 Matthew Bell1,2
Matthew Bell1,2 Stephen Gottschalk1*
Stephen Gottschalk1*- 1Department of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 2Graduate School of Biomedical Sciences, St. Jude Children’s Research Hospital, Memphis, TN, United States
Adoptive immunotherapy with T cells genetically modified to express chimeric antigen receptors (CARs) is a promising approach to improve outcomes for cancer patients. While CAR T cell therapy is effective for hematological malignancies, there is a need to improve the efficacy of this therapeutic approach for patients with solid tumors and brain tumors. At present, several approaches are being pursued to improve the antitumor activity of CAR T cells including i) targeting multiple antigens, ii) improving T cell expansion/persistence, iii) enhancing homing to tumor sites, and iv) rendering CAR T cells resistant to the immunosuppressive tumor microenvironment (TME). Augmenting signal 3 of T cell activation by transgenic expression of cytokines or engineered cytokine receptors has emerged as a promising strategy since it not only improves CAR T cell expansion/persistence but also their ability to function in the immunosuppressive TME. In this review, we will provide an overview of cytokine biology and highlight genetic approaches that are actively being pursued to augment cytokine signaling in CAR T cells.
Introduction
Despite recent advances in cancer treatment, patients with relapsed or refractory disease continue to have poor outcomes and novel approaches are needed. T cells that are genetically modified to express a chimeric antigen receptor (CAR) can kill chemotherapy-resistant tumor cells and therefore have the potential to improve outcomes and reduce treatment-related toxicity from conventional therapies (1, 2). CARs consist of four components: i) an extracellular antigen recognition domain, most commonly a single chain variable fragment (scFv), ii) structural components, such as hinge and transmembrane domains, iii) a costimulatory domain that provides signals to sustain CAR T cell effector functions, and iv) a CD3ζ activation domain (1–3) (Figure 1).
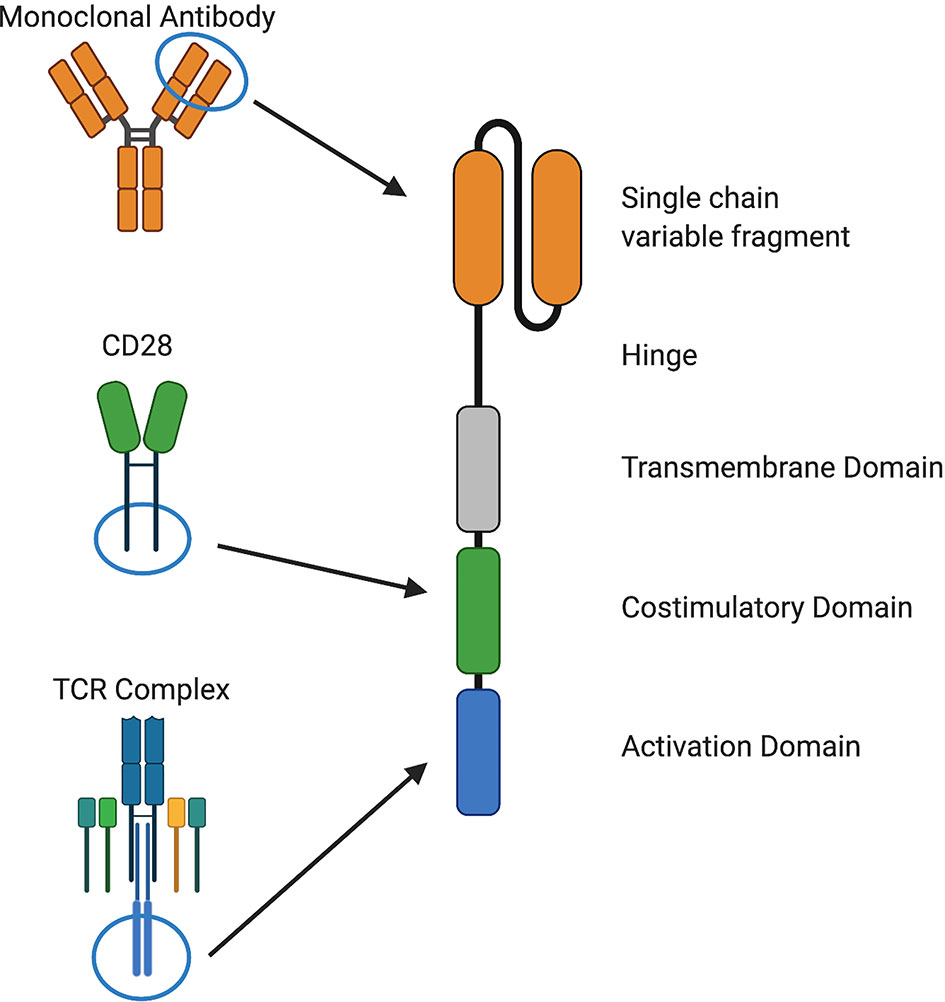
Figure 1 Components of chimeric antigen receptors. CARs recognize a cell surface antigen via a single chain variable fragment (scFv) from a monoclonal antibody or a ligand and signal through costimulatory domains derived from and CD28, 4-1BB, or other molecules and an activation domain derived from CD3ζ. Structural components, such as hinge and transmembrane domains derived from CD28, CD8α, or other molecules, are also important for CAR function.
CAR T cells targeting CD19 have shown significant overall response rates against CD19-positive leukemia and lymphoma (4–6), leading to their FDA approval in 2017. In addition, CAR T cells targeting BCMA, CD30, CD22, or CD20 expressed on hematological malignancies have also shown significant activity in clinical studies (7–10). However, a subset of patients does not achieve remission or relapses with antigen-positive disease due to suboptimal expansion or persistence of CAR T cells (11). CAR T cells for the treatment of solid tumors are also actively being explored, but they have shown less impressive clinical results (12–16), most likely due to a multitude of factors that limit CAR T cell activity. Previous preclinical studies have demonstrated that improvements in CAR design, such as optimizing scFvs, modification of structural components, or modulation of CAR signaling can improve the antitumor activity of CAR T cells (17–21). Likewise, additional genetic modification may be required to endow CAR T cells with potent and sustained effector function and to overcome the immunosuppressive tumor microenvironment (TME) to produce lasting benefits for cancer patients (22).
Physiological T cell activation requires three distinct signals for acquisition of effector function and formation of immunological memory. Signal 1 (activation) occurs via CD3ζ signal transduction following T cell receptor (TCR)-mediated antigen recognition. Signal 2 (costimulation) provides additional signals from CD28 or other molecules to augment signal 1. Finally, signal 3, mediated by cytokines, is required for optimal T cell proliferation, differentiation of naïve T cells into effector cells, and development of functional T cell memory (23–25) (Figure 2).
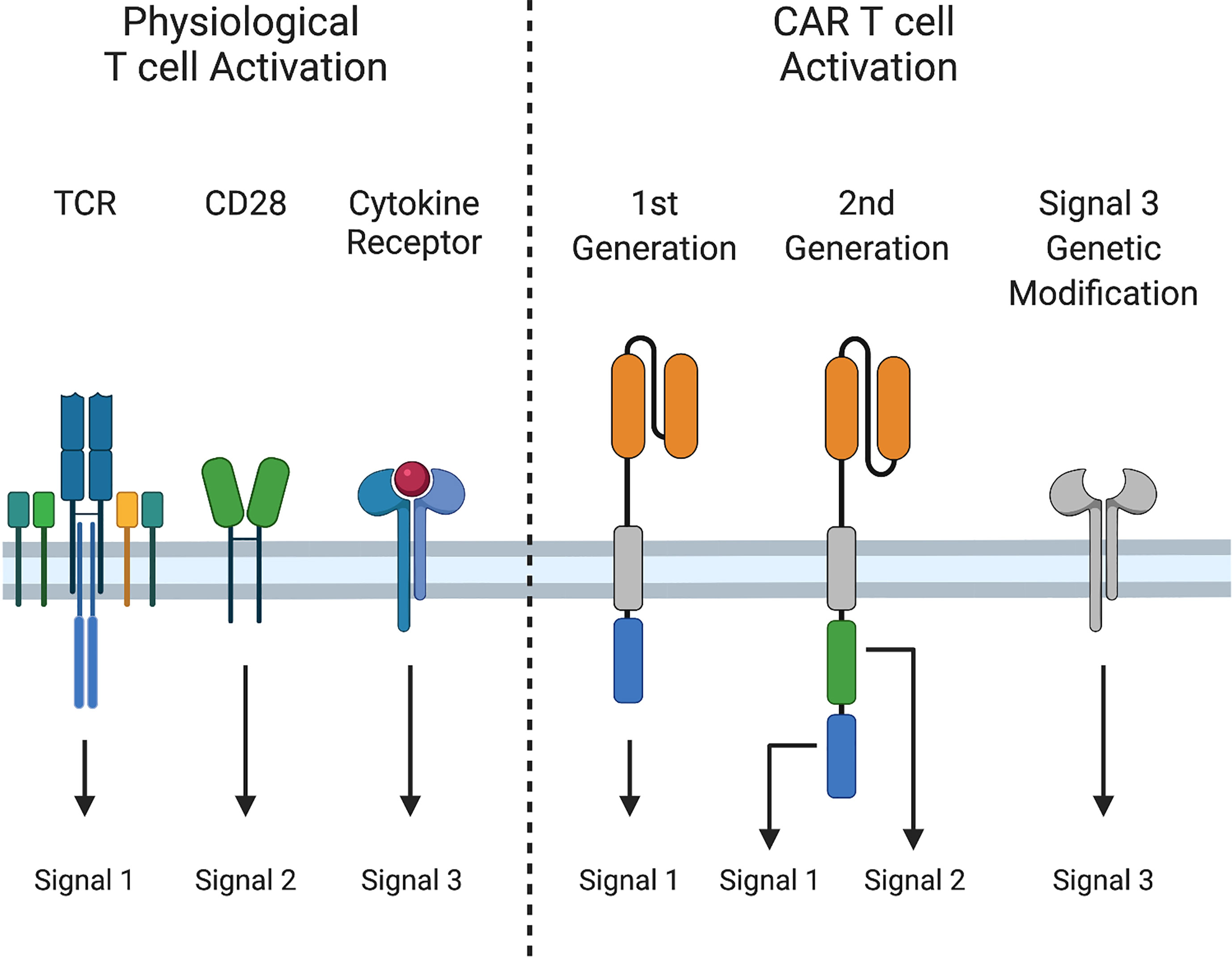
Figure 2 Signals 1, 2, and 3 in T cell activation. Physiological T cell activation and sustained effector function require i) Signal 1: peptide-MHC recognition and signaling through the T cell receptor (TCR), ii) Signal 2: costimulation through CD28, and iii) Signal 3: cytokine stimulation. First generation CARs provide only Signal 1, while second generation CARs provide Signals 1 and 2, and induce cytokine production (Signal 3). Signal 3 can be augmented by additional genetic modifications.
First generation CARs provide only signal 1, via CD3ζ. Second-generation CARs also provide signal 2, most often through CD28 or 4-1BB co-stimulation to sustain CAR T cell expansion following activation. Although activated CAR T cells produce cytokines, such as interleukin-2 (IL-2), production decreases after repeated exposure to tumor cells (26), and some cytokines that are important for T cell effector function, such as IL-12 and IL-15, are either produced at low levels are not at all by T cells (27, 28). Due to these limitations, investigators have engineered CAR T cells to augment signal 3. While incorporating cytokine receptor chains or JAK/STAT binding domains into CARs improves CAR T cell effector function (29, 30), most approaches to provide or modulate cytokine signaling have relied on transgenic expression of cytokines or cytokine receptors. In this review, we will provide a succinct overview of cytokine biology and highlight strategies to improve signal 3 in CAR T cells, including constitutive and inducible expression of cytokines and expression of native and engineered cytokine receptors.
Common Gamma Chain Cytokines and Their Receptors
Cytokine Biology
The common gamma chain family of cytokines – IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 – play critical roles in T cell differentiation, proliferation, and homeostasis. The receptors for these cytokines include the common gamma chain (γc) and a private receptor chain (IL-4Rα, IL-7Rα, IL-9Rα, IL-21R) except for IL-2 and IL-15 that share γc and IL-2Rβ. Additionally, the IL-2/15 receptor can associate with IL-2Rα (CD25) or IL-15Rα to form high affinity IL-2 or IL-15 receptors. Binding of cognate cytokines induces heterodimerization of γc with the private receptor chain to position inactive Janus kinase (JAK)1 and JAK3 in proximity where they trans-phosphorylate each other to become active. Activated JAK1 and JAK3 subsequently phosphorylate the receptor to provide phosphotyrosine binding sites for SH2-domain containing proteins. Some of the primary signaling molecules activated downstream of these cytokine receptors are members of the Signal Transducer and Activator of Transcription (STAT) family (Figure 3A). In the case of the IL-2 receptor, phosphorylation of STAT5A and STAT5B by JAK1/3 induces homo- or hetero-dimerization and immediate nuclear translocation to induce expression of several cell cycle and anti-apoptosis genes, including Bcl-2, Bcl-x, Pim-1, c-myc, and cyclin D2 (31, 32). Additionally, mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways activated downstream of the IL-2R play roles in proliferation and metabolic regulation that may potentiate STAT5 signaling (33), but each receptor varies in its ability to activate these pathways.
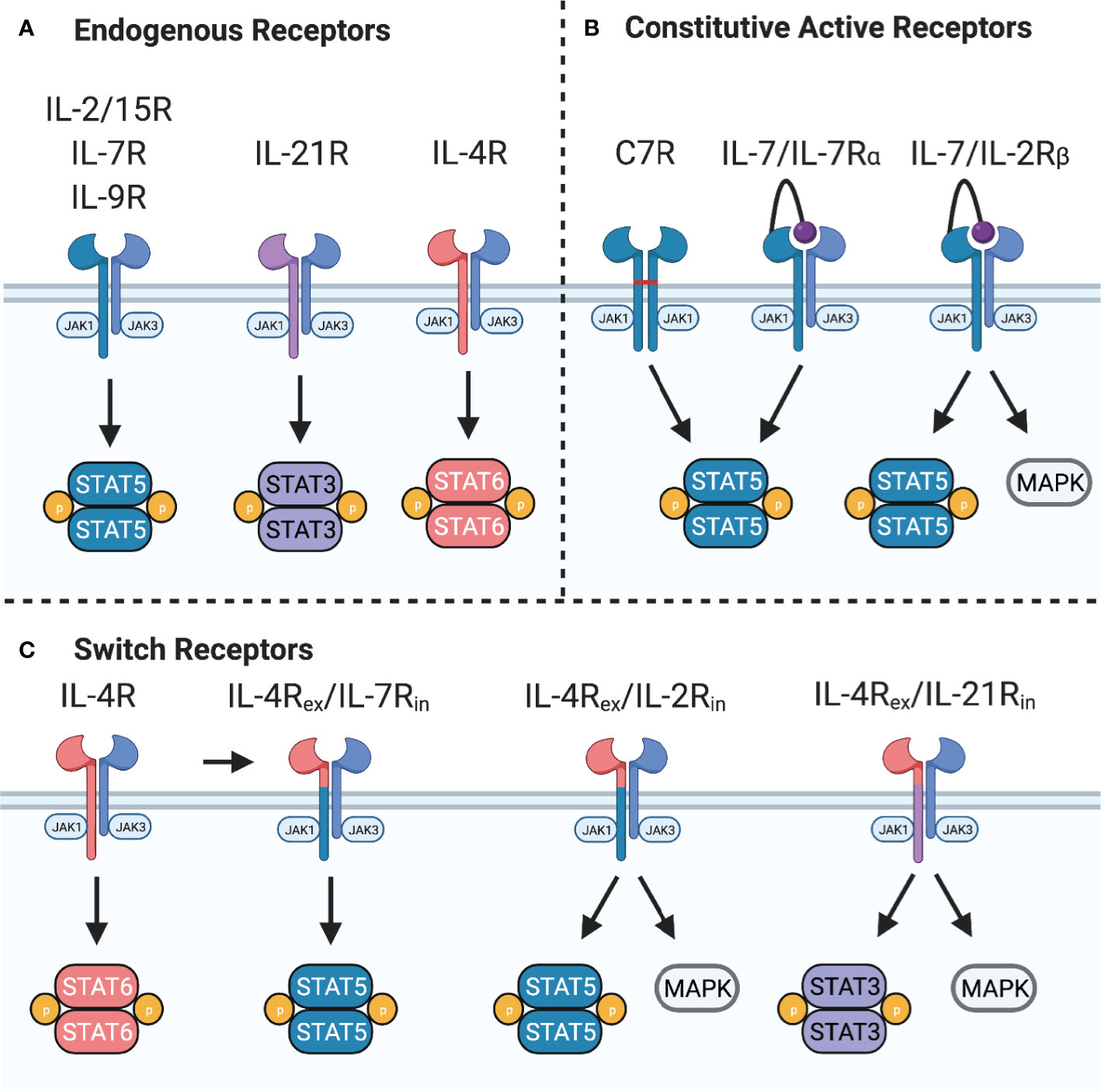
Figure 3 Common gamma chain cytokine signaling and synthetic receptors. (A) Common gamma chain cytokine receptors IL-2/15R, IL-7R, and IL-9R signal primarily through STAT5 while IL-21R signals primarily through STAT3. IL-4R signals through STAT6 to induce Th2 polarization. These receptors also activate PI3K and all except IL-7R activate MAPK pathways. (B) Constitutive active variants of IL-7R can provide constant STAT5 activation. C7R is formed from homodimerization of IL-7Rα chain mutants through a disulfide bridge in the transmembrane domain. A constitutively active IL-7R provides constant STAT5 and PI3K signaling by expression of IL-7 tethered to IL-7Rα. Switching the IL-7Rα intracellular domain with IL-2Rβ provides constant STAT5, PI3K, and MAPK signaling. (C) Switch receptors bind an immunosuppressive cytokine, such as IL-4, and convert it into a stimulatory signal. These receptors are composed of the IL-4Rα extracellular domain (IL-4Rex) fused to the IL-7Rα, IL-2Rβ, or IL-21R intracellular domain (IL-7Rin, IL-2Rin, or IL-21Rin).
An important aspect of γc receptor signaling is positive and negative regulation of the pathway to enhance or repress signaling circuits. For example, IL-2 upregulates expression of the high-affinity IL-2Rα chain, which can increase the sensitivity of T cells to IL-2, thus enhancing IL-2 signaling and T cell proliferation (34). Similarly, IL-4 induces expression of IL-4Rα, enforcing T helper (Th)2 polarization of CD4 T cells (35). Conversely, STAT5 signaling upregulates cytokine inducible SH2-containing protein (CISH) and suppressor of cytokine signaling (SOCS), which directly inhibit JAK activity and ubiquitinate the receptor complex, leading to proteasomal degradation (36).
While some of the γc cytokines can mediate similar signaling pathways and transcriptional programs (32, 37), physiological differences in cytokine signaling are mediated by competition for γc between the different receptors (38), variability in receptor expression on T cell subsets, a bias for signaling through different STAT molecules, and differences in activation of the MAPK and PI3K pathways (39, 40). The end result of γc cytokine receptor signaling is a transcriptional program that is influenced by activation of different STAT heterodimers, homodimers, and tetramers interacting with the chromatin landscape that differs in CD4 and CD8 T cells, as well as in naïve, memory, and exhausted T cells (41–43). Thus, future work to understand these transcriptional networks in CAR T cells could yield rational combinations to improve adoptive cell therapy for cancer.
Interleukin 4 and 9
So far, IL-4 and IL-9 in the context of adoptive cell therapies have been understudied. IL-4 can have anti-cancer properties (44, 45), but it is largely immunosuppressive (46). IL-4 is involved in the differentiation of naïve CD4 T cells into IL-4-producing Th2 cells, which have been associated with less antitumor activity than IFNγ-producing CD4 T cells (Th1) (47). Adoptive transfer of tumor-specific IL-4-producing cytotoxic CD8 T cells (Tc2) was also less effective in controlling tumor growth compared to IFNγ-producing CD8 T cells (Tc1) (48–50). Additionally, several cancer types express IL-4 (51, 52) and the IL-4R (53–55), which suggests a role in tumor progression.
IL-9 was originally described as a T cell growth factor involved in the Th2 response, but recent studies have shown that IL-9 producing T cells (Th9/Tc9) can cause tissue inflammation (56) and inhibit tumor growth by activating mast cells (57) or by indirectly attracting immature dendritic cells (DCs) and activated CD8 T cells to tumors (58). Activation of the endogenous immune system by Th9 cells was better able to control tumor growth than conventional Th1 cells (58). Similarly, Tc9 cells were found to be superior to Tc1 cells for adoptive cell transfer by differentiating into effector cells that can persist longer in vivo, resist exhaustion, and resist apoptosis, which allows them to better control tumor growth (59). While there have been few publications evaluating the utility of IL-9 in improving CAR T cell therapy, Th9/Tc9-polarized human CAR T cells were recently shown to have superior antitumor activity compared to conventional Th1/Tc1 CAR T cells (60). These IL-9 producing T cells have several characteristics of an ideal population of T cells for adoptive cell transfer – they are hyperproliferative, cytotoxic, and resistant to exhaustion – which allows them to eradicate established tumors (61). However, there is conflicting evidence about whether IL-9 is dispensable (60–62) or indispensable (57–59, 62, 63) for antitumor responses. Further work will be required to evaluate this unique T cell population for adoptive cell therapies and to delineate if provision of IL-9 signaling is a general approach to improve CAR T cells.
Interleukin 2 and 15
Early attempts to improve the antitumor activity of CAR T cells were based on the clinical use of IL-2 and IL-15 as anti-cancer immunotherapeutics, which as a monotherapy or in combination with adoptive transfer of tumor infiltrating lymphocytes (TILs) (64–66) had significant antitumor activity but were also associated with toxicity at higher doses (66–70). IL-2 and IL-15 promote immune cell proliferation and the transcription of anti-apoptotic proteins, but responsiveness to these two cytokines varies between Natural Killer (NK) cells and T cell subsets. The high-affinity IL-2 receptor is expressed by regulatory T cells (Tregs) and activated CD4 and CD8 T cells, though memory CD8 T cells and NK cells are also responsive to IL-2 to a lesser degree. Conversely, memory CD8 T cells and NK cells are most responsive to IL-15, while Tregs and naïve CD4 and CD8 T cells are not. Constitutive expression of IL-2 by 1st generation CD19-CAR T cells improved the tumor-free survival of mice with disseminated lymphoma compared to CAR T cells alone, although other γc cytokines were associated with greater improvements in survival and longer persistence of CAR T cells (71). However, the use of IL-2 has fallen out of favor due to the potential expansion of Tregs that can impede antitumor responses (72). Additionally, prolonged exposure to IL-2 promotes activation induced cell death (AICD) (73) and terminal differentiation of T cells into highly cytotoxic effector cells that can efficiently kill tumor cells, but are not able to sustain long-term antitumor activity in vivo that is associated with durable responses (74).
Constitutive expression of IL-15 improved the antitumor activity of CAR T cells specific for CD19, GPC-3, CLL-1, GD2 and IL-13Rα2 (75–79), likely due to a combination of greater expansion and persistence. Constitutive expression of IL-15 by first generation CD19-CAR T cells improved their in vivo antitumor activity and allowed cells to persist in mice for up to 110 days after tumor challenge (71). Notably, IL-15 expression maintained the greatest persistence of CD19-CAR T cells compared to other γc cytokines, and a subset of these persisting cells were memory-like T cells, a phenotype that is associated with sustained responses in patients treated with CAR T cells (80). The expansion or maintenance of memory T cell subsets, such as stem cell memory T cells (Tscm), by IL-15 (81) could contribute to enhanced CAR T cell expansion and persistence. In addition to transgenic expression of secretory IL-15, expressing a membrane-bound form of IL-15 is an alternative way to provide pro-survival signals to CAR T cells (82). This approach is currently under evaluation in a phase 1 trial of CD19-CAR T cells (Table 1).

Table 1 Selected CAR T cell clinical trials with additional modifications to enhance signal 3.
No studies have compared head-to-head the efficacy of membrane-bound IL-15 to secreted IL-15 in engineered T cells, but evidence from preclinical studies suggests that membrane-bound IL-15 may be advantageous in certain situations. In addition to tumor-infiltrating CAR T cells, other immune cells, such as NK cells, NKT cells, endogenous T cells, and innate lymphoid cells (83), are responsive to IL-15. Secreted IL-15 could theoretically induce proliferation in these cell types but in CD8 T cells, membrane-bound IL-15 presented in the context of IL-15Rα, in contrast to secreted IL-15, provided sustained signal transduction that likely contributes to maintenance of memory CD8 T cells (84). Similarly, membrane-bound IL-15 tethered to CD8α improved the survival of engineered NK cells to a greater extent than expression of secreted IL-15, although this effect was due to autocrine signaling (85). Membrane-bound IL-15 tethered to IL-15Rα also improved survival and maintained a greater number of stem cell memory-like CAR T cells following withdrawal of antigen, compared to CAR T cells treated with IL-15/IL-15Rα complex (82). Nonetheless, many preclinical studies support transgenic expression of secreted IL-15 to improve CAR T cell antitumor activity. Ongoing clinical trials with IL-15 engineered T cells and further preclinical work could inform the optimal mode of IL-15 delivery to enhance adoptive cell therapy for cancer.
Concerns of uncontrolled proliferation or toxicity of CAR T cells expressing γc cytokines (86) have spurred the development of inducible safety switches in combination with T cell stimulating cytokines. Inducible caspase 9 (iC9) is an engineered protein that can be activated by a chemical inducer of dimerization to initiate apoptotic cell death (87), and activation of iC9 in vivo resulted in rapid depletion of genetically modified cells in humans (88). Several preclinical studies have shown that co-expression of iC9 and IL-15 improves CAR T cell function and allows the selective depletion of genetically modified CAR T cells. Based on these preclinical studies, GD2-CAR T cells co-expressing iC9 and IL-15 are in early phase clinical evaluation (Table 1). In addition to CAR T cells, CAR natural killer T (NKT) cells have been genetically modified to express IL-15 (89). CAR.IL15 NKT cells maintained a population of central memory-like cells that were less prone to exhaustion and apoptosis, which translated into improved antitumor activity in vivo (90). Additionally, IL-15 protected CAR NKT cells from inhibition in the hypoxic TME (91). Based on these studies, the safety and efficacy of GD2-CAR NKT cells co-expressing IL-15 is currently being evaluated in pediatric patients with neuroblastoma (Table 1), and an interim analysis of the first three patients found that infusion of CAR.IL15 NKT cells is safe and associated with clinical benefit (92). In addition, NK cells expressing CD19-CARs, iC9 and IL-15 have been successfully evaluated in one early phase clinical study (93).
Interleukin 7
Subcutaneous administration of IL-7 induced dose-dependent increases in the number of circulating CD4 and CD8 T cells without causing serious adverse events, such as capillary leak syndrome, that were observed in patients treated with IL-2 or IL-15 (66, 69, 94). In addition to promoting homeostatic expansion of T cells, IL-7 has also been shown to increase TCR repertoire diversity by preferentially expanding naïve T cells and recent thymic emigrants, which could improve the formation of an anti-cancer immune response (95). Therefore, transgenic expression of IL-7 has been explored as an alternative method to improve CAR T cells without the limitations of IL-2. In fact, genetically modifying GD2-CAR virus-specific T cells to express IL-7 allowed them to kill target cells in the presence of inhibitory Tregs, while these cells were suppressed in the presence of IL-2 (96). Constitutive expression of IL-7 improved the antitumor activity of CD19-CAR T cells compared to CAR T cells expressing IL-2 or IL-15, possibly as a result of sustained target cell killing and T cell expansion, however these cells did not exhibit the long-term persistence observed with IL-15-expressing CD19-CAR T cells (71). IL-7 similarly improved CD20- and mesothelin-CAR T cells, but this effect required co-expression of C-C Motif Chemokine Ligand 19 (CCL19) (97). Expression of both IL-7 and CCL19 increased intra-tumoral infiltration of CAR T cells, endogenous T cells, and DCs, which achieved complete regression of established solid tumors and led to the formation of a T cell memory response via epitope spreading. CAR T cells engineered to express IL-7 with CCL19 or CCL21 also had improved antitumor activity due to enhanced CAR T cell proliferation and chemotaxis (98). These studies have been translated into clinical trials to evaluate the safety and efficacy of IL-7 and CCL19 expressing CAR T cells against lymphoma, multiple myeloma, and solid tumors (Table 1).
The greatest limitation to this approach is the downregulation of IL-7Rα as a result of sustained IL-7 signaling or TCR stimulation (99). To restore responsiveness to IL-7, T cells can be modified to constitutively express IL-7Rα (100), but prolonged exposure to IL-7 in IL-7Rα transgenic T cells can promote apoptosis through IFNγ-induced upregulation of Fas and FasL (101). Alternative approaches have been explored to provide the benefits of IL-7 signaling including expressing constitutive active cytokine receptors (102).
Interleukin 21
In contrast to other γc cytokines, IL-21 shows a preference for activation of STAT3 over STAT5 while also signaling through PI3K and MAPK pathways to mediate proliferation (39). Constitutive expression of IL-21 by 1st generation CD19-CAR T cells improved overall survival of mice with disseminated lymphoma (71). The greatest improvement in overall survival was seen in CAR T cells expressing IL-21, compared to CAR T cells expressing IL-2, IL-7, or IL-15, even though these cells expressed lower levels of Bcl-2, IFNγ, and TNFα. The improved antitumor activity could be due to maintenance of less differentiated effector memory T cell (TEM) subsets and/or by enhancing long-term persistence. In accordance with this observation, IL-21 has been shown to decrease terminal effector differentiation of CD8 T cells, as judged by expression of granzyme B and EOMES, in comparison to IL-2 treated CD8 T cells, which resulted in improved in vivo antitumor activity (103). IL-21 also improved the expansion and persistence of adoptively transferred CD8 T cells resulting in superior antitumor activity in syngeneic tumor models compared to IL-2 and IL-15 (104). While the efficacy of IL-21-driven STAT3 signaling in T cell immunotherapy is still being explored, STAT3 signaling has been correlated with improved CD19 CAR T cell treatment outcomes in chronic lymphocytic leukemia (80).
Constitutive Active Cytokine Receptors
An alternative strategy to enhance signal 3 of T cell activation is by transgenic expression of synthetic cytokine receptors (Figure 3B). For example, expression of a constitutively active IL-7 receptor (C7R) can overcome the downregulation of IL-7Rα due to negative feedback mechanisms and provide constant STAT5 signaling without a requirement for IL-7. Enhanced STAT5 signaling mediated by C7R was found to improve the in vitro and in vivo antitumor activity of GD2- and EphA2-CAR T cells (102) in an antigen-dependent fashion. C7R also increased tumor infiltration, expansion, and cytokine production of AXL-CAR T cells, but did not improve antitumor activity compared to the CAR alone (105). Clinical trials evaluating GD2-CAR T cells expressing C7R are in progress (Table 1).
Expression of IL-7Rα tethered to IL-7 is another way to provide cell-intrinsic IL-7 signaling (106). Since this is a modular platform for providing signal 3, the IL-7Rα intracellular domain can be replaced with other signaling domains. For example, investigators have expressed a chimeric IL-7Rα/IL-2Rβ receptor and IL-7 in CAR T cells to provide signal 3 and demonstrated that these engineered CAR T cells are resistant to TGFβ inhibition (107).
Chimeric Cytokine Receptors
Chimeric cytokine receptors or switch receptors, which convert one cytokine signal into another, are actively being explored to hijack immunosuppressive cytokines produced by tumor or tumor-associated cells to provide proliferative signals to CAR T cells (Figure 3C). An IL-4/IL-7 switch receptor, which binds IL-4 but activates IL-7 signaling pathways, allowed PSCA-CAR T cells to maintain their cytolytic and proliferative capabilities in vitro and improved in vivo antitumor activity (108). In an orthotopic breast cancer model, the IL-4/IL-7 switch receptor also improved the antitumor activity of 2nd generation MUC1-CAR T cells (109). Importantly, these cells were able to respond to tumor rechallenge at a distal site by proliferating and eliminating tumor cells in an IL-4-dependent manner. This approach has also been used to improve antitumor activity in the TME by converting immunosuppressive signals, such as TGFβ and IL-4, into separate stimulatory signals that improve tumor selectivity and CAR T cell potency (110). Expressing an IL-4/IL-2 switch receptor in MUC1- or PSMA-CAR T cells improved their cytolytic activity and proliferative capacity through increased STAT5 and ERK phosphorylation in the presence of IL-4 (111). Similarly, the receptor enhanced the antitumor activity and persistence of αvβ6 integrin-CAR T cells in an IL-4-dependent manner (112). An ongoing phase 1 clinical trial is investigating the utility of the IL-4/IL-2 switch receptor (4αβ) in CAR T cells for head and neck squamous cell carcinoma (Table 1).
While IL-4/IL-2 switch receptors convert STAT6 into STAT5 signals, IL-4/IL-21 switch receptors have been designed to convert STAT6 into STAT3 signals. This switch receptor improved cytolytic activity of GPC3-CAR T cells in the presence of IL-4, most likely by upregulating RORγt (113). Few studies have directly compared different cytokine switch receptors. However, comparison of IL-4/IL-7 and IL-4/IL-21 switch receptors in GPC3-CAR T cells demonstrated that in one solid tumor model, the IL-4/IL-21 switch receptor was superior (113). Lastly, TGFβ/IL-7 switch receptors have been expressed in PSMA-CAR T cells, improving their antitumor activity by upregulating AKT phosphorylation and Bcl-xL expression (114). In addition to tumor-derived cytokines that directly inhibit T cells, the TME can also produce cytokines for which T cells lack the corresponding receptor. For example, T cells lack the receptor for colony stimulating factor-1 (CSF-1). Engineering CAR T cells to express CSF-1R renders them responsive to CSF-1, which can promote chemotaxis, increase IFNγ production, and synergize with sub-optimal growth stimuli to enhance proliferation (115).
The orthologous IL-2 system is another way to modulate cytokine signaling in adoptively transferred cells. In order to limit the effects of the pleiotropic cytokine IL-2 to only select cell types, Sockolosky, et al. mutated IL-2 and IL-2Rβ (orthoIL-2 & orthoIL-2Rβ) such that the orthogonal cytokine/receptor pair can bind each other, but not the wild type IL-2 or IL-2Rβ (116). In vivo, orthoIL-2 could expand tumor-specific T cells expressing orthoIL-2Rβ to control tumor growth without the dose-limiting toxicity seen with high doses of IL-2. This approach could be adapted to drive other cytokine signaling pathways in engineered T cells without affecting endogenous immune cells.
Interleukin 12 Family Cytokines and Their Receptors
Cytokine Biology
The IL-12 family of cytokines, IL-12, IL-23, IL-27, and IL-35, have diverse roles in innate and adaptive immune responses, with IL-12 and IL-23 being pro-inflammatory, IL-27 having both pro- and anti-inflammatory effects, and IL-35 being anti-inflammatory (117) (Figure 4A). Due to the primarily anti-inflammatory roles of IL-27 and IL-35, only IL-12 and IL-23 have been explored in the context of adoptive cell therapies.
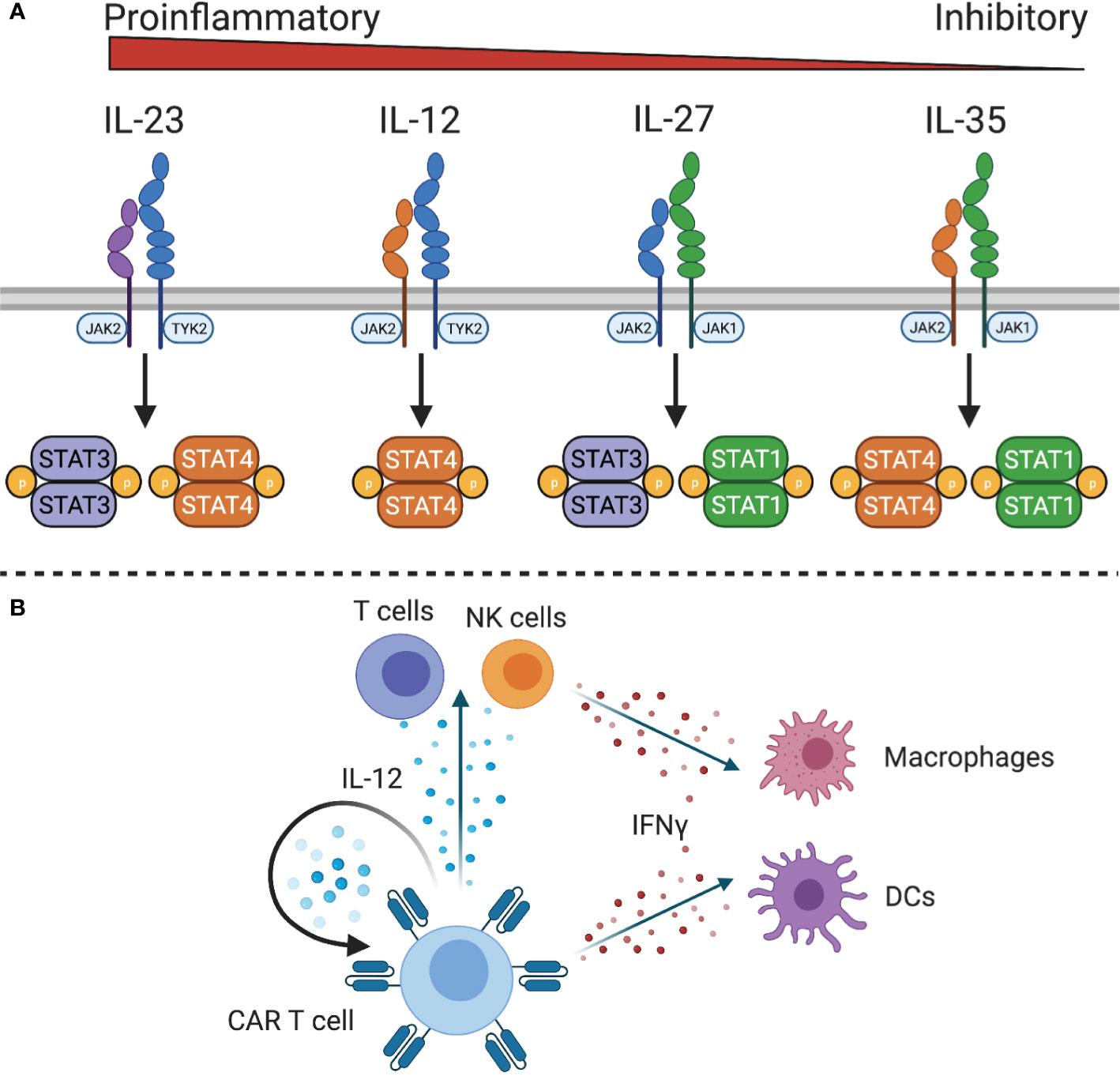
Figure 4 IL-12 family cytokine signaling. (A) The IL-12 family cytokine receptors signal through different STAT family members to exert proinflammatory or inhibitory functions. (B) IL-12 expressing CAR T cells can stimulate IL-12 responsive CAR T cells and endogenous T and NK cells. Downstream mediators of IL-12, such as IFNγ, can act on macrophages and dendritic cells to stimulate innate and adaptive immunity.
Because of the role of IL-12 in linking adaptive and innate immunity, it has been explored as a potential cytokine to initiate or sustain antitumor immune responses. IL-12 is a heterodimeric protein composed of IL-12p40 and IL-12p35 that is produced by antigen presenting cells in response to microbial stimulation (118). The IL-12 receptor (IL-12R) consists of two chains, IL-12Rβ1 and IL-12Rβ2, which signals through JAK2 and tyrosine kinase (TYK)2 to activate STAT4 and other STAT family members to a lesser degree. It is primarily expressed by activated T cells and NK cells (119) although functional IL-12 receptor expression has also been observed on DCs (120), B cells (121), and myeloid-derived suppressor cells (MDSCs) (122). In both T cells and NK cells, IL-12 enhances proliferation, cytotoxicity, and the production of high levels of IFNγ, TNFα, and GM-CSF (123, 124). Additionally, IL-12 controls Th1 differentiation by directly inducing production of IFNγ and upregulating expression of IL-12Rβ2 via IFNγ-induced expression of T-bet, the Th1 defining transcription factor. This leads to a positive feedback loop wherein IFNγ enhances IL-12 production by DCs and renders T cells more sensitive to IL-12 to reinforce a Th1 phenotype. In addition to increasing IFNγ production, IL-12 also promotes expression of immunostimulatory cytokines and chemokines, such as GM-CSF, C-C motif ligands, IP10 (CXCL10), and MIG (CXCL9) to recruit T cells, NK cells, and antigen presenting cells to the site of inflammation (28, 125, 126). This has implications for cancer immunity by recruiting and activating effector cells, reprogramming tumor-associated macrophages to a pro-inflammatory phenotype (127), increasing antigen processing and presentation, reprogramming immunosuppressive MDSCs (128), and inhibiting angiogenesis (129).
IL-23 is a functionally related heterodimeric cytokine that is composed of IL-23p19 and the shared IL-12p40 subunit. The IL-23 receptor is composed of IL-12Rβ2 and IL-23R, which signals through JAK2 and TYK2 to primarily activate STAT3 and STAT4. Similar to IL-12, IL-23 is produced by antigen presenting cells and plays an important role in T cell differentiation. During T cell activation in the presence of cytokines such as TGFβ, IL-1, or IL-6, IL-23 upregulates expression of the IL-23 receptor and RORγt to reinforce a Th17 phenotype but is not required for Th17 differentiation. In the context of tumor immunity, IL-23 may play a distinct role compared to IL-12. The antitumor effects of IL-12 have been demonstrated (130), but IL-23 can have pro or antitumor properties, depending on the concentration of IL-23 and cancer type (131, 132).
Interleukin 12
IL-12 has been used as a single agent immunotherapy and in combination with adoptive transfer of T cells to promote activation of tumor-specific T cells and differentiation into pro-inflammatory Th1/Tc1 cells. Systemic administration of IL-12 resulted in robust antitumor activity in preclinical models (133); however, clinical trials with recombinant human IL-12 were limited by modest efficacy and toxicity (134–137). Nonetheless, genetic modification of CAR T cells to secrete IL-12 is actively being explored. For example, modification of MUC16ecto-CAR T cells to constitutively secrete IL-12 enhanced IFNγ production and in vivo persistence, likely through an autocrine mechanism, leading to increased survival in an orthotopic xenograft model (138). Later work to define the interaction of tumor-associated immune cells and IL-12 producing CAR T cells confirmed that autocrine IL-12 signaling was necessary for antitumor activity by enhancing the effector function of CAR T cells, depleting tumor associated macrophages, and preventing tumor-mediated PD-L1 inhibition. Interestingly, IL-12 did not exert its effect in this model by recruiting endogenous T cells to the tumor or rely on host IFNγ producing cells (139). There are conflicting results about whether antitumor activity is primarily mediated by the effects of IL-12 on adoptively transferred T cells or host IL-12R expressing cells (128, 140). Nevertheless, a clinical study with MUC16ecto-CAR T cells expressing IL-12 is in progress (Table 1). Early results from this trial illustrate that IL-12 secreting MUC16ecto CAR T cells can be safely administered, although a high incidence of hemophagocytic lymphohistiocytosis/macrophage activation-like syndrome was observed when cells were administered after lymphodepleting chemotherapy (141).
The observation that constitutive expression of IL-12 by engineered T cells could promote T cell dysfunction (142–145) and toxicity in preclinical models (146) led to the design of vectors in which IL-12 expression is under the control of the Nuclear Factor of Activated T cell (NFAT) promoter (144). The NFAT promoter links IL-12 expression to T cell activation (144, 146), reducing systemic toxicity without decreasing antitumor activity (127, 146, 147). However, one clinical study with TILs modified to express NFAT-inducible IL-12 suggests that the NFAT promotor might not be sufficient to restrain IL-12 production to only activated TILs within the tumor (148). In addition to NFAT-controlled IL-12 expression, doxycycline-inducible IL-12 production is also actively being explored (149). While the autocrine effects of IL-12 contribute to improved antitumor activity of IL-12 CAR T cells, the importance of IL-12 in mustering an innate immune response cannot be overstated (Figure 4B). Endogenous T cells, NK cells, DCs, and MDSCs can respond to both IL-12 and the high levels of IFNγ produced by IL-12-responsive cells. One study found that inducible IL-12 allowed CAR T cells to eradicate antigen-positive tumor cells and prevent outgrowth of antigen negative tumor cells by recruiting and activating macrophages to produce TNFα and upregulate costimulatory molecules CD80/CD86 to enhance T cell responses (127). In another study, IL-12 was also shown to increase antigen processing and presentation (128), which could contribute to an endogenous antitumor immune response in solid tumors. Similarly, a study of IL-12 expressing VEGFR2-CAR T cells found that host IL-12R expressing cells, but not B or T cells, were required for the antitumor response (146). This activation of the innate immune system has the potential to produce lasting remissions for solid tumor patients, which is being evaluated in an ongoing phase I/II clinical trial of EGFR-specific CAR T cells with NFAT-inducible IL-12 to treat metastatic colorectal cancer (Table 1).
Additional approaches to recruit and activate innate immune cells to the TME are also under development. For example, CAR T cells engineered to secrete the DC growth factor Fms-like tyrosine kinase 3 ligand (Flt3L) were able to expand intra-tumoral conventional type 1 DCs (150). By engaging the endogenous immune system, Flt3L-CAR T cells, in conjunction with immune adjuvants, were able to mediate regression of established solid tumors and promote the formation of antitumor memory via epitope spreading. Approaches such as these will be necessary to overcome antigen heterogeneity and antigen-negative relapse in the treatment of solid tumors.
Interleukin 23
While there have been few studies using IL-23 in combination with cell-based therapies, recent work has suggested that this is an option to improve CAR T cell function in solid tumors. CAR/TCR signaling upregulates expression of IL-23R and IL-23p19, but not the second subunit of IL-23 – IL-12p40. Transduction of T cells to constitutively express IL-12p40 leads to activation-induced expression of IL-23, but not IL-12, and improves T cell proliferation. CAR T cells engineered to express IL-12p40 exhibit greater expansion, persistence, and antitumor activity in xenograft and syngeneic tumor models (151), which could be attributed to an IL-23-induced STAT3 and hypoxia inducible factor (HIF) gene signature. STAT3 signaling in CAR T cell products has been associated with improved responses in chronic lymphocytic leukemia patients (80) and modulation of STAT3 signaling has been used to improve CAR T cells in other preclinical models (29).
Interleukin 1 Superfamily of Cytokines
The IL-1 superfamily of cytokines – IL-1α, IL-1β, IL-33, IL-1 Receptor Antagonist, IL-18, IL-37, IL-36 Receptor Antagonist, IL-36α, IL-36β, IL-36γ, and IL-38 – plays important roles in innate and adaptive immunity, but only a subset of these cytokines has been evaluated in preclinical and clinical trials for their ability to elicit an antitumor immune response. For example, the role of IL-1β in cancer has been extensively studied, with different studies showing a role in either promoting or inhibiting tumorigenesis (152–155). Although IL-1β has the potential to improve adoptive cell therapy (156), IL-1β in conjunction with macrophage-derived nitric oxide and IL-6 plays a central role in cytokine release syndrome (CRS) (157, 158), a serious complication of CAR T cell therapy. However, other cytokines are emerging as anticancer mediators and their ability to improve CAR T cell therapy is currently under investigation.
Interleukin 18
IL-18 is a proinflammatory cytokine produced by macrophages, dendritic cells, epithelial cells, and other cell types that interacts with a heterodimeric receptor composed of IL-18Rα and IL-18Rβ expressed on NK cells and antigen-experienced T cells. IL-18 signals through MyD88 and NF-kB, and has been shown to have protumorigenic functions, such as promoting angiogenesis, metastasis, and proliferation (159), but it is thought to largely have antitumor activity due to its cooperation in the Th1 response (160). In conjunction with IL-12 or antigen stimulation, IL-18 induces the production of IFNγ and cytotoxic effector molecules by Th1 and CD8 T cells (Figure 5A) (161), resulting in activation of NK cells, macrophages, and other cell types. However, IL-18 signaling without concomitant inflammatory cytokines enhances Th2 responses, such as production of IL-4 and IL-13 (162, 163). Nonetheless, IL-18 has been tested in clinical trials for antitumor activity. While administration of IL-18 has been well tolerated (164), it showed no benefit in metastatic melanoma patients when given as a monotherapy (165).
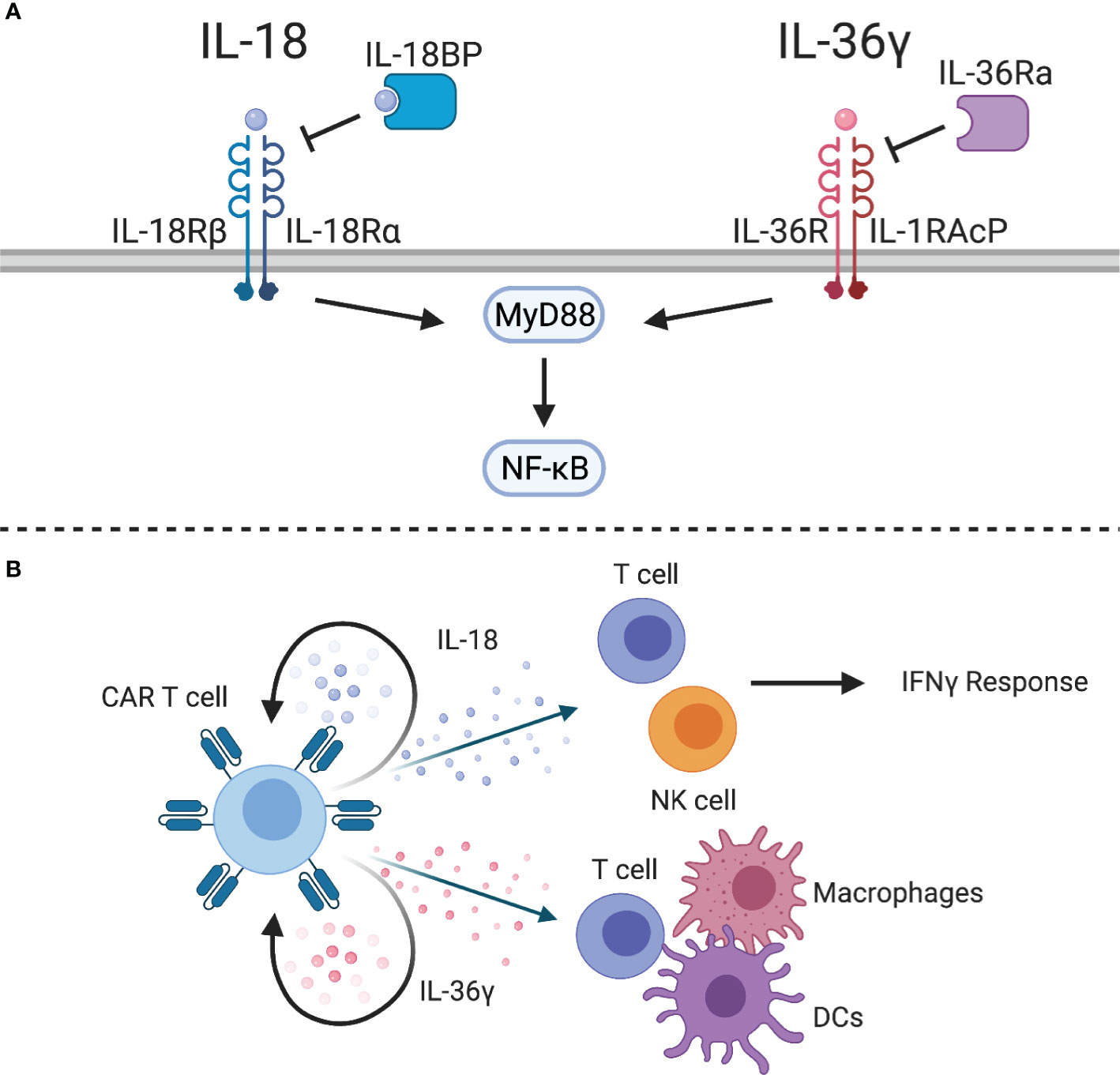
Figure 5 IL-1 superfamily cytokines. (A) The IL-1 superfamily cytokines, IL-18 and IL-36γ, signal through MyD88 to activate NF-kB. Inhibitory proteins, such as IL-18 binding protein (IL-18BP) or IL-36 receptor antagonist (IL-36Ra), can inhibit cytokine signaling. (B) IL-18-expressing CAR T cells can stimulate an IFNγ response from CAR T cells and endogenous T and NK cells. IL-36γ-expressing CAR T cells can provide autocrine signaling to enhance effector function and activate endogenous T cells, macrophages, and DCs.
Engineering human and murine CAR T cells to express IL-18 improved CAR/TCR-mediated proliferation, production of cytokines, and antitumor activity (166–169). One study highlighted the role of IL-18-secreting CD4 CAR T cells to promote expansion of CD8 CAR T cells (166). Another study highlighted that IL-18 induces a potent T cell effector subset characterized by a T-Bethigh FoxO1low phenotype (167). Similar to IL-12, NFAT-inducible IL-18 expression systems have also been explored to limit systemic side effects associated with constitutive IL-18 expression (167, 168). In syngeneic models, IL-18 was able to remodel the TME with increased numbers of CD8 T cells, NK cells, and activated antigen presenting cells resulting in induction of tumor-specific T cell responses via epitope spreading (169). This was mirrored by a reduced number of immunosuppressive dendritic cells, M2 macrophages, and Tregs (167, 169). A possible limitation of transgenic IL-18 expression is the recent discovery of an IL-18 binding protein (IL-18BP), an immune checkpoint, that inhibits IL-18 signaling; however, approaches to develop IL-18BP-resistant IL-18 are actively being explored (170). Lastly, a recent study has highlighted that it is feasible to design chimeric switch receptors that activate IL-18 signaling pathways in CAR T cells (171).
Interleukin 36
IL-36α, IL-36β, and IL-36γ are newly discovered members of the IL-1 superfamily that have shown potent antitumor activity in preclinical models (172, 173). These cytokines share a heterodimeric receptor, composed of IL-36R and IL-1RAcP, that signals through MyD88 and NF-kB (Figure 5A). Similar to IL-18, IL-36 cytokines can be inhibited by IL-36 receptor antagonist (IL-36Ra). While these cytokines have so far been understudied, they have the potential to improve adoptive cell therapy. For example, IL-36γ has been shown to transform the tumor microenvironment and mediate tumor-specific CD8 T cell responses (172). IL-36γ is produced by keratinocytes, epithelial cells, and immune cells, and exerts its functions on several cell types, including epithelial cells, macrophages, dendritic cells, and T cells (174). Engineering CAR T cells to express IL-36γ improves expansion and persistence, which results in improved antitumor activity compared to unmodified CAR T cells (175). While this effect was dependent on autocrine IL-36 signaling through MyD88 for initial tumor clearance, IL-36R is also abundantly expressed on myeloid cells. IL-36γ-expressing CAR T cells were able to enhance MHC class II and CD86 expression on splenic macrophages and DCs of tumor-bearing mice, which suggests that IL-36 plays a role in maturation of antigen presenting cells. Importantly, this induced antigen spreading as evidenced by tumor recognition by endogenous CD8 T cells. Therefore, IL-36γ-expressing CAR T cells could be a viable treatment option for solid tumor patients who often suffer from relapse related to antigen heterogeneity or antigen loss (Figure 5B).
Discussion
Transgenic expression of individual cytokines and/or cytokine receptors has improved the effector function of CAR T cells in preclinical models. Studies have highlighted that transgenic cytokines do not only enhance the antitumor activity of CAR T cells but also modulate other cells within the TME and are able to induce or enhance endogenous tumor-specific immune responses. However, safety concerns have also been raised. CRS is a potentially fatal complication of CAR T cell therapy and secondary genetic modifications that enhance CAR T cell function could exacerbate CRS. A major concern with γc cytokines (IL-2, IL-7, IL-15, and IL-21) is enhanced proliferation of infused CAR T cells leading to production of toxic levels of effector molecules IFNγ and TNFα. Since CAR T cell peak expansion correlates with CRS (176), approaches to improve CAR T cell expansion must be weighed against the risk of dose limiting toxicities. While IL-6 and IL-1 play central roles in CAR T cell-induced CRS, high levels of IFNγ are also observed (177), and any cytokine that enhances IFNγ production by CAR T cells or other immune cells could potentially increase the incidence and/or severity of CRS. IL-12 can directly augment IFNγ production by T cells and NK cells. Similarly, IL-18 can synergize with IL-12 or IL-15 to enhance IFNγ production. IL-23 and IL-36 have not been implicated in CRS, but they play central roles in inflammatory conditions in the gut and skin and could therefore potentially contribute to CRS if produced at high levels by adoptively transferred immune cells.
While early phase clinical testing of CAR T cells secreting cytokines or constitutively active cytokine receptors are in progress, there are opportunities to further enhance this approach. A physiological immune response requires coordination of different cytokines to i) initiate an inflammatory reaction, ii) amplify responses of multiple cell types, and iii) resolve inflammation. However, the majority of studies have so far only explored constitutive expression of a single cytokine. Thus, developing approaches that endow CAR T cells with the ability to express an array of cytokines at different stages of CAR T cell activation holds the promise to not only increase efficacy but also safety. As these approaches are being developed, not only xenograft but also immune competent models are needed to careful analyze how cytokine-secreting CAR T cells engage with endogenous immune cells. Based on the breath of preclinical data generated thus far, we are confident that ‘signal 3-enhanced’ CAR T cells have the potential to improve the currently limited antitumor activity of CAR T cells in early phase clinical studies for patients with solid tumors and brain tumors.
Author Contributions
MB and SG wrote, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by a grant from the National Cancer Institute 1F31CA250401-01A1, by a National Institutes of Health (NIH) grant R01NS106379 and the American Lebanese Syrian Associated Charities. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest
SG has patent applications in the field of T-cell and gene-modified T-cell therapy for cancer. He is a consultant for Catamaran Bio, Nektar Therapeutics, and TESSA Therapeutics, on the Scientific Advisory Board of Tidal, and a DSMB member of Immatics.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figures were created with BioRender (Biorender.com) for which we have license.
References
1. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. New Engl J Med (2018) 379(1):64–73. doi: 10.1056/NEJMra1706169
2. Srivastava S, Riddell SR. Engineering CAR-T Cells: Design Concepts. Trends Immunol (2015) 36(8):494–502. doi: 10.1016/j.it.2015.06.004
3. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and Development of Therapies Using Chimeric Antigen Receptor-Expressing T Cells. Immunol Rev (2014) 257(1):107–26. doi: 10.1111/imr.12131
4. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. New Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
5. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. New Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
6. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1-2 Trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7
7. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. New Engl J Med (2019) 380(18):1726–37. doi: 10.1056/NEJMoa1817226
8. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-Targeted CAR T Cells Induce Remission in B-ALL That Is Naive or Resistant to CD19-targeted CAR Immunotherapy. Nat Med (2018) 24(1):20–8. doi: 10.1038/nm.4441
9. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-Specific Adoptive Immunotherapy for Lymphoma Using a Chimeric Antigen Receptor With Both CD28 and 4-1BB Domains: Pilot Clinical Trial Results. Blood (2012) 119(17):3940–50. doi: 10.1182/blood-2011-10-387969
10. Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu MF, Ivanova A, et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J Clin Oncol: Off J Am Soc Clin Oncol (2020) 38(32):3794–804. doi: 10.1200/JCO.20.01342
11. Mueller KT, Maude SL, Porter DL, Frey N, Wood P, Han X, et al. Cellular Kinetics of CTL019 in Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia and Chronic Lymphocytic Leukemia. Blood (2017) 130(21):2317–25. doi: 10.1182/blood-2017-06-786129
12. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. Car T Cells Administered in Combination With Lymphodepletion and PD-1 Inhibition to Patients With Neuroblastoma. Mol Ther: J Am Soc Gene Ther (2017) 25(9):2214–24. doi: 10.1016/j.ymthe.2017.05.012
13. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor-Positive T Cells in Patients With Neuroblastoma. Blood (2011) 118(23):6050–6. doi: 10.1182/blood-2011-05-354449
14. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (Her2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol: Off J Am Soc Clin Oncol (2015) 33(15):1688–96. doi: 10.1200/JCO.2014.58.0225
15. Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of Metastatic Renal Cell Carcinoma (mRCC) With CAIX CAR-Engineered T-Cells-a Completed Study Overview. Biochem Soc Trans (2016) 44(3):951–9. doi: 10.1042/BST20160037
16. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The Clinical Efficacy of First-Generation Carcinoembryonic Antigen (CEACAM5)-Specific CAR T Cells is Limited by Poor Persistence and Transient Pre-Conditioning-Dependent Respiratory Toxicity. Cancer Immunol Immunother: CII (2017) 66(11):1425–36. doi: 10.1007/s00262-017-2034-7
17. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors is Decisive for In Vivo Antitumor Activity. Cancer Immunol Res (2015) 3(2):125–35. doi: 10.1158/2326-6066.CIR-14-0127
18. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin Cancer Res (2013) 19(12):3153–64. doi: 10.1158/1078-0432.CCR-13-0330
19. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC Locus With CRISPR/Cas9 Enhances Tumour Rejection. Nature (2017) 543(7643):113–7. doi: 10.1038/nature21405
20. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR Activation Potential Directs Alternative T Cell Fates and Therapeutic Potency. Nat Med (2019) 25(1):82–8. doi: 10.1038/s41591-018-0290-5
21. Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, et al. Single Residue in CD28-Costimulated CAR-T Cells Limits Long-Term Persistence and Antitumor Durability. J Clin Invest (2020) 130(6):3087–97. doi: 10.1172/JCI133215
22. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci Rep (2017) 7(1):737. doi: 10.1038/s41598-017-00462-8
23. Brocker T, Karjalainen K. Signals Through T Cell Receptor-Zeta Chain Alone are Insufficient to Prime Resting T Lymphocytes. JExpMed (1995) 181(5):1653–9. doi: 10.1084/jem.181.5.1653
24. Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M. Antigen-Dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. JExpMed (1998) 188(4):619–26. doi: 10.1084/jem.188.4.619
25. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I Ifns Provide a Third Signal to CD8 T Cells to Stimulate Clonal Expansion and Differentiation. J Immunol (2005) 174(8):4465–9. doi: 10.4049/jimmunol.174.8.4465
26. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov (2017) 7(11):1306–19. doi: 10.1158/2159-8290.CD-17-0263
27. Musso T, Calosso L, Zucca M, Millesimo M, Ravarino D, Giovarelli M, et al. Human Monocytes Constitutively Express Membrane-Bound, Biologically Active, and Interferon-Gamma-Upregulated Interleukin-15. Blood (1999) 93(10):3531–9. doi: 10.1182/blood.V93.10.3531.410k32_3531_3539
28. Trinchieri G. Interleukin-12 and the Regulation of Innate Resistance and Adaptive Immunity. Nat Rev Immunol (2003) 3(2):133–46. doi: 10.1038/nri1001
29. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A Novel Chimeric Antigen Receptor Containing a JAK-STAT Signaling Domain Mediates Superior Antitumor Effects. Nat Med (2018) 24(3):352–9. doi: 10.1038/nm.4478
30. Zhang H, Zhao H, He X, Xi F, Liu J. Jak-Stat Domain Enhanced MUC1-CAR-T Cells Induced Esophageal Cancer Elimination. Cancer Manag Res (2020) 12:9813–24. doi: 10.2147/CMAR.S264358
31. Lord JD, McIntosh BC, Greenberg PD, Nelson BH. The IL-2 Receptor Promotes Lymphocyte Proliferation and Induction of the C-Myc, bcl-2, and Bcl-X Genes Through the Trans-Activation Domain of Stat5. J Immunol (2000) 164(5):2533–41. doi: 10.4049/jimmunol.164.5.2533
32. Kovanen PE, Rosenwald A, Fu J, Hurt EM, Lam LT, Giltnane JM, et al. Analysis of Gamma C-Family Cytokine Target Genes. Identification of Dual-Specificity Phosphatase 5 (DUSP5) as a Regulator of Mitogen-Activated Protein Kinase Activity in Interleukin-2 Signaling. J Biol Chem (2003) 278(7):5205–13. doi: 10.1074/jbc.M209015200
33. Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, et al. Differential Effects of STAT5 and PI3K/AKT Signaling on Effector and Memory CD8 T-Cell Survival. Proc Natl Acad Sci USA (2010) 107(38):16601–6. doi: 10.1073/pnas.1003457107
34. Sereti I, Gea-Banacloche J, Kan MY, Hallahan CW, Lane HC. Interleukin 2 Leads to Dose-Dependent Expression of the Alpha Chain of the IL-2 Receptor on CD25-Negative T Lymphocytes in the Absence of Exogenous Antigenic Stimulation. Clin Immunol (2000) 97(3):266–76. doi: 10.1006/clim.2000.4929
35. Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, et al. Priming for T Helper Type 2 Differentiation by Interleukin 2-Mediated Induction of Interleukin 4 Receptor Alpha-Chain Expression. Nat Immunol (2008) 9(11):1288–96. doi: 10.1038/ni.1656
36. Alexander WS. Suppressors of Cytokine Signalling (SOCS) in the Immune System. Nat Rev Immunol (2002) 2(6):410–6. doi: 10.1038/nri818
37. Osinalde N, Sanchez-Quiles V, Akimov V, Guerra B, Blagoev B, Kratchmarova I. Simultaneous Dissection and Comparison of IL-2 and IL-15 Signaling Pathways by Global Quantitative Phosphoproteomics. Proteomics (2015) 15(2-3):520–31. doi: 10.1002/pmic.201400194
38. Gonnord P, Angermann BR, Sadtler K, Gombos E, Chappert P, Meier-Schellersheim M, et al. A Hierarchy of Affinities Between Cytokine Receptors and the Common Gamma Chain Leads to Pathway Cross-Talk. Sci Signal (2018) 11(524). doi: 10.1126/scisignal.aal1253
39. Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ. The Molecular Basis of IL-21-Mediated Proliferation. Blood (2007) 109(10):4135–42. doi: 10.1182/blood-2006-10-054973
40. Gadina M, Sudarshan C, Visconti R, Zhou YJ, Gu H, Neel BG, et al. The Docking Molecule Gab2 is Induced by Lymphocyte Activation and is Involved in Signaling by Interleukin-2 and Interleukin-15 But Not Other Common Gamma Chain-Using Cytokines. J Biol Chem (2000) 275(35):26959–66. doi: 10.1016/S0021-9258(19)61466-3
41. Rochman Y, Spolski R, Leonard WJ. New Insights Into the Regulation of T Cells by Gamma(C) Family Cytokines. Nat Rev Immunol (2009) 9(7):480–90. doi: 10.1038/nri2580
42. Huang W, August A. The Signaling Symphony: T Cell Receptor Tunes Cytokine-Mediated T Cell Differentiation. J Leukoc Biol (2015) 97(3):477–85. doi: 10.1189/jlb.1RI0614-293R
43. Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, et al. Stats Shape the Active Enhancer Landscape of T Cell Populations. Cell (2012) 151(5):981–93. doi: 10.1016/j.cell.2012.09.044
44. Gooch JL, Lee AV, Yee D. Interleukin 4 Inhibits Growth and Induces Apoptosis in Human Breast Cancer Cells. Cancer Res (1998) 58(18):4199–205.
45. Volpert OV, Fong T, Koch AE, Peterson JD, Waltenbaugh C, Tepper RI, et al. Inhibition of Angiogenesis by Interleukin 4. J Exp Med (1998) 188(6):1039–46. doi: 10.1084/jem.188.6.1039
46. Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, van Eden W, et al. Interleukin-4 Therapy of Psoriasis Induces Th2 Responses and Improves Human Autoimmune Disease. Nat Med (2003) 9(1):40–6. doi: 10.1038/nm804
47. Nishimura T, Nakui M, Sato M, Iwakabe K, Kitamura H, Sekimoto M, et al. The Critical Role of Th1-dominant Immunity in Tumor Immunology. Cancer Chemother Pharmacol (2000) 46 Suppl:S52–61. doi: 10.1007/PL00014051
48. Ye Z, Tang C, Xu S, Zhang B, Zhang X, Moyana T, et al. Type 1 CD8+ T Cells are Superior to Type 2 CD8+ T Cells in Tumor Immunotherapy Due to Their Efficient Cytotoxicity, Prolonged Survival and Type 1 Immune Modulation. Cell Mol Immunol (2007) 4(4):277–85.
49. Kemp RA, Ronchese F. Tumor-Specific Tc1, But Not Tc2, Cells Deliver Protective Antitumor Immunity. J Immunol (2001) 167(11):6497–502. doi: 10.4049/jimmunol.167.11.6497
50. Helmich BK, Dutton RW. The Role of Adoptively Transferred CD8 T Cells and Host Cells in the Control of the Growth of the EG7 Thymoma: Factors That Determine the Relative Effectiveness and Homing Properties of Tc1 and Tc2 Effectors. J Immunol (2001) 166(11):6500–8. doi: 10.4049/jimmunol.166.11.6500
51. Todaro M, Lombardo Y, Francipane MG, Alea MP, Cammareri P, Iovino F, et al. Apoptosis Resistance in Epithelial Tumors is Mediated by Tumor-Cell-Derived Interleukin-4. Cell Death Differ (2008) 15(4):762–72. doi: 10.1038/sj.cdd.4402305
52. Prokopchuk O, Liu Y, Henne-Bruns D, Kornmann M. Interleukin-4 Enhances Proliferation of Human Pancreatic Cancer Cells: Evidence for Autocrine and Paracrine Actions. Br J Cancer (2005) 92(5):921–8. doi: 10.1038/sj.bjc.6602416
53. Obiri NI, Siegel JP, Varricchio F, Puri RK. Expression of High-Affinity IL-4 Receptors on Human Melanoma, Ovarian and Breast Carcinoma Cells. Clin Exp Immunol (1994) 95(1):148–55. doi: 10.1111/j.1365-2249.1994.tb06029.x
54. Puri RK, Leland P, Kreitman RJ, Pastan I. Human Neurological Cancer Cells Express Interleukin-4 (IL-4) Receptors Which are Targets for the Toxic Effects of IL4-Pseudomonas Exotoxin Chimeric Protein. Int J Cancer (1994) 58(4):574–81. doi: 10.1002/ijc.2910580421
55. Conticello C, Pedini F, Zeuner A, Patti M, Zerilli M, Stassi G, et al. IL-4 Protects Tumor Cells From anti-CD95 and Chemotherapeutic Agents Via Up-Regulation of Antiapoptotic Proteins. J Immunol (2004) 172(9):5467–77. doi: 10.4049/jimmunol.172.9.5467
56. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 Inhibits TGF-Beta-Induced Foxp3+ T Cells and, Together With TGF-beta, Generates IL-9+ Il-10+ Foxp3(-) Effector T Cells. Nat Immunol (2008) 9(12):1347–55. doi: 10.1038/ni.1677
57. Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, et al. Robust Tumor Immunity to Melanoma Mediated by Interleukin-9-Producing T Cells. Nat Med (2012) 18(8):1248–53. doi: 10.1038/nm.2856
58. Lu Y, Hong S, Li H, Park J, Hong B, Wang L, et al. Th9 Cells Promote Antitumor Immune Responses In Vivo. J Clin Invest (2012) 122(11):4160–71. doi: 10.1172/JCI65459
59. Lu Y, Hong B, Li H, Zheng Y, Zhang M, Wang S, et al. Tumor-Specific IL-9-producing Cd8+ Tc9 Cells are Superior Effector Than Type-I Cytotoxic Tc1 Cells for Adoptive Immunotherapy of Cancers. Proc Natl Acad Sci USA (2014) 111(6):2265–70. doi: 10.1073/pnas.1317431111
60. Liu L, Bi E, Ma X, Xiong W, Qian J, Ye L, et al. Enhanced CAR-T Activity Against Established Tumors by Polarizing Human T Cells to Secrete Interleukin-9. Nat Commun (2020) 11(1):5902. doi: 10.1038/s41467-020-19672-2
61. Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, et al. Th9 Cells Represent a Unique Subset of CD4(+) T Cells Endowed With the Ability to Eradicate Advanced Tumors. Cancer Cell (2018) 33(6):1048–60.e7. doi: 10.1016/j.ccell.2018.05.004
62. Vegran F, Berger H, Boidot R, Mignot G, Bruchard M, Dosset M, et al. The Transcription Factor IRF1 Dictates the IL-21-Dependent Anticancer Functions of TH9 Cells. Nat Immunol (2014) 15(8):758–66. doi: 10.1038/ni.2925
63. Ma X, Bi E, Huang C, Lu Y, Xue G, Guo X, et al. Cholesterol Negatively Regulates IL-9-producing Cd8(+) T Cell Differentiation and Antitumor Activity. J Exp Med (2018) 215(6):1555–69. doi: 10.1084/jem.20171576
64. Rosenberg SA. Il-2: The First Effective Immunotherapy for Human Cancer. J Immunol (2014) 192(12):5451–8. doi: 10.4049/jimmunol.1490019
65. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients With Metastatic Melanoma. A Preliminary Rep N Engl J Med (1988) 319(25):1676–80. doi: 10.1056/NEJM198812223192527
66. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, Hyperproliferation, Activation of Natural Killer Cells and CD8 T Cells, and Cytokine Production During First-in-Human Clinical Trial of Recombinant Human interleukin-15 in Patients With Cancer. J Clin Oncol: Off J Am Soc Clin Oncol (2015) 33(1):74–82. doi: 10.1200/JCO.2014.57.3329
67. Conlon KC, Potter EL, Pittaluga S, Lee CR, Miljkovic MD, Fleisher TA, et al. IL15 by Continuous Intravenous Infusion to Adult Patients With Solid Tumors in a Phase I Trial Induced Dramatic NK-Cell Subset Expansion. Clin Cancer Res: Off J Am Assoc Cancer Res (2019) 25(16):4945–54. doi: 10.1158/1078-0432.CCR-18-3468
68. Miller JS, Morishima C, McNeel DG, Patel MR, Kohrt HEK, Thompson JA, et al. A First-in-Human Phase I Study of Subcutaneous Outpatient Recombinant Human IL15 (rhIL15) in Adults With Advanced Solid Tumors. Clin Cancer Res: Off J Am Assoc Cancer Res (2018) 24(7):1525–35. doi: 10.1158/1078-0432.CCR-17-2451
69. Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. Treatment of 283 Consecutive Patients With Metastatic Melanoma or Renal Cell Cancer Using High-Dose Bolus Interleukin 2. Jama (1994) 271(12):907–13. doi: 10.1001/jama.271.12.907
70. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-Dose Recombinant Interleukin 2 Therapy for Patients With Metastatic Melanoma: Analysis of 270 Patients Treated Between 1985 and 1993. J Clin Oncol: Off J Am Soc Clin Oncol (1999) 17(7):2105–16. doi: 10.1200/JCO.1999.17.7.2105
71. Markley JC, Sadelain M. IL-7 and IL-21 are Superior to IL-2 and IL-15 in Promoting Human T Cell-Mediated Rejection of Systemic Lymphoma in Immunodeficient Mice. Blood (2010) 115(17):3508–19. doi: 10.1182/blood-2009-09-241398
72. Ahmadzadeh M, Rosenberg SA. IL-2 Administration Increases CD4+ CD25(Hi) Foxp3+ Regulatory T Cells in Cancer Patients. Blood (2006) 107(6):2409–14. doi: 10.1182/blood-2005-06-2399
73. Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical Mechanisms of IL-2-Regulated Fas-Mediated T Cell Apoptosis. Immunity (1998) 8(5):615–23. doi: 10.1016/S1074-7613(00)80566-X
74. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of Full Effector Function In Vitro Paradoxically Impairs the In Vivo Antitumor Efficacy of Adoptively Transferred CD8+ T Cells. J Clin Invest (2005) 115(6):1616–26. doi: 10.1172/JCI24480
75. Chen Y, Sun C, Landoni E, Metelitsa LS, Dotti G, Savoldo B. Eradication of Neuroblastoma by T Cells Redirected With an Optimized GD2-specific Chimeric Antigen Receptor and interleukin-15. Clin Cancer Res: Off J Am Assoc Cancer Res (2019) 25(9):2915–24. doi: 10.1158/2326-6074.CRICIMTEATIAACR18-A025
76. Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, et al. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells But Results in Antigen Loss Variants. Cancer Immunol Res (2017) 5(7):571–81. doi: 10.1158/2326-6066.CIR-16-0376
77. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-Specific T Lymphocytes With interleukin-15 and a Suicide Gene to Enhance Their Anti-Lymphoma/Leukemia Effects and Safety. Leukemia (2010) 24(6):1160–70. doi: 10.1038/leu.2010.75
78. Batra SA, Rathi P, Guo L, Courtney AN, Fleurence J, Balzeau J, et al. Glypican-3-Specific Car T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity Against Hepatocellular Carcinoma. Cancer Immunol Res (2020) 8(3):309–20. doi: 10.1158/2326-6066.CIR-19-0293
79. Ataca Atilla P, McKenna MK, Tashiro H, Srinivasan M, Mo F, Watanabe N, et al. Modulating TNFalpha Activity Allows Transgenic IL15-Expressing Cll-1 Car T Cells to Safely Eliminate Acute Myeloid Leukemia. J Immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-001229
80. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
81. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. Il15 Enhances Car-T Cell Antitumor Activity by Reducing Mtorc1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol Res (2019) 7(5):759–72. doi: 10.1158/2326-6066.CIR-18-0466
82. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 Augments Antitumor Activity and Promotes a Stem-Cell Memory Subset in Tumor-Specific T Cells. Proc Natl Acad Sci USA (2016) 113(48):E7788–E97. doi: 10.1073/pnas.1610544113
83. Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, et al. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-Like T Cells. Cell (2016) 164(3):365–77. doi: 10.1016/j.cell.2016.01.002
84. Sato N, Patel HJ, Waldmann TA, Tagaya Y. The IL-15/IL-15Ralpha on Cell Surfaces Enables Sustained IL-15 Activity and Contributes to the Long Survival of CD8 Memory T Cells. Proc Natl Acad Sci USA (2007) 104(2):588–93. doi: 10.1073/pnas.0610115104
85. Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous Growth and Increased Cytotoxicity of Natural Killer Cells Expressing Membrane-Bound Interleukin-15. Blood (2014) 124(7):1081–8. doi: 10.1182/blood-2014-02-556837
86. Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, et al. Cytokine-Independent Growth and Clonal Expansion of a Primary Human CD8+ T-Cell Clone Following Retroviral Transduction With the IL-15 Gene. Blood (2007) 109(12):5168–77. doi: 10.1182/blood-2006-06-029173
87. Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An Inducible Caspase 9 Safety Switch for T-Cell Therapy. Blood (2005) 105(11):4247–54. doi: 10.1182/blood-2004-11-4564
88. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. New Engl J Med (2011) 365(18):1673–83. doi: 10.1056/NEJMoa1106152
89. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT Cells With Chimeric Antigen Receptor Provide a Novel Platform for Safe and Effective Cancer Immunotherapy. Blood (2014) 124(18):2824–33. doi: 10.1182/blood-2013-11-541235
90. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. Nkt Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced in Vivo Persistence and Antitumor Activity Against Neuroblastoma. Clin Cancer Res: Off J Am Assoc Cancer Res (2019) 25(23):7126–38. doi: 10.1158/1078-0432.CCR-19-0421
91. Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. Il-15 Protects NKT Cells From Inhibition by Tumor-Associated Macrophages and Enhances Antimetastatic Activity. J Clin Invest (2012) 122(6):2221–33. doi: 10.1172/JCI59535
92. Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT Cells in Patients With Relapsed or Refractory Neuroblastoma: An Interim Analysis. Nat Med (2020) 26(11):1686–90. doi: 10.1038/s41591-020-1074-2
93. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. New Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
94. Sportes C, Babb RR, Krumlauf MC, Hakim FT, Steinberg SM, Chow CK, et al. Phase I Study of Recombinant Human Interleukin-7 Administration in Subjects With Refractory Malignancy. Clin Cancer Res: Off J Am Assoc Cancer Res (2010) 16(2):727–35. doi: 10.1158/1078-0432.CCR-09-1303
95. Sportes C, Hakim FT, Memon SA, Zhang H, Chua KS, Brown MR, et al. Administration of rhIL-7 in Humans Increases In Vivo TCR Repertoire Diversity by Preferential Expansion of Naive T Cell Subsets. JExpMed (2008) 205(7):1701–14. doi: 10.1084/jem.20071681
96. Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, et al. Interleukin-7 Mediates Selective Expansion of Tumor-Redirected Cytotoxic T Lymphocytes (Ctls) Without Enhancement of Regulatory T-cell Inhibition. Clin Cancer Res: Off J Am Assoc Cancer Res (2014) 20(1):131–9. doi: 10.1158/1078-0432.CCR-13-1016
97. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat Biotechnol (2018) 36(4):346–51. doi: 10.1038/nbt.4086
98. Li Z, Luo H, Su J, Sun R, Sun Y, Wang Y, et al. Coexpression of IL-7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors Without Requiring Preconditioned Lymphodepletion. Clin Cancer Res: Off J Am Assoc Cancer Res (2020) 26(20):5494–505. doi: 10.1158/1078-0432.CCR-20-0777
99. Park JH, Yu Q, Erman B, Appelbaum JS, Montoya-Durango D, Grimes HL, et al. Suppression of IL7Ralpha Transcription by IL-7 and Other Prosurvival Cytokines: A Novel Mechanism for Maximizing IL-7-Dependent T Cell Survival. Immunity (2004) 21(2):289–302. doi: 10.1016/j.immuni.2004.07.016
100. Vera JF, Hoyos V, Savoldo B, Quintarelli C, Giordano Attianese GM, Leen AM, et al. Genetic Manipulation of Tumor-Specific Cytotoxic T Lymphocytes to Restore Responsiveness to IL-7. MolTher (2009) 17(5):880–8. doi: 10.1038/mt.2009.34
101. Kimura MY, Pobezinsky LA, Guinter TI, Thomas J, Adams A, Park JH, et al. IL-7 Signaling Must be Intermittent, Not Continuous, During CD8(+) T Cell Homeostasis to Promote Cell Survival Instead of Cell Death. Nat Immunol (2013) 14(2):143–51. doi: 10.1038/ni.2494
102. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive Signaling From an Engineered Il7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discovery (2017) 7(11):1238–47. doi: 10.1158/2159-8290.CD-17-0538
103. Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 Confer Opposing Differentiation Programs to CD8+ T Cells for Adoptive Immunotherapy. Blood (2008) 111(11):5326–33. doi: 10.1182/blood-2007-09-113050
104. Moroz A, Eppolito C, Li Q, Tao J, Clegg CH, Shrikant PA. Il-21 Enhances and Sustains CD8+ T Cell Responses to Achieve Durable Tumor Immunity: Comparative Evaluation of IL-2, Il-15, and IL-21. J Immunol (2004) 173(2):900–9. doi: 10.4049/jimmunol.173.2.900
105. Zhao Z, Li Y, Liu W, Li X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. BioMed Res Int (2020) 2020:4795171. doi: 10.1155/2020/4795171
106. Hunter MR, Prosser ME, Mahadev V, Wang X, Aguilar B, Brown CE, et al. Chimeric Gammac Cytokine Receptors Confer Cytokine Independent Engraftment of Human T Lymphocytes. Mol Immunol (2013) 56(1-2):1–11. doi: 10.1016/j.molimm.2013.03.021
107. Golumba-Nagy V, Kuehle J, Hombach AA, Abken H. CD28-Zeta CAR T Cells Resist TGF-Beta Repression Through IL-2 Signaling, Which can Be Mimicked by an Engineered Il-7 Autocrine Loop. Mol Ther: J Am Soc Gene Ther (2018) 26(9):2218–30. doi: 10.1016/j.ymthe.2018.07.005
108. Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol Ther: J Am Soc Gene Ther (2017) 25(1):249–58. doi: 10.1016/j.ymthe.2016.10.016
109. Bajgain P, Tawinwung S, D’Elia L, Sukumaran S, Watanabe N, Hoyos V, et al. Car T Cell Therapy for Breast Cancer: Harnessing the Tumor Milieu to Drive T Cell Activation. J Immunother Cancer (2018) 6(1):34. doi: 10.1186/s40425-018-0347-5
110. Sukumaran S, Watanabe N, Bajgain P, Raja K, Mohammed S, Fisher WE, et al. Enhancing the Potency and Specificity of Engineered T Cells for Cancer Treatment. Cancer Discovery (2018) 8(8):972–87. doi: 10.1158/2159-8290.CD-17-1298
111. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, et al. Selective Expansion of Chimeric Antigen Receptor-Targeted T-cells With Potent Effector Function Using Interleukin-4. JBiolChem (2010) 285(33):25538–44. doi: 10.1074/jbc.M110.127951
112. Whilding LM, Parente-Pereira AC, Zabinski T, Davies DM, Petrovic RMG, Kao YV, et al. Targeting of Aberrant Alphavbeta6 Integrin Expression in Solid Tumors Using Chimeric Antigen Receptor-Engineered T Cells. Mol Ther: J Am Soc Gene Ther (2017) 25(10):2427. doi: 10.1016/j.ymthe.2017.09.018
113. Wang Y, Jiang H, Luo H, Sun Y, Shi B, Sun R, et al. An IL-4/21 Inverted Cytokine Receptor Improving Car-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front Immunol (2019) 10:1691. doi: 10.3389/fimmu.2019.01691
114. Weimin S, Abula A, Qianghong D, Wenguang W. Chimeric Cytokine Receptor Enhancing PSMA-CAR-T Cell-Mediated Prostate Cancer Regression. Cancer Biol Ther (2020) 21(6):570–80. doi: 10.1080/15384047.2020.1739952
115. Lo AS, Taylor JR, Farzaneh F, Kemeny DM, Dibb NJ, Maher J. Harnessing the Tumour-Derived Cytokine, CSF-1, to Co-Stimulate T-cell Growth and Activation. Mol Immunol (2008) 45(5):1276–87. doi: 10.1016/j.molimm.2007.09.010
116. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective Targeting of Engineered T Cells Using Orthogonal IL-2 Cytokine-Receptor Complexes. Science (2018) 359(6379):1037–42. doi: 10.1126/science.aar3246
117. Vignali DA, Kuchroo VK. Il-12 Family Cytokines: Immunological Playmakers. Nat Immunol (2012) 13(8):722–8. doi: 10.1038/ni.2366
118. D’Andrea A, Rengaraju M, Valiante NM, Chehimi J, Kubin M, Aste M, et al. Production of Natural Killer Cell Stimulatory Factor (Interleukin 12) by Peripheral Blood Mononuclear Cells. J Exp Med (1992) 176(5):1387–98. doi: 10.1084/jem.176.5.1387
119. Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, et al. A Functional Interleukin 12 Receptor Complex is Composed of Two Beta-Type Cytokine Receptor Subunits. Proc Natl Acad Sci USA (1996) 93(24):14002–7. doi: 10.1073/pnas.93.24.14002
120. Grohmann U, Belladonna ML, Bianchi R, Orabona C, Ayroldi E, Fioretti MC, et al. Il-12 Acts Directly on DC to Promote Nuclear Localization of NF-kappaB and Primes DC for IL-12 Production. Immunity (1998) 9(3):315–23. doi: 10.1016/S1074-7613(00)80614-7
121. Airoldi I, Gri G, Marshall JD, Corcione A, Facchetti P, Guglielmino R, et al. Expression and Function of IL-12 and IL-18 Receptors on Human Tonsillar B Cells. J Immunol (2000) 165(12):6880–8. doi: 10.4049/jimmunol.165.12.6880
122. Steding CE, Wu ST, Zhang Y, Jeng MH, Elzey BD, Kao C. The Role of interleukin-12 on Modulating Myeloid-Derived Suppressor Cells, Increasing Overall Survival and Reducing Metastasis. Immunology (2011) 133(2):221–38. doi: 10.1111/j.1365-2567.2011.03429.x
123. Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, et al. Identification and Purification of Natural Killer Cell Stimulatory Factor (NKSF), a Cytokine With Multiple Biologic Effects on Human Lymphocytes. J Exp Med (1989) 170(3):827–45. doi: 10.1084/jem.170.3.827
124. Kubin M, Kamoun M, Trinchieri G. Interleukin 12 Synergizes With B7/CD28 Interaction in Inducing Efficient Proliferation and Cytokine Production of Human T Cells. J Exp Med (1994) 180:211–22. doi: 10.1084/jem.180.1.211
125. Tannenbaum CS, Tubbs R, Armstrong D, Finke JH, Bukowski RM, Hamilton TA. The CXC Chemokines IP-10 and Mig are Necessary for IL-12-mediated Regression of the Mouse RENCA Tumor. J Immunol (1998) 161(2):927–32.
126. Dajotoy T, Andersson P, Bjartell A, Lofdahl CG, Tapper H, Egesten A. Human Eosinophils Produce the T Cell-Attracting Chemokines MIG and IP-10 Upon Stimulation With IFN-Gamma. J Leukoc Biol (2004) 76(3):685–91. doi: 10.1189/jlb.0803379
127. Chmielewski M, Kopecky C, Hombach AA, Abken H. Il-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression. Cancer Res (2011) 71(17):5697–706. doi: 10.1158/0008-5472.CAN-11-0103
128. Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. Il-12 Triggers a Programmatic Change in Dysfunctional Myeloid-Derived Cells Within Mouse Tumors. JClinInvest (2011) 121(12):4746–57. doi: 10.1172/JCI58814
129. Voest EE, Kenyon BM, O’Reilly MS, Truitt G, D’Amato RJ, Folkman J. Inhibition of Angiogenesis In Vivo by Interleukin 12. J Natl Cancer Inst (1995) 87(8):581–6. doi: 10.1093/jnci/87.8.581
130. Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, et al. New Insights Into IL-12-mediated Tumor Suppression. Cell Death Differ (2015) 22(2):237–46. doi: 10.1038/cdd.2014.134
131. Li J, Zhang L, Zhang J, Wei Y, Li K, Huang L, et al. Interleukin 23 Regulates Proliferation of Lung Cancer Cells in a Concentration-Dependent Way in Association With the interleukin-23 Receptor. Carcinogenesis (2013) 34(3):658–66. doi: 10.1093/carcin/bgs384
132. Ngiow SF, Teng MW, Smyth MJ. A Balance of interleukin-12 and -23 in Cancer. Trends Immunol (2013) 34(11):548–55. doi: 10.1016/j.it.2013.07.004
133. Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, et al. Antitumor and Antimetastatic Activity of Interleukin 12 Against Murine Tumors. J Exp Med (1993) 178(4):1223–30. doi: 10.1084/jem.178.4.1223
134. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of Single-Dose interleukin-12 Exposure on Interleukin-12-associated Toxicity and Interferon-Gamma Production. Blood (1997) 90(7):2541–8. doi: 10.1182/blood.V90.7.2541
135. Hurteau JA, Blessing JA, DeCesare SL, Creasman WT. Evaluation of Recombinant Human interleukin-12 in Patients With Recurrent or Refractory Ovarian Cancer: A Gynecologic Oncology Group Study. Gynecol Oncol (2001) 82(1):7–10. doi: 10.1006/gyno.2001.6255
136. Lenzi R, Edwards R, June C, Seiden MV, Garcia ME, Rosenblum M, et al. Phase II Study of Intraperitoneal Recombinant Interleukin-12 (rhIL-12) in Patients With Peritoneal Carcinomatosis (Residual Disease < 1 Cm) Associated With Ovarian Cancer or Primary Peritoneal Carcinoma. J Trans Med (2007) 5:66. doi: 10.1186/1479-5876-5-66
137. Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, et al. Phase I Evaluation of Intravenous Recombinant Human Interleukin 12 in Patients With Advanced Malignancies. Clin Cancer Res: Off J Am Assoc Cancer Res (1997) 3(3):409–17.
138. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. Il-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors In Vivo. Oncoimmunology (2015) 4(3):e994446. doi: 10.4161/2162402X.2014.994446
139. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T Cells Enhance Antitumor Efficacy and Overcome the Tumor Microenvironment. Sci Rep (2017) 7(1):10541. doi: 10.1038/s41598-017-10940-8
140. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-Targeted T Cells Modified to Secrete IL-12 Eradicate Systemic Tumors Without Need for Prior Conditioning. Blood (2012) 119(18):4133–41. doi: 10.1182/blood-2011-12-400044
141. O’Cearbhaill RE, Park JH, Halton EF, Diamonte CR, Mead E, Lakhman Y, et al. A Phase I Clinical Trial of Autologous Chimeric Antigen Receptor (CAR) T Cells Genetically Engineered to Secrete IL-12 and to Target the MUC16ecto Antigen in Patients (Pts) With MUC16ecto+recurrent High-Grade Serous Ovarian Cancer (HGSOC). Gynecol Oncol (2020) 159:42. doi: 10.1016/j.ygyno.2020.06.089
142. Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, et al. Il-12 Upregulates TIM-3 Expression and Induces T Cell Exhaustion in Patients With Follicular B Cell non-Hodgkin Lymphoma. J Clin Invest (2012) 122(4):1271–82. doi: 10.1172/JCI59806
143. Meyaard L, Hovenkamp E, Otto SA, Miedema F. IL-12-Induced IL-10 Production by Human T Cells as a Negative Feedback for IL-12-Induced Immune Responses. J Immunol (1996) 156(8):2776–82.
144. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving Adoptive T Cell Therapy by Targeting and Controlling IL-12 Expression to the Tumor Environment. MolTher (2011) 19(4):751–9. doi: 10.1038/mt.2010.313
145. Fan H, Walters CS, Dunston GM, Tackey R. Il-12 Plays a Significant Role in the Apoptosis of Human T Cells in the Absence of Antigenic Stimulation. Cytokine (2002) 19(3):126–37. doi: 10.1006/cyto.2002.1958
146. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local Delivery of Interleukin-12 Using T Cells Targeting VEGF Receptor-2 Eradicates Multiple Vascularized Tumors in Mice. Clin Cancer Res (2012) 18(6):1672–83. doi: 10.1158/1078-0432.CCR-11-3050
147. Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, et al. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J Immunol (2019) 203(1):198–207. doi: 10.4049/jimmunol.1800033
148. Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-Infiltrating Lymphocytes Genetically Engineered With an Inducible Gene Encoding Interleukin-12 for the Immunotherapy of Metastatic Melanoma. Clin Cancer Res: Off J Am Assoc Cancer Res (2015) 21(10):2278–88. doi: 10.1158/1078-0432.CCR-14-2085
149. Alsaieedi A, Holler A, Velica P, Bendle G, Stauss HJ. Safety and Efficacy of Tet-regulated Il-12 Expression in Cancer-Specific T Cells. Oncoimmunology (2019) 8(3):1542917. doi: 10.1080/2162402X.2018.1542917
150. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive Cellular Therapy With T Cells Expressing the Dendritic Cell Growth Factor Flt3L Drives Epitope Spreading and Antitumor Immunity. Nat Immunol (2020) 21(8):914–26. doi: 10.1038/s41590-020-0676-7
151. Ma X, Shou P, Smith C, Chen Y, Du H, Sun C, et al. Interleukin-23 Engineering Improves CAR T Cell Function in Solid Tumors. Nat Biotechnol (2020) 38(4):448–59. doi: 10.1038/s41587-019-0398-2
152. North RJ, Neubauer RH, Huang JJ, Newton RC, Loveless SE. Interleukin 1-Induced, T Cell-Mediated Regression of Immunogenic Murine Tumors. Requirement for an Adequate Level of Already Acquired Host Concomitant Immunity. J Exp Med (1988) 168(6):2031–43. doi: 10.1084/jem.168.6.2031
153. Apte RN, Voronov E. Interleukin-1–a Major Pleiotropic Cytokine in Tumor-Host Interactions. Semin Cancer Biol (2002) 12(4):277–90. doi: 10.1016/S1044-579X(02)00014-7
154. Nakao S, Kuwano T, Tsutsumi-Miyahara C, Ueda S, Kimura YN, Hamano S, et al. Infiltration of COX-2-expressing Macrophages is a Prerequisite for IL-1 Beta-Induced Neovascularization and Tumor Growth. J Clin Invest (2005) 115(11):2979–91. doi: 10.1172/JCI23298
155. Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on Tumor-Associated Macrophages Promotes Lymphangiogenesis and Metastasis Via NLRP3/IL-1beta. J Exp Med (2017) 214(9):2695–713. doi: 10.1084/jem.20160392
156. Lee PH, Yamamoto TN, Gurusamy D, Sukumar M, Yu Z, Hu-Li J, et al. Host Conditioning With IL-1beta Improves the Antitumor Function of Adoptively Transferred T Cells. J Exp Med (2019) 216(11):2619–34. doi: 10.1084/jem.20181218
157. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. Car T Cell-Induced Cytokine Release Syndrome is Mediated by Macrophages and Abated by IL-1 Blockade. Nat Med (2018) 24(6):731–8. doi: 10.1038/s41591-018-0041-7
158. van der Stegen SJ, Davies DM, Wilkie S, Foster J, Sosabowski JK, Burnet J, et al. Preclinical In Vivo Modeling of Cytokine Release Syndrome Induced by ErbB-retargeted Human T Cells: Identifying a Window of Therapeutic Opportunity? J Immunol (2013) 191(9):4589–98. doi: 10.4049/jimmunol.1301523
159. Kang JS, Bae SY, Kim HR, Kim YS, Kim DJ, Cho BJ, et al. Interleukin-18 Increases Metastasis and Immune Escape of Stomach Cancer Via the Downregulation of CD70 and Maintenance of CD44. Carcinogenesis (2009) 30(12):1987–96. doi: 10.1093/carcin/bgp158
160. Hwang KS, Cho WK, Yoo J, Seong YR, Kim BK, Kim S, et al. Adenovirus-Mediated interleukin-18 Mutant In Vivo Gene Transfer Inhibits Tumor Growth Through the Induction of T Cell Immunity and Activation of Natural Killer Cell Cytotoxicity. Cancer Gene Ther (2004) 11(6):397–407. doi: 10.1038/sj.cgt.7700711
161. Tsutsui H, Nakanishi K, Matsui K, Higashino K, Okamura H, Miyazawa Y, et al. IFN-Gamma-Inducing Factor Up-Regulates Fas Ligand-Mediated Cytotoxic Activity of Murine Natural Killer Cell Clones. J Immunol (1996) 157(9):3967–73.
162. Hoshino T, Wiltrout RH, Young HA. Il-18 is a Potent Coinducer of IL-13 in NK and T Cells: A New Potential Role for IL-18 in Modulating the Immune Response. J Immunol (1999) 162(9):5070–7.
163. Yoshimoto T, Mizutani H, Tsutsui H, Noben-Trauth N, Yamanaka K, Tanaka M, et al. Il-18 Induction of IgE: Dependence on CD4+ T Cells, IL-4 and STAT6. Nat Immunol (2000) 1(2):132–7. doi: 10.1038/77811
164. Robertson MJ, Kirkwood JM, Logan TF, Koch KM, Kathman S, Kirby LC, et al. A Dose-Escalation Study of Recombinant Human interleukin-18 Using Two Different Schedules of Administration in Patients With Cancer. Clin Cancer Res: Off J Am Assoc Cancer Res (2008) 14(11):3462–9. doi: 10.1158/1078-0432.CCR-07-4740
165. Tarhini AA, Millward M, Mainwaring P, Kefford R, Logan T, Pavlick A, et al. A Phase 2, Randomized Study of SB-485232, rhIL-18, in Patients With Previously Untreated Metastatic Melanoma. Cancer (2009) 115(4):859–68. doi: 10.1002/cncr.24100
166. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of Antitumor Immunity by Human and Mouse Car T Cells Secreting Il-18. Cell Rep (2017) 20(13):3025–33. doi: 10.1016/j.celrep.2017.09.002
167. Chmielewski M, Abken H. Car T Cells Releasing Il-18 Convert to T-Bet(high) Foxo1(Low) Effectors That Exhibit Augmented Activity Against Advanced Solid Tumors. Cell Rep (2017) 21(11):3205–19. doi: 10.1016/j.celrep.2017.11.063
168. Zimmermann K, Kuehle J, Dragon AC, Galla M, Kloth C, Rudek LS, et al. Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-Gd2 CAR Expression and Inducible Cytokines. Cancers (Basel) (2020) 12(2). doi: 10.3390/cancers12020375
169. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and Through Activation of the Endogenous Immune System. Cell Rep (2018) 23(7):2130–41. doi: 10.1016/j.celrep.2018.04.051
170. Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, Rosen CE, et al. Il-18BP is a Secreted Immune Checkpoint and Barrier to IL-18 Immunotherapy. Nature (2020) 583(7817):609–14. doi: 10.1038/s41586-020-2422-6
171. Lange S, Sand LG, Bell M, Patil SL, Langfitt D, Gottschalk S. A Chimeric GM-CSF/IL18 Receptor to Sustain CAR T-Cell Function. Cancer Discov (2021). doi: 10.1158/2159-8290.CD-20-0896
172. Wang X, Zhao X, Feng C, Weinstein A, Xia R, Wen W, et al. IL-36gamma Transforms the Tumor Microenvironment and Promotes Type 1 Lymphocyte-Mediated Antitumor Immune Responses. Cancer Cell (2015) 28(3):296–306. doi: 10.1016/j.ccell.2015.07.014
173. Zhao X, Chen X, Shen X, Tang P, Chen C, Zhu Q, et al. IL-36beta Promotes Cd8(+) T Cell Activation and Antitumor Immune Responses by Activating Mtorc1. Front Immunol (2019) 10:1803. doi: 10.3389/fimmu.2019.01803
174. Bassoy EY, Towne JE, Gabay C. Regulation and Function of interleukin-36 Cytokines. Immunol Rev (2018) 281(1):169–78. doi: 10.1111/imr.12610
175. Li X, Daniyan AF, Lopez AV, Purdon TJ, Brentjens RJ. Cytokine IL-36gamma Improves CAR T-Cell Functionality and Induces Endogenous Antitumor Response. Leukemia (2020) 35(2):506–21. doi: 10.1038/s41375-020-0874-1
176. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome After CD19 Chimeric Antigen Receptor-Modified T-cell Therapy. Blood (2017) 130(21):2295–306. doi: 10.1182/blood-2017-06-793141
Keywords: CAR T cells, immunotherapy, T Cell therapy, cytokine receptors, cancer, cytokines
Citation: Bell M and Gottschalk S (2021) Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 12:684642. doi: 10.3389/fimmu.2021.684642
Received: 23 March 2021; Accepted: 11 May 2021;
Published: 04 June 2021.
Edited by:
Luis Alvarez-Vallina, Aarhus University, DenmarkReviewed by:
John Maher, King’s College London, United KingdomAvery Dexter Posey, Jr., University of Pennsylvania, United States
Copyright © 2021 Bell and Gottschalk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen Gottschalk, c3RlcGhlbi5nb3R0c2NoYWxrQHN0anVkZS5vcmc=