 Danyang Meng
Danyang Meng Man Luo1*
Man Luo1* Beibei Liu
Beibei Liu- 1Department of Neurology, Affiliated Hospital of Jiaxing University, Jiaxing, China
- 2Department of Central Laboratory, Affiliated Hospital of Jiaxing University, Jiaxing, China
C-type lectin-like receptor 2 (CLEC-2, also known as CLEC-1b) is expressed on platelets, Kupffer cells and other immune cells, and binds to various ligands including the mucin-like protein podoplanin (PDPN). The role of CLEC-2 in infection and immunity has become increasingly evident in recent years. CLEC-2 is involved in platelet activation, tumor cell metastasis, separation of blood/lymphatic vessels, and cerebrovascular patterning during embryonic development. In this review, we have discussed the role of CLEC-2 in thromboinflammation, and focused on the recent research.
Introduction
CLEC-2 (CLEC-1b) is a type II transmembrane receptor (1, 2) of the C-type lectin superfamily, which are characterized by one or more C-type lectin-like domains (CTLDs). Members of the C-type lectin superfamily are mainly involved in growth and development, respiration, blood coagulation, angiogenesis and inflammation (3). CLEC-2 regulates multiple physiological pathways by recognizing and binding to both endogenous and exogenous ligands (4–6). While the role of CLEC-2 in tumorigenesis (7, 8) and platelet activation (9) is well-established, its involvement in thrombosis is ambiguous. Recent studies have implicated CLEC-2 in the inflammatory response, and correlated the upregulation of CLEC-2 ligands in the inflamed tissues with vascular integrity (10), which further highlights its role in thrombosis. CLEC-2 and its ligands are the molecular bridge between platelets, immune cells and target cells, and a novel mechanistic link between inflammation and thrombosis. Therefore, CLEC-2 related pathways are potential therapeutic targets for thromboinflammation.
Molecular Structure and Cellular Distribution of CLEC-2
CLEC-2 is a platelet-activating type II transmembrane receptor with a molecular weight of ~32 kDa, and is highly expressed on megakaryocytes and platelets (1, 2). Originally known as snake venom protein receptor, CLEC-2 has a function similar to that of glycoprotein (GP) VI (GPVI) in activating Src (non-receptor tyrosine kinase) or Syk (spleen associated tyrosine kinase) upstream of phospholipase C (PLC) γ2 to trigger platelet aggregation (11–14). It comprises of a YXXL sequence, two conserved serine sequences at positions 21 and 27, and a partially conserved threonine sequence at position 9, of which YXXL is crucial for signal transduction (15). Binding of CLEC-2 to its cognate ligand triggers tyrosine phosphorylation of one intracytoplasmic YXXL motif, which activates the downstream semi-immunoreceptor tyrosine-based activation motif (ITAM) pathway (16). The semi-helical long loop region on the binding surface of CLEC-2 is variable compared with other parts, and binding of ligands can bring the cytoplasmic signal transduction domain of CLEC-2 closer to each other, thus promoting ligand induced dimerization (17). CLEC-2 binds to the tandem SH2 domain of Syk in a 2:1 stoichiometry based on its cytoplasmic tail phosphorylated peptide (18). Studies on transgenic mice have revealed that CLEC-2 is also expressed as relatively lower levels on Kupffer cells, sinusoidal endothelial cells (19), dendritic cells, macrophages (14), B lymphocytes and neutrophils induced during the inflammatory response (20). Lowe et al. demonstrated that the expression of CLEC-2 on neutrophils is likely the off-target effect of antibodies, and there are also statements the expression of CLEC-2 is probably limited to mice (20), whereas macrophages express CLEC-2 after phagocytosing platelets. Thus, the distribution of CLEC-2 has not been fully elucidated (5).
CLEC-2 and Its Ligands
The major exogenous ligands of CLEC-2 are the snake venom toxin rhodocytin and type 1 human immunodeficiency virus (HIV-1). Rhodocytin is a heterodimeric C-type lectin that induces platelet aggregation through CLEC-2 clustering (14). In addition, the CLEC-2 expressed on platelets captures HIV-1 particles and leads to its subsequent phagocytosis, which is the basis of the high levels of circulating HIV-1 in infected individuals (21). CLEC-2 may interact with the cytokines within the HIV-1 rather than directly with the envelope protein (Env) of the virus. The direct interaction between CLEC-2 and HIV-1 was considered, however, CLEC-2 lacks a known amino acid motiv to regulate calcium complexation and carbohydrate binding of C-type lectins (1). Therefore, the structure recognized by CLEC-2 on the cell surface and on the HIV-1 particle was not revealed (21). There is evidence that the interaction between HIV-1 and CLEC-2 is indirectly mediated by an endogenous ligand (4). Recent studies have also confirmed that CLEC-2 does not recognize soluble HIV-1 Env, and the results show that virion incorporation of podoplanin was required for efficient CLEC-2-dependent HIV-1 interactions with cell lines and platelets. The binding of CLEC-2 to HIV-1 is indirectly accomplished through podoplanin, and it was also found that primary T cells may express a hitherto unrecognized ligand of CLEC-2, which is integrated into the viral Env and promotes HIV-1 transmission (4). Other exogenous ligands of CLEC-2 include the sulfated polysaccharide fucoidan (22) and diesel exhaust particles (23), although the mechanisms underlying their interaction remain to be elucidated.
Podoplanin (PDPN), also known as GP38 or Aggrus, is an endogenous ligand of CLEC-2 (24) that was first identified in rat glomerular epithelial cells (25). Podoplanin is a type I transmembrane glycoprotein (26) that mediates venous thrombosis (27), extravascular platelet activation and inflammation in atherosclerosis (27, 28), and wound repair (29) upon binding to CLEC-2. Furthermore, the CLEC‐2/podoplanin axis also facilitates blood/lymphatic vessel separation during embryonic development (30), maintains the lymph node vascular integrity, and optimizes adaptive immune responses (31). A recent study showed that the smooth muscle calcium binding protein S100A13 is a potential ligand of CLEC-2, and activates platelets independent of podoplanin (32). In addition, the hemin produced during the turnover of red blood cells (RBCs) can also activate platelets through integrin platelet glycoprotein IIb/IIIa receptor (GPIIb/IIIa) or CLEC-2 at low concentrations, and induce platelet aggregation at high concentrations, as well CLEC-2 can maybe promote pro-caogulant platelets which express phosphatidylserine (33). The ligands of CLEC-2 are summarized in Figure 1.
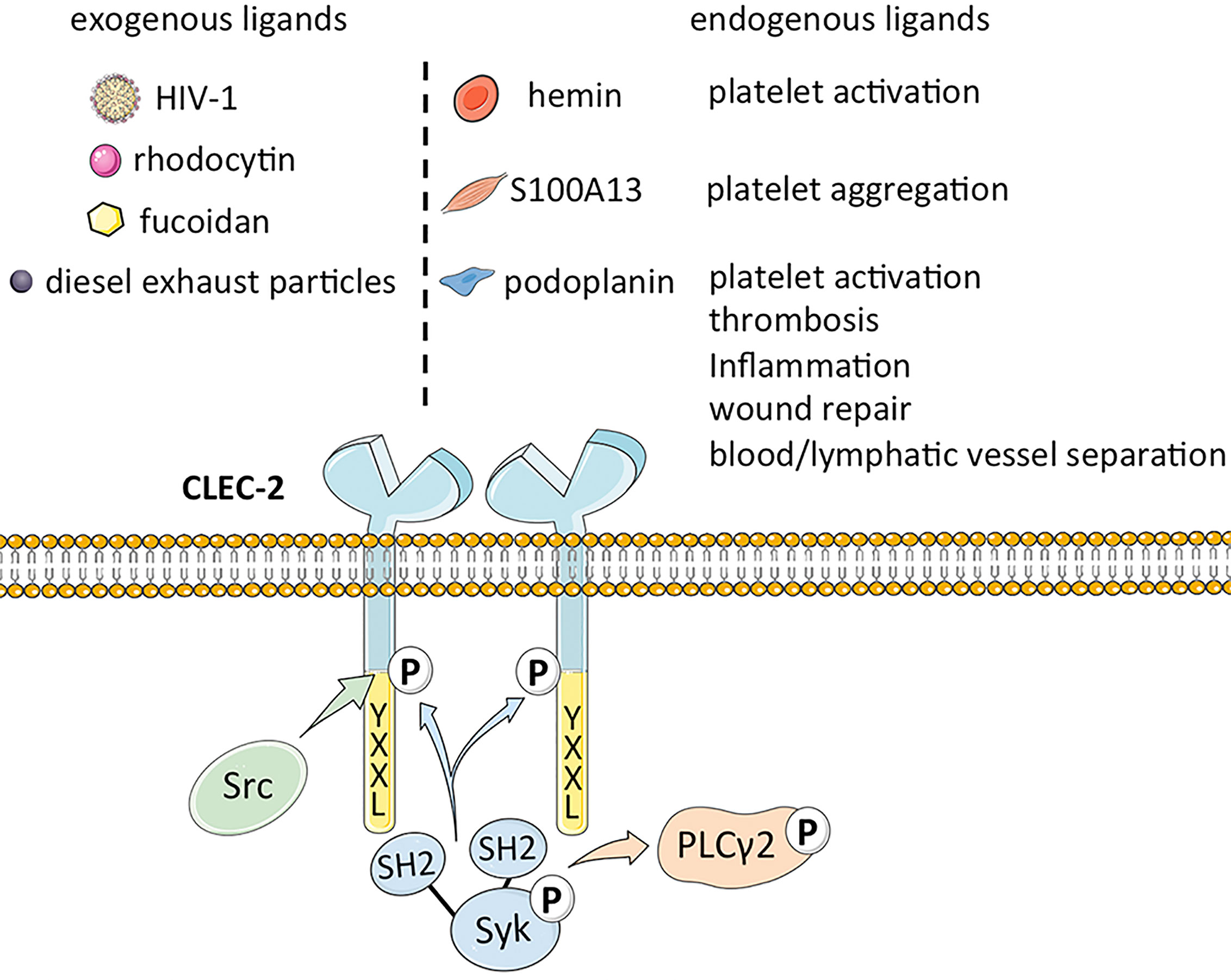
Figure 1 The structure of CLEC-2 and the function of its endogenous ligand.
CLEC-2 in Thrombosis
CLEC-2 is an important platelet activating receptor in the process of hemostasis and thrombosis (34–36). INU1, as an inhibitor of CLEC-2, can slightly prolong the tail bleeding time of mice, and CLEC-2 deficient platelets cannot form stable aggregates in vitro (34). In addition, the podoplanin/CLEC-2 axis regulates hemostasis and thrombosis (34–36) by promoting megakaryocyte proliferation and platelet formation (13, 19).
CLEC-2 and GPVI are two important receptors in platelet activation. HemITAM phosphorylation of CLEC-2 during the CLEC-2 process is mediated by the tyrosine kinase Syk, which is essential for signal transduction and downstream effector protein phosphorylation. Glycoprotein VI (GPVI), the center of platelet activation collagen receptor, has a pathway similar to CLEC-2, and its activation ultimately leads to platelet activation and thrombus growth. CLEC-2 may compensate for the lack of GPVI given that neither individuals with GPVI mutations nor the GPVI-knockout mice exhibit a hemorrhagic phenotype. Bender et al. showed that antibody-mediated blockade of either GPVI (JAQ1) or CLEC-2 (INU1) in mice down-regulated their expression and activity on the platelet surface, but did not affect bleeding, whereas simultaneous inactivation of both completely inhibited thrombus formation. The mutual compensatory action of the receptors indicates that targeted blocking of both can not only exert a strong anti-thrombotic effect but also impair normal hemostasis (36). In mice with suppressed Syk function, the increase in tail bleeding time was slight, reflecting the adhesion effect of CLEC-2 (37).
CLEC-2 plays a major role in process of thrombosis and a secondary role in normal hemostasis process. During wound healing, CLEC-2 may be useful in maintaining vascular integrity in the inflected skin only in the absence of GPVI (38). The ligand of CLEC-2 in the vascular system is not clear yet. Considering its undergo homophilic binding with submicromolar affinity, it is speculated that the endogenous ligand may be CLEC-2 itself (10, 35). It promotes the adhesion of platelets in vascular system, but in the case of inflammation, it plays a more important role in prevention of bleeding and thrombosis (10). There are corresponding changes in CLEC-2 in infectious and non-infectious diseases, especially in inflammatory reaction, CLEC-2 in platelets regulates the vascular integrity of acute inflammation (16), and its distribution varies with the anatomical location and inflammatory state of different immune cells (5), and there will be an up-regulation of CLEC-2 and its ligands (39, 40).
CLEC-2 in Thromboinflammation
Thromboinflammation refers to the coordinated activation of thrombotic and inflammatory responses that manifests in various diseases, and is a major cause of mortality and morbidity. In the following sections, the specific roles of CLEC-2 and its ligands in neurological diseases, atherosclerosis, deep vein thrombosis, infectious thrombosis and cancer thrombosis have been discussed.
CLEC-2 in Neurological Diseases
Platelets are central to the pathogenesis of ischemic stroke, which is characterized by a complex thromboinflammatory response triggered by the activation of platelets and immune cells, which destroys the blood-brain barrier (BBB) and leads to neuronal damage (41). GPVI plays an important role in the development of cerebral infarction by inducing an inflammatory response through the ITAM pathway (42, 43). Furthermore, the lectin-like oxidized low-density lipoprotein (LDL) receptor-1 (LOX-1) is elevated in the blood of patients with acute ischemic stroke and transient ischemic attack (44). The Syk cascade signal initiated by CLEC-2 is closely related to GPVI and belongs to C-type lectin receptor family with LOX-1. A study conducted on Chinese patients with acute ischemic stroke reported high levels of plasma CLEC-2, which was associated with poor prognosis and significantly increased risk of death (45). Other studies have also shown that plasma CLEC-2 is a predictor of cerebrovascular disease recurrence in patients with acute ischemic stroke (46). Podoplanin inhibitors mitigated the pathological changes after cerebral ischemia-reperfusion in a mouse model of middle cerebral artery occlusion-induced stroke, which indicates a potential role of the CLEC-2/podoplanin axis in thromboinflammation (47). In the same study, CLEC-2 and podoplanin were linked to the NLRP3 inflammasome (47). The above results were limited to cerebral artery embolization, and the role of CLEC-2 and its ligands in cerebral venous sinus thrombosis is still unclear.
Traumatic brain injury (TBI) is closely related to inflammation, hypercoagulation and apoptosis (48, 49). A significant proportion of patients with TBI have dysfunctional coagulation due to early platelet activation, leading to acute traumatic coagulation disease, which is systemic and affects many organs, arteries and veins (50–53). In addition, elevated plasma CLEC-2 levels in TBI patients correlate with poor prognosis, and can be used as a potential biomarker to evaluate disease severity and prognosis (54). However, in the mouse model, exogenous CLEC-2 appears to produce neuroprotective effects in TBI, improving brain edema, reversing blood-brain barrier damage and basement membrane degradation, and regulating inflammatory response, but does not improve the symptoms of neurological impairment in mice (55).
The underlying mechanisms of thromboinflammatory diseases induced by stroke and brain injury are not fully understood. GPVI and CLEC-2 expression in platelets is regulated by Src like adaptor (SLAP) and SLAP2, which are inactivated after cerebral ischemia, resulting in severe deterioration of neurological function after focal brain injury (56). Furthermore, the podoplanin/CLEC-2 axis has an endogenous role in the nervous system. Podoplanin is widely distributed in the central nervous system of mice from embryonic development to adulthood (57), and both podoplanin and CLEC-2 knockout mice show embryonic neurovascular development defects (9). In addition, podoplanin also plays an important role in neural progenitor cells proliferation and neuronal differentiation by interacting with the nerve growth factor (58). Podoplanin knockout mice show defects in cerebral neuron growth, synaptic plasticity and hippocampus-dependent learning and memory (59). Alzheimer disease (AD) patients have significantly higher levels of plasma CLEC-2 compared to those with mild cognitive impairment (MCI), which further underscores the role of GPIIb/IIIA-dependent platelet activation in cognitive diseases (60). Furthermore, podoplanin is overexpressed in the inflamed brain tissues of multiple sclerosis patients (61), indicating a specific role of podoplanin/CLEC-2 signaling in neuroinflammation. While a limited inflammatory response following brain injury promotes the repair of damaged tissues (62), unrestrained inflammation can lead to secondary damage and neuronal apoptosis (63). Podoplanin is up-regulated in the reactive astrocytes of mice with brain inflammation (64). Furthermore, rats injected with lipopolysaccharide (LPS) into their lateral ventricles exhibited neuroinflammation and upregulation of podoplanin in the neurons. Mechanistically, podoplanin promoted the re-entry of neurons into the cell cycle by altering the expression levels of cyclin D1 and cyclin-dependent kinase (CDK) 4, which triggered cell death and neuroinflammation (65). In addition to LPS, the pro-inflammatory cytokine TNF-α (Tumor necrosis factor-α) also upregulates podoplanin in the macrophages (66). However, it is unclear at present whether the podoplanin/CLEC-2-driven inflammatory changes following central nervous system injury are mediated through inflammasomes (NLRP3) or cytokines (IL-18, IL-1β, IL-6). Further studies are needed to explore the mechanisms underlying the thrombo-inflammatory responses induced by podoplanin and CLEC-2 using animal and cellular models.
CLEC-2 in Atherosclerosis
Atherosclerosis refers to the gradual buildup of fats, cholesterol and other substances in the artery walls that eventually forms a plaque. It is not only associated with acute cardiovascular events but also involved in the pathological process of thrombosis after stent implantation. Studies increasingly show that the formation of atherosclerotic plaques is an inflammatory process that involves endothelial cell activation and adhesion of macrophages to the vascular wall, which eventually release large amounts of chemokines and cytokines. Multiple factors such as hypertension, lipid metabolism disorder and delayed wound healing can induce atherosclerotic build-up. In the late stages of atherosclerosis, the plaques rupture at the lesion and promote thrombosis, leading to cardiovascular events.
Thrombosis caused by atherosclerosis is related to platelet aggregation. Case-control trials have shown that elevated plasma sCLEC-2 is an independent risk factor for CHD (67, 68). Furthermore, podoplanin expression in smooth muscle cells and macrophages increases with atherosclerotic progression, and further aggravates the injury (69). The smooth muscle cells also express S100A13, which activate platelets through CLEC-2 independent of podoplanin (32). It has been suggested that S100A13 plays a more important role than podoplanin in early atherosclerotic lesions, given that podoplanin is rare in early atherosclerotic lesions (32). Injuries to vascular endothelium lead to S100A13 exposure, triggering platelet aggregation, and plaque induced ischemia and hypoxia, which in turn leads to inflammation. S100A13 is distributed on the surface of atherosclerosis, and podoplanin is detected in the interior of advanced atherosclerosis, and the different localization of S100A13 and podoplanin may be related to the different effects (69). S100A13 aggravates plaque growth from the outside.
The role of internal podoplanin is controversial. CLEC-2/PDPN-mediated phagocytosis is important for the formation of atherosclerotic plaques in mice, but the distribution may be species-specific, as noted in the same study (20). Podoplanin is present inside of the advanced atherosclerotic lesions rather than on the surface of the lesion, which blocks access to CLEC-2, and podoplanin may play an important role in triggering plaque rupture. In addition, podoplanin expressed on the stromal fibroblasts can also promote cell migration and invasion (70, 71), and induce inflammatory changes (72–74), indicating that the podoplanin/CLEC-2 axis is closely related to atherosclerotic progression (69). Local hypoxia and inflammation in the atherosclerotic plaque induces VEGF-A expression in smooth muscle cells (75), macrophages and endothelial cells (76), which enhances platelet aggregation and plaque erosion via the podoplanin/CLEC-2 axis (77). Furthermore, pro-inflammatory factors released from the intima of the arterial lumen into the blood activate CLEC-2 on the platelets and may trigger thrombosis (78). Patients with stent implantation are perpetually at risk of thrombosis and therefore require long-term dual anti-platelet therapy. Mice with CLEC-2 deficiency require a lower dose of antiplatelet drugs without an increase in the risk of bleeding (36), Therefore, blocking CLEC-2 can prevent stent thrombosis and slow down the progression of atherosclerosis. In addition, studies show that podoplanin is a marker of myocardial injury, and inhibition of podoplanin can accelerate recovery after myocardial infarction (74). Taken together, CLEC-2 and its ligands are promising therapeutic targets for cardiovascular diseases.
CLEC-2 in Deep Venous Thrombosis
Deep venous thrombosis (DVT) is the formation of a blood clot in the deeper veins, usually in the lower extremities, following an aseptic inflammatory response (79) in the vascular endothelium. The endothelial cells in the intima of the blood vessels release Weibel-Palade bodies (WPB) containing cytokines, which mobilize platelets and white blood cells to the vessel wall, resulting in thrombosis (27, 80). The increase in blood pressure and turbulence caused by stenosis decreases the local blood flow, loosens the connection between vascular endothelial cells and increases vascular permeability. The resulting local hypoxia upregulates podoplanin in the vascular walls (81), which induces thrombosis through CLEC-2 (82). Thrombus formation is prevented in the absence of CLEC-2, which reduces platelet aggregation and white blood cell recruitment at the stenosis vessels. Podoplanin elevation after stenosis correlates with an increased risk of thrombosis, and further upregulation of podoplanin during thrombosis aggravates the condition. In addition, the weight and length of thrombus were significantly reduced in animals treated with the anti-podoplanin antibody (80). Up regulation of podoplanin and CLEC-2 is not only the cause of thrombosis, but also may be triggered by neutrophil-mediated inflammatory response. The reduced blood flow caused by stenosis and inducible factors produced by local hypoxia attract and activate neutrophils. In the early stage of DVT, multiple immune cells are recruited to the vessel wall, and platelets further enhance the inflammatory recruitment. Neutrophils recruited to the vascular wall release cytokines (83), further stabilize and promote the formation of thrombosis (27, 84). On the other hand, neutrophils also release metalloproteinases to further promote platelet aggregation and aggravate thrombosis (85). In this process, inflammation and thrombosis promote each other, leading to deep vein thrombosis (86).
Bruton’s tyrosine kinase (Btk) is an important non-receptor signaling kinase involved in platelet aggregation. Several Btk-dependent platelet aggregation pathways such as GPVI-activation by low collagen concentrations, FcgammaRIIA (an ITAM receptor) activation by cross-linking and VWF-stimulated GPIb activation are inhibited in human blood by low nanomolar IC50 concentrations of Btk-inhibitors (87). Besides, Btk is essential for platelet CLEC-2 (88). In heparin-induced thrombocytopenia, Btk inhibitors reduce CLEC-2- and GPIb-mediated platelet activation, monocyte interaction and activation, and neutrophil extracellular trap release (89). In addition, Btk inhibitors likely to reduce the microvascular and venous thrombosis in COVID-19 by blocking platelet CLEC-2, and well reduce thrombosis without an associated increase in bleeding (90), providing a new idea for the selective targeting of thrombotic inflammatory diseases.
CLEC-2 in Septic Thrombosis
The role of podoplanin/CLEC-2 in inflammatory thrombosis was studied using mouse typhoid model by linking infection, inflammation and thrombosis (91). Podoplanin expressed on macrophages can activate the toll like receptor 4 (TLR4) signaling pathway in vitro (92, 93). Hitchcock et al. further showed that up-regulation of podoplanin in CLEC-2-dependent thrombosis is associated with TLR4/Interferon (IFN)-γ-dependent inflammation in vivo, and CLEC-2 is a key participant in this pathway (82). Bacteria in the liver cause a large number of macrophages depending on TLR4 and IFN - γ to aggregate, and ultimately damage the vascular endothelium due to the combined effects of bacteremia, infection, inflammation and cell migration. The platelets extravasating from the blood vessels are exposed to monocytes and other cells overexpressing podoplanin, and are activated via CLEC-2. Thus, inflammation triggered by bacterial infection directly induces thrombosis through the CLEC-2 pathway, which can be inhibited by knocking out CLEC-2 on the platelets or with anti-podoplanin antibodies.
A study on the septicemia mouse model showed that platelets limited the severity of symptoms through CLEC-2 signaling independent of thrombosis. The protective effect of CLEC-2 in septicemia was partly mediated by its interaction with the podoplanin expressed on inflammatory macrophages, which limited the infiltration of immune cells into the infected site by controlling cytokine/chemokine secretion, and mitigated organ damage. In addition, podoplanin reduces bacteremia by mediating the role of inflammatory macrophages. Podoplanin expressed in inflammatory macrophages not only regulates platelet aggregation, but also regulates the secretion of TNF-α in macrophages. Podoplanin antibody, mAb 8.1.1, regulates inflammatory response after sepsis. In vitro, mAb 8.1.1 reduced TNF-α secretion. However, in vivo, although pro-inflammatory cytokines and chemokines increased, the injection of anti-podoplanin antibody regulated the inflammatory response and immune cell infiltration during sepsis. Platelet CLEC-2 restricts the symptoms of sepsis by controlling multiple factors, including monocyte/macrophage migration, inflammatory mediators, bacteremia and organ damage, suggesting a complex role of platelets in regulating innate immunity against infection (94).
Some bacteria actively dissociate the thrombus to spread in the bloodstream, which suggests that inflammation-driven thrombosis is a novel strategy for bacterial capture and clearance (95). However, studies on animal models show that thrombosis controls the inflammatory diffusion in the early stage of infection (82, 94). During inflammation, platelet activation and thrombosis may adversely affect the host by inducing disseminated intravascular coagulation (DIC), and extensive microthrombosis occurs between arterioles and venules. Furthermore, if the thrombus exceeds a certain size, it may rupture and lead to other complications such as cerebral artery infarction or pulmonary artery embolism.
CLEC-2 in Cancer Thrombosis
The interaction between CLEC-2 and podoplanin-expressing tumor cells promote angiogenesis, tumor growth and metastasis (96). Mice bearing lung tumors show extensive thrombosis, which can be alleviated by blocking CLEC-2. In addition, inhibition of CLEC-2 also reduced plasma cytokine levels, improved cachexia and prolonged survival of tumor-bearing mice (97). In the process of trying to deduce the mechanism of cancer-mediated inflammation, researchers found that podoplanin was up-regulated in the venous wall. Therefore, CLEC-2 may play an important role in tumor-induced thromboinflammation, and chronic long-term exposure to inflammatory cytokines induces podoplanin expression (98).
Conclusion
The podoplanin/CLEC-2 axis promotes aseptic and bacterial inflammation, maintains vascular endothelial integrity and protects against microthrombosis. Although CLEC-2 is a potential target of anti-inflammatory drugs, targeting the CLEC-2 pathway may affect neovascularization during infection. Therefore, CLEC-2 blockade should only be considered as a short-term option. To better understand these diseases, relevant information is summarized in Table 1. Through the study of many diseases, it is obvious that it is very important to find the balance of CLEC-2 and its ligand in the treatment and application of thromboinflammation. Whether the related findings of CLEC-2 in these thromboinflammatory diseases can be widely applied to other diseases, and whether the involved pathways can specifically target to improve the status of thromboinflammatory diseases are still the focus of our consideration and research. However, the important effects of CLEC-2 and its ligands on thrombosis and inflammation cannot be ignored, and the research on the mechanism of CLEC-2 and related drugs will still be the future research hotspot in this field.
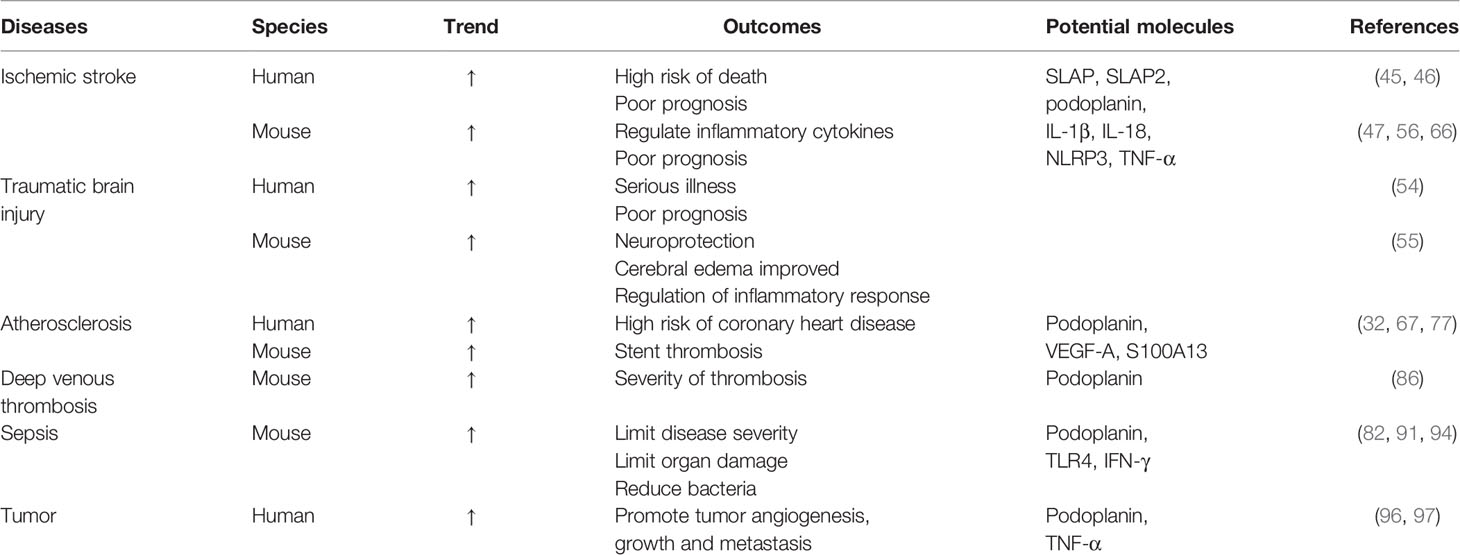
Table 1 CLEC-2 in thromboinflammation.
Author Contributions
DM drafted and edited the manuscript. ML and BL edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Key Supporting Discipline-Neurology (2019-fc-04) in Jiaxing, Jiaxing Public Welfare Research Program (2021AY30022), and the “Morning Star” project (2020-QMX-23) of the Affiliated Hospital of Jiaxing University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Professor Xu Xingshun from the First Affiliated Hospital of Soochow University for his support and critical evaluation of the manuscript. Figure 1 was created with the aid of Servier Medical Art (https://smart.servier.com).
References
1. Colonna M, Samaridis J, Angman L. Molecular Characterization of Two Novel C-type Lectin-Like Receptors, One of Which is Selectively Expressed in Human Dendritic Cells. Eur J Immunol (2000) 30(2):697–704. doi: 10.1002/1521-4141(200002)30:2<697::AID-IMMU697>3.0.CO;2-M
2. Sobanov Y, Bernreiter A, Derdak S, Mechtcheriakova D, Schweighofer B, Duchler M, et al. A Novel Cluster of Lectin-Like Receptor Genes Expressed in Monocytic, Dendritic and Endothelial Cells Maps Close to the NK Receptor Genes in the Human Nk Gene Complex. Eur J Immunol (2001) 31(12):3493–503. doi: 10.1002/1521-4141(200112)31:12<3493::AID-IMMU3493gt;3.0.CO;2-9
3. Brown GD, Willment JA, Whitehead L. C-Type Lectins in Immunity and Homeostasis. Nat Rev Immunol (2018) 18(6):374–89. doi: 10.1038/s41577-018-0004-8
4. Chaipan C, Steffen I, Tsegaye TS, Bertram S, Glowacka I, Kato Y, et al. Incorporation of Podoplanin Into HIV Released From Hek-293t Cells, But Not PBMC, is Required for Efficient Binding to the Attachment Factor Clec-2. Retrovirology (2010) 7:47. doi: 10.1186/1742-4690-7-47
5. Lowe KL, Navarro-Nunez L, Benezech C, Nayar S, Kingston BL, Nieswandt B, et al. The Expression of Mouse CLEC-2 on Leucocyte Subsets Varies According to Their Anatomical Location and Inflammatory State. Eur J Immunol (2015) 45(9):2484–93. doi: 10.1002/eji.201445314
6. Fu J, Xia L. CLEC-2 and Podoplanin, Partners Again. Blood (2016) 127(13):1629–30. doi: 10.1182/blood-2016-02-695445.
7. Suzuki-Inoue K. Essential In Vivo Roles of the Platelet Activation Receptor CLEC-2 in Tumour Metastasis, Lymphangiogenesis and Thrombus Formation. J Biochem (2011) 150(2):127–32. doi: 10.1093/jb/mvr079
8. Mir Seyed Nazari P, Riedl J, Pabinger I, Ay C. The Role of Podoplanin in Cancer-Associated Thrombosis. Thromb Res (2018) 164(Suppl 1):S34–s9. doi: 10.1016/j.thromres.2018.01.020
9. Lowe KL, Finney BA, Deppermann C, Hagerling R, Gazit SL, Frampton J, et al. Podoplanin and CLEC-2 Drive Cerebrovascular Patterning and Integrity During Development. Blood (2015) 125(24):3769–77. doi: 10.1182/blood-2014-09-603803
10. Rayes J, Watson SP, Nieswandt B. Functional Significance of the Platelet Immune Receptors GPVI and CLEC-2. J Clin Invest (2019) 129(1):12–23. doi: 10.1172/JCI122955
11. Huang TF, Liu CZ, Yang SH. Aggretin, a Novel Platelet-Aggregation Inducer From Snake (Calloselasma Rhodostoma) Venom, Activates Phospholipase C by Acting as a Glycoprotein Ia/Iia Agonist. Biochem J (1995) 309(Pt 3):1021–7. doi: 10.1042/bj3091021
12. Suzuki-Inoue K, Ozaki Y, Kainoh M, Shin Y, Wu Y, Yatomi Y, et al. Rhodocytin Induces Platelet Aggregation by Interacting With Glycoprotein Ia/Iia (Gpia/Iia, Integrin Alpha 2beta 1). Involvement of GPIa/IIa-associated Src and Protein Tyrosine Phosphorylation. J Biol Chem (2001) 276(2):1643–52. doi: 10.1074/jbc.M006191200
13. Suzuki-Inoue K, Fuller GL, Garcia A, Eble JA, Pohlmann S, Inoue O, et al. A Novel Syk-Dependent Mechanism of Platelet Activation by the C-type Lectin Receptor Clec-2. Blood (2006) 107(2):542–9. doi: 10.1182/blood-2005-05-1994
14. Chang CH, Chung CH, Hsu CC, Huang TY, Huang TF. A Novel Mechanism of Cytokine Release in Phagocytes Induced by Aggretin, a Snake Venom C-type Lectin Protein, Through CLEC-2 Ligation. J Thromb Haemostasis JTH (2010) 8(11):2563–70. doi: 10.1111/j.1538-7836.2010.04045.x
15. Hardy AT, Palma-Barqueros V, Watson SK, Malcor JD, Eble JA, Gardiner EE, et al. Significant Hypo-Responsiveness to GPVI and CLEC-2 Agonists in Pre-Term and Full-Term Neonatal Platelets and Following Immune Thrombocytopenia. Thromb Haemostasis (2018) 118(6):1009–20. doi: 10.1055/s-0038-1646924
16. Navarro-Nunez L, Langan SA, Nash GB, Watson SP. The Physiological and Pathophysiological Roles of Platelet Clec-2. Thromb Haemostasis (2013) 109(6):991–8. doi: 10.1160/th13-01-0060
17. Watson AA, Brown J, Harlos K, Eble JA, Walter TS, O’Callaghan CA. The Crystal Structure and Mutational Binding Analysis of the Extracellular Domain of the Platelet-Activating Receptor Clec-2. J Biol Chem (2007) 282(5):3165–72. doi: 10.1074/jbc.M610383200
18. Watson SP, Herbert JM, Pollitt AY. GPVI and CLEC-2 in Hemostasis and Vascular Integrity. J Thromb haemostasis JTH (2010) 8(7):1456–67. doi: 10.1111/j.1538-7836.2010.03875.x
19. Tamura S, Suzuki-Inoue K, Tsukiji N, Shirai T, Sasaki T, Osada M, et al. Podoplanin-Positive Periarteriolar Stromal Cells Promote Megakaryocyte Growth and Proplatelet Formation in Mice by CLEC-2. Blood (2016) 127(13):1701–10. doi: 10.1182/blood-2015-08-663708
20. Kerrigan AM, Dennehy KM, Mourão-Sá D, Faro-Trindade I, Willment JA, Taylor PR, et al. CLEC-2 is a Phagocytic Activation Receptor Expressed on Murine Peripheral Blood Neutrophils. J Immunol (2009) 182(7):4150–7. doi: 10.4049/jimmunol.0802808
21. Chaipan C, Soilleux EJ, Simpson P, Hofmann H, Gramberg T, Marzi A, et al. Dc-SIGN and CLEC-2 Mediate Human Immunodeficiency Virus Type 1 Capture by Platelets. J Virol (2006) 80(18):8951–60. doi: 10.1128/JVI.00136-06
22. Manne BK, Getz TM, Hughes CE, Alshehri O, Dangelmaier C, Naik UP, et al. Fucoidan is a Novel Platelet Agonist for the C-type Lectin-Like Receptor 2 (Clec-2). J Biol Chem (2013) 288(11):7717–26. doi: 10.1074/jbc.M112.424473
23. Alshehri OM, Montague S, Watson S, Carter P, Sarker N, Manne BK, et al. Activation of Glycoprotein VI (GPVI) and C-type Lectin-Like Receptor-2 (Clec-2) Underlies Platelet Activation by Diesel Exhaust Particles and Other Charged/Hydrophobic Ligands. Biochem J (2015) 468(3):459–73. doi: 10.1042/BJ20150192
24. Fuller GL, Williams JA, Tomlinson MG, Eble JA, Hanna SL, Pohlmann S, et al. The C-type Lectin Receptors CLEC-2 and Dectin-1, But Not Dc-Sign, Signal Via a Novel YXXL-Dependent Signaling Cascade. J Biol Chem (2007) 282(17):12397–409. doi: 10.1074/jbc.M609558200
25. Breiteneder-Geleff S, Matsui K, Soleiman A, Meraner P, Poczewski H, Kalt R, et al. Podoplanin, Novel 43-Kd Membrane Protein of Glomerular Epithelial Cells, is Down-Regulated in Puromycin Nephrosis. Am J Pathol (1997) 151(4):1141–52.
26. Quintanilla M, Montero-Montero L, Renart J, Martin-Villar E. Podoplanin in Inflammation and Cancer. Int J Mol Sci (2019) 20(3):707. doi: 10.3390/ijms20030707
27. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, Neutrophils, and Platelets Cooperate to Initiate and Propagate Venous Thrombosis in Mice In Vivo. J Exp Med (2012) 209(4):819–35. doi: 10.1084/jem.20112322
28. Schacht V, Ramirez MI, Hong YK, Hirakawa S, Feng D, Harvey N, et al. T1alpha/Podoplanin Deficiency Disrupts Normal Lymphatic Vasculature Formation and Causes Lymphedema. EMBO J (2003) 22(14):3546–56. doi: 10.1093/emboj/cdg342
29. Asai J, Hirakawa S, Sakabe J, Kishida T, Wada M, Nakamura N, et al. Platelets Regulate the Migration of Keratinocytes Via Podoplanin/CLEC-2 Signaling During Cutaneous Wound Healing in Mice. Am J Pathol (2016) 186(1):101–8. doi: 10.1016/j.ajpath.2015.09.007
30. Suzuki-Inoue K, Tsukiji N, Shirai T, Osada M, Inoue O, Ozaki Y. Platelet CLEC-2: Roles Beyond Hemostasis. Semin Thromb Hemostasis (2018) 44(2):126–34. doi: 10.1055/s-0037-1604090
31. Astarita JL, Cremasco V, Fu J. The CLEC-2-podoplanin Axis Controls the Contractility of Fibroblastic Reticular Cells and Lymph Node Microarchitecture. Nat Immunol (2015) 16(1):75–84. doi: 10.1038/ni.3035
32. Inoue O, Hokamura K, Shirai T, Osada M, Tsukiji N, Hatakeyama K, et al. Vascular Smooth Muscle Cells Stimulate Platelets and Facilitate Thrombus Formation Through Platelet Clec-2: Implications in Atherothrombosis. PLoS One (2015) 10(9):e0139357. doi: 10.1371/journal.pone.0139357
33. Bourne JH, Colicchia M, Di Y, Martin E, Slater A, Roumenina LT, et al. Heme Induces Human and Mouse Platelet Activation Through C-type-lectin-like Receptor-2. Haematologica (2021) 106(2):626–9. doi: 10.3324/haematol.2020.246488
34. May F, Hagedorn I, Pleines I, Bender M, Vogtle T, Eble J, et al. CLEC-2 is an Essential Platelet-Activating Receptor in Hemostasis and Thrombosis. Blood (2009) 114(16):3464–72. doi: 10.1182/blood-2009-05-222273
35. Suzuki-Inoue K, Inoue O, Ding G, Nishimura S, Hokamura K, Eto K, et al. Essential In Vivo Roles of the C-type Lectin Receptor Clec-2: Embryonic/Neonatal Lethality of CLEC-2-deficient Mice by Blood/Lymphatic Misconnections and Impaired Thrombus Formation of CLEC-2-deficient Platelets. J Biol Chem (2010) 285(32):24494–507. doi: 10.1074/jbc.M110.130575
36. Bender M, May F, Lorenz V, Thielmann I, Hagedorn I, Finney BA, et al. Combined In Vivo Depletion of Glycoprotein VI and C-type Lectin-Like Receptor 2 Severely Compromises Hemostasis and Abrogates Arterial Thrombosis in Mice. Arteriosclerosis thrombosis Vasc Biol (2013) 33(5):926–34. doi: 10.1161/atvbaha.112.300672
37. van Eeuwijk JM, Stegner D, Lamb DJ, Kraft P, Beck S, Thielmann I, et al. The Novel Oral Syk Inhibitor, Bl1002494, Protects Mice From Arterial Thrombosis and Thromboinflammatory Brain Infarction. Arteriosclerosis Thrombosis Vasc Biol (2016) 36(6):1247–53. doi: 10.1161/atvbaha.115.306883
38. Rayes J, Jadoui S, Lax S, Gros A, Wichaiyo S, Ollivier V, et al. The Contribution of Platelet Glycoprotein Receptors to Inflammatory Bleeding Prevention is Stimulus and Organ Dependent. Haematologica (2018) 103(6):e256–e8. doi: 10.3324/haematol.2017.182162
39. Fei M, Wang H, Zhou M, Deng C, Zhang L, Han Y. Podoplanin Influences the Inflammatory Phenotypes and Mobility of Microglia in Traumatic Brain Injury. Biochem Biophys Res Commun (2019) 523(2):361–7. doi: 10.1016/j.bbrc.2019.12.003
40. Sung PS, Hsieh SL. CLEC2 and CLEC5A: Pathogenic Host Factors in Acute Viral Infections. Front Immunol (2019) 10:2867. doi: 10.3389/fimmu.2019.02867
41. De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in Stroke Brain Damage. Stroke (2016) 47(4):1165–72. doi: 10.1161/STROKEAHA.115.011238
42. Al-Tamimi M, Gardiner EE, Thom JY, Shen Y, Cooper MN, Hankey GJ, et al. Soluble Glycoprotein VI is Raised in the Plasma of Patients With Acute Ischemic Stroke. Stroke (2011) 42(2):498–500. doi: 10.1161/strokeaha.110.602532
43. Wurster T, Poetz O, Stellos K, Kremmer E, Melms A, Schuster A, et al. Plasma Levels of Soluble Glycoprotein VI (sGPVI) are Associated With Ischemic Stroke. Platelets (2013) 24(7):560–5. doi: 10.3109/09537104.2012.746455
44. Skarpengland T, Skjelland M, Kong XY, Skagen K, Holm S, Otterdal K, et al. Increased Levels of Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 in Ischemic Stroke and Transient Ischemic Attack. J Am Heart Assoc (2018) 7(2):e006479. doi: 10.1161/jaha.117.006479
45. Zhang X, Zhang W, Wu X, Li H, Zhang C, Huang Z, et al. Prognostic Significance of Plasma Clec-2 (C-Type Lectin-Like Receptor 2) in Patients With Acute Ischemic Stroke. Stroke (2018), STROKEAHA118022563. doi: 10.1161/STROKEAHA.118.022563
46. Wu X, Zhang W, Li H, You S, Shi J, Zhang C, et al. Plasma CLEC-2 as a Predictor of Death and Vascular Events in Patients With Acute Ischemic Stroke. Eur J Neurol (2019) 26(10):1334–40. doi: 10.1111/ene.13984
47. Meng D, Ma X, Li H, Wu X, Cao Y, Miao Z. A Role of the Podoplanin-CLEC-2 Axis in Promoting Inflammatory Response After Ischemic Stroke in Mice. Neurotox Res (2021) 39(2):477–88. doi: 10.1007/s12640-020-00295-w
48. Kaur P, Sharma S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr Neuropharmacol (2018) 16(8):1224–38. doi: 10.2174/1570159X15666170613083606
49. Kelso ML, Gendelman HE. Bridge Between Neuroimmunity and Traumatic Brain Injury. Curr Pharm Design (2014) 20(26):4284–98. doi: 10.2174/13816128113196660653
50. Martin GE, Pugh AM, Moran R, Veile R, Friend LA, Pritts TA, et al. Microvesicles Generated Following Traumatic Brain Injury Induce Platelet Dysfunction Via Adenosine Diphosphate Receptor. J Trauma acute Care Surg (2019) 86(4):592–600. doi: 10.1097/TA.0000000000002171
51. Guillotte AR, Herbert JP, Madsen R, Hammer RD, Litofsky NS. Effects of Platelet Dysfunction and Platelet Transfusion on Outcomes in Traumatic Brain Injury Patients. Brain Injury (2018) 32(13-14):1849–57. doi: 10.1080/02699052.2018.1536805
52. Verni CC, Davila A Jr., Balian S, Sims CA, Diamond SL. Platelet Dysfunction During Trauma Involves Diverse Signaling Pathways and an Inhibitory Activity in Patient-Derived Plasma. J Trauma Acute Care Surg (2019) 86(2):250–9. doi: 10.1097/TA.0000000000002140
53. Skorko A, Mumford A, Thomas M, Pickering AE, Greenwood R, Griffiths E, et al. Platelet Dysfunction After Out of Hospital Cardiac Arrest. Results From Pohcar: A Prospective Observational, Cohort Study. Resuscitation (2019) 136:105–11. doi: 10.1016/j.resuscitation.2019.01.025
54. Guo M, Zhang H, Lv QW, Huang HB, Shen LJ. Higher Plasma C-Type Lectin-Like Receptor 2 Concentrations for Prediction of Higher Risk of 30-Day Mortality in Isolated Severe Blunt Traumatic Brain Injury. Clinica Chimica Acta; Int J Clin Chem (2019) 496:1–6. doi: 10.1016/j.cca.2019.06.014
55. Gao C, Wang H, Wang T, Luo C. Platelet Regulates Neuroinflammation and Restores Blood-Brain Barrier Integrity in a Mouse Model of Traumatic Brain Injury. Neurochem (2020) 154(2):190–204. doi: 10.1111/jnc.14983
56. Cherpokova D, Bender M. Slap/Slap2 Prevent Excessive Platelet (Hem)Itam Signaling in Thrombosis and Ischemic Stroke in Mice. Blood (2015) 125(1):185–94. doi: 10.1182/blood-2014-06-580597
57. Kotani M, Osanai T, Tajima Y, Kato H, Imada M, Kaneda H, et al. Identification of Neuronal Cell Lineage-Specific Molecules in the Neuronal Differentiation of P19 Ec Cells and Mouse Central Nervous System. J Neurosci Res (2002) 67(5):595–606. doi: 10.1002/jnr.10150
58. Kotani M, Tajima Y, Osanai T, Irie A, Iwatsuki K, Kanai-Azuma M, et al. Complementary DNA Cloning and Characterization of RANDAM-2, a Type I Membrane Molecule Specifically Expressed on Glutamatergic Neuronal Cells in the Mouse Cerebrum. J Neurosci Res (2003) 73(5):603–13. doi: 10.1002/jnr.10696
59. Cicvaric A, Yang J, Krieger S, Khan D, Kim EJ, Dominguez-Rodriguez M, et al. The Brain-Tumor Related Protein Podoplanin Regulates Synaptic Plasticity and Hippocampus-Dependent Learning and Memory. Ann Med (2016) 48(8):652–68. doi: 10.1080/07853890.2016.1219455
60. Stellos K, Panagiota V, Kogel A, Leyhe T, Gawaz M, Laske C. Predictive Value of Platelet Activation for the Rate of Cognitive Decline in Alzheimer’s Disease Patients. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2010) 30(11):1817–20. doi: 10.1038/jcbfm.2010.140
61. Chaitanya GV, Omura S, Sato F, Martinez NE, Minagar A, Ramanathan M, et al. Inflammation Induces Neuro-Lymphatic Protein Expression in Multiple Sclerosis Brain Neurovasculature. J Neuroinflamm (2013) 10:125. doi: 10.1186/1742-2094-10-125
62. Glezer I, Simard AR, Rivest S. Neuroprotective Role of the Innate Immune System by Microglia. Neuroscience (2007) 147(4):867–83. doi: 10.1016/j.neuroscience.2007.02.055
63. Block ML, Zecca L, Hong JS. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat Rev Neurosci (2007) 8(1):57–69. doi: 10.1038/nrn2038
64. Kolar K, Freitas-Andrade M, Bechberger JF, Krishnan H, Goldberg GS, Naus CC, et al. Podoplanin: A Marker for Reactive Gliosis in Gliomas and Brain Injury. J Neuropathol Exp Neurol (2015) 74(1):64–74. doi: 10.1097/nen.0000000000000150
65. Song Y, Shen J, Lin Y, Shen J, Wu X, Yan Y, et al. Up-Regulation of Podoplanin Involves in Neuronal Apoptosis in LPS-induced Neuroinflammation. Cell Mol Neurobiol (2014) 34(6):839–49. doi: 10.1007/s10571-014-0060-y
66. Kerrigan AM, Navarro-Nunez L, Pyz E, Finney BA, Willment JA, Watson SP, et al. Podoplanin-Expressing Inflammatory Macrophages Activate Murine Platelets Via Clec-2. J Thromb Haemostasis JTH (2012) 10(3):484–6. doi: 10.1111/j.1538-7836.2011.04614.x
67. Fei M, Xiang L, Chai X, Jin J, You T, Zhao Y, et al. Plasma Soluble C-Type Lectin-Like Receptor-2 is Associated With the Risk of Coronary Artery Disease. Front Med (2019) 14(1):81–90. doi: 10.1007/s11684-019-0692-x
68. Inoue O, Osada M, Nakamura J, Kazama F, Shirai T, Tsukiji N, et al. Soluble CLEC-2 is Generated Independently of ADAM10 and is Increased in Plasma in Acute Coronary Syndrome: Comparison With Soluble Gpvi. Int J Hematol (2019) 110(3):285–94. doi: 10.1007/s12185-019-02680-4
69. Hatakeyama K, Kaneko MK, Kato Y, Ishikawa T, Nishihira K, Tsujimoto Y, et al. Podoplanin Expression in Advanced Atherosclerotic Lesions of Human Aortas. Thromb Res (2012) 129(4):e70–6. doi: 10.1016/j.thromres.2012.01.003
70. Hoshino A, Ishii G, Ito T, Aoyagi K, Ohtaki Y, Nagai K, et al. Podoplanin-Positive Fibroblasts Enhance Lung Adenocarcinoma Tumor Formation: Podoplanin in Fibroblast Functions for Tumor Progression. Cancer Res (2011) 71(14):4769–79. doi: 10.1158/0008-5472.CAN-10-3228
71. Kawase A, Ishii G, Nagai K, Ito T, Nagano T, Murata Y, et al. Podoplanin Expression by Cancer Associated Fibroblasts Predicts Poor Prognosis of Lung Adenocarcinoma. Int J Cancer (2008) 123(5):1053–9. doi: 10.1002/ijc.23611
72. Groger M, Loewe R, Holnthoner W, Embacher R, Pillinger M, Herron GS, et al. Il-3 Induces Expression of Lymphatic Markers Prox-1 and Podoplanin in Human Endothelial Cells. J Immunol (2004) 173(12):7161–9. doi: 10.4049/jimmunol.173.12.7161
73. Suzuki H, Kato Y, Kaneko MK, Okita Y, Narimatsu H, Kato M. Induction of Podoplanin by Transforming Growth Factor-Beta in Human Fibrosarcoma. FEBS Lett (2008) 582(2):341–5. doi: 10.1016/j.febslet.2007.12.028
74. Cimini M, Garikipati VNS, de Lucia C, Cheng Z, Wang C, Truongcao MM, et al. Podoplanin Neutralization Improves Cardiac Remodeling and Function After Acute Myocardial Infarction. JCI Insight (2019) 5(15):e126967. doi: 10.1172/jci.insight.126967
75. Ferrara N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocrine Rev (2004) 25(4):581–611. doi: 10.1210/er.2003-0027
76. Chen YX, Nakashima Y, Tanaka K, Shiraishi S, Nakagawa K, Sueishi K. Immunohistochemical Expression of Vascular Endothelial Growth Factor/Vascular Permeability Factor in Atherosclerotic Intimas of Human Coronary Arteries. Arteriosclerosis Thrombosis Vasc Biol (1999) 19(1):131–9. doi: 10.1161/01.atv.19.1.131
77. Furukoji E, Yamashita A, Nakamura K, Hirai T, Asada Y. Podoplanin Expression on Endothelial Cells Promotes Superficial Erosive Injury and Thrombus Formation in Rat Carotid Artery: Implications for Plaque Erosion. Thromb Res (2019) 183:76–9. doi: 10.1016/j.thromres.2019.10.015
78. Siddiqi OK, Faxon DP. Very Late Stent Thrombosis: Current Concepts. Curr Opin Cardiol (2012) 27(6):634–41. doi: 10.1097/HCO.0b013e3283587c7e
79. Andre P, Denis CV, Ware J, Saffaripour S, Hynes RO, Ruggeri ZM, et al. Platelets Adhere to and Translocate on Von Willebrand Factor Presented by Endothelium in Stimulated Veins. Blood (2000) 96(10):3322–8. doi: 10.1182/blood.V96.10.3322.h8003322_3322_3328
80. Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, et al. Von Willebrand Factor-Mediated Platelet Adhesion is Critical for Deep Vein Thrombosis in Mouse Models. Blood (2011) 117(4):1400–7. doi: 10.1182/blood-2010-05-287623
81. Kolenda J, Jensen SS, Aaberg-Jessen C, Christensen K, Andersen C, Brunner N, et al. Effects of Hypoxia on Expression of a Panel of Stem Cell and Chemoresistance Markers in Glioblastoma-Derived Spheroids. J Neuro-Oncol (2011) 103(1):43–58. doi: 10.1007/s11060-010-0357-8
82. Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, et al. Inflammation Drives Thrombosis After Salmonella Infection Via CLEC-2 on Platelets. J Clin Invest (2015) 125(12):4429–46. doi: 10.1172/jci79070
83. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (New York NY) (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
84. Fuchs TA, Brill A, Wagner DD. Neutrophil Extracellular Trap (Net) Impact on Deep Vein Thrombosis. Arteriosclerosis Thrombosis Vasc Biol (2012) 32(8):1777–83. doi: 10.1161/ATVBAHA.111.242859
85. Lee RH, Bergmeier W. Platelet Immunoreceptor Tyrosine-Based Activation Motif (ITAM) and Hemitam Signaling and Vascular Integrity in Inflammation and Development. J Thromb Haemostasis JTH (2016) 14(4):645–54. doi: 10.1111/jth.13250
86. Payne H, Ponomaryov T, Watson SP. Mice With a Deficiency in CLEC-2 are Protected Against Deep Vein Thrombosis. Blood (2017) 129(14):2013–20. doi: 10.1182/blood-2016-09-742999
87. von Hundelshausen P, Siess W. Bleeding by Bruton Tyrosine Kinase-Inhibitors: Dependency on Drug Type and Disease. Cancers (Basel) (2021) 13(5):1103. doi: 10.3390/cancers13051103
88. Nicolson PLR, Nock SH, Hinds J, Garcia-Quintanilla L, Smith CW, Campos J, et al. Low Dose Btk Inhibitors Selectively Block Platelet Activation by CLEC-2. Haematologica (2020) 106(1):208–19. doi: 10.3324/haematol.2019.218545
89. von Hundelshausen P, Lorenz R, Siess W, Weber C. Vaccine-Induced Immune Thrombotic Thrombocytopenia (Vitt): Targeting Pathomechanisms With Bruton Tyrosine Kinase Inhibitors. Thromb Haemostasis (2021). doi: 10.1055/a-1481-3039
90. Nicolson PL, Welsh JD, Chauhan A, Thomas MR, Kahn ML, Watson SP. A Rationale for Blocking Thromboinflammation in COVID-19 With Btk Inhibitors. Platelets (2020) 31(5):685–90. doi: 10.1080/09537104.2020.1775189
91. Vazquez-Torres A, Vallance BA, Bergman MA, Finlay BB, Cookson BT, Jones-Carson J, et al. Toll-Like Receptor 4 Dependence of Innate and Adaptive Immunity to Salmonella: Importance of the Kupffer Cell Network. J Immunol (2004) 172(10):6202–8. doi: 10.4049/jimmunol.172.10.6202
92. Hou TZ, Bystrom J, Sherlock JP, Qureshi O, Parnell SM, Anderson G, et al. A Distinct Subset of Podoplanin (Gp38) Expressing F4/80+ Macrophages Mediate Phagocytosis and are Induced Following Zymosan Peritonitis. FEBS Lett (2010) 584(18):3955–61. doi: 10.1016/j.febslet.2010.07.053
93. Changming W, Xin L, Hua T, Shikun W, Qiong X, Zhigeng Z, et al. Monocytes can be Induced to Express Lymphatic Phenotypes. Lymphology (2011) 44(2):48–53.
94. Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, et al. The Podoplanin-CLEC-2 Axis Inhibits Inflammation in Sepsis. Nat Commun (2017) 8(1):2239. doi: 10.1038/s41467-017-02402-6
95. Engelmann B, Massberg S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat Rev Immunol (2013) 13(1):34–45. doi: 10.1038/nri3345
96. Takagi S, Sato S, Oh-hara T, Takami M, Koike S, Mishima Y, et al. Platelets Promote Tumor Growth and Metastasis Via Direct Interaction Between Aggrus/Podoplanin and CLEC-2. PLoS One (2013) 8(8):e73609. doi: 10.1371/journal.pone.0073609
97. Shirai T, Inoue O, Tamura S, Tsukiji N, Sasaki T, Endo H, et al. C-Type Lectin-Like Receptor 2 Promotes Hematogenous Tumor Metastasis and Prothrombotic State in Tumor-Bearing Mice. Blood (2017) 15(3):513–25. doi: 10.1111/jth.13604
Keywords: CLEC-2, podoplanin, thrombosis, inflammation, thromboinflammation
Citation: Meng D, Luo M and Liu B (2021) The Role of CLEC-2 and Its Ligands in Thromboinflammation. Front. Immunol. 12:688643. doi: 10.3389/fimmu.2021.688643
Received: 31 March 2021; Accepted: 24 May 2021;
Published: 09 June 2021.
Edited by:
Fabrice Cognasse, Groupe Sur L’immunité Des Muqueuses Et Agents Pathogènes (GIMAP), FranceReviewed by:
Julie Rayes, University of Birmingham, United KingdomPhilipp von Hundelshausen, Ludwig Maximilian University of Munich, Germany
Copyright © 2021 Meng, Luo and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Man Luo, TE1ST01BTkAxNjMuY29t; Beibei Liu, bGl1YmVpYmVpMDkyMEAxNjMuY29t