 Di Wu
Di Wu Anna Cline-Smith
Anna Cline-Smith Elena Shashkova
Elena Shashkova Ajit Perla
Ajit Perla Aditya Katyal
Aditya Katyal Rajeev Aurora
Rajeev Aurora- Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, St. Louis, MO, United States
Osteoporosis is the most prevalent metabolic bone disease that affects half the women in the sixth and seventh decade of life. Osteoporosis is characterized by uncoupled bone resorption that leads to low bone mass, compromised microarchitecture and structural deterioration that increases the likelihood of fracture with minimal trauma, known as fragility fractures. Several factors contribute to osteoporosis in men and women. In women, menopause – the cessation of ovarian function, is one of the leading causes of primary osteoporosis. Over the past three decades there has been growing appreciation that the adaptive immune system plays a fundamental role in the development of postmenopausal osteoporosis, both in humans and in mouse models. In this review, we highlight recent data on the interactions between T cells and the skeletal system in the context of postmenopausal osteoporosis. Finally, we review recent studies on the interventions to ameliorate osteoporosis.
Introduction
A great achievement of modern medicine is the increased lifespan of the human population. Unfortunately, the comorbidities of aging have created a large economic and health burden on society. The current challenge is to improve the healthspan and thus to reduce the burden. Osteoporosis is the most prevalent metabolic bone disease that affects half the women and one third of men, typically, in the sixth and seventh decade of life (1, 2). Osteoporosis is characterized by uncoupled bone resorption that leads to low bone mass, compromised microarchitecture and structural deterioration that increases the likelihood of fracture with minimal trauma. These fragility fractures lead to disproportionally high mortality rate and a drastic decline in quality of life for those affected.
Bone remodeling occurs th0roughout life and is a coordinated process to repair microfractures and maintain bone mass. Imbalances in the bone remodeling process underscore the pathophysiology of osteoporosis. Bone remodeling is a tightly coupled: resorption precedes formation and the amount of bone formed is balanced with the amount resorbed. Remodeling can be initiated by hormonal, environmental and nutritional factors (3). The major cell types involved in bone remodeling are bone resorbing osteoclasts (OC) and bone forming osteoblasts (OB). Over the last decade the bone-embedded osteocytes (Ocy) have also emerged as a key regulators. OC are multinucleated cells from the monocytic lineage whose differentiation depends on receptor activator of NF-κB (RANK) and its ligand (RANKL). OB differentiate from the mesenchymal stem cell (MSC) lineage and is regulated by several signaling pathways such as WNT/β-catenin and BMP. During remodeling the OC and OB form the bone remodeling unit (BRU). Ocy are stellate like cells enclosed within mineralized bone. They serve as mechanosensors within the bone and play a key regulatory role in bone homeostasis, directing and coordinating repair by regulating the BRU.
It was recognized nearly eight decades ago that involutional osteoporosis in postmenopausal women is mediated by loss of estrogen (E2) (4). The mechanism for how E2 loss leads to increased bone resorption has remained, despite intense focus of investigations (5). Decreased calcium absorption (6, 7), decline in renal function (8) and impaired vitamin D metabolism (9, 10) with aging and menopause. Over the past three decades there has been growing appreciation that the adaptive immune system plays a fundamental role in the development of postmenopausal osteoporosis (PMOP), both in humans and in mouse models. The recognition that T-cell derived cytokines affect bone has given rise to the field of osteoimmunology, a word was first coined in 2000 by Arron and Choi (11). There have been major advances in our understanding of the pro-resorptive effects of pro-inflammatory cytokine, in particular TNFα and interleukine IL-17A made by T cell. In this review, we highlight recent data on the interactions between T cells and the skeletal system in the context of PMOP. We also review recent studies on the interventions to ameliorate osteoporosis, with insights into immunomodulatory options. Finally, we highlight some questions that still remain unanswered.
Postmenopausal Osteoporosis
While dietary, lifestyle and other factors impact bone health (12, 13), in general there are two main reasons for the decline of bone mass. The skeletal system grows rapidly postnatally and through puberty and peak bone mass is attained by mid to late 20s (14). Both men and women gradually loss bone mass as they age (15) and the rate of loss varies by anatomical site (16). In addition to aging, loss of sex hormones and in particular estrogen (E2) contribute to skeletal homeostasis (5, 17, 18).
Sex hormones increase during puberty and are maintained during reproductive age. While testosterone (T) decreases linearly with age in men, women experience a sharp decline in E2 at menopause. Menopause, is the cessation of ovarian function, is one of the leading causes of primary osteoporosis. Early studies suggested that E2 directly regulates OC (19–22) and OB (23, 24) and its loss results in long lived OC and impaired OB leading to uncoupled bone resorption (25). Accordingly, PMOP has been traditionally regarded as an endocrinal, E2 deficiency mediated disease. While epidemiological observation suggest that E2 loss is responsible for osteoporosis in both sexes, the mechanism in males remains unclear. The role of estrogen and androgens on the bone have been extensively reviewed previously (25–27). In the next section, we will focus on the role of the immune system in the pathogenesis of PMOP.
The Crosstalk Between the Bone and the Immune System
In the past few decades, evidence has emerged supporting the notion that E2 loss promotes persistent inflammation that promote osteoporosis and perhaps other comorbidities. The mechanistic studies for linking E2 loss at menopause and activation of the T cells has come from ovariectomy (OVX) of rodents and key outcomes have been validated in human studies. In this section, we highlight recent advances in our understanding on how T cells and proinflammatory cytokines, namely TNFα and IL-17A, contribute to the pathogenesis of PMOP (Figure 1).
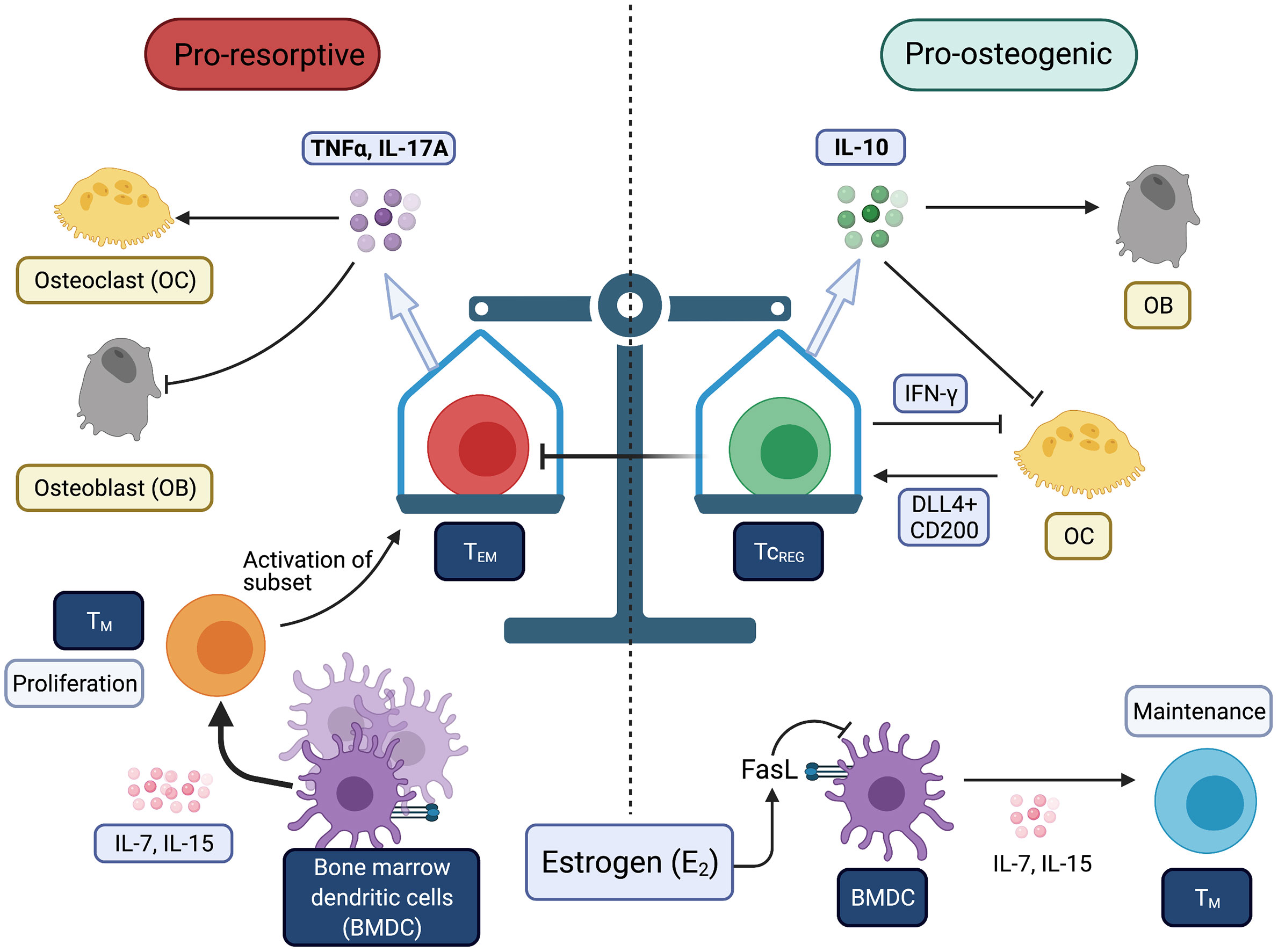
Figure 1 Balance between immunogenic and tolergenic states is linked with resorptive and osteogenic states of the bone. Chronic inflammation may be derived by increases in effector T-cells (TEFF) and effector memory T-cells (TEM). Bone is very sensitive to TNFα and IL-17A produced by T-cells. Estrogen (E2) prevents the conversion of TM to TEM. Tolerogenic T-cells (i.e., TREG and TcREG) promote bone formation through direct and indirect mechanisms. All Figures were created with (BioRender.com).
Inflammation Tips the Balance in Favor of Bone Resorption Through Osteoclasts
Takayanagi et al. were the first to report the bone-immune cross talk, demonstrating that T-cell produced IFN-γ can inhibit RANKL signaling during OC differentiation (28). Because Th1 are major producers of IFN-γ, inflammatory bone loss was thought as a Th1 mediated pathology. It was later demonstrated that Th17 cells are key drivers of bone erosion (29) and IL-17A is a potent promoter of bone destruction, particularly in the context of autoimmune pathologies (30–32). TNFα has also been shown to directly act on OC and its precursors in synergy with RANKL to promote osteoclastogenesis (33–36). Bone appears to be sensitive to T-cell derived cytokines even at distal anatomical sites. For instance, decline in bone mass is observed in patients with chronic HIV (37, 38), Hepatitis B and C (39) infections. Hepatic viruses also affect conversion of vitamin D3 to the metabolically active form calcitriol because they infect hepatocytes and affect calcium absorption in addition to increased IL-17A production by T-cells (40, 41). Increased prevalence of fracture are also observed in patients with rheumatoid arthritis (RA), inflammatory bowel disease (IBD) and chronic obstructive pulmonary disease (COPD) (42–45). The local cytokine milieu can contextually promote or protect against bone loss and the mechanism for how TNFα and IL-17A favor bone resorption via OC have been reviewed extensively (46, 47).
In the past decade, several studies showed that the immune system and inflammation play a critical pathogenic role in uncoupled bone loss in the context of E2 loss (30, 48–52). OVX of sexually mature mice that were T-cell deficient showed decreased bone loss, demonstrating that T-cells are required for promoting bone resorption (53–57). Recently, our lab has described a new pathway where E2 loss leads to chronic low-grade production of the proinflammatory cytokines TNFα and IL-17 by converting memory T-cells (TM) to effector TM (TEM). We showed that there us increased levels of IL-7 and IL-15. Both these cytokines are produced by bone marrow dendritic cells (BMDCs). E2 induces Fas ligand and apoptosis of BMDC and also TM. In the absence of E2, the BMDC become long-lived, which leads to higher IL-7 and IL-15 and to antigen-independent activation of a subset of TM to produce TNFα and IL-17A (58) (Figure 2). The canonical activation of TM and subsequent conversion to TEM are antigen-dependent (59), thus the activation observed by E2-loss does not follow the established paradigm. OVX of IL15RαΔT-cells mice, where TM cannot convert into TEM, did not result in bone loss (58). Our results uncovered another aspect of how E2 is anti-inflammatory, as it maintains TM homeostasis and limits their conversion to TEM in the absence of antigen.
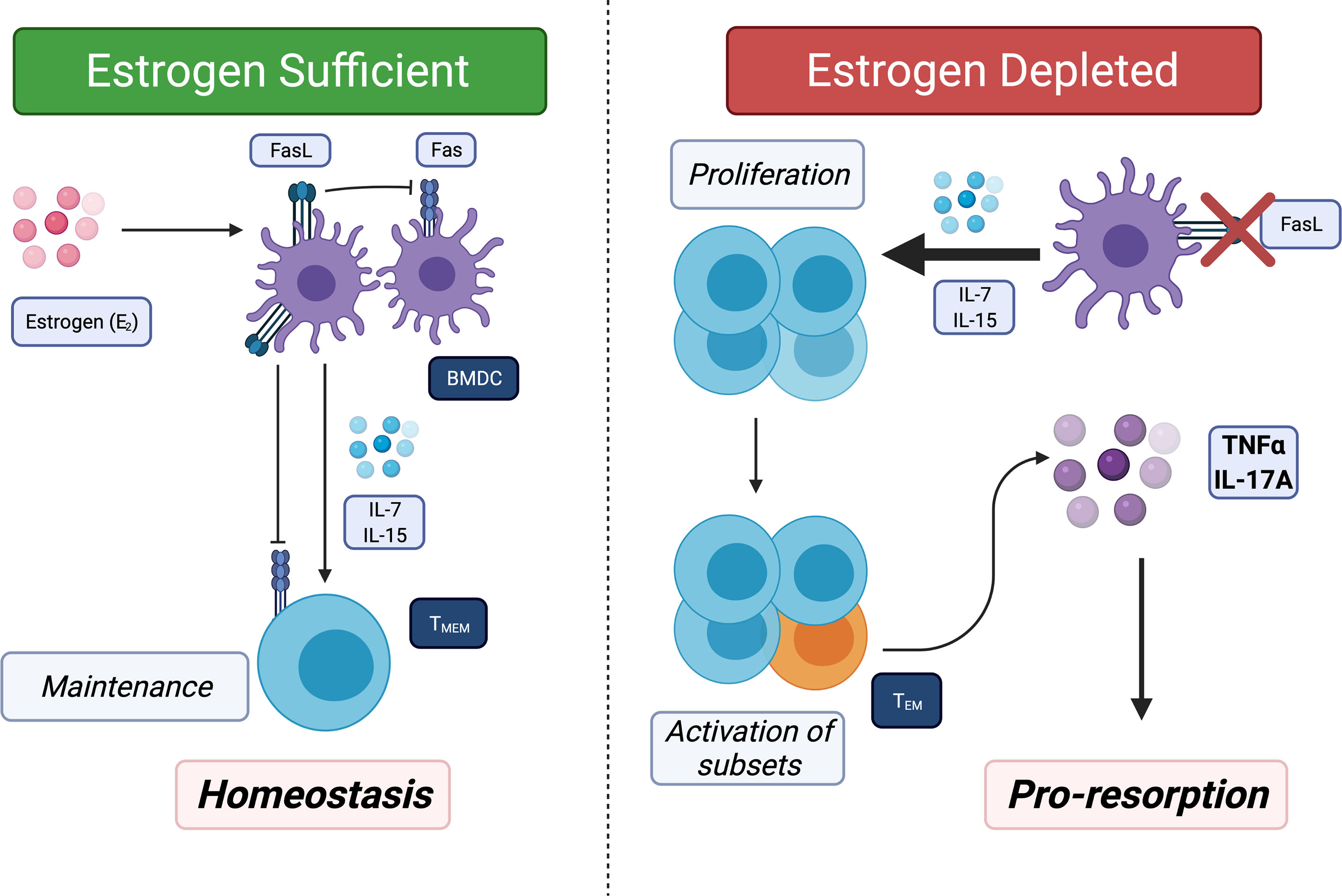
Figure 2 Estrogen (E2) regulates bone marrow resident memory T-cells (TBRM) homeostasis and E2 loss promotes conversion of TBRM to TEM. TM migrate to and take up long-term residence in the bone marrow. Left panel: Bone marrow resident dendritic cells (BMDC) secrete IL-7, IL-15 or both to promote survival of TM. In E2 replete females, BMDC have a short lifespan because E2 induces FasL in the BMDC. In addition, IL-15 induces Fas in proliferating TM in response to IL-7 and IL-15 thus maintain a homeostatic pool of TBRM. Right: In absence of E2, Fas ligand (FasL) is no longer induced leading to increased lifespans of BMDC and high concentrations of IL-7 and IL-15. In the presence of high IL-7 and IL-15 and absence of FasL, all TM proliferate and a subset (~5 to 10%) produce TNFα and IL-17A, which then promotes bone resorption and also limits bone formation.
The gut microbiome (GMB) plays an important role in regulating bone mass. A number of studies have shown an association between GBM and bone health in both animal models (60, 61) and in humans (13). Germfree (GF) mice have increased bone mass compared to conventionally raised (CONV-R) mice, and restoration of GBM normalized bone mass in GF mice (62). E2 loss increases gut permeability (63–65), which leads to increased priming and activation of inflammation in the gut mucosa, leading to the generation Th17 cells (66). GF mice do not lose bone post-OVX, and probiotics can prevent OVX-induced bone loss (67, 68). Recent studies have suggested GMB produce microbial metabolites that have regulatory function on distal organs, including the bone. GMB derived butyrate, polyamines and short-chain fatty acids have been shown to induce regulatory T cell (TREG) generation in the colon and be able to directly regulate the BRU (69–71). Thus, there appear to be several mechanisms by which GMB modulate bone health: first, GMB produce metabolites that directly modulate bone mass. Second GBM induce Th17 cell to promote bone loss or TREG to limit bone loss. Finally, GBM not only induce Th17 cells but many of these effector T-cells become TM. A subset of TM migrate to the bone marrow to become bone marrow resident TM (72). Thus prior exposures of pathogens and commensals are encoded in the TM. This may explain (at the population level) why only about half of postmenopausal women develop osteoporosis. Women who have more exposure to Th17 inducing microbes through their life would have a larger pool of TM that convert to TEM that produce IL-17 and TNFα postmenopause.
How Does Inflammation or Resolution of Inflammation Regulate Bone Mass?
The field has primarily focused on the effect of inflammation on OC as discussed above. How inflammation restrains bone formation is less well studied. Under coupled bone remodeling conditions, the increase in resorption is accompanied by recruitment of mesenchymal stromal cells (MSC) and their conversion to OB. However, in the presence of inflammatory cytokine (i.e., TNFα, IL-17 and IL-1β) this process appears to be impaired. Thus, bone formation lags behind bone resorption. Next, we discuss how proinflammatory cytokines effects the osteolineage.
Early in vitro culture studies showed that TNFα inhibited MSC to OB transition by regulating RUNX2 expression, a master transcription factor that commits MSC into osteogenic pathway. TNFα also targets Osterix (OSX; SP7) expression, a key transcription necessary for osteoblast maturation (73–75). Osteoblastogenesis is sensitive to glucose levels (76) and OB differentiation is regulated via mTOR pathway (77–80). While not clearly established in OB, there is precedence that TNFα regulates cellular metabolism in adipocytes and muscles cells (81–83). Since these three cell types all originate from MSC via different developmental pathway, it is likely that TNFα targets mTOR complexes in OB to alter cellular metabolism. Indeed, in vitro evidence demonstrated that TNFα can modulate autophagy and apoptosis via NF-κB signaling in OB (84–86) both of which are controlled by mTOR. While these results are controversial, there are reports indicating that IL-17A is able to affect MSC to OB differentiation as well as mature OB function and is summarized in a recent review (47). Understanding the effects of TNFα and IL-17A on OB will provide further insight into the imbalance between bone resorption and bone formation.
The effect of inflammation on Ocy is largely unknown. Ocy are regulators of bone homeostasis (87). Ocy produce RANKL that predominantly regulates osteoclastogenesis during remodeling (88, 89). In the presence of TNFα a much lower concentration of RANKL is needed to initiate osteoclastogenesis (90). Ablation of RANKL in Ocy via Dmp1-Cre protected against vertebral bone loss (91). TNFα and IL-17A can target Ocy to produce RANKL and thus contribute to increased resorption (92, 93). Furthermore, IL-17A can target Ocy to increase osteogenic differentiation of MSC in cooperation with OB (94). All evidence taken together, suggest that Ocy are at the center of BRU balance and regulate bone resorption and formation according to biological needs. OVX induced Ocy apoptosis (95) possibly via TNFα, IL-17A or both, suggesting that inflammation regulates bone health not just through OC or OB. Interestingly during lactation, where there is E2 loss triggers the same BMDC induced uncoupled bone resorption as OVX, Ocy have been shown to undergo a process called osteocytic osteolysis that promotes bone resorption to release calcium from cortical bone (96). Further investigation is need to understand the role Ocy play in PMOP.
It is clear that the skeletal system is exquisitely sensitive to chronic inflammation suggesting that the skeletal system is “a canary in the coal mine” – a sensor of overall persistent inflammatory burden. All currently available data is consistent with the observation that PMOP is mediated by inflammation. Specifically, E2 loss induces the conversion of bone marrow resident TM to TEM that secrete TNFα and IL-17A. These cytokines promote osteoclastogenesis and bone resorption and most likely also limit bone formation. However, the effect of TNFα and IL-17A on MSC, mature OB or Ocy is much less well understood. Future therapies for the treatment of PMOP should also address the underlying inflammation, which we will discuss in the following section.
Therapeutic Interventions
A number of therapies have been developed to treat osteoporosis in postmenopausal women. The traditional therapies fall into two classes: anti-resorptive and bone anabolics. Each has been used independently and in some clinical trials also used in combination.
Antiresorptive
The most commonly prescribed medication for osteoporosis are antiresorptives, notably bisphosphonates and denosumab. It has been reported that antiresorptives can interact with the immune system. Bisphosphonates have been shown to boost B-cell function and promote humoral immunity (97). Denosumab has been associated with increased risks of infection (98) and more recently, being investigated for oncology alongside immune checkpoint inhibitors (99). One drawback with this class of medications are potential severe adverse effects. Osteonecrosis of the jaw (ONJ) is observed in 1-3% of patients on anti-resorptive therapies with certain predisposing factors (i.e., after tooth extraction or in people with type 2 diabetes) (100–102). Atypical femoral fractures have also been reported in patients on bisphosphonates (103) while denosumab discontinuation have been associated with higher risk of vertebral fractures (104). Another disadvantage of anti-resorptive treatment is that there is a specific window where they are most effective. In addition, inhibition of bone resorption prevents bone remodeling and repair leading to effete bone that fractures from minimal trauma (105). As a result, while the patient may maintain BMD, it does not reflect that whether they have improved bone quality.
Anabolics
The second class of therapies are bone anabolics. A commonly used bone anabolic is teriparatide, derived from parathyroid hormone (PTH) (106). More recently romosozumab that targets sclerostin (product of the SOST gene) (107) has been approved as a bone anabolic biologic. Neutralization of sclerostin increases OB numbers and simultaneously suppresses bone resorption thus promoting bone formation. However, due to adaptive changes in bone and potential adverse effects with prolonged use, bone anabolic therapies are limited in their use (108–110), particularly in special populations (111). Antiresorptive therapies (RANKL blockade or bisphosphonates) in postmenopausal women did not increase bone mass in the SHOTZ clinical trial (112, 113) indicating that deficit in OB activity remains. Remarkably, while bone anabolic therapy improve bone mineral density, bone microarchitecture did not improve unless both antiresorptive and anabolic agents were combined (114–116). These results suggest that efficacy is obtained by using both antiresorptive (brakes on OC) and anabolics as accelerator. Since sclerostin is primarily (but not exclusively) produced by Ocy, the action and mechanism of action on Ocy remains to be understood.
All current therapies target the cells of the BRU, either to suppress resorption or to promote bone formation. Furthermore, the current therapies have shortcomings and adverse effects with prolonged use necessitating drug holidays (117).
Immunomodulatory Options
Therapeutic option that target the immune system has recently gained interested as treatment option for PMOP. Given that a number of inflammatory conditions lead to bone destruction, inhibition of specific cytokine signaling have also been used to disrupt the cell-cell signaling and to protect against bone destruction. Blockade of TNFα (118) and IL-17A (119) in mice have shown to prevent OVX-induced bone loss. Etanercept (anti-TNFα) have been used to treat PMOP patients and showed decreases in serum markers of bone resorption (120), which warrant further investigation whether it is safer and better than current therapies (121). A recent study showed that neutralization of IL-17A induces compensatory increase of other Th17 cytokines, including IL-17F, IL-22 and GM-CSF (122). While secukinumab (anti-IL-17A) has not been evaluated for the treatment of osteoporosis, this finding highlights the complex nature of targeting cytokines in preventing bone loss.
Probiotic supplementation can be considered as an immunomodulatory therapy, given the role of GMB in regulating bone health as mentioned in this review. Bone loss in OVX mice can be prevented with supplementation of probiotics (68). While randomized controlled trial were conducted (123) and showed reduction in bone loss in particular with Lactobacillus reuteri (124), these results should be interpreted with caution and further studies are needed to evaluate the benefits of probiotics.
Our laboratory discovered that OC are antigen presenting cells during bone resorption that express DLL4, CD200 and MHC I and induce naïve CD8+ T cells to become FoxP3+ regulatory T cells (TcREG) (Figure 3). TcREG express Eomes and T-bet, have increased the surface expression of CD25 and CTLA-4, and produce IFN-γ and IL-10 (125, 126). Bone resorbing OC induce TcREG which in turn suppress bone resorption, forming a negative feedback loop (127). TcREG are also immunosuppressive in addition to regulating OC (128). Both in vivo induction by low dose pulsed RANKL (pRANKL) and adoptive transfer of ex vivo generated TcREG suppressed bone resorption, TNFα and IL-17A levels and promoted bone formation (129) to ameliorate osteoporosis in OVX mice. In unpublished studies, IL-10 directly regulate OB at the gene expression level and OVX of IL-10 deficient mice were unresponsive to the anabolic effects of pRANKL. However, TcREG retained its ability to inhibit TNFα production in TEM. Induction of TcREG has two facets: first, regulatory T-cells promote a tolerogenic environment by reducing the overall inflammatory burden; second, induction of regulatory T-cells is not expected to immunocompromise the host, unlike targeting inflammatory cytokines with antibodies (i.e., TNF blockade) or by JAK inhibitors. Targeting inflammatory cytokines represses both chronic and acute inflammation and thus increases risk of opportunistic infections. In contrast, inducing antigen-specific regulatory T-cells can precisely target chronic inflammation. Furthermore, while cytokine blockade may slow disease progression, regulatory T-cells promote resolution of inflammation to restore immune homeostasis (130–132). Taken together, our observations indicate that the immune system plays a fundamental role in modulating bone homeostasis, able to tip the balance either in favor of uncoupled bone resorption or bone formation.
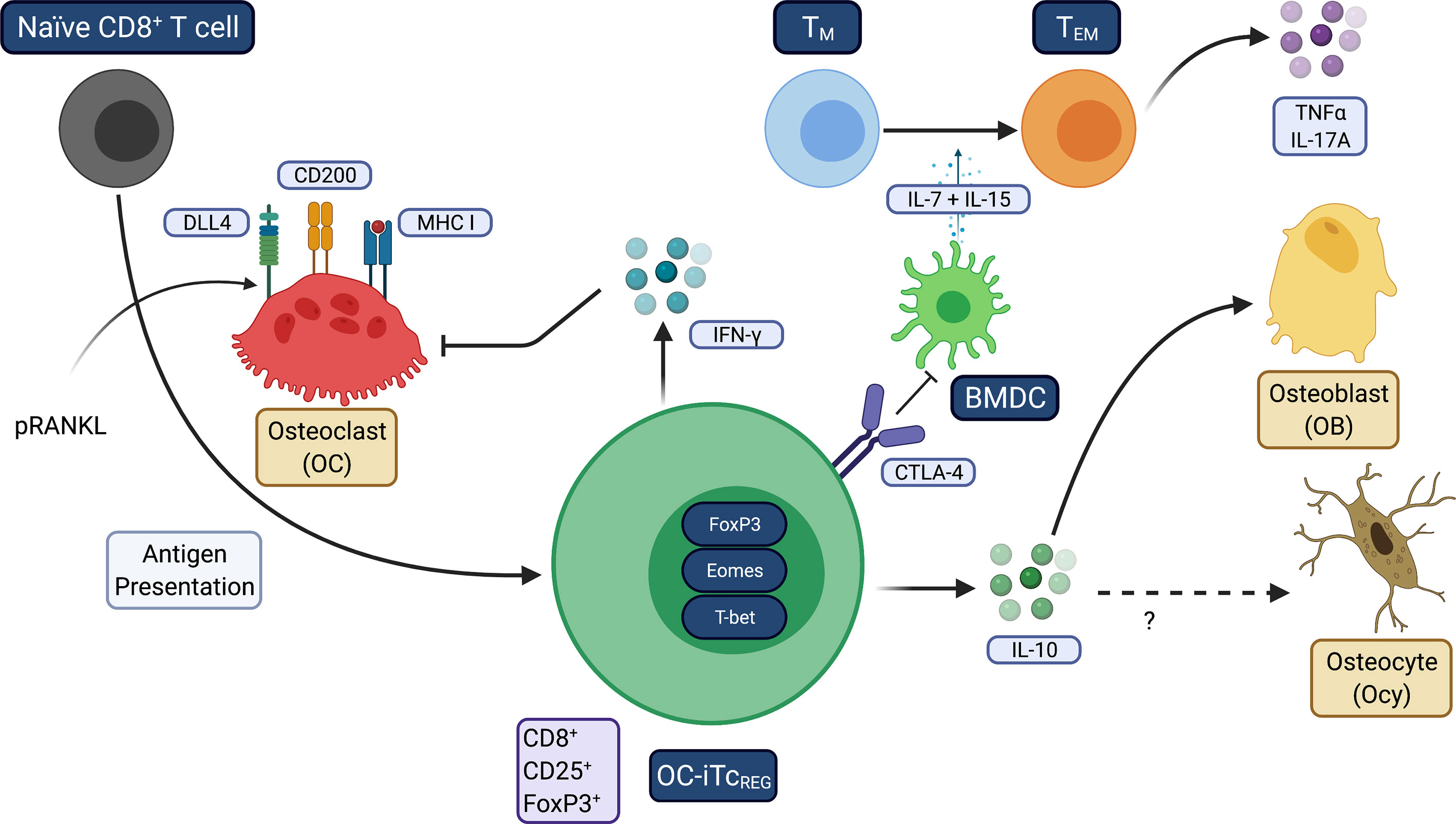
Figure 3 Osteoclasts induce tolerogenic TcREG. This figure summarizes how OC induce TcREG to promote restore homeostasis under inflammatory conditions. OC use three signals: antigen-loaded MHC I that ligates TCR on CD8 T-cells, CD200 (a costimulation molecule that activates NF-κB) and the Notch ligand DLL4 that engages Notch1 and Notch4 on the T-cells. TNFα and/or IL-17A downregulate DLL4 expression on OC. Treatment with pRANKL leads to increased expression DLL4. TcREG secrete IFN-γ that suppress osteoclastogenesis by degrading TRAF6 and also suppress resorption by mature OC. TcREG also secrete IL-10, which is required for the bone anabolic activity but not resolution of inflammation. IL-10 may also target Ocy to improve cortical bone mass. Resolution of inflammation appears to be mediated by CTLA4 expressed on TcREG.
Conclusion and Perspectives
In this review we highlighted how TM have emerged to be at the center of the pathophysiology of PMOP. These results demonstrate that E2 loss promotes inflammation leading to the acute phase bone erosion. We also underscore that none of the currently approved therapies for PMOP target inflammation but were intended to act directly on the BRU. The therapies also underscore that increasing bone mass may not be sufficient to reduce risk of fractures. How the proinflammatory cytokines TNFα, IL-17A and IL-1β leads to increased OC differentiation and increased resorption are well understood. In contrast, we do not have a similar level of understanding on the action of inflammation on OB and Ocy. We also highlighted how resolution of inflammation leads to increased bone formation. Additional studies are needed to understand the mechanism and the targets. In this context, better therapies will emerge from efforts to understand how Ocy sense bone quality and promote the repair process to produce bone that is resilient and less likely to fail. The correlation between bone mass (primarily by mineralization) to improving bone quality, with improved biomechanical properties needs to be further defined. Research in current decade is likely to provide new insights and mechanisms into the crosstalk. Revealing the mechanistic details on immune regulation on bone homeostasis will provide exciting new targets for therapies.
Author Contributions
RA conceived of the manuscript. DW and RA drafted the manuscript. AP and AK created figures and helped with edits. AC-S and ES provided literature search and edits. All authors were involved in scientific discussion of the review. All authors contributed to the article and approved the submitted version.
Funding
Research reported in this study was partially supported by National Institute of Arthritis and Musculoskeletal and Skin Disease of the NIH under Award Number RO1AR064821 and RO1AR068438. Washington University Musculoskeletal Research Core (NIH P30 AR057235) also partially supported this study.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Daniel Goering, Yiyi Zhang and Lizzie Geerling for contributing to additional unpublished experiments referenced herein.
References
1. Melton LJ. The Prevalence of Osteoporosis: Gender and Racial Comparison. Calcif Tissue Int (2001) 69(4):179. doi: 10.1007/s00223-001-1043-9
2. Wright NC, Looker AC, Saag KG, Curtis JR, Delzell ES, Randall S, et al. The Recent Prevalence of Osteoporosis and Low Bone Mass in the United States Based on Bone Mineral Density at the Femoral Neck or Lumbar Spine. J Bone Miner Res (2014) 29(11):2520–6. doi: 10.1002/jbmr.2269
3. Kenkre JS, Bassett J. The Bone Remodelling Cycle. Ann Clin Biochem (2018) 55(3):308–27. doi: 10.1177/0004563218759371
4. Albright F, Smith PH, Richardson AM. Postmenopausal Osteoprosis: its Clinical Feature. JAMA (1941) 116(22):2465–74. doi: 10.1001/jama.1941.02820220007002
5. Riggs BL, Khosla S, Melton LJ 3rd. A Unitary Model for Involutional Osteoporosis: Estrogen Deficiency Causes Both Type I and Type II Osteoporosis in Postmenopausal Women and Contributes to Bone Loss in Aging Men. J Bone Miner Res (1998) 13(5):763–73. doi: 10.1359/jbmr.1998.13.5.763
6. Fujita T. Calcium Intake, Calcium Absorption, and Osteoporosis. Calcif Tissue Int (1996) 58(4):215. doi: 10.1007/BF02508637
7. Avioli LV, McDonald JE, Lee SW. The Influence of Age on the Intestinal Absorption of 47-Ca Absorption in Post-Menopausal Osteoporosis. J Clin Invest (1965) 44(12):1960–7. doi: 10.1172/JCI105302
8. Ledger GA, Burritt MF, Kao PC, O’Fallon WM, Riggs BL, Khosla S. Role of Parathyroid Hormone in Mediating Nocturnal and Age-Related Increases in Bone Resorption. J Clin Endocrinol Metab (1995) 80(11):3304–10. doi: 10.1210/jcem.80.11.7593443
9. Eastell R, Yergey AL, Vieira NE, Cedel SL, Kumar R, Riggs BL. Interrelationship Among Vitamin D Metabolism, True Calcium Absorption, Parathyroid Function, and Age in Women: Evidence of an Age-Related Intestinal Resistance to 1,25-Dihydroxyvitamin D Action. J Bone Miner Res (1991) 6(2):125–32. doi: 10.1002/jbmr.5650060205
10. Francis RM, Peacock M, Taylor GA, Storer JH, Nordin BE. Calcium Malabsorption in Elderly Women With Vertebral Fractures: Evidence for Resistance to the Action of Vitamin D Metabolites on the Bowel. Clin Sci (Lond) (1984) 66(1):103–7. doi: 10.1042/cs0660103
11. Arron JR, Choi Y. Bone Versus Immune System. Nature (2000) 408(6812):535–6. doi: 10.1038/35046196
12. Aurora R. Confounding Factors in the Effect of Gut Microbiota on Bone Density. Rheumatology (Oxford) (2019) 58(12):2089–90. doi: 10.1093/rheumatology/kez347
13. Das M, Cronin O, Keohane DM, Cormac EM, Nugent H, Nugent M, et al. Gut Microbiota Alterations Associated With Reduced Bone Mineral Density in Older Adults. Rheumatology (Oxford) (2019) 58(12):2295–304. doi: 10.1093/rheumatology/kez302
14. Burr DB. Muscle Strength, Bone Mass, and Age-Related Bone Loss. J Bone Miner Res (1997) 12(10):1547–51. doi: 10.1359/jbmr.1997.12.10.1547
15. Bonnick SL. Osteoporosis in Men and Women. Clin Cornerstone (2006) 8(1):28–39. doi: 10.1016/s1098-3597(06)80063-3
16. Aerssens J, Boonen S, Joly J, Dequeker J. Variations in Trabecular Bone Composition With Anatomical Site and Age: Potential Implications for Bone Quality Assessment. J Endocrinol (1997) 155(3):411–21. doi: 10.1677/joe.0.1550411
18. Riggs BL, Khosla S, Atkinson EJ, Dunstan CR, Melton LJ 3rd. Evidence That Type I Osteoporosis Results From Enhanced Responsiveness of Bone to Estrogen Deficiency. Osteoporos Int (2003) 14(9):728–33. doi: 10.1007/s00198-003-1437-9
19. Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, et al. Estrogen Prevents Bone Loss Via Estrogen Receptor Alpha and Induction of Fas Ligand in Osteoclasts. Cell (2007) 130(5):811–23. doi: 10.1016/j.cell.2007.07.025
20. Oursler MJ, Landers JP, Riggs BL, Spelsberg TC. Oestrogen Effects on Osteoblasts and Osteoclasts. Ann Med (1993) 25(4):361–71. doi: 10.3109/07853899309147298
21. Oursler MJ, Osdoby P, Pyfferoen J, Riggs BL, Spelsberg TC. Avian Osteoclasts as Estrogen Target Cells. Proc Natl Acad Sci U S A (1991) 88(15):6613–7. doi: 10.1073/pnas.88.15.6613
22. Oursler MJ, Pederson L, Pyfferoen J, Osdoby P, Fitzpatrick L, Spelsberg TC. Estrogen Modulation of Avian Osteoclast Lysosomal Gene Expression. Endocrinology (1993) 132(3):1373–80. doi: 10.1210/endo.132.3.8440193
23. Kovacic N, Lukic IK, Grcevic D, Katavic V, Croucher P, Marusic A. The Fas/Fas Ligand System Inhibits Differentiation of Murine Osteoblasts But has a Limited Role in Osteoblast and Osteoclast Apoptosis. J Immunol (2007) 178(6):3379–89. doi: 10.4049/jimmunol.178.6.3379
24. Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, et al. Estrogen Protects Bone by Inducing Fas Ligand in Osteoblasts to Regulate Osteoclast Survival. EMBO J (2008) 27(3):535–45. doi: 10.1038/sj.emboj.7601984
25. Vanderschueren D, Gaytant J, Boonen S, Venken K. Androgens and Bone. Curr Opin Endocrinol Diabetes Obes (2008) 15(3):250–4. doi: 10.1097/MED.0b013e3282fe6ca9
26. Almeida M, Laurent MR, Dubois V, Claessens F, O’Brien CA, Bouillon R, et al. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol Rev (2017) 97(1):135–87. doi: 10.1152/physrev.00033.2015
27. Clarke BL, Khosla S. Physiology of Bone Loss. Radiol Clin North Am (2010) 48(3):483–95. doi: 10.1016/j.rcl.2010.02.014
28. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-Cell-Mediated Regulation of Osteoclastogenesis by Signalling Cross-Talk Between RANKL and IFN-Gamma. Nature (2000) 408(6812):600–5. doi: 10.1038/35046102
29. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 Functions as an Osteoclastogenic Helper T Cell Subset That Links T Cell Activation and Bone Destruction. J Exp Med (2006) 203(12):2673–82. doi: 10.1084/jem.20061775
30. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen Deficiency Induces the Differentiation of IL-17 Secreting Th17 Cells: A New Candidate in the Pathogenesis of Osteoporosis. PloS One (2012) 7(9):e44552. doi: 10.1371/journal.pone.0044552
31. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic Conversion of Foxp3+ T Cells Into TH17 Cells in Autoimmune Arthritis. Nat Med (2014) 20(1):62–8. doi: 10.1038/nm.3432
32. Zhao R, Wang X, Feng F. Upregulated Cellular Expression of IL-17 by CD4+ T-Cells in Osteoporotic Postmenopausal Women. Ann Nutr Metab (2016) 68(2):113–8. doi: 10.1159/000443531
33. Zhao B, Grimes SN, Li S, Hu X, Ivashkiv LB. TNF-Induced Osteoclastogenesis and Inflammatory Bone Resorption are Inhibited by Transcription Factor RBP-J. J Exp Med (2012) 209(2):319–34. doi: 10.1084/jem.20111566
34. Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor Necrosis Factor-α Induces Differentiation of and Bone Resorption by Osteoclasts. J Biol Chem (2000) 275(7):4858–64. doi: 10.1074/jbc.275.7.4858
35. Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor Necrosis Factor α Stimulates Osteoclast Differentiation by a Mechanism Independent of the Odf/Rankl–Rank Interaction. J Exp Med (2000) 191(2):275–86. doi: 10.1084/jem.191.2.275
36. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α Induces Osteoclastogenesis by Direct Stimulation of Macrophages Exposed to Permissive Levels of RANK Ligand. J Clin Invest (2000) 106(12):1481–8. doi: 10.1172/JCI11176
37. Moran CA, Weitzmann MN, Ofotokun I. Bone Loss in HIV Infection. Curr Treat Options Infect Dis (2017) 9(1):52–67. doi: 10.1007/s40506-017-0109-9
38. Ofotokun I, McIntosh E, Weitzmann MN. HIV: Inflammation and Bone. Curr HIV/AIDS Rep (2012) 9(1):16–25. doi: 10.1007/s11904-011-0099-z
39. Biver E, Calmy A, Rizzoli R. Bone Health in HIV and Hepatitis B or C Infections. Ther Adv Musculoskelet Dis (2017) 9(1):22–34. doi: 10.1177/1759720X16671927
40. Hoan NX, Tong HV, Song LH, Meyer CG, Velavan TP. Vitamin D Deficiency and Hepatitis Viruses-Associated Liver Diseases: A Literature Review. World J Gastroenterol (2018) 24(4):445–60. doi: 10.3748/wjg.v24.i4.445
41. Askari A, Naghizadeh MM, Homayounfar R, Shahi A, Afsarian MH, Paknahad A, et al. Increased Serum Levels of IL-17A and IL-23 are Associated With Decreased Vitamin D3 and Increased Pain in Osteoarthritis. PloS One (2016) 11(11):e0164757. doi: 10.1371/journal.pone.0164757
42. Blaschke M, Koepp R, Cortis J, Komrakova M, Schieker M, Hempel U, et al. IL-6, IL-1beta, and TNF-alpha Only in Combination Influence the Osteoporotic Phenotype in Crohn’s Patients Via Bone Formation and Bone Resorption. Adv Clin Exp Med (2018) 27(1):45–56. doi: 10.17219/acem/67561
43. Sapir-Koren R, Livshits G. Postmenopausal Osteoporosis in Rheumatoid Arthritis: The Estrogen Deficiency-Immune Mechanisms Link. Bone (2017) 103:102–15. doi: 10.1016/j.bone.2017.06.020
44. Klingberg E, Geijer M, Gothlin J, Mellstrom D, Lorentzon M, Hilme E, et al. Vertebral Fractures in Ankylosing Spondylitis are Associated With Lower Bone Mineral Density in Both Central and Peripheral Skeleton. J Rheumatol (2012) 39(10):1987–95. doi: 10.3899/jrheum.120316
45. Piodi LP, Poloni A, Ulivieri FM. Managing Osteoporosis in Ulcerative Colitis: Something New? World J Gastroenterol (2014) 20(39):14087–98. doi: 10.3748/wjg.v20.i39.14087
46. Zhao B. TNF and Bone Remodeling. Curr Osteoporos Rep (2017) 15(3):126–34. doi: 10.1007/s11914-017-0358-z
47. Tang M, Lu L, Yu X. Interleukin-17A Interweaves the Skeletal and Immune Systems. Front Immunol (2021) 11:625034(3841). doi: 10.3389/fimmu.2020.625034
48. Duque G, Huang DC, Dion N, Macoritto M, Rivas D, Li W, et al. Interferon-γ Plays a Role in Bone Formation In Vivo and Rescues Osteoporosis in Ovariectomized Mice. J Bone Miner Res (2011) 26(7):1472–83. doi: 10.1002/jbmr.350
49. Osta B, Benedetti G, Miossec P. Classical and Paradoxical Effects of TNF-α on Bone Homeostasis. Front Immunol (2014) 5(48):14–22. doi: 10.3389/fimmu.2014.00048
50. Tyagi AM, Mansoori MN, Srivastava K, Khan MP, Kureel J, Dixit M, et al. Enhanced Immunoprotective Effects by Anti-IL-17 Antibody Translates to Improved Skeletal Parameters Under Estrogen Deficiency Compared With Anti-RANKL and Anti-TNF-α Antibodies. J Bone Miner Res (2014) 29(9):1981–92. doi: 10.1002/jbmr.2228
51. Ginaldi L, De Martinis M, Ciccarelli F, Saitta S, Imbesi S, Mannucci C, et al. Increased Levels of Interleukin 31 (IL-31) in Osteoporosis. BMC Immunol (2015) 16(1):60. doi: 10.1186/s12865-015-0125-9
52. Du D, Zhou Z, Zhu L, Hu X, Lu J, Shi C, et al. TNF-Alpha Suppresses Osteogenic Differentiation of MSCs by Accelerating P2Y2 Receptor in Estrogen-Deficiency Induced Osteoporosis. Bone (2018) 117:161–70. doi: 10.1016/j.bone.2018.09.012
53. Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y, Qian WP, et al. Estrogen Deficiency Induces Bone Loss by Increasing T Cell Proliferation and Lifespan Through IFN-gamma-induced Class II Transactivator. Proc Natl Acad Sci U S A (2003) 100(18):10405–10. doi: 10.1073/pnas.1533207100
54. Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, et al. Estrogen Deficiency Induces Bone Loss by Enhancing T-Cell Production of TNF-Alpha. J Clin Invest (2000) 106(10):1229–37. doi: 10.1172/jci11066
55. Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, et al. Up-Regulation of TNF-Producing T Cells in the Bone Marrow: A Key Mechanism by Which Estrogen Deficiency Induces Bone Loss In Vivo. Proc Natl Acad Sci U S A (2001) 98(24):13960–5. doi: 10.1073/pnas.251534698
56. Roggia C, Tamone C, Cenci S, Pacifici R, Isaia GC. Role of TNF-alpha Producing T-Cells in Bone Loss Induced by Estrogen Deficiency. Minerva Med (2004) 95(2):125–32.
57. Weitzmann MN, Pacifici R. Estrogen Deficiency and Bone Loss: An Inflammatory Tale. J Clin Invest (2006) 116(5):1186–94. doi: 10.1172/JCI28550
58. Cline-Smith A, Axelbaum A, Shashkova E, Chakraborty M, Sanford J, Panesar P, et al. Ovariectomy Activates Chronic Low-Grade Inflammation Mediated by Memory T-Cells Which Promotes Osteoporosis in Mice. J Bone Miner Res (2020) 35(6):1174–87. doi: 10.1002/jbmr.3966
59. MacLeod MK, Kappler JW, Marrack P. Memory CD4 T Cells: Generation, Reactivation and Re-Assignment. Immunology (2010) 130(1):10–5. doi: 10.1111/j.1365-2567.2010.03260.x
60. Ohlsson C, Engdahl C, Fåk F, Andersson A, Windahl SH, Farman HH, et al. Probiotics Protect Mice From Ovariectomy-Induced Cortical Bone Loss. PloS One (2014) 9(3):e92368. doi: 10.1371/journal.pone.0092368
61. Britton RA, Irwin R, Quach D, Schaefer L, Zhang J, Lee T, et al. Probiotic L. Reuteri Treatment Prevents Bone Loss in a Menopausal Ovariectomized Mouse Model. J Cell Physiol (2014) 229(11):1822–30. doi: 10.1002/jcp.24636
62. Sjögren K, Engdahl C, Henning P, Lerner UH, Tremaroli V, Lagerquist MK, et al. The Gut Microbiota Regulates Bone Mass in Mice. J Bone Miner Res (2012) 27(6):1357–67. doi: 10.1002/jbmr.1588
63. Sabui S, Skupsky J, Kapadia R, Cogburn K, Lambrecht NW, Agrawal A, et al. Tamoxifen-Induced, Intestinal-Specific Deletion of Slc5a6 in Adult Mice Leads to Spontaneous Inflammation: Involvement of NF-kappaB, NLRP3, and Gut Microbiota. Am J Physiol Gastrointest Liver Physiol (2019) 317(4):G518–30. doi: 10.1152/ajpgi.00172.2019
64. Roomruangwong C, Carvalho AF, Geffard M, Maes M. The Menstrual Cycle may Not be Limited to the Endometrium But Also may Impact Gut Permeability. Acta Neuropsychiatr (2019) 31(6):294–304. doi: 10.1017/neu.2019.30
65. Rizzetto L, Fava F, Tuohy KM, Selmi C. Connecting the Immune System, Systemic Chronic Inflammation and the Gut Microbiome: The Role of Sex. J Autoimmun (2018) 92:12–34. doi: 10.1016/j.jaut.2018.05.008
66. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell (2009) 139(3):485–98. doi: 10.1016/j.cell.2009.09.033
67. Pacifici R. Bone Remodeling and the Microbiome. Cold Spring Harb Perspect Med (2018) 8:a031203 1–13. doi: 10.1101/cshperspect.a031203
68. Li JY, Chassaing B, Tyagi AM, Vaccaro C, Luo T, Adams J, et al. Sex Steroid Deficiency-Associated Bone Loss is Microbiota Dependent and Prevented by Probiotics. J Clin Invest (2016) 126(6):2049–63. doi: 10.1172/jci86062
69. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
70. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341(6145):569–73. doi: 10.1126/science.1241165
71. Chevalier C, Kieser S, Colakoglu M, Hadadi N, Brun J, Rigo D, et al. Warmth Prevents Bone Loss Through the Gut Microbiota. Cell Metab (2020) 32(4):575–90 e7. doi: 10.1016/j.cmet.2020.08.012
72. Pascutti MF, Geerman S, Collins N, Brasser G, Nota B, Stark R, et al. Peripheral and Systemic Antigens Elicit an Expandable Pool of Resident Memory CD8+ T Cells in the Bone Marrow. Eur J Immunol (2019) 49(6):853–72. doi: 10.1002/eji.201848003
73. Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, et al. Inhibition of Osteoblast Differentiation by Tumor Necrosis Factor-α*. Endocrinology (2000) 141(11):3956–64. doi: 10.1210/endo.141.11.7739
74. Gilbert L, He X, Farmer P, Rubin J, Drissi H, van Wijnen AJ, et al. Expression of the Osteoblast Differentiation Factor RUNX2 (Cbfa1/Aml3/Pebp2alpha A) is Inhibited by Tumor Necrosis Factor-Alpha. J Biol Chem (2002) 277(4):2695–701. doi: 10.1074/jbc.M106339200
75. Lu X, Gilbert L, He X, Rubin J, Nanes MS. Transcriptional Regulation of the Osterix (Osx, Sp7) Promoter by Tumor Necrosis Factor Identifies Disparate Effects of Mitogen-Activated Protein Kinase and NF Kappa B Pathways. J Biol Chem (2006) 281(10):6297–306. doi: 10.1074/jbc.M507804200
76. Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell (2015) 161(7):1576–91. doi: 10.1016/j.cell.2015.05.029
77. Chen J, Long F. mTORC1 Signaling Promotes Osteoblast Differentiation From Preosteoblasts. PloS One (2015) 10(6):e0130627. doi: 10.1371/journal.pone.0130627
78. Chen J, Holguin N, Shi Y, Silva MJ, Long F. mTORC2 Signaling Promotes Skeletal Growth and Bone Formation in Mice. J Bone Miner Res (2015) 30(2):369–78. doi: 10.1002/jbmr.2348
79. Fitter S, Matthews MP, Martin SK, Xie J, Ooi SS, Walkley CR, et al. mTORC1 Plays an Important Role in Skeletal Development by Controlling Preosteoblast Differentiation. Mol Cell Biol (2017) 37(7):e00668–16. doi: 10.1128/mcb.00668-16
80. Schaub T, Gurgen D, Maus D, Lange C, Tarabykin V, Dragun D, et al. mTORC1 and mTORC2 Differentially Regulate Cell Fate Programs to Coordinate Osteoblastic Differentiation in Mesenchymal Stromal Cells. Sci Rep (2019) 9(1):20071. doi: 10.1038/s41598-019-56237-w
81. Ciaraldi TP, Carter L, Mudaliar S, Kern PA, Henry RR. Effects of Tumor Necrosis Factor-α on Glucose Metabolism in Cultured Human Muscle Cells From Nondiabetic and Type 2 Diabetic Subjects**This Work was Supported by Funds From the American Diabetes Association, the Whittier Institute for Diabetes Research, Medical Research Service, the Department of Veterans Affairs, and the Veteran Affairs Medical Center-San Diego (to R.R.H.); Grant MO1-RR-00827 From the General Clinical Research Branch, Division of Research Resources, NIH, and the Medical Research Service, Department of Veterans Affairs and Veteran Affairs Medical Center-Little Rock (to P.A.K.); and NIH Grant Dk-39176. Endocrinology (1998) 139(12):4793–800. doi: 10.1210/endo.139.12.6368
82. Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. Tumor Necrosis Factor-α Induces Skeletal Muscle Insulin Resistance in Healthy Human Subjects Via Inhibition of Akt Substrate 160 Phosphorylation. Diabetes (2005) 54(10):2939–45. doi: 10.2337/diabetes.54.10.2939
83. Hauner H, Petruschke T, Russ M, Röhrig K, Eckel J. Effects of Tumour Necrosis Factor Alpha (Tnfα) on Glucose Transport and Lipid Metabolism of Newly-Differentiated Human Fat Cells in Cell Culture. Diabetologia (1995) 38(7):764–71. doi: 10.1007/s001250050350
84. Chen L, Bao J, Yang Y, Wang Z, Xia M, Tan J, et al. Autophagy was Involved in Tumor Necrosis Factor-Alpha-Inhibited Osteogenic Differentiation of Murine Calvarial Osteoblasts Through Wnt/beta-catenin Pathway. Tissue Cell (2020) 67:101401. doi: 10.1016/j.tice.2020.101401
85. Zheng LW, Wang WC, Mao XZ, Luo YH, Tong ZY, Li D. TNF-alpha Regulates the Early Development of Avascular Necrosis of the Femoral Head by Mediating Osteoblast Autophagy and Apoptosis Via the P38 MAPK/NF-kappaB Signaling Pathway. Cell Biol Int (2020) 44(9):1881–9. doi: 10.1002/cbin.11394
86. Zheng L, Wang W, Ni J, Mao X, Song D, Liu T, et al. Role of Autophagy in Tumor Necrosis Factor-Alpha-Induced Apoptosis of Osteoblast Cells. J Investig Med (2017) 65(6):1014–20. doi: 10.1136/jim-2017-000426
87. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for Osteocyte Regulation of Bone Homeostasis Through RANKL Expression. Nat Med (2011) 17(10):1231–4. doi: 10.1038/nm.2452
88. O’Brien CA, Nakashima T, Takayanagi H. Osteocyte Control of Osteoclastogenesis. Bone (2013) 54(2):258–63. doi: 10.1016/j.bone.2012.08.121
89. Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, Not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PloS One (2015) 10(9):e0138189. doi: 10.1371/journal.pone.0138189
90. Xing L, Schwarz EM, Boyce BF. Osteoclast Precursors, RANKL/RANK, and Immunology. Immunol Rev (2005) 208:19–29. doi: 10.1111/j.0105-2896.2005.00336.x
91. Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O’Brien CA. RANKL (Receptor Activator of NFkappaB Ligand) Produced by Osteocytes is Required for the Increase in B Cells and Bone Loss Caused by Estrogen Deficiency in Mice. J Biol Chem (2016) 291(48):24838–50. doi: 10.1074/jbc.M116.742452
92. Kim JH, Kim AR, Choi YH, Jang S, Woo GH, Cha JH, et al. Tumor Necrosis Factor-Alpha Antagonist Diminishes Osteocytic RANKL and Sclerostin Expression in Diabetes Rats With Periodontitis. PloS One (2017) 12(12):e0189702. doi: 10.1371/journal.pone.0189702
93. Li JY, Yu M, Tyagi AM, Vaccaro C, Hsu E, Adams J, et al. IL-17 Receptor Signaling in Osteoblasts/Osteocytes Mediates PTH-Induced Bone Loss and Enhances Osteocytic RANKL Production. J Bone Miner Res (2019) 34(2):349–60. doi: 10.1002/jbmr.3600
94. Liao C, Zhang C, Jin L, Yang Y. IL-17 Alters the Mesenchymal Stem Cell Niche Towards Osteogenesis in Cooperation With Osteocytes. J Cell Physiol (2020) 235(5):4466–80. doi: 10.1002/jcp.29323
95. Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, et al. Osteocyte Apoptosis and Control of Bone Resorption Following Ovariectomy in Mice. Bone (2010) 46(3):577–83. doi: 10.1016/j.bone.2009.11.006
96. Tsourdi E, Jähn K, Rauner M, Busse B, Bonewald LF. Physiological and Pathological Osteocytic Osteolysis. J Musculoskelet Neuronal Interact (2018) 18(3):292–303.
97. Tonti E, Jiménez de Oya N, Galliverti G, Moseman EA, Di Lucia P, Amabile A, et al. Bisphosphonates Target B Cells to Enhance Humoral Immune Responses. Cell Rep (2013) 5(2):323–30. doi: 10.1016/j.celrep.2013.09.004
98. Diker-Cohen T, Rosenberg D, Avni T, Shepshelovich D, Tsvetov G, Gafter-Gvili A. Risk for Infections During Treatment With Denosumab for Osteoporosis: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab (2020) 105(5):1641–58. doi: 10.1210/clinem/dgz322
99. Deligiorgi M V, Panayiotidis MI, Trafalis DT. Combining Immune Checkpoint Inhibitors With Denosumab: A New Era in Repurposing Denosumab in Oncology? J BUON (2020) 25(1):1–14.
100. Ruggiero SL. Bisphosphonate-Related Osteonecrosis of the Jaws. Compend Contin Educ Dent (2008) 29(2):96–8.
101. Kim J, Lee DH, Dziak R, Ciancio S. Bisphosphonate-Related Osteonecrosis of the Jaw: Current Clinical Significance and Treatment Strategy Review. Am J Dent (2020) 33(3):115–28.
102. Gupta M, Gupta N. Bisphosphonate Related Jaw Osteonecrosis. In: StatPearls. Treasure Island (FL): Stat Pearls Publishing (2020).
103. Jain TP, Thorn M. Atypical Femoral Fractures Related to Bisphosphonate Therapy. Indian J Radiol Imaging (2012) 22(3):178–81. doi: 10.4103/0971-3026.107178
104. Tripto-Shkolnik L, Rouach V, Marcus Y, Rotman-Pikielny P, Benbassat C, Vered I. Vertebral Fractures Following Denosumab Discontinuation in Patients With Prolonged Exposure to Bisphosphonates. Calcif Tissue Int (2018) 103(1):44–9. doi: 10.1007/s00223-018-0389-1
105. Novack DV, Teitelbaum SL. The Osteoclast: Friend or Foe? Annu Rev Pathol (2008) 3:457–84. doi: 10.1146/annurev.pathmechdis.3.121806.151431
106. Hodsman AB, Bauer DC, Dempster DW, Dian L, Hanley DA, Harris ST, et al. Parathyroid Hormone and Teriparatide for the Treatment of Osteoporosis: A Review of the Evidence and Suggested Guidelines for its Use. Endocr Rev (2005) 26(5):688–703. doi: 10.1210/er.2004-0006
107. Lim SY, Bolster MB. Profile of Romosozumab and its Potential in the Management of Osteoporosis. Drug Des Dev Ther (2017) 11:1221. doi: 10.2147/DDDT.S127568
108. Fixen C, Tunoa J. Romosozumab: A Review of Efficacy, Safety, and Cardiovascular Risk. Curr Osteoporos Rep (2021) 19(1):15–22. doi: 10.1007/s11914-020-00652-w
109. Saag KG, Zanchetta JR, Devogelaer JP, Adler RA, Eastell R, See K, et al. Effects of Teriparatide Versus Alendronate for Treating Glucocorticoid-Induced Osteoporosis: Thirty-Six-Month Results of a Randomized, Double-Blind, Controlled Trial. Arthritis Rheum (2009) 60(11):3346–55. doi: 10.1002/art.24879
110. Tashjian AH Jr., Gagel RF. Teriparatide [Human PTH(1-34)]: 2.5 Years of Experience on the Use and Safety of the Drug for the Treatment of Osteoporosis. J Bone Miner Res (2006) 21(3):354–65. doi: 10.1359/JBMR.051023
111. Brandenburg VM, Verhulst A, Babler A, D’Haese PC, Evenepoel P, Kaesler N. Sclerostin in Chronic Kidney Disease-Mineral Bone Disorder Think First Before You Block it! Nephrol Dial Transplant (2019) 34(3):408–14. doi: 10.1093/ndt/gfy129
112. Dempster DW, Zhou H, Recker RR, Brown JP, Recknor CP, Lewiecki EM, et al. Remodeling- and Modeling-Based Bone Formation With Teriparatide Versus Denosumab: A Longitudinal Analysis From Baseline to 3 Months in the AVA Study. J Bone Miner Res (2018) 33(2):298–306. doi: 10.1002/jbmr.3309
113. Dempster DW, Zhou H, Recker RR, Brown JP, Bolognese MA, Recknor CP, et al. A Longitudinal Study of Skeletal Histomorphometry at 6 and 24 Months Across Four Bone Envelopes in Postmenopausal Women With Osteoporosis Receiving Teriparatide or Zoledronic Acid in the SHOTZ Trial. J Bone Miner Res (2016) 31(7):1429–39. doi: 10.1002/jbmr.2804
114. Edwards WB, Simonian N, Haider IT, Anschel AS, Chen D, Gordon KE, et al. Effects of Teriparatide and Vibration on Bone Mass and Bone Strength in People With Bone Loss and Spinal Cord Injury: A Randomized, Controlled Trial. J Bone Miner Res (2018) 33(10):1729–40. doi: 10.1002/jbmr.3525
115. Tsai JN, Nishiyama KK, Lin D, Yuan A, Lee H, Bouxsein ML, et al. Effects of Denosumab and Teriparatide Transitions on Bone Microarchitecture and Estimated Strength: The DATA-Switch HR-pQCT Study. J Bone Miner Res (2017) 32(10):2001–9. doi: 10.1002/jbmr.3198
116. Eastell R, O’Neill TW, Hofbauer LC, Langdahl B, Reid IR, Gold DT, et al. Postmenopausal Osteoporosis. Nat Rev Dis Primers (2016) 2:16069. doi: 10.1038/nrdp.2016.69
117. Fink HA, MacDonald R, Forte ML, Rosebush CE, Ensrud KE, Schousboe JT, et al. Long-Term Drug Therapy and Drug Holidays for Osteoporosis Fracture Prevention: A Systematic Review. In: AHRQ Comparative Effectiveness Reviews. Rockville (MD): Agency for Health Research and Quality (AHRQ.gov) (2019).
118. Kimble RB, Bain S, Pacifici R. The Functional Block of TNF But Not of IL-6 Prevents Bone Loss in Ovariectomized Mice. J Bone Miner Res (1997) 12(6):935–41. doi: 10.1359/jbmr.1997.12.6.935
119. Deselm CJ, Takahata Y, Warren J, Chappel JC, Khan T, Li X, et al. IL-17 Mediates Estrogen-Deficient Osteoporosis in an Act1-dependent Manner. J Cell Biochem (2012) 113(9):2895–902. doi: 10.1002/jcb.24165
120. Charatcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL. Effect of Blockade of TNF-alpha and Interleukin-1 Action on Bone Resorption in Early Postmenopausal Women. J Bone Miner Res (2007) 22(5):724–9. doi: 10.1359/jbmr.070207
121. Kawai VK, Stein CM, Perrien DS, Griffin MR. Effects of Anti-Tumor Necrosis Factor α Agents on Bone. Curr Opin Rheumatol (2012) 24(5):576–85. doi: 10.1097/BOR.0b013e328356d212
122. Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, et al. The Cytokine IL-17a Limits Th17 Pathogenicity Via a Negative Feedback Loop Driven by Autocrine Induction of IL-24. Immunity (2020) 53(2):384–97.e5. doi: 10.1016/j.immuni.2020.06.022
123. Yu J, Cao G, Yuan S, Luo C, Yu J, Cai M. Probiotic Supplements and Bone Health in Postmenopausal Women: A Meta-Analysis of Randomised Controlled Trials. BMJ Open (2021) 11(3):e041393. doi: 10.1136/bmjopen-2020-041393
124. Nilsson AG, Sundh D, Bäckhed F, Lorentzon M. Lactobacillus Reuteri Reduces Bone Loss in Older Women With Low Bone Mineral Density: A Randomized, Placebo-Controlled, Double-Blind, Clinical Trial. J Intern Med (2018) 284(3):307–17. doi: 10.1111/joim.12805
125. Buchwald ZS, Kiesel JR, DiPaolo R, Pagadala MS, Aurora R. Osteoclast Activated Foxp3+ CD8+ T-Cells Suppress Bone Resorption In Vitro. PloS One (2012) 7(6):e38199–12. doi: 10.1371/journal.pone.0038199
126. Buchwald ZS, Yang C, Nellore S, Shashkova EV, Davis JL, Cline A, et al. A Bone Anabolic Effect of RANKL in a Murine Model of Osteoporosis Mediated Through FoxP3 +CD8 T Cells. J Bone Miner Res (2015) 30(8):1508–22. doi: 10.1002/jbmr.2472
127. Buchwald ZS, Aurora R. Osteoclasts and CD8 T Cells Form a Negative Feedback Loop That Contributes to Homeostasis of Both the Skeletal and Immune Systems. Clin Dev Immunol (2013) 2013:429373. doi: 10.1155/2013/429373
128. Buchwald ZS, Kiesel J, Yang C, DiPaolo R, Novack D, Aurora R. Osteoclast-Induced Foxp3+ CD8 T-Cells Limit Bone Loss in Mice. Bone (2013) 56:163–73. doi: 10.1016/j.bone.2013.05.024
129. Cline-Smith A, Gibbs J, Shashkova E, Buchwald ZS, Aurora R. Pulsed Low-Dose RANKL as a Potential Therapeutic for Postmenopausal Osteoporosis. JCI Insight (2016) 1(13):433–12. doi: 10.1172/jci.insight.88839
130. Chen Z, Bozec A, Ramming A, Schett G. Anti-Inflammatory and Immune-Regulatory Cytokines in Rheumatoid Arthritis. Nat Rev Rheumatol (2019) 15(1):9–17. doi: 10.1038/s41584-018-0109-2
131. Laumet G, Edralin JD, Chiang AC-A, Dantzer R, Heijnen CJ, Kavelaars A. Resolution of Inflammation-Induced Depression Requires T Lymphocytes and Endogenous Brain interleukin-10 Signaling. Neuropsychopharmacology (2018) 43(13):2597–605. doi: 10.1038/s41386-018-0154-1
Keywords: T cell, postmenopausal osteoporosis, estrogen loss, osteoimmunology, chronic inflammation
Citation: Wu D, Cline-Smith A, Shashkova E, Perla A, Katyal A and Aurora R (2021) T-Cell Mediated Inflammation in Postmenopausal Osteoporosis. Front. Immunol. 12:687551. doi: 10.3389/fimmu.2021.687551
Received: 29 March 2021; Accepted: 04 June 2021;
Published: 30 June 2021.
Edited by:
Massimo De Martinis, University of L’Aquila, ItalyReviewed by:
Kripa Elizabeth Cherian, Christian Medical College & Hospital, IndiaLuigi Gennari, University of Siena, Italy
Copyright © 2021 Wu, Cline-Smith, Shashkova, Perla, Katyal and Aurora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajeev Aurora, cmFqZWV2LmF1cm9yYUBoZWFsdGguc2x1LmVkdQ==