
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 28 May 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.687280
 Hirotaka Sakaguchi1
Hirotaka Sakaguchi1 Tatsuki Mizuochi1*
Tatsuki Mizuochi1* Masatoshi Haruta2Ryuta Takase1
Masatoshi Haruta2Ryuta Takase1 Shigeo Yoshida2
Shigeo Yoshida2 Yushiro Yamashita1
Yushiro Yamashita1 Ryuta Nishikomori1
Ryuta Nishikomori1Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a rare monogenic autosomal recessive disorder caused by mutation in the autoimmune regulator (AIRE) gene. Patients usually are diagnosed at ages between 5 and 15 years when they show 3 or more manifestations, most typically mucocutaneous candidiasis, Addison’s disease, and hypoparathyroidism. APECED-associated hepatitis (APAH) develops in only 10% to 40% of patients, with severity varying from subclinical chronic active hepatitis to potentially fatal acute liver failure (ALF). Ocular abnormalities are fairly common, most often keratopathy but sometimes retinopathy. Here we report a 2-year-old Japanese girl with an AIRE gene mutation who developed APAH with ALF, preceded by autoimmune retinopathy associated with anti-recoverin antibody before major symptoms suggested a diagnosis of APECED. Intravenous pulse methylprednisolone therapy followed by a corticosteroid combined with azathioprine treatment resolved ALF and achieved control of APAH. To our knowledge, our patient is the youngest reported to have ALF resulting from an AIRE gene mutation. Pulse methylprednisolone induction therapy followed by treatment with corticosteroid plus azathioprine may well be effective in other children with APAH and AIRE gene mutations.
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED; OMIM, 240300), also termed autoimmune polyendocrine syndrome type 1, is a rare monogenic autosomal recessive disease caused by mutations in the autoimmune regulator (AIRE) gene (1). APECED has been found to occur in approximately 1 in 90 000 to 200 000 individuals in most populations studied, mainly in European countries. The disease occurs more frequently in relatively isolated populations, being more prevalent among Iranian Jews (1:9000), Sardinians (1:14 000), Finns (1:25 000), and Slovenians (1:43 000). Norway (1:80 000), Poland (1:129 000), and France (1:500 000) show more typical frequencies of occurrence. In East Asia APECED is particularly infrequent; a rough estimate in Japan is 1:10 000 000 (2, 3).
AIRE participates in regulation of self-tolerance as T cells develop in the thymus. Defective function of AIRE encourages production of multiple anti-cytokine and organ-specific autoantibodies, leading to severe autoimmune disease that can damage multiple endocrine organs and other tissues. More than 20 different clinical manifestations have been associated with the disease; such high variability makes the course of disease unpredictable (4). Illness usually manifests during childhood, with additional clinical manifestations emerging later in life. APECED generally is diagnosed between the ages of 5 and 15 years (2). As a typical minimum, APECED patients manifest a triad of mucocutaneous candidiasis, Addison’s disease, and hypoparathyroidism. Patients may also exhibit autoimmune thyroiditis, Sjögren’s syndrome, premature ovarian/gonadal failure, anemia, diabetes mellitus, alopecia, vitiligo, gastritis, nail dystrophy, enamel hypoplasia, and hepatitis resembling autoimmune hepatitis (AIH), referred to as APECED-associated hepatitis (APAH) (5–7). APAH develops in 10% to 40% of patients with APECED, showing variable severity ranging from subclinical chronic active hepatitis to potentially fatal acute liver failure (ALF) (5–9). Ocular abnormalities are not infrequent; while keratopathy is most frequent, retinopathy may occur (1, 10–12). APECED is clinically defined based on the presence of at least 2 components of the classical triad; however, these 3 major criteria need not be met, especially when genetic testing demonstrates the mutation or minor criteria are met.
Here we report a 2-year-old Japanese girl with an AIRE gene mutation who developed ALF caused by APAH, as well as autoimmune retinopathy associated with anti-recoverin antibody that emerged before onset of major symptoms led to a diagnosis of APECED.
A previously healthy 2-year-old Japanese girl, the first child of healthy, non-consanguineous parents, presented with an episode of jaundice and vomiting. Growth and development were within the normal range, and no dysmorphic features were present. Hepatomegaly and jaundice were present. Neurologic findings were normal for age. Initial laboratory abnormalities included serum aspartate aminotransferase, 989 U/L (normal, <30); alanine aminotransferase, 1323 U/L (<30); gamma glutamyl transferase, 136 U/L (<40); total/direct bilirubin, 6.5/4.0 mg/dL (<1.2/<0.4); and immunoglobulin G, 2098 mg/dL (500 to 1280). Prothrombin time (INR) was 1.73 (0.75 to 1.15). Initial autoantibody screening, including antinuclear antibody (ANA), anti-smooth muscle antibody (SMA), and anti-liver kidney microsomal-1 antibody (LKM-1), was negative. Other causes of liver disease such as acute viral hepatitis and metabolic conditions were excluded by appropriate investigations. Abdominal magnetic resonance imaging (MRI) and ultrasonography showed a visible gallbladder and hepatomegaly; no choledochal cyst, bile duct dilation, tumor, or ascites was demonstrated. The patient was treated for ALF without hepatic encephalopathy, most likely caused by AIH. Intravenous pulse steroid therapy with methylprednisolone (pulse mPSL; 30 mg/kg/day for 3 days) was begun 2 days after admission. After pulse mPSL, we treated the patient with prednisolone (PSL, 1.5 mg/kg/day) and then added azathioprine (AZA; 1.2 to 2.0 mg/kg/day). As this combination therapy improved liver function, the PSL dose was tapered to 0.15 mg/kg/day (Table 1). After pulse mPSL had alleviated ALF, liver biopsy was performed. The specimen showed typical AIH findings including active interface hepatitis and predominance of plasma cells over lymphocytic inflammation. The patient was diagnosed with ALF as the cause of AIH; her score according to the system of the International Autoimmune Hepatitis Group was 20 points (13).
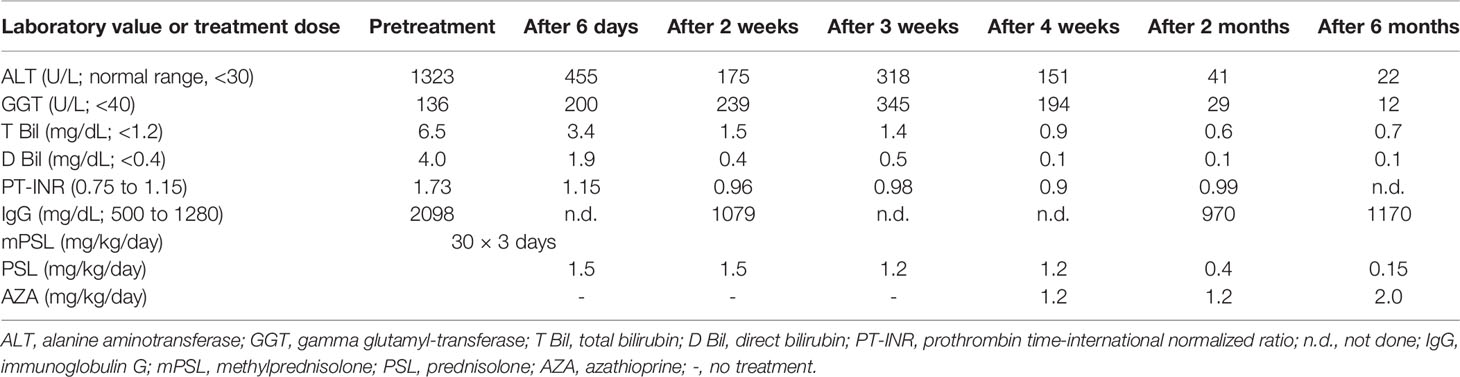
Table 1 Clinical course of laboratory results and treatment.
The patient was referred to an ophthalmologist for evaluation of possible adverse effects of long-term systemic corticosteroid treatment. Best-corrected visual acuity was no light perception in the right eye and 5/20 in the left eye, with eccentric fixation and exotropia in the right eye. Anterior slit-lamp examination had no remarkable findings. A dilated fundus examination showed vascular attenuation, optic disc pallor, and retinal pigmentary changes in both eyes (Figures 1A, B). Macular optical coherence tomography (OCT) demonstrated marked disruption of the external limiting membrane with an ellipsoid zone in both eyes, and small foveal cysts in the left eye (Figures 1C, D). Scotopic and photopic electroretinograms showed no recordable responses in either eye (Figures 1E, F). Ultra-widefield fundus autofluorescence examination showed a subtle ring of excessive autofluorescence in the parafoveal regions in both eyes, and ultra-widefield OCT demonstrated diffuse thinning of both retinas with loss of outer retinal structures, as reported in a previous diagnostic image presentation (12). Serologic tests were positive for anti-recoverin antibody and negative for anti-alpha enolase antibody. Based on these ocular and serologic findings, the patient was additionally diagnosed with bilateral autoimmune retinopathy. Systemic evaluation including MRI, ultrasonography, and serum tumor marker determinations such as alpha-fetoprotein, carbohydrate antigen 19-9, carcinoembryonic antigen, and squamous cell carcinoma-related antigen disclosed no underlying malignancy at the age of 3 years.
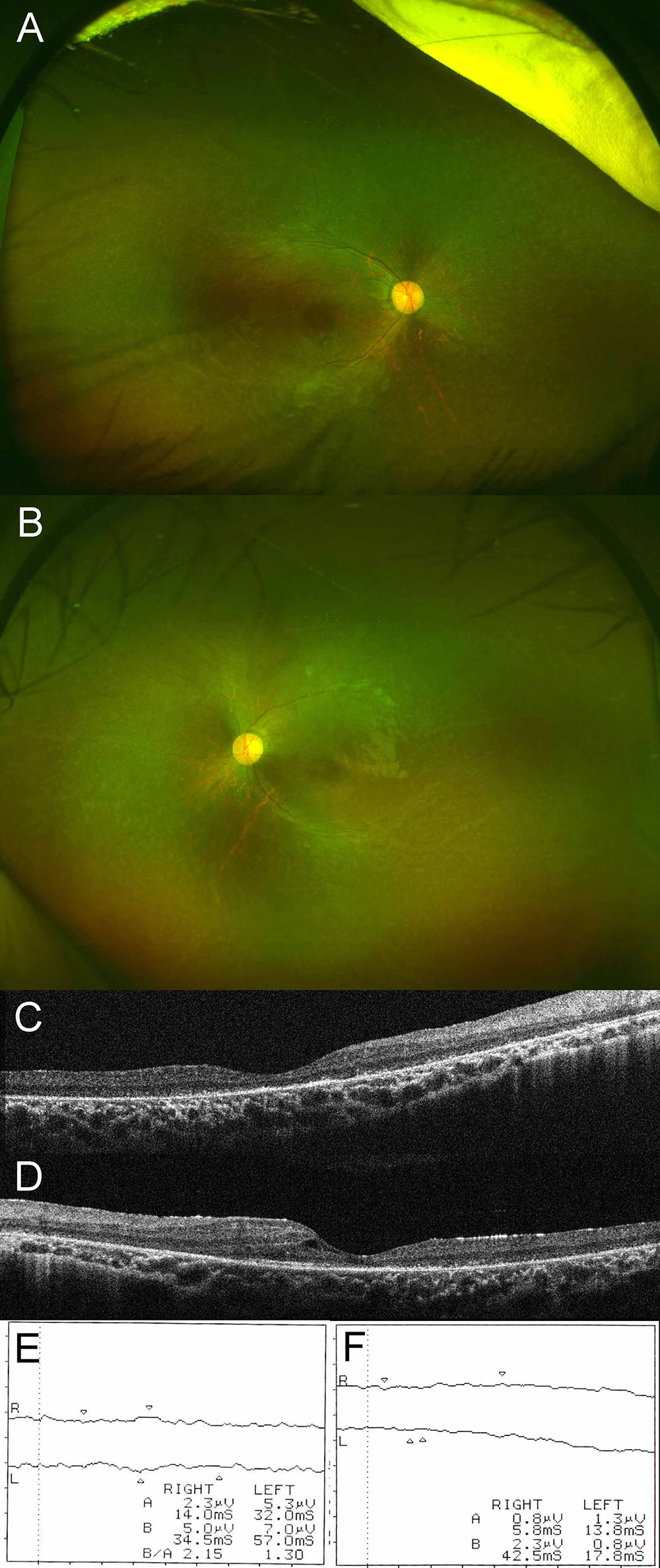
Figure 1 Ultra-widefield fundus photograph of right (A) and left (B) eyes. Dilated fundus examination disclosed vascular attenuation, optic disc pallor, and retinal pigmentary changes in both eyes. Macular optical coherence tomography of right (C) and left (D) eyes. Macular optical coherence tomography demonstrated marked disruption of the external limiting membrane and an ellipsoid zone in both eyes, with small foveal cysts in the left eye. Results of scotopic (E) and photopic (F) electroretinograms. Scotopic and photopic electroretinograms showed no recordable responses in either eye.
Since the patient had both AIH and autoimmune retinopathy, we suspected an inborn error of immunity. We therefore performed a 400-gene panel analysis for such inborn errors in the patient using next-generation sequencing, which identified a homozygous disease-causing variant in the AIRE gene (c.415C>T, p.139*). Each parent was found to be heterozygous for the variant. Additionally, the patient’s 1-year-old sister, who had been treated at another hospital for encephalopathy of unknown cause, was genetically tested and found to be homozygous for the same variant.
At this writing the patient was 3 years and 11 months old, and had no manifestations of APECED other than APAH and autoimmune retinopathy. Additionally, test results for serum autoantibodies typical of AIH such as ANA, SMA, and LKM-1 all were negative at that time.
Here we report a Japanese girl with AIRE gene mutation who presented at 2 years of age with ALF caused by APAH. The patient also was found to have an autoimmune retinopathy associated with anti-recoverin antibody. Pulse mPSL therapy followed by corticosteroid plus AZA treatment brought about recovery from ALF and control of APAH, respectively. The patient did not present with any of the conditions representing the classical clinical triad of APECED (mucocutaneous candidiasis, Addison’s disease, and hypoparathyroidism). In rare instances, other manifestations such as APAH and autoimmune retinopathy may precede the classical clinical symptoms as the presenting features of APECED.
APAH, which develops in 10% to 40% of patients with APECED, has been reported to cause ALF in some APECED cases (5–9). In a series of 68 patients with APECED from Finland, 12 had APAH including 2 who died of ALF at 7 and 17 years of age (5), while among 41 patients from Italy, 8 had APAH including 1 who died of ALF at 11 years of age (6). To our knowledge, our APECED patient is the youngest, at 2 years, to develop ALF caused by APAH. In Table 2, we summarize clinical, genetic, and treatment aspects of APAH patients with AIRE gene mutation in a reported case series (7) and in our patient. Compared with more common forms of AIH, APAH is characterized by an earlier onset of hepatitis and a lower positive rate for AIH-associated serum autoantibodies such as ANA, SMA, and LKM-1, as well as a more favorable response to immunomodulatory treatment. Available published information seems incomplete and inconclusive regarding treatment and outcome of patients with APAH. However, Zachou et al. (14) reported that high-dose intravenous corticosteroid was safe and effective for treatment of patients with acute severe AIH, while Sogo et al. (15) advocated pulse mPSL as a safe and effective therapy for children with AIH. Conventional treatment of childhood AIH consists of prednisolone at 2 mg/kg/day, to be gradually tapered over 4 to 8 weeks guided by declines in serum transaminases until a maintenance dose of 2.5 to 5 mg/day is reached (16). In general, AZA is added as a steroid-sparing agent when transaminase concentrations no longer respond to prednisolone alone or, rarely, when prednisolone produces serious adverse effects. AZA or 6-mercaptopurine with or without corticosteroids has been used in most APAH patients. Among the APAH patients in one report, 89% responded to the initial immunomodulatory regimen used to control hepatitis (7). In our present case, pulse mPSL was effective against ALF, and a combination of PSL and AZA provided effective induction and maintenance treatment in APAH. We would encourage a similar approach in APAH patients with ALF and an AIRE gene mutation.
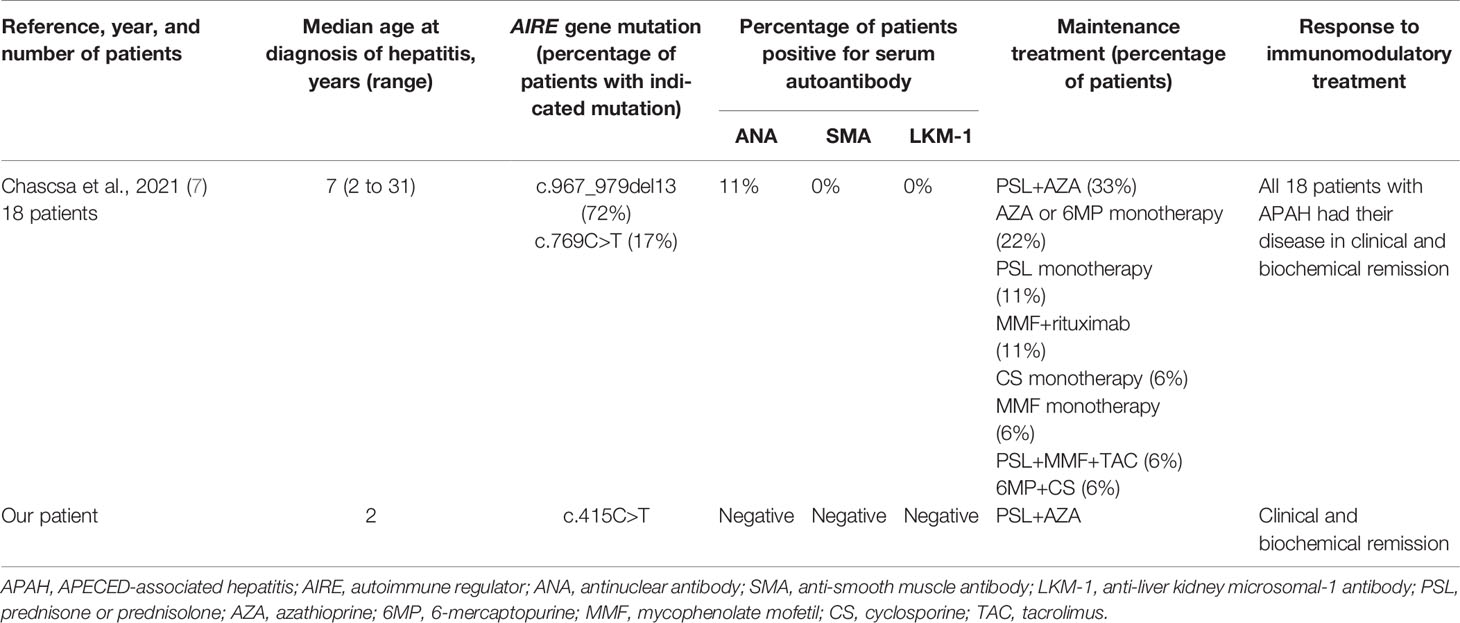
Table 2 Clinical, genetic, and treatment aspects of APAH patients with AIRE gene mutation in a reported series and in our patient.
Lankisch et al. (17) reported that among 25 children with AIH, AIRE gene analysis detected a heterozygous mutation in 4 patients, so even a heterozygous AIRE mutation might predispose to childhood AIH. We believe that the possibility of AIRE gene mutations should be investigated in children with early- onset AIH (e.g. under 6 years of age); if a pathogenic mutation is identified, one should treat the patient with a combination of corticosteroid and AZA-preceded by pulse mPSL if ALF is present.
Ocular complications previously reported in patients with APECED include autoimmune keratopathy and retinopathy (1, 10–12). Autoantibodies directed against lacrimal gland, cornea, or retina are believed to be responsible for the ocular manifestations of APECED. Bourgault et al. (11) reported 4 patients diagnosed with APECED who were found to have bilateral retinopathy and antiretinal antibodies. In addition to APAH, our 2-year-old patient with genetically confirmed APECED developed bilateral autoimmune retinopathy. She also appears to be among the youngest patients described as positive for anti-recoverin antibody (11). Although anti-recoverin antibody has been considered highly correlated with cancer (18, 19), systemic evaluation has disclosed no underlying malignancy in the present patient. Autoimmune retinopathy should be considered in the differential diagnosis of unusual retinal degeneration in patients with AIRE mutations, and prompt systemic work-up is needed to rule out occult malignancy in those positive for anti-recoverin antibody. APECED-associated retinal complications recently have drawn attention as a relatively common complication of this disorder; however, no effective treatment for APECED-associated retinopathy has been identified (10). Couturier et al. concluded that no treatment has shown efficacy against APECED-associated retinopathy (10). Bourgault et al. reported 3 patients who received different immunosuppressants during various stages of APECED-associated retinopathy; in all 3, retinopathy, vision, and/or electroretinographic findings continued to worsen despite immunosuppressive treatment (11).
Prevalence data for disease manifestations among patients with APECED in various countries and regions including Finland, Italy, Norway, Russia, North and South America, and Japan are summarized in Table 3 (5, 6, 9, 20–22). Among Japanese patients with APECED, Addison’s disease, hypoparathyroidism, autoimmune thyroiditis, nail dystrophy, and enamel hypoplasia are less prevalent than in patients from other countries, while type 1 diabetes is more prevalent.
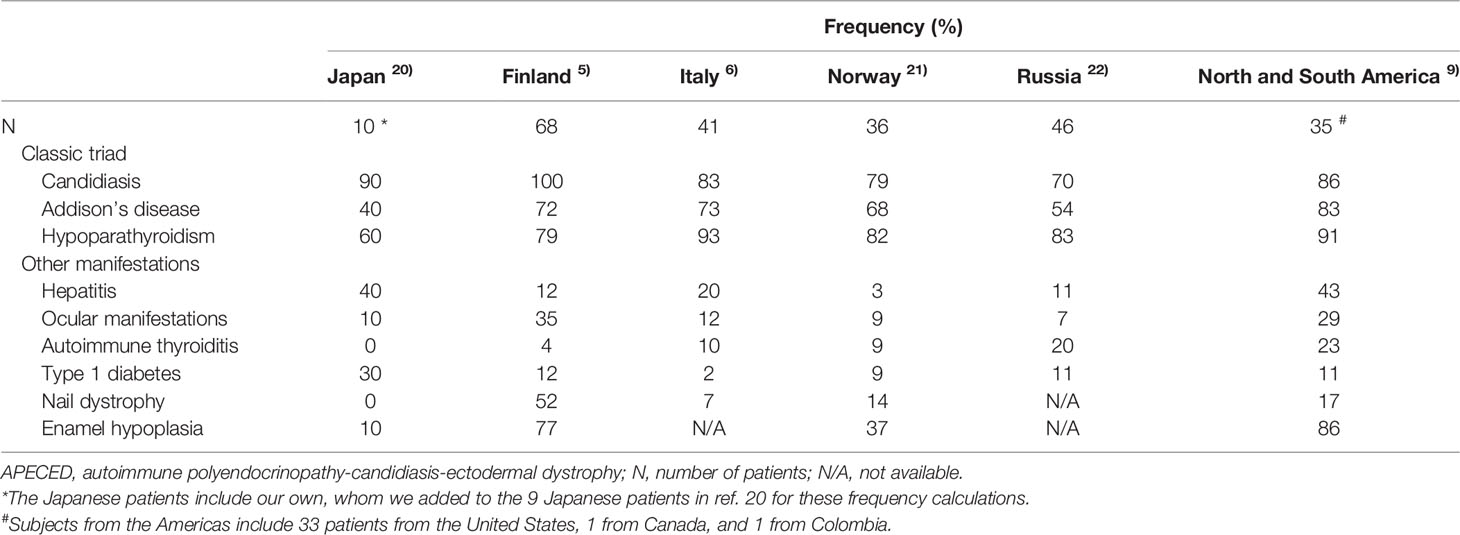
Table 3 Prevalence of disease manifestations in patients with APECED from various countries and regions.
In conclusion, we believe that our patient is the youngest reported to date to have ALF caused by an AIRE mutation. Pulse mPSL induction therapy followed by corticosteroid plus AZA was effective against ALF and APAH.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethical Committee of Kurume University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
HS and TM conceptualized and designed the study, collected data, drafted the initial manuscript, and reviewed and revised the manuscript. MH collected data, drafted the initial manuscript, and reviewed and revised the manuscript. RT performed genetic analysis and reviewed and revised the manuscript. SY collected data and reviewed and revised the manuscript. YY collected data and reviewed and revised the manuscript. RN performed genetic analysis, drafted the initial manuscript, and reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the patient and her parents for their cooperation. We also thank Drs. Miyuki Matsushita, Takaoki Yokochi, and Go Kawano at St. Mary’s Hospital, Osamu Ohara at Kazusa DNA Research Institute, and Ryosuke Yasuda, Kikumi Ushijima, Yoriko Watanabe, Saki Sakamoto, Hironori Kusano, and Takuya Tsuji at Kurume University for their collaboration.
AIH, autoimmune hepatitis; AIRE, autoimmune regulator; ALF, acute liver failure; ANA, antinuclear antibody; APAH, APECED-associated hepatitis; APECED, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; AZA, azathioprine; LKM-1, anti-liver kidney microsomal-1 antibody; mPSL, methylprednisolone; MRI, magnetic resonance imaging; OCT, optical coherence tomography; PSL, prednisolone; SMA, anti-smooth muscle antibody.
1. Husebye ES, Anderson MS, Kämpe O. Autoimmune Polyendocrine Syndromes. New Engl J Med (2018) 378(12):1132–41. doi: 10.1056/NEJMra1713301
2. Guo CJ, Leung PSC, Zhang W, Ma X, Gershwin ME. The Immunobiology and Clinical Features of Type 1 Autoimmune Polyglandular Syndrome (Aps-1). Autoimmun Rev (2018) 17(1):78–85. doi: 10.1016/j.autrev.2017.11.012
3. Sato K, Nakajima K, Imamura H, Deguchi T, Horinouchi S, Yamazaki K, et al. A Novel Missense Mutation of AIRE Gene in a Patient With Autoimmune Polyendocrinopathy, Candidiasis and Ectodermal Dystrophy (Apeced), Accompanied With Progressive Muscular Atrophy: Case Report and Review of the Literature in Japan. Endocrine J (2002) 49(6):625–33. doi: 10.1507/endocrj.49.625
4. Borchers J, Pukkala E, Mäkitie O, Laakso S. Patients With Apeced Have Increased Early Mortality Due to Endocrine Causes, Malignancies and Infections. J Clin Endocrinol Metab (2020) 105(6):e2207–13. doi: 10.1210/clinem/dgaa140
5. Ahonen P, Myllärniemi S, Sipilä I, Perheentupa J. Clinical Variation of Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) in a Series of 68 Patients. New Engl J Med (1990) 322(26):1829–36. doi: 10.1056/NEJM199006283222601
6. Betterle C, Greggio NA, Volpato M. Clinical Review 93: Autoimmune Polyglandular Syndrome Type 1. J Clin Endocrinol Metab (1998) 83(4):1049–55. doi: 10.1210/jcem.83.4.4682
7. Chascsa DM, Ferré EMN, Hadjiyannis Y, Alao H, Natarajan M, Quinones M, et al. Apeced-Associated Hepatitis: Clinical, Biochemical, Histological and Treatment Data From a Large, Predominantly American Cohort. Hepatol (Baltimore Md) (2021) 73(3):1088–104. doi: 10.1002/hep.31421
8. Smith D, Stringer MD, Wyatt J, O’Meara M, Davison S, Cheetham TD, et al. Orthotopic Liver Transplantation for Acute Liver Failure Secondary to Autoimmune Hepatitis in a Child With Autoimmune Polyglandular Syndrome Type 1. Pediatr Transplant (2002) 6(2):166–70. doi: 10.1034/j.1399-3046.2002.01063.x
9. Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined Clinical Features and Diagnostic Criteria in Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. JCI Insight (2016) 1(13):e88782. doi: 10.1172/jci.insight.88782
10. Couturier A, Brézin AP. Ocular Manifestations of Autoimmune Polyendocrinopathy Syndrome Type 1. Curr Opin Ophthalmol (2016) 27(6):505–13. doi: 10.1097/ICU.0000000000000306
11. Bourgault S, Baril C, Vincent A, Héon E, Ali A, MacDonald I, et al. Retinal Degeneration in Autoimmune Polyglandular Syndrome Type 1: A Case Series. Br J Ophthalmol (2015) 99(11):1536–42. doi: 10.1136/bjophthalmol-2014-305897
12. Haruta M, Tsuji T, Yoshida S. Ultra-Widefield OCT in Retinopathy of Autoimmune Polyendocrine Syndrome Type 1. Ophthalmol Retina (2021) 5(1):9. doi: 10.1016/j.oret.2020.06.007
13. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International Autoimmune Hepatitis Group Report: Review of Criteria for Diagnosis of Autoimmune Hepatitis. J Hepatol (1999) 31(5):929–38. doi: 10.1016/S0168-8278(99)80297-9
14. Zachou K, Arvaniti P, Azariadis K, Lygoura V, Gatselis NK, Lyberopoulou A, et al. Prompt Initiation of High-Dose I.V. Corticosteroids Seems to Prevent Progression to Liver Failure in Patients With Original Acute Severe Autoimmune Hepatitis. Hepatol Res Off J Japan Soc Hepatol (2019) 49(1):96–104. doi: 10.1111/hepr.13252
15. Sogo T, Fujisawa T, Inui A, Komatsu H, Etani Y, Tajiri H, et al. Intravenous Methylprednisolone Pulse Therapy for Children With Autoimmune Hepatitis. Hepatol Res Off J Japan Soc Hepatol (2006) 34(3):187–92. doi: 10.1016/j.hepres.2005.12.002
16. Mieli-Vergani G, Vergani D. Paediatric Autoimmune Liver Disease. Arch Dis childhood (2013) 98(12):1012–7. doi: 10.1136/archdischild-2013-303848
17. Lankisch TO, Mourier O, Sokal EM, Habes D, Lacaille F, Bridoux-Henno L, et al. Aire Gene Analysis in Children With Autoimmune Hepatitis Type I or II. J Pediatr Gastroenterol Nutr (2009) 48(4):498–500. doi: 10.1097/MPG.0b013e31818550de
18. Keltner JL, Thirkill CE. Cancer-Associated Retinopathy vs Recoverin-Associated Retinopathy. Am J Ophthalmol (1998) 126(2):296–302.
19. Adamus G. Antirecoverin Antibodies and Autoimmune Retinopathy. Arch Ophthalmol (Chicago Ill 1960) (2000) 118(11):1577–8. doi: 10.1001/archopht.118.11.1577
20. Yan Z, Gang X, Xie X, Gao Y, Li Z, Wang G. A Case Report and Literature Review: Identification of a Novel Aire Gene Mutation Associated With Autoimmune Polyendocrine Syndrome Type 1 in East Asians. Medicine (2020) 99(18):e20000. doi: 10.1097/MD.0000000000020000
21. Wolff AS, Erichsen MM, Meager A, Magitta NF, Myhre AG, Bollerslev J, et al. Autoimmune Polyendocrine Syndrome Type 1 in Norway: Phenotypic Variation, Autoantibodies, and Novel Mutations in the Autoimmune Regulator Gene. J Clin Endocrinol Metab (2007) 92(2):595–603. doi: 10.1210/jc.2006-1873
Keywords: AIRE, acute liver failure, autoimmune hepatitis, children, corticosteroid
Citation: Sakaguchi H, Mizuochi T, Haruta M, Takase R, Yoshida S, Yamashita Y and Nishikomori R (2021) AIRE Gene Mutation Presenting at Age 2 Years With Autoimmune Retinopathy and Steroid-Responsive Acute Liver Failure: A Case Report and Literature Review. Front. Immunol. 12:687280. doi: 10.3389/fimmu.2021.687280
Received: 29 March 2021; Accepted: 13 May 2021;
Published: 28 May 2021.
Edited by:
Satoshi Okada, Hiroshima University, JapanReviewed by:
Hidenori Ohnishi, Gifu University, JapanCopyright © 2021 Sakaguchi, Mizuochi, Haruta, Takase, Yoshida, Yamashita and Nishikomori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatsuki Mizuochi, bWl6dW9jaGlfdGF0c3VraUBrdXJ1bWUtdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.