 Yuyue Zuo
Yuyue Zuo Guo-Min Deng
Guo-Min Deng- Department of Rheumatology and Immunology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Bone erosion is one of the primary features of inflammatory arthritis and is caused by excessive differentiation and activation of osteoclasts. Fc gamma receptors (FcγRs) have been implicated in osteoclastogenesis. Our recent studies demonstrate that joint-deposited lupus IgG inhibited RANKL-induced osteoclastogenesis. FcγRI is required for RANKL-induced osteoclastogenesis and lupus IgG-induced signaling transduction. We reviewed the results of studies that analyzed the association between FcγRs and bone erosion in inflammatory arthritis. The analysis revealed the dual roles of FcγRs in bone destruction in inflammatory arthritis. Thus, IgG/FcγR signaling molecules may serve as potential therapeutic targets against bone erosion.
Introduction
Inflammatory arthritis is a group of diseases characterized by joint inflammation and bone damage. About 0.1% of adults develop inflammatory arthritis annually (1). Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by progressive synovitis and bone destruction, causing irreversible joint damage and disability (1–4). Bone erosion is the central hallmark of RA in ultrasonography identification (5, 6). Anti-citrullinated protein antibodies (ACPAs) are considered to be among the leading risk factors for bone destruction in RA (7). Ankylosing spondylitis (AS) and psoriatic arthritis (PsA) are other common inflammatory arthritis diseases with bone destruction (8, 9).
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by multi-organ tissue damage and high levels of autoantibodies in the serum (10). Arthritis is a common clinical manifestation with a prevalence of 69 to 95% in patients with SLE (11). However, only 4 to 6% of patients with SLE arthritis display bone erosion on plain radiographs (12–14). As to ACPA positive SLE patients, which are also called rhupus patients, they often overlapped clinical features and fulfilled American College of Rheumatology (ACR) criteria for RA classification (15, 16). It is still unclear why lupus arthritis without ACPA lacks bone destruction. Recently, FcγRs have been reported to exert a regulatory effect on osteoclastogenesis (17–25). Our recent study demonstrated that joint-deposited lupus IgG triggered arthritis without bone erosion in mice and lupus IgG inhibited osteoclastogenesis induced by receptor activator of nuclear factor kappa-B ligand (RANKL). FcγRI exerted an inhibitory effect of lupus IgG on RANKL-induced osteoclastogenesis (26). Our study suggests that FcγR could function as critical regulators of inflammatory arthritis.
Here, we review the published studies and demonstrate the association between the FcγR and bone erosion in inflammatory arthritis.
Fcγ Receptor Family
FcγRs are receptors for the constant (Fc) region of IgG; these are expressed widely on the surface of immune cells, including monocytes, macrophages, neutrophils, dendritic cells (DCs), B cells, natural killer cells, and mast cells. Four different classes of FcγRs have been identified in mice, namely FcγRI, FcγRIIB, FcγRIII, and FcγRIV (27–29). The human and primate FcγR classifications are more complex. Humans possess six classic FcγRs with different IgG binding capacity and downstream signaling pathways: FcγRI (CD64), FcγRIIA (CD32A), FcγRIIB (CD32B), FcγRIIC (CD32C), FcγRIIIA (CD16A), and FcγRIIIB (CD16B), which are encoded by genes FCGR1A, FCGR2A, FCGR2B, FCGR2C, FCGR3A, and FCGR3B, respectively (Figure 1).
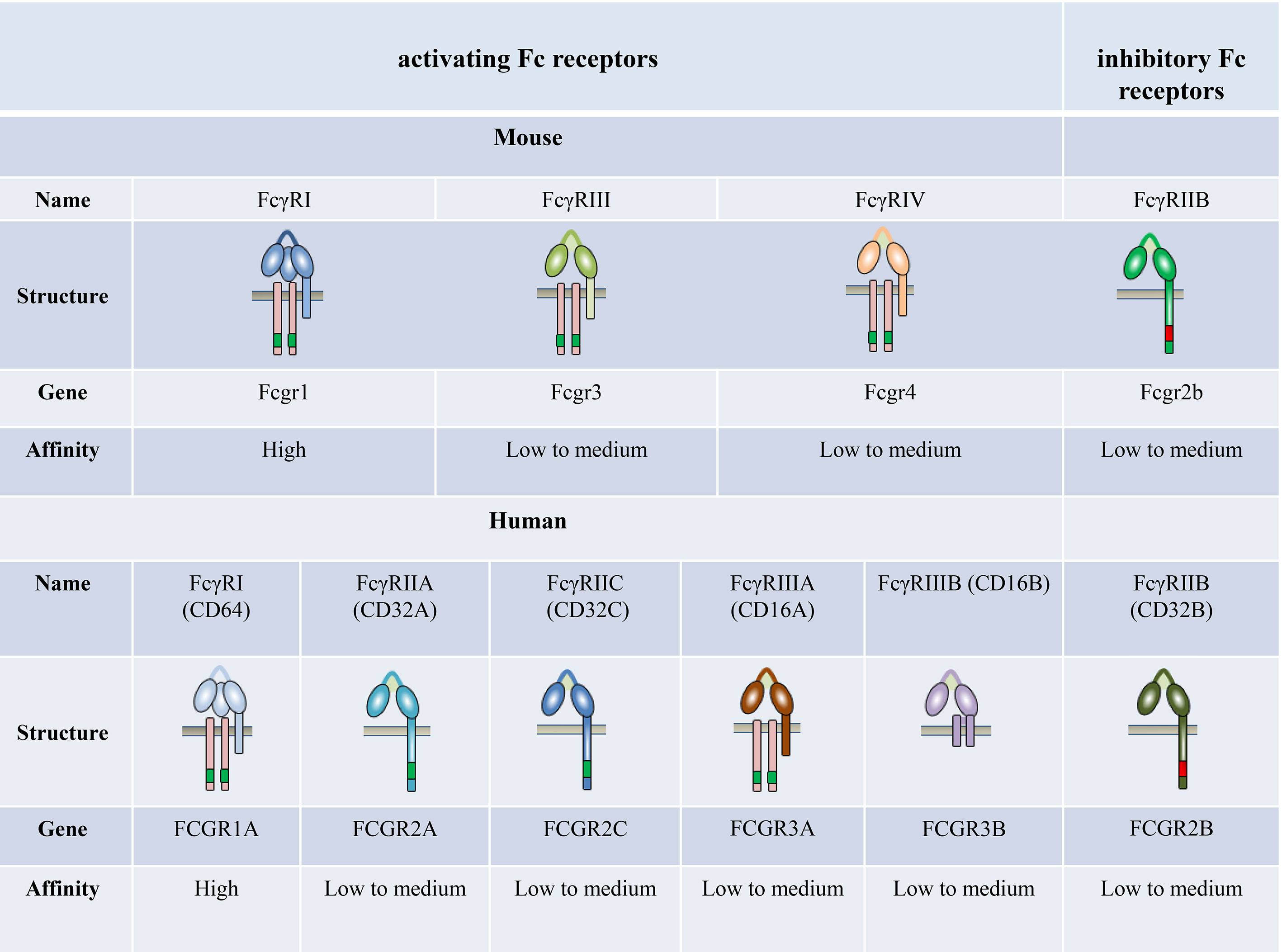
Figure 1 The family of classical Fc receptors for IgG. Schematic representations of FcγRs with respect to the cell membrane (brown bar), in complex with their respective signaling subunits. Mouse and humans have one high-affinity receptor, FcγRI; all other FcγRs have a low-to-medium affinity for the antibody Fc fragment.
The affinity of FcγRs for IgG depends on the type of FcγR and IgG isotypes (30–36). FcγRI is the only known high-affinity FcγR (108–109 M−1) with a restricted isotype specificity. In contrast, FcγRII and FcγRIII have a low affinity for IgG (about 106 M−1) with a broader isotype binding pattern (31, 32). FcγRIV is a novel receptor conserved across all mammalian species with an intermediate affinity (107 M−1) and restricted subclass specificity (29, 37). FcγRIIIA is engaged by IgG1 and IgG2, whereas FcγRI and FcγRIV are engaged by IgG2 only (35). The affinity of mouse FcγRs is significantly higher compared with their corresponding human FcγRs (36).
FcγRs are divided into activating and inhibitory receptors and coexpressed on the same cell (38). Activating FcγRs, including FcγRI and FcγRIII, contain an immunoreceptor tyrosine-based activation (ITAM) in intracellular structure and transmit their signals via the ITAM, which recruits spleen tyrosine kinase (Syk) (39). FcγRIIB is the only known inhibitory FcγR with an immunoreceptor tyrosine-based inhibitory motif (ITIM) in its intracytoplasmic domain (40). The phosphorylation of ITIM counteracts the signals mediated by activating FcγRs (41–43). FcγRIIB is expressed widely on B cells, macrophages, and mast cells and downregulates several cellular functions, such as B-cell activation and mast cell degranulation (44). The activating-to-inhibitory (A/I) ratio on the same cell acts as the specific checkpoint for the arrest or progression of an immune response. Surprisingly, when monomeric or low-affinity immune complexes bind to activating FcγRs, the normally activating ITAM domain cannot induce co-aggregation of activating receptors, thereby partially phosphorylating the ITAM domain. Thus, partial tyrosine phosphorylation of ITAM by Src family kinases may result in the recruitment of inhibitory SHIP. This is called inhibitory ITAM (ITAMi) signal and is important in maintaining immune homeostasis (45–47).
Unlike other activating FcγRs, FcγRII proteins do not require the common FcR γ-chain for stable expression or function. They all have signaling motifs in their intracellular cytoplasmic domains (48). All the above FcγRs are transmembrane glycoproteins, except for human FcγRIIIB, which is expressed on neutrophils and is a glycophosphatidylinositol (GPI)-anchored protein (49, 50). The mechanisms by which FcγRIIIB transduces signals are still unknown (51).
Fcγrs and Arthritis
During autoimmune diseases, such as RA and SLE, the autoantibodies and immune complexes cause inflammation via FcR aggregation (52). The altered expression of FcγRs on immune cells in the circulation and synovium of RA patients is the first indication of their involvement in inflammation (53–60). The absence of all FcγRs does not affect the number of osteoclast precursors or their osteoclastogenic potential. However, it reduces joint inflammation and bone erosion during inflammatory arthritis (61). FcγRIIB is particularly critical for maintaining the balance of an efficient inflammatory response or countering unwanted autoimmunity attacks. Multiple clinical studies have shown that FcγRIIB is a reliable biomarker for SLE susceptibility in different ethnic groups. FcγRIIB and its signaling pathways represent a vital checkpoint in peripheral and central tolerance and in controlling the development of autoreactive antibodies (62).
In addition to the altered expression of FcγRs, genetic variants associated with related single-nucleotide polymorphisms (SNPs) in populations with RA and lupus arthritis have been reported. Several genes encoding FcγRs that alter the affinity of FcγRs for IgGs have been described in several RA populations. In particular, some of these, such as the hFcγRIIa-R131 variant, which is related to an increased risk of developing RA, even influence the susceptibility to RA development and the response to treatment (63–70). In addition, an association between lupus arthritis and the FCGR2A as well as FCGR3A low copy number genotypes has been observed in Taiwan patients with SLE. The FCGR3A low copy number genotype was significantly enriched in patients with SLE having arthritis (71–73). Moreover, a meta-analysis revealed the association of the FcγRIIa-R131 allele with SLE, especially in African Americans, whereas the FcγRIIIa-F176 allele was associated with SLE in Caucasians and other groups (74). Furthermore, Tsang et al. demonstrated the association between low-affinity FcγR polymorphisms and susceptibility to SLE (75).
Studies using FcγR gene-deficient mice have greatly enhanced our understanding of the role of FcγRs in inflammatory arthritis (76, 77). The lack of activating FcγRs alleviates the disease severity in arthritis models (78–81). In different disease phases of inflammatory arthritis, the individual activating FcγRs have different significance (36, 61, 82–86). In the absence of FcγRI, FcγRIIB, and FcγRIIIA, FcγRIV is sufficient to induce arthritis alone (35). In contrast with activating FcγRs, the inhibitory FcγRIIB suppresses inflammation by inhibiting the activating signaling, as well as providing negative feedback on the production of autoantibodies by B cells (87–92).
Autoantibodies and their immune complexes play a central role in shaping a pro-inflammatory environment. Indeed, complexes of ACPA and rheumatoid factor (RF) induce the production of potent inflammatory cytokines (93–96). This effect is predominantly mediated by FcγR signaling on macrophages (51, 97). Tumor necrosis factor (TNF)-α, in combination with cytokines interleukin (IL)-4 and IL-13, downregulates FcγR-mediated function by decreasing the expression of activating FcγRs, suggesting that downregulated activating FcγRs might have an anti-inflammatory effect (98).
The Fc receptors on white blood cells are essential for effective phagocytosis of immune complexes and bacteria. Moreover, FcγRI is upregulated during infection. FcγRI (CD64) has previously been reported to distinguish systemic infections from inflammatory autoimmune diseases and viral infections. Patients without inflammatory and infectious conditions, such as osteoarthritis, have a lower level of neutrophil FcγRI than those with infections (99–104). Oppegaard et al. investigated the use of FcγRI in discerning septic arthritis from inflammatory joint disease and found that FcγRI is highly specific for infectious diseases, including septic arthritis. However, its sensitivity is poor in local infections (104). Although distinct meta-analyses have confirmed this, more large prospective studies need to be conducted to verify several cut-off values reported in the neutrophil FcγRI test in the clinical setting (105, 106).
Human and murine activating FcγRs are not functionally equivalent. A few studies performed in transgenic mice expressing human FcγRs examined their involvement in inflammatory arthritis (107). The results confirmed that the expression of the human FcγRIIA is associated with spontaneous autoimmune inflammation, with a crucial role in autoimmune diseases (92).
FcγR Role in Bone Erosion
Osteoclast Activation and Differentiation
Bone balance depends on a dynamic regulation of bone formation and resorption, which are predominantly mediated by osteoblasts and osteoclasts, respectively (108, 109). Enhanced osteoclast activity could result in severe bone destruction as exemplified in autoimmune inflammatory diseases such as RA, whereas defective osteoblast differentiation causes diseases with a high bone mass, including osteopetrosis. Osteoclasts are the only bone-resorbing cells and play a central role in bone erosion. Osteoclasts are derived from multinucleated progenitors of the monocyte/macrophage family and are the link between immune and bone systems. RANK and RANKL are critical factors that together regulate osteoclast functions. In addition, macrophage colony-stimulating factor (M-CSF) is an essential cytokine in osteoclastogenesis (109–111). RANKL is majorly secreted by osteoblasts, osteocytes, T cells, and endothelial cells. And osteocytes express a much higher amount of RANKL required for osteoclastogenesis than osteoblasts (112, 113). The most important negative regulator of RANKL is the decoy receptor osteoprotegerin (OPG), which inhibits osteoclastogenesis by preventing RANKL–RANK interaction. The RANKL–RANK–OPG system modulates bone homeostasis by regulating osteoclasts (114). Osteoblasts and osteocytes also produce OPG to suppress osteoclastogenesis by masking RANKL signaling (115, 116). RANKL initiates osteoclastogenesis by inducing nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1), via TNF receptor-associated factor 6 (TRAF6) and c-Fos pathways (117) (Figure 2). NFATc1 is the master transcription factor for pro-osteoclastogenic genes. In addition, several pro-inflammatory cytokines produced by innate immune cells and T cells, such as TNFα, IL-17, IL-1, and IL-6, stimulate osteoclastogenesis directly or indirectly (118).
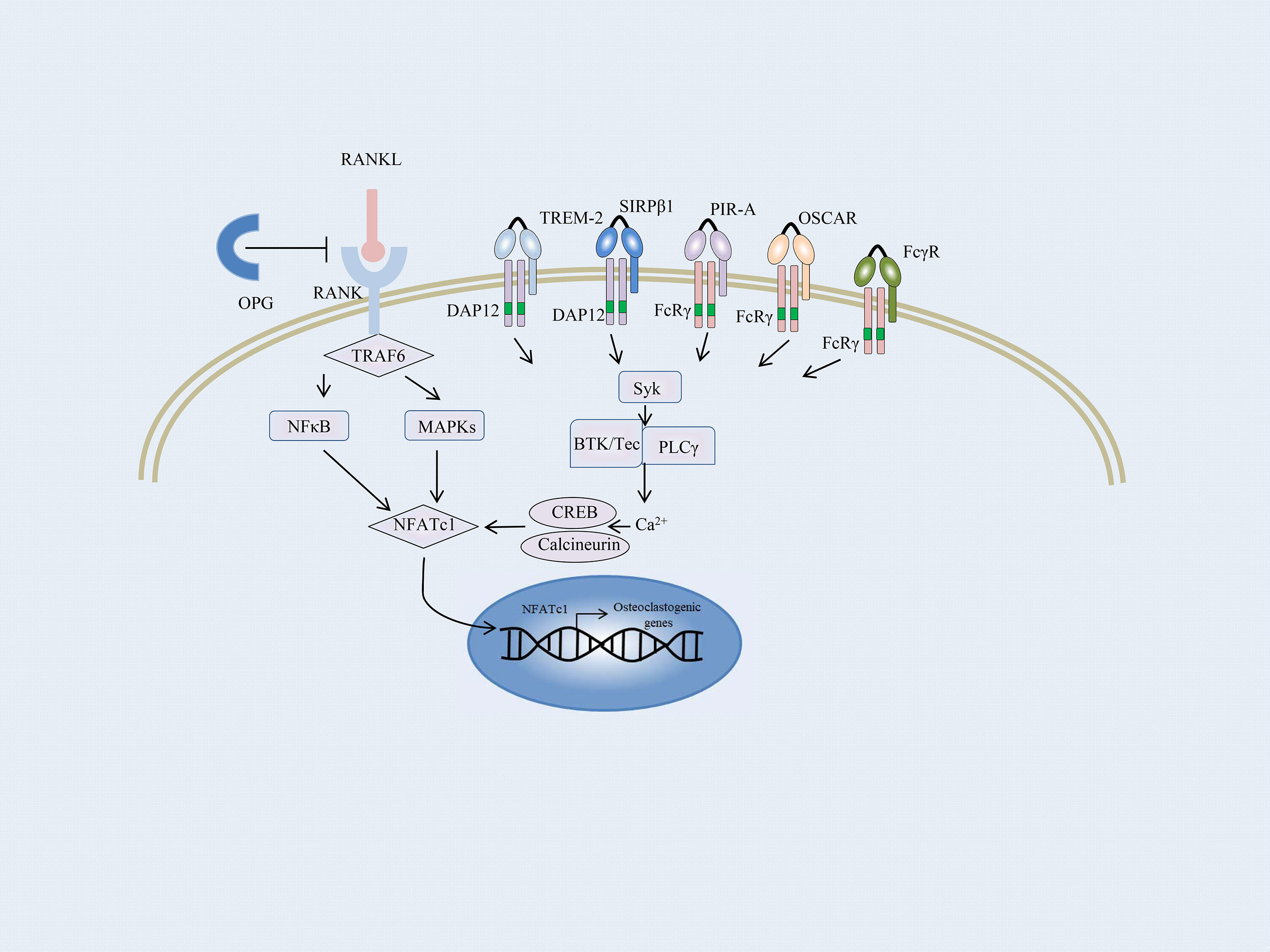
Figure 2 Overview of the osteoclast signaling network. A schematic representation of ITAM-mediated costimulatory signal in the RANKL-induced TRAF6 signaling pathway of osteoclast differentiation. In osteoclast precursors, phosphorylation of ITAM stimulated by immunoreceptors and RANKL–RANK interaction recruits the Syk family kinases, thus activating phospholipase Cγ (PLCγ), Bruton’s tyrosine kinase (BTK), as well as Tec kinases. They augment the calcium influx required for the activation of NFATc1. NFATc1 subsequently migrates to the nucleus, where it binds to its gene promoter and triggers an auto-amplifying feedback loop. Calcium signals in osteoclast precursors are provided by the ITAM-bearing proteins, Fc receptor γ subunit, and its functional analog DNAX activation protein of 12 kDa (DAP12). FcRγ-chain is associated with the immunoglobulin (Ig)-like receptors, such as osteoclast-associated receptor (OSCAR) and paired Ig-like receptor-A (PIR-A). DAP12 is associated with its signaling counterpart, triggering receptor expressed on myeloid cell-2 (TREM-2), and signal-regulatory protein β1 (SIRPβ1).
FcγRs and Osteoclastogenesis
Apart from the M-CSF and RANKL signaling, an ITAM costimulatory signal provided by the accessory protein for RANKL-RANK is required for osteoclastogenesis (119). Takayanagi et al. first reported that the activation of NFATc1 was insufficient for terminal differentiation of monocytes/macrophages into osteoclasts; calcium signals and calcineurin activation are essential for this process (117, 120). Calcium signals in myeloid cells are provided by the ITAM-bearing proteins, Fc receptor γ subunit, and its functional analog DNAX activation protein of 12 kDa (DAP12). Both the accessory proteins are intracellular adaptor molecules and play a crucial function in transducing the costimulatory signals for RANKL (121). Mice lacking the accessory proteins display a severe osteopetrotic phenotype with deficient osteoclast function (122).
FcRγ-chain is associated with immunoglobulin (Ig)-like receptors, such as osteoclast-associated receptor (OSCAR) and paired Ig-like receptor-A (PIR-A) (Figure 2). DAP12 is associated with its signaling counterpart, triggering receptor expressed on myeloid cell-2 (TREM-2), and signal-regulatory protein β1 (SIRPβ1), which are expressed on the cell membrane of osteoclast precursors and are essential for the communication between osteoclast precursors (33, 123). Activation of RANKL-RANK rapidly phosphorylates the ITAM motifs and recruits the protein kinase Syk, subsequently activating multiple downstream signaling cascades, such as phospholipase Cγ (PLCγ) and Bruton’s tyrosine kinase (BTK) as well as Tec kinases. They all enhance the effects of RANKL-signaling by augmenting the calcium influx required for the activation of NFATc1. NFATc1 subsequently migrates to the nucleus, where it binds to its gene promoter and triggers an auto-amplifying feedback loop (124, 125).
Osteoclasts and their precursors express FcγR (126), whereas FcγRI, FcγRIIB, and FcγRIIIA are significantly upregulated during human ex vivo osteoclastogenesis (127). Blocking of the FcR and deleting the FcγR gene reduce osteoclastogenesis stimulated by IgG complexes on osteoclast precursor cells (25). Although FcγRs are required for osteoclastogenesis and bone resorption in inflammatory disorders, their specific role in bone homeostasis is not completely understood.
FcγRs and Bone Erosion
Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis, suggesting the link between ITAM-harboring adaptors FcγRs and bone erosion (117). As an important link binding the bone system and immune system, FcγRs are not only receptors for the Fc portion of IgG but are also costimulatory molecules for RANKL-induced osteoclastogenesis (119, 121). Bone damage has been reported in seropositive RA patients before clinical disease onset, highlighting that osteoclastogenesis is independent of joint inflammation (128). This finding challenged the concept that inflammation is the primary trigger for bone erosion in inflammatory arthritis and indicated that bone loss might precede inflammation (129).
The expression of Fcγ receptors on osteoclast precursor cells and mature osteoclasts has been measured. FcγRI and FcγRIII are primarily expressed on human preosteoclasts, whereas the inhibitory FcγRIIB is majorly expressed on mature osteoclasts (127). Under physiological conditions, activating FcγRI and FcγRIV in mice does not have a major role in bone characteristics and osteoclast development (22). Bone homeostasis is not significantly different in mice with FcγRI or IV deficiency compared with wild mice (17). In addition, the deficiency of FcγRIIB does not affect osteoclastogenesis (23). Activating FcγRs transmit the positive signal. In contrast, FcγRIII functions as an inhibitory receptor in the differentiation of osteoclast precursor cells under physiological conditions. FcγRIII deprives the FcRγ subunit’s availability for other Ig-like receptors activating receptors, such as PIR-A and OSCAR, thus transmitting an ITAM-mediated inhibitory signal for osteoclastogenesis (130). Naive FcγRIII–/–mice have increased osteoclast numbers and an osteoporotic phenotype (22).
The relative importance of various FcγRs in osteoclastogenesis changes in the inflammatory arthritis microenvironment. Studies demonstrated that the stimulation of FcγRI and FcγRIV increases both osteoclast differentiation and function both in vitro and in vivo (22, 30). FcγRIII levels are increased, and FcγRIIB levels are decreased on bone marrow cells from mice with collagen-induced arthritis (CIA), indicating that FcγRIII induces osteoclastogenesis under inflammatory conditions (22). Furthermore, human RA patients with the FcγRIIIa-158V allele endure severe bone erosion compared with patients with the FcγRIIIa-158F allele (131, 132). Similarly, artificial crosslinking of FcγRI and FcγRIV leads to increased osteoclast differentiation without affecting their resorbing function in vitro (17). Osteoclast numbers and bone erosion were decreased in FcγRIV–/–mice compared with wild mice in a serum transfer model (17). FcγRIIB–/–mice spontaneously developed osteoporosis, which was reversed by an additional knockout of activating FcγRs (22).
De-sialylated IgGs binding to FcγRs with strong affinity have substantially high stimulatory effects on both murine and human osteoclasts (127, 133). In addition, IgGs were less sialylated during inflammation (22). Harre et al. confirmed that the interactions between immune complexes and osteoclasts were related to the degree of IgG sialylation, and only non-sialylated or low-sialylated immune complexes drive osteoclastogenesis. RA patients with low Fc sialylation levels of IgGs have significantly higher bone loss. The pro-osteoclastogenic effect of non-sialylated immune complexes is a common feature of all IgG antibodies (127). A recent study showed that in induced pluripotent stem cell derived mesenchymal stem cell (iMSCs), the sialylation degree of IgG determines the antibodies directed osteogenic potential by regulating immune responses and osteoclastogenesis (24),but desialylated IgG complexes do not affect arthritis-mediated bone loss (134).
Although the signaling of activating FcγRs mediated by immune complex increases osteoclast differentiation, different results exist for immune complex/FcγR on osteoclastogenesis and osteoclast function (Table 1). Previous studies demonstrated the immune complex-induced inhibition of osteoclastogenesis, which possibly acts via activating FcγRs (23, 139). This suggests that FcγRs may have dual roles in bone destruction in inflammatory arthritis. High levels of autoantibodies are a characteristic feature of SLE compared with other inflammatory arthritis (140, 141). The deposition of autoantibodies or immune complexes causes lupus nephritis (142), skin damage (143), splenomegaly (144), and damage to other organs. Lupus IgG can promote the differentiation of monocytes into DCs (145). These indicate that lupus autoantibodies may also play a protective role in bone destruction in inflammatory arthritis. Recently, our research results (26) demonstrated that joint-deposited lupus IgG induced arthritis without bone erosion by intraarticular injection of lupus IgG in mice. Monocytes/macrophages and their product TNFα are required for the development of lupus IgG-induced arthritis. To understand the mechanism of lupus IgG-induced arthritis with deficiency of bone erosion, we determined whether lupus IgG inhibited RANKL-induced osteoclastogenesis. We found that lupus IgG directly suppressed RANKL-induced osteoclastogenesis in a dose-dependent manner in vitro. The inhibitory effect of lupus IgG on osteoclastogenesis is related to timepoint in lupus IgG and RANKL treated macrophages. Deficiency of FcγRII and FcγRIII did not affect the inhibitory effect of lupus IgG on osteoclastogenesis, indicating that the inhibitory effect of lupus IgG on osteoclastogenesis is dependent on FcγRI. Lupus IgG and RANKL can downregulate the surface expression of FcγRI on bone marrow macrophages (20). Research results suggest that lupus IgG inhibits osteoclastogenesis by competitively occupying FcγRI on monocytes/macrophages and reducing RANKL signaling. The effect of activation or repression of RANKL-induced osteoclastogenesis depends on the extent of FcγRI occupancy by IgG. This protective mechanism explains non-destructive arthritis in SLE. In addition, it implies that FcγRI could be a therapeutic target for bone erosion in inflammatory arthritis.
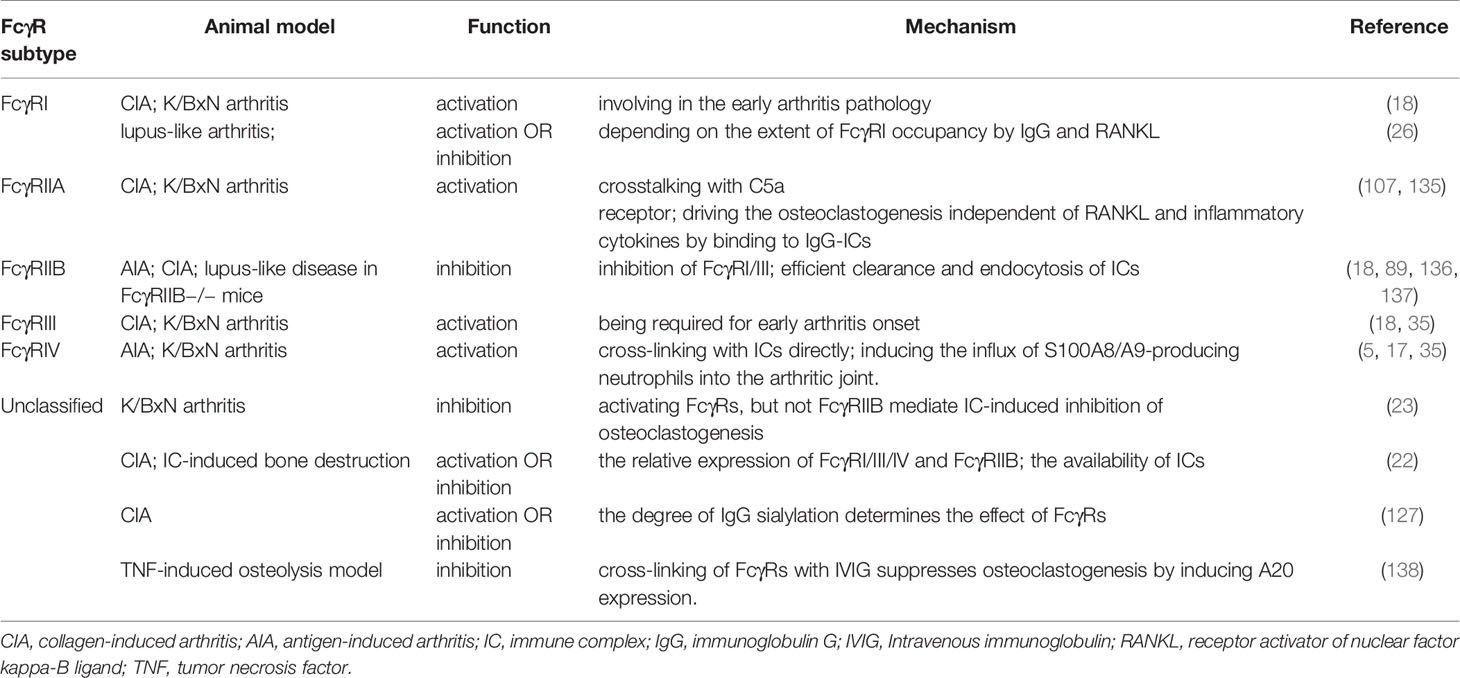
Table 1 Different roles of FcγRs in arthritis and bone destruction.
The deposition of ACPA is important for osteoclastogenesis in RA (146). Different studies have identified that ACPA prevalence is significantly increased in SLE patients with erosive arthritis (16). Recent studies have explored the direct effect of ACPA-mediated bone erosion. ACPA IgG together with their citrullinated antigens forms immune complexes that stimulate immune cells via their interaction with FcγRs (93, 147). By using polyclonal ACPAs purified from ACPA-containing serum of RA patients, Harre et al. provided the first validation that ACPAs can directly promote osteoclast differentiation and activation (7). ACPA IgG might affect osteoclastogenesis by the activation of Fc receptors on osteoclasts directly. IgG Fc sialylation is crucial for immune complex–osteoclast interactions (127). Besides, ACPA IgG is shown less sialylated than random IgG (148). There are other published papers regarding the detailed mechanisms of ACPA’s direct regulation, but the exact mechanism of ACPA’s direct effect on erosion remains unclear (149–152).
FcγR Immunotherapy
The crucial role of FcγRs in both inflammatory arthritis and bone erosion may offer a promising therapeutic target for bone destruction in inflammatory arthritis. One indirect mechanism involves the neutralization of autoimmune IgG Fc by soluble FcγRs, These drugs include the recombinant soluble FcγIIB receptor SM101 (NCT03851341) and monoclonal antibody targeted the receptors. For example, antagonistic monoclonal antibody against the hFcγRIIIA has been shown to be effective in a patient with immune thrombocytopenia (ITP) refractory to all conventional therapies (153). And human recombinant soluble FcγRIIB treatment could ameliorate collagen-induced arthritis by reducing immune complexes-stimulated inflammation and joint swelling (154). Besides, recombinant human soluble FcγRII was evaluated as an effective therapeutic strategy in inhibiting chronic murine lupus pathology (155).
Another mechanism involves the direct blocking of the IgG-binding site on FcγRs. Recombinant multimeric Fc fragments with a high affinity for FcγRs have been shown to be efficacious in animal models of RA, ITP, and graft-versus-host disease (GVHD) (156). These include PF-06755347 (NCT03275740), CSL730 (NCT04446000) and CSL777 (Preclinical) (157). However, nonspecific crosslinking of activating FcγRs could lead to undesired clinical adverse events, and monovalent antibody derivatives, such as Fab, may reduce severe clinical adverse events (158). Up to now, results of above molecules from clinical trials have been promising in autoimmune diseases, but further long-term data are needed (159, 160).
Intravenous immunoglobulin (IVIG) treatment is efficient in several different immune disorders (161, 162). IVIG consists predominantly of IgG and a small fraction of immune complexes. It exerts anti-inflammatory effects in both humans and animal models by its Fc but not Fab fragments (163). Besides, previous studies confirmed that IVIG could directly inhibits human osteoclastogenesis by suppressing the RANK signaling, the suppressive effect is partly mediated by IgG immune complexes contained within IVIG preparations (138). Our study showed that lupus IgG induced synovial inflammation but inhibited RANKL-induced osteoclastogenesis. The suppressive effect is mediated by the competitive occupation of FcγRI on monocytes/macrophages (26).
Conclusions and Future Perspectives
Bone erosions are remarkable features in inflammatory arthritis, such as RA, but not in lupus arthritis. Osteoclasts are major cells for bone erosions. Activating FcγR containing ITAM motifs is required for RANKL-induced osteoclastogenesis. FcγR can effectively regulate inflammatory arthritis and bone erosions. Based on published studies, we conclude that FcγR may have dual roles in osteoclastogenesis. The effect of activating and inhibiting osteoclastogenesis depends on the extent of FcγRI occupancy by IgG and RANKL, respectively. Specific IgG molecules or Fc fragments with a high affinity for FcγRI designed to occupy FcγRI may exert the inhibitory effect on bone erosion. The sialylation level of IgG Fc binding to FcγRs needs to be taken into account as well. A deeper understanding of FcγRs involved in physiological and pathological osteoclastogenesis will be valuable in identifying new targets and developing potential therapeutic strategies for inflammatory arthritis.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
YZ and G-MD designed the manuscript and figures. YZ and G-MD drafted the manuscript and approved the final version of the manuscript. G-MD revised the final version of the manuscript critically. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the initiating fund of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (G-MD, 02.03.2018-41).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer PZ declared a shared affiliation with the authors to the handling editor at the time of review.
References
1. Ledingham J, Snowden N, Ide Z. Diagnosis and Early Management of Inflammatory Arthritis. BMJ (2017) 358:j3248. doi: 10.1136/bmj.j3248
2. Firestein GS. Evolving Concepts of Rheumatoid Arthritis. Nature (2003) 423:356–61. doi: 10.1038/nature01661
3. Symmons D, Turner G, Webb R, Asten P, Barrett E, Lunt M, et al. The Prevalence of Rheumatoid Arthritis in the United Kingdom: New Estimates for a New Century. Rheumatol (Oxford) (2002) 41:793–800. doi: 10.1093/rheumatology/41.7.793
4. Lee DM, Weinblatt ME. Rheumatoid Arthritis. Lancet (2001) 358:903–11. doi: 10.1016/S0140-6736(01)06075-5
5. Di Ceglie I, Kruisbergen NNL, van den Bosch MHJ, van Lent P. Fc-gamma Receptors and S100A8/A9 Cause Bone Erosion During Rheumatoid Arthritis. Do They Act as Partners in Crime? Rheumatol (Oxford) (2019) 58:1331–43. doi: 10.1093/rheumatology/kez218
6. Cipolletta E, Smerilli G, Di Matteo A, Di Battista J, Di Carlo M, Grassi W, et al. The Sonographic Identification of Cortical Bone Interruptions in Rheumatoid Arthritis: A Morphological Approach. Ther Adv Musculoskelet Dis (2021) 13:1759720x211004326. doi: 10.1177/1759720x211004326
7. Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, et al. Induction of Osteoclastogenesis and Bone Loss by Human Autoantibodies Against Citrullinated Vimentin. J Clin Invest (2012) 122:1791–802. doi: 10.1172/JCI60975
8. Beringer A, Miossec P. Systemic Effects of IL-17 in Inflammatory Arthritis. Nat Rev Rheumatol (2019) 15:491–501. doi: 10.1038/s41584-019-0243-5
9. Crowson CS, Matteson EL, Myasoedova E, Michet CJ, Ernste FC, Warrington KJ, et al. The Lifetime Risk of Adult-Onset Rheumatoid Arthritis and Other Inflammatory Autoimmune Rheumatic Diseases. Arthritis Rheum J Clin Invest (2011) 63:633–9. doi: 10.1002/art.30155
10. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic Lupus Erythematosus. Nat Rev Dis Primers (2016) 2:16039. doi: 10.1038/nrdp.2016.39
11. Grossman JM. Lupus Arthritis. Best Pract Res Clin Rheumatol (2009) 23:495–506. doi: 10.1016/j.berh.2009.04.003
12. Mahmoud K, Zayat A, Vital EM. Musculoskeletal Manifestations of Systemic Lupus Erythmatosus. Curr Opin Rheumatol (2017) 29:486–92. doi: 10.1097/BOR.0000000000000421
13. Esdaile JM, Danoff D, Rosenthall L, Gutkowski A. Deforming Arthritis in Systemic Lupus Erythematosus. Ann Rheum Dis (1981) 40:124–6. doi: 10.1136/ard.40.2.124
14. Grigor R, Edmonds J, Lewkonia R, Bresnihan B, Hughes GR. Systemic Lupus Erythematosus. A Prospective Analysis. Ann Rheum Dis (1978) 37:121–8. doi: 10.1136/ard.37.2.121
15. Bouchedoub Y, Djenouhat K, Rachedi N, Babasaci R, Ould Ali L, Salah K, et al. Association of Markers of Rheumatoid Arthritis in Lupus. Is it a Rhupus? Annales biologie clinique (2020) 78:201–5. doi: 10.1684/abc.2020.1518
16. Budhram A, Chu R, Rusta-Sallehy S, Ioannidis G, Denburg JA, Adachi JD, et al. Anti-Cyclic Citrullinated Peptide Antibody as a Marker of Erosive Arthritis in Patients With Systemic Lupus Erythematosus: A Systematic Review and Meta-Analysis. Lupus (2014) 23:1156–63. doi: 10.1177/0961203314540967
17. Seeling M, Hillenhoff U, David JP, Schett G, Tuckermann J, Lux A, et al. Inflammatory Monocytes and Fcgamma Receptor IV on Osteoclasts Are Critical for Bone Destruction During Inflammatory Arthritis in Mice. Proc Natl Acad Sci USA (2013) 110:10729–34. doi: 10.1073/pnas.1301001110
18. Boross P, van Lent PL, Martin-Ramirez J, van der Kaa J, Mulder MH, Claassens JW, et al. Destructive Arthritis in the Absence of Both FcgammaRI and Fcgammariii. J Immunol (2008) 180:5083–91. doi: 10.4049/jimmunol.180.7.5083
19. Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, et al. Arthritis Critically Dependent on Innate Immune System Players. Immunity (2002) 16:157–68. doi: 10.1016/s1074-7613(02)00275-3
20. Kleinau S, Martinsson P, Heyman B. Induction and Suppression of Collagen-Induced Arthritis Is Dependent on Distinct Fcgamma Receptors. J Exp Med (2000) 191:1611–6. doi: 10.1084/jem.191.9.1611
21. van Lent PL, Grevers L, Lubberts E, de Vries TJ, Nabbe KC, Verbeek S, et al. Fcgamma Receptors Directly Mediate Cartilage, But Not Bone, Destruction in Murine Antigen-Induced Arthritis: Uncoupling of Cartilage Damage From Bone Erosion and Joint Inflammation. Arthritis Rheum (2006) 54:3868–77. doi: 10.1002/art.22253
22. Negishi-Koga T, Gober HJ, Sumiya E, Komatsu N, Okamoto K, Sawa S, et al. Immune Complexes Regulate Bone Metabolism Through FcRgamma Signalling. Nat Commun (2015) 6:6637. doi: 10.1038/ncomms7637
23. Grevers LC, de Vries TJ, Everts V, Verbeek JS, van den Berg WB. Van Lent Pl. Immune Complex-Induced Inhibition of Osteoclastogenesis is Mediated Via Activating But Not Inhibitory Fcgamma Receptors on Myeloid Precursor Cells. Ann Rheum Dis (2013) 72:278–85. doi: 10.1136/annrheumdis-2012-201568
24. Wu Q, Yang Y, Xie D, Li S, Liu Y, Shu L, et al. The Sialylation Profile of IgG Determines the Efficiency of Antibody Directed Osteogenic Differentiation of iMSCs by Modulating Local Immune Responses and Osteoclastogenesis. Acta biomaterialia (2020) 114:221–32. doi: 10.1016/j.actbio.2020.07.055
25. Kamohara A, Hirata H, Xu X, Shiraki M, Yamada S, Zhang JQ, et al. Igg Immune Complexes With Staphylococcus Aureus Protein A Enhance Osteoclast Differentiation and Bone Resorption by Stimulating Fc Receptors and TLR2. Int Immunol (2020) 32:89–104. doi: 10.1093/intimm/dxz063
26. Qiao W, Ding H, Zuo Y, Jiang L, Zhou J, Han X, et al. Lupus IgG Deposition Causes Arthritis But Inhibits Bone Destruction Through Competitive Occupation of FcgammaRI and Reduced RANKL Signalling. Clin Transl Immunol (2020) 9:e1174. doi: 10.1002/cti2.1174
27. Nimmerjahn F, Ravetch JV. Fcgamma Receptors as Regulators of Immune Responses. Nat Rev Immunol (2008) 8:34–47. doi: 10.1038/nri2206
28. Nimmerjahn F, Ravetch JV. Fc Gamma Receptors: Old Friends and New Family Members. Immunity (2006) 24:19–28. doi: 10.1016/j.immuni.2005.11.010
29. Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: A Novel FcR With Distinct IgG Subclass Specificity. Immunity (2005) 23:41–51. doi: 10.1016/j.immuni.2005.05.010
30. Nimmerjahn F, Ravetch JV. Divergent Immunoglobulin G Subclass Activity Through Selective Fc Receptor Binding. Science (2005) 310:1510–2. doi: 10.1126/science.1118948
31. Ravetch JV, Kinet JP. Fc Receptors. Annu Rev Immunol (1991) 9:457–92. doi: 10.1146/annurev.iy.09.040191.002325
32. Hulett MD, Hogarth PM. Molecular Basis of Fc Receptor Function. Adv Immunol (1994) 57:1–127. doi: 10.1016/s0065-2776(08)60671-9
33. Kim N, Takami M, Rho J, Josien R, Choi Y. A Novel Member of the Leukocyte Receptor Complex Regulates Osteoclast Differentiation. J Exp Med (2002) 195:201–9. doi: 10.1084/jem.20011681
34. Bruhns P. Properties of Mouse and Human IgG Receptors and Their Contribution to Disease Models. Blood (2012) 119:5640–9. doi: 10.1182/blood-2012-01-380121
35. Mancardi DA, Jonsson F, Iannascoli B, Khun H, Van Rooijen N, Huerre M, et al. Cutting Edge: The Murine High-Affinity IgG Receptor FcgammaRIV Is Sufficient for Autoantibody-Induced Arthritis. J Immunol (2011) 186:1899–903. doi: 10.4049/jimmunol.1003642
36. Nimmerjahn F, Lux A, Albert H, Woigk M, Lehmann C, Dudziak D, et al. FcgammaRIV Deletion Reveals Its Central Role for IgG2a and IgG2b Activity In Vivo. Proc Natl Acad Sci USA (2010) 107:19396–401. doi: 10.1073/pnas.1014515107
37. Mechetina LV, Najakshin AM, Volkova OY, Guselnikov SV, Faizulin RZ, Alabyev BY, et al. FCRL, a Novel Member of the Leukocyte Fc Receptor Family Possesses Unique Structural Features. Eur J Immunol (2002) 32:87–96. doi: 10.1002/1521-4141(200201)32:1<87::AID-IMMU87>3.0.CO;2-#
38. Ravetch JV, Lanier LL. Immune Inhibitory Receptors. Science (2000) 290:84–9. doi: 10.1126/science.290.5489.84
39. Daeron M. Fc Receptor Biology. Annu Rev Immunol (1997) 15:203–34. doi: 10.1146/annurev.immunol.15.1.203
40. Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, et al. Cytoplasmic Domain Heterogeneity and Functions of IgG Fc Receptors in B Lymphocytes. Science (1992) 256:1808–12. doi: 10.1126/science.1535455
41. Ono M, Bolland S, Tempst P, Ravetch JV. Role of the Inositol Phosphatase SHIP in Negative Regulation of the Immune System by the Receptor Fc(Gamma)RIIB. Nature (1996) 383:263–6. doi: 10.1038/383263a0
42. Daeron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor Tyrosine-Based Inhibition Motifs: A Quest in the Past and Future. Immunol Rev (2008) 224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x
43. Smith KG, Clatworthy MR. FcgammaRIIB in Autoimmunity and Infection: Evolutionary and Therapeutic Implications. Nat Rev Immunol (2010) 10:328–43. doi: 10.1038/nri2762
44. Takai T. Roles of Fc Receptors in Autoimmunity. Nat Rev Immunol (2002) 2:580–92. doi: 10.1038/nri856
45. Blank U, Launay P, Benhamou M, Monteiro RC. Inhibitory Itams as Novel Regulators of Immunity. Immunol Rev (2009) 232:59–71. doi: 10.1111/j.1600-065X.2009.00832.x
46. Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, et al. Identification of FcalphaRI as an Inhibitory Receptor That Controls Inflammation: Dual Role of FcRgamma Itam. Immunity (2005) 22:31–42. doi: 10.1016/j.immuni.2004.11.017
47. Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-Like Receptor Responses in the Absence of Signaling Adaptor DAP12. Nat Immunol (2005) 6:579–86. doi: 10.1038/ni1204
48. Li X, Ptacek TS, Brown EE, Edberg JC. Fcgamma Receptors: Structure, Function and Role as Genetic Risk Factors in SLE. Genes Immun (2009) 10:380–9. doi: 10.1038/gene.2009.35
49. Selvaraj P, Carpen O, Hibbs ML, Springer TA. Natural Killer Cell and Granulocyte Fc Gamma Receptor III (CD16) Differ in Membrane Anchor and Signal Transduction. J Immunol (1989) 143:3283–8.
50. Selvaraj P, Rosse WF, Silber R, Springer TA. The Major Fc Receptor in Blood has a Phosphatidylinositol Anchor and Is Deficient in Paroxysmal Nocturnal Haemoglobinuria. Nature (1988) 333:565–7. doi: 10.1038/333565a0
51. Steffen U, Schett G, Bozec A. How Autoantibodies Regulate Osteoclast Induced Bone Loss in Rheumatoid Arthritis. Front Immunol 4.71 (2019) 10:1483. doi: 10.3389/fimmu.2019.01483
52. Ben Mkaddem S, Benhamou M, Monteiro RC. Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front Immunol (2019) 10:811. doi: 10.3389/fimmu.2019.00811
53. Hepburn AL, Mason JC, Davies KA. Expression of Fcgamma and Complement Receptors on Peripheral Blood Monocytes in Systemic Lupus Erythematosus and Rheumatoid Arthritis. Rheumatol (Oxford) (2004) 43:547–54. doi: 10.1093/rheumatology/keh112
54. Amoruso A, Sola D, Rossi L, Obeng JA, Fresu LG, Sainaghi PP, et al. Relation Among Anti-Rheumatic Drug Therapy, CD14(+)CD16(+) Blood Monocytes and Disease Activity Markers (DAS28 and US7 Scores) in Rheumatoid Arthritis: A Pilot Study. Pharmacol Res (2016) 107:308–14. doi: 10.1016/j.phrs.2016.03.034
55. Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(Bright) CD16+ Monocyte Subset Is Expanded in Rheumatoid Arthritis and Promotes Expansion of the Th17 Cell Population. Arthritis Rheum (2012) 64:671–7. doi: 10.1002/art.33418
56. Tsukamoto M, Seta N, Yoshimoto K, Suzuki K, Yamaoka K, Takeuchi T. CD14(Bright)CD16+ Intermediate Monocytes are Induced by interleukin-10 and Positively Correlate With Disease Activity in Rheumatoid Arthritis. Arthritis Res Ther (2017) 19:28. doi: 10.1186/s13075-016-1216-6
57. Torsteinsdottir I, Arvidson NG, Hallgren R, Hakansson L. Monocyte Activation in Rheumatoid Arthritis (RA): Increased Integrin, Fc Gamma and Complement Receptor Expression and the Effect of Glucocorticoids. Clin Exp Immunol (1999) 115:554–60. doi: 10.1046/j.1365-2249.1999.00817.x
58. Matt P, Lindqvist U, Kleinau S. Elevated Membrane and Soluble Cd64: A Novel Marker Reflecting Altered Fcgammar Function and Disease in Early Rheumatoid Arthritis That Can be Regulated by Anti-Rheumatic Treatment. PloS One (2015) 10:e0137474. doi: 10.1371/journal.pone.0137474
59. Blom AB, Radstake TR, Holthuysen AE, Sloetjes AW, Pesman GJ, Sweep FG, et al. Increased Expression of Fcgamma Receptors II and III on Macrophages of Rheumatoid Arthritis Patients Results in Higher Production of Tumor Necrosis Factor Alpha and Matrix Metalloproteinase. Arthritis Rheum (2003) 48:1002–14. doi: 10.1002/art.10871
60. Magnusson SE, Engstrom M, Jacob U, Ulfgren AK, Kleinau S. High Synovial Expression of the Inhibitory FcgammaRIIb in Rheumatoid Arthritis. Arthritis Res Ther (2007) 9:R51. doi: 10.1186/ar2206
61. Di Ceglie I, Ascone G, Cremers NAJ, Sloetjes AW, Walgreen B, Vogl T, et al. Fcgamma Receptor-Mediated Influx of S100A8/A9-Producing Neutrophils as Inducer of Bone Erosion During Antigen-Induced Arthritis. Arthritis Res Ther4.14 (2018) 20:80. doi: 10.1186/s13075-018-1584-1
62. Jovanovic V, Dai X, Lim YT, Kemeny DM, MacAry PA. Fc Gamma Receptor Biology and Systemic Lupus Erythematosus. Int J Rheum Dis (2009) 12:293–8. doi: 10.1111/j.1756-185X.2009.01426.x
63. Kaifu T, Nakamura A. Polymorphisms of Immunoglobulin Receptors and the Effects on Clinical Outcome in Cancer Immunotherapy and Other Immune Diseases: A General Review. Int Immunol (2017) 29:319–25. doi: 10.1093/intimm/dxx041
64. Morgan AW, Griffiths B, Ponchel F, Montague BM, Ali M, Gardner PP, et al. Fcgamma Receptor Type IIIA is Associated With Rheumatoid Arthritis in Two Distinct Ethnic Groups. Arthritis Rheum (2000) 43:2328–34. doi: 10.1002/1529-0131(200010)43:10<2328::AID-ANR21>3.0.CO;2-Z
65. Morgan AW, Keyte VH, Babbage SJ, Robinson JI, Ponchel F, Barrett JH, et al. FcgammaRIIIA-158V and Rheumatoid Arthritis: A Confirmation Study. Rheumatol (Oxford) (2003) 42:528–33. doi: 10.1093/rheumatology/keg169
66. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F Polymorphism Influences the Binding of IgG by Natural Killer Cell Fc gammaRIIIa, Independently of the Fc gammaRIIIa-48L/R/H Phenotype. Blood (1997) 90:1109–14. doi: 10.1182/blood.V90.3.1109.1109_1109_1114
67. Sun Y, Mo L, Feng X, Yang D, Tan T, Zeng L, et al. Association of Fcgamma Receptor Type 2A and 3A Genotypes With Rheumatoid Arthritis in Chinese Population. Pharmacogenomics (2017) 18:255–64. doi: 10.2217/pgs-2016-0159
68. Lee YH, Bae SC, Song GG. FCGR2A, Fcgr3a, FCGR3B Polymorphisms and Susceptibility to Rheumatoid Arthritis: A Meta-Analysis. Clin Exp Rheumatol (2015) 33:647–54.
69. Avila-Pedretti G, Tornero J, Fernandez-Nebro A, Blanco F, Gonzalez-Alvaro I, Canete JD, et al. Variation at FCGR2A and Functionally Related Genes Is Associated With the Response to anti-TNF Therapy in Rheumatoid Arthritis. PloS One (2015) 10:e0122088. doi: 10.1371/journal.pone.0122088
70. Ben Mkaddem S, Hayem G, Jönsson F, Rossato E, Boedec E, Boussetta T, et al. Shifting Fcγriia-ITAM From Activation to Inhibitory Configuration Ameliorates Arthritis. J Clin Invest (2014) 124:3945–59. doi: 10.1172/jci74572
71. Chen JY, Wang CM, Ma CC, Luo SF, Edberg JC, Kimberly RP, et al. Association of a Transmembrane Polymorphism of Fcgamma Receptor IIb (FCGR2B) With Systemic Lupus Erythematosus in Taiwanese Patients. Arthritis Rheum (2006) 54:3908–17. doi: 10.1002/art.22220
72. Chen JY, Wang CM, Chang SW, Cheng CH, Wu YJ, Lin JC, et al. Association of FCGR3A and FCGR3B Copy Number Variations With Systemic Lupus Erythematosus and Rheumatoid Arthritis in Taiwanese Patients. Arthritis Rheumatol (2014) 66:3113–21. doi: 10.1002/art.38813
73. Ceccarelli F, Perricone C, Borgiani P, Ciccacci C, Rufini S, Cipriano E, et al. Genetic Factors in Systemic Lupus Erythematosus: Contribution to Disease Phenotype. J Immunol Res (2015) 2015:745647. doi: 10.1155/2015/745647
74. Lehrnbecher T, Foster CB, Zhu S, Leitman SF, Goldin LR, Huppi K, et al. Variant Genotypes of the Low-Affinity Fcgamma Receptors in Two Control Populations and a Review of Low-Affinity Fcgamma Receptor Polymorphisms in Control and Disease Populations. Blood (1999) 94:4220–32. doi: 10.1182/blood.V94.12.4220
75. Tsang ASMW, Nagelkerke SQ, Bultink IE, Geissler J, Tanck MW, Tacke CE, et al. Fc-Gamma Receptor Polymorphisms Differentially Influence Susceptibility to Systemic Lupus Erythematosus and Lupus Nephritis. Rheumatol (Oxford) (2016) 55:939–48. doi: 10.1093/rheumatology/kev433
76. el Bannoudi H, Ioan-Facsinay A, Toes RE. Bridging Autoantibodies and Arthritis: The Role of Fc Receptors. Curr Top Microbiol Immunol (2014) 382:303–19. doi: 10.1007/978-3-319-07911-0_14
77. Boross P, Verbeek JS. The Complex Role of Fcgamma Receptors in the Pathology of Arthritis. Springer Semin Immunopathol (2006) 28:339–50. doi: 10.1007/s00281-006-0049-9
78. Kagari T, Tanaka D, Doi H, Shimozato T. Essential Role of Fc Gamma Receptors in Anti-Type II Collagen Antibody-Induced Arthritis. J Immunol (2003) 170:4318–24. doi: 10.4049/jimmunol.170.8.4318
79. Kaplan CD, O’Neill SK, Koreny T, Czipri M, Finnegan A. Development of Inflammation in Proteoglycan-Induced Arthritis is Dependent on Fc Gamma R Regulation of the Cytokine/Chemokine Environment. J Immunol (2002) 169:5851–9. doi: 10.4049/jimmunol.169.10.5851
80. Blom AB, van Lent PL, van Vuuren H, Holthuysen AE, Jacobs C, van de Putte LB, et al. Fc Gamma R Expression on Macrophages is Related to Severity and Chronicity of Synovial Inflammation and Cartilage Destruction During Experimental Immune-Complex-Mediated Arthritis (ICA). Arthritis Res (2000) 2:489–503. doi: 10.1186/ar131
81. van Lent PL, van Vuuren AJ, Blom AB, Holthuysen AE, van de Putte LB, van de Winkel JG, et al. Role of Fc Receptor Gamma Chain in Inflammation and Cartilage Damage During Experimental Antigen-Induced Arthritis. Arthritis Rheum (2000) 43:740–52. doi: 10.1002/1529-0131(200004)43:4<740::AID-ANR4>3.0.CO;2-0
82. Corr M, Crain B. The Role of FcgammaR Signaling in the K/B X N Serum Transfer Model of Arthritis. J Immunol (2002) 169:6604–9. doi: 10.4049/jimmunol.169.11.6604
83. Díaz de Ståhl T, Andrén M, Martinsson P, Verbeek JS, Kleinau S. Expression of FcgammaRIII is Required for Development of Collagen-Induced Arthritis. Eur J Immunol (2002) 32:2915–22. doi: 10.1002/1521-4141(2002010)32:10<2915::Aid-immu2915>3.0.Co;2-4
84. Kaplan CD, Cao Y, Verbeek JS, Tunyogi-Csapo M, Finnegan A. Development of Proteoglycan-Induced Arthritis is Critically Dependent on Fcgamma Receptor Type III Expression. Arthritis Rheum (2005) 52:1612–9. doi: 10.1002/art.21030
85. Nabbe KC, Blom AB, Holthuysen AE, Boross P, Roth J, Verbeek S, et al. Coordinate Expression of Activating Fc Gamma Receptors I and III and Inhibiting Fc Gamma Receptor Type II in the Determination of Joint Inflammation and Cartilage Destruction During Immune Complex-Mediated Arthritis. Arthritis Rheum (2003) 48:255–65. doi: 10.1002/art.10721
86. van Lent PL, Nabbe K, Blom AB, Holthuysen AE, Sloetjes A, van de Putte LB, et al. Role of Activatory Fc Gamma RI and Fc Gamma RIII and Inhibitory Fc Gamma RII in Inflammation and Cartilage Destruction During Experimental Antigen-Induced Arthritis. Am J Pathol (2001) 159:2309–20. doi: 10.1016/s0002-9440(10)63081-7
87. Radstake TR, Franke B, Wenink MH, Nabbe KC, Coenen MJ, Welsing P, et al. The Functional Variant of the Inhibitory Fcgamma Receptor IIb (CD32B) is Associated With the Rate of Radiologic Joint Damage and Dendritic Cell Function in Rheumatoid Arthritis. Arthritis Rheum (2006) 54:3828–37. doi: 10.1002/art.22275
88. Nandakumar KS, Andren M, Martinsson P, Bajtner E, Hellstrom S, Holmdahl R, et al. Induction of Arthritis by Single Monoclonal IgG Anti-Collagen Type II Antibodies and Enhancement of Arthritis in Mice Lacking Inhibitory Fcgammariib. Eur J Immunol (2003) 33:2269–77. doi: 10.1002/eji.200323810
89. Yuasa T, Kubo S, Yoshino T, Ujike A, Matsumura K, Ono M, et al. Deletion of Fcgamma Receptor IIB Renders H-2(b) Mice Susceptible to Collagen-Induced Arthritis. J Exp Med (1999) 189:187–94. doi: 10.1084/jem.189.1.187
90. Yilmaz-Elis AS, Ramirez JM, Asmawidjaja P, van der Kaa J, Mus AM, Brem MD, et al. FcgammaRIIb on Myeloid Cells Rather Than on B Cells Protects From Collagen-Induced Arthritis. J Immunol (2014) 192:5540–7. doi: 10.4049/jimmunol.1303272
91. Brownlie RJ, Lawlor KE, Niederer HA, Cutler AJ, Xiang Z, Clatworthy MR, et al. Distinct Cell-Specific Control of Autoimmunity and Infection by Fcgammariib. J Exp Med (2008) 205:883–95. doi: 10.1084/jem.20072565
92. Tan Sardjono C, Mottram PL, van de Velde NC, Powell MS, Power D, Slocombe RF, et al. Development of Spontaneous Multisystem Autoimmune Disease and Hypersensitivity to Antibody-Induced Inflammation in Fcgamma Receptor IIa-transgenic Mice. Arthritis Rheum (2005) 52:3220–9. doi: 10.1002/art.21344
93. Anquetil F, Clavel C, Offer G, Serre G, Sebbag M. Igm and IgA Rheumatoid Factors Purified From Rheumatoid Arthritis Sera Boost the Fc Receptor- and Complement-Dependent Effector Functions of the Disease-Specific Anti-Citrullinated Protein Autoantibodies. J Immunol (2015) 194:3664–74. doi: 10.4049/jimmunol.1402334
94. Clavel C, Ceccato L, Anquetil F, Serre G, Sebbag M. Among Human Macrophages Polarised to Different Phenotypes, the M-CSF-oriented Cells Present the Highest Pro-Inflammatory Response to the Rheumatoid Arthritis-Specific Immune Complexes Containing ACPA. Ann Rheum Dis (2016) 75:2184–91. doi: 10.1136/annrheumdis-2015-208887
95. Laurent L, Clavel C, Lemaire O, Anquetil F, Cornillet M, Zabraniecki L, et al. Fcgamma Receptor Profile of Monocytes and Macrophages From Rheumatoid Arthritis Patients and Their Response to Immune Complexes Formed With Autoantibodies to Citrullinated Proteins. Ann Rheum Dis (2011) 70:1052–9. doi: 10.1136/ard.2010.142091
96. Machold KP, Stamm TA, Nell VP, Pflugbeil S, Aletaha D, Steiner G, et al. Very Recent Onset Rheumatoid Arthritis: Clinical and Serological Patient Characteristics Associated With Radiographic Progression Over the First Years of Disease. Rheumatol (Oxford) (2007) 46:342–9. doi: 10.1093/rheumatology/kel237
97. Clavel C, Nogueira L, Laurent L, Iobagiu C, Vincent C, Sebbag M, et al. Induction of Macrophage Secretion of Tumor Necrosis Factor Alpha Through Fcgamma Receptor IIa Engagement by Rheumatoid Arthritis-Specific Autoantibodies to Citrullinated Proteins Complexed With Fibrinogen. Arthritis Rheum (2008) 58:678–88. doi: 10.1002/art.23284
98. Liu Y, Masuda E, Blank MC, Kirou KA, Gao X, Park MS, et al. Cytokine-Mediated Regulation of Activating and Inhibitory Fc Gamma Receptors in Human Monocytes. J Leukoc Biol (2005) 77:767–76. doi: 10.1189/jlb.0904532
99. Allen E, Bakke AC, Purtzer MZ, Deodhar A. Neutrophil CD64 Expression: Distinguishing Acute Inflammatory Autoimmune Disease From Systemic Infections. Ann Rheum Dis (2002) 61:522–5. doi: 10.1136/ard.61.6.522
100. Hussein OA, El-Toukhy MA, El-Rahman HS. Neutrophil CD64 Expression in Inflammatory Autoimmune Diseases: Its Value in Distinguishing Infection From Disease Flare. Immunol Invest (2010) 39:699–712. doi: 10.3109/08820139.2010.491520
101. Fjaertoft G, Hakansson LD, Pauksens K, Sisask G, Venge P. Neutrophil Cd64 (FcgammaRI) Expression Is a Specific Marker of Bacterial Infection: A Study on the Kinetics and the Impact of Major Surgery. Scand J Infect Dis (2007) 39:525–35. doi: 10.1080/00365540601113693
102. Fjaertoft G, Pauksen K, Hakansson L, Xu S, Venge P. Cell Surface Expression of FcgammaRI (CD64) on Neutrophils and Monocytes in Patients With Influenza A, With and Without Complications. Scand J Infect Dis (2005) 37:882–9. doi: 10.1080/00365540500348929
103. Bournazos S, Wang TT, Dahan R, Maamary J, Ravetch JV. Signaling by Antibodies: Recent Progress. Annu Rev Immunol (2017) 35:285–311. doi: 10.1146/annurev-immunol-051116-052433
104. Oppegaard O, Skodvin B, Halse AK, Langeland N. CD64 as a Potential Biomarker in Septic Arthritis. BMC Infect Dis (2013) 13:278. doi: 10.1186/1471-2334-13-278
105. Li S, Huang X, Chen Z, Zhong H, Peng Q, Deng Y, et al. Neutrophil CD64 Expression as a Biomarker in the Early Diagnosis of Bacterial Infection: A Meta-Analysis. Int J Infect Dis (2013) 17:e12–23. doi: 10.1016/j.ijid.2012.07.017
106. Cid J, Aguinaco R, Sanchez R, Garcia-Pardo G, Llorente A. Neutrophil CD64 Expression as Marker of Bacterial Infection: A Systematic Review and Meta-Analysis. J Infect (2010) 60:313–9. doi: 10.1016/j.jinf.2010.02.013
107. Tsuboi N, Ernandez T, Li X, Nishi H, Cullere X, Mekala D, et al. Regulation of Human Neutrophil Fcgamma Receptor IIa by C5a Receptor Promotes Inflammatory Arthritis in Mice. Arthritis Rheum (2011) 63:467–78. doi: 10.1002/art.30141
108. Karsenty G, Wagner EF. Reaching a Genetic and Molecular Understanding of Skeletal Development. Dev Cell (2002) 2:389–406. doi: 10.1016/s1534-5807(02)00157-0
109. Teitelbaum SL, Ross FP. Genetic Regulation of Osteoclast Development and Function. Nat Rev Genet (2003) 4:638–49. doi: 10.1038/nrg1122
110. Boyle WJ, Simonet WS, Lacey DL. Osteoclast Differentiation and Activation. Nature (2003) 423:337–42. doi: 10.1038/nature01658
111. Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T Cells, Bone Loss, and Mammalian Evolution. Annu Rev Immunol (2002) 20:795–823. doi: 10.1146/annurev.immunol.20.100301.064753
112. Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, Not Osteoblasts or Lining Cells, Are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PloS One (2015) 10:e0138189. doi: 10.1371/journal.pone.0138189
113. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for Osteocyte Regulation of Bone Homeostasis Through RANKL Expression. Nat Med (2011) 17:1231–4. doi: 10.1038/nm.2452
114. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front Immunol (2014) 5:511. doi: 10.3389/fimmu.2014.00511
115. Terashima A, Takayanagi H. Overview of Osteoimmunology. Calcif Tissue Int (2018) 102:503–11. doi: 10.1007/s00223-018-0417-1
116. Compton JT, Lee FY. A Review of Osteocyte Function and the Emerging Importance of Sclerostin. J Bone Joint Surg Am (2014) 96:1659–68. doi: 10.2106/jbjs.M.01096
117. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev Cell (2002) 3:889–901. doi: 10.1016/s1534-5807(02)00369-6
118. Braun T, Zwerina J. Positive Regulators of Osteoclastogenesis and Bone Resorption in Rheumatoid Arthritis. Arthritis Res Ther (2011) 13:235. doi: 10.1186/ar3380
119. Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, et al. Costimulatory Signals Mediated by the ITAM Motif Cooperate With RANKL for Bone Homeostasis. Nature (2004) 428:758–63. doi: 10.1038/nature02444
120. Ivashkiv LB. Cross-Regulation of Signaling by ITAM-Associated Receptors. Nat Immunol (2009) 10:340–7. doi: 10.1038/ni.1706
121. Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, et al. The Immunomodulatory Adapter Proteins DAP12 and Fc Receptor Gamma-Chain (FcRgamma) Regulate Development of Functional Osteoclasts Through the Syk Tyrosine Kinase. Proc Natl Acad Sci USA (2004) 101:6158–63. doi: 10.1073/pnas.0401602101
122. Kaifu T, Nakahara J, Inui M, Mishima K, Momiyama T, Kaji M, et al. Osteopetrosis and Thalamic Hypomyelinosis With Synaptic Degeneration in DAP12-deficient Mice. J Clin Invest (2003) 111:323–32. doi: 10.1172/JCI16923
123. Paradowska-Gorycka A, Jurkowska M. Structure, Expression Pattern and Biological Activity of Molecular Complex TREM-2/DAP12. Hum Immunol (2013) 74:730–7. doi: 10.1016/j.humimm.2013.02.003
124. Tsukasaki M, Takayanagi H. Osteoimmunology: Evolving Concepts in Bone-Immune Interactions in Health and Disease. Nat Rev Immunol (2019) 19:626–42. doi: 10.1038/s41577-019-0178-8
125. Guerrini MM, Takayanagi H. The Immune System, Bone and RANKL. Arch Biochem Biophys (2014) 561:118–23. doi: 10.1016/j.abb.2014.06.003
126. Harre U, Keppeler H, Ipseiz N, Derer A, Poller K, Aigner M, et al. Moonlighting Osteoclasts as Undertakers of Apoptotic Cells. Autoimmunity (2012) 45:612–9. doi: 10.3109/08916934.2012.719950
127. Harre U, Lang SC, Pfeifle R, Rombouts Y, Fruhbeisser S, Amara K, et al. Glycosylation of Immunoglobulin G Determines Osteoclast Differentiation and Bone Loss. Nat Commun (2015) 6:6651. doi: 10.1038/ncomms7651
128. Kleyer A, Finzel S, Rech J, Manger B, Krieter M, Faustini F, et al. Bone Loss Before the Clinical Onset of Rheumatoid Arthritis in Subjects With Anticitrullinated Protein Antibodies. Ann Rheum Dis (2014) 73:854–60. doi: 10.1136/annrheumdis-2012-202958
129. Kleyer A, Schett G. Arthritis and Bone Loss: A Hen and Egg Story. Curr Opin Rheumatol (2014) 26:80–4. doi: 10.1097/BOR.0000000000000007
130. Onuora S. Osteoimmunology: IgG Immune Complexes Directly Regulate Bone Homeostasis. Nat Rev Rheumatol (2015) 11:257. doi: 10.1038/nrrheum.2015.51
131. Karsten CM, Kohl J. A Bone to Pick With Fc Gamma Receptors. Ann Transl Med (2015) 3:218. doi: 10.3978/j.issn.2305-5839.2015.07.11
132. Kastbom A, Ahmadi A, Soderkvist P, Skogh T. The 158V Polymorphism of Fc Gamma Receptor Type IIIA in Early Rheumatoid Arthritis: Increased Susceptibility and Severity in Male Patients (the Swedish TIRA Project). Rheumatol (Oxford) (2005) 44:1294–8. doi: 10.1093/rheumatology/kei010
133. Pagan JD, Kitaoka M, Anthony RM. Engineered Sialylation of Pathogenic Antibodies In Vivo Attenuates Autoimmune Disease. Cell (2018) 172:564–577 e513. doi: 10.1016/j.cell.2017.11.041
134. Sehic E, Westerlund A, Lagerquist MK, Lerner UH, Carlsten H, Henning P, et al. Immunoglobulin G Complexes Without Sialic Acids Enhance Osteoclastogenesis But do Not Affect Arthritis-Mediated Bone Loss. Scand J Immunol (2021) 93:e13009. doi: 10.1111/sji.13009
135. Zeng KQ, Gong FY, Pan XH, Miao J, Gong Z, Wang J, et al. IgG Immunocomplexes Drive the Differentiation of a Novel Subset of Osteoclasts Independent of RANKL and Inflammatory Cytokines. J Bone Miner Res (2021). doi: 10.1002/jbmr.4281
136. van Lent P, Nabbe KC, Boross P, Blom AB, Roth J, Holthuysen A, et al. The Inhibitory Receptor FcgammaRII Reduces Joint Inflammation and Destruction in Experimental Immune Complex-Mediated Arthritides Not Only by Inhibition of FcgammaRI/III But Also by Efficient Clearance and Endocytosis of Immune Complexes. Am J Pathol (2003) 163:1839–48. doi: 10.1016/s0002-9440(10)63543-2
137. Visitchanakun P, Saiworn W, Jongwattanapisan P, Leelahavanichkul A, Pisitkun P, Lotinun S. Lupus-Like Disease in FcγRIIB(-/-) Mice Induces Osteopenia. Sci Rep (2019) 9:17342. doi: 10.1038/s41598-019-53963-z
138. Lee MJ, Lim E, Mun S, Bae S, Murata K, Ivashkiv LB, et al. Intravenous Immunoglobulin (Ivig) Attenuates Tnf-Induced Pathologic Bone Resorption and Suppresses Osteoclastogenesis by Inducing A20 Expression. J Cell Physiol (2016) 231:449–58. doi: 10.1002/jcp.25091
139. MacLellan LM, Montgomery J, Sugiyama F, Kitson SM, Thummler K, Silverman GJ, et al. Co-Opting Endogenous Immunoglobulin for the Regulation of Inflammation and Osteoclastogenesis in Humans and Mice. Arthritis Rheum (2011) 63:3897–907. doi: 10.1002/art.30629
140. Tsokos GC. Systemic Lupus Erythematosus. A Disease With a Complex Pathogenesis. Lancet (2001) 358 Suppl:S65. doi: 10.1016/s0140-6736(01)07077-5
141. Salmon JE, Pricop L. Human Receptors for Immunoglobulin G: Key Elements in the Pathogenesis of Rheumatic Disease. Arthritis Rheum (2001) 44:739–50. doi: 10.1002/1529-0131(200104)44:4<739::AID-ANR129>3.0.CO;2-O
142. Zuniga R, Markowitz GS, Arkachaisri T, Imperatore EA, D’Agati VD, Salmon JE. Identification of IgG Subclasses and C-reactive Protein in Lupus Nephritis: The Relationship Between the Composition of Immune Deposits and FCgamma Receptor Type IIA Alleles. Arthritis Rheum (2003) 48:460–70. doi: 10.1002/art.10930
143. Deng GM, Tsokos GC. Pathogenesis and Targeted Treatment of Skin Injury in SLE. Nat Rev Rheumatol (2015) 11:663–9. doi: 10.1038/nrrheum.2015.106
144. Zhang Q, Xiang L, Zaman MH, Dong W, He G, Deng GM. Predominant Role of Immunoglobulin G in the Pathogenesis of Splenomegaly in Murine Lupus. Front Immunol (2019) 10:3020. doi: 10.3389/fimmu.2019.03020
145. Deng GM, Liu L, Kyttaris VC, Tsokos GC. Lupus Serum IgG Induces Skin Inflammation Through the TNFR1 Signaling Pathway. J Immunol (2010) 184:7154–61. doi: 10.4049/jimmunol.0902514
146. Malmstrom V, Catrina AI, Klareskog L. The Immunopathogenesis of Seropositive Rheumatoid Arthritis: From Triggering to Targeting. Nat Rev Immunol (2017) 17:60–75. doi: 10.1038/nri.2016.124
147. Auréal M, Machuca-Gayet I, Coury F. Rheumatoid Arthritis in the View of Osteoimmunology. Biomolecules (2020) 11:48. doi: 10.3390/biom11010048
148. Scherer HU, van der Woude D, Ioan-Facsinay A, l Bannoudi H, Trouw LA, Wang J, et al. Glycan Profiling of Anti-Citrullinated Protein Antibodies Isolated From Human Serum and Synovial Fluid. Arthritis Rheum (2010) 62:1620–9. doi: 10.1002/art.27414
149. Correction: Autoantibodies to Citrullinated Proteins Induce Joint Pain Independent of Inflammation Via a Chemokine-Dependent Mechanism. Ann Rheum Dis (2019) 78:865. doi: 10.1136/annrheumdis-2015-208094corr1
150. Wigerblad G, Bas DB, Fernades-Cerqueira C, Krishnamurthy A, Nandakumar KS, Rogoz K, et al. Autoantibodies to Citrullinated Proteins Induce Joint Pain Independent of Inflammation Via a Chemokine-Dependent Mechanism. Ann Rheum Dis (2016) 75:730–8. doi: 10.1136/annrheumdis-2015-208094
151. Correction: Identification of a Novel Chemokine-Dependent Molecular Mechanism Underlying Rheumatoid Arthritis-Associated Autoantibody-Mediated Bone Loss. Ann Rheum Dis (2019) 78:866. doi: 10.1136/annrheumdis-2015-208093corr1
152. Krishnamurthy A, Joshua V, Haj Hensvold A, Jin T, Sun M, Vivar N, et al. Identification of a Novel Chemokine-Dependent Molecular Mechanism Underlying Rheumatoid Arthritis-Associated Autoantibody-Mediated Bone Loss. Ann Rheum Dis (2016) 75:721–9. doi: 10.1136/annrheumdis-2015-208093
153. Clarkson SB, Bussel JB, Kimberly RP, Valinsky JE, Nachman RL, Unkeless JC. Treatment of Refractory Immune Thrombocytopenic Purpura With an anti-Fc Gamma-Receptor Antibody. N Engl J Med (1986) 314:1236–9. doi: 10.1056/nejm198605083141907
154. Magnusson SE, Andrén M, Nilsson KE, Sondermann P, Jacob U, Kleinau S. Amelioration of Collagen-Induced Arthritis by Human Recombinant Soluble Fcgammariib. Clin Immunol (2008) 127:225–33. doi: 10.1016/j.clim.2008.02.002
155. Werwitzke S, Trick D, Sondermann P, Kamino K, Schlegelberger B, Kniesch K, et al. Treatment of Lupus-Prone NZB/NZW F1 Mice With Recombinant Soluble Fc Gamma Receptor II (Cd32). Ann Rheum Dis (2008) 67:154–61. doi: 10.1136/ard.2006.068981
156. Jain A, Olsen HS, Vyzasatya R, Burch E, Sakoda Y, Mérigeon EY, et al. Fully Recombinant IgG2a Fc Multimers (Stradomers) Effectively Treat Collagen-Induced Arthritis and Prevent Idiopathic Thrombocytopenic Purpura in Mice. Arthritis Res Ther (2012) 14:R192. doi: 10.1186/ar4024
157. Zuercher AW, Spirig R, Baz Morelli A, Rowe T, Käsermann F. Next-generation Fc Receptor-Targeting Biologics for Autoimmune Diseases. Autoimmun Rev (2019) 18:102366. doi: 10.1016/j.autrev.2019.102366
158. Yu X, Lazarus AH. Targeting Fcγrs to Treat Antibody-Dependent Autoimmunity. Autoimmun Rev (2016) 15:510–2. doi: 10.1016/j.autrev.2016.02.006
159. Konstaninova TS, Leonidovna IV, Hellmann A, Kyrcz-Krzemien S, Tillmanns S, Sondermann P, et al. Interim Results From a Phase Ib/Iia Clinical Trial With the Soluble Fc-Gamma IIb Receptor SM101 for the Treatment of Primary Immune Thrombocytopenia. Blood (2012) 120:3388. doi: 10.1182/blood.V120.21.3388.3388
160. Tillmanns S, Kolligs C, D’Cruz DP, Doria A, Hachulla E, Voll RE, et al. SM101, a Novel Recombinant, Soluble, Human Fc Gamma IIB Receptor, in the Treatment of Systemic Lupus Erythematosus: Results of a Double-Blind, Placebo-Controlled Multicenter Study. Arthritis Rheumatol (2014) 66:S1238–8. doi: 10.1002/art.38914
161. Clynes R. Protective Mechanisms of IVIG. Curr Opin Immunol (2007) 19:646–51. doi: 10.1016/j.coi.2007.09.004
162. Nimmerjahn F, Ravetch JV. Anti-Inflammatory Actions of Intravenous Immunoglobulin. Annu Rev Immunol (2008) 26:513–33. doi: 10.1146/annurev.immunol.26.021607.090232
Keywords: FcγRs, autoantibodies, osteoclasts, bone erosion, inflammatory arthritis
Citation: Zuo Y and Deng G-M (2021) Fc Gamma Receptors as Regulators of Bone Destruction in Inflammatory Arthritis. Front. Immunol. 12:688201. doi: 10.3389/fimmu.2021.688201
Received: 30 March 2021; Accepted: 09 June 2021;
Published: 23 June 2021.
Edited by:
Naoki Iwamoto, Nagasaki University Hospital, JapanReviewed by:
Alakendu Ghosh, Dept. of Health & Family Welfare, IndiaPeng Zhang, Huazhong University of Science and Technology, China
Hyung-Mun Yun, Kyung Hee University, South Korea
Shoichi Fukui, Nagasaki University, Japan
Copyright © 2021 Zuo and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guo-Min Deng, Z21kZW5nQGh1c3QuZWR1LmNu