 Steven K. Yarmoska
Steven K. Yarmoska Ali M. Alawieh
Ali M. Alawieh Stephen Tomlinson
Stephen Tomlinson Kimberly B. Hoang
Kimberly B. Hoang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 October 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.689435
This article is part of the Research Topic Friend and Foe: Innate Neuro-Immune Interactions and Inflammation in Development, Homeostasis and Injury in the Central Nervous System View all 9 articles
The complement system is a highly conserved component of innate immunity that is involved in recognizing and responding to pathogens. The system serves as a bridge between innate and adaptive immunity, and modulation of the complement system can affect the entire host immune response to a foreign insult. Neoplastic diseases have been shown to engage the complement system in order to evade the immune system, gain a selective growth advantage, and co-opt the surrounding environment for tumor proliferation. Historically, the central nervous system has been considered to be an immune-privileged environment, but it is now clear that there are active roles for both innate and adaptive immunity within the central nervous system. Much of the research on the role of immunological modulation of neoplastic disease within the central nervous system has focused on adaptive immunity, even though innate immunity still plays a critical role in the natural history of central nervous system neoplasms. Here, we review the modulation of the complement system by a variety of neoplastic diseases of the central nervous system. We also discuss gaps in the current body of knowledge and comment on future directions for investigation.
The central nervous system (CNS) has been traditionally described as an immune-privileged environment. However, cumulative data over the past two decades have demonstrated that within the brain parenchyma as well as at the CNS endothelial surfaces, robust adaptive and innate immune responses can be elicited during both normal development and disease processes. In the context of neoplastic disease, inflammatory and immune mechanisms have been implicated in disease progression, response to systemic and local therapy, neurodegeneration and cerebral edema. The complement system, a component of the innate immunity, has been a major recent focus in the area of neuroscience given the role of complement proteins in early detection of stress signals, orchestrating both innate and adaptive responses and driving long-term neuroplasticity (1). In this work, we review the recent updates on the role of different complement components in the pathogenesis of neoplastic diseases of the CNS, both primary and metastatic, and the implications of this role in therapeutic interventions.
The complement system is an integral component of innate immunity. It was discovered in the late nineteenth century when it was shown to “complement” the ability of heat-stabile antibodies to kill bacteria (2). Similar to this seminal in vitro experiment, the in vivo complement system both directly responds to pathogens and indirectly recruits the adaptive immune system to assist in host defense (3). The complement system can be activated through one of three distinct pathways: the classical pathway, the lectin pathway, and the alternative pathway (4) (see Figure 1). The lectin pathway is triggered by recognition of conserved molecular patterns expressed on cells surfaces, whereas the classical pathway is triggered by antigen-bound IgG or IgM antibodies (5). The alternative pathway can be spontaneously activated on surfaces of foreign cells that lack complement regulators, and it also serves as an amplification loop for complement activation by other pathways (6). Through the sequential cleavage of unique zymogen proteins, all pathways converge at the cleavage and activation of complement protein C3 via the C3 convertase enzyme. Activation of protein C3 leads to the production of C3 opsonins, the anaphylatoxins C3a and C5a, and ultimately the membrane attack complex (MAC; C5b-9). Complement opsonins (C1q, C3b/d) deposit on surfaces of cells to tag them for phagocytosis and serve as activators of immune cells and microglia (7, 8). The complement anaphylatoxins have a multitude of proinflammatory effects including leukocyte recruitment, vasodilation, and the induction of mast cell degranulation and neutrophil oxidative burst. The MAC assembles in cell membranes, disrupting osmotic regulation and leading to cell lysis.
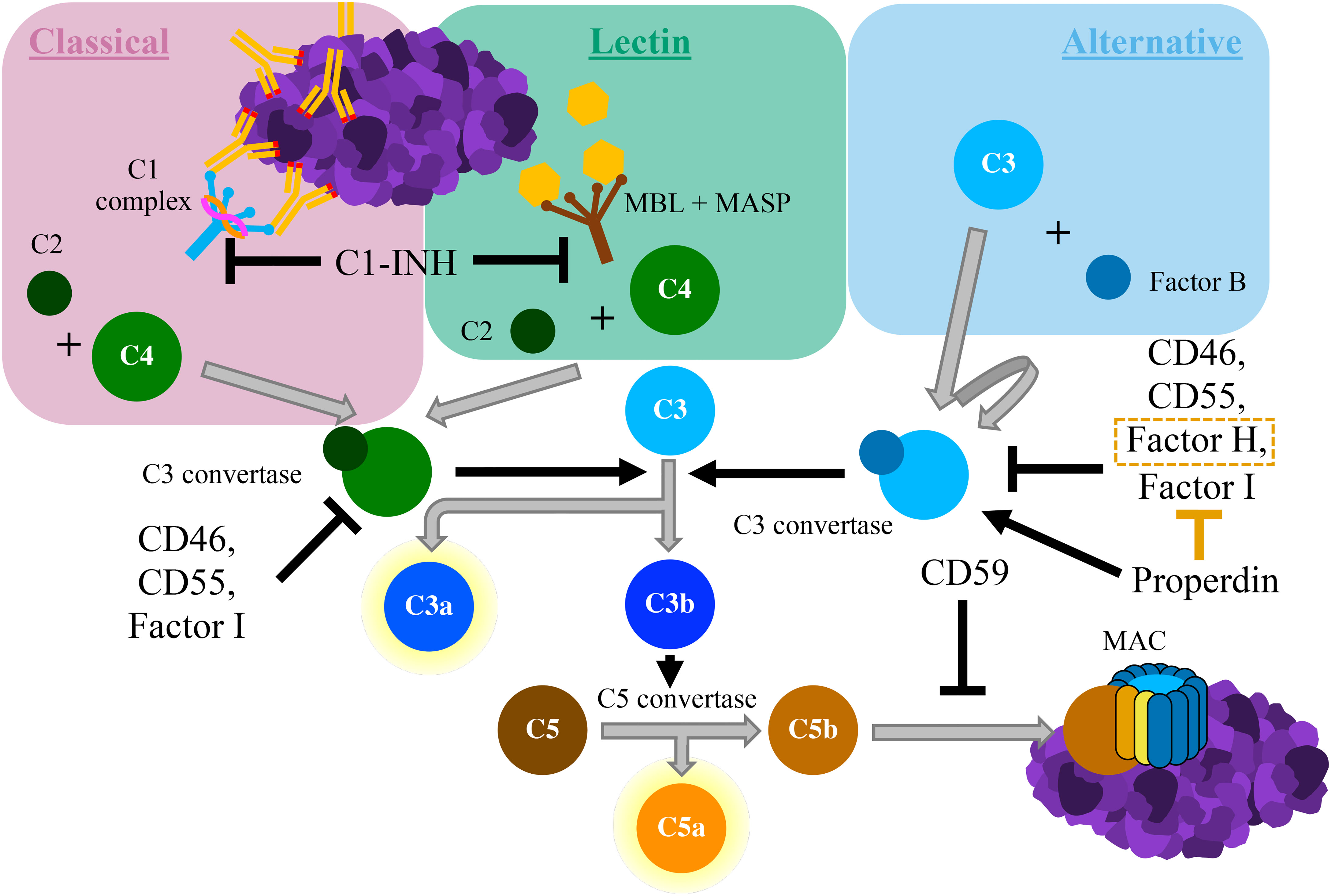
Figure 1 Schematic diagram of the complement cascade. The classical pathway begins with the binding of the C1 complex to antigen-antibody complexes containing IgG or IgM. The lectin pathway is triggered by the binding of mannose-binding lectin (MBL), which is bound to proteolytic MBL-associated serine proteases (MASPs), to carbohydrate moieties. Both of these pathways result in cleavage of C2 and C4 to form C3 convertase. The alternative pathway starts with the autoactivation of C3, typically via direct binding of C3 to pathogens. This creates a unique version of C3 convertase that involves the cleavage of Factor B All complement pathways converge with the cleavage of C3 into C3a and C3b. C3b associates with C3 convertase, which cleaves C5 into C5a and C5b. The anaphylatoxins C3a and C5a have potent proinflammatory effects. C5b deposits on cell surfaces, where it forms the membrane attack complex (MAC), which induces cell lysis. Various inhibitory proteins disrupt the complement cascade, as labelled within the figure above.
Complement opsonins also directly engage the development of B cell immunity via CD21 and CD35. On B cells, activation of these receptors lowers the threshold for B-cell receptor activation (9). On follicular dendritic cells, their expression leads to the retention of C3-coated particles for downstream presentation to B cells (10). Additionally, the complement system impacts T cell immunity. As in B cell immunity, C3 is thought to play a role in antigen presentation by marking foreign materials for APCs to phagocytose and present to T cells. However, studies on the role of C3 in the development of T cell immunity are equivocal (11–15), suggesting that this effect is antigen dependent. Murine studies have implicated C3a and C5a anaphylatoxins as chemoattractants that promote T cell recruitment either via direct effects on T cells or via indirect effects on the cytokine response of APCs (16, 17). Complement also provides negative feedback on the immune response through the induction of a regulatory phenotype in T cells through the co-stimulation of CD3 and CD46 by complement proteins C3b and C4b (3, 18).
A variety of mechanisms serve to regulate the complement system. The anaphylatoxins C3a and C5a are rapidly reduced via the removal of a C-terminal arginine, which dramatically reduces their biological activity (19). Similarly, C3b and C4b are cleaved by the serine protease Factor I into inactive metabolites with regards to complement activation, although the breakdown products still serve as opsonins. Several of the C3b breakdown products are bound by complement receptors (CR2, CR3, and CR4) that function within both the adaptive and innate immune systems (20). Factor H also accelerates the breakdown of C3 convertase in the alternative pathway (21). C3 convertase can be inhibited by the effects of decay acceleration factor (DAF; CD55) and C4 binding protein, as well. Upstream of the convergent complement components, C1 inhibitor (C1-INH) inactivates C1r and C1s as well as MASP2 (mannan-binding serine protease 2) (22), which are involved in the classical and lectin pathways, respectively. Downstream of the convergence of the complement pathways, formation of the MAC is negatively regulated by S protein, vimentin, and CD59 (4).
Properdin is the only known positive regulatory protein of the complement cascade (23). It binds to and stabilizes C3 convertase in the alternative pathway (24). Additionally, it inhibits the cleavage of C3b by Factor H (25, 26). A family of proteins structurally similar to Factor H, known as Factor H related proteins, also exist (27). However, the roles of these proteins within the canonical complement cascade are still under active investigation.
Circulating complement proteins can extravasate into the CNS when a pathologic inflammatory, infectious, or vascular process disrupts the integrity of the BBB (28). However, complement proteins can be found within the CSF of patients with an intact BBB, and recent evidence has shown that CNS-resident cells, including neurons and glial cells, are capable of producing nearly all complement proteins independent of serum derived factors (29). In vitro experiments demonstrated that astrocytes express complement proteins C2 through C9 and Factor B as well as regulatory proteins, such as Factor H and Factor I (30–32). Astrocytes have also been shown to express C5a receptor 1 (C5aR1), as well as the membrane regulatory proteins CD46, CD55, CD59, and CR1 (33). Similarly, both oligodendrocytes (34) and neurons (35, 36) have been shown to express complement proteins of the classical and alternative pathways. A subset of these genes are also expressed by microglia (37, 38).
In the healthy CNS, microglia recognize complement proteins C1q and C3 opsonins on synapses to be eliminated in the process of synaptic remodeling (39, 40). Low concentrations of MAC have also been shown to be protective for oligodendrocytes against apoptosis secondary to caspase-3 activation (41, 42). In pathologic states, the activation of complement proteins has been linked to the exacerbation of neuroinflammatory and neurodegenerative pathways in multiple sclerosis (43), stroke (1), and neurodegenerative diseases (44–46).
Neoplastic diseases within the CNS can modulate the expression and function of complement proteins in order to facilitate a pro-growth niche. The role of complement in cancer is complex, and evidence exists for both activation and inhibition of the complement cascade to result in tumorigenesis. Historically, due to the role of complement in immune surveillance (47, 48), it was thought that complement activation protected against neoplastic disease. However, more recent literature has demonstrated that complement activation within primary tumor microenvironments can enhance neoplastic growth (48, 49). The CNS provides a unique microenvironment of its own, and a diverse number of primary and secondary neoplasms can invade the CNS. Although the direct effects of complement on metastatic disease may be conserved regardless of their location, studies of complement modulation in tumors located outside the CNS may not directly translate to those located within the CNS when it comes to complement-dependent changes in the tumor microenvironment.
In this review, we discuss the role of different complement components in the pathology of CNS tumors and their implication for therapeutic interventions. A summary of the studies of complement modulation in CNS tumors, organized by disease state, can be found in Table 1. Particular focus will be placed on malignant neoplastic diseases that continue to have poor prognosis (e.g., glioma, metastasis, and leptomeningeal disease) where complement modulation may play a significant therapeutic role.
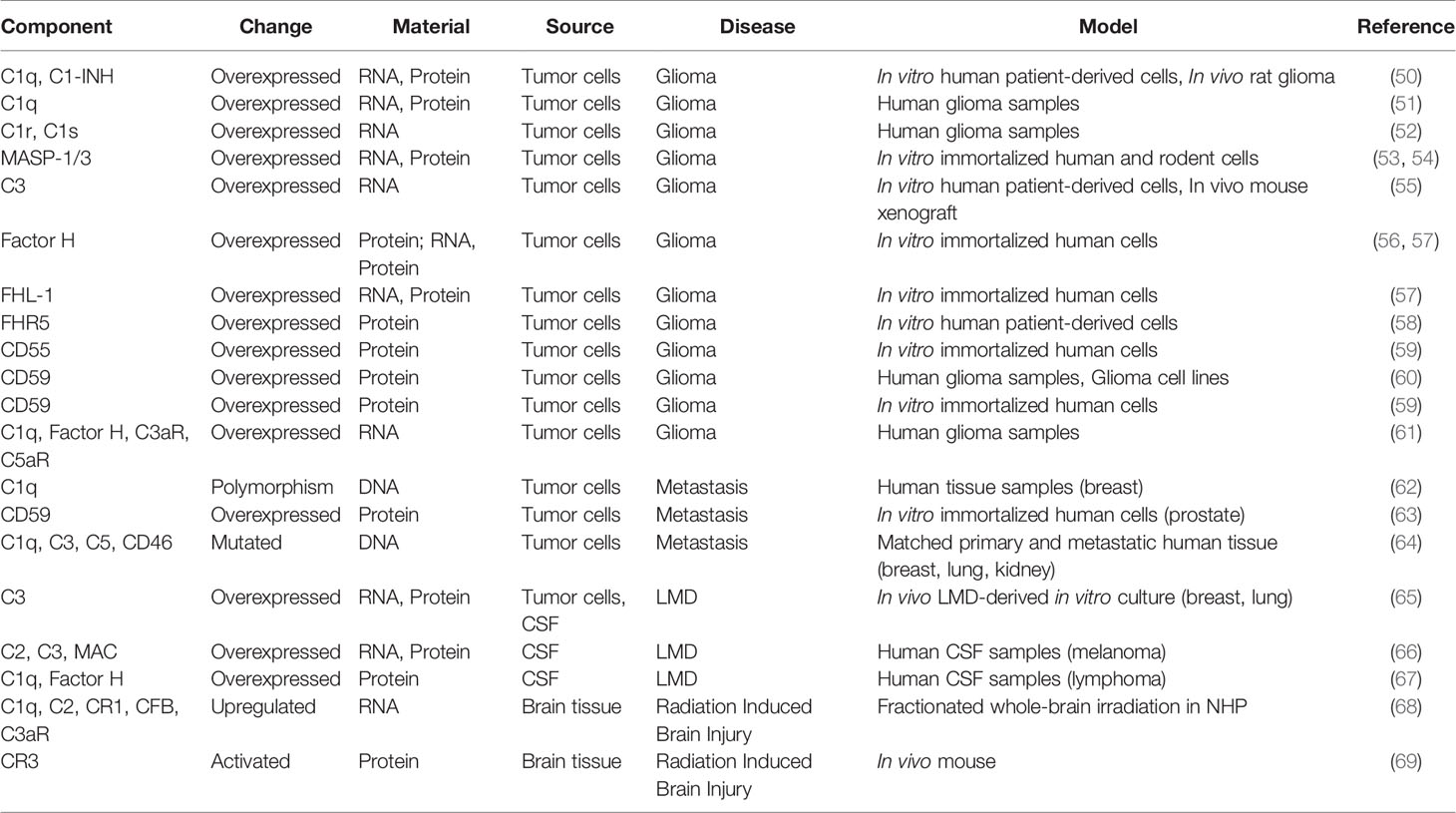
Table 1 Complement expression in brain neoplasms.
A “glioma” refers to any tumor that is thought to be of glial cell origin (70), including astrocytomas, oligodendrogliomas, ependymomas, and other rare or mixed subtypes within this family (71). High-dose radiation exposure and genetic syndromes (e.g., Li-Fraumeni syndrome, neurofibromatosis, tuberous sclerosis) are the only proven risk factors for the development of gliomas (70). Glioblastoma (i.e., WHO Grade IV astrocytoma) is the most common glioma and carries the worst prognosis. The specific inflammatory microenvironment of glioblastoma contributes to its aggressiveness, recurrence, and resistance to treatment (72). Although much of the research on immune modulation in glioma progression has focused on the adaptive immune system, innate immunity and complement also play roles in shaping the tumor microenvironment and orchestrating both adaptive and innate immune responses.
Consistent with the paradigm of immune evasion, current research on complement modulation in glioblastoma has focused on the classical pathway and the C1 complex. In both in-vitro and in-vivo models, glioblastoma was shown to upregulate the expression of the C1-INH protein that prevents the assembly of the C1 complex and inhibits the initiation of complement activation via the classical pathway (50). In synchrony with these findings, human glioblastoma tumor samples have been shown to demonstrate upregulation of the C1 complex proteins (C1q, C1s) as well as C1-INH protein (50). However, in complement pathology, mRNA upregulation does not necessarily correlate with function, given the need for proteolytic cleavage for complement activation. An increase in C1-INH effect results in upregulation of proximal classical pathway genes, which may explain the co-increase in expression of both C1q/C1s and C1-INH genes. Following these observations, the use of C1-INH as a treatment in a murine non-orthotopic model of glioblastoma demonstrated reduced tumor growth and prolonged host survival (73) (see Table 2 regarding preliminary targeted therapeutics). Serum analysis of rats in these studies showed a decrease in circulating IL-1β, one of the major proinflammatory cytokine downstream of the inflammasome complex that is produced by glioblastoma cells (77).
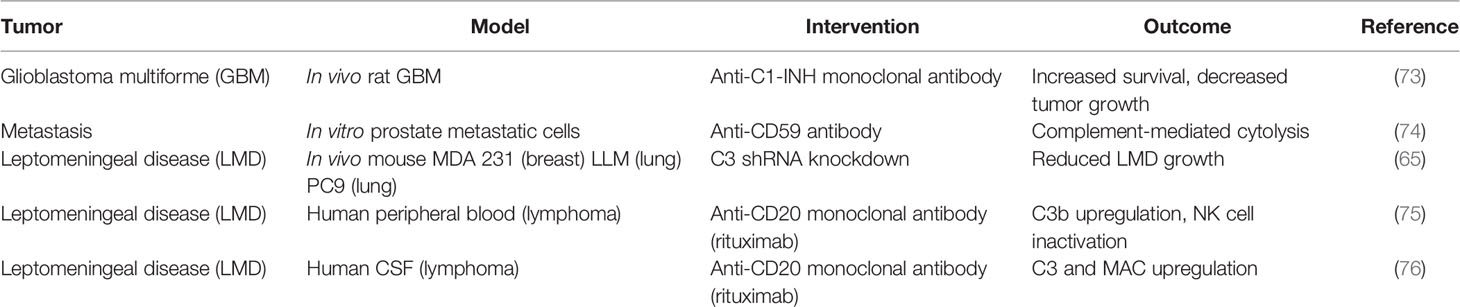
Table 2 Exogenous complement modulation as treatment in brain neoplasms.
In addition to its role as an initiator of complement activation, the C1 complex also includes the C1q protein. The C1q protein has an independent function of opsonization and clearance of apoptotic blebs, a process that is implicated in immune tolerance (78) and that has been described in the settings of autoimmunity (79, 80), angiogenesis (81), and tumorigenesis (80, 82). Although C1-INH binds to C1r and C1s, it does not bind to or inactivate C1q. Like C1r and C1s (52), C1q is also upregulated in response to the increased levels of C1-INH in glioblastoma (50). There is a greater than 4-fold change in C1q expression observed in human glioblastoma tissue samples (50), and genomic expression of C1q is positively correlated with unfavorable prognosis for patients in national glioma databases (51, 61). The tumorigenic effects of C1q are thought to be a product of both increased angiogenesis, promoting immune tolerance by clearance of antigenic products of tumor invasion, as well as direct promotion of adhesion and proliferation through integration within the extracellular matrix (82). C1q expression has been shown to activate canonical WNT signaling pathways (83), which have been shown to facilitate epithelial-to-mesenchymal transition of glioblastoma cells in vitro (84). Conversely, C1q has also been shown to inhibit tumor growth in models of breast (85), ovarian (86), and prostate cancers (87). It is thought that this inhibitory phenotype is secondary to increased tumor cell apoptosis. In the CNS, C1q is recognized by microglia during the process of synaptic pruning (39, 40), a mechanism that can also be carried out by astrocytes (88, 89). Additionally, C1q has been shown to induce a tumorigenic phenotype in tumor associated macrophages (90), a process that may extend to microglia (91, 92). Therefore, these findings suggest that the pro-growth and pro-survival effects of C1q on astroglial cells is preserved during malignant transformation as seen in primary brain tumors (i.e., gliomas). However, this may also explain why C1q has mostly a pro-apoptotic and tumor inhibitory effect on brain metastases as opposed to its effect on primary brain tumor cells.
The lectin pathway is initiated by the binding of mannose-binding lectin (MBL), which is structurally and functionally similar to C1q of the classical pathway (93). MBL predominantly binds to carbohydrate patterns, such as those found on pathogens (94–96) or IgM (97). When this binding occurs, the process activates MBL-associated serine proteases (MASPs) that subsequently cleave downstream complement proteins to propagate activation of the cascade (98). Three distinct MASPs have been described (98, 99), each of which has unique protease activity. In vitro studies of human and rat glioma cells have demonstrated their ability to secrete high levels of MASP-1 and MASP-3 (53, 54). It remains unclear whether there is a unique role for these proteases in disease progression or whether their upregulation is in response to upregulated C1-INH, which can also bind and inhibit the function of MASPs (72). Additional research is necessary to elucidate the role of MASP-1/3 in the natural history of glioma.
The alternative complement pathway plays a pivotal role in the amplification of complement activation and downstream effectors of complement by its feedforward effect on complement protein C3 activation. The alternative pathway is capable of promoting pathological levels of complement activation that may be able to evade the endogenous complement inhibitory proteins on surfaces of neurons and glia, as has been shown in ischemic and traumatic brain injury (100–103). Glioblastoma cells have demonstrated the ability to synthesize complement proteins, including C3 (55) and its receptor (61). Additionally, Factor H, which is expressed by astrocytes in the CNS, is a major fluid phase inhibitor of the alternative complement pathway. In vitro studies have demonstrated that glioblastoma cells can produce Factor H (56, 57), a result that has been supported by studies on ex vivo patient samples (61). Similarly, FH-like protein 1 (FHL-1) (57), a truncated form of Factor H that retains inhibitory activity against C3, is secreted by glioblastoma cells in vitro (21, 104). In addition to Factor H, glioblastoma cells were also shown to have upregulated expression of complement factor H related protein 5 (FHR5) (58). However, the physiological and pathological functions of the newly described FHR5 are still controversial (58, 105, 106).
Membrane-bound CD55 both prevents the assembly of and causes the dissociation of both C3 and C5 convertase (4, 107). Downstream of C3 and C5 activation, the cell-surface protein CD59 inhibits the formation of the terminal MAC by preventing C9 protein polymerization (108). Human-derived glioblastoma cell lines grown in vitro have been shown to have upregulated CD55 and CD59 (59, 60), which may be correlated with mutations in the p53 tumor suppressor gene (109). Expression of CD55 and CD59 could theoretically inhibit complement activation at the surface of growing GBM tumors in vivo, and CD59 overexpression has been observed in human glioma samples (60). However, lack of CD55 expression was found to be correlated with poor prognosis in solid tumors, such as breast cancer (110), and so further research is necessary to elucidate the effects of these proteins in glioblastoma at the organismal level. Moreover, the CD55 expression observed in GBM may be a multifactorial phenomenon.
Collectively, the current literature on the role of complement in glioblastoma support an overall tendency of the tumor to suppress the activation of the C3 protein and downstream effectors, while simultaneously preserving the functions of the C1q opsonins necessary for enhanced immune tolerance during tumor progression. This is in contrast to other primary cancers, such as cervical (111), breast (112), and lung (113), in which the complement system is activated in preclinical models to promote immune tolerance through the enhanced production of regulatory T cells (Tregs) and suppression of conventional T cells (114). Tregs are a major source of immune tolerance in glioblastoma, as Treg infiltration directly correlates to the WHO tumor grade (115), but the interplay between the complement system and Tregs within the context of glioma has yet to be conclusively elucidated.
Brain metastases are the most common type of intracranial tumor in adults (116). The most common sources of brain metastases are from primary breast, lung, and skin (i.e., melanoma) cancers (117). The “seed and soil” hypothesis defines metastasis to the brain, whereby circulating tumor cells attach to endothelial cells in brain vasculature, extravasate, and proliferate within the brain parenchyma (118). The majority of cancer cells that successfully extravasate into the brain parenchyma are ill-adapted to this microenvironment and subsequently die (119). However, this microenvironment imposes a selective pressure on migrating cancer cells that ultimately selects successful clones to seed the brain (120–122). A major component of this selective process is an ability of the metastasizing cell to evade the immune response (123). As a link between innate and adaptive immunity, complement modulation is one potential avenue for metastatic tumor cells to initiate this process.
Many metastatic cancers also present with thrombosis secondary to hypercoagulation (124). Modulation of the complement cascade is one potential factor contributing to this observed hypercoagulability in cancer (125–127). Pathway analysis has demonstrated a concomitant enrichment of proteins in the complement and coagulation pathways for brain metastases in breast, kidney, and lung cancer (64). Complement and coagulation are intrinsically related. For example, several complement proteins interact with tissue factor (TF; Factor III) and TF-bearing microparticles produced by tumor cells (128, 129). Hypercoagulation and thrombosis impact the metastatic process via dysregulation of the immune system. Peritumoral thrombosis has been shown to recruit inflammatory monocytes that promote metastatic tumor cell survival (130). Additionally, thrombi may induce a tumorigenic phenotype in neutrophils that helps to maintain a metastatic niche (131). Thrombin has also been shown to inhibit the expression of tumor suppressor genes within tumor cells (132). Furthermore, platelets can release growth factors that stimulate tumor growth, angiogenesis, and epithelial-to-mesenchymal transition (133, 134). Platelets have also demonstrated the ability to inhibit NK cell lysis of tumor cells, as well (135, 136). Moreover, it has been hypothesized that platelets, fibrin, and thrombin can shield circulating tumor cells from shear forces as they extravasate (127, 137). Clinically, anticoagulation has been shown to decrease the incidence of lung metastases (138, 139), but the effect of systemic anticoagulation on the development of brain metastasis remains unclear.
In this context, a single nucleotide polymorphism (SNP) within the C1q protein has been associated with an increased rate of breast cancer metastasis (62, 64). This association is even more pronounced for metastatic sites associate with hematogenous spread, such as the brain. The C1q protein is composed of six trimers of C1qA, C1qB, and C1qC chains (140). Among all of the known polymorphisms in these components, the only SNP within a coding region is at the 276th position of C1qA. Patients with either a homogenous or heterogeneous A-to-G polymorphism in the C1qA chain were found to have an increased risk of developing metastasis to bone, brain, or liver compared to patients without this polymorphism, even after adjusting for node and receptor status. Patients with the SNP also had a reduced time to develop these extranodal metastases. Previous studies have associated the “A” polymorphism with the development of systemic lupus erythematosus (141). This may imply that enhanced solid organ metastases with the “G” polymorphism is secondary to decreased immune activity, potentially due to reduced ability for the C1 complex to bind to and clear circulating tumor cells.
Similar to the case of glioma, malignant prostate cells have been shown to overexpress CD59 in vitro (63). Prostasomes are secretory vesicles produced by prostate cells, and they have been shown to contain the complement regulatory proteins CD46 and CD59 (142). In the case of prostate cancer, these prostasomes are secreted into the tumor microenvironment as opposed to the seminal plasma, which may contribute to the immune evasion and malignant potential of the growing tumor. A study by Babiker et al. demonstrated that prostasomes produced from prostate cancer cells contained a higher concentration of CD59 than those produced from benign prostate cells (63). Additionally, this study demonstrated that tumor-derived prostasomes were able to transfer CD59 to the surface of malignant cells in vitro. Prostasomes from brain-metastasis-derived DU145 cells exhibited the highest average CD59 content and one of the largest inhibitory effects on complement. These cells have been shown to be sensitive to complement-mediated cytolysis in the presence of a CD59-neutralizing antibody (74) (see Table 2), reinforcing the role of this molecular interaction. Follow-up studies are necessary to further elucidate the role of CD59 expression on the metastatic potential of prostate cells in vivo.
In CNS lymphomas, there is a growing body of evidence to support a role for complement in the pathogenesis of disease. Analysis of the CSF of patients with primary CNS lymphoma (PCNSL) demonstrates an upregulation of proteins associated with the innate immune system, including complement proteins C1q and Factor H (67). The upregulation of Factor H may contribute to overall suppression of the complement system and promotion of a tumorigenic microenvironment similar to studies in glioma, as discussed previously. However, the upregulation of C1q has been associated with a variety of downstream effects in different cancers and requires further exploration in PCNSL. Complement suppression being tumorigenic in PCNSL would be consistent with murine studies in non-CNS lymphoma that demonstrated attenuated tumor progression with complement activation (143).
Additionally, the intrathecal administration of rituximab, a monoclonal antibody against CD20 on B cells, has been shown to induce complement-dependent tumor cytotoxicity in PCNSL (76, 144) (see Table 2). In one clinical trial, complement proteins C3 and C5b-9 were found to be reproducibly upregulated within the CSF in response to the intrathecal administration of rituximab (76). Paradoxically, the constitutive activation of C3 within the CSF was correlated with a worse overall prognosis. The authors of this study hypothesize that this phenomenon may be due to NK cell inactivation by C3b, which has been previously demonstrated in the context of rituximab therapy (75). Alternatively, this may be related to immune dysregulation secondary to the role of C3a as a chemoattractant. Another more recent retrospective trial showed a correlation between hypocomplementemia and reduced progression-free and overall survival (145). These conflicting reports on the relationship between complement activation and PCNSL progression suggest a more nuanced role for complement in the natural history of disease, perhaps unique to different B cell lymphoma subtypes.
Leptomeningeal disease (LMD), also known as leptomeningeal metastasis, describes the spread of metastatic neoplastic disease into the arachnoid mater, pia mater, and CSF (146). Approximately 5 to 8 percent of all cancer patients will develop LMD (147), with a uniformly dismal prognosis. Common cancers to present with LMD include acute lymphoblastic leukemia (ALL), breast cancer, lung cancer, melanoma, and non-Hodgkin lymphoma (146). Seeding of the leptomeninges can occur either via preexisting metastases within the CNS or via the extension of a tumor mass that abuts the meninges (148). The exact molecular pathophysiology of LMD dissemination is poorly understood, but it is thought to abide by the general principles of metastatic invasion, which include the potential for immune invasion and vascular remodeling (149).
Work from Boire et al. has demonstrated that complement protein C3 is upregulated in metastatic tumor cells and facilitates leptomeningeal spread. In xenograft and syngeneic murine models of breast and lung cancer, tissue samples from LMD demonstrated significantly increased C3 expression compared to samples from the primary tumors (65). Knockdown of C3 expression with intrathecal short hairpin RNA (shRNA) dramatically reduced metastasis to the leptomeninges in these murine models (see Table 2). In naïve mice, administration of exogenous C3a led to increased extravasation of intravenously administered dextran to the CSF, which implies that tumor-produced C3a might disrupt the blood-CSF barrier in order to facilitate leptomeningeal spread. Moreover, mice deficient in the receptor for the C3a anaphylatoxin showed decreased growth of cancer cells inoculated directly into the leptomeningeal space, suggesting that C3a also primes the CSF for tumor invasion. Analysis of CSF samples from patients with LMD also demonstrated increased C3 expression, and decreases in C2, C3, and C4 within the CSF correlated with response to intrathecal treatment in these patients.
The role of complement in leptomeningeal melanoma (LMM) is also an area of active research, but recent data suggest that complement activation is implicated in the natural history of the disease. Pathway enrichment on CSF samples collected from patients with LMM demonstrated upregulation in various proteins involved in the complement cascade for patients who were poor responders to treatment (66). These proteins include C2, C3, and those that form the MAC. Studies in syngeneic murine models of melanoma have shown that the C3a receptor is necessary for the growth and spread of disease (150). Specifically, they demonstrated that interaction with this receptor disrupts neutrophil and CD4+ T cell responses within the tumor microenvironment. More research is necessary to further characterize the biomolecular changes that underly the development of LMM and how complement is modulated throughout the course of disease.
Radiation therapy (RT) remains a major component of the standard of care treatment for brain tumors. Despite significant improvement with the use of targeted and stereotactic radiosurgery to limit normal tissue toxicity, radiation necrosis (RN) remains a major adverse effect of RT. The incidence of RN has continued to increase given the more frequent use of adjuvant immunological therapy. RN is an undesirable, late exaggerated immune response to radiation-induced damage that results in progressive cerebral edema and peritumoral inflammation. Injury due to radiation can be further classified based on its spatial pattern (diffuse vs. focal) and the timing of injury in relation to treatment (acute, early delayed, and late) (151). Late radiation injury develops months to years after RT and is typically an irreversible and progressive phenomenon. Radiation exposure leads to endothelial cell injury, which manifests as dilated and thickened vessel walls. Along with astrocyte hyperplasia and hypertrophy, these changes are thought to comprise a “tissue injury unit” that precipitates eventual white matter necrosis (152, 153). Competing theories implicate oligodendrocyte damage and demyelination (154), autoimmune vasculitis (151), as well as changes in the fibrinolytic pathway (155) in the development of RN.
The role of complement in radiation-induced damage remains underexplored. Genetic studies in syngeneic mouse models of cancer and ex vivo human tumor samples have demonstrated that the C3a and C5a anaphylatoxins are upregulated in response to RT (156). In a murine model of melanoma, pretreatment of dexamethasone prior to RT reduced complement activation and reduced the efficacy of RT, as assessed by tumor volume measurements in the days post-RT (156). RT is known to engage the adaptive immune response (157, 158), so it is logical that elements of the complement cascade that bridge innate and adaptive immunity be engaged in this process.
As part of the stress response, RT also induces the exposure of damage-associated molecular patterns (DAMPs) and stress-related neo-epitopes following radiation-induced cell death. Exposure of these signals leads to a robust activation of the innate and adaptive immune system in the brain (101, 103, 159). Post-radiation inflammation likely involves locally activated and hematogenous-derived immune cells and components, given the effect of neoplastic pathology and radiation on blood brain barrier (BBB) integrity. The actual pathological pathways linking RT-induced cell stress, toxic edema, and cell death are still unknown. Consequently, there is a lack of targeted therapeutics to inhibit the initiation of this toxic neuroinflammation.
Prior work has demonstrated that exposure of DAMPs induces local activation of complement and deposition of C opsonins (i.e., C3b/C3d) in various disease models (160, 161). Relevant to malignancy and radiation exposure, prior work using a lymphoma model demonstrated that complement opsonins deposit locally after radiation injury and can be targeted by complement inhibitors to prevent the exacerbation of peritumoral inflammation (160). Studies in both rodents and nonhuman primates have demonstrated that complement proteins, including both opsonins and anaphylatoxins, as well as complement receptors are overexpressed and activated in the brain following radiation exposure (68, 69). Following its activation by radiation-induced stress, the complement system is then capable of self-amplification and robust activation of components of innate and adaptive immunity. This amplified activation is likely to promote worsening edema, mass effect, and neurodegeneration in peritumoral brain tissue.
Complement has been implicated in nearly all pathologies and disease of the CNS including vascular/ischemic stroke (1), autoimmune (162), and traumatic CNS pathologies (100). Despite the diverse pipeline of complement therapeutics developed over the past decade (159, 163), there are still limited complement inhibitors available to treat complement-related pathologies. To date, eculizumab, an anti-C5 antibody, along with its long-acting modification (i.e., Ultomiris) are the major success stories for complement based therapeutics. Initially approved to treat paroxysmal nocturnal hemoglobinuria and more recently approved for myasthenia gravis and AQp4-IgG positive neuromyelitis optica, anti-C5 acts systemically to inhibit complement activation at the level of the C5-convretase (163).
Although access to the BBB remains a major challenge in developing complement therapeutics, novel complement inhibitors that target different aspects of the cascade, C3 and C5 activation, are currently part of the treatment pipeline for CNS pathologies with age-related macular degeneration being the top targeted pathology. Although lampalizumab that targets the alternative pathway failed to show clinical benefit in macular degeneration, alternative inhibitors are still in different phases of clinical development and include: Compstatin (C3 inhibitor) along with its derivatives APL-1 and APL-2 (Apellis Pharmaceuticals), Mirococept (APT070, C3 inhibitor), PMX-53 (C5a receptor antagonist), C1-INH (inhibits classical and lectin pathways), Tesidolumab (inhibitor of C5 activation) among others (163).
In addition to the systemic therapeutics listed above, new generation of complement therapeutics are currently early in the translational spectrum and include tissue-targeted therapeutics that self-target to sites of active complement breakdown. In the setting of ischemic or traumatic brain injury, robust deposition of the complement breakdown product C3b/C3d provide a promising target to deliver therapeutics. As reviewed in (159), different generations of these targeted therapeutics have been described. Examples are complement receptor 2 (CR2) based inhibitors that constitute of fusion proteins of CR2 and inhibitors of one of different complement activation pathways; namely, CR2-fH, CR2-Crry and CR2-CD59 have been well studied in acute neurological pathologies such as stroke and traumatic brain injury. A similar approach include the tissue-targeted terminal pathway inhibitor CD59-2a-CRIg that uses the CRIg superfamily domains to target sites of C3 deposition and deliver complement inhibitor CD59 to the site of pathology. Finally, a different generation of targeted inhibitors applicable to CNS disease was designed using fusion proteins of single-chain variable fragments (scFv) of natural IgM antibodies that target stress-induced neo-epitopes fused to a complement inhibitor to allow its deliver to sites of active disease (159). This latter approach will have the added therapeutic value of inhibiting the antibody-based trigger of complement activation in addition to the targeted complement pathway.
Regarding complement modulation in the setting of CNS neoplastic disease, major frontiers in complement-targeted therapeutics in glioma are likely to include a focus on C1q-targeted therapeutics, given its consistent role in driving tumor progression, as well as C3-targeted therapeutics in the context of radiation to limit radiation-induced edema and degeneration allowing for maximal treatment dosing. Similarly, C3-targeted therapeutics (both C3 convertase inhibitors and C3a antagonists) are specifically attractive for metastatic disease given the role of C3 in driving edema after radiation, which is routinely used for these tumors, as well as the role of C3 in leptomeningeal spread. Systemic suppression of these complement components by therapeutics remains a major limitation due to limited bioavailability in the CNS and the risk of suppression of systemic defense mechanisms in a population known to be prone to infections. The use of tissue-targeted complement therapeutics [e.g., CR2-targeted or antibody targeted (159)] is one major avenue for overcoming these limitations.
As presented in this review, complement modulation has been implicated in the development and progression of brain and spine tumors as well as cell injury in the peritumoral environment. As CNS neoplasms are a heterogeneous group of diseases, it follows that the implicated complement mechanisms are similarly diverse. The current body of evidence with regards to glioma supports predominantly inhibitory changes, in contrast to more recent evidence in non-CNS tumors that characterize complement activation as tumorigenic, whereas data in brain metastases, LMD, and RN present a mixed picture with regards to complement activation and inhibition. The proposed pathways in this review suggest novel diagnostic and therapeutic targets for a population of patients notoriously difficult to treat, both at initial diagnosis and with recurrence. This work also supports the potential role for targeted complement modulation in CNS tumors and emphasizes the need for more translational and preclinical studies in this field.
SY, AA, and KH conceptualized the scope of the review. SY performed the literature review. SY and AA wrote the initial manuscript. AA, ST, and KH reviewed and edited. All authors contributed to the article and approved the submitted version.
SY is supported through grants from the National Institutes of Health (F30 CA216939, T32 GM008169). AA is supported through a grant from the Department of Veterans Affairs (I01 RX001141). KH is supported by a grant from the American Cancer Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Jocelyn Chow of Emory University for her assistance with the literature review process.
1. Alawieh A, Elvington A, Tomlinson S. Complement in the Homeostatic and Ischemic Brain. Front Immunol (2015) 6:417. doi: 10.3389/fimmu.2015.00417
2. Chaplin H. The Burgeoning History of the Complement System 1888-2005 Vol. 21. Washington DC: Immunohematology (2005). p. 85.
3. Carroll MC. The Complement System in Regulation of Adaptive Immunity. Nat Immunol (2004) 5:981–6. doi: 10.1038/ni1113
4. Sarma JV, Ward PA. The Complement System. Cell Tissue Res (2011) 343:227–35. doi: 10.1007/s00441-010-1034-0
5. Reid KB, Porter RR. The Proteolytic Activation Systems of Complement. Annu Rev Biochem (1981) 50:433–64. doi: 10.1146/annurev.bi.50.070181.002245
6. Lachmann P, Hughes-Jones N. Initiation of Complement Activation. In: . Springer Seminars in Immunopathology. Berlin, Germany: Springer (1984). p. 143–62.
7. Law S, Lichtenberg N, Levine R. Covalent Binding and Hemolytic Activity of Complement Proteins. Proc Natl Acad Sci (1980) 77:7194–8. doi: 10.1073/pnas.77.12.7194
8. Tack BF, Harrison RA, Janatova J, Thomas ML, Prahl JW. Evidence for Presence of an Internal Thiolester Bond in Third Component of Human Complement. Proc Natl Acad Sci (1980) 77:5764–8. doi: 10.1073/pnas.77.10.5764
9. Carter RH, Fearon DT. CD19: Lowering the Threshold for Antigen Receptor Stimulation of B Lymphocytes. Science (1992) 256:105–7. doi: 10.1126/science.1373518
10. Fang Y, Xu C, Fu Y-X, Holers VM, Molina H. Expression of Complement Receptors 1 and 2 on Follicular Dendritic Cells Is Necessary for the Generation of a Strong Antigen-Specific Igg Response. J Immunol (1998) 160:5273–9.
11. Fischer MB, Ma M, Goerg S, Zhou X, Xia J, Finco O, et al. Regulation of the B Cell Response to T-Dependent Antigens by Classical Pathway Complement. J Immunol (1996) 157:549–56.
12. Fischer MB, Ma M, Hsu NC, Carroll MC. Local Synthesis of C3 Within the Splenic Lymphoid Compartment can Reconstitute the Impaired Immune Response in C3-Deficient Mice. J Immunol (1998) 160:2619–25.
13. Da Costa XJ, Brockman MA, Alicot E, Ma M, Fischer MB, Zhou X, et al. Humoral Response to Herpes Simplex Virus Is Complement-Dependent. Proc Natl Acad Sci (1999) 96:12708–12. doi: 10.1073/pnas.96.22.12708
14. Ochsenbein AF, Pinschewer DD, Odermatt B, Carroll MC, Hengartner H, Zinkernagel RM. Protective T Cell–Independent Antiviral Antibody Responses are Dependent on Complement. J Exp Med (1999) 190:1165–74. doi: 10.1084/jem.190.8.1165
15. Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. Complement Component C3 Promotes T-Cell Priming and Lung Migration to Control Acute Influenza Virus Infection. Nat Med (2002) 8:373–8. doi: 10.1038/nm0402-373
16. Drouin SM, Corry DB, Hollman TJ, Kildsgaard J, Wetsel RA. Absence of the Complement Anaphylatoxin C3a Receptor Suppresses Th2 Effector Functions in a Murine Model of Pulmonary Allergy. J Immunol (2002) 169:5926–33. doi: 10.4049/jimmunol.169.10.5926
17. Drouin SM, Corry DB, Kildsgaard J, Wetsel RA. Cutting Edge: The Absence of C3 Demonstrates a Role for Complement in Th2 Effector Functions in a Murine Model of Pulmonary Allergy. J Immunol (2001) 167:4141–5. doi: 10.4049/jimmunol.167.8.4141
18. Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of Human CD4+ Cells With CD3 and CD46 Induces a T-Regulatory Cell 1 Phenotype. Nature (2003) 421:388–92. doi: 10.1038/nature01315
19. Kalant D, Cain SA, Maslowska M, Sniderman AD, Cianflone K, Monk PN. The Chemoattractant Receptor-Like Protein C5L2 Binds the C3a Des-Arg77/Acylation-Stimulating Protein. J Biol Chem (2003) 278:11123–9. doi: 10.1074/jbc.M206169200
20. Holers VM, Kulik L. Complement Receptor 2, Natural Antibodies and Innate Immunity: Inter-Relationships in B Cell Selection and Activation. Mol Immunol (2007) 44:64–72. doi: 10.1016/j.molimm.2006.07.003
21. de Córdoba SR, . Esparza-Gordillo J, . de Jorge EG, . Lopez-Trascasa M, . Sánchez-Corral P. The Human Complement Factor H: Functional Roles, Genetic Variations and Disease Associations. Mol Immunol (2004) 41:355–67. doi: 10.1016/j.molimm.2004.02.005
22. Davis AE III, Mejia P, Lu F. Biological Activities of C1 Inhibitor. Mol Immunol (2008) 45:4057–63. doi: 10.1016/j.molimm.2008.06.028
23. Chen JY, Cortes C, Ferreira VP. Properdin: A Multifaceted Molecule Involved in Inflammation and Diseases. Mol Immunol (2018) 102:58–72. doi: 10.1016/j.molimm.2018.05.018
24. Hourcade DE. The Role of Properdin in the Assembly of the Alternative Pathway C3 Convertases of Complement. J Biol Chem (2006) 281:2128–32. doi: 10.1074/jbc.M508928200
25. Medicus R, Götze O, Müller-Eberhard H. Alternative Pathway of Complement: Recruitment of Precursor Properdin by the Labile C3/C5 Convertase and the Potentiation of the Pathway. J Exp Med (1976) 144:1076–93. doi: 10.1084/jem.144.4.1076
26. Farries TC, Lachmann PJ, Harrison RA. Analysis of the Interactions Between Properdin, the Third Component of Complement (C3), and its Physiological Activation Products. Biochem J (1988) 252:47–54. doi: 10.1042/bj2520047
27. Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement Factor H Related Proteins (Cfhrs). Mol Immunol (2013) 56:170–80. doi: 10.1016/j.molimm.2013.06.001
28. Rus H, Cudrici C, David S, Niculescu F. The Complement System in Central Nervous System Diseases. Autoimmunity (2006) 39:395–402. doi: 10.1080/08916930600739605
29. Sano Y. Studies on the Complement System in Cerebrospinal Fluid. Acta Med Univ Kagoshima (1985) 27:1–9.
30. Levi-Strauss M, Mallat M. Primary Cultures of Murine Astrocytes Produce C3 and Factor B, Two Components of the Alternative Pathway of Complement Activation. J Immunol (1987) 139:2361–6.
31. Rus H, Kim L, Niculescu F, Shin M. Induction of C3 Expression in Astrocytes Is Regulated by Cytokines and Newcastle Disease Virus. J Immunol (1992) 148:928–33.
32. Barnum S. Complement Biosynthesis in the Central Nervous System. Crit Rev Oral Biol Med (1995) 6:132–46. doi: 10.1177/10454411950060020301
33. Singhrao SK, Neal JW, Rushmere NK, Morgan BP, Gasque P. Differential Expression of Individual Complement Regulators in the Brain and Choroid Plexus. Lab investigation; J Tech Methods Pathol (1999) 79:1247.
34. Hosokawa M, Klegeris A, Maguire J, McGeer PL. Expression of Complement Messenger Rnas and Proteins by Human Oligodendroglial Cells. Glia (2003) 42:417–23. doi: 10.1002/glia.10234
35. Singhrao SK, Neal JW, Rushmere NK, Morgan BP, Gasque P. Spontaneous Classical Pathway Activation and Deficiency of Membrane Regulators Render Human Neurons Susceptible to Complement Lysis. Am J Pathol (2000) 157:905–18. doi: 10.1016/S0002-9440(10)64604-4
36. Thomas A, Gasque P, Vaudry D, Gonzalez B, Fontaine M. Expression of a Complete and Functional Complement System by Human Neuronal Cells In Vitro. Int Immunol (2000) 12:1015–23. doi: 10.1093/intimm/12.7.1015
37. Gasque P, Fontaine M, Morgan BP. Complement Expression in Human Brain. Biosynthesis of Terminal Pathway Components and Regulators in Human Glial Cells and Cell Lines. J Immunol (1995) 154:4726–33.
38. Haga S, Ikeda K, Sato M, Ishii T. Synthetic Alzheimer Amyloid β/A4 Peptides Enhance Production of Complement C3 Component by Cultured Microglial Cells. Brain Res (1993) 601:88–94. doi: 10.1016/0006-8993(93)91698-R
39. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
40. Zabel MK, Kirsch WM. From Development to Dysfunction: Microglia and the Complement Cascade in CNS Homeostasis. Ageing Res Rev (2013) 12:749–56. doi: 10.1016/j.arr.2013.02.001
41. Soane L, Rus H, Niculescu F, Shin ML. Inhibition of Oligodendrocyte Apoptosis by Sublytic C5b-9 Is Associated With Enhanced Synthesis of Bcl-2 and Mediated by Inhibition of Caspase-3 Activation. J Immunol (1999) 163:6132–8.
42. Soane L, Cho H-J, Niculescu F, Rus H, Shin ML. C5b-9 Terminal Complement Complex Protects Oligodendrocytes From Death by Regulating Bad Through Phosphatidylinositol 3-Kinase/Akt Pathway. J Immunol (2001) 167:2305–11. doi: 10.4049/jimmunol.167.4.2305
43. Sellebjerg F, Jaliashvili I, Christiansen M, Garred P. Intrathecal Activation of the Complement System and Disability in Multiple Sclerosis. J Neurological Sci (1998) 157:168–74. doi: 10.1016/S0022-510X(98)00086-0
44. Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, et al. Complement Activation by Beta-Amyloid in Alzheimer Disease. Proc Natl Acad Sci (1992) 89:10016–20. doi: 10.1073/pnas.89.21.10016
45. Kawamata T, Akiyama H, Yamada T, McGeer P. Immunologic Reactions in Amyotrophic Lateral Sclerosis Brain and Spinal Cord Tissue. Am J Pathol (1992) 140:691.
46. Yang J, Bardes ESG, Moore JD, Brennan J, Powers MA, Kornbluth S. Control of Cyclin B1 Localization Through Regulated Binding of the Nuclear Export Factor CRM1. Genes Dev (1998) 12:2131–43. doi: 10.1101/gad.12.14.2131
47. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: A Key System for Immune Surveillance and Homeostasis. Nat Immunol (2010) 11:785–97. doi: 10.1038/ni.1923
48. Afshar-Kharghan V. The Role of the Complement System in Cancer. J Clin Invest (2017) 127:780–9. doi: 10.1172/JCI90962
49. Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in Cancer: Untangling an Intricate Relationship. Nat Rev Immunol (2018) 18:5. doi: 10.1038/nri.2017.97
50. Förnvik K, Maddahi A, Persson O, Osther K, Salford LG, Nittby Redebrandt H. C1-Inactivator is Upregulated in Glioblastoma. PloS One (2017) 12:e0183086. doi: 10.1371/journal.pone.0183086
51. Mangogna A, Belmonte B, Agostinis C, Zacchi P, Iacopino DG, Martorana A, et al. Prognostic Implications of the Complement Protein C1q in Gliomas. Front Immunol (2019) 10:2366. doi: 10.3389/fimmu.2019.02366
52. Gerber NK, Goenka A, Turcan S, Reyngold M, Makarov V, Kannan K, et al. Transcriptional Diversity of Long-Term Glioblastoma Survivors. Neuro-Oncology (2014) 16:1186–95. doi: 10.1093/neuonc/nou043
53. Kuraya M, Matsushita M, Endo Y, Thiel S, Fujita T. Expression of H-Ficolin/Hakata Antigen, Mannose-Binding Lectin-Associated Serine Protease (MASP)-1 and MASP-3 by Human Glioma Cell Line T98G. Int Immunol (2003) 15:109–17. doi: 10.1093/intimm/dxg008
54. Pagliara V, Parafati M, Adornetto A, White MC, Masullo M, Grimaldi M, et al. Dibutyryl Camp-or Interleukin-6-Induced Astrocytic Differentiation Enhances Mannose Binding Lectin (MBL)-Associated Serine Protease (MASP)-1/3 Expression in C6 Glioma Cells. Arch Biochem biophysics (2018) 653:39–49. doi: 10.1016/j.abb.2018.06.016
55. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science (2014) 344:1396–401. doi: 10.1126/science.1254257
56. Gasque P, Thomas A, Fontaine M, Morgan BP. Complement Activation on Human Neuroblastoma Cell Lines In Vitro: Route of Activation and Expression of Functional Complement Regulatory Proteins. J Neuroimmunol (1996) 66:29–40. doi: 10.1016/0165-5728(96)00015-X
57. Junnikkala S, Jokiranta T, Friese MA, Jarva H, Zipfel PF, Meri S. Exceptional Resistance of Human H2 Glioblastoma Cells to Complement-Mediated Killing by Expression and Utilization of Factor H and Factor H-Like Protein 1. J Immunol (2000) 164:6075–81. doi: 10.4049/jimmunol.164.11.6075
58. DeCordova S, Abdelgany A, Murugaiah V, Pathan AA, Nayak A, Walker T, et al. Secretion of Functionally Active Complement Factor H Related Protein 5 (FHR5) by Primary Tumour Cells Derived From Glioblastoma Multiforme Patients. Immunobiology (2019) 224:625–31. doi: 10.1016/j.imbio.2019.07.006
59. Fleurence J, Cochonneau D, Fougeray S, Oliver L, Geraldo F, Terme M, et al. Targeting and Killing Glioblastoma With Monoclonal Antibody to O-Acetyl GD2 Ganglioside. Oncotarget (2016) 7:41172. doi: 10.18632/oncotarget.9226
60. Mäenpää A, Junnikkala S, Hakulinen J, Timonen T, Meri S. Expression of Complement Membrane Regulators Membrane Cofactor Protein (CD46), Decay Accelerating Factor (CD55), and Protectin (CD59) in Human Malignant Gliomas. Am J Pathol (1996) 148:1139.
61. Di Jia SL, Li D, Xue H, Yang D, Liu Y. Mining TCGA Database for Genes of Prognostic Value in Glioblastoma Microenvironment. Aging (Albany NY) (2018) 10:592. doi: 10.18632/aging.101415
62. Racila E, Racila DM, Ritchie JM, Taylor C, Dahle C, Weiner GJ. The Pattern of Clinical Breast Cancer Metastasis Correlates With a Single Nucleotide Polymorphism in the C1qa Component of Complement. Immunogenetics (2006) 58:1–8. doi: 10.1007/s00251-005-0077-y
63. Babiker AA, Nilsson B, Ronquist G, Carlsson L, Ekdahl KN. Transfer of Functional Prostasomal CD59 of Metastatic Prostatic Cancer Cell Origin Protects Cells Against Complement Attack. Prostate (2005) 62:105–14. doi: 10.1002/pros.20102
64. Richichi C, Fornasari L, Melloni GE, Brescia P, Patanè M, Del Bene M, et al. Mutations Targeting the Coagulation Pathway Are Enriched in Brain Metastases. Sci Rep (2017) 7:1–6. doi: 10.1038/s41598-017-06811-x
65. Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massagué J. Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell (2017) 168:1101–13.e13. doi: 10.1016/j.cell.2017.02.025
66. Smalley I, Law V, Wyatt C, Evernden B, Fang B, Koomen JM, et al. Proteomic Analysis of CSF From Patients With Leptomeningeal Melanoma Metastases Identifies Signatures Associated With Disease Progression and Therapeutic Resistance. Clin Cancer Res (2020) 26:2163–75. doi: 10.1158/1078-0432.CCR-19-2840
67. Roy S, Josephson SA, Fridlyand J, Karch J, Kadoch C, Karrim J, et al. Protein Biomarker Identification in the CSF of Patients With CNS Lymphoma. J Clin Oncol: Off J Am Soc Clin Oncol (2008) 26:96. doi: 10.1200/JCO.2007.12.1053
68. Andrews RN, Dugan GO, Peiffer AM, Hawkins GA, Hanbury DB, Bourland JD, et al. White Matter is the Predilection Site of Late-Delayed Radiation-Induced Brain Injury in non-Human Primates. Radiat Res (2019) 191:217–31. doi: 10.1667/RR15263.1
69. Hinkle JJ, Olschowka JA, Love TM, Williams JP, O’Banion MK. Cranial Irradiation Mediated Spine Loss Is Sex-Specific and Complement Receptor-3 Dependent in Male Mice. Sci Rep (2019) 9:1–12. doi: 10.1038/s41598-019-55366-6
70. Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and Molecular Pathology of Glioma. Nat Clin Pract Neurol (2006) 2:494–503. doi: 10.1038/ncpneuro0289
71. Kleihues P. Tumors of the Nervous System. Pathology and genetics. In: World Health Organization International Classification of Tumors, IARC Press (2000). p. 72–81.
72. DeCordova S, Shastri A, Tsolaki AG, Yasmin H, Klein L, Singh SK, et al. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front Immunol (2020) 11:1402. doi: 10.3389/fimmu.2020.01402
73. Förnvik K, Ahlstedt J, Osther K, Salford LG, Redebrandt HN. Anti-C1-Inactivator Treatment of Glioblastoma. Oncotarget (2018) 9:37421. doi: 10.18632/oncotarget.26456
74. Jarvis GA, Li J, Hakulinen J, Brady KA, Nordling S, Dahiya R, et al. Expression and Function of the Complement Membrane Attack Complex Inhibitor Protectin (CD59) in Human Prostate Cancer. Int J Cancer (1997) 71:1049–55. doi: 10.1002/(SICI)1097-0215(19970611)71:6<1049::AID-IJC22>3.0.CO;2-7
75. Wang S-Y, Racila E, Taylor RP, Weiner GJ. NK-Cell Activation and Antibody-Dependent Cellular Cytotoxicity Induced by Rituximab-Coated Target Cells is Inhibited by the C3b Component of Complement. Blood J Am Soc Hematol (2008) 111:1456–63. doi: 10.1182/blood-2007-02-074716
76. Kadoch C, Li J, Wong VS, Chen L, Cha S, Munster P, et al. Complement Activation and Intraventricular Rituximab Distribution in Recurrent Central Nervous System Lymphoma. Clin Cancer Res (2014) 20:1029–41. doi: 10.1158/1078-0432.CCR-13-0474
77. Gauthier T, Hamou M-F, Monod L, Gallay P, de Tribolet N. Expression and Release of Interleukin-1 by Human Glioblastoma Cells In Vitro and In Vivo. Acta neurochirurgica (1993) 121:199–205. doi: 10.1007/BF01809276
78. Kouser L, Madhukaran SP, Shastri A, Saraon A, Ferluga J, Al-Mozaini M, et al. Emerging and Novel Functions of Complement Protein C1q. Front Immunol (2015) 6:317. doi: 10.3389/fimmu.2015.00317
79. Son M, Diamond B, Santiago-Schwarz F. Fundamental Role of C1q in Autoimmunity and Inflammation. Immunologic Res (2015) 63:101–6. doi: 10.1007/s12026-015-8705-6
80. Ghebrehiwet B, Hosszu K, Valentino A, Peerschke EI. The C1q Family of Proteins: Insights Into the Emerging non-Traditional Functions. Front Immunol (2012) 3:52. doi: 10.3389/fimmu.2012.00052
81. Bossi F, Tripodo C, Rizzi L, Bulla R, Agostinis C, Guarnotta C, et al. C1q as a Unique Player in Angiogenesis With Therapeutic Implication in Wound Healing. Proc Natl Acad Sci (2014) 111:4209–14. doi: 10.1073/pnas.1311968111
82. Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, et al. C1q Acts in the Tumour Microenvironment as a Cancer-Promoting Factor Independently of Complement Activation. Nat Commun (2016) 7:1–11. doi: 10.1038/ncomms10346
83. Naito AT, Sumida T, Nomura S, Liu M-L, Higo T, Nakagawa A, et al. Complement C1q Activates Canonical Wnt Signaling and Promotes Aging-Related Phenotypes. Cell (2012) 149:1298–313. doi: 10.1016/j.cell.2012.03.047
84. Kahlert UD, Maciaczyk D, Doostkam S, Orr BA, Simons B, Bogiel T, et al. Activation of Canonical WNT/β-Catenin Signaling Enhances In Vitro Motility of Glioblastoma Cells by Activation of ZEB1 and Other Activators of Epithelial-to-Mesenchymal Transition. Cancer Lett (2012) 325:42–53. doi: 10.1016/j.canlet.2012.05.024
85. Bandini S, Macagno M, Hysi A, Lanzardo S, Conti L, Bello A, et al. The non-Inflammatory Role of C1q During Her2/Neu-Driven Mammary Carcinogenesis. Oncoimmunology (2016) 5:e1253653. doi: 10.1080/2162402X.2016.1253653
86. Kaur A, Sultan SH, Murugaiah V, Pathan AA, Alhamlan FS, Karteris E, et al. Human C1q Induces Apoptosis in an Ovarian Cancer Cell Line via Tumor Necrosis Factor Pathway. Front Immunol (2016) 7:599. doi: 10.3389/fimmu.2016.00599
87. Hong Q, Sze C-I, Lin S-R, Lee M-H, He R-Y, Schultz L, et al. Complement C1q Activates Tumor Suppressor WWOX to Induce Apoptosis in Prostate Cancer Cells. PloS One (2009) 4:e5755. doi: 10.1371/journal.pone.0005755
88. DeWitt DA, Perry G, Cohen M, Doller C, Silver J. Astrocytes Regulate Microglial Phagocytosis of Senile Plaque Cores of Alzheimer’s Disease. Exp Neurol (1998) 149:329–40. doi: 10.1006/exnr.1997.6738
89. Konishi H, Okamoto T, Hara Y, Komine O, Tamada H, Maeda M, et al. Astrocytic Phagocytosis is a Compensatory Mechanism for Microglial Dysfunction. EMBO J (2020) 39:e104464. doi: 10.15252/embj.2020104464
90. Bohlson SS, O’Conner SD, Hulsebus HJ, Ho M-M, Fraser DA. Complement, C1q, and C1q-Related Molecules Regulate Macrophage Polarization. Front Immunol (2014) 5:402. doi: 10.3389/fimmu.2014.00402
91. Wurdinger T, Deumelandt K, van der Vliet HJ, Wesseling P, de Gruijl TD. Mechanisms of Intimate and Long-Distance Cross-Talk Between Glioma and Myeloid Cells: How to Break a Vicious Cycle. Biochim Biophys Acta (BBA)-Reviews Cancer (2014) 1846:560–75. doi: 10.1016/j.bbcan.2014.10.003
92. van der Vlis TB, Kros J, Mustafa D, van Wijck R, Ackermans L, van Hagen P, et al. The Complement System in Glioblastoma Multiforme. Acta Neuropathologica Commun (2018) 6:1–12. doi: 10.1186/s40478-018-0591-4
93. Bohlson SS, Fraser DA, Tenner AJ. Complement Proteins C1q and MBL are Pattern Recognition Molecules That Signal Immediate and Long-Term Protective Immune Functions. Mol Immunol (2007) 44:33–43. doi: 10.1016/j.molimm.2006.06.021
94. Anders EM, Hartley CA, Jackson DC. Bovine and Mouse Serum Beta Inhibitors of Influenza a Viruses are Mannose-Binding Lectins. Proc Natl Acad Sci (1990) 87:4485–9. doi: 10.1073/pnas.87.12.4485
95. Ji X, Gewurz H, Spear GT. Mannose Binding Lectin (MBL) and HIV. Mol Immunol (2005) 42:145–52. doi: 10.1016/j.molimm.2004.06.015
96. Choteau L, Parny M, Francois N, Bertin B, Fumery M, Dubuquoy L, et al. Role of Mannose-Binding Lectin in Intestinal Homeostasis and Fungal Elimination. Mucosal Immunol (2016) 9:767–76. doi: 10.1038/mi.2015.100
97. McMullen ME, Hart ML, Walsh MC, Buras J, Takahashi K, Stahl GL. Mannose-Binding Lectin Binds Igm to Activate the Lectin Complement Pathway In Vitro and In Vivo. Immunobiology (2006) 211:759–66. doi: 10.1016/j.imbio.2006.06.011
98. Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A Journey Through the Lectin Pathway of Complement—MBL and Beyond. Immunol Rev (2016) 274:74–97. doi: 10.1111/imr.12468
99. Yongqing T, Drentin N, Duncan RC, Wijeyewickrema LC, Pike RN. Mannose-Binding Lectin Serine Proteases and Associated Proteins of the Lectin Pathway of Complement: Two Genes, Five Proteins and Many Functions? Biochim Biophys Acta (BBA)-Proteins Proteomics (2012) 1824:253–62. doi: 10.1016/j.bbapap.2011.05.021
100. Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the Role of Complement in Triggering Neuroinflammation After Traumatic Brain Injury. J Neurosci (2018) 38:2519–32. doi: 10.1523/JNEUROSCI.2197-17.2018
101. Alawieh A, Langley EF, Tomlinson S. Targeted Complement Inhibition Salvages Stressed Neurons and Inhibits Neuroinflammation After Stroke in Mice. Sci Trans Med (2018) 10(441):eaao6459. doi: 10.1126/scitranslmed.aao6459
102. Alawieh A, Andersen M, Adkins DL, Tomlinson S. Acute Complement Inhibition Potentiates Neurorehabilitation and Enhances Tpa-Mediated Neuroprotection. J Neurosci (2018) 38:6527–45. doi: 10.1523/JNEUROSCI.0111-18.2018
103. Alawieh AM, Langley EF, Feng W, Spiotta AM, Tomlinson S. Complement-Dependent Synaptic Uptake and Cognitive Decline After Stroke and Reperfusion Therapy. J Neurosci (2020) 40:4042–58. doi: 10.1523/JNEUROSCI.2462-19.2020
104. Hellwage J, Jokiranta TS, Koistinen V, Vaarala O, Meri S, Zipfel PF. Functional Properties of Complement Factor H-Related Proteins FHR-3 and FHR-4: Binding to the C3d Region of C3b and Differential Regulation by Heparin. FEBS Lett (1999) 462:345–52. doi: 10.1016/S0014-5793(99)01554-9
105. McRae JL, Duthy TG, Griggs KM, Ormsby RJ, Cowan PJ, Cromer BA, et al. Human Factor H-Related Protein 5 has Cofactor Activity, Inhibits C3 Convertase Activity, Binds Heparin and C-Reactive Protein, and Associates With Lipoprotein. J Immunol (2005) 174:6250–6. doi: 10.4049/jimmunol.174.10.6250
106. De Jorge EG, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, et al. Dimerization of Complement Factor H-Related Proteins Modulates Complement Activation In Vivo. Proc Natl Acad Sci (2013) 110:4685–90. doi: 10.1073/pnas.1219260110
107. Lublin DM, Atkinson J. Decay-Accelerating Factor: Biochemistry, Molecular Biology, and Function. Annu Rev Immunol (1989) 7:35–58. doi: 10.1146/annurev.iy.07.040189.000343
108. Huang Y, Qiao F, Abagyan R, Hazard S, Tomlinson S. Defining the CD59-C9 Binding Interaction. J Biol Chem (2006) 281:27398–404. doi: 10.1074/jbc.M603690200
109. Pandya PH, Saadatzadeh M, Ding J, Bailey B, Ross S, Bijangi-Vishehsaraei K, et al. Complement Regulatory Protein Expression in Solid Tumors: Implications for Resistance to Antibody-Mediated Immunotherapy. AACR (2017) 77:4592. doi: 10.1158/1538-7445.AM2017-4592
110. Madjd Z, Durrant LG, Bradley R, Spendlove I, Ellis IO, Pinder SE. Loss of CD55 is Associated With Aggressive Breast Tumors. Clin Cancer Res (2004) 10:2797–803. doi: 10.1158/1078-0432.CCR-1073-03
111. Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell (2018) 34:561–78. doi: 10.1016/j.ccell.2018.09.003
112. Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, et al. Complement C5a Receptor Facilitates Cancer Metastasis by Altering T-Cell Responses in the Metastatic Niche. Cancer Res (2014) 74:3454–65. doi: 10.1158/0008-5472.CAN-14-0157
113. Kwak JW, Laskowski J, Li HY, McSharry MV, Sippel TR, Bullock BL, et al. Complement Activation via a C3a Receptor Pathway Alters CD4+ T Lymphocytes and Mediates Lung Cancer Progression. Cancer Res (2018) 78:143–56. doi: 10.1158/0008-5472.CAN-17-0240
114. Zhang R, Liu Q, Li T, Liao Q, Zhao Y. Role of the Complement System in the Tumor Microenvironment. Cancer Cell Int (2019) 19:1–12. doi: 10.1186/s12935-019-1027-3
115. Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJM, Wesseling P, et al. Prognostic Significance and Mechanism of Treg Infiltration in Human Brain Tumors. J Neuroimmunol (2010) 225:195–9. doi: 10.1016/j.jneuroim.2010.05.020
116. Tosoni A, Ermani M, Brandes AA. The Pathogenesis and Treatment of Brain Metastases: A Comprehensive Review. Crit Rev Oncol/Hematol (2004) 52:199–215. doi: 10.1016/j.critrevonc.2004.08.006
117. Steeg PS. Tumor Metastasis: Mechanistic Insights and Clinical Challenges. Nat Med (2006) 12:895–904. doi: 10.1038/nm1469
118. Fidler IJ, Yano S, . Zhang R-D, . Fujimaki T, . Bucana CD. The Seed and Soil Hypothesis: Vascularisation and Brain Metastases. Lancet Oncol (2002) 3:53–7. doi: 10.1016/S1470-2045(01)00622-2
119. Kienast Y, Von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-Time Imaging Reveals the Single Steps of Brain Metastasis Formation. Nat Med (2010) 16:116–22. doi: 10.1038/nm.2072
120. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH-F, Lee DJ, et al. Serpins Promote Cancer Cell Survival and Vascular Co-Option in Brain Metastasis. Cell (2014) 156:1002–16. doi: 10.1016/j.cell.2014.01.040
121. Louie E, Chen X, Coomes A, Ji K, Tsirka S, Chen E. Neurotrophin-3 Modulates Breast Cancer Cells and the Microenvironment to Promote the Growth of Breast Cancer Brain Metastasis. Oncogene (2013) 32:4064–77. doi: 10.1038/onc.2012.417
122. Boire A, Brastianos PK, Garzia L, Valiente M. Brain Metastasis. Nat Rev Cancer (2019) 20:1–8. doi: 10.1038/s41568-019-0220-y
123. Leibold AT, Monaco GN, Dey M. The Role of the Immune System in Brain Metastasis. Curr Neurobiol (2019) 10:33.
124. Buller H, Van Doormaal F, Van Sluis G, Kamphuisen P. Cancer and Thrombosis: From Molecular Mechanisms to Clinical Presentations. J Thromb Haemostasis (2007) 5:246–54. doi: 10.1111/j.1538-7836.2007.02497.x
125. Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction Between the Coagulation and Complement System. In: . Current Topics in Complement II. New York, NY: Springer (2008). p. 68–76.
126. Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and Coagulation: Strangers or Partners in Crime? Trends Immunol (2007) 28:184–92. doi: 10.1016/j.it.2007.02.006
127. Guglietta S, Rescigno M. Hypercoagulation and Complement: Connected Players in Tumor Development and Metastases. Semin Immunol (2016) 28:578–86. doi: 10.1016/j.smim.2016.10.011
128. Davila M, Amirkhosravi A, Coll E, Desai H, Robles L, Colon J, et al. Tissue Factor-Bearing Microparticles Derived From Tumor Cells: Impact on Coagulation Activation. J Thromb Haemostasis (2008) 6:1517–24. doi: 10.1111/j.1538-7836.2008.02987.x
129. Thomas G, Brill A, Mezouar S, Crescence L, Gallant M, Dubois C, et al. Tissue Factor Expressed by Circulating Cancer Cell-Derived Microparticles Drastically Increases the Incidence of Deep Vein Thrombosis in Mice. J Thromb Haemostasis (2015) 13:1310–9. doi: 10.1111/jth.13002
130. Gil-Bernabé AM, Ferjančič Š, Tlalka M, Zhao L, Allen PD, Im JH, et al. Recruitment of Monocytes/Macrophages by Tissue Factor-Mediated Coagulation is Essential for Metastatic Cell Survival and Premetastatic Niche Establishment in Mice. Blood (2012) 119:3164–75. doi: 10.1182/blood-2011-08-376426
131. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation Induced by C3ar-Dependent Netosis Drives Protumorigenic Neutrophils During Small Intestinal Tumorigenesis. Nat Commun (2016) 7:1–14. doi: 10.1038/ncomms11037
132. Villares GJ, Zigler M, Dobroff AS, Wang H, Song R, Melnikova VO, et al. Protease Activated Receptor-1 Inhibits the Maspin Tumor-Suppressor Gene to Determine the Melanoma Metastatic Phenotype. Proc Natl Acad Sci (2011) 108:626–31. doi: 10.1073/pnas.1006886108
133. Gay LJ, Felding-Habermann B. Contribution of Platelets to Tumour Metastasis. Nat Rev Cancer (2011) 11:123–34. doi: 10.1038/nrc3004
134. Labelle M, Begum S, Hynes RO. Direct Signaling Between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell (2011) 20:576–90. doi: 10.1016/j.ccr.2011.09.009
135. Bobek V, Boubelik M, Fišerová A, L’uptovcova M, Vannucci L, Kacprzak G, et al. Anticoagulant Drugs Increase Natural Killer Cell Activity in Lung Cancer. Lung Cancer (2005) 47:215–23. doi: 10.1016/j.lungcan.2004.06.012
136. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and Fibrin (Ogen) Increase Metastatic Potential by Impeding Natural Killer Cell–Mediated Elimination of Tumor Cells. Blood (2005) 105:178–85. doi: 10.1182/blood-2004-06-2272
137. Konstantopoulos K, McIntire LV. Effects of Fluid Dynamic Forces on Vascular Cell Adhesion. J Clin Invest (1996) 98:2661–5. doi: 10.1172/JCI119088
138. Agostino D, Cliffton EE, Girolami A. Effect of Prolonged Coumadin Treatment on the Production of Pulmonary Metastases in the Rat. Cancer (1966) 19:284–8. doi: 10.1002/1097-0142(196602)19:2<284::AID-CNCR2820190223>3.0.CO;2-0
139. Esumi N, Fan D, Fidler IJ. Inhibition of Murine Melanoma Experimental Metastasis by Recombinant Desulfatohirudin, a Highly Specific Thrombin Inhibitor. Cancer Res (1991) 51:4549–56.
140. Kishore U, Reid KB. C1q: Structure, Function, and Receptors. Immunopharmacology (2000) 49:159–70. doi: 10.1016/S0162-3109(00)80301-X
141. Racila D, Sontheimer C, Sheffield A, Wisnieski J, Racila E, Sontheimer R. Homozygous Single Nucleotide Polymorphism of the Complement C1QA Gene is Associated With Decreased Levels of C1q in Patients With Subacute Cutaneous Lupus Erythematosus. Lupus (2003) 12:124–32. doi: 10.1191/0961203303lu329oa
142. Rooney IA, Heuser JE, Atkinson JP. GPI-Anchored Complement Regulatory Proteins in Seminal Plasma. An Analysis of Their Physical Condition and the Mechanisms of Their Binding to Exogenous Cells. J Clin Invest (1996) 97:1675–86. doi: 10.1172/JCI118594
143. Elvington M, Huang Y, Morgan BP, Qiao F, Van Rooijen N, Atkinson C, et al. A Targeted Complement-Dependent Strategy to Improve the Outcome of Mab Therapy, and Characterization in a Murine Model of Metastatic Cancer. Blood J Am Soc Hematol (2012) 119:6043–51. doi: 10.1182/blood-2011-10-383232
144. Takami A, Hayashi T, Kita D, Nishimura R, Asakura H, Nakao S. Treatment of Primary Central Nervous System Lymphoma With Induction of Complement-Dependent Cytotoxicity by Intraventricular Administration of Autologous-Serum-Supplemented Rituximab. Cancer Sci (2006) 97:80–3. doi: 10.1111/j.1349-7006.2006.00138.x
145. Pan B-H, Kong Y-L, Wang L, Zhu H-Y, Li X-T, Liang J-H, et al. The Prognostic Roles of Hypogammaglobulinemia and Hypocomplementemia in Newly Diagnosed Diffuse Large B-Cell Lymphoma. Leukemia Lymphoma (2020) 62:291–9. doi: 10.1080/10428194.2020.1832673
146. Groves MD. Leptomeningeal Disease. Neurosurgery Clinics (2011) 22:67–78. doi: 10.1016/j.nec.2010.08.006
147. DeAngelis LM, DeAngelis LM, Posner JB. Neurologic Complications of Cancer. New York, NY: OUP USA (2009).
148. Balm M, Hammack J. Leptomeningeal Carcinomatosis: Presenting Features and Prognostic Factors. Arch Neurol (1996) 53:626–32. doi: 10.1001/archneur.1996.00550070064013
149. Chiang AC, Massagué J. Molecular Basis of Metastasis. N Engl J Med (2008) 359:2814–23. doi: 10.1056/NEJMra0805239
150. Nabizadeh JA, Manthey HD, Steyn FJ, Chen W, Widiapradja A, Akhir FNM, et al. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J Immunol (2016) 196:4783–92. doi: 10.4049/jimmunol.1600210
151. Giglio P, Gilbert MR. Cerebral Radiation Necrosis. Neurologist (2003) 9:180–8. doi: 10.1097/01.nrl.0000080951.78533.c4
152. Calvo W, Hopewell J, Reinhold H, Yeung T. Time-and Dose-Related Changes in the White Matter of the Rat Brain After Single Doses of X Rays. Br J Radiol (1988) 61:1043–52. doi: 10.1259/0007-1285-61-731-1043
153. Wiegel T, Hinkelbein W, Brock M, Hoell T. Pathophysiological Mechanisms Leading to the Development of Late Radiation-Induced Damage to the Central Nervous System. Controversies Neuro-Oncol (1999) 33:265–75. doi: 10.1159/000061239
154. Burger P, Boyko O. The Pathology of Central Nervous System Radiation Injury. In: Radiation Injury to the Nervous System, Raven Press (1991).
155. Sawaya R. The Fibrinolytic Enzymes in the Biology of Brain Tumors. In: Fibrinolysis and the Central Nervous System, Harnley and Belfus (1990). p. 106–26.
156. Surace L, Lysenko V, Fontana AO, Cecconi V, Janssen H, Bicvic A, et al. Complement is a Central Mediator of Radiotherapy-Induced Tumor-Specific Immunity and Clinical Response. Immunity (2015) 42:767–77. doi: 10.1016/j.immuni.2015.03.009
157. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, et al. Radiotherapy Promotes Tumor-Specific Effector CD8+ T Cells via Dendritic Cell Activation. J Immunol (2012) 189:558–66. doi: 10.4049/jimmunol.1200563
158. Brandmaier A, Formenti SC. The Impact of Radiation Therapy on Innate and Adaptive Tumor Immunity. Seminars Rad Oncol (2020) 30:139–44. doi: 10.1016/j.semradonc.2019.12.005
159. Alawieh A, Tomlinson S. Injury Site-Specific Targeting of Complement Inhibitors for Treating Stroke. Immunol Rev (2016) 274:270–80. doi: 10.1111/imr.12470
160. Elvington M, Scheiber M, Yang X, Lyons K, Jacqmin D, Wadsworth C, et al. Complement-Dependent Modulation of Antitumor Immunity Following Radiation Therapy. Cell Rep (2014) 8:818–30. doi: 10.1016/j.celrep.2014.06.051
161. Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, et al. The Indoleamine 2,3-Dioxygenase Pathway Controls Complement-Dependent Enhancement of Chemo-Radiation Therapy Against Murine Glioblastoma. J Immunother Cancer (2014) 2:21. doi: 10.1186/2051-1426-2-21
162. Chen M, Daha MR, Kallenberg CG. The Complement System in Systemic Autoimmune Disease. J Autoimmun (2010) 34:J276–86. doi: 10.1016/j.jaut.2009.11.014
Keywords: complement, brain, cancer, glioblastoma, glioma, metastasis, leptomeningeal, radiation
Citation: Yarmoska SK, Alawieh AM, Tomlinson S and Hoang KB (2021) Modulation of the Complement System by Neoplastic Disease of the Central Nervous System. Front. Immunol. 12:689435. doi: 10.3389/fimmu.2021.689435
Received: 31 March 2021; Accepted: 10 September 2021;
Published: 04 October 2021.
Edited by:
Jennifer K. Dowling, Royal College of Surgeons in Ireland, IrelandReviewed by:
Matthew Zabel, University of California, San Diego, United StatesCopyright © 2021 Yarmoska, Alawieh, Tomlinson and Hoang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberly B. Hoang, a2ltYmVybHkuYm9qYW5vd3NraS5ob2FuZ0BlbW9yeS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.