 Inhye E. Ahn
Inhye E. Ahn Jennifer R. Brown
Jennifer R. Brown
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 June 2021
Sec. B Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.687458
This article is part of the Research Topic New Insights on Bruton’s Tyrosine Kinase Inhibitors View all 6 articles
Targeting the B-cell receptor signaling pathway through BTK inhibition proved to be effective for the treatment of chronic lymphocytic leukemia (CLL) and other B-cell lymphomas. Covalent BTK inhibitors (BTKis) led to an unprecedented improvement in outcome in CLL, in particular for high-risk subgroups with TP53 aberration and unmutated immunoglobulin heavy-chain variable-region gene (IGHV). Ibrutinib and acalabrutinib are approved by the US Food and Drug Administration for the treatment of CLL and other B-cell lymphomas, and zanubrutinib, for patients with mantle cell lymphoma. Distinct target selectivity of individual BTKis confer differences in target-mediated as well as off-target adverse effects. Disease progression on covalent BTKis, driven by histologic transformation or selective expansion of BTK and PLCG2 mutated CLL clones, remains a major challenge in the field. Fixed duration combination regimens and reversible BTKis with non-covalent binding chemistry hold promise for the prevention and treatment of BTKi-resistant disease.
B cell receptor (BCR) signaling is an essential component of normal B cell development and malignant B cell survival (Figure 1). There are two types of BCR signaling: chronically activated BCR and tonic BCR. Activated BCR signaling is an antigen-dependent process utilizing the canonical nuclear factor-κB (NF-κB) pathway (1). Antigen binding by surface immunoglobulin initiates BCR signaling, resulting in coupling and autophosphorylation of the CD79A/CD79B heterodimer by Src family kinases (2). The phosphorylation of the immunoreceptor tyrosine-based activation motif recruits a cascade of signaling molecules. These include spleen tyrosine kinase (SYK), Bruton’s tyrosine kinase (BTK), phospholipase Cγ2 (PLCγ2), and protein kinase C, which lead to activation of NF-κB, phosphatidylinositol 3-kinase (PI3K) and ERK. Tonic BCR is an antigen-independent process that maintains B cell survival through PI3K-AKT-mTOR signaling rather than NF-κB (3, 4). PI3Kδ is a proximal component of the BCR signaling pathway involved in both tonic and chronic activated signaling.
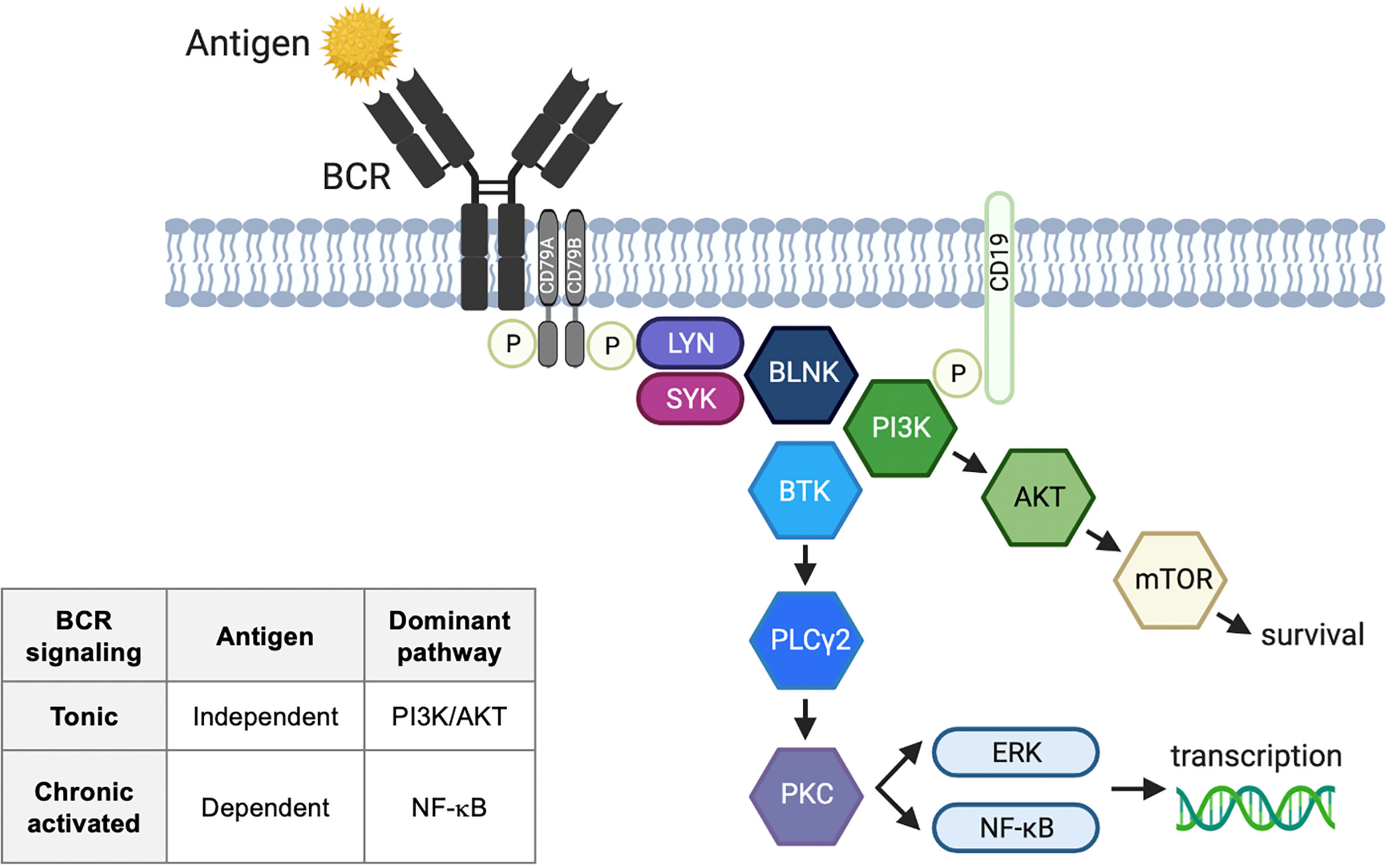
Figure 1 B cell receptor signaling pathway. Activated B cell receptor (BCR) signaling is an antigen-dependent process utilizing the canonical nuclear factor-κB (NF-κB) pathway. Antigen binding by surface immunoglobulin initiates BCR signaling, resulting in coupling and autophosphorylation of the CD79A/CD79B heterodimer by Src family kinases. The phosphorylation of the immunoreceptor tyrosine-based activation motifs recruits a cascade of signaling molecules. These include LYN tyrosine kinase (LYN), spleen tyrosine kinase (SYK), Bruton’s tyrosine kinase (BTK), phospholipase Cγ2 (PLCγ2), and protein kinase C (PKC), which lead to activation of NF-κB, phosphatidylinositol 3-kinase (PI3K) and ERK. Tonic BCR is an antigen-independent process that maintains B cell survival through PI3K-AKT-mTOR signaling rather than NF-κB. BLNK, B-cell linker; ERK, extracellular signal-regulated kinase; IgH, immunoglobulin heavy chain; IgL, immunoglobulin light chain; mTOR, mammalian target of rapamycin; P, phosphorylation.
The distinction between tonic and chronically activated BCR signaling is clinically relevant as it mirrors disease sensitivity to BTK inhibition. BTK inhibition alone is ineffective for the treatment of germinal center B diffuse large B-cell lymphoma (DLBCL) and Burkitt lymphoma with tonic BCR signaling, which require additional therapeutic targets such as SYK and CXCR4 (5–7). In contrast, lymphoma subtypes with activated BCR signaling are sensitive to BTK inhibitors (BTKis), and include chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), marginal zone lymphoma (MZL), and activated B cell (ABC) DLBCL. CLL and MCL have a non-genetic mechanism of BCR signaling where stereotyped BCRs and biased usage of unmutated immunoglobulin heavy-chain variable-region gene (IGHV) support the presence of cognate self-antigen leading to chronic BCR activation (8, 9). MZL also has a non-genetic mechanism of chronic BCR activation and is often associated with hepatitis C virus or Helicobacter pylori infection which provides a chronic antigenic stimulus to the BCR (10). ABC-DLBCL is characterized by genomic alterations of the BCR pathway and its downstream components (i.e. CD79A/B, CARD11) (11–13). Such mutations or amplification of genes in the BCR signaling pathway have not been reported in CLL (14) or MZL (1), and are rare in other indolent lymphomas (15, 16). Follicular lymphoma (FL) appears to have heterogeneous mechanisms of BCR activation. Although FL utilizes nonstereotyped BCR (17), studies have demonstrated antigen-mediated BCR signaling in a subset, not all, of FL (18, 19).
Of several approaches available for targeting the BCR pathway, BTK inhibition is the most popular and advanced in drug development. The U.S. Food and Drug Administration (FDA) has approved three BTKis for the treatment of B cell malignancies and graft-versus-host disease (GVHD): ibrutinib, acalabrutinib, and zanubrutinib. PI3K inhibition is another important BCR targeting strategy with four FDA-approved agents for the treatment of B cell malignancies (idelalisib, duvelisib, copanlisib, umbralisib). Isoform selectivity and potency of individual PI3K inhibitors contribute to observed differences in immune-mediated adverse events. The rates and severity of inflammatory toxicity, typically presenting as enteritis, hepatotoxicity, or pneumonitis, are relatively high with idelalisib. The rationale behind this observation is that idelalisib selectively inactivates the PI3K p110δ isoform in regulatory T cells thereby activating immune response (20, 21). Immune-mediated toxicity can be improved by less potent PI3Kδ inhibition via umbralisib, by targeting additional PI3K isoforms via copanlisib (pan-PI3K inhibitor), or in the relapsed/refractory setting where the immune effector function is downregulated (22). MALT1 inhibition is in the early stages of clinical development with no FDA-approved agent to date (23). We focus our discussion here on the successes and challenges of BTK inhibition in CLL, a malignant B cell disease with the most abundant data available on this topic.
Ibrutinib is the first-in-class, orally bioavailable, covalent BTKi. Ibrutinib binds to the cysteine 481 (C481) residue of BTK and irreversibly blocks phosphorylation of downstream kinases in the BCR signaling pathway (24). Since its discovery in 2007 (25), ibrutinib underwent rapid drug development leading to its initial FDA approval for the treatment of MCL in November 2013, followed by expansion of approved indications to include CLL in 2014 and other hematologic diseases thereafter (Table 1). The approved doses of ibrutinib are 420mg once daily for CLL, Waldenström’s macroglobulinemia (WM), and chronic GVHD, and 560mg for MCL and MZL. Both of these doses achieve sustained and complete BTK occupancy (>95%) (26). While ibrutinib is most potent against BTK (half maximal inhibitory concentration [IC50] 0.5nM), it can also inhibit other targets at lower potency such as EGFR (IC50 5.6nM), ErbB2 (IC50 9.4nM), ITK (IC50 10.7nM), and TEC (IC50 78nM) (24). Many of these unintended targets of ibrutinib have a conserved cysteine residue aligning with the C481 in BTK. Their inhibition is a proposed mechanism of ibrutinib toxicity. In particular, bleeding and cardiac arrhythmia are among the important side effects of ibrutinib, which are thought to be related to inhibition of TEC family kinases involved in platelet activation (27) and inhibition of C-terminal Src kinase expressed in cardiac tissue (28).
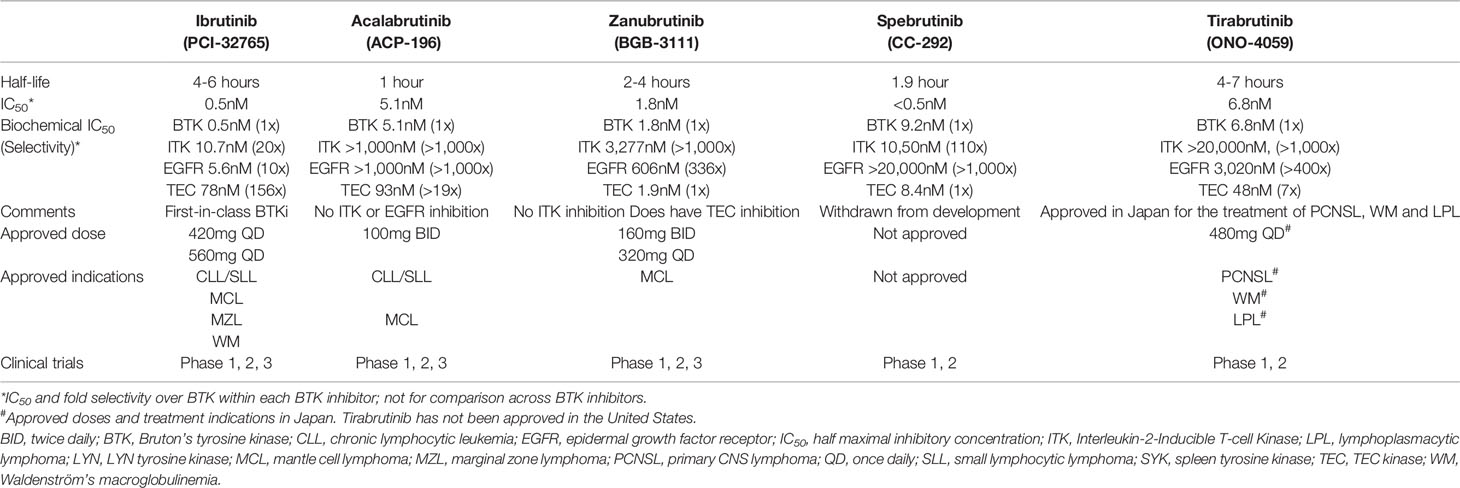
Table 1 Covalent BTK inhibitors.
To reduce toxicity and improve the tolerability of BTKis, alternative agents with more selective kinase inhibition profiles have been investigated. Acalabrutinib is a covalent BTKi with less potent inhibition of TEC compared to ibrutinib (IC50 93nM for acalabrutinib vs. 7nM for ibrutinib in a comparative recombinant kinase assay) and no EGFR or ITK inhibition (IC50 >1,000nM for both targets) (29). Acalabrutinib is approved for the treatment of CLL and MCL in the United States. For both diseases, acalabrutinib is dosed at 100mg PO twice daily. Studies have shown that the twice-daily dosing of acalabrutinib achieves the least variability at steady-state trough (29) and higher BTK occupancy (30) compared to 200mg given once daily.
In 2019, the FDA granted accelerated approval of zanubrutinib for the treatment of MCL. Zanubrutinib has less potent ITK and EGFR inhibition and a more favorable pharmacokinetic profile than ibrutinib (31). The twice daily dosing of zanubrutinib achieves 8-fold higher plasma drug exposure than ibrutinib and a longer half-life than acalabrutinib (4 vs. 1 hour), which effectively blocks the function of newly synthesized BTK protein as well as preexisting BTK irreversibly bound by zanubrutinib. A phase 1 study did show higher BTK occupancy with twice daily (>95%) than once daily dosing (89%) of zanubrutinib in lymphoid tissue (31). The same study, however, showed uniformly high BTK occupancy in blood and no difference in clinical outcome across once- and twice-daily dosing groups. Both doses of zanubrutinib are being marketed. Taken together, the positive correlation between pharmacokinetics and BTK inhibition in lymphoid tissue provides a theoretical basis for zanubrutinib being a better covalent BTKi than others. Longer follow-up coupled with correlative studies are needed to determine whether more effective blockade of BTK resynthesis translates to deeper and more durable response to zanubrutinib.
Spebrutinib and tirabrutinib are covalent BTKis without regulatory approval in the United States. Spebrutinib is less selective than other BTKis and was withdrawn from development due to lack of efficacy (32). Tirabrutinib is a selective BTKi approved in Japan for the treatment of primary CNS lymphoma, WM, and lymphoplasmacytic lymphoma (33, 34).
Despite the limited amount of high-quality evidence, emerging data indicate that selective BTKis may be safer and potentially more efficacious than ibrutinib. In the phase 3 ASPEN study for WM, the zanubrutinib arm achieved numerically higher rates of complete or very good partial responses (28%) than the ibrutinib arm (19%) without reaching statistical significance for the difference (35). Zanubrutinib was associated with a lower frequency and severity of bleeding and cardiovascular toxicities. The observed rate of atrial fibrillation, for instance, was 0.1 per 100 person-months with zanubrutinib compared to 1 per 100 person-months with ibrutinib. Indirect, prospective evidence further supports improved tolerability of second-generation BTKis. An open-label study of 33 CLL patients with ibrutinib intolerance reported 72% of the patients had tolerated acalabrutinib without having reoccurrences of ibrutinib-related adverse events (36). Two randomized studies ongoing in relapsed/refractory (R/R) CLL are expected to provide a direct comparison of first- and second-generation BTKis (ibrutinib vs. acalabrutinib, NCT02477696; ibrutinib vs. zanubrutinib, NCT03734016). Further research is needed to determine the comparative safety and efficacy of different BTKis including investigations in treatment-naïve (TN) CLL, long-term follow-up, and pharmacodynamic assessments of BTK and other targets.
BTKis pioneered the major shift in therapeutic approaches for CLL from chemoimmunotherapy to targeted therapy (Table 2). In randomized studies, single-agent ibrutinib demonstrated superior progression-free survival (PFS) and overall survival (OS) compared to conventional single-agent chemo- or immunotherapy in TN (55) and R/R CLL (42). In the TN setting, BTKi-containing regimens outperformed doublet or triplet chemoimmunotherapy regimens by improving PFS in four randomized studies (37–39, 49) and OS in one of the four studies (38).
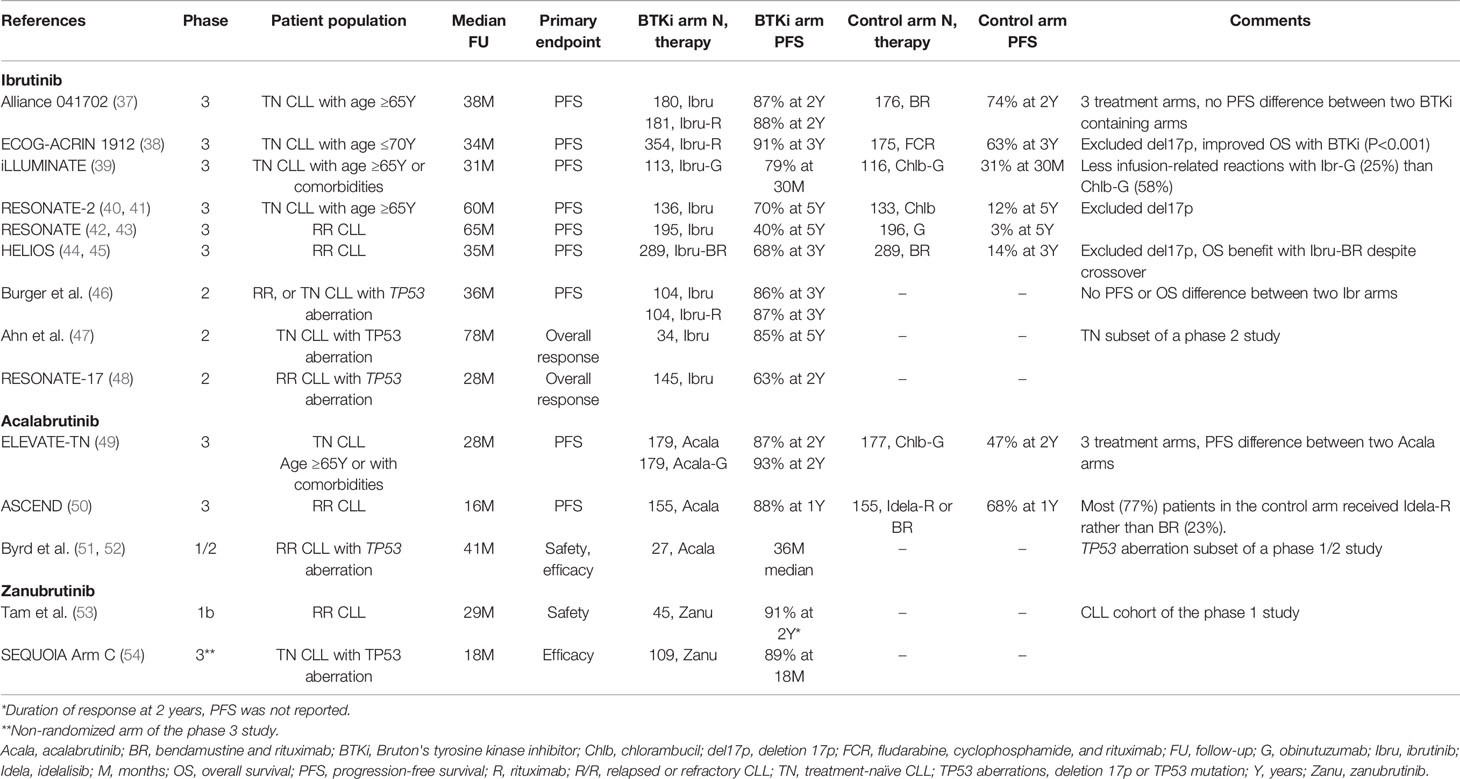
Table 2 Selected clinical trials testing covalent BTK inhibitors in CLL.
Patients who were classically considered to have high-risk characteristics benefit the most from BTKis (56, 57). TP53 aberration, referring to a mutation of the TP53 tumor suppressor gene or deletion of chromosome 17p where TP53 is encoded, is a strong negative prognostic marker in CLL (58). First-line treatment with an intensive chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab (FCR) reported median PFS of 15 months for patients with TP53 aberration as opposed to nearly 5 years for those without the aberration (48, 54). Treatment with BTKis substantially extended the survival of patients with TP53 aberration (48, 59) with the observed rates of 5-year PFS and OS being 70% and 85%, respectively, when ibrutinib was first-line therapy (47). Patients with unmutated IGHV, another high-risk genetic marker in CLL, also achieved superior PFS with BTKi-based therapy compared to chemoimmunotherapy (37–39, 49). However, such PFS benefit was not observed in the subgroup with mutated IGHV. Outcome data based on the IGHV mutation status should be interpreted with caution because of relatively short follow-up and small numbers of events at the time of analyses. Equally critical to consider is long-lasting remission—and possibly cure—observed after treatment with FCR in a subset of patients with mutated IGHV (60, 61). Chemoimmunotherapy remains an option for young and fit CLL patients with mutated IGHV who prefer a defined duration of treatment to avoid concerns of long-term toxicity, treatment adherence, and financial burden related to continuous BTKi therapy.
Although covalent BTKis provide an excellent disease control in most patients, BTK inhibition alone is insufficient to eradicate CLL or achieve deep responses. Undetectable minimal residual disease (U-MRD) with fewer than 1 CLL cell per 10,000 leukocytes is rarely observed with ibrutinib (46, 59) or acalabrutinib alone (<7%) (49). Depth of response marginally improves with prolonged therapy (33% reduction of circulating CLL cells with each additional year on ibrutinib) (59). To improve the efficacy of BTKis, multiple trials have tested the combination of BTKis with the following chemoimmunotherapy regimens: anti-CD20 monoclonal antibodies (mAbs) (37–39, 46, 49, 53, 62), fludarabine (63), bendamustine (64), FCR (65), fludarabine, cyclophosphamide plus obinutuzumab (66, 67), and bendamustine plus rituximab (BR) (44, 68). A few exceptional studies used conventional agents for an abbreviated period as part of sequential therapy (64, 66), debulking (63), or MRD clearance (67). The vast majority of the combination studies adopted up to six cycles of conventional therapy. Combination approaches did improve the rate of U-MRD up to 80-90% by adding triplet chemoimmunotherapy to ibrutinib (65–67), and a range of 5-35% by adding an anti-CD20 to a BTKi in TN CLL (37–39, 46, 49, 53, 62). Despite high rates of U-MRD, cytotoxic agents have fallen out of favor because of safety concerns related to hematologic toxicities and secondary myeloid neoplasms observed in 2-5% of long-term survivors treated with FCR (61, 69).
The combination of an anti-CD20 mAb and a BTKi is generally well tolerated, and its use is supported by U.S. prescriber information for ibrutinib and acalabrutinib. Nevertheless, it is unclear if mAbs add clinically meaningful benefit to BTK inhibition. Several thoughtfully designed randomized trials tackled this question and arrived at different conclusions. A randomized phase 2 study enrolling mostly R/R CLL patients showed no difference in PFS with or without the addition of rituximab to ibrutinib (46). A randomized phase 3 study in elderly TN CLL reached a similar conclusion (37). Nevertheless, trials testing newer generations of BTKis and anti-CD20 mAbs challenge previous observations. The GENUINE study showed significant PFS benefit with the addition of ublituximab to ibrutinib, and the PFS benefit was largely driven by high-risk patients with R/R disease and TP53 aberration (70). The ELEVATE-TN study also reported 50% reduction in risk of progression or death in patients treated with acalabrutinib plus obinutuzumab compared to those receiving acalabrutinib alone (49). Across four randomized trials testing ibrutinib- or acalabrutinib-based combinations, there was a greater risk reduction in studies adding obinutuzumab to a BTKi (hazard ratio [HR] for progression or death 0.08-0.15) (39, 49) than those adding rituximab to a BTKi (HR 0.26-0.51) (37, 38) in patients with unmutated IGHV. Although these data are limited as they are often exploratory or subgroup analyses of trials, nonetheless they raise an important question of whether BTK inhibition can be optimized by using specific drug combinations in selected patient populations.
Why did randomized trials testing the combination of anti-CD20 mAbs and BTKis draw contradicting conclusions? This is because effector mechanisms of mAbs vary by molecules and by BTKis used in combination. The newer generation of anti-CD20 mAbs exhibits improved effector function compared to rituximab and has been shown to circumvent mechanisms of rituximab failure in vitro. Obinutuzumab, a glycoengineered type 2 antibody, induces greater direct cell killing, greater antibody-dependent cellular cytotoxicity (ADCC), and less intra- and trans-cellular loss of target antigens compared to rituximab (71). Ublituximab is a glycoengineered type 1 antibody that targets a unique epitope of CD20 and has shown greater ADCC than rituximab in vitro (72). In support of these findings, two randomized studies conducted in context of chemoimmunotherapy favored obinutuzumab to be more efficacious than rituximab for the treatment of TN CLL (73, 74) and FL (75). These effector mechanisms of mAbs can be hampered by off target effects of ibrutinib but preserved with the use of more selective BTKis. In a preclinical study, a wide range of doses of ibrutinib, but not acalabrutinib, inhibited antibody-dependent cellular phagocytosis, a key therapeutic mechanism of any anti-CD20 mAb (76). Ibrutinib also downregulates CD20 expression via decreased NF-κB activity (77) and by reducing supportive chemokines from the microenvironment that are necessary for CD20 upregulation (78). No data specific to the dynamics of CD20 expression during treatment with second-generation BTKis have yet been generated.
Venetoclax, a BCL2 inhibitor, is one of the preferred partners to BTKis. Single-agent venetoclax can achieve U-MRD in bone marrow in 16% of R/R CLL (79). The rate of U-MRD in bone marrow increases to ~60% in TN CLL by combining venetoclax with either obinutuzumab (80) or ibrutinib (81). Early data from single-arm trials testing triplet therapy with a BTKi, venetoclax, and obinutuzumab reported further improvement in the proportion of patients achieving deep responses. In TN CLL, the rate of U-MRD in bone marrow was 67% with ibrutinib, venetoclax and obinutuzumab (82), 78% with acalabrutinib, venetoclax and obinutuzumab (83), and 84% with zanubrutinib, venetoclax and obinutuzumab (84). Simultaneous targeting of BCL2 and BTK with or without an anti-CD20 mAb has several advantages over single-agent approaches. Targeted combinations can achieve U-MRD in a substantial proportion of patients, enabling treatment cessation after a fixed period (NCT04608318, NCT03701282, NCT02950051) or based on MRD status (NCT04639362). Such modifications in treatment duration, which are being actively investigated in clinical trials, can potentially reduce long-term toxicities linked to continuous therapy. Moreover, a lead-in period with a BTKi and/or obinutuzumab can reduce risk of tumor lysis syndrome (TLS), a potentially fatal toxicity of venetoclax (82). BTKis can additionally reduce the rate and the severity of infusion-related reactions (IRR) associated with mAbs. The reported rate of IRR was 44% in patients treated with venetoclax and obinutuzumab (80) and 20% in a different study testing acalabrutinib, venetoclax and obinutuzumab (85). Similar findings were reported from a randomized study for WM, which demonstrated a significantly lower rate of IRR in patients receiving ibrutinib and rituximab (1%) than in those treated with ibrutinib alone (16%) (86).
Safety profiles of individual BTKis have shared features and differences depending on relative selectivity to BTK. Because ibrutinib has the largest and longest safety data available among BTKis, our discussion focuses on key non-hematologic toxicities originally identified from ibrutinib and highlights differences in safety profiles of BTKis.
BTKis increase the risk of bleeding by inhibiting platelet aggregation and adhesion (87). BTK has been experimentally shown to be necessary for collagen-induced and von Willebrand factor-dependent thrombus formation (88). Further, both BTK and TEC are independently involved in platelet activation through glycoprotein (GP) VI signaling (89). Treatment with ibrutinib can block the downstream GP VI signaling and subsequent platelet aggregation, although there is substantial inter-patient variability in the effects of ibrutinib on platelet functions (90, 91). Compared with ibrutinib, acalabrutinib is less effective at inhibiting GP VI signaling in vitro and overall weaker at inhibiting collagen-mediated platelet aggregation ex vivo (92). Nevertheless, acalabrutinib still impacts platelet aggregation in certain settings such as in the presence of concurrent anti-platelet therapy and samples with known sensitivity to ibrutinib in terms of platelet functions (92). In clinical trials, most bleeding was low grade presenting as contusion or petechiae. Major bleeding is uncommon with both ibrutinib (2-5%) (93) and acalabrutinib (2-5%) (49, 51). Concurrent administration of warfarin is generally avoided during treatment with BTKis because warfarin was an exclusion criterion for most trials after four patients on a phase 2 study developed subdural hematoma while taking ibrutinib and warfarin or aspirin (94). Direct oral anticoagulants, anti-platelet agents including aspirin and clopidogrel, and low molecular weight heparins can be used during treatment with BTKis (95). Dual anti-platelet therapy is generally avoided given very limited data on safety. Patients undergoing elective surgical procedures are recommended to interrupt BTKis for 3 to 7 days before and after the procedure to minimize the risk of post-operative bleeding.
Hypertension and atrial fibrillation (Afib) are the two most common cardiovascular toxicities of BTKis. In a retrospective analysis of 562 patients treated with ibrutinib, new or worsening hypertension affected 78% of the patients, occurred early in the treatment course (50% of the events occurred within 2 months of treatment initiation), and was associated with major cardiovascular events including Afib (96). Emergence of Afib during treatment with BTKi poses a particular challenge to clinicians. The complexity of care increases with the diagnosis of Afib as it requires cardiology consultation, assessment of the need for anticoagulation, and rate- and/or rhythm-controlling interventions. Reported incidences of Afib range from 7-13% in studies of ibrutinib (48, 93, 96) and 3-7% for acalabrutinib (49, 51, 97). This difference in incidences of Afib can be explained by the fact that ibrutinib, but not acalabrutinib, inhibits C-terminal Src kinase (CSK) expressed in cardiac tissue (28). In support of this hypothesis, CSK knock-out mice, as well as ibrutinib-treated mice with wild-type CSK, developed increased Afib, recapitulating observations from patients treated with ibrutinib. Results from ongoing randomized studies are eagerly awaited to determine the safety of second-generation BTKis in comparison with ibrutinib (NCT02477696, NCT03734016).
Opportunistic infection (OI) is an uncommon, yet important side effect of BTKis. Although variations in the use of antimicrobial prophylaxis make it difficult to determine the true risk of infection, a study by Rogers et al. reported the OI incidence rate of 1.9 per 100 person-years in a retrospective analysis of over 500 patients treated with BTKis (98). Invasive aspergillosis was the most common pathogen identified from this study (2% of the cohort), while others reported Pneumocystis jirovecii pneumonia in up to 3% of patients not on prophylaxis during BTKi therapy (99, 100). Other rare pathogens observed during BTKi therapy include atypical Mycobacterium spp., JC virus, and toxoplasmosis (98). Impaired immune surveillance during treatment with a BTKi is linked to the known role of BTK in macrophage toll-like receptor 9 activation (101). BTK deficient mice are unable to mount an immune response to fungus, indicating that BTKis control innate immunity (102).
Disease progression remains among the most common reasons for BTKi discontinuation in CLL. Long-term follow-up of patients treated with ibrutinib monotherapy reported 5-year PFS of 70-92% for first-line treatment with ibrutinib (40, 103), and 40-44% for relapsed CLL (43, 103). In addition to prior treatment status, risk of progression increases in the presence of high-risk genetic or biochemical markers at pre-treatment such as TP53 aberrations (56, 59), complex karyotype (104), increased ß-2 microglobulin, and elevated lactate dehydrogenase (105).
There are two types of disease progression on BTKis. First, CLL can histologically transform into a more aggressive type of lymphoma, a phenomenon termed Richter’s transformation (RT). Atypical B cells found in RT commonly have DLBCL-like immunophenotypes and less often present as Hodgkin-like lesions with Reed-Sternberg cells (106). Although mechanisms of histologic transformation are unclear, studies have identified enrichment of several notable molecular events in RT. There is a high prevalence of stereotyped BCR (70%) and biased usage of IGHV4-39 in patients with RT (107). Mutations of known driver genes in CLL (TP53, NOTCH1) and the CDKN2A/B cell cycle regulator are found more frequently in RT than CLL (108). RT is further characterized by complex copy number changes including 17p loss leading to TP53 aberration, gain/amplification of MYC on 8q, 9q loss resulting in haploinsufficiency of CDKN2A/2B, and 18p loss without a candidate gene (109). However, these genetic lesions are not unique to RT, raising the possibility of an additional non-genetic inducer of aggressive transformation. Akt-mediated transcriptional control and subsequent NOTCH activation have been recently proposed to have a role in RT, which is supported by increased AKT activation in primary RT samples and an accelerated lymphoma phenotype observed in the Eµ-TCL1 mouse model with constitutive Akt activation (110). Once RT develops during treatment with BTKis, most patients relapse shortly after or become refractory to alternative therapy including immune checkpoint inhibitors (111) and anthracycline-based chemoimmunotherapy (112, 113). Allogeneic stem cell transplant (114) and CD19 chimeric antigen receptor modified T-cell infusion (CAR T) (115) can offer durable remission in a minority of patients. Unfortunately, many patients with RT are deemed unsuitable for cell therapy because of comorbidities, low rates of remission after initial therapy, lack of stem cell donors, or limited access to cellular products.
The second and more commonly observed type of progression is CLL with secondary resistance to BTKis. Up to 80% of the patients with BTKi-resistant CLL carry BTK and/or PLCG2 mutations at the time of progression (Figure 2) (116–118). BTK mutations substitute the C481 residue with an alternative amino acid, most commonly serine, leading to the loss of a covalent bond between the drug and the kinase. The vast majority of PLCG2 mutations identified to date affect the N-terminal SH2 domain with an autoinhibitory function. Functional studies in CLL and autoimmune diseases demonstrated that a mutation or deletion of the SH2 domain can activate PLCγ2 and downstream BCR signaling (119–121). Acquisition of BTK and PLCG2 mutations can occur any time in the clinical course. Preexisting BTK or PLCG2 mutation is exceedingly rare and comprises a minor fraction of CLL, if any [allele frequency of 0.0002% in a report by Burger et al. (122)]. Under selective pressure of BTK inhibition, CLL undergoes linear or branching evolution with the latter giving rise to a multiclonal disease (Figure 3) (118). Multiclonality of BTKi-resistant CLL was clearly demonstrated by a single-cell analysis of a patient with four different PLCG2 mutations, which presented as distinct clonal subpopulations rather than coexisting mutations within the same cells (122).
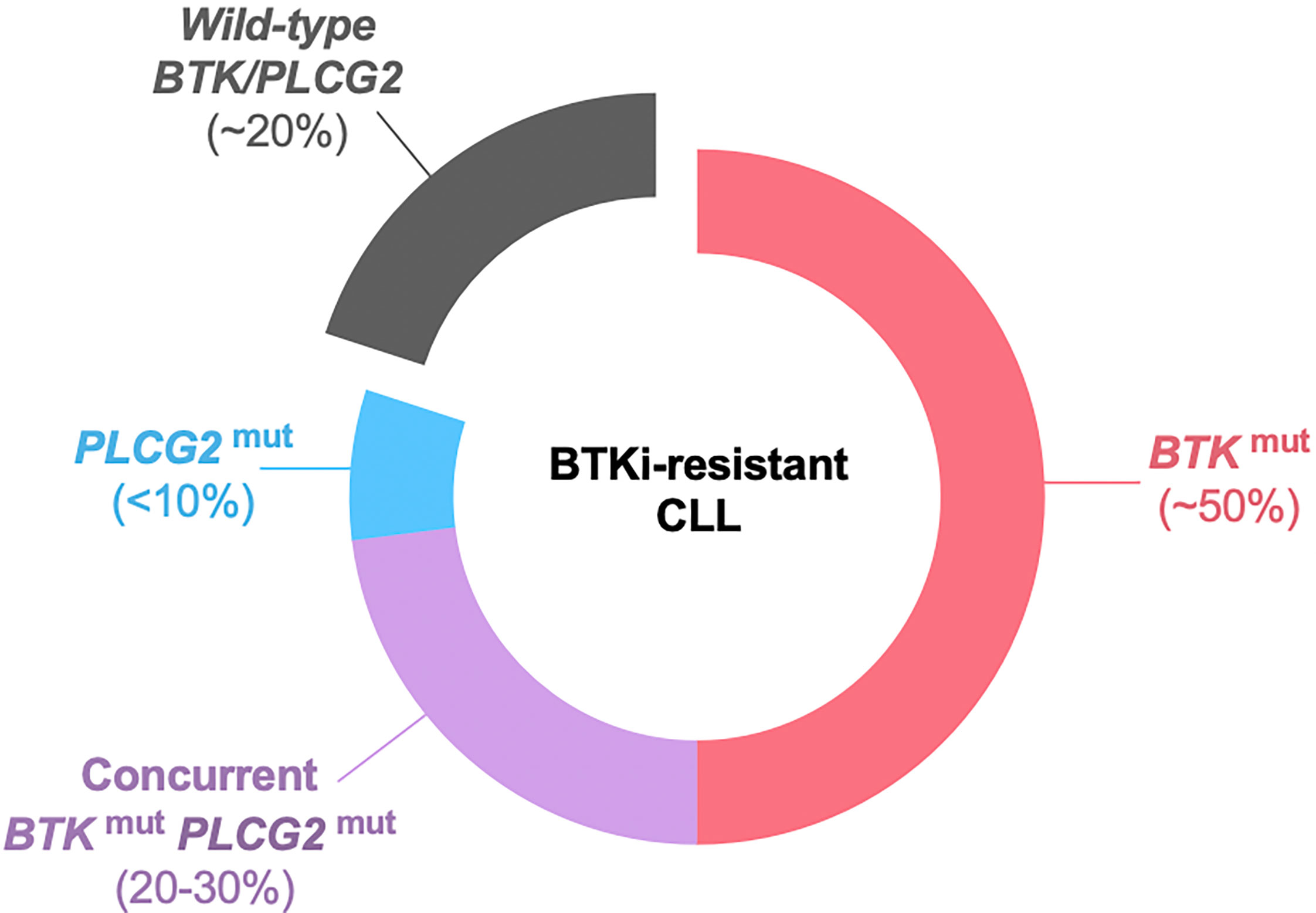
Figure 2 BTK and PLCG2 mutations in BTKi-resistant CLL. Approximately 20% of patients do not have detectable BTK or PLCG2 mutation at progression. BTK mutation is the most common mutation, found in half the patients as BTK mutation alone and in an additional 20-30% with coexisting PLCG2 mutation. Less than 10% of the patients have PLCG2 mutation alone. BTKi, Bruton’s tyrosine kinase inhibitor; mut, mutation.
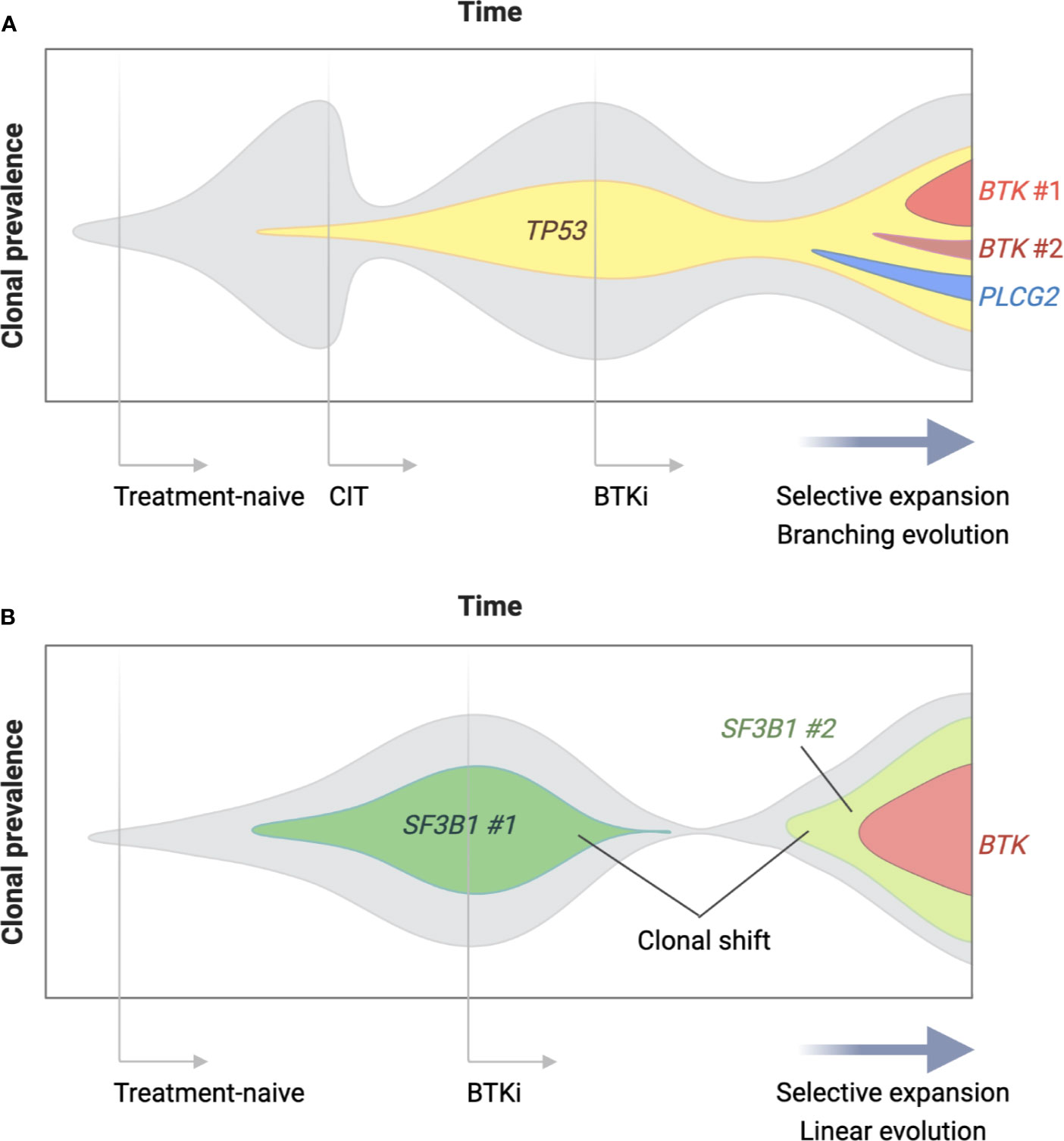
Figure 3 Clonal evolution of BTKi-resistant CLL. Clonal architecture of CLL changes over time under the selective pressure of treatment and in the presence of driver gene mutations. Panel (A) is a schematic representation of a patient who was treated with chemoimmunotherapy (CIT) as first-line and a BTK inhibitor (BTKi) as second-line therapy for CLL. Two lines of therapy selectively expanded a parental clone with a driver gene mutation (TP53 in this case), which became a parental clone of BTKi-resistant disease. Multiple BTK and PLCG2 mutations arose after branching evolution and were detectable at relatively low allele frequency at the time of progression. Panel (B) shows a patient who underwent a major shift in clonal dominance from one clone (SF3B1 mutation #1) to another (SF3B1 mutation #2) during treatment with a BTKi. Linear evolution of the emerging clone led to a single dominant BTK mutation at progression detectable at high allele frequency.
Several theoretical approaches can be considered to prevent the emergence of BTKi resistance. The simplest method is to stop BTKis after a defined period. Another approach is to combine multiple targeted agents with non-overlapping mechanisms of action as discussed previously. Several ongoing trials investigate time-limited, targeted agent-based combination therapy in CLL (NCT04608318, NCT03701282, NCT02950051). Critical to these approaches is the ability to monitor clonal evolution during and after treatment cessation in both investigational and control arms of these trials.
BTKi-resistant CLL demonstrates an aggressive clinical course in the absence of effective salvage therapy (123). Venetoclax can achieve initial responses (124). However, the responses are not durable, and most patients progress within 2 years. Novel combinations are being tested in BTKi-resistant CLL including ibrutinib plus venetoclax (NCT03943342, NCT03513562) and umbralisib plus ublituximab (NCT04149821). A phase 2 study of ibrutinib plus duvelisib in ibrutinib-resistant CLL was halted due to sudden death (NCT04209621). It will be of interest to determine the tolerability of novel combinations and the subsequent risk of clonal evolution in patients who became resistant to non-covalent BTKis.
Immune-directed therapy has an advantage over targeted agents as it can bypass resistance mechanisms intrinsic to tumor cells. In the modern era, allogeneic hematopoietic stem cell transplant has become safer and more accessible. Recent retrospective analyses of transplant outcomes in CLL reported 2-year PFS of 63% (125) and 3-year non-relapse mortality of 7% in patients who were previously treated with targeted agents (126). Treatment with CD19 CAR T can also achieve high rates of initial response in patients who failed ibrutinib (115). At present, none of the commercially available CD19 CAR T products are approved for the treatment of CLL. Further research is needed to define the durability of response to CAR T, minimize toxicities mediated by cytokine release, and improve access to adoptive cell therapy. Preclinical data indicate bispecific T-cell engager antibodies targeting CD3 and CD19 are efficacious against BTK/PLCG2 mutant CLL cells in vitro and in patient-derived xenograft models (127). Several bispecific antibodies have entered clinical development to test dual targeting of CD3xCD19 (blinatumomab, NCT02568553), CD3xCD20 (REGN1979, NCT02290951), and CD3xCD22 (JNJ-75348780, NCT04540796).
Retargeting of BTK with agents with reversible covalent binding chemistry is an attractive strategy for the treatment of CLL. BTK mutation, found in ~80% of patients with ibrutinib resistance, validates the importance of this kinase in CLL. Reversible BTKis can inhibit the kinase in the presence of BTK C481 mutation (128). Five reversible BTKis have entered clinical trials to date (Table 3). Of these, pirtobrutinib is most advanced in development and most specific to BTK with little to no effect on other targets. In a phase 1/2 study, the overall response rate to pirtobrutinib was 71% for CLL patients with BTK C481 mutation and 66% for those with wild-type BTK, indicating that clinical activity was independent of BTK mutation status (129). MK-1026 (formerly ARQ-531) differs from other reversible BTKis for its ability to inhibit SYK and LYN and indirectly inhibit MEK1/ERK. Intriguingly, MK-1026 inhibited downstream signaling of DT40 cell lines transfected with PLCG2 mutations, suggesting SYK/LYN inhibition has a role against PLCG2 mutant clones (128). A phase 1 study of MK-1026 is expected to complete soon (130) and to be followed by a phase 2 study in hematologic malignancies including CLL (NCT04728893). CG-806 is a dual inhibitor of BTK and FMS-like tyrosine kinase 3 with internal tandem duplication, a mutation found in 30% of patients with acute myeloid leukemia (136). A phase 1 study of CG-806 is ongoing in CLL and non-Hodgkin lymphomas (NCT03893682) (136) besides two additional studies in myeloid diseases. Fenebrutinib (132) and vecabrutinib (135) showed acceptable tolerability in phase 1 studies, but were withdrawn from development in B cell malignancies. Fenebrutinib continues to be tested in multiple sclerosis (NCT04586023, NCT04586010). Vecabrutinib was withdrawn due to insufficient evidence of activity limiting its advancement to a phase 2 study (NCT03037645).
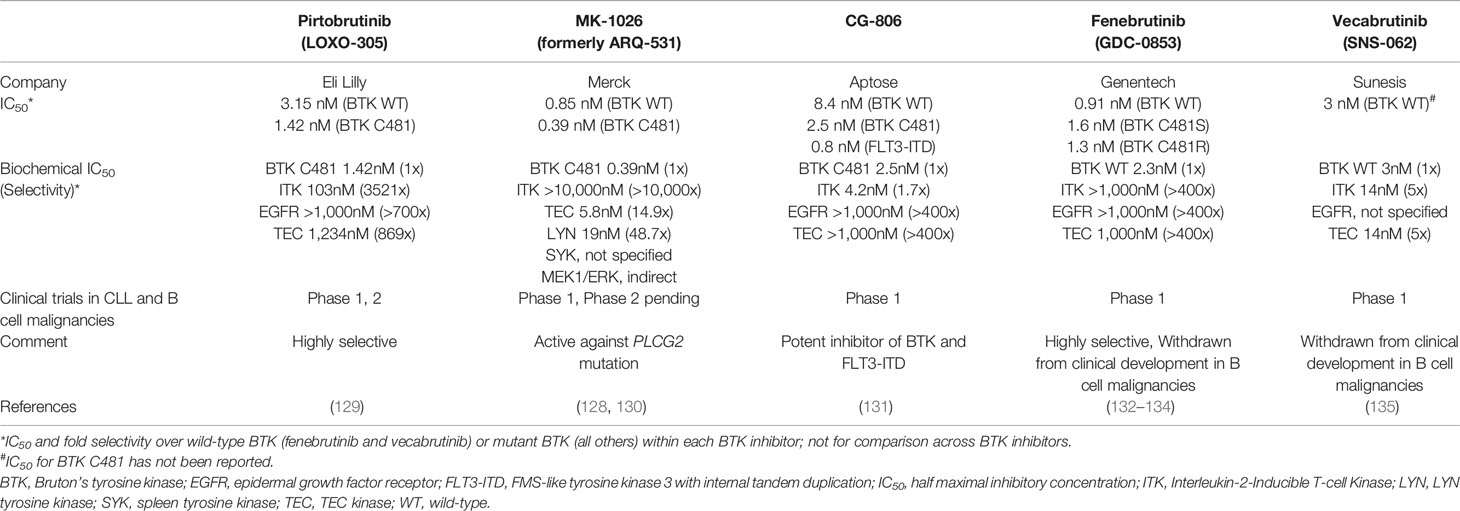
Table 3 Non-covalent BTK inhibitors.
Data accumulated from clinical trials of covalent BTKis identified research questions critical for further optimization of the BTK targeting strategy. First, a better distinction of safety and efficacy profiles of individual BTKis is anticipated through ongoing randomized trials. Second, novel targeted combinations can achieve deep response and enable treatment cessation upon attainment of U-MRD in CLL. What remains to be addressed is durability of response and long-term safety of novel combinations, randomized comparisons with approved BTKi- or BCL-2-based regimens, and clonal dynamics traced with sequential genomic testing. Lastly, treatments capable of preventing or overcoming resistance to covalent BTKis are urgently needed. Several non-covalent BTKis with activity against BTKi-resistant disease are under investigation, highlighting the importance of BTK as a therapeutic target in CLL.
IA and JB performed literature review and wrote the manuscript. All authors contributed to the article and approved the submitted version.
IA is funded by American Society of Hematology Scholar Award. JB is funded by NCI R01 CA 213442.
JB has served as a consultant for Abbvie, Acerta, Astra-Zeneca, Beigene, Catapult, Dynamo Therapeutics, Eli Lilly, Genentech/Roche, Juno/Celgene/Bristol Myers Squibb, Kite, Loxo, MEI Pharma, Nextcea, Novartis, Pfizer, Pharmacyclics, Rigel, Sunesis, TG Therapeutics; received research funding from Gilead, Loxo, SPARC, TG Therapeutics and Verastem; and served on data safety monitoring committees for Invectys.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic Active B-Cell-Receptor Signalling in Diffuse Large B-Cell Lymphoma. Nature (2010) 463(7277):88–92. doi: 10.1038/nature08638
2. Gauld SB, Dal Porto JM, Cambier JC. B Cell Antigen Receptor Signaling: Roles in Cell Development and Disease. Science (2002) 296(5573):1641–2. doi: 10.1126/science.1071546
3. Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 Kinase Signals BCR-Dependent Mature B Cell Survival. Cell (2009) 139(3):573–86. doi: 10.1016/j.cell.2009.08.041
4. Lam KP, Kuhn R, Rajewsky K. In Vivo Ablation of Surface Immunoglobulin on Mature B Cells by Inducible Gene Targeting Results in Rapid Cell Death. Cell (1997) 90(6):1073–83. doi: 10.1016/S0092-8674(00)80373-6
5. Chen L, Monti S, Juszczynski P, Ouyang J, Chapuy B, Neuberg D, et al. SYK Inhibition Modulates Distinct PI3K/AKT-Dependent Survival Pathways and Cholesterol Biosynthesis in Diffuse Large B Cell Lymphomas. Cancer Cell (2013) 23(6):826–38. doi: 10.1016/j.ccr.2013.05.002
6. Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt Lymphoma Pathogenesis and Therapeutic Targets From Structural and Functional Genomics. Nature (2012) 490(7418):116–20. doi: 10.1038/nature11378
7. Chen L, Ouyang J, Wienand K, Bojarczuk K, Hao Y, Chapuy B, et al. CXCR4 Upregulation is an Indicator of Sensitivity to B-Cell Receptor/PI3K Blockade and a Potential Resistance Mechanism in B-Cell Receptor-Dependent Diffuse Large B-Cell Lymphomas. Haematologica (2020) 105(5):1361–8. doi: 10.3324/haematol.2019.216218
8. Messmer BT, Albesiano E, Efremov DG, Ghiotto F, Allen SL, Kolitz J, et al. Multiple Distinct Sets of Stereotyped Antigen Receptors Indicate a Role for Antigen in Promoting Chronic Lymphocytic Leukemia. J Exp Med (2004) 200(4):519–25. doi: 10.1084/jem.20040544
9. Navarro A, Clot G, Royo C, Jares P, Hadzidimitriou A, Agathangelidis A, et al. Molecular Subsets of Mantle Cell Lymphoma Defined by the IGHV Mutational Status and SOX11 Expression Have Distinct Biologic and Clinical Features. Cancer Res (2012) 72(20):5307–16. doi: 10.1158/0008-5472.CAN-12-1615
10. Quinn ER, Chan CH, Hadlock KG, Foung SK, Flint M, Levy S. The B-Cell Receptor of a Hepatitis C Virus (HCV)-Associated Non-Hodgkin Lymphoma Binds the Viral E2 Envelope Protein, Implicating HCV in Lymphomagenesis. Blood (2001) 98(13):3745–9. doi: 10.1182/blood.V98.13.3745
11. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med (2018) 378(15):1396–407. doi: 10.1056/NEJMoa1801445
12. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated With Distinct Pathogenic Mechanisms and Outcomes. Nat Med (2018) 24(5):679–90. doi: 10.1038/s41591-018-0016-8
13. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 Mutations in Human Diffuse Large B Cell Lymphoma. Science (2008) 319(5870):1676–9. doi: 10.1126/science.1153629
14. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations Driving CLL and Their Evolution in Progression and Relapse. Nature (2015) 526(7574):525–30. doi: 10.1038/nature15395
15. Bartlett NL, Costello BA, LaPlant BR, Ansell SM, Kuruvilla JG, Reeder CB, et al. Single-Agent Ibrutinib in Relapsed or Refractory Follicular Lymphoma: A Phase 2 Consortium Trial. Blood (2018) 131(2):182–90. doi: 10.1182/blood-2017-09-804641
16. Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of Somatic Mutations and Clonal Evolution in Mantle Cell Lymphoma. Proc Natl Acad Sci USA (2013) 110(45):18250–5. doi: 10.1073/pnas.1314608110
17. Cha SC, Qin H, Kannan S, Rawal S, Watkins LS, Baio FE, et al. Nonstereotyped Lymphoma B Cell Receptors Recognize Vimentin as a Shared Autoantigen. J Immunol (2013) 190(9):4887–98. doi: 10.4049/jimmunol.1300179
18. Sachen KL, Strohman MJ, Singletary J, Alizadeh AA, Kattah NH, Lossos C, et al. Self-Antigen Recognition by Follicular Lymphoma B-Cell Receptors. Blood (2012) 120(20):4182–90. doi: 10.1182/blood-2012-05-427534
19. Dighiero G, Hart S, Lim A, Borche L, Levy R, Miller RA. Autoantibody Activity of Immunoglobulins Isolated From B-Cell Follicular Lymphomas. Blood (1991) 78(3):581–5. doi: 10.1182/blood.V78.3.581.bloodjournal783581
20. Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110delta Breaks Regulatory T-Cell-Mediated Immune Tolerance to Cancer. Nature (2014) 510(7505):407–11. doi: 10.1038/nature13444
21. Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, et al. Idelalisib Given Front-Line for Treatment of Chronic Lymphocytic Leukemia Causes Frequent Immune-Mediated Hepatotoxicity. Blood (2016) 128(2):195–203. doi: 10.1182/blood-2016-03-707133
22. Lampson BL, Brown JR. PI3Kdelta-Selective and PI3Kalpha/delta-combinatorial Inhibitors in Clinical Development for B-Cell Non-Hodgkin Lymphoma. Expert Opin Investig Drugs (2017) 26(11):1267–79. doi: 10.1080/13543784.2017.1384815
23. Dai B, Grau M, Juilland M, Klener P, Horing E, Molinsky J, et al. B-Cell Receptor-Driven MALT1 Activity Regulates MYC Signaling in Mantle Cell Lymphoma. Blood (2017) 129(3):333–46. doi: 10.1182/blood-2016-05-718775
24. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Blocks B-Cell Activation and Is Efficacious in Models of Autoimmune Disease and B-Cell Malignancy. Proc Natl Acad Sci USA (2010) 107(29):13075–80. doi: 10.1073/pnas.1004594107
25. Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem (2007) 2(1):58–61. doi: 10.1002/cmdc.200600221
26. Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton Tyrosine Kinase Inhibitor Ibrutinib (PCI-32765) has Significant Activity in Patients With Relapsed/Refractory B-Cell Malignancies. J Clin Oncol Off J Am Soc Clin Oncol (2013) 31(1):88–94. doi: 10.1200/JCO.2012.42.7906
27. Brown JR. How I Treat CLL Patients With Ibrutinib. Blood (2018) 131(4):379–86. doi: 10.1182/blood-2017-08-764712
28. Xiao L, Salem JE, Clauss S, Hanley A, Bapat A, Hulsmans M, et al. Ibrutinib-Mediated Atrial Fibrillation Attributable to Inhibition of C-Terminal Src Kinase. Circulation (2020) 142(25):2443–55. doi: 10.1161/CIRCULATIONAHA.120.049210
29. Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med (2016) 374(4):323–32. doi: 10.1056/NEJMoa1509981
30. Sun C, Nierman P, Kendall EK, Cheung J, Gulrajani M, Herman SEM, et al. Clinical and Biological Implications of Target Occupancy in CLL Treated With the BTK Inhibitor Acalabrutinib. Blood (2020) 136(1):93–105. doi: 10.1182/blood.2019003715
31. Tam CS, Trotman J, Opat S, Burger JA, Cull G, Gottlieb D, et al. Phase 1 Study of the Selective BTK Inhibitor Zanubrutinib in B-Cell Malignancies and Safety and Efficacy Evaluation in CLL. Blood (2019) 134(11):851–9. doi: 10.1182/blood.2019001160
32. Brown JR, Harb WA, Hill BT, Gabrilove J, Sharman JP, Schreeder MT, et al. Phase I Study of Single-Agent CC-292, a Highly Selective Bruton’s Tyrosine Kinase Inhibitor, in Relapsed/Refractory Chronic Lymphocytic Leukemia. Haematologica (2016) 101(7):e295–8. doi: 10.3324/haematol.2015.140806
33. Narita Y, Nagane M, Mishima K, Terui Y, Arakawa Y, Yonezawa H, et al. Phase I/II Study of Tirabrutinib, A Second-Generation Bruton’s Tyrosine Kinase Inhibitor, in Relapsed/Refractory Primary Central Nervous System Lymphoma. Neuro Oncol (2021) 23(1):122–33. doi: 10.1093/neuonc/noaa145
34. Munakata W, Ando K, Hatake K, Fukuhara N, Kinoshita T, Fukuhara S, et al. Phase I Study of Tirabrutinib (ONO-4059/GS-4059) in Patients With Relapsed or Refractory B-Cell Malignancies in Japan. Cancer Sci (2019) 110(5):1686–94. doi: 10.1111/cas.13983
35. Tam CS, Opat S, D’Sa S, Jurczak W, Lee HP, Cull G, et al. A Randomized Phase 3 Trial of Zanubrutinib vs Ibrutinib in Symptomatic Waldenstrom Macroglobulinemia: The ASPEN Study. Blood (2020) 136(18):2038–50. doi: 10.1182/blood.2020006844
36. Awan FT, Schuh A, Brown JR, Furman RR, Pagel JM, Hillmen P, et al. Acalabrutinib Monotherapy in Patients With Chronic Lymphocytic Leukemia Who Are Intolerant to Ibrutinib. Blood Adv (2019) 3(9):1553–62. doi: 10.1182/bloodadvances.2018030007
37. Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, et al. Ibrutinib Regimens Versus Chemoimmunotherapy in Older Patients With Untreated Cll. N Engl J Med (2018) 379(26):2517–28. doi: 10.1056/NEJMoa1812836
38. Shanafelt TD, Wang XV, Kay NE, Hanson CA, O’Brien S, Barrientos J, et al. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med (2019) 381(5):432–43. doi: 10.1056/NEJMoa1817073
39. Moreno C, Greil R, Demirkan F, Tedeschi A, Anz B, Larratt L, et al. Ibrutinib Plus Obinutuzumab Versus Chlorambucil Plus Obinutuzumab in First-Line Treatment of Chronic Lymphocytic Leukaemia (iLLUMINATE): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol (2019) 20(1):43–56. doi: 10.1016/S1470-2045(18)30788-5
40. Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, et al. Long-Term Efficacy and Safety of First-Line Ibrutinib Treatment for Patients With CLL/SLL: 5 Years of Follow-Up From the Phase 3 RESONATE-2 Study. Leukemia (2020) 34(3):787–98. doi: 10.1038/s41375-019-0602-x
41. O’Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, et al. Ibrutinib as Initial Therapy for Elderly Patients With Chronic Lymphocytic Leukaemia or Small Lymphocytic Lymphoma: An Open-Label, Multicentre, Phase 1b/2 Trial. Lancet Oncol (2014) 15(1):48–58. doi: 10.1016/S1470-2045(13)70513-8
42. Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib Versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N Engl J Med (2014) 371(3):213–23. doi: 10.1056/NEJMoa1400376
43. Munir T, Brown JR, O’Brien S, Barrientos JC, Barr PM, Reddy NM, et al. Final Analysis From RESONATE: Up to Six Years of Follow-Up on Ibrutinib in Patients With Previously Treated Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma. Am J Hematol (2019) 94(12):1353–63. doi: 10.1002/ajh.25638
44. Chanan-Khan A, Cramer P, Demirkan F, Fraser G, Silva RS, Grosicki S, et al. Ibrutinib Combined With Bendamustine and Rituximab Compared With Placebo, Bendamustine, and Rituximab for Previously Treated Chronic Lymphocytic Leukaemia or Small Lymphocytic Lymphoma (HELIOS): A Randomised, Double-Blind, Phase 3 Study. Lancet Oncol (2016) 17(2):200–11. doi: 10.1016/S1470-2045(15)00465-9
45. Fraser GAM, Chanan-Khan A, Demirkan F, Santucci Silva R, Grosicki S, Janssens A, et al. Final 5-Year Findings From the Phase 3 HELIOS Study of Ibrutinib Plus Bendamustine and Rituximab in Patients With Relapsed/Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. Leukemia lymphoma (2020) 61(13):3188–97. doi: 10.1080/10428194.2020.1795159
46. Burger JA, Sivina M, Jain N, Kim E, Kadia T, Estrov Z, et al. Randomized Trial of Ibrutinib vs Ibrutinib Plus Rituximab in Patients With Chronic Lymphocytic Leukemia. Blood (2019) 133(10):1011–9. doi: 10.1182/blood-2018-10-879429
47. Ahn IE, Tian X, Wiestner A. Ibrutinib for Chronic Lymphocytic Leukemia With TP53 Alterations. N Engl J Med (2020) 383(5):498–500. doi: 10.1056/NEJMc2005943
48. O’Brien S, Jones JA, Coutre SE, Mato AR, Hillmen P, Tam C, et al. Ibrutinib for Patients With Relapsed or Refractory Chronic Lymphocytic Leukaemia With 17p Deletion (RESONATE-17): A Phase 2, Open-Label, Multicentre Study. Lancet Oncol (2016) 17(10):1409–18. doi: 10.1016/S1470-2045(16)30212-1
49. Sharman JP, Banerji V, Fogliatto LM, Herishanu Y, Munir T, Walewska R, et al. Elevate TN: Phase 3 Study of Acalabrutinib Combined With Obinutuzumab (O) or Alone Vs O Plus Chlorambucil (Clb) in Patients (Pts) With Treatment-Naive Chronic Lymphocytic Leukemia (CLL). Blood (2019) 134. doi: 10.1182/blood-2019-128404
50. Ghia P, Pluta A, Wach M, Lysak D, Kozak T, Simkovic M, et al. Ascend: Phase III, Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol Off J Am Soc Clin Oncol (2020) 38(25):2849–61. doi: 10.1200/JCO.19.03355
51. Byrd JC, Wierda WG, Schuh A, Devereux S, Chaves JM, Brown JR, et al. Acalabrutinib Monotherapy in Patients With Relapsed/Refractory Chronic Lymphocytic Leukemia: Updated Phase 2 Results. Blood (2020) 135(15):1204–13. doi: 10.1182/blood.2018884940
52. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK With Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med (2013) 369(1):32–42. doi: 10.1056/NEJMoa1215637
53. Tam CS, Quach H, Nicol A, Badoux X, Rose H, Prince HM, et al. Zanubrutinib (BGB-3111) Plus Obinutuzumab in Patients With Chronic Lymphocytic Leukemia and Follicular Lymphoma. Blood Adv (2020) 4(19):4802–11. doi: 10.1182/bloodadvances.2020002183
54. Tam CS, Robak T, Ghia P, Kahl BS, Walker P, Janowski W, et al. Zanubrutinib Monotherapy for Patients With Treatment Naive Chronic Lymphocytic Leukemia and 17p Deletion. Haematologica (2020). doi: 10.3324/haematol.2020.259432
55. Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as Initial Therapy for Patients With Chronic Lymphocytic Leukemia. N Engl J Med (2015) 373(25):2425–37. doi: 10.1056/NEJMoa1509388
56. Brown JR, Hillmen P, O’Brien S, Barrientos JC, Reddy NM, Coutre SE, et al. Extended Follow-Up and Impact of High-Risk Prognostic Factors From the Phase 3 RESONATE Study in Patients With Previously Treated CLL/SLL. Leukemia (2018) 32(1):83–91. doi: 10.1038/leu.2017.175
57. Ahn IE, Tian X, Ipe D, Cheng M, Albitar M, Tsao LC, et al. Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia Treated With Ibrutinib: Development and Validation of a Four-Factor Prognostic Model. J Clin Oncol (2021) 39(6):576–85. doi: 10.1200/JCO.20.00979
58. Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic Aberrations and Survival in Chronic Lymphocytic Leukemia. N Engl J Med (2000) 343(26):1910–6. doi: 10.1056/NEJM200012283432602
59. Ahn IE, Farooqui MZH, Tian X, Valdez J, Sun C, Soto S, et al. Depth and Durability of Response to Ibrutinib in CLL: 5-Year Follow-Up of a Phase 2 Study. Blood (2018) 131(21):2357–66. doi: 10.1182/blood-2017-12-820910
60. Thompson PA, Tam CS, O’Brien SM, Wierda WG, Stingo F, Plunkett W, et al. Fludarabine, Cyclophosphamide, and Rituximab Treatment Achieves Long-Term Disease-Free Survival in IGHV-Mutated Chronic Lymphocytic Leukemia. Blood (2016) 127(3):303–9. doi: 10.1182/blood-2015-09-667675
61. Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, et al. Long-Term Remissions After FCR Chemoimmunotherapy in Previously Untreated Patients With CLL: Updated Results of the CLL8 Trial. Blood (2016) 127(2):208–15. doi: 10.1182/blood-2015-06-651125
62. Burger JA, Keating MJ, Wierda WG, Hartmann E, Hoellenriegel J, Rosin NY, et al. Safety and Activity of Ibrutinib Plus Rituximab for Patients With High-Risk Chronic Lymphocytic Leukaemia: A Single-Arm, Phase 2 Study. Lancet Oncol (2014) 15(10):1090–9. doi: 10.1016/S1470-2045(14)70335-3
63. Pleyer C, Tian X, Rampertaap S, Mu R, Soto S, Superata J, et al. A Phase II Study of Ibrutinib and Short-Course Fludarabine in Previously Untreated Patients With Chronic Lymphocytic Leukemia. Am J Hematol (2020) 95(11):E310–3. doi: 10.1002/ajh.25968
64. von Tresckow J, Cramer P, Bahlo J, Robrecht S, Langerbeins P, Fink AM, et al. Cll2-BIG: Sequential Treatment With Bendamustine, Ibrutinib and Obinutuzumab (GA101) in Chronic Lymphocytic Leukemia. Leukemia (2019) 33(5):1161–72. doi: 10.1038/s41375-018-0313-8
65. Davids MS, Brander DM, Kim HT, Tyekucheva S, Bsat J, Savell A, et al. Ibrutinib Plus Fludarabine, Cyclophosphamide, and Rituximab as Initial Treatment for Younger Patients With Chronic Lymphocytic Leukaemia: A Single-Arm, Multicentre, Phase 2 Trial. Lancet Haematol (2019) 6(8):e419–e28. doi: 10.1016/S2352-3026(19)30104-8
66. Jain N, Thompson PA, Burger JA, Ferrajoli A, Takahashi K, Estrov ZE, et al. Ibrutinib, Fludarabine, Cyclophosphamide, and Obinutuzumab (iFCG) for First-Line Treatment of IGHV-Mutated CLL and Without Del(17p)/mutated Tp53. Blood (2019) 134. doi: 10.1182/blood-2019-131939
67. Michallet AS, Dilhuydy MS, Subtil F, Rouille V, Mahe B, Laribi K, et al. Obinutuzumab and Ibrutinib Induction Therapy Followed by a Minimal Residual Disease-Driven Strategy in Patients With Chronic Lymphocytic Leukaemia (ICLL07 FILO): A Single-Arm, Multicentre, Phase 2 Trial. Lancet Haematol (2019) 6(9):e470–e9. doi: 10.1016/S2352-3026(19)30113-9
68. Brown JR, Barrientos JC, Barr PM, Flinn IW, Burger JA, Tran A, et al. The Bruton Tyrosine Kinase Inhibitor Ibrutinib With Chemoimmunotherapy in Patients With Chronic Lymphocytic Leukemia. Blood (2015) 125(19):2915–22. doi: 10.1182/blood-2014-09-585869
69. Benjamini O, Jain P, Trinh L, Qiao W, Strom SS, Lerner S, et al. Second Cancers in Patients With Chronic Lymphocytic Leukemia Who Received Frontline Fludarabine, Cyclophosphamide and Rituximab Therapy: Distribution and Clinical Outcomes. Leukemia lymphoma (2015) 56(6):1643–50. doi: 10.3109/10428194.2014.957203
70. Sharman JP, Brander DM, Mato AR, Kambhampati S, Burke JM, Lansigan F, et al. Ublituximab and Ibrutinib for Previously Treated Genetically High-Risk Chronic Lymphocytic Leukemia: Results of the GENUINE Phase 3 Study. J Clin Oncol (2017) 35(15). doi: 10.1200/JCO.2017.35.15_suppl.7504
71. Sharman JP. Targeting CD20: Teaching An Old Dog New Tricks. Hematology Am Soc Hematol Educ Program (2019) 2019(1):273–8. doi: 10.1182/hematology.2019000031
72. Le Garff-Tavernier M, Herbi L, de Romeuf C, Nguyen-Khac F, Davi F, Grelier A, et al. Antibody-Dependent Cellular Cytotoxicity of the Optimized Anti-CD20 Monoclonal Antibody Ublituximab on Chronic Lymphocytic Leukemia Cells With the 17p Deletion. Leukemia (2014) 28(1):230–3. doi: 10.1038/leu.2013.240
73. Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab Plus Chlorambucil in Patients With CLL and Coexisting Conditions. N Engl J Med (2014) 370(12):1101–10. doi: 10.1056/NEJMoa1313984
74. Goede V, Fischer K, Bosch F, Follows G, Frederiksen H, Cuneo A, et al. Updated Survival Analysis From the CLL11 Study: Obinutuzumab Versus Rituximab in Chemoimmunotherapy-Treated Patients With Chronic Lymphocytic Leukemia. Blood (2015) 126(23):1733. doi: 10.1182/blood.V126.23.1733.1733
75. Marcus R, Davies A, Ando K, Klapper W, Opat S, Owen C, et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N Engl J Med (2017) 377(14):1331–44. doi: 10.1056/NEJMoa1614598
76. Chu CC, Pinney JJ, Blick-Nitko SK, Baran AM, Peterson DR, Whitehead HE, et al. Ibrutinib Off-Target Inhibition Inhibits Antibody-Dependent Cellular Phagocytosis But Not Efferocytosis of Cll Cells. Blood (2020) 136(SUPPL 1):45. doi: 10.1182/blood-2020-139483
77. Skarzynski M, Niemann CU, Lee YS, Martyr S, Maric I, Salem D, et al. Interactions Between Ibrutinib and Anti-CD20 Antibodies: Competing Effects on the Outcome of Combination Therapy. Clin Cancer Res (2016) 22(1):86–95. doi: 10.1158/1078-0432.CCR-15-1304
78. Pavlasova G, Borsky M, Seda V, Cerna K, Osickova J, Doubek M, et al. Ibrutinib Inhibits CD20 Upregulation on CLL B Cells Mediated by the CXCR4/SDF-1 Axis. Blood (2016) 128(12):1609–13. doi: 10.1182/blood-2016-04-709519
79. Roberts AW, Ma S, Kipps TJ, Coutre SE, Davids MS, Eichhorst B, et al. Efficacy of Venetoclax in Relapsed Chronic Lymphocytic Leukemia is Influenced by Disease and Response Variables. Blood (2019) 134(2):111–22. doi: 10.1182/blood.2018882555
80. Fischer K, Al-Sawaf O, Bahlo J, Fink AM, Tandon M, Dixon M, et al. Venetoclax and Obinutuzumab in Patients With CLL and Coexisting Conditions. N Engl J Med (2019) 380(23):2225–36. doi: 10.1056/NEJMoa1815281
81. Jain N, Keating M, Thompson P, Ferrajoli A, Burger J, Borthakur G, et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med (2019) 380(22):2095–103. doi: 10.1056/NEJMoa1900574
82. Rogers KA, Huang Y, Ruppert AS, Abruzzo LV, Andersen BL, Awan FT, et al. Phase II Study of Combination Obinutuzumab, Ibrutinib, and Venetoclax in Treatment-Naïve and Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol (2020) 38(31):3626–37. doi: 10.1200/JCO.20.00491
83. Davids MS, Lampson BL, Tyekucheva S, Crombie JL, Ng S, Kim AI, et al. Updated Safety and Eicacy Results From a Phase 2 Study of Acalabrutinib, Venetoclax and Obinutuzumab (AVO) for Frontlinetreatment of Chronic Lymphocytic Leukemia (CLL). Blood (2020) 136(SUPPL 1):20–1. doi: 10.1182/blood-2020-139864
84. Soumerai J, Mato A, Carter J, Dogan A, Hochberg E, Barnes J, et al. Initial Results of a Multicenter, Investigatorinitiated Study of MRD Driven Time Limited Therapy With Zanubrutinib, Obinutuzumab and Venetoclax. J Clin Oncol (2020) 38(15). doi: 10.1200/JCO.2020.38.15_suppl.8006.
85. Lampson BL, Tyekucheva S, Crombie JL, Kim AI, Merryman RW, Lowney J, et al. Preliminary Safety and Efficacy Results From a Phase 2 Study of Acalabrutinib, Venetoclax and Obinutuzumab in Patients With Previously Untreated Chronic Lymphocytic Leukemia (Cll). Blood (2019) 134(1):32. doi: 10.1182/blood-2019-127506
86. Dimopoulos MA, Tedeschi A, Trotman J, Garcia-Sanz R, Macdonald D, Leblond V, et al. Phase 3 Trial of Ibrutinib Plus Rituximab in Waldenstrom’s Macroglobulinemia. N Engl J Med (2018) 378(25):2399–410. doi: 10.1056/NEJMoa1802917
87. Lipsky AH, Farooqui MZ, Tian X, Martyr S, Cullinane AM, Nghiem K, et al. Incidence and Risk Factors of Bleeding-Related Adverse Events in Patients With Chronic Lymphocytic Leukemia Treated With Ibrutinib. Haematologica (2015) 100(12):1571–8. doi: 10.3324/haematol.2015.126672
88. Liu J, Fitzgerald ME, Berndt MC, Jackson CW, Gartner TK. Bruton Tyrosine Kinase Is Essential for Botrocetin/VWF-Induced Signaling and GPIb-dependent Thrombus Formation In Vivo. Blood (2006) 108(8):2596–603. doi: 10.1182/blood-2006-01-011817
89. Atkinson BT, Ellmeier W, Watson SP. Tec Regulates Platelet Activation by GPVI in the Absence of Btk. Blood (2003) 102(10):3592–9. doi: 10.1182/blood-2003-04-1142
90. Kamel S, Horton L, Ysebaert L, Levade M, Burbury K, Tan S, et al. Ibrutinib Inhibits Collagen-Mediated But Not ADP-Mediated Platelet Aggregation. Leukemia (2015) 29(4):783–7. doi: 10.1038/leu.2014.247
91. Levade M, David E, Garcia C, Laurent PA, Cadot S, Michallet AS, et al. Ibrutinib Treatment Affects Collagen and Von Willebrand Factor-Dependent Platelet Functions. Blood (2014) 124(26):3991–5. doi: 10.1182/blood-2014-06-583294
92. Series J, Garcia C, Levade M, Viaud J, Sie P, Ysebaert L, et al. Differences and Similarities in the Effects of Ibrutinib and Acalabrutinib on Platelet Functions. Haematologica (2019) 104(11):2292–9. doi: 10.3324/haematol.2018.207183
93. Coutre SE, Byrd JC, Hillmen P, Barrientos JC, Barr PM, Devereux S, et al. Long-Term Safety of Single-Agent Ibrutinib in Patients With Chronic Lymphocytic Leukemia in 3 Pivotal Studies. Blood Adv (2019) 3(12):1799–807. doi: 10.1182/bloodadvances.2018028761
94. Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK With Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2013) 369(6):507–16. doi: 10.1056/NEJMoa1306220
95. Jones JA, Hillmen P, Coutre S, Tam C, Furman RR, Barr PM, et al. Use of Anticoagulants and Antiplatelet in Patients With Chronic Lymphocytic Leukaemia Treated With Single-Agent Ibrutinib. Br J Haematol (2017) 178(2):286–91. doi: 10.1111/bjh.14660
96. Dickerson T, Wiczer T, Waller A, Philippon J, Porter K, Haddad D, et al. Hypertension and Incident Cardiovascular Events Following Ibrutinib Initiation. Blood (2019) 134(22):1919–28. doi: 10.1182/blood.2019000840
97. Brown JR, Byrd JC, Ghia P, Sharman JP, Hillmen P, Stephens DM, et al. Pooled Analysis of Cardiovascular Events From Clinical Trialsevaluating Acalabrutinib Monotherapy in Patients With Chroniclymphocytic Leukemia (CLL). Blood (2020) 136(SUPPL 1):52–4. doi: 10.1182/blood-2020-134797
98. Rogers KA, Mousa L, Zhao Q, Bhat SA, Byrd JC, El Boghdadly Z, et al. Incidence of Opportunistic Infections During Ibrutinib Treatment for B-Cell Malignancies. Leukemia (2019) 33(10):2527–30. doi: 10.1038/s41375-019-0481-1
99. Ryan CE, Cheng MP, Issa NC, Brown JR, Davids MS. Pneumocystis Jirovecii Pneumonia and Institutional Prophylaxis Practices in CLL Patients Treated With BTK Inhibitors. Blood Adv (2020) 4(7):1458–63. doi: 10.1182/bloodadvances.2020001678
100. Ahn IE, Jerussi T, Farooqui M, Tian X, Wiestner A, Gea-Banacloche J. Atypical Pneumocystis Jirovecii Pneumonia in Previously Untreated Patients With CLL on Single-Agent Ibrutinib. Blood (2016) 128(15):1940–3. doi: 10.1182/blood-2016-06-722991
101. Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-Dependent Activation of a TLR9-BTK-Calcineurin-NFAT Pathway Co-Ordinates Innate Immunity to Aspergillus Fumigatus. EMBO Mol Med (2015) 7(3):240–58. doi: 10.15252/emmm.201404556
102. Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, et al. Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary Cns Lymphoma. Cancer Cell (2017) 31(6):833–43 e5. doi: 10.1016/j.ccell.2017.04.012
103. O’Brien S, Furman RR, Coutre S, Flinn IW, Burger JA, Blum K, et al. Single-Agent Ibrutinib in Treatment-Naive and Relapsed/Refractory Chronic Lymphocytic Leukemia: A 5-Year Experience. Blood (2018) 131(17):1910–9. doi: 10.1182/blood-2017-10-810044
104. Thompson PA, O’Brien SM, Wierda WG, Ferrajoli A, Stingo F, Smith SC, et al. Complex Karyotype is a Stronger Predictor Than Del(17p) for an Inferior Outcome in Relapsed or Refractory Chronic Lymphocytic Leukemia Patients Treated With Ibrutinib-Based Regimens. Cancer (2015) 121(20):3612–21. doi: 10.1002/cncr.29566
105. Soumerai JD, Ni A, Darif M, Londhe A, Xing G, Mun Y, et al. Prognostic Risk Score for Patients With Relapsed or Refractory Chronic Lymphocytic Leukaemia Treated With Targeted Therapies or Chemoimmunotherapy: A Retrospective, Pooled Cohort Study With External Validations. Lancet Haematol (2019) 6(7):e366–74. doi: 10.1016/S2352-3026(19)30085-7
106. Stephens DM, Boucher K, Kander E, Parikh SA, Parry EM, Shadman M, et al. Hodgkin Lymphoma Arising in Patients With Chronic Lymphocytic Leukemia: Outcomes From A Large Multi-Center Collaboration. Haematologica (2020). doi: 10.3324/haematol.2020.256388
107. Rossi D, Spina V, Cerri M, Rasi S, Deambrogi C, De Paoli L, et al. Stereotyped B-Cell Receptor is An Independent Risk Factor of Chronic Lymphocytic Leukemia Transformation to Richter Syndrome. Clin Cancer Res (2009) 15(13):4415–22. doi: 10.1158/1078-0432.CCR-08-3266
108. Fabbri G, Khiabanian H, Holmes AB, Wang J, Messina M, Mullighan CG, et al. Genetic Lesions Associated With Chronic Lymphocytic Leukemia Transformation to Richter Syndrome. J Exp Med (2013) 210(11):2273–88. doi: 10.1084/jem.20131448
109. Kadri S, Lee J, Fitzpatrick C, Galanina N, Sukhanova M, Venkataraman G, et al. Clonal Evolution Underlying Leukemia Progression and Richter Transformation in Patients With Ibrutinib-Relapsed CLL. Blood Adv (2017) 1(12):715–27. doi: 10.1182/bloodadvances.2016003632
110. Kohlhaas V, Blakemore SJ, Al-Maarri M, Nickel N, Pal M, Roth A, et al. Active Akt Signaling Triggers CLL Toward Richter Transformation Via Overactivation of Notch1. Blood (2021) 137(5):646–60. doi: 10.1182/blood.2020005734
111. Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in Patients With CLL and Richter Transformation or With Relapsed CLL. Blood (2017) 129(26):3419–27. doi: 10.1182/blood-2017-02-765685
112. Tsimberidou AM, Kantarjian HM, Cortes J, Thomas DA, Faderl S, Garcia-Manero G, et al. Fractionated Cyclophosphamide, Vincristine, Liposomal Daunorubicin, and Dexamethasone Plus Rituximab and Granulocyte-Macrophage-Colony Stimulating Factor (GM-CSF) Alternating With Methotrexate and Cytarabine Plus Rituximab and GM-CSF in Patients With Richter Syndrome or Fludarabine-Refractory Chronic Lymphocytic Leukemia. Cancer (2003) 97(7):1711–20. doi: 10.1002/cncr.11238
113. Rogers KA, Huang Y, Ruppert AS, Salem G, Stephens DM, Heerema NA, et al. A Single-Institution Retrospective Cohort Study of First-Line R-EPOCH Chemoimmunotherapy for Richter Syndrome Demonstrating Complex Chronic Lymphocytic Leukaemia Karyotype as An Adverse Prognostic Factor. Br J Haematol (2018) 180(2):259–66. doi: 10.1111/bjh.15035
114. Rozovski U, Benjamini O, Jain P, Thompson PA, Wierda WG, O’Brien S, et al. Outcomes of Patients With Chronic Lymphocytic Leukemia and Richter’s Transformation After Transplantation Failure. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33(14):1557–63. doi: 10.1200/JCO.2014.58.6750
115. Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With Cd19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol Off J Am Soc Clin Oncol (2017) 35(26):3010–20. doi: 10.1200/JCO.2017.72.8519
116. Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, et al. Ibrutinib Resistance in Chronic Lymphocytic Leukemia. N Engl J Med (2014) 370(24):2352–4. doi: 10.1056/NEJMc1402716
117. Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. Btk(C481s)-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J Clin Oncol Off J Am Soc Clin Oncol (2017) 35(13):1437–43. doi: 10.1200/JCO.2016.70.2282
118. Ahn IE, Underbayev C, Albitar A, Herman SE, Tian X, Maric I, et al. Clonal Evolution Leading to Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Blood (2017) 129(11):1469–79. doi: 10.1182/blood-2016-06-719294
119. Liu TM, Woyach JA, Zhong Y, Lozanski A, Lozanski G, Dong S, et al. Hypermorphic Mutation of Phospholipase C, Gamma2 Acquired in Ibrutinib-Resistant CLL Confers BTK Independency Upon B-Cell Receptor Activation. Blood (2015) 126(1):61–8. doi: 10.1182/blood-2015-02-626846
120. Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold Urticaria, Immunodeficiency, and Autoimmunity Related to PLCG2 Deletions. N Engl J Med (2012) 366(4):330–8. doi: 10.1056/NEJMoa1102140
121. Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A, et al. A Hypermorphic Missense Mutation in PLCG2, Encoding Phospholipase Cgamma2, Causes a Dominantly Inherited Autoinflammatory Disease With Immunodeficiency. Am J Hum Genet (2012) 91(4):713–20. doi: 10.1016/j.ajhg.2012.08.006
122. Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K, et al. Clonal Evolution in Patients With Chronic Lymphocytic Leukaemia Developing Resistance to BTK Inhibition. Nat Commun (2016) 7:11589. doi: 10.1038/ncomms11589
123. Jain P, Keating M, Wierda W, Estrov Z, Ferrajoli A, Jain N, et al. Outcomes of Patients With Chronic Lymphocytic Leukemia After Discontinuing Ibrutinib. Blood (2015) 125(13):2062–7. doi: 10.1182/blood-2014-09-603670
124. Jones JA, Mato AR, Wierda WG, Davids MS, Choi M, Cheson BD, et al. Venetoclax for Chronic Lymphocytic Leukaemia Progressing After Ibrutinib: An Interim Analysis of a Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol (2018) 19(1):65–75. doi: 10.1016/S1470-2045(17)30909-9
125. Roeker LE, Dreger P, Brown JR, Lahoud OB, Eyre TA, Brander DM, et al. Allogeneic Stem Cell Transplantation for Chronic Lymphocytic Leukemia in the Era of Novel Agents. Blood Adv (2020) 4(16):3977–89. doi: 10.1182/bloodadvances.2020001956
126. Kim HT, Shaughnessy CJ, Rai SC, Reynolds C, Ho VT, Cutler C, et al. Allogeneic Hematopoietic Cell Transplantation After Prior Targeted Therapy for High-Risk Chronic Lymphocytic Leukemia. Blood Adv (2020) 4(17):4113–23. doi: 10.1182/bloodadvances.2020002184
127. Robinson HR, Qi J, Cook EM, Nichols C, Dadashian EL, Underbayev C, et al. A CD19/CD3 Bispecific Antibody for Effective Immunotherapy of Chronic Lymphocytic Leukemia in the Ibrutinib Era. Blood (2018) 132(5):521–32. doi: 10.1182/blood-2018-02-830992
128. Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, et al. The BTK Inhibitor ARQ 531 Targets Ibrutinib-Resistant CLL and Richter Transformation. Cancer Discovery (2018) 8(10):1300–15. doi: 10.1158/2159-8290.CD-17-1409
129. Mato AR, Shah NN, Jurczak W, Cheah CY, Pagel JM, Woyach JA, et al. Pirtobrutinib in Relapsed or Refractory B-Cell Malignancies (BRUIN): A Phase 1/2 Study. Lancet (2021) 397(10277):892–901. doi: 10.1016/S0140-6736(21)00224-5
130. Woyach J, Stephens DM, Flinn IW, Bhat SA, Savage RE, Chai F, et al. Final Results of Phase 1, Dose Escalation Study Evaluating ARQ 531 in Patients With Relapsed or Refractory B-cell Lymphoid Malignancies. Blood (2019) 134. doi: 10.1182/blood-2019-127260
131. Bejar R, Zhang H, Rastgoo N, Benbatoul K, Jin Y, Thayer M, et al. Phase 1 a/B Dose Escalation Study of the Mutation agnosticBTK/FLT3 Inhibitor CG-806 in Patients With Relapsed or RefractoryCLL/SLL or Non-Hodgkin’s Lymphomas. Blood (2020) 136(SUPPL 1):35. doi: 10.1182/blood-2020-141495
132. Byrd JC, Smith S, Wagner-Johnston N, Sharman J, Chen AI, Advani R, et al. First-in-Human Phase 1 Study of the BTK Inhibitor GDC-0853 in Relapsed or Refractory B-Cell NHL and CLL. Oncotarget (2018) 9(16):13023–35. doi: 10.18632/oncotarget.24310
133. Reiff SD, Muhowski EM, Guinn D, Lehman A, Fabian CA, Cheney C, et al. Noncovalent Inhibition of C481S Bruton Tyrosine Kinase by GDC-0853: A New Treatment Strategy for Ibrutinib-Resistant CLL. Blood (2018) 132(10):1039–49. doi: 10.1182/blood-2017-10-809020
134. Crawford JJ, Johnson AR, Misner DL, Belmont LD, Castanedo G, Choy R, et al. Discovery of GDC-0853: A Potent, Selective, and Noncovalent Bruton’s Tyrosine Kinase Inhibitor in Early Clinical Development. J Med Chem (2018) 61(6):2227–45. doi: 10.1021/acs.jmedchem.7b01712
135. Allan JN, Patel K, Mato AR, Wierda WG, Ibarz JP, Choi MY, et al. Ongoing Results of a Phase 1b/2 Dose-Escalation and Cohort-Expansion Study of the Selective, Noncovalent, Reversible Bruton’s Tyrosine Kinase Inhibitor, Vecabrutinib, in B-Cell Malignancies. Blood (2019) 134. doi: 10.1097/01.HS9.0000562876.57990.65
136. Zhang H, Rastgoo N, Benbatoul K, Sheng S, Thayer M, Bejar R, et al. Early Clinical Findings From a Phase 1a/B Dose Escalation Trial to Evaluate the Safety and Tolerability of CG-806 in Patients With Relapsed or Refractory CLL/SLL or non-Hodgkin’s Lymphomas. Cancer Res (2020) 80(16 SUPPL). doi: 10.1158/1538-7445.AM2020-CT239
Keywords: Bruton’s tyrosine kinase, chronic lymphocytic leukemia, B-cell receptor signaling pathway, ibrutinib, acalabrutinib, zanubrutinib
Citation: Ahn IE and Brown JR (2021) Targeting Bruton’s Tyrosine Kinase in CLL. Front. Immunol. 12:687458. doi: 10.3389/fimmu.2021.687458
Received: 29 March 2021; Accepted: 18 May 2021;
Published: 23 June 2021.
Edited by:
Paolo Casali, University of Texas Health Science Center at San Antonio, United StatesReviewed by:
David A. Fruman, University of California, Irvine, United StatesCopyright © 2021 Ahn and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer R. Brown, amVubmlmZXJfYnJvd25AZGZjaS5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.