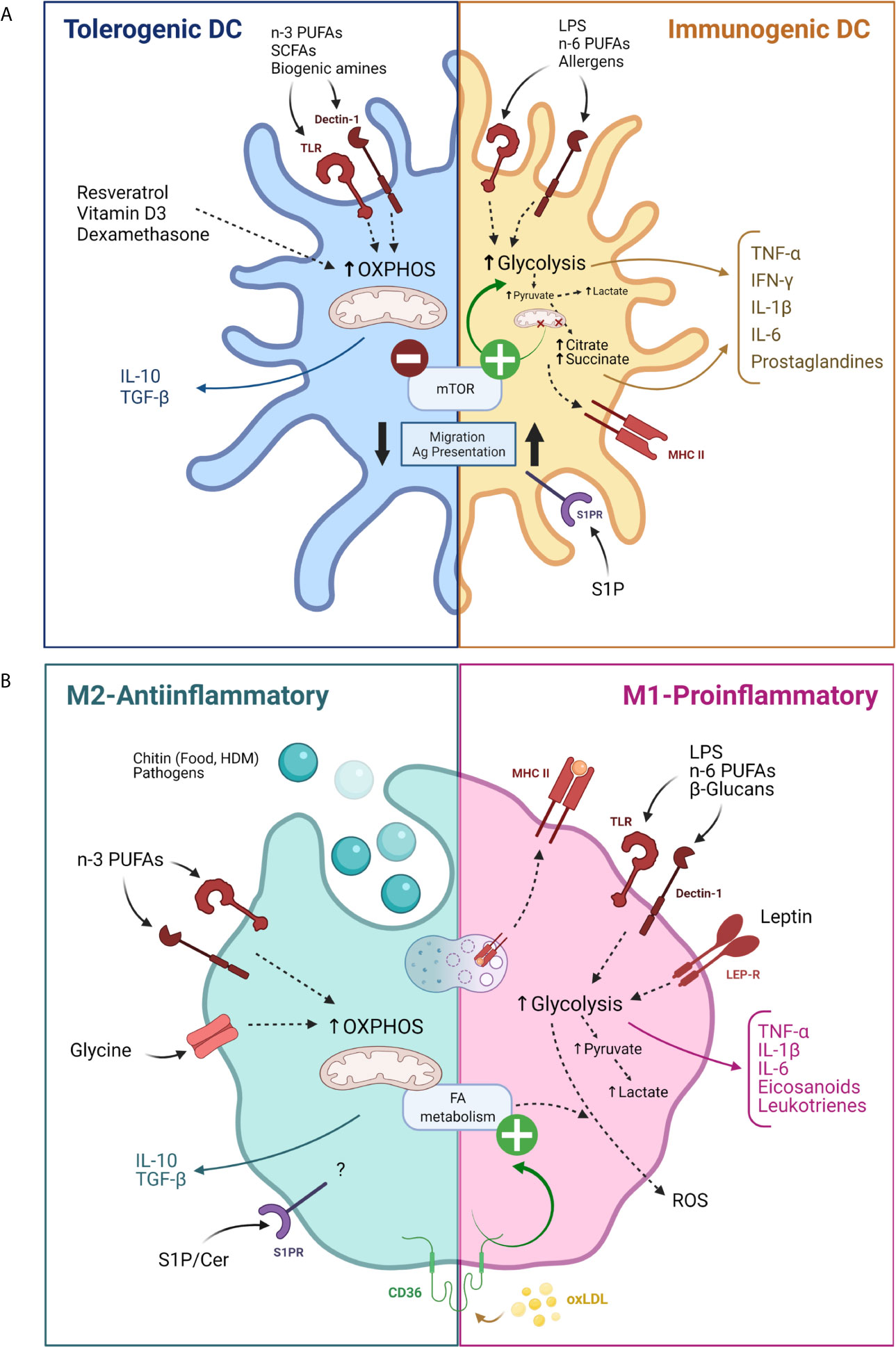
Figure 3 Modulation of antigen presenting cells activity by their metabolism. (A) Modulation of dendritic cells by their metabolism for tolerogenic (blue) and pro-inflammatory (yellow) functions. (B) Modulation of macrophages by their metabolism for M2 (green) or M1 (pink) differentiation. Cer, Ceramide; FA, Fatty acid; HDM, House dust mite; LEP-R, Leptin receptor; MHC, Major histocompatibility complex; oxLDL, Oxidized low density lipoprotein OXPHOS, Oxidative phosphorylation; PUFAs, Poly unsaturated fatty acids; ROS, Reactive oxygen species; SCFAs, Short chain fatty acids; S1P, Sphingosine 1 phosphate; SP1R, Sphingosine 1 phosphate receptor. Created with BioRender.com.
Macrophages respond to a variety of stimuli and can display a continuum of functional states that have been broadly classified between two functionally polarized extremes: M1 or pro-inflammatory macrophages and M2 or anti-inflammatory macrophages, each one associated with specific gene, protein, lipid signatures and metabolism requirements. Briefly, M1 macrophage functions mainly rely on aerobic glycolysis and succinate and citrate accumulation, while M2 macrophages properties depend on OXPHOS, enhanced FAO and a decreased glycolysis (106). Macrophage polarization can be redirected by modulation of metabolism in vitro (107).
Macrophages influence the response of allergen-specific T CD4+ helper as well as T CD8+ cytotoxic lymphocytes. Besides, murine macrophages produce histamine-releasing factor, which stimulates histamine release and IL-4 and IL-13 production from IgE-sensitized basophils and MCs (108).
Macrophages located at the oral and gut mucosa work as scavengers of damaged tissue and foreign material that is processed and presented to T cells. They also produce cytokines and chemokines that stimulate other cells such as fibroblasts (109, 110). In mice, oral and small intestine macrophages are key cells for tolerance as they exhibit an anti-inflammatory gene signature and produce IL-10, which maintains Treg activation (111, 112). Activation of macrophages via TLR-4 by a wide range of molecules is one of the main stimuli changing macrophage metabolism, function, and polarization towards M1-phenotype (113, 114). Via TLR-4 macrophages produce the typical M1-cytokines and lipid mediators such as PGD2, prostaglandin E2 (PGE2), and their metabolites (115). In addition, activation of the NLR family pyrin domain containing 3 (NLRP3) and other inflammasomes with subsequent IL-1β, IL-18, and pyroptosis, are tightly regulated by various metabolic cues (116). For example, PGE2, released by macrophages in response to TLR-4 activation and following initiation of arachidonic acid metabolism, limits macrophage function by inhibition of NLRP3 inflammasome activation (114).
Specific food-derived metabolites can also activate macrophages, such as the polysaccharide chitin, which is found in the cell walls of bacteria and fungi, mushrooms, and the exoskeleton of crustaceans and insects (117–119). Macrophages recognize chitin fragments through PRRs, mainly TLR-2 and Dectin-1, and, as consequence, they produce cytokines involved in inflammation and allergic responses. Also, macrophages of the lung and digestive tracts constitutively display chitinolytic enzymes which are increased in allergic conditions (120). Chitinase 3-like 1 (CHI3L1) protein levels are significantly higher in children with food allergy and ovalbumin (OVA)-sensitized mice than in healthy controls, and this has been linked to M2 macrophage polarization (121). Moreover, some chitinases from plant origin are a well-known group of food allergens (122). Another metabolite that modifies macrophage response is the amino acid Gly, which might prevent acute allergic response in cow’s milk allergy through the inhibition of macrophage inflammatory response (123). Additionally, ingested β-glucans bind to PRRs on macrophages and stimulate IL-12 and TNF-α cytokine production, which inhibits intestinal Th2-dependent allergies (124).
Several studies have demonstrated that airway macrophages (AMs) display altered metabolism in respiratory allergies, mainly via dysfunction in eicosanoid, glycolysis, and FA pathways (125). AMs degrade allergens through the secretion of specific enzymes, like chitinase, which neutralizes chitin (117). M1 and M2 may coexist in healthy and asthmatic lungs (126). Alveolar macrophages adapt their metabolism according to the required function. They utilize aerobic glycolysis to rapidly generate cytokines and ROS for pathogen defense but employ mitochondrial respiration to fuel inflammatory responses. Both macrophage subtypes are increased in asthmatic patients. M2 release IL-10 and IL-13, which promotes Th2 lymphocytes differentiation, as well as eosinophil recruitment (127). Likewise, they sustain and enhance inflammation initiated by Th2 cells, ILC2, eosinophils, and basophils (128). Mouse models suggest that M1 macrophages are beneficial in asthma by decreasing the recruitment of immune cells and inducing a pro-tolerogenic response. However, M1-associated features such as metabolic stress (ROS) and IFN‐γ and TNF‐α proteins are elevated in bronchoalveolar lavage and AMs of asthmatics patients (125, 129). These changes have also been related to an imbalance between oxidized and reduced forms of glutathione in AM (130). Additionally, increased production of the eicosanoid 5-hydroxyeicosatetraenoic acid (5- HETE) and leukotrienes B4 (LTB4) and E4 (LTE4) has been detected in AMs from asthmatic patients stimulated ex vivo (131). An interesting link between metabolism influence on macrophage functions in allergy is observed in obese patients with asthma, where there is a shift from M2 to M1 partly due to increased n-6 PUFAs and leptin levels found on these subjects (132). Both molecules trigger pro-inflammatory signaling pathways on macrophages and are involved in murine models of allergic inflammation (133–135).
Macrophage metabolic deregulation also plays a role in cutaneous allergic reactions. They accumulate in AD inflamed lesions where they interact closely with inflammatory immune cells, leading to the aggressive progression of severe AD (136). In lesional skin of AD patients, macrophages express higher levels of CD36+ than in normal skin (137), which is an M2-associated marker responsible for FA importation but whose signaling under dysregulated FA metabolism drives chronic inflammation (138). In contact dermatitis, macrophages present haptens to Th1 lymphocytes, which in turn produce IFN-γ that activates macrophage oxidative metabolism inducing skin damage (139). A summary of the aforementioned macrophage metabolism modulation can be found in Figure 3B.
Hence, metabolic alterations associated with allergic disorders impact APC immunomodulatory and inflammatory properties. In addition, allergens, PRR-ligands, cytokines, and lipids involved in allergic reactions affect the intracellular metabolism of APCs, determining their immunogenic or tolerogenic functions.
T lymphocytes are the essential part of the adaptive immune response. T cells can be divided into CD4+ and CD8+ populations, which have different functions. CD8+ cells constitute part of the immediate and memory cellular cytotoxic response in cancer and infectious diseases, whereas CD4+ cells are the main orchestrators of the adaptive immune response and antibody production. In allergic diseases, two subpopulations of CD4+ cells play major roles: Th2 cells, which express transcription factor GATA binding protein 3 (GATA3) and induce the production of IgE, and regulatory T cells (Tregs) which are the main inducers of tolerance towards the antigens. In the past years, it has been shown that the metabolic reprogramming of these cells has a critical influence on their function.
Th subsets rely mainly on glycolysis to quickly achieve great amounts of energy during a short span after activation (35, 37, 140, 141) whereas Tregs rely on OXPHOS as the energy supplier pathway to obtain a constant energy supply over longer periods of time (142, 143). Often, they use FA to fuel OXPHOS however the use of long-chain FAO to produce energy in any type of T cell has been shown to not be linked to their function (144).
An important requirement for T cell survival is IL-2. Oesterich et al. demonstrated that IL-2 might regulate CD4+ survival partially by regulating their metabolism. They showed that transcription factor B cell lymphoma 6 protein (Bcl-6) inhibited the expression of several glucose receptors and key enzymes from the glycolysis pathway such as HK2 and PKM2. This transcription factor was active only in a low IL-2 expression setting and was counteracted by Th1 transcription factor T-box transcription factor 21 (T-bet) (145). Moreover, it has been shown that when activated T cells are provided with co-stimulation and growth factors but are blocked from engaging glycolysis, their ability to produce IFN-β is markedly compromised (146). The importance of Bcl-6 for glycolysis was also shown in vitro and in a graft vs host disease (GVHD) mice animal model in which inhibition of glucose uptake led to preferential Treg differentiation instead of Th1 or Th17 (147). This differentiation could be a consequence of the accumulation of glycolysis intermediate subproduct PEP, which inhibits the reabsorption of intracellular calcium to the endoplasmic reticulum (ER) by suppressing the transporter sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) and thus making T cells more prone to activation (148). At the same time, glycolysis end-product lactate is able to modify T cell phenotype to Th17 in chronic inflammatory diseases by inhibiting the use of glycolysis to create pyruvate and activating the PPP. This process switches the uses of NADPH and citrate for FA synthesis which promotes phosphorylation of STAT3 and Th17 transcription factor RORyt (149). ACC1 plays a crucial role in this switch to the Th17 phenotype as its inhibition leads to blocking the glycolytic-lipogenic pathway and promotes differentiation to a Treg phenotype instead (150).
In the case of Treg cells, it has been recently shown that TLR ligands increase PI(3)K-Akt-mTORC1 signaling, glycolysis, and expression of GLUT1, impairing their suppressive functions (151). Moreover, airway Th2 cells which mediate allergic responses against house dust mite (HDM) have been shown to also rely on both glycolysis and lipid metabolism (152). Additionally, HIF-1α is a key factor to regulate T cell responses (152). Its deletion leads to an increase in Treg immunosuppression in the tumor microenvironment and inhibits their migratory capacities (153). Furthermore, complete loss of HIF-1α has been proved to lower the humoral response after carrier immunization and Th cytokine production, suggesting it is a possible target for allergen immunotherapy (154). Nevertheless, this process appears to differ in the gut as HIF-1α expression increased the number of Treg and their inhibitory functions in a colitis mouse model (155). As such, the intervention might not be effective in food allergy AIT.
Programmed cell death 1 (PD-1) and its ligand (PD1L) are key proteins in the control of immune responses and T cell fate (156–158). Patsoukis et al. have shown that PD-1 activation in T cells profoundly changes their metabolism by inhibiting glycolysis and glutaminolysis to promote FAO (159). At the same time, Palaskas et al. showed as well in a similar model an inhibition of de novo nucleoside phosphate synthesis which limits the proliferation of these cells. This phenotype could be replicated by inhibiting mTOR (160). These studies suggest new effects of the PD-1 and PDL1 axis to control inflammatory responses and could lead to the use of metabolic treatments to achieve immune suppression and tolerance as shown in allograft animal models (161).
Recent studies have shown that mitochondrial respiration and stability are also important for naïve T cells, which initiate mitochondrial biogenesis upon activation and undergo the synthesis of de novo nucleic acids using SHMT2 (162). Upon its knockdown, T cells engage in apoptosis. Milasta et al. observed that conditional ablation of mitochondrial interspace protein AIF in T cells inhibited their function. AIF did not impact the development nor increase cell death (163).
In addition, amino acids can potently control T cell functions. Arginine (Arg), an amino acid linked to NO production, is increased in asthmatic individuals (164, 165), and can influence in great manner T cells as a high intracellular concentration of Arg enhances T cell survival and function (166). Moreover, extracellular vesicles containing catabolic enzyme arginase-1 suppressed proliferation of both CD4+ and CD8+ cells, signaling the importance of this amino acid in T cell responses and suggesting a new potential drug target in asthmatic allergic diseases (167). Alanine (Ala) is also a key factor to exit quiescent state in naïve and restimulated T cells. Its deprivation leads to impaired functions, as activated T cells upregulate the expression of serine transporter and rely on this amino acid for protein biosynthesis (168). Another neutral amino acid with important functions is serine, which is required for correct glutathione metabolism and Treg suppressive functions (169, 170). Specific deletion of glutamate-cysteine ligase, a key enzyme in the biosynthesis of glutathione, led to an increase in intracellular serine, loss of expression of Foxp3, suppressive function, and increased mTOR signaling (169). A summary of the aforementioned metabolic reprogramming T cells can be found in Figure 4.
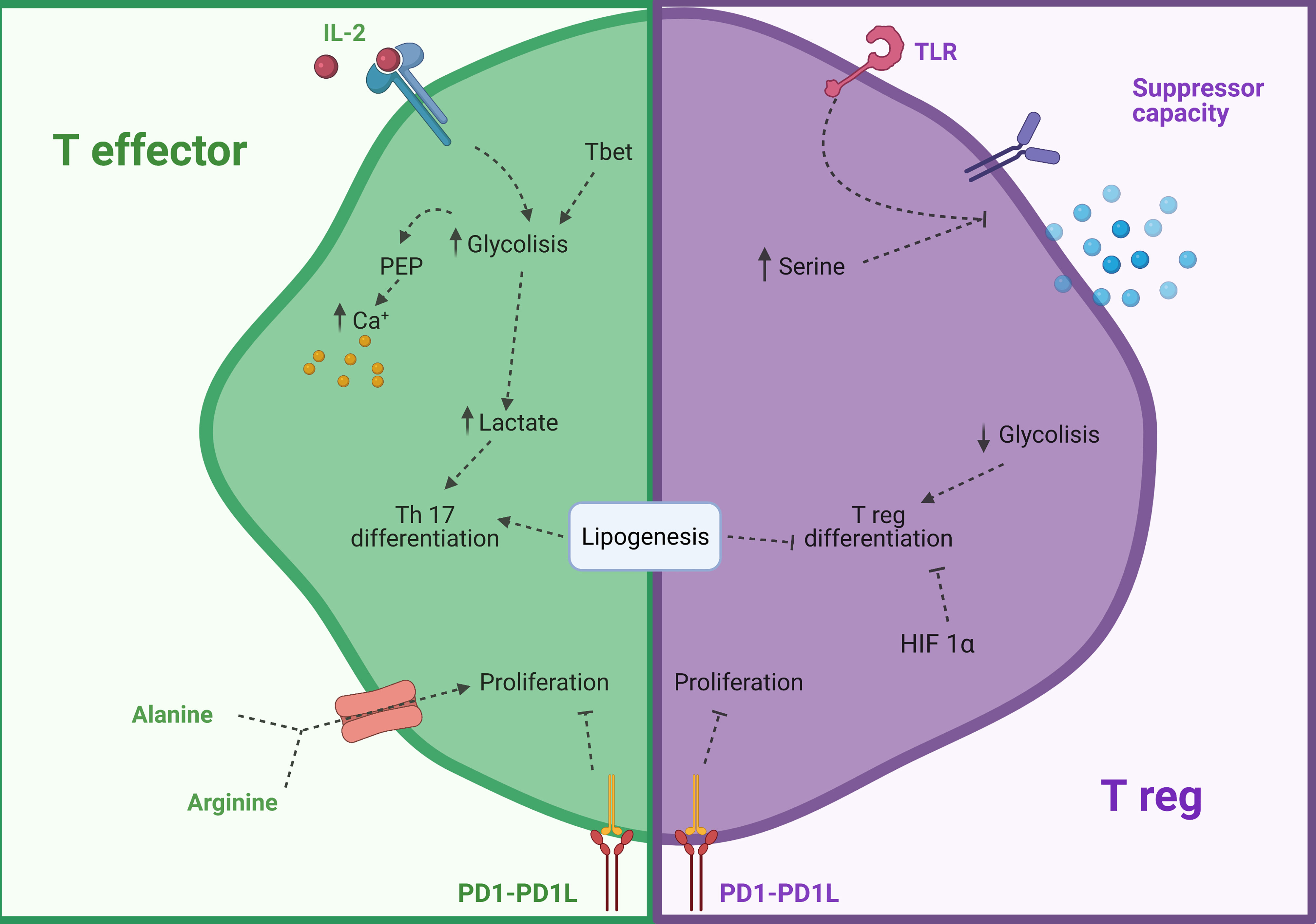
Figure 4 Modulation of CD4 T cells by their metabolism. In green modulation of T effector cells and in purple modulation of T reg cells. HIF 1β, Hypoxia inducible factor 1 β; PD-1, Programmed cell death protein 1; PD1L, Programmed cell death- ligand 1; PEP, Phosphoenol pyruvate; TLR, Toll-like receptor. Created with BioRender.com.
Due to the importance of immunometabolism in T cell differentiation and Treg function, further studies in the context of allergic inflammation are needed to explore its potential and translate it to new therapies.
Metabolic reprogramming of immune cells plays a pivotal role in different pathologies such as cancer, type 2 diabetes (T2D), obesity, and infectious diseases (171–175).
The ability of metabolic rewiring of tumor cells to support their growth is a hallmark of cancer and has been well known since the beginning of the XXth century, when the Warburg effect was initially discovered in tumoral cells (35, 36, 176). However, the Warburg effect has also been described in other cell types such as activated immune cells, signaling that metabolism may be extrapolated between different conditions or diseases (35, 36, 177–179). It is well known that tumoral cells modulate and escape the surveillance of immune cells surrounding them in the tumor microenvironment and in some cases by modifying their metabolism. For example, tumor-associated macrophages (TAMs) are abundant in most malignant tumor microenvironments and have specific immunological roles in cancer (180, 181). In a mouse model, lactic acid produced by tumoral cells could polarize TAMs towards an M2-like phenotype by a mechanism that depends on transcription factor HIF1α promoting cancer progression (182). Additionally, DCs activation depends on the use of citrate synthesized from pyruvate as a substrate for lipogenesis (86). Thus, lactate secretion by tumoral cells might also inhibit cross-presentation to T cells leading to immune evasion.
Other metabolites such as Arg also have been found to play a crucial role in the polarization between TAMs with an M1- or M2-like phenotype (TAM1 and TAM2, respectively). While TAM1s express inducible nitrate oxidase synthase (iNOS) producing high quantities of cytotoxic nitric oxide (NO) after stimulation by pro-inflammatory cytokines: TAM2s express arginase 1, which converts Arg to ornithine, promoting M2 polarization surrounding cancer cells (183). Competition for glucose between cancer cells and T cells has been shown to be critical in terms of determining whether tumors progress or regress (184). Following activation, T cells need to grow rapidly, proliferate, and generate cytokines to direct a functional immune response and thus, a metabolic switch is required (140, 141). Therefore, upon activation, the metabolic state of T cells resembles that of cancer cells. However, in the tumor microenvironment, low concentrations of glucose and direct immune suppression by the tumoral cells inhibit T cell activation (185).
Trp metabolism is also a key player in immune responses and cancer (186). Kynurenine, a metabolite derived from Trp, plays an important role in cancer, as it is absorbed by myeloid-derived suppressor cells (MDSCs) and inhibits the function of CD8+ T cells (187, 188). Kynurenine and other potential catabolic metabolites are endogenous ligands for AhR, a transcription factor that broadly modulates immunity. The activation of AhR by kynurenine can induce forkhead box p3 (Foxp3) expression in naïve T cells and prevent Th17 maturation, causing proliferation of Tregs (189, 190). Moreover, Trp depletion inhibits T cell proliferation, and indoleamine 2,3-dioxygenase 1 (IDO1) facilitates tumor evasion under immune surveillance. Additionally, activated T cells and B cells are found to possess increased Gln uptake, and Gln metabolism is critical for controlling reactive oxygen species (ROS) levels and for maintaining the balance between effector T cells (Teff) and Treg cells (191). Depletion of this metabolite blocks the proliferation and cytokine production of T cells (150). Recent studies have reported that Ser metabolism-related enzymes are induced upon naïve T cell activation and required for proliferation and survival. Specifically, they are required for de novo nucleotide biosynthesis (162, 192). Additionally, lipid metabolites such as cholesterol and prostaglandin E2 (PGE2) are extensively involved in the activation and function of immune cells (193).
Several studies have demonstrated that overnutrition is associated with low-grade, chronic inflammation increasing the risk of metabolic and cardiovascular disease and promoting autoreactivity (185). Specifically in T2D, the development of insulin resistance involves alterations of energy metabolism and inflammatory signals derived from immune cells (194). Furthermore, numerous studies have determined that T2D impairs the host defenses against pathogens including suppression of cytokine production, phagocytosis impairment, immune cells dysfunction, and incapacity to eradicate microorganisms (195, 196). Studies in mice models have shown that CD8+ T cell activation is one of the earliest events in the inflammatory response to obesity, preceding M1-like macrophage activation/infiltration in the adipose tissue (197). This effect is enhanced when elevated SFA levels are combined with hyperglycemia, as this combination appears to cause additive effects on β- cell proliferation (135). Finally, high quantities of ceramides and sphingomyelins in serum in diabetic patients induce inflammasome NLPR3 dependent activation of macrophages and increased IL-1β (198). Due to the impact of those molecules in asthmatic disease (114, 199–201), ceramides and sphingomyelins could be a potential biomarker for disease prognosis in allergic asthma.
Obesity is a co-morbidity characterized by higher levels of basal inflammatory state and associated with more severe asthma (202–204). This can be exemplified by a switch from an anti-inflammatory M2 phenotype to a pro-inflammatory M1 activation state of adipose tissue macrophages (ATM) during weight gain. Nonetheless, other studies have highlighted the heterogeneity and plasticity of ATM phenotypes in response to different metabolic stimuli such as high glucose or high insulin (205, 206). This plasticity is not limited to ATM, as alveolar macrophages in high insulin environments also showed defects in phagocytosis activity, highlighting the importance T2D might have on lung respiratory diseases (207).
In obesity, hypercholesterolemia leads to cholesterol accumulation in macrophages and other immune cells. The accumulation promotes inflammatory responses, including augmentation of TLR signaling, inflammasome activation, and the production of monocytes and neutrophils in the bone marrow and spleen (208–210). Inflammation in visceral adipose tissue (VAT) is a major driver of insulin resistance (211–213). Exposure of macrophages to very-low-density lipoproteins (VLDL) and SFAs promotes an M1-like phenotype by stimulating the secretion of proinflammatory cytokines (214, 215). Free FAs promote inflammation of proinflammatory macrophages in obese adipose tissue. These macrophages can also be distinguished from classically activated macrophages by their higher expression of genes involved in lipid metabolism (216). Obesity is also associated with reduced levels of Tregs (217).
In infection, a successful immune response relies on the ability of T cells not only to proliferate extensively and attain effector functions but also to form long-lived memory T cells that can respond again to future antigen encounters. FAO engagement is critical for the generation of memory CD8+ T cells, while mTOR signals dampen this development (218, 219). Memory CD8+ T cells possess a higher mitochondrial spare respiratory capacity than CD8+ Teffs and have greater mitochondrial mass than naive or activated T cells (220). Lipids such as FAs regulate inflammatory processes and are signaling molecules for the activation and function of macrophages, invariant Natural Killer T (iNKT) cells, and T cells (221). Moreover, mucosal-associated invariant T cells (MAIT) in obese patients present a defective mTOR functionality which leads to defective glycolysis and anti-microbial defense which could play a factor in the proinflammatory gut environment in these patients (222).
Finally, the interaction between microorganisms and their products with the immune cells can also modulate the immunometabolism. For example, antibiotic treatment promoted the overgrowth of Candida species in the gut of a mice model. This overgrowth resulted in increased levels of PGE2 in plasma which shifted macrophage polarization to M2 in the lung and thus enhancing susceptibility to allergic airway inflammation (223). Microbial products also play a role in immune system modulation as microbially-derived SCFAs can promote hematopoiesis and alter the gene expression profile of local macrophages (224, 225). Moreover, butyrate stimulates CD103+ DCs to produce higher levels of transforming growth factor-beta (TGF-β) which in turn promotes Treg differentiation (226). RA and dietary fiber also promote these CD103+ DC–Treg interactions (227). Finally, tissue-resident memory T (TRM) cells are key players to protect epithelial barrier tissues against pathogens (228). Mouse CD8+ TRM cells generated by viral infection differentially express high levels of several lipid uptake and intracellular transport molecules, including FABP4 and FABP5. The persistence of these CD8+ TRM cells in the skin was strongly diminished by inhibition of mitochondrial free FAO in vivo, suggesting that FABP4 and FABP5 have a critical role in the maintenance, longevity, and function of CD8+ TRM cells. As such, CD8+ TRM cells require the consumption of exogenous free FAs and their oxidative metabolism to persist in tissue and to mediate protective immunity (229).
In recent years different techniques have been used or adapted to study immunometabolism with a specific increase in the use of -omic techniques as they allow the acquisition and analysis of high amounts of data from the different levels of systems biology. The most used in immunology in recent years are genomics and transcriptomics. In the first sequencing studies, metabolic traits were used to define phenotypes (230). Due to the extensive databases, single-cell resolution, and broad applications, transcriptomics is a very popular technique, especially since the development of functional enrichment analysis which allowed to identify more genes and pathways related to cell metabolism. One such example is the study by Stevens et al. (74) who utilized single-cell RNA sequencing to compare the transcriptional profile of cells from patients with AERD and chronic rhinosinusitis with nasal polyps (CRSwNP) and could identify a novel lipid mediator,15-oxoicosatetraenoic acid (15-Oxo-ETE). Moreover, this technology can be used to obtain information about the impact of microbial metabolism as proposed by Singer et al. (231), who performed single-molecule real-time sequencing technology to identify phylogenetic microbial communities in high-resolution, enabling them to profile them and predict their metabolic potential.
Proteomics has also been extensively used in immunometabolism research. Proteomics is based on studying the proteic compartment of living cells. It provides more information than transcriptomics, since the key components controlling cellular metabolism are enzymes, and as such, their regulation can be done by post-translational modifications. Several studies have used proteomics for the study of immunometabolism. Tan et al. used it to study all the metabolic effects that are triggered on T cells upon TCR activation, from which OXPHOS was found to be critical for the T cells to proliferate and differentiate (232). Ron Harel et al. also showed that mitochondrial one-carbon metabolism was also crucial for T cell activation, survival, and adequate OXPHOS energy output (162).
The last -omic technique to investigate immunometabolism is metabolomics, the science advocated to the study of the metabolism in living organisms (233). Metabolomics enables the characterization of metabolites (endogenous small molecules) that are the intermediate or final products of biochemical reactions, revealing connections among different pathways that operate within a living cell (234). Currently, there is no single technique that detects the entire metabolome, as the metabolite repertoire encompasses wide concentration ranges and different physicochemical properties. However, high-throughput analytical techniques – such as mass spectrometry (MS) and nuclear magnetic resonance spectroscopy (NMR) – allow to detect high numbers of metabolites at the same time and give the structural information necessary for identification. Additionally, separation techniques – such as liquid chromatography, gas chromatography, and capillary electrophoresis – have been coupled with MS to increase the number of metabolites detected (235), and give complementary information. Metabolomics can be applied to any biological sample such as plasma, cells, or feces. This science can work following two approaches, non-targeted and targeted analysis. In the first one, the aim is to detect as many metabolites as possible in a single analysis in each sample, selecting afterward those statistically different metabolites between groups. This approach is hypothesis-free and leads to the discovery of promising metabolic changes to better understand the molecular mechanisms in the pathology. On the other hand, in targeted analysis, specific metabolites are selected based on previous knowledge, analyzed and, frequently, quantified. Usually, targeted analysis is applied to confirm and validate previous findings from the non-targeted analysis. These findings can constitute, after a proper validation, potential biomarkers used in the clinic for diagnosis or prognosis.
Immune cells have been analyzed by metabolomics. In a recent study, the characterization of cell subpopulations of mouse peritoneal macrophages after fluorescence-activated cell sorting (FACS) was carried out. The authors observed that FACS-treated cells have plasma membrane-derived metabolites causing inflammation, cell damage, stress, and specific changes in energy consumption-related metabolites (236). Additionally, metabolomics has also been used to characterize the metabolic profile of allergic patients, in which severe patients presented a characteristic pro-inflammatory profile (12). Furthermore, isotope tracing analysis using isotope-labeled metabolites – such as glucose or amino acids –can shed light on the flux of the metabolite inside specific pathways (237). These techniques can be used in combination with other omics for a full comprehensive study of the metabolism (166).
Finally, non -omic techniques can also be employed to study immunometabolism. Flow cytometry, a gold standard technique in immune phenotyping, has been recently used to study different metabolic proteins in immune cells at the single-cell level (238). This technique may be a good first step before approaching an -omic technique in which complex technical knowledge might be required. Another popular technique is the use of Seahorse extracellular flux analyses. These in vitro studies are based on the real-time measurement of different physical properties in the cell media to reflect the cellular metabolic activity – specifically, oxygen consumption to measure OXPHOS activity and medium acidification for glycolysis. As such, it allows to analyze the effects of different stimuli at the metabolic level. Numerous studies mentioned throughout this review have applied this technique (6, 86, 166, 184, 220).
All of these are the most extensively used techniques in immunometabolism and should give a good snapshot for new researchers into the field. A short summary of the techniques mentioned can be found in Table 1.
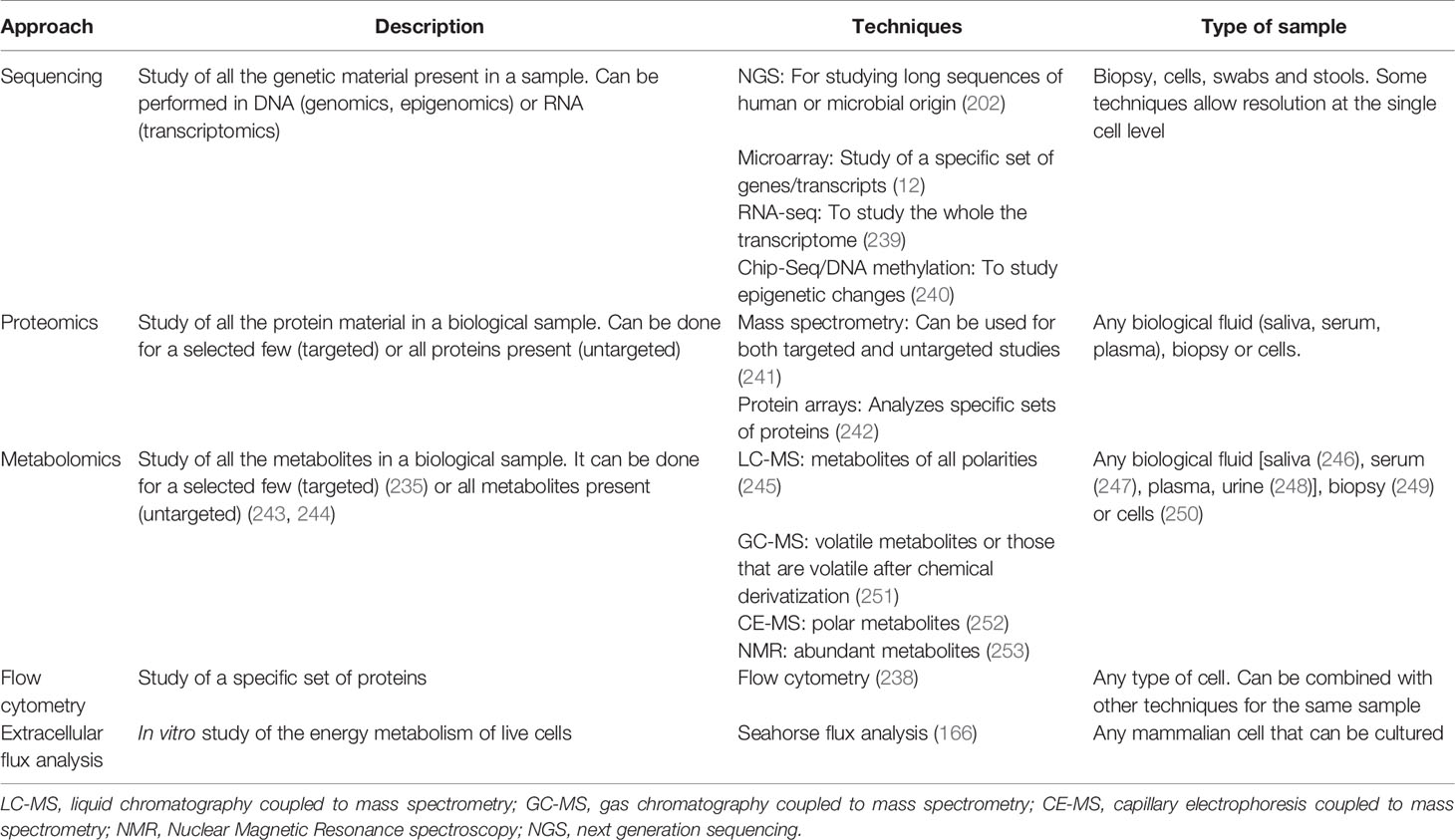
Table 1 Current approaches to study cell metabolism.
Cell metabolism actively participates in shaping immune responses in health and disease. Metabolic changes influence immune responses in various disorders, such as obesity, T2D, cancer, infectious diseases, and allergy. Metabolic dysregulation affects all cells in the body, with epithelium, macrophages, DCs, and T cells playing pivotal roles. Changes in metabolic profiles of immune cells affect the balance between immunogenic and tolerogenic phenotypes, subsequently participating in disease pathomechanism. The metabolic status of an individual is closely linked to nutrients availability. Excessive intake of SFAs might increase inflammation, especially in the presence of pre-activated immune cells related to the pathomechanism of the underlying disease. On the opposite, a diet rich in unsaturated n-3 FAs reduced the frequency of allergic diseases highlighting the importance of metabolism and diet in allergic diseases (254). Additionally, dietary habits also influence the metabolism of gut microbiota, which subsequently affects the health status of the host. Host–microbiota crosstalk modulates allergic inflammation not only in the gut but also in peripheral tissues, such as the lungs (224). The influence of metabolism on immune homeostasis in allergic diseases is not fully understood. However, a growing body of evidence suggests the pivotal role of immunometabolism in the regulation of mucosal barriers, altered pathways in antigen-presenting cells, and T cells in allergy. Understanding the complex effects of metabolism on immunological homeostasis is crucial for the prevention and treatment of allergic diseases. Metabolites are a direct link between the metabolic status of an individual and disease phenotype. Detection of metabolites is possible in various samples such as immune cells, serum, plasma, saliva, urine, stool, or tissue biopsies using metabolomics. The non-targeted analysis approach is the most used in exploratory studies because it can highlight the metabolic pathways involved under a condition. Complementarily, the targeted analysis is capable of validating the findings from the previous approach and thus lead to the determination of a more reliable biomarkers for a condition.
Regarding the coverage of metabolites, great efforts have been made to create robust methodologies to cover polar and lipid metabolites independently. In this sense, while analysis of TCA and glycolysis metabolites are a key part of the cellular metabolism, lipids (apart from other roles) are critical regulators of inflammation (255–257). Different studies have demonstrated the importance of lipid mediators in the development of allergy and other related diseases (258). In cells, lipids are participants in cell signaling events and have been associated with immune phenomena such as degranulation, chemotaxis, and sensitization (259). For example, leukotrienes are a class of immune-modulating eicosanoids that have emerged as useful clinical targets for the treatment of allergic diseases (260–262). Another class of lipids is sphingolipids, specifically sphingosine-1 phosphate, which has been closely linked to asthma and allergy progression (12). Other mechanisms such as fatty acid oxidation (FAO) have been observed increased in allergic airway inflammation in immune cells to support the production of cytokines, chemokines, and other factors important in the development of asthma (263).
The relevance of the omic sciences in the clinic relies on the idea that understanding the molecular mechanisms is crucial to improve diagnosis, prognosis, and therapeutic personalized medicine strategies.
Complementarily, several dedicated methodologies such as Seahorse flux analysis assays allow in-depth investigation of the metabolic status. All metabolomic approaches coupled with proteomics, genomics, transcriptomics, and epigenomics will allow for a better understanding of metabolism in allergic diseases.
The metabolism of immune cells is intimately linked to their differentiation and function. Immune cells under specific microenvironment changes display different metabolic responses. This metabolic reprogramming can be initiated not only by nutrient conditions but also by disease-related molecules –such as inflammatory molecules, pathogens, or allergens. In the present day, ongoing research is focused on understanding the immune cell metabolism in different conditions such as inflammation, cancer, obesity, T2D infectious diseases, and allergy, which could shed light on new therapeutic interventions.
JR-C contributed to parts 3, 6, 8, 9. AV contributed to parts 7, 8, and 10. EI contributed to part 5. MH contributed to parts 4 and 8. TB-T contributed to part 2, to figure 3 and to English proof correction. UR contributed to part 8 and 9. DB and MS contributed to parts 1, 9, and 10 and coordinated the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by Instituto de Salud Carlos III (project numbers PI19/00044 and PI18/01467) co-funded by European Regional Development Fund “Investing in your future” for the thematic network and co-operative research centers ARADyAL RD16/0006/0015 and by the Swiss National Science Foundation (SNF) grant nr 310030_189334 (to MS lab), and GlaxoSmithKline (GSK) scientific research grant (to MS lab). JR-C was supported by FPI-CEU predoctoral fellowship and a SEMP fellowship from University of Zurich. TB-T was supported by FPI-CEU predoctoral fellowship. AV is funded by a postdoctoral research fellowship from ARADyAL. MH and UR were supported by the SNF, GSK and SIAF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank all centers involved: Institute of Applied Molecular Medicine (IMMA, San Pablo CEU University, Madrid), Centre for Metabolomics and Bioanalysis (CEMBIO, San Pablo CEU University, Madrid) and the Swiss Institute of Allergy and Asthma Research (SIAF, Davos, Switzerland).

1. O’Neill LA, Kishton RJ, Rathmell J. A Guide to Immunometabolism for Immunologists. Nat Rev Immunol (2016) 16(9):553–65. doi: 10.1038/nri.2016.70
2. Drucker DJ. Diabetes, Obesity, Metabolism, and SARS-CoV-2 Infection: The End of the Beginning. Cell Metab (2021) 33(3):479–98. doi: 10.1016/j.cmet.2021.01.016
3. Radzikowska U, Ding M, Tan G, Zhakparov D, Peng Y, Wawrzyniak P, et al. Distribution of ACE2, CD147, CD26, and Other SARS-CoV-2 Associated Molecules in Tissues and Immune Cells in Health and in Asthma, COPD, Obesity, Hypertension, and COVID-19 Risk Factors. Allergy (2020) 75(11):2829–45. doi: 10.1111/all.14429
4. Sokolowska M, Lukasik ZM, Agache I, Akdis CA, Akdis D, Akdis M, et al. Immunology of COVID-19: Mechanisms, Clinical Outcome, Diagnostics, and Perspectives-A Report of the European Academy of Allergy and Clinical Immunology (EAACI). Allergy (2020) 75(10):2445–76. doi: 10.1111/all.14462
5. Aria H, Ghaedrahmati F, Ganjalikhani-Hakemi M. Cutting Edge: Metabolic Immune Reprogramming, Reactive Oxygen Species, and Cancer. J Cell Physiol (2021) 236(9):6168–89. doi: 10.1002/jcp.30303.
6. Klein Geltink RI, Edwards-Hicks J, Apostolova P, O’Sullivan D, Sanin DE, Patterson AE, et al. Metabolic Conditioning of CD8(+) Effector T Cells for Adoptive Cell Therapy. Nat Metab (2020) 2(8):703–16. doi: 10.1038/s42255-020-0256-z
7. Chen J, Zhang H, Zhou L, Hu Y, Li M, He Y, et al. Enhancing the Efficacy of Tumor Vaccines Based on Immune Evasion Mechanisms. Front Oncol (2020) 10:584367. doi: 10.3389/fonc.2020.584367
8. Kumar S, Singh SK, Rana B, Rana A. Tumor-Infiltrating CD8(+) T Cell Antitumor Efficacy and Exhaustion: Molecular Insights. Drug Discov Today (2021) 26(4):951–67. doi: 10.1016/j.drudis.2021.01.002
9. Pallet N, Fernández-Ramos AA, Loriot MA. Impact of Immunosuppressive Drugs on the Metabolism of T Cells. Int Rev Cell Mol Biol (2018) 341:169–200. doi: 10.1016/bs.ircmb.2018.05.009
10. Lund G, Brand S, Ramos T, Jimeno L, Boissy P, Vega F, et al. Strong and Frequent T-Cell Responses to the Minor Allergen Phl P 12 in Spanish Patients IgE-Sensitized to Profilins. Allergy (2018) 73(5):1013–21. doi: 10.1111/all.13351
11. Rosace D, Gomez-Casado C, Fernandez P, Perez-Gordo M, Dominguez MDC, Vega A, et al. Profilin-Mediated Food-Induced Allergic Reactions Are Associated With Oral Epithelial Remodeling. J Allergy Clin Immunol (2019) 143(2):681–90.e1. doi: 10.1016/j.jaci.2018.03.013
12. Obeso D, Mera-Berriatua L, Rodríguez-Coira J, Rosace D, Fernández P, Martín-Antoniano IA, et al. Multi-Omics Analysis Points to Altered Platelet Functions in Severe Food-Associated Respiratory Allergy. Allergy (2018) 73(11):2137–49. doi: 10.1111/all.13563
13. Agache I, Annesi-Maesano I, Bonertz A, Branca F, Cant A, Fras Z, et al. Prioritizing Research Challenges and Funding for Allergy and Asthma and the Need for Translational Research-The European Strategic Forum on Allergic Diseases. Allergy (2019) 74(11):2064–76. doi: 10.1111/all.13856
14. Torres MJ, Agundez J, Barber D, Bartra J, Davila I, Escribese MM, et al. The Spanish Multidisciplinary Research Network for Allergic Diseases. J Investig Allergol Clin Immunol (2020) 31(2):108–19. doi: 10.18176/jiaci.0629
15. Li S, Morita H, Sokolowska M, Tan G, Boonpiyathad T, Opitz L, et al. Gene Expression Signatures of Circulating Human Type 1, 2, and 3 Innate Lymphoid Cells. J Allergy Clin Immunol (2019) 143(6):2321–5. doi: 10.1016/j.jaci.2019.01.047
16. Tojima I, Matsumoto K, Kikuoka H, Hara S, Yamamoto S, Shimizu S, et al. Evidence for the Induction of Th2 Inflammation by Group 2 Innate Lymphoid Cells in Response to Prostaglandin D. Allergy (2019) 74(12):2417–26. doi: 10.1111/all.13974
17. Akdis CA, Arkwright PD, Brüggen MC, Busse W, Gadina M, Guttman-Yassky E, et al. Type 2 Immunity in the Skin and Lungs. Allergy (2020) 75(7):1582–605. doi: 10.1111/all.14318
18. Escribese MM, Gómez-Casado C, Barber D, Diaz-Perales A. Immune Polarization in Allergic Patients: Role of the Innate Immune System. J Investig Allergol Clin Immunol (2015) 25(4):251–8.
19. Tan HT, Hagner S, Ruchti F, Radzikowska U, Tan G, Altunbulakli C, et al. Tight Junction, Mucin, and Inflammasome-Related Molecules Are Differentially Expressed in Eosinophilic, Mixed, and Neutrophilic Experimental Asthma in Mice. Allergy (2019) 74(2):294–307. doi: 10.1111/all.13619
20. Maciag MC, Bartnikas LM, Sicherer SH, Herbert LJ, Young MC, Matney F, et al. A Slice of Food Protein-Induced Enterocolitis Syndrome (FPIES): Insights From 441 Children With FPIES as Provided by Caregivers in the International FPIES Association. J Allergy Clin Immunol Pract (2020) 8(5):1702–9. doi: 10.1016/j.jaip.2020.01.030
21. Galli SJ, Tsai M, Piliponsky AM. The Development of Allergic Inflammation. Nature (2008) 454(7203):445–54. doi: 10.1038/nature07204
22. Draijer C, Peters-Golden M. Alveolar Macrophages in Allergic Asthma: The Forgotten Cell Awakes. Curr Allergy Asthma Rep (2017) 17(2):12. doi: 10.1007/s11882-017-0681-6
23. Sawaguchi M, Tanaka S, Nakatani Y, Harada Y, Mukai K, Matsunaga Y, et al. Role of Mast Cells and Basophils in IgE Responses and in Allergic Airway Hyperresponsiveness. J Immunol (2012) 188(4):1809–18. doi: 10.4049/jimmunol.1101746
24. Jiménez-Saiz R, Chu DK, Mandur TS, Walker TD, Gordon ME, Chaudhary R, et al. Lifelong Memory Responses Perpetuate Humoral T. J Allergy Clin Immunol (2017) 140(6):1604–15.e5. doi: 10.1016/j.jaci.2017.01.018
25. Kraft S, Kinet JP. New Developments in FcepsilonRI Regulation, Function and Inhibition. Nat Rev Immunol (2007) 7(5):365–78. doi: 10.1038/nri2072
26. Sokolowska M, Rovati GE, Diamant Z, Untersmayr E, Schwarze J, Lukasik Z, et al. Current Perspective on Eicosanoids in Asthma and Allergic Diseases: EAACI Task Force Consensus Report, Part I. Allergy (2021) 76(1):114–30. doi: 10.1111/all.14295
27. Reber LL, Hernandez JD, Galli SJ. The Pathophysiology of Anaphylaxis. J Allergy Clin Immunol (2017) 140(2):335–48. doi: 10.1016/j.jaci.2017.06.003
28. Kay AB, Ali FR, Heaney LG, Benyahia F, Soh CP, Renz H, et al. Airway Expression of Calcitonin Gene-Related Peptide in T-Cell Peptide-Induced Late Asthmatic Reactions in Atopics. Allergy (2007) 62(5):495–503. doi: 10.1111/j.1398-9995.2007.01342.x
29. Samitas K, Carter A, Kariyawasam HH, Xanthou G. Upper and Lower Airway Remodelling Mechanisms in Asthma, Allergic Rhinitis and Chronic Rhinosinusitis: The One Airway Concept Revisited. Allergy (2018) 73(5):993–1002. doi: 10.1111/all.13373
30. Wilson JE. Isozymes of Mammalian Hexokinase: Structure, Subcellular Localization and Metabolic Function. J Exp Biol (2003) 206(Pt 12):2049–57. doi: 10.1242/jeb.00241
31. Tsai HJ, Wilson JE. Functional Organization of Mammalian Hexokinases: Characterization of Chimeric Hexokinases Constructed From the N- and C-Terminal Domains of the Rat Type I and Type II Isozymes. Arch Biochem Biophys (1995) 316(1):206–14. doi: 10.1006/abbi.1995.1029
32. Pilkis SJ, El-Maghrabi MR, Pilkis J, Claus TH, Cumming DA. Fructose 2,6-Bisphosphate. A New Activator of Phosphofructokinase. J Biol Chem (1981) 256(7):3171–4. doi: 10.1016/S0021-9258(19)69584-0
33. Pilkis SJ, El-Maghrabi MR, Pilkis J, Claus T. Inhibition of Fructose-1,6-Bisphosphatase by Fructose 2,6-Bisphosphate. J Biol Chem (1981) 256(8):3619–22. doi: 10.1016/S0021-9258(19)69494-9
34. Pilkis SJ, El-Maghrabi MR, McGrane MM, Pilkis J, Claus TH. The Role of Fructose 2,6-Bisphosphate in Regulation of Fructose-1,6-Bisphosphatase. J Biol Chem (1981) 256(22):11489–95. doi: 10.1016/S0021-9258(19)68427-9
35. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
37. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/S1074-7613(02)00323-0
38. Krebs HA. The History of the Tricarboxylic Acid Cycle. Perspect Biol Med (1970) 14(1):154–70. doi: 10.1353/pbm.1970.0001
39. Martínez-Reyes I, Chandel NS. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
40. Lehninger AL, Wadkins CL. Oxidative Phosphorylation. Annu Rev Biochem (1962) 31:47–78. doi: 10.1146/annurev.bi.31.070162.000403
41. Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol (2020) 37:101674. doi: 10.1016/j.redox.2020.101674
42. Osmundsen H, Bremer J, Pedersen JI. Metabolic Aspects of Peroxisomal Beta-Oxidation. Biochim Biophys Acta (1991) 1085(2):141–58. doi: 10.1016/0005-2760(91)90089-Z
43. Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, et al. Immune Responses in Healthy and Allergic Individuals Are Characterized by a Fine Balance Between Allergen-Specific T Regulatory 1 and T Helper 2 Cells. J Exp Med (2004) 199(11):1567–75. doi: 10.1084/jem.20032058
44. Ramsay RR, Gandour RD, van der Leij FR. Molecular Enzymology of Carnitine Transfer and Transport. Biochim Biophys Acta (2001) 1546(1):21–43. doi: 10.1016/S0167-4838(01)00147-9
45. Newsholme P, Procopio J, Lima MM, Pithon-Curi TC, Curi R. Glutamine and Glutamate–Their Central Role in Cell Metabolism and Function. Cell Biochem Funct (2003) 21(1):1–9. doi: 10.1002/cbf.1003
46. Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP, et al. Branched-Chain Amino Acid Catabolism Fuels Adipocyte Differentiation and Lipogenesis. Nat Chem Biol (2016) 12(1):15–21. doi: 10.1038/nchembio.1961
47. Victoni T, Barreto E, Lagente V, Carvalho VF. Oxidative Imbalance as a Crucial Factor in Inflammatory Lung Diseases: Could Antioxidant Treatment Constitute a New Therapeutic Strategy? Oxid Med Cell Longev (2021) 2021. doi: 10.1155/2021/6646923
48. Michaudel C, Mackowiak C, Maillet I, Fauconnier L, Akdis CA, Sokolowska M, et al. Ozone Exposure Induces Respiratory Barrier Biphasic Injury and Inflammation Controlled by IL-33. J Allergy Clin Immunol (2018) 142(3):942–58. doi: 10.1016/j.jaci.2017.11.044
49. Kast JI, McFarlane AJ, Głobińska A, Sokolowska M, Wawrzyniak P, Sanak M, et al. Respiratory Syncytial Virus Infection Influences Tight Junction Integrity. Clin Exp Immunol (2017) 190(3):351–9. doi: 10.1111/cei.13042
50. Liu J, Chen X, Dou M, He H, Ju M, Ji S, et al. Particulate Matter Disrupts Airway Epithelial Barrier via Oxidative Stress to Promote Pseudomonas Aeruginosa Infection. J Thorac Dis (2019) 11(6):2617–27. doi: 10.21037/jtd.2019.05.77
51. Anthony D, Papanicolaou A, Wang H, Seow HJ, To EE, Yatmaz S, et al. Excessive Reactive Oxygen Species Inhibit IL-17a(+) γδ T Cells and Innate Cellular Responses to Bacterial Lung Infection. Antioxid Redox Signal (2020) 32(13):943–56. doi: 10.1089/ars.2018.7716
52. Kao SS, Bassiouni A, Ramezanpour M, Finnie J, Chegeni N, Colella AD, et al. Proteomic Analysis of Nasal Mucus Samples of Healthy Patients and Patients With Chronic Rhinosinusitis. J Allergy Clin Immunol (2021) 147(1):168–78. doi: 10.1016/j.jaci.2020.06.037
53. Hristova M, Habibovic A, Veith C, Janssen-Heininger YM, Dixon AE, Geiszt M, et al. Airway Epithelial Dual Oxidase 1 Mediates Allergen-Induced IL-33 Secretion and Activation of Type 2 Immune Responses. J Allergy Clin Immunol (2016) 137(5):1545–56.e11. doi: 10.1016/j.jaci.2015.10.003
54. Boldogh I, Bacsi A, Choudhury BK, Dharajiya N, Alam R, Hazra TK, et al. ROS Generated by Pollen NADPH Oxidase Provide a Signal That Augments Antigen-Induced Allergic Airway Inflammation. J Clin Invest (2005) 115(8):2169–79. doi: 10.1172/JCI24422
55. Bleck B, Tse DB, Gordon T, Ahsan MR, Reibman J. Diesel Exhaust Particle-Treated Human Bronchial Epithelial Cells Upregulate Jagged-1 and OX40 Ligand in Myeloid Dendritic Cells via Thymic Stromal Lymphopoietin. J Immunol (2010) 185(11):6636–45. doi: 10.4049/jimmunol.1000719
56. Sunil VR, Vayas KN, Massa CB, Gow AJ, Laskin JD, Laskin DL. Ozone-Induced Injury and Oxidative Stress in Bronchiolar Epithelium Are Associated With Altered Pulmonary Mechanics. Toxicol Sci (2013) 133(2):309–19. doi: 10.1093/toxsci/kft071
57. Uchida M, Anderson EL, Squillace DL, Patil N, Maniak PJ, Iijima K, et al. Oxidative Stress Serves as a Key Checkpoint for IL-33 Release by Airway Epithelium. Allergy (2017) 72(10):1521–31. doi: 10.1111/all.13158
58. Jaffer OA, Carter AB, Sanders PN, Dibbern ME, Winters CJ, Murthy S, et al. Mitochondrial-Targeted Antioxidant Therapy Decreases Transforming Growth Factor-β-Mediated Collagen Production in a Murine Asthma Model. Am J Respir Cell Mol Biol (2015) 52(1):106–15. doi: 10.1165/rcmb.2013-0519OC
59. Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A, Calhoun WJ, et al. Superoxide Dismutase Inactivation in Pathophysiology of Asthmatic Airway Remodeling and Reactivity. Am J Pathol (2005) 166(3):663–74. doi: 10.1016/S0002-9440(10)62288-2
60. Li K, Li M, Li W, Yu H, Sun X, Zhang Q, et al. Airway Epithelial Regeneration Requires Autophagy and Glucose Metabolism. Cell Death Dis (2019) 10(12):1–14. doi: 10.1038/s41419-019-2111-2
61. Halldorsson S, Rohatgi N, Magnusdottir M, Choudhary KS, Gudjonsson T, Knutsen E, et al. Metabolic Re-Wiring of Isogenic Breast Epithelial Cell Lines Following Epithelial to Mesenchymal Transition. Cancer Lett (2017) 396:117–29. doi: 10.1016/j.canlet.2017.03.019
62. Rodriguez-Perez N, Schiavi E, Frei R, Ferstl R, Wawrzyniak P, Smolinska S, et al. Altered Fatty Acid Metabolism and Reduced Stearoyl-Coenzyme a Desaturase Activity in Asthma. Allergy (2017) 72(11):1744–52. doi: 10.1111/all.13180
63. Gally F, Kosmider B, Weaver MR, Pate KM, Hartshorn KL, Oberley-Deegan RE. FABP5 Deficiency Enhances Susceptibility to H1N1 Influenza A Virus-Induced Lung Inflammation. Am J Physiol-Lung Cell Mol Physiol (2013) 305(1):L64–72. doi: 10.1152/ajplung.00276.2012
64. Gally F, Chu HW, Bowler RP. Cigarette Smoke Decreases Airway Epithelial FABP5 Expression and Promotes Pseudomonas Aeruginosa Infection. PloS One (2013) 8(1):e51784. doi: 10.1371/journal.pone.0051784
65. Rao D, Perraud A-L, Schmitz C, Gally F. Cigarette Smoke Inhibits LPS-Induced FABP5 Expression by Preventing C-Jun Binding to the FABP5 Promoter. PloS One (2017) 12(5):e0178021. doi: 10.1371/journal.pone.0178021
66. Ng-Blichfeldt J-P, Schrik A, Kortekaas RK, Noordhoek JA, Heijink IH, Hiemstra PS, et al. Retinoic Acid Signaling Balances Adult Distal Lung Epithelial Progenitor Cell Growth and Differentiation. EBioMedicine (2018) 36:461–74. doi: 10.1016/j.ebiom.2018.09.002
67. Oliveira L, Teixeira FME, Sato MN. Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases. Mediators Inflammation (2018) 2018. doi: 10.1155/2018/3067126
68. Czarnewski P, Das S, Parigi SM, Villablanca EJ. Retinoic Acid and Its Role in Modulating Intestinal Innate Immunity. Nutrients (2017) 9(1):68. doi: 10.3390/nu9010068
69. Morita H, Kubo T, Rückert B, Ravindran A, Soyka MB, Rinaldi AO, et al. Induction of Human Regulatory Innate Lymphoid Cells From Group 2 Innate Lymphoid Cells by Retinoic Acid. J Allergy Clin Immunol (2019) 143(6):2190–201.e9. doi: 10.1016/j.jaci.2018.12.1018
70. Kobayashi S, Tayama S, Phung HT, Kagawa Y, Miyazaki H, Takahashi Y, et al. Fatty Acid-Binding Protein 5 Limits ILC2-Mediated Allergic Lung Inflammation in a Murine Asthma Model. Sci Rep (2020) 10(1):1–12. doi: 10.1038/s41598-020-73935-y
71. Hamberg M, Samuelsson B. Oxygenation of Unsaturated Fatty Acids by the Vesicular Gland of Sheep. J Biol Chem (1967) 242(22):5344–54. doi: 10.1016/S0021-9258(18)99434-2
72. Okuno T, Iizuka Y, Okazaki H, Yokomizo T, Taguchi R, Shimizu T. 12 (S)-Hydroxyheptadeca-5Z, 8E, 10E–Trienoic Acid Is a Natural Ligand for Leukotriene B4 Receptor 2. J Exp Med (2008) 205(4):759–66. doi: 10.1084/jem.20072329
73. Ishii Y, Saeki K, Liu M, Sasaki F, Koga T, Kitajima K, et al. Leukotriene B4 Receptor Type 2 (BLT2) Enhances Skin Barrier Function by Regulating Tight Junction Proteins. FASEB J (2016) 30(2):933–47. doi: 10.1096/fj.15-279653
74. Stevens WW, Staudacher AG, Hulse KE, Carter RG, Winter DR, Kato A, et al. Activation of the 15-Lipoxygenase Pathway in Aspirin-Exacerbated Respiratory Disease. J Allergy Clin Immunol (2020) 147(2):600–12. doi: 10.1164/ajrccm-conference.2020.201.1_MeetingAbstracts.A1262
75. Gutiérrez-Vázquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity (2018) 48(1):19–33. doi: 10.1016/j.immuni.2017.12.012
76. Chang YD, Li CH, Tsai CH, Cheng YW, Kang JJ, Lee CC. Aryl Hydrocarbon Receptor Deficiency Enhanced Airway Inflammation and Remodeling in a Murine Chronic Asthma Model. FASEB J (2020) 34(11):15300–13. doi: 10.1096/fj.202001529R
77. Xu T, Zhou Y, Qiu L, Do DC, Zhao Y, Cui Z, et al. Aryl Hydrocarbon Receptor Protects Lungs From Cockroach Allergen–Induced Inflammation by Modulating Mesenchymal Stem Cells. J Immunol (2015) 195(12):5539–50. doi: 10.4049/jimmunol.1501198
78. Walsh CJ, Guinane CM, O’Toole PW, Cotter PD. Beneficial Modulation of the Gut Microbiota. FEBS Lett (2014) 588(22):4120–30. doi: 10.1016/j.febslet.2014.03.035
79. Schilderink R, Verseijden C, Seppen J, Muncan V, Van Den Brink GR, Lambers TT, et al. The SCFA Butyrate Stimulates the Epithelial Production of Retinoic Acid via Inhibition of Epithelial HDAC. Am J Physiol-Gastrointest Liver Physiol (2016) 310(11):G1138–G46. doi: 10.1152/ajpgi.00411.2015
80. Tanaka H, Sirich TL, Meyer TW. Uremic Solutes Produced by Colon Microbes. Blood Purif (2015) 40(4):306–11. doi: 10.1159/000441578
81. Wypych TP, Pattaroni C, Perdijk O, Yap C, Trompette A, Anderson D, et al. Microbial Metabolism of L-Tyrosine Protects Against Allergic Airway Inflammation. Nat Immunol (2021) 22(3):279–86. doi: 10.1038/s41590-020-00856-3
82. Thorley AJ, Goldstraw P, Young A, Tetley TD. Primary Human Alveolar Type II Epithelial Cell CCL20 (Macrophage Inflammatory Protein-3α)–Induced Dendritic Cell Migration. Am J Respir Cell Mol Biol (2005) 32(4):262–7. doi: 10.1165/rcmb.2004-0196OC
83. Sun C-Y, Young G-H, Hsieh Y-T, Chen Y-H, Wu M-S, Wu V-C, et al. Protein-Bound Uremic Toxins Induce Tissue Remodeling by Targeting the EGF Receptor. J Am Soc Nephrol (2015) 26(2):281–90. doi: 10.1681/ASN.2014010021
84. Fu L, Fu S, Wang C, Xie M, Wang Y. Yogurt-Sourced Probiotic Bacteria Alleviate Shrimp Tropomyosin-Induced Allergic Mucosal Disorders, Potentially Through Microbiota and Metabolism Modifications. Allergol Int (2019) 68(4):506–14. doi: 10.1016/j.alit.2019.05.013
85. Collin M, Bigley V. Human Dendritic Cell Subsets: An Update. Immunology (2018) 154(1):3–20. doi: 10.1111/imm.12888
86. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-Driven Early Glycolytic Reprogramming via the Kinases TBK1-IKKε Supports the Anabolic Demands of Dendritic Cell Activation. Nat Immunol (2014) 15(4):323–32. doi: 10.1038/ni.2833
87. Svajger U, Obermajer N, Jeras M. Dendritic Cells Treated With Resveratrol During Differentiation From Monocytes Gain Substantial Tolerogenic Properties Upon Activation. Immunology (2010) 129(4):525–35. doi: 10.1111/j.1365-2567.2009.03205.x
88. Ferreira GB, Kleijwegt FS, Waelkens E, Lage K, Nikolic T, Hansen DA, et al. Differential Protein Pathways in 1,25-Dihydroxyvitamin D(3) and Dexamethasone Modulated Tolerogenic Human Dendritic Cells. J Proteome Res (2012) 11(2):941–71. doi: 10.1021/pr200724e
89. Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, et al. High Mitochondrial Respiration and Glycolytic Capacity Represent a Metabolic Phenotype of Human Tolerogenic Dendritic Cells. J Immunol (2015) 194(11):5174–86. doi: 10.4049/jimmunol.1303316
90. Bekkering S, Domínguez-Andrés J, Joosten LAB, Riksen NP, Netea MG. Trained Immunity: Reprogramming Innate Immunity in Health and Disease. Annu Rev Immunol (2021) 26(39):667–693. doi: 10.1146/annurev-immunol-102119-073855
91. Eljaszewicz A, Ruchti F, Radzikowska U, Globinska A, Boonpiyathad T, Gschwend A, et al. Trained Immunity and Tolerance in Innate Lymphoid Cells, Monocytes, and Dendritic Cells During Allergen-Specific Immunotherapy. J Allergy Clin Immunol (2020) 147(5):1865–77. doi: 10.1016/j.jaci.2020.08.042
92. Stagg AJ. Intestinal Dendritic Cells in Health and Gut Inflammation. Front Immunol (2018) 9:2883. doi: 10.3389/fimmu.2018.02883
93. Radzikowska U, Rinaldi AO, Çelebi Sözener Z, Karaguzel D, Wojcik M, Cypryk K, et al. The Influence of Dietary Fatty Acids on Immune Responses. Nutrients (2019) 11(12):2990. doi: 10.3390/nu11122990
94. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The Role of Short-Chain Fatty Acids in Health and Disease. Adv Immunol (2014) 121:91–119. doi: 10.1016/B978-0-12-800100-4.00003-9
95. Kull I, Bergström A, Lilja G, Pershagen G, Wickman M. Fish Consumption During the First Year of Life and Development of Allergic Diseases During Childhood. Allergy (2006) 61(8):1009–15. doi: 10.1111/j.1398-9995.2006.01115.x
96. van den Elsen LW, van Esch BC, Hofman GA, Kant J, van de Heijning BJ, Garssen J, et al. Dietary Long Chain N-3 Polyunsaturated Fatty Acids Prevent Allergic Sensitization to Cow’s Milk Protein in Mice. Clin Exp Allergy (2013) 43(7):798–810. doi: 10.1111/cea.12111
97. Venter C, Meyer RW, Nwaru BI, Roduit C, Untersmayr E, Adel-Patient K, et al. EAACI Position Paper: Influence of Dietary Fatty Acids on Asthma, Food Allergy, and Atopic Dermatitis. Allergy (2019) 74(8):1429–44. doi: 10.1111/all.13764
98. Linke M, Fritsch SD, Sukhbaatar N, Hengstschläger M, Weichhart T. Mtorc1 and Mtorc2 as Regulators of Cell Metabolism in Immunity. FEBS Lett (2017) 591(19):3089–103. doi: 10.1002/1873-3468.12711
99. Sinclair C, Bommakanti G, Gardinassi L, Loebbermann J, Johnson MJ, Hakimpour P, et al. mTOR Regulates Metabolic Adaptation of APCs in the Lung and Controls the Outcome of Allergic Inflammation. Science (2017) 357(6355):1014–21. doi: 10.1126/science.aaj2155
100. Park SJ, Im DS. Blockage of Sphingosine-1-Phosphate Receptor 2 Attenuates Allergic Asthma in Mice. Br J Pharmacol (2019) 176(7):938–49. doi: 10.1111/bph.14597
101. Idzko M, Hammad H, van Nimwegen M, Kool M, Müller T, Soullié T, et al. Local Application of FTY720 to the Lung Abrogates Experimental Asthma by Altering Dendritic Cell Function. J Clin Invest (2006) 116(11):2935–44. doi: 10.1172/JCI28295
102. Kleinjan A, van Nimwegen M, Leman K, Hoogsteden HC, Lambrecht BN. Topical Treatment Targeting Sphingosine-1-Phosphate and Sphingosine Lyase Abrogates Experimental Allergic Rhinitis in a Murine Model. Allergy (2013) 68(2):204–12. doi: 10.1111/all.12082
103. Gendron D, Lemay AM, Tremblay C, Lai LJ, Langlois A, Bernatchez É, et al. Treatment With a Sphingosine Analog After the Inception of House Dust Mite-Induced Airway Inflammation Alleviates Key Features of Experimental Asthma. Respir Res (2015) 16:7. doi: 10.1186/s12931-015-0180-z
104. Japtok L, Schaper K, Bäumer W, Radeke HH, Jeong SK, Kleuser B. Sphingosine 1-Phosphate Modulates Antigen Capture by Murine Langerhans Cells via the S1P2 Receptor Subtype. PloS One (2012) 7(11):e49427. doi: 10.1371/journal.pone.0049427
105. Reines I, Kietzmann M, Mischke R, Tschernig T, Lüth A, Kleuser B, et al. Topical Application of Sphingosine-1-Phosphate and FTY720 Attenuate Allergic Contact Dermatitis Reaction Through Inhibition of Dendritic Cell Migration. J Invest Dermatol (2009) 129(8):1954–62. doi: 10.1038/jid.2008.454
106. Mills EL, O’Neill LA. Reprogramming Mitochondrial Metabolism in Macrophages as an Anti-Inflammatory Signal. Eur J Immunol (2016) 46(1):13–21. doi: 10.1002/eji.201445427
107. Izquierdo E, Cuevas VD, Fernández-Arroyo S, Riera-Borrull M, Orta-Zavalza E, Joven J, et al. Reshaping of Human Macrophage Polarization Through Modulation of Glucose Catabolic Pathways. J Immunol (2015) 195(5):2442–51. doi: 10.4049/jimmunol.1403045
108. MacDonald SM. Histamine-Releasing Factors. Curr Opin Immunol (1996) 8(6):778–83. doi: 10.1016/S0952-7915(96)80004-5
109. Moutsopoulos NM, Konkel JE. Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol (2018) 39(4):276–87. doi: 10.1016/j.it.2017.08.005
110. Squier C, Brogden KA. Human Oral Mucosa: Development, Structure and Function. (2013). doi: 10.1002/9781118710470
111. Mazzini E, Massimiliano L, Penna G, Rescigno M. Oral Tolerance can be Established via Gap Junction Transfer of Fed Antigens From CX3CR1⁺ Macrophages to CD103⁺ Dendritic Cells. Immunity (2014) 40(2):248–61. doi: 10.1016/j.immuni.2013.12.012
112. Krause P, Morris V, Greenbaum JA, Park Y, Bjoerheden U, Mikulski Z, et al. IL-10-Producing Intestinal Macrophages Prevent Excessive Antibacterial Innate Immunity by Limiting IL-23 Synthesis. Nat Commun (2015) 6:7055. doi: 10.1038/ncomms8055
113. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab (2015) 21(1):65–80. doi: 10.1016/j.cmet.2014.12.005
114. Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation Through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J Immunol (2015) 194(11):5472–87. doi: 10.4049/jimmunol.1401343
115. Sokolowska M, Chen LY, Eberlein M, Martinez-Anton A, Liu Y, Alsaaty S, et al. Low Molecular Weight Hyaluronan Activates Cytosolic Phospholipase A2α and Eicosanoid Production in Monocytes and Macrophages. J Biol Chem (2014) 289(7):4470–88. doi: 10.1074/jbc.M113.515106
116. Bambouskova M, Potuckova L, Paulenda T, Kerndl M, Mogilenko DA, Lizotte K, et al. Itaconate Confers Tolerance to Late NLRP3 Inflammasome Activation. Cell Rep (2021) 34(10):108756. doi: 10.1016/j.celrep.2021.108756
117. Elieh Ali Komi D, Sharma L, Dela Cruz CS. Chitin and Its Effects on Inflammatory and Immune Responses. Clin Rev Allergy Immunol (2018) 54(2):213–23. doi: 10.1007/s12016-017-8600-0
118. Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The Role of Lung and Gut Microbiota in the Pathology of Asthma. Immunity (2020) 52(2):241–55. doi: 10.1016/j.immuni.2020.01.007
119. Lunjani N, Satitsuksanoa P, Lukasik Z, Sokolowska M, Eiwegger T, O’Mahony L. Recent Developments and Highlights in Mechanisms of Allergic Diseases: Microbiome. Allergy (2018) 73(12):2314–27. doi: 10.1111/all.13634
120. Rőszer T. Understanding the Mysterious M2 Macrophage Through Activation Markers and Effector Mechanisms. Mediators Inflamm (2015) 2015:816460. doi: 10.1155/2015/816460
121. Kim EG, Kim MN, Hong JY, Lee JW, Kim SY, Kim KW, et al. Chitinase 3-Like 1 Contributes to Food Allergy via M2 Macrophage Polarization. Allergy Asthma Immunol Res (2020) 12(6):1012–28. doi: 10.4168/aair.2020.12.6.1012
122. Leoni C, Volpicella M, Dileo M, Gattulli BAR, Ceci LR. Chitinases as Food Allergens. Molecules (2019) 24(11):2087. doi: 10.3390/molecules24112087
123. van Bergenhenegouwen J, Braber S, Loonstra R, Buurman N, Rutten L, Knipping K, et al. Oral Exposure to the Free Amino Acid Glycine Inhibits the Acute Allergic Response in a Model of Cow’s Milk Allergy in Mice. Nutr Res (2018) 58:95–105. doi: 10.1016/j.nutres.2018.07.005
124. Del Cornò M, Gessani S, Conti L. Shaping the Innate Immune Response by Dietary Glucans: Any Role in the Control of Cancer? Cancers (Basel) (2020) 12(1). doi: 10.3390/cancers12010155
125. Ogger PP, Byrne AJ. Macrophage Metabolic Reprogramming During Chronic Lung Disease. Mucosal Immunol (2020) 14(2):282–295. doi: 10.1038/s41385-020-00356-5
126. Hussell T, Bell TJ. Alveolar Macrophages: Plasticity in a Tissue-Specific Context. Nat Rev Immunol (2014) 14(2):81–93. doi: 10.1038/nri3600
127. Nagarkar DR, Bowman ER, Schneider D, Wang Q, Shim J, Zhao Y, et al. Rhinovirus Infection of Allergen-Sensitized and -Challenged Mice Induces Eotaxin Release From Functionally Polarized Macrophages. J Immunol (2010) 185(4):2525–35. doi: 10.4049/jimmunol.1000286
128. Wynn TA, Chawla A, Pollard JW. Macrophage Biology in Development, Homeostasis and Disease. Nature (2013) 496(7446):445–55. doi: 10.1038/nature12034
129. Moreira AP, Hogaboam CM. Macrophages in Allergic Asthma: Fine-Tuning Their Pro- and Anti-Inflammatory Actions for Disease Resolution. J Interferon Cytokine Res (2011) 31(6):485–91. doi: 10.1089/jir.2011.0027
130. Fitzpatrick AM, Teague WG, Burwell L, Brown MS, Brown LA. Program NNSAR. Glutathione Oxidation Is Associated With Airway Macrophage Functional Impairment in Children With Severe Asthma. Pediatr Res (2011) 69(2):154–9. doi: 10.1203/PDR.0b013e3182026370
131. Damon M, Chavis C, Daures JP, Crastes de Paulet A, Michel FB, Godard P. Increased Generation of the Arachidonic Metabolites LTB4 and 5-HETE by Human Alveolar Macrophages in Patients With Asthma: Effect In Vitro of Nedocromil Sodium. Eur Respir J (1989) 2(3):202–9.
132. Zhang L, Yin Y, Zhang H, Zhong W, Zhang J. Association of Asthma Diagnosis With Leptin and Adiponectin: A Systematic Review and Meta-Analysis. J Investig Med (2017) 65(1):57–64. doi: 10.1136/jim-2016-000127
133. Calixto MC, Lintomen L, Schenka A, Saad MJ, Zanesco A, Antunes E. Obesity Enhances Eosinophilic Inflammation in a Murine Model of Allergic Asthma. Br J Pharmacol (2010) 159(3):617–25. doi: 10.1111/j.1476-5381.2009.00560.x
134. Zheng H, Zhang X, Castillo EF, Luo Y, Liu M, Yang XO. Leptin Enhances TH2 and ILC2 Responses in Allergic Airway Disease. J Biol Chem (2016) 291(42):22043–52. doi: 10.1074/jbc.M116.743187
135. Riera-Borrull M, Cuevas VD, Alonso B, Vega MA, Joven J, Izquierdo E, et al. Palmitate Conditions Macrophages for Enhanced Responses Toward Inflammatory Stimuli via JNK Activation. J Immunol (2017) 199(11):3858–69. doi: 10.4049/jimmunol.1700845
136. Kiekens RC, Thepen T, Oosting AJ, Bihari IC, Van De Winkel JG, Bruijnzeel-Koomen CA, et al. Heterogeneity Within Tissue-Specific Macrophage and Dendritic Cell Populations During Cutaneous Inflammation in Atopic Dermatitis. Br J Dermatol (2001) 145(6):957–65. doi: 10.1046/j.1365-2133.2001.04508.x
137. Lonati A, Mommaas MA, Pasolini G, Lavazza A, Rowden G, De Panfilis G. Macrophages, But Not Langerhans Cell-Like Cells of Dendritic Lineage, Express the CD36 Molecule in Normal Human Dermis: Relevance to Downregulatory Cutaneous Immune Responses? J Invest Dermatol (1996) 106(1):96–101. doi: 10.1111/1523-1747.ep12328158
138. Chen Y, Yang M, Huang W, Chen W, Zhao Y, Schulte ML, et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ Res (2019) 125(12):1087–102. doi: 10.1161/CIRCRESAHA.119.315833
139. He D, Wu L, Kim HK, Li H, Elmets CA, Xu H. IL-17 and IFN-Gamma Mediate the Elicitation of Contact Hypersensitivity Responses by Different Mechanisms and Both Are Required for Optimal Responses. J Immunol (2009) 183(2):1463–70. doi: 10.4049/jimmunol.0804108
140. Kunkl M, Sambucci M, Ruggieri S, Amormino C, Tortorella C, Gasperini C, et al. CD28 Autonomous Signaling Up-Regulates C-Myc Expression and Promotes Glycolysis Enabling Inflammatory T Cell Responses in Multiple Sclerosis. Cells (2019) 8(6): 575. doi: 10.3390/cells8060575
141. Greiner EF, Guppy M, Brand K. Glucose Is Essential for Proliferation and the Glycolytic Enzyme Induction That Provokes a Transition to Glycolytic Energy Production. J Biol Chem (1994) 269(50):31484–90. doi: 10.1016/S0021-9258(18)31720-4
142. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
143. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-Like Receptor Signaling Balance T. Nat Immunol (2016) 17(12):1459–66. doi: 10.1038/ni.3577
144. Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab (2018) 28(3):504–15.e7. doi: 10.1016/j.cmet.2018.06.002
145. Oestreich KJ, Read KA, Gilbertson SE, Hough KP, McDonald PW, Krishnamoorthy V, et al. Bcl-6 Directly Represses the Gene Program of the Glycolysis Pathway. Nat Immunol (2014) 15(10):957–64. doi: 10.1038/ni.2985
146. Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
147. Li W, Qu G, Choi SC, Cornaby C, Titov A, Kanda N, et al. Targeting T Cell Activation and Lupus Autoimmune Phenotypes by Inhibiting Glucose Transporters. Front Immunol (2019) 10:833. doi: 10.3389/fimmu.2019.00833
148. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
149. Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing Cd4. Cell Metab (2019) 30(6):1055–74.e8. doi: 10.1016/j.cmet.2019.10.004.
150. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De Novo Fatty Acid Synthesis Controls the Fate Between Regulatory T and T Helper 17 Cells. Nat Med (2014) 20(11):1327–33. doi: 10.1038/nm.3704
151. Liu X, Mo W, Ye J, Li L, Zhang Y, Hsueh EC, et al. Regulatory T Cells Trigger Effector T Cell DNA Damage and Senescence Caused by Metabolic Competition. Nat Commun (2018) 9(1):249. doi: 10.1038/s41467-017-02689-5
152. Tibbitt CA, Stark JM, Martens L, Ma J, Mold JE, Deswarte K, et al. Single-Cell RNA Sequencing of the T Helper Cell Response to House Dust Mites Defines a Distinct Gene Expression Signature in Airway Th2 Cells. Immunity (2019) 51(1):169–84.e5. doi: 10.1016/j.immuni.2019.05.014
153. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. HIF-1α Is a Metabolic Switch Between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep (2019) 27(1):226–37.e4. doi: 10.1016/j.celrep.2019.03.029
154. Cho SH, Raybuck AL, Blagih J, Kemboi E, Haase VH, Jones RG, et al. Hypoxia-Inducible Factors in CD4(+) T Cells Promote Metabolism, Switch Cytokine Secretion, and T Cell Help in Humoral Immunity. Proc Natl Acad Sci U.S.A. (2019) 116(18):8975–84. doi: 10.1073/pnas.1811702116
155. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-Inducible Factor-1 Alpha-Dependent Induction of FoxP3 Drives Regulatory T-Cell Abundance and Function During Inflammatory Hypoxia of the Mucosa. Proc Natl Acad Sci USA (2012) 109(41):E2784–93. doi: 10.1073/pnas.1202366109
156. Yang ZZ, Novak AJ, Stenson MJ, Witzig TE, Ansell SM. Intratumoral CD4+CD25+ Regulatory T-Cell-Mediated Suppression of Infiltrating CD4+ T Cells in B-Cell non-Hodgkin Lymphoma. Blood (2006) 107(9):3639–46. doi: 10.1182/blood-2005-08-3376
157. Celada LJ, Kropski JA, Herazo-Maya JD, Luo W, Creecy A, Abad AT, et al. PD-1 Up-Regulation on CD4(+) T Cells Promotes Pulmonary Fibrosis Through STAT3-Mediated IL-17A and TGF-β1 Production. Sci Transl Med (2018) 10(460):eaar8356. doi: 10.1126/scitranslmed.aar8356
158. Zander RA, Obeng-Adjei N, Guthmiller JJ, Kulu DI, Li J, Ongoiba A, et al. PD-1 Co-Inhibitory and OX40 Co-Stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity. Cell Host Microbe (2015) 17(5):628–41. doi: 10.1016/j.chom.2015.03.007
159. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
160. Palaskas NJ, Garcia JD, Shirazi R, Shin DS, Puig-Saus C, Braas D, et al. Global Alteration of T-Lymphocyte Metabolism by PD-L1 Checkpoint Involves a Block of De Novo Nucleoside Phosphate Synthesis. Cell Discov (2019) 5:62. doi: 10.1038/s41421-019-0130-x
161. Lee CF, Lo YC, Cheng CH, Furtmüller GJ, Oh B, Andrade-Oliveira V, et al. Preventing Allograft Rejection by Targeting Immune Metabolism. Cell Rep (2015) 13(4):760–70. doi: 10.1016/j.celrep.2015.09.036
162. Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab (2016) 24(1):104–17. doi: 10.1016/j.cmet.2016.06.007
163. Milasta S, Dillon CP, Sturm OE, Verbist KC, Brewer TL, Quarato G, et al. Apoptosis-Inducing-Factor-Dependent Mitochondrial Function Is Required for T Cell But Not B Cell Function. Immunity (2016) 44(1):88–102. doi: 10.1016/j.immuni.2015.12.002
164. Malinovschi A, Janson C, Borres M, Alving K. Simultaneously Increased Fraction of Exhaled Nitric Oxide Levels and Blood Eosinophil Counts Relate to Increased Asthma Morbidity. J Allergy Clin Immunol (2016) 138(5):1301–8. doi: 10.1016/j.jaci.2016.01.044
165. Sato Y, Chibana K, Horigane Y, Uchida N, Masawa M, Koike R, et al. Comparison of Inducible Nitric Oxide Synthase mRNA Expression in Different Airway Portions and Association With Nitric Oxide Parameters From Patients With Asthma. Clin Exp Allergy (2019) 49(5):582–90. doi: 10.1111/cea.13344
166. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-Tumor Activity. Cell (2016) 167(3):829–42.e13. doi: 10.1016/j.cell.2016.09.031
167. Czystowska-Kuzmicz M, Sosnowska A, Nowis D, Ramji K, Szajnik M, Chlebowska-Tuz J, et al. Small Extracellular Vesicles Containing Arginase-1 Suppress T-Cell Responses and Promote Tumor Growth in Ovarian Carcinoma. Nat Commun (2019) 10(1):3000. doi: 10.1038/s41467-019-10979-3
168. Ron-Harel N, Ghergurovich JM, Notarangelo G, LaFleur MW, Tsubosaka Y, Sharpe AH, et al. T Cell Activation Depends on Extracellular Alanine. Cell Rep (2019) 28(12):3011–21.e4. doi: 10.1016/j.celrep.2019.08.034
169. Kurniawan H, Franchina DG, Guerra L, Bonetti L, Baguet LS, Grusdat M, et al. Glutathione Restricts Serine Metabolism to Preserve Regulatory T Cell Function. Cell Metab (2020) 31(5):920–36.e7. doi: 10.1016/j.cmet.2020.03.004
170. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity (2017) 46(4):675–89. doi: 10.1016/j.immuni.2017.03.019
171. Patel CH, Leone RD, Horton MR, Powell JD. Targeting Metabolism to Regulate Immune Responses in Autoimmunity and Cancer. Nat Rev Drug Discov (2019) 18(9):669–88. doi: 10.1038/s41573-019-0032-5
172. Moreno-Altamirano MMB, Kolstoe SE, Sánchez-García FJ. Virus Control of Cell Metabolism for Replication and Evasion of Host Immune Responses. Front Cell Infect Microbiol (2019) 9:95. doi: 10.3389/fcimb.2019.00095
173. Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque SM, et al. The Differential Roles of T Cells in Non-Alcoholic Fatty Liver Disease and Obesity. Front Immunol (2019) 10:82. doi: 10.3389/fimmu.2019.00082
174. O’Neill LA, Pearce EJ. Immunometabolism Governs Dendritic Cell and Macrophage Function. J Exp Med (2016) 213(1):15–23. doi: 10.1084/jem.20151570
175. Nonnenmacher Y, Hiller K. Biochemistry of Proinflammatory Macrophage Activation. Cell Mol Life Sci (2018) 75(12):2093–109. doi: 10.1007/s00018-018-2784-1
176. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
177. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection With Mycobacterium Tuberculosis Induces the Warburg Effect in Mouse Lungs. Sci Rep (2015) 5:18176. doi: 10.1038/srep18176
178. Traven A, Naderer T. Central Metabolic Interactions of Immune Cells and Microbes: Prospects for Defeating Infections. EMBO Rep (2019) 20(7):e47995. doi: 10.15252/embr.201947995
179. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
180. Sun L, Suo C, Li ST, Zhang H, Gao P. Metabolic Reprogramming for Cancer Cells and Their Microenvironment: Beyond the Warburg Effect. Biochim Biophys Acta Rev Cancer (2018) 1870(1):51–66. doi: 10.1016/j.bbcan.2018.06.005
181. Wenes M, Shang M, Di Matteo M, Goveia J, Martín-Pérez R, Serneels J, et al. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab (2016) 24(5):701–15. doi: 10.1016/j.cmet.2016.09.008
182. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
183. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine Metabolism in Myeloid Cells Shapes Innate and Adaptive Immunity. Front Immunol (2017) 8:93. doi: 10.3389/fimmu.2017.00093
184. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
185. Alwarawrah Y, Kiernan K, MacIver NJ. Changes in Nutritional Status Impact Immune Cell Metabolism and Function. Front Immunol (2018) 9:1055. doi: 10.3389/fimmu.2018.01055
186. Platten M, Wick W, Van den Eynde BJ. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res (2012) 72(21):5435–40. doi: 10.1158/0008-5472.CAN-12-0569
187. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity (2005) 22(5):633–42. doi: 10.1016/j.immuni.2005.03.013
188. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses Through IDO Expression and Correlate With Lymph Node Metastasis in Patients With Breast Cancer. J Immunol (2013) 190(7):3783–97. doi: 10.4049/jimmunol.1201449
189. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid Dendritic Cells From Mouse Tumor-Draining Lymph Nodes Directly Activate Mature Tregs via Indoleamine 2,3-Dioxygenase. J Clin Invest (2007) 117(9):2570–82. doi: 10.1172/JCI31911
190. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An Interaction Between Kynurenine and the Aryl Hydrocarbon Receptor can Generate Regulatory T Cells. J Immunol (2010) 185(6):3190–8. doi: 10.4049/jimmunol.0903670
191. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK During T Lymphocyte Activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
192. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab (2017) 25(2):482. doi: 10.1016/j.cmet.2017.01.014
193. Kalinski P. Regulation of Immune Responses by Prostaglandin E2. J Immunol (2012) 188(1):21–8. doi: 10.4049/jimmunol.1101029
194. Guzik TJ, Cosentino F. Epigenetics and Immunometabolism in Diabetes and Aging. Antioxid Redox Signal (2018) 29(3):257–74. doi: 10.1089/ars.2017.7299
195. Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R. Type 2 Diabetes and Its Impact on the Immune System. Curr Diabetes Rev (2020) 16(5):442–9. doi: 10.2174/1573399815666191024085838
196. American Diabetes Association. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care (2019) 42(Suppl 1):S13–28. doi: 10.2337/dc19-S002
197. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ Effector T Cells Contribute to Macrophage Recruitment and Adipose Tissue Inflammation in Obesity. Nat Med (2009) 15(8):914–20. doi: 10.1038/nm.1964
198. Szpigel A, Hainault I, Carlier A, Venteclef N, Batto AF, Hajduch E, et al. Lipid Environment Induces ER Stress, TXNIP Expression and Inflammation in Immune Cells of Individuals With Type 2 Diabetes. Diabetologia (2018) 61(2):399–412. doi: 10.1007/s00125-017-4462-5
199. Padrón-Morales J, Sanz C, Dávila I, Muñoz-Bellido F, Lorente F, Isidoro-García M. Polymorphisms of the IL12B, IL1B, and TNFA Genes and Susceptibility to Asthma. J Investig Allergol Clin Immunol (2013) 23(7):487–94.
200. Gordon EM, Yao X, Xu H, Karkowsky W, Kaler M, Kalchiem-Dekel O, et al. Apolipoprotein E Is a Concentration-Dependent Pulmonary Danger Signal That Activates the NLRP3 Inflammasome and IL-1β Secretion by Bronchoalveolar Fluid Macrophages From Asthmatic Subjects. J Allergy Clin Immunol (2019) 144(2):426–41.e3. doi: 10.1016/j.jaci.2019.02.027
201. Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Logun C, Alsaaty S, et al. Dysregulation of Lipidomic Profile and Antiviral Immunity in Response to Hyaluronan in Patients With Severe Asthma. J Allergy Clin Immunol (2017) 139(4):1379–83. doi: 10.1016/j.jaci.2016.09.031
202. Michalovich D, Rodriguez-Perez N, Smolinska S, Pirozynski M, Mayhew D, Uddin S, et al. Obesity and Disease Severity Magnify Disturbed Microbiome-Immune Interactions in Asthma Patients. Nat Commun (2019) 10(1):5711. doi: 10.1038/s41467-019-13751-9
203. Zhang X, Zheng J, Zhang L, Liu Y, Chen GP, Zhang HP, et al. Systemic Inflammation Mediates the Detrimental Effects of Obesity on Asthma Control. Allergy Asthma Proc (2018) 39(1):43–50. doi: 10.2500/aap.2018.39.4096
204. Forno E, Han YY, Mullen J, Celedón JC. Overweight, Obesity, and Lung Function in Children and Adults-A Meta-Analysis. J Allergy Clin Immunol Pract (2018) 6(2):570–81.e10. doi: 10.1016/j.jaip.2017.07.010
205. Restrepo BI, Twahirwa M, Rahbar MH, Schlesinger LS. Phagocytosis via Complement or Fc-Gamma Receptors Is Compromised in Monocytes From Type 2 Diabetes Patients With Chronic Hyperglycemia. PloS One (2014) 9(3):e92977. doi: 10.1371/journal.pone.0092977
206. Pavlou S, Lindsay J, Ingram R, Xu H, Chen M. Sustained High Glucose Exposure Sensitizes Macrophage Responses to Cytokine Stimuli But Reduces Their Phagocytic Activity. BMC Immunol (2018) 19(1):24. doi: 10.1186/s12865-018-0261-0
207. Ferracini M, Martins JO, Campos MR, Anger DB, Jancar S. Impaired Phagocytosis by Alveolar Macrophages From Diabetic Rats Is Related to the Deficient Coupling of LTs to the Fc Gamma R Signaling Cascade. Mol Immunol (2010) 47(11-12):1974–80. doi: 10.1016/j.molimm.2010.04.018
208. Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of Innate Immune Recognition of Bacteria Concentrate in Lipid Rafts and Facilitate Lipopolysaccharide-Induced Cell Activation. J Cell Sci (2002) 115(Pt 12):2603–11. doi: 10.1242/jcs.115.12.2603
209. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature (2010) 464(7293):1357–61. doi: 10.1038/nature08938
210. Tolani S, Pagler TA, Murphy AJ, Bochem AE, Abramowicz S, Welch C, et al. Hypercholesterolemia and Reduced HDL-C Promote Hematopoietic Stem Cell Proliferation and Monocytosis: Studies in Mice and FH Children. Atherosclerosis (2013) 229(1):79–85. doi: 10.1016/j.atherosclerosis.2013.03.031
211. Winer DA, Luck H, Tsai S, Winer S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab (2016) 23(3):413–26. doi: 10.1016/j.cmet.2016.01.003
212. Maurizi G, Della Guardia L, Maurizi A, Poloni A. Adipocytes Properties and Crosstalk With Immune System in Obesity-Related Inflammation. J Cell Physiol (2018) 233(1):88–97. doi: 10.1002/jcp.25855
213. Hotamisligil GS. Inflammation, Metaflammation and Immunometabolic Disorders. Nature (2017) 542(7640):177–85. doi: 10.1038/nature21363
214. Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased Inflammatory Properties of Adipose Tissue Macrophages Recruited During Diet-Induced Obesity. Diabetes (2007) 56(1):16–23. doi: 10.2337/db06-1076
215. Liu R, Nikolajczyk BS. Tissue Immune Cells Fuel Obesity-Associated Inflammation in Adipose Tissue and Beyond. Front Immunol (2019) 10:1587. doi: 10.3389/fimmu.2019.01587
216. Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW Jr. Obesity Activates a Program of Lysosomal-Dependent Lipid Metabolism in Adipose Tissue Macrophages Independently of Classic Activation. Cell Metab (2013) 18(6):816–30. doi: 10.1016/j.cmet.2013.11.001
217. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, But Not Obese, Fat Is Enriched for a Unique Population of Regulatory T Cells That Affect Metabolic Parameters. Nat Med (2009) 15(8):930–9. doi: 10.1038/nm.2002
218. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-Cell Memory by Modulating Fatty Acid Metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
219. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR Regulates Memory CD8 T-Cell Differentiation. Nature (2009) 460(7251):108–12. doi: 10.1038/nature08155
220. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development. Immunity (2012) 36(1):68–78. doi: 10.1016/j.immuni.2011.12.007
221. Di Cara F, Andreoletti P, Trompier D, Vejux A, Bülow MH, Sellin J, et al. Peroxisomes in Immune Response and Inflammation. Int J Mol Sci (2019) 20(16):3877. doi: 10.3390/ijms20163877
222. O’Brien A, Loftus RM, Pisarska MM, Tobin LM, Bergin R, Wood NAW, et al. Obesity Reduces Mtorc1 Activity in Mucosal-Associated Invariant T Cells, Driving Defective Metabolic and Functional Responses. J Immunol (2019) 202(12):3404–11. doi: 10.4049/jimmunol.1801600
223. Kim YG, Udayanga KG, Totsuka N, Weinberg JB, Núñez G, Shibuya A. Gut Dysbiosis Promotes M2 Macrophage Polarization and Allergic Airway Inflammation via Fungi-Induced PGE₂. Cell Host Microbe (2014) 15(1):95–102. doi: 10.1016/j.chom.2013.12.010
224. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut Microbiota Metabolism of Dietary Fiber Influences Allergic Airway Disease and Hematopoiesis. Nat Med (2014) 20(2):159–66. doi: 10.1038/nm.3444
225. Chang PV, Hao L, Offermanns S, Medzhitov R. The Microbial Metabolite Butyrate Regulates Intestinal Macrophage Function via Histone Deacetylase Inhibition. Proc Natl Acad Sci USA (2014) 111(6):2247–52. doi: 10.1073/pnas.1322269111
226. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341(6145):569–73. doi: 10.1126/science.1241165
227. Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, et al. Small Intestinal CD103+ Dendritic Cells Display Unique Functional Properties That Are Conserved Between Mice and Humans. J Exp Med (2008) 205(9):2139–49. doi: 10.1084/jem.20080414
228. Park CO, Kupper TS. The Emerging Role of Resident Memory T Cells in Protective Immunity and Inflammatory Disease. Nat Med (2015) 21(7):688–97. doi: 10.1038/nm.3883
229. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of Tissue-Resident Memory T Cells Requires Exogenous Lipid Uptake and Metabolism. Nature (2017) 543(7644):252–6. doi: 10.1038/nature21379
230. Gieger C, Geistlinger L, Altmaier E, Hrabé de Angelis M, Kronenberg F, Meitinger T, et al. Genetics Meets Metabolomics: A Genome-Wide Association Study of Metabolite Profiles in Human Serum. PloS Genet (2008) 4(11):e1000282. doi: 10.1371/journal.pgen.1000282
231. Badouin H, Gouzy J, Grassa CJ, Murat F, Staton SE, Cottret L, et al. The Sunflower Genome Provides Insights Into Oil Metabolism, Flowering and Asterid Evolution. Nature (2017) 546(7656):148–52. doi: 10.1038/nature22380
232. Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, et al. Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity (2017) 46(3):488–503. doi: 10.1016/j.immuni.2017.02.010
233. Villaseñor A, Rosace D, Obeso D, Pérez-Gordo M, Chivato T, Barbas C, et al. Allergic Asthma: An Overview of Metabolomic Strategies Leading to the Identification of Biomarkers in the Field. Clin Exp Allergy (2017) 47(4):442–56. doi: 10.1111/cea.12902
234. Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite Profiles and the Risk of Developing Diabetes. Nat Med (2011) 17(4):448–53. doi: 10.1038/nm.2307
235. González-Riano C, Dudzik D, Garcia A, Gil-de-la-Fuente A, Gradillas A, Godzien J, et al. Recent Developments Along the Analytical Process for Metabolomics Workflows. Anal Chem (2020) 92(1):203–26. doi: 10.1021/acs.analchem.9b04553
236. Binek A, Rojo D, Godzien J, Rupérez FJ, Nuñez V, Jorge I, et al. Flow Cytometry Has a Significant Impact on the Cellular Metabolome. J Proteome Res (2019) 18(1):169–81. doi: 10.1021/acs.jproteome.8b00472
237. Jang C, Chen L, Rabinowitz JD. Metabolomics and Isotope Tracing. Cell (2018) 173(4):822–37. doi: 10.1016/j.cell.2018.03.055
238. Ahl PJ, Hopkins RA, Xiang WW, Au B, Kaliaperumal N, Fairhurst AM, et al. Met-Flow, a Strategy for Single-Cell Metabolic Analysis Highlights Dynamic Changes in Immune Subpopulations. Commun Biol (2020) 3(1):305. doi: 10.1038/s42003-020-1027-9
239. Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic Inflammatory Memory in Human Respiratory Epithelial Progenitor Cells. Nature (2018) 560(7720):649–54. doi: 10.1038/s41586-018-0449-8
240. Xu CJ, Söderhäll C, Bustamante M, Baïz N, Gruzieva O, Gehring U, et al. DNA Methylation in Childhood Asthma: An Epigenome-Wide Meta-Analysis. Lancet Respir Med (2018) 6(5):379–88. doi: 10.1016/S2213-2600(18)30052-3
241. Neeland MR, Andorf S, Manohar M, Dunham D, Lyu SC, Dang TD, et al. Mass Cytometry Reveals Cellular Fingerprint Associated With IgE+ peanut Tolerance and Allergy in Early Life. Nat Commun (2020) 11(1):1091. doi: 10.1038/s41467-020-14919-4
242. Matsumoto N, Okochi M, Matsushima M, Kato R, Takase T, Yoshida Y, et al. Peptide Array-Based Analysis of the Specific IgE and IgG4 in Cow’s Milk Allergens and Its Use in Allergy Evaluation. Peptides (2009) 30(10):1840–7. doi: 10.1016/j.peptides.2009.07.005
243. Gertsman I, Barshop BA. Promises and Pitfalls of Untargeted Metabolomics. J Inherit Metab Dis (2018) 41(3):355–66. doi: 10.1007/s10545-017-0130-7
244. Gorrochategui E, Jaumot J, Lacorte S, Tauler R. Data Analysis Strategies for Targeted and Untargeted LC-MS Metabolomic Studies: Overview and Workflow. TrAC Trends Analyt Chem (2016) 82:425–42. doi: 10.1016/j.trac.2016.07.004
245. Want EJ, Masson P, Michopoulos F, Wilson ID, Theodoridis G, Plumb RS, et al. Global Metabolic Profiling of Animal and Human Tissues via UPLC-Ms. Nat Protoc (2013) 8(1):17–32. doi: 10.1038/nprot.2012.135
246. Gardner A, Carpenter G, So PW. Salivary Metabolomics: From Diagnostic Biomarker Discovery to Investigating Biological Function. Metabolites (2020) 10(2):47. doi: 10.3390/metabo10020047
247. Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, et al. The Human Serum Metabolome. PloS One (2011) 6(2):e16957. doi: 10.1371/journal.pone.0016957
248. Bouatra S, Aziat F, Mandal R, Guo AC, Wilson MR, Knox C, et al. The Human Urine Metabolome. PloS One (2013) 8(9):e73076. doi: 10.1371/journal.pone.0073076
249. Naz S, García A, Barbas C. Multiplatform Analytical Methodology for Metabolic Fingerprinting of Lung Tissue. Anal Chem (2013) 85(22):10941–8. doi: 10.1021/ac402411n
250. de Kivit S, Mensink M, Hoekstra AT, Berlin I, Derks RJE, Both D, et al. Stable Human Regulatory T Cells Switch to Glycolysis Following TNF Receptor 2 Costimulation. Nat Metab (2020) 2(10):1046–61. doi: 10.1038/s42255-020-00271-w
251. Garcia A, Barbas C. Gas Chromatography-Mass Spectrometry (GC-MS)-Based Metabolomics. Methods Mol Biol (2011) 708:191–204. doi: 10.1007/978-1-61737-985-7_11
252. Garcia A, Naz S, Barbas C. Metabolite Fingerprinting by Capillary Electrophoresis-Mass Spectrometry. Methods Mol Biol (2014) 1198:107–23. doi: 10.1007/978-1-4939-1258-2_8
253. Beckonert O, Keun HC, Ebbels TM, Bundy J, Holmes E, Lindon JC, et al. Metabolic Profiling, Metabolomic and Metabonomic Procedures for NMR Spectroscopy of Urine, Plasma, Serum and Tissue Extracts. Nat Protoc (2007) 2(11):2692–703. doi: 10.1038/nprot.2007.376
254. Miyake Y, Sasaki S, Tanaka K, Ohya Y, Miyamoto S, Matsunaga I, et al. Fish and Fat Intake and Prevalence of Allergic Rhinitis in Japanese Females: The Osaka Maternal and Child Health Study. J Am Coll Nutr (2007) 26(3):279–87. doi: 10.1080/07315724.2007.10719612
255. Barker-Tejeda TC, Villaseñor A, Gonzalez-Riano C, López-López Á, Gradillas A, Barbas C. In Vitro Generation of Oxidized Standards for Lipidomics. Application to Major Membrane Lipid Components. J Chromatogr A (2021) 1651:462254. doi: 10.1016/j.chroma.2021.462254
256. Villaseñor A, Godzien J, Barker-Tejeda TC, Gonzalez-Riano C, López-López Á, Dudzik D, et al. Analytical Approaches for Studying Oxygenated Lipids in the Search of Potential Biomarkers by LC-MS. TrAC Trends Analyt Chem (2021), 116367. doi: 10.1016/j.trac.2021.116367
257. Del Moral MG, Martínez-Naves E. The Role of Lipids in Development of Allergic Responses. Immune Netw (2017) 17(3):133–43. doi: 10.4110/in.2017.17.3.133
258. Hagemann PM, Nsiah-Dosu S, Hundt JE, Hartmann K, Orinska Z. Modulation of Mast Cell Reactivity by Lipids: The Neglected Side of Allergic Diseases. Front Immunol (2019) 10:1174. doi: 10.3389/fimmu.2019.01174
259. Schauberger E, Peinhaupt M, Cazares T, Lindsley AW. Lipid Mediators of Allergic Disease: Pathways, Treatments, and Emerging Therapeutic Targets. Curr Allergy Asthma Rep (2016) 16(7):48. doi: 10.1007/s11882-016-0628-3
260. Mullol J, Bousquet J, Bachert C, Canonica GW, Giménez-Arnau A, Kowalski ML, et al. Update on Rupatadine in the Management of Allergic Disorders. Allergy (2015) 70:1–24. doi: 10.1111/all.12531
261. Drazen JM, Israel E, O’Byrne PM. Treatment of Asthma With Drugs Modifying the Leukotriene Pathway. N Engl J Med (1999) 340(3):197–206. doi: 10.1056/NEJM199901213400306
262. Ducharme FM, Hicks GC. Anti-Leukotriene Agents Compared to Inhaled Corticosteroids in the Management of Recurrent and/or Chronic Asthma. Cochrane Database Syst Rev (2000) 3:Cd002314. doi: 10.1002/14651858.CD003133
Keywords: allergy, immunometabolism, immune cells, metabolic regulation, -omics
Citation: Rodriguez-Coira J, Villaseñor A, Izquierdo E, Huang M, Barker-Tejeda TC, Radzikowska U, Sokolowska M and Barber D (2021) The Importance of Metabolism for Immune Homeostasis in Allergic Diseases. Front. Immunol. 12:692004. doi: 10.3389/fimmu.2021.692004
Received: 07 April 2021; Accepted: 05 July 2021;
Published: 28 July 2021.
Edited by:
Cristobalina Mayorga, Universidad de Málaga, SpainReviewed by:
Isabel Varela-Nieto, Consejo Superior de Investigaciones Científicas (CSIC), SpainCopyright © 2021 Rodriguez-Coira, Villaseñor, Izquierdo, Huang, Barker-Tejeda, Radzikowska, Sokolowska and Barber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Milena Sokolowska, bWlsZW5hLnNva29sb3dza2FAc2lhZi51emguY2g=; Domingo Barber, ZG9taW5nby5iYXJiZXJoZXJuYW5kZXpAY2V1LmVz
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.