
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 May 2021
Sec. Inflammation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.682871
 Anna Kosyreva1,2*†
Anna Kosyreva1,2*† Dzhuliia Dzhalilova3†
Dzhuliia Dzhalilova3† Anastasia Lokhonina2,4Polina Vishnyakova2,4†
Anastasia Lokhonina2,4Polina Vishnyakova2,4† Timur Fatkhudinov2,5
Timur Fatkhudinov2,5Macrophages are cells that mediate both innate and adaptive immunity reactions, playing a major role in both physiological and pathological processes. Systemic SARS-CoV-2-associated complications include acute respiratory distress syndrome (ARDS), disseminated intravascular coagulation syndrome, edema, and pneumonia. These are predominantly effects of massive macrophage activation that collectively can be defined as macrophage activation syndrome. In this review we focus on the role of macrophages in COVID-19, as pathogenesis of the new coronavirus infection, especially in cases complicated by ARDS, largely depends on macrophage phenotypes and functionalities. We describe participation of monocytes, monocyte-derived and resident lung macrophages in SARS-CoV-2-associated ARDS and discuss possible utility of cell therapies for its treatment, notably the use of reprogrammed macrophages with stable pro- or anti-inflammatory phenotypes.
Since its initial outbreak in Wuhan (China) in 2019, the novel coronavirus infection has rapidly progressed into pandemic. The situation required massive counter measures with distinct signs of global emergency and drastic restrictions in human activities. Under the lack of efficient antivirals, fatality rates for the coronavirus disease of 2019 (COVID-19) are disastrously high. The main efforts in COVID-19 patient management are aimed at symptomatic and pathogenetic treatment, and effective new methods of dealing with the consequences are in great demand. In severe cases of COVID-19, the virus promotes cytokine storm with uncontrolled massive release of pro-inflammatory cytokines leading to acute respiratory distress syndrome (ARDS) and acute heart failure; these conditions are highly life-threatening and fraught with acquisition of secondary bacterial infections. Prevention of the cytokine storm in SARS-CoV-2-infected individuals remains extremely relevant, and safe means of such prevention stay among primary goals of medical research worldwide. As demonstrated, ARDS can be safely managed by cell therapy including the use of mesenchymal stromal cells (MSCs); the option can be enhanced by in vitro reprogramming of MSCs into anti-inflammatory macrophages (M2 macrophages) prior to administration. This review elucidates the role of macrophages in ARDS pathogenesis with a special focus on SARS-CoV-2-associated ARDS (COVID-19-ARDS). Identification of cellular and molecular mechanisms related to the role of macrophages in ARDS is highly relevant for the development of potential treatment strategies for COVID-19.
Severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) belongs to Coronaviridae family. Its particles contain single-stranded (+) RNA enclosed within a complex assembly of proteins responsible for entering a host cell and viral replication, including the membrane (M), envelope (E), nucleocapsid (N), and spike (S) proteins (1); S contains a receptor-binding domain that specifically recognizes angiotensin-converting enzyme 2 (ACE2) as its receptor; that is essential for penetration of the virus into alveolar epithelial cells (pneumocytes) type II which express ACE2 (2, 3). The interaction of S protein with ACE2 in lung epithelium promotes local enforcement of angiotensin II production and increase in bradykinin levels, leading to a surge of inflammatory response (4).
Moreover, the binding of SARS-CoV-2 S protein to ACE2 promotes formation of multinucleated syncytia (4, 5). S protein at the surface of infected cells effectively promotes their fusion with neighboring cells; the resulting syncytia may incorporate epithelial and myeloid cells (6). Although formation of syncytia in the lungs of patients with SARS-CoV-2 has been confirmed, its role in viral persistence and dissemination needs to be specified.
Clinical manifestations of COVID-19 begin on day 2–14 after a contact with infected carrier (day 5 on average) (7). The infection may lead to lethal outcome on day 6–41 after the first symptoms (day 14 on average). Duration, severity and lethality of the disease depend on the patient’s age and immune status (8, 9), the presence of chronic comorbidities (notably diabetes mellitus, chronic hypertension with vascular failure, chronic inflammatory conditions of the lungs, and malignant neoplasms) (10), genetic features, obesity status, and glucose levels (11, 12). For instance, in children with the age-related physiologically strong innate immunity clinical symptoms of COVID-19 are less pronounced than in adults (10). Severe obesity (BMI ≥ 35 kg/m2) has been associated with intensive care unit admission, whereas histories of heart disease and obesity (BMI ≥ 30 kg/m2) have been independently associated with the use of invasive mechanical ventilation (11).
Clinical manifestations of COVID-19 vary dramatically from asymptomatic carriage to acute respiratory distress syndrome; typical symptoms include fever, dry cough, breath shortness, myalgia, and fatigue (8, 13, 14). Severe coronavirus-associated pneumonia results from rapid replication of the virus in epithelial cells with their death accompanied by massive infiltration of macrophages and neutrophils to intra-alveolar septa and alveolar lumina. Extremely high production of pro-inflammatory mediators by the infiltrating immune cells leads to acute lung injury (ALI) and ARDS (15, 16). The pronounced inflammatory response in coronavirus infections can play both protective and destructive roles (15–17). Activation of immune cells provides elimination of the viruses but its excessive infiltration and functioning can lead to tissues injury. Massive release of pro-inflammatory mediators by infiltrating immune cells combined a direct cytotoxic effect of the virus on epithelial cells result in lung injury in SARS-CoV-infected patients (18).
SARS-CoV-2 is similar to other coronaviruses, such as SARS-CoV-1 and MERS-CoV (19, 20). Similarly with the fatal cases of SARS-CoV-1 and MERS-CoV infections, lung autopsies of patients who died of COVID-19 reveal extensive cellular infiltration with the predominance of macrophages. High levels of IFNγ, IL-6, IL-12, TGFβ, CCL2, CXCL9, CXCL10 and IL-8 are typical for both SARS-CoV-2- and SARS-CoV-1-associated fatal cases, with higher levels of IL-1β and lower levels of IL-10 for SARS-CoV-1.
For SARS and MERS coronaviruses, severity of the respiratory syndrome positively correlates with macrophage numbers (21, 22). Viral proteins obtained from SARS-CoV-1 stimulate the release of proinflammatory cytokines by peripheral blood monocytes (23). Macrophages and monocytes of peripheral blood can be activated by SARS-CoV-1 and recruit neutrophils and immune effectors (NK-, T-, and B-cells) to form early adaptive immunity (24, 25).
Given the important role of macrophages in SARS-CoV-2-induced immune responses, targeted modulation of their functional activity may provide a new approach for the management of patients with severe COVID-19.
Acute lung injury (ALI) rapidly progressing into ARDS is the major cause of high fatality in pneumonias, including SARS-CoV-2-associated pneumonia (2, 26, 27). Transition of ALI into ARDS results from diffuse alveolar damage (28), usually a consequence of excessive and uncontrolled systemic inflammatory reactions in response to lung injury caused by trauma, burn, pneumonia, sepsis, etc., violating endothelial and epithelial barriers (29). COVID-19-associated patterns of diffuse alveolar damage are typical for ARDS (30–32). ARDS develops in 42% of COVID-19 patients with pneumonia, or in 61–81% of intensive care patients with COVID-19 (33). The overwhelming prevalence of ARDS (90%) among fatal complications of COVID-19 was demonstrated on a sample of 2000 autopsies; other fatal complications included pulmonary embolism (6%) and sepsis (1.5%) (31). It should be noted that the prognosis for SARS-CoV-2-associated ARDS (COVID-19-ARDS) is worse than for ARDS of other genesis. In-hospital mortality for ARDS in general does not exceed 35.3% (33); for COVID-19-ARDS, it has been estimated 62% and reaches 65.7–94% for intensive care patients on ventilators. Risk factors for unfavorable prognosis include old age, the presence of comorbidities (hypertension, cardiovascular diseases, diabetes, renal failure), as well as reduced lymphocyte counts and elevated levels of D-dimer. Death from COVID-19-ARDS occurs mostly from respiratory failure either by itself (53%) or accompanied by cardiac failure (33%), and much less frequently from myocardial injury and/or circulatory failure (7%) (33).
At the initial stages of ALI, immune response is triggered by activation of antigen-presenting cells, including macrophages and dendritic cells, which facilitates production of pro-inflammatory cytokines (notably TNFα, IL-1, and IL-6), as well as prostaglandins and histamine (34, 35). These substances boost endothelial permeability and facilitate homing/migration of neutrophils, macrophages, and lymphocytes to the focus of inflammation, which exacerbates the injury and promotes ARDS. Accumulation of neutrophils in blood capillaries and inter-alveolar septa leads to intra-alveolar edema and avalanche ROS production which contribute to hypoxemia and critical deterioration of lung function (36). The increased permeability of microcirculatory vessels obliterates the air-blood barrier while promoting hemorrhages and fibrin deposition (37).
Analysis of the aforementioned sample of 2000 autopsies revealed similar pathological alterations in the affected lung tissues, differing only by the degree of expansion. These alterations can be described as diffuse alveolar damage with underlying microcirculatory failure (microangiopathy, thrombosis, and occasionally the destructive-productive vasculitis) and pulmonary hemorrhage. Such interstitial viral pneumonia with a pronounced vascular and hemorrhagic component represents the invariant morphological substrate of COVID-19-ARDS (31).
ARDS pathogenesis proceeds in several phases, most commonly defined as exudative phase, proliferative (rehabilitation) phase, and fibrotic (fibroproliferative) phase (38–40).
Exudative phase of ARDS is distinguished by diffuse alveolar damage with massive death of epithelial and endothelial cells. COVID-19 predominantly affects the respiratory system with only minor damage to other organs; it is assumed that SARS-CoV-2 has a negative effect on alveolar epithelial cells (pneumocytes) and endothelial cells (41). The massive death of pneumocytes severely violates gas exchange (42); concomitant damage to endothelium with the resulting increase in capillary permeability lead to perivascular and intra-alveolar edema with transudation of plasma proteins, notably fibrinogen B, to the alveoli. Early stages of ARDS pathogenesis are marked with massive activation and immigration of circulating neutrophils (43, 44). Edema-associated damage to pneumocytes type II results in decreased production of surfactant and increased production of inflammation mediators (e.g. pro-inflammatory cytokines IL-1β, IL-6, and IL-18) which stimulate homing of macrophages and neutrophils to the lungs by chemotaxis (45, 46). Endothelial dysfunction and disrupted oxygenation of pulmonary tissues leads to avalanche ROS production (47). Bacterial invasion, which is possible already in this phase, may lead to development of bacterial pneumonia and sepsis. Formation of hyaline membranes in the alveoli (indicative of ARDS) proceeds throughout this phase (5); the presence of multinucleated giant cells comprising markers of both macrophages and pneumocytes (48) indicates the scavenging involvement of macrophages.
Exudative phase of ARDS ends with the transition to proliferative (rehabilitation) phase marked by facile regeneration of pneumocytes and endothelium under conditions of suppressed pro-inflammatory cytokine production (49, 50). As neutrophils die massively by efferocytosis, macrophages accomplish debridement of the site by phagocytosis and their capacity for production of anti-inflammatory factors (e.g. IL-10 and TGF-β) increases (50). Phagocytosis is competitive with iNOS expression and stimulates Arg-1 expression thus inhibiting ROS production by macrophages (49).
The processes of prolonged recovery of lung tissue after infection are also considered a phase of ARDS, as 30–50% of adult patients with ARDS develop a full-scale inflammatory response leading to profound histoarchitectural alterations, development of fibrosing alveolitis with cystic restructuring, and overall deterioration of pulmonary function accompanied by abnormal activation of macrophages, fibrocytes, and fibroblasts (29, 51). Signs of pulmonary fibrosis can be clearly detected by computer tomography in about 17% of patients with confirmed COVID-19; additional fibrotic lesions with gradual replacement of epithelial cells by collagen and extracellular matrix (ECM) components can be formed during the post-pneumonia recovery (52, 53). At present, the true relationship between pronounced pulmonary fibrosis and prognosis in COVID-19 patients is debatable. Some evidence (52) associates the presence of fibrosis with favorable prognosis and stabilization of the patient’s condition. Nevertheless, the outcome of the inflammatory process certainly depends on the prevalence and progression of pulmonary fibrosis (54, 55).
Pulmonary fibrosis (sometimes marked as a delayed complication of ARDS, not a fibrotic phase of ARDS itself) results from the excessive proliferation of fibroblasts and excessive production of ECM components by these fibroblasts (39, 56). At this stage, patients need long-term respiratory support by mechanical ventilation, and the mortality rates are extremely high (56). At the tissue level, this phase is marked by release of various pro-fibrotic molecules (e.g. TGF-α, TGF-β, IL-1β, and the platelet-derived growth factor) which promote excessive deposition of fibronectin, collagens, and other ECM components (57). The fibro-proliferative response is facilitated by elevated expression of pro-angiogenic cytokines and growth factors by immune cells (e.g. MIP-2, angiopoietin 2, and VEGF) (58–61). Mechanical ventilation may exacerbate fibrosis progression by damaging the epithelium and interfering with surfactant production, ultimately promoting the ventilator-associated lung injury (62–65).
As macrophage participation is essential for every phase of ARDS development, these cells represent a potential target/tool for ARDS therapy.
Macrophages are cells mediating both innate and adaptive immunity; they play major roles in various physiological and pathological processes, and can be involved in tissue injury as either injury-inducing or repair-promoting. Exemplary recruited macrophage precursors, blood monocytes, originate in the bone marrow and constitute about 5–9% of peripheral blood leukocytes. Monocytes reside in circulation for 1–2 days; by the end of that period, monocytes either die by apoptosis or relocate from circulation into tissues where they further differentiate into macrophages (66). Resident macrophages such as alveolar macrophages (AMs), Kupffer cells, microglial cells, Langerhans cell, macrophages of splenic red pulp and peritoneal macrophages originate prenatally from the yolk sac and embryonic liver (67, 68).
In healthy humans, blood monocytes comprise three subpopulations including classical (CD14++CD16−, about 90%), non-classical (CD14+CD16++, about 5–10%), and intermediate monocytes (CD14++CD16+, the smallest subpopulation of 1–5%) (69). These subpopulations are distinguished by expression levels of chemokine receptors, as well as by phagocytic capacity and distribution in particular tissues under physiological conditions and/or in the course of inflammatory processes. CD14++ monocytes are considered mature; they have pronounced phagocytic activity and can produce reactive oxygen species (ROS) and cytokines upon Toll-like receptor (TLR) activation (70). CD16++ cells are engaged in synthesis of cytokines (predominantly pro-inflammatory) and do not produce ROS (69, 70). The role of intermediate subpopulation is unclear; given the high expression level of MHC-II on these monocytes, they are suggested involved in antigen presentation and T cell activation (71).
Some additional monocyte phenotypes have been characterized in recent studies based on gene and protein expression patterns (72–74). Under pathological conditions, the ratio between different monocyte subpopulations may be shifted; increased severity of inflammatory diseases is usually associated with increased proportions of non-classical and intermediate (CD16+) monocytes (75, 76).
Under physiological conditions, a considerable proportion of circulating monocytes spontaneously (under weak chemokine signaling) migrate into tissues and differentiate into resident macrophages which implement homeostatic and regulatory functions by scavenging dead cells and carrying out immune surveillance (77, 78). Under inflammatory conditions, monocytes become strongly attracted to the focus of inflammation where they differentiate into pro-inflammatory macrophages with enormously enhanced phagocytic and bactericidal activity. In comparison with the resident macrophages that survive in tissues for years, these pro-inflammatory macrophages are transient (survive for few weeks at most).
As a cell type, macrophages show high plasticity, i.e. they are capable of switching phenotypes (79, 80). The process of switching phenotypes and developing specific reactions to microenvironment by macrophages is called ‘polarization’. One of important factors directing macrophage polarization is local cytokine levels. Peripheral tissues in mammals (including humans, mice, and rats) were shown to comprise two functional subtypes of macrophages (Figure 1): the ‘classically’ activated pro-inflammatory macrophages (commonly designated M1) and the ‘alternatively’ activated anti-inflammatory macrophages (commonly designated M2); the distinction has been based on expression of specific markers and production of biologically active substances (81–86). M1 polarization is usually a response to pro-inflammatory cytokines, e.g. IFN-γ and TNFα, as well as pathogen-associated stimuli, e.g. lipopolysaccharide (LPS). M1 macrophages produce TNFα, IL-1, IL-6, IL-12, and IL-23, as well as chemokine CCL8, monocyte chemotactic protein 1 (MCP-1), macrophage inflammatory protein 2 (MIP-2), ROS, cyclooxygenase 2 (COX-2), CD16, and CD32. Expression of inducible NO-synthase (iNOS) by macrophages is a marker of M1 polarization in rodents. M1 macrophages are engaged in developing pro-inflammatory reactions, chemotaxis, production of free radicals, degradation of extracellular matrix, and antimicrobial and antitumor activities (38, 87).
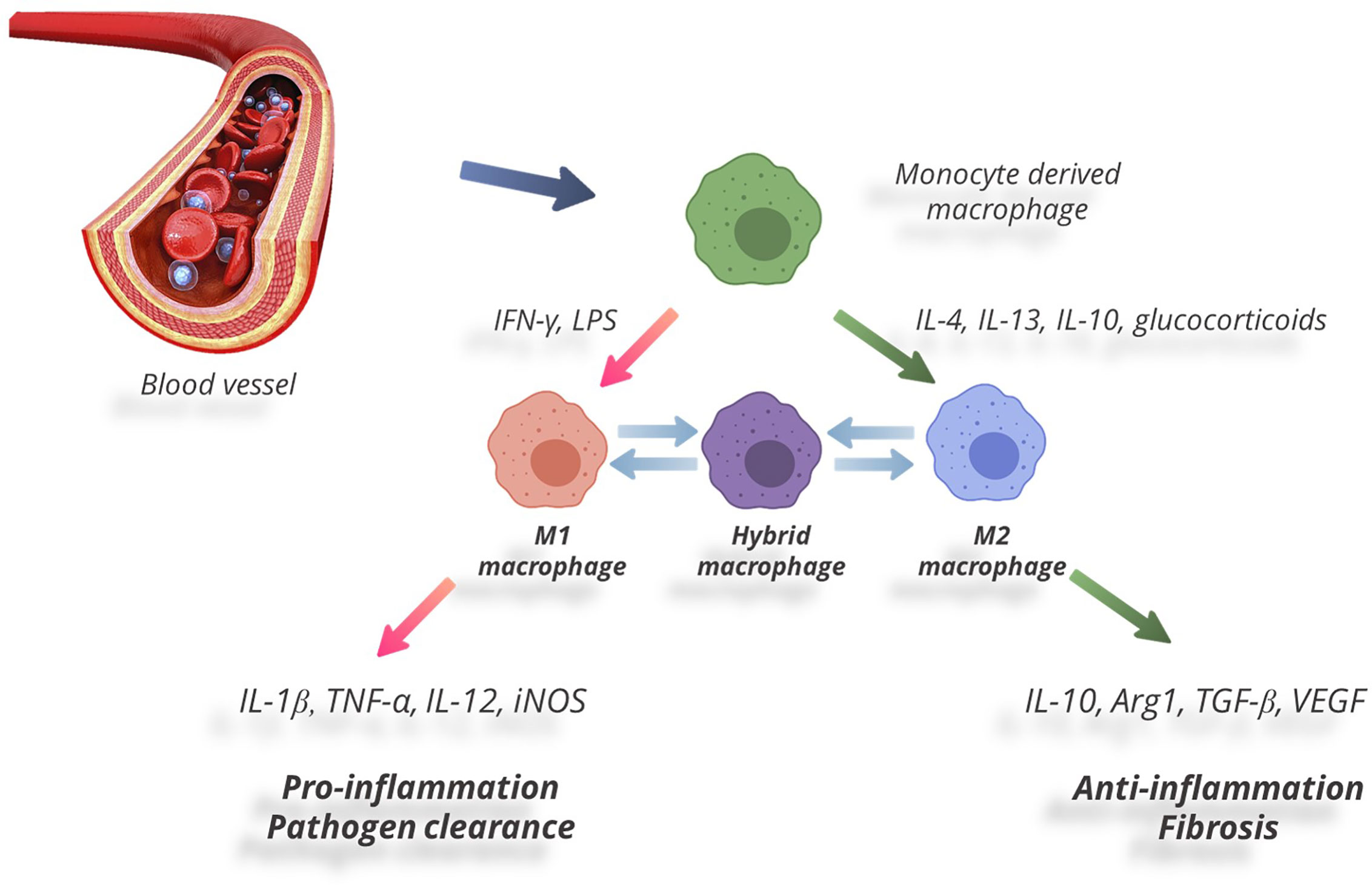
Figure 1 Two functional subtypes of macrophages.
The opposite type of macrophage polarization, M2, occurs in response to Th2 cytokines (IL-4, IL-13) and anti-inflammatory cytokines — notably IL-10 and transforming growth factor beta (TGF-β) (88). M2 macrophages are distinguished by high expression levels of IL-1 receptor antagonist (IL-1RA), arginase 1 (Arg-1), IL-10, TGF-β, CCL18, and low production of IL-12. M2 macrophages reduce inflammation by suppressing effector T cells; they promote wound healing and tissue repair by producing growth factors (VEGF, EGF, PDGF, IL-10). They also participate in Th2-mediated reactions and extracellular matrix remodeling (88–92). Under pathological conditions a shift in M1/M2 balance towards the anti-inflammatory М2 phenotype has been shown to facilitate regeneration and repair. Positive effects of M2 macrophages were demonstrated under conditions of injury, including skin wound healing in mice with diabetes (93), experimental spinal damage (94–96), cardiomyopathy in mice (97), prevention of atherosclerotic lesions in experimental models (98), and management of myocardial infarction in patients with diabetes (99, 100).
The M1/M2 paradigm has been lately criticized as too primitive. Recent data demonstrate that macrophages tend to present with a continuum of phenotypes even in the absence of inducing stimuli (101). The continuous nature of macrophage plasticity manifests itself under various physiological and pathological conditions (102). The switching between pro- and anti-inflammatory states has been reviewed in depth elsewhere (102, 103), with states of indistinct functionality lacking clear M1 or M2 markers conventionally classified as hybrid phenotypes (102, 104) (Figure 1). Despite the widespread opinion that the M1/M2 paradigm reflects in vitro extremization of the in vivo conditions, it still provides a good framework for many experimental findings (105–107).
As the major cytokine-producing and antigen-presenting cells, macrophages control effector T-cell homeostasis, promote T-cell priming, and induce Th17 cell differentiation (108). In total, macrophages mediate innate immune responses directly and make a crucial contribution to the effector phase of adaptive immune response (109). Infection of macrophages by SARS-CoV-2 impairs adaptive immune responses against the virus (110).
Two distinct populations of lung macrophages (LMs) are both of mixed origin, derived prenatally from the yolk sac and fetal liver and postnatally from the bone marrow (111, 112). The first and most abundant of them, alveolar macrophages (AMs), is associated with the epithelial walls of alveoli (113).
AMs express peroxisome proliferator-activated receptor gamma (PPARγ, a regulator of lipid metabolism) under the action of granulocyte-macrophage colony-stimulating factor (GM-CSF) (114); their main function is phagocytosis and surfactant catabolism (115). AMs are further divided into two subpopulations by their functional state and sources of origin: long-lived resident AMs and recruited AMs (39). The long-lived resident AMs are uniform, quiescent, and immunosuppressive; they mainly present with M2 phenotypes (39, 116). Resident AMs are sentinels at the tissue/air interface; they implement immune surveillance and maintain tissue homeostasis (38, 116, 117). They are counterbalanced by peripheral blood monocytes recruited from circulation to the alveoli in response to certain stimuli (39); these monocytes differentiate into ‘recruited’ M1 AMs (117). Accumulating evidence suggests that both resident and recruited AMs are deeply involved in ARDS (39, 40).
The second population comprises interstitial macrophages (IMs), which account for 30–40% of LMs (113). In contrast to AMs, which differentiate from circulating monocytes during inflammation only (118), IMs can be classified as resident monocyte-derived macrophages (113). IMs have been associated with tissue remodeling and repair, antigen presentation, and modulation of dendritic cell functions (113, 119).
Recently, a new IM subpopulation of Nerve- and Airway-associated Macrophages (NAMs) has been characterized in mice (120). NAMs are distinct from other lung-resident macrophage subsets; they express higher levels of MHCII, Cx3cr1, C1q, C3ar1, and F13a1 and lower levels of Lyve1 (Lymphatic vessel endothelial hyaluronan receptor 1) than other macrophage populations (120). Involved in maintenance of immune and tissue homeostasis, NAMs also regulate infection-induced inflammation through secretion of immunosuppressive factors such as IL-10.
A minor population of LMs is constituted by intravascular macrophages (111). As shown by Evren et al. (121), lung vasculature contains not only patrolling CD14loCD16+ and extravasating CD14+ monocytes but also intravascular macrophages. Despite populating the same anatomical niche, intravascular macrophages are distinct from blood monocytes in terms of morphology, phenotype, and gene expression. Intravascular LMs are functionally related to red pulp macrophages of the spleen and Kupffer cells of the liver; they preferentially express genes involved in red blood cell turnover and iron metabolism (CD163, HMOX1, FTL, SLC40A1, SLC48A1) and genes related to leukocyte-endothelium adhesion (121).
As other tissue macrophages both AMs and IMs can be divided into two functional phenotypes, that is microenvironment-depended: M1 macrophage and M2 macrophage (122, 123). Mitsi et al. (124) demonstrated that resident AMs have mostly hybrid phenotypes, i.e. express M1 and M2 markers simultaneously, and that such hybrid phenotypes of resident AMs are stable in human populations independently of geographic habitat. Specht at al (101). used single-cell proteomic analysis to demonstrate co-expression of M1 and M2 signatures in cultured AMs in the absence of polarizing stimuli. The existence of stable hybrid M1/M2 phenotypes is clearly beyond the conventional (original) views on macrophage polarization. Such hybrid phenotypes may enable rapid adaptation of resident AMs to the demands of their volatile microenvironment by switching between M1-and M2-specific functions in response to particular stimuli.
Macrophages and monocytes are the first immune cells to interact with viral pathogens; they can be infected and may become instrumental for spreading of the virus (125). They can contribute to virus dissemination by interaction with other populations of cells through direct cell-to-cell contacts. Macrophages and monocytes actively migrate to the sites of damage while producing chemokines and cytokines that play important roles in immunity reactions and inflammatory processes. Such behaviors can be employed by viruses for spreading throughout the body to various tissues and organs including the brain. The penetration of infected monocytes through blood-tissue barriers, notably the ‘Trojan horse’ delivery of viral particles into the central nervous system, is common for human immunodeficiency virus (HIV) (126–128), human herpes virus (HCV) (129), human cytomegalovirus (HCMV) (125) and Japanese encephalitis virus (JEV) (130) infections. It provides stable viral pools capable of sudden reinforcement upon optimal conditions for their reactivation.
Viruses have to come over the particular barriers to interact with macrophage or monocyte in order to infect and replicate in these cells. Based on inner genetic program, in three days of circulation in blood flow, monocytes have to either follow the apoptosis pathway or differentiate into tissue macrophages (125). In such case, viruses demonstrate their ultimate potential to modulate and moderate cells to avoid degradation and instead replicate in the organism effectively. They decline apoptosis route and prolong cell’s life span thanks to apoptotic pathways regulation (NFkB and PI3K), which suggest microRNAs involvement (131, 132) and the mitochondrial pathway modulation (133). They provide cell polarization changes, disguise viral receptors from cell surface and effect on cytokine/chemokine expression in order to elude from the immune response. They also provide the influence on cells motility in order to make the viral distribution through the body. This can facilitate the disorders, chronic inflammation and further predisposition to the cognitive pathologies, multiple sclerosis, inflammatory demyelinating disease, precancerous conditions and cancer (134).
Although macrophages and monocytes of peripheral blood are capable of delivering viruses to organs and tissues (135), it has been shown that these cells are infected by SARS-CoV-1 non-productively, i.e. the virion RNA is synthesized, but the virus is produced non- or almost non-infectious (136). Nevertheless, the macrophage/monocyte responses are central to SARS pathogenesis (137, 138). It should be noted that, while SARS-CoV-1 infection in mononuclear phagocytes is abortive, MERS-CoV can replicate in monocytes, macrophages, and dendritic cells (16).
Abrupt and rapidly progressing clinical manifestations of COVID-19, peaking at 7–10 days after the onset, have been identified with the long-known phenomenon of cytokine storm (139). The systemic reaction, which results from massive macrophage activation and has been accordingly designated ‘macrophage activation syndrome’ (MAS), plays the central role in SARS-CoV-2-associated complications including ARDS, disseminated intravascular coagulation syndrome (DICS), edema, and pneumonia (2, 26, 27, 140). As demonstrated by Huang et al. (141), COVID-19 patients (notably those admitted to intensive care units) have elevated blood levels of IL-1β, IL-2, IL-7, IL-9, IL-10, IL-17, G-CSF, GM-CSF, IFN-γ, TNF-α, CXCL8, CXCL10, MCP1, MIP1A, and MIP1B. Although blood counts of monocytes/macrophages in COVID-19 patients often stay within the reference range, proportion of activated cells among them is dramatically elevated.
Although studies devoted to the role of macrophages in COVID-19 are few, considerable efforts have been focused on phenotypes and counts of blood monocytes. Zhou et al. (142) studied a cohort of 33 hospitalized patients diagnosed with COVID-19 and observed that the counts of CD16+ monocytes (including non-classical CD14+CD16++ and intermediate pro-inflammatory CD14++CD16+ subpopulations with high expression of IL-6) were significantly increased, especially in patients who developed ARDS. Specifically, relative counts of intermediate monocytes in patients with severe COVID-19 were increased to 45% of all blood monocytes (compared with 1–5% in healthy individuals). This finding highlights the important role of monocytes in generalization of the inflammatory reactions inflicted by the new coronavirus infection.
Zhang et al. (143) performed a detailed study of expression profiles for the intermediate pro-inflammatory CD14++CD16+ subpopulation of blood monocytes in a cohort of 28 COVID-19 patients. The authors observed expression of both pro-inflammatory (CD80+) and anti-inflammatory (CD163+, CD206+) differentiation markers by these cells, as well as their positivity for pan-macrophage marker CD68+. These results indicate that, under given conditions of acute progression of the disease, monocytes differentiate into macrophages while still in the circulation. This newly identified population of circulating monocytes/macrophages produces IL-6, TNFα, and IL-10 at high levels, which could effectively promote the cytokine storm. The counts of such cells were the highest in hospitalized patients with severe COVID-19 and positively correlated with the length of stay in intensive care units. Consistently with other studies, the counts of non-classical and intermediate monocytes in COVID-19 patients were heavily increased, against a decrease in the classical monocyte counts (143). In monocytes, SARS-CoV-2 stimulated the default transcriptional program (enriched with M2-type genes), as CD163 expression in SARS-CoV-2-infected monocytes was significantly higher, whereas HLA-DR expression was decreased, as compared with non-infected controls (144).
As stated before, viral particles of SARS-CoV-2 provide no productive infection in mononuclear phagocytes (144). Moreover, Liu et al. (145) revealed no significant differences in surface expression of CD80, CD86, CD72, and PD-L1 on monocytes between COVID-19-convalescent individuals and matched SARS-CoV-2-unexposed individuals, indicating the lack of long-term impact of SARS-CoV-2 on monocytes (145). However, another study demonstrates that SARS-CoV2 triggers mitochondrial ROS production in monocytes, which induces stabilization of hypoxia-inducible factor-1α (HIF-1α) and consequently promotes glycolysis (12). The elevated glucose levels enhance SARS-CoV-2 replication in monocytes and induce cytokine expression, thus directly inhibiting T cell response and reducing epithelial cell survival (12).
Immune scoring of COVID-19 lung biopsies reveal massive myeloid infiltration, specifically by monocytes, M1 macrophages, and neutrophils (146), which confirms the predominance of these cells in severe COVID-19. Insignificant lymphoid scores for such samples suggest a submerged status of adaptive immunity in severe COVID-19 (147). By using single-cell transcriptomic analysis, Shaath et al. (148) show that bronchoalveolar lavage fluid (BALF) macrophages of patients with severe COVID-19 display pro-inflammatory gene expression signatures; the functional categories include inflammatory response and chemotaxis of myeloid cells, phagocytes, and granulocytes.
Liao et al. (149) used the emerging single-cell RNA sequencing (scRNA-seq) and single-cell TCR sequencing to characterize BALF cells isolated from COVID-19 patients. The results indicate that monocyte-derived macrophages, which replace AMs and predominate among macrophage lineages in the severely damaged lung, are highly inflammatory and potent chemokine producers. Massive clonal expansion of CD8+ T cells in the lungs of patients with mild course of the disease is consistent with the notion on critical importance of rapid and robust adaptive immune response for the control of COVID-19. The leading role of adaptive immunity in COVID-19 is also emphasized in a recent study by Grant (150) demonstrating persistent enrichment of the alveolar space with T cells and monocytes in the majority of patients with SARS-CoV-2 infection.
There is increasing evidence that ARDS pathogenesis depends on macrophages, including resident AMs and transient monocytes/macrophages recruited from the blood (49, 151). AMs and IMs play crucial roles in ARDS, with the timing and degree of macrophage polarization determining severity of the disease and its outcome (152).
High COVID-19 morbidity and mortality in patients with diabetes, chronic obstructive pulmonary disease (COPD), or congestive heart failure (CHF) have been associated with increased counts of AMs in BALF (153). Increased AM counts in BALF have been also observed in patients with COPD, correlating with the disease severity (154), in mice following protracted exposure to diesel exhaust particles (155), in aged rodents (156), in mice subjected to heart failure following augmented hypertension (157), and in models of dilated cardiomyopathy combined with exercise (158). Furthermore, experimental aging and diabetic state was associated with altered distributions of AM phenotypes and their reduced bactericidal capacity (159–161). Accordingly, increased susceptibility to severe COVID-19 can be associated with increased content of AMs that might facilitate homing of COVID-19 by their abundant expression of ACE2.
In severe COVID-19, AMs expand to alveolar cavities in a mixed infiltrate with neutrophils and lymphocytes (147). The SARS-CoV-2-induced aggregation of AMs is evident throughout the acute inflammatory response (147, 162, 163). At the early phase of ARDS resident AMs are highly prone to M1 polarization mediated by activation of TLRs or other pattern recognition receptors (PRRs). Macrophages generally express various types of PRRs and are capable of recognizing viral pathogen-associated molecular patterns (PAMPs) (164, 165). The resulting M1-polarized AMs act as a first line of defense from pathogenic factors including viruses, bacteria, and endotoxins (166–168). M1-polarized AMs release ROS and a variety of pro-inflammatory cytokines including IL-1β, IL-6, IL-18, MCP-1, MIP-2, and TNFα (45, 169, 170). The production of pro-inflammatory cytokines by AMs stimulates immigration of neutrophils from circulation into inter-alveolar septa and alveolar space (39, 171). Administration of natural or synthetic inhibitors of M1 polarization at this stage may allow significant reduction in ALI-associated mortality; attempts to identify and use such substances in ALI/ARDS experimental models have been reported (172–176). Administration of methylprednisolone to mice has been shown to direct AM polarization towards M2 phenotype which helps to preserve oxygenation function of the lungs in ARDS (177). These findings indicate that M1-polarized AMs and their microenvironments play a principal role in ALI/ARDS progression. At the same time, despite their distinct pro-inflammatory behaviors, M1-polarized AMs can express amphiregulin (a potent ligand of the epidermal growth factor receptor) which protects the epithelial barrier, inhibits expression of pro-inflammatory cytokines, and reduces the degree of inflammation; the effect has been demonstrated using a lipopolysaccharide-induced ALI model (178, 179). No data on possible alterations of amphiregulin expression in COVID-19 are available as yet.
A link between COVID-19 severity and repression of the PPARγ complex by AMs, recently demonstrated by Desterke et al. (146), confirms the impact of this macrophage dysregulation in COVID-19 as well as in other viral infections (180–182). In patients with critical manifestation of the disease, the aggregation and activation of AMs may be directly related to the cytokine storm.
In addition to their pro-inflammatory properties, AMs can exert anti-inflammatory action. Depletion of AMs during inflammation also reduces the recruitment of other leukocytes to the lungs (118, 164, 183, 184). During the resolution phase of pulmonary inflammation, AMs debride apoptotic cells by phagocytosis. This efferocytosis process competes with the production of pro-inflammatory cytokines and chemokines through secretion of transforming growth factor beta (TGF-β), prostaglandin E2 (PGE2), and platelet-activating factor (PAF) by AMs (185). The massive infiltration and activation of AMs in COVID-19 may represent a shift from classically activated phenotype (M1) towards alternatively activated phenotype (M2) (147). In ARDS, particularly in the case of antibody-dependent enhancement, such shifts may contribute to inflammatory injuries and fibrosis of respiratory tracts (186). It has been suggested that SARS-CoV-2 can enter AMs via the interaction between S protein and ACE2 receptor, given high levels of S protein binding and high receptor density of ACE2 on the surface of LMs (142, 147).
The newly described IM subpopulation of NAMs represents a prospective target for ARDS therapy. These cells express strong regulatory gene profiles and play an important role in regulating pulmonary inflammation in vivo. NAMs have been shown to proliferate robustly after influenza infection and activation with the polyinosinic:polycytidylic acid [Poly(I:C)]; in their absence the inflammatory response is augmented, which results in excessive production of pro-inflammatory cytokines and innate immune cell infiltration (120). Viral infection in the absence of NAMs is associated with an excess of pro-inflammatory cytokines and chemokines such as IL-6, CCL2, CCL3, and CCL5, eventually leading to massive lung damage and death (187).
A minor population of intravascular LMs is apparently involved in early stages of ARDS pathogenesis; these cells contribute to vascular injury by excessively aggregating in lung capillaries and producing pro-inflammatory factors in response to PAMPs (121, 188).
Inflammatory reactions in the lungs are invariably supported by immigration of recruited macrophages (21, 22). In ARDS, peripheral blood monocytes accumulate in the lungs to invade pulmonary tissues where they differentiate into M1 macrophages (50, 117).
At later stages of ARDS, M1 macrophages facilitate ECM degradation by producing matrix metalloproteinases (MMPs) and various chemokines, e.g. CXCL10 (189, 190), which support the reversal of fibrosis. M1 macrophages may also induce the expression of MMP-13 and MMP-3 by myofibroblasts (190, 191). M2 macrophages produce anti-inflammatory cytokines and tissue inhibitors of metalloproteinases (TIMPs) which interfere with excessive ECM remodeling and promote its deposition (190, 192). For instance, myofibroblasts can be induced to deposit ECM complexes upon co-cultivation with M2 macrophages (190). The presence of M2 macrophages in pulmonary lesions indicates progression of fibrosis; sustained expression of IL-4, IL-13, TGF-β, and Arg-1 may facilitate collagen deposition (193, 194). The degree of myofibroblast activation and ECM deposition, and accordingly the degree of resulting fibrosis, depend on the balance of M1- and M2-polarized macrophages among other factors of local microenvironment in pulmonary lesions.
Thus, ARDS pathogenesis involves both the recruited pro-inflammatory monocytic macrophages and the anti-inflammatory (or hybrid) resident AMs. Cell therapies involving M2 polarization/reprogramming are highly relevant for the treatment of acute pulmonary inflammation. An enforced shift in macrophage polarization towards anti-inflammatory phenotypes may alleviate ARDS symptoms (49, 195) and save lives.
There are no 100% effective therapies for ARDS, especially for SARS-CoV-2-associated ARDS; the treatment largely remains symptomatic. Increased survival with ARDS has been achieved over the last decade through the use of mechanical ventilation in moderate and severe cases (196).
In this regard, emerging therapies that can target mechanisms of damage, maintain or enhance defenses of the body against pathogens, and facilitate lung recovery are of great interest. The possibility of regulating macrophage polarization by switching it from M1 to M2 and vice versa (so-called repolarization, or reprogramming) may provide new therapeutic strategies for a wide range of autoimmune and inflammatory diseases including COVID-19 (38, 86, 197–199).
The relevance of research on macrophages for their subsequent use as a target or agent of cell therapy is undeniable. A prospect of regulating cellular homeostasis under pathological conditions by changing the phenotypes of macrophages in damaged tissues and organs, and using reprogramming approaches both in situ and ex vivo (followed by reverse transplantation of modified macrophages) is most obvious. However, precise mechanisms that govern macrophage polarization remain underexplored. Macrophage polarization is a finely regulated process that involves a wide network of signaling pathways with an abundance of transcription factors and other major regulators. The shifts in M1/M2 balance may occur as a consequence of suppressed M1 polarization or enhanced M2 polarization (or both simultaneously, or vice versa); moreover, some macrophages may remain non-polarized. Enforced M1 polarization of macrophages can stimulate inflammation and cell death, thus providing a useful tool for the treatment of proliferative disorders, notably tumors (200, 201). Enforced M2 polarization of macrophages will have the opposite effect. Increased proportion of M2 macrophages can stimulate regeneration, angiogenesis and ECM remodeling, thus providing a useful tool for the treatment of inflammatory disorders. Obtaining a stable M2-polarized line of macrophages represents a highly relevant goal for transplantology (202).
The number of articles about the ARDS therapy by directed polarization of macrophages towards the desired phenotype is insufficient and all published articles refer to recent years (39, 40, 195, 203–205). The objective of manipulating the phenotype in the exudative phase of the ARDS, when macrophages are predominantly proinflammatory, is an attractive strategy for preventing the manifestations of the acute phase of the disease. Thus, in the work of Wang and colleagues, researchers developed P12 gold nanoparticles coated with hexapeptides with the sequence CLPFFD and investigated their ability to influence the phenotype of alveolar macrophages in LPS-induced mice model of ALI (203). They demonstrated that P12 promoted a significant decrease of M1 cytokines levels (IL-12p40 and IFN-γ) and increase of M2 cytokines levels (IL-10, IL-4, IL-13) in the BALF. In vitro experiments showed that P12 nanoparticles reduce the LPS-induced growth in pro-inflammatory cytokines in a culture of bone marrow-derived macrophage. As the main hypotheses for the action of P12 particles coated with hexapeptides, the authors suggest the prevention of excessive activation of macrophages at the early stage of ALI/ARDS by inhibiting TLR4 signaling pathways, as well as reprogramming inflammatory macrophages into anti-inflammatory ones, promoting the repair and arrest of inflammation (203).
Another work investigated the specific type of neutrophil death, neutrophil extracellular traps (NETs), and their relationship with macrophage polarization (204). Neutrophils are the first immune cells to arrive at the site of inflammation. The release of NETs, followed by the ejection of DNA strands to trap pathogens, is characteristic of ALI. Song et al. (204) show that NET levels in ARDS patients correlate positively with pro-inflammatory macrophage markers. Using an LPS-induced mouse model of ALI, the authors demonstrate that the use of NET inhibitors (NE inhibitor and PAD4 inhibitor) reduce the number of neutrophils in BALF and mitigate M1-like macrophage polarization, as indicated by reduced levels of pro-inflammatory markers iNOS, CD11c, and CD54 after experimental administration of NET inhibitors. The M1-polarizing effects of NETs have been also confirmed in vitro.
Pro- and anti-inflammatory polarization of macrophages has been associated with alterations in their metabolic status: M1 macrophages produce ATP mainly by glycolysis, while M2 macrophages use aerobic respiration (206). Liu et al. suggested intraperitoneal administration of α-ketoglutarate (α-KG) as the means for reprogramming alveolar macrophages in LPS-induced ALI/ARDS model (205). At first, the authors demonstrated that α-KG induced anti-inflammatory properties in vitro: α-KG significantly decreased the expression of IL-1β, IL-6, and TNFα and increased the intracellular ATP level in M1-polarized MH-S cells. In the ALI/ARDS murine model, α-KG inhibited the LPS-induced elevated serum levels of inflammatory cytokines IL-6 and IL-12, as well as the expression of pro-inflammatory genes (IL-1β, IL-6, TNFα) triggered by LPS, while stimulating the expression of M2 marker genes (Arg1 and Mrc1) in the lungs. Taken together, these data indicate that α-KG attenuates the LPS-induced ALI/ARDS by regulating macrophage polarization in vivo.
Specifically for ARDS, LMs represent a promising therapeutic target. Effective control over their activation and polarization will help to deter excessive inflammation and limit tissue damage. The potential of targeted macrophage polarization approaches for ARDS treatment was demonstrated in a number of preclinical studies (173–177, 207). On the other hand, unrestrained anti-inflammatory polarization may contribute to fibrosis development and increase susceptibility to secondary infections (208, 209). The articles presented in this section authors did not provide data on the negative effects of the obtained anti-inflammatory polarization of macrophages in LPS-induced ALI/ARDS models, however, such effects should always be taken into account. Probably, the use of transient modification like small noncoding RNAs or chemical agents with controlled factor release is a preferable technology.
One of the most effective methods for obtaining a specific knockout is the use of CRISPR/Cas9 complexes, which can be used to stabilize reprogrammed M1/M2 macrophages. A successful example is provided by knockout of ubiquitin-specific proteinase USP18 involved in IFN signaling in macrophages (210). However, selection of appropriate targets for CRISPR/Cas9 complexes represents a difficult task due to the complexity of metabolic and signaling pathways in macrophages during inflammatory processes. By now, two candidate target genes with differential expression in pro- and anti-inflammatory macrophages have been identified, encoding cytochrome b-245 (CYBB) and 7-dehydrocholesterol reductase (DHCR7) (211). Modification of these genes may provide macrophages with stable pro- and anti-inflammatory phenotypes as a potential tool for ARDS and COVID-19 cell therapy.
To summarize, pathogenesis of the new coronavirus infection, especially in cases aggravated by ARDS, depends on phenotypes and functionalities of monocytes, monocyte-derived and resident macrophages in lung tissues. The ratio of pro- and anti-inflammatory macrophages at the sites of injury strongly influences the course and severity of the pathological process in lungs, and this balance represents a potential therapeutic target. The choice of macrophage reprogramming strategy should account for a particular phase of the disease (early or late) in which the treatment is supposed to be administered. For instance, in the early phase of ARDS, resident and recruited AMs are polarized predominantly as pro-inflammatory, consistently with the acute inflammation of lung tissues characteristic of this phase. Therefore, mitigation of this phase would require reprogramming of macrophages towards anti-inflammatory phenotype. By contrast, the next phase of ARDS is marked by massive conversion of both the resident and recruited lung macrophages from pro-inflammatory into anti-inflammatory phenotype. The final phase depends on a fine balance between pro-inflammatory and anti-inflammatory macrophage phenotypes, as both of them are important and special for this phase: pro-inflammatory macrophages facilitate ECM degradation thus limiting fibrosis (preventing excessive fibrosis and ultimately promoting the reversal of fibrosis), whereas anti-inflammatory macrophages express anti-inflammatory cytokines and tissue inhibitors of metalloproteinases thus supporting fibrosis. The degree of ECM deposition and severity of pulmonary fibrosis in this phase strongly depend on the phenotype and functioning of macrophages.
Thus, the use of cell therapy with reprogrammed macrophages for the treatment of ARDS may be successful in COVID-19. Switching macrophage polarization from pro-inflammatory to anti-inflammatory phenotype in the early phase of ARDS can alleviate the enormous production of pro-inflammatory cytokines thereby preventing a cytokine storm and reducing mortality in patients with coronavirus infections. Efficient techniques of macrophage modification (safe and otherwise suitable for clinical purposes) are under way; the approaches include transient genetic modification of macrophages to knock down specific genes responsible for polarization. Convincing preliminary results in vitro and in vivo were obtained with the use of macrophage cell lines, which are more accessible and easier to modify than primary cultures (212). Continuous improvement of existing methods for delivery of genetic constructs into cells will certainly make it possible to modify primary cultures of macrophages, including isolated blood monocytes, for autologous transplantations (Figure 2).
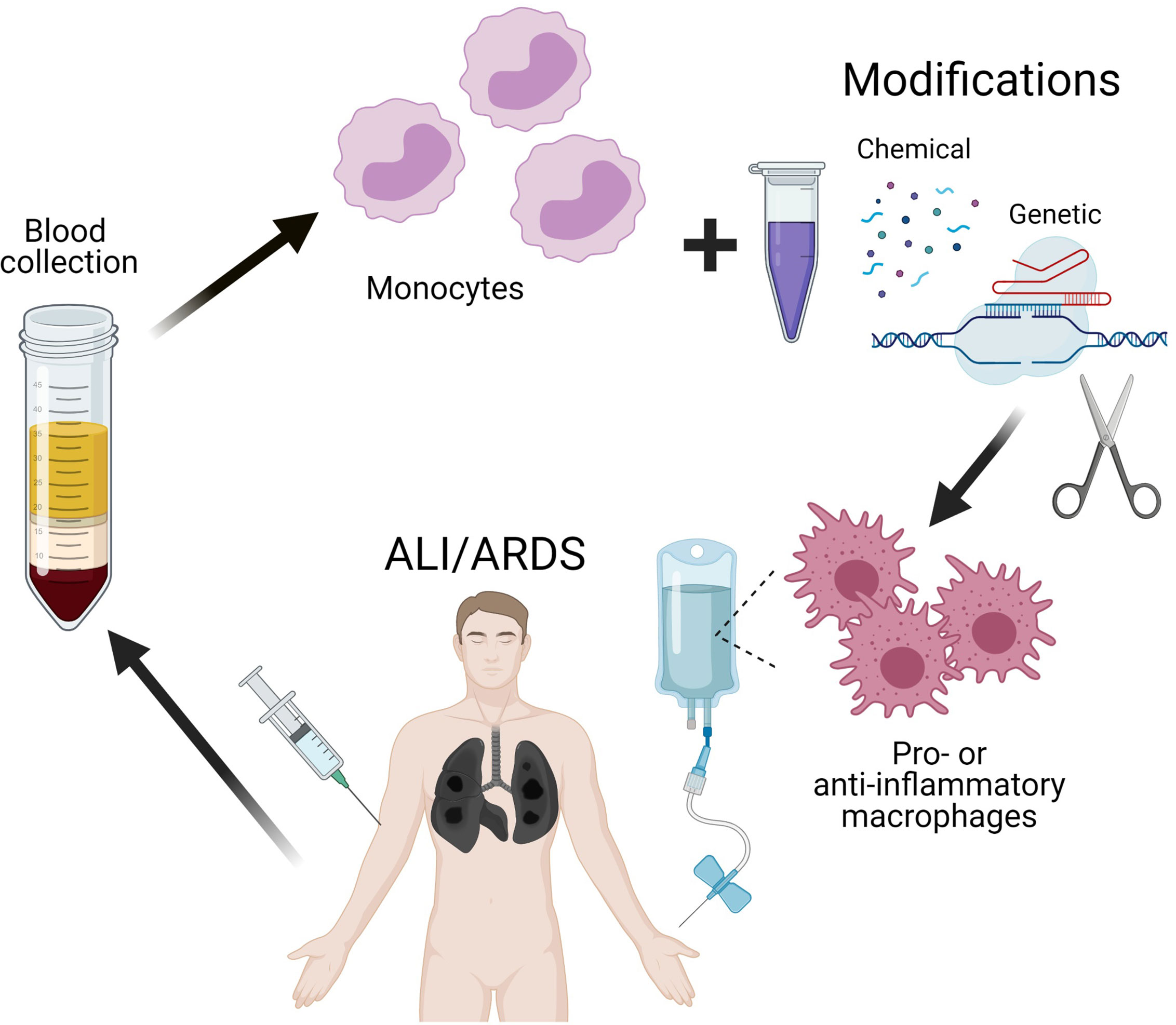
Figure 2 Scheme of perspective therapy of ALI/ARDS by reprogramming of macrophages.
AK and TF contributed to conception and design of the research. DD, AL, and PV collected the literature data. DD and AK wrote the first draft of the manuscript. AL and PV wrote sections of the manuscript. All authors discussed and commented the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Russian Foundation for Basic Research (grant number 20-04-60027) and the RUDN University Strategic Academic Leadership Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Pandey A, Nikam AN, Shreya AB, Mutalik SP, Gopalan D, Kulkarni S, et al. Potential Therapeutic Targets for Combating SARS-Cov-2: Drug Repurposing, Clinical Trials and Recent Advancements. Life Sci (2020) 256:117883. doi: 10.1016/j.lfs.2020.117883
2. Prompetchara E, Ketloy C, Palaga T. Immune Responses in COVID-19 and Potential Vaccines: Lessons Learned From SARS and MERS Epidemic. Asian Pacific J Allergy Immunol (2020) 38:1–9. doi: 10.12932/AP-200220-0772
3. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T-cells and Inflammatory Monocytes Incite Inflammatory Storms in Severe Covid-19 Patients. Natl Sci Rev (2020) 7:998–1002. doi: 10.1093/nsr/nwaa041
4. Imai Y, Kuba K, Penninger JM. The Discovery of Angiotensin-Converting Enzyme 2 and its Role in Acute Lung Injury in Mice. Exp Physiol (2008) 93:543–8. doi: 10.1113/expphysiol.2007.040048
5. World Health Organization, Mission China Joint. Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (Covid-19). WHO-China Jt Mission Coronavirus Dis 2019 (2020). Available at: https://www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report.pdf.
6. Buchrieser J, Dufloo J, Hubert M, Monel B, Planas D, Rajah MM, et al. Syncytia Formation by SARS-CoV-2-infected Cells. EMBO J (2020) 39:e106267. doi: 10.15252/embj.2020106267
7. Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, et al. The Incubation Period of Coronavirus Disease 2019 (Covid-19) From Publicly Reported Confirmed Cases: Estimation and Application. Ann Intern Med (2020) 172:577–82. doi: 10.7326/M20-0504
8. Rothan HA, Byrareddy SN. The Epidemiology and Pathogenesis of Coronavirus Disease (Covid-19) Outbreak. J Autoimmun (2020) 109:102433. doi: 10.1016/j.jaut.2020.102433
9. Wang W, Tang J, Wei F. Updated Understanding of the Outbreak of 2019 Novel Coronavirus (2019-nCoV) in Wuhan, China. J Med Virol (2020) 92:441–7. doi: 10.1002/jmv.25689
10. Fisher D, Heymann D. Q&a: The Novel Coronavirus Outbreak Causing Covid-19. BMC Med (2020) 18:57. doi: 10.1186/s12916-020-01533-w
11. Kalligeros M, Shehadeh F, Mylona EK, Benitez G, Beckwith CG, Chan PA, et al. Association of Obesity With Disease Severity Among Patients With Coronavirus Disease 2019. Obesity (2020) 28:1200–4. doi: 10.1002/oby.22859
12. Codo AC, Davanzo GG, Monteiro L de B, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor Sars-Cov-2 Infection and Monocyte Response Through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab (2020) 32:437–46.e5. doi: 10.1016/j.cmet.2020.07.007
13. Sarzi-Puttini P, Giorgi V, Sirotti S, Marotto D, Ardizzone S, Rizzardini G, et al. Covid-19, Cytokines and Immunosuppression: What can We Learn From Severe Acute Respiratory Syndrome? Clin Exp Rheumatol (2020) 38:337–42.
14. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (Covid-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA - J Am Med Assoc (2020) 323:1239–42. doi: 10.1001/jama.2020.2648
15. D́elia RV, Harrison K, Oyston PC, Lukaszewski RA, Clark GC. Targeting the “Cytokine Storm” for Therapeutic Benefit. Clin Vaccine Immunol (2013) 20:319–27. doi: 10.1128/CVI.00636-12
16. Channappanavar R, Perlman S. Pathogenic Human Coronavirus Infections: Causes and Consequences of Cytokine Storm and Immunopathology. Semin Immunopathol (2017) 39:529–39. doi: 10.1007/s00281-017-0629-x
17. Yazdanpanah F, Hamblin MR, Rezaei N. The Immune System and COVID-19: Friend or Foe? Life Sci (2020) 256:117900. doi: 10.1016/j.lfs.2020.117900
18. Perlman S, Dandekar AA. Immunopathogenesis of Coronavirus Infections: Implications for SARS. Nat Rev Immunol (2005) 5:917–27. doi: 10.1038/nri1732
19. Zhong NS, Zheng BJ, Li YM, Poon LLM, Xie ZH, Chan KH, et al. Epidemiology and Cause of Severe Acute Respiratory Syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet (2003) 362:1353–8. doi: 10.1016/S0140-6736(03)14630-2
20. Assiri A, Al-Tawfiq JA, Al-Rabeeah AA, Al-Rabiah FA, Al-Hajjar S, Al-Barrak A, et al. Epidemiological, Demographic, and Clinical Characteristics of 47 Cases of Middle East Respiratory Syndrome Coronavirus Disease From Saudi Arabia: A Descriptive Study. Lancet Infect Dis (2013) 13:752–61. doi: 10.1016/S1473-3099(13)70204-4
21. Franks TJ, Chong PY, Chui P, Galvin JR, Lourens RM, Reid AH, et al. Lung Pathology of Severe Acute Respiratory Syndrome (Sars): A Study of 8 Autopsy Cases From Singapore. Hum Pathol (2003) 34:743–8. doi: 10.1016/S0046-8177(03)00367-8
22. Nicholls JM, Poon LLM, Lee KC, Ng WF, Lai ST, Leung CY, et al. Lung Pathology of Fatal Severe Acute Respiratory Syndrome. Lancet (2003) 361:1773–8. doi: 10.1016/S0140-6736(03)13413-7
23. Dosch SF, Mahajan SD, Collins AR. Sars Coronavirus Spike Protein-Induced Innate Immune Response Occurs Via Activation of the NF-κb Pathway in Human Monocyte Macrophages In Vitro. Virus Res (2009) 142:19–27. doi: 10.1016/j.virusres.2009.01.005
24. He L, Ding Y, Zhang Q, Che X, He Y, Shen H, et al. Expression of Elevated Levels of Pro-Inflammatory Cytokines in SARS-CoV-infected Ace2+ Cells in SARS Patients: Relation to the Acute Lung Injury and Pathogenesis of SARS. J Pathol (2006) 210:288–97. doi: 10.1002/path.2067
25. He L, Ding Y, Wang W, Zhang Q, Zhang J, Geng J, et al. Expression of Immune Cells and Their Roles in the Involved Tissues of SARS Patients. Di Yi Jun Yi Da Xue Xue Bao (2003) 23:774–6, 780.
26. İnandıklıoğlu N, Akkoc T. Immune Responses to SARS-CoV, Mers-CoV and SARS-Cov-2. In: Advances in Experimental Medicine and Biology. (2020) 1288:5–12. doi: 10.1007/5584_2020_549
27. Li X, Geng M, Peng Y, Meng L, Lu S. Molecular Immune Pathogenesis and Diagnosis of COVID-19. J Pharm Anal (2020) 10:102–8. doi: 10.1016/j.jpha.2020.03.001
28. Castillo R, Carrasco Loza R, Romero-Dapueto C. Pathophysiological Approaches of Acute Respiratory Distress Syndrome: Novel Bases for Study of Lung Injury. Open Respir Med J (2015) 9:83–91. doi: 10.2174/1874306401509010083
29. Yang CY, Chen CS, Yiang GT, Cheng YL, Yong SB, Wu MY, et al. New Insights Into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome. Int J Mol Sci (2018) 19:588. doi: 10.3390/ijms19020588
30. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological Study of the 2019 Novel Coronavirus Disease (Covid-19) Through Postmortem Core Biopsies. Mod Pathol (2020) 33:1007–14. doi: 10.1038/s41379-020-0536-x
31. Zayratyants ZOV, Malyavin MAG, Samsonova SMV, Chernyaev CAL, Mishnev MOD, Mikhaleva MLM, et al. Pathomorphological Changes in Lungs in Case of COVID-19: Clinical and Therapeutic Parallels. Therapy (2020) 5(2020):35–46. doi: 10.18565/therapy.2020.5.35-46
32. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological Findings of COVID-19 Associated With Acute Respiratory Distress Syndrome. Lancet Respir Med (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
33. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
34. Remick DG. Pathophysiology of Sepsis. Am J Pathol (2007) 170:1435–44. doi: 10.2353/ajpath.2007.060872
35. Kendrick SFW, Jones DEJ. Mechanisms of Innate Immunity in Sepsis. In: Baudouin SV, editor. Sepsis. Competency-Based Critical Care. London: Springer. (2009) p. 5–10. doi: 10.1007/978-1-84628-939-2_2
36. Wheeler AP, Bernard GR. Acute Lung Injury and the Acute Respiratory Distress Syndrome: A Clinical Review. Lancet (2007) 369:1553–64. doi: 10.1016/S0140-6736(07)60604-7
37. Matuschak GM, Lechner AJ. Acute Lung Injury and the Acute Respiratory Distress Syndrome: Pathophysiology and Treatment. Mo Med (2010) 107:252–8.
38. Aggarwal NR, King LS, D’Alessio FR. Diverse Macrophage Populations Mediate Acute Lung Inflammation and Resolution. Am J Physiol - Lung Cell Mol Physiol (2014) 306:L709–25. doi: 10.1152/ajplung.00341.2013
39. Huang X, Xiu H, Zhang S, Zhang G. The Role of Macrophages in the Pathogenesis of Ali/Ards. Mediators Inflammation (2018) 2018:1264913. doi: 10.1155/2018/1264913
40. Chen X, Tang J, Shuai W, Meng J, Feng J, Han Z. Macrophage Polarization and its Role in the Pathogenesis of Acute Lung Injury/Acute Respiratory Distress Syndrome. Inflammation Res (2020) 69:883–95. doi: 10.1007/s00011-020-01378-2
41. Li X, Ma X. Acute Respiratory Failure in COVID-19: Is it “Typical” Ards? Crit Care (2020) 24:198. doi: 10.1186/s13054-020-02911-9
42. Spinelli E, Mauri T, Beitler JR, Pesenti A, Brodie D. Respiratory Drive in the Acute Respiratory Distress Syndrome: Pathophysiology, Monitoring, and Therapeutic Interventions. Intensive Care Med (2020) 46:606–18. doi: 10.1007/s00134-020-05942-6
43. Haddad IY, Ingbar DH, Panoskaltsis-Mortari A, Blazar BR. Activated Alveolar Macrophage-Derived Nitric Oxide Predicts the Development of Lung Damage After Marrow Transplantation in Mice. Chest (1999) 116(1 Suppl):37S. doi: 10.1378/chest.116.suppl_1.37S
44. Wood C, Kataria V, Modrykamien AM. The Acute Respiratory Distress Syndrome. Baylor Univ Med Cent Proc (2020) 33:357–65. doi: 10.1080/08998280.2020.1764817
45. Bhatia M, Zemans RL, Jeyaseelan S. Role of Chemokines in the Pathogenesis of Acute Lung Injury. Am J Respir Cell Mol Biol (2012) 46:566–72. doi: 10.1165/rcmb.2011-0392TR
46. Tyrrell C, McKechnie SR, Beers MF, Mitchell TJ, McElroy MC. Differential Alveolar Epithelial Injury and Protein Expression in Pneumococcal Pneumonia. Exp Lung Res (2012) 38:266–76. doi: 10.3109/01902148.2012.683321
47. Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of Oxidants and Redox Signaling in the Pathogenesis of Acute Respiratory Distress Syndrome. Antioxidants Redox Signal (2008) 10:739–53. doi: 10.1089/ars.2007.1940
48. Weiss SR, Leibowitz JL. Coronavirus Pathogenesis. In: Advances in Virus Research. (2011) 81 p. 85–164. doi: 10.1016/B978-0-12-385885-6.00009-2
49. Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary Macrophage Subpopulations in the Induction and Resolution of Acute Lung Injury. Am J Respir Cell Mol Biol (2012) 47:417–26. doi: 10.1165/rcmb.2012-0090OC
50. Herold S, Gabrielli NM, Vadász I. Novel Concepts of Acute Lung Injury and Alveolar-Capillary Barrier Dysfunction. Am J Physiol - Lung Cell Mol Physiol (2013) 305:L665–81. doi: 10.1152/ajplung.00232.2013
51. Sharp C, Millar AB, Medford ARL. Advances in Understanding of the Pathogenesis of Acute Respiratory Distress Syndrome. Respiration (2015) 89:420–34. doi: 10.1159/000381102
52. Pan Y, Guan H, Zhou S, Wang Y, Li Q, Zhu T, et al. Initial CT Findings and Temporal Changes in Patients With the Novel Coronavirus Pneumonia (2019-Ncov): A Study of 63 Patients in Wuhan, China. Eur Radiol (2020) 30:3306–9. doi: 10.1007/s00330-020-06731-x
53. Ye Z, Zhang Y, Wang Y, Huang Z, Song B. Chest CT Manifestations of New Coronavirus Disease 2019 (Covid-19): A Pictorial Review. Eur Radiol (2020) 30:4381–9. doi: 10.1007/s00330-020-06801-0
54. Pan F, Ye T, Sun P, Gui S, Liang B, Li L, et al. Time Course of Lung Changes At Chest CT During Recovery From Coronavirus Disease 2019 (Covid-19). Radiology (2020) 295:715–21. doi: 10.1148/radiol.2020200370
55. Kong W, Agarwal PP. Chest Imaging Appearance of COVID-19 Infection. Radiol Cardiothorac Imaging (2020) 2:e200028. doi: 10.1148/ryct.2020200028
56. Mason C, Dooley N, Griffiths M. Acute Respiratory Distress Syndrome. Clin Med (2016) 16:s66–70. doi: 10.7861/clinmedicine.16-6-s66
57. Burnham EL, Janssen WJ, Riches DWH, Moss M, Downey GP. The Fibroproliferative Response in Acute Respiratory Distress Syndrome: Mechanisms and Clinical Significance. Eur Respir J (2014) 43:276–85. doi: 10.1183/09031936.00196412
58. Keane MP, Belperio JA, Moore TA, Moore BB, Arenberg DA, Smith RE, et al. Neutralization of the CXC Chemokine, Macrophage Inflammatory Protein-2, Attenuates Bleomycin-Induced Pulmonary Fibrosis. J Immunol (1999) 162:5511–8.
59. Hamada N, Kuwano K, Yamada M, Hagimoto N, Hiasa K, Egashira K, et al. Anti-Vascular Endothelial Growth Factor Gene Therapy Attenuates Lung Injury and Fibrosis in Mice. J Immunol (2005) 175:1224–31. doi: 10.4049/jimmunol.175.2.1224
60. Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, et al. Excess Circulating Angiopoietin-2 may Contribute to Pulmonary Vascular Leak in Sepsis in Humans. PloS Med (2006) 3:356–70. doi: 10.1371/journal.pmed.0030046
61. Ou XM, Li WC, Liu DS, Li YP, Wen FQ, Feng YL, et al. Vegfr-2 Antagonist Su5416 Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice. Int Immunopharmacol (2009) 9:70–9. doi: 10.1016/j.intimp.2008.10.002
62. Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A, et al. Ventilation With Lower Tidal Volumes as Compared With Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. N Engl J Med (2000) 342:1301–8. doi: 10.1056/nejm200005043421801
63. Tremblay LN, Miatto D, Hamid Q, Govindarajan A, Slutsky AS. Injurious Ventilation Induces Widespread Pulmonary Epithelial Expression of Tumor Necrosis Factor-α and Interleukin-6 Messenger Rna. Crit Care Med (2002) 30:1693–700. doi: 10.1097/00003246-200208000-00003
64. Tremblay LN, Slutsky AS. Ventilator-Induced Lung Injury: From the Bench to the Bedside. Intensive Care Med (2006) 32:24–33. doi: 10.1007/s00134-005-2817-8
65. Pelosi P, Rocco PR. Effects of Mechanical Ventilation on the Extracellular Matrix. Intensive Care Med (2008) 34:631–9. doi: 10.1007/s00134-007-0964-9
66. Hume DA, Irvine KM, Pridans C. The Mononuclear Phagocyte System: The Relationship Between Monocytes and Macrophages. Trends Immunol (2019) 40:98–112. doi: 10.1016/j.it.2018.11.007
67. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-Resident Macrophages Originate From Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature (2015) 518:547–51. doi: 10.1038/nature13989
68. Haldar M, Murphy KM. Origin, Development, and Homeostasis of Tissue-Resident Macrophages. Immunol Rev (2014) 262:25–35. doi: 10.1111/imr.12215
69. Ziegler-Heitbrock L. Monocyte Subsets in Man and Other Species. Cell Immunol (2014) 289:135–9. doi: 10.1016/j.cellimm.2014.03.019
70. Mildner A, Marinkovic G, Jung S. Murine Monocytes: Origins, Subsets, Fates, and Functions. In: Myeloid Cells in Health and Disease: A Synthesis. (2016) (5) p. 141–53. doi: 10.1128/9781555819194.ch9
71. Wong KL, Tai JJ-Y, Wong W-C, Han H, Sem X, Yeap W-H, et al. Gene Expression Profiling Reveals the Defining Features of the Classical, Intermediate, and Nonclassical Human Monocyte Subsets. Blood (2011) 118:e16–31. doi: 10.1182/blood-2010-12-326355
72. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-Cell RNA-seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science (2017) 356:eaah4573. doi: 10.1126/science.aah4573
73. Hamers AAJ, Dinh HQ, Thomas GD, Marcovecchio P, Blatchley A, Nakao CS, et al. Human Monocyte Heterogeneity as Revealed by High-Dimensional Mass Cytometry. Arterioscler Thromb Vasc Biol (2019) 39:25–36. doi: 10.1161/ATVBAHA.118.311022
74. Wolf AA, Yáñez A, Barman PK, Goodridge HS. The Ontogeny of Monocyte Subsets. Front Immunol (2019) 10:1642. doi: 10.3389/fimmu.2019.01642
75. Kapellos TS, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley ER, et al. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front Immunol (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
76. Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical Monocytes in Health and Disease. Annu Rev Immunol (2019) 37:439–56. doi: 10.1146/annurev-immunol-042617-053119
77. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-Resident Macrophages Self-Maintain Locally Throughout Adult Life With Minimal Contribution From Circulating Monocytes. Immunity (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
78. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages Under Homeostasis. Immunity (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
79. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
80. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage Plasticity and Polarization in Tissue Repair and Remodelling. J Pathol (2013) 229:176–85. doi: 10.1002/path.4133
81. Cassetta L, Cassol E, Poli G. Macrophage Polarization in Health and Disease. ScientificWorldJournal (2011) 11:2391–402. doi: 10.1100/2011/213962
82. Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage Polarization and Plasticity in Health and Disease. Immunol Res (2012) 53:11–24. doi: 10.1007/s12026-012-8291-9
83. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
84. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
85. Sica A, Erreni M, Allavena P, Porta C. Macrophage Polarization in Pathology. Cell Mol Life Sci (2015) 72:4111–26. doi: 10.1007/s00018-015-1995-y
86. Patel U, Rajasingh S, Samanta S, Cao T, Dawn B, Rajasingh J. Macrophage Polarization in Response to Epigenetic Modifiers During Infection and Inflammation. Drug Discovery Today (2017) 22:186–93. doi: 10.1016/j.drudis.2016.08.006
87. Fukui S, Iwamoto N, Takatani A, Igawa T, Shimizu T, Umeda M, et al. M1 and M2 Monocytes in Rheumatoid Arthritis: A Contribution of Imbalance of M1/M2 Monocytes to Osteoclastogenesis. Front Immunol (2018) 8:1958. doi: 10.3389/fimmu.2017.01958
88. Gordon S, Martinez FO. Alternative Activation of Macrophages: Mechanism and Functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
89. Braga TT, Agudelo JSH, Camara NOS. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front Immunol (2015) 6:602. doi: 10.3389/fimmu.2015.00602
90. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MPJ, Donners MMPC. Anti-Inflammatory M2, But Not Pro-Inflammatory M1 Macrophages Promote Angiogenesis In Vivo. Angiogenesis (2014) 17:109–18. doi: 10.1007/s10456-013-9381-6
91. Wang N, Liang H, Zen K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
92. Zhang YH, He M, Wang Y, Liao AH. Modulators of the Balance Between M1 and M2 Macrophages During Pregnancy. Front Immunol (2017) 8:120. doi: 10.3389/fimmu.2017.00120
93. Okizaki Si, Ito Y, Hosono K, Oba K, Ohkubo H, Amano H, et al. Suppressed Recruitment of Alternatively Activated Macrophages Reduces Tgf-β1 and Impairs Wound Healing in Streptozotocin-Induced Diabetic Mice. BioMed Pharmacother (2015) 70:317–25. doi: 10.1016/j.biopha.2014.10.020
94. Gensel JC, Zhang B. Macrophage Activation and its Role in Repair and Pathology After Spinal Cord Injury. Brain Res (2015) 1619:1–11. doi: 10.1016/j.brainres.2014.12.045
95. Guerrero AR, Uchida K, Nakajima H, Watanabe S, Nakamura M, Johnson WEB, et al. Blockade of Interleukin-6 Signaling Inhibits the Classic Pathway and Promotes an Alternative Pathway of Macrophage Activation After Spinal Cord Injury in Mice. J Neuroinflamm (2012) 9:40. doi: 10.1186/1742-2094-9-40
96. Nakajima H, Uchida K, Guerrero AR, Watanabe S, Sugita D, Takeura N, et al. Transplantation of Mesenchymal Stem Cells Promotes an Alternative Pathway of Macrophage Activation and Functional Recovery After Spinal Cord Injury. J Neurotrauma (2012) 29:1614–25. doi: 10.1089/neu.2011.2109
97. Urbina P, Singla DK. Bmp-7 Attenuates Adverse Cardiac Remodeling Mediated Through M2 Macrophages in Prediabetic Cardiomyopathy. Am J Physiol - Hear Circ Physiol (2014) 307:H762–72. doi: 10.1152/ajpheart.00367.2014
98. Singla DK, Singla R, Wang J. Bmp-7 Treatment Increases M2 Macrophage Differentiation and Reduces Inflammation and Plaque Formation in Apo E-/-Mice. PloS One (2016) 11:e0147897. doi: 10.1371/journal.pone.0147897
99. Singla DK, Singla RD, Abdelli LS, Glass C. Fibroblast Growth Factor-9 Enhances M2 Macrophage Differentiation and Attenuates Adverse Cardiac Remodeling in the Infarcted Diabetic Heart. PloS One (2015) 10:e0120739. doi: 10.1371/journal.pone.0120739
100. Singla DK, Johnson TA, Tavakoli Dargani Z. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells (2019) 8:1224. doi: 10.3390/cells8101224
101. Specht H, Emmott E, Petelski AA, Huffman RG, Perlman DH, Serra M, et al. Single-Cell Proteomic and Transcriptomic Analysis of Macrophage Heterogeneity Using Scope2. Genome Biol (2021) 22:50. doi: 10.1186/s13059-021-02267-5
102. Palma A, Jarrah AS, Tieri P, Cesareni G, Castiglione F. Gene Regulatory Network Modeling of Macrophage Differentiation Corroborates the Continuum Hypothesis of Polarization States. Front Physiol (2018) 9:1659. doi: 10.3389/fphys.2018.01659
103. Atri C, Guerfali FZ, Laouini D. Role of Human Macrophage Polarization in Inflammation During Infectious Diseases. Int J Mol Sci (2018) 19:1801. doi: 10.3390/ijms19061801
104. O’Carroll C, Fagan A, Shanahan F, Carmody RJ. Identification of a Unique Hybrid Macrophage-Polarization State Following Recovery From Lipopolysaccharide Tolerance. J Immunol (2014) 192:427–36. doi: 10.4049/jimmunol.1301722
105. Palmieri EM, Gonzalez-Cotto M, Baseler WA, Davies LC, Ghesquière B, Maio N, et al. Nitric Oxide Orchestrates Metabolic Rewiring in M1 Macrophages by Targeting Aconitase 2 and Pyruvate Dehydrogenase. Nat Commun (2020) 11:698. doi: 10.1038/s41467-020-14433-7
106. Li L, Lv G, Wang B, Kuang L. Xist/Mir-376c-5p/OPN Axis Modulates the Influence of Proinflammatory M1 Macrophages on Osteoarthritis Chondrocyte Apoptosis. J Cell Physiol (2020) 235:281–93. doi: 10.1002/jcp.28968
107. Yang SJ, Chen YY, Hsu CH, Hsu CW, Chang CY, Chang JR, et al. Activation of M1 Macrophages in Response to Recombinant Tb Vaccines With Enhanced Antimycobacterial Activity. Front Immunol (2020) 11:1298. doi: 10.3389/fimmu.2020.01298
108. Muraille E, Leo O, Moser M. Th1/Th2 Paradigm Extended: Macrophage Polarization as an Unappreciated Pathogen-Driven Escape Mechanism? Front Immunol (2014) 5:603. doi: 10.3389/fimmu.2014.00603
109. Janeway CA, Travers P, Walport M. Immunobiology - NCBI Bookshelf, in: Hypersensitivity Diseases (2001). Garl Sci. Available at: https://www.ncbi.nlm.nih.gov/books/NBK10757/ (Accessed April 11, 2021).
110. Jafarzadeh A, Chauhan P, Saha B, Jafarzadeh S, Nemati M. Contribution of Monocytes and Macrophages to the Local Tissue Inflammation and Cytokine Storm in COVID-19: Lessons From SARS and MERS, and Potential Therapeutic Interventions. Life Sci (2020) 257:118102. doi: 10.1016/j.lfs.2020.118102
111. Evren E, Ringqvist E, Willinger T. Origin and Ontogeny of Lung Macrophages: From Mice to Humans. Immunology (2020) 160:126–38. doi: 10.1111/imm.13154
112. Chiang AJ, Thanabalasuriar A, Boo CC. Proteomics: An Advanced Tool to Unravel the Role of Alveolar Macrophages in Respiratory Diseases. Int J Biochem Cell Biol (2021) 134:105966. doi: 10.1016/j.biocel.2021.105966
113. Tan SYS, Krasnow MA. Developmental Origin of Lung Macrophage Diversity. Dev (2016) 143:1318–27. doi: 10.1242/dev.129122
114. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The Cytokine Tgf-β Promotes the Development and Homeostasis of Alveolar Macrophages. Immunity (2017) 47:903–912.e4. doi: 10.1016/j.immuni.2017.10.007
115. Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. Gm-CSF Regulates Alveolar Macrophage Differentiation and Innate Immunity in the Lung Through Pu. 1. Immun (2001) 15:557–67. doi: 10.1016/S1074-7613(01)00218-7
116. Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct Macrophage Subpopulations Characterize Acute Infection and Chronic Inflammatory Lung Disease. J Immunol (2012) 189:946–55. doi: 10.4049/jimmunol.1200660
117. Short KR, Kroeze EJBV, Fouchier RAM, Kuiken T. Pathogenesis of Influenza-Induced Acute Respiratory Distress Syndrome. Lancet Infect Dis (2014) 14:57–69. doi: 10.1016/S1473-3099(13)70286-X
118. Maus UA, Janzen S, Wall G, Srivastava M, Blackwell TS, Christman JW, et al. Resident Alveolar Macrophages are Replaced by Recruited Monocytes in Response to Endotoxin-Induced Lung Inflammation. Am J Respir Cell Mol Biol (2006) 35:227–35. doi: 10.1165/rcmb.2005-0241OC
119. Aguzzi A, Barres BA, Bennett ML. Microglia: Scapegoat, Saboteur, or Something Else? Science (2013) 339:156–61. doi: 10.1126/science.1227901
120. Ural BB, Yeung ST, Damani-Yokota P, Devlin JC, de Vries M, Vera-Licona P, et al. Identification of a Nerve-Associated, Lung-Resident Interstitial Macrophage Subset With Distinct Localization and Immunoregulatory Properties. Sci Immunol (2020) 5:eaax8756. doi: 10.1126/sciimmunol.aax8756
121. Evren E, Ringqvist E, Tripathi KP, Sleiers N, Rives IC, Alisjahbana A, et al. Distinct Developmental Pathways From Blood Monocytes Generate Human Lung Macrophage Diversity. Immunity (2021) 54:259–75. doi: 10.1016/j.immuni.2020.12.003
122. Hu G, Christman JW. Editorial: Alveolar Macrophages in Lung Inflammation and Resolution. Front Immunol (2019) 10:2275. doi: 10.3389/fimmu.2019.02275
123. Abassi Z, Knaney Y, Karram T, Heyman SN. The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front Immunol (2020) 11:1312. doi: 10.3389/fimmu.2020.01312
124. Mitsi E, Kamng’ona R, Rylance J, Solórzano C, Jesus Reiné J, Mwandumba HC, et al. Human Alveolar Macrophages Predominately Express Combined Classical M1 and M2 Surface Markers in Steady State. Respir Res (2018) 19:66. doi: 10.1186/s12931-018-0777-0
125. Stevenson EV, Collins-McMillen D, Kim JH, Cieply SJ, Bentz GL, Yurochko AD. Hcmv Reprogramming of Infected Monocyte Survival and Differentiation: A Goldilocks Phenomenon. Viruses (2014) 6:782–807. doi: 10.3390/v6020782
126. Liu Y, Tang X, McArthur J, Scott J, Gartner S. Analysis of Human Immunodežciency Virus Type 1 Evidence for Monocyte Trafžcking Into Brain (2000). Available at: www.jneurovirol.com (Accessed April 10, 2021).
127. Fischer-Smith T, Rappaport J. Evolving Paradigms in the Pathogenesis of HIV-1-associated Dementia. Expert Rev Mol Med (2005) 7:1–26. doi: 10.1017/S1462399405010239
128. Herbein G, Varin A. The Macrophage in HIV-1 Infection: From Activation to Deactivation? Retrovirology (2010) 7:33. doi: 10.1186/1742-4690-7-33
129. Wilkinson J, Radkowski M, Eschbacher JM, Laskus T. Activation of Brain Macrophages/Microglia Cells in Hepatitis C Infection. Gut (2010) 59:1394–400. doi: 10.1136/gut.2009.199356
130. Dutta K, Mishra MK, Nazmi A, Kumawat KL, Basu A. Minocycline Differentially Modulates Macrophage Mediated Peripheral Immune Response Following Japanese Encephalitis Virus Infection. Immunobiology (2010) 215:884–93. doi: 10.1016/j.imbio.2009.12.003
131. Qin Z, Freitas E, Sullivan R, Mohan S, Bacelieri R, Branch D, et al. Upregulation of xCT by KSHV-encoded Micrornas Facilitates Kshv Dissemination and Persistence in an Environment of Oxidative Stress. PloS Pathog (2010) 6:e1000742. doi: 10.1371/journal.ppat.1000742
132. Chan G, Nogalski MT, Bentz GL, Smith MS, Parmater A, Yurochko AD. Pi3k-Dependent Upregulation of Mcl-1 by Human Cytomegalovirus Is Mediated by Epidermal Growth Factor Receptor and Inhibits Apoptosis in Short-Lived Monocytes. J Immunol (2010) 184:3213–22. doi: 10.4049/jimmunol.0903025
133. Fernández Larrosa PN, Croci DO, Riva DA, Bibini M, Luzzi R, Saracco M, et al. Apoptosis Resistance in HIV-1 Persistently-Infected Cells is Independent of Active Viral Replication and Involves Modulation of the Apoptotic Mitochondrial Pathway. Retrovirology (2008) 5:19. doi: 10.1186/1742-4690-5-19
134. Murakami M, Hirano T. The Molecular Mechanisms of Chronic Inflammation Development. Front Immunol (2012) 3:323. doi: 10.3389/fimmu.2012.00323
135. Shao H, Lan D, Duan Z, Liu Z, Min J, Zhang L, et al. Upregulation of Mitochondrial Gene Expression in PBMC From Convalescent Sars Patients. J Clin Immunol (2006) 26:546–54. doi: 10.1007/s10875-006-9046-y
136. Li L, Wo J, Shao J, Zhu H, Wu N, Li M, et al. Sars-Coronavirus Replicates in Mononuclear Cells of Peripheral Blood (Pbmcs) From Sars Patients. J Clin Virol (2003) 28:239–44. doi: 10.1016/S1386-6532(03)00195-1
137. Li Q, Verma IM. Nf-κb Regulation in the Immune System. Nat Rev Immunol (2002) 2:725–34. doi: 10.1038/nri910
138. Hsueh PR, Chen PJ, Hsiao CH, Yeh SH, Cheng WC, Wang JL, et al. Patient Data, Early SARS Epidemic, Taiwan. Emerg Infect Dis (2004) 10:489–93. doi: 10.3201/eid1003.030571
139. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should We Stimulate or Suppress Immune Responses in COVID-19? Cytokine and Anti-Cytokine Interventions. Autoimmun Rev (2020) 19:102567. doi: 10.1016/j.autrev.2020.102567
140. Med Sci TJ, Tufan A, Avanoğlu Güler A, Matucci-cerinic M. Turkish Journal of Medical Sciences COVID-19, Immune System Response, Hyperinflammation and Repurposing Antirheumatic Drugs. Turk J Med Sci (2020) 50:620–32. doi: 10.3906/sag-2004-168
141. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
142. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
143. Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, et al. Frontline Science: Covid-19 Infection Induces Readily Detectable Morphologic and Inflammation-Related Phenotypic Changes in Peripheral Blood Monocytes. J Leukoc Biol (2021) 109:13–22. doi: 10.1002/JLB.4HI0720-470R
144. Boumaza A, Gay L, Mezouar S, Bestion E, Diallo AB, Michel M, et al. Monocytes and Macrophages, Targets of SARS-CoV-2: The Clue for Covid-19 Immunoparalysis. J Infect Dis (2021) jiab044. doi: 10.1093/infdis/jiab044
145. Liu J, Yang X, Wang H, Li Z, Deng H, Liu J, et al. The Analysis of the Long-Term Impact of SARS-CoV-2 on the Cellular Immune System in Individuals Recovering From Covid-19 Reveals a Profound Nkt Cell Impairment. medRxiv (2020) 12(2):e00085–21. doi: 10.1101/2020.08.21.20179358
146. Desterke C, Turhan AG, Bennaceur-Griscelli A, Griscelli F. Pparγ Cistrome Repression During Activation of Lung Monocyte-Macrophages in Severe Covid-19. iScience (2020) 23:101611. doi: 10.1016/j.isci.2020.101611
147. Wang C, Xie J, Zhao L, Fei X, Zhang H, Tan Y, et al. Alveolar Macrophage Dysfunction and Cytokine Storm in the Pathogenesis of Two Severe COVID-19 Patients. EBioMedicine (2020) 57:102833. doi: 10.1016/j.ebiom.2020.102833
148. Shaath H, Vishnubalaji R, Elkord E, Alajez NM. Single-Cell Transcriptome Analysis Highlights a Role for Neutrophils and Inflammatory Macrophages in the Pathogenesis of Severe Covid-19. Cells (2020) 9:2374. doi: 10.3390/cells9112374
149. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With Covid-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
150. Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Guzman ER, Abbott DA, et al. Alveolitis in Severe SARS-Cov-2 Pneumonia is Driven by Self-Sustaining Circuits Between Infected Alveolar Macrophages and T Cells. bioRxiv (2020) 2020:08.05.238188. doi: 10.1101/2020.08.05.238188
151. Lomas-Neira J, Chung CS, Perl M, Gregory S, Biffl W, Ayala A. Role of Alveolar Macrophage and Migrating Neutrophils in Hemorrhage-Induced Priming for ALI Subsequent to Septic Challenge. Am J Physiol - Lung Cell Mol Physiol (2006) 290:L51–8. doi: 10.1152/ajplung.00028.2005
152. Gill SE, Yamashita CM, Veldhuizen RAW. Lung Remodeling Associated With Recovery From Acute Lung Injury. Cell Tissue Res (2017) 367:495–509. doi: 10.1007/s00441-016-2521-8
153. Perico L, Benigni A, Remuzzi G. Should COVID-19 Concern Nephrologists? Why and to What Extent? The Emerging Impasse of Angiotensin Blockade. Nephron (2020) 144:213–21. doi: 10.1159/000507305
154. Agustí A, Hogg JC. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N Engl J Med (2019) 381:1248–56. doi: 10.1056/nejmra1900475
155. Ribeiro Júnior G, de Souza Xavier Costa N, Belotti L, dos Santos Alemany AA, Amato-Lourenço LF, da Cunha PG, et al. Diesel Exhaust Exposure Intensifies Inflammatory and Structural Changes Associated With Lung Aging in Mice. Ecotoxicol Environ Saf (2019) 170:314–23. doi: 10.1016/j.ecoenv.2018.11.139
156. Veldhuizen RAW, McCaig LA, Pape C, Gill SE. The Effects of Aging and Exercise on Lung Mechanics, Surfactant and Alveolar Macrophages. Exp Lung Res (2019) 45:113–22. doi: 10.1080/01902148.2019.1605633
157. Wang H, Kwak D, Fassett J, Hou L, Xu X, Burbach BJ, et al. Cd28/B7 Deficiency Attenuates Systolic Overload-Induced Congestive Heart Failure, Myocardial and Pulmonary Inflammation, and Activated T Cell Accumulation in the Heart and Lungs. Hypertension (2016) 68:688–96. doi: 10.1161/HYPERTENSIONAHA.116.07579
158. Sugihara M, Odagiri F, Suzuki T, Murayama T, Nakazato Y, Unuma K, et al. Usefulness of Running Wheel for Detection of Congestive Heart Failure in Dilated Cardiomyopathy Mouse Model. PloS One (2013) 8:e55514. doi: 10.1371/journal.pone.0055514
159. Martinez N, Ketheesan N, West K, Vallerskog T, Kornfeld H. Impaired Recognition of Mycobacterium Tuberculosis by Alveolar Macrophages From Diabetic Mice. J Infect Dis (2016) 214(11):1629–37. doi: 10.1093/infdis/jiw436
160. Lafuse WP, Rajaram MVS, Wu Q, Moliva JI, Torrelles JB, Turner J, et al. Identification of an Increased Alveolar Macrophage Subpopulation in Old Mice That Displays Unique Inflammatory Characteristics and Is Permissive to Mycobacterium Tuberculosis Infection. J Immunol (2019) 203:2252–64. doi: 10.4049/jimmunol.1900495
161. Vance J, Santos A, Sadofsky L, Morice A, Cervantes J. Effect of High Glucose on Human Alveolar Macrophage Phenotype and Phagocytosis of Mycobacteria. Lung (2019) 197:89–94. doi: 10.1007/s00408-018-0181-z
162. Zeng Z, Xu L, Xie XY, Yan HL, Xie BJ, Xu WZ, et al. Pulmonary Pathology of Early-Phase Covid-19 Pneumonia in a Patient With a Benign Lung Lesion. Histopathology (2020) 77:823–31. doi: 10.1111/his.14138
163. Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY. Pulmonary Pathology of Early-Phase 2019 Novel Coronavirus (Covid-19) Pneumonia in Two Patients With Lung Cancer. J Thorac Oncol (2020) 15:700–4. doi: 10.1016/j.jtho.2020.02.010
164. Swiecki M, Colonna M. Type I Interferons: Diversity of Sources, Production Pathways and Effects on Immune Responses. Curr Opin Virol (2011) 1:463–75. doi: 10.1016/j.coviro.2011.10.026
165. Higgins DM, Sanchez-Campillo J, Rosas-Taraco AG, Higgins JR, Lee EJ, Orme IM, et al. Relative Levels of M-CSF and GM-CSF Influence the Specific Generation of Macrophage Populations During Infection With Mycobacterium Tuberculosis. J Immunol (2008) 180:4892–900. doi: 10.4049/jimmunol.180.7.4892
166. Wilson MR, Choudhury S, Goddard ME, O’Dea KP, Nicholson AG, Takata M. High Tidal Volume Upregulates Intrapulmonary Cytokines in an In Vivo Mouse Model of Ventilator-Induced Lung Injury. J Appl Physiol (2003) 95:1385–93. doi: 10.1152/japplphysiol.00213.2003
167. Beck-Schimmer B, Schwendener R, Pasch T, Reyes L, Booy C, Schimmer RC. Alveolar Macrophages Regulate Neutrophil Recruitment in Endotoxin-Induced Lung Injury. Respir Res (2005) 6:61. doi: 10.1186/1465-9921-6-61
168. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive Neutrophils and Neutrophil Extracellular Traps Contribute to Acute Lung Injury of Influenza Pneumonitis. Am J Pathol (2011) 179:199–210. doi: 10.1016/j.ajpath.2011.03.013
169. Vergadi E, Vaporidi K, Theodorakis EE, Doxaki C, Lagoudaki E, Ieronymaki E, et al. Akt2 Deficiency Protects From Acute Lung Injury Via Alternative Macrophage Activation and Mir-146a Induction in Mice. J Immunol (2014) 192:394–406. doi: 10.4049/jimmunol.1300959
170. Laskin DL, Malaviya R, Laskin JD. Role of Macrophages in Acute Lung Injury and Chronic Fibrosis Induced by Pulmonary Toxicants. Toxicol Sci (2019) 168:287–301. doi: 10.1093/toxsci/kfy309
171. Silva-Santos JEDA, Santos-Silva MC, De Queiroz Cunha F, Assreuy J. The Role of ATP-sensitive Potassium Channels in Neutrophil Migration and Plasma Exudation. J Pharmacol Exp Ther (2002) 300:946–51. doi: 10.1124/jpet.300.3.946
172. Pinheiro NM, Santana FPR, Almeida RR, Guerreiro M, Martins MA, Caperuto LC, et al. Acute Lung Injury is Reduced by the A7nAChR Agonist PNU-282987 Through Changes in the Macrophage Profile. FASEB J (2017) 31:320–32. doi: 10.1096/fj.201600431R
173. Bittencourt-Mernak MI, Pinheiro NM, Santana FPR, Guerreiro MP, Saraiva-Romanholo BM, Grecco SS, et al. Prophylactic and Therapeutic Treatment With the Flavonone Sakuranetin Ameliorates LPS-Induced Acute Lung Injury. Am J Physiol - Lung Cell Mol Physiol (2017) 312:L217–30. doi: 10.1152/ajplung.00444.2015
174. Wang Y, Xu Y, Zhang P, Ruan W, Zhang L, Yuan S, et al. Smiglaside A Ameliorates LPS-induced Acute Lung Injury by Modulating Macrophage Polarization Via Ampk-Pparγ Pathway. Biochem Pharmacol (2018) 156:385–95. doi: 10.1016/j.bcp.2018.09.002
175. Wang J, Li R, Peng Z, Zhou W, Hu B, Rao X, et al. Gts-21 Reduces Inflammation in Acute Lung Injury by Regulating M1 Polarization and Function of Alveolar Macrophages. Shock (2019) 51:389–400. doi: 10.1097/SHK.0000000000001144
176. Zhuo Y, Li D, Cui L, Li C, Zhang S, Zhang Q, et al. Treatment With 3,4-Dihydroxyphenylethyl Alcohol Glycoside Ameliorates Sepsis-Induced ALI in Mice by Reducing Inflammation and Regulating M1 Polarization. BioMed Pharmacother (2019) 116:109012. doi: 10.1016/j.biopha.2019.109012
177. Tu Gw, Shi Y, Zheng Yj, Ju Mj, He H, Ma Gg, et al. Glucocorticoid Attenuates Acute Lung Injury Through Induction of Type 2 Macrophage. J Transl Med (2017) 15:181. doi: 10.1186/s12967-017-1284-7
178. Dolinay T, Kaminski N, Felgendreher M, Kim HP, Reynolds P, Watkins SC, et al. Gene Expression Profiling of Target Genes in Ventilator-Induced Lung Injury. Physiol Genomics (2006) 26:68–75. doi: 10.1152/physiolgenomics.00110.2005
179. Xu Y, Meng C, Liu G, Yang D, Fu L, Zhang M, et al. Classically Activated Macrophages Protect Against Lipopolysaccharide-Induced Acute Lung Injury by Expressing Amphiregulin in Mice. Anesthesiology (2016) 124:1086–99. doi: 10.1097/ALN.0000000000001026
180. Josset L, Belser JA, Pantin-Jackwood MJ, Chang JH, Chang ST, Belisle SE, et al. Implication of Inflammatory Macrophages, Nuclear Receptors, and Interferon Regulatory Factors in Increased Virulence of Pandemic 2009 H1N1 Influenza A Virus After Host Adaptation. J Virol (2012) 86:7192–206. doi: 10.1128/jvi.00563-12
181. Huang S, Zhu B, Cheon IS, Goplen NP, Jiang L, Zhang R, et al. Ppar-γ in Macrophages Limits Pulmonary Inflammation and Promotes Host Recovery Following Respiratory Viral Infection. J Virol (2019) 93:e00030–19. doi: 10.1128/jvi.00030-19
182. Huang S, Jiang L, Cheon IS, Sun J. Targeting Peroxisome Proliferator-Activated Receptor-Gamma Decreases Host Mortality After Influenza Infection in Obese Mice. Viral Immunol (2019) 32:161–9. doi: 10.1089/vim.2019.0016
183. Lee YJ, Moon C, Lee SH, Park HJ, Seoh JY, Cho MS, et al. Apoptotic Cell Instillation After Bleomycin Attenuates Lung Injury Through Hepatocyte Growth Factor Induction. Eur Respir J (2012) 40:424–35. doi: 10.1183/09031936.00096711
184. Franke-Ullmann G, Pförtner C, Walter P, Steinmüller C, Lohmann-Matthes ML, Kobzik L. Characterization of Murine Lung Interstitial Macrophages in Comparison With Alveolar Macrophages In Vitro. J Immunol (1996) 157:3097–104.
185. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages That Have Ingested Apoptotic Cells in Vitro Inhibit Proinflammatory Cytokine Production Through Autocrine/Paracrine Mechanisms Involving Tgf-β, PGE2, and PAF. J Clin Invest (1998) 101:890–8. doi: 10.1172/JCI1112
186. Hussell T, Bell TJ. Alveolar Macrophages: Plasticity in a Tissue-Specific Context. Nat Rev Immunol (2014) 14:81–93. doi: 10.1038/nri3600
187. Martinez-Pomares L, Gordon S. Cd169 + Macrophages At the Crossroads of Antigen Presentation. Trends Immunol (2012) 33:66–70. doi: 10.1016/j.it.2011.11.001
188. Longworth KE. The Comparative Biology of Pulmonary Intravascular Macrophages. Front Biosci (1997) 2:d232–41. doi: 10.2741/A186
189. Lupher ML, Gallatin WM. Regulation of Fibrosis by the Immune System. Adv Immunol (2006) 89:245–88. doi: 10.1016/S0065-2776(05)89006-6
190. Strieter RM. What Differentiates Normal Lung Repair and Fibrosis? Inflammation, Resolution of Repair, and Fibrosis. Proc Am Thoracic Soc, (2008) 5(3):305–10. doi: 10.1513/pats.200710-160DR
191. Sinha P, Clements VK, Ostrand-Rosenberg S. Interleukin-13-Regulated M2 Macrophages in Combination With Myeloid Suppressor Cells Block Immune Surveillance Against Metastasis. Cancer Res (2005) 65:11743–51. doi: 10.1158/0008-5472.CAN-05-0045
192. Tsoutsou PG, Gourgoulianis KI, Petinaki E, Germenis A, Tsoutsou AG, Mpaka M, et al. Cytokine Levels in the Sera of Patients With Idiopathic Pulmonary Fibrosis. Respir Med (2006) 100:938–45. doi: 10.1016/j.rmed.2005.06.016
193. Mora AL, Torres-González E, Rojas M, Corredor C, Ritzenthaler J, Xu J, et al. Activation of Alveolar Macrophages Via the Alternative Pathway in Herpesvirus-Induced Lung Fibrosis. Am J Respir Cell Mol Biol (2006) 35:466–73. doi: 10.1165/rcmb.2006-0121OC
194. Duru N, Wolfson B, Zhou Q. Mechanisms of the Alternative Activation of Macrophages and non-Coding RNAs in the Development of Radiation-Induced Lung Fibrosis. World J Biol Chem (2016) 7:231. doi: 10.4331/wjbc.v7.i4.231
195. Dong H, Li J, Lv Y, Zhou Y, Wang G, Hu S, et al. Comparative Analysis of the Alveolar Macrophage Proteome in ALI/ARDS Patients Between the Exudative Phase and Recovery Phase. BMC Immunol (2013) 14:25. doi: 10.1186/1471-2172-14-25
196. Matthay MA, Ware LB, Zimmerman GA. The Acute Respiratory Distress Syndrome. J Clin Invest (2012) 122:2731–40. doi: 10.1172/JCI60331
197. Huang X, Xiu H, Zhang S, Zhang G. The Role of Macrophages in the Pathogenesis of Ali/Ards. Mediators Inflammation (2018) 2018:1264913. doi: 10.1155/2018/1264913
198. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage Polarization and Function With Emphasis on the Evolving Roles of Coordinated Regulation of Cellular Signaling Pathways. Cell Signal (2014) 26:192–7. doi: 10.1016/j.cellsig.2013.11.004
199. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage Plasticity, Polarization, and Function in Health and Disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
200. Weagel E, Smith C, Liu PG, Robison R, O’Neill K. Macrophage Polarization and Its Role in Cancer. J Clin Cell Immunol (2015) 06:4–11. doi: 10.4172/2155-9899.1000338
201. Yin M, Shen J, Yu S, Fei J, Zhu X, Zhao J, et al. Tumor-Associated Macrophages (Tams): A Critical Activator in Ovarian Cancer Metastasis. Onco Targets Ther (2019) 12:8687–99. doi: 10.2147/OTT.S216355
202. Brown BN, Ratner BD, Goodman SB, Amar S, Badylak SF. Macrophage Polarization: An Opportunity for Improved Outcomes in Biomaterials and Regenerative Medicine. Biomaterials (2012) 33:3792–802. doi: 10.1016/j.biomaterials.2012.02.034
203. Song C, Li H, Li Y, Dai M, Zhang L, Liu S, et al. Nets Promote Ali/Ards Inflammation by Regulating Alveolar Macrophage Polarization. Exp Cell Res (2019) 382:111486. doi: 10.1016/j.yexcr.2019.06.031
204. Liu M, Chen Y, Wang S, Zhou H, Feng D, Wei J, et al. α-Ketoglutarate Modulates Macrophage Polarization Through Regulation of Pparγ Transcription and Mtorc1/P70s6k Pathway to Ameliorate Ali/Ards. Shock (2020) 53:103–13. doi: 10.1097/SHK.0000000000001333
205. Kumar V. Macrophages: The Potent Immunoregulatory Innate Immune Cells. In: Bhat KH, editor. Macrophage Activation - Biology and Disease. London: IntechOpen. (2019) doi: 10.5772/intechopen.88013
206. D’Alessio FR, Craig JM, Singer BD, Files DC, Mock JR, Garibaldi BT, et al. Enhanced Resolution of Experimental Ards Through Il-4-Mediated Lung Macrophage Reprogramming. Am J Physiol - Lung Cell Mol Physiol (2016) 310:L733–46. doi: 10.1152/ajplung.00419.2015
207. Wang L, Zhang H, Sun L, Gao W, Xiong Y, Ma A, et al. Manipulation of Macrophage Polarization by Peptide-Coated Gold Nanoparticles and its Protective Effects on Acute Lung Injury. J Nanobiotechnology (2020) 18:38. doi: 10.1186/s12951-020-00593-7
208. Hou J, Shi J, Chen L, Lv Z, Chen X, Cao H, et al. M2 Macrophages Promote Myofibroblast Differentiation of LR-MSCs and are Associated With Pulmonary Fibrogenesis 11 Medical and Health Sciences 1102 Cardiorespiratory Medicine and Haematology. Cell Commun Signal (2018) 16:89. doi: 10.1186/s12964-018-0300-8
209. Nicol MQ, Dutia BM. The Role of Macrophages in Influenza A Virus Infection. Future Virol (2014) 9:847–62. doi: 10.2217/fvl.14.65
210. Taylor JP, Cash MN, Santostefano KE, Nakanishi M, Terada N, Wallet MA. Crispr/Cas9 Knockout of USP18 Enhances Type I Ifn Responsiveness and Restricts Hiv-1 Infection in Macrophages. J Leukoc Biol (2018) 103:1225–40. doi: 10.1002/JLB.3MIA0917-352R
211. Gerrick KY, Gerrick ER, Gupta A, Wheelan SJ, Yegnasubramanian S, Jaffee EM. Transcriptional Profiling Identifies Novel Regulators of Macrophage Polarization. PloS One (2018) 13:e0208602. doi: 10.1371/journal.pone.0208602
Keywords: M1/M2 macrophages, inflammation, ARDS, cells therapy, SARS-CoV-2
Citation: Kosyreva A, Dzhalilova D, Lokhonina A, Vishnyakova P and Fatkhudinov T (2021) The Role of Macrophages in the Pathogenesis of SARS-CoV-2-Associated Acute Respiratory Distress Syndrome. Front. Immunol. 12:682871. doi: 10.3389/fimmu.2021.682871
Received: 19 March 2021; Accepted: 22 April 2021;
Published: 10 May 2021.
Edited by:
Vanessa Pinho, Federal University of Minas Gerais, BrazilReviewed by:
Luciana Padua Tavares, Harvard Medical School, United StatesCopyright © 2021 Kosyreva, Dzhalilova, Lokhonina, Vishnyakova and Fatkhudinov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Kosyreva, a29zeXJldmEuYUBsaXN0LnJ1
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.